 Zhenhua Sun*†
Zhenhua Sun*† Chaohui Wang
Chaohui Wang- Department of Thyroid and Breast Surgery, Affiliated Hospital of Jiangsu University, Zhenjiang, China
The majority of patients with thyroid cancer can attain a favorable prognosis with a comprehensive treatment program based on surgical treatment. However, the current treatment options for advanced thyroid cancer are still limited. In recent years, chimeric antigen receptor-modified T-cell (CAR-T) therapy has received widespread attention in the field of oncology treatment. It has achieved remarkable results in the treatment of hematologic tumors. However, due to the constraints of multiple factors, the therapeutic efficacy of CAR-T therapy for solid tumors, including thyroid cancer, has not yet met expectations. This review outlines the fundamental structure and treatment strategies of CAR-T cells, provides an overview of the advancements in both preclinical investigations and clinical trials focusing on targets associated with CAR-T cell therapy in treating thyroid cancer, and discusses the challenges and solutions to CAR-T cell therapy for thyroid cancer. In conclusion, CAR-T cell therapy is a promising therapeutic approach for thyroid cancer, and we hope that our review will provide a timely and updated study of CAR-T cell therapy for thyroid cancer to advance the field.
1 Background
Thyroid cancer is the most common malignant tumor of the endocrine system, and its incidence has been increasing significantly year by year; it has become the malignant tumor with the highest incidence rate among women in certain regions (1). The main tissue types are papillary thyroid carcinoma(PTC), follicular thyroid carcinoma (FTC), anaplastic thyroid cancer (ATC), and medullary thyroid cancer (MTC) (2). Due to the expression of sodium iodide symporter (NIS) and thyroid stimulating hormone (TSH)-dependent growth pattern, radioactive iodine 131 (131 I) therapy and TSH suppression therapy are effective in most differentiated thyroid cancers. However, treatment options for recurrent/metastatic differentiated thyroid cancer, poorly differentiated/undifferentiated thyroid cancer, and medullary thyroid carcinoma are still insufficient (3).
CAR-T is one of the methods of adoptive cell transfer therapy (ACT); its main principle is to isolate T cells from patients, use genetic engineering technology to insert a chimeric antigen receptor (CAR) into the T cells that can recognize tumor cells and activate the T cells at the same time, and then infuse the expanded CAR-T cells back into the patients and attack the target cells expressing the relevant antigens without relying on the major histocompatibility complex (MHC) (4). As an active medication, CAR-T cell immunotherapy has significantly advanced the treatment of cancer, especially hematologic malignancies. In August 2017, the U.S. Food and Drug Administration (FDA) approved Novartis’ Kymriah, the world’s first CAR-T cell therapy product for the treatment of hematologic malignancies, for the treatment of refractory and relapsed B-cell acute lymphoblastic leukemia (B-ALL) patients (5). The successful application in hematologic malignancies has driven extensive research on CAR-T cell therapy in refractory and relapsed solid malignancies. Several basic studies and clinical trials have demonstrated that CAR-T cell therapy has significantly progressed in thyroid cancer (6). This comprehensive review will present the most recent advancements in CAR-T therapy for thyroid cancer, covering topics such as the biological foundation of CAR-T, ongoing clinical studies, obstacles encountered, and potential solutions to these obstacles.
2 Principles of CAR-T therapy and basic structure of CAR-T
T cells depend on the attachment of T cell receptor (TCR) on their surface to antigens presented by MHC molecules on the cell membrane for identifying various cells (7). However, tumor cells evade T cell recognition and killing by reducing or losing MHC expression (8). Conventional ACT approaches, such as tumor-infiltrating lymphocyte (TIL) therapy and T-cell receptor-engineered T-cell (TCR-T) therapy, which recognize only MHC-delivered antigens, may be limited by down-regulation or mutation of MHC molecules of the tumor cells, thus evading immune surveillance, which has certain limitations in the clinic (9, 10). To overcome the limitations of MHC, a promising approach is to modify T cells using CAR to acquire specificity for specific antigenic epitopes, thus enhancing the antigen recognition and activation function of T cells (11). Compared with natural T-cell surface receptors, CAR confers HLA-independent recognition of tumor antigens on T cells, which traditional cellular overlay therapies have unmatched advantages. The U.S. FDA has approved six CAR-T cell therapy medications to treat hematologic cancers (Table 1) (18).
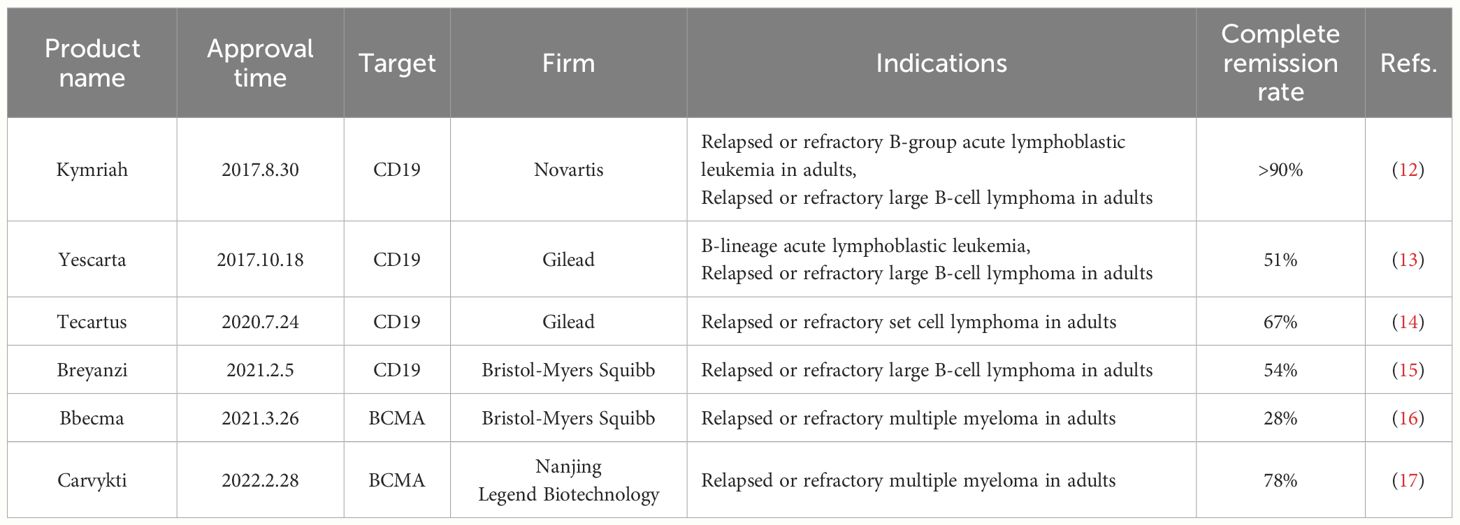
Table 1 FDA approves CAR-T therapeutics.
The typical structure of CAR consists of four parts (Figure 1A), including the extracellular antigen recognition structural domain, the spacer region, the transmembrane structural domain, and the intracellular signaling structural region (19). The extracellular antigen recognition structural domain is the single chain variable fragment (ScFv), which is the key part of CAR-T to recognize tumor antigen targets and can recognize tumor-associated antigen (TAA) or tumor-specific antigen (TSA). The spacer region, also known as the hinge region, together with ScFv, forms the extracellular structural domain. The transmembrane domain is considered the most important structural feature and generally consists of dimeric membrane proteins that anchor the CAR to the T cell and connect to the intracellular signaling domain (20, 21).
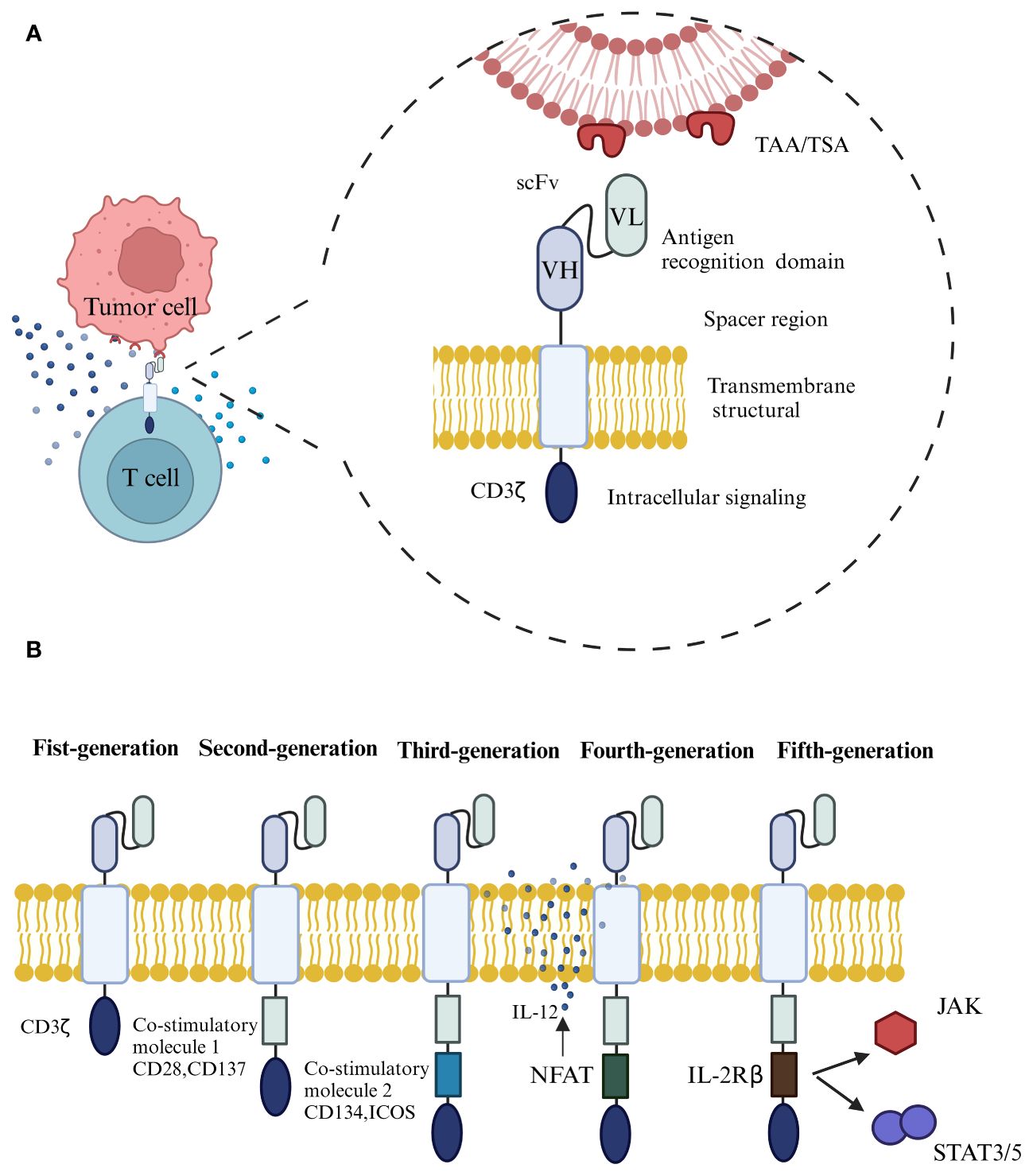
Figure 1 (A) The typical structure of CAR consists of four parts, including the extracellular antigen recognition structural domain, the spacer region, the transmembrane structural domain, and the intracellular signaling structural region. (B) The development of CAR through the generations, from the first to the fifth generation.
Depending on the design of intracellular signaling structural domains for different purposes, CAR has undergone an evolution from the first to the fifth generation (Figure 1B) (22). The extracellular structural domain scFv of first-generation CARs is connected to the intracellular signaling motif CD3ζ through a transmembrane structural domain, which cannot transduce proliferative signals and induce cytokine production due to the absence of co-stimulatory molecule expression, rendering the T-cells unable to increase continuously in vivo, The therapeutic effect of tumor-killing is not apparent (23). The second-generation CARs add a co-stimulatory structural domain to its predecessor by adding a co-stimulatory molecule such as CD28, CD137, or an inducible co-stimulatory molecule (ICOS), which allows the T cells to proliferate and release cytokines even in the absence of exogenous co-stimulatory molecules, which can increase the immune memory effect and killing activity (24). Third-generation CARs contain two co-stimulatory structural domains, CD28, CD137, CD134, and ICOS, designed to further enhance signaling capabilities and anti-tumor responses (25, 26). However, it has been shown that the third-generation CART cell-killing activity did not gain significant enhancement, probably because the activation signal generated by a single co-stimulatory molecule in T lymphocytes has reached the threshold, and simply adding co-stimulatory structural domains in quantity will not further enhance the activation effect of CAR on T cells (27). The fourth generation of CAR, also called T cells redirected for universal cytokine-mediated killing (TRUCKs) (28), introduces the activated intra-nuclear factor of T cells (NFAT) based on the previous generation, which can be used to promote the further activation of CAR-T cells in the tumor microenvironment through the production of a series of cytokines, such as interleukin-12 (IL-12), or to add suicide genes or chemokine receptor structures on the structure of CAR-T cells to avoid off-target effects and increase the infiltration of T cells in tumor tissues (29), thus achieving enhanced killing effects on solid tumors. The addition of suicide genes or chemokine receptors to the CAR-T structure can help to avoid off-target effects and increase the infiltration of T-cells in tumor tissues, thus enhancing the killing effect of solid tumors. Based on the second-generation CAR structure, the fifth-generation CAR incorporates co-stimulatory domains that trigger additional signaling pathways, such as the IL-2 receptor β-chain fragment (IL-2Rβ). When the CAR-T cells target tumor antigens, activating the receptor specific to the antigen can activate the downstream JAK-STAT signaling pathway, increasing T-cell proliferation, survival, and anti-tumor activity. While a thorough assessment of the safety and effectiveness of fifth-generation CARs is necessary, their promising potential for advancement has been demonstrated (4, 30).
3 CAR-T treatment process
CAR-T cell therapy undergoes the following processes (Figure 2A): First, blood is extracted from the patient and non-specific T cells are isolated, the isolated T cells are enriched and activated, and then CAR gene transfer is performed using a viral or non-viral vector system that inserts CARs on the surface of the T cells that recognize relevant tumor antigens (31). The reconstructed T cells are cultured and expanded in vitro, and finally, the screened and quality-controlled CAR-T cells are infused back into the patient’s body to fight against tumor cells (32). Even though this type of autologous CAR-T cells can successfully evade rejection, the procedure not only requires an extensive preparation period but also comes with a high price tag.”Off-the-shelf” CAR-T cells made from allogeneic T cells are a new alternative (Figure 2B) (33, 34). The process begins with the isolation of T lymphocytes from a healthy donor, followed by the transfer and expansion of the CAR gene into the T cells via a viral vector, and finally, quality control, packaging, and frozen storage. Theoretically, allogeneic CAR-T cells are similar to “medicines” and can be used by patients at any time. However, it also faces challenges such as immune rejection and low durability (35).
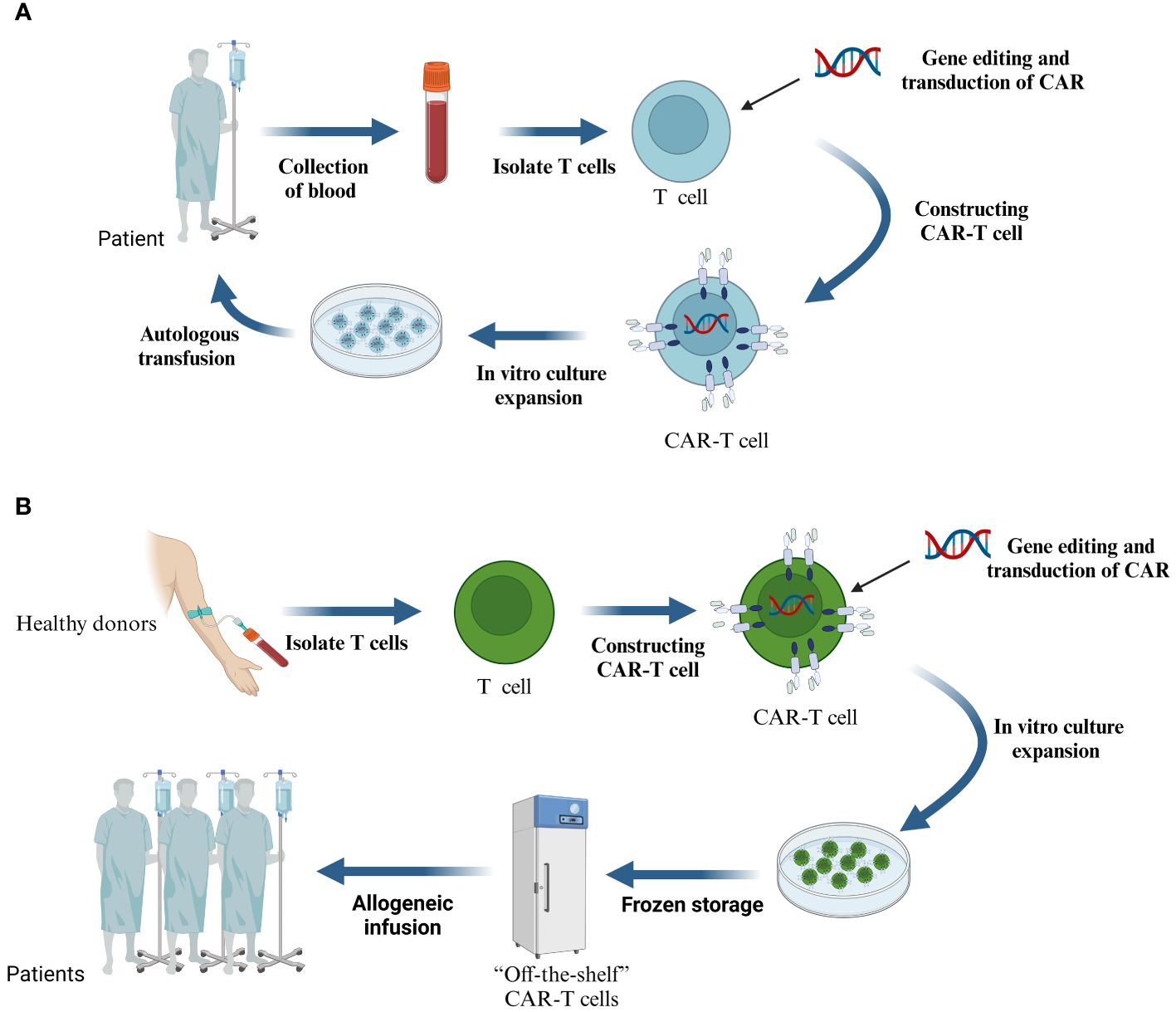
Figure 2 (A) The process of treating malignant tumors with CAR-T cells prepared from autologous T cells. (B) CAR-T cells are prepared from T cells of allogeneic origin.
4 Targeting of CAR-T cells for the treatment of thyroid cancer
Tumor antigens can be classified into two groups based on their specificity: TSAs and TAAs (36). Since solid tumors often lack TSA, we have to choose TAA as the target in CAR-T cell design, and solid tumor TAA is expressed on the surface of both tumor cells and normal tissue cells in different degrees. Although this CAR-T target design mode has a certain tumor-killing effect, it is also prone to an “off-target effect”, resulting in serious and long-lasting damage to normal tissues and organs. This “off-target effect” is the main difficulty limiting CAR-T therapy’s clinical application for solid tumors (37). In past research on CAR-T therapy for solid tumors, we tried to avoid tissue-specific antigens in the target screening to avoid the damage of CAR-T cells to normal tissues. Currently, the main clinical treatment strategy for thyroid cancer is to surgically remove the tumor and normal thyroid tissues, followed by oral thyroid hormone replacement therapy (38). Based on this treatment strategy, in selecting CAR-T targets for thyroid cancer, tissue-specific targets expressed on the surfaces of both normal and cancerous thyroid cells can be selected, such as thyroid stimulating hormone receptor (TSHR) and glial-derived neurotrophic factor receptor α4 (GDNFRα4, GFRα4).In addition, non-tissue-specific targets such as carcinoembryonic antigen (CEA), intercellular cell adhesion molecule-1 (ICAM-1), and B7-H3 (CD26), a member of the B7 family of immune checkpoint proteins, are also the main research directions of the current CAR-T cell therapy for thyroid cancer. In the following sections, we will discuss in detail the pre-clinical studies and selected clinical trials of these targets (Table 2).
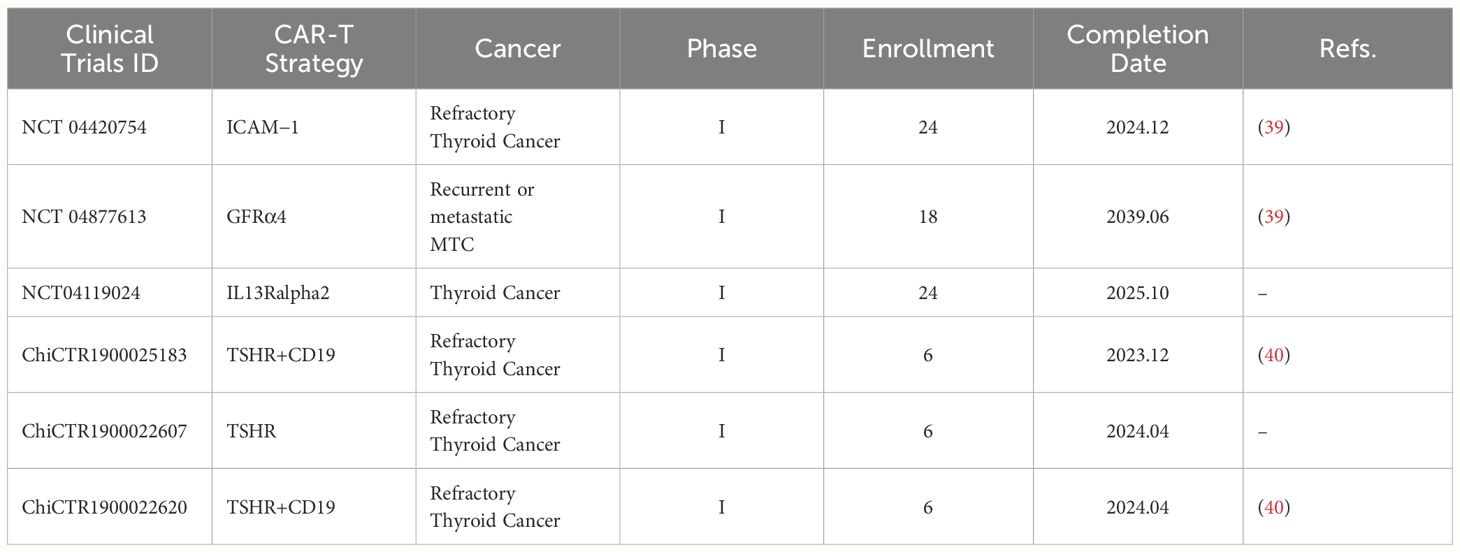
Table 2 Ongoing clinical trials of CAR-T therapy for different targets in thyroid cancer.
4.1 TSHR
TSHR is expressed on the basolateral membrane of thyroid follicular cells and is a member of the transmembrane protein superfamily, G-protein-coupled receptors (41). The development of thyroid cells and their differentiation, as well as the production and release of thyroid hormones, are controlled by TSHR through its interaction with thyrotropin. Research conducted in the past has demonstrated the presence of TSHR in both healthy thyroid tissue and thyroid tumors (42).
Li et al. reported for the first time that TSHR showed good efficacy and safety as a target for CAR-T cell therapy in DTC (40). The researchers first validated the expression of TSHR in tissues and found that TSHR was highly expressed in DTC and low in ATC. Meanwhile, TSHR expression remained high in cervical lymph nodes and refractory thyroid cancer tissues, while it was largely absent in other normal tissues. Given that long-term culture may lead to the transformation of existing differentiated thyroid cancer cell lines into TSHR-deficient types, the researchers constructed TSHR-highly expressing cell lines for in vitro experiments. By using pre-existing TSHR antibodies, the researchers screened for scFv capable of targeting TSHR and obtained TSHR CAR-T cells by lentiviral transfection of T cells. After the successful construction, no significant change in the CD4+/CD8+ cell ratio was observed, indicating that the CAR structure was not significantly toxic to the autoimmune system, and the excellent killing effect of CAR-T cells on target cells was evaluated. The research team further established a thyroid cancer model in NSG mice to validate the anti-tumor effect of TSHR CAR-T cells. The outcomes demonstrated that in mice receiving TSHR-CAR-T cell therapy, the tumor volume was considerably decreased. After treatment, the TSHR CAR-T cells increased in vivo on days 20 and 34. By monitoring the behavior of the mice and examining their major organs, no significant toxic reactions were observed. Based on this study, a phase I clinical trial entitled “Clinical Study of the Safety and Efficacy of dPD-1 (TSHR+CD19) CART Cells in the Treatment of Relapsed Refractory Thyroid Cancer” has been conducted, which has been completed, and the results have not yet been reported.
2022 Ding et al. reported a clinical case of TSHR+CD19 dual-targeted CAR-T cells to treat one case of recurrent refractory thyroid cancer (43). A female patient with poorly differentiated thyroid cancer received a TSHR+CD19 CAR-T cell infusion. Imaging on day 30 after infusion suggested no tumor progression and partial remission occurred on day 90. Dynamic monitoring of the patient’s in vivo CAR-T cell proliferation revealed that the number of CAR-T cells continued to increase after infusion, reaching a peak on day six and remaining detectable for three months. The patient died of a lung infection on day 141 post-infusion, but the efficacy of thyroid cancer treatment was still evaluated as a partial remission (PR) before the patient’s death. Overall, TSHR+CD19CAR-T cells expanded well in vivo in humans and have promising efficacy with aggressive control of adverse effects.
4.2 ICAM−1
ICAM-1, a cell surface glycoprotein and adhesion receptor, belongs to the immunoglobulin superfamily and is found on various cells. It plays a crucial role in controlling the migration of white blood cells from the bloodstream to inflammatory areas. It involves essential physiological processes such as cell signaling and activation, immune response, and inflammatory response. Some studies have shown that ICAM-1 expression is increased in a variety of tumors, including thyroid, pancreatic, and breast cancers, and is involved in biological processes such as tumor invasion and metastasis (44, 45). Previous studies have shown that CAR-T cells constructed against overexpressed ICAM-1 have excellent anti-tumor efficacy and safety and thus are expected to be a target for tumor CAR-T therapy (46–48).
In 2017, Min et al. reported the first study of CAR-T therapy for thyroid cancer utilizing ICAM-1 as a target (49). In vitro experiments, ICAM-1 CAR-T cells demonstrated efficient killing effects on ICAM-1-expressing papillary thyroid cancer and undifferentiated cancer cell lines. As verified by the metastatic tumor animal model constructed in NSG mice, ICAM-1, CAR-T significantly inhibited the growth of tumor cells and exhibited durable anti-tumor activity, prolonging the survival of mice. In addition, the killing effect of autologous ICAM-1 CAR-T on thyroid malignant tumor cells in vivo and in vitro was verified. Studies in cytotoxicity revealed that ICAM-1 CAR-T had no significant killing effect on non-tumor cells and that endogenously expressed ICAM-1 had no significant adverse effect on the autoimmune system. After infusing ICAM-1 CAR-T cells into immunodeficient mice, no significant damage was observed to essential tissues such as lungs and livers. In summary, CAR-T cell therapy for refractory and recurrent thyroid cancer has significant potential with ICAM-1 as a target.
In follow-up studies, Min and his team optimized the production process of ICAM-1 CAR-T cells, improved the cell structure, and conducted an in-depth evaluation of their efficacy and safety. This ultimately provided sufficient preparation for developing phase I clinical trials (50). In 2020, AffyImmune Therapeutics, Inc. of the U.S. is conducting a Phase I clinical trial of ICAM-1 CAR-T cells for the treatment of patients with relapsed/refractory poorly-differentiated thyroid cancer and patients with BRAF wild-type undifferentiated thyroid to evaluate the safety and tolerability of its product AIC100, the trial is expected to last until June 2024. Reports from this preclinical trial indicated that AIC100 amplified well in ATC and PTC patients, demonstrated an excellent safety profile, and effectively killed tumors (51, 52).
4.3 GFRα4
GFRα4 is a member of the glial-derived neurotrophic factor (GDNF) family of receptors alpha expressed in normal and malignant medullary thyroid cells in humans. When GFRα4 binds specifically to the GDNF family ligand persephin (PSPN), it induces phosphorylation of RET proteins, activating downstream signaling pathways and exerting its physiological effects (53). Given the restricted expression profile of GFRα4 in MTC (54), it may be a potential specific and relevant antigenic target for CAR-T treatment of MTC.
A study by Bhoj et al. reported that GFRα4 expression in humans is restricted to parafollicular thyroid cells and was detected in the tumors of all MTC patients tested (55). Due to the lack of specific monoclonal antibodies, the investigators performed a structural screen of scFv against GFRα4 isoforms and successfully obtained GFRα4 CAR-T cells by lentiviral transfection of T cells. In vitro experiments verified the specificity of the CAR structure binding to GFRα4 and observed that cells expressing the CAR structure could be activated by MTC cells. Further studies showed that the GFRα4 CAR-T had a killing effect on MTC cell lines and leukemia cell lines with high expression of GFRα4 and secreted IL-2 and interferon γ (IFN-γ). In in vivo experiments, the researchers found that GFRα4 CAR-T cells could effectively inhibit tumor growth in the constructed MTC cell line NSG mouse xenograft animal model. However, GFRα4 CAR-T cells expanded to a greater extent, possibly related to off-target effects. In terms of in vivo toxicity, GFRα4 CAR-T cells exhibited toxicity against the skin of NSG mice, but no significant toxicity manifestation was observed in other tissues and organs. Further studies showed that, unlike mouse epithelial cells, human epithelial cells, as well as other cells except for MTC, could not activate GFRα4 CAR-T cells. The skin toxicity may result from the off-target response of CAR to unknown antigens in mice but not human keratinocytes. Subsequent studies humanized scFv and validated its specific binding to GFRα4 as well as its efficacy in vitro and in vivo and skin toxicity (56). In 2021, the University of Pennsylvania initiated a Phase I clinical trial of single-stranded scFv CAR-T cells targeting GFRα4 for the treatment of recurrent or metastatic MTC to evaluate the safety and feasibility of the product and to find the maximum safe dose, with a trial cut-off date expected to be June 2039 (39).
4.4 CEA
CEA is a tumor-associated antigen, initially thought to be a proteoglycan complex present in colon cancer and normal embryonic intestines, and later shown to be widely present in digestive tumors of endodermal origin. Its expression is positively correlated with the malignancy of the cancer. It is also found in trace amounts in the blood of ordinary people, so CAR-T therapy targeting CEA needs to consider its possible adverse effects (57). CAR-T cell therapy targeting CEA has demonstrated significant efficacy in early studies and clinical trials against metastatic liver cancer, colorectal cancer, and other tumors (58). Erickson et al. (59)reported the design and synthesis of a CAR-T cell targeting CEA for targeted therapy against MTC. The results of in vitro experiments clarified the binding ability of CEA CAR-T cells to their target. They confirmed that these cells had a significant killing effect on CEA-expressing positive MTC cells but not CEA-negative ones. In addition, high levels of IFN-γ, tumor necrosis factor (TNF), and IL-2 expression also demonstrated the practical killing effect of CEA CAR-T cells on target cells. The killing ability of CEA CAR-T cells against CEA-expressing positive MTC cells was further validated by an animal model of MTC established in NSG mice. However, its safety still needs to be further evaluated. No relevant clinical trials for CEA CAR-T cell therapy against MTC have been registered.
4.5 B7-H3
B7-H3, an immunological checkpoint protein belonging to the B7 family, is expressed significantly in a range of primary malignancies and exhibits restricted expression in normal tissues (60). As a co-stimulatory molecule, B7-H3 provides a second signal in T-cell proliferation and activation. However, its effects on T-cells appear to have multiple effects. Recent research indicates that B7-H3 plays a key role in immune evasion, tumor spread, angiogenesis, and resistance to treatment in cancers. This suggests that targeting B7-H3 could be a promising strategy for tumor immunotherapy, particularly in MTC (61). The utilization of the pan-cancer antigen B7-H3 as a target for CAR-T cells has demonstrated effective preclinical activity in the treatment of a variety of malignant tumors. Duan et al. designed a CAR-T cell targeting B7-H3 (62), which tandemly linked fragment of antigen binding (Fab) and natural TCR intracellular signaling structural domains to form a novel Fab CAR, which recognizes MHC-independent tumor antigens and mimics the natural activation process of endogenous TCR. This design effectively addresses the problem of premature T-cell depletion. The study findings indicated that Fab CAR-T cells could efficiently target cancerous cells of the human follicular thyroid and induce cytotoxic effects. Unfortunately, the experiment was not evaluated for safety in animal studies. Further animal experiments are needed to evaluate the safety of Fab CAR-T cells, which will help to comprehensively evaluate their efficacy and potential application prospects.
4.6 Other potential targets
In addition to the targets mentioned above, no additional studies on CAR-T cell therapy for thyroid cancer have been reported. Based on the principles of CAR construction (63), we speculate that pan-tumor markers such as NIS, thyroglobulin(Tg), thyroid peroxidase(TPO), proto-oncogene tyrosine-protein kinase receptors(RET), calcitonin(CT), protein kinase B(PKB), telomerase reverse transcriptase (TERT), and the kinetochore element Ndc80 could be potential targets for thyroid cancer immunotherapy (59, 64). However, further preliminary studies and clinical trials need to validate these speculations.
5 Challenges and strategies for CAR-T cell therapy for thyroid cancer
Despite demonstrating potential in both preclinical research and clinical trials as a treatment for thyroid cancer, the application of CAR-T cell therapy continues to encounter numerous obstacles. These challenges mainly include the lack of specific antigens and tumor heterogeneity, deficient infiltration of CAR-T cells within tumors, immunosuppression of the tumor microenvironment (TME), and toxic side effects of CAR-T cells (65, 66). To address these issues, researchers have proposed strategies to promote further CAR-T cell therapy’s use in treating solid tumors.
5.1 Specific antigen deficiency and tumor heterogeneity
A key factor in immune cell therapy’s remarkable success in treating hematologic tumors is its ability to target well-defined, uniformly expressed, and tumor cell-specific antigens. In contrast, in solid tumor therapy, fewer TSAs are present, mostly TAA, and these antigens are not only expressed in tumor cells but can also be found in normal tissues, thus increasing the risk of off-targeting (36). In addition, the heterogeneity of expression intensity and distribution exhibited by solid tumor antigens in the immune microenvironment makes it challenging to ensure efficacy even when ideal targets are identified (67, 68). In addition to searching for and developing new targets with higher specificity, the lack of specific antigens can be addressed by modifying CAR-T cells to improve their ability to recognize tumor antigens. Wendell A Lim’s team implanted the synNotch system in CAR-T cells. Under the regulation of synNotch, CARs recognizing the relevant TAA will only be expressed on T cells migrating into tumors and will not attack cells in normal tissues, improving the specificity of CAR-T recognition of antigens (66). In addition, various traditional epigenetic modulators have been shown to modulate antigen density, thereby increasing the sensitivity of CAR-T to antigen recognition. For example, Decitabine, a DNA methylation transferase inhibitor, can enhance the specific recognition and killing effect of MUC1 CAR-T by upregulating the expression of MUC1 antigen on pancreatic cancer cells (69). Various strategies for modifying CARs have been developed to target antigenic heterogeneity in solid tumors, one of which is the simultaneous targeting of multiple tumor antigens to provide a higher level of antigen recognition ability for infused immune cells, such as dual-targeted tandem CARs recognizing EpCAM and ICAM-1 (70), and triple-targeted CARs targeting three antigens, namely, HER2, IL-13Rα2, and EphA2, at the same time (71). Secondly, EGFRvIII -CAR-T cells that secrete bispecific T-cell engager (BiTE) targeting EGFR can circumvent toxicity and improve anti-tumor efficacy against heterogeneous glioblastoma (72). These approaches have shown better efficacy in preclinical models.
5.2 Inefficient transit of CAR-T cells in thyroid cancer therapy
Infiltration of immune cells into the tumor site is a prerequisite for their effector function. The highly abnormal vascular and stromal structures in the microenvironment of solid tumors are thought to be the main factors hindering the infiltration of CAR-T cells. Tumor vasculature usually exhibits an irregular shape with varying degrees of collapse, whereas the tumor stroma is much denser. These two constitute a physical barrier resulting in brutal penetration of CAR-T cells (73). In addition, some solid tumors inhibit the secretion of specific chemokines. The interaction of chemokines with their receptors promotes the migration of T cells to the tumor microenvironment (74). Concurrently, CAR-T cells also lack pertinent surface receptors that correspond to the chemokines released by solid tumors, resulting in inadequate CAR-T cell infiltration at tumor sites. Recent studies targeting chemokines have opened up new possibilities for CAR-T cell infiltration of solid tumors. Overexpression of chemokines by pretreatment of tumors with chemotherapeutic agents or modification of CAR-T cells can increase endogenous immune cell infiltration at the tumor site (75). Similarly, modification of the chemokine receptors CXCR1/CXCR2/CXCR5 to the surface of CAR-T cells can increase cellular transport within the tumor and anti-tumor efficacy (76–78). Modification of CAR-T cells by strategies targeting the tumor stroma, such as using ECM-degrading agents such as acetyl heparinase or fibroblast activating proteins (79, 80), enhances their ability to penetrate physical barriers and thus increases aggregation in solid tumors. In addition, multidisciplinary cross-fertilization opens up new ideas to break the dilemma of insufficient immune cell infiltration in the tumor microenvironment, such as intratumoral injection (81), implantable scaffolds (82), and special biomaterials delivery (83), etc., which can enable the infused immune cells to be delivered to the tumor site more efficiently and effectively and target the tumor as well as reduce the off-target effect.
5.3 Immunosuppression of the tumor microenvironment
TME is a microenvironment that promotes the growth of cancer cells and consists of tumor cells, inflammatory cells, fibroblasts, mesenchymal tissues, and various cytokines. CAR-T cells can reach their destination directly after entering the blood circulation for hematologic tumors (74). However, for solid tumors, the tumor microenvironment not only has a physical barrier to inhibit T-cell infiltration but also contains a variety of immunosuppressive cells and immune checkpoints (84). This environment predisposes CAR-T cells to poor migration and persistence, impaired cellular function, and cellular exhaustion, thus failing to kill tumor cells effectively (85). Low-dose chemotherapy, a primary pretreatment strategy in present-day clinical CAR-T therapy, can potentially alter the tumor’s immune microenvironment. This modification can heighten CAR-T effectiveness through the removal of immunosuppressive cells (86, 87). In addition, studies have shown that CAR-T efficacy can be improved by modifying CARs to release immunostimulatory cytokines (88) or by interfering with immunosuppressive cytokines and inhibitory signaling pathways to counteract TME-induced immunosuppression (89). Immune checkpoints present an alternative solution for assisting CAR-T cells in overcoming the immunosuppressive conditions within solid tumors (90). Combining programmed death 1(PD-1) blockers with CAR-T cells that target ICAM1 enhances the efficacy of eliminating ICAM1-expressing thyroid tumor cells compared to using only CAR-T therapy (91). In addition to combining immune checkpoint inhibitors, it is also possible to construct CAR with anti-programmed cell death-Ligand 1 (PD-L1) scFv sequences (92), which are CAR-T cells that kill tumor cells while also blocking the PD-1/PD-L1 signaling pathway to release the immunosuppressive state.
5.4 CAR-T cell therapy-related toxicity
While CAR-T cell therapy has demonstrated revolutionary potential in cancer treatment, its elevated toxicity rates have hindered its widespread adoption as a primary clinical intervention, with the main types of toxicity being targeted tumor toxicity, non-tumor-targeted toxicity, off-target toxicity, and neurotoxicity (93). In addition, the FDA’s latest announcement states that all approved CAR-T cell therapy products targeting BCMA or CD19 carry a malignant risk of secondary T-lymphoblastic neoplasms (94). However, this risk has not been reported in solid tumors. Several approaches have been developed in recent years to address the issue of toxicity, including modification of CAR-T cell structure, modification of CAR-transduced T cells, implementation of a CAR “off switch,” or introduction of a suicide gene. By modifying the modular structure of CAR-T cells, it is possible to reduce their affinity for antigens, thus avoiding targeting normal tissues, and at the same time regulating the secretion of cytokines and reducing the immunogenicity of CAR, to achieve the purpose of controlling the toxicity of CAR-T cell therapy (95–97). Mitigation of CAR-T cell toxicity can also be achieved by implementing an “off switch” or introducing a suicide gene that selectively reduces genetically modified cells upon the occurrence of an adverse event by treatment with a secondary inducer, e.g., by inducing caspase 9 (iCasp9) and CD20 as a safety switch to remove CAR-T cells by rituximab treatment. Shikinumab treatment removes CAR-T cells, thereby reducing their toxicity (98). Another promising approach involves using the tyrosine kinase inhibitor Dasatinib to reversibly activate T cells by inhibiting proximal TCR signaling kinases (99). T-cell activation is ensured while reducing toxicity.
6 Conclusion and outlook
Currently, immunotherapy is not yet used as a conventional treatment for thyroid cancer. Still, for those patients who are in advanced stages of the disease and for whom other treatments have not been effective, novel therapeutic regimens, represented by CAR-T, may offer a new avenue for them and clinical practitioners. The purpose of this article is to review the development of CAR-T cell therapy for thyroid cancer from basic research to clinical trials. In preclinical studies, CAR-T cells constructed with antigens such as TSHR, ICAM-1, GFRα4, B7-H3, and CEA demonstrated anti-tumor effects. Still, only three antigens have entered clinical trials, and some relevant results have not yet been published. Further, fundamental studies and clinical trials are required to refine the CAR architecture for improved T cell activation, recognition specificity, anti-tumor activity, and safety control to achieve a reliable cure for solid tumors such as thyroid cancer. Finding optimal signaling and co-stimulation regions to enhance CAR-T therapy efficacy is crucial. In addition, there is a need to overcome the immunosuppressive tumor microenvironment with additional modifications to CAR-modified T cells. Establish standard clinical protocols, including patient pretreatment, cytokine support, and other potential combination therapies (100, 101). Through these efforts, we expect to make reliable advancements in CAR-T cell therapy in thyroid cancer and provide better treatment options for patients.
Author contributions
ZS: Writing – original draft, Writing – review & editing. CW: Writing – original draft, Writing – review & editing. YZ: Writing – original draft. QL: Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by a grant from the Medical Research Project of Jiangsu Provincial Health Commission (Grant No. Z2019029).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. (2023) 73:17–48. doi: 10.3322/caac.21763
2. Baloch ZW, Asa SL, Barletta JA, Ghossein RA, Juhlin CC, Jung CK, et al. Overview of the 2022 WHO classification of thyroid neoplasms. Endocr Pathol. (2022) 33:27–63. doi: 10.1007/s12022-022-09707-3
3. Filetti S, Durante C, Hartl DM, Leboulleux S, Locati LD, Newbold K, et al. ESMO Clinical Practice Guideline update on the use of systemic therapy in advanced thyroid cancer. Ann Oncol. (2022) 33:674–84. doi: 10.1016/j.annonc.2022.04.009
4. Labanieh L, Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature. (2023) 614:635–48. doi: 10.1038/s41586-023-05707-3
5. Mullard A. FDA approves first CAR T therapy. Nat Rev Drug Discovery. (2017) 16:669. doi: 10.1038/nrd.2017.196
6. French JD, Haugen BR. Thyroid cancer: CAR T cell therapy - potential in advanced thyroid cancer? Nat Rev Endocrinol. (2018) 14:10–1. doi: 10.1038/nrendo.2017.160
7. Rossjohn J, Gras S, Miles JJ, Turner SJ, Godfrey DI, McCluskey J. T cell antigen receptor recognition of antigen-presenting molecules. Annu Rev Immunol. (2015) 33:169–200. doi: 10.1146/annurev-immunol-032414-112334
8. Ramos CA, Heslop HE, Brenner MK. CAR-T cell therapy for lymphoma. Annu Rev Med. (2016) 67:165–83. doi: 10.1146/annurev-med-051914-021702
9. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. (2012) 12:269–81. doi: 10.1038/nri3191
10. Li X, Zhu Y, Yi J, Deng Y, Lei B, Ren H. Adoptive cell immunotherapy for breast cancer: harnessing the power of immune cells. J Leukoc Biol. (2024) 115:866–81. doi: 10.1093/jleuko/qiad144
11. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. (2018) 379:64–73. doi: 10.1056/NEJMra1706169
12. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
13. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
14. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. (2020) 382:1331–42. doi: 10.1056/NEJMoa1914347
15. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. (2020) 396:839–52. doi: 10.1016/s0140-6736(20)31366-0
16. Munshi NC, Anderson LD Jr., Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. (2021) 384:705–16. doi: 10.1056/NEJMoa2024850
17. Mi JQ, Zhao W, Jing H, Fu W, Hu J, Chen L, et al. Open-label study of ciltacabtagene autoleucel, an anti-B-cell maturation antigen chimeric antigen receptor-T-cell therapy, in chinese patients with relapsed/refractory multiple myeloma (CARTIFAN-1). J Clin Oncol. (2023) 41:1275–84. doi: 10.1200/jco.22.00690
18. Cappell KM, Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. (2023) 20:359–71. doi: 10.1038/s41571-023-00754-1
19. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discovery. (2013) 3:388–98. doi: 10.1158/2159-8290.Cd-12-0548
20. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. (2018) 359:1361–5. doi: 10.1126/science.aar6711
21. Larkin HD. CAR T-cell therapy shows early promise for gastrointestinal cancer. Jama. (2022) 327:2182. doi: 10.1001/jama.2022.9511
22. Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. (2021) 21:145–61. doi: 10.1038/s41568-020-00323-z
23. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci United States America. (1989) 86:10024–8. doi: 10.1073/pnas.86.24.10024
24. Milone MC, Bhoj VG. The pharmacology of T cell therapies. Mol Ther Methods Clin Dev. (2018) 8:210–21. doi: 10.1016/j.omtm.2018.01.010
25. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. (2010) 18:413–20. doi: 10.1038/mt.2009.210
26. Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol. (2004) 172:104–13. doi: 10.4049/jimmunol.172.1.104
27. Ramello MC, Benzaïd I, Kuenzi BM, Lienlaf-Moreno M, Kandell WM, Santiago DN, et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci Signal. (2019) 12. doi: 10.1126/scisignal.aap9777
28. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
29. Tang L, Pan S, Wei X, Xu X, Wei Q. Arming CAR-T cells with cytokines and more: Innovations in the fourth-generation CAR-T development. Mol Ther. (2023) 31:3146–62. doi: 10.1016/j.ymthe.2023.09.021
30. Daei Sorkhabi A, Mohamed Khosroshahi L, Sarkesh A, Mardi A, Aghebati-Maleki A, Aghebati-Maleki L, et al. The current landscape of CAR T-cell therapy for solid tumors: Mechanisms, research progress, challenges, and counterstrategies. Front Immunol. (2023) 14:1113882. doi: 10.3389/fimmu.2023.1113882
31. Li C, Mei H, Hu Y. Applications and explorations of CRISPR/Cas9 in CAR T-cell therapy. Brief Funct Genomics. (2020) 19:175–82. doi: 10.1093/bfgp/elz042
32. Feins S, Kong W, Williams EF, Milone MC, Fraietta JA. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am J Hematol. (2019) 94:S3–s9. doi: 10.1002/ajh.25418
33. Ghassemi S, Durgin JS, Nunez-Cruz S, Patel J, Leferovich J, Pinzone M, et al. Rapid manufacturing of non-activated potent CAR T cells. Nat BioMed Eng. (2022) 6:118–28. doi: 10.1038/s41551-021-00842-6
34. Qasim W. Genome-edited allogeneic donor "universal" chimeric antigen receptor T cells. Blood. (2023) 141:835–45. doi: 10.1182/blood.2022016204
35. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. 'Off-the-shelf' allogeneic CAR T cells: development and challenges. Nat Rev Drug Discovery. (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
36. Abbott RC, Cross RS, Jenkins MR. Finding the keys to the CAR: identifying novel target antigens for T cell redirection immunotherapies. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21020515
37. Flugel CL, Majzner RG, Krenciute G, Dotti G, Riddell SR, Wagner DL, et al. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat Rev Clin Oncol. (2023) 20:49–62. doi: 10.1038/s41571-022-00704-3
38. Chen DW, Lang BHH, McLeod DSA, Newbold K, Haymart MR. Thyroid cancer. Lancet. (2023) 401:1531–44. doi: 10.1016/s0140-6736(23)00020-x
39. Hu C, Liu M, Li Y, Zhao Y, Sharma A, Liu H, et al. Recent advances and future perspectives of CAR-T cell therapy in head and neck cancer. Front Immunol. (2023) 14:1213716. doi: 10.3389/fimmu.2023.1213716
40. Li H, Zhou X, Wang G, Hua D, Li S, Xu T, et al. CAR-T cells targeting TSHR demonstrate safety and potent preclinical activity against differentiated thyroid cancer. J Clin Endocrinol Metab. (2022) 107:1110–26. doi: 10.1210/clinem/dgab819
41. Szkudlinski MW, Fremont V, Ronin C, Weintraub BD. Thyroid-stimulating hormone and thyroid-stimulating hormone receptor structure-function relationships. Physiol Rev. (2002) 82:473–502. doi: 10.1152/physrev.00031.2001
42. Wang ZF, Liu QJ, Liao SQ, Yang R, Ge T, He X, et al. Expression and correlation of sodium/iodide symporter and thyroid stimulating hormone receptor in human thyroid carcinoma. Tumori. (2011) 97:540–6. doi: 10.1177/030089161109700420
43. Ding J, Li D, Liu X, Hei H, Sun B, Zhou D, et al. Chimeric antigen receptor T-cell therapy for relapsed and refractory thyroid cancer. Exp Hematol Oncol. (2022) 11:59. doi: 10.1186/s40164-022-00311-z
44. Qiu Z, Wang Y, Zhang Z, Qin R, Peng Y, Tang W, et al. Roles of intercellular cell adhesion molecule-1 (ICAM-1) in colorectal cancer: expression, functions, prognosis, tumorigenesis, polymorphisms and therapeutic implications. Front Oncol. (2022) 12:1052672. doi: 10.3389/fonc.2022.1052672
45. Bui TM, Wiesolek HL, Sumagin R. ICAM-1: A master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J Leukoc Biol. (2020) 108:787–99. doi: 10.1002/jlb.2mr0220-549r
46. Park S, Shevlin E, Vedvyas Y, Zaman M, Park S, Hsu YS, et al. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep. (2017) 7:14366. doi: 10.1038/s41598-017-14749-3
47. Wei H, Wang Z, Kuang Y, Wu Z, Zhao S, Zhang Z, et al. Intercellular adhesion molecule-1 as target for CAR-T-cell therapy of triple-negative breast cancer. Front Immunol. (2020) 11:573823. doi: 10.3389/fimmu.2020.573823
48. Jung M, Yang Y, McCloskey JE, Zaman M, Vedvyas Y, Zhang X, et al. Chimeric antigen receptor T cell therapy targeting ICAM-1 in gastric cancer. Mol Ther Oncolytics. (2020) 18:587–601. doi: 10.1016/j.omto.2020.08.009
49. Min IM, Shevlin E, Vedvyas Y, Zaman M, Wyrwas B, Scognamiglio T, et al. CAR T therapy targeting ICAM-1 eliminates advanced human thyroid tumors. Clin Cancer Res. (2017) 23:7569–83. doi: 10.1158/1078-0432.Ccr-17-2008
50. Vedvyas Y, McCloskey JE, Yang Y, Min IM, Fahey TJ, Zarnegar R, et al. Manufacturing and preclinical validation of CAR T cells targeting ICAM-1 for advanced thyroid cancer therapy. Sci Rep. (2019) 1):10634. doi: 10.1038/s41598-019-46938-7
51. Hsu JM, Hofe Ev, Hsu Y-M, Shieh J-H, Chaekal O-K, Guarneri D, et al. Phase I study of AIC100 in relapsed and/or refractory advanced thyroid cancer and anaplastic thyroid cancer. J Clin Oncol. (2022) 40:6093–3. doi: 10.1200/JCO.2022.40.16_suppl.6093
52. Srour SA, Cabanillas ME, Gupta S, Zafereo ME, Lu Y, Dadu R, et al. Safety and early efficacy results of phase 1 study of affinity tuned and trackable AIC100 CAR T cells in ICAM-1 positive relapsed and/or refractory advanced poorly differentiated and anaplastic thyroid cancers. J Clin Oncol. (2023) 41:6095–5. doi: 10.1200/JCO.2023.41.16_suppl.6095
53. Fielder GC, Yang TW, Razdan M, Li Y, Lu J, Perry JK, et al. The GDNF family: A role in cancer? Neoplasia. (2018) 20:99–117. doi: 10.1016/j.neo.2017.10.010
54. Lindahl M, Poteryaev D, Yu L, Arumae U, Timmusk T, Bongarzone I, et al. Human glial cell line-derived neurotrophic factor receptor alpha 4 is the receptor for persephin and is predominantly expressed in normal and Malignant thyroid medullary cells. J Biol Chem. (2001) 276:9344–51. doi: 10.1074/jbc.M008279200
55. Bhoj VG, Nunez-Cruz S, Zhou K, Arhontoulis D, Feldman M, Mansfield K, et al. Abstract 2295: GDNF family receptor alpha 4 (GFRa4)-targeted adoptive T-cell immunotherapy for medullary thyroid carcinoma. Cancer Res. (2016) 76:2295–5. doi: 10.1158/1538-7445.Am2016-2295
56. Bhoj VG, Li L, Parvathaneni K, Zhang Z, Kacir S, Arhontoulis D, et al. Adoptive T cell immunotherapy for medullary thyroid carcinoma targeting GDNF family receptor alpha 4. Mol Ther Oncolytics. (2021) 20:387–98. doi: 10.1016/j.omto.2021.01.012
57. Holzinger A, Abken H. CAR T cells targeting solid tumors: carcinoembryonic antigen (CEA) proves to be a safe target. Cancer immunol immunother CII. (2017) 66:1505–7. doi: 10.1007/s00262-017-2045-4
58. Zhao Y, Zhu X, Shen J, Xu Y, Yang Z, Qi Y, et al. "Armed" CEA CAR-T with a SIRPγ-CD28 chimeric co-receptor to exhibit the enhanced antitumor activity in preclinical study of colorectal cancer [Meeting Abstract]. J Clin Oncol. (2023) 41.
59. Erickson TA, Shih YP, Fass J, Jang M, Tran E. T cells engineered to express immunoreceptors targeting the frequently expressed medullary thyroid cancer antigens calcitonin, CEA, and RET M918T. Thyroid. (2022) 32:789–98. doi: 10.1089/thy.2022.0020
60. Zhou WT, Jin WL. B7-H3/CD276: an emerging cancer immunotherapy. Front Immunol. (2021) 12:701006. doi: 10.3389/fimmu.2021.701006
61. Hińcza-Nowak K, Kowalik A, Walczyk A, Pałyga I, Gąsior-Perczak D, Płusa A, et al. CD276 as a candidate target for immunotherapy in medullary thyroid cancer. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms241210019
62. Duan H, Huang H, Jing G. An antibody fab fragment-based chimeric antigen receptor could efficiently eliminate human thyroid cancer cells. J Cancer. (2019) 10:1890–5. doi: 10.7150/jca.30163
63. Hong M, Clubb JD, Chen YY. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell. (2020) 38:473–88. doi: 10.1016/j.ccell.2020.07.005
64. Hung S-H, Henderson Y, Pan K, Chiu Y, Singh S, Chen Y, et al. Abstract PO-061: Utilizing T cell receptor-based therapy to treat anaplastic thyroid cancer. Clin Cancer Res. (2023) 29:PO–061-PO-061. doi: 10.1158/1557-3265.Aacrahns23-po-061
65. Yan T, Zhu L, Chen J. Current advances and challenges in CAR T-Cell therapy for solid tumors: tumor-associated antigens and the tumor microenvironment. Exp Hematol Oncol. (2023) 12:14. doi: 10.1186/s40164-023-00373-7
66. Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. (2021) 12:81. doi: 10.1186/s13287-020-02128-1
67. Kailayangiri S, Altvater B, Wiebel M, Jamitzky S, Rossig C. Overcoming heterogeneity of antigen expression for effective CAR T cell targeting of cancers. Cancers (Basel). (2020) 12. doi: 10.3390/cancers12051075
68. Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discovery. (2018) 8:1219–26. doi: 10.1158/2159-8290.Cd-18-0442
69. Anurathapan U, Chan RC, Hindi HF, Mucharla R, Bajgain P, Hayes BC, et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol Ther. (2014) 22:623–33. doi: 10.1038/mt.2013.262
70. Yang Y, McCloskey JE, Yang H, Puc J, Alcaina Y, Vedvyas Y, et al. Bispecific CAR T cells against epCAM and inducible ICAM-1 overcome antigen heterogeneity and generate superior antitumor responses. Cancer Immunol Res. (2021) 9:1158–74. doi: 10.1158/2326-6066.Cir-21-0062
71. Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. (2018) 20:506–18. doi: 10.1093/neuonc/nox182
72. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. (2019) 37:1049–58. doi: 10.1038/s41587-019-0192-1
73. Sackstein R, Schatton T, Barthel SR. T-lymphocyte homing: an underappreciated yet critical hurdle for successful cancer immunotherapy. Lab Invest. (2017) 97:669–97. doi: 10.1038/labinvest.2017.25
74. Hou AJ, Chen LC, Chen YY. Navigating CAR-T cells through the solid-tumour microenvironment. Nat Rev Drug Discovery. (2021) 20:531–50. doi: 10.1038/s41573-021-00189-2
75. Hong M, Puaux AL, Huang C, Loumagne L, Tow C, Mackay C, et al. Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res. (2011) 71:6997–7009. doi: 10.1158/0008-5472.Can-11-1466
76. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. (2019) 10:4016. doi: 10.1038/s41467-019-11869-4
77. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. (2018) 36:346–51. doi: 10.1038/nbt.4086
78. Luo H, Su J, Sun R, Sun Y, Wang Y, Dong Y, et al. Coexpression of IL7 and CCL21 increases efficacy of CAR-T cells in solid tumors without requiring preconditioned lymphodepletion. Clin Cancer Res. (2020) 26:5494–505. doi: 10.1158/1078-0432.Ccr-20-0777
79. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. (2014) 2:154–66. doi: 10.1158/2326-6066.Cir-13-0027
80. Newick K, O'Brien S, Sun J, Kapoor V, Maceyko S, Lo A, et al. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase A localization. Cancer Immunol Res. (2016) 4:541–51. doi: 10.1158/2326-6066.Cir-15-0263
81. Donovan LK, Delaidelli A, Joseph SK, Bielamowicz K, Fousek K, Holgado BL, et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat Med. (2020) 26:720–31. doi: 10.1038/s41591-020-0827-2
82. Liao Z, Jiang J, Wu W, Shi J, Wang Y, Yao Y, et al. Lymph node-biomimetic scaffold boosts CAR-T therapy against solid tumor. Natl Sci Rev. (2024) 11:nwae018. doi: 10.1093/nsr/nwae018
83. Long J, Wang Y, Jiang X, Ge J, Chen M, Zheng B, et al. Nanomaterials boost CAR-T therapy for solid tumors. Adv Healthc Mater. (2024) 14:e2304615. doi: 10.1002/adhm.202304615
84. Liu G, Rui W, Zhao X, Lin X. Enhancing CAR-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cell Mol Immunol. (2021) 18:1085–95. doi: 10.1038/s41423-021-00655-2
85. Liu Z, Zhou Z, Dang Q, Xu H, Lv J, Li H, et al. Immunosuppression in tumor immune microenvironment and its optimization from CAR-T cell therapy. Theranostics. (2022) 12:6273–90. doi: 10.7150/thno.76854
86. Hegde M, DeRenzo CC, Zhang H, Mata M, Gerken C, Shree A, et al. Expansion of HER2-CAR T cells after lymphodepletion and clinical responses in patients with advanced sarcoma [Meeting Abstract]. J Clin Oncol. (2017) 35. doi: 10.1200/JCO.2017.35.15_suppl.10508
87. Guo Y, Feng K, Liu Y, Wu Z, Dai H, Yang Q, et al. Phase I study of chimeric antigen receptor-modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clin Cancer Res. (2018) 24:1277–86. doi: 10.1158/1078-0432.Ccr-17-0432
88. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. (2015) 4:e994446. doi: 10.4161/2162402x.2014.994446
89. Wilkie S, Burbridge SE, Chiapero-Stanke L, Pereira AC, Cleary S, van der Stegen SJ, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem. (2010) 285:25538–44. doi: 10.1074/jbc.M110.127951
90. Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. (2019) 36:471–82. doi: 10.1016/j.ccell.2019.09.006
91. Gray KD, McCloskey JE, Vedvyas Y, Kalloo OR, Eshaky SE, Yang Y, et al. PD1 blockade enhances ICAM1-directed CAR T therapeutic efficacy in advanced thyroid cancer. Clin Cancer Res. (2020) 26:6003–16. doi: 10.1158/1078-0432.Ccr-20-1523
92. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. (2018) 36:847–56. doi: 10.1038/nbt.4195
93. Sun S, Hao H, Yang G, Zhang Y, Fu Y. Immunotherapy with CAR-modified T cells: toxicities and overcoming strategies. J Immunol Res. (2018) 2018:2386187. doi: 10.1155/2018/2386187
94. Suran M. FDA adds boxed warning to CAR T-cell therapies, but says benefits outweigh risks of secondary cancers. Jama. (2024) 331:818–20. doi: 10.1001/jama.2024.1011
95. Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. (2019) 25:947–53. doi: 10.1038/s41591-019-0421-7
96. Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal. (2018) 11. doi: 10.1126/scisignal.aat6753
97. Sommermeyer D, Hill T, Shamah SM, Salter AI, Chen Y, Mohler KM, et al. Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia. (2017) 31:2191–9. doi: 10.1038/leu.2017.57
98. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. (2014) 124:1277–87. doi: 10.1182/blood-2014-01-545020
99. Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med. (2019) 11. doi: 10.1126/scitranslmed.aau5907
100. Al-Haideri M, Tondok SB, Safa SH, Maleki AH, Rostami S, Jalil AT, et al. CAR-T cell combination therapy: the next revolution in cancer treatment. Cancer Cell Int. (2022) 22:365. doi: 10.1186/s12935-022-02778-6
Keywords: CAR-T cell therapy, thyroid cancer, biological basis, clinical application, immunotherapy
Citation: Sun Z, Wang C, Zhao Y and Ling Q (2024) CAR-T cell therapy in advanced thyroid cancer: from basic to clinical. Front. Immunol. 15:1411300. doi: 10.3389/fimmu.2024.1411300
Received: 02 April 2024; Accepted: 27 May 2024;
Published: 07 June 2024.
Edited by:
Lei Tao, Fudan University, ChinaReviewed by:
Thanh Huong Phung, Hanoi University of Pharmacy, VietnamWilliam J. Magner, University at Buffalo, United States
Copyright © 2024 Sun, Wang, Zhao and Ling. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenhua Sun, Ymp4aDEwMEAxNjMuY29t
†These authors have contributed equally to this work