 Jiajie Jing1†
Jiajie Jing1† Yuan Ma1†Ziwen Xie1Bingyan Wang1Yueming Chen1Enjie Chi1Jiadong Wang1Kejin Zhang1Zhujun Wang2*Sisi Li1*
Yuan Ma1†Ziwen Xie1Bingyan Wang1Yueming Chen1Enjie Chi1Jiadong Wang1Kejin Zhang1Zhujun Wang2*Sisi Li1*- 1Department of Clinical Medicine, Hangzhou City University School of Medicine, Hangzhou, China
- 2Department of Pediatrics, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, China
Acute lymphoblastic leukemia (ALL) is a prevalent malignancy affecting the hematopoietic system, encompassing both B-cell ALL (B-ALL) and T-cell ALL (T-ALL). T-ALL, characterized by the proliferation of T-cell progenitors in the bone marrow, presents significant treatment challenges, with patients often experiencing high relapse rates and poor long-term survival despite advances in chemotherapy and hematopoietic stem cell transplantation (HSCT). This review explores the pathogenesis and traditional treatment strategies of T-ALL, emphasizing the promising potential of chimeric antigen receptor (CAR) technology in overcoming current therapeutic limitations. CAR therapy, leveraging genetically modified immune cells to target leukemia-specific antigens, offers a novel and precise approach to T-ALL treatment. The review critically analyzes recent developments in CAR-T and CAR-NK cell therapies, their common targets, optimization strategies, clinical outcomes, and the associated challenges, providing a comprehensive overview of their clinical prospects in T-ALL treatment.
1 Introduction
T-cell acute lymphoblastic leukemia (T-ALL) predominantly originates from early T-cell progenitors, constituting 25% and 15% of adult and pediatric acute leukemias, respectively. The pathogenesis of T-ALL is intricate, and therapeutic options for T-ALL are comparatively restricted when compared to B-cell acute lymphoblastic leukemia (B-ALL) (1). Presently, newly diagnosed T-ALL patients commonly undergo high-risk multi-agent chemotherapy regimens lasting 2-3 years, with or without concurrent cranial radiotherapy. Despite novel drug combinations with chemotherapy have moderately improved the prognosis of T-ALL, achieving an 80% complete remission (CR) rate, over 50% of patients experience disease relapse and develop drug resistance. Even with allogeneic hematopoietic stem cell transplantation (allo-HSCT), the 5-year disease-free survival rate (5-DFS) for adult T-ALL remains below 30%, with approximately 30% cumulative relapse rate observed in pediatric T-ALL. The long-term survival rate for relapsed patients is less than 15% (2). HSCT stands as the sole potential curative option available at present (3). A pivotal prognostic factor for T-ALL is the presence of Minimal Residual Disease (MRD) after the induction and consolidation therapy phases. MRD denotes the persistence of a small number of leukemia cells in the patient’s body post-treatment, potentially predisposing to relapse (4). Common MRD markers used include flow cytometry markers (e.g., CD3, CD7, CD4, CD8, CD1a) and molecular markers (e.g., TCR gene rearrangements, NOTCH1 mutations) (5, 6).
The Chimeric Antigen Receptor (CAR) represents a revolutionary immunotherapy approach that uses genetic engineering to enable immune cells to specifically identify and eradicate tumor cells. This methodology revolves introducing receptor genes into immune cells via gene editing techniques, enable them to recognize tumor-associated antigens (TAAs). These modified cells are then expanded in vitro and administered to patients, facilitating the precise targeting and efficient elimination of tumor cells by the immune system. Currently, CAR immunotherapy predominantly harnesses T cells and NK cells as effector cells, with some partial involvement of B cells activated through cytokine networks. Particularly in hematologic malignancies, CAR-T immunotherapy, which employs T cells as effector cells, has demonstrated remarkable efficacy (7).
2 Pathogenesis and clinical treatment of T-ALL
2.1 Pathogenesis of T-ALL
T-ALL arises from the abnormal proliferation of T-lineage precursor cells within the bone marrow, influenced significantly by aberrant signaling pathways. The NOTCH, IL7-IL7R-JAK-STAT, and PI3K-Akt-mTOR pathways are critically implicated in T-ALL. Dysregulation in these pathways disrupts normal cellular processes, including metabolism, proliferation, differentiation, and apoptosis, ultimately leading to the development of T-ALL. These disruptions also play a significant role in contributing to drug resistance. For instance, NOTCH1 mutations, found in about 60% of T-ALL cases, lead to uninhibited signaling that promotes leukemic growth. These mutations can involve the disruption of the NOTCH1 gene itself or inactivating mutations of its negative regulators like FBXW7 (8–10). Additionally, NOTCH1 negatively regulates the tumor suppressor gene P53 in T-ALL through various mechanisms, such as inhibiting P53 activation, phosphorylation, and nuclear localization via eIF4E, leading to drug resistance in malignant cells. Mutations in the IL-7R and JAK-STAT pathway components are identified in 20-30% of T-ALL cases, promoting oncogenic activations that exacerbate the disease progression and mediate resistance to glucocorticoids (11, 12). The PI3K-Akt-mTOR pathway, often activated by the loss of tumor suppressor PTEN, is another critical factor of T-ALL pathogenesis, promoting malignant cell survival and resistance to therapy (13, 14). Epigenetic changes, such as DNA methylation and histone modifications, and dysregulation of apoptosis-related genes (e.g., TP53, BCL-2) further reduce sensitivity to chemotherapy. Genetic predispositions and environmental factors, such as exposure to ionizing radiation or carcinogenic chemicals, also contribute to the risk of developing T-ALL (15). Due to these complex pathogenic mechanisms, T-ALL is notably less responsive to chemotherapy than B-ALL. Targeted therapies developed from these insights, including inhibitors of γ-secretase, cyclin-dependent kinases, and mTOR, have shown promise in preclinical and clinical studies (3) (Figure 1).
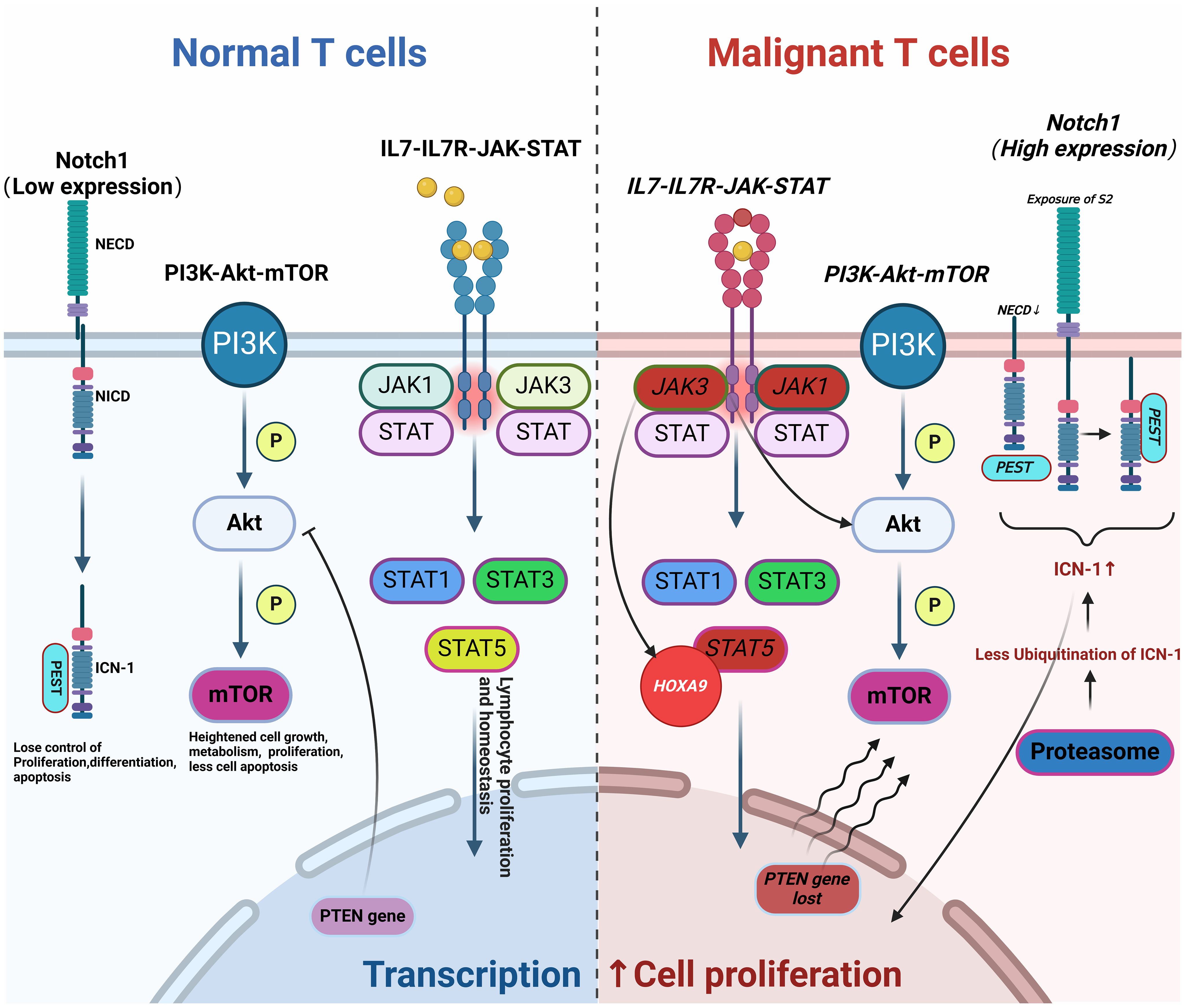
Figure 1. Pathogenesis of T-ALL. Abnormal changes in molecular pathways (such as NOTCH, IL7-IL7R-JAK-STAT, and PI3K-Akt-mTOR) lead to aberrant T cell proliferation and differentiation, resulting in disease onset, progression, and drug resistance (Malignant mutations are indicated in italics). This figure was drawn using Biorender.
2.2 Traditional clinical treatment
The management of T-ALL primarily revolves around combination chemotherapy, which is essential for induction, consolidation, maintenance therapy, and Central Nervous System (CNS) prophylaxis. The standard regimen includes drugs such as L-Asparaginase, Cyclophosphamide, Glucoroticoid and Vincristine. Intensive chemotherapy regimens are often employed to achieve effective induction and reduce relapse rates. To address CNS involvement, high-dose methotrexate and triple intrathecal therapy have proven effective in improving remission rates and managing CNS prophylaxis (16, 17). For high-risk patients, allo-HSCT is considered the most effective curative approach, particularly following initial intensive chemotherapy. Clinical trials have demonstrated that allo-HSCT provides superior outcomes compared to conventional chemotherapy or autologous transplantation, especially in standard-risk patients during the first CR (18, 19).
3 Studies on CAR technology in the treatment of T-ALL
CAR technology, which involves the modification of immune cells, has represented a significant breakthrough in the treatment of hematological malignancies. Notably, it has demonstrated remarkable success in treating drug-resistant B-ALL and B-cell non-Hodgkin lymphoma (NHL) (20).This technology has received approval from the US Food and Drug Administration (FDA) for clinical application (21–23). A CAR construct comprises three main domains: an extracellular domain, a transmembrane domain, and an intracellular activation fragment. The extracellular domain’s antigen-binding segment is responsible for precisely recognizing specific antigens on tumor cell surfaces, thereby facilitating targeted therapy. Meanwhile, the intracellular activation fragment activates effector cell killing functions upon antigen recognition. Different intracellular activation domains exhibit varying activation effects. The most commonly used activation domain is CD3ζ, which contains immunoreceptor tyrosine-based activation motifs (ITAMs) that initiate T-cell activation and proliferation upon antigen binding (24). CD3ζ alone can trigger cytotoxic responses but is often paired with co-stimulatory domains to enhance efficacy and persistence. FcRγ, an activation domain derived from the Fc receptor, is used in some CAR-NK cell therapies to initiate signaling cascades similar to CD3ζ in T-cells, thereby activating NK cells and inducing cytotoxicity against target cells (24, 25). Co-stimulatory domains such as CD28, 4-1BB (CD137), OX40 (CD134), and 2B4 (CD244) play crucial roles in enhancing CAR-T and CAR-NK cell therapies. CD28 provides essential secondary signals that enhance T-cell activation, proliferation, and cytokine production, but it may be associated with higher risks of CRS. 4-1BB enhances the survival and persistence of CAR-T cells by promoting anti-apoptotic signaling pathways, often showing longer persistence in vivo and lower incidences of severe cytokine release syndrome (CRS) compared to CD28-based CARs (26). OX40 enhances T-cell expansion and survival by activating the NF-κB pathway, increasing T-cell proliferation and resistance to apoptosis, thus improving the efficacy and durability of CAR-T cell responses (27), predominantly used in CAR-NK cell therapies, promotes NK cell activation and enhances cytotoxicity against target cells, sustaining NK cell responses and improving overall therapeutic efficacy (28). The choice of intracellular activation and co-stimulatory domains significantly impacts the efficacy, persistence, and safety profile of CAR-T and CAR-NK cell therapies. Understanding these differences allows for the optimization of CAR designs tailored to specific clinical needs. The following discussion will highlight therapeutic strategies that employ CAR technology in the treatment of T-ALL.
3.1 CAR-T therapy for T-ALL
CAR-T cell therapy involves genetically engineering a patient’s own T cells to target and destroy tumor cells (Figure 2). The primary challenge in developing CAR-T therapy for T-ALL is the presence of target antigens on both malignant and non-malignant T cells and effector cells. This dual expression can result in several complications (1): Damage to Normal T Cells: CAR-T cells may inadvertently attack normal T cells, leading to the depletion of healthy T cells and potentially causing severe immunodeficiency, posing a life-threatening risk (29) (2). Fratricide:Fratricide refers to CAR-T cells attacking each other due to the expression of the same target antigen. For example, in therapies targeting CD38 for T-ALL, CAR-T cells may also express CD38, leading to mutual destruction (30) (3). Contamination by malignant T Cells: During the isolation of therapeutic T cells, there is a risk of malignant T cells being mixed in and subsequently engineered into malignant CAR-T cells. Given these challenges, the progress of CAR-T cell therapy development for T-ALL has been hindered (31).
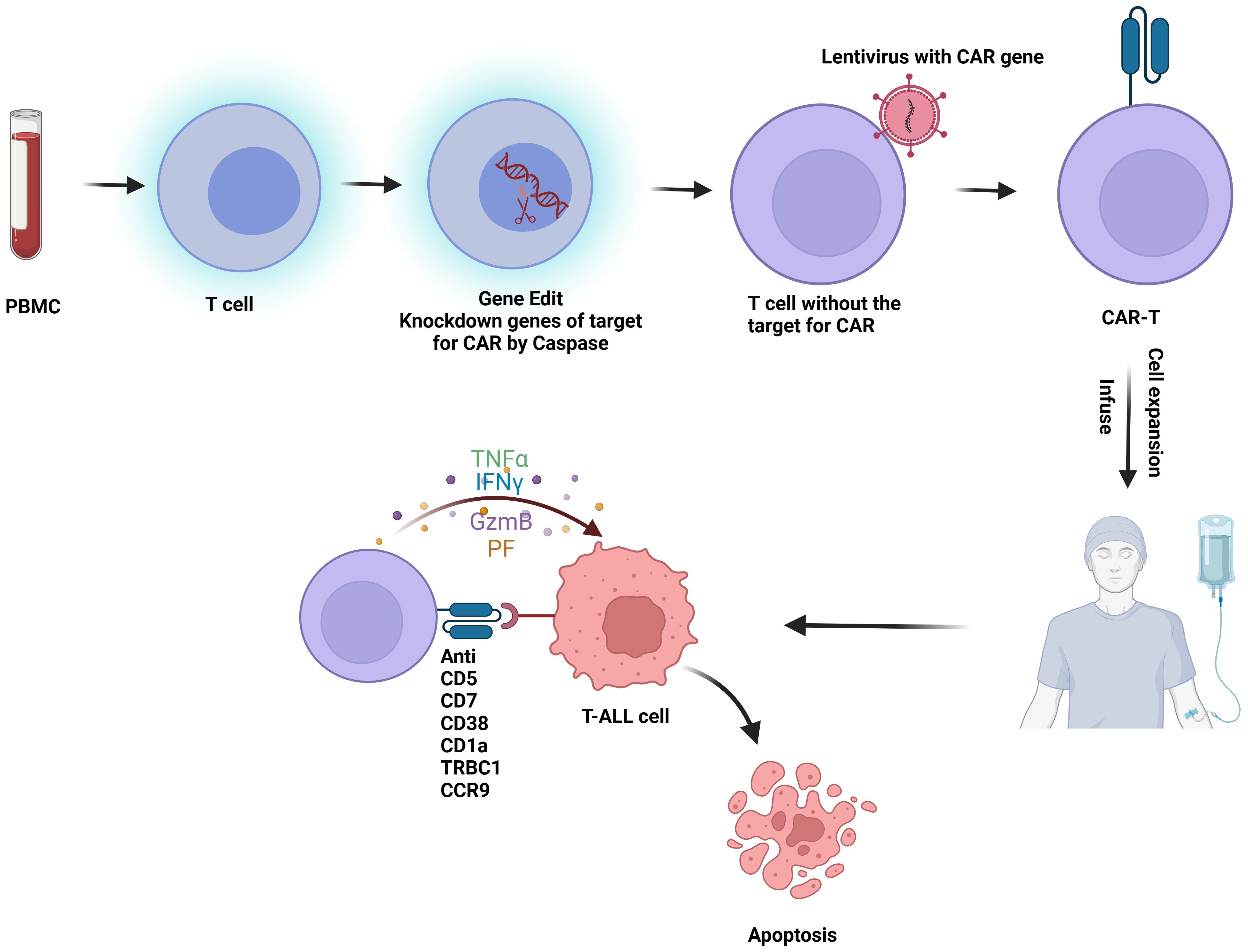
Figure 2. Flowchart of CAR-T therapy for T-ALL: T cells are first isolated from peripheral blood mononuclear cells (PBMC). These T cells undergo gene editing to silence the CAR-targeting antigen gene. Lentiviral vectors are then used to introduce the CAR construct into the T cells, creating CAR-T cells. Once engineered, the CAR-T cells are expanded and infused back into the patient, where they target and kill malignant T cells in vivo. This figure was drawn using Biorender.
3.1.1 CD5
CD5 is a characteristic surface marker found on malignant T cells, present in approximately 80% of cases of T-ALL and T-cell lymphomas (32). The Maksim’s group engineered a CD5-targeting CAR-T construct that includes an extracellular anti-CD5 single-chain variable fragment (scFv) derived from clone H65, an IgG Fc transmembrane domain, and intracellular segments from CD28 and TCRζ chains. Notably, CAR expression led to the down-regulation of CD5 on CAR-T cells, effectively reducing fratricide and enhancing in vivo expansion. Co-culture assays with CD5-positive T-ALL cell lines such as Jurkat, CCRF, and MOLT4 showed that CD5 CAR-T cells eliminated over 95% of target cells within 48 hours, with no detectable target cells after 7 days, indicating a high specificity and cytotoxic capability towards malignant T cells (33).
Further development by Dai’s group introduced fully human CD5-targeting heavy-chain variable (FHVH) domains to create a biepitopic CAR, termed FHVH3/VH1, which includes FHVH1 and FHVH3 domains that bind to different epitopes of the CD5 antigen. Additionally, they employed CRISPR-Cas9 technology to produce CD5 knockout (CD5KO) CAR-T cells, which reduces fratricide among the CD5 CAR-T population. This strategy not only enhanced the CD5 binding affinity of CAR-T cells but also significantly improved their therapeutic efficacy against CD5-expressing T-cell malignancies (34).
Moreover, integrating insights from the most recent advances, such as the development of a CH2CH3 hinge region that enhances the cytotoxicity of CD5 CAR-T cells (35) to optimize the efficacy and durability of CD5 CAR-T therapies. These enhancements not only bolster the CAR constructs’ direct anticancer activities but also refine their persistence in the hostile tumor microenvironment.
Overall, the evolving landscape of CD5 CAR-T cell research points to a promising future for the treatment of T-cell malignancies, with ongoing innovations aimed at improving efficacy, safety, and treatment accessibility (36).
3.1.2 CD7
CD7 is highly expressed on the surface of T-ALL cells, making it a potent target for T-ALL therapy. Although CD7 is also found on NK cells and normal T cells, research in CD7 knockout mice has shown normal immune development, indicating that targeting CD7 does not impairing the essential functions of T and NK cells (37–41).
Diogo’s group used CRISPR-Cas9 to knock out CD7 expression in CAR-T cells, developed second-generation CAR-T cells incorporating CD28 and CD3ζ as intracellular signaling domains. These CD7 CAR-T cells demonstrated high efficacy in eliminating various T-ALL cell lines (Jurkat, CCRF-CEM, MOLT-4, Hut 78, SupT1, and Raji), as well as primary T-ALL cells, and exhibited therapeutic effects in mouse models of T-ALL (42). Jiang’s group employed a novel technique using EF1α to specifically disrupt the CD7 site on CAR-T cells. This approach aims to prevent fratricide, enhancing both the specificity and safety of CAR-T cell therapies (43). Conversely, Peihua Lu’s group adopted a different approach by employing a natural selection method to mask or sequence CD7 molecules on the cell surface using a specialized scFv, developing a novel CD7-targeting CAR (NS7CAR) -T cell. This method, though less invasive than gene editing, might not completely eliminate the risk of fratricide as the masking could be incomplete under certain physiological conditions. In a phase I trial (NCT04572308), NS7CAR was administered to 20 patients with refractory/relapsed T-ALL (r/r T-ALL) and T-cell lymphoblastic lymphoma (T-LBL). Nineteen patients achieved MRD negativity at Day28, attained complete remission, and became eligible for allo-HSCT (44). Additionally, Zhang’s group engineered CD7-targeted CAR-T cells that blocked CD7 expression by coupling CD7 nanoantibody VHH6 to an ER/Golgi-retention motif peptide. In a phase I clinical trial (NCT04004637), eight patients with r/r T-ALL received this treatment, achieving a CR rate of 87.5% at 3 months post-infusion, with detectable CAR-T cells persisting for up to 270 days (45).
Notably, Wong’s group introduced the (protein expression blocker) PEBL technology, which provides a groundbreaking solution to the problem of fratricide in CD7-targeted CAR-T therapy. By engineering a PEBL that retains CD7 within the Golgi apparatus, PEBL effectively prevents CD7 from being presented on the cell surface, thereby safeguarding CAR-T cells from fratricide. This approach not only maintains the efficacy of the CAR-T cells but also improves their survival and persistence in the patient’s body. The PEBL technology offers significant advantages over previous methods, including its non-invasive nature and the avoidance of potential gene editing complications. It represents a safer alternative that could potentially be applied to other CAR-T therapies targeting antigens expressed on the T cells themselves. A phase I is ongoing with these CAR T-cells in children and adults with relapse/refractory T-ALL in Singapore (CARTALL, NCT05043571). However, the scalability of this technology and its adaptability to other antigens require further investigation (46).
3.1.3 C-C chemokine receptor type 9
CCR9, a G protein-coupled receptor with a 7-transmembrane structure, is expressed on approximately 5% of normal circulating T and B cells (47). Recent studies have demonstrated a significant upregulation of CCR9 in r/r T-ALL cases, compared to its minimal expression on normal T cells. This differential expression profile marks CCR9 as a viable target for CAR-T cell therapy for T-ALL. Paul’s team knocked out CCR9 of T cells and developed CCR9-targeted CAR-T cells. These CAR-T cells demonstrated strong cytotoxicity against both T-ALL cell lines and primary T-ALL blasts in vitro, confirming their potent anti-leukemic properties. Further exploration into the functionality of these CCR9-targeted CAR-T cells revealed significant therapeutic efficacy in preclinical studies, including standardized mouse models with T-ALL cell lines and patient-derived xenograft (PDX) models representing high tumor burden in r/r T-ALL (48). These studies confirmed that targeting CCR9 can lead to substantial reduction of tumor burden without causing significant toxicity to the normal T cell population.
3.1.4 CD38
CD38, a cell surface glycoprotein part of the ribocylase family, is crucial in various cellular functions including cell migration, receptor-mediated adhesion, extracellular metabolism, and intracellular Ca2+ signaling. Its expression is significantly elevated in multiple myeloma (MM), NHL,T-ALL, and NK/T cell tumors, yet remains low in normal lymphocytes, bone marrow cells, and some non-hematopoietic tissues (49, 50). Importantly, CD38 is not present in multipotent hematopoietic stem cells, which are essential for bone marrow recovery, making it an attractive target for the treatment of various hematologic malignancies (51).
Wang’s group engineered T cells from PBMCs to express an anti-CD38 scFv fragment, controlled by an EF1α promoter and enhanced with co-stimulatory domains from 4-1BB and CD3ζ, developing a CD38 CAR-T. It demonstrated significant targeted killing capabilities against MM, B cell lymphoma, T-ALL, and NK/T cell lymphoma both in vitro and in vivo (52). Glisovic-Aplenc’s group further confirmed these findings, demonstrating the effectiveness of CD38 CAR-T in treating primary hematological malignant cells (53).
In 2023, Liao’s group introduced an innovative approach by integrating CD38-specific CARs into the CD38 gene locus itself, driven by different promoters. This novel approach led to the creation of both CAR-T and CAR-NK cells that exhibited robust anti-tumor activities. The use of native CD38 promoters for expression control potentially enhances the physiological relevance and tumor specificity of these CAR-modified cells, minimizing off-target effects and improving safety profiles (54).
3.1.5 CD1a
CD1a is predominantly expressed on cortical T-ALL (coT-ALL) cells, but is absent on CD34-positive progenitor cells and mature T cells. This distinction provides a unique therapeutic advantage by minimizing off-target cytotoxic effects, significantly reducing the risk of damaging non-malignant hematopoietic cells (55–57).
Sanchez-Martinez’s group developed CD1a-targeting CAR-T cells, demonstrating strong and specific cytotoxicity when co-cultured with coT-ALL cell lines and primary lymphoid cells derived from coT-ALL patients. In NSG mouse xenograft models generated using Jurkat cells or T-ALL patient-derived tumor cells, it demonstrated significant anti-leukemic efficacy, with a marked reduction in tumor burden and prolonged survival in treated mice. These promising outcomes underscore the therapeutic potential of CD1a CAR-T cells and advocate for their further clinical development for this challenging leukemia subtype (58).
3.1.6 TRBC1
Expression of T-cell receptor beta chain constant domain1 (TRBC1) and T cell receptor beta chain constant domain 2 (TRBC2) are mutually exclusive, with TRBC1 T cells comprising 25% to 47% of the T-cell population in healthy individuals. Maciocia’s group has made significant advancements in immunotherapy by developing a CAR-T therapy targeting the TRBC1. They engineered CAR-T cells that selectively recognize and eliminate TRBC1-expressing cells, both normal and malignant, while preserving TRBC2 positive cells, maintaining essential immune functions. This selective targeting strategy is advantageous over nonselective methods that deplete the entire T-cell population, thereby avoiding severe immunosuppression (59). However, a challenge identified by Zhang’s group highlights the complexity of CAR-T cell production: the anti-TRBC1 CAR gene might unintentionally transfer to malignant T cells during manufacturing, leading to a self-binding scenario where the CAR interacts with TRBC1 on malignant cells, thus shielding them from cytotoxic action (60). This underscores the need for meticulous control measures to prevent cross-contamination and ensure the purity of therapeutic cells.
In the evolving landscape of CAR-T cell therapy, it is essential to fully explore these technologies in larger, more diverse patient populations. Addressing challenges related to manufacturing, regulatory approval, and cost-effectiveness is equally important. Investigating PEBL and similar technologies in clinical trials will provide valuable insights into their long-term safety and efficacy, potentially revolutionizing treatment for T-ALL and other T-cell malignancies. Continuous innovations, like the integration of CAR constructs into specific gene loci demonstrated by Liao’s team, offer more regulated expression and could reduce the risk of cytokine release syndrome and other side effects. This advancement points to a future where CAR therapies are finely tuned for maximum efficacy and minimal toxicity. Targeting strategies focused on CD3, CCR7, CD4, and CD8 molecules have shown initial efficacy, broadening the scope of therapeutic targets. However, despite these advancements, challenges such as non-targeted cell damage and fratricide highlight the need for ongoing optimization. These developments underscore the dynamic and promising future of CAR cell therapy, aiming for greater precision and effectiveness in treating T-ALL and other T-cell malignancies (61).
3.2 CAR-NK therapy for T-ALL
NK cells, also known as innate lymphoid cells, play a crucial role in the immune system by directly eliminating tumor cells without requiring specific activation. Unlike cytotoxic T cells, NK cells are not constrained by major histocompatibility complex (MHC) recognition and do not trigger the release of inflammatory cytokines such as IL-1 and IL-6 during killing, thereby avoiding the induction of CRS. In comparison to CAR-T cells, CAR-engineered NK cells offer several advantages, including potent anti-tumor activity, reduced toxicity and side effects, and a broader availability of cell sources (62). Similar to CAR-T therapy, the targeted molecules for CAR-NK cells include CD38, CD7, and CD5, etc. (Figure 3).
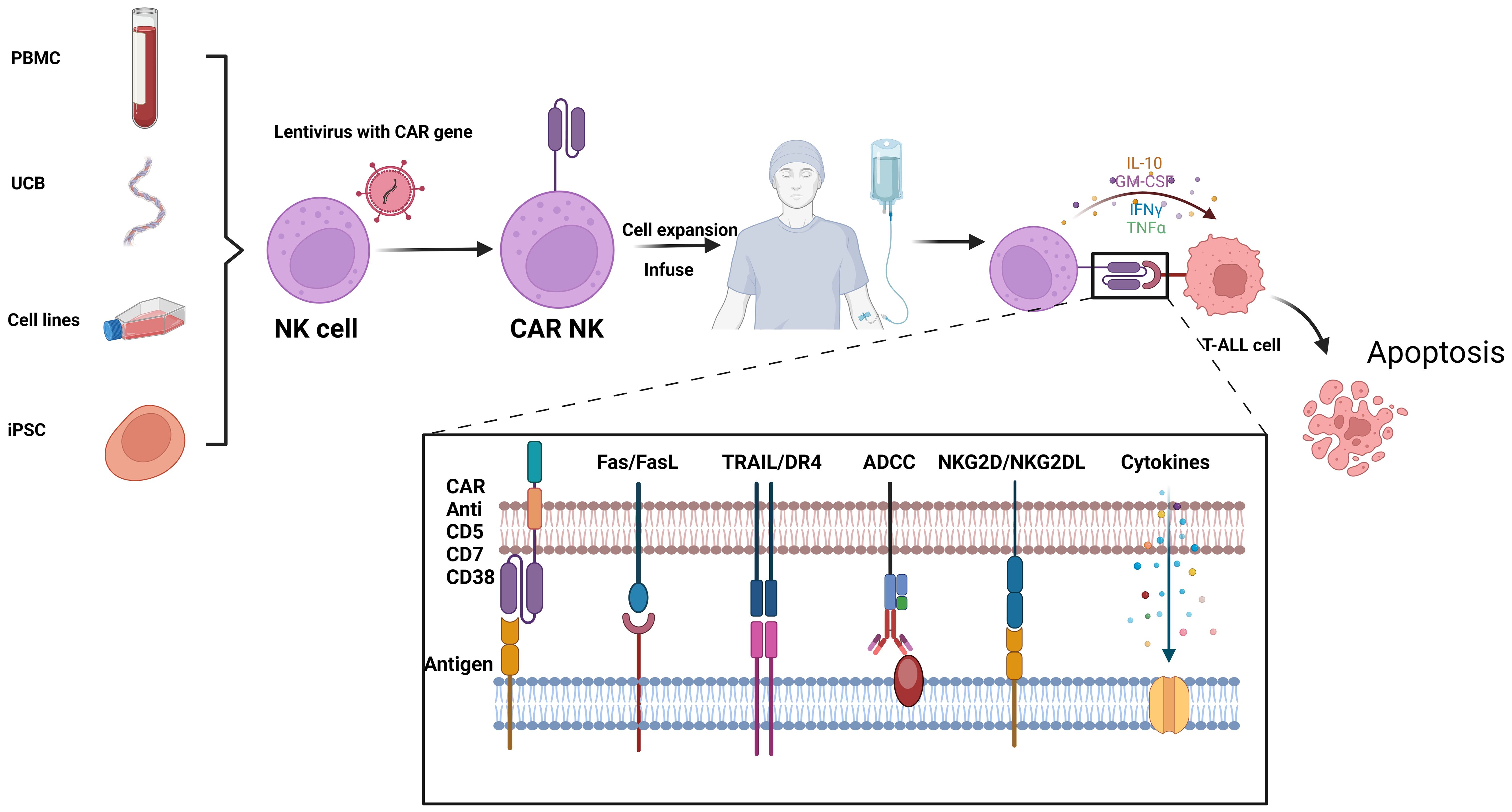
Figure 3. Flowchat and mechanistic diagram of CAR-NK therapy for T-ALL: NK cells from various sources are first isolated. Using lentiviral vectors, these NK cells are infected to create CAR-NK cells. Once engineered, the CAR-NK cells are expanded and infused back into the patient. In vivo, they target and kill malignant T cells through multiple mechanisms, including CAR-mediated targeting, Fas/FasL interactions, TRAIL/DR4 pathways, antibody-dependent cellular cytotoxicity (ADCC), and NKG2D/NKG2DL interactions. This figure was drawn using Biorender.
3.2.1 CD38
As previously mentioned, Liao’s group integrated CD38-specific CARs into the CD38 gene locus, creating CAR-NK cells with native CD38 promoters that demonstrated robust anti-tumor activities and enhanced specificity while minimizing off-target effects. To address CD38 expression on normal NK cells and prevent fratricide, they employed a “two-in-one” CRISPR/Cas9 strategy, giving CAR-NK cells anti-fratricide capabilities. This allowed efficient elimination of primary T-ALL cells, showing the significant therapeutic effects (54). Furthermore, Clara’s group utilized CRISPR/Cas9 technology to disrupt CD38 expression in ex vivo expanded NK cells and integrated high-affinity CD16 into the edited CD38 locus. The integration of CD16 is based on its role as a high-affinity Fc receptor, which allows NK cells to bind to the Fc region of antibodies attached to target cells. This binding facilitates antibody-dependent cellular cytotoxicity (ADCC), enhancing the NK cells’ ability to identify and destroy malignant cells. This innovative approach not only circumvented NK cell autologous attack but also significantly enhanced antitumor efficacy by improving targeting and elimination of leukemic cells (63).
Looking forward, the potential of CD38 CAR-NK cells, with their targeted integration and controlled expression facilitated by CRISPR/Cas9, offers a promising alternative to conventional CAR therapies. However, the real-world efficacy of these cells requires comprehensive clinical trials to assess their stability, persistence, and effectiveness across various tumor environments.
3.2.2 CD5
CD5 is recognized as a critical characteristic marker of T-ALL cells and is expressed on nearly all normal T cells, yet is absent on NK cells. This distinct expression profile enables the utilization of CD5 CAR-NK cell immunotherapy to bypass the fratricide effects often seen with CAR-T therapies. Xu’s group harnessed NK-92 cells as a platform for CD5-CAR modification, engineering anti-CD5 CAR constructs that incorporated different costimulatory domains: one with the T-cell-associated 4-1BB (BB.z) activating receptor and another with the NK cell-associated 2B4 (2B4.z) activating receptor. In vitro studies showed that both BB.z-NK and 2B4.z-NK cells exhibited targeted cytotoxicity against CD5-positive malignant cell lines, primary malignant cells, and normal T cells. In a T-ALL mouse xenograft model, both types of CAR-NK cells demonstrated significant anti-tumor effects, with 2B4.z-NK cells displaying enhanced anti-CD5 activity compared to BB.z-NK cells (64).
Further analysis by Voynova’s group compared the efficacy of CD3/CD5-directed CAR-NK and CAR-T therapies in T-ALL. Their findings highlighted that NK cells, when used as effector cells, effectively reduced the fratricide effect and addressed complications related to T cell aplasia and long-term immunodeficiency resulting from targeting molecules expressed on healthy T cells. CD3/CD5-directed CAR-NK cells do not cause long-term immunodeficiency due to their distinct biological characteristics and shorter lifespan compared to T cells. Unlike T cells, NK cells have a reduced risk of prolonged immune suppression and do not undergo extensive clonal expansion or persistence, minimizing their impact on long-term immune function. In contrast, CAR-T cells, being part of the adaptive immune system, can persist and expand over time, potentially leading to sustained depletion of target cells and subsequent immunodeficiency. Voynova’s group observed that although CD3 CAR-NK cells demonstrated robust efficacy against lymphoma cells in vitro, their performance in xenograft tumor models was compromised due to significant down-regulation of target antigens on malignant cells after the infusion of CD3 CAR-NK cells (65). In contrast, therapies directed towards CD5 present a potentially safer and more effective alternative. The addition of DAP10 co-stimulation imparts memory-like features to CAR-NK cells targeting CD5, enhancing their efficacy and persistence. This design significantly bolsters the therapeutic potential of CD5 CAR-NK cells as a treatment for T-ALL (66, 67).
3.2.3 CD7
Zhu’s group developed CD7 CAR-NK-92MI cells, which exhibited significant cytotoxicity against CD7-positive hematological malignancies, including T-ALL, AML, and T-cell lymphoma, compared to their non-modified counterparts. These modified NK cells exhibited increased secretion of cytokines (e.g., IL-2, IFN-γ, granzyme B) play a critical role in mediating immune responses and enhancing cytotoxic activities (68). Jiang’s group developed a CD7-targeting CAR-NK by integrating the CAR directly into the CD7 gene locus of NK cells, using EF1α as the intracellular activation segment. This approach prevents the elimination of NK cells caused by their own CD7 expression, effectively addressing issues related to cell fratricide. These CD7-specific CAR-NK cells exhibit promising anti-tumor activity with reduced risk of off-target effects and enhanced tumor specificity (43).
4 Clinical studies of CAR technology in T-ALL
The clinical efficacy of CAR-T therapy in treating hematological malignancies has been well-established, with many related clinical trials currently underway. The number of trials investigating CAR-T cell therapy for T-ALL is steadily increasing, reflecting the rapid advancements in this field. Although CAR-NK therapy is under investigation for AML and solid tumors, its potential for treating T-ALL remains underexplored. While there is a comprehensive list of registered clinical trials for CAR-T cell therapy targeting T-ALL on the ClinicalTrials.gov website (refer to Table 1), there are no registered trials specifically focusing on CAR-NK cells for T-ALL. This highlights the emerging interest in CAR-NK cell therapy, necessitating further research to understand its potential in treating T-ALL.
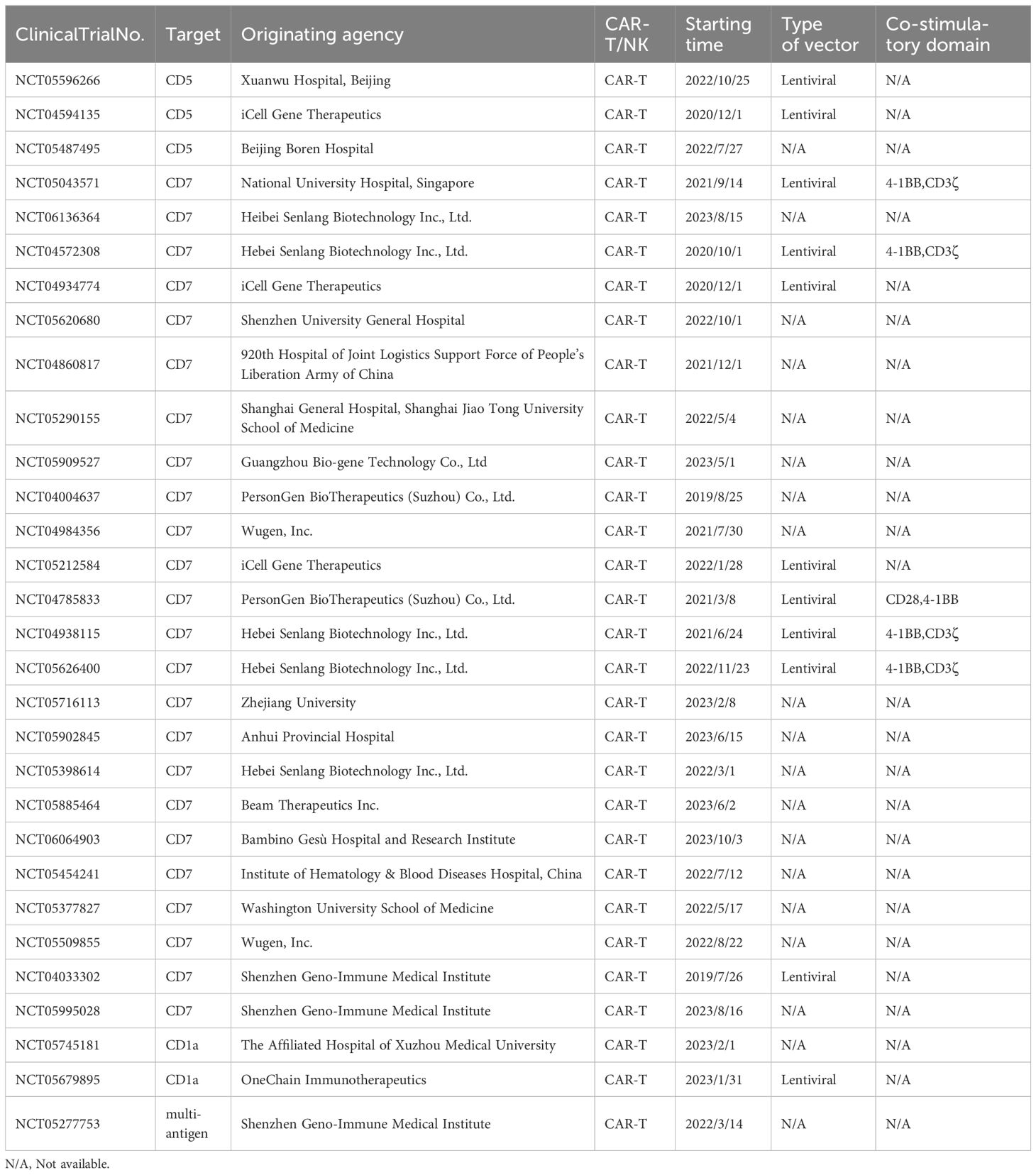
Table 1. Clinical study of chimeric antigen receptor technology in the treatment of T-ALL (Data sources: ClinicalTrials.gov).
Although CAR-T cell therapy has shown positive effects in clinical trials of T-ALL, serious side effects may exist. For example, CRS, immune effector cell-associated neurotoxicity syndrome (ICANS) and the accompanying organ damage have become important reasons that prevent the clinical application of CAR-T (69). Among them, CRS is often described as the most serious complication of CAR-T therapy and poses a great threat to life (70, 71). The clinical features of CRS are high fever, sinus tachycardia, hypotension, hypoxia, cardiac dysfunction and other organ dysfunction (22, 70, 72–77). It is generally believed that CRS is started by excessive secretion of proinflammatory cytokines, mainly IFN-γ and TNF-α, when CAR-T cells are cytotoxic to tumor cells. Inflammatory cytokines further activate the interaction of natural killer cells, endothelial cells, monocytes/macrophages and dendritic cells, and further increase the secretion of cytokines, including IL-6, IL-1, IL-5, IL-10, etc., through an ever-amplifying cascade, eventually resulting in a severe systemic inflammatory response (73). Available clinical trial data indicate that the first symptom of CRS is often fever (75, 78, 79), which usually occurs hours to days after CAR-T cell infusion. After first fever, patients often exhibit circulatory and respiratory disorders, such as sinus tachycardia, low blood pressure, and hypoxia. Subsequently, nephrotoxicity and hepatotoxicity are common manifestations of organ toxicity (70). Due to the rapid onset, for acute and severe CRS, medical staff may need to firstly give corresponding emergency strategies, such as vasopressors to relieve hypotension, rather than giving cytokine antagonists only (80). Hypoxia caused by capillary leak syndrome caused by CRS can be fatal in some cases. In critical cases, mechanical ventilation should be adopted decisively (81–83).
Compared with CRS, ICANS has a slightly better prognosis. In most cases, ICANS is completely reversible (84), but fatal cerebral edema and epilepsy still occur, suggesting that ICANS is still a serious complication of CAR-T therapy, which needs extensive attention (85, 86). The onset cycle of ICANS was variable, ranging from day 2 (77) to week 4 (87) after CAR-T infusion, suggesting that long-term, scheduled observation of neurotoxicity may be needed after CAR-T treatment. ICANS beyond the CNS, delirium, hallucinations, cognitive impairment, tremor, ataxia, speech disorders, nerve palsy, focal motor or sensory dysfunction, myoclonus, sleepiness, dull, or seizures (88, 89) were observed in clinical trials. There is now clear evidence that ICANS is closely related to systemic cytokine levels and CRS exacerbation. Among them, IFN-γ, IL-15, IL-6, IL-10, GM-CSF, IL-2, IL-2Rα, IL-1RA, and CXCL10 are considered to be associated with ICANS. However, the pathophysiological link remains unclear, as not all patients with severe CRS develop ICANS, and not all patients with ICANS experience CRS (84). Therefore, it is more accurate to describe ICANS as a process that is associated with the onset of CRS but operates independently of it (88). The present studies have pointed out that, in addition to clinical necessary symptomatic first aid measures, favorable effects have also been reported for blockers targeting cytokines. For example, IL-1 blockers can alleviate the occurrence of macrophage-mediated CRS and can reduce the severity of CAR-T cell-related neurotoxicity (90). Tocilizumab and/or single-dose corticosteroids have a good remission effect in CRS caused by CAR-T, but long-term use is prone to develop drug resistance (88). In addition to drug therapy to alleviate CRS, modifying the intracellular signaling domains of CAR-T cells can also achieve this goal. Studies have shown that the toxicity rate of CAR-T containing 4-1BB is higher than that of CD28-containing CAR-T (91).
Graft-versus-host disease (GVHD) is a severe immune response that occurs when donor CAR-T cells attack the recipient’s tissues, typically seen in allogeneic CAR-T cell therapies. This condition arises when T lymphocytes from the allogeneic donor graft react against the recipient’s antigens after transplantation, potentially leading to severe complications. To mitigate GVHD, strategies include using autologous CAR-T cells or modifying CAR constructs to reduce immune responses (92, 93).
Currently, the application of CAR-T cell therapy in T-ALL is in its early stages. Initial clinical trials and studies have shown promising early response rates, including complete remission in r/r T-ALL patients. However, long-term follow-up data remains limited, with most studies only covering periods of several months to a few years, making it premature to draw conclusions on long-term efficacy and safety. While some patients maintain prolonged disease-free survival after initial remission, relapse does occur, indicating the need for further research. Numerous clinical trials are ongoing globally, and as more patients are treated and followed up, more comprehensive long-term data will emerge. Thus, while early results are encouraging, more research and long-term data are necessary to make definitive conclusions about the long-term outcomes of CAR-T therapy in T-ALL (44, 94, 95).
5 Comparison of different CAR technologies in the treatment of T-ALL
CAR-T and CAR-NK therapies each have unique strengths and challenges, particularly in treating T-ALL. CAR-T therapies are celebrated for their potent antitumor efficacy and have demonstrated remarkable results in clinical settings. However, major clinical challenges such as CRS, neurotoxicity, GVHD and off-target effects limit their application and efficacy. The intricate individualized preparation process of CAR-T cells further complicates their widespread use (96–98).
CAR-NK cell therapy also presents unique advantages. CAR-NK cells can be derived from allogeneic sources and used as off-the-shelf therapeutics without the risk of inducing GVHD, offering a versatile and potentially safer treatment option. NK cells naturally possess an ability to eliminate tumor cells via diverse activating receptors, and when coupled with the specific tumor-killing effect mediated by CARs, this provides a dual mechanism for effective tumor therapy (99). Additionally, CAR-NK cells are associated with fewer and less severe instances of CRS due to their distinct activation pathways. The development of off-the-shelf CAR-NK cells also presents economic benefits, reducing the need for individual patient cell harvesting and customization (96–98). The diversity in the sources of NK cells—such as peripheral blood, umbilical cord blood, human embryonic stem cells (hESCs), induced pluripotent stem cells (iPSCs), and NK cell lines like NK-92—streamlines the preparation process and mitigates costs associated with therapy (100). However, the persistence and expansion of CAR-NK cells in vivo remain challenges that need to be addressed to enhance their therapeutic efficacy.
In conclusion, the ongoing comparative analysis and refinement of CAR-T and CAR-NK platforms are crucial for enhancing their therapeutic index. This involves advancing the design and functionality of CAR constructs and deepening our understanding of the tumor microenvironment to support the efficacy of these engineered cells. As the field progresses, integrating insights from clinical trials and translational research will be essential to optimize the use of CAR-based therapies in oncology, ensuring they deliver the most effective and safest treatments possible.
6 Conclusion and Outlook
This review has explored the pathogenesis and conventional treatment modalities of T-ALL, emphasizing the latest advancements in CAR technology for its management. While CAR-T cell therapy has demonstrated remarkable efficacy, its application is often limited by potential side effects. In contrast, CAR-NK cell therapy offers unique advantages, such as improved safety and the potential for off-the-shelf use. The continuous and rapid advancements in CAR-T and CAR-NK technology encompass both improvements in the technology itself and its combination with other approaches. Moreover, dual-antigen activated synNotch CARs have shown significant advantages in both solid tumors and hematological malignancies, offering new strategies for better tumor targeting and reducing off-target effects (101, 102).
In summary, the evolution of CAR technology instills renewed optimism for the precise treatment of T-ALL. While challenges persist, ongoing technological innovations and optimizations are expected to enhance the efficacy and safety of CAR-based immunotherapy for T-ALL patients. Future research should focus on refining CAR design, target selection, and managing associated side effects to maximize therapeutic benefits while minimizing adverse reactions. This concerted effort aims to establish CAR therapies as precise therapeutic tools with substantial promise for T-ALL management.
Author contributions
JJ: Writing – original draft, Writing – review & editing. YM: Writing – review & editing. ZX: Investigation, Writing – review & editing. BW: Data curation, Writing – review & editing. YC: Data curation, Writing – review & editing. EC: Writing – review & editing. JW: Writing – review & editing. KZ: Writing – review & editing. ZW: Funding acquisition, Supervision, Writing – review & editing. SL: Funding acquisition, Supervision, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Zhejiang Medical and Health Science and Technology Plan Project (No. 2022KY1051), the Scientific Research Foundation of Zhejiang University City College (J-202103) and Natural Science Foundation of Hubei Province of China (No: 2024AFB644).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. (2015) 373:1541–52. doi: 10.1056/NEJMra1400972
2. Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer. (2016) 16:494–507. doi: 10.1038/nrc.2016.63
3. Raetz EA, Teachey DT. T-cell acute lymphoblastic leukemia. Hematol Am Soc Hematol Educ Program. (2016) 2016:580–88. doi: 10.1182/asheducation-2016.1.580
4. Ekpa QL, Akahara PC, Anderson AM, Adekoya OO, Ajayi OO, Alabi PO, et al. A review of acute lymphocytic leukemia (ALL) in the pediatric population: evaluating current trends and changes in guidelines in the past decade. Cureus. (2023) 15:e49930. doi: 10.7759/cureus.49930
5. Kruse A, Abdel-Azim N, Kim HN, Ruan Y, Phan V, Ogana H, et al. Minimal residual disease detection in acute lymphoblastic leukemia. Int J Mol Sci. (2020) 21(3):1054. doi: 10.3390/ijms21031054
6. Singh N, Agrawal N, Sood R, Vishwakarma G, Kumar D, Dhanda S, et al. T-ALL minimal residual disease using a simplified gating strategy and its clinico-hematologic correlation: A single center experience from north India. Indian J Hematol Blood Transfus. (2019) 35:707–10. doi: 10.1007/s12288-019-01106-9
7. Maalej KM, Merhi M, Inchakalody VP, Mestiri S, Alam M, Maccalli C, et al. CAR-cell therapy in the era of solid tumor treatment: current challenges and emerging therapeutic advances. Mol Cancer. (2023) 22:20. doi: 10.1186/s12943-023-01723-z
8. Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, et al. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. (1991) 66:649–61. doi: 10.1016/0092-8674(91)90111-B
9. Sulis ML, Williams O, Palomero T, Tosello V, Pallikuppam S, Real PJ, et al. NOTCH1 extracellular juxtamembrane expansion mutations in T-ALL. Blood. (2008) 112:733–40. doi: 10.1182/blood-2007-12-130096
10. You MJ, Medeiros LJ, Hsi ED. T-lymphoblastic leukemia/lymphoma. Am J Clin Pathol. (2015) 144:411–22. doi: 10.1309/AJCPMF03LVSBLHPJ
11. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. (2021) 6:402. doi: 10.1038/s41392-021-00791-1
12. Zi FM, Ye LL, Zheng JF, Cheng J, Wang QM. Using JAK inhibitor to treat cytokine release syndrome developed after chimeric antigen receptor T cell therapy for patients with refractory acute lymphoblastic leukemia: A case report. Med (Baltimore). (2021) 100:e25786. doi: 10.1097/md.0000000000025786
13. Cheng Z, Yi Y, Xie S, Yu H, Peng H, Zhang G. The effect of the JAK2 inhibitor TG101209 against T cell acute lymphoblastic leukemia (T-ALL) is mediated by inhibition of JAK-STAT signaling and activation of the crosstalk between apoptosis and autophagy signaling. Oncotarget. (2017) 8:106753–63. doi: 10.18632/oncotarget.22053
14. Roberts KG, Gu Z, Payne-Turner D, McCastlain K, Harvey RC, Chen IM, et al. High frequency and poor outcome of philadelphia chromosome-like acute lymphoblastic leukemia in adults. J Clin Oncol. (2017) 35:394–401. doi: 10.1200/jco.2016.69.0073
15. Schmidt JA, Hornhardt S, Erdmann F, Sánchez-García I, Fischer U, Schüz J, et al. Risk factors for childhood leukemia: radiation and beyond. Front Public Health. (2021) 9:805757. doi: 10.3389/fpubh.2021.805757
16. Schrappe M, Valsecchi MG, Bartram CR, Schrauder A, Panzer-Grümayer R, Möricke A, et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood. (2011) 118:2077–84. doi: 10.1182/blood-2011-03-338707
17. Winter SS, Dunsmore KP, Devidas M, Wood BL, Esiashvili N, Chen Z, et al. Improved survival for children and young adults with T-lineage acute lymphoblastic leukemia: results from the children's oncology group AALL0434 methotrexate randomization. J Clin Oncol. (2018) 36:2926–34. doi: 10.1200/jco.2018.77.7250
18. Cornelissen JJ, van der Holt B, Verhoef GE, van't Veer MB, van Oers MH, Schouten HC, et al. Myeloablative allogeneic versus autologous stem cell transplantation in adult patients with acute lymphoblastic leukemia in first remission: a prospective sibling donor versus no-donor comparison. Blood. (2009) 113:1375–82. doi: 10.1182/blood-2008-07-168625
19. Goldstone AH, Richards SM, Lazarus HM, Tallman MS, Buck G, Fielding AK, et al. In adults with standard-risk acute lymphoblastic leukemia, the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients: final results of the International ALL Trial (MRC UKALL XII/ECOG E2993). Blood. (2008) 111:1827–33. doi: 10.1182/blood-2007-10-116582
20. Zhang X, Zhu L, Zhang H, Chen S, Xiao Y. CAR-T cell therapy in hematological Malignancies: current opportunities and challenges. Front Immunol. (2022) 13:927153. doi: 10.3389/fimmu.2022.927153
21. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. (2018) 359:1361–65. doi: 10.1126/science.aar6711
22. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
23. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discovery. (2013) 3:388–98. doi: 10.1158/2159-8290.Cd-12-0548
24. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. (2020) 17:147–67. doi: 10.1038/s41571-019-0297-y
25. Diwanji N, Getts D, Wang Y. Chimeric antigen cytotoxic receptors for in vivo engineering of tumor-targeting NK cells. ImmunoHorizons. (2024) 8:97–105. doi: 10.4049/immunohorizons.2300099
26. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
27. Zhang H, Li F, Cao J, Wang X, Cheng H, Qi K, et al. A chimeric antigen receptor with antigen-independent OX40 signaling mediates potent antitumor activity. Sci Transl Med. (2021) 13(578):eaba7308. doi: 10.1126/scitranslmed.aba7308
28. Hombach AA, Rappl G, Abken H. Arming cytokine-induced killer cells with chimeric antigen receptors: CD28 outperforms combined CD28–OX40 “Super-stimulation. Mol Ther. (2013) 21:2268–77. doi: 10.1038/mt.2013.192
29. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) 11:69. doi: 10.1038/s41408-021-00459-7
30. Jogalekar MP, Rajendran RL, Khan F, Dmello C, Gangadaran P, Ahn BC. CAR T-Cell-Based gene therapy for cancers: new perspectives, challenges, and clinical developments. Front Immunol. (2022) 13:925985. doi: 10.3389/fimmu.2022.925985
31. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. 'Off-the-shelf' allogeneic CAR T cells: development and challenges. Nat Rev Drug Discovery. (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
32. Pui CH, Behm FG, Crist WM. Clinical and biologic relevance of immunologic marker studies in childhood acute lymphoblastic leukemia. Blood. (1993) 82:343–62. doi: 10.1182/blood.V82.2.343.343
33. Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell Malignancies. Blood. (2015) 126:983–92. doi: 10.1182/blood-2015-02-629527
34. Dai Z, Mu W, Zhao Y, Jia X, Liu J, Wei Q, et al. The rational development of CD5-targeting biepitopic CARs with fully human heavy-chain-only antigen recognition domains. Mol Ther. (2021) 29:2707–22. doi: 10.1016/j.ymthe.2021.07.001
35. Wu H, Yu Y, Zhao Y, Liu W, Liu Z, Zhang G, et al. A C(H)2C(H)3 hinge region enhances the cytotoxicity of anti-CD5 CAR-T cells targeting T cell acute lymphoblastic leukemia. Int Immunopharmacol. (2023) 124:110904. doi: 10.1016/j.intimp.2023.110904
36. Hill LC, Rouce RH, Wu MJ, Wang T, Ma R, Zhang H, et al. Antitumor efficacy and safety of unedited autologous CD5.CAR T cells in relapsed/refractory mature T-cell lymphomas. Blood. (2024) 143:1231–41. doi: 10.1182/blood.2023022204
37. Bonilla FA, Kokron CM, Swinton P, Geha RS. Targeted gene disruption of murine CD7. Int Immunol. (1997) 9:1875–83. doi: 10.1093/intimm/9.12.1875
38. Kim MY, Cooper ML, Jacobs MT, Ritchey JK, Hollaway J, Fehniger TA, et al. CD7-deleted hematopoietic stem cells can restore immunity after CAR T cell therapy. JCI Insight. (2021) 6(16):e149819. doi: 10.1172/jci.insight.149819
39. Lee DM, Staats HF, Sundy JS, Patel DD, Sempowski GD, Scearce RM, et al. Immunologic characterization of CD7-deficient mice. J Immunol. (1998) 160:5749–56. doi: 10.4049/jimmunol.160.12.5749
40. Rabinowich H, Pricop L, Herberman RB, Whiteside TL. Expression and function of CD7 molecule on human natural killer cells. J Immunol. (1994) 152:517–26. doi: 10.4049/jimmunol.152.2.517
41. Wang S, Zhang R, Zhong K, Guo W, Tong A. An anti-CD7 antibody-drug conjugate target showing potent antitumor activity for T-lymphoblastic leukemia (T-ALL). Biomolecules. (2024) 14(1):106. doi: 10.3390/biom14010106
42. Gomes-Silva D, Srinivasan M, Sharma S, Lee CM, Wagner DL, Davis TH, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell Malignancies. Blood. (2017) 130:285–96. doi: 10.1182/blood-2017-01-761320
43. Jiang J, Chen J, Liao C, Duan Y, Wang Y, Shang K, et al. Inserting EF1α-driven CD7-specific CAR at CD7 locus reduces fratricide and enhances tumor rejection. Leukemia. (2023) 37:1660–70. doi: 10.1038/s41375-023-01948-3
44. Lu P, Liu Y, Yang J, Zhang X, Yang X, Wang H, et al. Naturally selected CD7 CAR-T therapy without genetic manipulations for T-ALL/LBL: first-in-human phase 1 clinical trial. Blood. (2022) 140:321–34. doi: 10.1182/blood.2021014498
45. Zhang M, Chen D, Fu X, Meng H, Nan F, Sun Z, et al. Autologous nanobody-derived fratricide-resistant CD7-CAR T-cell therapy for patients with relapsed and refractory T-cell acute lymphoblastic leukemia/lymphoma. Clin Cancer Res. (2022) 28:2830–43. doi: 10.1158/1078-0432.Ccr-21-4097
46. Png YT, Vinanica N, Kamiya T, Shimasaki N, Coustan-Smith E, Campana D. Blockade of CD7 expression in T cells for effective chimeric antigen receptor targeting of T-cell Malignancies. Blood Adv. (2017) 1:2348–60. doi: 10.1182/bloodadvances.2017009928
47. Wurbel MA, Malissen M, Guy-Grand D, Meffre E, Nussenzweig MC, Richelme M, et al. Mice lacking the CCR9 CC-chemokine receptor show a mild impairment of early T- and B-cell development and a reduction in T-cell receptor gammadelta(+) gut intraepithelial lymphocytes. Blood. (2001) 98:2626–32. doi: 10.1182/blood.v98.9.2626
48. Maciocia PM, Wawrzyniecka PA, Maciocia NC, Burley A, Karpanasamy T, Devereaux S, et al. Anti-CCR9 chimeric antigen receptor T cells for T-cell acute lymphoblastic leukemia. Blood. (2022) 140:25–37. doi: 10.1182/blood.2021013648
49. Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y, et al. CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discovery. (2018) 8:1156–75. doi: 10.1158/2159-8290.Cd-17-1033
50. van de Donk N, Usmani SZ. CD38 antibodies in multiple myeloma: mechanisms of action and modes of resistance. Front Immunol. (2018) 9:2134. doi: 10.3389/fimmu.2018.02134
51. Laubach JP, Tai YT, Richardson PG, Anderson KC. Daratumumab granted breakthrough drug status. Expert Opin Investig Drugs. (2014) 23:445–52. doi: 10.1517/13543784.2014.889681
52. Wang X, Yu X, Li W, Neeli P, Liu M, Li L, et al. Expanding anti-CD38 immunotherapy for lymphoid Malignancies. J Exp Clin Cancer Res. (2022) 41:210. doi: 10.1186/s13046-022-02421-2
53. Glisovic-Aplenc T, Diorio C, Chukinas JA, Veliz K, Shestova O, Shen F, et al. CD38 as a pan-hematologic target for chimeric antigen receptor T cells. Blood Adv. (2023) 7:4418–30. doi: 10.1182/bloodadvances.2022007059
54. Liao C, Wang Y, Huang Y, Duan Y, Liang Y, Chen J, et al. CD38-specific CAR integrated into CD38 locus driven by different promoters causes distinct antitumor activities of T and NK cells. Adv Sci (Weinh). (2023) 10:e2207394. doi: 10.1002/advs.202207394
55. Chancellor A, Gadola SD, Mansour S. The versatility of the CD1 lipid antigen presentation pathway. Immunology. (2018) 154:196–203. doi: 10.1111/imm.12912
56. Niehues T, Kapaun P, Harms DO, Burdach S, Kramm C, Körholz D, et al. A classification based on T cell selection-related phenotypes identifies a subgroup of childhood T-ALL with favorable outcome in the COALL studies. Leukemia. (1999) 13:614–7. doi: 10.1038/sj.leu.2401382
57. van Grotel M, Meijerink JP, van Wering ER, Langerak AW, Beverloo HB, Buijs-Gladdines JG, et al. Prognostic significance of molecular-cytogenetic abnormalities in pediatric T-ALL is not explained by immunophenotypic differences. Leukemia. (2008) 22:124–31. doi: 10.1038/sj.leu.2404957
58. Sánchez-Martínez D, Baroni ML, Gutierrez-Agüera F, Roca-Ho H, Blanch-Lombarte O, González-García S, et al. Fratricide-resistant CD1a-specific CAR T cells for the treatment of cortical T-cell acute lymphoblastic leukemia. Blood. (2019) 133:2291–304. doi: 10.1182/blood-2018-10-882944
59. Maciocia PM, Wawrzyniecka PA, Philip B, Ricciardelli I, Akarca AU, Onuoha SC, et al. Targeting the T cell receptor β-chain constant region for immunotherapy of T cell Malignancies. Nat Med. (2017) 23:1416–23. doi: 10.1038/nm.4444
60. Zhang C, Palashati H, Rong Z, Lin N, Shen L, Liu Y, et al. Pre-depletion of TRBC1+ T cells promotes the therapeutic efficacy of anti-TRBC1 CAR-T for T-cell Malignancies. Mol Cancer. (2020) 19:162. doi: 10.1186/s12943-020-01282-7
61. Rasaiyaah J, Georgiadis C, Preece R, Mock U, Qasim W. TCRαβ/CD3 disruption enables CD3-specific antileukemic T cell immunotherapy. JCI Insight. (2018) 3(13):e99442. doi: 10.1172/jci.insight.99442
62. Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer. (1975) 16:230–9. doi: 10.1002/ijc.2910160205
63. Clara JA, Levy ER, Reger R, Barisic S, Chen L, Cherkasova E, et al. High-affinity CD16 integration into a CRISPR/Cas9-edited CD38 locus augments CD38-directed antitumor activity of primary human natural killer cells. J Immunother Cancer. (2022) 10(2):e003804. doi: 10.1136/jitc-2021-003804
64. Xu Y, Liu Q, Zhong M, Wang Z, Chen Z, Zhang Y, et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell Malignancies. J Hematol Oncol. (2019) 12:49. doi: 10.1186/s13045-019-0732-7
65. Sgro C. Side-effects of a monoclonal antibody, muromonab CD3/orthoclone OKT3: bibliographic review. Toxicology. (1995) 105:23–9. doi: 10.1016/0300-483X(95)03123-W
66. Basar R, Daher MDM, Uprety N, Ensley E, Nunez Cortes AK, Acharya S, et al. DAP10 co-stimulation imparts memory-like features to CD5 targeting cord blood derived CAR-NK cells. Blood. (2023) 142:2089–89. doi: 10.1182/blood-2023-187665
67. Voynova E, Hawk N, Flomerfelt FA, Telford WG, Gress RE, Kanakry JA, et al. Increased activity of a NK-specific CAR-NK framework targeting CD3 and CD5 for T-cell leukemias. Cancers (Basel). (2022) 14(3):524. doi: 10.3390/cancers14030524
68. Zhu XY, Liu X, Wang XB, Wang AY, Wang M, Liu NN, et al. [Killing effect of A CD7 chimeric antigen receptor-modified NK-92MI cell line on CD7-positive hematological Malignant cells]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. (2020) 28:1367–75. doi: 10.19746/j.cnki.issn.1009-2137.2020.04.049
69. Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer. (2018) 6:56. doi: 10.1186/s40425-018-0343-9
70. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. (2015) 385:517–28. doi: 10.1016/s0140-6736(14)61403-3
71. Neelapu SS. Managing the toxicities of CAR T-cell therapy. Hematol Oncol. (2019) 37 Suppl 1:48–52. doi: 10.1002/hon.2595
72. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. (2016) 127:3321–30. doi: 10.1182/blood-2016-04-703751
73. Cosenza M, Sacchi S, Pozzi S. Cytokine release syndrome associated with T-cell-based therapies for hematological Malignancies: pathophysiology, clinical presentation, and treatment. Int J Mol Sci. (2021) 22(14):7652. doi: 10.3390/ijms22147652
74. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
75. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
76. Shah D, Soper B, Shopland L. Cytokine release syndrome and cancer immunotherapies - historical challenges and promising futures. Front Immunol. (2023) 14:1190379. doi: 10.3389/fimmu.2023.1190379
77. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/jci85309
78. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. (2014) 124:188–95. doi: 10.1182/blood-2014-05-552729
79. Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. (2016) 8:355ra116. doi: 10.1126/scitranslmed.aaf8621
80. Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discovery. (2016) 6:664–79. doi: 10.1158/2159-8290.Cd-16-0040
81. Kochenderfer JN, Somerville RPT, Lu T, Shi V, Bot A, Rossi J, et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J Clin Oncol. (2017) 35:1803–13. doi: 10.1200/jco.2016.71.3024
82. Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 results of ZUMA-1: A multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. (2017) 25:285–95. doi: 10.1016/j.ymthe.2016.10.020
83. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
84. Gust J, Ponce R, Liles WC, Garden GA, Turtle CJ. Cytokines in CAR T cell-associated neurotoxicity. Front Immunol. (2020) 11:577027. doi: 10.3389/fimmu.2020.577027
85. Grupp S, Hu Z-H, Zhang Y, Keating A, Pulsipher MA, Philips C, et al. Tisagenlecleucel chimeric antigen receptor (CAR) T-cell therapy for relapsed/refractory children and young adults with acute lymphoblastic leukemia (ALL): real world experience from the center for international blood and marrow transplant research (CIBMTR) and cellular therapy (CT) registry. Blood. (2019) 134:2619–19. doi: 10.1182/blood-2019-129279
86. Pasquini MC, Locke FL, Herrera AF, Siddiqi T, Ghobadi A, Komanduri KV, et al. Post-marketing use outcomes of an anti-CD19 chimeric antigen receptor (CAR) T cell therapy, axicabtagene ciloleucel (Axi-cel), for the treatment of large B cell lymphoma (LBCL) in the United States (US). Blood. (2019) 134:764–64. doi: 10.1182/blood-2019-124750
87. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. (2018) 15:47–62. doi: 10.1038/nrclinonc.2017.148
88. Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. (2019) 34:45–55. doi: 10.1016/j.blre.2018.11.002
89. Schubert ML, Schmitt M, Wang L, Ramos CA, Jordan K, Müller-Tidow C, et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol. (2021) 32:34–48. doi: 10.1016/j.annonc.2020.10.478
90. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. (2018) 24:731–38. doi: 10.1038/s41591-018-0041-7
91. Li S, Zhang J, Wang M, Fu G, Li Y, Pei L, et al. Treatment of acute lymphoblastic leukaemia with the second generation of CD19 CAR-T containing either CD28 or 4-1BB. Br J Haematol. (2018) 181:360–71. doi: 10.1111/bjh.15195
92. Broers AEC, de Jong CN, Bakunina K, Hazenberg MD, van Marwijk Kooy M, de Groot MR, et al. Posttransplant cyclophosphamide for prevention of graft-versus-host disease: results of the prospective randomized HOVON-96 trial. Blood Adv. (2022) 6:3378–85. doi: 10.1182/bloodadvances.2021005847
93. Shiratori S, Sugita J, Ota S, Kasahara S, Ishikawa J, Tachibana T, et al. Low-dose anti-thymocyte globulin for GVHD prophylaxis in HLA-matched allogeneic peripheral blood stem cell transplantation. Bone Marrow Transplant. (2021) 56:129–36. doi: 10.1038/s41409-020-0985-3
94. Hu Y, Li J, Ni F, Yang Z, Gui X, Bao Z, et al. CAR-T cell therapy-related cytokine release syndrome and therapeutic response is modulated by the gut microbiome in hematologic Malignancies. Nat Commun. (2022) 13:5313. doi: 10.1038/s41467-022-32960-3
95. Hu Y, Zhou Y, Zhang M, Zhao H, Wei G, Ge W, et al. Genetically modified CD7-targeting allogeneic CAR-T cell therapy with enhanced efficacy for relapsed/refractory CD7-positive hematological Malignancies: a phase I clinical study. Cell Res. (2022) 32:995–1007. doi: 10.1038/s41422-022-00721-y
96. Hu Y, Tian ZG, Zhang C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol Sin. (2018) 39:167–76. doi: 10.1038/aps.2017.125
97. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. (2018) 23:181–92.e5. doi: 10.1016/j.stem.2018.06.002
98. Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol Ther. (2017) 25:1769–81. doi: 10.1016/j.ymthe.2017.06.012
99. Mehta RS, Rezvani K. Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer. Front Immunol. (2018) 9:283. doi: 10.3389/fimmu.2018.00283
100. Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine. (2020) 59:102975. doi: 10.1016/j.ebiom.2020.102975
101. Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. (2021) 13(591):eabe7378. doi: 10.1126/scitranslmed.abe7378
Keywords: T-ALL, pathogenesis, chimeric antigen receptor, CAR-T, CAR-NK
Citation: Jing J, Ma Y, Xie Z, Wang B, Chen Y, Chi E, Wang J, Zhang K, Wang Z and Li S (2024) Acute T-cell lymphoblastic leukemia: chimeric antigen receptor technology may offer a new hope. Front. Immunol. 15:1410519. doi: 10.3389/fimmu.2024.1410519
Received: 01 April 2024; Accepted: 15 July 2024;
Published: 13 August 2024.
Edited by:
Luca Lo Nigro, Azienda Ospedaliero Universitaria Policlinico - San Marco, ItalyReviewed by:
Sarah Tettamanti, Fondazione Matilde Tettamanti Menotti De Marchi, ItalyThomas Stiehl, RWTH Aachen University, Germany
Copyright © 2024 Jing, Ma, Xie, Wang, Chen, Chi, Wang, Zhang, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sisi Li, bGlzaXNpQGh6Y3UuZWR1LmNu; Zhujun Wang, d2FuZ3podWp1bjIwMDlAMTI2LmNvbQ==
†These authors share first authorship