 Ying Li1†
Ying Li1† Zhenhua Hu
Zhenhua Hu Xiaoyan Wu
Xiaoyan Wu- 1Department of Pediatrics, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Department of Health and Nursing, Nanfang College of Sun Yat-sen University, Guangzhou, China
- 3Zhongshan Institute for Drug Discovery, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Zhongshan, China
- 4Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China
- 5University of Chinese Academy of Sciences, Beijing, China
Chimeric antigen receptor-T (CAR-T) cell therapy has made remarkable strides in treating hematological malignancies. However, the widespread adoption of CAR-T cell therapy is hindered by several challenges. These include concerns about the long-term and complex manufacturing process, as well as efficacy factors such as tumor antigen escape, CAR-T cell exhaustion, and the immunosuppressive tumor microenvironment. Additionally, safety issues like the risk of secondary cancers post-treatment, on-target off-tumor toxicity, and immune effector responses triggered by CAR-T cells are significant considerations. To address these obstacles, researchers have explored various strategies, including allogeneic universal CAR-T cell development, infusion of non-activated quiescent T cells within a 24-hour period, and in vivo induction of CAR-T cells. This review comprehensively examines the clinical challenges of CAR-T cell therapy and outlines strategies to overcome them, aiming to chart pathways beyond its current Achilles heels.
1 Introduction
In recent years, chimeric antigen receptor-T (CAR-T) cell therapy has emerged as a pivotal immunotherapeutic approach, profoundly reshaping the treatment landscape of hematological malignancies. Engineered synthetic receptors, CAR-T cells empower T cells to selectively recognize and eliminate tumor cells independent of the major histocompatibility complex (MHC) (1, 2). Since the first FDA approval of a CAR-T cell product in 2017 (3), this therapy has witnessed rapid expansion in hematologic malignancies. As of now, six CAR-T cell products have received FDA approval, achieving remarkable complete remission (CR) rates of up to 80% in certain relapsed or refractory (R/R) B-cell malignancies (Table 1) (14, 15), while hundreds of CAR-T cell therapies are currently undergoing clinical trials (16, 17). Additionally, CAR-T cell therapy has shown notable success in treating autoimmune diseases, offering a 100% drug-free alternative to systemic lupus erythematosus in clinical trials (18, 19). The high remission rates and broad application across various diseases underscore the significance of CAR-T cell therapy as a pivotal tool in combating disease and reducing mortality rates (20, 21).
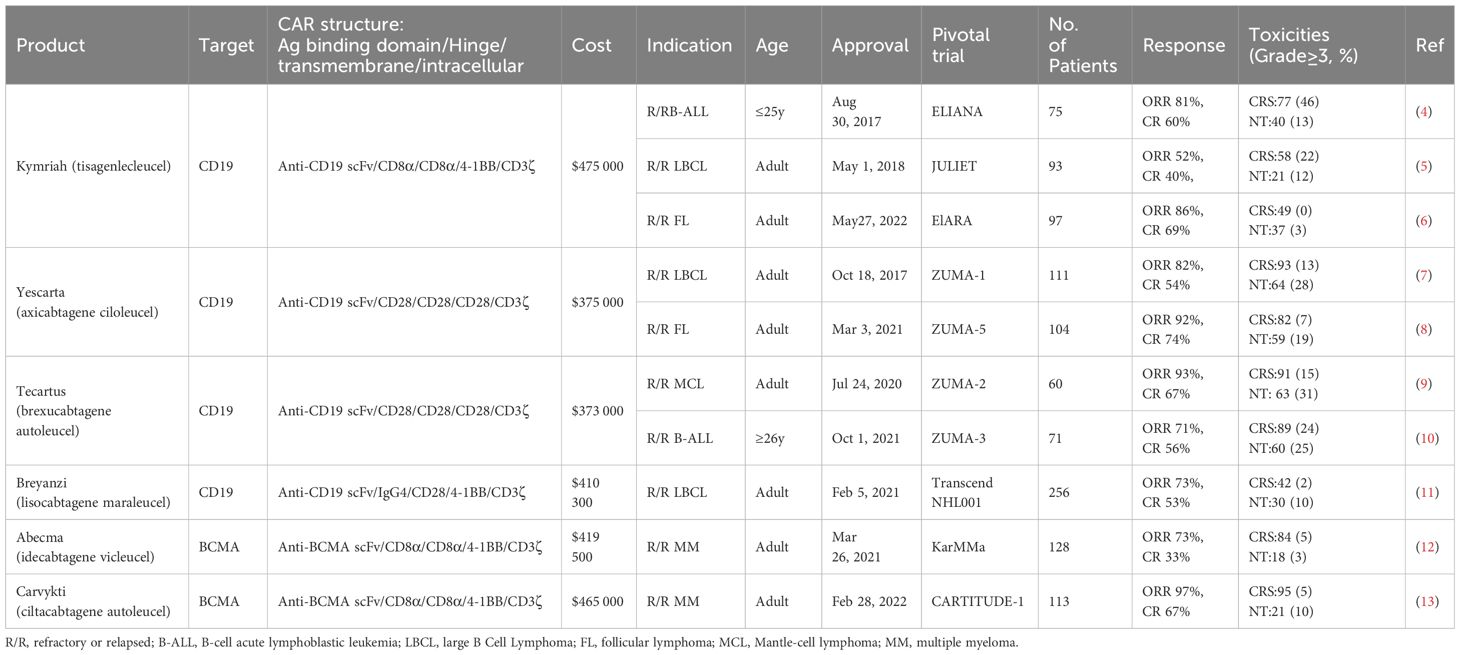
Table 1 FDA-approved CAR T-cell products.
However, despite significant achievements, the widespread application of CAR-T cell therapy encounters numerous challenges. Firstly, the individualized customization and labor-intensive manufacturing process of CAR-T cells result in high costs and prolonged production cycles, limiting patient affordability and treatment accessibility (22–24). Moreover, efficacy concerns such as tumor antigen modulation, CAR-T cell persistence, and the immunosuppressive tumor microenvironment (TME) contribute to initial resistance or relapse in some patients (25–27). Additionally, safety issues including the risk of secondary cancers post-treatment, on-target off-tumor toxicity, and immune effector responses triggered by CAR-T cell activation further impede broad adoption (28–30). Specifically, the FDA requires the addition of warning information regarding the risk of secondary cancers post-treatment to the label of CAR-T cell products, introducing a new Achilles’ heel to CAR-T cell therapy.
To tackle these challenges, various strategies have been explored. These include constructing allogeneic CAR-T cells with enhanced potency and safety through leveraging the multiple gene editing functions of CRISPR-Cas9 and base editing (31, 32), in vivo induction of CAR-T cells by nanocarriers and optimized lentiviral vectors (33, 34), and the rapid production of potent CAR-T cells employing the FasTCAR platform or the MASTER scaffolds (35, 36). These efforts aim to provide more economically viable, efficacious, and secure therapeutic alternatives. In this review, we comprehensively scrutinize the clinical challenges associated with CAR-T cell therapy and provide an overview of potential strategies to overcome these obstacles. Our aim is to chart pathways for CAR-T cell therapy to navigate beyond its current limitations.
2 Achilles heels of CAR T-cell therapy
As depicted in Figure 1, the Achilles’ heels of CAR T-cell therapy, which currently hinder its wider efficacy and acceptance in clinical practice, encompass resistance to CAR-T cell therapy, safety concerns, and manufacturing intricacies (26, 37, 38). One major issue is tumor antigen escape, where cancer cells mutate or downregulate the target antigen recognized by CAR-T cells, leading to treatment resistance and disease relapse (37). Additionally, CAR-T cell exhaustion, characterized by decreased efficacy and persistence of infused cells over time, poses a significant challenge to long-term therapeutic success (39). On-target off-tumor toxicity is another concern, as CAR-T cells may inadvertently target healthy tissues expressing the target antigen, leading to adverse effects (29). Furthermore, the development of secondary T-cell malignancies following CAR-T cell treatment, though rare, underscores the need for continued vigilance regarding long-term safety outcomes (40). The toxicities associated with CRS and ICANS further complicate CAR-T therapy, requiring careful management to mitigate potentially life-threatening complications (41). Moreover, the complex and lengthy manufacturing processes involved in producing personalized CAR-T cell products limit their scalability and accessibility to patients (42). Addressing these Achilles’ heels is crucial for enhancing the overall efficacy, safety, and feasibility of CAR T-cell therapy in clinical settings.
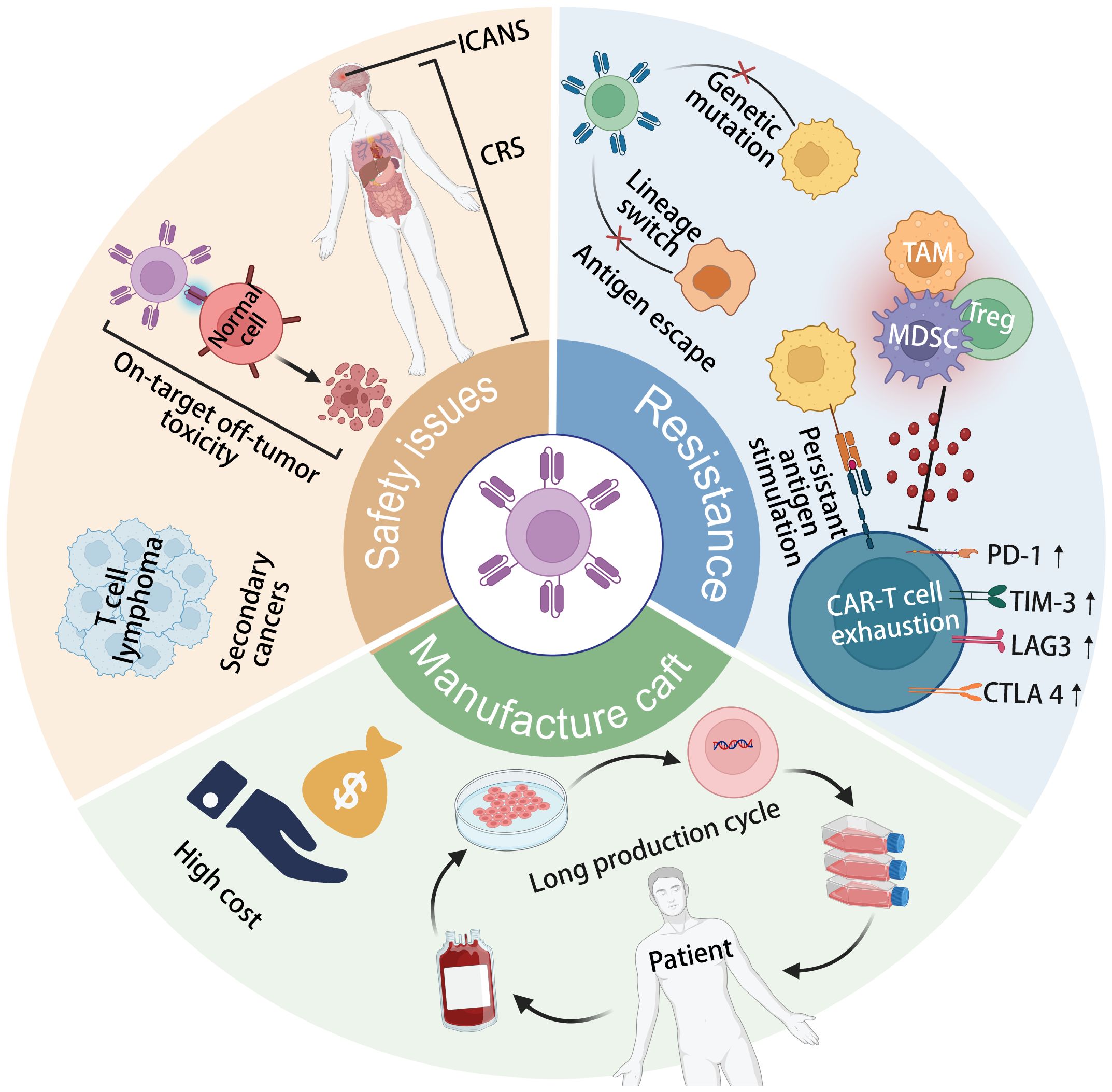
Figure 1 The current limitations of CAR-T cell therapy include tumor antigen escape, CAR-T cell exhaustion, secondary T-cell malignancies following treatment, cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) toxicity, on-target off-tumor toxicity, and complex, long-term manufacturing processes.
2.1 Tumor antigen escape
While CAR-T cell therapy has demonstrated unprecedented response rates, not all patients benefit from it, and a significant percentage experience relapses (43). Antigen escape stands out as the most common cause of relapse in CD19-positive B-cell malignancies following CAR-T cell therapy. Published data indicate that CD19-negative relapse occurs in 7-25% of B-cell acute lymphoblastic leukemia (B-ALL) cases and approximately 30% of large B-cell lymphoma (LBCL) cases in patients treated with CD19 CAR therapy (4, 44–46). Mechanisms underlying CD19 loss have been extensively investigated, including CD19 gene mutations or splice variants, abnormal processing or trafficking of CD19 due to CD81 expression deficiency, and lineage marker switching from the lymphoid to the myeloid lineage (46, 47). Zah et al. developed a CD19-CD20 CAR and demonstrated its efficacy in preventing the spontaneous emergence of CD19-negative tumor cell variants in immune-deficient mice. In contrast to CD19, loss of B cell maturation antigen (BCMA) appears to be infrequent following anti-BCMA CAR-T cell therapy and has only been reported in a few studies (48). For instance, only 4% (3 out of 71) of patients in a phase II clinical trial using Idecabtagene Vicleucel for multiple myeloma (MM) treatment developed BCMA expression loss (12). Deletion and mutation of the biallelic TNFRSF17 gene encoding BCMA have been identified as the main mechanisms causing BCMA loss (49–51). To mitigate the risk of relapse and failed responses attributed to tumor antigen escape, it may be necessary to conduct more precise screening of the patient’s genetic spectrum before initiating a new CAR-T cell therapy (52).
2.2 CAR-T cell exhaustion
CAR-T cell exhaustion, a significant factor contributing to CAR-T cell resistance, often leads to antigen-positive relapse and results from various factors. Firstly, the differentiation status of T cells is crucial for maintaining the functional persistence of CAR-T cells. Previous studies have shown that less differentiated T cells have greater expansion potential and prolonged persistence compared to fully differentiated effector T cells (53). Most current CAR-T cell products are autologous, and due to factors, such as the presence of tumors, prior cytotoxic treatments, and prolonged ex vivo cultivation, these cells often exhibit an exhausted phenotype characterized by excessive differentiation (54, 55). Additionally, immunosuppressive components of the tumor immune microenvironment, including regulatory T cells, myeloid-derived suppressor cells, tumor-associated macrophages (TAMs), and immunosuppressive ligands, also contribute to CAR-T cell exhaustion (25, 56, 57). Furthermore, tonic CAR signaling transduction, mainly associated with the costimulatory domain, plays a key role in CAR-T cell exhaustion. Insufficient signaling compromises cell persistence, while excessive signaling leads to exhaustion (58). For example, compared to 4-1BB, CD28 CAR-T cells demonstrate faster and stronger cellular effector functions, but this rapid and intense signal transduction can induce CAR-T cell exhaustion, thereby limiting persistence (59–61). Achieving potent and sustained activity against specific tumor targets requires careful selection of optimal CAR designs. Recent studies have underscored the importance of positively charged plaques (PCPs) on CAR in mediating CAR aggregation at the antigen-binding domain surface, thereby facilitating sustained CAR signaling (62, 63). Regulating PCPs offers a means to optimize CAR-T cell function. For CARs with high sustained signaling, such as GD2.CAR and CSPG4.CAR, reducing PCPs during in vitro expansion or enhancing ionic strength in culture can diminish spontaneous CAR activation and mitigate CAR-T cell exhaustion. Conversely, for CARs with weak tonic signaling like CD19.CAR, augmenting PCPs on the CAR surface can enhance in vivo persistence and anti-tumor efficacy (62). Recently, the same team developed CAR-Toner, an artificial intelligence (AI)-based PCP score calculator and optimizer (64). Taking the camel single-domain nanobody (VHH) targeting the acute myeloid leukemia (AML) tumor-associated antigen CLL1 as an example, the authors optimized the CAR design using CAR-Toner to systematically reduce their PCP scores. The results showed that an intermediate tonic signaling strength optimally benefits CAR-T cell function. As an AI-based tool, CAR-Toner is capable of not only conducting PCP calculations but also offering optimization recommendations for PCP scores. This groundbreaking tool is anticipated to catalyze progress in the field of CAR-T design and significantly contribute to the advancement of AI-driven CAR-T design.
Various strategies have emerged to combat CAR-T cell exhaustion, such as blocking exhaustion-promoting signaling pathways, inhibiting downstream effectors, alleviating immunosuppression within the tumor microenvironment (TME), and converting inhibitory signals into stimulatory ones (52, 61). When coupled with AI-based tools, these approaches facilitate the development of more potent CAR designs, thus augmenting the efficacy of CAR-T cell therapy (65).
2.3 Secondary T-cell malignancies following treatment
Recent evidence indicates a concerning association between CAR-T cell therapy and the development of secondary T-cell malignancies, prompting regulatory measures by the FDA to enhance safety oversight (66, 67). By the close of 2023, 22 reported cases have documented the emergence of T-cell cancers subsequent to CAR-T treatment. Notably, in three instances, the CAR transgene was identified within the malignant clone, strongly implicating the therapy in the genesis of T-cell cancer (28). Presently, CAR-T products approved by regulatory agencies employ T cells engineered via viral transduction to convey the genetic construct. However, the use of current retroviral vectors still harbors potential risks of oncogenesis through genomic integration or related mechanisms. For example, lentiviral vector constructs, while integrating into the genome in a semi-random manner, display an affinity for genomic regions characterized by active gene expression, thereby heightening the risk of insertional oncogenesis (68). To mitigate these risks in the future, strategies such as precision targeting of CAR construct insertion to specific genomic loci or the utilization of transient CAR mRNA delivery to the cytoplasm offer promising avenues for enhancing safety in CAR-T therapy (68).
2.4 CRS and ICANS toxicity trigger by CAR-T cells
While CAR-T cell therapy holds tremendous promise in the therapeutic realm of hematologic malignancies, the associated potential life-threatening toxicities remain a significant concern. Cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) emerge as the two most common adverse events during CAR-T cell therapy (Table 1), attributed to the overactivation of CAR-T cells and the massive release of cytokines (69–71). CRS typically manifests a few days after the initial infusion, with symptoms ranging from mild flu-like manifestations such as fever, fatigue, chills, and muscle pain to severe life-threatening complications including shock, hypotension, coagulation abnormalities, and multi-organ dysfunction (72, 73). On the other hand, ICANS typically occurs 1-3 weeks post-CAR-T cell infusion, characterized by symptoms such as aphasia, delirium, focal neurological deficits, tremors, seizures, or life-threatening cerebral edema (74, 75). The severity of CRS and ICANS is categorized from grade I to grade IV, depending on factors such as CAR structure, CAR-T cell dosage, tumor burden, treatment targets, and patients’ individual characteristics. Most patients receiving CAR-T cell therapy are likely to experience varying degrees of CRS, with nearly half developing ICANS (Table 1). Among these patients, 10%-40% may experience severe toxicity reactions (≥ grade 3), necessitating admission to the intensive care unit (ICU) for life support and the use of additional medications such as the interleukin (IL)-6 receptor antagonist tocilizumab (76–78). Blocking cytokine networks or optimizing the structural design of CARs to reduce toxicity are potential strategies to mitigate CRS and ICANS triggered by CAR-T cells (79, 80).
2.5 On-target off-tumor toxicity
Given that most CAR-T cell target antigens are not exclusively tumor-specific and are also expressed in normal cells, CAR-T cells may inadvertently cause damage to normal tissue and organs while targeting tumors (81). Long-term follow-up data indicate that on-target off-tumor toxicity, leading to B-cell aplasia and hypogammaglobulinemia, are the most common long-term adverse reactions following treatment with anti-CD19 CAR-T cells (14). Although these side effects can be managed through sequential intravenous immunoglobulin, the long-term repeated infusions may escalate treatment costs. Additionally, cytopenia, infections, and tumor lysis syndrome are also common side effects of CAR-T cell therapy for hematologic malignancies. While proper management and intervention can control these toxicities, the resulting disease and economic burden are significant factors limiting the widespread adoption of CAR-T cell therapy (77, 82). Additionally, multiple-targeting CAR-T cells are designed to mitigate the impact of on-target off-tumor effects. Intensive research has centered on developing diverse protein-based logic-circuit strategies to enhance the specificity of CAR T cell activation and cytotoxicity towards tumor cells (83–85). Additionally, locoregional administration of CAR T cells, aimed at concentrating antitumor activity within the tumor microenvironment, may offer a potential solution to mitigate off-tumor toxicity (86–88).
2.6 Complexed and long-term manufacturing process
A major challenge of CAR-T cell therapy is its high cost and lengthy manufacturing process. Most FDA-approved and clinical trial CAR-T cell products are autologous, requiring personalized customization involving complex processes like T cell isolation, activation, CAR gene transduction, expansion, and reinfusion (89). This labor-intensive process occurs in specialized facilities, adding to expenses and time. Viral vector preparation, crucial for CAR transfection, also contributes to costs and production delays (90). Production of viral vectors requires at least 2 weeks, meeting cGMP standards and extensive safety testing (91–93). The cost of a single dose of approved CAR-T cell products ranges from $373,000 to $475,000 (Table 1), with a production cycle of 2 to 4 weeks (Figure 2), posing economic burdens and logistical challenges for patients (94). With advancements in related technologies, it is imperative to establish increasingly standardized and simplified manufacturing processes to mitigate the costs associated with CAR-T therapy (22, 95).
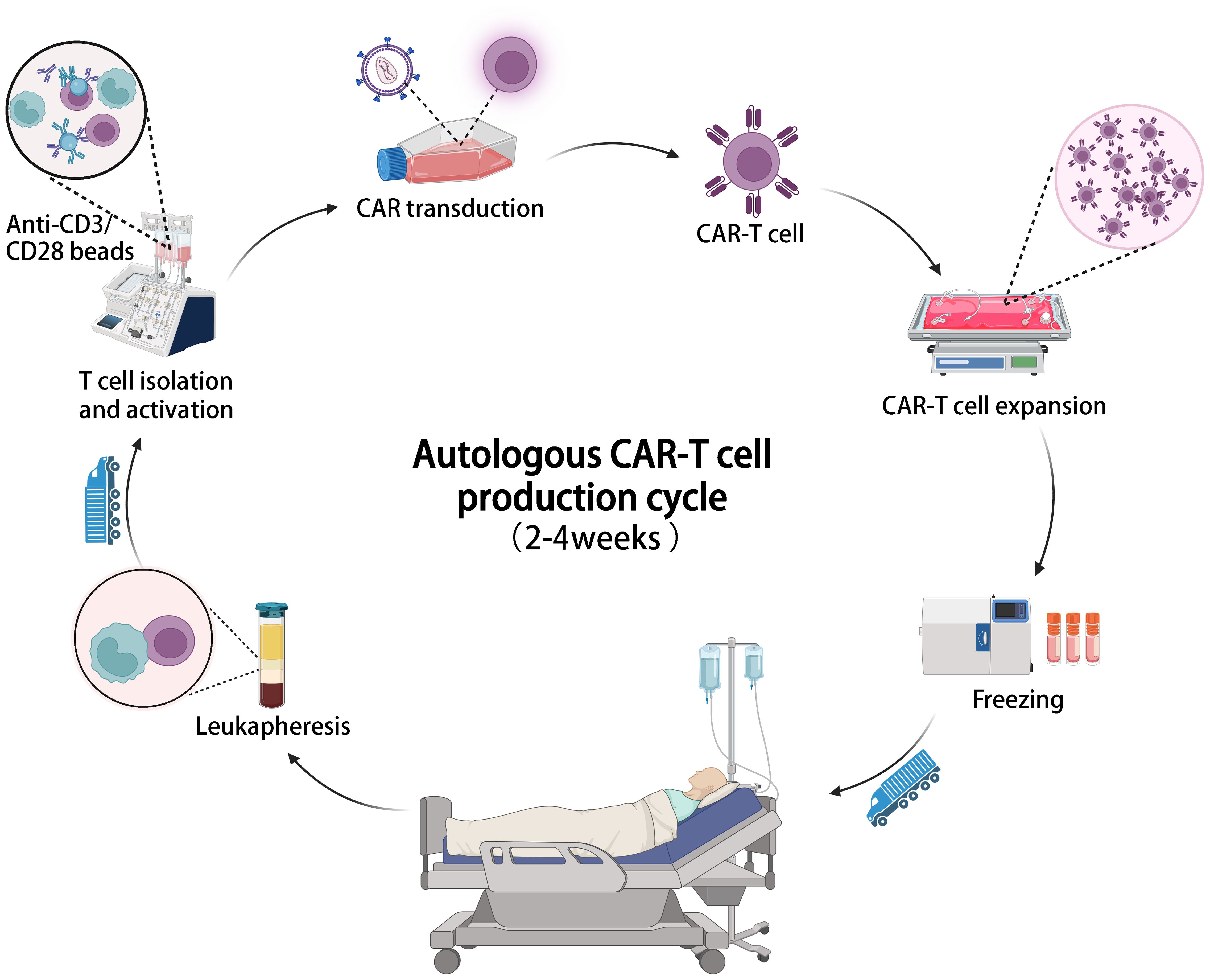
Figure 2 The illustration of the entire manufacturing process of autologous CAR-T cell therapy, which typically takes 2-4 weeks from cell collection to infusion.
3 New paradigms for CAR-T cell therapy beyond current Achilles heels
3.1 Allogeneic universal CAR-T cells
Allogeneic CAR-T cells sourced from healthy donors offer advantages such as scalable production, lower manufacturing costs, and immediate availability (96). As illustrated in Figure 3, allogeneic universal CAR-T cells represent off-the-shelf cell production. They are undergoing testing in numerous clinical trials, with promising outcomes documented in Table 2. For instance, a meta-analysis showed CR rates of 70% in R/R ALL and 52% in non-Hodgkin lymphoma (NHL) with CD19 CAR-T cells derived from healthy donors (97). However, allogeneic CAR-T cells face challenges like T-cell receptor (TCR)-mediated graft-versus-host disease (GVHD) and human leukocyte antigen (HLA) related host-versus-graft (HvG) response due to their allogeneic nature (96). Targeted knockout of TCR, HLA, and related molecules using gene editing techniques is an effective strategy. Various gene editing tools have been explored, including zinc finger nucleases (ZFNs) (98), transcription activator-like effector nucleases (TALENs) (99), and CRISPR-Cas9 (100). CRISPR-Cas9 stands out for its versatility, allowing for simultaneous editing of multiple genes. It has been extensively used in allogeneic CAR-T cells, targeting TCR and HLA class I molecules, and guiding CAR insertion to specific loci, enhancing CAR-T cell potency and stability (101). However, CRISPR-Cas9 editing has limitations such as off-target editing and large-scale genomic rearrangements. Base editing technology, a precise gene editing tool based on CRISPR, offers new possibilities for overcoming these challenges without inducing double-stranded DNA breaks (DSBs) (102, 103). Adenine base editors (ABEs) and cytosine base editors (CBEs) enable the conversion of specific DNA bases without DSBs (104). Recent studies have shown the feasibility of highly specific knockout of T cell genes using base editing, paving the way for the development of allogeneic CAR-T cells with enhanced safety and efficacy (105). While allogeneic CAR-T cell therapy holds promise, ongoing clinical trials are essential to further evaluate its safety and efficacy. Longer-term follow-up data from these trials will provide valuable insights into the durability of responses and the potential for adverse events. Continued research efforts are necessary to optimize allogeneic CAR-T cell therapies and address any challenges that may arise, ultimately advancing their clinical utility in treating various malignancies (106).
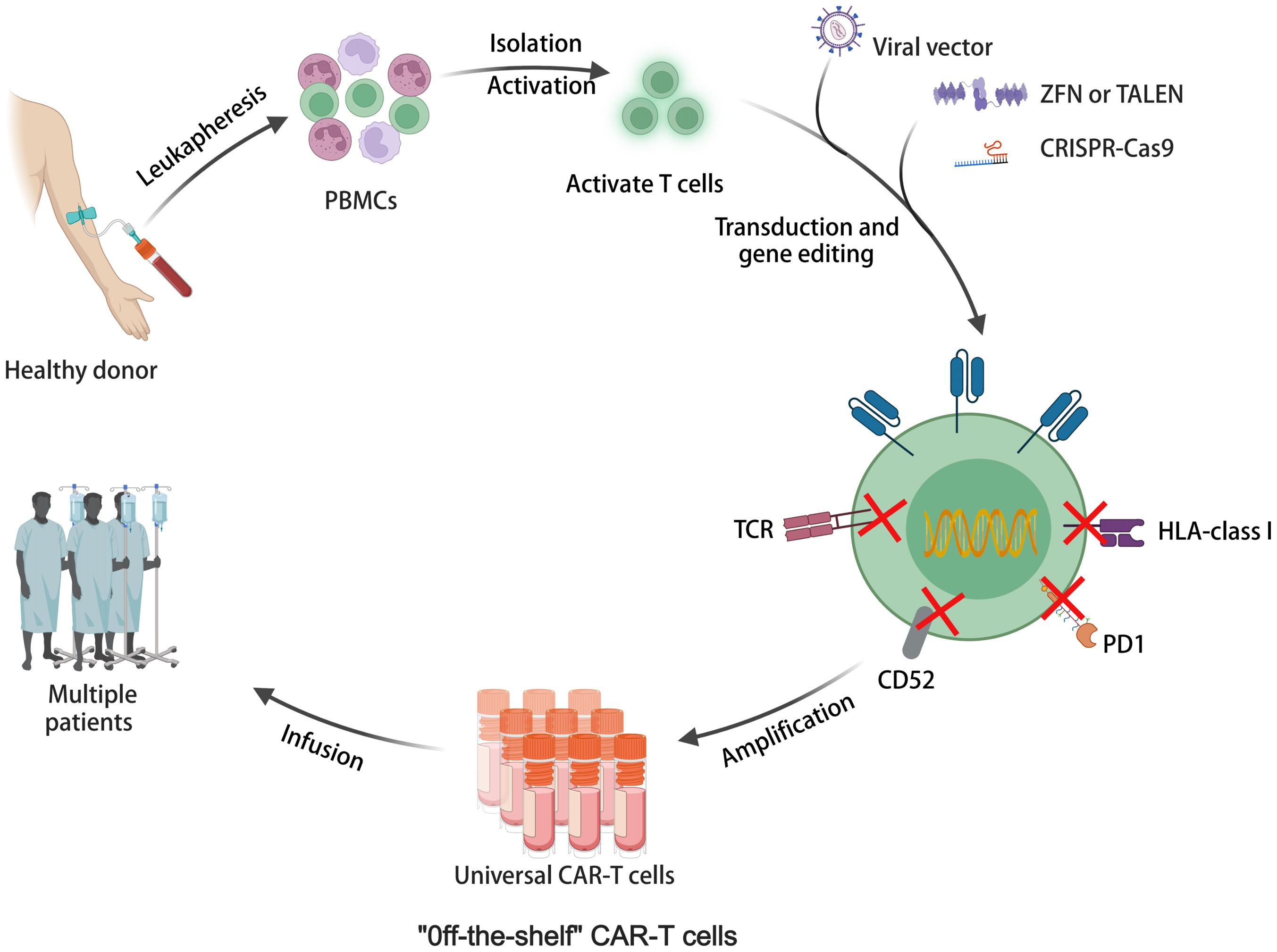
Figure 3 The illustration of allogeneic universal CAR-T cell therapy for off-the-shelf cell production.
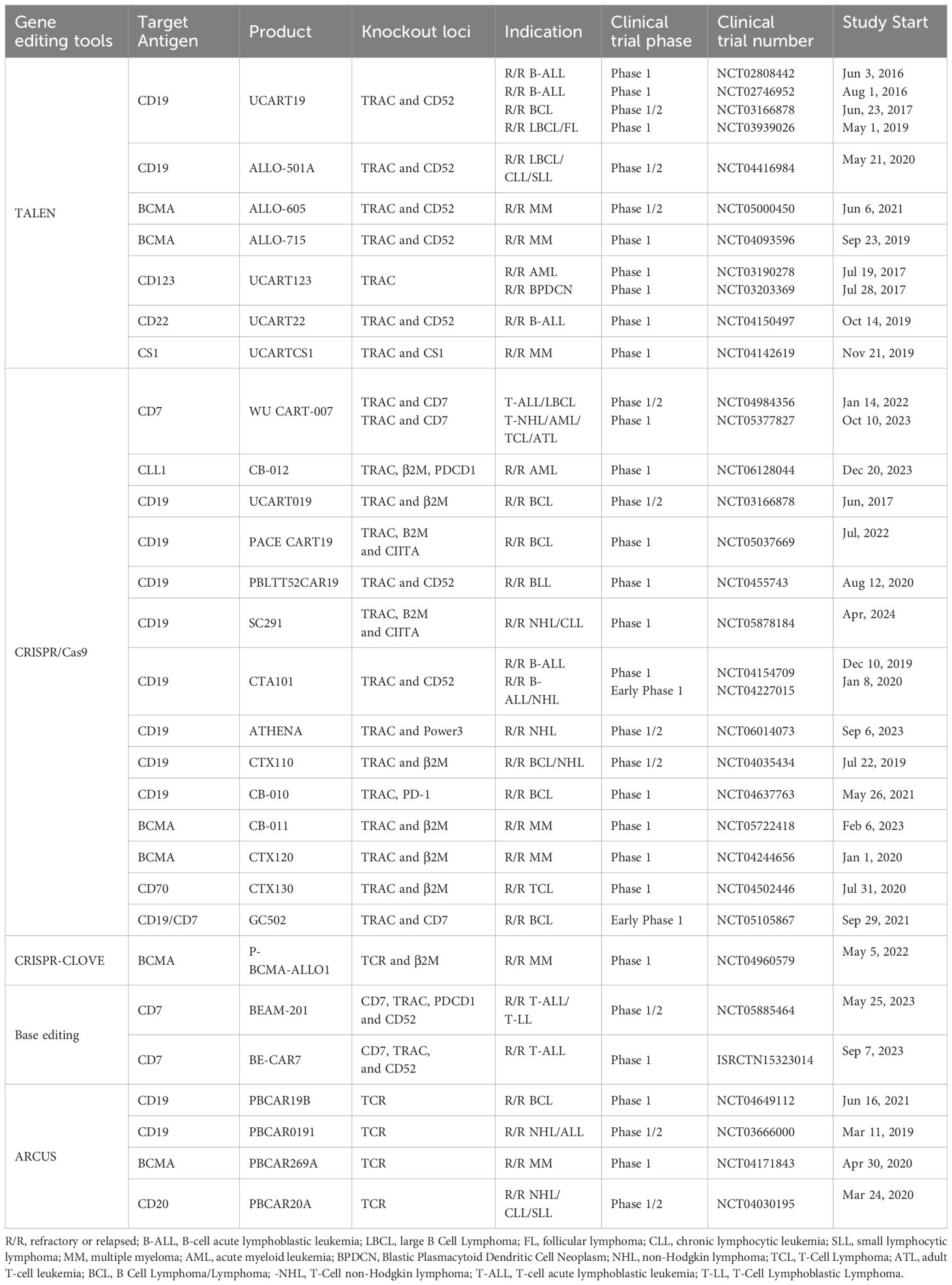
Table 2 Currently registered clinical trials of genome-edited allogeneic CAR-cell products.
3.2 Rapid manufacturing of autologous CAR-T cells
While significant progress has been made in the exploration of allogeneic and in vivo CAR-T cells, they remain in continuous proof-of-concept stages without clinical approval. Therefore, enhancing product quality, refining manufacturing processes, and reducing self-production time are crucial to address the challenges faced by autologous CAR-T cells. Shortening the ex vivo culture time has been shown to yield CAR-T cells with improved effector function and reduced production costs (54). Several promising methods for rapid CAR-T cell production have been explored, such as the FasTCAR platform by Gracell Biotechnologies, which produces CAR-T cells within a day and has demonstrated efficacy in preclinical and clinical evaluations for R/R B-ALL (35). Additionally, researchers at the University of Pennsylvania have successfully prepared functional CAR-T cells within 24 hours by directly transducing non-activated quiescent T cells (by lentivirus vector), extending the survival of tumor-bearing mice (107). Agarwalla et al. have developed an implantable Multifunctional Alginate Scaffold for T Cell Engineering and Release (MASTER), which integrates T cell activation, reprogramming, and in vivo expansion, reducing manufacturing time to 1 day. CAR-T cells produced using the MASTER scaffold have shown promising anti-tumor activity in mouse xenograft models of lymphoma (36). These rapidly manufactured CAR-T cells exhibit superior anti-tumor activity and greater persistence compared to conventional CAR-T cells, potentially offering a more cost-effective approach.
3.3 In vivo induced CAR-T cell therapy
Allogeneic universal CAR-T cells, while offering solutions to immune rejection through gene editing, also raise safety concerns. A previous clinical trial of allogeneic CAR-T therapy was halted by the FDA due to the emergence of chromosomal abnormalities (108). Although investigations suggested the abnormality wasn’t related to gene editing, safety concerns persist. A potential solution lies in direct CAR-T cell generation in patients via a universally applicable medicinal product containing the CAR gene. The illustration of in vivo generation of CAR-T cells is depicted in Figure 4 (109). This approach’s combination of simplicity, speed, and cost-effectiveness makes it an attractive option for CAR-T cell therapy. By leveraging the body’s natural processes, in vivo CAR-T cell generation eliminates the requirement for ex vivo cell manipulation and lengthy manufacturing processes. This streamlined method not only reduces production time but also lowers associated costs, potentially improving the accessibility of CAR-T cell therapy to a broader patient population. Various vector platforms are under exploration, including lentiviral vectors (LVs) and nanoparticles (NPs) (110, 111). Lentiviral vectors (LVs) stand as the predominant choice for ex vivo CAR-T cell transduction, boasting stable gene integration, high transduction efficiency, and a broad host range. Pioneering work by Buchholz et al. introduced the pseudotyping of vectors with modified envelope proteins of Nipah virus (Niv), enabling specific targeting of CD8 by fusing the envelope protein to a CD8-specific single-chain variable fragment (scFv) (112). Their study demonstrated that CD8-LV could directly generate human CD19-CAR-T cells in NGS mice in vivo, showcasing potent anti-tumor activity. Furthermore, Buchholz et al. developed Niv-LV targeting CD3 and CD4, capable of directly generating functional CAR-T cells in mice (113). Despite LVs’ high transduction efficiency, the potential risk of insertional mutagenesis remains a concern (114). In recent years, researchers have explored virus-like particles (VLPs) as a novel vector. VLPs retain viral proteins without containing a packaged genome, combining viral vector targeting specificity with the transient delivery advantages of non-viral vectors. Hamilton et al. demonstrated the generation of gene-edited CAR-T cells in vivo by packaging Cas9 RNPs into retroviral VLPs, offering a new direction for future in vivo CAR-T cell generation, albeit with potentially lower in vivo transduction efficiency compared to LVs (115). Nanoparticles (NPs) have garnered significant attention as gene delivery vehicles due to their low immunogenicity, cost-effectiveness, and customizable production (116). Unlike the complex machinery of viruses, gene delivery using NPs relies on the physicochemical properties of the particles and payloads, offering advantages such as payload flexibility and ease of modification (117). The two primary types of nanocarriers used in CAR-T cell development are lipid- and polymer-based NPs. Numerous preclinical studies have successfully employed these carriers to deliver CAR gene-containing DNA or mRNA into T cells in vivo, leading to the on-site generation of CAR-T cells and effective cytotoxic functions in small animal models. For instance, Matthias T. Stephan’s team designed polymer nanoparticles encapsulating CAR DNA and mRNA (118, 119), resulting in anti-tumor efficacy comparable to CAR-T cells prepared using traditional lentiviral vectors. In addition to antibody-targeted nanocarriers, Daniel J. Siegwart’s team recently introduced a Selective Organ Targeting (SORT) LNP capable of delivering mRNA to the spleen and locally generating CAR-T cells in a controlled manner (120). The research into in vivo generation of CAR-T cells has the potential to transform CAR-T cell therapy into a widely adopted pharmaceutical treatment (109). This advancement could significantly enhance clinical compliance and substantially reduce costs (121). Amid the FDA’s recent warning regarding secondary cancer risks following CAR-T cell infusion treatments, the application of in vivo editing to transiently modify CAR-T cells using mRNA offers a promising, potentially safer, and cost-effective solution to address the existing challenges associated with CAR-T cell therapy (122). By harnessing AI-based tools to refine CAR design, the advancement of in vivo CAR-T cell development stands poised to accelerate, offering a more streamlined and effective approach toward overcoming the current challenges in CAR-T cell therapy (123, 124).
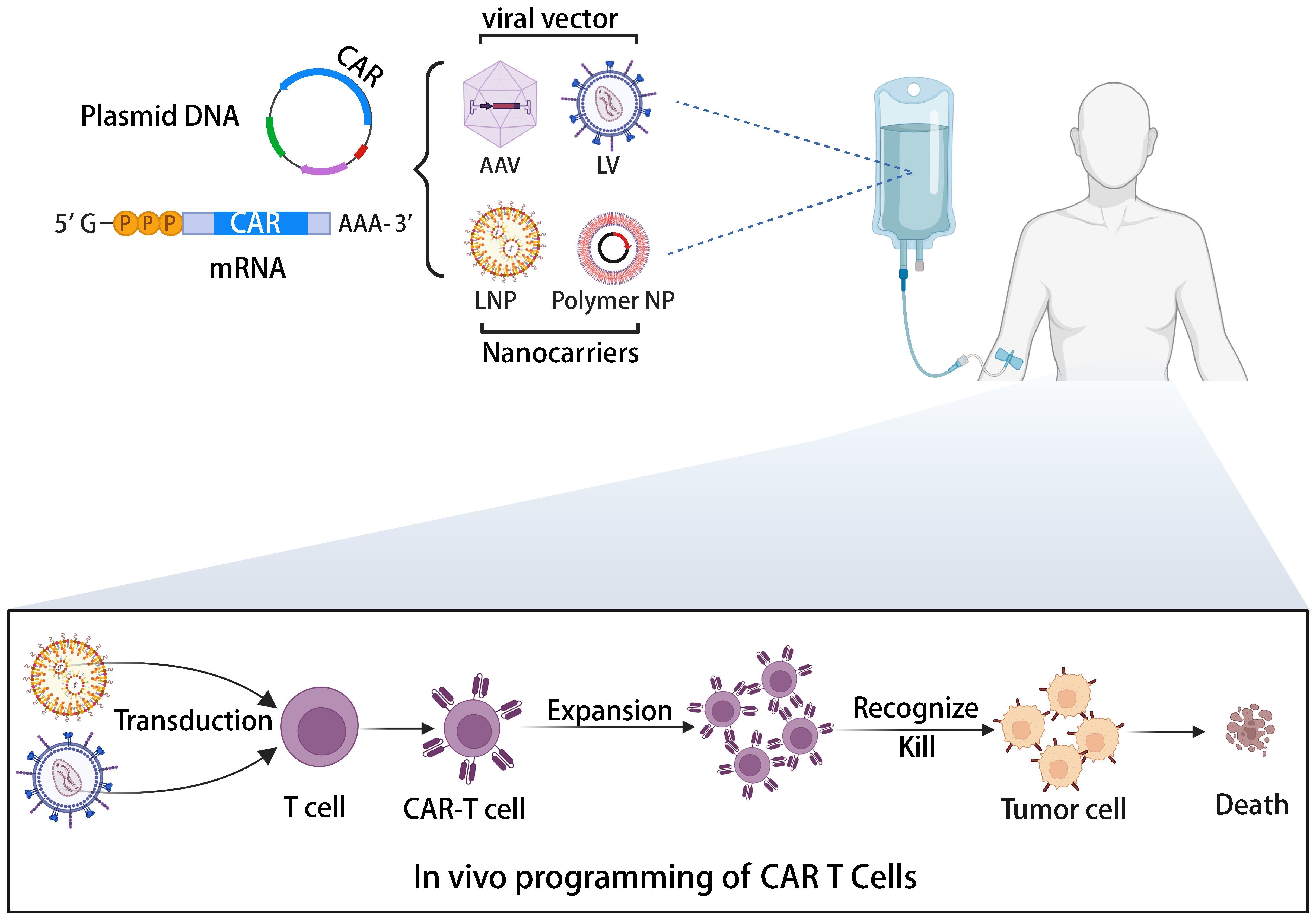
Figure 4 The illustration of the in vivo induction of CAR-T cells utilizing viral vectors or non-viral nanocarriers.
4 Conclusions and prospects
CAR-T therapies have demonstrated unprecedented efficacy in the treatment of hematologic malignancies. However, their widespread application is impeded by high costs, lengthy preparation time, safety concerns, and limited effectiveness. To overcome these obstacles, a revolution in gene editing tools and delivery vectors is necessary to establish new paradigms for CAR-T cell therapy that surpass current Achilles heels. With the ongoing refinement of gene editing tools and delivery vectors, highly potent and super safe CAR-T cells are poised to become widely utilized in clinical settings akin to conventional living drugs in the future. The expansion and exploration of these technologies are opening new possibilities for CAR-T cell therapy, offering patients more efficient, secure, and affordable therapeutic options.
Author contributions
YL: Investigation, Software, Writing – original draft. ZH: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. YYL: Funding acquisition, Methodology, Supervision, Writing – review & editing. XW: Conceptualization, Funding acquisition, Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study is supported by the National Natural Science Foundation of China 82104252, Shanghai Pujiang Program 21PJ1415600, Guangdong Basic and Applied Basic Research Foundation 2022A1515010491, Zhongshan Municipal Bureau of Science and Technology, and the foundation from Hubei Provincial Department of Science and Technology Project 2023BCB026.
Acknowledgments
Thank you to all the authors who provided comments and assistance in the conception and writing of this review.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lyu L, Feng Y, Chen X, Hu Y. The global chimeric antigen receptor T (CAR-T) cell therapy patent landscape. Nat Biotechnol. (2020) 38:1387–94. doi: 10.1038/s41587-020-00749-8
2. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discovery. (2013) 3:388–98. doi: 10.1158/2159-8290.CD-12-0548
3. Braendstrup P, Levine BL, Ruella M. The long road to the first FDA-approved gene therapy: chimeric antigen receptor T cells targeting CD19. Cytotherapy (2020). 22:57–69. doi: 10.1016/j.jcyt.2019.12.004
4. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. New Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
5. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. New Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
6. Fowler NH, Dickinson M, Dreyling M, Martinez-Lopez J, Kolstad A, Butler J, et al. Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: the phase 2 ELARA trial. Nat Med. (2022) 28:325–32. doi: 10.1038/s41591-021-01622-0
7. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. (2019) 20:31–42. doi: 10.1016/S1470-2045(18)30864-7
8. Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol. (2022) 23:91–103. doi: 10.1016/S1470-2045(21)00591-X
9. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. New Engl J Med. (2020) 382:1331–42. doi: 10.1056/NEJMoa1914347
10. Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet (London England). (2021) 398:491–502. doi: 10.1016/S0140-6736(21)01222-8
11. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet (London England). (2020) 396:839–52. doi: 10.1016/S0140-6736(20)31366-0
12. Munshi NC, Anderson LD Jr., Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. New Engl J Med. (2021) 384:705–16. doi: 10.1056/NEJMoa2024850
13. Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet (London England). (2021) 398:314–24. doi: 10.1016/S0140-6736(21)00933-8
14. Cappell KM, Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. (2023) 20:359–71. doi: 10.1038/s41571-023-00754-1
15. Haradhvala NJ, Leick MB, Maurer K, Gohil SH, Larson RC, Yao N, et al. Distinct cellular dynamics associated with response to CAR-T therapy for refractory B cell lymphoma. Nat Med. (2022) 28:1848–59. doi: 10.1038/s41591-022-01959-0
16. Schroeder BA, Jess J, Sankaran H, Shah NN. Clinical trials for chimeric antigen receptor T-cell therapy: lessons learned and future directions. Curr Opin Hematol. (2022) 29:225–32. doi: 10.1097/MOH.0000000000000723
17. Wang V, Gauthier M, Decot V, Reppel L, Bensoussan D. Systematic review on CAR-T cell clinical trials up to 2022: academic center input. Cancers. (2023) 15:1003. doi: 10.3390/cancers15041003
18. Schett G, Mackensen A, Mougiakakos D. CAR T-cell therapy in autoimmune diseases. Lancet (London England). (2023) 402:2034–44. doi: 10.1016/S0140-6736(23)01126-1
19. Müller F, Taubmann J, Bucci L, Wilhelm A, Bergmann C, Völkl S, et al. CD19 CAR T-cell therapy in autoimmune disease - A case series with follow-up. New Engl J Med. (2024) 390:687–700. doi: 10.1056/NEJMoa2308917
20. Baker DJ, Arany Z, Baur JA, Epstein JA, June CH. CAR T therapy beyond cancer: the evolution of a living drug. Nature. (2023) 619:707–15. doi: 10.1038/s41586-023-06243-w
21. Baker DJ, June CH. CAR T therapy extends its reach to autoimmune diseases. Cell. (2022) 185:4471–3. doi: 10.1016/j.cell.2022.10.026
22. Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. (2017) 4:92–101. doi: 10.1016/j.omtm.2016.12.006
23. Abou-El-Enein M, Elsallab M, Feldman SA, Fesnak AD, Heslop HE, Marks P, et al. Scalable manufacturing of CAR T cells for cancer immunotherapy. Blood Cancer Discovery. (2021) 2:408–22. doi: 10.1158/2643-3230.BCD-21-0084
24. Watanabe N, Mo F, McKenna MK. Impact of manufacturing procedures on CAR T cell functionality. Front Immunol. (2022) 13:876339. doi: 10.3389/fimmu.2022.876339
25. Ruella M, Korell F, Porazzi P, Maus MV. Mechanisms of resistance to chimeric antigen receptor-T cells in haematological Malignancies. Nat Rev Drug Discovery. (2023) 22:976–95. doi: 10.1038/s41573-023-00807-1
26. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) 11:69. doi: 10.1038/s41408-021-00459-7
27. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. (2019) 16:372–85. doi: 10.1038/s41571-019-0184-6
28. Verdun N, Marks P. Secondary cancers after chimeric antigen receptor T-cell therapy. New Engl J Med. (2024) 390:584–6. doi: 10.1056/NEJMp2400209
29. Zhang Y, Li Y, Cao W, Wang F, Xie X, Li Y, et al. Single-cell analysis of target antigens of CAR-T reveals a potential landscape of “On-target, off-tumor toxicity”. Front Immunol. (2021) 12:799206. doi: 10.3389/fimmu.2021.799206
30. Jain T, Olson TS, Locke FL. How I treat cytopenias after CAR T-cell therapy. Blood. (2023) 141:2460–9. doi: 10.1182/blood.2022017415
31. Hu Y, Zhou Y, Zhang M, Ge W, Li Y, Yang L, et al. CRISPR/Cas9-engineered universal CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia. Clin Cancer Res. (2021) 27:2764–72. doi: 10.1158/1078-0432.CCR-20-3863
32. Chiesa R, Georgiadis C, Syed F, Zhan H, Etuk A, Gkazi SA, et al. Base-edited CAR7 T cells for relapsed T-cell acute lymphoblastic leukemia. New Engl J Med. (2023) 389:899–910. doi: 10.1056/NEJMoa2300709
33. Rurik JG, Tombácz I, Yadegari A, Méndez Fernández PO, Shewale SV, Li L, et al. CAR T cells produced in vivo to treat cardiac injury. Sci (New York NY). (2022) 375:91–6. doi: 10.1126/science.abm0594
34. Pfeiffer A, Thalheimer FB, Hartmann S, Frank AM, Bender RR, Danisch S, et al. In vivo generation of human CD19-CAR T cells results in B-cell depletion and signs of cytokine release syndrome. EMBO Mol Med. (2018) 10:e9158. doi: 10.15252/emmm.201809158
35. Yang J, He J, Zhang X, Li J, Wang Z, Zhang Y, et al. Next-day manufacture of a novel anti-CD19 CAR-T therapy for B-cell acute lymphoblastic leukemia: first-in-human clinical study. Blood Cancer J. (2022) 12:104. doi: 10.1038/s41408-022-00694-6
36. Agarwalla P, Ogunnaike EA, Ahn S, Froehlich KA, Jansson A, Ligler FS, et al. Bioinstructive implantable scaffolds for rapid in vivo manufacture and release of CAR-T cells. Nat Biotechnol. (2022) 40:1250–8. doi: 10.1038/s41587-022-01245-x
37. Yan T, Zhu L, Chen J. Current advances and challenges in CAR T-Cell therapy for solid tumors: tumor-associated antigens and the tumor microenvironment. Exp Hematol Oncol. (2023) 12:14. doi: 10.1186/s40164-023-00373-7
38. Dagar G, Gupta A, Masoodi T, Nisar S, Merhi M, Hashem S, et al. Harnessing the potential of CAR-T cell therapy: progress, challenges, and future directions in hematological and solid tumor treatments. J Trans Med. (2023) 21:449. doi: 10.1186/s12967-023-04292-3
39. Gumber D, Wang LD. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. EBioMedicine. (2022) 77:103941. doi: 10.1016/j.ebiom.2022.103941
40. Levine BL, Pasquini MC, Connolly JE, Porter DL, Gustafson MP, Boelens JJ, et al. Unanswered questions following reports of secondary Malignancies after CAR-T cell therapy. Nat Med. (2024) 30:338–41. doi: 10.1038/s41591-023-02767-w
41. Jain MD, Smith M, Shah NN. How I treat refractory CRS and ICANS after CAR T-cell therapy. Blood. (2023) 141:2430–42. doi: 10.1182/blood.2022017414
42. Blache U, Popp G, Dünkel A, Koehl U, Fricke S. Potential solutions for manufacture of CAR T cells in cancer immunotherapy. Nat Commun. (2022) 13:5225. doi: 10.1038/s41467-022-32866-0
43. Chong EA, Ruella M, Schuster SJ. Five-year outcomes for refractory B-cell lymphomas with CAR T-cell therapy. N Engl J Med. (2021) 384:673–4. doi: 10.1056/NEJMc2030164
44. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
45. Plaks V, Rossi JM, Chou J, Wang L, Poddar S, Han G, et al. CD19 target evasion as a mechanism of relapse in large B-cell lymphoma treated with axicabtagene ciloleucel. Blood. (2021) 138:1081–5. doi: 10.1182/blood.2021010930
46. Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discovery. (2018) 8:1219–26. doi: 10.1158/2159-8290.CD-18-0442
47. Braig F, Brandt A, Goebeler M, Tony H-P, Kurze A-K, Nollau P, et al. Resistance to anti-CD19/CD3 BiTE in acute lymphoblastic leukemia may be mediated by disrupted CD19 membrane trafficking. Blood. (2017) 129:100–4. doi: 10.1182/blood-2016-05-718395
48. Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. (2016) 128:1688–700. doi: 10.1182/blood-2016-04-711903
49. Da Vià MC, Dietrich O, Truger M, Arampatzi P, Duell J, Heidemeier A, et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat Med. (2021) 27:616–9. doi: 10.1038/s41591-021-01245-5
50. Samur MK, Fulciniti M, Aktas Samur A, Bazarbachi AH, Tai Y-T, Prabhala R, et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat Commun. (2021) 12:868. doi: 10.1038/s41467-021-21177-5
51. Lee H, Ahn S, Maity R, Leblay N, Ziccheddu B, Truger M, et al. Mechanisms of antigen escape from BCMA- or GPRC5D-targeted immunotherapies in multiple myeloma. Nat Med. (2023) 29:2295–306. doi: 10.1038/s41591-023-02491-5
52. Goodman DB, Azimi CS, Kearns K, Talbot A, Garakani K, Garcia J, et al. Pooled screening of CAR T cells identifies diverse immune signaling domains for next-generation immunotherapies. Sci Trans Med. (2022) 14:eabm1463. doi: 10.1126/scitranslmed.abm1463
53. Chan JD, Lai J, Slaney CY, Kallies A, Beavis PA, Darcy PK. Cellular networks controlling T cell persistence in adoptive cell therapy. Nat Rev Immunol. (2021) 21:769–84. doi: 10.1038/s41577-021-00539-6
54. Ghassemi S, Nunez-Cruz S, O’Connor RS, Fraietta JA, Patel PR, Scholler J, et al. Reducing ex vivo culture improves the antileukemic activity of chimeric antigen receptor (CAR) T cells. Cancer Immunol Res. (2018) 6:1100–9. doi: 10.1158/2326-6066.CIR-17-0405
55. Finney OC, Brakke HM, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest. (2019) 129:2123–32. doi: 10.1172/JCI125423
56. Desantis V, Savino FD, Scaringella A, Potenza MA, Nacci C, Frassanito MA, et al. The leading role of the immune microenvironment in multiple myeloma: A new target with a great prognostic and clinical value. J Clin Med. (2022) 11:2513. doi: 10.3390/jcm11092513
57. Autio M, Leivonen S-K, Brück O, Karjalainen-Lindsberg M-L, Pellinen T, Leppä S. Clinical impact of immune cells and their spatial interactions in diffuse large B-cell lymphoma microenvironment. Clin Cancer Res. (2022) 28:781–92. doi: 10.1158/1078-0432.CCR-21-3140
58. Hotblack A, Straathof K. Fine-tuning CARs for best performance. Cancer Cell. (2022) 40:11–3. doi: 10.1016/j.ccell.2021.12.010
59. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
60. Guedan S, Madar A, Casado-Medrano V, Shaw C, Wing A, Liu F, et al. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J Clin Invest. (2020) 130:3087–97. doi: 10.1172/JCI133215
61. Kouro T, Himuro H, Sasada T. Exhaustion of CAR T cells: potential causes and solutions. J Transl Med. (2022) 20:239. doi: 10.1186/s12967-022-03442-3
62. Chen J, Qiu S, Li W, Wang K, Zhang Y, Yang H, et al. Tuning charge density of chimeric antigen receptor optimizes tonic signaling and CAR-T cell fitness. Cell Res. (2023) 33:341–54. doi: 10.1038/s41422-023-00789-0
63. Wang H, Huang Y, Xu C. Charging CAR by electrostatic power. Immunol Rev. (2023) 320:138–46. doi: 10.1111/imr.13232
64. Qiu S, Chen J, Wu T, Li L, Wang G, Wu H, et al. CAR-Toner: an AI-driven approach for CAR tonic signaling prediction and optimization. Cell Res. (2024) 1–3. doi: 10.1038/s41422-024-00936-1
65. Hort S, Herbst L, Bäckel N, Erkens F, Niessing B, Frye M, et al. Toward rapid, widely available autologous CAR-T cell therapy - artificial intelligence and automation enabling the smart manufacturing hospital. Front Med. (2022) 9:913287. doi: 10.3389/fmed.2022.913287
66. Furlow B. FDA investigates risk of secondary lymphomas after CAR-T immunotherapy. Lancet Oncol. (2024) 25:21. doi: 10.1016/S1470-2045(23)00631-9
67. The Lancet Haematology. Balancing the risks and benefits of CAR T-cell therapy. Lancet Haematol. (2024) 11:e169. doi: 10.1016/S2352-3026(24)00037-1
68. Mátrai J, Chuah MK, VandenDriessche T. Recent advances in lentiviral vector development and applications. Mol Therapy: J Am Soc Gene Ther. (2010) 18:477–90. doi: 10.1038/mt.2009.319
69. Gust J, Ponce R, Liles WC, Garden GA, Turtle CJ. Cytokines in CAR T cell-associated neurotoxicity. Front Immunol. (2020) 11:577027. doi: 10.3389/fimmu.2020.577027
70. Chohan KL, Siegler EL, Kenderian SS. CAR-T cell therapy: the efficacy and toxicity balance. Curr Hematol Malig Rep. (2023) 18:9–18. doi: 10.1007/s11899-023-00687-7
71. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. (2018) 15:47–62. doi: 10.1038/nrclinonc.2017.148
72. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. (2014) 124:188–95. doi: 10.1182/blood-2014-05-552729
73. Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer. (2018) 6:56. doi: 10.1186/s40425-018-0343-9
74. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discovery. (2018) 8:958–71. doi: 10.1158/2159-8290.CD-17-1319
75. Grant SJ, Grimshaw AA, Silberstein J, Murdaugh D, Wildes TM, Rosko AE, et al. Clinical presentation, risk factors, and outcomes of immune effector cell-associated neurotoxicity syndrome following chimeric antigen receptor T cell therapy: A systematic review. Transplant Cell Ther. (2022) 28:294–302. doi: 10.1016/j.jtct.2022.03.006
76. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. (2019) 25:625–38. doi: 10.1016/j.bbmt.2018.12.758
77. Shimabukuro-Vornhagen A, Böll B, Schellongowski P, Valade S, Metaxa V, Azoulay E, et al. Critical care management of chimeric antigen receptor T-cell therapy recipients. CA Cancer J Clin. (2022) 72:78–93. doi: 10.3322/caac.21702
78. Schmidts A, Wehrli M, Maus MV. Toward better understanding and management of CAR-T cell-associated toxicity. Annu Rev Med. (2021) 72:365–82. doi: 10.1146/annurev-med-061119-015600
79. Sterner RC, Sterner RM. Immune effector cell associated neurotoxicity syndrome in chimeric antigen receptor-T cell therapy. Front Immunol. (2022) 13:879608. doi: 10.3389/fimmu.2022.879608
80. Mucha SR, Rajendram P. Management and prevention of cellular-therapy-related toxicity: early and late complications. Curr Oncol (Toronto Ont). (2023) 30:5003–23. doi: 10.3390/curroncol30050378
81. Hombach AA, Abken H. Shared target antigens on cancer cells and tissue stem cells: go or no-go for CAR T cells? Expert Rev Clin Immunol. (2017) 13:151–5. doi: 10.1080/1744666X.2016.1221763
82. Miao L, Zhang Z, Ren Z, Li Y. Reactions related to CAR-T cell therapy. Front Immunol. (2021) 12:663201. doi: 10.3389/fimmu.2021.663201
83. Flugel CL, Majzner RG, Krenciute G, Dotti G, Riddell SR, Wagner DL, et al. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat Rev Clin Oncol. (2023) 20:49–62. doi: 10.1038/s41571-022-00704-3
84. Savanur MA, Weinstein-Marom H, Gross G. Implementing logic gates for safer immunotherapy of cancer. Front Immunol. (2021) 12:780399. doi: 10.3389/fimmu.2021.780399
85. Lajoie MJ, Boyken SE, Salter AI, Bruffey J, Rajan A, Langan RA, et al. Designed protein logic to target cells with precise combinations of surface antigens. Sci (New York NY). (2020) 369:1637–43. doi: 10.1126/science.aba6527
86. Sagnella SM, White AL, Yeo D, Saxena P, van Zandwijk N, Rasko JEJ. Locoregional delivery of CAR-T cells in the clinic. Pharmacol Res. (2022) 182:106329. doi: 10.1016/j.phrs.2022.106329
87. Brown CE, Hibbard JC, Alizadeh D, Blanchard MS, Natri HM, Wang D, et al. Locoregional delivery of IL-13Rα2-targeting CAR-T cells in recurrent high-grade glioma: a phase 1 trial. Nat Med. (2024) 30:1001–12. doi: 10.1038/s41591-024-02928-5
88. Vitanza NA, Ronsley R, Choe M, Henson C, Breedt M, Barrios-Anderson A, et al. Locoregional CAR T cells for children with CNS tumors: Clinical procedure and catheter safety. Neoplasia (New York NY). (2023) 36:100870. doi: 10.1016/j.neo.2022.100870
89. Vormittag P, Gunn R, Ghorashian S, Veraitch FS. A guide to manufacturing CAR T cell therapies. Curr Opin Biotechnol. (2018) 53:164–81. doi: 10.1016/j.copbio.2018.01.025
90. Moretti A, Ponzo M, Nicolette CA, Tcherepanova IY, Biondi A, Magnani CF. The past, present, and future of non-viral CAR T cells. Front Immunol. (2022) 13:867013. doi: 10.3389/fimmu.2022.867013
91. Dai X, Mei Y, Cai D, Han W. Standardizing CAR-T therapy: Getting it scaled up. Biotechnol Adv. (2019) 37:239–45. doi: 10.1016/j.biotechadv.2018.12.002
92. Poorebrahim M, Sadeghi S, Fakhr E, Abazari MF, Poortahmasebi V, Kheirollahi A, et al. Production of CAR T-cells by GMP-grade lentiviral vectors: latest advances and future prospects. Crit Rev Clin Lab Sci. (2019) 56:393–419. doi: 10.1080/10408363.2019.1633512
93. Labbé RP, Vessillier S, Rafiq QA. Lentiviral vectors for T cell engineering: clinical applications, bioprocessing and future perspectives. Viruses. (2021) 13:1528. doi: 10.3390/v13081528
94. Hernandez I, Prasad V, Gellad WF. Total costs of chimeric antigen receptor T-cell immunotherapy. JAMA Oncol. (2018) 4:994–6. doi: 10.1001/jamaoncol.2018.0977
95. Arcangeli S, Falcone L, Camisa B, De Girardi F, Biondi M, Giglio F, et al. Next-generation manufacturing protocols enriching T(SCM) CAR T cells can overcome disease-specific T cell defects in cancer patients. Front Immunol. (2020) 11:1217. doi: 10.3389/fimmu.2020.01217
96. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discovery. (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
97. Al-Mansour M, Al-Foheidi M, Ibrahim E. Efficacy and safety of second-generation CAR T-cell therapy in diffuse large B-cell lymphoma: A meta-analysis. Mol Clin Oncol. (2020) 13:33. doi: 10.3892/mco.2020.2103
98. Martínez Bedoya D, Dutoit V, Migliorini D. Allogeneic CAR T cells: an alternative to overcome challenges of CAR T cell therapy in glioblastoma. Front Immunol. (2021) 12:640082. doi: 10.3389/fimmu.2021.640082
99. Jo S, Das S, Williams A, Chretien AS, Pagliardini T, Le Roy A, et al. Endowing universal CAR T-cell with immune-evasive properties using TALEN-gene editing. Nat Commun. (2022) 13:3453. doi: 10.1038/s41467-022-30896-2
100. Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer. (2022) 21:78. doi: 10.1186/s12943-022-01559-z
101. Wei W, Chen ZN, Wang K. CRISPR/Cas9: A powerful strategy to improve CAR-T cell persistence. Int J Mol Sci. (2023) 24:12317. doi: 10.3390/ijms241512317
102. Diorio C, Murray R, Naniong M, Barrera L, Camblin A, Chukinas J, et al. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood. (2022) 140:619–29. doi: 10.1182/blood.2022015825
103. Georgiadis C, Rasaiyaah J, Gkazi SA, Preece R, Etuk A, Christi A, et al. Base-edited CAR T cells for combinational therapy against T cell Malignancies. Leukemia. (2021) 35:3466–81. doi: 10.1038/s41375-021-01282-6
104. Jeong YK, Song B, Bae S. Current status and challenges of DNA base editing tools. Mol Therapy: J Am Soc Gene Ther. (2020) 28:1938–52. doi: 10.1016/j.ymthe.2020.07.021
105. Schmidt R, Ward CC, Dajani R, Armour-Garb Z, Ota M, Allain V, et al. Base-editing mutagenesis maps alleles to tune human T cell functions. Nature. (2024) 625:805–12. doi: 10.1038/s41586-023-06835-6
106. Chattaraj A, Rehman MEU, Khan I, Franco D, Ibrahim A, Khanam R, et al. Safety and efficacy of allogeneic CAR-T cells in B-cell Malignancies: A systematic review and meta-analysis. J Clin Oncol. (2022) 40:e19530–0. doi: 10.1200/JCO.2022.40.16_suppl.e19530
107. Ghassemi S, Durgin JS, Nunez-Cruz S, Patel J, Leferovich J, Pinzone M, et al. Rapid manufacturing of non-activated potent CAR T cells. Nat BioMed Eng. (2022) 6:118–28. doi: 10.1038/s41551-021-00842-6
108. Xin T, Cheng L, Zhou C, Zhao Y, Hu Z, Wu X. In-vivo induced CAR-T cell for the potential breakthrough to overcome the barriers of current CAR-T cell therapy. Front Oncol. (2022) 12:809754. doi: 10.3389/fonc.2022.809754
109. Wakao R, Fukaya-Shiba A. In vivo CAR T cells and targeted gene delivery: A theme for the Pharmaceuticals and Medical Devices Agency Science Board to address. Front Med. (2023) 10:1141880. doi: 10.3389/fmed.2023.1141880
110. Michels KR, Sheih A, Hernandez SA, Brandes AH, Parrilla D, Irwin B, et al. Preclinical proof of concept for VivoVec, a lentiviral-based platform for in vivo CAR T-cell engineering. J Immunother Cancer. (2023) 11:e006292. doi: 10.1136/jitc-2022-006292
111. Billingsley MM, Gong N, Mukalel AJ, Thatte AS, El-Mayta R, Patel SK, et al. In Vivo mRNA CAR T Cell Engineering via Targeted Ionizable Lipid Nanoparticles with Extrahepatic Tropism. Small (Weinheim an der Bergstrasse Germany). (2024) 20:e2304378. doi: 10.1002/smll.202304378
112. Agarwal S, Hanauer JDS, Frank AM, Riechert V, Thalheimer FB, Buchholz CJ. In vivo generation of CAR T cells selectively in human CD4(+) lymphocytes. Mol Therapy: J Am Soc Gene Ther. (2020) 28:1783–94. doi: 10.1016/j.ymthe.2020.05.005
113. Frank AM, Braun AH, Scheib L, Agarwal S, Schneider IC, Fusil F, et al. Combining T-cell-specific activation and in vivo gene delivery through CD3-targeted lentiviral vectors. Blood Adv. (2020) 4:5702–15. doi: 10.1182/bloodadvances.2020002229
114. Gurumoorthy N, Nordin F, Tye GJ, Wan Kamarul Zaman WS, Ng MH. Non-integrating lentiviral vectors in clinical applications: A glance through. Biomedicines. (2022) 10:107. doi: 10.3390/biomedicines10010107
115. Hamilton JR, Chen E, Perez BS, Sandoval Espinoza CR, Kang MH, Trinidad M, et al. In vivo human T cell engineering with enveloped delivery vehicles. Nat Biotechnol. (2024). doi: 10.1038/s41587-023-02085-z
116. Mirza Z, Karim S. Nanoparticles-based drug delivery and gene therapy for breast cancer: Recent advancements and future challenges. Semin Cancer Biol. (2021) 69:226–37. doi: 10.1016/j.semcancer.2019.10.020
117. Hald Albertsen C, Kulkarni JA, Witzigmann D, Lind M, Petersson K, Simonsen JB. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv Drug Deliv Rev. (2022) 188:114416. doi: 10.1016/j.addr.2022.114416
118. Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D, et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol. (2017) 12:813–20. doi: 10.1038/nnano.2017.57
119. Parayath NN, Stephan SB, Koehne AL, Nelson PS, Stephan MT. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat Commun. (2020) 11:6080. doi: 10.1038/s41467-020-19486-2
120. Álvarez-Benedicto E, Tian Z, Chatterjee S, Orlando D, Kim M, Guerrero ED, et al. Spleen SORT LNP generated in situ CAR T cells extend survival in a mouse model of lymphoreplete B cell lymphoma. Angewandte Chemie (International Ed English). (2023) 62:e202310395. doi: 10.1002/anie.202310395
121. Michels A, Ho N, Buchholz CJ. Precision medicine: In vivo CAR therapy as a showcase for receptor-targeted vector platforms. Mol Therapy: J Am Soc Gene Ther. (2022) 30:2401–15. doi: 10.1016/j.ymthe.2022.05.018
122. Wu J, Wu W, Zhou B, Li B. Chimeric antigen receptor therapy meets mRNA technology. Trends Biotechnol. (2024) 42:228–40. doi: 10.1016/j.tibtech.2023.08.005
123. Bujak J, Kłęk S, Balawejder M, Kociniak A, Wilkus K, Szatanek R, et al. Creating an innovative artificial intelligence-based technology (TCRact) for designing and optimizing T cell receptors for use in cancer immunotherapies: protocol for an observational trial. JMIR Res Protoc. (2023) 12:e45872. doi: 10.2196/45872
Keywords: chimeric antigen receptor, immunotherapy, allogeneic universal CAR-T cells, in vivo CAR-T cell, Achilles heels
Citation: Li Y, Hu Z, Li Y and Wu X (2024) Charting new paradigms for CAR-T cell therapy beyond current Achilles heels. Front. Immunol. 15:1409021. doi: 10.3389/fimmu.2024.1409021
Received: 29 March 2024; Accepted: 18 April 2024;
Published: 01 May 2024.
Edited by:
Maurizio Chiriva-Internati, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Haopeng Wang, ShanghaiTech University, ChinaHiroki Torikai, University of Texas MD Anderson Cancer Center, United States
Copyright © 2024 Li, Hu, Li and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenhua Hu, aHV6aGhAbmZ1LmVkdS5jbg==; Yuanyuan Li, bGl5dWFueXVhbkBzaW1tLmFjLmNu; Xiaoyan Wu, eHd1QGh1c3QuZWR1LmNu
†These authors share first authorship