
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 03 June 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1407992
This article is part of the Research Topic Emerging Strategies to Overcome Current Roadblocks in CAR T Cell Immunotherapy View all 7 articles
 Antonio Maria Alviano1†
Antonio Maria Alviano1† Marta Biondi1,2
Marta Biondi1,2 Erica Grassenis1
Erica Grassenis1 Andrea Biondi1
Andrea Biondi1 Marta Serafini1,2*
Marta Serafini1,2* Sarah Tettamanti1
Sarah Tettamanti1Although adoptive transfer of chimeric antigen receptor (CAR)-engineered T cells has achieved unprecedented response rates in patients with certain hematological malignancies, this therapeutic modality is still far from fulfilling its remarkable potential, especially in the context of solid cancers. Antigen escape variants, off-tumor destruction of healthy tissues expressing tumor-associated antigens (TAAs), poor CAR-T cell persistence, and the occurrence of functional exhaustion represent some of the most prominent hurdles that limit CAR-T cell ability to induce long-lasting remissions with a tolerable adverse effect profile. In this review, we summarize the main approaches that have been developed to face such bottlenecks, including the adapter CAR (AdCAR) system, Boolean-logic gating, epitope editing, the modulation of cell-intrinsic signaling pathways, and the incorporation of safety switches to precisely control CAR-T cell activation. We also discuss the most pressing issues pertaining to the selection of co-stimulatory domains, with a focus on strategies aimed at promoting CAR-T cell persistence and optimal antitumor functionality.
● CAR-T cell therapy has significantly transformed immuno-oncology; however, there are still several challenges that need to be addressed.
● Adapter CARs and “OR” logic-gating allow to improve CAR-T cell activity against heterogeneous tumors.
● “AND” logic-gating strategies and ON/OFF switches can be implemented to enhance CAR-T cell safety.
● Careful selection of co-stimulatory domains and targeted manipulations of intracellular signaling pathways allow for the optimization of CAR-T cell functionality.
The outstanding results achieved by immunotherapy in patients with malignant neoplasms have fueled a growing interest in this approach to cancer treatment, which aims to harness the potential of the immune system to target neoplastic diseases. Among passive immunotherapeutic approaches, chimeric antigen receptor (CAR)-T cell therapy has emerged as an extremely powerful tool, particularly in the context of relapsed/refractory (R/R) B-cell malignancies and multiple myeloma, leading to the approval of six CAR-T cell products (as of early 2024) by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) (1, 2). CAR-T cells are genetically engineered T lymphocytes expressing artificial receptors, which typically combine the single-chain variable fragment (scFv) derived from a monoclonal antibody (mAb) with the intracellular signaling machinery of T cells. The goal is to elicit T-cell effector functions and cytotoxic activity upon the recognition of the selected target antigen (3). While the intracellular portion of first-generation CARs consists solely of the T-cell receptor (TCR) CD3ζ signaling domain, second- and third-generation CARs also contain one or two co-stimulatory domains, respectively (4).
Even though CAR-T cell therapy has significantly transformed immuno-oncology, its broader application to various hematological malignancies and solid tumors has encountered many obstacles. These challenges include, among others, tumor heterogeneity, lack of cancer-specific antigens (with the subsequent risk of on-target-off-tumor toxicities), and limited CAR-T cell expansion and persistence (5, 6).
Therefore, several strategies have been proposed to prevent antigen escape, improve CAR-T selectivity for malignant cells, and promote their long-term persistence (7).
A high degree of genomic instability is a hallmark of cancer, leading to the continuous accumulation of genetic and epigenetic alterations throughout the natural history of the disease (8). The emergence of genetically and phenotypically distinct malignant subclones can have a crucial role in tumor invasiveness, dissemination, and resistance to treatment. In this context, the selective pressure imposed by conventional single-targeted CAR-T cells could favor the development of antigen-negative neoplastic subpopulations, resulting in disease relapse. Clinical data regarding patients with B-cell malignancies treated with CD19.CAR-T cells indicate that up to 50% of them relapse within 12 months of initial infusion, and that antigen loss, downregulation or modulation represent pivotal mechanisms underlying treatment failure (9). To overcome these drawbacks, a considerable body of research has been focusing on next-generation engineering approaches, such as the adapter CAR (AdCAR) platform and the “OR” logic-gating system, allowing for the simultaneous or sequential targeting of different antigens by CAR-T cells (Figure 1).
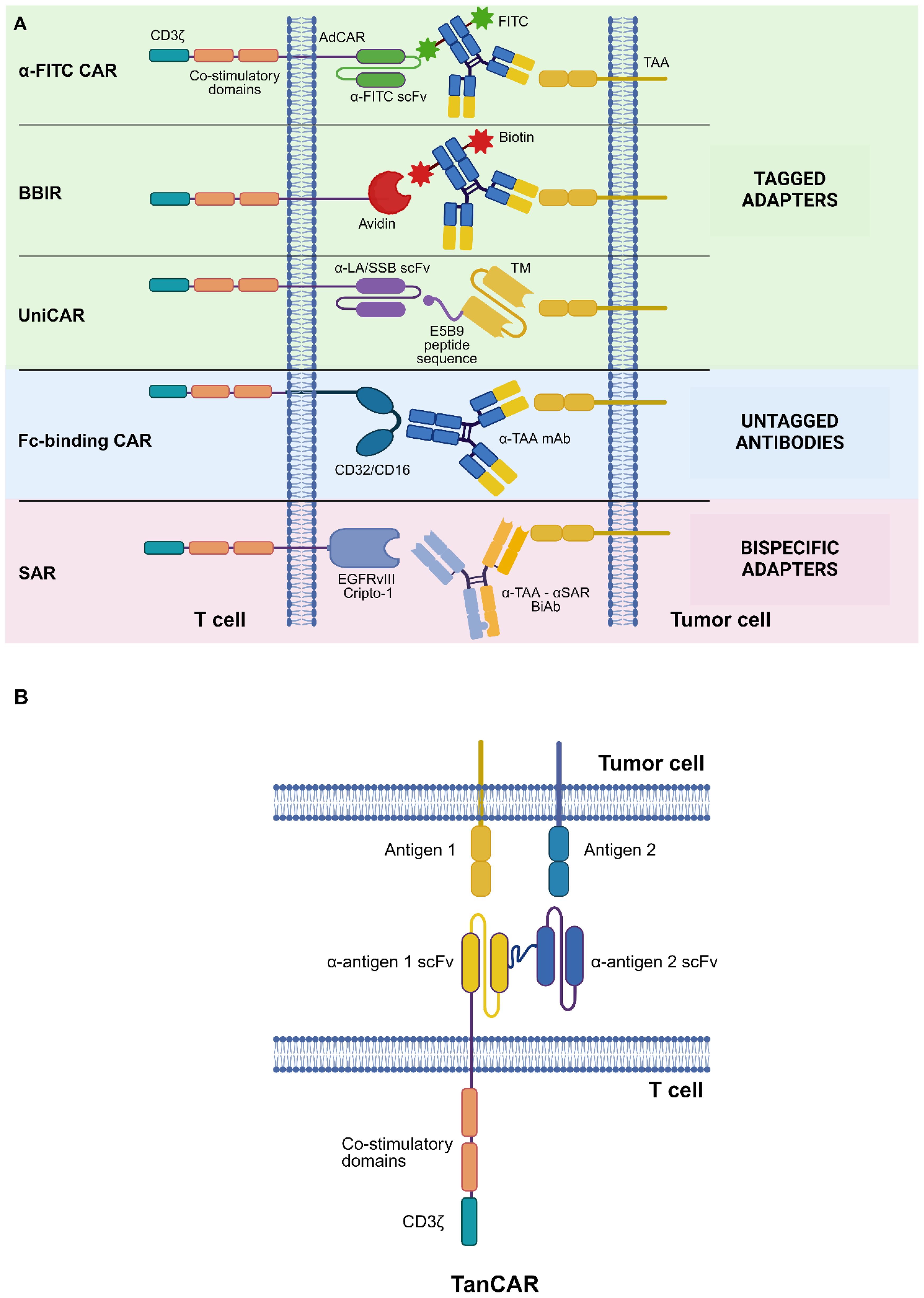
Figure 1 CAR design strategies to tackle tumor heterogeneity. (A) Adapter CAR (AdCAR) platform. Shown are tagged adapters, untagged antibodies (Abs), and bispecific adapters. (B) “OR” logic-gating: TanCAR system. See Glossary for abbreviations. Modified from Refs. 7, 10. Figure created with BioRender.com.
In the adapter CAR (AdCAR) system, T lymphocytes are transduced with a second- or third-generation CAR whose antigen-binding domain recognizes an exogenous adapter molecule (AM) rather than a membrane protein expressed on target cells. Adapters are bifunctional molecules with distinct binding domains, which allow them to bridge target antigen-expressing cells with AdCAR-T cells (10) (Figure 1A).
A key advantage of the AdCAR platform lies in the possibility of targeting multiple antigens with a single CAR-T cell product. The significant degree of flexibility of this therapeutic strategy allows to rapidly redirect the specificity of CAR-T cells towards a different target antigen by simply switching the AM. This approach is considerably more cost-effective and time-saving compared to re-isolating T lymphocytes, transducing them with a newly designed CAR, and performing a second CAR-T cell infusion. Another favorable characteristic of this design is that it allows to implement treatment-free intervals in the therapeutic protocol by simply withholding the AM. This, in turn, safeguards T-lymphocytes from exhaustion induced by chronic stimulation without the need for CAR-T cell depletion strategies.
Moreover, the AdCAR platform is precisely controllable, since the clearance of the AM leads to the cessation of CAR-T cell activity. In addition, modulating the concentration of the AM at the tumor site allows to fine-tune the magnitude of CAR-T cell response. Thus, it is possible to achieve satisfactory tumor control while avoiding untoward effects associated with T-cell hyperactivation (e.g., cytokine release syndrome [CRS]). In this regard, the AdCAR system can be considered a form of safety switch (specifically, a “killing switch”), allowing regulated CAR-T cell activation (11). Other types of switches specifically conceived to enhance CAR-T cell safety will be discussed in the following sections.
Potential limitations of the AdCAR approach are related to the fact that AdCAR-T cells are functionally inert in the absence of the corresponding AMs. For this reason, the AM concentration at the tumor site needs to be maintained above the threshold required for an efficient CAR-T cell – tumor cell bridging for an adequate amount of time. Moreover, some of the extracellular domains that can be incorporated into the AdCAR structure, particularly avidin and streptavidin, are immunogenic. Lastly, the use of Fc-glycoengineered antibodies (Abs) as AMs has been associated with the occurrence of CRS (12).
Three main classes of AMs are recognized, namely tagged adapters, untagged immunoglobulins (Igs), and bispecific adapters (10).
Tagged adapters can be labeled with various molecules, each of which allows them to bind the extracellular domain of an aptly designed AdCAR.
Anti-fluorescein isothiocyanate (FITC) AdCAR-T cells in combination with FITC-labeled rituximab demonstrated a remarkable antitumor efficacy in an immunocompetent mouse model of rituximab-resistant B-cell non-Hodgkin lymphoma (NHL) (13).
Similarly, it has been shown that FITC-labeled trastuzumab allows to redirect AdCAR-T cells against epidermal growth factor receptor 2 (HER2)-expressing breast cancer cells, resulting in the eradication of established tumors in xenograft mouse models (14).
Biotinylated adapters have been employed in conjunction with AdCARs whose antigen-binding domain consists of avidin or streptavidin: biotin-binding immune receptor (BBIR) T cells in combination with biotinylated anti-mesothelin (MSLN) Abs have been shown to possess a potent antitumor activity against MSLN+ ovarian cancer, both in vitro and in vivo (15).
Nixdorf and colleagues designed and tested a biotin-directed AdCAR in combination with biotinylated anti-CD33, anti-human C-type lectin-like molecule-1 (CLL-1), or anti-CD123 AMs against acute myeloid leukemia (AML) cell lines and primary AML (pAML), both in vitro and in vivo. AdCAR-T cells demonstrated a specific cytotoxic activity when co-cultured with AML cell lines and pAML cells. Interestingly, their cytolytic potential and later effector functions were markedly improved by the intermittent rather than continuous stimulation with AMs, possibly due to a transcriptional reprogramming mechanism. This reprogramming resulted in an enhanced expression of memory-related genes during periods of functional rest, with the subsequent re-expression of activation markers upon restimulation. Furthermore, the sequential administration of AMs of different specificities was found to significantly improve tumor control by AdCAR-T cells in a long-term co-culture with pAML blasts, underscoring the potential of the AdCAR strategy to target heterogeneous tumors (16).
Target modules (TMs) are AMs tagged with a short peptide sequence, such as the E5B9 epitope of the La/SS-B nuclear protein, which is recognized by an AdCAR bearing an anti-La scFv as its extracellular domain: the corresponding AdCAR platform, known as the “UniCAR” system, has shown promise in preclinical models of various malignancies, including AML, B-cell NHL, disialoganglioside GD2+ neuroblastoma, and sialyl-Tn (STn)+ breast cancer (17–22).
A further development of the “UniCAR” platform involves the use of a trimeric TM consisting of an antigen-binding domain, the E5B9 epitope, and a single-chain format of the 4-1BB ligand (4-1BBL). The latter is the natural ligand of the 4-1BB co-stimulatory receptor, which plays a crucial role in eliciting the “second signal” required for T-cell activation. The incorporation of the 4-1BBL into the structure of TMs provides UniCAR-T cells with transient co-stimulation at the tumor site, potentially boosting their effector functions. Indeed, a preclinical study focusing on AML has shown that trimeric TMs directed against CD123 (TM123-4-1BBL) improve the persistence and long-term cytolytic capabilities of UniCAR-T cells as compared to conventional TM123 modules both in vitro and in vivo. An additional advantage of trimeric TMs was identified in their prolonged terminal plasma half-life, due to their higher hydrodynamic volume as compared to TM123 modules. This, in turn, could allow for more sustained anticancer responses by UniCAR-T cells. Of note, co-culture experiments with the MOLM-13 AML line showed that a significant cytokine secretion by UniCAR-T cells is elicited only at concentrations of the TM123-4-1BBL module much higher than those required for tumor cell killing. This could have important safety implications, as UniCAR-T cells might eradicate bulky tumors without causing cytokine-mediated adverse effects (23).
Interestingly, tagged adapters can also be employed to selectively modulate the functional properties of AdCAR-T cells without affecting bystander (CAR-) cells. Luo and colleagues demonstrated that a FITC-labeled toll-like receptor-7 agonist (FITC-TLR7a) can restore the antitumor activity and proliferative capacities of exhausted FITC.CAR-T cells both in vitro and in vivo without significant toxicities. The proposed mechanism involves internalization of the FITC-TLR7a – CAR complex via receptor-mediated endocytosis. This is followed by TLR-7 activation in the endosomal compartment of AdCAR-T cells (24).
Untagged Abs can function as AMs for AdCARs bearing the extracellular portion of FcγRIII (CD16) or FcγRII (CD32) as their antigen-binding domain (25).
CD16-based AdCAR-T cells engineered to express a high-affinity CD16 variant (CD16V-BB-ζ T lymphocytes) were shown to be highly cytolytic against the Daudi lymphoma cell line or primary chronic lymphocytic leukemia (CLL) cells in the presence of rituximab; against osteosarcoma or neuroblastoma cells in the presence of an anti-GD2 mAb; or against HER2-expressing stomach and breast cancer cells in combination with trastuzumab. These results were also validated in vivo, where CD16V-BB-ζ T cells exerted remarkable antitumor effects in immunodeficient mice engrafted with the NB1691 neuroblastoma cell line (in the presence of an anti-GD2 mAb) or with the Daudi cell line (in combination with rituximab) (26).
Besides modifying the CD16 sequence, it is possible to employ glycoengineered Abs with increased CD16 affinity as AMs. Rataj and colleagues demonstrated that the Fc-modified anti-CD20 Ab obinutuzumab allows to boost the activation and antitumor functionality of CD16.CAR-T cells against the CD20+ Raji lymphoma cell line when compared to a non-glycoengineered anti-CD20 Ab recognizing the same epitope. Similarly, in a preclinical model of malignant melanoma, improved tumor cell killing was observed when combining CD16-CAR-T cells with the glycoengineered anti-melanoma-associated chondroitin sulfate proteoglycan (MCSP) Ab LC007 as compared to the corresponding wild-type (WT) Ab (12).
Bispecific adapters are artificial molecules designed to simultaneously bind multiple target antigens. They generally consist of bispecific antibodies (BiAbs) bearing two distinct scFvs, one of which recognizes a given tumor-associated antigen (TAA) and the other binds to a suitable AdCAR.
In the synthetic agonist receptor (SAR) platform, T lymphocytes are transduced with AdCARs bearing the ECD of the Cripto-1 embryonic antigen or the EGFR variant III (EGFRvIII, which is not expressed in normal tissues). Engineered cells are then administered in combination with bispecific adapters that simultaneously bind a designated TAA and either Cripto-1 or EGFRvIII. It has been shown that SAR-T cells, in conjunction with an anti-epithelial cell adhesion molecule (EpCAM) x anti-EGFRvIII BiAb, mediate potent cytotoxic effects upon EpCAM+ tumor cell encounter in vitro. Moreover, the administration of SAR-T cells together with an anti-MSLN x anti-EGFRvIII BiAb was found to significantly prolong the survival of immunodeficient mice engrafted with MSLN+ pancreatic cancer cells, outperforming conventional MSLN.CAR-T cells (27).
Given that osteosarcoma cells frequently overexpress folate receptors, the NCT05312411 phase I trial is evaluating the use of second-generation anti-FITC AdCAR-T cells in combination with FITC-conjugated folate as a treatment strategy for R/R osteosarcoma in young adults (28).
The NCT04230265 dose-escalating phase I trial is evaluating the UniCAR-T-CD123 system AVC-101, developed by AvenCell Therapeutics (Cambridge, MA, USA), as a treatment strategy for adult patients with heavily-pretreated, R/R AML or blastic plasmacytoid dendritic cell neoplasm (BPDCN) (29, 30). This platform takes advantage of a universal CAR that recognizes a CD123-specific TM to redirect UniCAR-T cells towards CD123+ targets. Preliminary results indicated an acceptable safety profile and a meaningful clinical activity of this UniCAR system, with two complete remissions with incomplete hematologic recovery (CRi) and a partial remission (PR) observed among the three patients who completed treatment (31). In subsequent reports, an encouraging clinical efficacy was observed, with an objective response rate (ORR) of 53% in the first 19 AML patients who completed the full treatment course (32, 33).
The UniCAR system is also being tested in solid tumors: the NCT04633148 dose-escalating phase I trial is currently recruiting patients with prostate-specific membrane antigen (PSMA)+ prostate cancer to evaluate the safety and efficacy of the PSMA-directed UniCAR platform AVC-102 (also developed by AvenCell Therapeutics) (34).
The CD16-BB-ζ AdCAR-T cell system ACTR087, developed by Cogent Biosciences (Cambridge, MA, USA), has been evaluated in combination with rituximab in patients with R/R B-cell NHL unresponsive to rituximab monotherapy (NCT02776813). In all dose levels (DLs) tested, an ORR of 50% was reported, with an adverse event profile consisting of two cases of severe immune effector cell-associated neurotoxicity syndrome (ICANS) and four cases of severe CRS, leading to the death of three patients (35, 36).
Another CD16-based AdCAR-T cell product (ACTR707, also developed by Cogent Biosciences) has been evaluated in conjunction with trastuzumab in patients with HER2-expressing advanced solid tumors, showing no dose-limiting adverse events (NCT03680560) (37).
Of note, the use of Fc-binding AdCAR-T cells should be avoided in patients with hypergammaglobulinemia, due to the risk of adverse events stemming from T-cell hyperactivation.
The application of Boolean-logic gating to CAR-T cell design has been envisioned as a promising strategy to address tumor heterogeneity as well as to improve CAR-T cell safety (as will be discussed in the next sections) (38). Regarding the first point, the “OR” logic gate allows to target a heterogeneous tumor population and to retain CAR-T cell efficacy even in the face of clonal escape or antigen-negative relapse.
Among “OR” logic-gated platforms, the tandem CAR (TanCAR) system entails the engineering of T lymphocytes to express a CAR with two distinct scFvs connected in tandem via a flexible linker. This strategy allows to trigger a T-cell response upon the engagement of either of the two targeted antigens. Notably, the simultaneous binding of both target antigens elicits an exponentially more potent response by TanCAR-T cells, pointing towards a synergistic effect of double-binding on their cytotoxic capabilities (39) (Figure 1B). This bivalency boosts CAR signal strength, possibly resulting in a superior antitumor functionality as compared to, e.g., the AdCAR system.
However, an excessive CAR signal strength could promote CAR-T cell exhaustion, limit their persistence, and increase the risk of adverse events. In addition, this next-generation CAR design is not suitable for targeting TAAs with a significant degree of expression on normal tissues, due to the risk of intolerable off-tumor toxicities. Other limitations are related to the size of the TanCAR transgene, which could exceed the cargo capacity of lentiviral vectors used for transduction, as well as the possibility of cross-linking events involving the light and heavy chain variable (VL-VH) domains of a given TanCAR (39).
In a study by Zah and colleagues, CD19 x CD20 TanCAR-T cells outperformed single-targeted CD19.CAR-T cells in terms of cytotoxic activity after co-culture with WT (CD19+CD20+) or CD19-knockout (KO) Raji cells. TanCAR-T cells retained their effector functions even after repeated antigen stimulation. Moreover, they showed remarkable levels of tumor control in immunodeficient mice engrafted with WT or a combination of WT plus CD19- Raji cells (40).
In solid tumors, Grada et al. tested CD19 x HER2 TanCAR-T cells against the HER2+ Daoy medulloblastoma cell line modified to artificially express the CD19 antigen. The induction of CD19 expression on target cells resulted in an exponential enhancement in TanCAR-T cell killing and interferon gamma (IFN-γ) secretion. Importantly, TanCAR-T cells retained their cytolytic capabilities in the context of HER2 downregulation on target cells. In a xenograft mouse model of medulloblastoma, CD19 x HER2 TanCAR-T cells significantly prolonged the survival of treated mice, particularly after the induction of CD19 expression on target cells (41).
Moreover, patient-derived TanCAR-T cells redirected against HER2 and the α2 subunit of the interleukin (IL)-13 receptor (HER2 x IL13Rα2 TanCAR-T cells) were shown to induce higher levels of cytolysis against autologous glioblastoma multiforme (GBM) cells compared to single targeted HER2.CAR- or IL13Rα2.CAR-T cells, dual CAR-T cells co-expressing the anti-HER2 and anti- IL13Rα2 CARs, or a mixture of HER2.CAR- and IL13Rα2.CAR-T cells (pooled CAR-T cells). TanCAR-T cells also showed a significantly enhanced cytokine production and a more sustained antitumor activity after co-culture with the U373 GBM cell line. Notably, concomitant HER2 and IL13Rα2 binding was found to induce a synergistic effect on cytokine production by TanCAR-T cells. Mice engrafted with GBM cells and treated with TanCAR-T cells had a significantly prolonged survival compared to those receiving single-targeted CAR-T cells. This next-generation CAR platform also allowed to counteract antigen escape. Indeed, tumors relapsing after TanCAR-T cell infusion did not show complete antigen loss, but rather maintained low levels of expression of both the HER2 and IL13Rα2 antigens (42).
Interestingly, TanCARs can be repurposed for the simultaneous targeting of malignant cells and elements of the tumor microenvironment (TME). This could be especially useful to disrupt the complex interplay between tumor cells and bystander elements, which has been shown to contribute to cancer growth and invasiveness (43). Alberti et al. designed CD33 x CD146 TanCAR-cytokine-induced killer (CIK) cells to concomitantly target CD33+ AML blasts and CD146+ bone marrow (BM) mesenchymal stromal cells (44). The aim was to counteract the pro-leukemic and immunosuppressive effects of the BM niche mediated, at least in part, by this specific stromal component (45–47).
As for the clinical translation of the TanCAR system, the NCT03019055 phase I/Ib study evaluated CD20 x CD19 TanCAR-T cells in subjects with R/R CD19+ or CD20+ B-cell cancers. Investigators reported an overall response rate of 82% at day 28 from initial infusion, with a 64% CR rate. Among the 12 patients who received the CAR-T cell dose chosen for expansion, the overall response rate was 100%, with 92% of them achieving CRs. Even in primary refractory cases or in those who experienced a subsequent relapse, no evidence of CD19 loss was found. Observed toxicities included grade 3-4 neurotoxicity (in 3 out of 22 patients) and grade 3-4 CRS (in 1 individual) (48).
The NCT03614858 trial assessed the safety and efficacy of CD19 x CD22 TanCAR-T cells in patients with R/R B-ALL. The reported 1 – and 2-year overall survival (OS) rates were 76.2% and 73.8%, respectively. 12 out of 47 patients suffered leukemia relapse, but antigen loss was observed in only 2 of these cases (who relapsed with CD19-CD22+ disease) (49).
To date, the identification of tumor-specific antigens (TSAs) suitable for CAR-T cell targeting has largely remained elusive. Indeed, many TSAs have an intracellular localization, so potential tumor neoepitopes are mainly presented on the cancer cell surface in association with human leukocyte antigen (HLA) molecules (50). Unfortunately, being HLA-unrestricted, conventional CAR-T cells can only recognize cell surface antigens not associated with HLA proteins.
A potential solution could be offered by TCR-CARs or TCR-like CARs. TCR-CARs are composed of a TCR variable (TCRv) domain targeting a peptide-HLA (pHLA) complex, linked to a hinge/transmembrane (H/T) region and an intracytoplasmic tail shared with conventional CARs. TCR-like CARs have the same structure, albeit with an anti-pHLA scFv instead of an anti-pHLA TCRv. This allows to avoid possible rearrangements between the native and artificial TCR αβ chains, abating the risk of autoimmune toxicities resulting from the generation of autoreactive TCRv fragments (51).
However, it should be noted that most neoepitopes are different between patients, requiring personalized scFv or TCRv designs for each individual patient. Moreover, HLA molecules are frequently downregulated by malignant cells as an immune escape mechanism, hindering their recognition by TCR – or TCR-like CARs (52).
In the next sections, we will focus on on-target-off-tumor toxicities caused by conventional CAR-T cells targeting TAAs. A notable example is the prolonged B-cell aplasia observed in patients treated with CD19.CAR-T cells, due to the expression of CD19 on normal B lymphocytes. While this specific toxicity is manageable with Ig replacement therapy, the same does not hold true in the context of other hematological as well as solid tumors. For instance, targeting the CD33 myeloid marker in AML can produce an intolerable myelosuppression, making it necessary to proceed to allogeneic hematopoietic stem cell transplantation (alloHSCT) in all treated patients (53). Severe adverse events attributable to off-tumor effects were also reported in clinical trials involving patients with metastatic solid tumors: evidence of hepatotoxicity was observed in a clinical study evaluating carbonic anhydrase-IX (CAIX).CAR-T cells for the treatment of metastatic renal cell carcinoma. This was attributed to off-tumor effects against CAIX+ bile duct epithelial cells (54). Moreover, a patient with lung metastases from colon cancer suffered fatal acute respiratory distress syndrome (ARDS) after the infusion of HER2.CAR-T cells, due to off-tumor effects against HER2dim lung epithelial cells (55).
Therefore, considerable research efforts have been directed towards improving CAR-T cell safety profile (Figure 2).
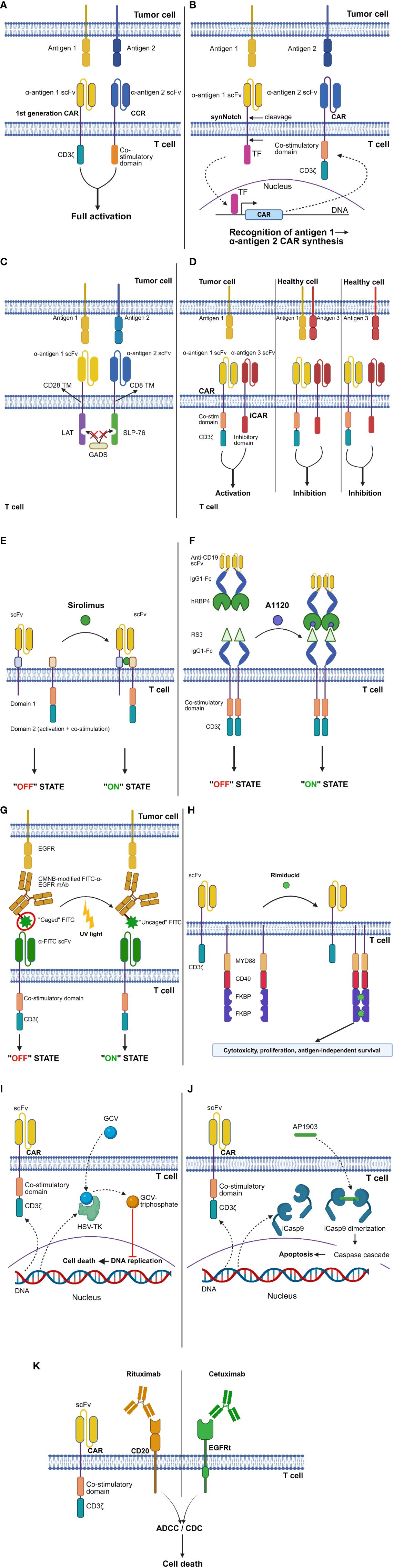
Figure 2 CAR design strategies to improve safety. (A) “AND” logic-gating: “combinatorial antigen recognition with balanced signaling”. (B) “AND” logic-gating: synthetic Notch (synNotch) platform. (C) “AND” logic-gating: logic-gated intracellular network (LINK) system. (D) “NOT” logic-gating: inhibitory CAR (iCAR) strategy. (E) ON-switches: dimerizing agent-regulated immunoreceptor complex (DARIC) platform. (F) ON-switches: human retinol binding protein 4 (hRBP4)-RS3 switch. (G) ON-switches: light-controlled switch. (H) ON-switches: GoCAR-T® platform. (I) OFF-switches: Herpes simplex virus thymidine kinase (HSV-TK) switch. (J) OFF-switches: inducible caspase 9 (iCasp9) switch. (K) OFF-switches: truncated epidermal growth factor receptor (EGFRt)-cetuximab and CD20-rituximab switches. See Glossary for abbreviations. Modified from Refs. 7, 56–60. Figure created with BioRender.com.
A relatively straightforward approach could entail the induction of a transient CAR expression by delivering the corresponding transgene via mRNA electroporation (61). The key advantage of a short-lived CAR expression lies in the rapid termination of any possible off-tumor effects. In addition, RNA CAR-T cell manufacturing is more rapid and cost-effective compared to viral vector- or transposon-based approaches. This technique also allows to transduce T lymphocytes with multiple constructs, thereby broadening their specificity. Due to the transient nature of CAR expression, a drawback of this transfection approach could lie in the induction of weaker and less sustained antitumor effects by RNA CAR-T cells. However, this could potentially be overcome by repeated CAR-T cell administration.
Disappointing results in terms of efficacy have been reported in a phase I trial of mRNA-engineered CD123.CAR-T cells in patients with R/R CD123+ AML (NCT02623582) (62). This resulted in the early termination of the study and prompted the investigators to employ a lentiviral system to achieve a stable CD123 CAR expression. Therefore, a separate clinical study involving lentivirally transduced CD123.CAR-T cells has been planned.
In another trial, transient responses were observed in subjects with R/R Hodgkin lymphoma (HL) treated with mRNA-electroporated CD19.CAR-T cells, without severe toxicities (NCT02277522) (63).
Lastly, in a phase I study of mRNA-engineered MSLN.CAR-T cells for the treatment of metastatic ductal adenocarcinoma of the pancreas (NCT04809766), disease stabilization was observed in 2 out of 6 treated patients. The total metabolic active tumor volume substantially decreased in one patient, with a radiological CR in liver metastases even in the absence of effects on the primary tumor. No cases of CRS or dose-limiting toxicities were reported (64).
As mentioned above, logic-gating can also be exploited to improve the safety profile of CAR-T cells. Specifically, the “AND” – and “NOT” gating strategies have been developed to mitigate on-target-off-tumor toxicities (7, 11, 39).
In a 2013 study, Kloss and colleagues introduced a novel approach called “combinatorial antigen recognition with balanced signaling” (38). This method involves splitting the T-cell signaling and co-stimulatory domains, typically combined in cis in the traditional CAR design, into 2 separate chimeric receptors. The latter target different antigens and are delivered together to T lymphocytes, often in a single construct. One of these receptors is usually a first-generation CAR, while the other is termed a “chimeric co-stimulatory receptor” (CCR). The CCR is characterized by an intracellular portion comprising only co-stimulatory domains, such as CD28 and 4-1BB. The aim is to elicit a full-blown cytotoxic response by CAR-T cells only against double-positive but not single antigen-expressing cells, due to the lack of the “second signal” required for CAR-T cell activation in the latter scenario (Figure 2A). However, the achievement of true cancer specificity requires a low-affinity scFv as the extracellular domain of the first-generation CAR, since CARs bearing only the CD3ζ intracellular moiety are still able to elicit T-cell cytolytic functions. In the above-mentioned study, CAR-T cells simultaneously expressing a first-generation, high-affinity anti-CD19 CAR and an anti-PSMA CCR eradicated not only double positive (CD19+PSMA+) cancers, but also CD19+PSMA- ones. Instead, the administration of CAR-T cells with an affinity-tuned anti-PSCA CAR and an anti-PSMA CCR resulted in the eradication of only double-positive (PSCA+PSMA+) prostate cancer cells. Interestingly, neoplasms expressing only the antigen recognized by the low-affinity first-generation CAR (PSCA+PSMA-) were spared even in mice concomitantly engrafted with double positive (PSCA+PSMA+) tumor cells in a separate anatomical location. This confirms that T-cell activation remains limited to the double-positive tumor niche (38).
Perriello et al. exploited the “AND”-gating approach in AML by engineering CIK cells with a CD33.CCR and a low-affinity IL-3 zetakine recognizing the CD123 antigen. This “AND”-gated product was highly cytolytic against AML cell lines in vitro and significantly prolonged the survival of immunodeficient mice engrafted with the KG-1 AML cell line. No significant toxicities against CD33+CD123+ hematopoietic stem cells (HSCs) and CD123+ endothelial cells were observed in vitro (65).
The synthetic Notch (synNotch) platform is a modular “AND”-gating strategy based on the basic framework of Notch signaling (66). Upon the recognition of a specific target molecule via its antigen-binding portion, the synNotch receptor undergoes cleavage in its intracellular domain, releasing a transcription factor that promotes the synthesis of a CAR directed against a different antigen (Figure 2B). This allows to prevent T-cell activation against tissues expressing either one of the two target antigens, while a complete cytotoxic response is elicited upon the encounter of double-positive tumor cells. Morsut et al. first demonstrated the feasibility of replacing the extracellular and intracellular portions of the WT Notch receptor with specific protein domains to drive novel intercellular signaling cascades (67). The same research group showed that primary T cells equipped with an anti-green fluorescent protein (GFP) synNotch receptor, whose intracellular domain can induce the transcription of an anti-CD19 CAR, undergo activation only after co-culture with target cells expressing both the designated antigens. This spatially controlled CAR synthesis was confirmed in vivo by generating xenograft mouse models simultaneously bearing WT (CD19+GFP-) and modified (CD19+GFP+) Daudi tumor cells (in separate body sites). Engineered T cells showed evidence of CAR expression in the CD19+GFP+ but not in the CD19+GFP- Daudi tumor. Compared to non-transduced T lymphocytes, synNotch-CAR-T cells significantly prolonged the survival of mice with CD19+GFP+ but not CD19+GFP- tumors. Due to the relatively short half-life of CAR expression after cessation of the synNotch signal, CAR-T cells primed by GFP+ tumors did not show any significant cytotoxic activity against single-positive CD19 tumors established in a separate body site. Thus, in vivo T-cell activation remained confined to the double-positive tumor (68).
synNotch receptors can also be designed to recognize peptide-HLA complexes, thereby allowing engineered T cells to respond to intracellular antigens. Williams et al. transduced primary T cells with an anti-HLA-A2 – α-fetoprotein (AFP) synNotch controlling the transcription of an anti-HER2 CAR. This synthetic circuit allowed for the selective killing of double-positive but not of single-positive targets (69).
A potential limitation of the synNotch system, which is shared with other “AND” logic-gating strategies, is that the downregulation of either or both target antigens could allow tumors to escape CAR-T cell-mediated killing. However, the same research group proved that the synNotch platform can be used to design more complex cellular recognition circuits. Specifically, they conceived a 3-input “AND”-gating system, designed either in series or in parallel. The in-series circuit was obtained by engineering T cells with an anti-EGFR synNotch activating the synthesis of an anti-hepatocyte growth factor receptor (anti-MET) synNotch, in turn controlling the transcription of a HER2.CAR. As for the in-parallel circuit, the first (anti-EGFR) synNotch activated the expression of a secreted adapter composed of an anti-HER2 scFv linked to a peptide neo-epitope (PNE), while the second (anti-MET) synNotch induced the synthesis of a PNE.CAR. Whereas the in-series circuit induced the selective killing of only triple-positive targets, the in-parallel circuit demonstrated a cytotoxic activity also against dual-positive EGFR+HER2+ cells. This was ascribed to antigen-independent activation of the anti-MET synNotch, leading to the synthesis of the PNE.
Lastly, they showed that it is possible to combine different forms of logic gating in a single cellular recognition circuit. To this end, they engineered T cells with an anti-GFP synNotch receptor activating the expression of a conventional CD19.CAR, in combination with an anti-HER2 synNotch receptor controlling the synthesis of the proapoptotic factor truncated BH3-interacting domain death agonist (tBID). This “AND-NOT” circuit spared HER2+ target cells irrespective of their GFP and/or CD19 expression, due to the activation of the so-called OFF-Notch. HER2-GFP-CD19+ target cells were also spared, since the absence of the GFP antigen prevented CD19.CAR synthesis. On the other hand, HER2-GFP+CD19+ targets were efficiently killed (69).
A peculiar form of “AND”-gating strategy recently proposed by Tousley et al. is the Logic-gated Intracellular NetworK (LINK) system (Figure 2C). This approach is unique in that it leverages intracellular mediators involved in the TCR signaling cascade (i.e., linker for activation of T cells [LAT] and SH2 domain-containing leukocyte protein of 76 kDa [SLP-76]) to design CARs devoid of the CD3ζ intracellular domain. Specifically, T lymphocytes were engineered with two chimeric receptors directed against separate antigens and bearing either the LAT or the SLP-76 intracellular domains, with the aim to completely abolish CAR-T cell activation against single antigen-expressing targets. However, T lymphocytes continued to respond to single-positive cells due to the heterodimerization of LAT and SLP-76 mediated by the GRB2-related adaptor downstream of Shc (GADS) protein. Therefore, targeted mutations were strategically introduced into the LAT and SLP-76 coding sequences to disrupt GADS binding, resulting in an absolute degree of tumor-selective killing. The LINK platform was found to outperform other “AND” logic-gating systems, such as the synNotch strategy, in terms of both in vivo antitumor activity and safety. Indeed, undesired toxicities were observed in mice treated with synNotch – or conventional CAR-T cells but not in those receiving LINK-CAR-T cells (56).
The NCT06186401 phase I trial is being conducted to assess the safety and efficacy of synNotch CAR-T cells in patients with EGFRvIII+ recurrent GBM. Specifically, this platform consists of an anti-EGFRvIII synNotch receptor activating the transcription of a TanCAR directed against IL13Rα2 and ephrin type-A receptor 2 (EphA2) (70). The rationale behind this study is based on preclinical research indicating that IL13Rα2 x EphA2 TanCAR-T cells show significantly greater antitumor activity compared to single-targeted IL13Rα2.CAR – or EphA2.CAR-T cells, both in vitro and in murine xenograft models (71).
Under physiological conditions, T-cell activation is tightly controlled by a balance between stimulatory and inhibitory signals. The latter derive, for example, from immune checkpoint receptors like programmed cell death protein 1 (PD-1) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) (72). On these grounds, a “NOT”-gating strategy has been proposed that entails the engineering of T cells with a conventional CAR plus a separate chimeric receptor, known as “inhibitory CAR” (iCAR). The latter recognizes a different antigen and bears the intracellular portion of the PD-1 or CTLA-4 molecule in its cytoplasmic tail. In this way, the engagement of the iCAR ligand (I) results in CAR-T cell inhibition, even in cases where the antigen targeted by the conventional CAR (A) is present on the same target cell. CAR-T cell activation is thus observed only against A+I- tissues (Figure 2D). A remarkable drawback of the iCAR approach is that it only allows to generate monospecific cellular products. This, in turn, poses the risk of antigen-negative tumor relapses.
Fedorov and colleagues demonstrated that the artificial expression of an anti-CD19 CAR and an anti-PSMA iCAR in primary T cells significantly impairs their cytotoxic potential, cytokine secretion, and proliferative abilities in a co-culture with CD19+PSMA+ artificial antigen-presenting cells (AAPCs). On the other hand, no detrimental effects on iCAR-T cell effector functions are seen against targets lacking the antigen recognized by the iCAR (i.e., CD19+PSMA- AAPCs). This selective inhibition of CAR-T cell function was confirmed in a xenograft mouse model, where iCAR-T cells eradicated CD19+PSMA- but not double-positive B-cell tumors. Of note, iCAR engagement did not permanently impair the functional properties of engineered T lymphocytes: even after being challenged with CD19+PSMA+ AAPCs, iCAR-T cells showed similar levels of cytotoxicity, cytokine production, and proliferation as compared to conventional CD19.CAR-T cells in a subsequent co-culture with CD19+PSMA- AAPCs (73).
The “NOT”-gating approach has also been employed in the setting of AML, allowing to target the leukemic stem cell (LSC) marker CD93 (74) while sparing CD93+ healthy endothelial cells. Richards et al. engineered T cells to express a conventional CD93.CAR in combination with anti-CD19 iCARs. CD93.CAR-T cells bearing a PD-1-based iCAR showed a significantly lower cytokine secretion compared to CD93.CAR-T cells when challenged with CD93+ endothelial cells modified to co-express CD19. Importantly, these “NOT”-gated CAR-T cells demonstrated preserved cytokine release upon co-culture with the THP-1 AML line (75).
Other “NOT”-gated platforms leverage the loss of heterozygosity (LOH) for the HLA-A*02 antigen, which frequently occurs in malignant cells but not in healthy tissues. For instance, the A2B694 “NOT” logic-gated platform (developed by A2 Biotherapeutics, Inc., Westlake Village, CA, USA) is composed of a third generation MSLN.CAR and an anti-HLA-A*02 iCAR bearing the leukocyte immunoglobulin-like receptor (LIR)-1 intracellular domain. This allows for the selective eradication of MSLN+ tumor cells showing HLA-A*02 LOH, while sparing MSLN+ healthy tissues with preserved HLA-A*02 expression (76).
The basic iCAR design was recently improved by Bangayan and colleagues, who designed iCARs bearing multiple inhibitory domains in their cytoplasmic tail [e.g., PD-1 – PD-1, PD-1 – leukocyte-associated immunoglobulin-like receptor-1 (LAIR-1)]. This novel platform, known as “DiCAR”, outperformed a PD-1-based iCAR system in terms of efficiency. It also produced faster inhibition kinetics, significantly reducing the delay in T-cell inhibition classically observed with first-generation iCARs (77).
As for the clinical translation of “NOT”-gating approaches, the NCT04981119 observational study was conducted to screen subjects with advanced colorectal, pancreatic, or non-small cell lung cancer for HLA-A*02 LOH (78). The aim was to identify patients eligible for a subsequent interventional study (NCT06051695) assessing the above-mentioned A2B694 platform for the treatment of these malignancies. As of early 2024, the NCT06051695 trial is in the recruitment phase (79).
Another strategy to improve the safety of cellular immunotherapies consists in controlling CAR-T cell activation status via the incorporation of safety switches. Specifically, ON-switches promote CAR-T cell transition from an inactive towards a reactive state in a precisely controllable fashion. In contrast, OFF-switches allow for the termination of CAR-T cell activity in the event of serious toxicities. Several classes of safety switches have been developed. Here, we will focus mainly on small molecule-based systems.
The dimerizing agent-regulated immunoreceptor complex (DARIC) is an ON-switch system whereby the intracellular portion and antigen-binding region of conventional CARs are split into 2 separate proteins co-expressed by engineered T cells (Figure 2E). Therefore, even in the presence of target antigen-expressing cells, CAR-T cells remain functionally inert. The supplementation of the mammalian target of rapamycin (mTOR) inhibitor sirolimus results in the dimerization of the two DARIC proteins, restoring CAR-T cell responsiveness (“ON” state). In the presence of sirolimus, CD19.DARIC-CAR-T cells were shown to mediate potent antigen-dependent cytolysis against the Nalm-6 ALL line both in vitro and in vivo. Only minimal cytotoxicity was observed against the same ALL line in the absence of sirolimus. Of note, the cytotoxicity kinetics were found to be similar between CD19.DARIC-CAR-T cells and conventional CD19.CAR-T cells (57).
Zajc and colleagues designed an ON-switch system composed of human retinol binding protein 4 (hRBP4) in combination with a binder derived from an archaeal protein (RS3). In this system, the binding affinity between its 2 components is greatly increased in the presence of the small molecule A1120. The authors exploited this ON-switch platform to finely regulate CAR-T cell activation: primary T cells were transduced with a CAR whose extracellular portion consisted of the above-mentioned binder linked to an IgG1-Fc spacer domain. The other component of the system (“chain II”) was the hRBP4 protein linked to an anti-CD19 scFv (Figure 2F). In the concomitant presence of chain II and A1120, ON-switch CAR-T cells were found to be comparable to conventional CD19.CAR-T cells in terms of cytokine release and antigen-specific cytotoxicity against the Nalm-6 ALL line. However, when A1120 was not added to the co-culture, the antileukemic activity and cytokine production of ON-switch CAR-T cells became similar to those of control T cells, even in the presence of chain II (58).
Besides small molecule-controlled systems, light-operated switches have also been developed. Kobayashi et al. covalently modified FITC-labeled anti-EGFR mAbs with 5-carboxymethoxy-2-nitrobenzyl (CMNB) caging groups, to hinder FITC recognition by FITC.CAR-T cells (Figure 2G). The latter effectively killed EGFR+ targets in the presence of uncaged FITC-anti-EGFR mAbs. On the other hand, they failed to exert significant cytotoxicity and to release effector cytokines when combined with CMNB-modified (“caged”) FITC-anti-EGFR mAbs.
UV light exposure restored FITC.CAR-T cell reactivity against EGFR+ cells in the presence of CMNB-modified mAbs, thus acting as an inducible and controllable switch. Importantly, no cellular damage due to UV light was reported (59).
The applicability of this ON-switch system could be hindered by poor tissue penetrance of UV light. However, this limitation could be overcome by employing advanced light-delivering devices such as optical-lens microneedle arrays (80).
The GoCAR-T® platform employs a peculiar type of ON-switch, whereby T lymphocytes are transduced with a first-generation CAR and an inducible MyD88/CD40 (iMC) molecule. The latter is a membrane protein composed of truncated CD40 and truncated MyD88 fused with 2 FK506 binding protein (FKBP) domains, which induce iMC dimerization in the presence of the small molecule rimiducid (Figure 2H). This platform is unique in that GoCAR-T cells are not completely dependent on the ON-switch to exert their effector functions (since they express a fully functional first-generation CAR). Rather, the ON-switch boosts GoCAR-T cell activation by providing them with co-stimulatory signals.
Foster et al. demonstrated that iMC dimerization results in enhanced proliferation, persistence, and antitumor efficacy of PSCA.CAR-T cells against PSCA+ pancreatic cancer in vivo. In addition, iMC activation was shown to induce antigen-independent CAR-T cell survival. This could be especially useful, e.g., to promote CAR-T cell engraftment in the context of a low tumor burden or to prolong CAR-T cell persistence after tumor eradication (60).
Similarly to AdCAR-T cells, a potential limitation of ON-switches is that the concentration of the switch activator in the tumor niche needs to be maintained above a certain threshold. In addition, it should be noted that the activation kinetics of the various ON-switches are different. Therefore, a delayed ON-switch system engagement could potentially hinder effective tumor control.
The safety and efficacy of the BPX-601 anti-PSCA GoCAR-T® platform (developed by Bellicum Pharmaceuticals, Inc., Houston, TX, USA) have been evaluated in patients with metastatic prostate cancer (NCT02744287). Rimiducid administration was found to induce CAR-T cell expansion in the peripheral blood of the 8 treated patients, accompanied by a rapid rise of serum cytokine levels. Early reports indicated encouraging results in terms of biological activity, with 1 PR and 3 cases of stable disease. As for the safety profile of this platform, CRS occurred in all patients, while ICANS was observed in 2 subjects. In addition, one case of neutropenic sepsis with possible hemophagocytic lymphohistiocytosis was reported (81). However, the subsequent occurrence of grade IV CRS in a patient enrolled in the study led to the discontinuation of the trial. The company also halted enrollment in a separate trial evaluating the BPX-603 anti-HER2 GoCAR-T® platform in patients with advanced HER2+ solid tumors (NCT04650451) (82, 83).
Among OFF-switches, the Herpes simplex virus thymidine kinase (HSV-TK) system allows to achieve CAR-T cell depletion upon ganciclovir (GCV) administration. The underlying mechanism involves GCV phosphorylation by HSV-TK, leading to the generation of GCV-triphosphate nucleotide analogues that interfere with DNA synthesis (Figure 2I). This suicide switch has mainly been evaluated in the context of donor lymphocyte infusion to patients who relapsed after T cell-depleted BM transplantation, with the aim to control possible graft-versus-host disease (GvHD) (84). Despite its validated efficacy, however, it should be noted that this T-cell depleting strategy has a relatively slow onset of action. Also, HSV-TK is potentially immunogenic.
A safety switch with no immunogenicity and a more rapid onset of action is the inducible caspase 9 (iCasp9) system, which leverages the intrinsic apoptotic pathway (85). Specifically, the iCasp9 gene encodes a fusion molecule which also contains a ligand-binding portion. In this way, it is possible to induce Casp9 dimerization and CAR-T cell apoptosis upon the intravenous administration of the AP1903 ligand (86) (Figure 2J).
Minagawa and colleagues showed that the retroviral transduction of activated T cells (ATCs) with a construct encoding the iCasp9 and an anti-CD33 CAR allows to efficiently redirect T lymphocytes against CD33+ AML cells, including pAML samples. Importantly, a sizeable proportion of iCasp9-CD33.CAR-T cells was depleted following the supplementation of an iCasp9 activator, with only negligible effects on untransduced ATCs or CD33.CAR-ATCs. The degree of iCasp9-CAR-T cell depletion was nearly complete following the co-supplementation of BCL inhibitors or the alkylating agent mafosfamide (87).
Hoyos et al. demonstrated that iCasp9-CD19.CAR-T cells are efficiently depleted upon supplementation of a small molecule activator of iCasp9. Moreover, in vivo experiments revealed that these modified T lymphocytes are significantly superior to CD19.CAR-T cells in terms of antitumor activity and expansion potential upon antigen encounter (88).
CAR-T cell depletion can also be achieved by leveraging Ab-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). The CD20-rituximab and truncated epidermal growth factor receptor (EGFRt)-cetuximab suicide switches represent two possible ways of implementing this safety strategy. Specifically, T cells are transduced with a construct containing the desired CAR and either the CD20 or the modified EGFR gene. This allows to induce apoptotic cell death (via ADCC) or complement-mediated lysis (via CDC) of engineered T lymphocytes by administering an anti-CD20 or anti-EGFR mAb, respectively (Figure 2K).
Philip et al. showed that T lymphocytes transduced with a suicide gene combining epitopes from the CD20 and CD34 antigens are selectively eliminated in vivo upon rituximab administration (89).
Key advantages offered by the CD20 and EGFRt switches are the rapid elimination of CAR-T cells and the possibility of employing clinically available mAbs. However, these systems are heavily dependent on the biodistribution of the corresponding mAbs. Their clinical translation could also be hindered by on-target toxicities (e.g., against CD20+ healthy cells) and other adverse events associated with mAb administration (90).
An in vitro comparison of different suicide gene systems revealed that the iCasp9, CD20, and HSV-TK platforms have similar efficacy in achieving T-cell depletion. However, there were significant differences in the kinetics of cell death induction, with the first 2 switches reaching full effect much faster than the HSV-TK system (91).
A shared limitation of suicide switches is that they are reactive rather than preventative measures, since they can only be deployed after the onset of undesired toxicities. Moreover, these systems deplete CAR-T cells irrespective of their therapeutic efficacy, since they do not allow to discriminate between desirable antitumor responses and unwanted off-tumor effects. Consequently, the timing of suicide switch activation becomes a crucial consideration for clinicians, adding an additional layer of complexity to therapeutic decision-making.
Among the clinical trials involving suicide gene switches, the NCT03016377 phase I/II study assessed the safety and efficacy of autologous iCasp9-CD19.CAR-T cells in patients with R/R B-cell malignancies. In this trial, the administration of rimiducid, a protein dimerizer that acts as an iCasp9 activator, was found to alleviate steroid- and tocilizumab-refractory ICANS (92).
iCasp9-CD19.CAR-T cells were also evaluated in an academic, phase I/II trial enrolling pediatric patients with B-ALL or B-cell NHL. CRS occurred in 58.8% of patients, but reached grade 3 only in one subject, and no life-threatening adverse events requiring iCasp9 safety switch activation were noted (93).
Del Bufalo et al. performed an academic, phase I/II trial (NCT03373097) to evaluate iCasp9-CAR-T cells redirected against disialoganglioside GD2 in children and young adults with R/R, high-risk neuroblastoma. The incidence of CRS was 74% (20 of 27 patients), but most cases were mild and safety switch activation was needed only in a single subject (94).
The NCT02028455 phase I/II trial assessed the use of CD19.CAR-T cells expressing the EGFRt safety gene in 45 children and young adults with R/R B-ALL. No cases of acute, life-threatening toxicities requiring cetuximab administration were reported (95).
In the NCT02159495 phase I study evaluating EGFRt-CD123.CAR-T cells in patients with CD123+ R/R AML or BPDCN, no cases of severe CRS or neurotoxicity requiring cetuximab administration were observed (96).
Even if the expression of a given membrane protein is shared between healthy and cancerous cells (as is the case for TAAs), the latter could still express a different isoform of said protein. This could be due to specific post-translational modifications (e.g., glycosylation) occurring in malignant but not in healthy tissues. On these premises, some research groups have focused on the identification of tumor-specific membrane protein glycoforms that could allow to selectively target neoplastic cells without causing damage to vital tissues. This could represent a promising strategy to design safer cellular immunotherapies without having to resort to complex logic-gating or suicide gene systems.
In several cancer types, the membrane mucin MUC1 is frequently expressed as the Tn (GalNAca1-O-Ser/Thr)-glycoform, which is absent in healthy tissues. On these grounds, Posey Jr. and colleagues demonstrated that CAR-T cells redirected against Tn-MUC1 induce the lysis of Tn-MUC1+ cells while sparing various MUC1+ healthy human tissues in vitro. Their efficacy was confirmed in a xenograft model of T-cell leukemia, where Tn-MUC1.CAR-T cells significantly prolonged the survival of mice when compared to unmanipulated T lymphocytes or CD19.CAR-T cells. Interestingly, Tn-MUC1.CAR-T cells were able to distinguish Tn-MUC1+ from Tn-MUC1- cells within the same co-culture well, thus confirming the absence of off-target effects.
The shared expression of the Tn-MUC1 glycoform across several cancer types could enable Tn-MUC1.CAR-T cells to recognize and kill multiple different tumors while maintaining a favorable safety profile. Indeed, they were found to outperform unmanipulated T lymphocytes and CD19.CAR-T cells in terms of antitumor activity in a xenograft mouse model of pancreatic cancer (97).
The NCT05239143 phase I trial enrolling patients with advanced epithelial tumors is evaluating the safety and efficacy of the P-MUC1C-ALLO1 platform (developed by Poseida Therapeutics, Inc., Kansas City, MO, USA). The latter is an allogeneic CAR-T cell product directed against the MUC1-C epitope, which is selectively expressed in carcinomas of the lung, esophagus, colon, breast, and ovary. Early reports indicated a favorable safety profile of this platform, with no dose-limiting toxicities, GvHD, or CRS observed in the first 3 treated patients (98).
A novel and potentially revolutionary approach to prevent off-tumor effects consists in ablating the expression or modifying the structure of the CAR target antigen in healthy cells. This could be especially useful in the context of hematological malignancies, where the modification of HSCs could allow to combine CAR-T cell therapy with HSCT in the absence of on-target myelotoxicity against healthy hematopoietic precursors.
Kim and colleagues exploited the clustered regularly interspaced short palindromic repeats (CRISPR) – CRISPR-associated protein 9 (Cas9) technology to delete the CD33 gene from normal human HSC, with the aim to make them resistant to CD33-directed therapies. The authors demonstrated that primary human CD34+ cells (i.e., immature hematopoietic progenitors) with disrupted CD33 expression are comparable to control human HSCs in terms of engraftment and differentiation capabilities when inoculated into immunodeficient mice. CD33-KO cells did not show any alterations in terms of morphology or functional capabilities (e.g., cytokine secretion, generation of reactive oxygen species, phagocytosis, etc.), both in vitro and in vivo. Also, no changes in gene expression profile or in the differentiation pattern elicited by the exposure to external factors were noted. These findings were validated in a non-human primate model, where modified HSCs showed preserved engraftment potential and established a morphologically normal hematopoietic process in the BM. Moreover, CD33 KO effectively shielded modified HSCs from CD33.CAR-T cells in a human xenograft model. The absence of bystander toxicities was confirmed by inoculating engrafted mice with the CD33+ MOLM-14 AML cell line: CD33.CAR-T cells were able to achieve tumor control in mice engrafted with either CD33-KO – or control HSCs, but caused off-leukemia toxicities only in the latter scenario (99).
Humbert et al. obtained similar results by selectively ablating CD33 exon 2, which codes for the V-set Ig-like domain that is the target of CD33-directed therapies. CD33ΔE2 human HSCs showed normal engraftment and differentiation potential in a murine xenograft model and were resistant to the toxic effects of the anti-CD33 Ab-drug conjugate gemtuzumab ozogamicin (100).
More recently, researchers have managed to incorporate non-synonymous mutations in epitopes of interest to hinder their recognition by CAR-T cells while preserving the function of the corresponding protein (an approach referred to as “epitope editing”). This could allow to target essential molecules without having to abrogate their expression on healthy HSCs.
Marone et al. generated HSCs expressing a modified but fully functional IL-3 receptor alpha-chain (CD123), with the aim to shield them from CD123.CAR-T cells. CD123 is expressed in most AML cases, including on the surface of LSCs. However, this molecule plays a crucial role in the biology of normal HSCs, thus discouraging a KO approach to alleviate on-target toxicities. On the basis of in silico mutagenesis studies, the authors engineered two specific amino acid substitutions into HSCs isolated from healthy donors (HDs). In colony-forming assays, epitope-edited HSCs demonstrated comparable differentiation and colony forming potential as WT HSCs. In contrast, CD123-KO HSCs showed a significant competitive disadvantage. Edited HSCs also maintained their engraftment capabilities and long-term repopulating potential when engrafted into immunodeficient mice. Finally, in co-culture assays involving the MOLM-14 AML line and engineered HSCs (either CD123 epitope-edited or CD123-KO), CD123.CAR-T cells depleted malignant cells as well as CD123hi unedited HSCs and CD123lo elements within the CD123-KO HSC population. On the other hand, epitope-edited HSCs were resistant to CAR-T cell cytolysis and retained their expression of the modified CD123 molecule (101).
Casirati et al. optimized a base-editing strategy to introduce desired amino acid variants into the CD123, FMS-like tyrosine kinase 3 (FLT3), and KIT molecules. The aim was to shield these molecules from therapeutic mAbs without altering their function. Epitope-edited HSCs were shown to be fully functional in vitro and in vivo. In addition, they were resistant to FLT3.CAR-T cells, allowing for the selective elimination of patient-derived AML xenografts (102).
Lastly, since the CD45 antigen is expressed by all nucleated cells of hematopoietic origin (including their malignant counterparts), Wellhausen and colleagues envisioned the use of CD45-epitope editing of HSCs to develop a universal CAR-T cell therapy for blood cancers with no off-tumor toxicities (103).
In all the previously mentioned studies, specificity analyses (e.g., computational off-target prediction) confirmed the safe engineering of HSCs (i.e., no off-target genotoxic effects were detected). However, several concerns still exist regarding gene editing-based cellular therapies. For instance, their manufacturing is highly complex, requiring HSC isolation and genetic manipulation in addition to the conventional CAR-T cell manufacturing process. Therefore, the future clinical translation of these approaches could be hindered by an excessive burden of preparatory procedures imposed on patients, as well as longer manufacturing time and an increased risk of production failures. This, in turn, could have a significant impact on treatment costs and could prevent some patients from receiving CAR-T cell infusions due to disease progression during the prolonged bridging period.
As discussed in the previous sections, broadening CAR-T cell specificity and avoiding off-tumor effects are critical for the development of safe and effective cellular immunotherapies. Another aspect that cannot be overlooked entails the optimization of CAR-T cell functional properties to achieve complete and long-lasting tumor control. In this context, several strategies have been proposed, including specific modifications to the basic CAR structure and the modulation of intracellular signaling pathways involved in T-cell antitumor responses.
CAR-T cell functional properties are critically dependent on CAR design, as evidenced by the profound differences between CD28 – and 4-1BB – co-stimulated CAR-T cell products in terms of cytolytic activity, cytokine production, memory differentiation, persistence, and metabolic profile. Preclinical studies comparing the antitumor potential of CARs with either a CD28 or a 4-1BB co-stimulatory moiety have yielded mixed results, due in part to the confounding effect associated with the use of different H/T domains (104).
A substantial body of evidence indicates an almost complete overlap between downstream protein phosphorylation events that follow the activation of either CD28 – or 4-1BB-co-stimulated CARs (105). However, some studies highlighted considerable differences in terms of the specific intracellular CAR interaction partners and their relative abundance when comparing CD28 – and 4-1BB – based constructs (106). Therefore, further investigations are needed to more precisely dissect the complex network of pathways that relay CAR-initiated signals. Anyhow, it is widely accepted that phosphorylation events occur more rapidly and with increased magnitude upon the engagement of CD28 co-stimulated CARs (105). This, in turn, is reflected in a more rapid tumor clearance by CD28 – co-stimulated CAR-T cells, both in vitro and in vivo (107, 108).
Bulk RNA sequencing (RNA-seq) has revealed unique transcriptional programs in 4-1BB – vs CD28-CAR-T cells. These differences are observed not only upon T-cell stimulation through the CAR, but also in the resting state, indicating a “tonic signaling signature”. Specifically, 4-1BB – based CAR-T cells tend to be enriched for MHC class II and fatty acid oxidation genes, along with marker genes typical of CD8 central memory (TCM)-like cells. CAR engagement amplifies the above-mentioned differences and results also in the upregulation of several cytokine cascades in 4-1BB-CAR-T cells, such as the tumor necrosis factor (TNF)-α, IL-21, and IFN-γ pathways. On the other hand, activated 4-1BB-CAR-T cells downregulate the expression of the PDCD1 gene, resulting in a lower PD-1 surface expression. They also undergo a T helper (Th)1 polarization program, while activated CD28-based products tend to show a Th2 phenotype. The resultant differences in cytokine secretion could influence the risk of CRS after CAR-T cell administration (109).
As for in vivo persistence, 4-1BB-CAR-T cells generally exhibit longer-lasting anticancer activity compared to CD28 co-stimulated products. This may be due to a slower tumor eradication process, leading to extended interactions between CAR-T and malignant cells. Another factor contributing to the enhanced persistence of 4-1BB-CAR-T cells is their relative resistance to functional exhaustion during prolonged antigen stimulation. This is evidenced by their increased cytokine production and reduced expression of exhaustion markers (e.g., lymphocyte-activation gene 3 [LAG-3], T-cell immunoglobulin and mucin domain 3 [TIM-3]) as compared to CD28-CAR-T cells (110) (Figure 3).
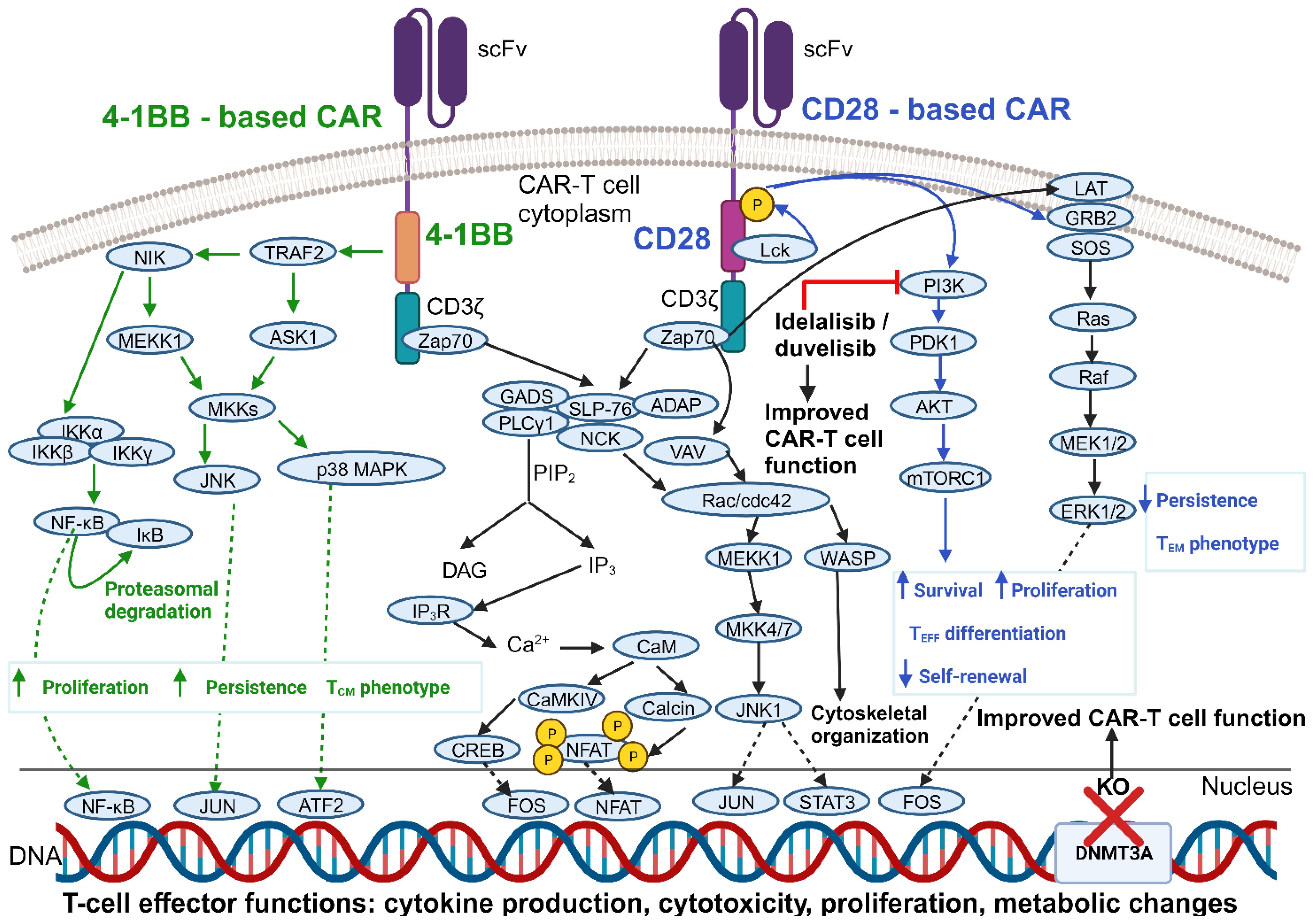
Figure 3 Comparison of 4-1BB – vs CD28 – co-stimulated CARs and improvements in CAR-T cell functionality obtained by modulating key intracellular mediators. Shown are the main signaling cascades elicited upon CAR engagement. Only the signaling mechanisms most strongly associated with a given co-stimulatory domain have been reported in its proximity (and color-coded accordingly), although it should be noted that 4-1BB – and CD28 – based CARs likely signal through almost identical pathways. Also shown are strategies to improve CAR-T cell function by leveraging intracellular signaling pathways and gene expression programs. See Glossary for abbreviations. Modified from Ref. 111. Figure created with BioRender.com.
To this day, there is insufficient clinical evidence to establish the superiority of either 4-1BB – or CD28 – co-stimulated CARs in terms of safety and anticancer efficacy. This is due, at least in part, to differences in several other factors besides the co-stimulatory domains (e.g., CAR design, CAR-T cell manufacturing processes, conditioning chemotherapy protocols, etc.) that complicate possible comparisons between different clinical trials (104).
Among FDA- and EMA-approved products targeting CD19, tisagenlecleucel (tisa-cel) and lisocabtagene maraleucel (liso-cel) are lentivirally-transduced and contain a 4-1BB co-stimulatory domain, but differ in their CAR H/T domains and T-cell expansion protocols (112). On the other hand, the CD28 – co-stimulated products, namely brexucabtagene autoleucel (brexu-cel) and axicabtagene ciloleucel (axi-cel), are both retrovirally-transduced, contain identical H/T domains, and undergo the same manufacturing process. Moreover, lymphodepleting chemotherapy preceding 4-1BB – or CD28-CAR-T cell administration consists in the same cyclophosphamide-fludarabine combination regimen, except in the case of tisa-cel.
Bearing in mind the limitations outlined above, the CR rates and remission duration observed in R/R B-ALL patients treated with 4-1BB – or CD28 – co-stimulated CD19.CAR-T cells are similar. Interestingly, however, consolidative alloHSCT confers an event-free survival (EFS) benefit only in subjects treated with 4-1BB – based products. In the case of R/R B-cell NHL, 4-1BB-based liso-cel and CD28-based axi-cel are associated with comparable CR and 12-month progression-free survival (PFS) rates, which are superior to those achieved with 4-1BB – based tisa-cel.
4-1BB – and CD28 – co-stimulated CAR-T cells also differ in terms of their safety profile, since the occurrence of CRS and ICANS has been reported more frequently in clinical studies evaluating CD28-based products (104, 112–122) (Table 1).
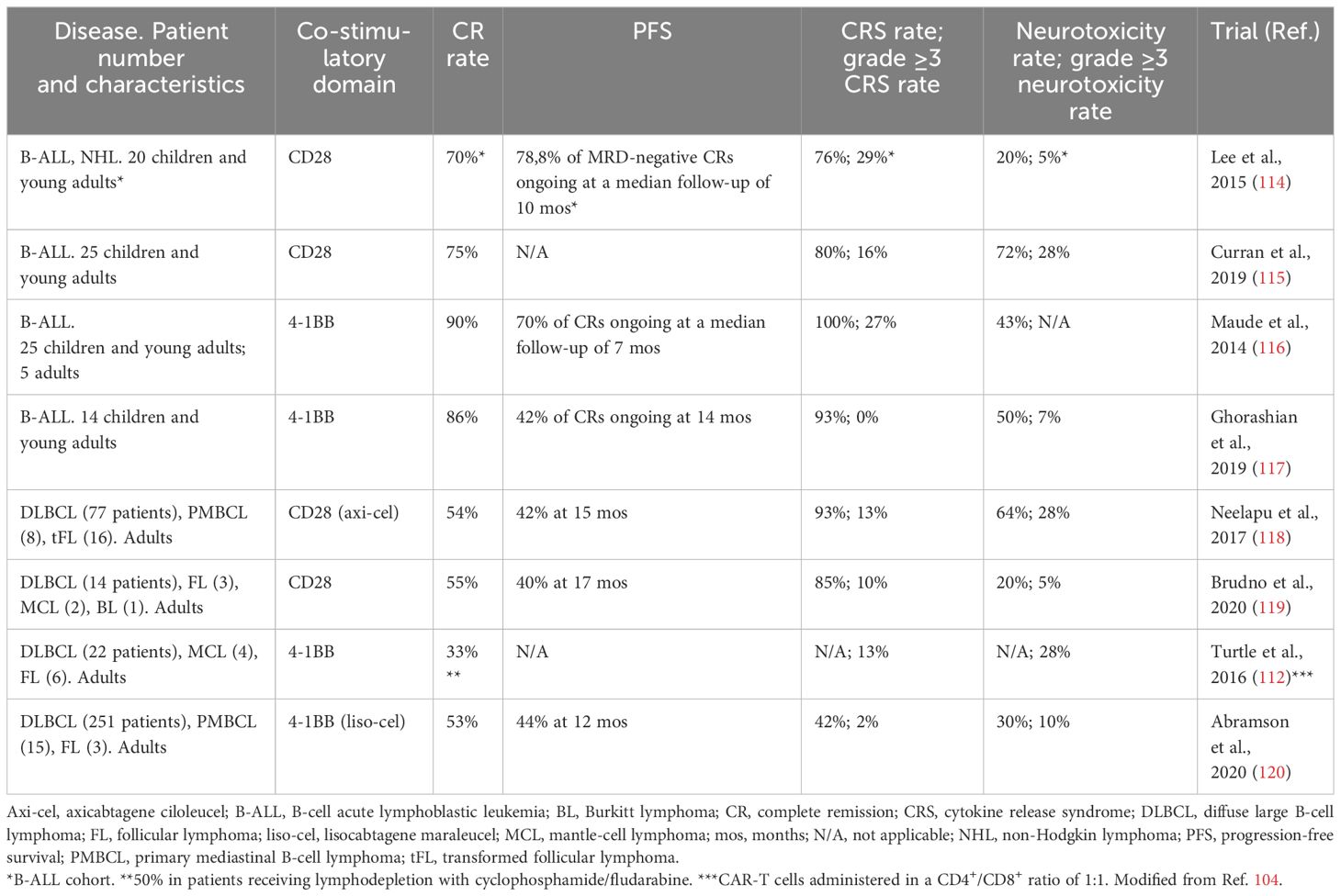
Table 1 Efficacy and safety of CD28 - or 4-1BB - based CD19. CAR-T cells in select trials enrolling patients with B-cell malignancies.
Optimal CAR-T cell performance can only be achieved when target antigen density exceeds a certain threshold. Therefore, manipulating the antigen density requirement for efficient CAR-T cell responses has emerged as a promising strategy to boost CAR-T cell functionality. Alternatively, CAR-T cell reactivity could be enhanced by inducing target antigen upregulation on the surface of malignant cells.
Majzner et al. investigated the impact of the CAR H/T region on the antigen density threshold for CAR-T cell activation. They showed that the presence of a CD28 – instead of a CD8α-H/T domain significantly enhances the antitumor function of 4-1BB-based CD19.CAR-T cells against CD19lo B-cell leukemia. Indeed, H/T-modified 4-1BB-CAR-T cells performed similarly to CD28-based products, with the remarkable advantage of a more durable persistence. This improvement in 4-1BB-CAR-T cell function against antigenlo targets was confirmed in murine models of solid tumors, including neuroblastoma and osteosarcoma. The underlying mechanism was found to involve an increased CAR clustering and recruitment of downstream signaling molecules upon antigen encounter (123). These findings could allow to counteract one of the most prevalent resistance mechanisms exploited by cancer cells, namely antigen modulation resulting in antigenlo relapse.
Priming cancer cells with the hypomethylating drugs decitabine (DAC) or azacitidine (AZA) could represent a relatively straightforward way to promote CAR target antigen upregulation, thus enhancing CAR-T cell antitumor activity (124).
Li et al. showed that pretreatment of B-cell NHL lines with DAC at concentrations that have negligible effects on malignant cell viability results in a significantly increased CD19 expression. This, in turn, rendered malignant cells more susceptible to CD19.CAR-T cell cytolysis. Importantly, no significant impairment in CAR-T cell functional properties was reported after DAC exposure (125).
A similar strategy was employed by El Khawanky and colleagues, who demonstrated that AZA treatment increases CD123 expression on the surface of AML cell lines and renders them more immunogenic. This, in turn, facilitates their lysis by CD123.CAR-T cells. AZA administration was also found to influence the subpopulation composition of CAR-T cells infused into AML xenograft mouse models. Specifically, the authors reported an increase in the proportion of CTLA-4- CAR-T cells, which were found to possess superior functional capabilities in terms of malignant cell killing and cytokine production. This ultimately led to a significant prolongation of the survival of AZA-pretreated mice compared with the control group (126).
Similarly, AZA was found to increase CD70 expression on the surface of AML cells, both in vitro and in vivo. AZA pretreatment of AML-bearing mice before CD70.CAR-T cell administration resulted in enhanced tumor control and improved survival compared to non-pretreated mice (127).
The NCT03196830 phase II trial assessed the role of a DAC-based lymphodepletion regimen preceding CD19 x CD22 TanCAR-T cell infusion in patients with R/R diffuse large B-cell lymphoma (DLBCL). Investigators reported a CR rate of 63,6% at a median follow-up of 10.9 months, and a 2-year OS of 54.3%. Toxicities were mostly mild and reversible (128).
A growing body of evidence indicates that leveraging T-lymphocyte intrinsic signaling pathways and gene expression programs could represent another way to enhance CAR-T cell functionality (Figure 3).
The phosphoinositide 3-kinase (PI3K) signaling pathway is a master regulator of T-cell activation upon antigen encounter, promoting the proliferation, survival, and differentiation of activated immune cells (129).
Previous research has shown that the PI3K inhibitor idelalisib can modulate T-cell differentiation in patients with B-cell NHL (130). Idelalisib also improved the persistence and antitumor function of adoptively transferred CD8+ T cells in vivo (131, 132). On this basis, investigators have explored the use of PI3K inhibitors to induce a more favorable differentiation profile and enhance the antitumor function of CAR-T cells. Funk and colleagues showed that supplementation of the PI3Kδ/γ inhibitor duvelisib (Duv) to CAR-T cell cultures induces epigenetic modifications characteristic of stem-like cells and upregulates co-stimulatory receptor – and TCR-activated signaling pathways. This translates into an improved cytolytic potential of Duv-CAR-T cells in vitro and in vivo. Indeed, in a murine CLL model, CD19.Duv-CAR-T cells demonstrated superior expansion, persistence, tumor control, and resistance to exhaustion as compared to conventional CAR-T cells (133).
Similar findings were reported by Petersen et al., who showed that idelalisib supplementation improves the expansion potential of CD5.CAR-T cells and boosts their antitumor functionality against lymphoma cells in vitro and in vivo (134).
A key limitation of CAR-T cell therapy, especially in solid tumors, is their functional exhaustion due to chronic antigen stimulation. This dysfunctional state appears to be subjected, at least in part, to epigenetic regulation. Zebley and colleagues showed that, after infusion into B-ALL patients, CD19.CAR-T cells acquire DNA methylation programs linked to functional exhaustion. These programs are maintained even in the absence of sustained antigenic stimulation. As a result, CAR-T cell ability to respond to a subsequent tumor relapse is impaired. Therefore, modulating DNA methylation programs could represent a promising strategy to promote sustained tumor control by CAR-T cells (135).
DNA methyltransferase 3A (DNMT3A) is a key enzyme in the establishment of de novo DNA methylation programs (136). Given that Dnmt3a deletion in mouse T cells can prevent their exhaustion during chronic viral infections (137), researchers have explored the possibility of deleting DNMT3A in CAR-T cells to preserve their antitumor functionality.
In an in vitro model of chronic stimulation developed by Prinzing et al., CAR-T cells lacking DNMT3A (DNMT3A KO) showed enhanced antigen-specific expansion compared to conventional CAR-T cells. Their cytokine response upon repeated antigen encounter was similar to that of control CAR-T cells, except for a remarkable IL-10 upregulation. This cytokine was found to support DNMT3A KO CAR-T cell survival and proliferation during chronic antigen exposure by limiting sustained co-stimulatory signaling. As a result, DNMT3A KO CAR-T cells showed preserved antigen-specific cytolysis upon repeated antigen stimulation. They also displayed a reduced expression of the TIM-3 exhaustion marker and a predominantly TCM phenotype. The favorable properties of DNMT3A KO CAR-T cells reported in vitro translated to an improved and more sustained antitumor efficacy in murine models of glioma and osteosarcoma (138).
CAR-T cells have reshaped the treatment landscape of B-cell malignancies, offering new hope for individuals with an otherwise dismal prognosis. At present, however, not all patients benefit from this therapeutic modality to the same degree, owing to limitations of the specific CAR design employed or to functional defects of adoptively transferred cells.
By introducing rational modifications into the CAR design, researchers have managed to improve CAR-T cell selectivity towards malignant tissues and mitigate the risk of toxicities associated with the targeting of TAAs. The choice of specific co-stimulatory domains, the manipulation of healthy hematopoietic cells targeted by CAR-T cell activity, and the modulation of T-lymphocyte intrinsic intracellular signaling cascades represent additional advances in the field.
Nonetheless, there are still several outstanding issues which need to be addressed, especially in the context of solid tumors. Such bottlenecks pertain not only to the intrinsic properties of CAR-T cells, but also involve their complex interactions with malignant cells and other immunosuppressive elements of the TME (139). Another key area of improvement could be related to the optimization of bridging and conditioning regimens preceding CAR-T cell infusion, to provide a more favorable environment for CAR-T cell engraftment (140).
As we continue to develop our understanding of the factors underlying therapeutic success, we will manage to design safer and more effective cellular immunotherapies. The anticipated goal is to induce long-lasting remissions, and possibly cure, in an ever-growing number of patients.
AA: Conceptualization, Writing – original draft, Writing – review & editing. MB: Writing – review & editing. EG: Writing – review & editing. AB: Supervision, Writing – review & editing, Funding acquisition, Resources. MS: Writing – review & editing, Conceptualization, Funding acquisition, Resources, Supervision. ST: Writing – review & editing, Supervision, Conceptualization, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from AIRC 5×1000 Immunity in Cancer Spreading and Metastasis (grant 21147), AIRC IG 2022 (grant 27507), the Ministero della Salute Research grant RF-2021-12374120, and PRIN 2021-NAZ- 0033.
AB serves in the scientific advisory board SAB of Galapagos.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Mitra A, Barua A, Huang L, Ganguly S, Feng Q, He B. From bench to bedside: the history and progress of CAR T cell therapy. Front Immunol. (2023) 14:1188049. doi: 10.3389/fimmu.2023.1188049
2. Jommi C, Bramanti S, Pani M, Ghirardini A, Santoro A. CAR T-cell therapies in Italy: patient access barriers and recommendations for health system solutions. Front Pharmacol. (2022) 13:915342. doi: 10.3389/fphar.2022.915342
3. Fischer JW, Bhattarai N. CAR-T cell therapy: mechanism, management, and mitigation of inflammatory toxicities. Front Immunol. (2021) 12:693016. doi: 10.3389/fimmu.2021.693016
4. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
5. Marofi F, Achmad H, Bokov D, Abdelbasset WK, Alsadoon Z, Chupradit S, et al. Hurdles to breakthrough in CAR T cell therapy of solid tumors. Stem Cell Res Ther. (2022) 13:140. doi: 10.1186/s13287-022-02819-x
6. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. (2016) 127:3321–30. doi: 10.1182/blood-2016-04-703751
7. Yu S, Yi M, Qin S, Wu K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol Cancer. (2019) 18:125. doi: 10.1186/s12943-019-1057-4
8. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
9. Minutolo NG, Hollander EE, Powell DJ Jr. The emergence of universal immune receptor T cell therapy for cancer. Front Oncol. (2019) 9:176. doi: 10.3389/fonc.2019.00176
10. Arndt C, Fasslrinner F, Loureiro LR, Koristka S, Feldmann A, Bachmann M. Adaptor CAR platforms—Next generation of T cell-based cancer immunotherapy. Cancers. (2020) 12:1302. doi: 10.3390/cancers12051302
11. Lu L, Xie M, Yang B, Zhao Wb, Cao J. Enhancing the safety of CAR-T cell therapy: Synthetic genetic switch for spatiotemporal control. Sci Adv. (2024) 10:eadj6251. doi: 10.1126/sciadv.adj6251
12. Rataj F, Jacobi SJ, Stoiber S, Asang F, Ogonek J, Tokarew N, et al. High-affinity CD16-polymorphism and Fc-engineered antibodies enable activity of CD16-chimeric antigen receptor-modified T cells for cancer therapy. Br J Cancer. (2019) 120:79–87. doi: 10.1038/s41416-018-0341-1
13. Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, et al. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res. (2012) 18:6436–45. doi: 10.1158/1078-0432.CCR-12-1449
14. Cao Y, Rodgers DT, Du J, Ahmad I, Hampton EN, Ma JSY, et al. Design of switchable chimeric antigen receptor T cells targeting breast cancer. Angew Chem Int Ed Engl. (2016) 55:7520–4. doi: 10.1002/anie.201601902
15. Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, et al. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. (2012) 72:1844–52. doi: 10.1158/0008-5472.CAN-11-3890
16. Nixdorf D, Sponheimer M, Berghammer D, Engert F, Bader U, Philipp N, et al. Adapter CAR T cells to counteract T-cell exhaustion and enable flexible targeting in AML. Leukemia. (2023) 37:1298–310. doi: 10.1038/s41375-023-01905-0
17. Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, et al. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. (2016) 6:e458–8. doi: 10.1038/bcj.2016.61
18. Bachmann D, Aliperta R, Bergmann R, Feldmann A, Koristka S, Arndt C, et al. Retargeting of UniCAR T cells with an in vivo synthesized target module directed against CD19 positive tumor cells. Oncotarget. (2017) 9:7487–500. doi: 10.18632/oncotarget.23556
19. Mitwasi N, Feldmann A, Bergmann R, Berndt N, Arndt C, Koristka S, et al. Development of novel target modules for retargeting of UniCAR T cells to GD2 positive tumor cells. Oncotarget. (2017) 8:108584–603. doi: 10.18632/oncotarget.21017
20. Loureiro LR, Feldmann A, Bergmann R, Koristka S, Berndt N, Arndt C, et al. Development of a novel target module redirecting UniCAR T cells to Sialyl Tn-expressing tumor cells. Blood Cancer J. (2018) 8:1–6. doi: 10.1038/s41408-018-0113-4
21. Albert S, Arndt C, Koristka S, Berndt N, Bergmann R, Feldmann A, et al. From mono- to bivalent: improving theranostic properties of target modules for redirection of UniCAR T cells against EGFR-expressing tumor cells in vitro and in vivo. Oncotarget. (2018) 9:25597–616. doi: 10.18632/oncotarget.25390
22. Feldmann A, Arndt C, Bergmann R, Loff S, Cartellieri M, Bachmann D, et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget. (2017) 8:31368–85. doi: 10.18632/oncotarget.15572
23. Meyer JE, Loff S, Dietrich J, Spehr J, Jurado Jiménez G, von Bonin M, et al. Evaluation of switch-mediated costimulation in trans on universal CAR-T cells (UniCAR) targeting CD123-positive AML. Oncoimmunol. (2021) 10:1945804. doi: 10.1080/2162402X.2021.1945804
24. Luo Q, Napoleon JV, Liu X, Zhang B, Zheng S, Low PS. Targeted rejuvenation of exhausted chimeric antigen receptor T cells regresses refractory solid tumors. Mol Cancer Res. (2022) 20:823–33. doi: 10.1158/1541-7786.MCR-21-0711
25. Caratelli S, Sconocchia T, Arriga R, Coppola A, Lanzilli G, Lauro D, et al. FCγ Chimeric receptor-engineered T cells: methodology, advantages, limitations, and clinical relevance. Front Immunol. (2017) 8:457. doi: 10.3389/fimmu.2017.00457
26. Kudo K, Imai C, Lorenzini P, Kamiya T, Kono K, Davidoff AM, et al. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. (2014) 74:93–103. doi: 10.1158/0008-5472.CAN-13-1365
27. Karches CH, Benmebarek MR, Schmidbauer ML, Kurzay M, Klaus R, Geiger M, et al. Bispecific antibodies enable synthetic agonistic receptor-transduced T cells for tumor immunotherapy. Clin Cancer Res. (2019) 25:5890–900. doi: 10.1158/1078-0432.CCR-18-3927
28. Albert CM, Pinto NR, Taylor M, Wilson A, Rawlings-Rhea S, Huang W, et al. ENLIGHten-01: A phase 1 study of fluorescein-specific (FITC-E2)-CAR T cells in combination with folate-fluorescein (UB-TT170) for osteosarcoma. JCO. (2023) 41:TPS11581–TPS11581. doi: 10.1200/JCO.2023.41.16_suppl.TPS11581
29. Koedam J, Wermke M, Ehninger A, Cartellieri M, Ehninger G. Chimeric antigen receptor T-cell therapy in acute myeloid leukemia. Curr Opin Hematol. (2022) 29:74. doi: 10.1097/MOH.0000000000000703
30. AvenCell Europe GmbH. Multicenter, Open-label, Adaptive Design Phase I Trial With Genetically Modified T-cells Carrying Universal Chimeric Antigen Receptors (UniCAR02-T) in Combination With CD123 Target Module (TM123) for the Treatment of Patients With Hematologic and Lymphatic Malignancies Positive for CD123 (2023). Available online at: https://clinicaltrials.gov/study/NCT04230265 (Accessed November 15, 2023).
31. Wermke M, Kraus S, Ehninger A, Bargou RC, Goebeler ME, Middeke JM, et al. Proof of concept for a rapidly switchable universal CAR-T platform with UniCAR-T-CD123 in relapsed/refractory AML. Blood. (2021) 137:3145–8. doi: 10.1182/blood.2020009759
32. Ehninger G, Kraus S, Sala E, Metzelder SK, Vucinic V, Fiedler W, et al. Phase 1 dose escalation study of the rapidly switchable universal CAR-T therapy unicar-T-CD123 in relapsed/refractory AML. Blood. (2022) 140:2367–8. doi: 10.1182/blood-2022-168877
33. Wermke M, Metzelder S, Kraus S, Sala E, Vucinic V, Fiedler W, et al. Updated results from a phase I dose escalation study of the rapidly-switchable universal CAR-T therapy uniCAR-T-CD123 in relapsed/refractory AML. Blood. (2023) 142:3465. doi: 10.1182/blood-2023-177867
34. AvenCell Europe GmbH. Multicenter, Open-label, Adaptive Design Phase I Trial With Genetically Modified T-cells Carrying Universal Chimeric Antigen Receptors (UniCAR02-T) in Combination With PSMA Peptide Target Module (TMpPSMA) for the Treatment of Patients With Progressive Disease After Standard Systemic Therapy in Cancers With Positive PSMA Marker (2023). Available online at: https://clinicaltrials.gov/study/NCT04633148 (Accessed November 20, 2023).
35. Cogent Biosciences, Inc. Phase 1 Study of ACTR087, Autologous T Lymphocytes Expressing Antibody Coupled T-cell Receptors (CD16V-41BB-CD3ζ), in Combination With Rituximab, in Subjects With Relapsed or Refractory CD20-Positive B-Cell Lymphoma (2020). Available online at: https://clinicaltrials.gov/study/NCT02776813 (Accessed November 21, 2023)
36. Munoz J, Jaglowski S, McKinney MS, Isufi I, Stiff PJ, Sachs J, et al. A phase 1 study of ACTR087 in combination with rituximab, in subjects with relapsed or refractory CD20-positive B-cell lymphoma. Blood. (2019) 134:244. doi: 10.1182/blood-2019-123810
37. Cogent Biosciences, Inc. A Phase 1 Study of an Autologous ACTR T Cell Product in Combination With Trastuzumab, a Monoclonal Antibody, in Subjects With HER2-Positive Advanced Malignancies (2020). Available online at: https://clinicaltrials.gov/study/NCT03680560 (Accessed November 21, 2023).
38. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. (2013) 31:71–5. doi: 10.1038/nbt.2459
39. Han X, Wang Y, Wei J, Han W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J Hematol Oncol. (2019) 12:1–10. doi: 10.1186/s13045-019-0813-7
40. Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by Malignant B cells. Cancer Immunol Res. (2016) 4:498–508. doi: 10.1158/2326-6066.CIR-15-0231
41. Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, et al. TanCAR: A novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids. (2013) 2:e105. doi: 10.1038/mtna.2013.32
42. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest. (2016) 126:3036–52. doi: 10.1172/JCI83416
43. Lorusso G, Rüegg C. The tumor microenvironment and its contribution to tumor evolution toward metastasis. Histochem Cell Biol. (2008) 130:1091–103. doi: 10.1007/s00418-008-0530-8
44. Alberti G, Arsuffi C, Pievani A, Salerno D, Mantegazza F, Dazzi F, et al. Engineering tandem CD33xCD146 CAR CIK (cytokine-induced killer) cells to target the acute myeloid leukemia niche. Front Immunol. (2023) 14:1192333. doi: 10.3389/fimmu.2023.1192333
45. Korn C, Méndez-Ferrer S. Myeloid Malignancies and the microenvironment. Blood. (2017) 129:811–22. doi: 10.1182/blood-2016-09-670224
46. Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. (2008) 2:141–50. doi: 10.1016/j.stem.2007.11.014
47. Di Ianni M, Del Papa B, De Ioanni M, Moretti L, Bonifacio E, Cecchini D, et al. Mesenchymal cells recruit and regulate T regulatory cells. Exp Hematol. (2008) 36:309–18. doi: 10.1016/j.exphem.2007.11.007
48. Shah NN, Johnson BD, Schneider D, Zhu F, Szabo A, Keever-Taylor CA, et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell Malignancies: a phase 1 dose escalation and expansion trial. Nat Med. (2020) 26:1569–75. doi: 10.1038/s41591-020-1081-3
49. Cui W, Zhang XY, Li Z, Dai HP, Yin J, Cui QY, et al. Long-term follow-up of tandem CD19/CD22 CAR T-Cells in r/r B-ALL patients with high-risk features. Am J Hematol. (2023) 98:E338–40. doi: 10.1002/ajh.27076
50. Smith CC, Selitsky SR, Chai S, Armistead PM, Vincent BG, Serody JS. Alternative tumour-specific antigens. Nat Rev Cancer. (2019) 19:465–78. doi: 10.1038/s41568-019-0162-4
51. Poorebrahim M, Mohammadkhani N, Mahmoudi R, Gholizadeh M, Fakhr E, Cid-Arregui A. TCR-like CARs and TCR-CARs targeting neoepitopes: an emerging potential. Cancer Gene Ther. (2021) 28:581–9. doi: 10.1038/s41417-021-00307-7
52. Hazini A, Fisher K, Seymour L. Deregulation of HLA-I in cancer and its central importance for immunotherapy. J Immunother Cancer. (2021) 9:e002899. doi: 10.1136/jitc-2021-002899
53. Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJD, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. (2015) 29:1637–47. doi: 10.1038/leu.2015.52
54. Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. (2013) 21:904–12. doi: 10.1038/mt.2013.17
55. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
56. Tousley AM, Rotiroti MC, Labanieh L, Rysavy LW, Kim WJ, Lareau C, et al. Co-opting signalling molecules enables logic-gated control of CAR T cells. Nature. (2023) 615:507–16. doi: 10.1038/s41586-023-05778-2
57. Leung WH, Gay J, Martin U, Garrett TE, Horton HM, Certo MT, et al. Sensitive and adaptable pharmacological control of CAR T cells through extracellular receptor dimerization. JCI Insight. (2019) 5:e124430. doi: 10.1172/jci.insight.124430
58. Zajc CU, Dobersberger M, Schaffner I, Mlynek G, Pühringer D, Salzer B, et al. A conformation-specific ON-switch for controlling CAR T cells with an orally available drug. Proc Natl Acad Sci USA. (2020) 117:14926–35. doi: 10.1073/pnas.1911154117
59. Kobayashi A, Nobili A, Neier SC, Sadiki A, Distel R, Zhou ZS, et al. Light-controllable binary switch activation of CAR T cells. ChemMedChem. (2022) 17:e202100722. doi: 10.1002/cmdc.202100722
60. Foster AE, Mahendravada A, Shinners NP, Chang WC, Crisostomo J, Lu A, et al. Regulated expansion and survival of chimeric antigen receptor-modified T cells using small molecule-dependent inducible myD88/CD40. Mol Ther. (2017) 25:2176–88. doi: 10.1016/j.ymthe.2017.06.014
61. Irving M, Lanitis E, Migliorini D, Ivics Z, Guedan S. Choosing the right tool for genetic engineering: clinical lessons from chimeric antigen receptor-T cells. Hum Gene Ther. (2021) 32:1044–58. doi: 10.1089/hum.2021.173
62. Cummins KD, Frey N, Nelson AM, Schmidt A, Luger S, Isaacs RE, et al. Treating relapsed / refractory (RR) AML with biodegradable anti-CD123 CAR modified T cells. Blood. (2017) 130:1359. doi: 10.1182/blood.V130.Suppl_1.1359.1359
63. Svoboda J, Rheingold SR, Gill SI, Grupp SA, Lacey SF, Kulikovskaya I, et al. Nonviral RNA chimeric antigen receptor-modified T cells in patients with Hodgkin lymphoma. Blood. (2018) 132:1022–6. doi: 10.1182/blood-2018-03-837609
64. Beatty GL, O’Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology. (2018) 155:29–32. doi: 10.1053/j.gastro.2018.03.029
65. Perriello VM, Rotiroti MC, Pisani I, Galimberti S, Alberti G, Pianigiani G, et al. IL-3-zetakine combined with a CD33 costimulatory receptor as a dual CAR approach for safer and selective targeting of AML. Blood Adv. (2023) 7:2855–71. doi: 10.1182/bloodadvances.2022008762
66. Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. (2006) 7:678–89. doi: 10.1038/nrm2009
67. Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, et al. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell. (2016) 164:780–91. doi: 10.1016/j.cell.2016.01.012
68. Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell. (2016) 164:770–9. doi: 10.1016/j.cell.2016.01.011
69. Williams JZ, Allen GM, Shah D, Sterin IS, Kim KH, Garcia VP, et al. Precise T cell recognition programs designed by transcriptionally linking multiple receptors. Science. (2020) 370:1099–104. doi: 10.1126/science.abc6270
70. Okada H. Phase 1 Study of Autologous Anti-EGFRvIII synNotch Receptor Induced Anti-EphA2/IL-13R alpha2 CAR (E-SYNC) T Cells in Adult Participants With EGFRvIII+ Glioblastoma (2024). Available online at: https://clinicaltrials.gov/study/NCT06186401 (Accessed May 2, 2024)
71. Muhammad N, Wang R, Li W, Zhang Z, Chang Y, Hu Y, et al. A novel TanCAR targeting IL13Rα2 and EphA2 for enhanced glioblastoma therapy. Mol Ther Oncolytics. (2022) 24:729–41. doi: 10.1016/j.omto.2022.02.012
72. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. (2005) 25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
73. Fedorov VD, Themeli M, Sadelain M. PD-1– and CTLA-4–based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. (2013) 5:215ra172–215ra172. doi: 10.1126/scitranslmed.3006597
74. Iwasaki M, Liedtke M, Gentles AJ, Cleary ML. CD93 marks a non-quiescent human leukemia stem cell population and is required for development of MLL-rearranged acute myeloid leukemia. Cell Stem Cell. (2015) 17:412–21. doi: 10.1016/j.stem.2015.08.008
75. Richards RM, Zhao F, Freitas KA, Parker KR, Xu P, Fan A, et al. NOT-gated CD93 CAR T cells effectively target AML with minimized endothelial cross-reactivity. Blood Cancer Discovery. (2021) 2:648–65. doi: 10.1158/2643-3230.BCD-20-0208
76. Molina J, Tokatlian T, Wang J, Sharma S, Manry D, Naradikian M, et al. 263 A2B694, an autologous logic-gated cell therapy targeting mesothelin. J Immunother Cancer. (2022) 10. doi: 10.1136/jitc-2022-SITC2022.0263
77. Bangayan NJ, Wang L, Burton Sojo G, Noguchi M, Cheng D, Ta L, et al. Dual-inhibitory domain iCARs improve the efficiency of the AND-NOT gate CAR T strategy. Proc Natl Acad Sci USA. (2023) 120:e2312374120. doi: 10.1073/pnas.2312374120
78. Simeone DM, Hecht JR, Smith C, Fakih M, Welling T, Garon E, et al. 636 BASECAMP-1: A master prescreening study to identify patients with high-risk or metastatic solid tumors with HLA loss of heterozygosity (LOH) in preparation for Tmod CAR T-cell therapy trials. J Immunother Cancer. (2023) 11. doi: 10.1136/jitc-2023-SITC2023.0636
79. A2 Biotherapeutics Inc. A Seamless Phase 1/2 Study to Evaluate the Safety and Efficacy of A2B694, an Autologous Logic-gated Tmod™ CAR T, in Heterozygous HLA-A*02 Adults With Recurrent Unresectable, Locally Advanced, or Metastatic Solid Tumors That Express MSLN and Have Lost HLA-A*02 Expression (2024). Available online at: https://clinicaltrials.gov/study/NCT06051695 (Accessed May 2, 2024).
80. Kim M, An J, Kim KS, Choi M, Humar M, Kwok SJJ, et al. Optical lens-microneedle array for percutaneous light delivery. BioMed Opt Express. (2016) 7:4220–7. doi: 10.1364/BOE.7.004220
81. Stein MN, Teply BA, Gergis U, Strickland D, Senesac J, Bayle H, et al. Early results from a phase 1, multicenter trial of PSCA-specific GoCAR T cells (BPX-601) in patients with metastatic castration-resistant prostate cancer (mCRPC). JCO. (2023) 41:140–0. doi: 10.1200/JCO.2023.41.6_suppl.140
82. Bellicum Pharmaceuticals. A Phase 1/2, Open-Label, Multicenter, Non-Randomized, Safety and Activity Study of HER2-Targeted Dual Switch CAR-T Cells (BPX-603) In Subjects With Previously Treated Advanced HER2-Positive Solid Tumors (2023). Available online at: https://clinicaltrials.gov/study/NCT04650451 (Accessed May 2, 2024)
83. OncLive. BPX-601 and BPX-603 Trials Discontinued in Advanced Solid Tumors (2023). Available online at: https://www.onclive.com/view/bpx-601-and-bpx-603-trials-discontinued-in-advanced-solid-tumors (Accessed May 2, 2024)
84. Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. (1997) 276:1719–24. doi: 10.1126/science.276.5319.1719
85. Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. (2019) 20:175–93. doi: 10.1038/s41580-018-0089-8
86. Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. (2014) 5:235. doi: 10.3389/fphar.2014.00235
87. Minagawa K, Jamil MO, AL-Obaidi M, Pereboeva L, Salzman D, Erba HP, et al. In vitro pre-clinical validation of suicide gene modified anti-CD33 redirected chimeric antigen receptor T-cells for acute myeloid leukemia. PloS One. (2016) 11:e0166891. doi: 10.1371/journal.pone.0166891
88. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. (2010) 24:1160–70. doi: 10.1038/leu.2010.75
89. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. (2014) 124:1277–87. doi: 10.1182/blood-2014-01-545020
90. Jones BS, Lamb LS, Goldman F, Di Stasi A. Improving the safety of cell therapy products by suicide gene transfer. Front Pharmacol. (2014) 5:254. doi: 10.3389/fphar.2014.00254
91. Marin V, Cribioli E, Philip B, Tettamanti S, Pizzitola I, Biondi A, et al. Comparison of different suicide-gene strategies for the safety improvement of genetically manipulated T cells. Hum Gene Ther Methods. (2012) 23:376–86. doi: 10.1089/hgtb.2012.050
92. Foster MC, Savoldo B, Lau W, Rubinos C, Grover N, Armistead P, et al. Utility of a safety switch to abrogate CD19.CAR T-cell–associated neurotoxicity. Blood. (2021) 137:3306–9. doi: 10.1182/blood.2021010784
93. Del Bufalo F, Quintarelli C, De Angelis B, Caruana I, Sinibaldi M, Vinti L, et al. Academic, phase I/II trial on T cells expressing a second generation, CD19-specific chimeric antigen receptor (CAR) and inducible caspase 9 safety switch for the treatment of B-cell precursor acute lymphoblastic leukemia (BCP-ALL) and B-cell non-hodgkin lymphoma (B-NHL) in children. Blood. (2019) 134:1341. doi: 10.1182/blood-2019-129821
94. Del Bufalo F, De Angelis B, Caruana I, Del Baldo G, De Ioris MA, Serra A, et al. GD2-CART01 for relapsed or refractory high-risk neuroblastoma. N Engl J Med. (2023) 388:1284–95. doi: 10.1056/NEJMoa2210859
95. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. (2017) 129:3322–31. doi: 10.1182/blood-2017-02-769208
96. Budde LE, Song J, Real MD, Kim Y, Toribio C, Wood B, et al. Abstract PR14: CD123CAR displays clinical activity in relapsed/refractory (r/r) acute myeloid leukemia (AML) and blastic plasmacytoid dendritic cell neoplasm (BPDCN): Safety and efficacy results from a phase 1 study. Cancer Immunol Res. (2020) 8:PR14–4. doi: 10.1158/2326-6074.TUMIMM18-PR14
97. Posey AD, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, et al. Engineered CAR T cells targeting the cancer-associated tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity. (2016) 44:1444–54. doi: 10.1016/j.immuni.2016.05.014
98. Henry J, Oh D, Eskew J, Baranda J, Rivera IIR, Dumbrava E, et al. 728 Phase 1 study of P-MUC1C-ALLO1 allogeneic CAR-T cells in patients with epithelial-derived cancers. J Immunother Cancer. (2022) 10. doi: 10.1136/jitc-2022-SITC2022.0728
99. Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. (2018) 173:1439–1453.e19. doi: 10.1016/j.cell.2018.05.013
100. Humbert O, Laszlo GS, Sichel S, Ironside C, Haworth KG, Bates OM, et al. Engineering resistance to CD33-targeted immunotherapy in normal hematopoiesis by CRISPR/Cas9-deletion of CD33 exon 2. Leukemia. (2019) 33:762–808. doi: 10.1038/s41375-018-0277-8
101. Marone R, Landmann E, Devaux A, Lepore R, Seyres D, Zuin J, et al. Epitope-engineered human hematopoietic stem cells are shielded from CD123-targeted immunotherapy. J Exp Med. (2023) 220:e20231235. doi: 10.1084/jem.20231235
102. Casirati G, Cosentino A, Mucci A, Salah Mahmoud M, Ugarte Zabala I, Zeng J, et al. Epitope editing enables targeted immunotherapy of acute myeloid leukaemia. Nature. (2023) 621:404–14. doi: 10.1038/s41586-023-06496-5
103. Wellhausen N, O’Connell RP, Lesch S, Engel NW, Rennels AK, Gonzales D, et al. Epitope base editing CD45 in hematopoietic cells enables universal blood cancer immune therapy. Sci Transl Med. (2023) 15:eadi1145. doi: 10.1126/scitranslmed.adi1145
104. Cappell KM, Kochenderfer JN. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat Rev Clin Oncol. (2021) 18:715–27. doi: 10.1038/s41571-021-00530-z
105. Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal. (2018) 11:eaat6753. doi: 10.1126/scisignal.aat6753
106. Ramello MC, Benzaïd I, Kuenzi BM, Lienlaf-Moreno M, Kandell WM, Santiago DN, et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci Signal. (2019) 12:eaap9777. doi: 10.1126/scisignal.aap9777
107. Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. (2015) 28:415–28. doi: 10.1016/j.ccell.2015.09.004
108. Honikel MM, Olejniczak SH. Co-stimulatory receptor signaling in CAR-T cells. Biomolecules. (2022) 12:1303. doi: 10.3390/biom12091303
109. Boroughs AC, Larson RC, Marjanovic ND, Gosik K, Castano AP, Porter CBM, et al. A distinct transcriptional program in human CAR T cells bearing the 4-1BB signaling domain revealed by scRNA-seq. Mol Ther. (2020) 28:2577–92. doi: 10.1016/j.ymthe.2020.07.023
110. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
111. Cell Signaling Technology. CAR Signaling Networks (2020). Available online at: https://www.cellsignal.com/pathways/car-signaling-networks (Accessed December 9, 2023)
112. Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. (2016) 8:355ra116. doi: 10.1126/scitranslmed.aaf8621
113. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
114. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
115. Curran KJ, Margossian SP, Kernan NA, Silverman LB, Williams DA, Shukla N, et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood. (2019) 134:2361–8. doi: 10.1182/blood.2019001641
116. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
117. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. (2019) 25:1408–14. doi: 10.1038/s41591-019-0549-5
118. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
119. Brudno JN, Lam N, Vanasse D, wei SY, Rose JJ, Rossi J, et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat Med. (2020) 26:270–80. doi: 10.1038/s41591-019-0737-3
120. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. (2020) 396:839–52. doi: 10.1016/S0140-6736(20)31366-0
121. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
122. Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. (2019) 25:947–53. doi: 10.1038/s41591-019-0421-7
123. Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discovery. (2020) 10:702–23. doi: 10.1158/2159-8290.CD-19-0945
124. Harrer DC, Dörrie J, Schaft N. CARs and drugs: pharmacological ways of boosting CAR-T-cell therapy. Int J Mol Sci. (2023) 24:2342. doi: 10.3390/ijms24032342
125. Li S, Xue L, Wang M, Qiang P, Xu H, Zhang X, et al. Decitabine enhances cytotoxic effect of T cells with an anti-CD19 chimeric antigen receptor in treatment of lymphoma. Onco Targets Ther. (2019) 12:5627–38. doi: 10.2147/OTT.S198567
126. El Khawanky N, Hughes A, Yu W, Myburgh R, Matschulla T, Taromi S, et al. Demethylating therapy increases anti-CD123 CAR T cell cytotoxicity against acute myeloid leukemia. Nat Commun. (2021) 12:6436. doi: 10.1038/s41467-021-26683-0
127. Leick MB, Silva H, Scarfò I, Larson R, Choi BD, Bouffard AA, et al. Non-cleavable hinge enhances avidity and expansion of CAR-T cells for acute myeloid leukemia. Cancer Cell. (2022) 40:494–508.e5. doi: 10.1016/j.ccell.2022.04.001
128. Qu C, Zou R, Wang P, Zhu Q, Kang L, Ping N, et al. Decitabine-primed tandem CD19/CD22 CAR-T therapy in relapsed/refractory diffuse large B-cell lymphoma patients. Front Immunol. (2022) 13:969660. doi: 10.3389/fimmu.2022.969660
129. Murter B, Kane LP. Control of T lymphocyte fate decisions by PI3K signaling. F1000Res. (2020) 9:F1000 Faculty Rev–1171. doi: 10.12688/f1000research.26928.1
130. Patton DT, Garden OA, Pearce WP, Clough LE, Monk CR, Leung E, et al. Cutting edge: the phosphoinositide 3-kinase p110δ Is critical for the function of CD4+CD25+Foxp3+ Regulatory T cells. J Immunol. (2006) 177:6598–602. doi: 10.4049/jimmunol.177.10.6598
131. Bowers JS, Majchrzak K, Nelson MH, Aksoy BA, Wyatt MM, Smith AS, et al. PI3Kδ Inhibition enhances the antitumor fitness of adoptively transferred CD8+ T cells. Front Immunol. (2017) 8:1221. doi: 10.3389/fimmu.2017.01221
132. Poorebrahim M, Melief J, Pico de Coaña Y, L Wickström S, Cid-Arregui A, Kiessling R. Counteracting CAR T cell dysfunction. Oncogene. (2021) 40:421–35. doi: 10.1038/s41388-020-01501-x
133. Funk CR, Wang S, Chen KZ, Waller A, Sharma A, Edgar CL, et al. PI3Kδ/γ inhibition promotes human CART cell epigenetic and metabolic reprogramming to enhance antitumor cytotoxicity. Blood. (2022) 139:523–37. doi: 10.1182/blood.2021011597
134. Petersen CT, Hassan M, Morris AB, Jeffery J, Lee K, Jagirdar N, et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kδ inhibitors and VIP antagonists. Blood Adv. (2018) 2:210–23. doi: 10.1182/bloodadvances.2017011254
135. Zebley CC, Brown C, Mi T, Fan Y, Alli S, Boi S, et al. CD19-CAR T cells undergo exhaustion DNA methylation programming in patients with acute lymphoblastic leukemia. Cell Rep. (2021) 37:110079. doi: 10.1016/j.celrep.2021.110079
136. Chen BF, Chan WY. The de novo DNA methyltransferase DNMT3A in development and cancer. Epigenetics. (2014) 9:669–77. doi: 10.4161/epi.28324
137. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell. (2017) 170:142–157.e19. doi: 10.1016/j.cell.2017.06.007
138. Prinzing B, Zebley CC, Petersen CT, Fan Y, Anido AA, Yi Z, et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci Transl Med. (2021) 13:eabh0272. doi: 10.1126/scitranslmed.abh0272
139. Fonkoua LAK, Sirpilla O, Sakemura R, Siegler EL, Kenderian SS. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol Ther Oncolytics. (2022) 25:69–77. doi: 10.1016/j.omto.2022.03.009
Keywords: CAR-T cells, adapter CARs (AdCARs), tandem CARs (TanCARs), logic-gated CARs, safety switches, epitope editing, antigen density
Citation: Alviano AM, Biondi M, Grassenis E, Biondi A, Serafini M and Tettamanti S (2024) Fully equipped CARs to address tumor heterogeneity, enhance safety, and improve the functionality of cellular immunotherapies. Front. Immunol. 15:1407992. doi: 10.3389/fimmu.2024.1407992
Received: 27 March 2024; Accepted: 15 May 2024;
Published: 03 June 2024.
Edited by:
Claudia Arndt, Helmholtz Center Dresden-Rossendorf, GermanyReviewed by:
Alexander Kamb, A2 Biotherapeutics, Inc., United StatesCopyright © 2024 Alviano, Biondi, Grassenis, Biondi, Serafini and Tettamanti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Serafini, c2VyYWZpbmltNzJAZ21haWwuY29t
†Present address: Antonio Maria Alviano, Department of Medicine and Surgery, Pathology, IRCCS Fondazione San Gerardo dei Tintori, University of Milano-Bicocca, Milan, Italy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.