 José Miguel Rodríguez-Frade
José Miguel Rodríguez-Frade Luis Ignacio González-Granado
Luis Ignacio González-Granado César A. Santiago4
César A. Santiago4 Mario Mellado
Mario Mellado
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 05 July 2024
Sec. Primary Immunodeficiencies
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1406532
This article is part of the Research Topic Advancing Research, Clinical Recognition, and Targeted Therapies for WHIM Syndrome View all 4 articles
Heterozygous autosomal dominant mutations in the CXCR4 gene cause WHIM syndrome, a severe combined immunodeficiency disorder. The mutations primarily affect the C-terminal region of the CXCR4 chemokine receptor, specifically several potential phosphorylation sites critical for agonist (CXCL12)-mediated receptor internalization and desensitization. Mutant receptors have a prolonged residence time on the cell surface, leading to hyperactive signaling that is responsible for some of the symptoms of WHIM syndrome. Recent studies have shown that the situation is more complex than originally thought, as mutant WHIM receptors and CXCR4 exhibit different dynamics at the cell membrane, which also influences their respective cellular functions. This review examines the functional mechanisms of CXCR4 and the impact of WHIM mutations in both physiological and pathological conditions.
The elucidation of the CXCR4 gene and its impact on human immunology followed several key discoveries in the 1990s and the early 2000s. The CXCR4 gene was first identified and characterized in the 1990s. Initial efforts mapped the gene to chromosome 2q21 using isotopic in situ hybridization (1, 2). The genomic structure of the CXCR4 gene was determined by Wegner et al. in 1998 (3). Key elements in the CXCR4 promoter include a TATA box, a nuclear respiratory factor-1 (NRF-1) site, and two GC boxes, which are critical for the regulation of gene expression.
CXCR4 is a unique polypeptide organized into seven transmembrane α-helices that interact with heterotrimeric G proteins to activate intracellular signaling pathways. Originally described as an orphan G protein-coupled receptor (GPCR), CXCR4 was later found to facilitate HIV-1 fusion with target cells (4). CXCR4 is a homeostatic receptor that is widely expressed in both embryonic and adult tissues (5) and has a unique chemokine ligand, CXCL12 (6), although it can also bind other ligands including CXCL14 (7), migration inhibitory factor (MIF) (8) and HMGB1, which forms complexes with CXCL12 (9). Studies in mice deficient in CXCR4 (Cxcr4-/-) have demonstrated significant impairments in hematopoiesis, and in the development of the nervous and cardiovascular systems. The importance of the CXCR4/CXCL12 axis is underscored not only by the embryonic lethality of both CXCR4- and CXCL12-deficient mice, but also by its high degree of conservation throughout evolution (10).
The CXCR4 protein was first crystallized in 2010 and five independent crystal structures of CXCR4 bound to a small molecule antagonist and a cyclic peptide were reported (11). These studies revealed a consistent homodimer interface involving helices V and VI. Recently, a comparative structural and phylogenetic analysis of CXCR4 sequences from 30 mammalian species revealed a complex evolutionary history with multiple gene duplication and loss events, along with the identification of key functional domains and amino acid residues conserved across species (12).
Similar to other chemokine receptors, CXCR4 plays a pivotal role in leukocyte trafficking and arrest in specific anatomical niches under both homeostatic and pathological conditions (13). Indeed, the CXCL12/CXCR4 axis is essential for both adaptive and innate immune responses, as well as for the organization and maintenance of the bone marrow (BM) (14). Importantly, CXCR4 is critical for the migration, homing and survival of hematopoietic stem cells (HSC) in the BM (15–18). CXCL12 is primarily produced by perivascular mesenchymal stem cells and is essential for HSC quiescence and retention in the BM (19). During the establishment of antigen-presenting cell-T cell contacts, CXCR4, together with other chemokine receptors, is recruited to the peripheral supramolecular activation cluster where it contributes to integrin activation, necessary for the formation of a productive immunological synapse and correct T cell activation (20). Furthermore, B cell organization in the germinal centers of lymph nodes is dependent on CXCR4 expression, and CXCR4-deficient B cells are excluded from the germinal center dark zone (21).
CXCR4 is also expressed in non-hematopoietic tissues including lung, liver, kidney, gastrointestinal tract, adrenal gland, ovary and brain. Conditional Cxcr4-/- mouse models have demonstrated the importance of CXCR4 in regulating the development of the central nervous system (22), as well as the development of the vasculature in the gastrointestinal tract (23) and kidney (24).
Importantly, CXCR4 and CCR5, another chemokine receptor, serve with CD4 as primary co-receptors for HIV-1 entry into target cells (25). Data suggest that viral use of CXCR4 correlates with immunodeficiency and progression to AIDS (26, 27).
Although CXCR4 is primarily considered a homeostatic receptor, its expression can be modulated in various pathological situations. For example, CXCR4 is overexpressed in many tumor types, including breast (28), lung (29) ovarian (30), prostate (31), colon (32), melanoma (33) and neuroblastoma (34). Increased CXCR4 expression in metastatic lesions correlates with tumor progression and with preferential metastatic sites of the primary tumor (28, 35, 36). Studies in mice have shown that CXCR4 blockade inhibits cancer cell dissemination and metastasis in several cancer models (37, 38). The CXCL12/CXCR4 axis is also involved in tumor growth, tumor cell-microenvironment interactions (39), vasculogenesis and angiogenesis (40).
Inflammation is another mechanism involved in the modulation of CXCR4. Transforming growth factor-beta 1 (TGF-β1) (41), vascular endothelial growth factor (VEGF) (42) and basic fibroblast growth factor (bFGF) (43) have all been reported to upregulate CXCR4 expression, whereas cytokines such as IL-5 (44), interferon-alpha (IFN-α) and interferon-gamma (IFN-γ) (45) have the opposite effect. In addition, activation of CXCR4 in macrophages after LPS stimulation suppresses the expression of inflammatory cytokines by blocking MAPK and NF-kB signaling pathways (46). Taken together, these findings support the involvement of CXCR4 in the development and progression of immunodeficiency, inflammatory diseases and cancer and highlight its potential as a therapeutic target.
Heterozygous autosomal dominant mutations in the CXCR4 gene cause WHIM syndrome (47, 48) a severe combined immunodeficiency disorder characterized by increased susceptibility to human papillomavirus pathogenesis, resulting in warts, condyloma acuminata and carcinomas. Patients with WHIM syndrome often present with neutropenia, a common symptom in several primary immunodeficiencies (Table 1) (49, 50), B cell lymphopenia, hypogammaglobulinemia, recurrent infections and myelokathexis characterized by myeloid hyperplasia and an overabundance of mature senescent neutrophils in the BM (48). Some patients also have developmental defects of the cardiovascular, genitourinary and nervous systems (51), although only the cardiovascular defects appear to be clinically significant (52–54). Moreover, decreases in bone mineral density and bone defects leading to osteoporosis have been reported in a WHIM mouse model (55). Some defects in T cell activation in WHIM syndrome may also be associated with a compromised stability of the immunological synapse formed during T cell-APC engagement (56, 57). While CXCR4 gain-of-function variants are the most common cause of WHIM syndrome, a proportion of patients remain undiagnosed. While some patients harboring CXCR2 mutations show myelokathexis or neutropenia, the absence of other features of WHIM syndrome, indicate that this CXCR2 deficiencies have characteristics distinct from those of WHIM (58, 59).
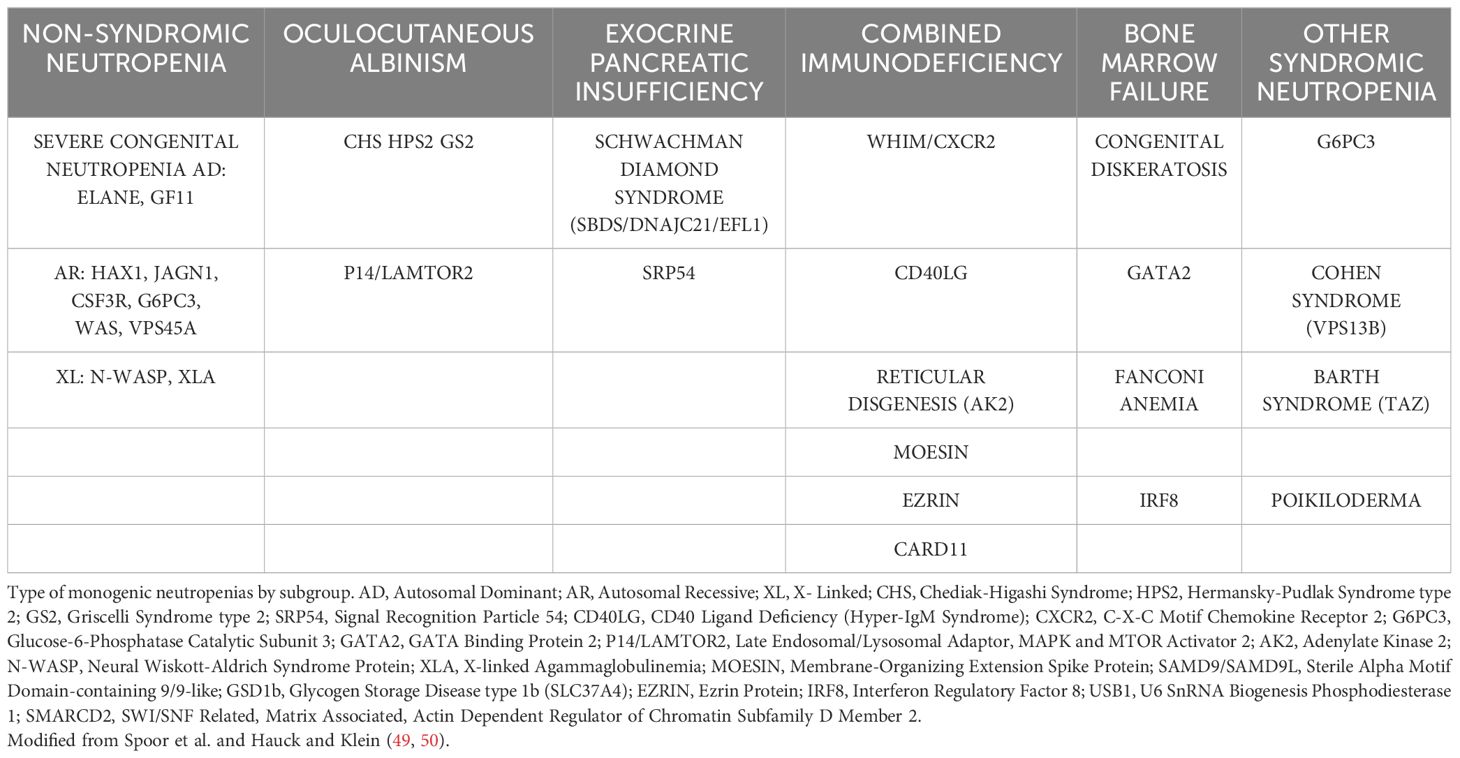
Table 1 Differential diagnosis of WHIM syndrome.
Little is known about the contribution of WHIM mutations to HIV-1 infection, with the exception of one report claiming similar or even lower susceptibility to infection (48) or about the role of WHIM expression in inflammation (60). Paradoxically, WHIM syndrome behaves as a relatively benign immunodeficiency. Indeed, the majority of infections are usually not invasive or life-threatening, and patients survive into adulthood, in part due to the ability of the host response to acute infections to mobilize leukocytes into the circulation. Patients with WHIM do not show a clear genotype-phenotype correlation. In fact, there is considerable phenotypic variability between patients with the same genotype, even within the same family (61, 62). However, they have several unmet needs, such as long-term immunoglobulin replacement therapy and the management of periodontal disease, which affects up to 63.6% of patients with moderate or severe periodontitis (63).
At the molecular level, WHIM syndrome is caused by heterozygous gain-of-function mutations at the C-terminal end of CXCR4 that affect key residues involved in receptor phosphorylation and desensitization (64), explaining the associated hyperactivation of downstream signaling and the retention of leukocytes in the BM, causing robust neutropenia (65). Consistent with this, mice with a myeloid lineage-restricted deletion of CXCR4 also exhibit marked neutrophilia (66). These data indicate that CXCR4 has a dual role in neutrophil homeostasis, regulating both neutrophil release from BM and clearance from blood. CXCR4 expression increases in senescent neutrophils and blocking anti-CXCR4 antibodies abolish neutrophil homing to BM (67, 68). In addition, the p.H323fs329X mutation in CXCR2, the receptor involved in neutrophil egress from BM, is also associated with a similar reduction in circulating neutrophils (58, 67). The severe neutropenia, triggered by delayed neutrophil egress into the circulation and enhanced neutrophil homing to BM, determines the known susceptibility to bacterial and viral infections associated with these patients. Several of the mutations described in WHIM syndrome are also found in patients with Waldenström’s macroglobulinemia, a rare B-cell lymphoma (69) characterized by lymphoplasmacytic infiltrates in the BM, lymph nodes and spleen, often associated with the presence of high IgM titers in the blood (70).
For decades, G-CSF was the only drug available to increase neutrophil and lymphocyte counts in patients with WHIM syndrome. Initial results from phase 1 trials of plerixafor, a selective CXCR4 inhibitor, were reported in 2014 and 2019 in open-label studies (71). Recently, results of a phase 3 crossover randomized controlled trials (RCT) of 19 patients treated with either plerixafor or G-CSF over 12 months became available. While plerixafor was not superior to G-CSF in reducing the overall infection severity score (the primary endpoint), it did result in wart regression and hematological improvement (72).
By contrast, patients with WHIM syndrome participating in a phase 2 trial of mavorixafor (400 mg quaque die, administered orally, a more convenient option than subcutaneous infusion) not only had increased total white blood cell, neutrophil and lymphocyte counts, but also showed reduced annualized infection rates and reduced wart numbers in those treated for six months or more. Results from the phase 3 trial confirmed positive outcomes in terms of the primary endpoints: time (hours) above the absolute neutrophil count (ANC) threshold ≥500/μL, time (hours) above the lymphocyte threshold, and reduced infection frequency, severity and duration (73).
A functional cure of WHIM syndrome has been reported in a patient following chromothriptic deletion of the abnormal CXCR4 gene in hematopoietic stem cells, suggesting the potential of gene editing as a future therapeutic approach (74). Novel CRISPR-Cas9 base editing techniques, including cytosine base editors (CBEs) and adenine base editors (ABEs), along with prime editing, might offer curative treatment without the known risks of hematopoietic stem cell transplantation (HSCT). In a global cohort of 66 patients, the only patient who died had undergone HSCT. Notably, the CRISPR-Cas9 approach successfully corrected the disease in a WHIM mouse model (75).
CXCR4 signaling is finely coordinated by physical receptor interactions with multiple proteins (G proteins, G protein receptor kinases [GRKs] and β-arrestins, filamin A, cofilin, etc) (76, 77). CXCL12 binding to CXCR4 triggers Gαi activation, although signaling involving Gα12-13 and Gαq has also been described (78, 79). One report has linked the induction of Gαq by the HIV-1 envelope glycoprotein through CXCR4 to viral entry (80). These initial signaling events trigger the activation of multiple signaling pathways, including those associated with Src, PI3K, PLC, PKC and MAPK (81).
Binding of CXCL12 to CXCR4 triggers rapid phosphorylation within its 45 amino acid serine/threonine (Ser/Thr)-rich region, primarily at the distal C-terminus. Phosphorylation is critical for agonist (CXCL12)-mediated receptor internalization (82, 83) and degradation (84) and depends on the activity of GRK2 (85, 86), GRK3 (87) and GRK6 (88). GRK-mediated phosphorylation of residues along the C-terminus of CXCR4 facilitates the recruitment and activation of β-arrestins, which mediate receptor desensitization and internalization. It has also been reported that CXCR4 and other chemokine receptors show constitutive activity and internalization in the absence of β-arrestins (89–91), an effect that is attributed to PKC (82). In addition, β-arrestins have a scaffolding role (92) linking the activated receptor to the actin cytoskeleton via several actin-binding proteins (i.e., FLNA and cofilin) (93, 94). This allows the transmission of the CXCL12-mediated conformational changes in CXCR4 to drive signal transduction that triggers the cytoskeletal rearrangements required for cell polarization and the formation of a leading edge, and to sustain productive chemokine-directed cell migration (95). These observations are also consistent with the stabilization of different β-arrestin conformations on the receptor depending on the phosphorylation residues present in the C-tail of the receptors (96).
Previous studies have identified 26 autosomal dominant mutations in CXCR4 associated with WHIM syndrome (64, 97). Most of these mutations affect the C-terminal receptor tail and are unlikely to affect the rest of the quaternary structure, indicating the importance of the C-terminal region in CXCR4 signaling (Figure 1). It is therefore not surprising that GRK-mediated phosphorylation of Ser/Thr residues, β−arrestin coupling and receptor desensitization and internalization are affected by C-terminal mutations. Indeed, WHIM mutants show impaired receptor internalization and degradation, resulting in prolonged receptor residence time at the cell membrane, which in turn contributes to the gain-of-function properties of these receptors and their hyperactive signaling nature compared with wild-type CXCR4 (Figure 2). Further investigation is, however, required to explore the full impact of WHIM mutations on G protein recruitment and activation (97). In vitro assays have shown that CXCL12-induced 35S-GTPγS binding to activated Gαi-containing membranes from cells expressing comparable levels of wild-type CXCR4, CXCR4S338X or CXCR4R334X, the most common mutations observed in WHIM patients, results in increased coupling efficiency and potency of the mutant receptors (98–100). In addition to the defects in β-arrestin coupling, the stronger association of G proteins to mutant receptors might also contribute to the heightened responsiveness of WHIM mutant CXCR4 to CXCL12.
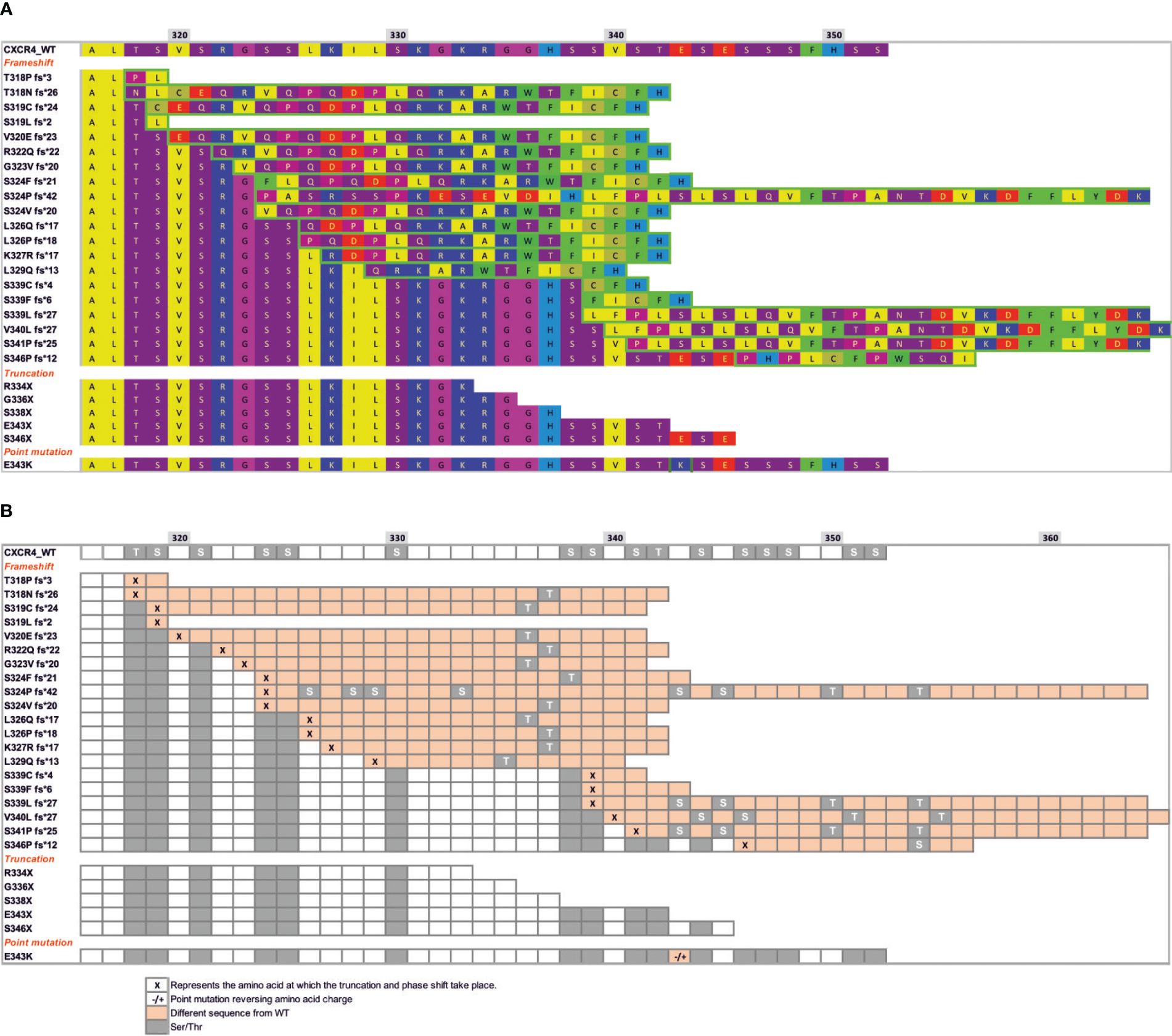
Figure 1 Mutations at the C-terminal end of CXCR4 WHIM receptors lead to different potential changes in the Ser/Thr phosphorylation pattern. (A) Sequence of CXCR4 residues (318–352) showing the three different types of mutations that occur in WHIM syndrome: frameshift, truncation or nonsense and missense point mutations. Hydrophobic residues are shown in yellow, bulky hydrophobic residues in green, hydrogen donors in purple, and positively and negatively charged residues in blue and red, respectively. Residues framed in green show a different sequence from wild-type CXCR4, resulting from a frameshift or point mutation. (B) Scheme showing the pattern of phosphorylation sites present in helix 8 and in the c-terminal region of CXCR4, shown as grey colored squares. Changes in the sequence following a frameshift or point mutation (-/+) are shown in orange boxes following the sequence from (A). Specific residues where the frameshift occurs are shown as ‘x’ in orange boxes.
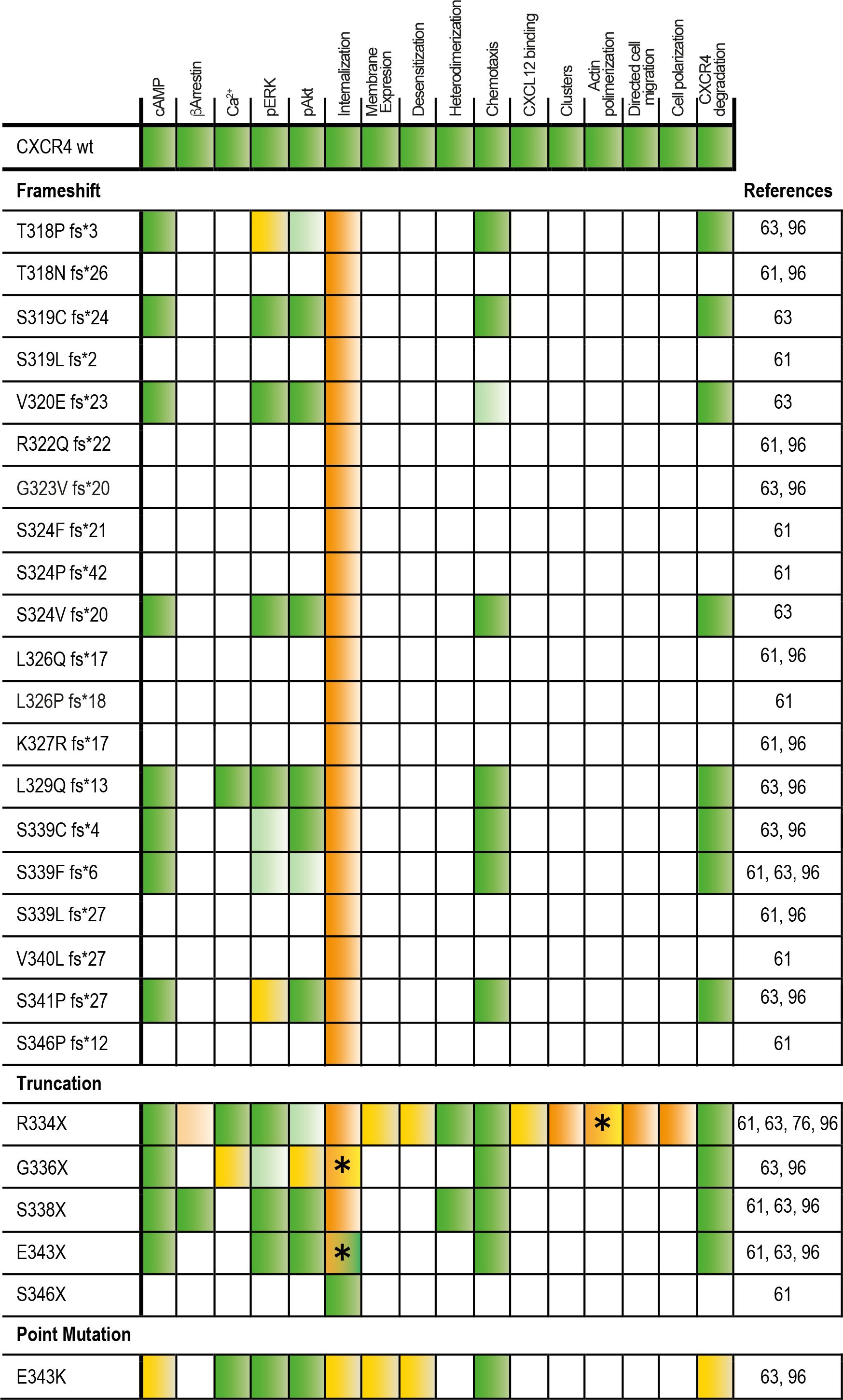
Figure 2 Response to CXCL12 of the different WHIM mutants as compared with the wild-type receptor. An increase in the response to CXCL12 compared with the wild-type receptor is indicated by green boxes (light green indicates a weak but significant response). A decrease in the response is indicated by red boxes (light red indicates a weak but significant decrease). No effect of the mutation on CXCL12 responses is shown in yellow. White boxes indicate missing information. Different results depending on the concentration or the cell line used in the study are indicated by an asterisk (*).
The most common mutation in WHIM syndrome is a truncation at Arg334 (R334X) (101) which, similar to the CXCR4S338X truncation, promotes increased CXCL12-mediated signaling, G protein interactions and ERK and AKT activation, and decreased GRK6 associations, β-arrestin2 interactions and impaired receptor internalization (98–105). Both mutants are also associated with prolonged CXCL12-mediated F-actin polymerization (99). Similarly, the E343K point mutation results in increased receptor signaling, decreased Ser/Thr phosphorylation and impaired receptor internalization (106, 107), although receptor internalization was only affected at low CXCL12 concentrations (63). The case of CXCR4E343K is quite interesting, as this receptor has a full length C-terminus with complete phosphorylation sites and no change in all potential phosphorylation sites. However, the negative charge of the 343 site at the receptor tail is essential for CXCR4 function, and changing the charge through an amino acid substitution dysregulates the signaling events downstream of G proteins. Cells overexpressing CXCR4E343D have no functional changes compared with wild-type CXCR4, whereas cells overexpressing CXCR4E343K, CXCR4E343R or CXCR4E343A show increased cell migration, prolonged phosphorylation of ERK1/2, p38, JNK1/2/3 and increased activation of PI3K/AKT/NF-κB signal pathway, that is, all these mutants reproduce the in vitro signaling events associated with WHIM syndrome (108). By contrast, the CXCR4E343X and CXCR4S341P fs*25 mutants are partially internalized at high doses of CXCL12 (63). Taken together, these data suggest that the mutant CXCR4-dependent changes in signaling may reflect differences in the symptoms experienced by patients, or may even be involved in the differential penetrance of this syndrome between patients. Defects in receptor internalization show a strong correlation with the severity of neutropenia and lymphopenia in patients with WHIM syndrome, as well as with their susceptibility to recurrent infections (64). Some studies also associate AKT hyperactivation with reduced IgA levels in blood and decreased T cell counts (64), a finding common in activated PI3K delta syndrome, which is characterized by constitutively active AKT signaling (109), and a reduction in T cell numbers has been associated with strong inhibition of cAMP release (64). These are, nonetheless, expected consequences, as these CXCR4 variants are gain-off-function receptors due to their lack of internalization.
Interestingly, some CXCR4 mutations associated with the WHIM phenotype are not consistent with a gain-of-function phenotype. For example, CXCR4T318P fs*3 mutants show impaired β-arrestin recruitment with decreased ERK1/2 phosphorylation and calcium mobilization, resulting in reduced cell migration in response to CXCL12 (110), whereas CXCR4E343K and CXCR4G336X mutants do not affect ligand-mediated internalization (97). CXCR4E343K has a full-length C-terminus and the same number of Ser/Thr phosphorylation sites as wild-type CXCR4, but still functions as a gain-of-function mutant (108). CXCR4S338X retains its interaction with β-arrestins, but is not internalized and induces stronger ERK phosphorylation and cell chemotaxis than wild-type CXCR4 (97). The S339F fs*6 mutation enhances agonist-driven signaling, decreases Ser/Thr phosphorylation, β-arrestin binding and endocytosis and increases basal degradation (111). While some mutations have been clearly linked to defects in the phosphorylation of specific residues required for efficient receptor endocytosis and degradation, others appear to be less essential, supporting the involvement of additional factors to explain the phenotype associated with this syndrome.
During the review process of this manuscript a a novel heterozygous CXCR4 variant (c.250G>C; D84H) localized at a highly conserved position in the transmembrane domain of the receptor outside the C-terminus has been described (112). The patient-derived peripheral blood mononuclear cells carrying this mutation, and in vitro cellular assays show decreased CXCR4 internalization and increased chemotaxis in response to CXCL12, similar to known CXCR4WHIM, but also revealed unique features of CXCR4D84H signaling as shows impaired cAMP inhibition and Ca2+ mobilization and does not show enhancement in pAKT or pERK levels as the other WHIM variants do. These findings are consistent with molecular dynamics simulations that show disruption of the Na+ binding pocket by D84H, resulting in collapse of the hydrophobic gate above and destabilization of the inactive state of CXCR4.
Collectively, these data suggest that WHIM syndrome is molecularly more complex than originally thought, as different mutations on receptors and different effects on CXCL12-mediated functions can lead to similar cellular phenotypes.
Until relatively recently, GPCRs were thought to be monomeric entities that transiently interact with a G protein, promoting its dissociation into Gα and Gβγ subunits. Biophysical and biochemical studies of rhodopsin, the first purified GPCR, supported the concept that GPCRs are monomeric (113). Accordingly, a single activated receptor was thought to sequentially activate multiple G proteins in a simple ternary model of ligand/receptor/G protein complexes, which was considered sufficient to explain the functions triggered by ligand binding under equilibrium conditions (114). The model included two receptor populations: an inactive receptor and an active receptor. Ligand binding and G proteins cooperatively promoted the transition to the active form, initiating downstream signaling (115). In addition, both rhodopsin and β2 adrenergic receptors remained functional when entrapped as monomers in nanolipid disks, and were able to bind their corresponding ligands and activate G proteins (113, 116).
Over the past decade, however, structural and spectroscopic studies have revealed that GPCRs occupy a continuum of conformational states that progressively facilitate G protein activation (117, 118). The receptor is maintained in an inactive conformation by interhelical ionic locks that act as molecular switches, and ligand binding increase the conformational heterogeneity of the receptors, flipping these molecular switches to facilitate receptor activation (119). This is also true for CXCR4, where CXCL12 binding induces conformational changes in the transmembrane domains. A two-step binding site model has been proposed for the interaction between CXCL12 and CXCR4 (120, 121). In the first step, the central body of the chemokine interacts with the N-terminal end of CXCR4 in the chemokine recognition site 1 (CRS1), allowing optimal orientation of CXCL12 on the receptor. This allows the N-terminus to enter the receptor, which in a second step facilitates its binding to the CRS2 region (122). This second interaction occurs between the first two N-terminal residues of the ligand and a group of residues mainly found in CRS2 (120). In addition, eight residues in the transmembrane segments TMVI and TMVII link the conformational changes in the transmembrane regions of CXCR4 to the residues involved in signal initiation. This is a critical event for signal transduction, as the residues are part of the microswitch that allows G protein coupling. Notably, residues F248 to V242 in TMVI are in contact with almost all the conserved motifs critical for signaling in GPCRs, including the CWxP motif in TMVI, NPxxY in TMVII, DRY in TMIII, and Y(x)5KL in TMV. These residues play a role in controlling the transition between active and inactive receptor states by allowing helix and side-chain translation, as described in studies using mutational strategies of this hydrophobic bridge in different receptors (123–125). The existence of multiple receptor conformational states, each capable of differentially binding ligand and G protein, suggests the need for a continuum model of ternary complex formation. In addition, several studies have highlighted the role of G protein nucleotide states in the kinetics of ligand binding and receptor conformation (126–128), necessitating the inclusion of G protein activation states in GPCR signaling models. Using a computational approach, a recent report predicted receptor self-associations and designed CXCR4 dimers with different quaternary structures and signaling properties. The authors designed CXCR4 oligomers that activated Gi, but not all recruited β-arrestins (129) supporting the presence of multiple CXCR4 conformations at the cell membrane.
The ability of GPCRs, including chemokine receptors, to homo- and heterodimerize has been well characterized (130). Evidence from a variety of experimental approaches, including co-immunoprecipitation, cross-linking assays, resonance energy transfer technologies, functional complementation experiments and advanced light microscopy techniques, confirms that GPCRs form both homo- and heterodimers (130). These receptor complexes add additional layers of complexity that also modulate cell responses (131, 132). Crystallographic studies of CXCR4 confirm the existence of dimeric conformations, with the implicated residues mainly located at the extracellular portion of helices V and VI in the case of the CXCR4:IT1t complex, and at the base of helices III and IV in the case of CXCR4:CVX15 (11, 133). However, it is important to note that in these studies, and similarly to the case of the β2-adrenergic receptor (134, 135) and the A2A adenosine receptor (136), the strategy used a T4 lysozyme fusion inserted between TMV and TMVI on the cytoplasmic side of the CXCR4 and a thermostabilizing L1253.41W mutation to stabilize the receptor (11, 133). Thus, it was an artifactual strategy to stabilize the conformations, which could influence the results and conclusions obtained. The regions involved in these interactions differ from those previously described in models of GPCR dimerization, where the contacting residues were assigned to helices I and IV (137, 138). The functional implications of these differences for the CXCR4 life cycle remain unclear, but as there is a low sequence identity in the dimerization region between dimerizing GPCRs the data may represent a feature specific to CXCR4.
Chemokine receptor complexes, including CXCR4 and CCR5, appear to form during their synthesis and maturation, and clusters of CXCR4 and CCR5 can be detected in small trans-Golgi vesicles (139). While dimerization may not be required for functional coupling of the GPCR to heterotrimeric G-proteins per se, in some cases mutant chemokine receptors that cannot dimerize show a reduced ability to induce cell migration, suggesting that these complexes might be functionally relevant (140, 141). Homo- and heterodimerization processes add complexity to the biology of these receptors and affect their functionality. CXCR4 has been shown to constitutively dimerize (142, 143) and to form heterodimers with other GPCRs, including other chemokine receptors (144–146).
The crystal structure of CXCR4 has shown that the receptor exists as a homodimer (11), suggesting that both wild-type and WHIM mutant forms may coexist as independent monomers, homodimers and/or heterodimers, in cells from patients. In vitro studies using FRET and BRET have revealed WHIM homodimers and heterodimerization between some WHIM mutants and CXCR4, providing a molecular mechanism to explain the dominant-negative role of these mutants (77). Although CXCR4 and WHIM alleles are likely to be co-expressed, the stoichiometry of the different complexes could vary between different cell types and patients, potentially contributing to the observed phenotypic heterogeneity of the disease (101). Some recent data in immortalized Jurkat cells expressing CXCR4R334X alone or with wild-type CXCR4 have shown varying levels of impact on signaling cascades (63). Furthermore, the contribution of potential heterodimers between WHIM mutants and other chemokine receptors, including ACKR3, or even with other GPCRs, as demonstrated for CXCR4 (144, 147) may also be relevant in WHIM syndrome and needs to be addressed.
Recent findings using total internal reflection fluorescence microscopy (TIRF-M) have shown that, in addition to the ligand-mediated conformational changes that activate G proteins and influence receptor dimerization, CXCL12 induces CXCR4 nanoclustering. This oligomerization process is associated with conformational changes on the receptor that are required for full activation of the signaling cascade (148, 149). G-protein activation, ERK1/2 and PI3K phosphorylation occur normally in CXCR4 mutants that are unable to nanocluster in the presence of CXCL12 (77, 149) but correct cell polarization, leading edge formation and ligand-mediated directed cell migration (77) require CXCL12-mediated receptor nanoclustering. These findings thus add another layer of complexity because chemokine receptors, like other GPCRs, are dynamic structures embedded in the lipid bilayer of the cell membrane. Receptor nanoclustering is a CXCL12-mediated process that is very limited or absent in unstimulated cells and requires the transduction of conformational changes through the transmembrane helical domains (122), ultimately leading to G protein interaction and signaling. Treatment with pertussis toxin abolishes CXCL12-mediated receptor nanoclustering, suggesting that the process requires ligand binding and receptor activation. Furthermore, the scaffolding role of β-arrestins is critical for proper actin dynamics and receptor nanoclustering, as cells lacking β-arrestin1 have defects in actin dynamics, as well as impaired CXCL12-mediated CXCR4 nanoclustering and weakened cell migration towards CXCL12 gradients (77, 150). Similarly, treatment of CXCR4-expressing cells with latrunculin A, an inhibitor of actin polymerization (149), results in defective CXCL12-mediated nanoclustering and loss of directed cell migration. This evidence thus suggests an active role for the actin cytoskeleton in regulating receptor nanoclustering.
While data are not available for all WHIM CXCR4 mutants, a recent TIRF-M evaluation of the conformation and dynamics of CXCR4R334X at the cell membrane indicates that this mutant is unable to nanocluster in the presence of CXCL12 and blocks CXCR4 nanoclustering when both receptors are co-expressed (heterozygosis). Although CXCR4R334X behaves as a gain-of-function mutant, increasing MAPK and PI3K activation, and promoting stronger chemotaxis when compared with wild-type CXCR4, it fails to promote directed cell migration to CXCL12. Primary T cells expressing CXCR4R334X show CXCL12-mediated polarization but exhibit multiple actin-rich protrusions, suggesting defects in leading edge formation (77). These defects are attributed to insufficient actin cytoskeleton remodeling due to inadequate β-arrestin1 activation when CXCR4R334X is co-expressed. As a consequence, the balance between activated and deactivated cofilin is disrupted and cells fail to reorganize their actin cytoskeleton in the presence of CXCL12. In addition, the receptors exhibit free diffusion at the cell membrane and receptor nanoclustering is lost, consistent with the inability of cells to sense CXCL12 gradients despite the presence of CXCR4R334X homodimers and heterodimers with CXCR4 (77).
Differences in nanoclustering between CXCR4 and CXCR4R334X are unlikely due to the different internal structures of the two receptors. Although it remains to be formally evaluated, we speculate that WHIM mutants and CXCR4 share identical transmembrane structures and the same residues in the cluster that mediate chemokine engagement, signal initiation, propagation and microswitch activation. Differences in the receptors are likely to involve β-arrestin binding and/or activation, which subsequently affect actin cytoskeleton dynamics. For example, although the internal structure of the transmembrane domains is very similar, the absence of 19 residues in the C-terminal end of CXCR4R334X dramatically alters the phosphorylation pattern induced by GRK proteins, which is known to provide a readable barcode for β-arrestin association and function (Figures 1A, B) (64). Under such conditions, the association of β-arrestins, as well as the internalization processes, the proper dynamics of the actin cytoskeleton and the ability of the cells to sense the chemoattractant gradient, are altered.
From a structural perspective, WHIM mutations may initially compromise the integrity and thus the environment of the intracellular helix 8. This small fragment has been demonstrated to form an amphipathic helix in numerous GPCRs and is critical for stabilizing effective intracellular signaling after ligand binding (151). The generation of helix 8 and C-terminal models for wild-type CXCR4 and WHIM mutants using Alphafold2 (152) has revealed a wealth of different possible conformations and newly acquired secondary structure motifs that may shed light on the impact of a defective C-terminus on CXCR4 function (Figure 3). The WHIM mutant models show remarkable differences from the wild-type structure, particularly after a frameshift, where a new sequence is added to the C-terminus. Changes in the integrity of helix 8 can occur, disrupting the folding of the alpha-helix (Figure 3), as in the T318P fs*3, L319 fs*24, V320E fs*23 mutants. The addition of new secondary structural motifs may affect both the interaction of the receptor with other proteins and its own oligomerization. In addition, in many of the frameshift mutants, residues that were previously susceptible to phosphorylation either disappeared or shifted their position, altering the pattern of GRK-mediated phosphorylation and thus the barcode used for β-arrestin binding and activation.
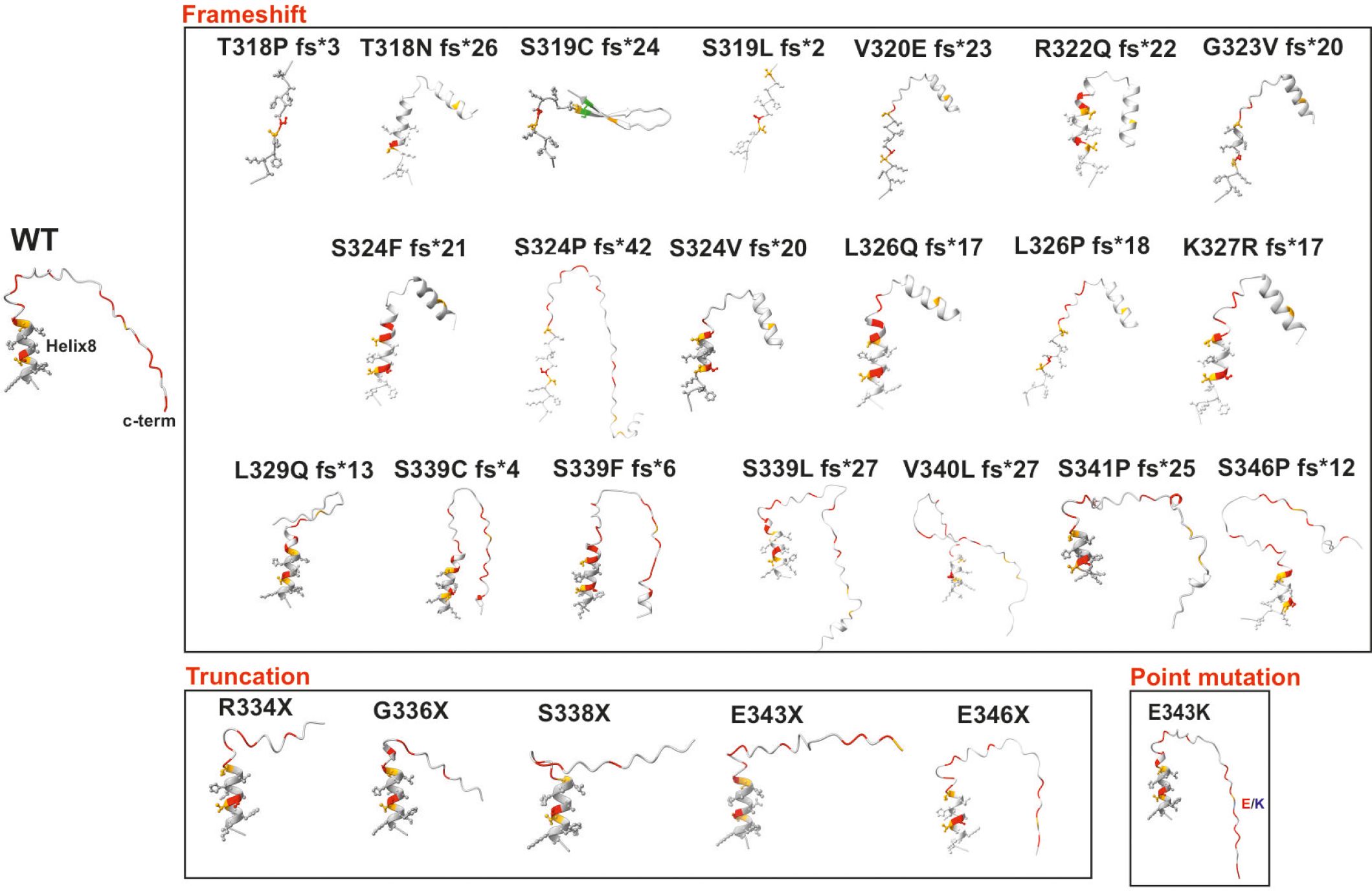
Figure 3 Alphafold2-generated models of helix 8 and C-terminus of wild-type CXCR4 and WHIM syndrome mutants. Models of wild-type CXCR4 helix 8 and the C-terminus end of different mutants associated with WHIM syndrome were generated to analyze the theoretical changes in both the helix and the predicted intrinsically disordered region at the C-terminus. The three different types of mutations present in WHIM syndrome are grouped by black rectangles. The structures are shown as ribbons, with residues belonging to helix 8 shown as spheres and sticks. Serine residues are colored red, threonine residues orange, cysteine residues green and lysine residues in the E343K mutant blue.
Recent years have seen remarkable progress in understanding the function of chemokine receptors, including receptor structures, interaction with ligand(s), signaling pathways activated and interactions with other membrane proteins. These breakthroughs have defined chemokines as a highly complex family with multiple possible functions depending on the local microenvironment. When analyzing the functionality of WHIM mutants and their association with specific phenotypes in patients, it is crucial to consider the quaternary conformation of the receptors, their interaction with other chemokine receptors (dimers, oligomers) and with other membrane proteins (CD4, tetraspanins, other GPCRs, etc), membrane lipids, and with signaling molecules (G proteins, GRKs, β-arrestins, etc.). Their dysfunction cannot be attributed solely to their lack of internalization and/or degradation.
JR: Writing – original draft, Writing – review & editing. LG: Writing – review & editing. CS: Writing – original draft, Writing – review & editing. MM: Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the Spanish Ministry of Science and Innovation (PID2020-114980RB-I00 supported by MCIN/AEI/10.13039/501100011033/FEDER, UE) to MM and (PID2022-140651NB-I00 supported by MCIN/AEI/10.13039/501100011033/FEDER, UE) to CS and from the Biomedicine Program of the Regional Government of Comunidad de Madrid, Spain (S2022/BMD-7274 RETAR-A-COVID-CM) to MM.
We thank the members of the Chemokine signaling group in the Centro Nacional de Biotecnología/CSIC for their excellent work that has inspired this review.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Federsppiel B, Melhado IG, Duncan AMV, Delaney A, Schappert K, Clark-Lewis I, et al. Molecular cloning of the cDNA and chromosomal localization of the gene for a putative seven-transmembrane segment (7-TMS) receptor isolated from human spleen. Genomics. (1993) 16:707–12. doi: 10.1006/geno.1993.1251
2. Herzog H, Hort YJ, Shine J, Selbie LA. Molecular cloning, characterization, and localization of the human homolog to the reported bovine NPY Y3 receptor: lack of NPY binding and activation. DNA Cell Biol. (1993) 12:465–71. doi: 10.1089/dna.1993.12.465
3. Wegner SA, Ehrenberg PK, Chang G, Dayhoff DE, Sleeker AL, Michael NL. Genomic organization and functional characterization of the chemokine receptor CXCR4, a major entry co-receptor for human immunodeficiency virus type 1. J Biol Chem. (1998) 273:4754–60. doi: 10.1074/jbc.273.8.4754
4. Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. (1996) 272:872–7. doi: 10.1126/science.272.5263.872
5. Juarez J, Bendall L, Bradstock K. Chemokines and their receptors as therapeutic targets: the role of the SDF-1/CXCR4 axis. Curr Pharm Des. (2004) 10:1245–59. doi: 10.2174/1381612043452640
6. Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana- Seisdedos F, et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature. (1996) 382:833–5. doi: 10.1038/382833a0
7. Hara T, Tanegashima K. CXCL14 antagonizes the CXCL12-CXCR4 signaling axis. Biomol Concepts. (2014) 5:167–73. doi: 10.1515/bmc-2014-0007
8. Rajasekaran D, Gröning S, Schmitz C, Zierow S, Drucker N, Bakou M, et al. Macrophage migration inhibitory factor-CXCR4 receptor interactions: EVIDENCE FOR PARTIAL ALLOSTERIC AGONISM IN COMPARISON WITH CXCL12 CHEMOKINE. J Biol Chem. (2016) 291:15881–95. doi: 10.1074/jbc.M116.717751
9. Schiraldi M, Raucci A, Muñoz LM, Livoti E, Celona B, Venereau E, et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med. (2012) 209:551–63. doi: 10.1084/jem.20111739
10. Nomiyama H, Osada N, Yoshie O. A family tree of vertebrate chemokine receptors for a unified nomenclature. Dev Comp Immunol. (2011) 35:705–15. doi: 10.1016/j.dci.2011.01.019
11. Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Sci (1979) . (2010) 330:1066–1071. doi: 10.2210/pdb3oe0/pdb
12. Naheed F, Mumtaz R, Shabbir S, Jamil A, Asif AR, Rahman A, et al. Structural and phylogenetic analysis of CXCR4 protein reveals new insights into its role in emerging and re-emerging diseases in mammals. Vaccines (Basel). (2023) 11:671. doi: 10.3390/vaccines11030671
13. Cambier S, Gouwy M, Proost P. The chemokines CXCL8 and CXCL12: molecular and functional properties, role in disease and efforts towards pharmacological intervention. Cell Mol Immunol. (2023) 20:217–51. doi: 10.1038/s41423-023-00974-6
14. Nie Y, Han YC, Zou YR. CXCR4 is required for the quiescence of primitive hematopoietic cells. J Exp Med. (2008) 205:777–83. doi: 10.1084/jem.20072513
15. Wright DE, Bowman EP, Wagers AJ, Butcher EC, Weissman IL. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J Exp Med. (2002) 195:1145–54. doi: 10.1084/jem.20011284
16. Broxmeyer HE, Cooper S, Kohli L, Hangoc G, Lee Y, Mantel C, et al. Transgenic expression of stromal cell-derived factor-1/CXC chemokine ligand 12 enhances myeloid progenitor cell survival/antiapoptosis in vitro in response to growth factor withdrawal and enhances myelopoiesis in vivo. J Immunol. (2003) 170:421–9. doi: 10.4049/jimmunol.170.1.421
17. Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. (2005) 201:1307–18. doi: 10.1084/jem.20041385
18. Lapidot T, Kollet O. The essential roles of the chemokine SDF-1 and its receptor CXCR4 in human stem cell homing and repopulation of transplanted immune-deficient NOD/SCID and NOD/SCID/B2m(null) mice. Leukemia. (2002) 16:1992–2003. doi: 10.1038/sj.leu.2402684
19. Singh P, Mohammad KS, Pelus LM. CXCR4 expression in the bone marrow microenvironment is required for hematopoietic stem and progenitor cell maintenance and early hematopoietic regeneration after myeloablation. Stem Cells. (2020) 38:849–59. doi: 10.1002/stem.3174
20. Cascio G, Martín-Cófreces NB, Rodríguez-Frade JM, López-Cotarelo P, Criado G, Pablos JL, et al. CXCL12 regulates through JAK1 and JAK2 formation of productive immunological synapses. J Immunol. (2015) 194:5509–19. doi: 10.4049/jimmunol.1402419
21. Allen CDC, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. (2004) 5:943–52. doi: 10.1038/ni1100
22. Cash-Padgett T, Sawa A, Jaaro-Peled H. Increased stereotypy in conditional Cxcr4 knockout mice. Neurosci Res. (2016) 105:75–9. doi: 10.1016/j.neures.2015.10.001
23. Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, et al. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature. (1998) 393:591–4. doi: 10.1038/31261
24. Takabatake Y, Sugiyama T, Kohara H, Matsusaka T, Kurihara H, Koni PA, et al. The CXCL12 (SDF-1)/CXCR4 axis is essential for the development of renal vasculature. J Am Soc Nephrol. (2009) 20:1714–23. doi: 10.1681/ASN.2008060640
25. Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol. (1999) 17:657–700. doi: 10.1146/annurev.immunol.17.1.657
26. Tersmette M, de Goede RE, Al BJ, Winkel IN, Gruters RA, Cuypers HT, et al. Differential syncytium-inducing capacity of human immunodeficiency virus isolates: frequent detection of syncytium-inducing isolates in patients with acquired immunodeficiency syndrome (AIDS) and AIDS-related complex. J Virol. (1988) 62:2026–32. doi: 10.1128/jvi.62.6.2026-2032.1988
27. Zhu T, Mo H, Wang N, Nam DS, Cao Y, Koup RA, et al. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science. (1993) 261:1179–81. doi: 10.1126/science.8356453
28. Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. (2001) 410:50–6. doi: 10.1038/35065016
29. Qiu L, Xu Y, Xu H, Yu B. The clinicopathological and prognostic value of CXCR4 expression in patients with lung cancer: a meta-analysis. BMC Cancer. (2022) 22:1–13. doi: 10.1186/s12885-022-09756-1
30. Hall JM, Korach KS. Stromal cell-derived factor 1, a novel target of estrogen receptor action, mediates the mitogenic effects of estradiol in ovarian and breast cancer cells. Mol Endocrinol. (2003) 17:792–803. doi: 10.1210/me.2002-0438
31. Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK, et al. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. (2002) 62:1832–7. Available at: https://aacrjournals.org/cancerres/article/62/6/1832/509740/Use-of-the-Stromal-Cell-derived-Factor-1-CXCR4. (Accessed November 10, 2022).
32. Lv S, Yang Y, Kwon S, Han M, Zhao F, Kang H, et al. The association of CXCR4 expression with prognosis and clinicopathological indicators in colorectal carcinoma patients: A meta-analysis. Histopathology. (2014) 64:701–12. doi: 10.1111/his.12321
33. Scala S, Giuliano P, Ascierto PA, Ieranò C, Franco R, Napolitano M, et al. Human melanoma metastases express functional CXCR4. Clin Cancer Res. (2006) 12:2427–33. doi: 10.1158/1078-0432.CCR-05-1940
34. Geminder H, Sagi-Assif O, Goldberg L, Meshel T, Rechavi G, Witz IP, et al. A possible role for CXCR4 and its ligand, the CXC chemokine stromal cell-derived factor-1, in the development of bone marrow metastases in neuroblastoma. J Immunol. (2001) 167:4747–57. doi: 10.4049/jimmunol.167.8.4747
35. Fingleton B. Molecular targets in metastasis: Lessons from genomic approaches. Cancer Genomics Proteomics. (2007) 4:211–22.
36. Bohn OL, Nasir I, Brufsky A, Tseng GC, Bhargava R, MacManus K, et al. Biomarker profile in breast carcinomas presenting with bone metastasis. Int J Clin Exp Pathol. (2010) 3:139.
37. Kim SY, Lee CH, Midura BV, Yeung C, Mendoza A, Hong SH, et al. Inhibition of the CXCR4/CXCL12 chemokine pathway reduces the development of murine pulmonary metastases. Clin Exp Metastasis. (2008) 25:201–11. doi: 10.1007/s10585-007-9133-3
38. Porvasnik S, Sakamoto N, Kusmartsev S, Eruslanov E, Kim WJ, Cao W, et al. Effects of CXCR4 antagonist CTCE-9908 on prostate tumor growth. Prostate. (2009) 69:1460–9. doi: 10.1002/pros.21008
39. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. (2005) 121:335–48. doi: 10.1016/j.cell.2005.02.034
40. Liang Z, Brooks J, Willard M, Liang K, Yoon Y, Kang S, et al. CXCR4/CXCL12 axis promotes VEGF-mediated tumor angiogenesis through Akt signaling pathway. Biochem Biophys Res Commun. (2007) 359:716–22. doi: 10.1016/j.bbrc.2007.05.182
41. Buckley CD, Amft N, Bradfield PF, Pilling D, Ross E, Arenzana-Seisdedos F, et al. Persistent induction of the chemokine receptor CXCR4 by TGF-beta 1 on synovial T cells contributes to their accumulation within the rheumatoid synovium. J Immunol. (2000) 165:3423–9. doi: 10.4049/jimmunol.165.6.3423
42. Zagzag D, Lukyanov Y, Lan L, Ali MA, Esencay M, Mendez O, et al. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: implications for angiogenesis and glioma cell invasion. Lab Invest. (2006) 86:1221–32. doi: 10.1038/labinvest.3700482
43. Salcedo R, Wasserman K, Young HA, Grimm MC, Howard OMZ, Anver MR, et al. Vascular endothelial growth factor and basic fibroblast growth factor induce expression of CXCR4 on human endothelial cells: In vivo neovascularization induced by stromal-derived factor-1alpha. Am J Pathol. (1999) 154:1125–35. doi: 10.1016/S0002-9440(10)65365-5
44. Iikura M, Miyamasu M, Yamaguchi M, Kawasaki H, Matsushima K, Kitaura M, et al. Chemokine receptors in human basophils: inducible expression of functional CXCR4. J Leukoc Biol. (2001) 70:113–20. doi: 10.1189/jlb.70.1.113
45. Nagase H, Miyamasu M, Yamaguchi M, Imanishi M, Tsuno NH, Matsushima K, et al. Cytokine-mediated regulation of CXCR4 expression in human neutrophils. J Leukoc Biol. (2002) 71:711–7. doi: 10.1189/jlb.71.4.711
46. Tian X, Xie G, Xiao H, Ding F, Bao W, Zhang M. CXCR4 knockdown prevents inflammatory cytokine expression in macrophages by suppressing activation of MAPK and NF-κB signaling pathways. Cell Biosci. (2019) 9:55. doi: 10.1186/s13578-019-0315-x
47. Kawai T, Malech HL. WHIM syndrome: Congenital immune deficiency disease. Curr Opin Hematol. (2009) 16:20–26. doi: 10.1097/MOH.0b013e32831ac557
48. Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F, et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. (2003) 34:70–4. doi: 10.1038/ng1149
49. Spoor J, Farajifard H, Rezaei N. Congenital neutropenia and primary immunodeficiency diseases. Crit Rev Oncol Hematol. (2019) 133:149–62. doi: 10.1016/j.critrevonc.2018.10.003
50. Hauck F, Klein C. Pathogenic mechanisms and clinical implications of congenital neutropenia syndromes. Curr Opin Allergy Clin Immunol. (2013) 13:596–606. doi: 10.1097/ACI.0000000000000014
51. Galli J, Pinelli L, Micheletti S, Palumbo G, Notarangelo LD, Lougaris V, et al. Cerebellar involvement in warts Hypogammaglobulinemia immunodeficiency myelokathexis patients: neuroimaging and clinical findings. Orphanet J Rare Dis. (2019) 14. doi: 10.1186/s13023-019-1030-8
52. Taniuchi S, Yamamoto A, Fujiwara T, Hasui M, Tsuji S, Kobayashi Y. Dizygotic twin sisters with myelokathexis: mechanism of its neutropenia - PubMed. Am J Hematol . (1999) 62:106–111. doi: 10.1002/(ISSN)1096-8652
53. Badolato R, Dotta L, Tassone L, Amendola G, Porta F, Locatelli F, et al. Tetralogy of fallot is an uncommon manifestation of warts, hypogammaglobulinemia, infections, and myelokathexis syndrome. J Pediatr. (2012) 161:763–5. doi: 10.1016/j.jpeds.2012.05.058
54. Beaussant Cohen S, Fenneteau O, Plouvier E, Rohrlich PS, Daltroff G, Plantier I, et al. Description and outcome of a cohort of 8 patients with WHIM syndrome from the French Severe Chronic Neutropenia Registry. Orphanet J Rare Dis. (2012) 7:71. doi: 10.1186/1750-1172-7-71
55. Anginot A, Nguyen J, Abou Nader Z, Rondeau V, Bonaud A, Kalogeraki M, et al. WHIM Syndrome-linked CXCR4 mutations drive osteoporosis. Nat Commun. (2023) 14:2058. doi: 10.1038/s41467-023-37791-4
56. Kallikourdis M, Trovato AE, Anselmi F, Sarukhan A, Roselli G, Tassone L, et al. The CXCR4 mutations in WHIM syndrome impair the stability of the T-cell immunologic synapse. Blood. (2013) 122:666–73. doi: 10.1182/blood-2012-10-461830
57. Roselli G, Martini E, Lougaris V, Badolato R, Viola A, Kallikourdis M. CXCL12 mediates aberrant costimulation of B lymphocytes in warts, hypogammaglobulinemia, infections, myelokathexis immunodeficiency. Front Immunol. (2017) 8:1068. doi: 10.3389/fimmu.2017.01068
58. Auer PL, Teumer A, Schick U, O’Shaughnessy A, Lo KS, Chami N, et al. Rare and low-frequency coding variants in CXCR2 and other genes are associated with hematological traits. Nat Genet. (2014) 46:629–34. doi: 10.1038/ng.2962
59. Marin-Esteban V, Youn J, Beaupain B, Jaracz-Ros A, Barlogis V, Fenneteau O, et al. Biallelic CXCR2 loss-of-function mutations define a distinct congenital neutropenia entity. Haematologica. (2022) 107:765–9. doi: 10.3324/haematol.2021.279254
60. Gallego C, Vétillard M, Calmette J, Roriz M, Marin-Esteban V, Evrard M, et al. CXCR4 signaling controls dendritic cell location and activation at steady state and in inflammation. Blood. (2021) 137:2770–84. doi: 10.1182/blood.2020006675
61. Dotta L, Notarangelo LD, Moratto D, Kumar R, Porta F, Soresina A, et al. Long-term outcome of WHIM syndrome in 18 patients: high risk of lung disease and HPV-related Malignancies. J Allergy Clin Immunol Pract. (2019) 7:1568–77. doi: 10.1016/j.jaip.2019.01.045
62. Geier CB, Ellison M, Cruz R, Pawar S, Leiss-Piller A, Zmajkovicova K, et al. Disease progression of WHIM syndrome in an international cohort of 66 pediatric and adult patients. J Clin Immunol. (2022) 42:1748–65. doi: 10.1007/s10875-022-01312-7
63. Brenchley L, McDermott DH, Gardner PJ, Silva LM, Gao JL, Cho E, et al. Periodontal disease in patients with WHIM syndrome. J Clin Periodontol. (2024) 51:464–73. doi: 10.1111/jcpe.13940
64. Zmajkovicova K, Pawar S, Maier-Munsa S, Maierhofer B, Wiest I, Skerlj R, et al. Genotype–phenotype correlations in WHIM syndrome: a systematic characterization of CXCR4 WHIM variants. Genes Immun. (2022) 23:196–204. doi: 10.1038/s41435-022-00181-9
65. De Filippo K, Rankin SM. CXCR4, the master regulator of neutrophil trafficking in homeostasis and disease. Eur J Clin Invest. (2018) 48:e12949. doi: 10.1111/eci.12949
66. Eash KJ, Means JM, White DW, Link DC. CXCR4 is a key regulator of neutrophil release from the bone marrow under basal and stress granulopoiesis conditions. Blood. (2009) 113:4711–9. doi: 10.1182/blood-2008-09-177287
67. Martin C, Burdon PCE, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. (2003) 19:583–93. doi: 10.1016/S1074-7613(03)00263-2
68. Suratt BT, Petty JM, Young SK, Malcolm KC, Lieber JG, Nick JA, et al. Role of the CXCR4/SDF-1 chemokine axis in circulating neutrophil homeostasis. Blood. (2004) 104:565–71. doi: 10.1182/blood-2003-10-3638
69. Okolo ON, Johnson AC, Yun S, Arnold SJ, Anwer F. Rare transformation to double hit lymphoma in Waldenstrom’s macroglobulinemia. Immunotherapy. (2017) 9:709–14. doi: 10.2217/imt-2017-0027
70. Chiang MY, Radojcic V, Maillard I. Oncogenic Notch signaling in T-cell and B-cell lymphoproliferative disorders. Curr Opin Hematol. (2016) 23:362–70. doi: 10.1097/MOH.0000000000000254
71. McDermott DH, Liu Q, Velez D, Lopez L, Anaya-O’Brien S, Ulrick J, et al. A phase 1 clinical trial of long-term, low-dose treatment of WHIM syndrome with the CXCR4 antagonist plerixafor. Blood. (2014) 123:2308–16. doi: 10.1182/blood-2013-09-527226
72. McDermott DH, Velez D, Cho E, Cowen EW, DiGiovanna JJ, Pastrana DV, et al. A phase III randomized crossover trial of plerixafor versus G-CSF for treatment of WHIM syndrome. J Clin Invest. (2023) 133:e164918. doi: 10.1172/JCI164918
73. Badolato R, Alsina L, Azar A, Bertrand Y, Bolyard AA, Dale DC, et al. Phase 3 randomized trial of mavorixafor, CXCR4 antagonist, in WHIM syndrome. Blood. (2024). doi: 10.1182/blood.2023022658
74. McDermott DH, Gao JL, Liu Q, Siwicki M, Martens C, Jacobs P, et al. Chromothriptic cure of WHIM syndrome. Cell. (2015) 160:686–99. doi: 10.1016/j.cell.2015.01.014
75. Gao JL, Owusu-Ansah A, Yang A, Yim E, McDermott DH, Jacobs P, et al. CRISPR/Cas9-mediated Cxcr4 disease allele inactivation for gene therapy in a mouse model of WHIM syndrome. Blood. (2023) 142:23–32. doi: 10.1182/blood.2022019142
76. Gómez-Moutón C, Fischer T, Peregil RM, Jiménez-Baranda S, Stossel TP, Nakamura F, et al. Filamin A interaction with the CXCR4 third intracellular loop regulates endocytosis and signaling of WT and WHIM-like receptors. Blood. (2015) 125:1116–25. doi: 10.1182/blood-2014-09-601807
77. García-Cuesta EM, Rodríguez-Frade JM, Gardeta SR, D’Agostino G, Martínez P, Palacios BS, et al. Altered CXCR4 dynamics at the cell membrane impairs directed cell migration in WHIM syndrome patients. Proc Natl Acad Sci U.S.A. (2022) 119:e2119483119. doi: 10.1073/pnas.2119483119
78. Yagi H, Tan W, Dillenburg-Pilla P, Armando S, Amornphimoltham P, Simaan M, et al. A synthetic biology approach reveals a CXCR4-G13-rho signaling axis driving transendothelial migration of metastatic breast cancer cells. Sci Signal. (2011) 4:ra60. doi: 10.1126/SCISIGNAL.2002221
79. Park C, Lee JW, Kim K, Seen DS, Jeong JY, Huh WK. Simultaneous activation of CXC chemokine receptor 4 and histamine receptor H1 enhances calcium signaling and cancer cell migration. Sci Rep. (2023) 13:1894. doi: 10.1038/s41598-023-28531-1
80. Harmon B, Ratner L. Induction of the Galpha(q) signaling cascade by the human immunodeficiency virus envelope is required for virus entry. J Virol. (2008) 82:9191–205. doi: 10.1128/JVI.00424-08
81. Busillo JM, Benovic JL. Regulation of CXCR4 signaling. Biochim Biophys Acta Biomembr. (2007) 1768:952–63. doi: 10.1016/j.bbamem.2006.11.002
82. Signoret N, Oldridge J, Pelchen-Matthews A, Klasse PJ, Tran T, Brass LF, et al. Phorbol esters and SDF-1 induce rapid endocytosis and down modulation of the chemokine receptor CXCR4. J Cell Biol. (1997) 139:651–64. doi: 10.1083/jcb.139.3.651
83. Orsini MJ, Parent JL, Mundell SJ, Benovic JL. Trafficking of the HIV coreceptor CXCR4. Role of arrestins and identification of residues in the c-terminal tail that mediate receptor internalization. J Biol Chem. (1999) 274:31076–86. doi: 10.1074/jbc.274.43.31076
84. Marchese A, Benovic JL. Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J Biol Chem. (2001) 276:45509–12. doi: 10.1074/jbc.C100527200
85. Cheng ZJ, Zhao J, Sun Y, Hu W, Wu YL, Cen B, et al. beta-arrestin differentially regulates the chemokine receptor CXCR4-mediated signaling and receptor internalization, and this implicates multiple interaction sites between beta-arrestin and CXCR4. J Biol Chem. (2000) 275:2479–85. doi: 10.1074/jbc.275.4.2479
86. Jiménez-Sainz MC, Murga C, Kavelaars A, Jurado-Pueyo M, Krakstad BF, Heijnen CJ, et al. G protein-coupled receptor kinase 2 negatively regulates chemokine signaling at a level downstream from G protein subunits. Mol Biol Cell. (2006) 17:25–31. doi: 10.1091/mbc.e05-05-0399
87. Balabanian K, Levoye A, Klemm L, Lagane B, Hermine O, Harriague J, et al. Leukocyte analysis from WHIM syndrome patients reveals a pivotal role for GRK3 in CXCR4 signaling. J Clin Invest. (2008) 118:1074–84. doi: 10.1172/JCI33187
88. Fong AM, Premont RT, Richardson RM, Yu YRA, Lefkowitz RJ, Patel DD. Defective lymphocyte chemotaxis in beta-arrestin2- and GRK6-deficient mice. Proc Natl Acad Sci U.S.A. (2002) 99:7478–83. doi: 10.1073/PNAS.112198299
89. Fraile-Ramos A, Kohout TA, Waldhoer M, Marsh M. Endocytosis of the viral chemokine receptor US28 does not require beta-arrestins but is dependent on the clathrin-mediated pathway. Traffic. (2003) 4:243–53. doi: 10.1034/J.1600-0854.2003.00079.X
90. Bauer A, Madela J, Berg C, Daugvilaite V, Gurka S, Mages HW, et al. Rat cytomegalovirus-encoded γ-chemokine vXCL1 is a highly adapted, species-specific agonist for rat XCR1-positive dendritic cells. J Cell Sci. (2019) 133:jcs236190. doi: 10.1242/JCS.236190
91. Spiess K, Bagger SO, Torz LJ, Jensen KHR, Walser AL, Kvam JM, et al. Arrestin-independent constitutive endocytosis of GPR125/ADGRA3. Ann N Y Acad Sci. (2019) 1456:186–99. doi: 10.1111/nyas.14263
92. Peterson YK, Luttrell LM. The diverse roles of arrestin scaffolds in g protein–coupled receptor signaling. Pharmacol Rev. (2017) 69:256–97. doi: 10.1124/pr.116.013367
93. Zoudilova M, Min J, Richards HL, Carter D, Huang T, DeFea KA. β-arrestins scaffold cofilin with chronophin to direct localized actin filament severing and membrane protrusions downstream of protease-activated receptor-2. J Biol Chem. (2010) 285:14318–29. doi: 10.1074/jbc.M109.055806
94. Min J, DeFea K. β-arrestin-dependent actin reorganization: Bringing the right players together at the leading edge. Mol Pharmacol. (2011) 80:760–768. doi: 10.1124/mol.111.072470
96. Zhuo Y, Crecelius JM, Marchese A. G protein-coupled receptor kinase phosphorylation of distal C-tail sites specifies βarrestin1-mediated signaling by chemokine receptor CXCR4. J Biol Chem. (2022) 298:102351. doi: 10.1016/j.jbc.2022.102351
97. Milanesi S, Locati M, Borroni EM. Aberrant CXCR4 signaling at crossroad of WHIM syndrome and Waldenstrom’s macroglobulinemia. Int J Mol Sci. (2020) 21:1–15. doi: 10.3390/ijms21165696
98. Lagane B, Chow KYC, Balabanian K, Levoye A, Harriague J, Planchenault T, et al. CXCR4 dimerization and 2-arrestin mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood. (2008) 112:34–44. doi: 10.1182/blood-2007-07-102103
99. Balabanian K, Lagane B, Pablos JL, Laurent L, Planchenault T, Verola O, et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood. (2005) 105:2449–57. doi: 10.1182/blood-2004-06-2289
100. Balabanian K, Brotin E, Biajoux V, Bouchet-Delbos L, Lainey E, Fenneteau O, et al. Proper desensitization of CXCR4 is required for lymphocyte development and peripheral compartmentalization in mice. Blood. (2012) 119:5722–30. doi: 10.1182/blood-2012-01-403378
101. Heusinkveld LE, Majumdar S, Gao JL, McDermott DH, Murphy PM. WHIM syndrome: from pathogenesis towards personalized medicine and cure. J Clin Immunol. (2019) 39:532–556. doi: 10.1007/s10875-019-00665-w
102. McCormick PJ, Segarra M, Gasperini P, Gulino AV, Tosato G. Impaired recruitment of Grk6 and beta-Arrestin 2 causes delayed internalization and desensitization of a WHIM syndrome-associated CXCR4 mutant receptor. PloS One. (2009) 4:e8102. doi: 10.1371/journal.pone.0008102
103. Levy E, Reger R, Segerberg F, Lambert M, Leijonhufvud C, Baumer Y, et al. Enhanced bone marrow homing of natural killer cells following mRNA transfection with gain-of-function variant CXCR4R334X. Front Immunol. (2019) 10. doi: 10.3389/fimmu.2019.01262
104. Freitas C, Wittner M, Nguyen J, Rondeau V, Biajoux V, Aknin ML, et al. Lymphoid differentiation of hematopoietic stem cells requires efficient Cxcr4 desensitization. J Exp Med. (2017) 214:2023–40. doi: 10.1084/jem.20160806
105. Cao Y, Hunter ZR, Liu X, Xu L, Yang G, Chen J, et al. The WHIM-like CXCR4(S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom’s Macroglobulinemia. Leukemia. (2015) 29:169–76. doi: 10.1038/leu.2014.187
106. Liu Q, Chen H, Ojode T, Gao X, Anaya-O’Brien S, Turner NA, et al. WHIM syndrome caused by a single amino acid substitution in the carboxy-tail of chemokine receptor CXCR4. Blood. (2012) 120:181–9. doi: 10.1182/blood-2011-12-395608
107. Mueller W, Schütz D, Nagel F, Schulz S, Stumm R. Hierarchical organization of multi-site phosphorylation at the CXCR4 C terminus. PloS One. (2013) 8:e64975. doi: 10.1371/journal.pone.0064975
108. Wang L, Xiong Q, Li P, Chen G, Tariq N, Wu C. The negative charge of the 343 site is essential for maintaining physiological functions of CXCR4. BMC Mol Cell Biol. (2021) 22:8. doi: 10.1186/s12860-021-00347-9
109. Singh A, Joshi V, Jindal AK, Mathew B, Rawat A. An updated review on activated PI3 kinase delta syndrome (APDS). Genes Dis. (2019) 7:67–74. doi: 10.1016/j.gendis.2019.09.015
110. Kumar R, Milanesi S, Szpakowska M, Dotta L, Silvestre DD, Trotta AM, et al. Reduced G protein signaling despite impaired internalization and β-arrestin recruitment in patients carrying a CXCR4Leu317fsX3 mutation causing WHIM syndrome. JCI Insight. (2023) 8:e145688. doi: 10.1172/jci.insight.145688
111. Luo J, De Pascali F, Richmond GW, Khojah AM, Benovic JL. Characterization of a new WHIM syndrome mutant reveals mechanistic differences in regulation of the chemokine receptor CXCR4. J Biol Chem. (2022) 298:101551. doi: 10.1016/j.jbc.2021.101551
112. Zmajkovicova K, Pawar S, Sharapova SO, Geier CB, Wiest I, Nguyen C, et al. A novel transmembrane CXCR4 variant that expands the WHIM genotype-phenotype paradigm. Blood Adv. (2024). doi: 10.1182/bloodadvances.2023011875
113. Whorton MR, Bokoch MP, Rasmussen SGF, Huang B, Zare RN, Kobilka B, et al. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci U.S.A. (2007) 104:7682–7. doi: 10.1073/PNAS.0611448104
114. De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled β-adrenergic receptor. J Biol Chem. (1980) 255:7108–17. doi: 10.1016/S0021-9258(20)79672-9
115. Park PS-H. Ensemble of G protein-coupled receptor active states. Curr Med Chem. (2012) 19:1146–54. doi: 10.2174/092986712799320619
116. Whorton MR, Jastrzebska B, Park PSH, Fotiadis D, Engel A, Palczewski K, et al. Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J Biol Chem. (2008) 283:4387–94. doi: 10.1074/jbc.M703346200
117. Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, et al. Activation mechanism of the β 2-adrenergic receptor. Proc Natl Acad Sci U.S.A. (2011) 108:18684–9. doi: 10.1073/pnas.1110499108
118. Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, et al. The dynamic process of β2-adrenergic receptor activation. Cell. (2013) 152:532–42. doi: 10.1016/j.cell.2013.01.008
119. Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, et al. Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell. (2015) 161:1101–11. doi: 10.1016/j.cell.2015.04.043
120. Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, et al. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. (1997) 16:6996–7007. doi: 10.1093/emboj/16.23.6996
121. Gupta SK, Pillarisetti K, Thomas RA, Aiyar N. Pharmacological evidence for complex and multiple site interaction of CXCR4 with SDF-1α: Implications for development of selective CXCR4 antagonists. Immunol Lett. (2001) 78:29–34. doi: 10.1016/S0165-2478(01)00228-0
122. Wescott MP, Kufareva I, Paes C, Goodman JR, Thaker Y, Puffer BA, et al. Signal transmission through the CXC chemokine receptor 4 (CXCR4) transmembrane helices. Proc Natl Acad Sci. (2016) 113:9928–33. doi: 10.1073/pnas.1601278113
123. Deupi X, Standfuss J. Structural insights into agonist-induced activation of G-protein-coupled receptors. Curr Opin Struct Biol. (2011) 21:541–51. doi: 10.1016/j.sbi.2011.06.002
124. Greasley PJ, Fanelli F, Rossier O, Abuin L, Cotecchia S. Mutagenesis and modelling of the alpha(1b)-adrenergic receptor highlight the role of the helix 3/helix 6 interface in receptor activation. Mol Pharmacol. (2002) 61:1025–32. doi: 10.1124/mol.61.5.1025
125. Han X, TaChado SD, Koziel H, Boisvert WA. Leu128(3.43) (l128) and Val247(6.40) (V247) of CXCR1 are critical amino acid residues for g protein coupling and receptor activation. PloS One. (2012) 7:e42765. doi: 10.1371/journal.pone.0042765
126. Xiao JY, Ruiz GV, Whorton MR, Rasmussen SGF, DeVree BT, Deupi X, et al. The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc Natl Acad Sci U.S.A. (2009) 106:9501–6. doi: 10.1073/pnas.0811437106
127. Devree BT, Mahoney JP, Vélez-Ruiz GA, Rasmussen SGF, Kuszak AJ, Edwald E, et al. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature. (2016) 535:182–6. doi: 10.1038/nature18324
128. Gregorio GG, Masureel M, Hilger D, Terry DS, Juette M, Zhao H, et al. Single-molecule analysis of ligand efficacy in β2AR-G-protein activation. Nature. (2017) 547:68–73. doi: 10.1038/nature22354
129. Paradis JS, Feng X, Murat B, Jefferson RE, Sokrat B, Szpakowska M, et al. Computationally designed GPCR quaternary structures bias signaling pathway activation. Nat Commun. (2022) 13:6826. doi: 10.1038/s41467-022-34382-7
130. Kasai RS, Kusumi A. Single-molecule imaging revealed dynamic GPCR dimerization. Curr Opin Cell Biol. (2014) 27:78–86. doi: 10.1016/j.ceb.2013.11.008
131. Maurice P, Kamal M, Jockers R. Asymmetry of GPCR oligomers supports their functional relevance. Trends Pharmacol Sci. (2011) 32:514–20. doi: 10.1016/j.tips.2011.05.006
132. Sleno R, Hébert TE. The dynamics of GPCR oligomerization and their functional consequences. Int Rev Cell Mol Biol. (2018) 338:141–71. doi: 10.1016/bs.ircmb.2018.02.005
133. Qin L, Kufareva I, Holden L, Wang C, Zheng Y, Zhao C, et al. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Sci (1979). (2014) 301:24322–7.
134. Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Foon ST, Kobilka TS, et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Sci (1979). (2007) 318:1258–65. doi: 10.1126/science.1150577
135. Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EYT, et al. A specific cholesterol binding site is established by the 2.8 Å Structure of the human β2-adrenergic receptor. Structure. (2008) 16:897–905. doi: 10.1016/j.str.2008.05.001
136. Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. (2008) 322:1211–7. doi: 10.1126/science.1164772
137. Fanelli F, De Benedetti PG. Inactive and active states and supramolecular organization of GPCRs: insights from computational modeling. J Comput Aided Mol Des. (2006) 20:449–61. doi: 10.1007/s10822-006-9064-0
138. Vohra S, Chintapalli S V, Illingworth CJR, Reeves PJ, Mullineaux PM, Clark HSX, et al. Computational studies of family A and family B GPCRs. Biochem Soc Trans. (2007) 35:749–54. doi: 10.1042/BST0350749
139. Singer II, Scott S, Kawka DW, Chin J, Daugherty BL, DeMartino JA, et al. CCR5, CXCR4, and CD4 are clustered and closely apposed on microvilli of human macrophages and T cells. J Virol. (2001) 75:3779–90. doi: 10.1128/JVI.75.8.3779-3790.2001
140. Mellado M, Rodríguez-Frade JM, Vila-Coro AJ, Fernández S, De Ana AM, Jones DR, et al. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. (2001) 20:2497–507. doi: 10.1093/emboj/20.10.2497
141. Milligan G. G protein-coupled receptor hetero-dimerization: contribution to pharmacology and function. Br J Pharmacol. (2009) 158:5–14. doi: 10.1111/j.1476-5381.2009.00169.x
142. Vila-Coro AJ, Rodríguez‐Frade JM, De Ana AM, Moreno‐Ortíz MC, Martínez AC, Mellado M, et al. The chemokine SDF-lα triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. (1999) 13:1699–710.
143. Babcock GJ, Farzan M, Sodroski J. Ligand-independent dimerization of CXCR4, a principal HIV-1 coreceptor. J Biol Chem. (2003) 278:3378–85. doi: 10.1074/jbc.M210140200
144. Pello OM, Martínez-Muñoz L, Parrillas V, Serrano A, Rodríguez-Frade JM, Toro MJ, et al. Ligand stabilization of CXCR4/δ-opioid receptor heterodimers reveals a mechanism for immune response regulation. Eur J Immunol. (2008) 38:537–49. doi: 10.1002/eji.200737630
145. Martinez-Munoz L, Barroso R, Dyrhaug SY, Navarro G, Lucas P, Soriano SF, et al. CCR5/CD4/CXCR4 oligomerization prevents HIV-1 gp120IIIB binding to the cell surface. Proc Natl Acad Sci. (2014) 111:E1960–9. doi: 10.1073/pnas.1322887111
146. Sohy D, Parmentier M, Springael JY. Allosteric transinhibition by specific antagonists in CCR2/CXCR4 heterodimers. J Biol Chem. (2007) 282:30062–9. doi: 10.1074/jbc.M705302200
147. Sierro F, Biben C, Martínez-Muñoz L, Mellado M, Ransohoff RM, Li M, et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci U.S.A. (2007) 104:14759–64. doi: 10.1073/PNAS.0702229104
148. Gardeta SR, García-Cuesta EM, D’Agostino G, Soler Palacios B, Quijada-Freire A, Lucas P, et al. Sphingomyelin depletion inhibits CXCR4 dynamics and CXCL12-mediated directed cell migration in human T cells. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.925559
149. Martínez-Muñoz L, Rodríguez-Frade JM, Barroso R, Sorzano CÓS, Torreño-Pina JA, Santiago CA, et al. Separating actin-dependent chemokine receptor nanoclustering from dimerization indicates a role for clustering in CXCR4 signaling and function. Mol Cell. (2018) 70:106–119.e10. doi: 10.1016/j.molcel.2018.02.034
150. D’Agostino G, Artinger M, Locati M, Perez L, Legler DF, Bianchi ME, et al. β-arrestin1 and β-arrestin2 are required to support the activity of the CXCL12/HMGB1 heterocomplex on CXCR4. Front Immunol. (2020) 11. doi: 10.3389/FIMMU.2020.550824
151. Verzijl D, Pardo L, Van Dijk M, Gruijthuijsen YK, Jongejan A, Timmerman H, et al. Helix 8 of the viral chemokine receptor ORF74 directs chemokine binding. J Biol Chem. (2006) 281:35327–35. doi: 10.1074/jbc.M606877200
Keywords: CXCR4, CXCL12, WHIM syndrome, receptor conformations, signaling pathways, β-arrestins
Citation: Rodríguez-Frade JM, González-Granado LI, Santiago CA and Mellado M (2024) The complex nature of CXCR4 mutations in WHIM syndrome. Front. Immunol. 15:1406532. doi: 10.3389/fimmu.2024.1406532
Received: 25 March 2024; Accepted: 20 June 2024;
Published: 05 July 2024.
Edited by:
Françoise Bachelerie, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Saul Oswaldo Lugo Reyes, National Institute of Pediatrics, MexicoCopyright © 2024 Rodríguez-Frade, González-Granado, Santiago and Mellado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mario Mellado, bW1lbGxhZG9AY25iLmNzaWMuZXM=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.