 Géssica Sabrine Braga Barbosa1,2
Géssica Sabrine Braga Barbosa1,2 Precil Diego Miranda de Menezes Neves1,2*
Precil Diego Miranda de Menezes Neves1,2* Sara Mohrbacher1,3André Néder Ramires Abdo4Lívia Barreira Cavalcante5Yara de Menezes5
Sara Mohrbacher1,3André Néder Ramires Abdo4Lívia Barreira Cavalcante5Yara de Menezes5 Victor Augusto Hamamoto Sato1,3Érico de Souza Oliveira1,3Leonardo Victor Barbosa Pereira1,3Alessandra Martins Bales1,3Marcella Martins Frediani1,3
Victor Augusto Hamamoto Sato1,3Érico de Souza Oliveira1,3Leonardo Victor Barbosa Pereira1,3Alessandra Martins Bales1,3Marcella Martins Frediani1,3 Pedro Renato Chocair1,3Américo Lourenço Cuvello-Neto1,3
Pedro Renato Chocair1,3Américo Lourenço Cuvello-Neto1,3- 1Nephrology and Dialysis Center, Oswaldo Cruz German Hospital, São Paulo, Brazil
- 2Nephrology Division, University of São Paulo School of Medicine, São Paulo, Brazil
- 3Internal Medicine Service, Oswaldo Cruz German Hospital, São Paulo, Brazil
- 4Hematology Service, Oswaldo Cruz German Hospital, São Paulo, Brazil
- 5Pathology Service, Oswaldo Cruz German Hospital, São Paulo, Brazil
Introduction: Kimura’s disease (KD) is a rare chronic inflammatory disorder characterized by subcutaneous lymphoid hyperplasia with peripheral eosinophilia. Kidney involvement is reported in 15%–18% of adult patients with KD, in many cases as nephrotic syndrome. We present a case of overlapping membranous nephropathy and IgA nephropathy associated with KD.
Case report: A 27-year-old man was admitted with a history of bilateral leg edema for the last 2 months and concomitant progressive increase of cervical mass and fever. Laboratory findings were as follows: peripheral leukocyte count, 10,080/mm³; eosinophils, 3,200/mm³ (31.7%); serum creatinine, 0.83 mg/dL; and eGFR: 140 mL/min per 1.73 m2. Urinalysis revealed the presence of hematuria and proteinuria and the following results: 24-h proteinuria, 12.9 g; serum albumin, 1.3 g/dL; and elevated IgE level, 750 kU/L. Serologies for hepatitis B, hepatitis C, HIV, and VDRL were all negative. Complement C3 and C4 levels were normal. No monoclonal protein was detected in blood and urine. Parasite infestation was discarded. A biopsy of the cervical lymph node revealed eosinophilic lymphoid hyperplasia, suggesting KD. A kidney biopsy revealed findings consistent with the overlapping of membranous nephropathy with IgA nephropathy. The patient was treated for KD with prednisone 1 mg/kg/d with progressive dose tapering and posterior association of methotrexate 15 mg/week. A renin–angiotensin system inhibitor was prescribed for nephrotic syndrome. The cervical mass regressed, and proteinuria achieved partial remission, with an increase in serum albumin level and normalization of eosinophils and IgE levels.
Conclusion: Although uncommon, kidney involvement must be considered in patients with KD. Glomerular diseases are the most frequent form of kidney injury.
Introduction
Kimura’s disease (KD) is a rare chronic inflammatory disorder characterized by lymphoid hyperplasia with peripheral eosinophilia. It was first described by Kim and Szeto in 1937 and coined in 1948 by Kimura, a Japanese pathologist who detailed the pathological features of the disease (1). KD usually affects young men (male-to-female ratio, 4:1) in their second to third decade of life, and it is more common in East and Southeast Asia (2–4). KD etiology is unknown, but some associated risk factors are autoimmune diseases, allergy, cancer, and parasite infestation (5). The classic presentation of KD involves painless soft tissue swelling, mainly in the head and neck, as well as a single or multiple papules, nodules, and/or masses. Regional lymph nodes and salivary glands are usually affected, and the most common involved site is the periauricular area. Peripheral blood analysis reveals eosinophilia and an increase in serum IgE levels. The histologic aspect of lymph node in KD depicts angiolymphoid hyperplasia and intense eosinophilic infiltration (2, 5).
Kidney disease is the most common finding regarding systemic manifestations, affecting 15%–18% of adult patients with KD (3). Renal manifestation ranges from asymptomatic urinalysis abnormalities to chronic kidney disease. Proteinuria is reported in 12%–16% of patients with KD (6) and nephrotic syndrome is reported in approximately 60% of kidney involvement in KD (7). Kidney biopsy may present different histologic presentations such as membranous nephropathy (MN), IgA nephropathy (IgAN), minimal change disease (MCD), focal segmental glomerulosclerosis (FSGS), and membranoproliferative glomerulonephritis (MPGN) (3).
Herein, we report a case of overlapping MN and IgAN in a patient with KD.
Case report
A 27-year-old Brazilian man was admitted complaining of increasing cervical mass and fever for the last 2 months, which had been recently associated with concomitant progressive bilateral leg edema. The patient denied fever, sweating, weight loss, contact with animals, use of medication, pruritus, or skin lesions. Physical examination upon admission revealed a patient with anasarca, as well as a bilateral cervical mass, more prominent on the left, with enlarged submental lymph nodes. The cervical mass was painless and there was no evidence of phlogistic signs. The spleen and liver were not palpable in the abdominal examination.
A cervical computerized tomography (CT) evidenced multiple confluents and enlarged lymph nodes in the left cervical (Figure 1A) and supraclavicular areas. Those lymph nodes exhibited high tracer uptake in the PET-CT scan (Figure 1B).
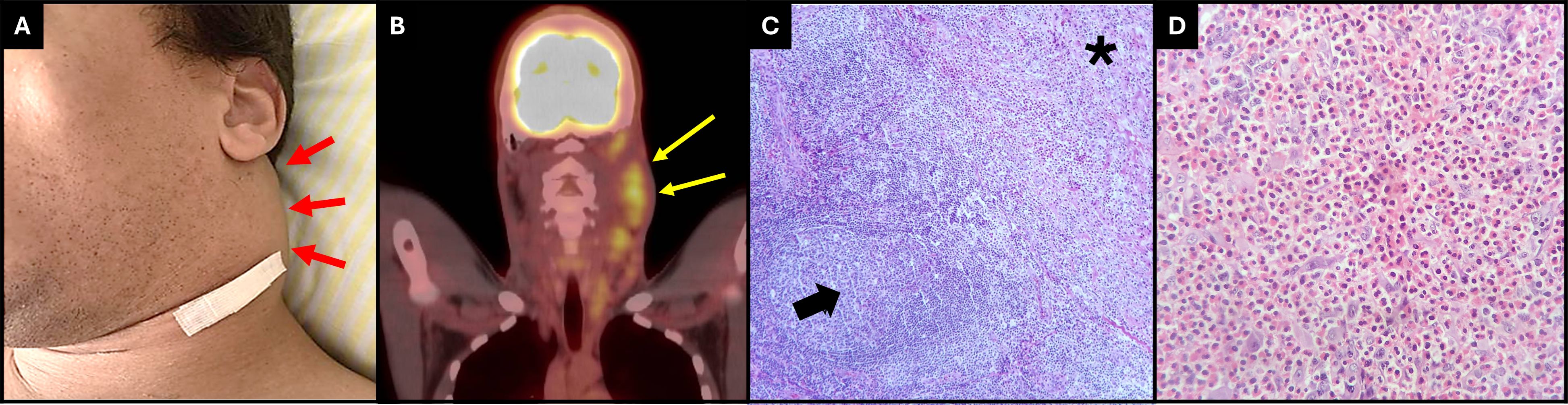
Figure 1 (A) Picture revealing the patient’s left neck mass (red arrows) (B) PET-CT scan reveals a high uptake of tracer in left cervical lymphonodes. (yellow arrows). (C) Lymph node showing zone of eosinophil-rich polimormorphic infiltrate, proliferation of reactive endothelium, and interstitial collagen deposition in a nodular fashion (black asterisk). The rest of the nodal parenchyma reveals numerous reactive lymphoid follicles (black arrow) (HE, 100×). (D) High magnification of the eosinophil-rich polymorphic infiltrate (HE, 400×).
Laboratory workup showed the following: hemoglobin, 14.4 g/dL; leukocyte, 10,080/mm³; neutrophils, 3,630/mm3; lymphocytes, 2,340/mm3; monocytes, 1,070/mm3; and eosinophils, 3,200/mm³. His serum creatinine was 0.83 mg/dL [estimated glomerular filtration rate (eGFR) by CKD-EPI: 140 mL/min per 1.73 m2], without electrolytic or acid–base disorders. Urinalysis revealed hematuria and proteinuria, with a 24-h proteinuria of 12.9 g. Metabolic investigation revealed the following: serum albumin, 1.3 g/dL; total cholesterol, 434 mg/dL; low-density lipoprotein, 320 mg/dL; high-density lipoprotein, 79 mg/dL; serum triglycerides, 177 mg/dL; IgE, 750 kU/L; IgA, 358 mg/dL; IgG, 324 mg/dL; and IgM, 73 mg/dL. The normal ranges of laboratory tests are presented in Supplementary Table 1. Serologies for hepatitis B, hepatitis C, HIV, and VDRL were all negative. Complement C3 and C4 levels were normal. No monoclonal protein was detected in blood or urine. Autoimmune panel (antinuclear antibody, rheumatoid factor, and antineutrophil cytoplasmic antibodies) was negative. Parasite infestation was ruled out. The patient presented IgG+ for cytomegalovirus and Epstein–Barr virus, and had negative serologies for Toxoplasma and rubeola. Serum anti-PLA2R was negative. Ultrasound evaluation showed kidneys with normal size and echographic aspects.
A cervical lymph node biopsy revealed proliferation of polymorphic cell consisting of plasma cells, mature lymphocytes (Figure 1C), and a dense infiltrate of eosinophils (Figure 1D). There was a proliferation of vascular structures at the periphery of the lymph node with dilated lumens, irregular contours, and thin walls, sometimes containing some red blood cells within it, with some smooth muscle fibers and lined by endothelium of flattened cells, without atypia. Immunohistochemistry was positive for CD15 in the dense eosinophil infiltrate, positive for CD3 and CD20 in rare lymphocytes, and negative for CD30, cytokeratins AE1/AE3, CD1a, and CD21. Bone marrow biopsy detected no proliferative process. The association of clinical and histological findings was consistent with KD.
A kidney biopsy was performed in the scenario of nephrotic syndrome. Light microscopy revealed the expansion of the mesangial matrix and glomerular basement membrane (Figure 2A) thickness with spike-like projections (Figure 2B). Immunofluorescence staining was global and diffuse positive for IgA (+2/+3) in the mesangium (Figure 2C), for IgG (+2/+3) (Figure 2D) and C3 (+2/+3) in capillary walls, and for kappa and lambda (+2/+3) in capillary walls and the mesangium. Immunohistochemistry highlighted a positive global and diffuse staining for PLA2R (Figure 2E) and IgG4 (Figure 2F) on capillary walls. Electron microscopy revealed electron-dense deposits in the subepithelial and mesangial region (Figures 2G, H). These findings were consistent with the overlapping of MN grade I/II associated with IgAN (MEST-C: M:0, E:0, S:0, T:0, C:0).
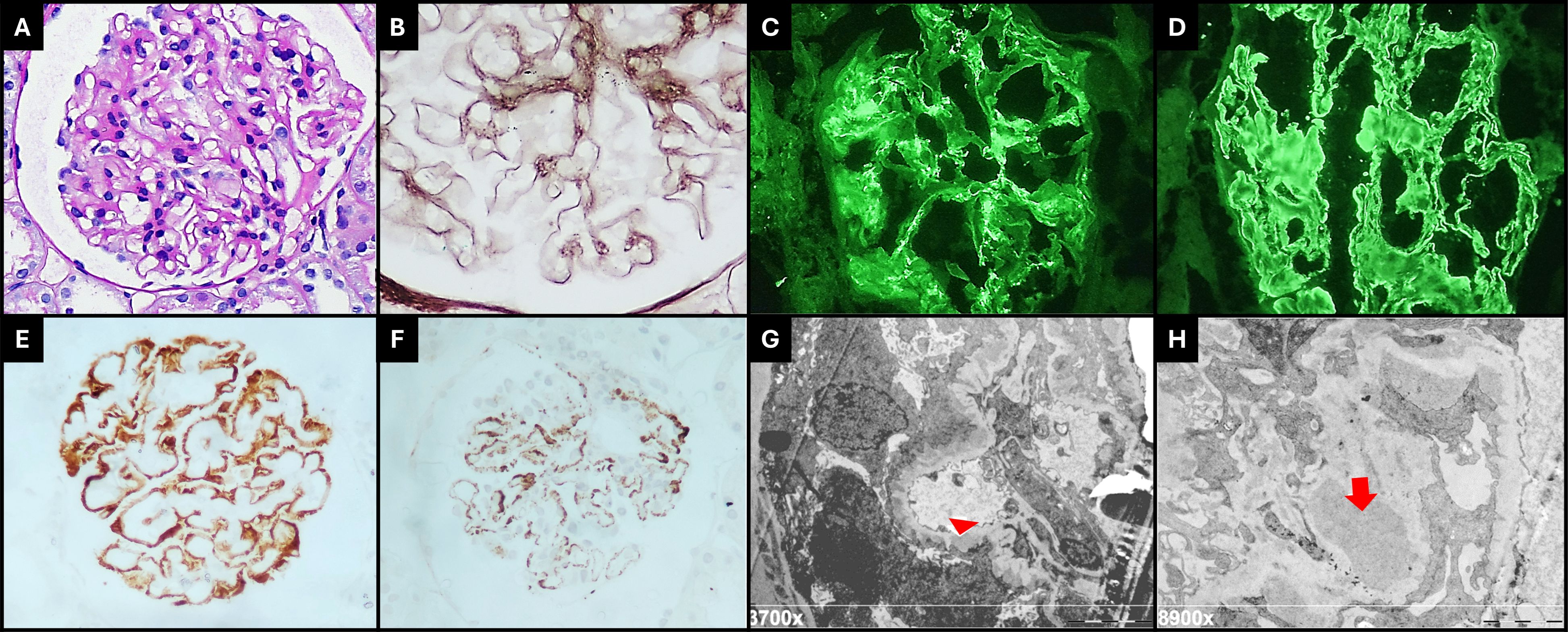
Figure 2 (A) Glomeruli show mild mesangial cell hipercellularity and matrix expansion (PAS, 400×). (B) The capillary walls are slightly thickened, and spikes are visible on Jones silver stain (400× + zoom). Immunofluorescence microscopy revealed diffuse granular deposits of IgA on mesangium and paramesangium [(C), 400×], and IgG was diffusely positive on capillary walls, with a membranous pattern [(D), 400×]. Immunohistochemistry staining for PLA2R [(E), 400×] and IgG4 [(F), 400×] were positive on capillary walls with a global and diffuse distribution. Electron microscopy shows subepithelial deposits (red arrow head) with surrounding basement membrane reaction, global podocyte foot process effacement [(G), 3,700×], large electron-dense mesangial deposits (red arrow), and increased matrix [(H), 8,900×].
The patient was treated for KD with prednisone 1 mg/kg/d with progressive dose tapering and posterior association of methotrexate 15 mg/week. A renin–angiotensin system inhibitor was prescribed for nephrotic syndrome. The cervical mass reduced drastically, and the patient reached partial nephrotic syndrome remission. After a 3-year follow-up, his laboratory findings were as follows: Cr, 0.79 mg/dL; eGFR, 118 mL/min/1.73 m2; 24-h proteinuria, 1.8 g; serum albumin, 3.7 g/dL; and normal eosinophil and IgE levels.
Discussion
KD is usually characterized by painless subcutaneous nodules in the head and neck. The diagnosis is confirmed by biopsy, and lymph node histology reveals angiolymphatic hyperplasia and intense eosinophilic infiltration. The pathogenesis of KD remains undetermined, but there is evidence of T-cell immunity dysregulation associated with allergy, parasite infections, vaccination, IgG4-related disease, and eosinophil and IgE disorders. Previous studies have shown that type 2 helper T (Th2) cytokines are elevated in patients with KD (1, 4, 8).
Kidney involvement is well documented in association with KD, especially nephrotic syndrome. Proteinuria usually appeared simultaneously with skin lesions, but in some cases, it can appear months or even years before (9). The pathogenesis of nephropathy seems to be related to alteration in the immune system, since nephrotic syndrome usually responds to immunosuppressive therapy (10). Another hypothesis is the existence of common etiological factors for both KD and glomerular lesion. Infectious agents or toxins may alter T-cell immunoregulation or induce an IgE-mediated type I hypersensitivity. This process results in the release of cytokines, which increases the permeability of glomerular basement membrane in glomeruli, causing proteinuria (3). Renal histology is variable and includes MN, IgAN, MCD, FSGS, and MPGN, with MN being the most common presentation (3, 11, 12). Other kidney disorders include interstitial lesions, such as eosinophil infiltration, and renal insufficiency (3, 11). We performed a narrative review from literature searching for the association of glomerular diseases and KD. Table 1 summarizes clinical cases and treatment of KD associated with MN or IgAN.
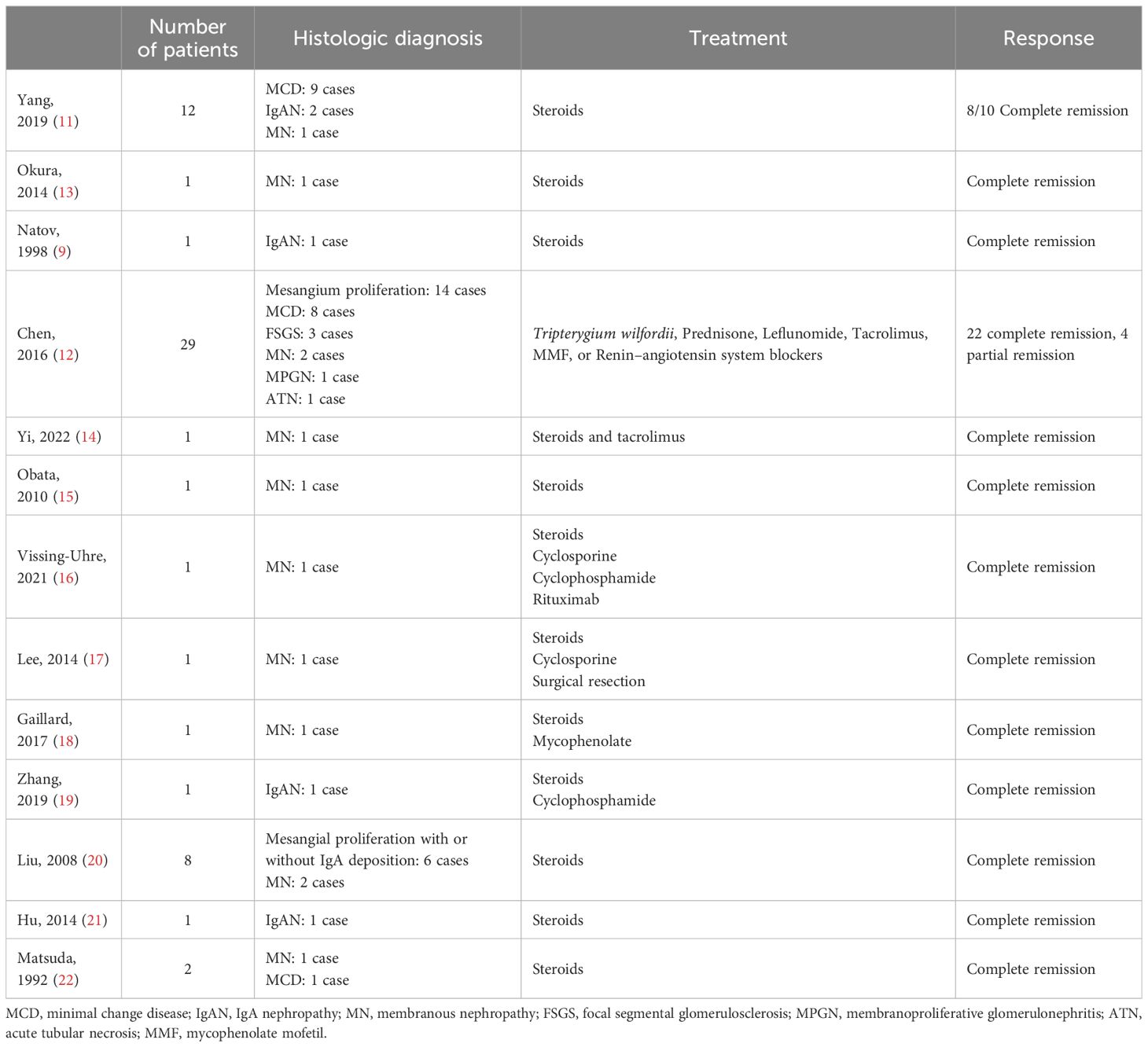
Table 1 Glomerular diseases related to Kimura’s disease, treatment, and therapeutic response.
The present case illustrates an overlapping of MN and IgAN in a patient with KD. The occurrence of overlapping MN and IgA is rare in the literature and, to the best of our knowledge, has not been previously reported. IgAN is the most common primary glomerular disease worldwide, with a major prevalence in young patients. On the other hand, MN is more frequent in patients at age ≥40 years and may frequently be associated with systemic diseases. The main kidney manifestation of IgAN is hematuria, while MN is characterized by proteinuria and nephrotic syndrome. The histology of IgAN is characterized by IgA immunocomplex deposits in the mesangium, and proliferative forms occur in many cases. The histology of MN presents thickening of the glomerular basement membrane due to subepithelial granular IgG deposits associated with M-type phospholipase A2 receptor antibody (anti-PLA2R) in most primary subtype cases. The patients with overlap syndrome show features of both diseases (23). The incidence of MN–IgAN overlap is rare, with onset during middle age and predominantly affecting male patients. It is reported that MN–IgAN presents a higher proteinuria level, including nephrotic syndrome, compared to isolated IgA, and a similar (or less) frequency of nephrotic syndrome to MN. The galactose-deficient IgA1 levels are low in the MN–IgAN overlap or similar compared to IgAN and plasma anti-PLA2R positivity, which is less frequent than in MN (23–25).
A study on histological features showed that MN–IgAN exhibits significantly increased mesangial cell proliferation, matrix expansion, and inflammatory cell infiltration, with higher proportions of arteriole hyalinosis and lesions when compared to MN alone (26). MN–IgAN treatment in the literature is variable, which is similar to MN management, including renin–angiotensin system inhibitors for mild cases and corticosteroids plus cyclophosphamide or mycophenolate and/or calcineurin inhibitors for severe cases. The prognosis of MN–IgAN seems to be significantly better than IgAN, but outcomes have similar results when compared to MN (23–25).
MN is the most common histological pattern in patients with KD and proteinuria (3). Although it could be considered as secondary MN, there are reports with anti-PLA2R subepithelial deposition in these cases, typically of idiopathic MN (13). As MN is well documented in KD, it is difficult to consider two independent diseases occurring at the same time. KD seems to be associated with immunological disorder and MN is mediated by immunocomplexes. The production of antibodies against PLA2R antigens can be part of immune system dysregulation due to KD. On the other hand, the association between IgAN and KD is extremely rare, with few cases reported in the literature (9, 11). Considering that both KD and IgAN are more common in the Asian population, it would be possible to explain the independent occurrence of these diseases. However, the response of nephrotic syndrome and IgAN related to corticosteroids raises the hypothesis of immune dysregulation secondary to KD (9).
There is no consensus about the treatment for KD with kidney involvement. Corticosteroids have been the main and initial treatment for skin lesions and nephrotic syndrome. However, other immunosuppressive therapies are being used, including cyclosporin, mycophenolate, tacrolimus, rituximab, and cyclophosphamide. Other drugs have been used for KD treatment such as methotrexate and mepolizumab (27, 28). Irradiation and surgical removal have already been used for masses and subcutaneous nodules (3). Recurrence is common and proteinuria/nephrotic syndrome can relapse before or after skin lesions (9). Our case was managed with corticosteroids, renin–angiotensin system inhibition, and methotrexate with posterior prednisone dose reduction, progressing to partial response of proteinuria and complete cervical mass regression, without recurrence.
In conclusion, although uncommon, renal involvement as a glomerular disease must be considered in patients with KD.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethics Committee - Hospital Alemão Oswaldo Cruz. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
GB: Data curation, Writing – original draft. PN: Conceptualization, Investigation, Supervision, Writing – original draft. SM: Conceptualization, Formal analysis, Investigation, Supervision, Writing – review & editing. AA: Formal analysis, Investigation, Supervision, Writing – review & editing. LC: Investigation, Methodology, Writing – original draft. YM: Formal analysis, Methodology, Writing – original draft. VS: Investigation, Writing – review & editing. ÉO: Investigation, Writing – review & editing. LP: Investigation, Writing – review & editing. AB: Investigation, Writing – review & editing. MF: Investigation, Writing – review & editing. PC: Investigation, Supervision, Writing – review & editing. AC: Investigation, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1404954/full#supplementary-material
References
1. Liu Y, Liu S, Xu J, Xu X, Wang M. An unusual case of systemic lymphadenopathy - kimura’s disease. J Inflammation Res. (2023) 16:701–5. doi: 10.2147/JIR.S397470
2. Brito MT, Baptista D, Pereira E, Fonseca E, Almeida JS. Kimura’s disease: A literature review based on a clinical case. Cureus. (2023) 5(12):e50463. doi: 10.7759/cureus.50463
3. Wang DY, Mao JH, Zhang Y, Gu WZ, Zhao SA, Chen YF, et al. Kimura disease: A case report and review of the Chinese literature. Nephron Clin Pract. (2008) 111:c55–61. doi: 10.1159/000178980
4. Lee C-C, Yu K-H, Chan T-M. Kimura’s disease: A clinicopathological study of 23 cases. Front Med (Lausanne). (2022) 9. doi: 10.3389/fmed.2022.1069102
5. Sun Q-F, Xu D-Z, Pan S-H, Ding JG, Xue ZQ, Miao CS, et al. Kimura disease: review of the literature. Intern Med J. (2008) 38:668–72. doi: 10.1111/j.1445-5994.2008.01711.x
6. Balwani MR, Bawankule CP, Pasari A, Tolani P, Vakil S, Yadav R. Minimal change disease and Kimura’s disease responding to tacrolimus therapy. Saudi J Kidney Dis Transpl. (2019) 30:254–7. doi: 10.4103/1319-2442.252921
7. Whelan TV, Maher JF, Kragel P, Dysart N, Dannenhoffer R, Prager L. Nephrotic syndrome associated with kimura’s disease. Am J Kidney Dis. (1988) 11:353–6. doi: 10.1016/S0272-6386(88)80143-4
8. Han Q, Han J, Wang W, Gao J, Qiao Y, Jia J, et al. Case report: Kimura’s disease with minimal degenerative glomerulopathy without eosinophil infiltration responds to mycophenolate mofetil treatment. Front Med (Lausanne). (2023) 9. doi: 10.3389/fmed.2022.1069553
9. Natov S. Relapsing nephrotic syndrome in a patient with Kimura’s disease and IgA glomerulonephritis. Nephrol Dialysis Transplant. (1998) 13:2358–63. doi: 10.1093/ndt/13.9.2358
10. Chartapisak W, Opastirakul S. Steroid-resistant nephrotic syndrome associated with kimura’s disease. Am J Nephrol. (2002) 22:381–4. doi: 10.1159/000065232
11. Yang S, Wang J, Chen Y, Jiang Z, Zhong T, Dong Y, et al. Concurrent kidney glomerular and interstitial lesions associated with kimura’s disease. Nephron. (2019) 143:92–9. doi: 10.1159/000501638
12. Chen Y, Wang J, Xu F, Zeng C, Liu Z. Clinicopathological features and prognosis of Kimura’s disease with renal involvement in Chinese patients. Clin Nephrol. (2016) 85:332–9. doi: 10.5414/CN108749
13. Okura T, Miyoshi K, Irita J, Enomoto D, Nagao T, Kukida M, et al. Kimura’s disease associated with membranous nephropathy with IgG4 and phospholipase A2 receptor-positive staining of the glomerular basement membrane. Intern Med. (2014) 53:1435–40. doi: 10.2169/internalmedicine.53.2042
14. Yi F, Jiang J, Li J, Lan L. A case of membranous nephropathy with Kimura’s disease. Clin Nephrol. (2022) 97:183–7. doi: 10.5414/CN110358
15. Obata Y, Furusu A, Nishino T, Ichinose H, Ohnita A, Iwasaki K. Membranous nephropathy and Kimura’s disease manifesting a hip mass. A case report with literature review. Intern Med. (2010) 49:1405–9. doi: 10.2169/internalmedicine.49.2977
16. Vissing-Uhre R, Hansen A, Frevert S, Hansen D. Rituximab treatment in a patient with kimura disease and membranous nephropathy: case report. Case Rep Nephrol Dial. (2021) 11:116–23. doi: 10.1159/000515644
17. Lee S, Yi YJ, Ah Jo H, Huh H, Kim KH, Ki Kim D, et al. Remission of secondary membranous nephropathy in a patient with Kimura disease after surgical resection. Kidney Res Clin Pract. (2014) 33:157–60. doi: 10.1016/j.krcp.2014.06.002
18. Gaillard J, Rotman S, Girardet C, Spertini F. Successful therapy with mycophenolic acid in a membranous glomerulonephritis due to Kimura disease. Clin Nephrol. (2017) 88:221–5. doi: 10.5414/CN109086
19. Zhang W, Preeatum A, Liu C. Kimura’s disease associated with IgA nephropathy: A case report. Exp Ther Med. (2019) 17:1837–40. doi: 10.3892/etm.2018.7103
20. Liu C, Hu W, Chen H, Tang Z, Zeng C, Liu Z, et al. Clinical and pathological study of Kimura’s disease with renal involvement. J Nephrol. (2008) 21:517–25.
21. Hu Y, Wang R, Lv X. Kimura disease associated with IgA nephropathy. Kaohsiung J Med Sci. (2014) 30:213–4. doi: 10.1016/j.kjms.2013.02.003
22. Matsuda O, Makiguchi K, Ishibashi K, Chida Y, Ida T, Matsuda K, et al. Long-term effects of steroid treatment on nephrotic syndrome associated with Kimura’s disease and a review of the literature. Clin Nephrol. (1992) 37:119–23.
23. He J-W, Cui D-F, Zhou X-J, Chen P, Li Y, Zhang X, et al. Concurrent igA nephropathy and membranous nephropathy, is it an overlap syndrome? Front Immunol. (2022) 13:846323. doi: 10.3389/fimmu.2022.846323.
24. Chen P, Shi S-F, Qu Z, Zhao N, Xie XF, Lv JC, et al. Characteristics of patients with coexisting IgA nephropathy and membranous nephropathy. Ren Fail. (2018) 40:213–8. doi: 10.1080/0886022X.2018.1455591
25. Qin Y, Yu Z, Wu H, Wang A, Wang F, Wang D, et al. Prognostic factors affecting long-term outcomes in patients with concurrent IgA nephropathy and membranous nephropathy. Heliyon. (2024) 10:e23436. doi: 10.1016/j.heliyon.2023.e23436
26. Chen X, Chen Y, Shi K, Lv Y, Tong H, Zhao G, et al. Comparison of prognostic, clinical, and renal histopathological characteristics of overlapping idiopathic membranous nephropathy and IgA nephropathy versus idiopathic membranous nephropathy. Sci Rep. (2017) 7:11468. doi: 10.1038/s41598-017-11838-1
27. Ma H. Treatment of Kimura’s disease with oral corticosteroid and methotrexate. Bras Dermatol. (2020) 95:115–7. doi: 10.1016/j.abd.2019.03.006
Keywords: Kimura’s disease, nephrotic syndrome, membranous nephropathy, IgA nephropathy, kidney biopsy
Citation: Barbosa GSB, Neves PDMM, Mohrbacher S, Abdo ANR, Cavalcante LB, Menezes Y, Sato VAH, Oliveira ÉS, Pereira LVB, Bales AM, Frediani MM, Chocair PR and Cuvello-Neto AL (2024) Overlap of membranous nephropathy and IgA nephropathy in a patient with Kimura’s disease: a case report and literature review. Front. Immunol. 15:1404954. doi: 10.3389/fimmu.2024.1404954
Received: 21 March 2024; Accepted: 21 June 2024;
Published: 12 July 2024.
Edited by:
Chris Wincup, King’s College Hospital NHS Foundation Trust, United KingdomCopyright © 2024 Barbosa, Neves, Mohrbacher, Abdo, Cavalcante, Menezes, Sato, Oliveira, Pereira, Bales, Frediani, Chocair and Cuvello-Neto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Precil Diego Miranda de Menezes Neves, cHJlY2lsbWVkNjFAeWFob28uY29tLmJy