
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 07 May 2024
Sec. Molecular Innate Immunity
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1404846
 Mackenzie E. Turner1,2†
Mackenzie E. Turner1,2† Jingru Che1†
Jingru Che1† Gabriel J. M. Mirhaidari1,3
Gabriel J. M. Mirhaidari1,3 Catherine C. Kennedy1Kevin M. Blum1,3
Catherine C. Kennedy1Kevin M. Blum1,3 Sahana Rajesh1Jacob C. Zbinden1
Sahana Rajesh1Jacob C. Zbinden1 Christopher K. Breuer1Cameron A. Best1,2‡
Christopher K. Breuer1Cameron A. Best1,2‡ Jenny C. Barker1,4*‡
Jenny C. Barker1,4*‡Lysosomes and lysosome related organelles (LROs) are dynamic organelles at the intersection of various pathways involved in maintaining cellular hemostasis and regulating cellular functions. Vesicle trafficking of lysosomes and LROs are critical to maintain their functions. The lysosomal trafficking regulator (LYST) is an elusive protein important for the regulation of membrane dynamics and intracellular trafficking of lysosomes and LROs. Mutations to the LYST gene result in Chédiak-Higashi syndrome, an autosomal recessive immunodeficiency characterized by defective granule exocytosis, cytotoxicity, etc. Despite eight decades passing since its initial discovery, a comprehensive understanding of LYST’s function in cellular biology remains unresolved. Accumulating evidence suggests that dysregulation of LYST function also manifests in other disease states. Here, we review the available literature to consolidate available scientific endeavors in relation to LYST and discuss its relevance for immunomodulatory therapies, regenerative medicine and cancer applications.
In 1943, a pediatrician discovered a strange, previously uncharacterized disease. The pediatrician described the patients’ peripheral blood smears as containing abnormally large, distended granules, a first-in-man observation (1, 2). Clinicians in Cuba and Japan corroborated these findings, reporting similar childhood cases unified by three hallmark features: oculocutaneous albinism, granule abnormality, and death in early childhood (3, 4). This disease came to be known as Chédiak-Higashi Syndrome (CHS) (5, 6). In the ensuing decades, the lysosomal trafficking regulator (LYST) has emerged as the gene underlying CHS. While LYST initially garnered attention due to its connection to CHS, researchers identified LYST as a pivotal player in cellular and immune functions. Discovering CHS marked the beginning of decades of efforts to understand LYST’s role in biology and eventually its implications for human health and disease.
For decades following the landmark discovery, the scientific community was puzzled by this abnormal disease. While no one understood the etiology of disease, it was clear that CHS caused immune dysfunction and granule anomalies that warranted further investigation. These investigations eventually led to identifying the enlarged intracellular inclusions as being lysosomal in origin (7–9). Following the discovery of CHS, similar phenotypes were reported in animal models (Table 1). Many seminal publications that provide insights into LYST molecular function involve the mouse homolog of LYST, beige, which has a phenotype that closely resembles human CHS. This model is discussed in more detail in section III. Animal models of LYST mutations. Human samples and animal models provided tools to examine the consequences of CHS on immune function and molecular biology, but efforts were limited by the unidentified cause of CHS. It was not until 1996, forty years after the first description of CHS, that researchers identified and cloned the causative human LYST gene (19, 20), which consists of 53 exons and is located on chromosome 1q42.1-1q42.2 (21).
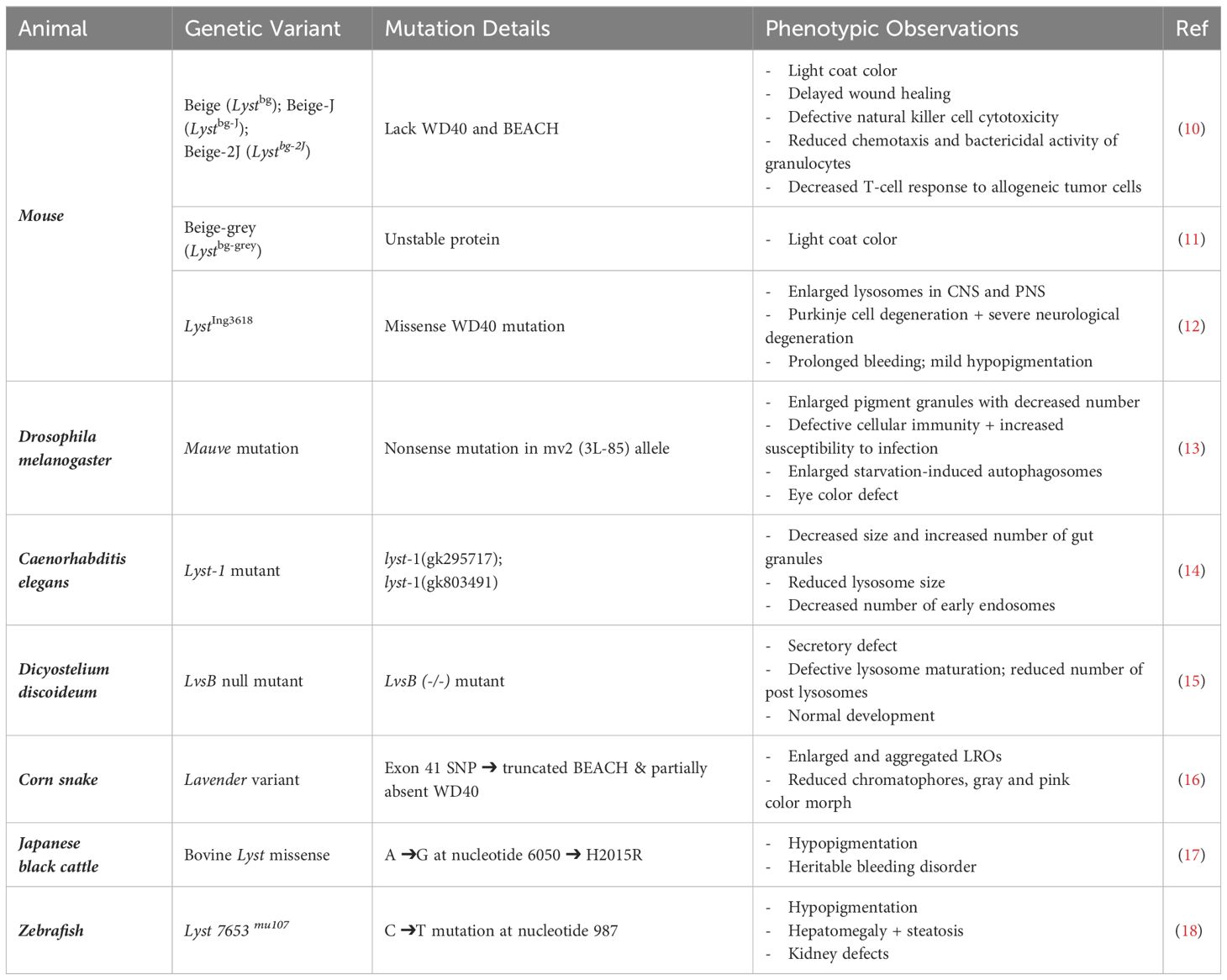
Table 1 Summary of the model organisms available for lyst research.
After identifying the causative gene, researchers began dedicating efforts to understanding a place for LYST in cellular biology. Some groups focused on clarifying whether LYST causes granule abnormalities through fusion or fission processes (8, 22–28). Other groups were interested in potential binding partners of LYST that could suggest how LYST is involved in membrane dynamics and vesicle trafficking (29, 30). The journey to understanding LYST has extended into the 21st century. With time, more tools have become available for studying LYST function. Recently, sequencing and structural analysis provided a more detailed molecular understanding of the LYST gene and protein structure (31, 32). These advancements provided more clues for its involvement in a myriad of cellular processes. Over the past decade alone, LYST has been linked to human health outside of CHS through connections to cancer and wound healing (33–40).
Despite longstanding interest in investigating LYST, a complete understanding of LYST biology is yet to be established. Here, we provide a comprehensive literature review to consolidate available scientific endeavors in relation to LYST, which may be of great value to the basic science and clinical community.
Following the initial discovery of the CHS, researchers published additional reports discussing clinical cases. The connection of LYST to CHS prompted further efforts to understand LYST function, which ultimately revealed its involvement in cellular biology and human health outside of CHS-related conditions. This autosomal recessive disorder presents along a clinical spectrum characterized by hypopigmentation, recurrent bacterial infections, developmental delays, and coagulation defects (41–43). Over 80% of patients reach the “accelerated phase” of the disease, wherein they develop life-threatening hemophagocytic lymphohistiocytosis (HLH), a hyper-inflammatory disorder associated with recurrent viral infections. This phenomenon represents the most common cause of death in patients with CHS (1, 2). Hematopoietic stem cell transplant is the only available curative treatment for CHS. Case studies demonstrated its potential in correcting immunologic complications and extending lifespan into adulthood (44). Yet, patients who survive into adulthood develop progressive central and peripheral neurodegeneration. A paucity of literature exists surrounding CHS, and while the exact prevalence is unknown, there have been fewer than 500 reported cases worldwide since its discovery (45, 46).
Animal models are critical for gaining further insight into the pathophysiology of LYST mutations. The most investigated animal models are murine equivalents of CHS. A radiation-induced mutation resulted in the first described Lyst-mutant mouse, deemed the beige (bg or Lystbg) mouse for its silvery-grey coat coloration compared to C57BL/6J wild-type (WT) mice (47, 48). The spontaneous mutation of beige allele is caused by either i. 3-bp deletion of a single amino acid (isoleucine) within WD40 domain, or ii. a LINE1 element insertion, resulting in a truncated Lyst protein lacking both WD40 and BEACH domains (49). The genetic details are more thoroughly described in section VII toward a molecular understanding of LYST. Today, researchers use the beige-J (Lystbg-J) strain, which is a spontaneous re-mutation virtually identical to the originally reported Lystbg mice (10). Lystbg-J and the less commonly used Lystbg-2J mice, are phenotypically identical to the original Lystbg mouse (10). Murine beige-grey mutant (Lystbg-grey) mice carry a chemically induced Lyst mutation (discovered in an ENU mutation screen) that results in an unstable Lyst protein caused by Exon 25 skipping (11). Thus, unlike Lystbg and Lystbg-J mice, the beige-grey mutation leaves the WD40 and BEACH domains intact (11). Notably, all Lyst-mutant mice exhibit phenotypes commonly seen in human CHS patients, including abnormal pigmentation of coat color comparable to oculocutaneous albinism in human, immunodeficiency associated with abnormal or defective NK cells, and enlarged granules (Table 1). The differential inclusion and/or exclusion of WD40 repeats, the BEACH domain, and Exon 25 result in a variety of Lyst-mutant transcripts and a spectrum of unstable Lyst proteins, potentially mirroring the spectrum of disease severity observed in human CHS patients.
A murine missense mutation (LystIng3618) located in Exon 48 of the WD40 domain caused a progressive neurodegenerative phenotype, and the mouse lacked the immunodeficiency observed in beige mice (12). LystIng3618/LystIng3618 mice display progressive Purkinje cell degeneration and impaired neurological functions during aging compared to WT mice (12). By 18 months, the LystIng3618 mice have enlarged lysosomes and almost no detectable neuronal Purkinje cells. This phenotype resembles the mild, atypical forms of CHS, characterized by early neurodevelopmental issues in childhood followed by progressive neurodegeneration in adulthood (50). Many other animal models possessing CHS-equivalent diseases resulting from Lyst mutations have also been reported (Table 1), including mink (51, 52), cat (53–55), fox (56, 57), Wagyu cattle (58), corn snake (16), and rat (59).
Together, the beige mutant mice and related strains present a compelling experimental paradigm wherein the relative contributions of intact WD40 or WD40/BEACH domains to Lyst-dependent mechanisms could be interrogated in vivo to better understand LYST function in humans. We anticipate that these models could not only be used for understanding CHS pathology but also for investigating defective vesicular trafficking.
Much of what is known about LYST comes from the cell type-specific manifestations of LYST dysfunction observed in disease. LYST underlies the vesicle trafficking responsible for mobilizing specialized LROs that are central to host defense. In parallel with characterization of Lyst-dependent molecular mechanisms, investigations into the cell type-specific mechanisms demonstrated the extensive manifestations of defective endolysosomal trafficking. Enlarged granules have been observed in LYST-mutant NK cells (60), cytotoxic T cells (61), B cells and helper T cells, neurons, neutrophils/granulocytes, monocytes/macrophages, fibroblasts, platelets, and pigment-producing cells. Available evidence regarding each cell type is discussed in the following section, a graphical overview of which is provided in Figure 1.
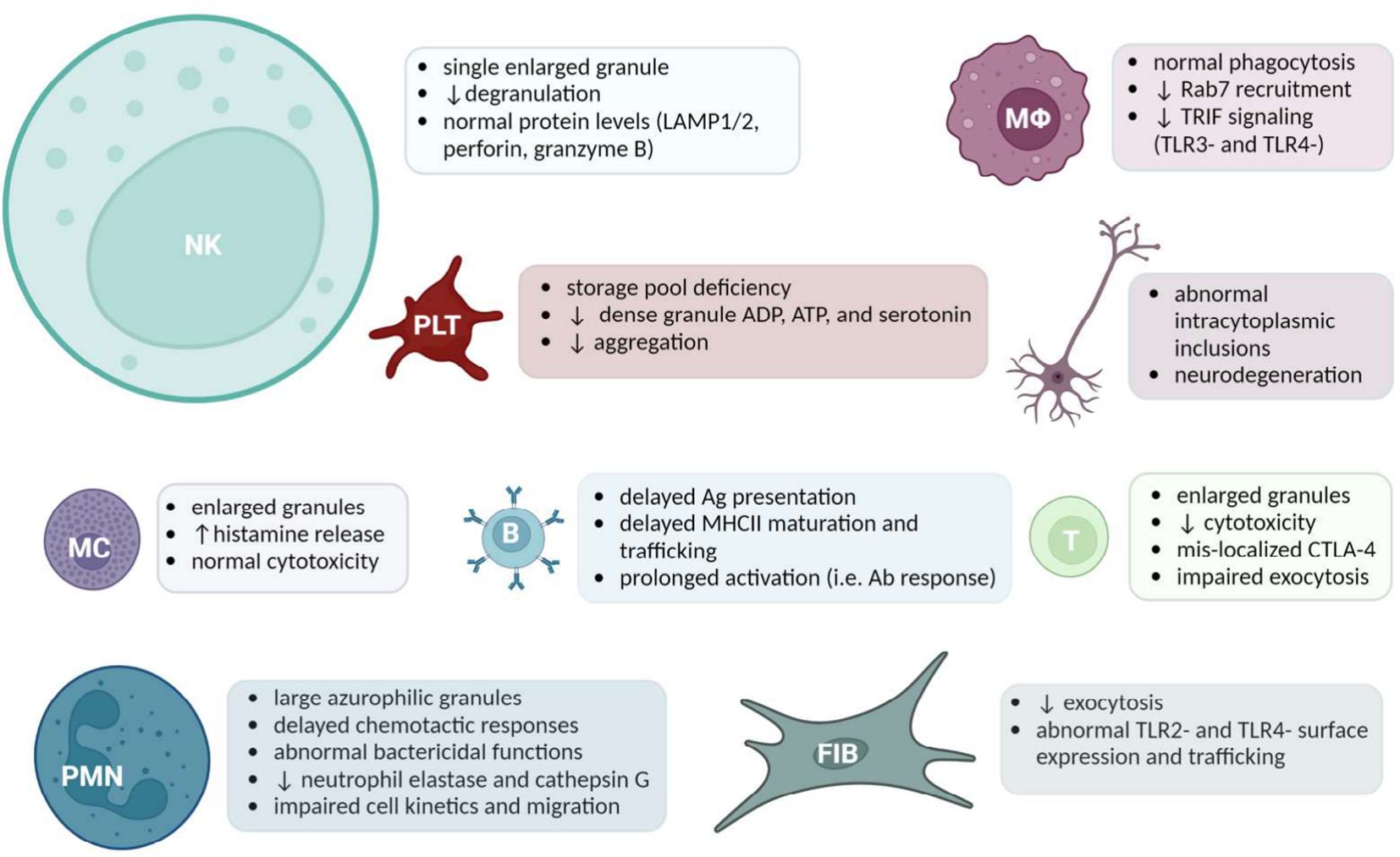
Figure 1 A Summary of Cell Specific LYST-Deficient Phenotypes. Acronyms (from left to right): Natural killer (NK) cells, Macrophages (MΦ), Platelets (PLT), cells of the nervous system, Mast cells (MC), B cells, Cytotoxic T cells, Polymorphonuclear leukocytes (PMN), Fibroblasts (FIB). The size of the cell reflects the amount of literature available. Created with BioRender.com.
Defective natural killer (NK) cell function is a common feature of CHS. NK cells are a lymphocyte subset critical to the innate host defense against tumors and microbial infections (62). The cytotoxic function of NK cells is accomplished via secretory lysosome exocytosis. In this process, cytotoxic proteins are released from the NK cell secretory lysosome, or lytic granule, resulting in the death of the target cell through interruption of membrane integrity and inducing apoptosis (62, 63). Interestingly, NK cells are present in normal quantities in CHS patients and the LYST mutation did not impact their ability to recognize and bind target cells in vitro (61). Studies involving CHS patient-derived cells suggested that LYST may modulate the exocytosis of secretory lysosomes (64). However, LYST-deficient granules contained a relatively normal number of lysosome-associated proteins and lytic protein activity, despite their enlarged morphology (60). Chiang et al. reported that enlarged granules failed to polarize to the immune synapse and release their cytotoxic cargo (61). To further resolve the impaired transportation of enlarged granules, Gil-Krzewska et al. investigated the actin meshwork as a potential mechanistic barrier to polarization (60). They observed that the cellular actin meshwork is not permissive for enlarged granules in LYST-deficient cells, since lytic granules must navigate the tangled actin network at the immunologic synapse to be secreted onto target cells (60). The authors concluded that the cortical actin meshwork may prohibit the exocytosis of LYST-mutated granules in NK cells. Unresponsive NK cells increase the patient’s susceptibility to contracting viral infections (Figure 1) which are known to accelerate CHS to HLH.
Cytotoxic T lymphocytes (CTLs) have an important role in the adaptive immune response, recognizing antigens presented in MHC class I molecules (65). Similar to NK cells, activated CTLs kill target cells through the release of perforin and granzyme from lytic granules (66). LYST deficiency reduces the normal cytotoxicity of CTLs and enlarges their granules (61, 67, 68).
Compared to CHS NK cells, Lyst-mutant CTLs contain a greater quantity of relatively smaller yet still abnormally enlarged LROs. This suggests that these smaller CTL granules may more efficiently navigate cellular obstacles. However, in vitro assays revealed a marked deficiency of granule exocytosis and decreased cytotoxicity (61). As described previously, while entanglement in the cytoskeleton may impact the exocytosis of enlarged LROs, data from CHS CTLs suggest that the mechanism of defective vesicular transport is likely more complex (69).
Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), a major T cell regulator, functions by inhibiting the activation of T cells. Although CTLA-4 is mainly stored in intracellular vesicles, cell membrane expression is highly regulated by endocytosis and trafficking through a secretory lysosome pathway. An earlier study revealed that CHS patients have impaired intracellular trafficking of CTLA-4 in the T cells, which results in defective cell-surface expression of CTLA-4 (69). This abnormal regulation of cytotoxic T cells likely contributes to the accelerated phase of CHS (69).
Although efforts to define the nature and extent of the LYST protein’s role in B and T helper cell (Th cells) biology have provided some insights regarding the kinetics of adaptive immune system activation in CHS, recent mechanistic investigations are lacking. CHS CD4+ T cells had shown decreased infection compared to WT CD4+ T cells upon HIV-1 exposure, which primarily spreads through cell-to-cell contact involving virion fusion with cell membrane (70). Further analysis indicated that HIV-1 spread is restricted by deficient LYST function in CHS T cells.
B cells are dependent on endosomal trafficking pathways to internalize and process antigens for major histocompatibility complex class II (MHCII) mediated surface presentation (71). LYST mutations slow the kinetics of antigen presentation and MHCII maturation and transport (72, 73). In beige B cells, one study observed delayed mobilization of the endocytosed antigen-B cell receptor (Ag-BCR) to lysosomes, which led to the suboptimal presentation of BCR-targeted antigen to T cells in vitro (73). The authors describe how a delay in antigen presentation results in prolonged B cell activation, even in the presence of low antigen levels (73). Sustained B cell engagement may reinforce pro-proliferative signaling cascades, and when uncontrolled, lead to a constant state of lymphoproliferation as demonstrated in the “accelerated phase” resulting in HLH (74, 75). Indeed, HLH is caused by defective vesicle trafficking-dependent immune cell apoptosis, leading to marked overactivation of host defenses, bone marrow infiltration, pancytopenia, liver dysfunction, and hepatosplenomegaly often requiring cytotoxic immunosuppression or stem cell transplant.
While CHS is primarily described by severe immunological defects, neurologic impairments are also commonly observed among mild to late-onset CHS patients. Patients affected by this condition may experience developmental impairment (learning disabilities), dementia, progressive neuronal degeneration (especially among those surviving to adulthood), seizures, sensory deficit, and weakness (50).
A case study published in 1994 reported a 39-year-old CHS patient who developed Parkinsonian features along with dementia. Single-photon emission computerized tomography (SPECT) revealed severe neuronal degeneration in the cortex, basal ganglia, brainstem, and spinal cord (9). Sung et al. reported abnormal intracytoplasmic inclusions in various CHS patient cells including neurons, astrocytes, epithelial cells of the choroid plexus, satellite cells of the dorsal spinal ganglia, and Schwann cells (76). A comparable phenotype has also been described in mice. As mentioned in the III. Animal models of LYST mutations section, the LystIng3618 mouse (Table 1) exhibits a neurodegenerative phenotype without immunological defects (12). This represents the only immune-competent Lyst-mutant murine model reported to date, offering a window into the relatively unexplored biology of CHS patients with neurological impairments. The effects of the LYST mutation on the central nervous system requires further exploration.
Neutrophils are a subset of granulocytes critical to the detection and elimination of microbes and foreign bodies. Neutrophils circulate in a quiescent state until activated by cytokines and growth factors released from sentinel cells such as endothelial cells (77, 78). Once activated, neutrophils degranulate, or release stored pro-inflammatory mediators and/or microbicidal proteins, from either: i. primary or azurophilic, ii. secondary or specific, iii. tertiary, or iv. secretory vesicle granular subset (79). Neutrophils isolated from CHS patients demonstrated delayed chemotactic responses, large and irregular azurophilic granules, and abnormal bactericidal functions (80). Azurophilic granules contain potent hydrolytic enzymes that allow the neutrophil to confer bactericidal and cytotoxic properties (81). CHS patients were deficient in neutrophil elastase and cathepsin G (82). The deficiency of azurophilic components may be partially responsible for the microbicidal defect observed in LYST-deficient cells. The presence of the enlarged, abnormal granules in CHS neutrophils also impaired cell kinetics and prohibited proper migration (25, 83). While the observed neutrophil defect does not appear to impact signal recognition, LYST mutations among CHS patients likely interfere with vesicular trafficking pathways. Such pathways direct granulocyte migration including cytokine release by macrophages or signaling by endothelial cells (83–85). Neutrophils represent a prototypical LYST-dependent cell type, wherein interrupted antimicrobial function due to abnormal phagolysosomes renders patients and laboratory animals alike susceptible to bacterial infections. The disruption of normal antimicrobial function likely results in recurrent infections of CHS patients. The correlation between abnormal neutrophil function and clinical manifestations raises an intriguing possibility that recent advances in neutrophil biology and therapeutics could yield targeted immunomodulatory therapies.
Fibroblasts have an essential role in wound healing, tissue regrowth, and regulating immune responses through secretion and remodeling of extracellular matrix (ECM) (86).. CHS and beige fibroblasts contain a relatively reduced quantity of abnormally enlarged lysosomes (22, 87). With their wide availability, ease of culture, and well-characterized LRO biology, experiments in cultured fibroblasts provided much of the literature surrounding the biogenesis of enlarged lysosomes in LYST mutants. Other defining features characteristic of LYST-deficient fibroblasts include reduced exocytosis capacity and viability in response to membrane injury (64). Restoring LYST expression in beige fibroblasts reverses the enlarged lysosomal phenotype and rescues exocytosis, suggesting a possible link between lysosome size and cell function in fibroblasts (64). The baseline hyperactive inflammatory gene transcription was observed in CHS fibroblasts. However, the cells failed to initiate an appropriate immune response following lipopolysaccharide (LPS) challenge, indicating an inability to respond to an inflammatory stimulus. This defect was attributed to abnormal expression of toll-like-receptors 2 (TLR2) and TLR4 or aberrant intracellular processing or membrane localization of these receptors (88). This observation is similar to findings in innate and adaptive immune cell types, specifically NK cells, neutrophils, macrophages, and T cells. However, Westphal et al. noted that LYST-mutant dendritic cells and macrophages exhibit normal activation of MAPKs upon TLR4 stimulation, demonstrating that LYST likely does not interact with the MyD88 pathway (89). This observed dysfunction correlates with impaired TLR3- and TLR4-induced IRF3 phosphorylation, suggesting that TRIF pathway dysregulation is responsible for abnormal trafficking in LYST-mutant innate immune cells (89). This finding does not discredit Wang et al., but rather suggests that LYST likely exerts cell-type specific effects. Fibroblasts also provided the foundation for investigating LYST in conditions beyond CHS. Cutaneous wound healing studies showed that LYST is an important regulator of secretory exocytosis by fibroblasts, which confer deficits in MCP-1, IGF-1, and IGFBP-2 secretion when LYST is mutated (40). Such insights into fibroblast behavior underscore the complexity of LYST’s impact, paving the way for broader implications in various physiological processes and pathological conditions.
Monocytes, macrophages, and their products comprise the population of cells responsible for debris clearance, tissue remodeling, and aspects of host defense. These cells exhibit diverse functions by undergoing differential polarization into various tissue-specific phenotypes, ranging from resident macrophages to recruited pro-inflammatory monocytic precursors (90). Differential activation of macrophages into classical M1 or alternative M2 types occurs via Th1/PAMP and Th2 signaling, respectively. These prototypical phagocytes, similar to neutrophils discussed above, exhibit marked functional deficiencies due to their reliance upon the endolysosomal network and LRO’s in the context of LYST mutations.
Recent insights reveal that macrophage phagocytosis relies on lysosomal degradation and recycling (91). Furthermore, there is a newfound understanding of the dependence of monocyte/macrophage signal transduction on intracellular membrane and endosomal trafficking events. Despite normal monocyte phagocytosis in both CHS patients and beige mice (92, 93), Lyst-mutant monocyte-derived granulocytes are still at an increased risk of bacterial infection (92).
Interestingly, studies indicate a delayed antitumor cytostatic and cytotoxic activity of beige macrophages against lung carcinoma during the initial 24-48 hours of exposure (93). These findings corroborate the notion that Lyst mutations interrupt, rather than prevent, trafficking of the phagolysosome. Recent findings suggest that Lyst mediates phagosome maturation and pro-inflammatory pathways in a Trif (TLR3 and TLR4)- dependent manner (89). Specifically, beige-J cells are not able to recruit Rab7 during late stage endolysosomal maturation, subsequently interrupting Trif-mediated pro-inflammatory signaling pathways (89).
Platelets are important homeostatic and thrombotic regulatory cells. Following vascular insult, these cells often mark the first step of the wound healing cascade (94). Platelets contain LROs such as dense, alpha, and lysosomal granules. The LROs store effector molecules required for platelet aggregation, activation, paracrine signaling, and protein degradation (95). CHS patients classically present with platelet storage pool deficiencies resulting in bleeding tendency (96, 97). Though circulating platelet levels are normal, LYST mutations inhibit exocytosis of platelet dense granules. Bleeding tendency is a hallmark of the clinical presentation of CHS observed across species (17). Beige mice bleed excessively due to reduced ADP, ATP, and serotonin within platelet dense granules (98–100). Further study of beige platelets revealed an impaired ability to aggregate in response to collagen, thrombin, and phorbol-12-myristate 13-acetate stimulation, which the authors attributed to platelet storage deficiency (101). These results underscore the impact of dysregulated platelet granule exocytosis in the pathophysiology of CHS.
Melanocytes are melanin-producing cells primarily located in the epidermis, cochlea, and iris (102). Melanosomes are specialized LROs that synthesize and contain the pigment melanin. Following maturation, melanosomes are anchored and transported to the cell periphery via microtubules, eventually transferring to adjacent keratinocytes from melanocyte dendrites (103). Studies reported enlarged and disorganized melanosomes in CHS melanocytes (104, 105). Tyrosinase catalyzes the production of melanin, and, along with beta-glucuronidase, is understood to be abnormally trafficked in cultured CHS melanocytes (105). Additionally, reports indicate reduced melanin quantity and uneven pigment distribution in the mouse retinal pigment epithelium, leading to eventual retinal detachment (106). This phenomenon is attributed to the accumulation of photoreceptor outer segment phagosomes, coinciding with elevated levels of cathepsins, MMP-3, and markers of oxidative stress (106).
Interestingly, pigment granule biogenesis in Drosophila is paired with machinery of lysosomal protein delivery (107). This evidence establishes a molecular explanation for commonly reported oculocutaneous albinism of CHS patients and the LYST-dependent melanosome/lysosome events required for normal skin, hair, and eye pigment distribution.
Mast cells actively mediate innate and adaptive immunity (108) by releasing histamine and other chemical mediators to modulate acute allergic inflammation (109). Dysfunction of mast cells is responsible for chronic allergic and inflammatory disorders, autoimmune diseases, and cancers (110). Large granules were observed in the peritoneal, dermal, and bone marrow derived mast cells of beige mice (11, 111, 112). Other studies demonstrated that, compared to WT rat mast cells, beige rat mast cells released relatively greater amounts of histamine, which appeared to cause granule enlargement (113, 114). In contrast to the secretory defect observed in other cell types, subsequent mast cell investigations demonstrated normal mast cell cytotoxicity in response to TNF-α stimulation in beige mast cells despite their enlarged granules (113). Mast cells have received relatively little attention in the LYST literature, perhaps due to their auxiliary role in the progression of CHS. However, mast cells represent an additional model system to study LYST function. The modulation of Lyst activity in mast cells could provide a promising alternative to current mast-cell targeted therapies. Specifically, mast cells have recently been implicated in the progression of inflammation-mediated neurodegeneration (115) and may play an underappreciated role in the progressive neurodegeneration observed in CHS survivors.
Initial and continued pursuits toward understanding the effect of LYST mutations focused on the pathognomonic hallmark described at the initial discovery of CHS: stark changes in the morphology and function of secretory lysosomes.
Peripheral blood leukocytes from CHS patients underwent extensive examination using light microscopy and various staining techniques. Initially, the enlarged bodies in peripheral blood neutrophils were thought to be giant peroxidase granules, Döhle bodies, and azurophilic clumps (4, 5, 116). Subsequent technological advancements in electron microscopy and immunofluorescence led to the discovery that the enlarged bodies were, in fact, lysosomes, supported by the morphological characteristics, localization, and acid phosphatase activity (7–9). Further investigation extended this understanding to beige and CHS fibroblasts, revealing acid phosphatase-positive intracellular inclusions containing the lysosomal markers, alpha-2-macroglobulin and rhodamine (23, 24, 117, 118). Lysosomal origin was also confirmed in various cell types including beige osteoclasts, gastric chief cells, parietal cells, and renal proximal convoluted tubal cells with enlarged granules (119–121). Recent studies consistently affirmed the identity of the enlarged inclusions using specific lysosomal markers including lysosomal-associated membrane protein-1 (LAMP-1), LAMP-2, and Rab7 (40, 64, 87, 122–124).
Lysosomes are dynamic organelles that operate at the intersection of the endocytic and secretory pathways. Lysosomes originate from transport vesicles budded from the trans-Golgi network, and these vesicles periodically fuse with endosomes to degrade material. Early endosomes are specialized compartments that receive material from the extracellular environment through processes like endocytosis. Early endosomes transition into late endosomes, marked by a conversion of the small GTPase Rab5 to Rab7 (125, 126). Late endosomes fuse directly with lysosomes, forming a hybrid organelle that acts as a site for degradation. Lysosome populations reform from these hybrid organelles. Lysosomes undergo fusion with one another, other intracellular compartments, or target membranes facilitated by docking proteins, including SNAREs, Rab GTPases, and Rab effector proteins (127). Lysosomes are critical for macromolecular degradation and recycling, intracellular protein and vesicle trafficking, and cellular signaling (127). Lysosome related organelles (LROs) are similar to lysosomes, and some cells utilize LROs for lysosomal functions (128). While the LROs originate from the endolysosomal system, certain cells including neutrophils and melanocytes harbor specialized LROs such as azurophilic granules and melanosomes.
While the mechanisms of lysosome biogenesis are well-defined, the role of LYST in these processes remains incompletely understood. Lysosome numbers and size are maintained by a steady state of fission and fusion events (129). Dysregulation of fusion and fission manifests as morphological changes to lysosomes and LROs. Fusion with endosomes increases lysosome size by incorporating vesicle content and membrane material (28), while fission leads to fragmentation and size reduction. LYST is hypothesized to affect lysosomes and LROs by either i. limiting the fusion or ii. promoting the fission of endolysosomal organelles (23, 24, 26).
The first hypothesis is that the enlarged lysosomes arise from excessive fusion events (8, 22–26). Time course tracking of lysosomes in cultured CHS cytotoxic T lymphocytes demonstrated that over the course of 9 days, normal-sized lysosomes clustered around the nucleus and aggregated to form the characteristic enlarged structures found in LYST mutant cells (68). The greatest support for LYST’s potential inhibitory effect on lysosome fusion came from investigations into the Dictyostelium ortholog of LYST: large volume sphere B (lvsB). In lvsB-null cells, the endolysosomal fusion and phagosome-phagosome fusion events were significantly increased as measured using a fusion assay (130). Further investigation found that lvsB antagonized the activity of the DdRab14 protein, a GTPase with pro-fusion activity (124). While the lvsB-mutant cells could not prevent the fusion of lysosomes with post-lysosomes, they did not affect the fusion of post-lysosomes with early endosomes (124). Post-lysosomes are secretory vesicles destined for exocytosis, indicating that the role of lsvB in inhibiting lysosome fusion is specific to certain cellular compartments and does not affect the fusion events involving early endosomes. This suggests a nuanced and targeted regulatory mechanism where lsvB selectively modulates lysosomal fusion processes, highlighting the complexity of intracellular trafficking and secretion pathways in the context of lvsB and DdRab14 protein activity. LYST-related fusion/fission dynamics were further explored by comparing lsvB null cells with two well characterized fission defect mutants, µ3-null and WASH-null cells. lsvB-null cells exhibited increased multi-particulate phagosomes compared to the fission defect mutants and WT cells. Tracking lysosome maturation with latex beads also showed a deceleration in particle transfer in the IvsB null cells. Ultimately, only lysosome fusion with post-lysosomes was impacted by absence of lsvB (124). In a follow-up examination, IvsB mutant revealed significantly enlarged lysosomes and decreased post-lysosomes. However, the authors were unable to claim whether fusion dysregulation caused the enlarged lysosomes (15, 131).
A competing model establishes LYST and its homologs as a positive regulator of fission events, the second hypothesis for LYST’s effects on lysosomes. Experiments in murine fibroblasts demonstrated that Lyst overexpression led to the reversal of the enlarged lysosomal phenotype, yielding smaller-than-normal lysosomes with no observed effects on lysosomal fusion (27). Since fusion was unimpacted, overexpressing murine Lyst may have corrected the diseased phenotype by promoting fission events. Beige bone marrow derived macrophages were treated with acetic acid to fragment lysosomes. Upon withdrawal, beige lysosomes re-fused with no delay compared to WT cells. Then, CHS and beige fibroblasts were treated with vacuolin-1 to cause lysosomal swelling. The swelled lysosomes in beige fibroblasts recovered slower compared to WT fibroblasts, yet overexpression of Lyst increased lysosomal size recovery (27). Findings from these experiments led the authors to conclude that mutations in LYST impact lysosomal fission, not fusion (28). Similar findings are also observed in a C. Elegans study, in which disrupting the LYST ortholog, Lyst-1, resulted in decreased lysosome size. To determine if this was attributable to defects in lysosome fusion, the authors created double knockouts for Lyst-1 and Cup-5, which is a known lysosomal fission regulator necessary for limiting lysosome size. In Cup-5 mutants, disrupting genes known to be involved in lysosomal fusion suppressed lysosome enlargement (73). The Lyst-1/Cup-5 double mutant, however, did not experience a reduction in lysosome size, leading authors to conclude that Lyst-1 was not involved in lysosome fusion (14).
While other studies contributed additional insights into LYST-associated abnormal granules, they have not definitively concluded whether LYST mediates lysosome morphology through fusion or fission events. In the context of CHS, cytotoxic T lymphocytes exhibited abnormal localization of necessary effectors for lytic granule exocytosis, Muncl3-4, Rab27a, and Slp3, within enlarged intracellular organelles. Overexpression of these effectors restored degranulation in the CHS mutant cells, indicating that LYST mutations impaired the maturation of pre-secretory granules to secretory granules. However, the specific influence on lysosome fusion or fission events remained undetermined (123). In a Drosophila study investigating a LYST ortholog mutation, Mauve, researchers found that the enlarged lysosomal related organelle in Mauve-mutants resulted from uncontrolled late phagosome homotypic fusion prior to fusing with lysosomes. Ultimately, the data could not distinguish whether this was due to LYST regulation of fusion or fission events (13).
The story of LYST and its place in cellular biology is enigmatic, characterized by extended gaps between publications. The compromised immune system that is characteristic of Chédiak-Higashi Syndrome is associated with enlarged lysosomes in various immune cells. While LYST mutations do not prevent activation of immune cells, the functional defects seem to be caused by impaired polarization of these enlarged lysosomes or granules to the immunological synapse (61, 132).
To explain the impaired polarization, some groups detailed apparent defects in microtubule (MT) organization in LYST-mutant cells. Oliver and colleagues investigated the formation of concanavalin caps on polymorphonuclear leukocytes (PMNs) and differences in capping behavior in WT and LYST-mutant mice (133). The study revealed that when various cell types are treated with concanavalin A (conA), lectin aggregates into one region of the cell, thereby forming a cap. Colchicine is an anti-mitotic agent that is known to disrupt microtubule polymerization and assembly, which is necessary for vesicle trafficking. PMNs from WT mice only cap with ConA after colchicine treatment, whereas PMNs from Lyst-mutant mice cap spontaneously. The similarity between LYST-mutant PMNs to colchicine-treated cells suggests a potential link between the mutant LYST phenotype and microtubule instability or dysfunction. Interestingly, this spontaneous surface cap formation was not unique to mice, as further studies indicated the abnormality in human peripheral blood PMNs from Chédiak-Higashi patients. This abnormality is linked to impaired microtubule function, which could be due to issues in 3’,5’ cyclic guanosine monophosphate (cGMP) generation. Indeed, treatment with cGMP decreased cap formation in LYST-mutant cells.
The investigation surrounding the role of LYST in microtubule function gained momentum with the discovery that beige mice exhibited an impaired mu-opioid and kappa-opioid receptor-mediated analgesic response to morphine (134, 135). This was one of the few studies that explored the role of LYST in neurological systems. The study discovered that impaired response to morphine was independent of receptor number or binding affinity, indicating the strain-dependency of the impaired response to morphine (136). The authors speculated that this was a consequence of disrupted membrane-related microtubular function in beige mice. Beige mice were given cholinergic agonists, which restored the analgesic response to morphine (137). This serves as the only evidence that the beige-specific impaired response to morphine was related to microtubular dysfunction. More recent studies provided definitive evidence that Mauve was necessary for microtubule nucleation during cell division (122).
While some studies support that the LYST mutations adversely affect microtubule function, other groups found intact MT function in addition to normal number, length, and distribution (138, 139). Despite normal MT function, both large WT lysosomes and abnormally enlarged lysosomes in LYST-mutant cells were unable to migrate through the cytoplasm due to their size (140). Thus, a new hypothesis emerged postulating that lysosome and granule dysfunction arose not from microtubule (MT) impairment but potentially from alterations in lysosomal motility due to abnormally enlarged size. This thought prompted further investigations into the intricate molecular mechanisms underlying LYST mutations and their broader implications in cellular processes.
Researchers further explored the role of LYST in lysosomal membrane dynamics. SNAREs (Soluble N-ethylmaleimide-Sensitive Factor Attachment Protein Receptors), are key proteins involved in membrane fusion processes, facilitating the docking and fusion of vesicles with target membranes. Some studies suggested that the connection between LYST and SNARE complex assembly and function could lead to abnormal lysosome morphology and lysosomal dysfunction observed in LYST-mutants. Yeast two-hybrid screening revealed several potential LYST binding partners including calmodulin (CaM), casein kinase II (CK2), 14-3-3, and Hepatocyte growth factor regulated tyrosine kinase substrate (HRS) (29). These proteins are well described members of the SNARE complex responsible for mediating cellular membrane fusion events such as exocytosis, vesicle transportation, and signal transduction (Table 2) (30). It is worth noting that additional experiments are necessary since the authors were unable to validate the binding partners in their study.
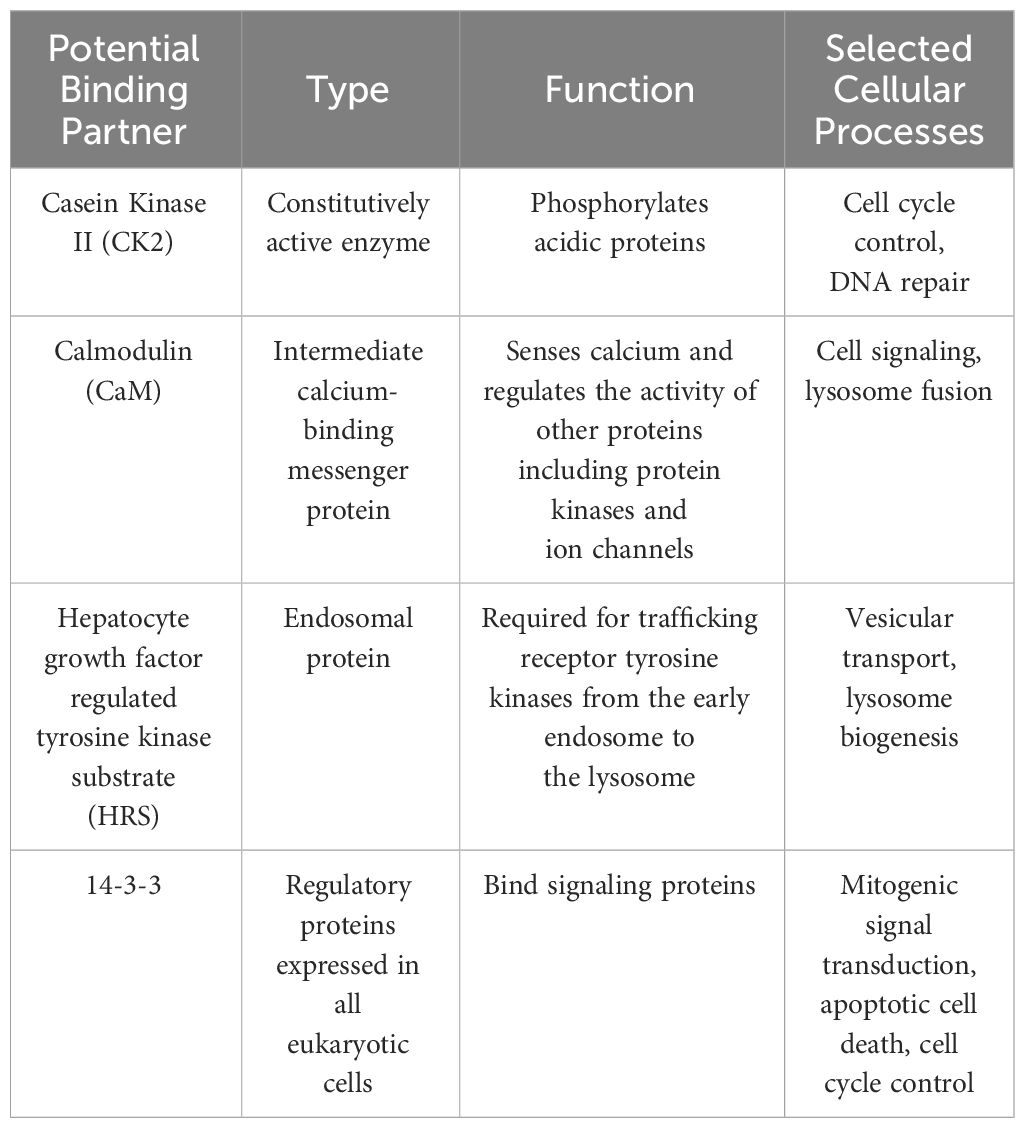
Table 2 Potential LYST binding partners.
The leading hypothesis was that LYST acts as a scaffolding protein facilitating critical interactions between SNARE complex proteins through regulation of Synaptotagmin and Protein Kinase C (PKC):
Initially discovered in neurons, synaptotagmin is a ubiquitously expressed calcium sensing protein that mediates exocytosis. There are 17 different synaptotagmin isoforms in mice and humans (141, 142). Synaptotagmin VII mediates Ca2+-dependent lysosome exocytosis in fibroblasts, macrophages, and neurons (143–145). Phosphorylation of synaptotagmin VII by regulatory proteins modulates its Ca2+ sensing exocytic activity (146). Notably, CK2 has been shown to phosphorylate Synaptotagmin I at a site that is highly conserved across isoforms (147). Additionally, CaM seems to be required by synaptotagmin VII for vesicle trafficking to the plasma membrane (148). Beyond vesicle trafficking, synaptotagmin-mediated calcium-dependent exocytosis is also responsible for plasma membrane repair (149) (127). As LYST may act as a scaffolding protein for synaptotagmin-regulating proteins, these connections may explain the impaired plasma membrane repair and defective lysosomal polarization following plasma membrane injury observed in LYST-mutant cells (64).
To further explain LYST involvement in vesicle docking and fusion machinery, it is necessary to consider other vesicle fusion proteins that interact with LYST’s potential binding partners, 14-3-3 and CK2. PKC is known to promote CK2 function, and PKC function is facilitated, in part, through interactions with 14-3-3 (150). Interestingly, beige cells naturally exhibited rapid downregulation of PKC activity. Beige NK cell cytotoxicity was improved upon treatment with an agent that prevents PKC breakdown (151). Additionally, this “PKC protecting” agent reversed the enlarged lysosome phenotype in beige cells; however, complete inhibition of PKC resulted in an enlarged lysosome phenotype similar to beige cells. PKC downregulation is potentially attributable to high levels of ceramide- a sphingolipid in beige fibroblasts which may cause the formation of enlarged granules (152). The relation of PKC downregulation to LYST-mutant phenotypes may loosely corroborate the relationship between LYST function and microtubules. Inhibiting PKC activity in WT cells enhanced ConA capping, similar to the abnormal spontaneous capping observed in beige cells (153). It is possible that PKC function is tied to cap formation, and that the downregulation of PKC activity leads to the abnormal capping in LYST-mutant cells. Despite the compelling case for PKC contributing to LYST-mutant phenotypes, some studies provide evidence against the link between PKC activity and enlarged lysosomes in LYST-mutant fibroblasts (154). Yet, it cannot be ruled out that while the coupling of PKC to enlarged lysosomes may not be universal, PKC isotypes may affect lysosome formation in other cell types, including macrophages and PMNs.
LYST is a member of the class of “BEACH domain-containing proteins” (BDCPs) (31). The Beige and Chédiak-Higashi (BEACH) domain is a highly conserved region first identified within the LYST protein but present in all other BDCPs. BDCPs regulate a variety of cellular functions, such as synapse formation regulated by neurobeachin (NBEA) and granule size regulated by beige-like anchor protein (LBRA) and LYST (31). Other BDCPs are also involved in vesicle trafficking and membrane dynamics in humans (31). Mutations in BDCPs cause several complications including epilepsy (NBEA mutation), grey platelet syndrome (NBEAL2 mutation), and rare autoimmune diseases (LRBA mutation) (155–157). Current consensus suggests that BDCPs, including LYST, act as cytosolic scaffolding proteins that facilitate membrane events involved with vesicular trafficking (31).
Recent advancement in molecular technologies further promoted understanding of the molecular aspect of LYST. The LYST protein is comprised of 3801 amino acids (158), but alternative splicing yields multiple transcript variants (documented in RefSeq). Generally, LYST contains an ARM/HEAT domain followed by a ConA-like lectin domain, a pleckstrin homology-like (PH-like) domain, a highly conserved BEACH domain, and finally, C-terminal WD40 repeats (31, 159).
Even though current literature lacks studies into the biological functions of each domain, protein structural analysis revealed possible functions related to each domain. ARM/HEAT repeats have been shown to mediate vesicle trafficking and lipid binding (132, 160, 161). The PH domain has a central fold associated with membrane binding to specific proteins (162). The crystal structure of the 130-residue segment N-terminal to the BEACH domain reveals a backbone fold with a PH domain and an extensive interface between BEACH and PH domains (31). Consistent with these structural analyses, protein binding assays demonstrated that the PH and BEACH domains strongly interact with one another in vitro and in silico (32). LYST contains an 8-15 amino acid insertion in the BEACH domain that differentiates its function from the other BDCPs (31, 163). Interestingly, a Lyst-specific BEACH function that determines lysosome and granule size likely exists. In mouse cells, protein truncation of only the Lyst-BEACH C-terminal fragment, but not the Nbea-BEACH C-terminal fragment, led to reversible lysosome enlargement (31, 163).
The parallel advancements in murine reagents and DNA sequencing contributed much of the genotype and phenotype correlations reported to date. A comprehensive literature review was conducted, involving a systematic search of peer-reviewed articles and case reports associated with “LYST” and “Chédiak-Higashi Syndrome” through the PubMed database (https://pubmed.ncbi.nlm.nih.gov/advanced/). We analyzed 29 case reports related to CHS, involving 61 patients (Figure 2, Supplementary Table 1) to explore the potential connection between LYST mutant genotypes and clinical manifestations of the disease. The genetic investigations suggest a link between the nature of the mutant LYST gene and the severity of the disease. For instance, it was observed that nonsense mutations, which typically result in the production of a truncated LYST protein, are associated with more severe phenotypes and often lead to infant mortality. In contrast, missense mutations tend to be linked to milder phenotypes or a later onset of the disease. A recent study by Morimoto and colleagues found comparable connections between the type of mutation and disease severity, providing additional support for genotype-phenotype correlations (164).
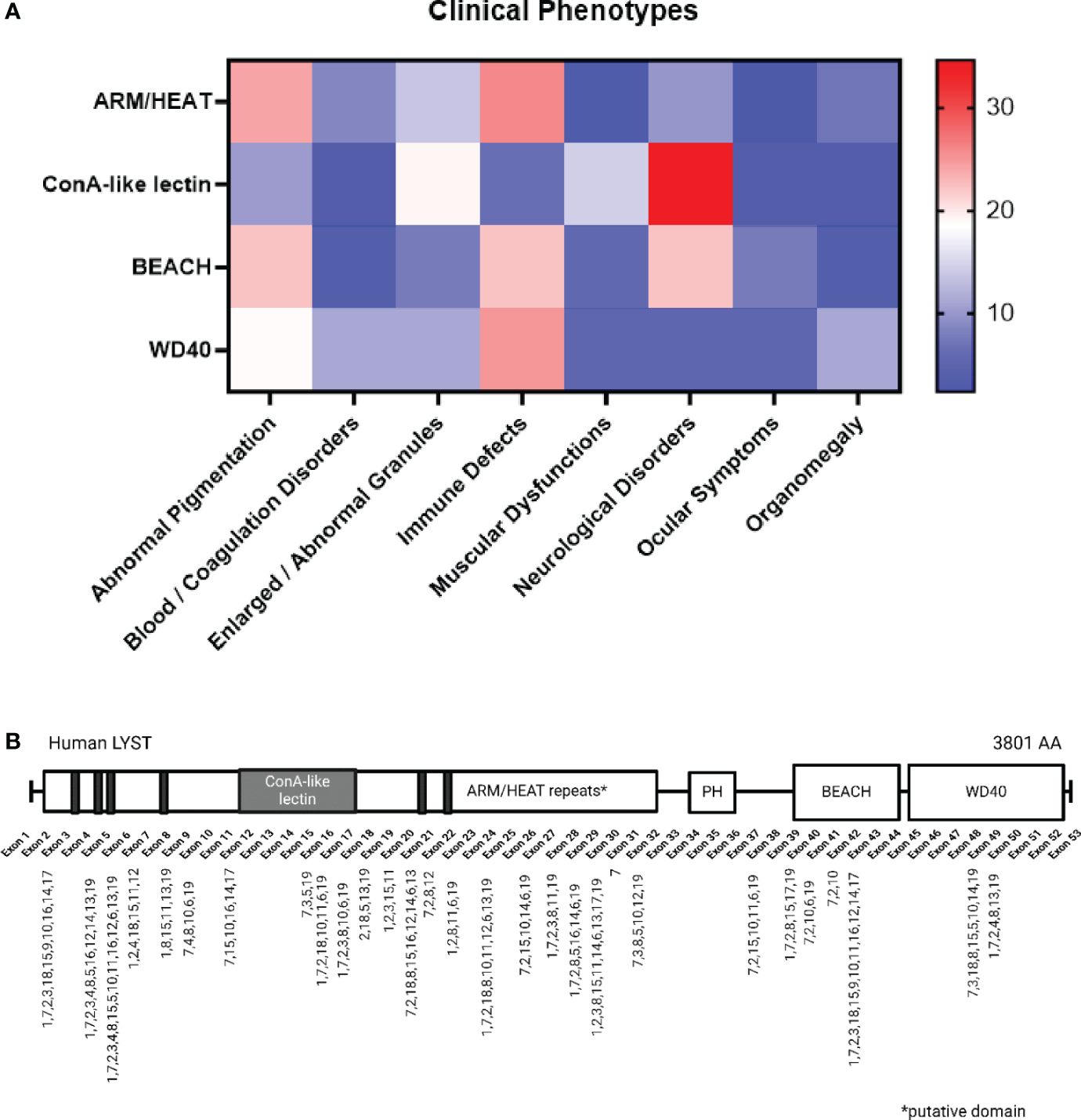
Figure 2 LYST Mutation and Correlation with Clinical Phenotypes. (A) The color gradient indicates the relative frequencies of specific disease phenotypes associated with each domain (e.g. Immune Defects account for 22.22% of cases with BEACH domain mutations). The 19 major phenotypes are categorized into eight groups and presented alphabetically, including: Abnormal Pigmentation (1. silver gray hair, 2. cutaneous albinism/partial albinism, 3. ocular albinism), Blood/Coagulation Disorders (4. coagulopathy/platelet dysfunctions, 5. isolated cytopenia, 6. pancytopenia), 7. Enlarged/Abnormal Granules, Immune Defects (8. fever/rash/lymphadenopathy, 9. oral infection/periodontitis, 10. defective/absent NK cell activity, 11. CMV/EBV seropositive, 12. respiratory infection, 13. recurrent infections), Muscular Dysfunction (14. motor dysfunction/myopathy/hypotonia), Neurological Disorders (15. GDD/intellectual disability/dementia, 16. neuropathy, 17. atrophic MRI brain), 18. Ocular Symptoms (i.e. myopia), and 19. Organomegaly (i.e. hepato/splenomegaly). (B) The mutation loci and their associated phenotypes are indicated on a schematic representation of the LYST protein spanning from exon 1 to exon 53, with a note indicating putative domains. Created with BioRender.com.
Further analysis indicates that the location of the mutated nucleotide(s) with respect to protein structure, in addition to mutation type, more closely correlates with disease phenotype (Figure 2). For example, mutations occurring within the ARM/HEAT repeats are associated with albinism, while neurological disorders are connected to mutations in the ConA-like lectin domain. Nonsense mutations within the WD40 domain are likely contributors to severe immune dysfunctions in CHS, leading to the accelerated phase, as depicted in Figure 2. Notably, ARM/HEAT mutations have been reported more frequently than mutations in other domains due to the domain’s larger size. However, a lack of data regarding the BEACH and WD40 domains may introduce potential inaccuracies in the interpretation of their functions.
Furthermore, it is important to highlight that the precise location of the ARM/HEAT repeats has not been definitively confirmed, despite proposed by Nagle et al. suggesting that these repeats may extend across a substantial helical region (31).
LYST was recently established as a gene of interest in patients with acute myeloid leukemia (AML), colorectal cancer, epithelial ovarian cancer, multiple myeloma, pulmonary carcinosarcoma, and sporadic chordoma (33–39). The association of LYST mutations with various forms of cancer is a chance discovery stemming from next generation sequencing analysis. Currently, the trend seems to be that downregulation or truncation of LYST protein leads to cancer progression. Downregulation of LYST was associated with low survival of AML patients, but in calcitonin receptor-like receptor (CALCRL) knockdown AML cell lines, LYST was upregulated (33). CALCRL deficient cells have been shown to be resistant to chemotherapy. The authors postulated that CALCRL downregulates LYST expression, which enlarges and stabilizes lysosomes and enhances drug resistance. In patients with sporadic chordoma, many patients carried truncating mutations in LYST. The authors explained that lysosomes were recently determined to be important for notochordal development, which could explain why defective LYST led to chordoma development. LYST also localized within copy number aberration regions in patients with multiple myeloma (39). Silencing LYST expression using anti-LYST siRNA inhibited proliferation and induced apoptosis in multiple myeloma cells (34). Both missense and nonsense LYST mutations were associated with pulmonary carcinosarcoma (38). Currently, studies are correlative but not mechanistic; authors appear to be divided on whether the connection of LYST to cancer is a spurious correlation or if LYST mutations truly drive cancer progression.
Chédiak-Higashi Syndrome (CHS) is a rare and life-threatening disorder, particularly affecting children. Despite identifying the causative gene decades ago, there is no comprehensive understanding of the cellular and molecular mechanisms underlying LYST function. The limited therapeutic options, primarily hematopoietic stem cell transplant, highlight the urgent need for more effective treatments to halt disease progression and prevent childhood mortality.
Recent studies employing various LYST-mutant organisms not only provided opportunities to study CHS, but also contributed to the understanding of the cell-specific role of LYST in endolysosomal trafficking and the immune response. Animal and clinical research highlights how LYST mutations manifest clinically in the setting of CHS, demonstrating its importance in immunity, neurological functions, pigmentation, and blood coagulation. Generally, cells from affected systems exhibit abnormal LRO morphology, impaired polarization, and decreased intracellular mobility, leading to reduced exocytosis of granules and protein trafficking.
While existing literature lacks unified theories on how LYST affects cellular processes, previous research proposes intriguing mechanisms contributing to the consistently observed enlarged lysosomes in CHS. This review provides a summary of the potential role of LYST in microtubule dynamics, granule fusion and fission events, and membrane docking facilitated by SNARE complex proteins. The current body of literature is divided on whether LYST impacts granule size through fusion or fission. Some studies suggest upregulated fusion events causing abnormal morphology, while others argue for insufficient fission as the culprit. The enlarged lysosomes and LROs do not fulfill their biological functions, and some studies suggest that the observed dysfunction involves changes in microtubule dynamics that impair proper vesicle transportation. Contradictory findings indicate intact microtubule function and suggest that the abnormally enlarged lysosomes physically cannot transit the actin cytoskeleton.
Sequencing technology enhanced our comprehension of the molecular structure and potential biological function of LYST by identifying genetic mutations. Recently, 11 novel disease-causing LYST mutations in the BEACH domain were identified using targeted Sanger sequencing, thereby expanding the scope and depth of LYST research (164). These findings provided valuable insight into mutation pathogenicity, laying the groundwork for potential CHS therapies and next-generation immuno-modulatory treatments.
Further investigation into the mRNA and protein sequences of LYST and lesser-explored members of BDCPs, such as WD repeat domain 81 (WDR81), is necessary to clarify the sequences and the precise locations of their domains. LYST and other BDCPs have diverse cellular functions, including cell cycle control, transcription regulation, apoptosis, and vesicle trafficking (31).
Knowing how LYST exerts its effects on a cellular and molecular scale holds significance not only for the development of CHS therapies but for other diseases involving cell regulation and vesicle trafficking. LYST mutations appear to be associated with certain cancer types, yet its potential role in cancer is not yet understood. Cutaneous wound healing studies revealed LYST’s crucial involvement in regulating secretory exocytosis by fibroblasts, which sheds light on the broader impact of LYST function (56, 57). Moreover, LYST attenuated immunomodulation combined with tissue engineering may improve transplant performance. While in the early stage of investigation, the importance of LYST in immune signaling suggests that LYST may influence the foreign body response to transplanted material. Thus, targeted alteration and temporary modulation of LYST function could offer immune privilege to recipients and protect donor tissues from overactive immune responses and rejection. The SCID/bg mouse model, where the Lyst beige mutation is introduced in SCID mice, has improved xenotransplantation compatibility (165). Recent advancements in tissue engineering, such as Tissue Engineered Vascular Grafts (TEVG) attracted attention due to the capacity for growth and remodeling (166). Despite their success, clinical studies have identified stenosis as a primary limitation. The immunodeficient SCID/bg mouse model demonstrated a drastic improvement in TEVGs patency (166). In the realm of regenerative medicine, LYST immunomodulation emerges as a promising target, aligning with the evolving paradigm of “immune interactive” strategies over traditional “immune evasive” approaches.
The discoveries presented herein underscore the potential for LYST as a tool for understanding the intersection of vesicle trafficking and cell cycle regulation and harnessing these findings to inform immune-modulatory strategies.
MT: Conceptualization, Data curation, Formal analysis, Investigation, Writing – original draft, Writing – review & editing. JC: Conceptualization, Data curation, Formal analysis, Investigation, Writing – original draft, Writing – review & editing. GM: Investigation, Writing – original draft, Writing – review & editing. CK: Writing – review & editing. KB: Investigation, Writing – original draft, Writing – review & editing. SR: Writing – review & editing. JZ: Writing – original draft, Writing – review & editing. ChB: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. CaB: Conceptualization, Data curation, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing. JB: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health through 1R01HL157491-01 awarded to ChB, F31HL1145962 supporting CaB, and 3R01HL157491-02S1 supporting MT.
ChB and CaB are co-founders of Lyst Therapeutics, LLC Columbus, OH and did not receive support for the present work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1404846/full#supplementary-material
1. Sánchez-Guiu I, Antón AI, García-Barberá N, Navarro-Fernández J, Martínez C, Fuster JL, et al. Chediak–Higashi syndrome: description of two novel homozygous missense mutations causing divergent clinical phenotype. Eur J Haematol. (2014) 92:49–58. doi: 10.1111/ejh.12203
2. Baguez-Cesar A. Neutoropenia cronica Maligna familiar con granulaciones atipicas de los leucocitos. Bol Soc Cubbana Pediatr. (1943) 15:900–22. Undefined.
3. Chediak M, Chediak B, Fleites O, Hernandez A. Presentation of a case of Cooley’s anemia in a Cuban boy; first report in Cuba. Bol Liga Contra Cancer Havana. (1952) 27:20–6.
4. Higashi O. Congenital gigantism of peroxidase granules; the first case ever reported of qualitative abnormity of peroxidase. Tohoku J Exp Med. (1954) 59:315–32. doi: 10.1620/tjem.59.315
5. Sato A. Chediak and higashi’s disease: probable identity of A new leucocytal anomaly (Chediak) and congenital gigantism of peroxidase granules (Higashi). Tohoku J Exp Med. (1955) 61:201–10. doi: 10.1620/tjem.61.201
6. Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol. Gen. Meta. (1999) 68(2):283–303. doi: 10.1006/mgme.1999.2927
7. Padgett GA, Reiquam CW, Gorham JR, Henson JB, O’Mary CC. Comparative studies of the Chediak-Higashi syndrome. Am J Pathol. (1967) 51:553–71.
8. White JG. The Chediak-Higashi syndrome: cytoplasmic sequestration in circulating leukocytes. Blood. (1967) 29:435–51. doi: 10.1182/blood.V29.4.435.435
9. Cohn ZA, Hirsch JG. The isolation and properties of the specific cytoplasmic granules of rabbit polymorphonuclear leucocytes. J Exp Med. (1960) 112:983–1004. doi: 10.1084/jem.112.6.983
11. Runkel F, Büssow H, Seburn KL, Cox GA, Ward DMV, Kaplan J, et al. Grey, a novel mutation in the murine Lyst gene, causes the beige phenotype by skipping of exon 25. Mamm Genome. (2006) 17:203–10. doi: 10.1007/s00335-005-0015-1
12. Rudelius M, Osanger A, Kohlmann S, Augustin M, Piontek G, Heinzmann U, et al. A missense mutation in the WD40 domain of murine Lyst is linked to severe progressive Purkinje cell degeneration. Acta Neuropathol. (2006) 112:267–76. doi: 10.1007/s00401-006-0092-6
13. Rahman M, Haberman A, Tracy C, Ray S, Krämer H. Drosophila mauve mutants reveal a role of LYST homologs late in the maturation of phagosomes and autophagosomes. Traffic. (2012) 13:1680–92. doi: 10.1111/tra.12005
14. Barrett A, Hermann GJ. A caenorhabditis elegans homologue of LYST functions in endosome and lysosome-related organelle biogenesis. Traffic. (2016) 17:515–35. doi: 10.1111/tra.12381
15. Charette SJ, Cosson P. A LYST/beige homolog is involved in biogenesis of Dictyostelium secretory lysosomes. J Cell Sci. (2007) 120:2338–43. doi: 10.1242/jcs.009001
16. Ullate-Agote A, Burgelin I, Debry A, Langrez C, Montange F, Peraldi R, et al. Genome mapping of a LYST mutation in corn snakes indicates that vertebrate chromatophore vesicles are lysosome-related organelles. Proc Natl Acad Sci U S A. (2020) 117:26307–17. doi: 10.1073/pnas.2003724117
17. Kunieda T, Nakagiri M, Takami M, Ide H, Ogawa H. Cloning of bovine LYST gene and identification of a missense mutation associated with Chediak-Higashi syndrome of cattle. Mamm Genome. (1999) 10:12. doi: 10.1007/s003359901181
18. Kim SH, Wu SY, Baek JI, Choi SY, Su Y, Flynn CR, et al. A post-developmental genetic screen for zebrafish models of inherited liver disease. PloS One. (2015) 10. doi: 10.1371/journal.pone.0125980
19. Fukai K, Oh J, Karim MA, Moore KJ, Kandil HH, Ito H, et al. Homozygosity mapping of the gene for Chediak-Higashi syndrome to chromosome 1q42-q44 in a segment of conserved synteny that includes the mouse beige locus (bg). Am J Hum Genet. (1996) 59:620–4.
20. Barrat FJ, Auloge L, Pastural E, Lagelouse RD, Vilmer E, Cant AJ, et al. Genetic and physical mapping of the Chediak-Higashi syndrome on chromosome 1q42-43. Am J Hum Genet. (1996) 59:625–32.
21. Nagle DL, Karim MA, Woolf EA, Holmgren L, Bork P, Misumi DJ, et al. Identification and mutation analysis of the complete gene for Chediak–Higashi syndrome. Nat Genet. (1996) 14:307–11. doi: 10.1038/ng1196-307
22. Oliver JM, Zurier RB. Correction of characteristic abnormalities of microtubule function and granule morphology in Chediak-Higashi syndrome with cholinergic agonists. J Clin Invest. (1976) 57:1239–47. doi: 10.1172/JCI108392
23. Willingham MC, Spicer SS, Vincent RA. The origin and fate of large dense bodies in beige mouse fibroblasts. Lysosomal fusion and exocytosis. Exp Cell Res. (1981) 136:157–68. doi: 10.1016/0014-4827(81)90047-1
24. Vincent RA, Spicer SS. Giant dense bodies in fibroblasts cultured from beige mice with the Chédiak-Higashi syndrome. Am J Pathol. (1981) 105:270–8.
25. White JG, Clawson CC. The Chédiak-Higashi syndrome; the nature of the giant neutrophil granules and their interactions with cytoplasm and foreign particulates. I. Progressive enlargement of the massive inclusions in mature neutrophils. II. Manifestations of cytoplasmic injury and sequestration. III. Interactions between giant organelles and foreign particulates. Am J Pathol. (1980) 98:151.
26. Rausch PG, Pryzwansky KB, Spitznagel JK. Immunocytochemical identification of azurophilic and specific granule markers in the giant granules of Chediak-Higashi neutrophils. New Engl J Med. (1978) 298:693–8. doi: 10.1056/NEJM197803302981301
27. Perou CM, Leslie JD, Green W, Liangtao Li H, Ward DMV, Kaplan J. The Beige/Chediak-Higashi syndrome gene encodes a widely expressed cytosolic protein. J Biol Chem. (1997) 272:29790–4. doi: 10.1074/jbc.272.47.29790
28. Durchfort N, Verhoef S, Vaughn MB, Shrestha R, Adam D, Kaplan J, et al. The enlarged lysosomes in beige j cells result from decreased lysosome fission and not increased lysosome fusion. Traffic. (2012) 13:108–19. doi: 10.1111/j.1600-0854.2011.01300.x
29. Tchernev VT, Mansfield TA, Giot L, Kumar AM, Nandabalan K, Li Y, et al. The Chediak-Higashi protein interacts with SNARE complex and signal transduction proteins. Mol Med. (2002) 8:56–64. doi: 10.1007/BF03402003
30. Han J, Pluhackova K, Böckmann RA. The multifaceted role of SNARE proteins in membrane fusion. Front Physiol. (2017) 8:5. doi: 10.3389/fphys.2017.00005
31. Cullinane AR, Schäffer AA, Huizing M. The BEACH is hot: A LYST of emerging roles for BEACH-domain containing proteins in human disease. Traffic. (2013) 14:749–66. doi: 10.1111/tra.12069
32. Jogl G, Shen Y, Gebauer D, Li J, Wiegmann K, Kashkar H, et al. Crystal structure of the BEACH domain reveals an unusual fold and extensive association with a novel PH domain. EMBO J. (2002) 21:4785–95. doi: 10.1093/emboj/cdf502
33. Tarpey PS, Behjati S, Young MD, Martincorena I, Alexandrov LB, Farndon SJ, et al. The driver landscape of sporadic chordoma. Nat Commun. (2017) 8:890. doi: 10.1038/s41467-017-01026-0
34. Pau Ni IB, Ching N, Fakiruddin SK, Moon Nian L, Zakaria Z. Small interfering RNA-mediated silencing of nicotinamide phosphoribosyltransferase (NAMPT) and lysosomal trafficking regulator (LYST) induce growth inhibition and apoptosis in human multiple myeloma cells: A preliminary study. Bosn J Basic Med Sci. (2016) 16(4):268–75. doi: 10.17305/bjbms.2016.1568
35. Zhao Q, Wang F, Chen YX, Chen S, Yao YC, Zeng ZL, et al. Comprehensive profiling of 1015 patients’ exomes reveals genomic-clinical associations in colorectal cancer. Nat Commun. (2022) 13:2342. doi: 10.1038/s41467-022-30062-8
36. Li L, Zheng H, Huang Y, Huang C, Zhang S, Tian J, et al. DNA methylation signatures and coagulation factors in the peripheral blood leucocytes of epithelial ovarian cancer. Carcinogenesis. (2017) 38:797–805. doi: 10.1093/carcin/bgx057
37. Huang Z, Zhang H, Xing C, Zhang L, Zhu H, Deng Z, et al. Identification and validation of CALCRL-associated prognostic genes in acute myeloid leukemia. Gene. (2022) 809:146009. doi: 10.1016/j.gene.2021.146009
38. Li F, Hu S, Kong K, Cao P, Han P, Deng Y, et al. Next-generation sequencing analysis identified genomic alterations in pathological morphologies of 3 cases of pulmonary carcinosarcoma. Onco Targets Ther. (2020) 13:7963–72. doi: 10.2147/OTT.S264617
39. Ivyna Bong PN, Ng CC, Lam KY, Megat Baharuddin PJN, Chang KM, Zakaria Z. Identification of novel pathogenic copy number aberrations in multiple myeloma: the Malaysian context. Mol Cytogenet. (2014) 7:24. doi: 10.1186/1755-8166-7-24
40. Zbinden MS JC, Mirhaidari GJ M, Blum KM, Musgrave BS AJ, Reinhardt JW, Breuer CK, et al. The lysosomal trafficking regulator is necessary for normal wound healing. (2021). doi: 10.1111/wrr.12984
41. Jordison Boxer G, Holmsen H, Robkin L, Bang NU, Boxer LA, Baehner RL. Abnormal platelet function in Chediak-Higashi syndrome. Br J Haematol. (1977) 35:521–33. doi: 10.1111/j.1365-2141.1977.tb00618.x
42. Stegmaier OC, Schneider LA. Chediak-Higashi syndrome: Dermatologic manifestations. Arch Dermatol. (1965) 91:1–9. doi: 10.1001/archderm.1965.01600070007001
43. Uyama E, Hirano T, Ito K, Nakashima H, Sugimoto M, Naito M, et al. Adult Chédiak-Higashi syndrome presenting as parkinsonism and dementia. Acta Neurol Scand. (2009) 89:175–83. doi: 10.1111/j.1600-0404.1994.tb01657.x
44. Eapen M, DeLaat CA, Baker KS, Cairo MS, Cowan MJ, Kurtzberg J, et al. Hematopoietic cell transplantation for Chediak-Higashi syndrome. Bone Marrow Transplant. (2007) 39:411–5. doi: 10.1038/sj.bmt.1705600
45. Kaplan J, De Domenico I, Ward DM. Chediak-Higashi syndrome. Curr Opin Hematol. (2008) 15:22–9. doi: 10.1097/MOH.0b013e3282f2bcce
48. Davisson MT, Lewis SE. Chromosome aberrations associated with induced mutations: effect on mapping new mutations. Biol Mamm germ Cell mutagenesis. (1990), 195–206.
49. Perou CM, Moore KJ, Nagle DL, Misumi DJ, Woolf EA, McGrail SH, et al. Identification of the murine beige gene by YAC complementation and positional cloning. Nat Genet. (1996) 13:303–9. doi: 10.1038/ng0796-303
50. Introne WJ, Westbroek W, Cullinane AR, Groden CA, Bhambhani V, Golas GA, et al. Neurologic involvement in patients with atypical Chediak-Higashi disease. Neurology. (2016) 86:1320–8. doi: 10.1212/WNL.0000000000002551
51. Anistoroaei R, Krogh AK, Christensen K. A frameshift mutation in the LYST gene is responsible for the Aleutian color and the associated Chédiak-Higashi syndrome in American mink. Anim Genet. (2013) 44:178–83. doi: 10.1111/j.1365-2052.2012.02391.x
52. Leader RW, Padgett GA, Gorham JR. Studies of abnormal leukocytes bodies in the mink. Blood. (1963) 22:477–84. doi: 10.1182/blood.V22.4.477.477
53. Kahraman MM, Prieur DJ. Chediak-Higashi syndrome: Prenatal diagnosis by fetal blood examination in the feline model of the disease. Am J Med Genet. (1989) 32:325–9. doi: 10.1002/ajmg.1320320310
54. Kramer JW, Davis WC, Prieur DJ. The Chediak-Higashi syndrome of cats. Lab Invest. (1977) 36:554–62.
55. Prieur DJ, Collier LL. Inheritance of the Chediak-Higashi syndrome in cats. J Hered. (1981) 72:175–7. doi: 10.1093/oxfordjournals.jhered.a109467
56. Fagerland JA, Hagemoser WA, Ireland WP. Ultrastructure and stereology of leukocytes and platelets of normal foxes and a fox with a Chediak-Higashi-like syndrome. Vet Pathol. (1987) 24:164–9. doi: 10.1177/030098588702400210
57. Nes N, Lium B, Braend M, Sjaastad O. A Chediak-Higashi-like syndrome in Arctic blue foxes. Finsk Veterinaertidsskrift. (1983), 89–313.
58. Yamakuchi H, Agaba M, Hirano T, Hara K, Todoroki J, Mizoshita K, et al. Chediak-Higashi syndrome mutation and genetic testing in Japanese black cattle (Wagyu). Anim Genet. (2000) 31:13–9. doi: 10.1046/j.1365-2052.2000.00586.x
59. Nishimura M, Inoue M, Nakano T, Nishikawa T, Miyamoto M, Kobayashi T, et al. Beige rat: a new animal model of Chediak-Higashi syndrome. Blood. (1989) 74:270–3. doi: 10.1182/blood.V74.1.270.270
60. Gil-Krzewska A, Saeed MB, Oszmiana A, Fischer ER, Lagrue K, Gahl WA, et al. An actin cytoskeletal barrier inhibits lytic granule release from natural killer cells in patients with Chediak-Higashi syndrome. J Allergy Clin Immunol. (2018) 142:914–927.e6. doi: 10.1016/j.jaci.2017.10.040
61. Chiang SCC, Wood SM, Tesi B, Akar HH, Al-Herz W, Ammann S, et al. Differences in Granule Morphology yet Equally Impaired Exocytosis among Cytotoxic T Cells and NK Cells from Chediak-Higashi Syndrome Patients. Front Immunol. (2017) 8. doi: 10.3389/fimmu.2017.00426
62. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. (2008) 9:503–10. doi: 10.1038/ni1582
63. Krzewski K, Coligan JE. Human NK cell lytic granules and regulation of their exocytosis. Front Immunol. (2012) 3:335. doi: 10.3389/fimmu.2012.00335
64. Huynh C, Roth D, Ward DM, Kaplan J, Andrews NW. Defective lysosomal exocytosis and plasma membrane repair in Chediak-Higashi/beige cells. Proc Natl Acad Sci U.S.A. (2004) 101:16795–800. doi: 10.1073/pnas.0405905101
65. Kievits F, Wijffels J, Lokhorst W, Ivanyi P. Recognition of xeno-(HLA, SLA) major histocompatibility complex antigens by mouse cytotoxic T cells is not H-2 restricted: a study with transgenic mice. Proc Natl Acad Sci U.S.A. (1989) 86:617–20. doi: 10.1073/pnas.86.2.617
66. Matti U, Pattu V, Halimani M, Schirra C, Krause E, Liu Y, et al. Synaptobrevin2 is the v-SNARE required for cytotoxic T-lymphocyte lytic granule fusion. Nat Commun. (2013) 4:1439. doi: 10.1038/ncomms2467
67. Baetz K, Isaaz S, Griffiths GM. Loss of cytotoxic T lymphocyte function in Chediak-Higashi syndrome arises from a secretory defect that prevents lytic granule exocytosis. J Immunol. (1995) 154:6122–31. doi: 10.4049/jimmunol.154.11.6122
68. Stinchcombe JC, Page LJ, Griffiths GM. Secretory lysosome biogenesis in cytotoxic T lymphocytes from normal and syndrome patients. Traffic. (2000) 1:435–44. doi: 10.1034/j.1600-0854.2000.010508.x
69. Barrat FJ, Le Deist F, Benkerrou M, Bousso P, Feldmann J, Fischer A, et al. Defective CTLA-4 cycling pathway in Chediak-Higashi syndrome: A possible mechanism for deregulation of T lymphocyte activation. Proc Natl Acad Sci U.S.A. (1999) 96:8645–50. doi: 10.1073/pnas.96.15.8645
70. Jolly C, Welsch S, Michor S, Sattentau QJ. The regulated secretory pathway in CD4(+) T cells contributes to human immunodeficiency virus type-1 cell-to-cell spread at the virological synapse. PloS Pathog. (2011) 7. doi: 10.1371/journal.ppat.1002226
71. Jiang W, Adler LN, Macmillan H, Mellins ED. Synergy between B cell receptor/antigen uptake and MHCII peptide editing relies on HLA-DO tuning. Sci Rep. (2019) 9(1):1439. doi: 10.1038/s41598-019-50455-y
72. Faigle W, Raposo G, Tenza D, Pinet V, Vogt AB, Kropshofer H, et al. Deficient peptide loading and MHC class II endosomal sorting in a human genetic immunodeficiency disease: the Chediak-Higashi syndrome. J Cell Biol. (1998) 141:1121–34. doi: 10.1083/jcb.141.5.1121
73. Chatterjee P, Tiwari RK, Rath S, Bal V, George A. Modulation of antigen presentation and B cell receptor signaling in B cells of beige mice. J Immunol. (2012) 188:2695–702. doi: 10.4049/jimmunol.1101527
74. Donahue AC, Fruman DA. Proliferation and survival of activated B cells requires sustained antigen receptor engagement and phosphoinositide 3-kinase activation. J Immunol. (2003) 170:5851–60. doi: 10.4049/jimmunol.170.12.5851
75. Nargund AR, Madhumathi DS, Premalatha CS, Rao CR, Appaji L, Lakshmidevi V. Accelerated phase of syndrome mimicking lymphoma–a case report. J Pediatr Hematol Oncol. (2010) 32:e223–6. doi: 10.1097/MPH.0b013e3181e62663
76. Sung JH, Meyers JP, Stadlan EM, Cowen D, Wolf A. Neuropathological changes in Chediak-Higashi disease. J Neuropathol Exp Neurol. (1969) 28:86–118. doi: 10.1097/00005072-196901000-00005
77. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
78. Masgrau-Alsina S, Sperandio M, Rohwedder I. Neutrophil recruitment and intracellular vesicle transport: A short overview. Eur J Clin Invest. (2020) 50. doi: 10.1111/eci.13237
79. Sheshachalam A, Srivastava N, Mitchell T, Lacy P, Eitzen G. Granule protein processing and regulated secretion in neutrophils. Front Immunol. (2014) 5:448. doi: 10.3389/fimmu.2014.00448
80. Wolff SM. The Chediak-Higashi syndrome: studies of host defenses. Ann Intern Med. (1972) 76:293–306. doi: 10.7326/0003-4819-76-2-293
81. Jena P, Mohanty S, Mohanty T, Kallert S, Morgelin M, Lindstrøm T, et al. Azurophil granule proteins constitute the major mycobactericidal proteins in human neutrophils and enhance the killing of mycobacteria in macrophages. PloS One. (2012) 7. doi: 10.1371/journal.pone.0050345
82. Ganz T, Metcalf JA, Gallin JI, Boxer LA, Lehrer RI. Microbicidal/cytotoxic proteins of neutrophils are deficient in two disorders: Chediak-Higashi syndrome and “specific” granule deficiency. J Clin Invest. (1988) 82:552–6. doi: 10.1172/JCI113631
83. Clark RA, Kimball HR. Defective granulocyte chemotaxis in the Chediak-Higashi syndrome. J Clin Invest. (1971) 50:2645. doi: 10.1172/JCI106765
84. Nauseef WM, Borregaard N. Neutrophils at work. Nat Immunol. (2014) 15:602–11. doi: 10.1038/ni.2921
85. Wilson BJ, Allen JL, Caswell PT. Vesicle trafficking pathways that direct cell migration in 3D matrices and in vivo. Traffic. (2018) 19:899. doi: 10.1111/tra.12605
86. Bainbridge P. Wound healing and the role of fibroblasts. J Wound Care. (2013) 22:407–12. doi: 10.12968/jowc.2013.22.8.407
87. Holland P, Torgersen ML, Sandvig K, Simonsen A. LYST Affects Lysosome Size and Quantity, but not Trafficking or Degradation Through Autophagy or Endocytosis. Traffic. (2014) 15:1390–405. doi: 10.1111/tra.12227
88. Wang L, Kantovitz KR, Cullinane AR, Nociti FH, Foster BL, Roney JC, et al. Skin fibroblasts from individuals with Chediak-Higashi Syndrome (CHS) exhibit hyposensitive immunogenic response. Orphanet J Rare Dis. (2014) 9:212. doi: 10.1186/s13023-014-0212-7
89. Westphal A, Cheng W, Yu J, Grassl G, Krautkrämer M, Holst O, et al. Lysosomal trafficking regulator Lyst links membrane trafficking to toll-like receptor–mediated inflammatory responses. J Exp Med. (2017) 214:227. doi: 10.1084/jem.20141461
90. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. Functional differentiation. Front Immunol. (2014) 5. doi: 10.3389/fimmu.2014.00514
91. Wong CO, Gregory S, Hu H, Chao Y, Sepúlveda VE, He Y, et al. Lysosomal degradation is required for sustained phagocytosis of bacteria by macrophages. Cell Host Microbe. (2017) 21:719–730.e6. doi: 10.1016/j.chom.2017.05.002
92. Root RK, Rosenthal AS, Balestra DJ. Abnormal bactericidal, metabolic, and lysosomal functions of Chediak-Higashi Syndrome leukocytes. J Clin Invest. (1972) 51:649–65. doi: 10.1172/JCI106854
93. Mahoney KH, Morse SS, Morahan PS. Macrophage functions in beige (Chédiak-Higashi syndrome) mice. Cancer Res. (1980) 40:3934–9.
94. Wagner DD, Burger PC. Platelets in inflammation and thrombosis. Arterioscler Thromb Vasc Biol. (2003) 23:2131–7. doi: 10.1161/01.ATV.0000095974.95122.EC
95. Holinstat M. Normal platelet function. Cancer Metastasis Rev. (2017) 36:195–8. doi: 10.1007/s10555-017-9677-x
96. Costa JL, Fauci AS, Wolff SM. A platelet abnormality in the Chediak-Higashi syndrome of man. Blood. (1976) 48:517–20. doi: 10.1182/blood.V48.4.517.517
97. Rendu F, Breton-Gorius J, Lebret M, Klebanoff C, Buriot D, Griscelli C, et al. Evidence that abnormal platelet functions in human Chédiak-Higashi syndrome are the result of a lack of dense bodies. Am J Pathol. (1983) 111:307–14.
98. Holland JM. Serotonin deficiency and prolonged bleeding in beige mice. Proc Soc Exp Biol Med. (1976) 151:32–9. doi: 10.3181/00379727-151-39137
99. Lampugnani MG, Buczko W, Ceci A, Mennini A, de Gaetano G. Normal serotonin uptake by blood platelets and brain synaptosomes but selective impairment of platelet serotonin storage in mice with Chediack-Higashi syndrome. Life Sci. (1986) 38:2193–8. doi: 10.1016/0024-3205(86)90571-0
100. Reddington M, Novak EK, Hurley E, Medda C, McGarry MP, Swank RT. Immature dense granules in platelets from mice with platelet storage pool disease. Blood. (1987) 69:1300–6. doi: 10.1182/blood.V69.5.1300.1300
101. Pratt HL, Carroll RC, Jones JB, Lothrop CD. Platelet aggregation, storage pool deficiency, and protein phosphorylation in mice with Chediak-Higashi syndrome. Am J Vet Res. (1991) 52:945–50. doi: 10.2460/ajvr.1991.52.06.945
102. Yamaguchi Y, Hearing VJ. Melanocytes and their diseases. Cold Spring Harb Perspect Med. (2014) 4. doi: 10.1101/cshperspect.a017046
103. Fukuda M. Multiple roles of VARP in endosomal trafficking: rabs, retromer components and R- SNARE VAMP7 meet on VARP. Traffic. (2016) 17:709–19. doi: 10.1111/tra.12406
104. Shiflett SL, Kaplan J, Ward DMV. Chediak-Higashi Syndrome: a rare disorder of lysosomes and lysosome related organelles. Pigment Cell Res. (2002) 15:251–7. doi: 10.1034/j.1600-0749.2002.02038.x
105. Zhao H, Boissy YL, Abdel-Malek Z, King RA, Nordlund JJ, Boissy RE. On the analysis of the pathophysiology of Chediak-Higashi syndrome. Defects expressed by cultured melanocytes. Lab Invest. (1994) 71:25–34.
106. Ji X, Zhao L, Umapathy A, Fitzmaurice B, Wang J, Williams DS, et al. Deficiency in Lyst function leads to accumulation of secreted proteases and reduced retinal adhesion. PloS One. (2022) 17. doi: 10.1371/journal.pone.0254469
107. Lloyd V, Ramaswami M, Krämer H. Not just pretty eyes: Drosophila eye-colour mutations and lysosomal delivery. Trends Cell Biol. (1998) 8:257–9. doi: 10.1016/S0962-8924(98)01270-7
108. da Silva EZM, Jamur MC, Oliver C. Mast cell function: a new vision of an old cell. J Histochem Cytochem. (2014) 62:698–738. doi: 10.1369/0022155414545334
109. Galli SJ, Nakae S, Tsai M. Mast cells in the development of adaptive immune responses. Nat Immunol. (2005) 6:135–42. doi: 10.1038/ni1158
110. Rao KN, Brown MA. Mast cells: multifaceted immune cells with diverse roles in health and disease. Ann N Y Acad Sci. (2008) 1143:83–104. doi: 10.1196/annals.1443.023
111. Chi EY, Lagunoff D. Abnormal mast cell granules in the beige (Chédiak-Higashi syndrome) mouse. J Histochem Cytochem. (1975) 23:117–22. doi: 10.1177/23.2.46876
112. Kiyoi T, Liu S, Sahid MNA, Shudou M, Ogasawara M, Mogi M, et al. Morphological and functional analysis of beige (Chèdiak-Higashi syndrome) mouse mast cells with giant granules. Int Immunopharmacol. (2019) 69:202–12. doi: 10.1016/j.intimp.2019.01.053
113. Jippo-Kanemoto T, Kasugai T, Yamatodani A, Ushio H, Mochizuki T, Tohya K, et al. Sb1upernormal histamine release and normal cytotoxic activity of beige (Chédiak-Higashi syndrome) rat mast cells with giant granules. Int Arch Allergy Immunol. (1993) 100:99–106. doi: 10.1159/000236395
114. Hammel I, Lagunoff D, Galli SJ. Regulation of secretory granule size by the precise generation and fusion of unit granules. J Cell Mol Med. (2010) 14:1904–16. doi: 10.1111/j.1582-4934.2010.01071.x
115. Jones MK, Nair A, Gupta M. Mast cells in neurodegenerative disease. Front Cell Neurosci. (2019) 13:171. doi: 10.3389/fncel.2019.00171
116. Efrati P, Jonas W. Chediak’s anomaly of leukocytes in Malignant lymphoma associated with leukemic manifestations: case report with necropsy. Blood. (1958) 13:1063–73. doi: 10.1182/blood.V13.11.1063.1063
117. Danes BS, Bearn AG. Cell culture and the Chediak-Higashi syndrome. Lancet. (1967) 2:65–7. doi: 10.1016/S0140-6736(67)92059-4
118. Penner JD, Prieur DJ, Opitz JM, Reynolds JF. A comparative study of the lesions in cultured fibroblasts of humans and four species of animals with Chediak-Higashi syndrome. Am J Med Genet. (1987) 28:445–54. doi: 10.1002/ajmg.1320280222
119. Ash P, Loutit JF, Townsend KM. Giant lysosomes, a cytoplasmic marker in osteoclasts of beige mice. J Pathol. (1980) 130:237–45. doi: 10.1002/path.1711300405
120. Prieur DJ, Davis WC, Padgett GA. Defective function of renal lysosomes in mice with the Chediak-Higashi syndrome. Am J Pathol. (1972) 67:227–36.
121. Sato A, Spicer SS. An ultrastructural and cytochemical investigation of the development of inclusions in gastric chief cells and parietal cells of mice with the Chediak-Higashi syndrome. Lab Invest. (1981) 44:288–99.
122. Lattao R, Rangone H, Llamazares S, Glover DM. Mauve/LYST limits fusion of lysosome-related organelles and promotes centrosomal recruitment of microtubule nucleating proteins. Dev Cell. (2021) 56:1000–1013.e6. doi: 10.1016/j.devcel.2021.02.019
123. Sepulveda FE, Burgess A, Heiligenstein X, Goudin N, Ménager MM, Romao M, et al. LYST controls the biogenesis of the endosomal compartment required for secretory lysosome function. Traffic. (2015) 16:191–203. doi: 10.1111/tra.12244
124. Kypri E, Falkenstein K, De Lozanne A. Antagonistic control of lysosomal fusion by Rab14 and the Lyst-related protein LvsB. Traffic. (2013) 14:599–609. doi: 10.1111/tra.12058
125. Scott CC, Vacca F, Gruenberg J. Endosome maturation, transport and functions. Semin Cell Dev Biol. (2014) 31:2–10. doi: 10.1016/j.semcdb.2014.03.034
126. Stoorvogel W, Strous GJ, Geuze HJ, Oorschot V, Schwartzt AL. Late endosomes derive from early endosomes by maturation. Cell. (1991) 65:417–27. doi: 10.1016/0092-8674(91)90459-C
127. Trivedi PC, Bartlett JJ, Pulinilkunnil T. Lysosomal biology and function: modern view of cellular debris bin. Cells. (2020) 9(5):1131. doi: 10.3390/cells9051131
128. Bowman SL, Bi-Karchin J, Le L, Marks MS. The road to lysosome-related organelles: Insights from Hermansky-Pudlak syndrome and other rare diseases. Traffic. (2019) 20:404–35. doi: 10.1111/tra.12646
129. Saffi GT, Botelho RJ. Lysosome fission: planning for an exit. Trends Cell Biol. (2019) 29:635–46. doi: 10.1016/j.tcb.2019.05.003
130. Harris E, Wang N, Wu WL, Weatherford A, De Lozanne A, Cardelli J. Dictyostelium LvsB mutants model the lysosomal defects associated with Chediak-Higashi syndrome. Mol Biol Cell. (2002) 13:656–69. doi: 10.1091/mbc.01-09-0454
131. Charette SJ, Cosson P. Altered composition and secretion of lysosome-derived compartments in Dictyostelium AP-3 mutant cells. Traffic. (2008) 9:588–96. doi: 10.1111/j.1600-0854.2008.00706.x
132. Gil-Krzewska A, Wood SM, Murakami Y, Nguyen V, Chiang SCC, Cullinane AR, et al. Chediak-Higashi syndrome: Lysosomal trafficking regulator domains regulate exocytosis of lytic granules but not cytokine secretion by natural killer cells. J Allergy Clin Immunol. (2015) 137:1165–77. doi: 10.1016/j.jaci.2015.08.039
133. Oliver JM. Impaired microtubule function correctable by cyclic GMP and cholinergic agonists in the Chediak-Higashi syndrome. Am J Pathol. (1976) 85:395–418.
134. Mathiasen JR, Raffa RB, Vaught JL. C57BL/6J-bgJ (beige) mice: differential sensitivity in the tail flick test to centrally administered mu- and delta-opioid receptor agonists. Life Sci. (1987) 40:1989–94. doi: 10.1016/0024-3205(87)90288-8
135. Muraki T, Oike N, Shibata Y, Nomoto T. Analgesic effect of mu- and kappa-opioid agonists in beige and CXBK mice. J Pharm Pharmacol. (1991) 43:210–2. doi: 10.1111/j.2042-7158.1991.tb06669.x
136. Raffa RB, Baldy WJ, Shank RP, Mathiasen JR, Vaught JL. [3H][D-Ala2,NMePhe4,Gly-ol5]-enkephalin (mu-opioid) binding in beige-J mice. Peptides (NY). (1988) 9:637–42. doi: 10.1016/0196-9781(88)90176-3
137. Raffa RB, Mathiasen JR, Vaught JL. Restoration of analgesic response of C57BL/6J-bgJ (beige-J) mice to morphine by carbachol. Eur J Pharmacol. (1987) 141:507–10. doi: 10.1016/0014-2999(87)90575-9
138. Ostlund RE, Tucker RW, Leung JT, Okun N, Williamson JR. The cytoskeleton in Chediak-Higashi syndrome fibroblasts. Blood. (1980) 56:806–11. doi: 10.1182/blood.V56.5.806.bloodjournal565806
139. Frankel FR, Tucker RW, Bruce J, Stenberg R. Fibroblasts and macrophages of mice with the Chediak-Higashi-like syndrome have microtubules and actin cables. J Cell Biol. (1978) 79:401–8. doi: 10.1083/jcb.79.2.401
140. Perou CM, Kaplan J. Chediak-Higashi syndrome is not due to a defect in microtubule-based lysosomal mobility. J Cell Sci. (1993) 106:99–107. doi: 10.1242/jcs.106.1.99
141. Brose N, Petrenko AG, Südhof TC, Jahn R. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. (1992) 256:1021–5. doi: 10.1126/science.1589771
142. Wolfes AC, Dean C. The diversity of synaptotagmin isoforms. Curr Opin Neurobiol. (2020) 63:198–209. doi: 10.1016/j.conb.2020.04.006
143. Martinez I, Chakrabarti S, Hellevik T, Morehead J, Fowler K, Andrews NW. Synaptotagmin VII regulates Ca(2+)-dependent exocytosis of lysosomes in fibroblasts. J Cell Biol. (2000) 148:1141–9. doi: 10.1083/jcb.148.6.1141
144. Czibener C, Sherer NM, Becker SM, Pypaert M, Hui E, Chapman ER, et al. Ca2+ and synaptotagmin VII-dependent delivery of lysosomal membrane to nascent phagosomes. J Cell Biol. (2006) 174:997–1007. doi: 10.1083/jcb.200605004
145. Arantes RME, Andrews NW. A role for synaptotagmin VII-regulated exocytosis of lysosomes in neurite outgrowth from primary sympathetic neurons. J Neurosci. (2006) 26:4630–7. doi: 10.1523/JNEUROSCI.0009-06.2006
146. Wu B, Wei S, Petersen N, Ali Y, Wang X, Bacaj T, et al. Synaptotagmin-7 phosphorylation mediates GLP-1-dependent potentiation of insulin secretion from β-cells. Proc Natl Acad Sci U S A. (2015) 112:9996–10001. doi: 10.1073/pnas.1513004112
147. Davletov BA, Südhof TC. A single C2 domain from synaptotagmin I is sufficient for high affinity Ca2+/phospholipid binding. J Biol Chem. (1993) 268:26386–90. doi: 10.1016/S0021-9258(19)74326-9
148. Dolai S, Xie L, Zhu D, Liang T, Qin T, Xie H, et al. Synaptotagmin-7 functions to replenish insulin granules for exocytosis in human islet β-cells. Diabetes. (2016) 65:1962–76. doi: 10.2337/db15-1436
149. Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. (2001) 106:157–69. doi: 10.1016/S0092-8674(01)00421-4
150. Pennington K, Chan T, Torres M, Andersen J. The dynamic and stress-adaptive signaling hub of 14-3-3: emerging mechanisms of regulation and context-dependent protein–protein interactions. Oncogene. (2018) 37:5587–604. doi: 10.1038/s41388-018-0348-3
151. Ito M, Sato A, Tanabe F, Ishida E, Takami Y, Shigeta S. The thiol proteinase inhibitors improve the abnormal rapid down-regulation of protein kinase C and the impaired natural killer cell activity in (Chediak-Higashi syndrome) beige mouse. Biochem Biophys Res Commun. (1989) 160:433–40. doi: 10.1016/0006-291X(89)92451-0
152. Tanabe F, Cui SH, Ito M. Abnormal down-regulation of PKC is responsible for giant granule formation in fibroblasts from CHS (beige) mice–a thiol proteinase inhibitor, E-64-d, prevents giant granule formation in beige fibroblasts. J Leukoc Biol. (2000) 67:749–55. doi: 10.1002/jlb.67.5.749
153. Sato A, Tanabe F, Ito M, Ishida E, Shigeta S. Thiol proteinase inhibitors reverse the increased protein kinase C down-regulation and concanavalin A cap formation in polymorphonuclear leukocytes from Chediak-Higashi syndrome (Beige) mouse. J Leukoc Biol. (1990) 48:377–81. doi: 10.1002/jlb.48.5.377
154. Möhlig H, Mathieu S, Thon L, Frederiksen MC, Ward DM, Kaplan J, et al. The WD repeat protein FAN regulates lysosome size independent from abnormal downregulation/membrane recruitment of protein kinase C. Exp Cell Res. (2007) 313:2703–18. doi: 10.1016/j.yexcr.2007.04.020
155. Mulhern MS, Stumpel C, Stong N, Brunner HG, Bier L, Lippa N, et al. NBEA: Developmental disease gene with early generalized epilepsy phenotypes. Ann Neurol. (2018) 84:788–95. doi: 10.1002/ana.25350
156. Lo RW, Li L, Leung R, Pluthero FG, Kahr WHA. NBEAL2 (Neurobeachin-like 2) is required for retention of cargo proteins by α-granules during their production by megakaryocytes. Arterioscler Thromb Vasc Biol. (2018) 38:2435–47. doi: 10.1161/ATVBAHA.118.311270
157. Gunay-Aygun M, Falik-Zaccai TC, Vilboux T, Zivony-Elboum Y, Gumruk F, Cetin M, et al. NBEAL2 is mutated in gray platelet syndrome and is required for biogenesis of platelet α-granules. Nat Genet. (2011) 43:732–4. doi: 10.1038/ng.883
158. Karim MA, Nagle DL, Kandil HH, Bürger J, Moore KJ, Spritz RA. Mutations in the Chediak-Higashi syndrome gene (CHS1) indicate requirement for the complete 3801 amino acid CHS protein. Hum Mol Genet. (1997) 6:1087–9. doi: 10.1093/hmg/6.7.1087
159. Ji X, Chang B, Naggert JK, Nishina PM. Lysosomal trafficking regulator (LYST). Adv Exp Med Biol. (2016) 854:745–50. doi: 10.1007/978-3-319-17121-0_99
160. Striegl H, Andrade-Navarro MA, Heinemann U. Armadillo motifs involved in vesicular transport. PloS One. (2010) 5. doi: 10.1371/journal.pone.0008991
161. Peifer M, Berg S, Reynolds AB. A repeating amino acid motif shared by proteins with diverse cellular roles. Cell. (1994) 76:789–91. doi: 10.1016/0092-8674(94)90353-0
162. Gebauer D, Li J, Jogl G, Shen Y, Myszka DG, Tong L. Crystal structure of the PH-BEACH domains of human LRBA/BGL. Biochemistry. (2004) 43:14873–80. doi: 10.1021/bi049498y
163. Ward DMV, Shiflett SL, Huynh D, Vaughn MB, Prestwich G, Kaplan J. Use of expression constructs to dissect the functional domains of the CHS/beige protein: Identification of multiple phenotypes. Traffic. (2003) 4:403–15. doi: 10.1034/j.1600-0854.2003.00093.x
164. Morimoto M, Nicoli ER, Kuptanon C, Roney JC, Serra-Vinardell J, Sharma P, et al. Spectrum of LYST mutations in Chediak-Higashi syndrome: a report of novel variants and a comprehensive review of the literature. J Med Genet. (2023) 61(3):212–23. doi: 10.1136/jmg-2023-109420
165. Shibata S, Asano T, Ogura A, Hashimoto N, Hayakawa JI, Uetsuka K, et al. SCID-bg mice as xenograft recipients. Lab Anim. (1997) 31:163–8. doi: 10.1258/002367797780600107
Keywords: lysosomes, vesicle traffic, Chédiak-Higashi syndrome, cancer, wound healing, immunotherapy, LYST, beige
Citation: Turner ME, Che J, Mirhaidari GJM, Kennedy CC, Blum KM, Rajesh S, Zbinden JC, Breuer CK, Best CA and Barker JC (2024) The lysosomal trafficking regulator “LYST”: an 80-year traffic jam. Front. Immunol. 15:1404846. doi: 10.3389/fimmu.2024.1404846
Received: 21 March 2024; Accepted: 17 April 2024;
Published: 07 May 2024.
Edited by:
Paras K. Anand, Imperial College London, United KingdomReviewed by:
Gael Ménasché, Institut National de la Santé et de la Recherche Médicale, FranceCopyright © 2024 Turner, Che, Mirhaidari, Kennedy, Blum, Rajesh, Zbinden, Breuer, Best and Barker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jenny Barker, amVubnkuYmFya2VyQG5hdGlvbndpZGVjaGlsZHJlbnMub3Jn
†These authors contributed equally to this work and share first authorship
‡These authors contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.