 Fahd Alhamdan
Fahd Alhamdan Ganchimeg Bayarsaikhan
Ganchimeg Bayarsaikhan Koichi Yuki
Koichi Yuki- 1Department of Anesthesiology, Critical Care and Pain Medicine, Cardiac Anesthesia, Boston Children’s Hospital, Boston, MA, United States
- 2Department of Anesthesia and Immunology, Harvard Medical School, Boston, MA, United States
- 3Broad Institute of MIT and Harvard, Cambridge, MA, United States
Immune system recognizes invading microbes at both pathogen and antigen levels. Toll-like receptors (TLRs) play a key role in the first-line defense against pathogens. Major functions of TLRs include cytokine and chemokine production. TLRs share common downstream signaling pathways with other receptors. The crosstalk revolving around TLRs is rather significant and complex, underscoring the intricate nature of immune system. The profiles of produced cytokines and chemokines via TLRs can be affected by other receptors. Integrins are critical heterodimeric adhesion molecules expressed on many different cells. There are studies describing synergetic or inhibitory interplay between TLRs and integrins. Thus, we reviewed the crosstalk between TLRs and integrins. Understanding the nature of the crosstalk could allow us to modulate TLR functions via integrins.
Introduction
Immune cells are mounted with a number of pattern recognition receptors (PRRs) that recognize foreign pathogens. Microbial components are main targets for host immune cells to use for the recognition of microbes, and Toll-like receptors (TLRs) are one of major PRRs and evolutionarily ancient mediators for innate host defense (1, 2). Other PRRs include RIG-I-like receptors (RLRs), Nod-like receptors (NLRs), and C-type lectin receptors (CLRs) (3). So far 10 human TLRs (TLR1-TLR10) and 12 mouse TLRs (TLR1–9, TLR11–13) are identified (4). They are expressed on the plasma membrane or the endocytic vesicles.
Among all the TLRs, TLR4 has been studied most extensively. TLR4 mainly recognizes lipopolysaccharide (LPS) of Gram-negative bacteria (5, 6). To demonstrate its function, TLR4 binds to adaptor protein MD-2 to form TLR4-MD-2 complex (7). TLR4-MD-2 complex binds to LPS, then forms a dimer to activate intracellular signaling cascade. Other TLRs also form dimers (homodimer or heterodimer) to be functional. TLR2 recognizes peptidoglycan, lipopeptide, and lipoprotein of Gram-positive bacteria in concert with TLR1 or TLR6 (8, 9). TLR3 recognizes double-stranded RNA (dsRNA) (10). TLR5 recognizes bacterial flagellin (11). TLR7 and TLR8 recognize single-stranded RNA (ssRNA) (12–14). TLR9 recognizes bacterial and viral CpG DNA motifs (15, 16). The recognition of microbial pathogens by TLRs induces the activation of intracellular signaling pathways, resulting in the production of inflammatory cytokines, type I interferon, and chemokines. TLRs also induce the upregulation of costimulatory molecules on dendritic cells (DCs) (17). TLR10 is the latest human TLR to be discovered, and its ligand is still unclear (18). In contrast to TLR1–9, TLR10 demonstrates anti-inflammatory response (19, 20). While it is known to respond to influenza virus infection (21), this TLR still requires more extensive work in the future.
In addition to recognizing exogenous ligands derived from microbes, TLRs interact with endogenous molecules released from damaged tissues or dead cells (22). For example, high mobility group box 1 (HMGB1) is a nonhistone nuclear protein (23) and can bind to TLR2, TLR4, and TL9. The list of ligands for each TLR is listed in Table 1. The location of each TLR is also shown.
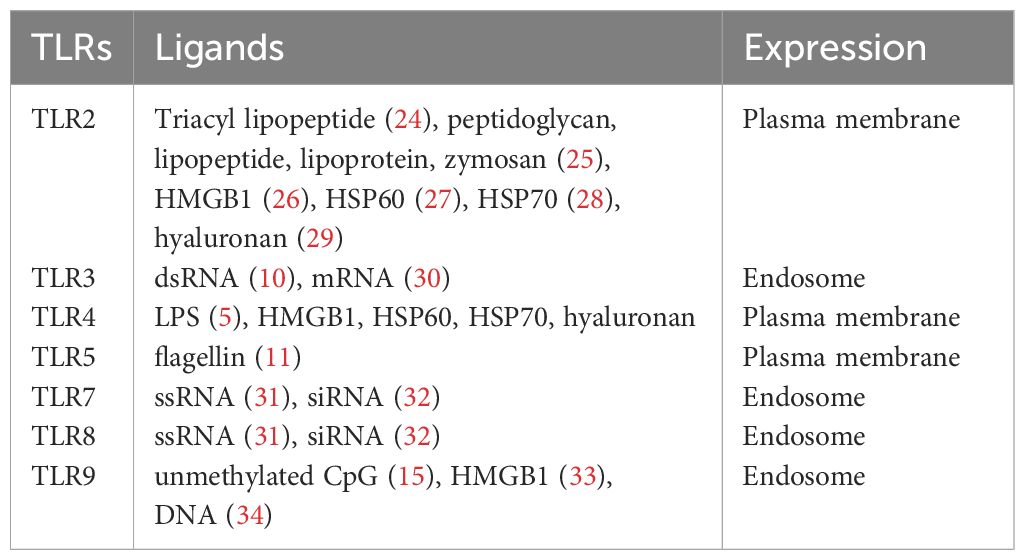
Table 1 List of TLRs, their location and ligands.
The production of pro-inflammatory mediators such as cytokines and chemokines is one of the major TLR functions. Those inflammatory mediators would help regulating the immune system (35). For example, TLR2 and TLR4 are recognized by various ligands (Table 1). However, inflammatory response by different TLR2 ligands may not be the same. The involvement of non TLR receptors can provide a more tailored, specific response to TLRs. Integrins are critical adhesion molecules involved in many biological processes and play an important role in TLR crosstalk. Thus, we will first describe TLR signaling pathways. Then we will examine the role of integrins as regulators of TLR functions.
TLRs signaling pathways
TLRs induce intracellular pro-inflammatory signaling events via myeloid differentiation primary response protein 88 (MyD88) and/or Toll/IL-1R (TIR) domain-containing adaptor inducing interferon (TRIF) (Figure 1). Here we focus on describing pro-inflammatory signaling pathways via MyD88 and/or TIR for TLR1–9. The dimerization of TLRs triggers signaling events.
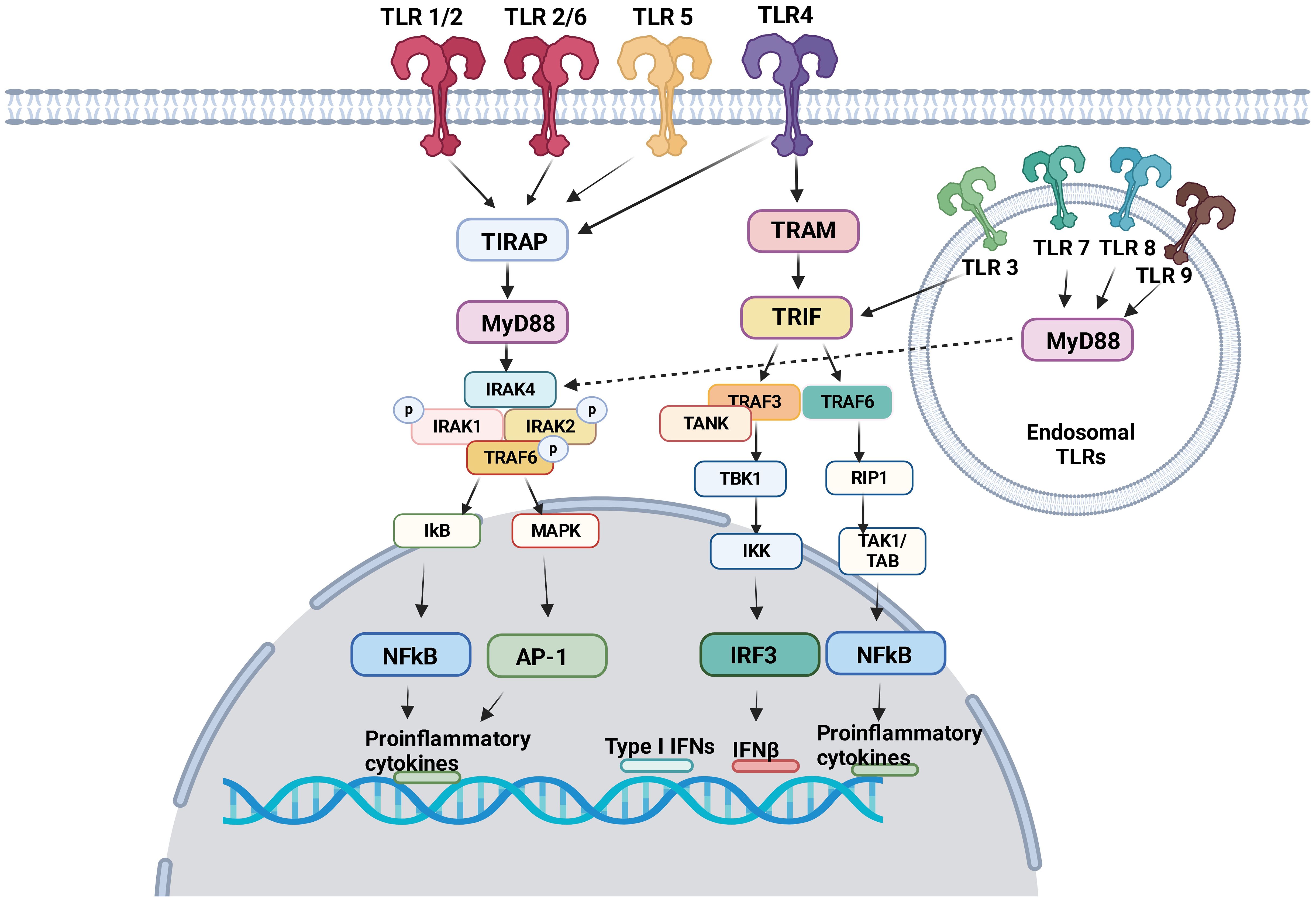
Figure 1 TLR signaling pathway TLR1, TLR2, TLR4, TLR5 and TLR6 are expressed on the cell surface. TLR3, TLR7, TLR8 and TLR9 are expressed in the endosome. Following ligand engagement, TLRs are dimerized, and interact either with MyD88 or TRIF. MyD88 signaling pathways involve NFκB and AP-1, both of which induces pro-inflammatory cytokines. Via TRIF, IRF3 and NFκB induces interferons and pro-inflammatory cytokines, respectively.
MyD88 signaling pathway
TIR domains are essential components of the innate immune system (36). The proximal events of TLR-mediated intracellular signaling are initiated by the interaction of TIR-domain of TLRs with TIR-domain-containing cytosolic adaptors and MyD88 is a central adaptor protein for TLRs. With the exception of TLR3, all TLRs mediate the downstream signaling pathway via MyD88 (37). The association of TLRs with MyD88 recruits the members of the interleukin-1 receptor associated kinase (IRAK) family, forming MyD88-IRAK-4 complex. This recruits IRAK-1 and IRAK-2, leading to the phosphorylation of IRAKs and interaction with tumor necrosis factor receptor associated factor 6 (TRAF6). TRAF6 induces the activation of transforming growth factor-β activated kinase 1 (TAK-1), thereby I-κB (IκB) and mitogen-activated protein kinase (MAPK). The activation of IκB and MAPK results in nuclear factor kappa B (NF-κB) and activator protein 1 (AP-1)-mediated gene transcription (38, 39). IRAK activation also stimulates interferon-regulatory factor (IRF) such as IRF7 (40–42) and activates the gene transcription of type I IFN (43). As a result, pro-inflammatory cytokines including tumor necrosis factor (TNF), interleukin (IL)-1, IL-6, IL-12, and interferon (IFN)-α are produced (44).
TRIF signaling pathway
TRIF was identified as MyD88-independent pathway (alternative pathway). TRIF is recruited to TLR3 and TLR4. TRAF activation recruits TRAF6 and TRAF3. TRAF6 recruits receptor interacting protein 1 (RIP1). The subsequent activation of TGF-β-activated kinase 1 (TAK1)/TAK1-binding proteins (TABs) leads to the activation of NFκB and IFN-β promoter (45) to express pro-inflammatory cytokines and type I interferons. TRIF also activates TANK-binding kinase 1 (TBK1) and inhibitor of NF-κB kinase (IKK). Subsequently interferon regulatory factor 3 (IRF3) is activated and negatively regulates the activation of NF-κB and IFN-β promoter (46).
Integrins
Integrins are α/β heterodimeric cell adhesion molecules that mediate cell-to-cell and cell-to-matrix interactions (47, 48). They are type I membrane glycoproteins with large extracellular domains, single transmembrane domains, and relatively short intracellular tails. The head of the large extracellular domain serves for ligand binding. To date, 18 α- and 8 β-subunits have been identified that combine to form at least 24 distinct α/β heterodimers. The list of integrins with representative ligands is included in Table 2. Integrins on the membrane (the outside) can receive signals triggered by non-integrin receptors via intracellular signaling (inside-out signaling) and vice versa (outside-in signaling) (Figure 2) (52).
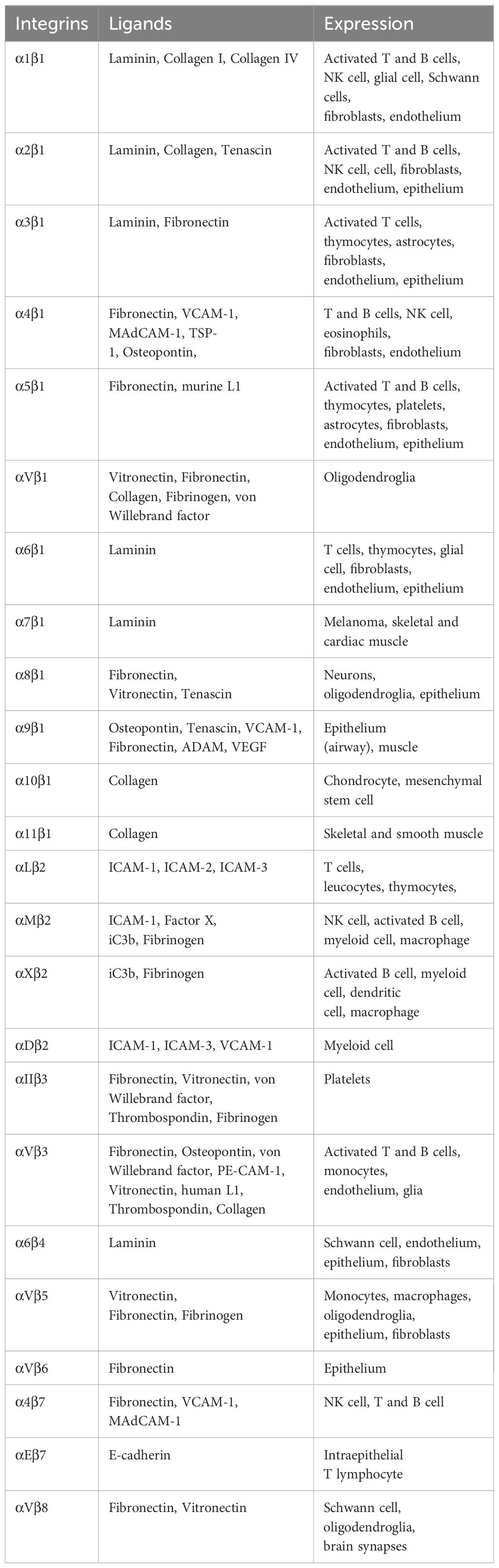
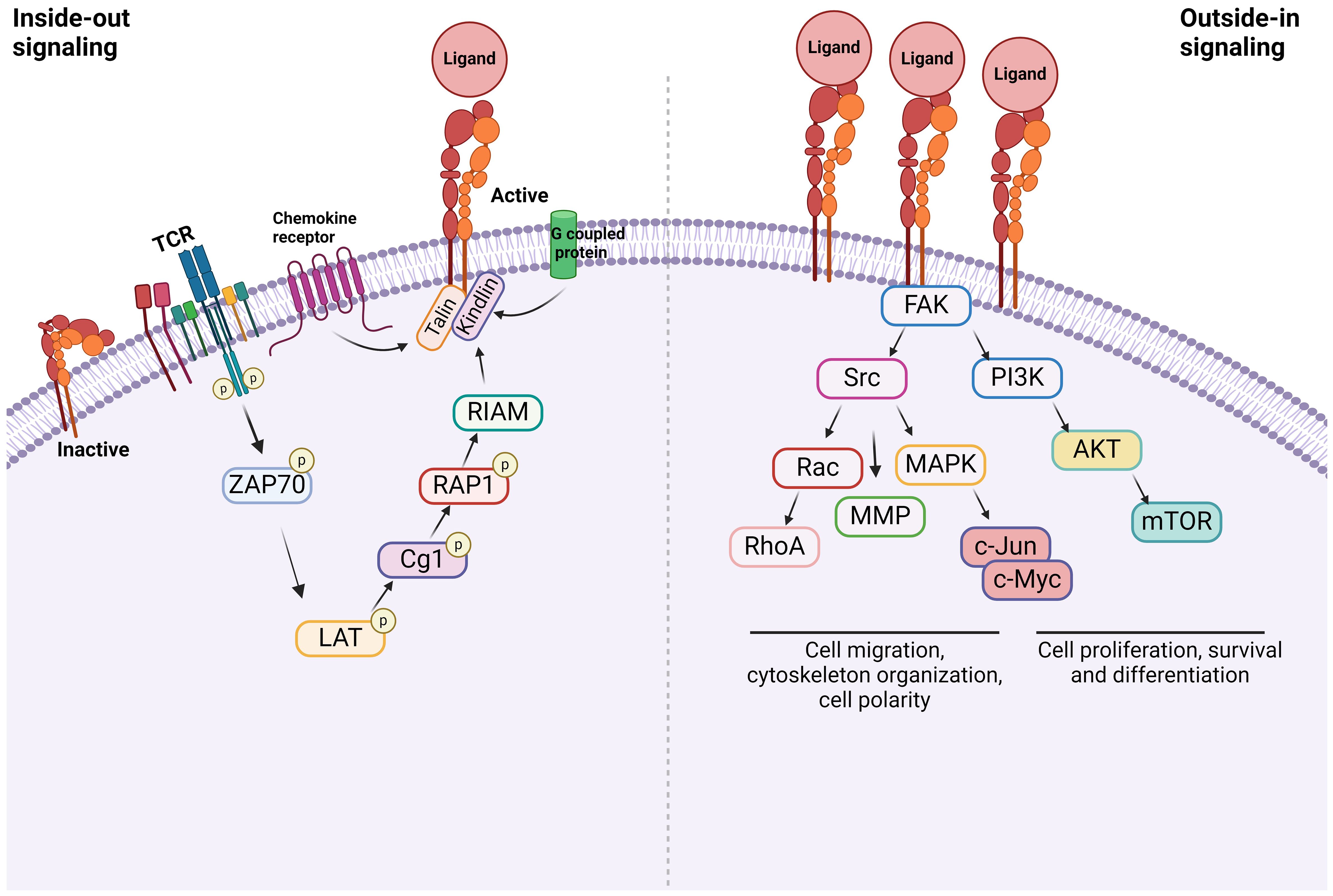
Figure 2 Integrin signaling Inside-out signal: Integrins are in an inactive conformation at baseline. However, the activation of receptors such as GPCRs, chemokine receptors, and TCR induces a cascade of events within the cells. The example shown here is via TCR. At the end, talin along with kindlin bind to β subunit of integrins, inducing its conformational change, which triggers the structural change of α subunit, allowing the integrin to bind to its ligand. Outside-in signal: Integrins that bind to their ligands cause cytoskeletal changes via focal adhesion molecules including focal adhesion kinase (FAK), leading to cell proliferation, survival, differentiation, and migration.
Inside-out signal
Inside-out signal is initiated by non-integrin receptors such as G protein-coupled receptors (GPCRs), selectins, and chemokine receptors (49, 53, 54). Their signals are transmitted to activate integrins. Integrin αLβ2 was extensively studied on inside-out signal in the setting of T cells (55, 56). The activation of T cell receptor (TCR) and tyrosine kinase Lck leads to the phosphorylation of ZAP-70 kinase. This triggers the phosphorylation of LAT adaptor protein and the activation of phospholipase Cg1. This ultimately activates the small G protein ras-related protein-1 (Rap1). Rap1 binds to Rap-1 interacting adaptor molecule (RIAM). These events trigger the binding of talin and kindlin to β2 subunit, which induces the conformational change of αLβ2 into its active form (57). Although the binding of talin alone to integrin can activate it, its potency is extremely weak, supporting the critical role of kindlin in this process (58). The activation of αLβ2 results in its binding with ligands including intercellular adhesion molecule-1 (ICAM-1). What kind of molecules each integrin uses for inside-out signal and whether the same integrin uses different combination of molecules depending on cell type would be an important research area.
Outside-in signal
Upon the inside-out activation, an integrin binds to a specific ligand. However, for the integrin to tightly bind to its ligand to mediate cell adhesion and migration, its cytoplasmic domains must be anchored to the cytoskeleton (59, 60). When the integrin binds to its ligand, it triggers the assembly of large protein complexes known as focal adhesions by incorporating a variety of molecules including cytoskeletal proteins and signaling molecules. Linking the integrin to the actin cytoskeleton promotes firm cell adhesion, cell spreading, migration and proliferation (57). Talin and kindlin serve as seed proteins to recruit proteins and initiate focal adhesion assembly (61). In case of αLβ2, the binding of ICAM-1 induces the activation/deactivation of kinases and phosphatases, leading to the cytoskeletal remodeling for the fine-tuning of effector functions such as T cell migration (62). Interestingly, this outside-in signal can be modified by the heterotrimeric guanine nucleotide-binding protein (G protein) Gα13. GPCRs activate Gα13, triggering its interaction with β integrin to regulate the outside-in signal (53).
Integrin-TLR crosstalk
β1 integrin
β1 integrin receptors regulate numerous functions, including cell adhesion, migration, differentiation, growth, and survival. β1 integrin subfamily consists of 12 α-chains that non-covalently bind to β1 chain (CD29) (49, 63). They can be categorized by their binding characteristics such as Arg-Gly-Asp (RGD)-binding integrins (αvβ1, α8β1, and α5β1), Leu-Asp-Val (LDV)-binding integrins (α4β1 and α9β1), collagen-binding integrins through triple helical GFOGER sequence in major collagens (α1β1, α2β1, α10β1, and α11β1), and laminin-binding integrins which includes both non-α Inserted (I) domain-containing integrins (α3β1, α6β1 and α7β1) and αI domain-containing integrins (α10β1, α2β1, and α1β1) (64). The key downstream signaling molecules of integrins include focal adhesion kinase (FAK), AKT, MAPK, Src-family protein tyrosine kinases, and integrin-linked kinase (ILK) (65). Integrins regulate intracellular signal transduction cascades that control differentiation, proliferation, and survival. Upon binding to fibronectin, collagen, and laminin, β1 integrin induces cell adhesion and migration that is extensively observed in pancreatic cancer models. Blockade or knockdown of β1 on cancer cells resulted better prognosis by reducing tumor growth and metastasis (66), which makes β1 integrin as an attractive therapeutic target. β1 integrins, in particular α9β1 has been reported to induce Th17 cell promoting cytokines in dendritic cells and macrophages in synergy with TLR2 and TLR4 through ERK pathway, that developed functional Th17 cells and arthritis (67). In addition to that, upon engaging with extracellular matrix (ECM) or other ligands, they initiate signaling pathways that can either reinforce or inhibit the activity of other receptors through negative or positive feedback loops. Interactions of α3β1 and α4β1 with TLRs have reported in several studies, which will be discussed in the following sections.
i. α3β1
α3β1 is expressed extensively on nearly all types of cells. It binds to a wide range of ligands with or without classical RGD integrin-binding motifs (68). α3β1 integrin serves as a receptor for collagen (type I and VI), laminin (α1β1γ1), laminin-5 (α3β3γ2), laminin-10 (α5β1γ1), laminin-11 (α5β2γ1), fibronectin, entactin, nidegon, and thrombospondin-1 with high specificities and affinities (69). Integrins are often targeted by bacterial and viral pathogens to adhere to and invade host cells. β1 integrins are particularly prone to their targets (70). β1 integrins serve as receptors for bacterial surface proteins including invasin and FimH (71, 72). α3β1 binds to BBB07 expressed on Borrelia burgdorferi (B. burgdorferi), the causative microbe of Lyme disease (73). BBB07 also serves as a TLR2 ligand. By ligation to the same ligand by both α3β1 and TLR2/1, human macrophages manifested enhanced pro-inflammatory responses to bacterial components.
α3β1 also mediates the endocytosis of TLR2 ligand Pam3CSK4, thereby facilitating its recognition by TLR2/1 within the endosome (74). This leads to the recruitment of adaptor molecules such as MyD88 by TLR2/1, eventually activating NF-κB signaling pathway and inducing the production of pro-inflammatory cytokines such as IL-6 (56). In murine macrophages, the endosomal activation of TLR2/1 induces IFN-β (75). This endocytosis mediated by α3β1 was observed for both live bacteria and bacterial proteins.
The impact on TLR2 mediated signaling via α3β1 is cell type-dependent (76). As in macrophages, α3β1 and TLR2/1 crosstalk selectively enhances IL-6 and IL-10 production by neutrophils in the setting of sepsis. However, neutrophils do not produce TNF production. Activated neutrophils release laminin (77) which bind to α3β1 on their cell surface, and increase the phosphorylation of FAK, but not Syk. This is responsible for the aforementioned profiles of pro-inflammatory cytokines by neutrophils (78). Activated FAK feeds into the MyD88-dependent TLR signaling. It is not certain about the presence of direct interaction between α3β1 and TLR2/1 on the neutrophils, but it is suggested that they may interact transiently within the lipid rafts upon activation since both of them localize there during activation (79, 80).
ii. α4β1
α4β1, also referred to as very late antigen-4 (VLA-4), is expressed on most leukocytes. It plays a crucial role in cell homing, trafficking, differentiation, activation, and survival. The ligands of this receptor include ECM protein fibronectin and the vascular cell adhesion molecule-1 (VCAM-1), which are expressed on endothelial cells (81). α4β1 binding site to fibronectin contains the tripeptide sequence Leu-Asp-Val (LDV) and is located in the alternatively spliced connecting segment 1 (CS-1) region, while VCAM-1 is recognized through the sequence Ile-Asp-Ser (IDS) (82). The domain called extra domain A (EDA) within fibronectin activates TLR4 (83). Thus, fibronectin severs as a ligand for both α4β1 and TLR4 (84). α4β1 was shown to function as a co-receptor for TLR4 in fibroblasts. Blockade of α4β1 or TLR4 or knockdown of α4 subunit in fibroblasts resulted in a decreased production of pro-inflammatory cytokines such as TNF and IL-10 (85).
β2 integrin
β2 integrins consist of four members- αLβ2 (CD11a/CD18, lymphocyte function-associated antigen-1), αMβ2 (CD11b/CD18, macrophage-1 antigen, complement receptor 3), αXβ2 (CD11c/CD18, p150.95, complement receptor 4), and αDβ2 (CD11d/CD18). αLβ2 is ubiquitously expressed on all leukocytes, while αMβ2, αXβ2, and αDβ2 are mainly expressed on myeloid cells at different levels (86). αLβ2 binds to intercellular adhesion molecule (ICAM)-1~5 that can be found on the surface of other cells. αMβ2 has broad versatility, having over 40 known binding partners, such as ICAMs, iC3b, fibrinogen, RAGE (receptor for advanced glycation end products), and CD40L (87). αMβ2 and αXβ2 share several ligands as including iC3b, ICAM-1 and fibrinogen, but their binding sites on the same ligand are not exactly the same (88). αDβ2 also binds to multiple ligands, encompassing extracellular matrix-associated proteins like fibronectin, fibrinogen, vitronectin, and plasminogen as well as ICAM-1 (89). Reactive oxygen species (ROS) produced by the ligation of TLR2 and TLR5 induced rapid β2-integrin activation on myelomonocytes, and promoted leukocyte adhesion, suggesting that TLRs collaborate with one another (90). CD18 (β2) knockout (KO) macrophages and DCs produced higher level of IL-12p40 and IL-6 in response to TLR2, TLR4 and TLR9 stimulation, and higher level of type I interferon in response to TLR4 stimulation (91), suggesting that β2 integrins modulate TLR response. Further investigation of β2 ablation showed NF-κB and p38 MAPK pathway activations were involved in these processes (91, 92). Among β2 integrins, the interplay between αMβ2 and TLRs is well studied, which will be discussed further.
i. αMβ2
αMβ2 is highly expressed on macrophages, DCs, monocytes, granulocytes, and mature or activated NK cells (93). It regulates TLR signaling positively or negatively, depending on cell types and inflammatory status.
TLR4 KO neutrophils reduced αMβ2 activation, but not αLβ2 or αXβ2, suggesting that TLR4 would selectively facilitate the activation of αMβ2 on neutrophils. TLR4-mediated αMβ2 induction involved the activation of transcription factors NF-κB and c-Jun (94). αMβ2 can affect several TLRs. Upon in vivo challenge with TLR ligand stimulations (LPS, poly(I:C), and CpG) pro-inflammatory cytokines (TNF, IL-6, IL-10, and IFN-β) were greatly increased in the serum of CD11b (αM) KO mice (39). Higher level of pro-inflammatory cytokines in the serum was observed in CD11b KO mice during methicillin-resistant Staphylococcus aureus (MRSA) (95) and Escherichia coli (E.coli)<i> (96) </i>infection. Bacterial loads were higher in CD11b KO mice following MRSA and E. coli infections. In contrast, CD11b KO mice demonstrated better clearance of L. monocytogenes following its infection, despite higher serum TNF and IL-6 levels were detected (95). The difference in the phenotype may be because TNF induces apoptosis of certain bacteria (97). In case of MRSA and E.coli infection, TLR4 ligation activated αMβ2 on macrophages by inside-out signaling through PI3K and RapL pathway, which negatively looped back TLR4 signaling (Figure 3) (96). Outside-in signaling activated Src-Syk and promoted degradation of MyD88 and TRIF (Figure 3). This feedback loop in macrophages may control balance of both TLR4 and αMβ2 signaling pathways since their uncontrolled activation can cause harmful pathogenesis. Of note, syk is typically associated with other receptors such as C-type lectin receptors (CLRs). To make complicated further, resident macrophages or bone marrow derived macrophages from CD11b KO mice showed similar level of pro-inflammatory cytokines and activation status upon LPS stimulation, thus suggesting that the interplay of αMβ2 with TLR4 was not involved in steady-state macrophages (96). Thus, the interplay between TLRs and αMβ2 may be dictated by cell types and their cellular state. In fact, the lack of αMβ2 in DCs resulted in decreased pro-inflammatory cytokines and reduced MyD88-dependent phosphorylation of p38, Erk1/2, JNK, and IκBα in response to LPS stimulation (96). Upon stimulation with LPS, αMβ2 was clustered in DCs and co-localized with CD14, which has been shown important for TLR4 endocytosis, suggesting that αMβ2 was a part of TLR4 endocytosis. Furthermore, CD11b KO in DCs impaired RANTES production in LPS induced TRIF–mediated signaling in the endosome (44). Unlike TLR4, αMβ2 in DCs negatively regulated TLR9 signaling by selectively reducing IL-12p70 production, which was possibly regulated by upregulated miR-146. The consequence of IL-12p70 production affected poor cross-priming of DCs to cytotoxic T lymphocyte (CTL) response (98). TLR3 and αMβ2 interplay has been reported on NK cells. KO and neutralization of αMβ2 enhanced cytotoxic function of NK cells in response to TLR3 stimulation and limited acute liver infection (93). αMβ2 deficiency impaired the activation of MAPK/JNK pathway, suggesting that it inhibited TLR3 mediated activation of NK cells (99). Inside-out activation of αMβ2 by TLR2 in association with CD14 was reported in monocytes during the infection of Porphyromonas gingivalis, a pathogen implicated in chronic periodontitis and atherosclerosis. The activation of αMβ2 induced adhesion and recruitment of monocytes to the site of the infection (100). This recruited inflammatory monocytes can be beneficial to control infection, but uncontrolled accumulation results a tissue destruction. Although current data are all based on either αMβ2 or TLR KO system or depletion by neutralizing antibodies, the studies suggested a possible indirect interplay between αMβ2 and TLR2 (100).
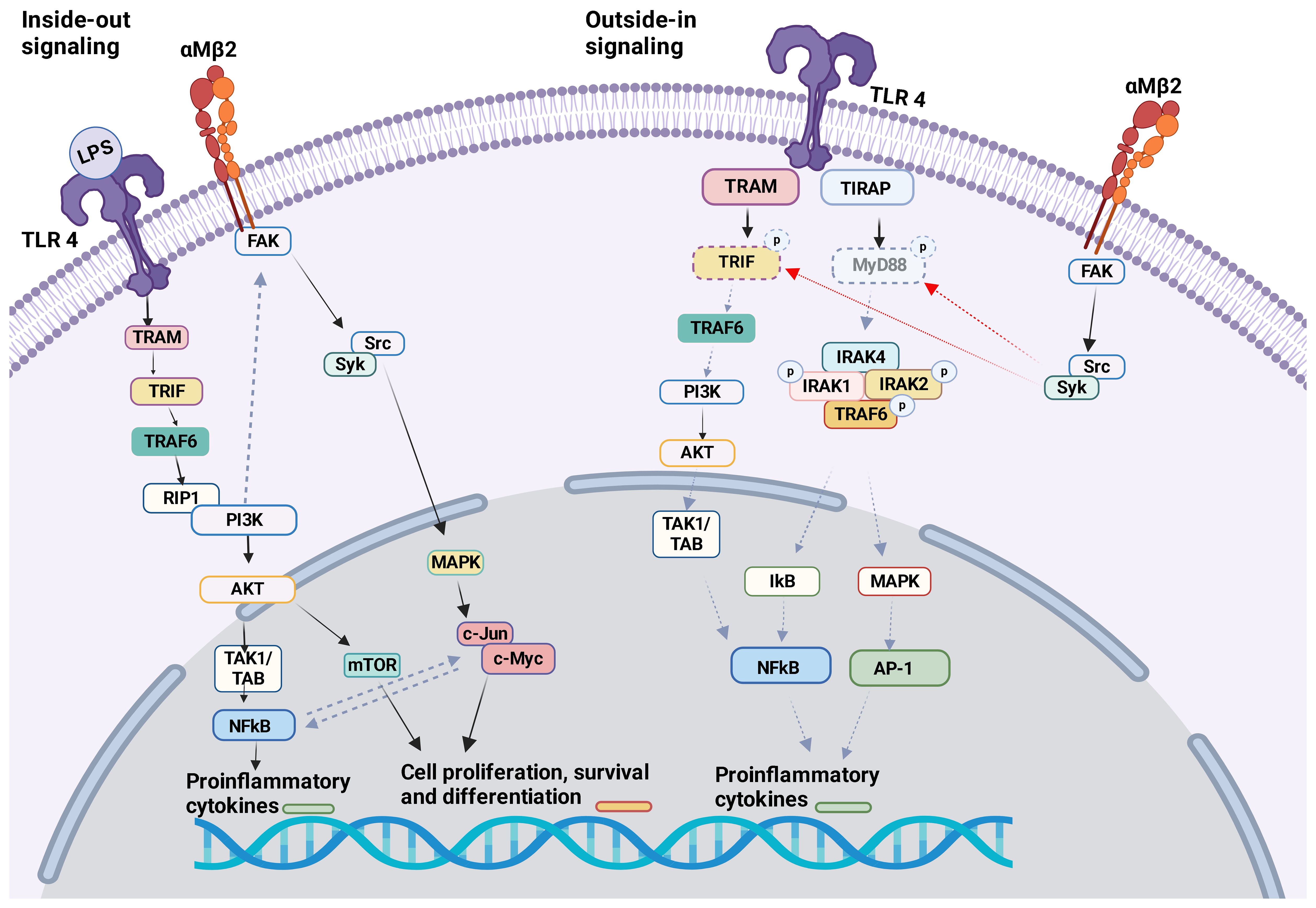
Figure 3 αMβ2 and TLR4 crosstalk The crosstalk between αMβ2 and TLR4 is shown.Inside-out signal: LPS binding to TLR4 induces the activation of many molecules including PI3K. PI3K facilitates the activation of αMβ2 intracellular adaptor proteins, therefore αMβ2 itself. Outside-in signal: Activated αMβ2 communicates with Src/Syk, which facilitates the degradation of MyD88 and TRIF. This will attenuate TLR4 activation signal. Of note, Syk is typically associated with other receptors like C-type lectin receptors (CLRs).
While most studies examined the interaction between TLRs and αMβ2 by inhibiting or deleting αMβ2, some studied by activating it. αMβ2 activation by leukadherin-1 (LA1), its allosteric agonist, protected mice from pathological injuries and reduced the mortality induced by LPS (101). αMβ2 activation by LA-1 inhibited M1 macrophage response to LPS both in vivo and in vitro. Although it is not clear whether LA-1 facilitated a direct interaction between αMβ2 and TLR4 on macrophages, it induced an endocytosis of both αMβ2 and TLR4 and prevented LPS binding to TLR4 (101). While it prevented an excessive activation of TLR4 signaling pathway and pro-inflammatory response in macrophages, LA-1 pretreatment induced pro-inflammatory cytokines in DCs, suggesting that the effect of LA-1 could be cell type-dependent (101). It is worth noting that the expression levels of αMβ2 on macrophages and DCs are different (102), which may be in part responsible for the different effect of LA-1 on these two cell types.
A recent study showed that CD11b deficiency of donor non-classical monocytes increased CXCL2 production and exacerbated primary graft dysfunction in lung transplantation model (103). High mobility group box 1 (HMGB1), a DAMP released from dying cells, activates TLR4 in nonclassical monocytes. It was released from the donor lungs with primary graft dysfunction. Interestingly, HMGB1 stimulation induced lower CXCR2 production by TLR4 single KO or TLR2/TLR4 double KO monocytes, but not TLR2 single KO. It is uncertain whether αMβ2 interacts directly with TLR2 or TLR4, however, αMβ2 agonist LA-1 prevented primary graft dysfunction, suggesting that αMβ2 might facilitate TLR endocytosis.
Although all the evidence supports the presence of crosstalk between αMβ2 and TLRs, several questions still need to be answered. Do αMβ2 and TLRs bind to the same ligands? In fact, several studies reported that αMβ2 binds viral dsRNA (104), and bacterial toxins (105) and LPS (106–108). So far the interaction between ligands and αMβ2 has been shown in vitro. Therefore, it will be critical to determine if reported ligands for αMβ2 are in fact relevant in vivo. If so, it is not known whether both TLRs and αMβ2 bind a ligand at the same time. If they do, which signaling should be activated first? Does ligand binding avidity and affinity affect downstream signal? Or do they limit activation? If they don’t, is there ligand binding competition between TLR and αMβ2? Would it be possible that the rest of β2 integrin members interplay with TLRs? This may be very possible since extracellular part of αXβ2 has over 80% of sequence homology to αMβ2 and about 50% of homology in intracellular tail, for example (109, 110). Furthermore, LPS also binds to caspase-11 intracellularly (111), which makes the crosstalk complicated. β2 integrin members may also collaborate each other to inhibit or induce TLR response, since β2 integrin members are expressed upon activation of cells, especially on myeloid cells. They may function synergistically. Apparently, the proposed crosstalk between αMβ2 and TLRs depend on cell types, but does ligand binding affinity or avidity affect crosstalk? For example, high-affinity ligand binding affects the degree of up- or downstream signal? There might be a rivalry between the ligands. It is interesting to know whether these crosstalks depend on the timing of activation or not. A previous study showed αMβ2 on dendritic cells was activated through inside-out signaling by TLR4 (112) that was necessary for αMβ2-induced phagocytosis but not affected αXβ2, suggesting a bidirectional action between αMβ2 and TLR4.
αV integrin
αV integrin also known as CD51 or MSK8, is a transmembrane protein that is involved in cell adhesion, migration, and signaling (113). αV integrin forms heterodimers with various β integrin subunits such as β1, β3, β5, β6, and β8. Together they designate a various array of receptors to bind to specific ligands in the extracellular matrix (ECM) including fibronectin, vitronectin, fibrinogen, and osteopontin, enabling cells to adhere and respond to their surrounding environment (114, 115).
In addition to adhesion, αV integrin promotes the activation of a multitude of signaling pathways, primarily the FAK pathway (116). The phosphorylation of FAK will in turn recruit Src kinases, phosphoinositide 3-kinase (PI3K) subunit p85, or phospholipase (PL)Cγ and stimulate the signaling cascades of Ras/Erk, PI3k/Akt, and Crk/Dock180/Rac. These pathways contribute to cell survival, proliferation, differentiation, and migration, emphasizing the multifaceted role of integrin αV.
The significance of αV integrin is not only linked with normal physiological functions. Dysregulation of αV integrin function has been associated with a variety of pathological conditions, including cancer, metastasis, angiogenesis, and wound healing (115). Additionally, αV integrin is involved in vascular remodeling and fibrosis (117).
i. αVβ3
αVβ3 is a multifaceted integrin due to its expression on a plethora of cell types and its ability to bind to many extracellular ligands. Through recognizing Arg-Gly-Asp (RGD) motif, αVβ3 binds to extracellular matrix proteins such as vitronectin, fibronectin, fibrinogen, and von Willebrand factor (48, 118). It can also serve as a receptor of some viruses for their entry into target cells (119). The αVβ3-TLRs cooperation has been described in several studies; Plasma membrane TLR4, TLR5 and endosomal TLR3 activated epithelial cells via NF-kB signaling pathway in response to viral and bacterial pathogen-associated molecular pattern molecules (PAMPs) (120). αVβ3 further enhanced their NF-κB activation. αVβ3 also positively orchestrated TLR2 signaling by facilitating a recruitment of the adaptor MyD88 to TLR2 (121). This mechanism was driven by a physical interaction of both αVβ3 and TLR2 with herpes simplex virus (HSV). This leads to NF-κB activation and the production of various mediators including IFN-α, IFN-β, IL-2, and IL-10 in response to the viral infection. Another type of αVβ3-TLR2 interplay has been attested in a different study, in which αVβ3 was shown serve as a co-sensor for bacterial lipopeptide (BLP) to be detected by TLR2 (122). The molecular mechanism mediating TLR2 activation was through the recognition of BLP by vitronectin on human monocytes. The TLR2-αVβ3 complex interaction was entirely dissociated following the completion of BLP stimulation. This further confirmed the physical link between αVβ3 and TLR2 in recognizing invading pathogens and initiating a synergistic response. The collaboration between αVβ3 and TLRs was also described in bacterial infection. In a murine cecal ligation and puncture (CLP)-induced sepsis and in a LPS-stimulated macrophage cell model, αVβ3 positively regulated TLR4 signaling in peritoneal macrophages (123). The deficiency of αVβ3 attenuated TLR4 activation. This effect appears to be mediated by CD14 expression, as αVβ3 deficiency inhibited CD14 expression. The deleterious impact of the αVβ3 -CD14-TLR4 crosstalk was caused by the release of a variety of pro-inflammatory cytokines. Therefore, CD61 (β3) KO mice exhibited higher survival rates and were more resistant to septic organ injury. A similar study revealed that thw previous crosstalk was mediated by WNT1 inducible secreted protein 1 (WISP1) (124). Ligation of WISP1 to αVβ3 synergistically enhanced TLR4-mediated TNF synthesis in LPS treated peritoneal macrophage.
ii. αVβ5
Similar to αVβ3, αVβ5 serves as a receptor for vitronectin (125). αVβ5 mediates phagocytosis of apoptotic cells and promotes angiogenesis and wound healing (126). The interaction of αVβ5 with TLR4 during infection was illustrated in a murine two hit-model of CLP and mechanical ventilation (MV)-induced lung injury (127). TLR4 KO mice showed better survival and less lung injury compared to wild type (WT) mice. αVβ5 regulated vascular permeability in both ventilator-induced lung injury (VILI) (128) and CLP (129). In line with this knowledge, neutralizing antibodies against αVβ5 partially attenuated lung injury. In this model, peritoneal macrophages increased the expression of αVβ5 in response to TLR4 activation. The connection between αVβ5 and TLR4 contributed to the exacerbations of the CLP-MV lung injury model.
iii. αVβ6
αVβ6 is expressed mainly on epithelial cells and involved in wound healing (130). Excessive production of αVβ6 leads to lung fibrosis and cancer (131). Activation of transforming growth factor-β1 (TGF-β1) represents the key role of αVβ6 (131, 132). In line with this, influenza infection stimulated TLR3 and further induced αVβ6-dependent TGF-β1 activation in epithelial cells (132). TLR3- αVβ6 crosstalk converged on the RhoA kinase that was activated by TLR3. RhoA kinase was further required to activate TGF-β1 via αVβ6. This suggests that the crosstalk was through a signaling pathway rather than a direct physical interaction between TLR3 and αVβ6. Blocking αVβ6 seemed to have no effect on the viral entry to the epithelial cells or the replication of viral genes. The biological consequences of TGF-β1 activation via αVβ6-TLR3 axis were epithelial cell death and accumulation of collagen in mouse lungs, which in turn promoted fibrosis. Another adverse effect of αVβ6 during influenza infection of lung epithelium was the suppression of type I IFN response (133). The IFN antiviral response was mainly mediated by endosomal TLR7. αVβ6 activated lysosomal autophagy machinery to remove TLR7, leading to the suppression of TLR7-mediated IFN signaling against Influenza infection. Opposite to αVβ3, αVβ6 seemed to have no physical interaction with TLRs.
Conclusion
Without doubt, TLRs regulate major signaling pathways to modulate the degree of inflammation. While TLRs crosstalk is not exclusively restricted to integrins as complement system has been shown to intercommunicate with TLRs in the host immunity during infection (134), we highlighted ones involving integrins here. As there are a number of signaling pathways to regulate inflammation, it is not surprising that crosstalk system involving integrins has been established to coordinate inflammatory responses as we examined (135). Underhill has proposed several possibilities why the crosstalk has evolved; 1) To provide robust response against invading microbes. 2) Compensation against genetic diversity in host population, 3) Multiple receptors can facilitate a more tailored, specific response (136). The idea of “a more tailored, specific response” is very fascinating, because innate immune cells, which usually express TLRs predominantly, are rather considered promiscuous and relatively non-specific compared to adaptive immunity. Further understanding the role of crosstalks between TLRs and integrins would allow us to understand very complex system that innate immunity has developed and intervene if indicated.
Author contributions
FA: Writing – original draft. GB: Writing – original draft. KY: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work is in part supported by R01GM148392 (KY).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Leulier F, Lemaitre B. Toll-like receptors–taking an evolutionary approach. Nat Rev Genet. (2008) 9:165–78. doi: 10.1038/nrg2303
2. Yuki K, Koutsogiannaki S. Pattern recognition receptors as therapeutic targets for bacterial, viral and fungal sepsis. Int Immunopharmacol. (2021) 98:107909. doi: 10.1016/j.intimp.2021.107909
3. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
4. Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. (2005) 17:1–14. doi: 10.1093/intimm/dxh186
5. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. (1998) 282:2085–8. doi: 10.1126/science.282.5396.2085
6. Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. (1999) 162:3749–52. doi: 10.4049/jimmunol.162.7.3749
7. Okuno T, Koutsogiannaki S, Hou L, Bu W, Ohto U, Eckenhoff RG, et al. Volatile anesthetics isoflurane and sevoflurane directly target and attenuate Toll-like receptor 4 system. FASEB J. (2019) 33:14528–41. doi: 10.1096/fj.201901570R
8. Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. (1999) 11:443–51. doi: 10.1016/s1074–7613(00)80119–3
9. Takeuchi O, Kawai T, Mühlradt PF, Morr M, Radolf JD, Zychlinsky A, et al. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol. (2001) 13:933–40. doi: 10.1093/intimm/13.7.933
10. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. (2001) 413:732–8. doi: 10.1038/35099560
11. Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. (2001) 410:1099–103. doi: 10.1038/35074106
12. Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. . Nat Immunol. (2002) 3:196–200. doi: 10.1038/ni758
13. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. (2004) 303:1529–31. doi: 10.1126/science.1093616
14. Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol. (2002) 3:499. doi: 10.1038/ni0602–499
15. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. (2000) 408:740–5. doi: 10.1038/35047123
16. Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, et al. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. (2004) 21:107–19. doi: 10.1016/j.immuni.2004.06.007
17. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. (2004) 5:987–95. doi: 10.1038/ni1112
18. Fore F, Budipranama M, Destiawan RA. TLR10 and its role in immunity. Handb Exp Pharmacol. (2022) 276:161–74. doi: 10.1007/164_2021_541
19. Mourits VP, Arts RJW, Novakovic B, Matzaraki V, de Bree LCJ, Koeken VACM, et al. The role of Toll-like receptor 10 in modulation of trained immunity. Immunology. (2020) 159:289–97. doi: 10.1111/imm.13145
20. Jiang S, Li X, Hess NJ, Guan Y, Tapping RI. TLR10 is a negative regulator of both myD88-dependent and -independent TLR signaling. J Immunol. (2016) 196:3834–41. doi: 10.4049/jimmunol.1502599
21. Lee SM, Kok KH, Jaume M, Cheung TK, Yip TF, Lai JC, et al. Toll-like receptor 10 is involved in induction of innate immune responses to influenza virus infection. Proc Natl Acad Sci U.S.A. (2014) 111:3793–8. doi: 10.1073/pnas.1324266111
22. Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med. (2010) 14:2592–603. doi: 10.1111/j.1582-4934.2010.01127.x
23. Chen R, Kang R, Tang D. The mechanism of HMGB1 secretion and release. Exp Mol Med. (2022) 54:91–102. doi: 10.1038/s12276-022-00736-w
24. Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, et al. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. (2002) 169:10–4. doi: 10.4049/jimmunol.169.1.10
25. Lee BL, Barton GM. Trafficking of endosomal Toll-like receptors. Trends Cell Biol. (2014) 24:360–9. doi: 10.1016/j.tcb.2013.12.002
26. Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PloS Med. (2009) 6:e10. doi: 10.1371/journal.pmed.1000010
27. Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Häcker H, et al. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem. (2001) 276:31332–9. doi: 10.1074/jbc.M103217200
28. Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. (2002) 277:15028–34. doi: 10.1074/jbc.M200497200
29. Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. (2005) 11:1173–9. doi: 10.1038/nm1315
30. Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. (2004) 279:12542–50. doi: 10.1074/jbc.M310175200
31. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. (2004) 303:1526–9. doi: 10.1126/science.1093620
32. Sioud M. Innate sensing of self and non-self RNAs by Toll-like receptors. Trends Mol Med. (2006) 12:167–76. doi: 10.1016/j.molmed.2006.02.004
33. Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. (2007) 8:487–96. doi: 10.1038/ni1457
34. Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A. Activation of autoreactive B cells by CpG dsDNA. Immunity. (2003) 19:837–47. doi: 10.1016/s1074–7613(03)00323–6
35. Duan T, Du Y, Xing C, Wang HY, Wang RF. Toll-like receptor signaling and its role in cell-mediated immunity. Front Immunol. (2022) 13:812774. doi: 10.3389/fimmu.2022.812774
36. Nimma S, Gu W, Maruta N, Li Y, Pan M, Saikot FK, et al. Structural evolution of TIR-domain signalosomes. Front Immunol. (2021) 12:784484. doi: 10.3389/fimmu.2021.784484
37. Falck-Hansen M, Kassiteridi C, Monaco C. Toll-like receptors in atherosclerosis. Int J Mol Sci. (2013) 14:14008–23. doi: 10.3390/ijms140714008
38. Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. (2002) 4:E131–136. doi: 10.1038/ncb0502-e131
39. Whitmarsh AJ, Davis RJ. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med (Berl). (1996) 74:589–607. doi: 10.1007/s001090050063
40. Lin R, Mamane Y, Hiscott J. Multiple regulatory domains control IRF-7 activity in response to virus infection. J Biol Chem. (2000) 275:34320–7. doi: 10.1074/jbc.M002814200
41. Marie I, Smith E, Prakash A, Levy DE. Phosphorylation-induced dimerization of interferon regulatory factor 7 unmasks DNA binding and a bipartite transactivation domain. Mol Cell Biol. (2000) 20:8803–14. doi: 10.1128/MCB.20.23.8803–8814.2000
42. Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. (2004) 5:1061–8. doi: 10.1038/ni1118
43. Smits EL, Ponsaerts P, Berneman ZN, Van Tendeloo VF. The use of TLR7 and TLR8 ligands for the enhancement of cancer immunotherapy. Oncologist. (2008) 13:859–75. doi: 10.1634/theoncologist.2008–0097
44. Zheng C, Chen J, Chu F, Zhu J, Jin T. Inflammatory role of TLR-myD88 signaling in multiple sclerosis. Front Mol Neurosci. (2019) 12:314. doi: 10.3389/fnmol.2019.00314
45. Xu YR, Lei CQ. TAK1-TABs complex: A central signalosome in inflammatory responses. Front Immunol. (2020) 11:608976. doi: 10.3389/fimmu.2020.608976
46. Zhao X, Huo R, Yan X, Xu T. IRF3 negatively regulates toll-like receptor-mediated NF-kappaB signaling by targeting TRIF for degradation in teleost fish. Front Immunol. (2018) 9:867. doi: 10.3389/fimmu.2018.00867
47. Shimaoka M, Takagi J, Springer TA. Conformational regulation of integrin structure and function. Annu Rev Biophys Biomol Struct. (2002) 31:485–516. doi: 10.1146/annurev.biophys.31.101101.140922
48. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. (2002) 110:673–87. doi: 10.1016/s0092–8674(02)00971–6
49. Pang X, He X, Qiu Z, Zhang H, Xie R, Liu Z, et al. Targeting integrin pathways: mechanisms and advances in therapy. Signal Transduct Target Ther. (2023) 8:1. doi: 10.1038/s41392–022-01259–6
50. Harburger DS, Calderwood DA. Integrin signaling at a glance. J Cell Sci. (2009) 122:159–63. doi: 10.1242/jcs.018093
51. Park EJ, Myint PK, Ito A, Appiah MG, Darkwah S, Kawamoto E, et al. Integrin-ligand interactions in inflammation, cancer, and metabolic disease: insights into the multifaceted roles of an emerging ligand irisin. Front Cell Dev Biol. (2020) 8:588066. doi: 10.3389/fcell.2020.588066
52. Wrighton KH. Cell adhesion: the 'ins' and 'outs' of integrin signaling. Nat Rev Mol Cell Biol. (2013) 14:752. doi: 10.1038/nrm3708
53. Shen B, Delaney MK, Du X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr Opin Cell Biol. (2012) 24:600–6. doi: 10.1016/j.ceb.2012.08.011
54. Sun H, Liu J, Zheng Y, Pan Y, Zhang K, Chen J. Distinct chemokine signaling regulates integrin ligand specificity to dictate tissue-specific lymphocyte homing. Dev Cell. (2014) 30:61–70. doi: 10.1016/j.devcel.2014.05.002
55. Gahmberg CG, Gronholm M. How integrin phosphorylations regulate cell adhesion and signaling. Trends Biochem Sci. (2022) 47:265–78. doi: 10.1016/j.tibs.2021.11.003
56. Springer TA, Dustin ML. Integrin inside-out signaling and the immunological synapse. Curr Opin Cell Biol. (2012) 24:107–15. doi: 10.1016/j.ceb.2011.10.004
57. Zhu L, Plow EF, Qin J. Initiation of focal adhesion assembly by talin and kindlin: A dynamic view. Protein Sci. (2021) 30:531–42. doi: 10.1002/pro.4014
58. Lu F, Zhu L, Bromberger T, Yang J, Yang Q, Liu J, et al. Mechanism of integrin activation by talin and its cooperation with kindlin. Nat Commun. (2022) 13:2362. doi: 10.1038/s41467-022-30117-w
59. Qin J, Vinogradova O, Plow EF. Integrin bidirectional signaling: a molecular view. PloS Biol. (2004) 2:e169. doi: 10.1371/journal.pbio.0020169
60. Giancotti FG, Ruoslahti E. Integrin signaling. Science. (1999) 285:1028–32. doi: 10.1126/science.285.5430.1028
61. Horton ER, Byron A, Askari JA, Ng DHJ, Millon-Frémillon A, Robertson J, et al. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat Cell Biol. (2015) 17:1577–87. doi: 10.1038/ncb3257
62. Verma NK, Kelleher D. Not just an adhesion molecule: LFA-1 contact tunes the T lymphocyte program. J Immunol. (2017) 199:1213–21. doi: 10.4049/jimmunol.1700495
63. Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell (1992) 69:11–25. doi: 10.1016/0092-8674(92)90115-S
64. Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J Cell Sci. (2006) 119:3901–3. doi: 10.1242/jcs.03098
65. Humphries JD, Chastney MR, Askari JA, Humphries MJ. Signal transduction via integrin adhesion complexes. Curr Opin Cell Biol. (2019) 56:14–21. doi: 10.1016/j.ceb.2018.08.004
66. Grzesiak JJ, Tran Cao HS, Burton DW, Kaushal S, Vargas F, Clopton P, et al. Knockdown of the beta(1) integrin subunit reduces primary tumor growth and inhibits pancreatic cancer metastasis. Int J Cancer. (2011) 129:2905–15. doi: 10.1002/ijc.25942
67. Kanayama M, Morimoto J, Matsui Y, Ikesue M, Danzaki K, Kurotaki D, et al. alpha9beta1 integrin-mediated signaling serves as an intrinsic regulator of pathogenic Th17 cell generation. J Immunol. (2011) 187:5851–64. doi: 10.4049/jimmunol.1101524
68. Hemler ME, Elices MJ, Chan BM, Zetter B, Matsuura N, Takada Y. Multiple ligand binding functions for VLA-2 (alpha 2 beta 1) and VLA-3 (alpha 3 beta 1) in the integrin family. Cell Differ Dev. (1990) 32:229–38. doi: 10.1016/0922-3371(90)90035-U
69. Nishiuchi R, Takagi J, Hayashi M, Ido H, Yagi Y, Sanzen N, et al. Ligand-binding specificities of laminin-binding integrins: a comprehensive survey of laminin-integrin interactions using recombinant alpha3beta1, alpha6beta1, alpha7beta1 and alpha6beta4 integrins. Matrix Biol. (2006) 25:189–97. doi: 10.1016/j.matbio.2005.12.001
70. Dupuy AG, Caron E. Integrin-dependent phagocytosis: spreading from microadhesion to new concepts. J Cell Sci. (2008) 121:1773–83. doi: 10.1242/jcs.018036
71. Isberg RR, Leong JM. Multiple beta 1 chain integrins are receptors for invasin, a protein that promotes bacterial penetration into mammalian cells. Cell. (1990) 60:861–71. doi: 10.1016/0092-8674(90)90099-Z
72. Eto DS, Jones TA, Sundsbak JL, Mulvey MA. Integrin-mediated host cell invasion by type 1-piliated uropathogenic Escherichia coli. PloS Pathog. (2007) 3:e100. doi: 10.1371/journal.ppat.0030100
73. Behera AK, Durand E, Cugini C, Antonara S, Bourassa L, Hildebrand E, et al. Borrelia burgdorferi BBB07 interaction with integrin alpha3beta1 stimulates production of pro-inflammatory mediators in primary human chondrocytes. Cell Microbiol. (2008) 10:320–31. doi: 10.1111/j.1462–5822.2007.01043.x
74. Marre ML, Petnicki-Ocwieja T, DeFrancesco AS, Darcy CT, Hu LT. Human integrin alpha(3)beta(1) regulates TLR2 recognition of lipopeptides from endosomal compartments. PloS One. (2010) 5:e12871. doi: 10.1371/journal.pone.0012871
75. Dietrich N, Lienenklaus S, Weiss S, Gekara NO. Murine toll-like receptor 2 activation induces type I interferon responses from endolysosomal compartments. PloS One. (2010) 5:e10250. doi: 10.1371/journal.pone.0010250
76. Behera AK, Hildebrand E, Uematsu S, Akira S, Coburn J, Hu LT. Identification of a TLR-independent pathway for Borrelia burgdorferi-induced expression of matrix metalloproteinases and inflammatory mediators through binding to integrin alpha 3 beta 1. J Immunol. (2006) 177:657–64. doi: 10.4049/jimmunol.177.1.657
77. Wondimu Z, Geberhiwot T, Ingerpuu S, Juronen E, Xie X, Lindbom L. An endothelial laminin isoform, laminin 8 (alpha4beta1gamma1), is secreted by blood neutrophils, promotes neutrophil migration and extravasation, and protects neutrophils from apoptosis. Blood. (2004) 104:1859–66. doi: 10.1182/blood-2004–01-0396
78. Lerman YV, Lim K, Hyun YM, Falkner KL, Yang H, Pietropaoli AP, et al. Sepsis lethality via exacerbated tissue infiltration and TLR-induced cytokine production by neutrophils is integrin alpha3beta1-dependent. Blood. (2014) 124:3515–23. doi: 10.1182/blood-2014–01-552943
79. Triantafilou M, Morath S, Mackie A, Hartung T, Triantafilou K. Lateral diffusion of Toll-like receptors reveals that they are transiently confined within lipid rafts on the plasma membrane. J Cell Sci. (2004) 117:4007–14. doi: 10.1242/jcs.01270
80. Leitinger B, Hogg N. The involvement of lipid rafts in the regulation of integrin function. J Cell Sci. (2002) 115:963–72. doi: 10.1242/jcs.115.5.963
81. Jackson D. Alpha 4 integrin antagonists. Curr Pharm Des. (2002) 8:1229–53. doi: 10.2174/1381612023394737
82. Newham P CS, Seddon GN, Schofield NR, Rees A, Edwards RM, Jones EY, et al. Alpha4 integrin binding interfaces on VCAM-1 and MAdCAM-1. Integrin binding footprints identify accessory binding sites that play a role in integrin specificity. J Biol Chem. (1997) 272:19429–40. doi: 10.1074/jbc.272.31.19429
83. Gondokaryono SP, Ushio H, Niyonsaba F, Hara M, Takenaka H, Jayawardana ST, et al. The extra domain A of fibronectin stimulates murine mast cells via toll-like receptor 4. J Leukoc Biol. (2007) 82:657–65. doi: 10.1189/jlb.1206730
84. Shinde AV KR, Peters JH, Sekiguchi K, Van De Water L, McKeown-Longo PJ. The α4β1 integrin and the EDA domain of fibronectin regulate a profibrotic phenotype in dermal fibroblasts. Matrix Biol. (2015) 41:26–35. doi: 10.1016/j.matbio.2014.11.004
85. Kelsh-Lasher RM, Ambesi A, Bertram C, McKeown-Longo PJ. Integrin alpha4beta1 and TLR4 cooperate to induce fibrotic gene expression in response to fibronectin's EDA domain. J Invest Dermatol. (2017) 137:2505–12. doi: 10.1016/j.jid.2017.08.005
86. Tan SM. The leucocyte beta2 (CD18) integrins: the structure, functional regulation and signaling properties. Biosci Rep. (2012) 32:241–69. doi: 10.1042/BSR20110101
87. Schittenhelm L, Hilkens CM, Morrison VL. beta(2) integrins as regulators of dendritic cell, monocyte, and macrophage function. Front Immunol. (2017) 8:1866. doi: 10.3389/fimmu.2017.01866
88. Xu S, Wang J, Wang JH, Springer TA. Distinct recognition of complement iC3b by integrins alpha(X)beta(2) and alpha(M)beta(2). Proc Natl Acad Sci U.S.A. (2017) 114:3403–8. doi: 10.1073/pnas.1620881114
89. Yakubenko VP, Yadav SP, Ugarova TP. Integrin alphaDbeta2, an adhesion receptor up-regulated on macrophage foam cells, exhibits multiligand-binding properties. Blood. (2006) 107:1643–50. doi: 10.1182/blood-2005–06-2509
90. Chung KJ, Mitroulis I, Wiessner JR, Zheng YY, Siegert G, Sperandio M, et al. A novel pathway of rapid TLR-triggered activation of integrin-dependent leukocyte adhesion that requires Rap1 GTPase. Mol Biol Cell. (2014) 25:2948–55. doi: 10.1091/mbc.E14–04-0867
91. Yee NK, Hamerman JA. beta(2) integrins inhibit TLR responses by regulating NF-kappaB pathway and p38 MAPK activation. Eur J Immunol. (2013) 43:779–92. doi: 10.1002/eji.201242550
92. Wang L, Gordon RA, Huynh L, Su X, Park Min KH, Han J, et al. Indirect inhibition of Toll-like receptor and type I interferon responses by ITAM-coupled receptors and integrins. Immunity. (2010) 32:518–30. doi: 10.1016/j.immuni.2010.03.014
93. Zhang M, Han Y, Han C, Xu S, Bao Y, Chen Z, et al. The beta2 integrin CD11b attenuates polyinosinic:polycytidylic acid-induced hepatitis by negatively regulating natural killer cell functions. Hepatology. (2009) 50:1606–16. doi: 10.1002/hep.23168
94. Zhou X, Gao XP, Fan J, Liu Q, Anwar KN, Frey RS, et al. LPS activation of Toll-like receptor 4 signals CD11b/CD18 expression in neutrophils. Am J Physiol Lung Cell Mol Physiol. (2005) 288:L655–662. doi: 10.1152/ajplung.00327.2004
95. Sim H, Jeong D, Kim HI, Pak S, Thapa B, Kwon HJ, et al. CD11b deficiency exacerbates methicillin-resistant staphylococcus aureus-induced sepsis by upregulating inflammatory responses of macrophages. Immune Netw. (2021) 21:e13. doi: 10.4110/in.2021.21.e13
96. Ling GS, Bennett J, Woollard KJ, Szajna M, Fossati-Jimack L, Taylor PR, et al. Integrin CD11b positively regulates TLR4-induced signaling pathways in dendritic cells but not in macrophages. Nat Commun. (2014) 5:3039. doi: 10.1038/ncomms4039
97. Gao LY, Kwaik YA. The modulation of host cell apoptosis by intracellular bacterial pathogens. Trends Microbiol. (2000) 8:306–13. doi: 10.1016/s0966–842x(00)01784–4
98. Bai Y, Qian C, Qian L, Ma F, Hou J, Chen Y, et al. Integrin CD11b negatively regulates TLR9-triggered dendritic cell cross-priming by upregulating microRNA-146a. J Immunol. (2012) 188:5293–302. doi: 10.4049/jimmunol.1102371
99. Zhang X, Kimura Y, Fang C, Zhou L, Sfyroera G, Lambris JD, et al. Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood. (2007) 110:228–36. doi: 10.1182/blood-2006–12-063636
100. Harokopakis E, Albzreh MH, Martin MH, Hajishengallis G. TLR2 transmodulates monocyte adhesion and transmigration via Rac1- and PI3K-mediated inside-out signaling in response to Porphyromonas gingivalis fimbriae. J Immunol. (2006) 176:7645–56. doi: 10.4049/jimmunol.176.12.7645
101. Yao X, Dong G, Zhu Y, Yan F, Zhang H, Ma Q, et al. Leukadherin-1-mediated activation of CD11b inhibits LPS-induced pro-inflammatory response in macrophages and protects mice against endotoxic shock by blocking LPS-TLR4 interaction. Front Immunol. (2019) 10:215. doi: 10.3389/fimmu.2019.00215
102. Sandor N, Lukácsi S, Ungai-Salánki R, Orgován N, Szabó B, Horváth R, et al. CD11c/CD18 dominates adhesion of human monocytes, macrophages and dendritic cells over CD11b/CD18. PloS One. (2016) 11:e0163120. doi: 10.1371/journal.pone.0163120
103. Querrey M, Chiu S, Lecuona E, Wu Q, Sun H, Anderson M, et al. CD11b suppresses TLR activation of nonclassical monocytes to reduce primary graft dysfunction after lung transplantation. J Clin Invest. (2022) 132:e157262. doi: 10.1172/JCI157262
104. Zhou H, Liao J, Aloor J, Nie H, Wilson BC, Fessler MB, et al. CD11b/CD18 (Mac-1) is a novel surface receptor for extracellular double-stranded RNA to mediate cellular inflammatory responses. J Immunol. (2013) 190:115–25. doi: 10.4049/jimmunol.1202136
105. Trstenjak N, Milić D, Graewert MA, Rouha H, Svergun D, Djinović-Carugo K, et al. Molecular mechanism of leukocidin GH-integrin CD11b/CD18 recognition and species specificity. Proc Natl Acad Sci U.S.A. (2020) 117:317–27. doi: 10.1073/pnas.1913690116
106. Wright SD, Jong MT. Adhesion-promoting receptors on human macrophages recognize Escherichia coli by binding to lipopolysaccharide. J Exp Med. (1986) 164:1876–88. doi: 10.1084/jem.164.6.1876
107. Ingalls RR, Monks BG, Savedra R Jr, Christ WJ, Delude RL, Medvedev AE, et al. CD11/CD18 and CD14 share a common lipid A signaling pathway. J Immunol. (1998) 161:5413–20. doi: 10.4049/jimmunol.161.10.5413
108. Wright SD, Levin SM, Jong MT, Chad Z, Kabbash LG. CR3 (CD11b/CD18) expresses one binding site for Arg-Gly-Asp-containing peptides and a second site for bacterial lipopolysaccharide. J Exp Med. (1989) 169:175–83. doi: 10.1084/jem.169.1.175
109. Corbi AL, Kishimoto TK, Miller LJ, Springer TA. The human leukocyte adhesion glycoprotein Mac-1 (complement receptor type 3, CD11b) alpha subunit. Cloning, primary structure, and relation to the integrins, von Willebrand factor and factor B. J Biol Chem. (1988) 263:12403–11. doi: 10.1016/S0021-9258(18)37770-6
110. Ross GD, Reed W, Dalzell JG, Becker SE, Hogg N. Macrophage cytoskeleton association with CR3 and CR4 regulates receptor mobility and phagocytosis of iC3b-opsonized erythrocytes. J Leukoc Biol. (1992) 51:109–17. doi: 10.1002/jlb.51.2.109
111. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. (2014) 514:187–92. doi: 10.1038/nature13683
112. Lukacsi S, Gerecsei T, Balázs K, Francz B, Szabó B, Erdei A, et al. The differential role of CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in the adherence, migration and podosome formation of human macrophages and dendritic cells under inflammatory conditions. PloS One. (2020) 15:e0232432. doi: 10.1371/journal.pone.0232432
113. Wang H, Chen H, Jiang Z, Lin Y, Wang X, Xiang J, et al. Integrin subunit alpha V promotes growth, migration, and invasion of gastric cancer cells. Pathol Res Pract. (2019) 215:152531. doi: 10.1016/j.prp.2019.152531
114. Cheuk IW, Siu MT, Ho JC, Chen J, Shin VY, Kwong A. ITGAV targeting as a therapeutic approach for treatment of metastatic breast cancer. Am J Cancer Res. (2020) 10:211–23.
116. Ruegg C, Mariotti A. Vascular integrins: pleiotropic adhesion and signaling molecules in vascular homeostasis and angiogenesis. Cell Mol Life Sci. (2003) 60:1135–57. doi: 10.1007/s00018–003-2297–3
117. Li Z, Belozertseva E, Parlakian A, Bascetin R, Louis H, Kawamura Y. Smooth muscle alpha(v) integrins regulate vascular fibrosis via CD109 downregulation of TGF-beta signaling. Eur Heart J Open. (2023) 3:oead010. doi: 10.1093/ehjopen/oead010
118. Ruoslahti E, Pierschbacher MD. Arg-Gly-Asp: a versatile cell recognition signal. Cell. (1986) 44:517–8. doi: 10.1016/0092-8674(86)90259-X
119. Parry C, Bell S, Minson T, Browne H. Herpes simplex virus type 1 glycoprotein H binds to alphavbeta3 integrins. J Gen Virol. (2005) 86:7–10. doi: 10.1099/vir.0.80567–0
120. Casiraghi C, Gianni T, Campadelli-Fiume G. alphavbeta3 Integrin Boosts the Innate Immune Response Elicited in Epithelial Cells through Plasma Membrane and Endosomal Toll-Like Receptors. J Virol. (2016) 90:4243–8. doi: 10.1128/JVI.03175–15
121. Gianni T, Campadelli-Fiume G. The epithelial alphavbeta3-integrin boosts the MYD88-dependent TLR2 signaling in response to viral and bacterial components. PloS Pathog. (2014) 10:e1004477. doi: 10.1371/journal.ppat.1004477
122. Gerold G, Abu Ajaj K, Bienert M, Laws HJ, Zychlinsky A, de Diego JL. A Toll-like receptor 2-integrin beta3 complex senses bacterial lipopeptides. via vitronectin. Nat Immunol. (2008) 9:761–8. doi: 10.1038/ni.1618
123. Chen Z, Wang S, Chen Y, Shao Z, Yu Z, Mei S, et al. Integrin beta3 modulates TLR4-mediated inflammation by regulation of CD14 expression in macrophages in septic condition. Shock. (2020) 53:335–43. doi: 10.1097/SHK.0000000000001383
124. Chen Z, Ding X, Jin S, Pitt B, Zhang L, Billiar T, et al. WISP1-alphavbeta3 integrin signaling positively regulates TLR-triggered inflammation response in sepsis induced lung injury. Sci Rep. (2016) 6:28841. doi: 10.1038/srep28841
125. Borland G, Edkins AL, Acharya M, Matheson J, White LJ, Allen JM, et al. alphavbeta5 integrin sustains growth of human pre-B cells through an RGD-independent interaction with a basic domain of the CD23 protein. J Biol Chem. (2007) 282:27315–26. doi: 10.1074/jbc.M609335200
126. Wu Y, Singh S, Georgescu MM, Birge RB. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J Cell Sci. (2005) 118:539–53. doi: 10.1242/jcs.01632
127. Ding X, Tong Y, Jin S, Chen Z, Li T, Billiar TR, et al. Mechanical ventilation enhances extrapulmonary sepsis-induced lung injury: role of WISP1-alphavbeta5 integrin pathway in TLR4-mediated inflammation and injury. Crit Care. (2018) 22:302. doi: 10.1186/s13054–018-2237–0
128. Su G, Hodnett M, Wu N, Atakilit A, Kosinski C, Godzich M, et al. Integrin alphavbeta5 regulates lung vascular permeability and pulmonary endothelial barrier function. Am J Respir Cell Mol Biol. (2007) 36:377–86. doi: 10.1165/rcmb.2006-0238OC
129. Su G, Atakilit A, Li JT, Wu N, Luong J, Chen R, et al. Effective treatment of mouse sepsis with an inhibitory antibody targeting integrin alphavbeta5. Crit Care Med. (2013) 41:546–53. doi: 10.1097/CCM.0b013e3182711b1e
130. Breuss JM, Gallo J, DeLisser HM, Klimanskaya IV, Folkesson HG, Pittet JF, et al. Expression of the beta 6 integrin subunit in development, neoplasia and tissue repair suggests a role in epithelial remodeling. J Cell Sci 108 ( Pt. (1995) 6):2241–51. doi: 10.1242/jcs.108.6.2241
131. Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. (1999) 96:319–28. doi: 10.1016/s0092–8674(00)80545–0
132. Jolly L, Stavrou A, Vanderstoken G, Meliopoulos VA, Habgood A, Tatler AL, et al. Influenza promotes collagen deposition via alphavbeta6 integrin-mediated transforming growth factor beta activation. J Biol Chem. (2014) 289:35246–63. doi: 10.1074/jbc.M114.582262
133. Smith M, Meliopoulos V, Tan S, Bub T, Brigleb PH, Sharp B, et al. The beta6 Integrin Negatively Regulates TLR7-Mediated Epithelial Immunity via Autophagy During Influenza A Virus Infection. bioRxiv. (2023). doi: 10.1101/2023.08.28.555098
134. Hajishengallis G, Lambris JD. More than complementing Tolls: complement-Toll-like receptor synergy and crosstalk in innate immunity and inflammation. Immunol Rev. (2016) 274:233–44. doi: 10.1111/imr.12467
135. Mohan S, Gupta D. Crosstalk of toll-like receptors signaling and Nrf2 pathway for regulation of inflammation. BioMed Pharmacother. (2018) 108:1866–78. doi: 10.1016/j.biopha.2018.10.019
Keywords: toll-like receptor, β1 integrin, β2 integrin, αV integrin, crosstalk
Citation: Alhamdan F, Bayarsaikhan G and Yuki K (2024) Toll-like receptors and integrins crosstalk. Front. Immunol. 15:1403764. doi: 10.3389/fimmu.2024.1403764
Received: 19 March 2024; Accepted: 24 May 2024;
Published: 10 June 2024.
Edited by:
Jeremy P. McAleer, Marshall University, United StatesReviewed by:
Pranita Sarangi, Indian Institute of Technology Roorkee, IndiaCarla Guenther, Osaka University, Japan
Copyright © 2024 Alhamdan, Bayarsaikhan and Yuki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Koichi Yuki, a29pY2hpLnl1a2lAY2hpbGRyZW5zLmhhcnZhcmQuZWR1
†These authors have contributed equally to this work