 David Ngai
David Ngai Santosh R. Sukka
Santosh R. Sukka Ira Tabas
Ira Tabas- 1Department of Medicine, Columbia University Irving Medical Center, New York, NY, United States
- 2Department of Pathology and Cell Biology, Columbia University Irving Medical Center, New York, NY, United States
- 3Department of Physiology, Columbia University Irving Medical Center, New York, NY, United States
The interplay between myeloid cells and T-lymphocytes is critical to the regulation of host defense and inflammation resolution. Dysregulation of this interaction can contribute to the development of chronic inflammatory diseases. Important among these diseases is atherosclerosis, which refers to focal lesions in the arterial intima driven by elevated apolipoprotein B-containing lipoproteins, notably low-density lipoprotein (LDL), and characterized by the formation of a plaque composed of inflammatory immune cells, a collection of dead cells and lipids called the necrotic core, and a fibrous cap. As the disease progresses, the necrotic core expands, and the fibrous cap becomes thin, which increases the risk of plaque rupture or erosion. Plaque rupture leads to a rapid thrombotic response that can give rise to heart attack, stroke, or sudden death. With marked lowering of circulating LDL, however, plaques become more stable and cardiac risk is lowered—a process known as atherosclerosis regression. A critical aspect of both atherosclerosis progression and regression is the crosstalk between innate (myeloid cells) and adaptive (T-lymphocytes) immune cells. Myeloid cells are specialized at clearing apoptotic cells by a process called efferocytosis, which is necessary for inflammation resolution. In advanced disease, efferocytosis is impaired, leading to secondary necrosis of apoptotic cells, inflammation, and, most importantly, defective tissue resolution. In regression, efferocytosis is reawakened aiding in inflammation resolution and plaque stabilization. Here, we will explore how efferocytosing myeloid cells could affect T-cell function and vice versa through antigen presentation, secreted factors, and cell-cell contacts and how this cellular crosstalk may contribute to the progression or regression of atherosclerosis.
1 Introduction
Appropriate regulation of inflammation and its resolution is critical for host defense and maintaining tissue homeostasis. Communication between the innate arm of immunity, particularly myeloid cells, notably macrophages and dendritic cells, and the adaptive arm of immunity, notably T-lymphocytes, is essential. Dysregulation of myeloid-T-lymphocyte crosstalk can contribute to the pathogenesis and exacerbation of chronic inflammatory diseases such as atherosclerosis (1). Atherosclerosis is characterized by the formation of a fibro-fatty plaque in the arterial wall consisting of a necrotic core made up of deposited lipids and dead cells, immune cells including myeloid cells and T-lymphocytes, and a fibrous cap overlaying the necrotic core (2). Atherosclerosis progression is driven by elevated circulating LDL levels that cause increased lipid deposition and inflammatory cell infiltration into the plaque (2). Atherosclerosis regression on the other hand is a clinically relevant phenomenon that occurs with marked lowering of circulating LDL, which promotes inflammation resolution, reduced necrotic core size, and thickening/strengthening of the fibrous cap (3).
Macrophages and dendritic cells play key roles in atherosclerosis progression and regression. One critical role is related to the process by which apoptotic cells (ACs) are cleared by phagocytic cells such as macrophages and dendritic cells, called efferocytosis (4, 5). Efferocytosis prevents secondary necrosis of ACs and subsequent inflammation and contributes to the production of pro-resolving mediators, which can feedback to further enhance efferocytosis (efferocytosis-resolution cycle) (4, 5). As atherosclerosis progresses, the efferocytosis-resolution cycle becomes defective, characterized by increased inflammation, enhanced necrosis, and thinning of the protective fibrous cap of lesions (4, 5). In humans, these changes lead to plaques that are “unstable”, meaning they increase the risk for plaque rupture or erosion, acute luminal thrombosis, and major adverse cardiovascular events, notably myocardial infarction, stroke, and sudden cardiac death (4, 5). On the other hand, the efferocytosis-resolution cycle is reawakened during atherosclerosis regression, a process in which marked lipid lowering leads to atherosclerotic plaque stabilization through necrotic core reduction and fibrous cap thickening and, in humans, lower the risk of coronary artery disease (6–8).
There is also substantial evidence indicating that T-cells from the adaptive immune system play important roles in atherosclerosis pathogenesis (9–12). Under homeostatic conditions and during an immune response, myeloid cells and T-cells communicate with each other (13–15), with macrophages and dendritic cells serving as a critical bridge between the innate and adaptive immune systems through antigen presentation, secreted mediators, and cell-cell contact (13–15). T-cells can also feedback through secreted factors and cell-cell contact to regulate myeloid cell functions (16, 17).
While the roles of myeloid cells in atherosclerosis are varied and complex, we focus here on the key issue of how efferocytosis by lesional macrophages and dendritic cells can affect both plaque progression and regression through crosstalk with T-cells. Crosstalk between efferocytic myeloid cells and T-cells can promote a pro-resolving milieu that dampens chronic inflammation and promotes plaque stabilization, while failure of this process can result in the exacerbation of atherosclerosis and attenuation of regression. In this review, we will explore three critical aspects of this interaction and how it may affect atherosclerosis progression and regression: 1) the role of antigen cross-presentation by myeloid cells of AC-derived antigens during efferocytosis and its effect on T-cells; 2) the role of secreted cytokines, such as transforming growth factor-β1 (TGF-β1) and interleukin-10 (IL-10), and metabolites, such as lactate and polyamines in the crosstalk between efferocytic myeloid cells and T-cells; and 3) the role of cell-cell contacts through membrane proteins, including the immune checkpoint proteins programmed death ligand 1 (PD-L1), programmed cell death protein 1 (PD-1), and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) in regulating efferocyte-T-cell crosstalk.
2 The role of myeloid cell antigen presentation
Myeloid cells are adept at a process called antigen presentation. Proteins taken up by myeloid cells through efferocytosis and other endocytic pathways are degraded, and peptides (antigens) are loaded onto major histocompatibility complex (MHC) molecules to present to adaptive immune cells such as T-cells. The recognition of presented antigens by T-cells through the engagement of the T-cell receptor (TCR) can either trigger immune activation towards the antigen or induce tolerance toward self-antigens (18, 19). Disruption of tolerance mechanisms can result in aberrant presentation of self-antigens to auto-reactive T-cells and a lack of restraint on T-cell activation, leading to autoimmunity. Efferocytosis helps facilitate tolerance by preventing self-antigens derived from ACs from triggering an autoimmune response and chronic inflammation (20). Accordingly, an impairment in efferocytosis, which is observed in advanced atherosclerosis, can result in an accumulation of uncleared self-antigen thus inducing autoimmunity (4, 21). In late atherosclerosis, defective efferocytosis can be attributed in part to the loss of cell surface efferocytic receptors, e.g., through the cleavage of the efferocytosis receptor, MER proto-oncogene tyrosine kinase (MerTK) (22). A direct example of autoimmunity triggered by impaired efferocytosis comes from a study showing that genetically deleting the efferocytic receptor CD300f in mice enhanced self-antigen presentation by dendritic cells, contributing to memory T-cell expansion and autoimmunity (23). Loss of tolerance due to impaired efferocytosis is likely relevant to atherosclerosis, which displays hallmarks of autoimmunity such as anti-apolipoprotein B autoantibodies and CD4+ T-cells (24, 25). This topic will be discussed in more detail in the following sections.
Antigen presentation is initiated when internalized proteins are degraded by lysosomes or by the proteasome and loaded onto MHC class I or II molecules and presented to a corresponding T-cell receptor (TCR). The proliferation and activation of T-cells can be initiated when TCRs on a CD4+ or CD8+ T-cell surpass the threshold binding affinity of a peptide presented on MHC class II or class I, respectively (26, 27). During efferocytosis, AC degradation generates self-antigens, but efferocytosing myeloid cells can restrain autoimmunity by suppressing the presentation of these antigens. The mechanism involves the secretion of pro-resolving mediators by efferocytes that (a) lower co-stimulatory signals needed for full activation of T-cells, e.g., those that mediate T-cell CD28 interaction with myeloid CD80/86; and (b) promote the formation of self-antigen-specific forkhead box P3 (FoxP3)+ regulatory T-cells (Treg cells), which suppress auto-reactive effector T-cells, dampen inflammation, and promote tissue repair (28–30). In atherosclerosis, FoxP3+ Treg cells are athero-protective (31) and are critical to atherosclerosis regression (32). In dendritic cells, AC-binding and efferocytosis mediated by MerTK triggers the suppression of nuclear factor kappa B (NFκB) signaling to prevent the full maturation of these cells (33). This process results in low expression of MHC, co-stimulatory molecules like CD80, and the pro-inflammatory cytokine tumor necrosis factor-alpha (TNF-α), all of which inhibit the presentation of AC-derived self-antigens and prevent the activation of auto-reactive T-cells (33). Pro-resolving mediators from efferocytes, such as TGF-β1 and IL-10, can induce Treg cells, which feedback to suppress CD80/86 and stop effector T-cell activity (34).
In atherosclerosis, antigen-presentation by myeloid cells to T-cells contributes to chronic inflammation by inducing effector CD4+ T-cells and the release of pro-inflammatory cytokines, e.g., IFN-γ and TNF-α, by activated T-cells (35). These cytokines in turn enhance the uptake of oxidized LDL (oxLDL) and minimally modified LDL by lesional macrophages, which promotes disease progression (36). Furthermore, pro-inflammatory cytokines such as IFN-γ and TNF-α are known to augment antigen presentation by myeloid cells, thus further activating pro-atherogenic effector T-cells (37, 38). Efferocytosis still occurs in inflammatory progressing atherosclerotic lesions, albeit reduced and with impaired induction of pro-resolution signaling. Thus, it is possible that in atherosclerosis progression, the suppression of antigen presentation and co-stimulatory molecules by efferocytes is impaired. For example, although antigenic peptides generated during efferocytosis can bypass being loaded onto MHC class II by the activity of Ras-related protein Rab-17 (Rab17) to avoid antigen presentation and activation of auto-reactive CD4+ T-cells (39), stimulation of toll-like receptors (TLRs) such as TLR4 can re-direct antigens to be loaded onto MHC molecules for antigen presentation (40–42). Thus, oxLDL, which accumulates in atherosclerotic plaques, can upregulate TLR4 expression and signal through TLR4 (43, 44), and other pro-inflammatory signals in atherosclerosis may enhance the presentation of self-peptide by efferocytosing myeloid cells to T-cells. Furthermore, chronic inflammation in atherosclerosis promotes the maturation of dendritic cells and expression of co-stimulatory molecules to enhance activation of self-reactive T-cells (45). Interestingly, MHC class II whole-body knockout in ApoE-/- mice, designed to inhibit antigen presentation to CD4+ T-cells, worsened atherosclerosis despite reduced total CD4+ T-cells and circulating pro-inflammatory cytokines (46). This result may be related to the finding that antigen presentation by MHC class II of plaque antigens to Treg cells is critical for immunosuppression (47), suggesting that a loss of Treg cell activity is more consequential than reduced antigen presentation to effector CD4+ T-cells in atherosclerosis progression (46). Because these experiments were performed in whole-body MHC class II knockout mice and most cells can present antigen on MHC class II, it would be interesting to observe the effect on atherosclerosis when MHC class II is knocked out solely in myeloid antigen-presenting cells (46). In this context, mice with dendritic cell-specific knockout of the TLR signaling adaptor, myeloid differentiation primary response 88 (MyD88), attenuated dendritic cell maturation, antigen presentation to Treg cells, Treg cell maturation, and most importantly, worsened atherosclerosis (48). Thus, in this model of atherosclerosis, defective Treg cell development trumped the benefits of reducing effector T-cells (48).
3 The role of secreted factors on myeloid-lymphocyte crosstalk
As mentioned above, both efferocytes and activated T-lymphocytes can produce a variety of cytokines and lipid-derived specialized pro-resolving mediators (SPMs). In addition, efferocytes release metabolites derived from the degradation of ACs and from shifts in cellular metabolism, and these molecules could have immunomodulatory effects on both the efferocyte and T-cells. The role of secreted molecules in efferocytosing myeloid cell-lymphocyte crosstalk is summarized in Figure 1.
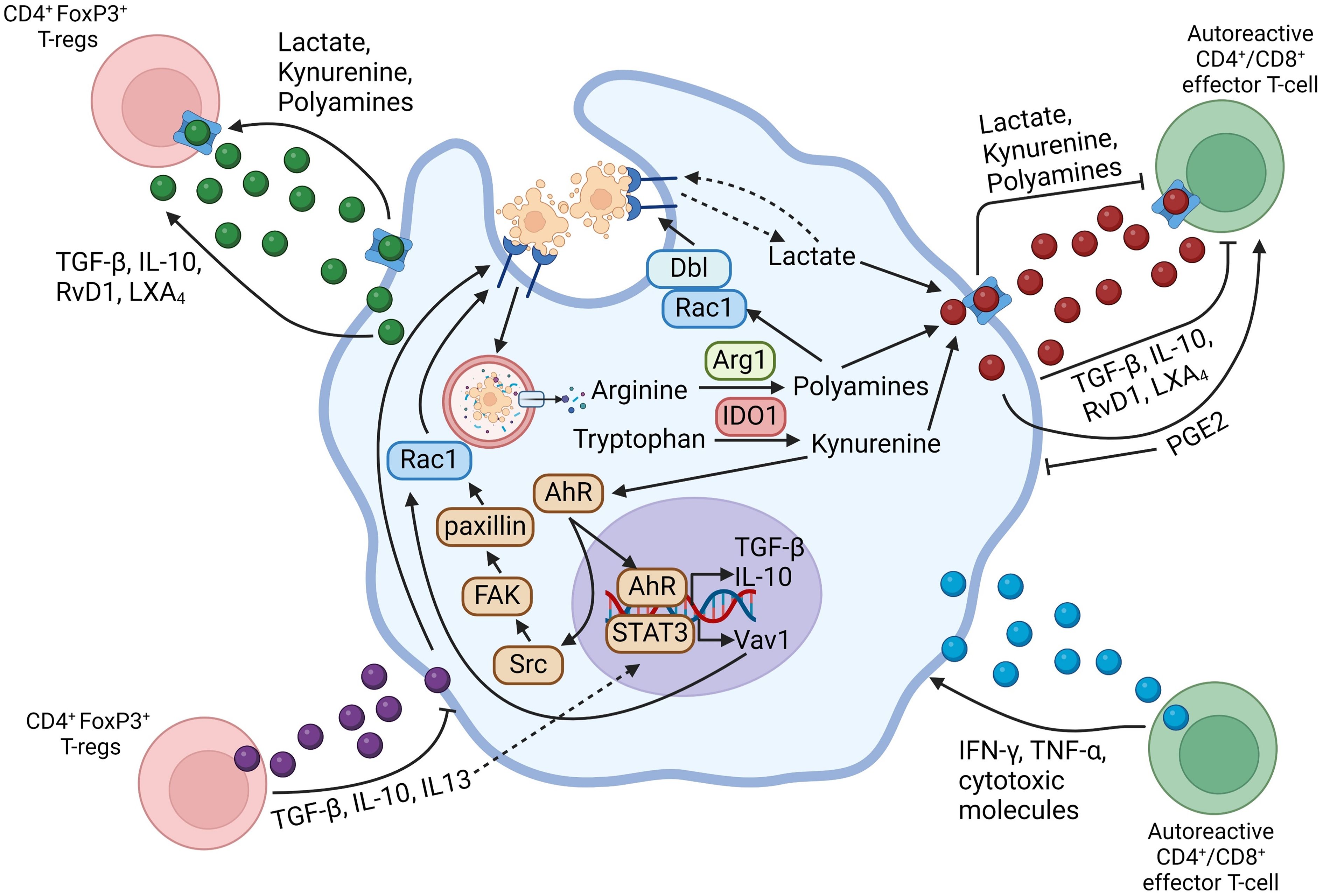
Figure 1 Schematic summarizing efferocytosing myeloid cell-T-lymphocyte crosstalk through secreted mediators. Arrows going from the myeloid cell-secreted molecule to the T-lymphocyte indicate that the secreted mediator activates the functions of the indicated T-lymphocyte subset, whereas flat-ended lines indicate an inhibitory function of the secreted molecule towards the indicated T-lymphocyte subset. Arrows going from the T-lymphocyte-secreted molecule towards the myeloid cell indicate a pro-inflammatory function on the myeloid cell, whereas flat-ended lines indicate a pro-resolving function of the secreted molecule on the myeloid cell. Figure generated with BioRender.com.
3.1 Cytokines
Efferocytosis of ACs promotes the secretion of pro-resolving cytokines such as TGF-β1 and IL-10. TGF-β1 is a key signal in driving the polarization of CD4+ CD25+ T-cells to FoxP3+ Treg cells by inducing FoxP3 expression and stabilization (49). In the thymus, negative selection and subsequent thymocyte apoptosis drives enhanced thymic FoxP3+ Treg cell differentiation due to TGF-β1 production resulting from efferocytosis by thymic myeloid cells (50). IL-10 can also maintain FoxP3 expression and promote the immunosuppressive activity of FoxP3+ Treg cells (51). TGF-β1 and IL-10 also play a role in the suppression of the pro-inflammatory TH1 effector response (52, 53). TGF-β1 can inhibit the expression of T-bet and IRF1, which are lineage-specific transcription factors for pro-inflammatory TH1 cells, through the activity of the protein tyrosine phosphatase Src homology region 2 domain-containing phosphatase-1 (Shp-1) (54), and IL-10 plays a critical role in restraining TH1 cell activity (53). Alloreactive/autoreactive CD4+ T-cells pre-treated with TGF-β1 and IL-10 are converted into Treg cells, preventing an aberrant T-cell response, while increased Treg cell numbers help maintain immunosuppression and generate a pro-resolving environment (55, 56). Thus attenuated TGF-β1 and IL-10 accumulation in atherosclerotic lesions due to impaired efferocytosis may result in a lower threshold to T-cell activation, thus resulting in pro-atherosclerotic autoimmunity and excessive T-cell activity. However, the actions of TGF-β1 are complex. On the one hand, TGF-β1 can enhance the activation and proliferation of TH1 cells, but TGF-β1 can also enhance IL-10 production by TH1 cells to limit this effect (57). As another example, TGF-β1 can suppress naïve CD8+ cytotoxic T-cells (58, 59), though TGF-β1 supports the activation and survival of memory or effector CD8+ T-cells (58). This dichotomy suggests that TGF-β1 prevents aberrant CD8+ T-cell activation, but will support a CD8+ response in clones that under normal conditions are appropriately responsive to a threat. In later stages of atherosclerosis, CD8+ cytotoxic T-cells that show specificity to a limited set of plaque-associated antigens remain and constitute a large proportion of plaque immune cells, likely due to impaired suppression of allo/auto-reactive CD8+ T-cells (60). CD8+ T-cells then contribute to necrotic core growth and plaque destabilization by the cytotoxic killing of cells in the plaque (61) and by secreting pro-inflammatory cytokines such as IFN-γ (62).
Cytokines secreted by T-cells can also feedback to enhance efferocytosis and generate a pro-resolving phenotype in myeloid cells, such as FoxP3+ Treg cells secreting pro-resolving TGF-β1 and IL-10 (63). Both TGF-β1 and IL-10 signal to macrophages to enhance efferocytosis and polarize macrophages towards a pro-resolving phenotype (64, 65). Thus, this could be a mechanism by which efferocytosing myeloid cells drive a positive feedback loop by activating FoxP3+ Treg cells with TGF-β1 and IL-10, which further enhances TGF-β1 and IL-10 levels through Treg cells. In addition to TGF-β1 and IL-10, FoxP3+ Treg cells could also promote efferocytosis by IL-13 (17). IL-13 signaling in macrophages stimulates IL-10 production by macrophages, which then signals in an autocrine/paracrine manner leading to signal transducer and activator of transcription 3 (STAT3)-mediated upregulation of the small Rho GTPase Vav1 to promote actin mobilization through Ras-related C3 botulinum toxin substrate 1 (Rac1) and enhanced efferocytosis (17).
3.2 Metabolites
Efferocytosing macrophages rewire their metabolism by enhancing glycolysis and fatty acid oxidation, and increase the expression and activity of enzymes that process metabolic cargo like amino acids released from the degradation of ACs. Metabolites released by macrophages can modulate the survival, proliferation, differentiation, and activity of T-cells.
3.2.1 Lactate
Recent studies have shown that efferocytosis causes a transient increase in aerobic glycolysis and lactate production in macrophages, which is distinct from the prolonged nature of these responses in inflammatory macrophages (66–68). Efferocytosis promotes glycolysis through phosphorylation and activation of the kinase PFKFB2 and increased glucose uptake by stabilizing GLUT1 (67). Efferocytosis-induced lactate can be secreted into the extracellular space by the lactate transporter SLC16A1 to signal to neighboring cells (66–68). Secreted lactate regulates the differentiation, proliferation, and activity of multiple T-cell subsets (69). FoxP3+ Treg cells efficiently utilise lactate rather than glucose as a fuel source by shuttling lactate into the TCA cycle, which sustains the immunosuppressive functions of Treg cells and promotes their expansion (70). Lactate was found to also promote the expression of FoxP3 in naïve T-cells resulting in their polarization to FoxP3+ Treg cells (71). Pro-inflammatory effector T-cells, however, are inhibited from proliferation by lactate due to a reduction in the NAD+/NADH ratio, which promotes reductive stress (72). Under conditions of chronic inflammation, such as in a mouse model of rheumatoid arthritis where lactate accumulates in the microenvironment, CD4+ effector T-cells metabolically adapt by upregulating the lactate transporter, SLC5A12 (73). This process exacerbates inflammation by (a) increasing lactate import, which promotes the expression of the transcriptional regulator of the TH17 lineage, RORγT; (b) boosting the production of the pro-inflammatory cytokine IL-17 though PKM2 dimer-STAT3 signaling and enhanced fatty acid synthesis, which further enhances TH17 polarization (73). These findings also indicate that the duration of glycolytic lactate production and concentration of lactate can influence T-cell polarization and activity. A more transient increase in glycolysis by efferocytic macrophages may support FoxP3+ Treg cells and suppress effector T-cells to dampen inflammation, while chronic inflammation may result in metabolic adaptations by effector T-cells that lead to the induction of pro-inflammatory signals.
In murine atherosclerosis, myeloid-specific knockout of GLUT1 in Western diet-fed Ldlr-/- mice resulted in expanded necrotic cores and accumulation of ACs due in part from reductions in efferocytosis resulting from impaired glycolysis and lactate production (66). Moreover, the circulating lactate concentration in humans correlates with atherosclerosis severity (74). Thus, it will be interesting to determine in the future if perturbations in glycolytic metabolism and lactate production affect the activity of effector T-cells and the differentiation of Treg cells in atherosclerotic plaques.
3.2.2 Polyamines
Polyamines are a class of metabolites derived from the enzymatic conversion of the amino acid L-arginine. L-arginine is converted by the enzymes Arginase 1/2 to L-ornithine, which is subsequently converted to either L-proline to support collagen synthesis or other polyamine species including putrescine, spermidine, and spermine, through ornithine decarboxylase 1 (ODC1) and other downstream enzymes (75). Efferocytosing macrophages increase intracellular L-arginine, which is derived from degraded ACs, and convert it to polyamines such as ornithine and putrescine (76). Putrescine generated after the uptake of a first AC was found to enhance continuing efferocytosis by increasing the expression of the GTPase Dbl to activate Rac1 and subsequent actin remodeling for second AC internalization (76). Other studies have also identified that efferocytosing macrophages can enhance the import of spermidine and spermine to suppress pro-inflammatory cytokine production (e.g. IL-1β and IL-6) in LPS-treated macrophages (77).
Arginine and polyamines can be exported by amino acid and diamine transporters such as SLC3A2 (78). T-cells have the capacity to metabolize arginine to polyamines and import polyamines to modulate their behavior. In vitro supplementation of T-cells with spermidine enhanced FoxP3 expression and enhanced polarization of T-cells to Treg cells (79). Administration of L-arginine or spermidine to mice increased Treg cell content in vivo (79). Spermidine and its conversion to hypusine was also shown to be necessary for maintaining T-cell lineage fidelity through hypusination of the translation elongation factor eIF5A, which is necessary for chromatin remodeling to ensure proper T-cell lineage commitment (80). ODC1 KO in CD4+ T-cells resulted in dysregulated expression of lineage-specific transcription factors and cytokines in several T-helper cell subsets, and adoptive transfer of ODC1 KO CD4+ T-cells into Rag1-/- mice exacerbated inflammation and disease progression in a colitis model (80). These data suggest that polyamines regulate CD4+ T-cell pro-inflammatory signaling in inflammatory diseases. By blocking both polyamine biosynthesis through ODC1 KO and polyamine uptake through the polyamine mimetic AMXT 1501 to fully deplete intracellular polyamines, T-cell proliferation and differentiation toward TH1 and TH17 was inhibited, while differentiation towards Treg cells was increased (81). In a mouse experimental autoimmune encephalopathy (EAE) model of autoimmune disease, a combination of ODC1 KO donor T-cells and daily injections of AMXT 1501 almost completely suppressed disease progression (81). ODC1 expression is distinct between different T-cell subsets, suggesting that variations in polyamine synthesis guide and maintain lineage fidelity rather than triggering CD4+ T-cell polarization towards a particular lineage (80). In CD8+ T-cells, polyamines can support immunosuppression. Spermidine was shown to prevent TCR clustering on the cell membrane by cholesterol depletion to inhibit CD8+ T-cell activation (82). Furthermore, spermidine and hypusination of eIF5A in effector CD8+ T-cells prevented the formation of tissue-resident memory CD8+ T-cells and reduced the production of the pro-inflammatory cytokines TNF-α and IFN-γ (83).
In atherosclerosis, macrophage-specific deletion of arginase-1 in Ldlr-/- mice did not affect atherosclerosis progression, but it significantly attenuated atherosclerosis regression as assessed by an increase in plaque area and thinning of the fibrous cap (76). Given that arginase-1 is upregulated in pro-resolving/efferocytic macrophages, this may explain the importance of arginase-1 and polyamine production in regressing lesions, which are characterized by enhanced efferocytosis. Overexpression of arginase-1 in macrophages resulted in the suppressed production of pro-inflammatory cytokines such as TNF-α and IL-6 in vitro and in vivo in the atherosclerotic plaque (84). Both IL-6 and TNF-α can contribute to the proliferation of pro-atherogenic T-cell subsets including CD4+ TH1 and TH17 cells (85, 86), suggesting that macrophage arginase-1, perhaps through ornithine-derived polyamines, may contribute to ameliorating atherosclerosis progression by suppressing these T-cell subsets from differentiating and expanding. In atherosclerosis progression, direct supplementation of the polyamine putrescine to the drinking water was able to reduce lesion area and necrotic core area while increasing fibrous cap thickness (76). This was found to be due in part to enhanced continual efferocytosis from putrescine-mediated upregulation of the small GTPase Dbl (76). However, an additional mechanism could be that putrescine and other polyamines produced from AC-derived arginine can also play a role in increasing FoxP3+ Treg cell activity and reducing effector T-cells to attenuate atherosclerosis progression and enhance atherosclerosis regression.
3.2.3 Tryptophan-derived metabolites
The amino acid tryptophan can be converted into several downstream bioactive metabolites by myeloid cells. Tryptophan is converted by indoleamine 2,3-dioxygenase 1 (IDO1) to kynurenine, which is the central metabolite to produce other tryptophan-derived molecules. Efferocytosing macrophages can utilise tryptophan derived from the degradation of ACs to generate downstream metabolites. Kynurenine is a key activator of the transcription factor, aryl hydrocarbon receptor (AhR), leading to its release from chaperone proteins that keep AhR in the cytoplasm and its nuclear translocation to bind to gene promoters at aryl hydrocarbon responsive elements (AHREs) (87). AhR activity is known to enhance pro-resolution signaling in macrophages (88). In efferocytosing macrophages, activation of AhR increased the production of pro-resolving mediators such as TGF-β1 and IL-10 and suppressed the production of pro-inflammatory cytokines such as TNF-α and IL-1β (88). In a mouse model of lupus, AhR inhibition with the antagonist CH223191 was found to exacerbate autoimmunity (88).
Metabolites such as kynurenine can be exported and imported through transporters such as SLC7A5 (LAT-1), SLC7A11, or SLC36A4 (PAT-4) by myeloid cells and T-cells (89–91). Kynurenine and related metabolites play an important role in the differentiation and activity of T-cells. Macrophage-derived tryptophan metabolites can suppress the proliferation of activated T-cells (92), and the uptake of extracellular IDO1-dependent tryptophan metabolites by CD8+ T-cells was found to be necessary for their proliferation following activation (90). In the regulation of T-cell differentiation, kynurenine can be imported to activate AhR and induce a pro-resolving phenotype by enhancing the Treg cell-specific transcription factor, FoxP3 (93, 94). However, some evidence suggests that AhR can promote both Treg cell and pro-inflammatory TH17 differentiation depending on the context (95).
Tryptophan-derived metabolites play a role in the pathogenesis of atherosclerosis. In mice, IDO1 activity in dendritic cells and Treg cell activity are mutually supportive in reducing atherosclerosis (96). This suggests a role of secreted tryptophan-derived metabolites from dendritic cells to enhance Treg cell pro-resolving functions and a role of Treg cells in increasing the production of tryptophan-derived metabolites (96). In human atherosclerosis, a decrease in the expression of enzymes within the plaque that are necessary to generate AhR ligands is correlated with an unstable plaque and exacerbation of atherosclerosis (97).
3.3 Lipid mediators
Efferocytosis promotes the production and secretion of a group of lipid pro-resolving mediators called SPMs, which are synthesized from arachidonic acid (AA) and docosahexaenoic acid (DHA). Pro-inflammatory lipid mediators can also be generated by the same precursors depending on the activity of downstream enzymes, and the ratio of pro-resolving:inflammatory mediators is a determinant of whether inflammation resolution and tissue repair versus inflammatory tissue damage occurs. Prominent SPMs produced by efferocytosing myeloid cells include resolvin D1 (RvD1), lipoxin A4 (LXA4), and prostaglandin E2 (PGE2) (64, 98, 99). Although PGE2 signaling has been shown to have pro-resolving effects in the context of efferocytosis, it has also been shown to have a pro-inflammatory effect in other contexts (64, 100, 101). RvD1, LXA4, and PGE2 have been shown to act on other myeloid cells in an autocrine-paracrine manner to promote a pro-resolving and pro-efferocytic phenotype (64, 100, 102–105). Efferocytosis-induced PGE2 can also act on dendritic cells to inhibit their maturation and antigen presentation capacity, which prevents the activation and proliferation of T-cells in autoimmunity (106). SPMs are also able to mediate pro-resolving signaling in T-cells. RvD1 and LXA4 signaling can enhance T-cell differentiation to Treg cells while suppressing the differentiation to pro-inflammatory TH1 and TH17 cells (107, 108). However, PGE2 signaling through the EP2 and EP4 receptors resulted in the inhibition of TGF-β-mediated FoxP3 expression in naïve T-cells, which blocked Treg cell differentiation, and enhanced pro-inflammatory TH1 and TH17 differentiation (109–111). Thus, PGE2 seems to have antagonistic effects suppressing pro-inflammatory polarization in macrophages while supporting a pro-inflammatory phenotype in T-cells.
In atherosclerosis, SPM levels are known to decrease in both mice and humans (112, 113). Specifically, a decrease in the ratio of SPMs to pro-inflammatory lipid mediators is associated with plaque instability and plaque progression (112, 113). Increasing this ratio by exogenous administration of RvD1 was found to enhance features of plaque stability in mice (112). As mentioned above, the effects of PGE2 are complex, and this applies to atherosclerosis (114, 115), i.e., the relative balance or its ability to promote a pro-resolving phenotype in myeloid cells and to polarize T cells toward a TH1/TH17 cell versus Treg cell phenotypes. In addition, PGE2 may have effects on other cell types within the plaque that influence atherosclerosis progression or regression.
4 The role of cell-cell contacts between efferocytes and T-cells
Macrophages can affect T cells through cell-cell contact involving interaction between the checkpoint proteins on the two cell types, notably PD-L1 and PD-1, and cytotoxic T-lymphocyte associated protein 4 (CTLA-4). The role of cell-cell contacts in efferocytosing myeloid cell-lymphocyte crosstalk is summarized in Figure 2. PD-L1 is expressed more ubiquitously including on macrophages and dendritic cells, whereas PD-1 is primarily expressed on T-cells (116). Interaction between PD-L1 and PD-1 can suppress the activation of effector CD4+ and CD8+ T-cells while promoting the differentiation of naïve CD4+ T-cells to FoxP3+ Treg cells and sustaining Treg cell activity, in part by upregulating and sustaining FoxP3 expression (16, 117). Through MerTK-p38-STAT3 signaling, macrophages upregulate PD-L1 upon binding ACs (118). Moreover, PD-1 expression on FoxP3+ Treg cells and cytotoxic CD8+ T-cells, and PD-L1 expression on macrophages can be upregulated by secreted factors from efferocytosing macrophages such as lactate (119), TGF-β1 (120), and tryptophan-derived metabolites (91).
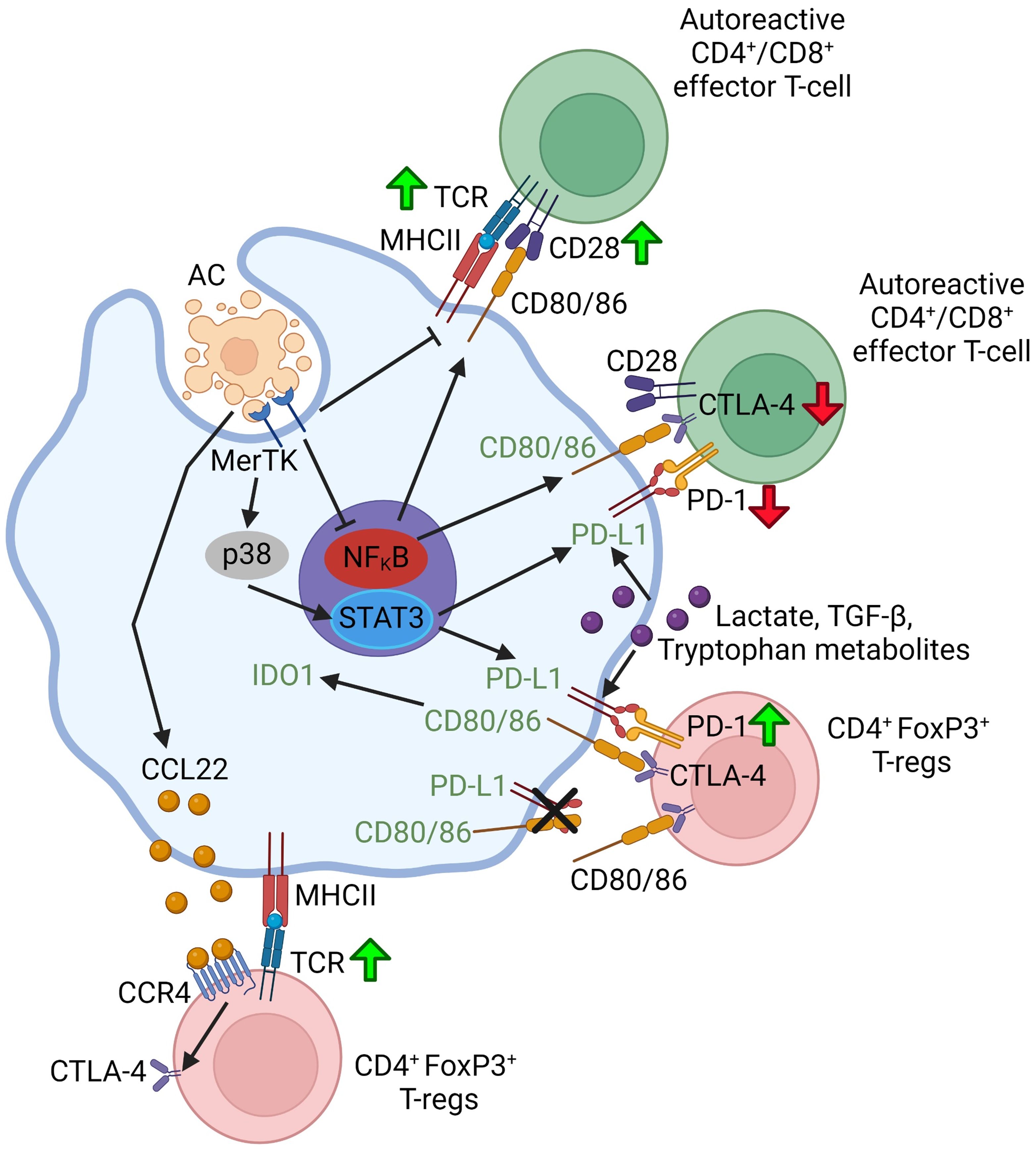
Figure 2 Schematic summarizing efferocytosing myeloid cell-T-lymphocyte crosstalk through cell-cell contact. Green up-arrows indicate that a particular molecular interaction activates the function of the indicated T-lymphocyte subset or promotes the differentiation of naïve T-lymphocytes to that subset. Red down-arrows in T-cells indicate an inhibition of function or differentiation. Green font in the myeloid cell indicate that a particular molecular interaction promotes a pro-resolving myeloid cell phenotype or function. Figure generated with BioRender.com.
There is also evidence that efferocytosis by myeloid cells may also induce the expression of another immune checkpoint protein, CTLA-4, on Treg cells. CTLA-4 is a cell surface protein that is expressed principally on T-cells, but it can also be expressed by other immune and non-immune cell types such as myeloid cells and tumor cells. CTLA-4 expression is induced and sustained on Treg cells and memory T-cells following the interaction of the T-cell receptor (TCR) and CD28 on activated T cells with the MHC-peptide complex and CD80/86 on antigen-presenting cells, such as macrophages and dendritic cells (121). CTLA-4 suppresses signaling downstream of the TCR to shut off T-cell proliferation and the T-cell response in effector T-cells (122, 123). On Treg cells, CTLA-4 expression is critical to their tolerogenic and immunosuppressive function in several ways (124–126). Firstly, CTLA-4 can compete with CD28 for binding to CD80/86 on antigen-presenting cells to reduce T-cell activity (127). Secondly, CTLA-4 can mediate “transendocytosis” of CD80/CD86, which is the process by which Treg cells engulf and remove CD80/86 from the antigen-presenting cell (125, 128, 129). This degradation can prevent CD28 co-stimulation as well as disrupt cis-CD80-PD-L1 dimer interactions on the antigen presenting cell (125, 128, 129). CD80 dimerization with PD-L1 prevents PD-1 on other cell types such as T-cells from interacting with PD-L1 to induce tolerogenic signaling, and thus disruption of the dimers allows for restoration of PD-L1-PD-1 signaling (125). Thirdly, administration of CTLA-4-Ig, which mimics CTLA-4 binding and its interaction with macrophage CD80/86, can suppress macrophage pro-inflammatory cytokine production and promote phenotypic switching to a pro-resolving phenotype (130, 131). Although not directly shown using Treg cells, this may be a potential mechanism for Treg cell-mediated inflammation resolution. Finally, Treg cell CTLA-4 can induce dendritic cells to express IDO1 and increase tryptophan catabolism in a CD80/86-dependent manner (132). This increase in IDO1 (a) enhances the secretion of kynurenine and other tryptophan-derived metabolites by dendritic cells to modulate efferocytosing myeloid cells and T-cells as described previously; and (b) suppresses antigen presentation by dendritic cells (132). In the context of efferocytosis, CD169+ splenic marginal zone macrophages upregulated the secretion of CCL22, the chemotactic ligand for CCR4, following the uptake of apoptotic thymocytes in vitro (133). In vivo, injection of ACs into mice upregulated CCL22 secretion by CD169+ splenic macrophages, and Treg cells were found to migrate into the spleen and increase CTLA-4 expression in a CCR4-dependent manner (133). These findings suggest that CCL22-CCR4 signaling or signaling by another secreted factor produced by efferocytosing macrophages, can increase CTLA-4 expression on Treg cells. Moreover, CTLA-4 can be upregulated in T cells, including Treg cells, by pro-resolving lactate and TGF-β (120, 134), but more work is needed to understand how CTLA-4 expression is affected by efferocytosing myeloid cells and how T-cell CTLA-4 signals back to efferocytosing myeloid cells.
In murine models of atherosclerosis progression, whole-body knockouts of either PD-L1 or PD-1 resulted in an exacerbation of atherosclerosis progression as measured by plaque area (135, 136). This corresponded with an increase in the number of macrophages, CD4+ T-cells, and CD8+ T-cells (135, 136). In vitro, myeloid cells from PD-L1 KO mice could more readily activate T-cells following antigen presentation, and T-cells from PD-1 KO mice were more inflammatory and cytotoxic in the case of CD8+ T-cells (135, 136). Interestingly, hypercholesterolemia increased PD-L1 expression on myeloid cells and increased PD-1 expression on T-cells, suggesting a possible compensatory response in which PD-L1-PD-1 interaction restrains the inflammatory response during atherosclerosis progression (135, 136). In this context, cancer patients who receive blocking antibodies targeting PD-L1 or PD-1 have an aggravation of atherosclerotic cardiovascular disease (137).
CTLA-4 was also found to have an anti-atherogenic effect in mice and humans (138–141). CTLA-4 blocking antibodies, similar to PD-L1/PD-1 blocking antibodies, are used to treat cancer, which is associated with an increased risk of coronary artery disease (138, 141). In Western diet-fed Ldlr-/- mice, CTLA-4 blockade increased effector CD4+ and CD8+ T-cells in the blood, spleen, and plaque, increased plaque size, and decreased features of plaque stability based on increased necrotic core size and thinner fibrous caps (139). Conversely, transgenic T-cell overexpression of CTLA-4 resulted in an attenuation of atherosclerotic plaque size in the aortic root, with a reduced accumulation of macrophages and effector T-cells in the plaque (140). Furthermore, dendritic cell maturation was inhibited as determined by reduced CD80/86 expression, although this could at least in part be a result of enhanced transendocytosis of CD80/86 by Treg cells (140).
5 Conclusion
Both myeloid cells and T-cells play important roles in affecting the plaque microenvironment in atherosclerosis progression and regression by modulating inflammation and other immune processes. Significant crosstalk occurs between myeloid cells and T-cells to influence the phenotype and behavior of both groups of immune cells. Efferocytosis by myeloid cells is a critical component of inflammation resolution and tissue repair, affecting the metabolism, secretome, and gene expression of macrophages and dendritic cells. These changes in efferocytes can lead to altered interactions between myeloid cells and T-cells and phenotypic alterations in T-cells. T-cells can also feedback to regulate efferocytes, thus contributing to the perpetuation or disruption of the efferocytosis-resolution cycle. In atherosclerosis, impaired efferocytosis exacerbates inflammation and increased plaque destabilization, whereas improved efferocytosis in atherosclerosis regression reversed these changes. Although the pro-tolerogenic function of myeloid cell efferocytosis and its associated downstream signals, e.g., secreted lactate or kynurenine, are beneficial in the context of chronic inflammation and atherosclerosis progression, this process can also contribute to immunosuppression in cancer, leading to cancer cell evasion of the immune system (142–145). Thus, conceptualizing methods to target the crosstalk between efferocytosing myeloid cells and T-cells for the treatment of atherosclerosis must also consider that excessive immunosuppression may result in impaired anti-tumor immunity. Thus, a deeper understanding of efferocyte-T-cell crosstalk is crucial to better understand how to achieve a balance between pro-inflammatory signaling and pro-resolution signaling. Future analysis of publicly available datasets or using tools such as CellChat to analyse cell-cell interactions could be used to identify candidate signaling pathways to target to better achieve the balance (146).
In this review, we introduce mechanisms by which efferocyte-T-cell crosstalk through antigen presentation, secreted factors, and cell-cell contact by ligand-receptor interactions could contribute to the benefits of efferocytosis on inflammation resolution and plaque stability. Much remains to be discovered about this interplay, and further work in this area has the potential to provide new insights into the pathogenesis of atherosclerosis and to suggest novel therapeutic targets.
Author contributions
DN: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. SS: Investigation, Writing – review & editing. IT: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by an American Heart Association Postdoctoral Fellowship (24POST1192241 to DN) and NIH/NHLBI grant R35-HL145228 (to IT).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hansson GK, Libby P, Schönbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. (2002) 91:281–91. doi: 10.1161/01.res.0000029784.15893.10
2. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. (2019) 5:56. doi: 10.1038/s41572-019-0106-z
3. Barrett TJ. Macrophages in atherosclerosis regression. Arterioscler Thromb Vasc Biol. (2020) 40:20–33. doi: 10.1161/ATVBAHA.119.312802
4. Kojima Y, Weissman IL, Leeper NJ. The role of efferocytosis in atherosclerosis. Circulation. (2017) 135:476–89. doi: 10.1161/CIRCULATIONAHA.116.025684
5. Yurdagul A, Doran AC, Cai B, Fredman G, Tabas IA. Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front Cardiovasc Med. (2017) 4:86. doi: 10.3389/fcvm.2017.00086
6. Tabas I, Bornfeldt KE. Intracellular and intercellular aspects of macrophage immunometabolism in atherosclerosis. Circ Res. (2020) 126:1209–27. doi: 10.1161/CIRCRESAHA.119.315939
7. Pencina MJ, Pencina KM, Lloyd-Jones D, Catapano AL, Thanassoulis G, Sniderman AD. The expected 30-year benefits of early versus delayed primary prevention of cardiovascular disease by lipid lowering. Circulation. (2020) 142:827–37. doi: 10.1161/CIRCULATIONAHA.120.045851
8. Li Y, Deng S, Liu B, Yan Y, Du J, Jing X, et al. The effects of lipid-lowering therapy on coronary plaque regression: a systematic review and meta-analysis. Sci Rep. (2021) 11:7999. doi: 10.1038/s41598-021-87528-w
9. Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. (2020) 17:387–401. doi: 10.1038/s41569-020-0352-5
10. Lacy M, Bürger C, Shami A, Ahmadsei M, Winkels H, Nitz K, et al. Cell-specific and divergent roles of the CD40L-CD40 axis in atherosclerotic vascular disease. Nat Commun. (2021) 12:3754. doi: 10.1038/s41467-021-23909-z
11. Gisterå A, Klement ML, Polyzos KA, Mailer RKW, Duhlin A, Karlsson MCI, et al. Low-density lipoprotein-reactive T cells regulate plasma cholesterol levels and development of atherosclerosis in humanized hypercholesterolemic mice. Circulation. (2018) 138:2513–26. doi: 10.1161/CIRCULATIONAHA.118.034076
12. Mallat Z, Binder CJ. The why and how of adaptive immune responses in ischemic cardiovascular disease. Nat Cardiovasc Res. (2022) 1:431–44. doi: 10.1038/s44161-022-00049-1
13. Hoebe K, Janssen E, Beutler B. The interface between innate and adaptive immunity. Nat Immunol. (2004) 5:971–4. doi: 10.1038/ni1004-971
14. Bonomo AC, Pinto-Mariz F, Riederer I, Benjamim CF, Butler-Browne G, Mouly V, et al. Crosstalk between innate and T cell adaptive immunity with(in) the muscle. Front Physiol. (2020) 11:573347. doi: 10.3389/fphys.2020.573347
15. Sun L, Wang X, Saredy J, Yuan Z, Yang X, Wang H. Innate-adaptive immunity interplay and redox regulation in immune response. Redox Biol. (2020) 37:101759. doi: 10.1016/j.redox.2020.101759
16. Diskin B, Adam S, Cassini MF, Sanchez G, Liria M, Aykut B, et al. PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat Immunol. (2020) 21:442–54. doi: 10.1038/s41590-020-0620-x
17. Proto JD, Doran AC, Gusarova G, Yurdagul A, Sozen E, Subramanian M, et al. Regulatory T cells promote macrophage efferocytosis during inflammation resolution. Immunity. (2018) 49:666–677.e6. doi: 10.1016/j.immuni.2018.07.015
18. Hopp AK, Rupp A, Lukacs-Kornek V. Self-antigen presentation by dendritic cells in autoimmunity. Front Immunol. (2014) 5:55. doi: 10.3389/fimmu.2014.00055
19. Tai Y, Wang Q, Korner H, Zhang L, Wei W. Molecular mechanisms of T cells activation by dendritic cells in autoimmune diseases. Front Pharmacol. (2018) 9:642. doi: 10.3389/fphar.2018.00642
20. Suurmond J, Diamond B. Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest. (2015) 125:2194–202. doi: 10.1172/JCI78084
21. Singh B, Li K, Cui K, Peng Q, Cowan DB, Wang DZ, et al. Defective efferocytosis of vascular cells in heart disease. Front Cardiovasc Med. (2022) 9:1031293. doi: 10.3389/fcvm.2022.1031293
22. Cai B, Thorp EB, Doran AC, Sansbury BE, Daemen MJ, Dorweiler B, et al. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J Clin Invest. (2017) 127:564–8. doi: 10.1172/JCI90520
23. Tian L, Choi SC, Lee HN, Murakami Y, Qi CF, Sengottuvelu M, et al. Enhanced efferocytosis by dendritic cells underlies memory T-cell expansion and susceptibility to autoimmune disease in CD300f-deficient mice. Cell Death Differ. (2016) 23:1086–96. doi: 10.1038/cdd.2015.161
24. Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res. (2019) 124:315–27. doi: 10.1161/CIRCRESAHA.118.313591
25. McLeod O, Silveira A, Fredrikson GN, Gertow K, Baldassarre D, Veglia F, et al. Plasma autoantibodies against apolipoprotein B-100 peptide 210 in subclinical atherosclerosis. Atherosclerosis. (2014) 232:242–8. doi: 10.1016/j.atherosclerosis.2013.11.041
26. Stone JD, Chervin AS, Kranz DM. T-cell receptor binding affinities and kinetics: impact on T-cell activity and specificity. Immunology. (2009) 126:165–76. doi: 10.1111/j.1365-2567.2008.03015.x
27. Au-Yeung BB, Zikherman J, Mueller JL, Ashouri JF, Matloubian M, Cheng DA, et al. A sharp T-cell antigen receptor signaling threshold for T-cell proliferation. Proc Natl Acad Sci U.S.A. (2014) 111:E3679–88. doi: 10.1073/pnas.1413726111
28. Georgiev P, Charbonnier LM, Chatila TA. Regulatory T cells: the many faces of foxp3. J Clin Immunol. (2019) 39:623–40. doi: 10.1007/s10875-019-00684-7
29. Charaix J, Borelli A, Santamaria JC, Chasson L, Giraud M, Sergé A, et al. Recirculating Foxp3+ regulatory T cells are restimulated in the thymus under Aire control. Cell Mol Life Sci. (2022) 79:355. doi: 10.1007/s00018-022-04328-9
30. Schildknecht A, Brauer S, Brenner C, Lahl K, Schild H, Sparwasser T, et al. FoxP3+ regulatory T cells essentially contribute to peripheral CD8+ T-cell tolerance induced by steady-state dendritic cells. Proc Natl Acad Sci U.S.A. (2010) 107:199–203. doi: 10.1073/pnas.0910620107
31. Mallat Z, Gojova A, Brun V, Esposito B, Fournier N, Cottrez F, et al. Induction of a regulatory T cell type 1 response reduces the development of atherosclerosis in apolipoprotein E-knockout mice. Circulation. (2003) 108:1232–7. doi: 10.1161/01.CIR.0000089083.61317.A1
32. Sharma M, Schlegel MP, Afonso MS, Brown EJ, Rahman K, Weinstock A, et al. Regulatory T cells license macrophage pro-resolving functions during atherosclerosis regression. Circ Res. (2020) 127:335–53. doi: 10.1161/CIRCRESAHA.119.316461
33. Sen P, Wallet MA, Yi Z, Huang Y, Henderson M, Mathews CE, et al. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-kappaB activation in dendritic cells. Blood. (2007) 109:653–60. doi: 10.1182/blood-2006-04-017368
34. Chattopadhyay G, Shevach EM. Antigen-specific induced T regulatory cells impair dendritic cell function via an IL-10/MARCH1-dependent mechanism. J Immunol. (2013) 191:5875–84. doi: 10.4049/jimmunol.1301693
35. Koltsova EK, Garcia Z, Chodaczek G, Landau M, McArdle S, Scott SR, et al. Dynamic T cell-APC interactions sustain chronic inflammation in atherosclerosis. J Clin Invest. (2012) 122:3114–26. doi: 10.1172/JCI61758
36. Angelovich TA, Hearps AC, Jaworowski A. Inflammation-induced foam cell formation in chronic inflammatory disease. Immunol Cell Biol. (2015) 93:683–93. doi: 10.1038/icb.2015.26
37. Zhou F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int Rev Immunol. (2009) 28:239–60. doi: 10.1080/08830180902978120
38. Hallermalm K, Seki K, Wei C, Castelli C, Rivoltini L, Kiessling R, et al. Tumor necrosis factor-alpha induces coordinated changes in major histocompatibility class I presentation pathway, resulting in increased stability of class I complexes at the cell surface. Blood. (2001) 98:1108–15. doi: 10.1182/blood.v98.4.1108
39. Yin C, Kim Y, Argintaru D, Heit B. Rab17 mediates differential antigen sorting following efferocytosis and phagocytosis. Cell Death Dis. (2016) 7:e2529. doi: 10.1038/cddis.2016.431
40. Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. (2006) 440:808–12. doi: 10.1038/nature04596
41. Nair-Gupta P, Baccarini A, Tung N, Seyffer F, Florey O, Huang Y, et al. TLR signals induce phagosomal MHC-I delivery from the endosomal recycling compartment to allow cross-presentation. Cell. (2014) 158:506–21. doi: 10.1016/j.cell.2014.04.054
42. Yin C, Heit B. Cellular responses to the efferocytosis of apoptotic cells. Front Immunol. (2021) 12:631714. doi: 10.3389/fimmu.2021.631714
43. Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. (2001) 104:3103–8. doi: 10.1161/hc5001.100631
44. Yang K, Wang X, Liu Z, Lu L, Mao J, Meng H, et al. Oxidized low-density lipoprotein promotes macrophage lipid accumulation via the toll-like receptor 4-Src pathway. Circ J. (2015) 79:2509–16. doi: 10.1253/circj.CJ-15-0345
45. Liu J, Zhang X, Cheng Y, Cao X. Dendritic cell migration in inflammation and immunity. Cell Mol Immunol. (2021) 18:2461–71. doi: 10.1038/s41423-021-00726-4
46. Wigren M, Rattik S, Yao Mattisson I, Tomas L, Grönberg C, Söderberg I, et al. Lack of ability to present antigens on major histocompatibility complex class II molecules aggravates atherosclerosis in apoE. Circulation. (2019) 139:2554–66. doi: 10.1161/CIRCULATIONAHA.118.039288
47. McLachlan JB, Catron DM, Moon JJ, Jenkins MK. Dendritic cell antigen presentation drives simultaneous cytokine production by effector and regulatory T cells in inflamed skin. Immunity. (2009) 30:277–88. doi: 10.1016/j.immuni.2008.11.013
48. Subramanian M, Thorp E, Hansson GK, Tabas I. Treg-mediated suppression of atherosclerosis requires MYD88 signaling in DCs. J Clin Invest. (2013) 123:179–88. doi: 10.1172/JCI64617
49. Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. (2005) 201:1061–7. doi: 10.1084/jem.20042276
50. Konkel JE, Jin W, Abbatiello B, Grainger JR, Chen W. Thymocyte apoptosis drives the intrathymic generation of regulatory T cells. Proc Natl Acad Sci U.S.A. (2014) 111:E465–73. doi: 10.1073/pnas.1320319111
51. Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. (2009) 10:1178–84. doi: 10.1038/ni.1791
52. Ishigame H, Zenewicz LA, Sanjabi S, Licona-Limón P, Nakayama M, Leonard WJ, et al. Excessive Th1 responses due to the absence of TGF-β signaling cause autoimmune diabetes and dysregulated Treg cell homeostasis. Proc Natl Acad Sci U.S.A. (2013) 110:6961–6. doi: 10.1073/pnas.1304498110
53. Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. (1989) 170:2081–95. doi: 10.1084/jem.170.6.2081
54. Park IK, Shultz LD, Letterio JJ, Gorham JD. TGF-beta1 inhibits T-bet induction by IFN-gamma in murine CD4+ T cells through the protein tyrosine phosphatase Src homology region 2 domain-containing phosphatase-1. J Immunol. (2005) 175:5666–74. doi: 10.4049/jimmunol.175.9.5666
55. Chen ZM, O’Shaughnessy MJ, Gramaglia I, Panoskaltsis-Mortari A, Murphy WJ, Narula S, et al. IL-10 and TGF-beta induce alloreactive CD4+CD25- T cells to acquire regulatory cell function. Blood. (2003) 101:5076–83. doi: 10.1182/blood-2002-09-2798
56. Zheng SG, Wang JH, Gray JD, Soucier H, Horwitz DA. Natural and induced CD4+CD25+ cells educate CD4+CD25- cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J Immunol. (2004) 172:5213–21. doi: 10.4049/jimmunol.172.9.5213
57. Huss DJ, Winger RC, Peng H, Yang Y, Racke MK, Lovett-Racke AE. TGF-beta enhances effector Th1 cell activation but promotes self-regulation via IL-10. J Immunol. (2010) 184:5628–36. doi: 10.4049/jimmunol.1000288
58. Filippi CM, Juedes AE, Oldham JE, Ling E, Togher L, Peng Y, et al. Transforming growth factor-beta suppresses the activation of CD8+ T-cells when naive but promotes their survival and function once antigen experienced: a two-faced impact on autoimmunity. Diabetes. (2008) 57:2684–92. doi: 10.2337/db08-0609
59. Smith LK, Boukhaled GM, Condotta SA, Mazouz S, Guthmiller JJ, Vijay R, et al. Interleukin-10 directly inhibits CD8+ T cell function by enhancing N-glycan branching to decrease antigen sensitivity. Immunity. (2018) 48:299–312.e5. doi: 10.1016/j.immuni.2018.01.006
60. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. (2019) 25:1576–88. doi: 10.1038/s41591-019-0590-4
61. Kyaw T, Winship A, Tay C, Kanellakis P, Hosseini H, Cao A, et al. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in apoE-deficient mice. Circulation. (2013) 127:1028–39. doi: 10.1161/CIRCULATIONAHA.112.001347
62. Cochain C, Koch M, Chaudhari SM, Busch M, Pelisek J, Boon L, et al. CD8+ T cells regulate monopoiesis and circulating ly6C-high monocyte levels in atherosclerosis in mice. Circ Res. (2015) 117:244–53. doi: 10.1161/CIRCRESAHA.117.304611
63. O’Garra A, Vieira P. Regulatory T cells and mechanisms of immune system control. Nat Med. (2004) 10:801–5. doi: 10.1038/nm0804-801
64. Ampomah PB, Cai B, Sukka SR, Gerlach BD, Yurdagul A, Wang X, et al. Macrophages use apoptotic cell-derived methionine and DNMT3A during efferocytosis to promote tissue resolution. Nat Metab. (2022) 4:444–57. doi: 10.1038/s42255-022-00551-7
65. Zhang S, Weinberg S, DeBerge M, Gainullina A, Schipma M, Kinchen JM, et al. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab. (2019) 29:443–456.e5. doi: 10.1016/j.cmet.2018.12.004
66. Morioka S, Perry JSA, Raymond MH, Medina CB, Zhu Y, Zhao L, et al. Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature. (2018) 563:714–8. doi: 10.1038/s41586-018-0735-5
67. Schilperoort M, Ngai D, Katerelos M, Power DA, Tabas I. PFKFB2-mediated glycolysis promotes lactate-driven continual efferocytosis by macrophages. Nat Metab. (2023) 5(3):431-44. doi: 10.1038/s42255-023-00736-8
68. Ngai D, Schilperoort M, Tabas I. Efferocytosis-induced lactate enables the proliferation of pro-resolving macrophages to mediate tissue repair. Nat Metab. (2023) 5(12):2206-91. doi: 10.1038/s42255-023-00921-9
69. Comito G, Iscaro A, Bacci M, Morandi A, Ippolito L, Parri M, et al. Lactate modulates CD4+ T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene. (2019) 38:3681–95. doi: 10.1038/s41388-019-0688-7
70. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature. (2021) 591:645–51. doi: 10.1038/s41586-020-03045-2
71. Gu J, Zhou J, Chen Q, Xu X, Gao J, Li X, et al. Tumor metabolite lactate promotes tumorigenesis by modulating MOESIN lactylation and enhancing TGF-β signaling in regulatory T cells. Cell Rep. (2022) 40:111122. doi: 10.1016/j.celrep.2022.111122
72. Quinn WJ, Jiao J, TeSlaa T, Stadanlick J, Wang Z, Wang L, et al. Lactate limits T cell proliferation via the NAD(H) redox state. Cell Rep. (2020) 33:108500. doi: 10.1016/j.celrep.2020.108500
73. Pucino V, Certo M, Bulusu V, Cucchi D, Goldmann K, Pontarini E, et al. Lactate buildup at the site of chronic inflammation promotes disease by inducing CD4+ T cell metabolic rewiring. Cell Metab. (2019) 30:1055–1074.e8. doi: 10.1016/j.cmet.2019.10.004
74. Shantha GP, Wasserman B, Astor BC, Coresh J, Brancati F, Sharrett AR, et al. Association of blood lactate with carotid atherosclerosis: the Atherosclerosis Risk in Communities (ARIC) Carotid MRI Study. Atherosclerosis. (2013) 228:249–55. doi: 10.1016/j.atherosclerosis.2013.02.014
75. Morris SM Jr. Enzymes of arginine metabolism. J Nutr. (2004) 134:2743S–2747S;discussion 2765S-2767S. doi: 10.1093/jn/134.10.2743S
76. Yurdagul A Jr., Subramanian M, Wang X, Crown SB, Ilkayeva OR, Darville L, et al. Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab. (2020) 31:518–533.e10. doi: 10.1016/j.cmet.2020.01.001
77. McCubbrey AL, McManus SA, McClendon JD, Thomas SM, Chatwin HB, Reisz JA, et al. Polyamine import and accumulation causes immunomodulation in macrophages engulfing apoptotic cells. Cell Rep. (2022) 38:110222. doi: 10.1016/j.celrep.2021.110222
78. Uemura T, Yerushalmi HF, Tsaprailis G, Stringer DE, Pastorian KE, Hawel L, et al. Identification and characterization of a diamine exporter in colon epithelial cells. J Biol Chem. (2008) 283:26428–35. doi: 10.1074/jbc.M804714200
79. Carriche GM, Almeida L, Stüve P, Velasquez L, Dhillon-LaBrooy A, Roy U, et al. Regulating T-cell differentiation through the polyamine spermidine. J Allergy Clin Immunol. (2021) 147:335–348.e11. doi: 10.1016/j.jaci.2020.04.037
80. Puleston DJ, Baixauli F, Sanin DE, Edwards-Hicks J, Villa M, Kabat AM, et al. Polyamine metabolism is a central determinant of helper T cell lineage fidelity. Cell. (2021) 184:4186–4202.e20. doi: 10.1016/j.cell.2021.06.007
81. Wu R, Chen X, Kang S, Wang T, Gnanaprakasam JR, Yao Y, et al. De novo synthesis and salvage pathway coordinately regulate polyamine homeostasis and determine T cell proliferation and function. Sci Adv. (2020) 6:eabc4275. doi: 10.1126/sciadv.abc4275
82. Hibino S, Eto S, Hangai S, Endo K, Ashitani S, Sugaya M, et al. Tumor cell-derived spermidine is an oncometabolite that suppresses TCR clustering for intratumoral CD8+ T cell activation. Proc Natl Acad Sci U.S.A. (2023) 120:e2305245120. doi: 10.1073/pnas.2305245120
83. Elmarsafawi AG, Hesterberg RS, Fernandez MR, Yang C, Darville LN, Liu M, et al. Modulating the polyamine/hypusine axis controls generation of CD8+ tissue-resident memory T cells. JCI Insight. (2023) 8:e169308. doi: 10.1172/jci.insight.169308
84. Wang XP, Zhang W, Liu XQ, Wang WK, Yan F, Dong WQ, et al. Arginase I enhances atherosclerotic plaque stabilization by inhibiting inflammation and promoting smooth muscle cell proliferation. Eur Heart J. (2014) 35:911–9. doi: 10.1093/eurheartj/eht329
85. Li B, Jones LL, Geiger TL. IL-6 promotes T cell proliferation and expansion under inflammatory conditions in association with low-level RORγt expression. J Immunol. (2018) 201:2934–46. doi: 10.4049/jimmunol.1800016
86. Alam MS, Otsuka S, Wong N, Abbasi A, Gaida MM, Fan Y, et al. TNF plays a crucial role in inflammation by signaling via T cell TNFR2. Proc Natl Acad Sci U.S.A. (2021) 118:e2109972118. doi: 10.1073/pnas.2109972118
87. Kaiser H, Parker E, Hamrick MW. Kynurenine signaling through the aryl hydrocarbon receptor: Implications for aging and healthspan. Exp Gerontol. (2020) 130:110797. doi: 10.1016/j.exger.2019.110797
88. Shinde R, Hezaveh K, Halaby MJ, Kloetgen A, Chakravarthy A, da Silva Medina T, et al. Apoptotic cell-induced AhR activity is required for immunological tolerance and suppression of systemic lupus erythematosus in mice and humans. Nat Immunol. (2018) 19:571–82. doi: 10.1038/s41590-018-0107-1
89. Sinclair LV, Neyens D, Ramsay G, Taylor PM, Cantrell DA. Single cell analysis of kynurenine and System L amino acid transport in T cells. Nat Commun. (2018) 9:1981. doi: 10.1038/s41467-018-04366-7
90. Fiore A, Zeitler L, Russier M, Groß A, Hiller MK, Parker JL, et al. Kynurenine importation by SLC7A11 propagates anti-ferroptotic signaling. Mol Cell. (2022) 82:920–932.e7. doi: 10.1016/j.molcel.2022.02.007
91. Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, et al. Tumor-repopulating cells induce PD-1 expression in CD8+ T cells by transferring kynurenine and ahR activation. Cancer Cell. (2018) 33:480–494.e7. doi: 10.1016/j.ccell.2018.02.005
92. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. (1999) 189:1363–72. doi: 10.1084/jem.189.9.1363
93. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. (2010) 185:3190–8. doi: 10.4049/jimmunol.0903670
94. Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol. (2010) 11:846–53. doi: 10.1038/ni.1915
95. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. (2008) 453:65–71. doi: 10.1038/nature06880
96. Forteza MJ, Polyzos KA, Baumgartner R, Suur BE, Mussbacher M, Johansson DK, et al. Activation of the regulatory T-cell/indoleamine 2,3-dioxygenase axis reduces vascular inflammation and atherosclerosis in hyperlipidemic mice. Front Immunol. (2018) 9:950. doi: 10.3389/fimmu.2018.00950
97. Baumgartner R, Berg M, Matic L, Polyzos KP, Forteza MJ, Hjorth SA, et al. Evidence that a deviation in the kynurenine pathway aggravates atherosclerotic disease in humans. J Intern Med. (2021) 289:53–68. doi: 10.1111/joim.13142
98. Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. (2012) 120:e60–72. doi: 10.1182/blood-2012-04-423525
99. Cai B, Thorp EB, Doran AC, Subramanian M, Sansbury BE, Lin CS, et al. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc Natl Acad Sci U.S.A. (2016) 113:6526–31. doi: 10.1073/pnas.1524292113
100. Meriwether D, Jones AE, Ashby JW, Solorzano-Vargas RS, Dorreh N, Noori S, et al. Macrophage COX2 mediates efferocytosis, resolution reprogramming, and intestinal epithelial repair. Cell Mol Gastroenterol Hepatol. (2022) 13:1095–120. doi: 10.1016/j.jcmgh.2022.01.002
101. Sheppe AEF, Kummari E, Walker A, Richards A, Hui WW, Lee JH, et al. PGE2 augments inflammasome activation and M1 polarization in macrophages infected with salmonella typhimurium and yersinia enterocolitica. Front Microbiol. (2018) 9:2447. doi: 10.3389/fmicb.2018.02447
102. Börgeson E, Johnson AM, Lee YS, Till A, Syed GH, Ali-Shah ST, et al. Lipoxin A4 attenuates obesity-induced adipose inflammation and associated liver and kidney disease. Cell Metab. (2015) 22:125–37. doi: 10.1016/j.cmet.2015.05.003
103. Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol. (2000) 164:1663–7. doi: 10.4049/jimmunol.164.4.1663
104. Rymut N, Heinz J, Sadhu S, Hosseini Z, Riley CO, Marinello M, et al. Resolvin D1 promotes efferocytosis in aging by limiting senescent cell-induced MerTK cleavage. FASEB J. (2020) 34:597–609. doi: 10.1096/fj.201902126R
105. Titos E, Rius B, González-Périz A, López-Vicario C, Morán-Salvador E, Martínez-Clemente M, et al. Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J Immunol. (2011) 187:5408–18. doi: 10.4049/jimmunol.1100225
106. Pujol-Autonell I, Ampudia RM, Planas R, Marin-Gallen S, Carrascal J, Sanchez A, et al. Efferocytosis promotes suppressive effects on dendritic cells through prostaglandin E2 production in the context of autoimmunity. PloS One. (2013) 8:e63296. doi: 10.1371/journal.pone.0063296
107. Chiurchiù V, Leuti A, Dalli J, Jacobsson A, Battistini L, Maccarrone M, et al. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci Transl Med. (2016) 8:353ra111. doi: 10.1126/scitranslmed.aaf7483
108. Gao Y, Min K, Zhang Y, Su J, Greenwood M, Gronert K. Female-specific downregulation of tissue polymorphonuclear neutrophils drives impaired regulatory T cell and amplified effector T cell responses in autoimmune dry eye disease. J Immunol. (2015) 195:3086–99. doi: 10.4049/jimmunol.1500610
109. Boniface K, Bak-Jensen KS, Li Y, Blumenschein WM, McGeachy MJ, McClanahan TK, et al. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med. (2009) 206:535–48. doi: 10.1084/jem.20082293
110. Yao C, Hirata T, Soontrapa K, Ma X, Takemori H, Narumiya S. Prostaglandin E2 promotes Th1 differentiation via synergistic amplification of IL-12 signalling by cAMP and PI3-kinase. Nat Commun. (2013) 4:1685. doi: 10.1038/ncomms2684
111. Goepp M, Crittenden S, Zhou Y, Rossi AG, Narumiya S, Yao C. Prostaglandin E2 directly inhibits the conversion of inducible regulatory T cells through EP2 and EP4 receptors via antagonizing TGF-β signaling. Immunology. (2021) 164:777–91. doi: 10.1111/imm.13417
112. Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. (2016) 7:12859. doi: 10.1038/ncomms12859
113. Viola JR, Lemnitzer P, Jansen Y, Csaba G, Winter C, Neideck C, et al. Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ Res. (2016) 119:1030–8. doi: 10.1161/CIRCRESAHA.116.309492
114. Wang W, Liang M, Wang L, Bei W, Rong X, Xu J, et al. Role of prostaglandin E2 in macrophage polarization: Insights into atherosclerosis. Biochem Pharmacol. (2023) 207:115357. doi: 10.1016/j.bcp.2022.115357
115. Gomez I, Foudi N, Longrois D, Norel X. The role of prostaglandin E2 in human vascular inflammation. Prostaglandins Leukot Essent Fatty Acids. (2013) 89:55–63. doi: 10.1016/j.plefa.2013.04.004
116. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. (2017) 8:561. doi: 10.3389/fphar.2017.00561
117. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. (2009) 206:3015–29. doi: 10.1084/jem.20090847
118. Lin J, Xu A, Jin J, Zhang M, Lou J, Qian C, et al. MerTK-mediated efferocytosis promotes immune tolerance and tumor progression in osteosarcoma through enhancing M2 polarization and PD-L1 expression. Oncoimmunology. (2022) 11:2024941. doi: 10.1080/2162402X.2021.2024941
119. Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin YT, Togashi Y, et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. (2022) 40:201–218.e9. doi: 10.1016/j.ccell.2022.01.001
120. Bao S, Jiang X, Jin S, Tu P, Lu J. TGF-β1 induces immune escape by enhancing PD-1 and CTLA-4 expression on T lymphocytes in hepatocellular carcinoma. Front Oncol. (2021) 11:694145. doi: 10.3389/fonc.2021.694145
121. Jago CB, Yates J, Câmara NO, Lechler RI, Lombardi G. Differential expression of CTLA-4 among T cell subsets. Clin Exp Immunol. (2004) 136:463–71. doi: 10.1111/j.1365-2249.2004.02478.x
122. Kim GR, Choi JM. Current understanding of cytotoxic T lymphocyte antigen-4 (CTLA-4) signaling in T-cell biology and disease therapy. Mol Cells. (2022) 45:513–21. doi: 10.14348/molcells.2022.2056
123. Brunner MC, Chambers CA, Chan FK, Hanke J, Winoto A, Allison JP. CTLA-4-Mediated inhibition of early events of T cell proliferation. J Immunol. (1999) 162:5813–20. doi: 10.4049/jimmunol.162.10.5813
124. Sojka DK, Hughson A, Fowell DJ. CTLA-4 is required by CD4+CD25+ Treg to control CD4+ T-cell lymphopenia-induced proliferation. Eur J Immunol. (2009) 39:1544–51. doi: 10.1002/eji.200838603
125. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci U.S.A. (2021) 118:e2023739118. doi: 10.1073/pnas.2023739118
126. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. (2008) 322:271–5. doi: 10.1126/science.1160062
127. Pentcheva-Hoang T, Egen JG, Wojnoonski K, Allison JP. B7-1 and B7-2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity. (2004) 21:401–13. doi: 10.1016/j.immuni.2004.06.017
128. Kennedy A, Waters E, Rowshanravan B, Hinze C, Williams C, Janman D, et al. Differences in CD80 and CD86 transendocytosis reveal CD86 as a key target for CTLA-4 immune regulation. Nat Immunol. (2022) 23:1365–78. doi: 10.1038/s41590-022-01289-w
129. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. (2011) 332:600–3. doi: 10.1126/science.1202947
130. Cutolo M, Soldano S, Montagna P, Sulli A, Seriolo B, Villaggio B, et al. CTLA4-Ig interacts with cultured synovial macrophages from rheumatoid arthritis patients and downregulates cytokine production. Arthritis Res Ther. (2009) 11:R176. doi: 10.1186/ar2865
131. Cutolo M, Soldano S, Gotelli E, Montagna P, Campitiello R, Paolino S, et al. CTLA4-Ig treatment induces M1-M2 shift in cultured monocyte-derived macrophages from healthy subjects and rheumatoid arthritis patients. Arthritis Res Ther. (2021) 23:306. doi: 10.1186/s13075-021-02691-9
132. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. (2003) 4:1206–12. doi: 10.1038/ni1003
133. Ravishankar B, Shinde R, Liu H, Chaudhary K, Bradley J, Lemos HP, et al. Marginal zone CD169+ macrophages coordinate apoptotic cell-driven cellular recruitment and tolerance. Proc Natl Acad Sci U.S.A. (2014) 111:4215–20. doi: 10.1073/pnas.1320924111
134. Ding R, Yu X, Hu Z, Dong Y, Huang H, Zhang Y, et al. Lactate modulates RNA splicing to promote CTLA-4 expression in tumor-infiltrating regulatory T cells. Immunity. (2024) 57(3):528-40.e6. doi: 10.1016/j.immuni.2024.01.019
135. Gotsman I, Grabie N, Dacosta R, Sukhova G, Sharpe A, Lichtman AH. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J Clin Invest. (2007) 117:2974–82. doi: 10.1172/JCI31344
136. Bu DX, Tarrio M, Maganto-Garcia E, Stavrakis G, Tajima G, Lederer J, et al. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler Thromb Vasc Biol. (2011) 31:1100–7. doi: 10.1161/ATVBAHA.111.224709
137. Dong M, Yu T, Tse G, Lin Z, Lin C, Zhang N, et al. PD-1/PD-L1 blockade accelerates the progression of atherosclerosis in cancer patients. Curr Probl Cardiol. (2023) 48:101527. doi: 10.1016/j.cpcardiol.2022.101527
138. Suero-Abreu GA, Zanni MV, Neilan TG. Atherosclerosis with immune checkpoint inhibitor therapy: evidence, diagnosis, and management. JACC CardioOncol. (2022) 4:598–615. doi: 10.1016/j.jaccao.2022.11.011
139. Poels K, van Leent MMT, Reiche ME, Kusters PJH, Huveneers S, de Winther MPJ, et al. Antibody-mediated inhibition of CTLA4 aggravates atherosclerotic plaque inflammation and progression in hyperlipidemic mice. Cells. (2020) 9:1987. doi: 10.3390/cells9091987
140. Matsumoto T, Sasaki N, Yamashita T, Emoto T, Kasahara K, Mizoguchi T, et al. Overexpression of cytotoxic T-lymphocyte-associated antigen-4 prevents atherosclerosis in mice. Arterioscler Thromb Vasc Biol. (2016) 36:1141–51. doi: 10.1161/ATVBAHA.115.306848
141. Drobni ZD, Alvi RM, Taron J, Zafar A, Murphy SP, Rambarat PK, et al. Association between immune checkpoint inhibitors with cardiovascular events and atherosclerotic plaque. Circulation. (2020) 142:2299–311. doi: 10.1161/CIRCULATIONAHA.120.049981
142. Astuti Y, Raymant M, Quaranta V, Clarke K, Abudula M, Smith O, et al. Efferocytosis reprograms the tumor microenvironment to promote pancreatic cancer liver metastasis. Nat Cancer. (2024). doi: 10.1038/s43018-024-00731-2
143. Zhou Y, Yao Y, Deng Y, Shao A. Regulation of efferocytosis as a novel cancer therapy. Cell Commun Signal. (2020) 18:71. doi: 10.1186/s12964-020-00542-9
144. Li Z, Wang Q, Huang X, Yang M, Zhou S, Fang Z, et al. Lactate in the tumor microenvironment: A rising star for targeted tumor therapy. Front Nutr. (2023) 10:1113739. doi: 10.3389/fnut.2023.1113739
145. Jennings MR, Munn D, Blazeck J. Immunosuppressive metabolites in tumoral immune evasion: redundancies, clinical efforts, and pathways forward. J Immunother Cancer. (2021) 9:e003013. doi: 10.1136/jitc-2021-003013
Keywords: efferocytosis, myeloid, T-lymphocytes, crosstalk, inflammation, atherosclerosis, resolution
Citation: Ngai D, Sukka SR and Tabas I (2024) Crosstalk between efferocytic myeloid cells and T-cells and its relevance to atherosclerosis. Front. Immunol. 15:1403150. doi: 10.3389/fimmu.2024.1403150
Received: 18 March 2024; Accepted: 17 May 2024;
Published: 30 May 2024.
Edited by:
Klaus Ley, Augusta University, United StatesReviewed by:
Catherine Hedrick, Augusta University, United StatesJohnna Francis Varghese, Harvard Medical School, United States
Copyright © 2024 Ngai, Sukka and Tabas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Ngai, ZGhuMjExMUBjdW1jLmNvbHVtYmlhLmVkdQ==; Ira Tabas, aWF0MUBjdW1jLmNvbHVtYmlhLmVkdQ==