 Zhanpeng Chen
Zhanpeng Chen Xingxing Kong
Xingxing Kong Xiaomin Wang
Xiaomin Wang Shuihua Lu
Shuihua Lu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 30 May 2024
Sec. Microbial Immunology
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1402024
Mycobacterium tuberculosis (Mtb) is an intracellular pathogen capable of adapting and surviving within macrophages, utilizing host nutrients for its growth and replication. Cholesterol is the main carbon source during the infection process of Mtb. Cholesterol metabolism in macrophages is tightly associated with cell functions such as phagocytosis of pathogens, antigen presentation, inflammatory responses, and tissue repair. Research has shown that Mtb infection increases the uptake of low-density lipoprotein (LDL) and cholesterol by macrophages, and enhances de novo cholesterol synthesis in macrophages. Excessive cholesterol is converted into cholesterol esters, while the degradation of cholesterol esters in macrophages is inhibited by Mtb. Furthermore, Mtb infection suppresses the expression of ATP-binding cassette (ABC) transporters in macrophages, impeding cholesterol efflux. These alterations result in the massive accumulation of cholesterol in macrophages, promoting the formation of lipid droplets and foam cells, which ultimately facilitates the persistent survival of Mtb and the progression of tuberculosis (TB), including granuloma formation, tissue cavitation, and systemic dissemination. Mtb infection may also promote the conversion of cholesterol into oxidized cholesterol within macrophages, with the oxidized cholesterol exhibiting anti-Mtb activity. Recent drug development has discovered that reducing cholesterol levels in macrophages can inhibit the invasion of Mtb into macrophages and increase the permeability of anti-tuberculosis drugs. The development of drugs targeting cholesterol metabolic pathways in macrophages, as well as the modification of existing drugs, holds promise for the development of more efficient anti-tuberculosis medications.
Tuberculosis (TB) is a chronic infectious disease caused by Mycobacterium tuberculosis (Mtb). According to the World Health Organization, TB is the second leading cause of death from a single infectious agent worldwide in 2022, surpassed only by coronavirus disease 2019 (COVID-19). It is estimated that approximately one-quarter of the global population is infected with Mtb which typically leads to pulmonary tuberculosis. Without proper treatment, the mortality rate among TB patients can reach up to 50% (1). Mtb primarily invades the respiratory tract and spreads through airborne droplets to infect the lung tissues of the host. As a highly adaptable pathogenic bacterium, Mtb is capable of evading host immune system attacks through various mechanisms and establishing infection within host macrophages. Macrophages are critical components of the body’s immune system with the ability to engulf and degrade pathogens, thus playing a role in the clearance of infections. However, Mtb can adapt and survive within macrophages, utilizing host nutrients to provide energy and materials for its growth and replication (2).
Cholesterol is the main carbon source during the infection process of Mtb. Cholesterol is crucial for the optimal growth and persistence of Mtb. It serves as a critical growth substrate for Mtb within phagosomes and promotes the formation of foamy macrophages and necrotic granulomas. Mtb can utilize cholesterol as the sole carbon source for energy synthesis and membrane lipid production, leading to the generation of virulence lipids in its cell wall. Genes involved in cholesterol metabolism play a vital role in Mtb infection, and inhibition of the expression of these genes suppresses Mtb growth (3–5). Upon infection by Mtb, macrophages exhibit inhibition of cholesterol ester hydrolysis, facilitation of cholesterol esterification, hyperactivation of cholesterol synthesis metabolism, augmentation of cholesterol intake, and reduction of cholesterol efflux, which ultimately results in cholesterol accumulation. The excessive accumulation of cholesterol forms characteristic lipid droplet structures, promoting the formation of foam-like macrophages (6–9). The formation of foam cells not only suppresses the immune response of host cells but also helps Mtb achieve immune evasion and sustained survival, leading to the development of tuberculosis granulomas, tissue cavitation, and systemic dissemination of Mtb, which in turn contribute to the adverse progression of TB (10–12). Furthermore, cholesterol in macrophages can be converted into oxidized cholesterol which exhibits activity against Mtb infection (13).
In this review, we summarize the existing research findings, elucidate the significant role of cholesterol in macrophage function, and explore how Mtb regulates cholesterol metabolism pathways in macrophages, including cholesterol uptake, synthesis, esterification, hydrolysis, oxidation, and efflux. It would help to understand the pathological evolution process of TB and provide an important theoretical basis for the developing of new treatment strategies and vaccines, aiming to achieve the challenging goal of TB elimination by 2035.
Macrophages, as immunocytes, play crucial roles in maintaining tissue homeostasis, host defense, and tissue repair in various physiological and pathological conditions. Cholesterol, a major lipid component of eukaryotic cell membranes, is vital for maintaining the stability of macrophage membranes and prominent impacts macrophage functions (14, 15). Cholesterol is a double-edged sword. On the one hand, it is indispensable for phagocytosis, antigen presentation, and other important functions of macrophages. On the other hand, excessive cholesterol accumulation impairs the tissue repair ability of macrophages and promotes foam cell formation.
Macrophages play a key role in clearing pathogens from the body through phagocytosis. Studies have shown that cholesterol on the macrophage membrane acts as a signaling molecule, mediating macrophage phagocytosis of Mtb and facilitating Mtb entry into macrophages (16). Thus, cholesterol serves as a bridge for macrophage-mediated pathogen clearance. As an essential component of the cell membrane, cholesterol not only mediates the phagocytosis of macrophages but also strengthens its antigen presentation function. A study was conducted to evaluate the role of cholesterol in macrophage phagocytosis and antigen presentation by treating macrophages with a cholesterol biosynthesis inhibitor, and the effects were observed at different time points. The results indicated that cholesterol depletion inhibits the intracellular transport of major histocompatibility complex class I (MHC I) antigens, thereby reducing the surface expression level of MHC I molecules on macrophages (17). Another study also found that increased cholesterol levels may enhance antigen presentation by upregulating the expression of human leukocyte antigen class II (HLA-D) region products on antigen-presenting cells (18). It is well known that macrophages adopt different phenotypes, mainly divided into pro-inflammatory M1 macrophages and anti-inflammatory M2 macrophages, upon phagocytosis of pathogens or exposure to antigens. Accumulation of cholesterol in macrophages mainly promotes the generation of M1 macrophages, which enhance macrophage inflammatory responses. It has been demonstrated that Mtb infection promotes cholesterol accumulation in macrophages, which may further activate the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, leading to increased production of cytokines such as interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α, as well as reactive oxygen species (ROS) (14, 19). In addition, cholesterol not only serves as a pro-inflammatory mediator but also constitutes an important structural domain for catalyzing immune responses. Studies have shown that cholesterol-rich domains on the membrane regulate immune responses by modulating the nuclear transport of interferon-gamma (IFN-γ) and interferon gamma receptor 1(IFNGR-1), as well as the activation of signal transducer and activator of transcription 1 alpha (STAT1α) (20). On the basis of the inflammatory phenotype, macrophages also exhibit a pathological phenotype known as foam cell phenotype, which plays a crucial role in the development of TB. Research has demonstrated that Mtb promotes its survival by reprogramming the metabolism of host macrophages, leading to the accumulation of cholesterol as a carbon source for its own survival. Simultaneously, Mtb-mediated cholesterol accumulation further promotes the formation of foam cells (6, 21). Foam cells serve as a sanctuary for Mtb survival and are also a hallmark of TB lesions. Foam cells formed during Mtb infection are not only key participants in granuloma formation and tissue cavitation, but they also further facilitate systemic dissemination of Mtb upon their departure from the primary granuloma (10–12, 22). Macrophages not only possess phagocytic and immune response functions but also play a role in tissue repair. It is well-known that tissue repair requires recruitment of a large number of macrophages. Studies have shown that cholesterol can regulate cell stiffness and motility, and excessive cholesterol accumulation inhibits the chemotactic migration of macrophages to wounds, thereby impairing wound healing (23). Interestingly, Mtb-infection can induce the production of oxidised cholesterol, thereby enhancing the host’s immune response to Mtb-infection (24). In conclusion, cholesterol plays a crucial role in macrophage pathogen engulfment, antigen presentation, and inflammatory responses. It is closely associated with the development of TB, and also affects the tissue repair function of macrophages (Figure 1).
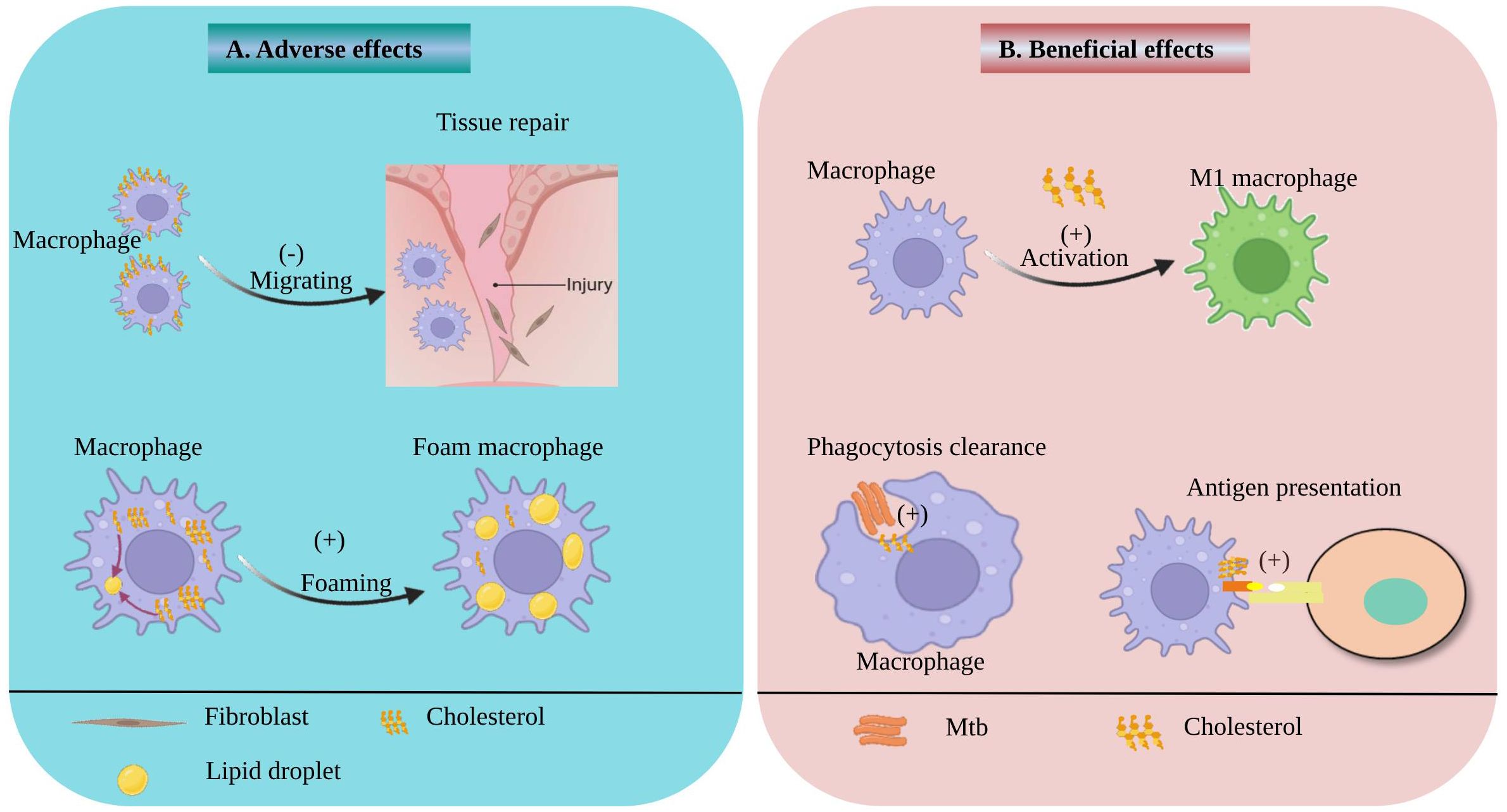
Figure 1 Beneficial and adverse effects of Cholesterol on macrophage. (A) the adverse effects of cholesterol on macrophage. The accumulation of cholesterol on macrophage membranes can inhibit macrophage migration and impede tissue repair. Cholesterol accumulation in macrophages promotes the formation of lipid droplets and foam cells. (B) the beneficial effects of cholesterol on macrophage. Cholesterol also can facilitate the activation of macrophages into M1 macrophages and promote macrophage antigen presentation. In addition, membrane cholesterol can enhance phagocytosis of Mtb by macrophages.
Cholesterol can exist in the form of free cholesterol, but it is more commonly transported to tissues throughout the body, including macrophages, in the form of low-density lipoprotein (LDL). Macrophages uptake cholesterol and its lipoproteins through a family of low-density lipoprotein receptor (LDLR) proteins expressed on cell membranes. Low-density lipoprotein receptor-related protein 2 (Lrp2), a 600 kDa giant membrane glycoprotein, belongs to the LDLR protein family and is primarily responsible for mediating the uptake of intracellular cholesterol. Lrp2 expression was found to be remarkably increased in macrophages infected with H37Rv, and it is positively correlated with the accumulation of intracellular cholesterol (7). Cluster of differentiation 36 (CD36), a transmembrane glycoprotein belonging to the LDLR protein family, forms a channel through the cell membrane and is responsible for mediating the absorption of lipid molecules. Tuberculous pleural effusion has been shown to induce massive accumulation of cholesterol in macrophages and markedly enriches the fluorescence intensity of CD36 on the cell membrane (25). Further evidence from a guinea pig model of Mtb infection confirms the accumulation of oxidized low-density lipoprotein (OxLDL) in extrapulmonary granulomas and pulmonary macrophages of TB guinea pigs. Moreover, macrophages increase the surface expression of scavenger receptor CD36 and lectin-like oxidized low-density lipoprotein receptor-1 (LOX1), promoting the uptake of cholesterol and LDL (26). LOX1 is a membrane receptor protein belonging to the C-type lectin family involved in the absorption and transport of LDL. A study was conducted to analyze granuloma samples from human TB. Transcriptomic analysis revealed a significant upregulation of genes involved in cholesterol and other lipid synthesis, and immunohistochemistry confirmed the high expression of these lipid synthesis proteins in human TB granulomas. Thin-layer chromatography and mass spectrometry analyses revealed that granulomas and foam cells were rich in cholesterol, cholesterol esters, and triglycerides, with LDL being a probable major source of these lipids (9).
In summary, cholesterol is primarily taken up by macrophages in the form of LDL in the body. Macrophages uptake cholesterol and LDL through the expression of LDLR family proteins, such as Lrp2, LOX1, and CD36. Although animal experiments and human samples provide some evidence, further clinical research is needed to validate these findings, especially in observing the accumulation of cholesterol and LDL and the expression changes of receptors in the human body during Mtb infection. Besides, other potential receptors involved in the uptake of cholesterol and LDL, as well as their roles in the development of the disease, can be explored to provide new therapeutic strategies for the prevention and treatment of TB (Figure 2).
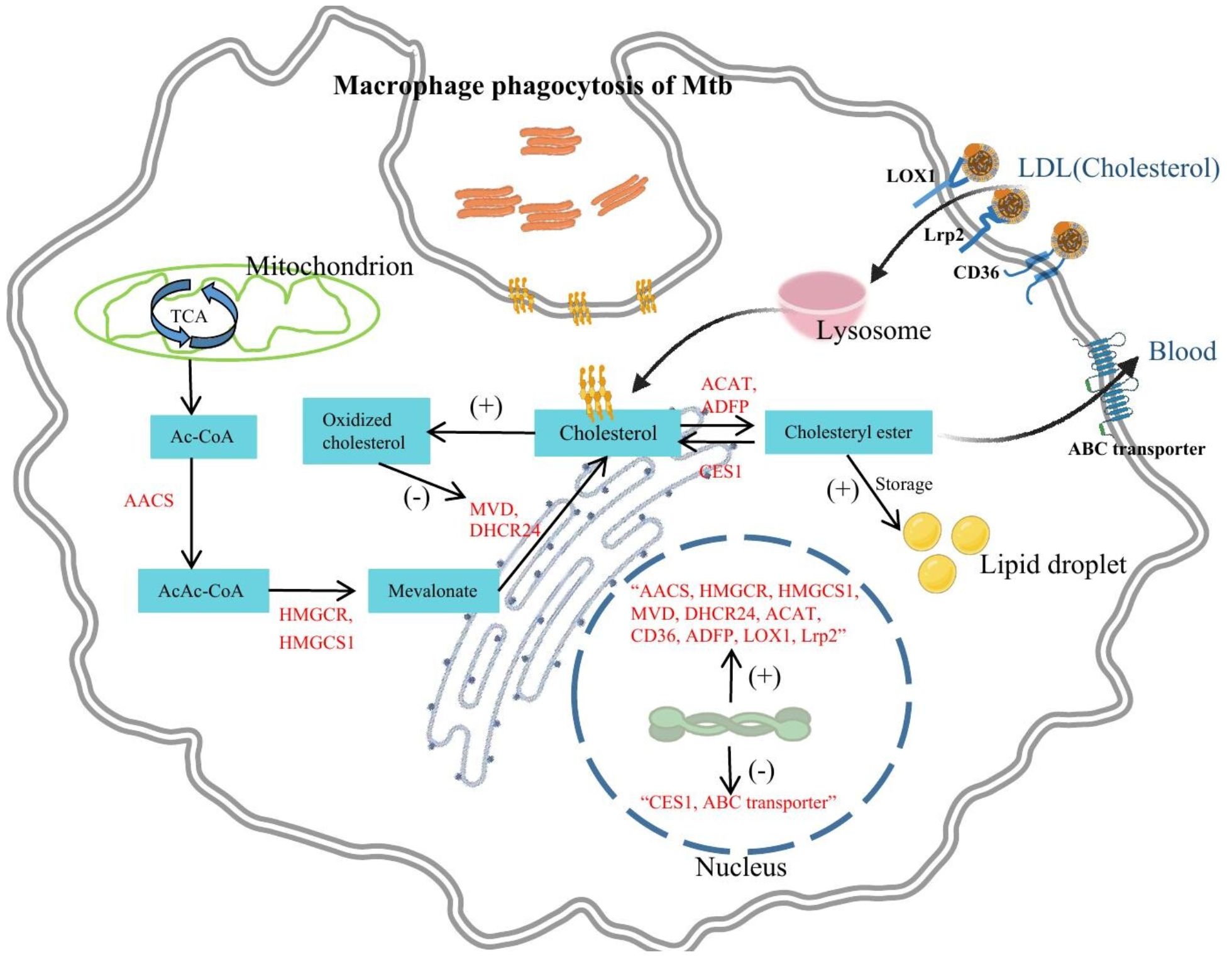
Figure 2 The effects of Mtb infection on macrophage cholesterol metabolism. Mtb infection significantly impacts macrophage cellular processes. It increases the uptake of LDL and cholesterol through upregulation of receptors like Lrp2, CD36, and LOX1. Additionally, Mtb infection stimulates de novo cholesterol synthesis by upregulating enzymes like AACS, HMGCS1, HMGCR, MVD, and DHCR24. This excess cholesterol is converted into cholesterol esters, contributing to lipid droplet formation facilitated by upregulation of ADFP and ACAT. Cholesterol ester degradation is hindered due to downregulation of CES1. Mtb infection also suppresses ABC transporters, impeding cholesterol efflux. These alterations lead to cholesterol accumulation, ultimately promoting the formation of foam cells. Furthermore, cholesterol in macrophages may also be converted into oxidized cholesterol which exhibits anti-Mtb activity.
Multiple transcriptomic studies have revealed that Mtb infection leads to upregulation of cholesterol synthesis-related enzyme gene expression in macrophages, including acetoacetyl-coenzyme A synthetase (AACS), 3-hydroxy-3-methylglutaryl- coenzyme A synthase 1 (HMGCS1), 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), mevalonate diphosphate decarboxylase (MVD), and 3β-hydroxysterol Δ24-reductase (DHCR24) (7, 27). AACS is a cytoplasmic enzyme involved in fatty acid biosynthesis that catalyzes the coupling of acetyl-coenzyme A (CoA) to produce acetoacetyl-CoA, which serves as the starting substrate for cholesterol synthesis (28). Therefore, the promotion of AACS gene expression by Mtb may contribute to de novo cholesterol synthesis. HMGCS1 is a key enzyme in the mevalonate pathway of cholesterol biosynthesis, catalyzing the conversion of acetoacetyl-CoA to 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA), which is further reduced to mevalonic acid by HMGCR, a rate-limiting enzyme in cholesterol synthesis that regulates cholesterol production (29). HMGCR and HMGCS1 play pivotal roles in macrophage function. According to previous research, HMGCR knockout was found to significantly inhibit the migration, proliferation, and survival of macrophages. However, another study found that increased expression of HMGCR induces excessive cholesterol synthesis, resulting in decreased membrane fluidity and suppressed phagocytic activity of macrophages (30, 31). When HMGCS1 activity is excessively elevated, it can cause the accumulation of hydroxymethylglutaryl-CoA in macrophages, which result in cell necrosis. Necrosis is a unique form of cell death characterized by cell membrane rupture, cytoplasmic leakage, and organelle damage. In macrophages, cell death induced by HMGCS1 via necrosis pathway exerts irreversible effects on the physiological function of macrophages, thereby impacting the normal immune system function (32). Furthermore, HMGCR and HMGCS1 are key genes in promoting macrophage foam cell formation. Increased activity of HMGCR and HMGCS1 contributes to excessive activation of the cholesterol synthesis pathway, which in turn leads to macrophage lipid droplet formation (33). Therefore, Mtb-induced overexpression of HMGCR and HMGCS1 may inhibit phagocytic activity of macrophages, promote cholesterol accumulation and foam cell formation, and even cause macrophage death.
MVD catalyzes the conversion of mevalonate diphosphate to isopentenyl diphosphate, which is an early step in cholesterol biosynthesis (34). MVD and the mevalonate pathway are crucial for macrophage survival and play an important role in the synthesis and release of inflammatory mediators, contributing to pro-inflammatory responses of macrophages (35). DHCR24, also known as 3β-hydroxysterol Δ24-reductase, is a human flavin adenine dinucleotide (FAD)-dependent oxidoreductase that catalyzes the final step of cholesterol synthesis, converting desmosterol to cholesterol (36). In addition to HMGCR and HMGCS1, DHCR24 is also a key gene in promoting macrophage foam cell formation and possesses antioxidant functions that effectively inhibit oxidative stress (33, 37). Consequently, Mtb infection of macrophages may elevate immune inflammation levels, inhibit oxidative stress, and induce cholesterol accumulation as well as foam cell formation by upregulating MVD and DHCR24 expression. Despite some progress has been made in current studies, most of them have been conducted using in vitro cell culture models, and these findings need to be further validated in vivo. Animal models or clinical samples can provide a better understanding of the impact of Mtb infection on cholesterol anabolism in macrophages (Figure 2).
Accumulation of cholesterol esters (CE) stored in cytoplasmic lipid droplets is a key feature of foam cells, which are central to the development of atherosclerosis and tuberculosis (38). When macrophages are infected with Mtb, not only does it promote cholesterol accumulation, but it also promotes the conversion of cholesterol to cholesterol esters within macrophages, thereby facilitating the formation of lipid droplets and foam cells. Animal pathology studies have shown that macrophages isolated from granulomatous lungs of tuberculosis mice contain large amounts of cholesterol esters, whereas the ester content in resident peritoneal macrophages of normal mice is very low (39). Metabolomics studies of human lung tissue have revealed that large amounts of cholesterol, cholesteryl esters, and triglycerides accumulate in the granulomas of patients with TB. All of these studies provide ample evidence that cholesterol in Mtb-infected macrophages is converted to cholesterol esters for storage. Further mechanistic exploration has revealed that Mtb, especially its cell wall surface lipid trehalose dimycolate, upregulates the expression of adipose differentiation-related protein (ADRP), thereby promoting cholesterol esterification and lipid droplet formation (9). Acyl-CoA: cholesterol acyltransferase (ACAT) is a key enzyme catalyzing cholesterol esterification in macrophages, and it has been found that tuberculosis pleural effusion (TB-PE) induces the expression of ACAT, promoting cholesterol esterification in macrophages. When ACAT is inhibited, the levels of cholesterol esters and triglycerides in TB-PE-treated macrophages are significantly reduced (25). Accordingly, it can be concluded from the existing studies that Mtb and its cell wall surface lipid virulence factors enhance the conversion of cholesterol to cholesterol esters in macrophages by upregulating the expression of ACAT and ADFP, thereby facilitating the formation of lipid droplets and foam cells. Currently, there are still some research gaps and directions that need further exploration. Firstly, the detailed mechanisms underlying the accumulation of cholesterol esters in Mtb-infected macrophages have not been fully understood, including how Mtb regulates the expression and activity of ACAT. In the future, more animal models and patient samples should be utilized to further explore the mechanisms of the accumulation of cholesterol esters in Mtb-infected macrophages (Figure 2).
Only non-esterified or free cholesterol (FC) can efflux from the cell to the extracellular space, thus the excessively accumulated cholesterol esters need to be effectively degraded by cells to prevent pathological progression. Cholesterol esters are hydrolyzed by cholesterol esterase within the cells, resulting in the formation of free cholesterol and fatty acids (38). Cholesterol ester hydrolysis contributes to the reduction of lipid load in macrophages, diminishing inflammation and related diseases, and is crucial for macrophage function (40). Inhibition of cholesterol ester hydrolysis leads to dysregulation of cholesterol content in microdomains, which prolongs the activation of immune cell signaling pathways and promotes immune cell proliferation, infiltration, and foam cell formation (41). A study using transcriptomic analysis and in vitro validation demonstrated that macrophage receptor with collagenous structure (MARCO) and collectin sub-family member 12 (COLEC12), which are involved in phagocytosis, as well as carboxylesterase 1 (CES1), the cholesterol ester hydrolase, were downregulated in alveolar macrophages (AMs) from TB patients compared to controls, indicating impaired cholesterol ester degradation and reduced free cholesterol, leading to excessive accumulation of cholesterol esters and adverse progression of pulmonary TB (8). Further research can further validate the key role of CES1 in TB progression through protein detection and functional experiments, and explore the specific mechanisms by which Mtb regulates the downregulation of macrophage CES1 using cell and animal models (Figure 2).
In addition to cholesterol esters, cholesterol can also be converted into oxidized cholesterol forms in macrophages, with 25-hydroxycholesterol(25HC)and 1,25 dihydroxyvitamin D (1,25(OH)2D) being two typical oxidized cholesterol derivatives. Mtb infection induces macrophage activation, and activated macrophages are a rich source of oxidized cholesterol (42–45). In macrophage models of Mtb infection, oxidized cholesterol typically exerts an antimicrobial effect. Studies have shown that the production of 25HC is significantly increased in activated macrophages, which in turn inhibits cholesterol biosynthesis and accumulation, while cholesterol accumulation promotes the growth of Mtb (21, 46). An increase in 25HC concentrations was shown in lungs of mice infected with Mtb (24). Furthermore, 7α,25-dihydroxycholesterol can increase Mtb phagocytosis and reduces intracellular Mtb growth in primary human monocytes (47). Multiple studies have demonstrated the potent anti-Mtb infection activity of 1,25(OH)2D (48). 1,25(OH)2D inhibits Mtb growth by suppressing cholesterol and lipid droplet formation (49). Additionally, 1,25(OH)2D promotes the production of antimicrobial peptides and the maturation of phagolysosomes in macrophages, thereby enhancing an effective cellular immune response against Mtb (13, 50). In conclusion, Mtb infection may promote the conversion of cholesterol into oxidized cholesterol in macrophages, and oxidized cholesterol plays a promoting role in macrophage-mediated anti-Mtb infection. Future research needs to explore the underlying mechanisms and target points by which Mtb infection promotes the production of oxidized cholesterol, as well as develop new therapeutic strategies for anti-Mtb infection based on oxidized cholesterol (Figure 2).
Even though cholesterol esters can be degraded to cholesterol, macrophages themselves are unable to degrade cholesterol. Therefore, when cholesterol accumulates excessively in macrophages, it ultimately needs to be eliminated from the cells through efflux. ATP-binding cassette (ABC) transporters are a class of transmembrane proteins that utilize ATP hydrolysis to mediate cholesterol efflux, including genes such as ABCA1, ABCG1, and ABCA5 (51). In vitro studies have shown that infection of macrophages with the Mtb strain H37Ra inhibits the expression of ABCG1 and promotes the formation of lipid bodies and foam cells. In contrast, overexpression of ABCG1 in macrophages through lentiviral transduction can significantly reverse foam cell formation (52). Researchers have constructed a macrophage model infected with Bacille Calmette-Guérin (BCG) and found that BCG infection inhibit the expression of macrophage ABCG1, ABCA1, and ABCA5, promoting intracellular cholesterol accumulation and further inducing macrophage autophagy to suppress BCG infection (53). Further animal experiments have provided more compelling evidence, as it was found that the expression of ABCA1 and ABCG1, which are involved in cholesterol efflux, was significantly down-regulated in peritoneal macrophages and lung tissue sections of mice infected with H37Ra (7). In conclusion, Mtb infection has a direct regulatory effect on cholesterol efflux in macrophages by downregulating the expression of ABC transporters, inhibiting cholesterol efflux, and promoting cholesterol accumulation and foam cell formation. Current research mainly focuses on in vitro experiments and animal experiments, and further human-related studies, including clinical research and proteomic studies, are needed to assess the impact of Mtb infection on ABC transporters and cholesterol metabolism, as well as to explore the relevant molecular mechanisms. Additionally, while overexpression of ABC transporters can reverse foam cell formation, further studies are needed to investigate their potential therapeutic efficacy in the treatment of TB (Figure 2).
Foam cells are lipid-rich macrophages that serve as a hallmark of granulomas and tuberculous lesions in TB. Consistent with the discussion in the previous section, Mtb infection promotes the accumulation of lipids such as cholesterol in macrophages, leading to the formation of foam macrophages (54). Foam cells exhibit various functions in tuberculosis. They possess anti-apoptotic properties, as evidenced by the upregulation of anti-apoptotic protein expression, which prevents cell death (55, 56). Additionally, foam cells exhibit dormancy capabilities. Research has shown that Mtb remains in a non-replicating state within foam cells, facilitating persistent infection (57). Furthermore, foam cells exert anti-inflammatory functions in TB. Mtb-infected foam cells decrease secretion of TNF-α and IL-1α, increase secretion of Transforming Growth Factor (TGF)-β, and inhibit effector T cells through inducing higher levels of nitric oxide (58, 59). Foam cells not only undergo functional changes but also play a role in promoting TB progression. Firstly the lipid-rich content of foam cells provides abundant nutrients and a stable environment for intracellular growth of Mtb (11). Moreover, foam cells play a central role in the development, maintenance, and dissemination of TB granulomas. Following Mtb infection, alveolar macrophages migrate to the interstitium and induce an inflammatory response, that leads to immune cell infiltration. These infiltrating immune cells surround foam cells which serve as reservoirs for Mtb, resulting in the formation of TB granulomas (60–62). The necrotic area surrounding the lipid-rich core is referred to as caseation. Foam cells promote the formation of the necrotic core by releasing their abundant content of triglycerides into the caseum, leading to proteinaceous transformation and enlargement of granulomas, ultimately causing progressive destruction of lung tissue and loss of pulmonary function (63). Foam cell death or departure from the primary granuloma generates secondary granulomas, facilitating the systemic dissemination of Mtb (11, 64).
Not only macrophage foam formation, but also macrophage phenotypes play a crucial role in the progression of tuberculosis. During the early stages of Mtb infection, macrophages differentiate into M1 macrophages (65). M1 macrophages exhibit the Warburg effect, promoting pro-inflammatory responses and enhancing ROS production, which helps restrict Mtb survival (66–68). Over time, the major virulence factor of Mtb, ESAT-6, converts M1 macrophages to M2 macrophages (69). M2 macrophages secrete IL-10 and TGF-β, inhibiting antimicrobial immunity and promoting sustained Mtb survival (70, 71). Thus, further investigation into the transition process from M1 to M2 macrophages induced by Mtb can reveal potential immune targets for TB. We should also pay particular attention to and explore the role of cholesterol metabolism during this transitional process.
During the occurrence and development of TB, Mtb infection exerts multiple impacts on the cholesterol metabolism in macrophages. At first, Mtb infection promotes the uptake of LDL and cholesterol accumulation through receptors expressed on macrophages, such as Lrp2, LOX1, and CD36. Secondly, Mtb infection leads to upregulation of genes involved in cholesterol synthesis enzymes (AACS, HMGCS1, HMGCR, MVD, and DHCR24), enhancing cholesterol synthesis. Also, Mtb infection promotes the conversion of cholesterol to cholesterol esters, increasing lipid droplet accumulation. However, Mtb infection may inhibit the expression of CES1, which result in excessive accumulation of cholesterol esters and the formation of adverse foam cells. Furthermore, Mtb infection inhibits the expression of ABC transporters in macrophages, impeding cholesterol efflux and further exacerbating the generation of adverse foam cells. While oxidized cholesterol demonstrates good activity in combating Mtb infection, the mechanisms by which Mtb infection impacts oxidized cholesterol generation remain unclear. In summary, the dysregulation of cholesterol metabolism in macrophages caused by Mtb infection is an important feature in the development of TB, and it is of great significance for a deeper understanding of the pathogenesis of TB and the search for therapeutic strategies.
Currently, there are drug developments targeting the cholesterol metabolism pathway for the treatment of TB. For example, statins reduce cholesterol levels in macrophages by inhibiting HMGCR, thereby reducing the entry of Mtb into macrophages and improving the permeability of anti-tuberculosis drugs (72). Although negative results exist in certain situations, statin therapy as an adjunctive treatment has demonstrated improvements in the treatment outcomes of TB patients in clinical practice (11). Therefore, it is promising to develop effective anti-tuberculosis drugs by repurposing existing drugs. GSK2556286 is a novel anti-tuberculosis drug targeting cholesterol metabolism, which inhibits cholesterol catabolism by acting on adenosine nucleotide cyclase, thereby suppressing the intracellular and extracellular growth of Mtb (73). In the early stages of clinical trials, it has demonstrated the potential to shorten TB treatment and can be considered as a potential substitute for pretomanid or linezolid in the BPaL regimen. Moreover, it does not exhibit cross-resistance with known anti-tuberculosis drugs (74, 75). 1,25(OH)2D exhibits excellent anti-tuberculosis activity, and related clinical trials have shown that vitamin D may help minimize excessive tissue damage during active TB (76). Therefore, further investigation and application of oxidized cholesterol are warranted. For detailed information on the association between lipid metabolism and anti-tuberculosis drugs, please refer to another review article (77). In the future, we should not only further validate and explore the regulatory mechanisms of Mtb infection on cholesterol metabolism in macrophages, but also focus on the key targets of cholesterol metabolism pathways during Mtb infection to design efficient anti-tuberculosis drugs.
CZ: Conceptualization, Writing – original draft, Writing – review & editing. XK: Writing – review & editing. QM: Writing – review & editing. JC: Writing – review & editing. YZ: Writing – review & editing. HL: Writing – review & editing. XW: Conceptualization, Supervision, Writing – review & editing. SL: Conceptualization, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Shenzhen High-level Hospital Construction Fund (No. G2022061), the National Nature Science Foundation of China (Grant Numbers 32394014 and 32260883) and the Special Fund of Shenzhen Central-leading-local Scientific and Technological Foundation (No. LCYX20220620105200001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Organization WH. Global tuberculosis report 2023 (2023). Available online at: https://www.who.int/publications/i/item/9789240083851.
2. Bo H, Moure UAE, Yang Y, Pan J, Li L, Wang M, et al. Mycobacterium tuberculosis-macrophage interaction: Molecular updates. Front Cell Infect Microbiol. (2023) 13:1062963. doi: 10.3389/fcimb.2023.1062963
3. Ho NA, Dawes SS, Crowe AM, Casabon I, Gao C, Kendall SL, et al. The structure of the transcriptional repressor kstR in complex with coA thioester cholesterol metabolites sheds light on the regulation of cholesterol catabolism in mycobacterium tuberculosis. J Biol Chem. (2016) 291:7256–66. doi: 10.1074/jbc.M115.707760
4. Wilburn KM, Fieweger RA, VanderVen BC. Cholesterol and fatty acids grease the wheels of Mycobacterium tuberculosis pathogenesis. Pathog Dis. (2018) 76(2):fty021. doi: 10.1093/femspd/fty021
5. Yang M, Lu R, Guja KE, Wipperman MF, St Clair JR, Bonds AC, et al. Unraveling cholesterol catabolism in mycobacterium tuberculosis: chsE4-chsE5 α(2)β(2) acyl-coA dehydrogenase initiates β-oxidation of 3-oxo-cholest-4-en-26-oyl coA. ACS Infect Dis. (2015) 1:110–25. doi: 10.1021/id500033m
6. Laval T, Chaumont L, Demangel C. Not too fat to fight: The emerging role of macrophage fatty acid metabolism in immunity to Mycobacterium tuberculosis. Immunol Rev. (2021) 301:84–97. doi: 10.1111/imr.12952
7. Prakhar P, Bhatt B, Lohia GK, Shah A, Mukherjee T, Kolthur-Seetharam U, et al. G9a and Sirtuin6 epigenetically modulate host cholesterol accumulation to facilitate mycobacterial survival. PloS Pathog. (2023) 19:e1011731. doi: 10.1371/journal.ppat.1011731
8. Lavalett L, Rodriguez H, Ortega H, Sadee W, Schlesinger LS, Barrera LF. Alveolar macrophages from tuberculosis patients display an altered inflammatory gene expression profile. Tuberculosis (Edinb). (2017) 107:156–67. doi: 10.1016/j.tube.2017.08.012
9. Kim MJ, Wainwright HC, Locketz M, Bekker LG, Walther GB, Dittrich C, et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med. (2010) 2:258–74. doi: 10.1002/emmm.201000079
10. Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol. (2009) 10:943–8. doi: 10.1038/ni.1781
11. Shim D, Kim H, Shin SJ. Mycobacterium tuberculosis infection-driven foamy macrophages and their implications in tuberculosis control as targets for host-directed therapy. Front Immunol. (2020) 11:910. doi: 10.3389/fimmu.2020.00910
12. Riaz SM, Bjune GA, Wiker HG, Sviland L, Mustafa T. Mycobacterial antigens accumulation in foamy macrophages in murine pulmonary tuberculosis lesions: Association with necrosis and making of cavities. Scand J Immunol. (2020) 91:e12866. doi: 10.1111/sji.12866
13. Bah SY, Dickinson P, Forster T, Kampmann B, Ghazal P. Immune oxysterols: Role in mycobacterial infection and inflammation. J Steroid Biochem Mol Biol. (2017) 169:152–63. doi: 10.1016/j.jsbmb.2016.04.015
14. Aguilar-Ballester M, Herrero-Cervera A, Vinué Á, Martínez-Hervás S, González-Navarro H. Impact of cholesterol metabolism in immune cell function and atherosclerosis. Nutrients. (2020) 12(7):2021. doi: 10.3390/nu12072021
15. Yan J, Horng T. Lipid metabolism in regulation of macrophage functions. Trends Cell Biol. (2020) 30:979–89. doi: 10.1016/j.tcb.2020.09.006
16. Gatfield J, Pieters J. Essential role for cholesterol in entry of mycobacteria into macrophages. Science. (2000) 288:1647–50. doi: 10.1126/science.288.5471.1647
17. Rao M, Peachman KK, Alving CR, Rothwell SW. Depletion of cellular cholesterol interferes with intracellular trafficking of liposome-encapsulated ovalbumin. Immunol Cell Biol. (2003) 81:415–23. doi: 10.1046/j.1440-1711.2003.01192.x
18. Hughes DA, Townsend PJ, Haslam PL. Enhancement of the antigen-presenting function of monocytes by cholesterol: possible relevance to inflammatory mechanisms in extrinsic allergic alveolitis and atherosclerosis. Clin Exp Immunol. (1992) 87:279–86. doi: 10.1111/j.1365-2249.1992.tb02988.x
19. Martens GW, Vallerskog T, Kornfeld H. Hypercholesterolemic LDL receptor-deficient mice mount a neutrophilic response to tuberculosis despite the timely expression of protective immunity. J Leukoc Biol. (2012) 91:849–57. doi: 10.1189/jlb.0311164
20. Subramaniam PS, Johnson HM. Lipid microdomains are required sites for the selective endocytosis and nuclear translocation of IFN-gamma, its receptor chain IFN-gamma receptor-1, and the phosphorylation and nuclear translocation of STAT1alpha. J Immunol. (2002) 169:1959–69. doi: 10.4049/jimmunol.169.4.1959
21. Brzostek A, Pawelczyk J, Rumijowska-Galewicz A, Dziadek B, Dziadek J. Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. J Bacteriol. (2009) 191:6584–91. doi: 10.1128/jb.00488-09
22. Davis JM, Ramakrishnan L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell. (2009) 136:37–49. doi: 10.1016/j.cell.2008.11.014
23. Zhao C, Yang Q, Tang R, Li W, Wang J, Yang F, et al. DNA methyltransferase 1 deficiency improves macrophage motility and wound healing by ameliorating cholesterol accumulation. NPJ Regener Med. (2023) 8:29. doi: 10.1038/s41536-023-00306-2
24. Ngo MD, Bartlett S, Bielefeldt-Ohmann H, Foo CX, Sinha R, Arachchige BJ, et al. A blunted GPR183/oxysterol axis during dysglycemia results in delayed recruitment of macrophages to the lung during mycobacterium tuberculosis infection. J Infect Dis. (2022) 225:2219–28. doi: 10.1093/infdis/jiac102
25. Genoula M, Franco Marín JL, Dupont M, Kviatcovsky D, Milillo A, Schierloh P, et al. Formation of foamy macrophages by tuberculous pleural effusions is triggered by the interleukin-10/signal transducer and activator of transcription 3 axis through ACAT upregulation. Front Immunol. (2018) 9:459. doi: 10.3389/fimmu.2018.00459
26. Palanisamy GS, Kirk NM, Ackart DF, Obregón-Henao A, Shanley CA, Orme IM, et al. Uptake and accumulation of oxidized low-density lipoprotein during Mycobacterium tuberculosis infection in Guinea pigs. PloS One. (2012) 7:e34148. doi: 10.1371/journal.pone.0034148
27. Rienksma RA, Suarez-Diez M, Mollenkopf HJ, Dolganov GM, Dorhoi A, Schoolnik GK, et al. Comprehensive insights into transcriptional adaptation of intracellular mycobacteria by microbe-enriched dual RNA sequencing. BMC Genomics. (2015) 16:34. doi: 10.1186/s12864-014-1197-2
28. Bergstrom JD. The lipogenic enzyme acetoacetyl-CoA synthetase and ketone body utilization for denovo lipid synthesis, a review. J Lipid Res. (2023) 64:100407. doi: 10.1016/j.jlr.2023.100407
29. Cerqueira NM, Oliveira EF, Gesto DS, Santos-Martins D, Moreira C, Moorthy HN, et al. Cholesterol biosynthesis: A mechanistic overview. Biochemistry. (2016) 55:5483–506. doi: 10.1021/acs.biochem.6b00342
30. Sakai K, Nagashima S, Wakabayashi T, Tumenbayar B, Hayakawa H, Hayakawa M, et al. Myeloid HMG-coA (3-hydroxy-3-methylglutaryl-coenzyme A) reductase determines atherosclerosis by modulating migration of macrophages. Arterioscler Thromb Vasc Biol. (2018) 38:2590–600. doi: 10.1161/atvbaha.118.311664
31. Mukherjee M, Basu Ball W, Das PK. Leishmania donovani activates SREBP2 to modulate macrophage membrane cholesterol and mitochondrial oxidants for establishment of infection. Int J Biochem Cell Biol. (2014) 55:196–208. doi: 10.1016/j.biocel.2014.08.019
32. Jiang M, Cui H, Liu Z, Zhou X, Zhang L, Cao L, et al. The role of amino acid metabolism of tumor associated macrophages in the development of colorectal cancer. Cells. (2022) 11(24):4106. doi: 10.3390/cells11244106
33. Song Z, Lv S, Wu H, Qin L, Cao H, Zhang B, et al. Identification of foam cell biomarkers by microarray analysis. BMC Cardiovasc Disord. (2020) 20:211. doi: 10.1186/s12872-020-01495-0
34. La Y, Wong W, Peng K, Tian Z, Pan J, Sun R, et al. Decreased imiquimod-induced psoriasis-like skin inflammation in a novel mvd(F250S/+) knock-in mouse model. Inflammation. (2023) 46:1575–86. doi: 10.1007/s10753-023-01828-z
35. Larson-Casey JL, Murthy S, Ryan AJ, Carter AB. Modulation of the mevalonate pathway by akt regulates macrophage survival and development of pulmonary fibrosis. J Biol Chem. (2014) 289:36204–19. doi: 10.1074/jbc.M114.593285
36. Shen Y, Zhou J, Nie K, Cheng S, Chen Z, Wang W, et al. Oncogenic role of the SOX9-DHCR24-cholesterol biosynthesis axis in IGH-BCL2+ diffuse large B-cell lymphomas. Blood. (2022) 139:73–86. doi: 10.1182/blood.2021012327
37. Yao M, Li F, Xu L, Ma L, Zhang S. 24-Dehydrocholesterol Reductase alleviates oxidative damage-induced apoptosis in alveolar epithelial cells via regulating Phosphatidylinositol-3-Kinase/Protein Kinase B activation. Bioengineered. (2022) 13:155–63. doi: 10.1080/21655979.2021.2011634
38. Ghosh S, Zhao B, Bie J, Song J. Macrophage cholesteryl ester mobilization and atherosclerosis. Vascul Pharmacol. (2010) 52:1–10. doi: 10.1016/j.vph.2009.10.002
39. Kondo E, Kanai K. Accumulation of cholesterol esters in macrophages incubated with mycobacteria in vitro. Jpn J Med Sci Biol. (1976) 29:123–37. doi: 10.7883/yoken1952.29.123
40. Ghosh S. Macrophage cholesterol homeostasis and metabolic diseases: critical role of cholesteryl ester mobilization. Expert Rev Cardiovasc Ther. (2011) 9:329–40. doi: 10.1586/erc.11.16
41. Sorci-Thomas MG, Thomas MJ. Microdomains, inflammation, and atherosclerosis. Circ Res. (2016) 118:679–91. doi: 10.1161/circresaha.115.306246
42. Spann NJ, Glass CK. Sterols and oxysterols in immune cell function. Nat Immunol. (2013) 14:893–900. doi: 10.1038/ni.2681
43. Talat N, Perry S, Parsonnet J, Dawood G, Hussain R. Vitamin d deficiency and tuberculosis progression. Emerg Infect Dis. (2010) 16:853–5. doi: 10.3201/eid1605.091693
44. Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science. (2014) 345:679–84. doi: 10.1126/science.1254790
45. Bauman DR, Bitmansour AD, McDonald JG, Thompson BM, Liang G, Russell DW. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc Natl Acad Sci US. (2009) 106:16764–9. doi: 10.1073/pnas.0909142106
46. Lu H, Talbot S, Robertson KA, Watterson S, Forster T, Roy D, et al. Rapid proteasomal elimination of 3-hydroxy-3-methylglutaryl-CoA reductase by interferon-γ in primary macrophages requires endogenous 25-hydroxycholesterol synthesis. Steroids. (2015) 99:219–29. doi: 10.1016/j.steroids.2015.02.022
47. Bartlett S, Gemiarto AT, Ngo MD, Sajiir H, Hailu S, Sinha R, et al. GPR183 regulates interferons, autophagy, and bacterial growth during mycobacterium tuberculosis infection and is associated with TB disease severity. Front Immunol. (2020) 11:601534. doi: 10.3389/fimmu.2020.601534
48. Selvaraj P. Vitamin D, vitamin D receptor, and cathelicidin in the treatment of tuberculosis. Vitam Horm. (2011) 86:307–25. doi: 10.1016/b978-0-12-386960-9.00013-7
49. Salamon H, Bruiners N, Lakehal K, Shi L, Ravi J, Yamaguchi KD, et al. Cutting edge: Vitamin D regulates lipid metabolism in Mycobacterium tuberculosis infection. J Immunol. (2014) 193:30–4. doi: 10.4049/jimmunol.1400736
50. Anand PK, Kaul D, Sharma M. Synergistic action of vitamin D and retinoic acid restricts invasion of macrophages by pathogenic mycobacteria. J Microbiol Immunol Infect. (2008) 41:17–25.
51. Wang N, Westerterp M. ABC transporters, cholesterol efflux, and implications for cardiovascular diseases. Adv Exp Med Biol. (2020) 1276:67–83. doi: 10.1007/978-981-15-6082-8_6
52. Ye Y, Liu J, Guo Y, Gao Y, Rao J, Su R, et al. PPARγ Ameliorates Mycobacterium tuberculosis H37Ra-Induced Foamy Macrophage Formation via the ABCG1-Dependent Cholesterol Efflux Pathway in THP-1 Macrophages. Front Microbiol. (2022) 13:829870. doi: 10.3389/fmicb.2022.829870
53. Zheng X, Li M, Chen Q, Ma B, Nie X, Liu Y, et al. Ror2-mediated cholesterol accumulation regulates autophagic activity within BCG-infected macrophages. Microb Pathog. (2022) 167:105564. doi: 10.1016/j.micpath.2022.105564
54. Agarwal P, Gordon S, Martinez FO. Foam cell macrophages in tuberculosis. Front Immunol. (2021) 12:775326. doi: 10.3389/fimmu.2021.775326
55. Ordway D, Henao-Tamayo M, Orme IM, Gonzalez-Juarrero M. Foamy macrophages within lung granulomas of mice infected with Mycobacterium tuberculosis express molecules characteristic of dendritic cells and antiapoptotic markers of the TNF receptor-associated factor family. J Immunol. (2005) 175:3873–81. doi: 10.4049/jimmunol.175.6.3873
56. Agarwal P, Combes TW, Shojaee-Moradie F, Fielding B, Gordon S, Mizrahi V, et al. Foam cells control mycobacterium tuberculosis infection. Front Microbiol. (2020) 11:1394. doi: 10.3389/fmicb.2020.01394
57. Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PloS Pathog. (2008) 4:e1000204. doi: 10.1371/journal.ppat.1000204
58. Nabeshima S, Nomoto M, Matsuzaki G, Kishihara K, Taniguchi H, Yoshida S, et al. T-Cell hyporesponsiveness induced by activated macrophages through nitric oxide production in mice infected with Mycobacterium tuberculosis. Infect Immun. (1999) 67:3221–6. doi: 10.1128/iai.67.7.3221-3226.1999
59. Hernandez-Pando R, Orozco H, Arriaga K, Sampieri A, Larriva-Sahd J, Madrid-Marina V. Analysis of the local kinetics and localization of interleukin-1 alpha, tumour necrosis factor-alpha and transforming growth factor-beta, during the course of experimental pulmonary tuberculosis. Immunology. (1997) 90:607–17. doi: 10.1046/j.1365-2567.1997.00193.x
60. Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, et al. Alveolar macrophages provide an early mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe. (2018) 24:439–446.e4. doi: 10.1016/j.chom.2018.08.001
61. Ehlers S, Schaible UE. The granuloma in tuberculosis: dynamics of a host-pathogen collusion. Front Immunol. (2012) 3:411. doi: 10.3389/fimmu.2012.00411
62. Orme IM, Basaraba RJ. The formation of the granuloma in tuberculosis infection. Semin Immunol. (2014) 26:601–9. doi: 10.1016/j.smim.2014.09.009
63. Guerrini V, Gennaro ML. Foam cells: one size doesn’t fit all. Trends Immunol. (2019) 40:1163–79. doi: 10.1016/j.it.2019.10.002
64. McClean CM, Tobin DM. Macrophage form, function, and phenotype in mycobacterial infection: lessons from tuberculosis and other diseases. Pathog Dis. (2016) 74(7):ftw068. doi: 10.1093/femspd/ftw068
65. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med. (2018) 215:1135–52. doi: 10.1084/jem.20172020
66. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. (2014) 511:99–103. doi: 10.1038/nature13489
67. Shi L, Salamon H, Eugenin EA, Pine R, Cooper A, Gennaro ML. Infection with Mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Sci Rep. (2015) 5:18176. doi: 10.1038/srep18176
68. Braverman J, Sogi KM, Benjamin D, Nomura DK, Stanley SA. HIF-1α Is an essential mediator of IFN-γ-dependent immunity to mycobacterium tuberculosis. J Immunol. (2016) 197:1287–97. doi: 10.4049/jimmunol.1600266
69. Refai A, Gritli S, Barbouche MR, Essafi M. Mycobacterium tuberculosis virulent factor ESAT-6 drives macrophage differentiation toward the pro-inflammatory M1 phenotype and subsequently switches it to the anti-inflammatory M2 phenotype. Front Cell Infect Microbiol. (2018) 8:327. doi: 10.3389/fcimb.2018.00327
70. Abdalla AE, Lambert N, Duan X, Xie J. Interleukin-10 family and tuberculosis: an old story renewed. Int J Biol Sci. (2016) 12:710–7. doi: 10.7150/ijbs.13881
71. Warsinske HC, Pienaar E, Linderman JJ, Mattila JT, Kirschner DE. Deletion of TGF-β1 increases bacterial clearance by cytotoxic T cells in a tuberculosis granuloma model. Front Immunol. (2017) 8:1843. doi: 10.3389/fimmu.2017.01843
72. Davuluri KS, Singh AK, Singh AV, Chaudhary P, Raman SK, Kushwaha S, et al. Atorvastatin potentially reduces mycobacterial severity through its action on lipoarabinomannan and drug permeability in granulomas. Microbiol Spectr. (2023) 11:e0319722. doi: 10.1128/spectrum.03197-22
73. Brown KL, Wilburn KM, Montague CR, Grigg JC, Sanz O, Pérez-Herrán E, et al. Cyclic AMP-mediated inhibition of cholesterol catabolism in mycobacterium tuberculosis by the novel drug candidate GSK2556286. Antimicrob Agents Chemother. (2023) 67:e0129422. doi: 10.1128/aac.01294-22
74. Nuermberger EL, Martínez-Martínez MS, Sanz O, Urones B, Esquivias J, Soni H, et al. GSK2556286 is a novel antitubercular drug candidate effective in vivo with the potential to shorten tuberculosis treatment. Antimicrob Agents Chemother. (2022) 66:e0013222. doi: 10.1128/aac.00132-22
75. Li SY, Tyagi S, Soni H, Betoudji F, Converse PJ, Mdluli K, et al. Bactericidal and sterilizing activity of novel regimens combining bedaquiline or TBAJ-587 with GSK2556286 and TBA-7371 in a mouse model of tuberculosis. Antimicrob Agents Chemother. (2024) 68:e0156223. doi: 10.1128/aac.01562-23
76. Selvaraj P, Harishankar M, Afsal K. Vitamin D: Immuno-modulation and tuberculosis treatment. Can J Physiol Pharmacol. (2015) 93:377–84. doi: 10.1139/cjpp-2014-0386
Keywords: MTB, TB, cholesterol metabolism, foam cell, macrophages, lipid droplet, oxidized cholesterol
Citation: Chen Z, Kong X, Ma Q, Chen J, Zeng Y, Liu H, Wang X and Lu S (2024) The impact of Mycobacterium tuberculosis on the macrophage cholesterol metabolism pathway. Front. Immunol. 15:1402024. doi: 10.3389/fimmu.2024.1402024
Received: 16 March 2024; Accepted: 14 May 2024;
Published: 30 May 2024.
Edited by:
Igor Kramnik, Boston University, United StatesReviewed by:
Debora Decote-Ricardo, Federal Rural University of Rio de Janeiro, BrazilCopyright © 2024 Chen, Kong, Ma, Chen, Zeng, Liu, Wang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaomin Wang, d3htX3ptdUAxNjMuY29t; Shuihua Lu, bHVzaHVpaHVhNjZAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.