 Clara Machado Campolim1,2†
Clara Machado Campolim1,2† Bianca Camilo Schimenes3†
Bianca Camilo Schimenes3† Mariana Matera Veras4
Mariana Matera Veras4 Young-Bum Kim2
Young-Bum Kim2 Patricia Oliveira Prada1,5,6*
Patricia Oliveira Prada1,5,6*- 1Department of Internal Medicine, School of Medical Science, State University of Campinas (UNICAMP), Campinas, SP, Brazil
- 2Division of Endocrinology, Diabetes, and Metabolism, Department of Medicine, Beth Israel Deaconess Medical Center, and Harvard Medical School, Boston, MA, United States
- 3School of Applied Sciences, State University of Campinas (UNICAMP), Limeira, SP, Brazil
- 4Laboratory of Environmental and Experimental Pathology LIM05, Department of Pathology, School of Medicine, University of São Paulo (USP), São Paulo, SP, Brazil
- 5Obesity and Comorbidities Research Center, Campinas, SP, Brazil
- 6Department of Structural and Functional Biology, Institute of Biology (IB), University of Campinas, Campinas, SP, Brazil
Air pollution is an urgent concern linked to numerous health problems in low- and middle-income countries, where 92% of air pollution-related deaths occur. Particulate matter 2.5 (PM2.5) is the most harmful component of air pollutants, increasing inflammation and changing gut microbiota, favoring obesity, type 2 diabetes, and Alzheimer’s Disease (AD). PM2.5 contains lipopolysaccharides (LPS), which can activate the Toll-like receptor 4 (TLR4) signaling pathway. This pathway can lead to the release of pro-inflammatory markers, including interleukins, and suppressor of cytokine signaling-3 (SOCS3), which inhibits leptin action, a hormone that keeps the energy homeostasis. Leptin plays a role in preventing amyloid plaque deposition and hyperphosphorylation of tau-protein (p-tau), mechanisms involved in the neurodegeneration in AD. Approximately 50 million people worldwide are affected by dementia, with a significant proportion living in low—and middle-income countries. This number is expected to triple by 2050. This mini-review focuses on the potential impact of PM2.5 exposure on the TLR4 signaling pathway, its contribution to leptin resistance, and dysbiosis that exacerbates the link between obesity and AD.
1 Introduction
Air pollution is one of the significant environmental risks associated with morbidity and premature death (1). This concern is even more urgent in low- and middle-income countries, where 92% of air pollution-related deaths occur (1, 2). One of the most harmful components of air pollutants is particulate matter, classified according to its aerodynamic diameter, whose size determines its distribution and potential health impacts.
The classes of particles based on their aerodynamic diameter typically include particulate matter 2.5 (PM2.5) - particles with a diameter of 2.5 micrometers or less (3) - which can remain suspended in the air for extended periods. Its toxicity relies on its concentration and composition. Their surface area-to-volume ratio makes them more effective at absorbing other toxic substances in the air. Considering the toxicity of PM2.5, The World Health Organization’s Air Quality Concentration Guideline established in 2005 that the mean concentrations of PM2.5 should not surpass 10 µg/m3 per year. However, data from 2019 indicated that 90% of the population consistently faced concentrations overcoming the guidelines, resulting in fatalities and illnesses. Thus, in 2021, the guideline set a new goal of 5 µg/m3 annually (2).
In urban areas, the primary source of PM2.5 is vehicular emissions, composed of sulfates, nitrates, ammonia, carbons, hydrogen, lipopolysaccharides (LPS), metals, and water (4, 5). PM2.5 primarily enters the body through inhalation via the upper airways. It reaches the lungs, and the immune system recognizes it, causing local inflammation to spread to the systemic circulation and tissues (6–9). In addition, PM2.5 might directly access the central nervous system (CNS) via the nasal epithelium and the olfactory bulb, inducing neuroinflammation (10, 11). The inflammation caused by PM2.5 exposure could be the link between air pollution exposure (PM2.5) and obesity, which is a disorder that, in turn, increases an individual’s risk of developing dementia.
This mini-review will focus on the implications of PM2.5 exposure, particularly its potential to activate the immune system in the brain areas that regulate feeding, cognition, memory, and behavior. This activation could contribute to the pathology of obesity and Alzheimer’s disease (AD). Furthermore, we will explore the possible involvement of leptin in this mechanism.
2 PM2.5 infiltration into the central nervous system causing inflammation
PM2.5 exposure caused tissue-like damage and pro-inflammatory responses in the hippocampus, cortex, and hypothalamus (12–16), potential areas involved in the pathophysiology of obesity and dementia. In vivo and in vitro studies have demonstrated that long- and short-term exposure to PM2.5 increases inflammatory markers such as tumor necrosis factor-alpha (TNF-α), interleukin 1 beta (IL-1β), interleukin 6 (IL-6), Toll-like receptor 4 (TLR4), and nuclear factor–kappa B (NFkB) in different brain areas (12, 17, 18).
Since PM2.5 may contain LPS (19, 20), a component of gram-negative bacteria membranes, and a primary agonist of TLR4, our immune cells could recognize it as a pathogen after it crosses the blood-brain barrier (BBB). TLR4 is a receptor from the innate immune system. Its activation recruited myeloid differentiation factor 88 (Myd88). This association leads to the inhibitor kappa B kinase (ikkB) phosphorylation, releasing NFkB to the cell nucleus to start the production and secretion of pro-inflammatory proteins (21, 22).
Studies have shown that six months of PM2.5 exposure increased the expression of pro-inflammatory genes and activated the Ikk/NF-κB pathway in the arcuate nucleus of the hypothalamus (ARC) of control mice (13), an area related to feeding regulation. Evidence demonstrated that PM2.5 exposure led to microglia activation by upregulating ionized calcium-binding adaptor molecule 1 (Iba-1) expression in the hypothalamus, hippocampus, and cortex (23–25). One of our recent studies revealed that maternal exposure to PM2.5 induces hypothalamic inflammation in offspring (16), suggesting that in addition to the direct exposure damage seen in other studies, prenatal exposure to PM2.5 also induces inflammation (15, 16, 18, 24). Diesel exhaust (DE) is also a significant component of PM2.5, and its acute exposure increased interleukin one alpha (IL-1α), IL-1β, interleukin 3 (IL-3), IL-6, and TNF-α levels in the olfactory bulb and hippocampus, in addition to activate microglia in the hippocampus of control mice (26).
Besides obesity (24) and AD (27), PM2.5 led to other conditions involving neuroinflammation, including Ischemic Stroke (28), Parkinson’s disease (29), and Type 2 Diabetes Mellitus (T2DM) (30, 31). Therefore, PM2.5 might trigger systemic inflammation and elevated levels of pro-inflammatory cytokines in the CNS, thereby being a risk factor for metabolic diseases and neurodegenerative disorders.
3 The effects of air pollution in the development of Alzheimer’s disease
AD is a progressive neurodegenerative disease that primarily affects memory, cognition, and behavior and gradually worsens over time. While specific medications can alleviate some symptoms, there is currently no cure for AD (32). The accumulation of amyloid beta (Aβ) plaques and hyperphosphorylated tau-protein (p-tau) in the brain are characteristics of AD (33, 34). In 2018, Alzheimer’s Disease International estimated that approximately 50 million people worldwide were affected by dementia, with a significant proportion living in low— and middle-income countries. This number is expected to triple by 2050 (35).
The primary risk factors for developing AD are typically over 65 and carrying at least one apolipoprotein E (APOE) ϵ4 allele. In the preclinical phase, alterations in neurons, microglia, and astroglia contribute to the progression of the disease before cognitive impairments become noticeable. Neuroinflammation, vascular changes, aging, and non-efficient astroglial cell waste clearance play crucial roles in this cellular disease landscape (35).
The presence of Aβ aggregates in the brain initiates a response from microglia, which releases harmful elements like nitric oxide (NO), reactive oxygen species (ROS), pro-inflammatory cytokines, and chemokines, causing neuron damage. These microglia triggered by Aβ are often co-localized with amyloid plaques in AD. In this state, microglia may contribute to the hyperphosphorylation of p-tau (36, 37).
The activation of microglia TLR4 by Aβ aggregates facilitates its clearance and function as a natural protected event. However, accumulating Aβ aggregates due to self-sustaining cycles generating Aβ diminishes TLR4 capacity, overwhelming the body’s natural mechanisms for resolving Aβ aggregates (38, 39). Therefore, the protective effect of TLR4 is present in the initial stages of aggregation, but prolonged TLR4 activation may contribute to Aβ deposition (40).
Stewart et al. (41), proposed that Aβ interacts with the TLR4 through a CD36-TLR4-TLR6 complex. This mechanism involves the recognition of Aβ by cluster of differentiation 36 (CD36), which then initiates signaling through TLR4 and toll-like receptor 6 (TLR6), activating downstream inflammatory pathways. The CD36-TLR4-TLR6 complex increases the IL-1β expression, a cytokine prominently found in AD plaques (41).
IL-1β is a maker of neuroinflammation and could be triggered by PM2.5 (42). As discussed, constant exposure to PM2.5, which contains LPS, might cause prolonged TLR4 activation (19, 20), ultimately contributing to Aβ deposition. Evidence suggests that PM2.5 exposure could be a risk factor for AD (10, 25, 43).
In a study by Lee et al. (10), AD-model mice exposed to PM2.5 for 3 months (11.38 μg/m3 mean concentration) showed an increase in p-tau levels at olfactory bulb, as well as MDA, an oxidative stress biomarker, in the hippocampus and olfactory bulb. These effects indicate the meaningful pathway through which PM2.5 enters and propagates oxidative damage. The olfactory bulb could be the starting point for the damage caused by PM2.5, and the lack of damage to other brain regions or biomarkers could be due to low-level exposure (10). A study with an AD mouse model (APP/PS1) found that exposure to PM2.5 at 25.8 μg/m3 for 3 months significantly increased Aβ plaque density in the hippocampus. This result suggests that chronic exposure to PM2.5 could worsen AD pathology (25).
Furthermore, Ning et al. (44) found that 1 month of exposure to PM2.5 (3 mg/kg) led to impaired learning and memory in young mice (4 weeks) after performing the Morris water maze test (44). Another study revealed that exposure to PM2.5 (3 mg/kg) for one month was enough to raise tau hyper-phosphorylation levels in the cortex of middle-aged mice. Remarkably, these elevated levels returned to baseline after the cessation of PM2.5 exposure (43). Collectively, these observations suggest that chronic PM2.5 exposure could be a crucial AD risk factor.
4 PM2.5-induced leptin resistance: implications for Alzheimer’s disease and obesity
Leptin is an anorexigenic hormone secreted by adipocytes in proportion to their mass that regulates satiety and energy expenditure through a molecular signaling cascade. This cascade involves binding to its receptor and subsequently activates the phosphorylation of signal transducer and activator of transcription 3 (STAT3). STAT3 translocates to the nucleus cell and stimulates the transcription of Pomc neurons while reducing Agrp/Npy secretion from neurons on the ARC, increasing satiety and energy expenditure (45–47). Besides the neurons of the hypothalamus, leptin receptor long isoform (LepRb) is present in the hippocampus, cortex, cerebellum, brainstem, and thalamus (48, 49). In these areas, leptin is involved in motivation, reproduction, growth, learning, and memory (50–52).
A suppressor of cytokine signaling-3 (SOCS3) is a physiologic leptin signaling inhibitor typically expressed with leptin signaling. However, in the context of obesity and leptin resistance with the activation of the TLR4/Myd88/NFkB axis, an enhanced SOCS3 expression occurs, causing a pathologic leptin signaling inhibition (45, 53). PM2.5’s LPS content might activate the TLR4 signaling pathway in the various brain regions, including the hypothalamus, hippocampus, and cortex (40, 45, 54). This effect increases several pro-inflammatory cytokines and SOCS3 production, consequently causing leptin resistance (Figure 1) (21, 22, 45, 53). Therefore, unhealthy obesity is considered a state of low-grade systemic and hypothalamic inflammation, which could impair neuronal function (55, 56). Leptin resistance contributes to an obese state and promotes the formation of Aβ plaques and hyperphosphorylation of p-tau, which ultimately leads to the onset and progression of AD (Figure 1) (57–60).
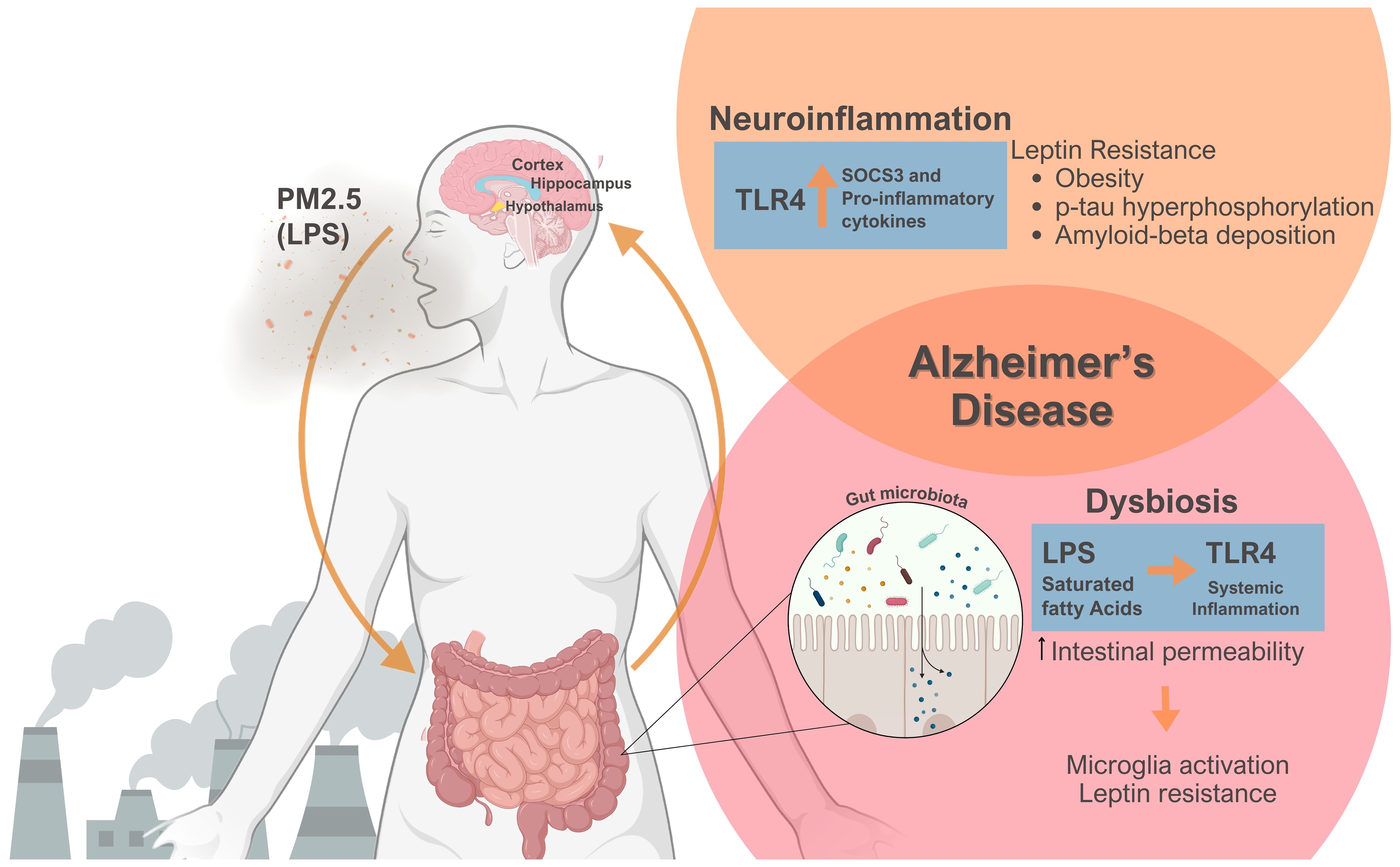
Figure 1 Emphasizes the role of PM2.5 exposure on the gut-brain axis, culminating in leptin resistance, obesity, and Alzheimer’s disease. PM2.5-derived Lipopolysaccharide (LPS) can engage Toll-like receptor 4 (TLR4) signaling, thereby triggering neuroinflammation via the production of pro- inflammatory cytokines and suppressor of cytokine signaling-3 (SOCS3), a physiological inhibitor of signal transducer and activator of transcription 3 (STAT3) phosphorylation leading to leptin resistance. Obesity and the lack of leptin action heightened tau-protein phosphorylation and amyloid-beta deposition. Simultaneously, PM2.5 exposure causes dysbiosis, changing the composition of gut microbiota. Combined with a poor diet, LPS, and Saturated Fatty Acids could provoke systemic inflammation, thereby increasing intestinal permeability. This scenario could activate microglia and worsen leptin resistance. Significantly, PM2.5 induces neuroinflammation and changes the gut microbiota, two phenomena crucial in Alzheimer’s disease pathogenesis. Figure created using BioRender.com.
Rodents with obesity displayed elevated circulating leptin concentrations. Despite higher leptin levels, the anorexigenic effect is lacking, suggesting leptin resistance (61, 62). Although age and genetics are well-established risk factors in AD pathogenesis, the link between leptin resistance, obesity, and the development of AD has been the subject of several studies (63–65).
Interestingly, individuals with AD had lower plasma leptin levels. Over 50% of subjects with mild cognitive impairment showed lower leptin levels than the control group (66). The APP/PS1 mouse model of AD had similar lower leptin levels (67). Together, these studies suggest that circulating leptin levels may play a role in cognitive impairment.
Not only are lower leptin levels associated with AD, but patients with AD exhibit increased leptin levels in the cerebrospinal fluid (CSF) and hippocampus while concurrently showing a decrease in its receptor, suggesting leptin resistance. Furthermore, a reduction in leptin receptor expression is evident even in cases where leptin levels are the same between groups (68, 69). The hippocampus is a complex brain structure that plays an essential role in learning, memory, cognition, and synaptic plasticity (70). It is AD’s first and primary affected area (69). In addition to the reduction of the leptin receptor expression in the hippocampus of AD rodent experimental models, such as old Tg2576, apoE4, and APP/PS1 mice, the STAT3 phosphorylation is also impaired in this area, as reviewed by McGregor et al. (48).
In control mice, leptin increases glutamate receptors long-term potentiation (LTP), improving excitatory synaptic transmission and decreasing long-term depression (LTD). This modification suggests that leptin influences the brain’s fundamental process underlying learning and memory. In APP/PS1 mice with lower leptin levels, supplementation with leptin improves the harmful effects of Aβ on hippocampal LTP and LTD, restoring normal synaptic function, increasing synaptic density, and ameliorating memory deficits (68).
In vivo and in vitro studies showed leptin injection attenuates Aβ toxic levels in hippocampus neuronal cells (57, 71). Other studies showed that activating the leptin signaling pathway in the AD mouse model decreases tau phosphorylation in the hippocampus, indicating that leptin has a possible protective effect (72, 73). Leptin protects against AD by reducing the expression of the β and γ-secretase enzymes. The β and γ-secretase enzymes made the amyloid precursor protein (APP) cleavage, assembling Aβ toxic structures. Therefore, they promote the initiation of amyloid plaque deposition and tau hyperphosphorylation, leading to neurodegeneration (57, 58). Together, the studies suggest that normal leptin signaling and action are beneficial to avoid AD progression. Human and mouse models of AD displayed lower leptin levels or leptin resistance, in which leptin has an incomplete effect.
In summary, elevated leptin levels contribute to reduced signaling response in obesity, leading to leptin resistance. In contrast, a lack of leptin alters essential brain functions currently under investigation for their potential impact on AD development, particularly obesity. In both obesity and AD scenarios, insufficient leptin action is a marked risk factor for neurodegenerative progression. Scientific research seeks to understand how leptin influences AD pathology and whether it holds promise as a therapeutic target.
5 The overlapping aspects of Alzheimer’s disease and obesity
AD and obesity share several environmental risk factors, such as a sedentary lifestyle, consuming a high-fat and high-sugar diet, and increased stress due to lack of sleep (74–76). Obesity in midlife is associated with cognitive reduction and memory and verbal and spatial ability impairments afterward (77–79). Exposure to lower education, poor nutrition, and family stressors during early years increases the risk of cognitive impairment and dementia in later life (80, 81).
Age and genetics are independent risk factors contributing to AD pathogenesis development (82). Individuals who develop obesity in midlife have a 33% higher incidence of AD (83). Neurodegenerative diseases often display obesity as a comorbidity in neurodegenerative diseases, as they share biomolecular mechanisms that can lead to brain damage. Both conditions show increased levels of pro-inflammatory cytokines, including IL-1β, IL-6, TNF-α, and leptin resistance. These conditions trigger neurodegeneration through neuroinflammation (12, 80, 84–86).
In addition to inflammation, obese individuals with elevated oxidative stress and mitochondrial dysfunction have an increased risk of developing AD (87, 88). Notably, elevated levels of APP in adipose tissue and of Aβ in the bloodstream have also been identified as significant contributors to this heightened (87, 89, 90).
In the review by Flores-Cordero et al. (33), they highlight that obesity resulting from high-fat diets (HFD) increases the risk of dementia, affecting cognitive abilities such as memory, attention, and executive functions, with neuroinflammation playing a crucial role. Studies on male Wistar rats fed with HFD displayed impaired memory. In contrast, assessments of rats fed with a high-fructose-high-coconut oil diet using the Morris water maze task revealed hippocampal-dependent learning and memory problems. These impairments were followed by molecular changes related to inflammation, stress, and central insulin resistance (33).
Similarly, a recent study by da Cruz Rodrigues et al. (91) revealed that obese mice aged 32 weeks lacking Low density lipoprotein receptor-related protein 1 (LRP1) in GABAergic neurons demonstrated impaired performance in learning and memory tasks, including the Water-T Maze and Spatial Novelty Y Maze compared to mice at 16 weeks of age. The aging mice exhibited diminished locomotor activity compared to their control counterparts. Also, these mice showed higher leptin levels and neurodegeneration indicators. These findings suggest that obesity may significantly influence cognitive function age-dependently (91).
In summary, the studies point to increased inflammation, Aβ and APP elevated levels, and leptin resistance as the sharing characteristics between obesity and AD. These elements contribute to neuroinflammation and neurodegeneration, which are associated with the development of AD.
6 PM2.5 exposure and gut microbiota dynamics: insights into obesity and Alzheimer’s disease
The gut microbiota is another aspect influencing obesity and AD pathology, potentially affected by environmental factors like PM2.5 and dietary choices.
A healthy gut microbiota shows significant diversity and variability in microorganisms (92, 93), a crucial regulator of energy balance. The alteration, known as dysbiosis, is associated with obesity, T2DM, colorectal cancer, and AD (94, 95).
Animal studies reveal that environmental pollutants notably disrupt the composition of the gut microbiota (96–98). Microbes ingested with PM2.5 induce inflammatory responses in local immune cells, increase intestinal permeability, and alter the gut environment, promoting the growth of specific microbial strains adapted to an inflammatory milieu (99).
Transplanting the gut microbiota from obese mice into germ-free animals increases fat mass, indicating that obese animals have a specific gut microbiota that promotes excess body weight accumulation (100, 101). Several diet factors might also change the gut microbiota, contributing to obesity induction. High-fat and high-sucrose diets are often deficient in essential vitamins and minerals and can induce adverse metabolic effects (102). A high-fat diet and a cafeteria diet led to leptin resistance and dysbiosis, which, in turn, altered the gut microbiota composition, increasing the intestinal permeability to LPS uptake. As a result, LPS and saturated fatty acids from the diet can activate TLR4, initiating systemic inflammation (Figure 1) (102, 103).
Studies on germ-free (GF) mice emphasize the role of gut microbiota in modulating immune responses and gene expression patterns associated with inflammation, including the microglia activation in the brain (95, 104, 105). Microglia from GF mice showed immature profiles and impaired immune responses compared to conventionally raised mice. In particular, transcription and survival factors usually suppressed in mature microglia were increased in GF mice. Recolonization of GF mice with fecal samples from AD patients activates microglia more significantly than samples from healthy donors (95, 106).
Evidence suggests that the development of AD may originate in the gut and then spread to the brain, reinforcing the idea of the gut-brain axis (107–110). Aβ1–42 oligomers injected into the gastric wall of mice migrate from the intestine to the brain. Additionally, bacterial strains such as Escherichia coli and Salmonella enterica produce extracellular amyloid fibers, which can initiate immune responses and enhance the formation of neuronal amyloid in the brain (95, 111, 112).
In summary, PM2.5 impacts obesity and AD by influencing the gut microbiota. Dysbiosis and nutrient-deficient diets lead to leptin resistance and exacerbated intestinal permeability (Figure 1), thereby triggering systemic inflammation. Additionally, the gut microbiota influences immune responses, including microglia function. Given the importance of the connection between the gut and AD, interventions targeting the gut microbiome are crucial in managing both obesity and AD.
7 Major conclusions
Our mini-review discusses the increasing prevalence of AD and obesity as global health challenges, exploring the impact of PM2.5 exposure on these illnesses. We examine evidence suggesting that immune system activation leads to leptin resistance, a critical facet in these conditions. Additionally, we discuss the need for further research on PM2.5-induced inflammation, neurodegeneration, and the protective role of leptin on AD pathogenesis. Finally, we highlight the urgent action to reduce air pollution and its associated health risks.
Author contributions
CC: Conceptualization, Writing – original draft. BS: Writing – original draft. MV: Writing – review & editing. Y-BK: Writing – review & editing. PP: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico), Brazil: 402941/2022-5 (PP), and 200568/2022-1 (CC); CNPq - INCT (Instituto Nacional de Ciencia e Tecnologia de Obesidade e Diabetes), Brazil: 573856/2008-7 (PP) and INCT-FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo), São Paulo, Brazil: 465693/2014-8 (PP); FAPESP: 2017/18498-6 (PP), and 2017/11518- 1 (CC), and National Institutes of Health (R01DK129946, R01AG080842 to Y-BK). Grant CNPq 311576/2022-2 (MMV).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fuller R, Landrigan PJ, Balakrishnan K, Bathan G, Bose-O'Reilly S, Brauer M, et al. Pollution and health: a progress update. Lancet Planet Health. (2022) 6:e535–47. doi: 10.1016/S2542–5196(22)00090–0
2. World Health Organization. Global air quality guidelines: Particulate matter (PM2.5 and PM10), ozone, nitrogen dioxide, sulfur dioxide and carbon monoxide. Geneva:World Health Organization. License: CC BYNCSA 3.0 IGO (2021).
3. Polichetti G, Cocco S, Spinali A, Trimarco V, Nunziata A. Effects of particulate matter (PM(10), PM(2.5) and PM(1)) on the cardiovascular system. Toxicology. (2009) 261:1–8. doi: 10.1016/j.tox.2009.04.035
4. He M, Ichinose T, Yoshida S, Ito T, He C, Yoshida Y, et al. PM2.5-induced lung inflammation in mice: Differences of inflammatory response in macrophages and type II alveolar cells. J Appl Toxicol. (2017) 37:1203–18. doi: 10.1002/jat.3482
5. Yariwake VY, Torres JI, Dos Santos ARP, Freitas SCF, De Angelis K, Farhat SCL, et al. Chronic exposure to PM2.5 aggravates SLE manifestations in lupus-prone mice. Part Fibre Toxicol. (2021) 18:15. doi: 10.1186/s12989–021-00407–0
6. Riva DR, Magalhães CB, Lopes AA, Lanças T, Mauad T, Malm O, et al. Low dose of fine particulate matter (PM2.5) can induce acute oxidative stress, inflammation and pulmonary impairment in healthy mice. Inhal Toxicol. (2011) 23:257–67. doi: 10.3109/08958378.2011.566290
7. Glencross DA, Ho TR, Camiña N, Hawrylowicz CM, Pfeffer PE. Air pollution and its effects on the immune system. Free Radic Biol Med. (2020) 151:56–68. doi: 10.1016/j.freeradbiomed.2020.01.179
8. Serafini MM, Maddalon A, Iulini M, Galbiati V. Air pollution: possible interaction between the immune and nervous system? Int J Environ Res Public Health. (2022) 19:16037. doi: 10.3390/ijerph192316037
9. Li D, Li Y, Li G, Zhang Y, Li J, Chen H. Fluorescent reconstitution on deposition of PM2.5 in lung and extrapulmonary organs. Proc Natl Acad Sci U S A. (2019) 116:2488–93. doi: 10.1073/pnas.1818134116
10. Lee SH, Chen YH, Chien CC, Yan YH, Chen HC, Chuang HC, et al. Three month inhalation exposure to low-level PM2.5 induced brain toxicity in an Alzheimer's disease mouse model. PloS One. (2021) 16:e0254587. doi: 10.1371/journal.pone.0254587
11. Kang YJ, Tan HY, Lee CY, Cho H. An air particulate pollutant induces neuroinflammation and neurodegeneration in human brain models. Adv Sci (Weinh). (2021) 8:e2101251. doi: 10.1002/advs.202101251
12. Fonken LK, Xu X, Weil ZM, Chen G, Sun Q, Rajagopalan S, et al. Air pollution impairs cognition, provokes depressive-like behaviors and alters hippocampal cytokine expression and morphology. Mol Psychiatry. (2011) 16:987–95, 973. doi: 10.1038/mp.2011.76
13. Ying Z, Xu X, Bai Y, Zhong J, Chen M, Liang Y, et al. Long-term exposure to concentrated ambient PM2.5 increases mouse blood pressure through abnormal activation of the sympathetic nervous system: a role for hypothalamic inflammation. Environ Health Perspect. (2014) 122:79–86. doi: 10.1289/ehp.1307151
14. Ku T, Li B, Gao R, Zhang Y, Yan W, Ji X, et al. NF-κB-regulated microRNA-574–5p underlies synaptic and cognitive impairment in response to atmospheric PM2.5 aspiration. Part Fibre Toxicol. (2017) 14:34. doi: 10.1186/s12989–017-0215–3
15. Chao MW, Yang CH, Lin PT, Yang YH, Chuang YC, Chung MC, et al. Exposure to PM2.5 causes genetic changes in fetal rat cerebral cortex and hippocampus. Environ Toxicol. (2017) 32:1412–25. doi: 10.1002/tox.22335
16. Zordão OP, Campolim CM, Yariwake VY, Castro G, Ferreira CKO, Santos A, et al. Maternal exposure to air pollution alters energy balance transiently according to gender and changes gut microbiota. Front Endocrinol (Lausanne). (2023) 14:1069243. doi: 10.3389/fendo.2023.1069243
17. Woodward NC, Levine MC, Haghani A, Shirmohammadi F, Saffari A, Sioutas C, et al. Toll-like receptor 4 in glial inflammatory responses to air pollution in vitro and in vivo. J Neuroinflamm. (2017) 14:84. doi: 10.1186/s12974–017-0858-x
18. Wang J, Huang J, Wang L, Chen C, Yang D, Jin M, et al. Urban particulate matter triggers lung inflammation via the ROS-MAPK-NF-κB signaling pathway. J Thorac Dis. (2017) 9:4398–412. doi: 10.21037/jtd.2017.09.135
19. Yoda Y, Tamura K, Shima M. Airborne endotoxin concentrations in indoor and outdoor particulate matter and their predictors in an urban city. Indoor Air. (2017) 27:955–64. doi: 10.1111/ina.12370
20. Chen X, Deng T, Huo T, Dong F, Deng J. MiR-140–5p/TLR4 /NF-κB signaling pathway: Crucial role in inflammatory response in 16HBE cells induced by dust fall PM2.5. Ecotoxicol Environ Saf. (2021) 208:111414. doi: 10.1016/j.ecoenv.2020.111414
21. Fu H, Liu X, Li W, Zu Y, Zhou F, Shou Q, et al. PM2.5 exposure induces inflammatory response in macrophages via the TLR4/COX-2/NF-κB pathway. Inflammation. (2020) 43:1948–58. doi: 10.1007/s10753–020-01269-y
22. Squillace S, Niehoff ML, Doyle TM, Green M, Esposito E, Cuzzocrea S, et al. Sphingosine-1-phosphate receptor 1 activation in the central nervous system drives cisplatin-induced cognitive impairment. J Clin Invest. (2022) 132:e157738. doi: 10.1172/JCI157738
23. Xu MX, Ge CX, Li Q, Lou DS, Hu LF, Sun Y, et al. Fisetin nanoparticles protect against PM2.5 exposure-induced neuroinflammation by down-regulation of astrocytes activation related NF-κB signaling pathway. J Funct Foods. (2020) 65:103716. doi: 10.1016/j.jff.2019.103716
24. Campolim CM, Weissmann L, Ferreira CKO, Zordão OP, Dornellas APS, de Castro G, et al. Short-term exposure to air pollution (PM2.5) induces hypothalamic inflammation, and long-term leads to leptin resistance and obesity via Tlr4/Ikbke in mice. Sci Rep. (2020) 10:10160. doi: 10.1038/s41598–020-67040–3
25. Sahu B, Mackos AR, Floden AM, Wold LE, Combs CK. Particulate matter exposure exacerbates amyloid-β Plaque deposition and gliosis in APP/PS1 mice. J Alzheimers Dis. (2021) 80:761–74. doi: 10.3233/JAD-200919
26. Cole TB, Coburn J, Dao K, Roqué P, Chang YC, Kalia V, et al. Sex and genetic differences in the effects of acute diesel exhaust exposure on inflammation and oxidative stress in mouse brain. Toxicology. (2016) 374:1–9. doi: 10.1016/j.tox.2016.11.010
27. Yang L, Wan W, Yu C, Xuan C, Zheng P, Yan J. Associations between PM2.5 exposure and Alzheimer's Disease prevalence Among elderly in eastern China. Environ Health. (2022) 21:119. doi: 10.1186/s12940–022-00937-w
28. Danesh Yazdi M, Wang Y, Di Q, Wei Y, Requia WJ, Shi L, et al. Long-term association of air pollution and hospital admissions among medicare participants using a doubly robust additive model. Circulation. (2021) 143:1584–96. doi: 10.1161/CIRCULATIONAHA.120.050252
29. Wang Y, Li C, Zhang X, Kang X, Li Y, Zhang W, et al. Exposure to PM2.5 aggravates Parkinson's disease via inhibition of autophagy and mitophagy pathway. Toxicology. (2021) 456:152770. doi: 10.1016/j.tox.2021.152770
30. Lao XQ, Guo C, Chang LY, Bo Y, Zhang Z, Chuang YC, et al. Long-term exposure to ambient fine particulate matter (PM2.5) and incident type 2 diabetes: a longitudinal cohort study. Diabetologia. (2019) 62:759–69. doi: 10.1007/s00125–019-4825–1
31. Liu C, Bai Y, Xu X, Sun L, Wang A, Wang TY, et al. Exaggerated effects of particulate matter air pollution in genetic type II diabetes mellitus. Part Fibre Toxicol. (2014) 11:27. doi: 10.1186/1743–8977-11–27
32. Neha, Wali Z, Pinky, Hattiwale SH, Jamal A, Parvez S. GLP-1/Sigma/RAGE receptors: An evolving picture of Alzheimer's disease pathology and treatment. Ageing Res Rev. (2024) 93:102134. doi: 10.1016/j.arr.2023.102134
33. Flores-Cordero JA, Pérez-Pérez A, Jiménez-Cortegana C, Alba G, Flores-Barragán A, Sánchez-Margalet V. Obesity as a risk factor for dementia and alzheimer's disease: the role of leptin. Int J Mol Sci. (2022) 23:5202. doi: 10.3390/ijms23095202
34. Haberzettl P, Jin L, Riggs DW, Zhao J, O'Toole TE, Conklin DJ. Fine particulate matter air pollution and aortic perivascular adipose tissue: Oxidative stress, leptin, and vascular dysfunction. Physiol Rep. (2021) 9:e14980. doi: 10.14814/phy2.14980
35. Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, et al. Alzheimer's disease. Lancet. (2021) 397:1577–90. doi: 10.1016/S0140–6736(20)32205–4
36. Doorn KJ, Goudriaan A, Blits-Huizinga C, Bol JG, Rozemuller AJ, Hoogland PV, et al. Increased amoeboid microglial density in the olfactory bulb of Parkinson's and Alzheimer's patients. Brain Pathol. (2014) 24:152–65. doi: 10.1111/bpa.12088
37. Combs CK, Karlo JC, Kao SC, Landreth GE. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. (2001) 21:1179–88. doi: 10.1523/JNEUROSCI.21–04-01179.2001
38. Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer's disease. Cell Physiol Biochem. (2007) 20:947–56. doi: 10.1159/000110455
39. Kuhla A, Ludwig SC, Kuhla B, Münch G, Vollmar B. Advanced glycation end products are mitogenic signals and trigger cell cycle reentry of neurons in Alzheimer's disease brain. Neurobiol Aging. (2015) 36:753–61. doi: 10.1016/j.neurobiolaging.2014.09.025
40. Huang NQ, Jin H, Zhou SY, Shi JS, Jin F. TLR4 is a link between diabetes and Alzheimer's disease. Behav Brain Res. (2017) 316:234–44. doi: 10.1016/j.bbr.2016.08.047
41. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. (2010) 11:155–61. doi: 10.1038/ni.1836
42. Wang BR, Shi JQ, Ge NN, Ou Z, Tian YY, Jiang T, et al. PM2.5 exposure aggravates oligomeric amyloid beta-induced neuronal injury and promotes NLRP3 inflammasome activation in an in vitro model of Alzheimer's disease. J Neuroinflamm. (2018) 15:132. doi: 10.1186/s12974–018-1178–5
43. Gao R, Ku T, Ji X, Zhang Y, Li G, Sang N. Abnormal energy metabolism and tau phosphorylation in the brains of middle-aged mice in response to atmospheric PM2.5 exposure. J Environ Sci (China). (2017) 62:145–53. doi: 10.1016/j.jes.2017.06.037
44. Ning X, Li B, Ku T, Guo L, Li G, Sang N. Comprehensive hippocampal metabolite responses to PM2.5 in young mice. Ecotoxicol Environ Saf. (2018) 165:36–43. doi: 10.1016/j.ecoenv.2018.08.080
45. Baldini G, Phelan KD. The melanocortin pathway and control of appetite-progress and therapeutic implications. J Endocrinol. (2019) 241:R1–R33. doi: 10.1530/JOE-18–0596
46. Friedman JM. Leptin and the endocrine control of energy balance. Nat Metab. (2019) 1:754–64. doi: 10.1038/s42255–019-0095-y
47. Obradovic M, Sudar-Milovanovic E, Soskic S, Essack M, Arya S, Stewart AJ, et al. Leptin and obesity: role and clinical implication. Front Endocrinol (Lausanne). (2021) 12:585887. doi: 10.3389/fendo.2021.585887
48. McGregor G, Harvey J. Food for thought: Leptin regulation of hippocampal function and its role in Alzheimer's disease. Neuropharmacology. (2018) 136:298–306. doi: 10.1016/j.neuropharm.2017.09.038
49. Hamilton K, Harvey J. The neuronal actions of leptin and the implications for treating Alzheimer's disease. Pharm (Basel). (2021) 14:52. doi: 10.3390/ph14010052
50. Fulton S, Pissios P, Manchon RP, Stiles L, Frank L, Pothos EN, et al. Leptin regulation of the mesoaccumbens dopamine pathway. Neuron. (2006) 51:811–22. doi: 10.1016/j.neuron.2006.09.006
51. Morrison CD, Daniel JA, Hampton JH, Buff PR, McShane TM, Thomas MG, et al. Luteinizing hormone and growth hormone secretion in ewes infused intracerebroventricularly with neuropeptide Y. Domest Anim Endocrinol. (2003) 24:69–80. doi: 10.1016/s0739–7240(02)00206–0
52. Hamilton K, Harvey J. Leptin regulation of hippocampal synaptic function in health and disease. Vitam Horm. (2021) 115:105–27. doi: 10.1016/bs.vh.2020.12.006
53. Lubis AR, Widia F, Soegondo S, Setiawati A. The role of SOCS-3 protein in leptin resistance and obesity. Acta Med Indones. (2008) 40:89–95.
54. Woodward NC, Haghani A, Johnson RG, Hsu TM, Saffari A, Sioutas C, et al. Prenatal and early life exposure to air pollution induced hippocampal vascular leakage and impaired neurogenesis in association with behavioral deficits. Transl Psychiatry. (2018) 8:261. doi: 10.1038/s41398–018-0317–1
55. Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. (2012) 122:153–62. doi: 10.1172/JCI59660
56. Hotamisligil GS. Inflammation and metabolic disorders. Nature. (2006) 444:860–7. doi: 10.1038/nature05485
57. Niedowicz DM, Studzinski CM, Weidner AM, Platt TL, Kingry KN, Beckett TL, et al. Leptin regulates amyloid β production via the γ-secretase complex. Biochim Biophys Acta. (2013) 1832:439–44. doi: 10.1016/j.bbadis.2012.12.009
58. Marwarha G, Raza S, Meiers C, Ghribi O. Leptin attenuates BACE1 expression and amyloid-β genesis via the activation of SIRT1 signaling pathway. Biochim Biophys Acta. (2014) 1842:1587–95. doi: 10.1016/j.bbadis.2014.05.015
59. Doherty GH, Beccano-Kelly D, Yan SD, Gunn-Moore FJ, Harvey J. Leptin prevents hippocampal synaptic disruption and neuronal cell death induced by amyloid β. Neurobiol Aging. (2013) 34:226–37. doi: 10.1016/j.neurobiolaging.2012.08.003
60. Platt TL, Beckett TL, Kohler K, Niedowicz DM, Murphy MP. Obesity, diabetes, and leptin resistance promote tau pathology in a mouse model of disease. Neuroscience. (2016) 315:162–74. doi: 10.1016/j.neuroscience.2015.12.011
61. Pan WW, Myers MG Jr. Leptin and the maintenance of elevated body weight. Nat Rev Neurosci. (2018) 19:95–105. doi: 10.1038/nrn.2017.168
62. Crujeiras AB, Carreira MC, Cabia B, Andrade S, Amil M, Casanueva FF. Leptin resistance in obesity: An epigenetic landscape. Life Sci. (2015) 140:57–63. doi: 10.1016/j.lfs.2015.05.003
63. Maioli S, Lodeiro M, Merino-Serrais P, Falahati F, Khan W, Puerta E, et al. Alzheimer's Disease Neuroimaging Initiative. Alterations in brain leptin signalling in spite of unchanged CSF leptin levels in Alzheimer's disease. Aging Cell. (2015) 14:122–9. doi: 10.1111/acel.12281
64. Xu WL, Atti AR, Gatz M, Pedersen NL, Johansson B, Fratiglioni L. Midlife overweight and obesity increase late-life dementia risk: a population-based twin study. Neurology. (2011) 76:1568–74. doi: 10.1212/WNL.0b013e3182190d09
65. Marwarha G, Dasari B, Prabhakara JP, Schommer J, Ghribi O. β-Amyloid regulates leptin expression and tau phosphorylation through the mTORC1 signaling pathway. J Neurochem. (2010) 115:373–84. doi: 10.1111/j.1471–4159.2010.06929.x
66. Johnston JM, Hu WT, Fardo DW, Greco SJ, Perry G, Montine TJ, et al. Alzheimer’s Disease Neuroimaging Initiative. Low plasma leptin in cognitively impaired ADNI subjects: gender differences and diagnostic and therapeutic potential. Curr Alzheimer Res. (2014) 11:165–74. doi: 10.2174/1567205010666131212114156
67. Fewlass DC, Noboa K, Pi-Sunyer FX, Johnston JM, Yan SD, Tezapsidis N. Obesity-related leptin regulates Alzheimer's Abeta. FASEB J. (2004) 18:1870–8. doi: 10.1096/fj.04–2572com
68. Lloret A, Monllor P, Esteve D, Cervera-Ferri A, Lloret MA. Obesity as a risk factor for alzheimer's disease: implication of leptin and glutamate. Front Neurosci. (2019) 13:508. doi: 10.3389/fnins.2019.00508
69. Bonda DJ, Stone JG, Torres SL, Siedlak SL, Perry G, Kryscio R, et al. Dysregulation of leptin signaling in Alzheimer disease: evidence for neuronal leptin resistance. J Neurochem. (2014) 128:162–72. doi: 10.1111/jnc.12380
70. Anand KS, Dhikav V. Hippocampus in health and disease: An overview. Ann Indian Acad Neurol. (2012) 15:239–46. doi: 10.4103/0972–2327.104323
71. Calió ML, Mosini AC, Marinho DS, Salles GN, Massinhani FH, Ko GM, et al. Leptin enhances adult neurogenesis and reduces pathological features in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis. (2021) 148:105219. doi: 10.1016/j.nbd.2020.105219
72. Greco SJ, Bryan KJ, Sarkar S, Zhu X, Smith MA, Ashford JW, et al. Leptin reduces pathology and improves memory in a transgenic mouse model of Alzheimer's disease. J Alzheimers Dis. (2010) 19:1155–67. doi: 10.3233/JAD-2010–1308
73. Liu J, Liu Z, Zhang Y, Yin F. Leptin signaling plays a critical role in the geniposide-induced decrease of tau phosphorylation. Acta Biochim Biophys Sin (Shanghai). (2015) 47:1018–22. doi: 10.1093/abbs/gmv106
74. Bakrania K, Edwardson CL, Khunti K, Bandelow S, Davies MJ, Yates T. Associations between sedentary behaviors and cognitive function: cross-sectional and prospective findings from the UK biobank. Am J Epidemiol. (2018) 187:441–54. doi: 10.1093/aje/kwx273
75. Gomes Gonçalves N, Vidal Ferreira N, Khandpur N, Martinez Steele E, Bertazzi Levy R, Andrade Lotufo P, et al. Association between consumption of ultraprocessed foods and cognitive decline. JAMA Neurol. (2023) 80:142–50. doi: 10.1001/jamaneurol.2022.4397
76. Yaffe K, Laffan AM, Harrison SL, Redline S, Spira AP, Ensrud KE, et al. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA. (2011) 306:613–9. doi: 10.1001/jama.2011.1115
77. Wolf PA, Beiser A, Elias MF, Au R, Vasan RS, Seshadri S. Relation of obesity to cognitive function: importance of central obesity and synergistic influence of concomitant hypertension. The Framingham Heart Study. Curr Alzheimer Res. (2007) 4:111–6. doi: 10.2174/156720507780362263
78. Karlsson IK, Gatz M, Arpawong TE, Dahl Aslan AK, Reynolds CA. The dynamic association between body mass index and cognition from midlife through late-life, and the effect of sex and genetic influences. Sci Rep. (2021) 11:7206. doi: 10.1038/s41598–021-86667–4
79. Chuang YF, An Y, Bilgel M, Wong DF, Troncoso JC, O'Brien RJ, et al. Midlife adiposity predicts earlier onset of Alzheimer's dementia, neuropathology and presymptomatic cerebral amyloid accumulation. Mol Psychiatry. (2016) 21:910–5. doi: 10.1038/mp.2015.129
80. Flores-Dorantes MT, Díaz-López YE, Gutiérrez-Aguilar R. Environment and gene association with obesity and their impact on neurodegenerative and neurodevelopmental diseases. Front Neurosci. (2020) 14:863. doi: 10.3389/fnins.2020.00863
81. Wang XJ, Xu W, Li JQ, Cao XP, Tan L, Yu JT. Early-life risk factors for dementia and cognitive impairment in later life: A systematic review and meta-analysis. J Alzheimers Dis. (2019) 67:221–9. doi: 10.3233/JAD-180856
82. Silva MVF, Loures CMG, Alves LCV, de Souza LC, Borges KBG, Carvalho MDG. Alzheimer's disease: risk factors and potentially protective measures. J BioMed Sci. (2019) 26:33. doi: 10.1186/s12929–019-0524-y
83. Zhuang QS, Meng L, Wang Z, Shen L, Ji HF. Associations between obesity and Alzheimer's disease: multiple bioinformatic analyses. J Alzheimers Dis. (2021) 80:271–81. doi: 10.3233/JAD-201235
84. Morys F, Dadar M, Dagher A. Association between midlife obesity and its metabolic consequences, cerebrovascular disease, and cognitive decline. J Clin Endocrinol Metab. (2021) 106:e4260–74. doi: 10.1210/clinem/dgab135
85. Someya Y, Tamura Y, Kaga H, Sugimoto D, Kadowaki S, Suzuki R, et al. Sarcopenic obesity is associated with cognitive impairment in community-dwelling older adults: The Bunkyo Health Study. Clin Nutr. (2022) 41:1046–51. doi: 10.1016/j.clnu.2022.03.017
86. Wang L, Wei LY, Ding R, Feng Y, Li D, Li C, et al. Predisposition to Alzheimer's and age-related brain pathologies by PM2.5 exposure: perspective on the roles of oxidative stress and TRPM2 channel. Front Physiol. (2020) 11:155. doi: 10.3389/fphys.2020.00155
87. Carvalho C, Cardoso S, Correia SC, Santos RX, Santos MS, Baldeiras I, et al. Metabolic alterations induced by sucrose intake and Alzheimer's disease promote similar brain mitochondrial abnormalities. Diabetes. (2012) 61:1234–42. doi: 10.2337/db11–1186
88. Tucsek Z, Toth P, Sosnowska D, Gautam T, Mitschelen M, Koller A, et al. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: effects on expression of genes involved in beta-amyloid generation and Alzheimer's disease. J Gerontol A Biol Sci Med Sci. (2014) 69:1212–26. doi: 10.1093/gerona/glt177
89. Picone P, Di Carlo M, Nuzzo D. Obesity and Alzheimer's disease: Molecular bases. Eur J Neurosci. (2020) 52:3944–50. doi: 10.1111/ejn.14758
90. Lee YH, Martin JM, Maple RL, Tharp WG, Pratley RE. Plasma amyloid-beta peptide levels correlate with adipocyte amyloid precursor protein gene expression in obese individuals. Neuroendocrinology. (2009) 90:383–90. doi: 10.1159/000235555
91. da Cruz Rodrigues KC, Kim SC, Uner AA, Hou ZS, Young J, Campolim C, et al. LRP1 in GABAergic neurons is a key link between obesity and memory function. Mol Metab. (2024) 84:101941. doi: 10.1016/j.molmet.2024.101941
92. Franzosa EA, Huang K, Meadow JF, Gevers D, Lemon KP, Bohannan BJ, et al. Identifying personal microbiomes using metagenomic codes. Proc Natl Acad Sci U S A. (2015) 112:E2930–8. doi: 10.1073/pnas.1423854112
93. Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. (2012) 486:207–14. doi: 10.1038/nature11234
94. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. (2008) 457:480–4. doi: 10.1038/nature07540
95. Megur A, Baltriukienė D, Bukelskienė V, Burokas A. The microbiota-gut-brain axis and alzheimer's disease: neuroinflammation is to blame? Nutrients. (2020) 13:37. doi: 10.3390/nu13010037
96. Wang J, Yan Y, Si H, Li J, Zhao Y, Gao T, et al. The effect of real-ambient PM2.5 exposure on the lung and gut microbiomes and the regulation of Nrf2. Ecotoxicol Environ Saf. (2023) 254:114702. doi: 10.1016/j.ecoenv.2023.114702
97. Ran Z, An Y, Zhou J, Yang J, Zhang Y, Yang J, et al. Subchronic exposure to concentrated ambient PM2.5 perturbs gut and lung microbiota as well as metabolic profiles in mice. Environ pollut. (2021) 272:115987. doi: 10.1016/j.envpol.2020.115987
98. Wang W, Zhou J, Chen M, Huang X, Xie X, Li W, et al. Exposure to concentrated ambient PM2.5 alters the composition of gut microbiota in a murine model. Part Fibre Toxicol. (2018) 15:17. doi: 10.1186/s12989–018-0252–6
99. Wu Y, Pei C, Wang X, Wang M, Huang D, Wang F, et al. Effect of probiotics on nasal and intestinal microbiota in people with high exposure to particulate matter ≤ 2.5 μm (PM2.5): a randomized, double-blind, placebo-controlled clinical study. Trials. (2020) 21. doi: 10.1186/s13063-020-04759-4
100. Cox AJ, West NP, Cripps AW. Obesity, inflammation, and the gut microbiota. Lancet Diabetes Endocrinol. (2015) 3:207–15. doi: 10.1016/S2213–8587(14)70134–2
101. Sonnenburg JL, Bäckhed F. Diet–microbiota interactions as moderators of human metabolism. Nature. (2016) 535:56–64. doi: 10.1038/nature18846
102. Malesza IJ, Malesza M, Walkowiak J, Mussin N, Walkowiak D, Aringazina R, et al. High-fat, western-style diet, systemic inflammation, and gut microbiota: A narrative review. Cells. (2021) 10:3164. doi: 10.3390/cells10113164
103. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. (2006) 116:3015–25. doi: 10.1172/JCI28898
104. Cryan JF, O'Riordan KJ, Cowan CSM, Sandhu KV, Bastiaanssen TFS, Boehme M, et al. The microbiota-gut-brain axis. Physiol Rev. (2019) 99:1877–2013. doi: 10.1152/physrev.00018.2018
105. Erny D, Hrabě de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. (2015) 18:965–77. doi: 10.1038/nn.4030
106. Chen C, Liao J, Xia Y, Liu X, Jones R, Haran J, et al. Gut microbiota regulate Alzheimer's disease pathologies and cognitive disorders via PUFA-associated neuroinflammation. Gut. (2022) 71:2233–52. doi: 10.1136/gutjnl-2021–326269
107. Wang X, Wang BR, Zhang XJ, Xu Z, Ding YQ, Ju G. Evidences for vagus nerve in maintenance of immune balance and transmission of immune information from gut to brain in STM-infected rats. World J Gastroenterol. (2002) 8:540–5. doi: 10.3748/wjg.v8.i3.540
108. Wastyk HC, Fragiadakis GK, Perelman D, Dahan D, Merrill BD, Yu FB, et al. Gut-microbiota-targeted diets modulate human immune status. Cell. (2021) 184:4137–53. doi: 10.1016/j.cell.2021.06.019
109. Wang Y, Li N, Yang JJ, Zhao DM, Chen B, Zhang GQ, et al. Probiotics and fructo-oligosaccharide intervention modulate the microbiota-gut brain axis to improve autism spectrum reducing also the hyper-serotonergic state and the dopamine metabolism disorder. Pharmacol Res. (2020) 157:104784. doi: 10.1016/j.phrs.2020.104784
110. Needham BD, Funabashi M, Adame MD, Wang Z, Boktor JC, Haney J, et al. A gut-derived metabolite alters brain activity and anxiety behaviour in mice. Nature. (2022) 602:647–53. doi: 10.1038/s41586–022-04396–8
111. Quigley EMM. Microbiota-brain-gut axis and neurodegenerative diseases. Curr Neurol Neurosci Rep. (2017) 17:94. doi: 10.1007/s11910–017-0802–6
Keywords: air pollution, particulate matter 2.5, leptin, neuroinflammation, TLR4, obesity, Alzheimer’s disease
Citation: Campolim CM, Schimenes BC, Veras MM, Kim Y-B and Prada PO (2024) Air pollution accelerates the development of obesity and Alzheimer’s disease: the role of leptin and inflammation - a mini-review. Front. Immunol. 15:1401800. doi: 10.3389/fimmu.2024.1401800
Received: 16 March 2024; Accepted: 29 May 2024;
Published: 12 June 2024.
Edited by:
Changwan Hong, Pusan National University, Republic of KoreaReviewed by:
Yang-Ding Lin, The University of Texas Health Science Center at San Antonio, United StatesCopyright © 2024 Campolim, Schimenes, Veras, Kim and Prada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patricia Oliveira Prada, cHByYWRhQHVuaWNhbXAuYnI=
†These authors have contributed equally to this work and share first authorship