
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 14 May 2024
Sec. Molecular Innate Immunity
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1398935
This article is part of the Research Topic A New Perspective in Immune Polymorphism (The HLA, KIR, and LILR genes) View all 9 articles
 Kouyuki Hirayasu1,2,3,4*
Kouyuki Hirayasu1,2,3,4* Seik-Soon Khor5,6
Seik-Soon Khor5,6 Yosuke Kawai5Mihoko Shimada5Yosuke Omae5Gen Hasegawa3Yuko Hashikawa7Hiromu Tanimoto4Jun Ohashi8
Yosuke Kawai5Mihoko Shimada5Yosuke Omae5Gen Hasegawa3Yuko Hashikawa7Hiromu Tanimoto4Jun Ohashi8 Kazuyoshi Hosomichi9
Kazuyoshi Hosomichi9 Atsushi Tajima10Hiroyuki Nakamura11
Atsushi Tajima10Hiroyuki Nakamura11 Minoru Nakamura12,13,14,15
Minoru Nakamura12,13,14,15 Katsushi Tokunaga5
Katsushi Tokunaga5 Rikinari Hanayama1,3,4,7
Rikinari Hanayama1,3,4,7 Masao Nagasaki15,16
Masao Nagasaki15,16Leukocyte immunoglobulin (Ig)-like receptors (LILRs) on human chromosome 19q13.4 encode 11 immunoglobulin superfamily receptors, exhibiting genetic diversity within and between human populations. Among the LILR genes, the genomic region surrounding LILRB3 and LILRA6 has yet to be fully characterized due to their significant sequence homology, which makes it difficult to differentiate between them. To examine the LILRB3 and LILRA6 genomic region, a tool named JoGo-LILR CN Caller, which can call copy number from short-read whole genome sequencing (srWGS) data, was applied to an extensive international srWGS dataset comprising 2,504 samples. During this process, a previously unreported loss of both LILRB3 and LILRA6 was detected in three samples. Using long-read sequencing of these samples, we have discovered a novel large deletion (33,692 bp) in the LILRB3 and LILRA6 genomic regions in the Japanese population. This deletion spanned three genes, LILRB3, LILRA6, and LILRB5, resulting in LILRB3 exons 12-13 being located immediately downstream of LILRB5 exons 1-12 with the loss of LILRA6, suggesting the potential expression of a hybrid gene between LILRB5 and LILRB3 (LILRB5-3). Transcription and subsequent translation of the LILRB5-3 hybrid gene were also verified. The hybrid junction was located within the intracellular domain, resulting in an LILRB5 extracellular domain fused to a partial LILRB3 intracellular domain with three immunoreceptor tyrosine-based inhibitory motifs (ITIMs), suggesting that LILRB5-3 acquired a novel signaling function. Further application of the JoGo-LILR tool to srWGS samples suggested the presence of the LILRB5-3 hybrid gene in the CEU population. Our findings provide insight into the genetic and functional diversity of the LILR family.
Leukocyte immunoglobulin-like receptors (LILRs), which belong to the immunoglobulin superfamily, are found in primates, but not in rodents, which have an analogous receptor system such as paired Ig-like receptors (PIR) (1). The human LILR multigene family is encoded by human chromosome 19q13.4, and consists of five inhibitory LILRBs (LILRB1, LILRB2, LILRB3, LILRB4, and LILRB5), five activating LILRAs (LILRA1, LILRA2, LILRA4, LILRA5, and LILRA6), one secretory protein LILRA3, and two pseudogenes (LILRP1 and LILRP2). Inhibitory LILRs possess a long cytoplasmic domain containing immunoreceptor tyrosine-based inhibitory motifs (ITIMs) for the transmission of inhibitory signals. In contrast, activating LILRs possess a positively charged arginine residue in their transmembrane domains that associates with the FcRγ-chain, which contains immunoreceptor tyrosine-based activating motifs (ITAM) for the transmission of activation signals. LILRs recognize both endogenous and exogenous ligands. For instance, LILRB1 and LILRB2 can recognize self-molecules, such as human leukocyte antigen (HLA) class I molecules (2–4), potentially preventing self-responses. In particular, LILRB1 recognizes the conserved α3 domain of HLA class I in a β2-microglobulin (β2m) -dependent manner, while LILRB2 additionally exhibits recognition of the β2m-free heavy chain of HLA class I molecules (5). In either way, the recognition site is different from that of T cell receptors (TCRs), which specifically recognize the α1 and α2 domains of HLA class I. However, the inhibitory functions of LILRBs can be exploited by pathogens for immune evasion through the recognition of exogenous ligands from pathogens (6, 7). Currently, the role of LILRA activation is not fully understood. One potential function may be the recognition of non-self-ligands for immune activation, as demonstrated by LILRA2, which recognizes microbially cleaved immunoglobulins to detect invading pathogens (8). The expression patterns of the LILR family vary by gene and are found not only in immune cells but also in non-immune cells, such as neurons (9), suggesting that the LILR family may also play important roles in non-immune functions.
Numerous genetic variants have been discovered in LILR genes thus far (10). LILRB1 and LILRB2 have moderately non-synonymous single nucleotide polymorphisms (SNPs), whereas LILRB4, LILRB5, LILRA1, LILRA2, LILRA4, and LILRA5 appear to be relatively conserved with some functional SNPs. LILRA3 has null alleles detected exclusively in Northeast Asians (11, 12). Among the LILR genes, LILRB3 and LILRA6 exhibit the highest allelic diversity. In pairwise comparisons, LILRB3 and LILRA6 alleles exhibit significantly elevated ratios of non-synonymous to synonymous substitutions, indicating natural selection (13–16). Genome-wide association studies have revealed several associations with the LILR genomic region, such as serum levels of creatine kinase and lactate dehydrogenase (LILRB5) (17, 18), prostate cancer (LILRA3) (19), and Takayasu arteritis (LILRB3/LILRA3) (20, 21).
The LILR genomic region also exhibits extensive copy number variation (CNV). LILRA3 has 0-2 copies per individual (22), and is one of the most differentiated genes in the human genome (11, 12, 23). LILRA3 CNV can be explained by a single 6.7 kb deletion. In contrast, LILRA6 displays a broad range of CNVs, ranging from 0 - 6 copies per individual, indicating the presence of both gene duplications and deletions (13–15, 23). However, the genomic structure of the LILRA6 gene has not yet been fully elucidated. LILRA6 shares a high degree of homology with the adjacent gene LILRB3 within their shared 5 kbp region. Consequently, short-read sequencing technology cannot distinguish between LILRA6 and LILRB3 due to the long-range homologous region, which makes it difficult to determine the genomic structure. To address this, we aimed to clarify the genomic structure of the LILRA6 CNVs using the JoGo-LILR CN Caller, a tool engineered to delineate the genomic structure of LILRB3 and LILRA6 copy numbers from srWGS data. During this process, we unexpectedly discovered a novel large 33,692 bp deletion that results in the hybrid gene LILRB5-LILRB3.
The research ethics committee of Kanazawa University, Kyushu University, and National Center for Global Health and Medicine reviewed and approved this study. All participants provided written informed consent.
The original 2,504 unrelated samples from the 1kGP were used as a discovery cohort, and their srWGS data were downloaded from the International Genome Sample Resource website (https://www.internationalgenome.org/data-portal/data-collection/30x-grch38). Genomic DNA and RNA were obtained from the blood samples of volunteers belonging to Kanazawa University and residents of Shika town, where the cohort study was being conducted (24). Shika town is located on the Noto Peninsula in Ishikawa Prefecture, Japan. Total of 1,310 samples from an independent Shika cohort were used as a validation cohort in the Japanese population. Genomic DNA was extracted from the blood samples using the QIAamp DNA Blood Mini or Maxi Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions, or extracted by SRL Inc. (Tokyo, Japan). Peripheral blood mononuclear cells (PBMCs) were isolated from blood samples by density gradient centrifugation using Ficoll-Paque PLUS (GE Healthcare, Pittsburgh, PA, USA). Subsequently, RNA was extracted from the PBMCs using the FastGene RNA Premium Kit (NIPPON Genetics, Tokyo, Japan) in accordance with the manufacturer’s instructions. First-strand cDNA synthesis was performed using a PrimeScript II 1st strand cDNA Synthesis Kit (TaKaRa, Shiga, Japan) in accordance with the manufacturer’s instructions. Genomic DNA from the HapMap JPT samples was obtained from the Coriell Cell Repository. Among the three samples (NA12413, NA18959, and NA19087) identified with JoGo-LILR CN Caller, NA18959, and NA19087 were used for long-read sequencing technology and NA12413 was used for JoGo-LILR-trio analysis.
DNA was extracted from the immortalized cell line of GM18959 and GM19087 obtained from the Coriell Cell Repository (Camden, NJ, USA; corresponding to NA18959 and NA19087, respectively). The DNA was subjected to long-read whole genome sequencing (lrWGS) on Sequel IIe (Pacific Biosciences (PacBio)) with the standard high-fidelity (HiFi) sequencing protocol. For the subreads generated in Sequel IIe, the CCS tool was used to generate unmapped HiFi BAM files. The unmapped BAM files were mapped to the GRCh38 reference assembly (without decoy and alt contigs) using Minimap2 (ver. 2.23) (25) and created aligned BAM files. For the aligned BAM file of each sample, the aligned HiFi reads from chr19: 54,180,000 to 54,280,000 were extracted. For NA18959 and NA19087, a total of 122 and 116 reads mapped to chr19: 54,180,000 to 54,280,000 (LILRB5-LILRA6-LILRB3 region) were obtained, respectively. To examine the long deletions in the HiFi reads, the reads were re-aligned to the GRCh38 reference assembly using Minimap2 with the option “-ax splice:hq” (Supplementary Figures 1, 2), which can capture long deletions. For local de novo assembly of the extracted reads, HiFiasm (ver. 0.16.1) was also applied (26). The assembled contigs were aligned by Minimap2 with the option “-r 500,40000 -ax map-hifi” to allow long deletion alignment. The re-aligned and de novo assembled results were inspected on Integrative Genomics Viewer (IGV) (ver. 2.16.2) to analyze the deletion haplotype and the base-pair resolution length (Supplementary Figures 1, 2).
The breakpoint of the 33,692 bp deletion was verified using Sanger sequencing. Briefly, polymerase chain reaction (PCR) was performed on genomic DNA to amplify a 6,612 bp product spanning from LILRB5 3’UTR to LILRB3 exon 13 using primers (LILRB5 forward primer:5’-CCTGCACAGCTGAGTCCAGT-3’ and LILRB3 reverse primer:5’-TTAGTCATCTTTGAGTCAGGTGAG-3’). The PacI (aataatttaattaa) and NotI (aataatgcggccgc) restriction sites were added to the 5’ sequences of the LILRB5 forward and LILRB3 reverse primers, respectively, to allow for gene cloning. Following treatment of the PCR products with ExoSAP-IT Express PCR Cleanup Reagents (Thermo Fisher Scientific, Waltham, MA, USA), direct sequencing was performed using specific primers (Supplementary Table 1). Sanger sequencing data were analyzed using Sequencher software (Gene Codes Corporation). Multiple sequence alignments were performed using the Clustal Omega software. The copy numbers of LILRB3 and LILRA6 were quantified using genomic DNA and a QIAcuity Digital PCR System (Qiagen, Hilden, Germany) in accordance with the manufacturer’s instructions. Digital PCR primer/probe sets were used as previously described (14). Individuals with a single copy of LILRB3 were considered carriers of the LILRB5-3 hybrid gene.
To identify the LILRB5 isoforms and LILRB5-3 hybrid gene, PCR was performed with cDNA templates using TaKaRa Ex Premier DNA Polymerase (TaKaRa, Japan) with the following primers: 5′-CCTGCACAGCTGAGTCCAGT-3′ for the LILRB5 forward primer, and 5′-TTAGTCATCTTTGAGTCAGGTGAG-3′ for the LILRB3 reverse primer. The PCR conditions consisted of initial denaturation at 94 °C for 1 min, followed by 40 cycles of denaturation at 98 °C for 10 s, annealing at 62.5 °C for 15 s, and extension at 68 °C for 2 min using a VeritiPro Thermal Cycler (Thermo Fisher Scientific). For cDNA cloning, the PCR products were inserted into the pMXs-puro retroviral expression vector according to standard cloning methods. For PCR-direct sequencing, PCR products treated with ExoSAP-IT Express PCR Cleanup Reagents (Thermo Fisher Scientific) were subjected to Sanger sequencing. cDNA clones that were consistent with the PCR-direct sequencing data were regarded as the major isoforms.
The JoGo-LILR CN Caller was employed to extract LILRB3-LILRA6 CNs from srWGS data. The JoGo-LILR CN Caller is available on the Japanese Open Genome and Omics Portal (https://jogo.csml.org/public/JoGo-LILR). This tool operates by processing BAM or CRAM files aligned to the GRCh38DH reference assembly. It then calculates normalized CNs for specific regions within the LILRB3 and LILRA6 genes (termed LILRB3core and LILRA6core) and their gene body regions (named LILRB3+LILRA6). The current implementation uses CNVnator (version 0.4.1) (27) to calculate the normalized CN values.
For the normalized CN values, the JoGo-LILR tool generated the LILR cluster plot with LILRB3+LILRA6 on the x-axis and LILRB3core/LILRA6core on the y-axis. For the plot, joint CN calling was applied after clustering to the dataset. The tool is particularly adept at stably calling CNs even in datasets with a small sample size, ranging from a single sample to several hundreds. For this purpose, the 1000 Genomes Project (1kGP) samples are typically employed as a background dataset, providing a comprehensive reference for comparison. This practice is instrumental in enhancing the accuracy and reliability of CN calling across diverse sample sizes.
For the detection of the LILRB5-3 hybrid gene, JoGo-LILR was applied to CRAM files from 2,504 non-pedigree and two pedigree samples from the 1kGP (Supplementary Table 2) (28). To establish a Japanese CN profile for the cohorts from the southwest region of Japan (SW-JP) and northeast region of Japan (NE-JP), we analyzed 348 quality control (QC)-passed samples from the Tokyo healthy control dataset and 180 samples from volunteers at the National Hospital Organization Nagasaki Medical Center, Nagasaki, Japan, adhering to a cnv2 ratio QC metric of >0.85. In this process, the 1kGP samples were again utilized as a background reference for joint CN calling.
Finally, the visualization of the LILRB3-LILRA6 genomic region by short-read mapping was conducted using IGV (ver. 2.6.2) (29).
The JoGo-LILR-trio algorithm is a specialized tool used for accurately predicting LILRB3-LILRA6 CN types in trio genotypes. It operates on a probabilistic model that assesses all potential combinations of these CN types in both parents. The primary objective of the JoGo-LILR-trio algorithm is to identify pairs of haploid LILRB3-LILRA6 CN types that align with Mendelian inheritance patterns evident in their offspring. This algorithm is particularly adept at handling situations where multiple parental CN type combinations are plausible. In such cases, JoGo-LILR-trio employs a strategic approach to select the most likely combination that would result in the observed CN types in the children. In this study, the trio dataset comprising individuals NA12413, NA12412, and NA12485 was utilized.
To assess the cell surface expression of LILRB5, plasmids encoding LILRB5 and the LILRB5-3 hybrid gene were transiently co-transfected with green fluorescent protein (GFP) into 293T cells using PEI Max (Polyscience). GFP-positive cells were then identified by flow cytometry, as previously described (15). The cell surface expression of LILRB5 and LILRB5-3 was analyzed by flow cytometry using an anti-LILRB5 antibody (clone 395239, R&D Systems, Minneapolis, MN, USA) and MACSQuant 10 (Miltenyi Biotec, Germany).
Since LILRB3 and LILRA6 exhibit a high degree of homology across a 5 kbp region encompassing the promoter and extracellular domains, usual analysis of srWGS cannot be properly mapped to a reference genome (Figure 1A). In contrast, the JoGo-LILR CN Caller called the LILRB3-LILRA6 CNs with the LILR cluster plot and estimated the probable pair of allelic LILRB3-LILRA6 CN type from srWGS (Figure 1B, Supplementary Table 3). As depicted in Figure 1B, nine distinct clusters were clearly illustrated in the two-dimensional representation of the sum of LILRB3 and LILRA6 CNs versus LILRB3/LILRA6 CN ratios. The main clusters near the coordinates (3, 2), (4, 1), (5, 0.67), and (6, 0.5) indicate 1, 2, 3, and 4 copies of LILRA6 with 2 copies of LILRB3, respectively. As noted in previous reports, the LILRA6 copy number ranges from 0 to 6 in most individuals, with the exception of one individual who exhibited seven LILRA6 copies (Figure 1B, Supplementary Table 3). The JoGo-LILR CN Caller also estimated the existence of a previously unreported LILRB3-missing haplotype in the three samples surrounding the coordinate (2, 1), NA12413, NA18959, and NA19087 with high probability (Supplementary Table 4).
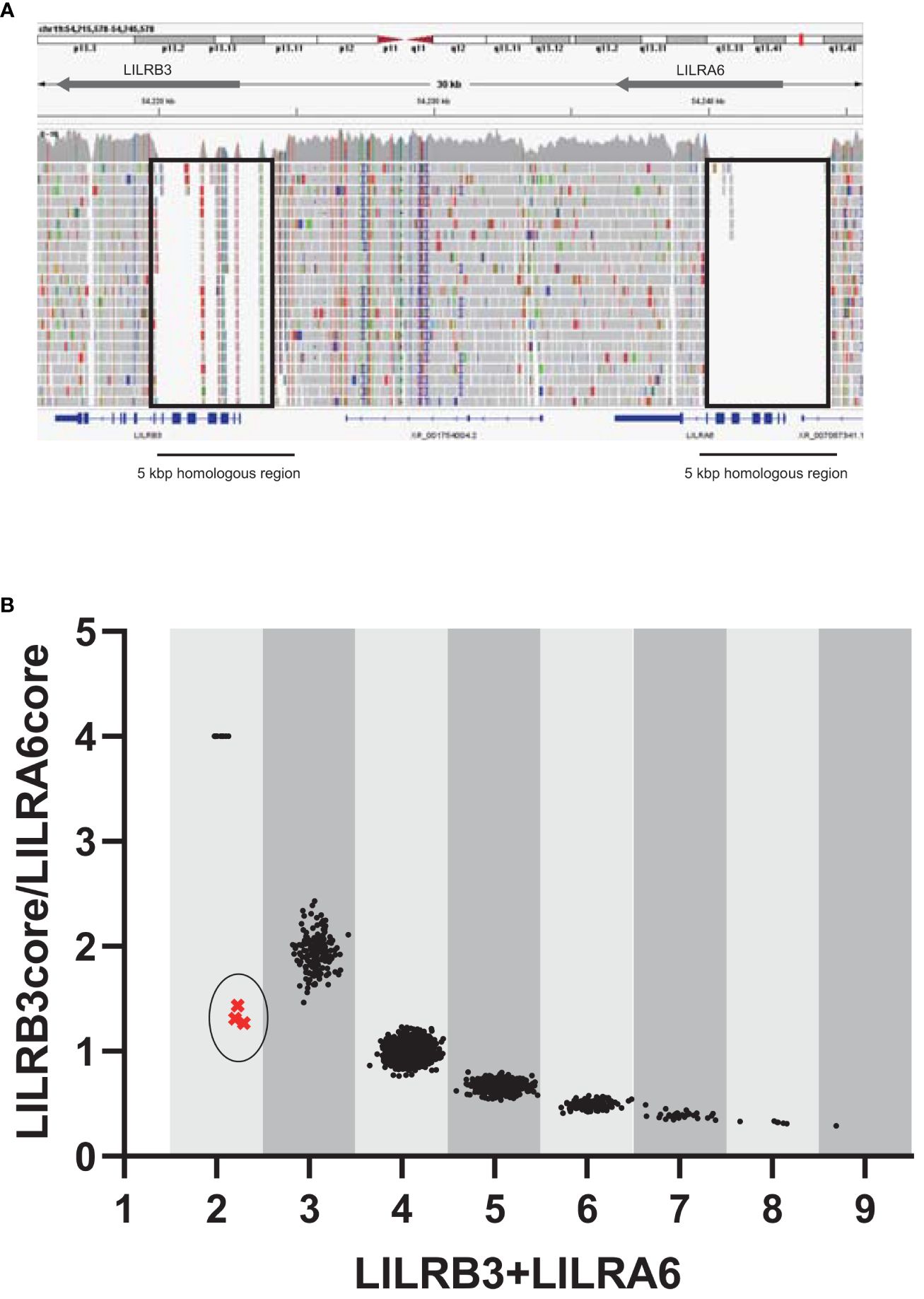
Figure 1 Estimation of the copy numbers within the LILRB3-LILRA6 genomic region using pre-existing short-read sequencing data. (A) Visualization of short-read mapping to the LILRB3-LILRA6 genomic region using the Integrative Genomics Viewer (IGV). One example of the individual NA18954 is shown. The block indicates the unmapped region with a low mapping quality (mapping quality threshold = 30). (B) Two-dimensional representation of LILRB3 and LILRA6 copy numbers (CN) in populations worldwide (n = 2,504). Each individual is represented by a single dot. The vertical and horizontal axes represent the LILRB3/LILRA6 CN ratio and the sum of LILRB3 and LILRA6 copies, respectively. When LILRA6 shows zero copy, the ratio of LILRB3/LILRA6 becomes infinite. To visualize these dots on the graph, the LILRB3/LILRA6 ratio corresponding to LILRA6 zero copy was arbitrarily set to 4. The coordinate (x=2, y=4) indicates two copies of LILRB3 in the absence of LILRA6. The circled cluster represents the individuals with the estimated haplotype that is missing LILRB3.
The analysis using srWGS is limited by only providing an overview of the structure of the LILRB3 and LILRA6 genomic region. For a more detailed examination at the base pair level, we employed real-time HiFi lrWGS on two Japanese-in-Tokyo (JPT) DNA samples, specifically NA18959 and NA19087. The lrWGS analysis revealed a novel large deletion (33,692 bp) carried by two individuals (Supplementary Figures 1, 2). This 33,692 bp deletion spanned the LILRB3, LILRA6, and LILRB5 genes (Figure 2A). This type of deletion is the largest ever reported and has not yet been detected in the LILR genomic region. Individuals carrying this deletion also had two copies of the LILRA3 6.7 kb deletion, indicating that the haplotype containing the 33,692 bp deletion has the fewest LILR genes. Thus, we carried out a detailed validation of the 33,692 bp deletion. After analyzing the region around the 33,692 bp deletion breakpoint using Sanger sequencing, we observed the presence of a 33,692 bp deletion eliminating exons 1–11 of LILRB3; LILRA6; and exon 13 of LILRB5 (Figure 2B, Supplementary Figure 3). The 33,692 bp deletion left exons 1–12 of LILRB5 and exons 12–13 of LILRB3 intact, resulting in the hybrid gene structure of LILRB5 and LILRB3 (hereafter referred to as “LILRB5-3”). Examination of exon 8 of both LILRB3 and LILRA6 by digital PCR revealed that two individuals (NA18959 and NA19087) carrying the 33,692 bp deletion lacked a single copy of both genes, which was consistent with the sequence data (Figure 2C). These data suggest that the 33,692 bp deletion results in the hybrid gene LILRB5-3 and the deletion of LILRB3 and LILRA6.
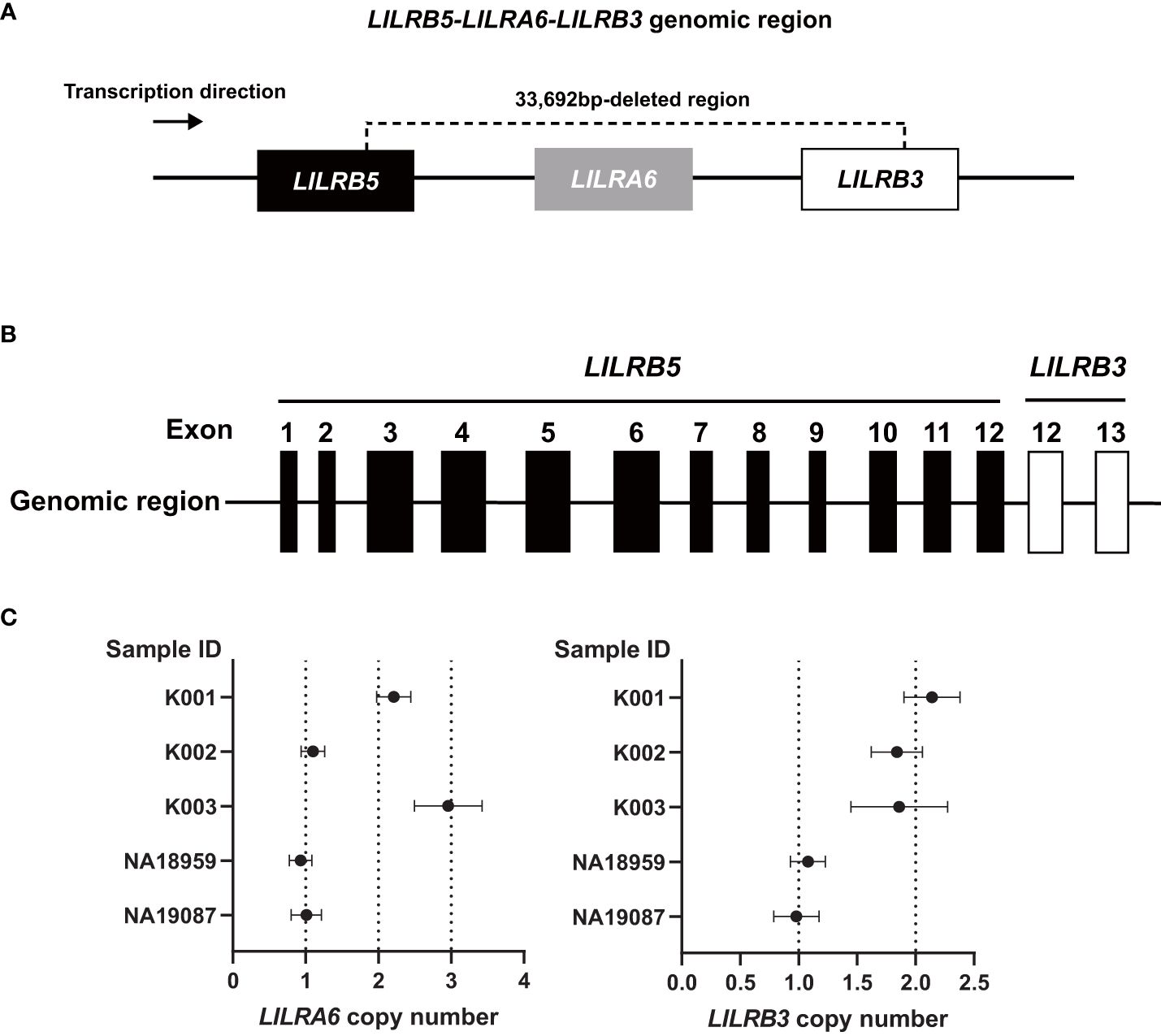
Figure 2 The 33,692 bp deletion removes LILRB3 exons 1-11, the entire LILRA6 gene, and LILRB5 exon 13. (A) Schematic diagram of the 33,692 bp deletion. (B) The 33,692 bp deletion results in the LILRB5-3 hybrid gene comprising LILRB5 exons 1-12 and LILRB3 exons 12-13. (C) LILRA6 and LILRB3 copy numbers of five individuals determined by digital PCR. The vertical and horizontal axes represent the sample ID and LILRA6 copy number (left panel) or LILRB3 copy number (right panel), respectively. The error bar indicates a 95% confidence interval. Individuals K001, K002, and K003 are examples of two, one, and three copies of LILRA6, respectively, while all three individuals possess two LILRB3 copies. Carriers of the 33692 bp deletion (NA18959, and NA19087) possess a single copy of both LILRA6 and LILRB3.
To validate the existence of the hybrid gene LILRB5-3 in the other Japanese samples, we subjected 1,310 samples from an independent Shika cohort study in Japan to digital PCR analysis (Supplementary Table 5) using the DNA extracted from whole blood. The analysis detected a single copy of both LILRB3 and LILRA6 in four samples. These results confirmed that the existence of the large deletion did not originate from the structural variation acquired in the lymphoblastoid cell lines in 1KGP.
To ascertain the frequencies of the LILRB5-3 hybrid gene within the Japanese population, we conducted an analysis using the JoGo-LILR CN Caller. Our dataset included 180 srWGS samples from SW-JP and 348 samples from NE-JP (Supplementary Figures 4, 5, Supplementary Tables 6, 7). Among these, two samples from NE-JP were identified as carrying the hybrid gene, while none from SW-JP exhibited this genetic feature. Notably, the Japanese samples in the 1kGP project was also catalogued in NE-JP (Tokyo).
The presence of the LILRB5-3 hybrid gene has been definitively confirmed in the Japanese population. This genetic feature was not detected in the other East Asian samples from the 1kGP, which included 93 CDX (Dai Chinese), 103 CHB (Han Chinese), 105 CHS (Southern Han Chinese), and 99 KHV (Kinh Vietnamese) individuals (Supplementary Table 3). However, the Korean population was not represented in the 1kGP dataset. Therefore, we examined 70 QC srWGS data from the Korean Personal Genome Project (KPGP) using JoGo-LILR CN Caller (Supplementary Figure 6, Supplementary Table 8). Our analysis revealed the absence of the LILRB5-3 hybrid gene in these Korean samples. Since the LILRB5-3 hybrid gene appears to be rare in the Japanese population, additional sequencing is needed to definitively determine its presence or absence in the other regions of East Asia.
Among a total of 2,504 unrelated samples analyzed, only one individual, NA12413 from the CEU cohort of European descent, was identified with a single CN for both LILRA6 and LILRB3, aside from two samples in the JPT cohort. This individual, NA12413, along with NA12412 and NA12485, were annotated as a father, mother, and child, respectively. All these srWGS data were available as pedigree dataset in 1kGP project (28). Utilizing the JoGo-LILR-trio tool, which is designed to predict the most likely pair of allelic LILRB3-LILRA6 CN types in offspring, we analyzed this trio’s srWGS data. The analysis revealed that the child, NA12485, possesses one copy of both LILRB3 and LILRA6 genes and is estimated to have a genotype with zero copies of both LILRB3-LILRA6 on one allele and a single copy of both LILRB3-LILRA6 on the other allele (JoGo-LILR CN Caller value (x=2.3, y=1.4)). This observation suggests that the LILRB5-3 hybrid gene was transmitted from the father (NA12413) to the child (NA12485), and provides robust support for the existence of the hybrid gene in the CEU population.
To verify the expression of the LILRB5-3 hybrid gene, PCR-direct sequencing and cDNA cloning were conducted on Japanese individuals with and without the 33,692 bp deletion. Two primary LILRB5 isoforms, long and short, were identified in individuals without deletions through cDNA cloning of LILRB5 transcripts. The long isoform contained three ITIMs, whereas the short isoform contained two ITIMs (Figure 3A). This difference is due to alternative splicing of the splice acceptor site of intron 12. In contrast, a chimeric gene composed of LILRB5 and LILRB3 was detected in a Japanese individual with a deletion using LILRB5 forward and LILRB3 reverse primers (Figure 3B). The amplicon was approximately 2 kb in size, which was consistent with the expected size of the exonic region of LILRB5-3 (Figure 3B). cDNA cloning and sequencing identified two isoforms of a chimeric gene composed of LILRB5 and LILRB3, providing evidence that LILRB5-3 was transcribed at the mRNA level (Figure 3C). These two isoforms differ by three bp at the acceptor sites of exons 8 and 12 of LILRB5 and LILRB3, respectively. This resulted in a disparity between the two amino acids that were not located within any functional domains or motifs. The hybrid junction between LILRB5 and LILRB3 was located within the intracellular domain and fused the extracellular domain of LILRB5 with the intracellular domains containing partial LILRB3. The original number of ITIMs in LILRB3 was four, whereas LILRB5-3 contained three of the four LILRB3 ITIMs (Figure 4A). This suggests that LILRB5-3 acquired a novel signaling pathway.
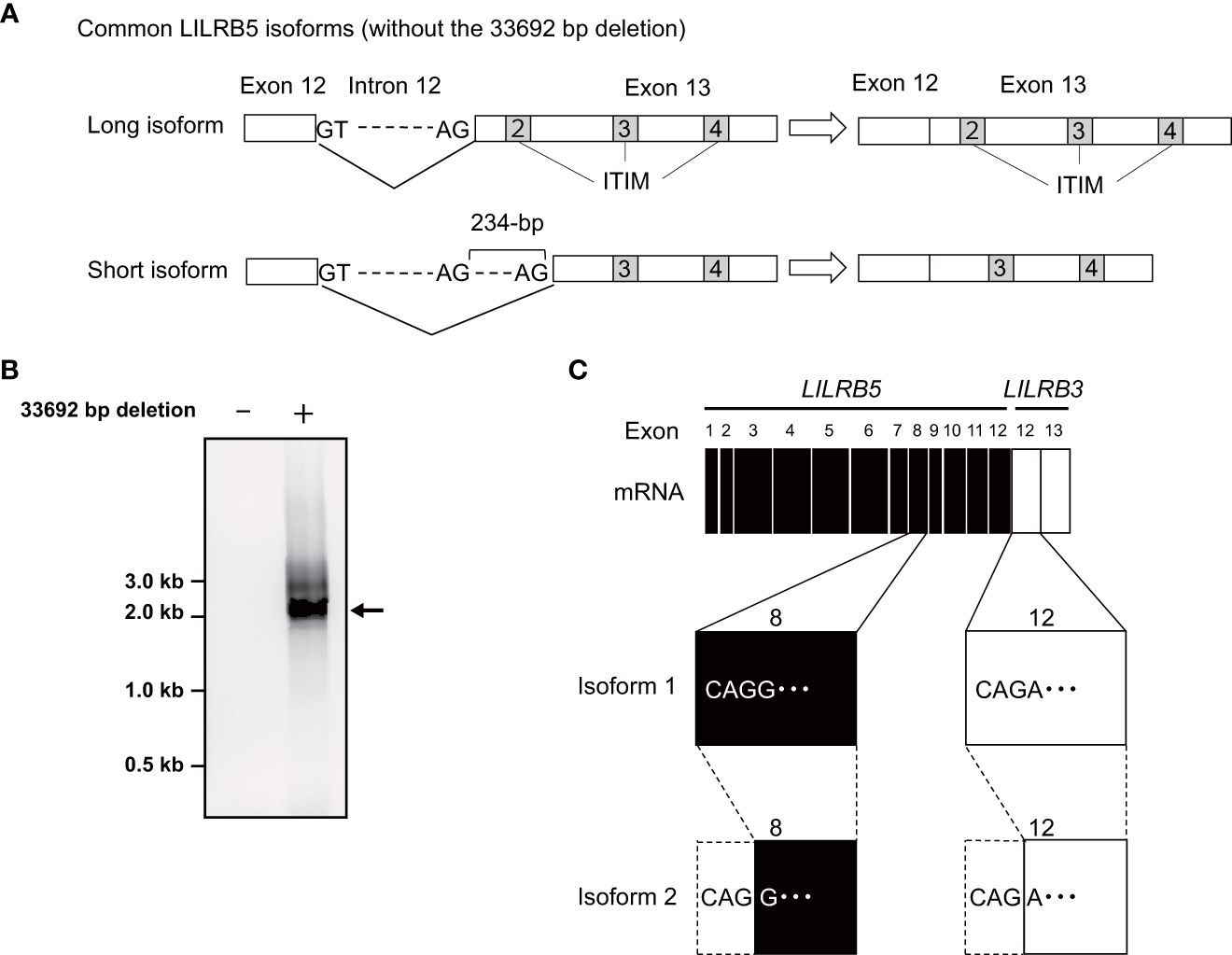
Figure 3 Identification of the LILRB5 and LILRB5-3 hybrid isoforms. (A) A schematic diagram of long and short isoforms of LILRB5 identified from non-carriers of the 33692 bp deletion. The short isoform lacks 234-bp of exon 13 containing a second immunoreceptor tyrosine-based inhibitory motif (ITIM), compared with the long isoform. (B) An RT-PCR analysis, utilizing a forward primer positioned at LILRB5 exon 1 and a reverse primer positioned at LILRB3 exon 13, amplified the expected size of the LILRB5-3 hybrid gene in the carrier (indicated as +), but not in the non-carrier (indicated as -). The arrow indicates the expected PCR product of the LILRB5-3 hybrid gene. (C) cDNA cloning of the PCR products identified two alternatively spliced isoforms of the LILRB5-3 hybrid gene.
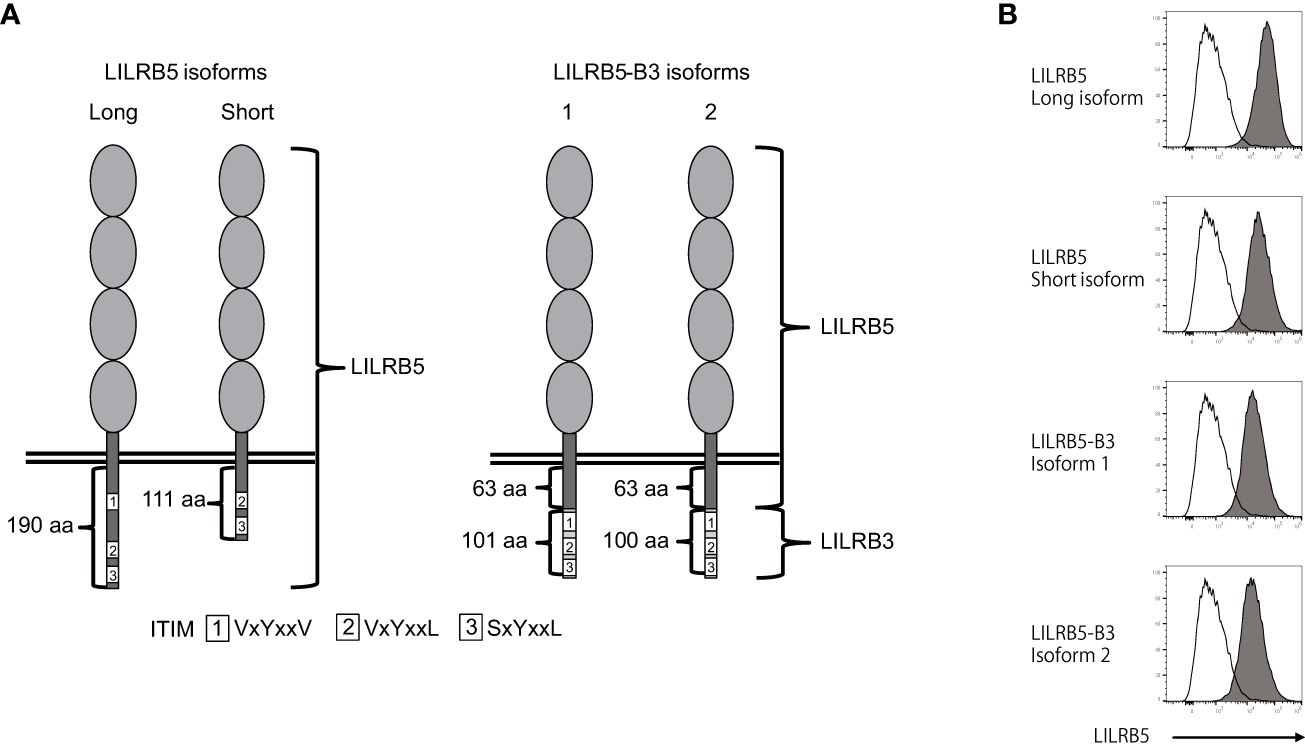
Figure 4 The LILRB5-3 hybrid can be expressed at the protein level. (A) A schematic diagram of the protein products of long and short LILRB5 isoforms in addition to LILRB5-3 isoforms 1 and 2. The four elliptical structures in the extracellular region indicate Ig-like domains. The aa indicates amino acid. (B) Plasmids encoding the full-length of long and short LILRB5 isoforms, as well as LILRB5-3 isoforms 1 and 2, were transiently co-transfected with green fluorescent protein (GFP) into 293T cells. The transfectants were subsequently stained with an anti-LILRB5 antibody (clone 395239), and the GFP-positive cells were analyzed. Open and closed histograms represent mock and LILRB5 transfectants, respectively. A shift of cell populations from the left (open histogram) to the right (closed histogram) indicates cell surface expression of LILRB5 and LILRB5-3.
There exists an example wherein the killer cell Ig-like receptor, KIR3DL1*004, lacks cell surface expression but exhibits intracellular retention (30). Therefore, we investigated whether LILRB5-3 can be displayed on the cell surface. To this end, we transfected the identified LILRB5 and LILRB5-3 genes into 293T cells and stained the transfectants with an anti-LILRB5 antibody. Flow cytometry confirmed the detection of the LILRB5-3 proteins on the cell surface, demonstrating that LILRB5-3 can be expressed at the cell-surface protein level (Figure 4B). These data suggest that LILRB5-3 may have a novel function in carriers with the 33,692 bp deletion.
In this study, we identified a novel large 33,692 bp deletion in the LILRA6 genomic region using long-read sequencing technology. The 33,692 bp deletion, which lacks both LILRB3 and LILRA6, is the largest reported deletion within the LILR genomic region. The 33,692 bp deletion was unique in generating the hybrid gene of LILRB5 and LILRB3. It is estimated that the breakpoints of the 33,692 bp deletion are located within intron 12 of the LILRB5 gene and intron 11 of the LILRB3 gene (Supplementary Figure 3). These two introns share a high degree of homology, with 269 base pairs of complete identity. These observations suggest that non-allelic homologous recombination occurred at this site. Similar hybrid genes are also found within the killer cell Ig-like receptor (KIR) gene cluster, which lies in close proximity to the LILR region on chromosome 19 (31). In particular, the hybrid gene between KIR2DL1 and KIR3DL2 has arisen by a deletion spanning the intermediate and telomeric regions (32). The region of recombination encompasses a 129 bp sequence, which is identically found in both KIR2DL1 and KIR3DL2 alleles, a similar pattern observed in LILRB5-3. However, the limitation of this study is that the precise mechanism underlying the 33,692 bp deletion remains uncertain. Specifically, whether the 33,692 bp deletion results from intra- or inter-chromosomal recombination remains elusive due to the lack of the haplotype data around the LILRB5-LILRA6-LILRB3 genomic region in human populations (Figure 5). Haplotyping of these regions would provide valuable insights into the potential mechanisms driving this substantial genetic alteration.
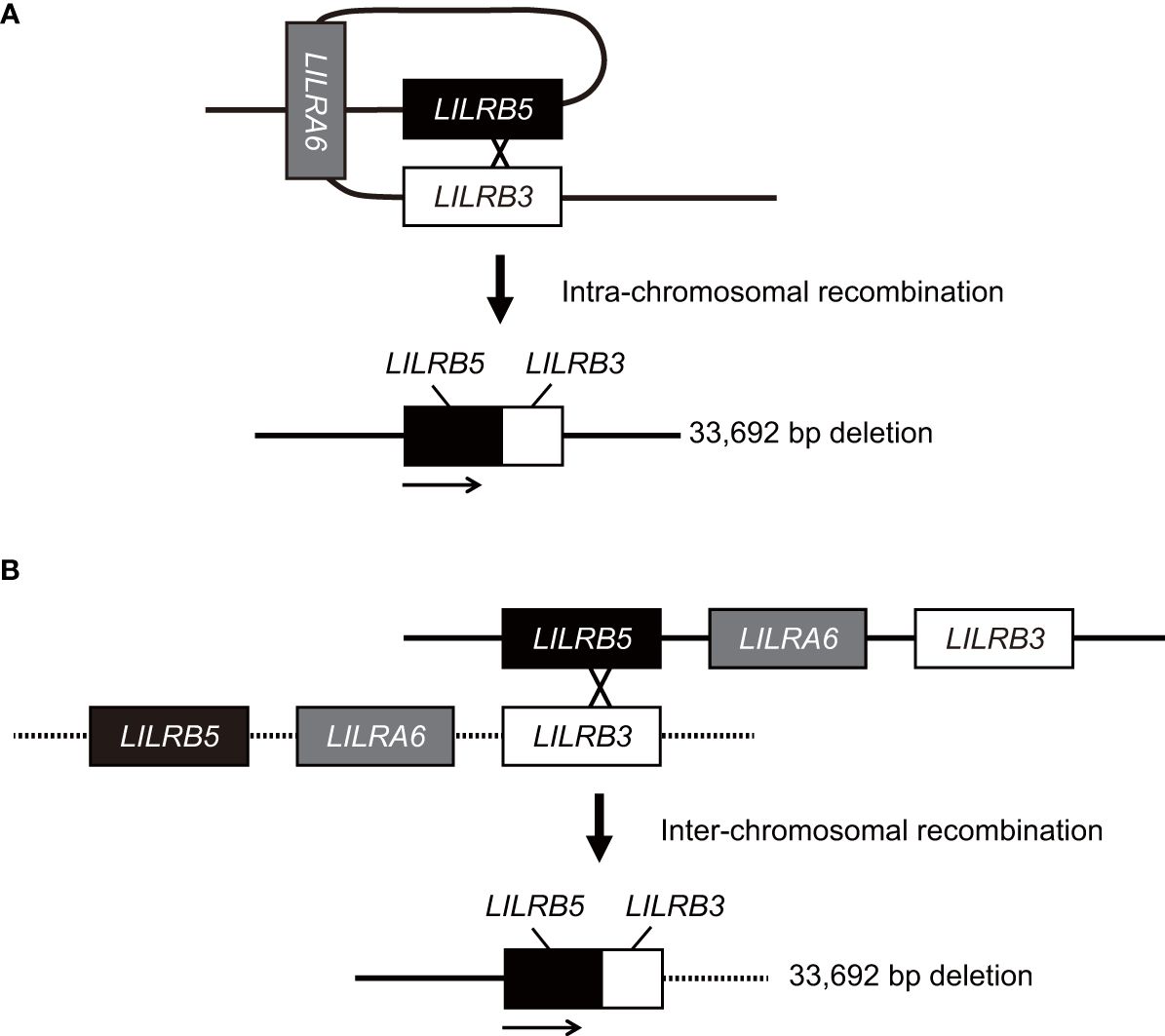
Figure 5 Model for the generation of the LILRB5-3 hybrid gene. The LILRB5-3 hybrid gene may have been generated by the non-allelic homologous recombination of misaligned LILRB3 and LILRB5 during meiosis. Two putative mechanisms that underlie the LILRB5-3 hybrid gene are intra- (A) and inter-chromosomal homologous recombination (B). Solid and dashed lines indicate one, and another chromosomes, respectively. Intra-chromosomal recombination preserves the identical haplotype.
Recently, a thorough comparative analysis between the primate LILR genomic loci was conducted, revealing that the LILR region remained largely conserved throughout primate evolution (33). However, the LILRA6 ortholog was lacking in orangutans and common marmosets. Due to limited sample sizes, it is currently unclear whether the absence of LILRA6 in orangutans and common marmosets represents CNV. Future analyses of non-human primates will uncover the evolutionary dynamics of the LILR CNVs in primates.
The presence of the LILRB5-3 hybrid transcript was confirmed in an individual carrying the hybrid gene but was not detected in a non-carrier. The LILRB5-3 hybrid gene produces a protein that contains the extracellular and transmembrane domains of LILRB5, a fragment of the LILRB5 intracellular region devoid of ITIMs, and a portion of the LILRB3 intracellular region that contains the second, third, and fourth ITIMs of LILRB3. This suggests that the hybrid receptor recognizes ligands through the LILRB5 extracellular domain, whereas receptor signaling is mediated by previously unobserved motifs. Although HLA class I-free heavy chains have been reported to be ligands for LILRB5, the physiological significance of this interaction is still not well understood (34). In contrast, the novel signaling motif of the hybrid gene, which contains three instead of four LILRB3 ITIMs, may represent a new function. The intracellular domain of LILRB3 can be divided functionally into two distinct regions: one responsible for binding TRAF2 and the other for binding SHP-1 (35). The intracellular domain of LILRB5-3 lacks the TRAF2 binding site of LILRB3 due to replacement by the LILRB5 intracellular domain. In contrast, the LILRB5-3 intracellular domain still retains the SHP-1 binding site of LILRB3. Consequently, LILRB5-3 shows a new hybrid motif that combines the LILRB5 and LILRB3 intracellular regions. Therefore, the function of the hybrid gene needs to be confirmed experimentally.
We have applied JoGo-LILR CN Caller to call LILRA6 CNs from pre-existing srWGS data. The tool was capable of accurately detecting not only the candidate 33,692 bp deletion that contains the LILRB5-3 hybrid gene, but also multiple copies of LILRA6 in populations worldwide. Therefore, the tool is a useful tool for disease-association studies. Individuals with the 33,692 bp deletion were more frequent in the Japanese population than in other populations worldwide. Genome-wide association studies have indicated that the LILRB5 SNP is significantly associated with creatine kinase levels (17, 18), and therefore, the 33,692 bp deletion may also be linked to a particular phenotype in the Japanese population. In addition, the haplotype of the LILRB5-3 hybrid gene also lacks LILRA3. Since LILRA3 deletion is associated with Takayasu arteritis in the Japanese population (21), it is conceivable that the combination of the LILRB5-3 hybrid gene and the LILRA3 deletion may be useful for further classification of the pathogenesis of Takayasu arteritis.
Given that the region with high homology between LILRB3 and LILRA6 is approximately 5 kb in length, it is difficult to distinguish between LILRB3 and LILRA6 using SNP-based genotyping and short-read sequencing technology. We previously showed that 1 KG data contains a significant number of genotyping errors due to the utilization of short-read sequencing technology (15). Therefore, regions with high homology will become increasingly accessible using long-read sequencing technology. The deleted fragment contains both LILRB3 and LILRA6, which are paired inhibitory and activating receptors, respectively. However, it remains unclear how they function cooperatively. Future functional studies and large-scale disease association studies will provide insight into the immunological significance of this large deletion within the human population.
The data presented in the study are deposited in the DDBJ/EMBL/GenBank repository, accession number LC785381 (LILRB5 long isoform); LC785382 (LILRB5 short isoform); LC785383 (LILRB5-3 isoform 1); LC785384 (LILRB5-3 isoform 2); and LC785385 (LILRB5-3 genomic sequence).
The studies involving humans were approved by the research ethics committee of Kanazawa University, Kyushu University, and National Center for Global Health and Medicine. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
KHi: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. S-SK: Conceptualization, Investigation, Methodology, Project administration, Resources, Software, Writing – original draft, Writing – review & editing. YK: Investigation, Methodology, Project administration, Resources, Software, Writing – original draft, Writing – review & editing. MS: Data curation, Methodology, Resources, Writing – original draft, Writing – review & editing. YO: Data curation, Methodology, Resources, Writing – original draft, Writing – review & editing. GH: Investigation, Writing – original draft, Writing – review & editing. YH: Investigation, Writing – original draft, Writing – review & editing. HT: Investigation, Writing – original draft, Writing – review & editing. JO: Resources, Writing – original draft, Writing – review & editing. KHo: Resources, Writing – original draft, Writing – review & editing. AT: Resources, Writing – original draft, Writing – review & editing. HN: Resources, Writing – original draft, Writing – review & editing. MiN: Resources, Writing – original draft, Writing – review & editing. KT: Writing – original draft, Writing – review & editing, Project administration, Resources, Supervision. RH: Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing. MaN: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by JSPS KAKENHI grant number JP23H02714 (KHi), JP21H02681 (MaN); AMED under grant number JP20gm6010021 (KHi), JP23wm0225036 (KHi), JP20ek0109492 (MaN), JP21wm0425009 (MaN), JP20ek0109485 (MaN), JP21ek0109548 (MaN), JP23ek0109675 (MaN), JP23ek0109672 (MaN); JST NBDC Grant Number JPMJND2302 (MaN), and the Naito Foundation (KHi). This work was partially supported by the “Joint Usage/Research Center for Interdisciplinary Large-scale Information Infrastructures” and “High Performance Computing Infrastructure” in Japan (Project ID: jh200047-NWH, jh210018-NWH, jh220014, and jh230016).
The authors would like to thank all the workers engaged in the Shika Study for their contributions. Part of the computational resource is provided by the infrastructure of Omics Science Center Secure Information Analysis System, Medical Institute of Bioregulation at Kyushu University. This work was partly performed in the Medical Research Center Initiative for High Depth Omics of the Medical Institute of Bioregulation, Kyushu University. We would like to thank Editage (www.editage.jp) for English language editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1398935/full#supplementary-material
1. Redondo-García S, Barritt C, Papagregoriou C, Yeboah M, Frendeus B, Cragg MS, et al. Human leukocyte immunoglobulin-like receptors in health and disease. Front Immunol. (2023) 14:1282874. doi: 10.3389/fimmu.2023.1282874
2. Colonna M, Navarro F, Bellon T, Llano M, Garcia P, Samaridis J, et al. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J Exp Med. (1997) 186:1809–18. doi: 10.1084/jem.186.11.1809
3. Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L, et al. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity. (1997) 7:273–82. doi: 10.1016/S1074-7613(00)80529-4
4. Borges L, Hsu ML, Fanger N, Kubin M, Cosman D. A family of human lymphoid and myeloid Ig-like receptors, some of which bind to MHC class I molecules. J Immunol. (1997) 159:5192–6. doi: 10.4049/jimmunol.159.11.5192
5. Shiroishi M, Kuroki K, Rasubala L, Tsumoto K, Kumagai I, Kurimoto E, et al. Structural basis for recognition of the nonclassical MHC molecule HLA-G by the leukocyte Ig-like receptor B2 (LILRB2/LIR2/ILT4/CD85d). Proc Natl Acad Sci USA. (2006) 103:16412–7. doi: 10.1073/pnas.0605228103
6. Chapman TL, Heikeman AP, Bjorkman PJ. The inhibitory receptor LIR-1 uses a common binding interaction to recognize class I MHC molecules and the viral homolog UL18. Immunity. (1999) 11:603–13. doi: 10.1016/S1074-7613(00)80135-1
7. Saito F, Hirayasu K, Satoh T, Wang CW, Lusingu J, Arimori T, et al. Immune evasion of Plasmodium falciparum by RIFIN via inhibitory receptors. Nature. (2017) 552:101–5. doi: 10.1038/nature24994
8. Hirayasu K, Saito F, Suenaga T, Shida K, Arase N, Oikawa K, et al. Microbially cleaved immunoglobulins are sensed by the innate immune receptor LILRA2. Nat Microbiol. (2016) 1:16054. doi: 10.1038/nmicrobiol.2016.54
9. Kim T, Vidal GS, Djurisic M, William CM, Birnbaum ME, Garcia KC, et al. Human lilrB2 is a β-amyloid receptor and its murine homolog pirB regulates synaptic plasticity in an alzheimer’s model. Science. (2013) 341:1399–404. doi: 10.1126/science.1242077
10. Hirayasu K, Arase H. Functional and genetic diversity of leukocyte immunoglobulin-like receptor and implication for disease associations. J Hum Genet. (2015) 60:703–8. doi: 10.1038/jhg.2015.64
11. Hirayasu K, Ohashi J, Kashiwase K, Takanashi M, Satake M, Tokunaga K, et al. Long-term persistence of both functional and non-functional alleles at the leukocyte immunoglobulin-like receptor A3 (LILRA3) locus suggests balancing selection. Hum Genet. (2006) 119:436–43. doi: 10.1007/s00439-006-0152-y
12. Hirayasu K, Ohashi J, Tanaka H, Kashiwase K, Ogawa A, Takanashi M, et al. Evidence for natural selection on leukocyte immunoglobulin-like receptors for HLA class I in Northeast Asians. Am J Hum Genet. (2008) 82:1075–83. doi: 10.1016/j.ajhg.2008.03.012
13. Bashirova AA, Apps R, Vince N, Mochalova Y, Yu XG, Carrington M. Diversity of the human LILRB3/A6 locus encoding a myeloid inhibitory and activating receptor pair. Immunogenetics. (2014) 66:1–8. doi: 10.1007/s00251-013-0730-9
14. López-Álvarez MR, Jones DC, Jiang W, Traherne JA, Trowsdale J. Copy number and nucleotide variation of the LILR family of myelomonocytic cell activating and inhibitory receptors. Immunogenetics. (2014) 66:73–83. doi: 10.1007/s00251-013-0742-5
15. Hirayasu K, Sun J, Hasegawa G, Hashikawa Y, Hosomichi K, Tajima A, et al. Characterization of LILRB3 and LILRA6 allelic variants in the Japanese population. J Hum Genet. (2021) 66:739–48. doi: 10.1038/s10038-021-00906-0
16. Bashirova AA, Kasprzak W, O’hUigin C, Carrington M. Distinct frequency patterns of LILRB3 and LILRA6 allelic variants in Europeans. Immunogenetics. (2023) 75:263–7. doi: 10.1007/s00251-022-01286-1
17. Dubé MP, Zetler R, Barhdadi A, Brown AM, Mongrain I, Normand V, et al. CKM and LILRB5 are associated with serum levels of creatine kinase. Circulation: Cardiovasc Genet. (2014) 7:880–6. doi: 10.1161/CIRCGENETICS.113.000395
18. Kristjansson RP, Oddsson A, Helgason H, Sveinbjornsson G, Arnadottir GA, Jensson BO, et al. Common and rare variants associating with serum levels of creatine kinase and lactate dehydrogenase. Nat Commun. (2016) 7:10572. doi: 10.1038/ncomms10572
19. Xu J, Mo Z, Ye D, Wang M, Liu F, Jin G, et al. Genome-wide association study in Chinese men identifies two new prostate cancer risk loci at 9q31.2 and 19q13.4. Nat Genet. (2012) 44:1231–5. doi: 10.1038/ng.2424
20. Renauer PA, Saruhan-Direskeneli G, Coit P, Adler A, Aksu K, Keser G, et al. Identification of susceptibility loci in IL6, RPS9/LILRB3, and an intergenic locus on chromosome 21q22 in takayasu arteritis in a genome-wide association study. Arthritis Rheumatol. (2015) 67:1361–8. doi: 10.1002/art.39035
21. Terao C, Yoshifuji H, Matsumura T, Naruse TK, Ishii T, Nakaoka Y, et al. Genetic determinants and an epistasis of LILRA3 and HLA-B*52 in Takayasu arteritis. PNAS. (2018) 115:13045–50. doi: 10.1073/pnas.1808850115
22. Torkar M, Haude A, Milne S, Beck S, Trowsdale J, Wilson MJ. Arrangement of the ILT gene cluster: a common null allele of the ILT6 gene results from a 6.7-kbp deletion. Eur J Immunol. (2000) 30:3655–62. doi: 10.1002/1521-4141(200012)30:12<3655::AID-IMMU3655>3.0.CO;2-Y
23. Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, et al. Diversity of human copy number variation and multicopy genes. Science. (2010) 330:641–6. doi: 10.1126/science.1197005
24. Nguyen TTT, Tsujiguchi H, Kambayashi Y, Hara A, Miyagi S, Yamada Y, et al. Relationship between vitamin intake and depressive symptoms in elderly Japanese individuals: differences with gender and body mass index. Nutrients. (2017) 9:1319. doi: 10.3390/nu9121319
25. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. (2018) 34:3094–100. doi: 10.1093/bioinformatics/bty191
26. Cheng H, Concepcion GT, Feng X, Zhang H, Li H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. (2021) 18:170–5. doi: 10.1038/s41592-020-01056-5
27. Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. (2011) 21:974–84. doi: 10.1101/gr.114876.110
28. Byrska-Bishop M, Evani US, Zhao X, Basile AO, Abel HJ, Regier AA, et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell. (2022) 185:3426–3440.e19. doi: 10.1016/j.cell.2022.08.004
29. Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. (2011) 29:24–6. doi: 10.1038/nbt.1754
30. Pando MJ, Gardiner CM, Gleimer M, McQueen KL, Parham P. The protein made from a common allele of KIR3DL1 (3DL1*004) is poorly expressed at cell surfaces due to substitution at positions 86 in Ig domain 0 and 182 in Ig domain 1. J Immunol. (2003) 171:6640–9. doi: 10.4049/jimmunol.171.12.6640
31. Traherne JA, Martin M, Ward R, Ohashi M, Pellett F, Gladman D, et al. Mechanisms of copy number variation and hybrid gene formation in the KIR immune gene complex. Hum Mol Genet. (2010) 19:737–51. doi: 10.1093/hmg/ddp538
32. Roe D, Vierra-Green C, Pyo C-W, Eng K, Hall R, Kuang R, et al. Revealing complete complex KIR haplotypes phased by long-read sequencing technology. Genes Immun. (2017) 18:127–34. doi: 10.1038/gene.2017.10
33. Storm L, Bruijnesteijn J, de Groot NG, Bontrop RE. The genomic organization of the LILR region remained largely conserved throughout primate evolution: implications for health and disease. Front Immunol. (2021) 12:716289. doi: 10.3389/fimmu.2021.716289
34. Zhang Z, Hatano H, Shaw J, Nordkamp MO, Jiang G, Li D, et al. The leukocyte immunoglobulin-like receptor family member LILRB5 binds to HLA-class I heavy chains. PloS One. (2015) 10:e0129063. doi: 10.1371/journal.pone.0129063
Keywords: LILR, LILRB3, LILRA6, LILRB5, deletion, long-read sequencing, copy number variation, inhibitory receptor
Citation: Hirayasu K, Khor SS, Kawai Y, Shimada M, Omae Y, Hasegawa G, Hashikawa Y, Tanimoto H, Ohashi J, Hosomichi K, Tajima A, Nakamura H, Nakamura M, Tokunaga K, Hanayama R and Nagasaki M (2024) Identification of the hybrid gene LILRB5-3 by long-read sequencing and implication of its novel signaling function. Front. Immunol. 15:1398935. doi: 10.3389/fimmu.2024.1398935
Received: 11 March 2024; Accepted: 10 April 2024;
Published: 14 May 2024.
Edited by:
Payam Behzadi, Islamic Azad University, IranReviewed by:
Aruna Pal, West Bengal University of Animal and Fishery Sciences, IndiaCopyright © 2024 Hirayasu, Khor, Kawai, Shimada, Omae, Hasegawa, Hashikawa, Tanimoto, Ohashi, Hosomichi, Tajima, Nakamura, Nakamura, Tokunaga, Hanayama and Nagasaki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kouyuki Hirayasu, aGlyYXlhc3VAbWVkLmthbmF6YXdhLXUuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.