Abstract
Autoimmune rheumatic diseases comprise a group of immune-related disorders characterized by non-organ-specific inflammation. These diseases include systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), ankylosing spondylitis (AS), gout, among others. Typically involving the hematologic system, these diseases may also affect multiple organs and systems. The pathogenesis of autoimmune rheumatic immune diseases is complex, with diverse etiologies, all associated with immune dysfunction. The current treatment options for this type of disease are relatively limited and come with certain side effects. Therefore, the urgent challenge remains to identify novel therapeutic targets for these diseases. Sterol regulatory element-binding proteins (SREBPs) are basic helix-loop-helix-leucine zipper transcription factors that regulate the expression of genes involved in lipid and cholesterol biosynthesis. The expression and transcriptional activity of SREBPs can be modulated by extracellular stimuli such as polyunsaturated fatty acids, amino acids, glucose, and energy pathways including AKT-mTORC and AMP-activated protein kinase (AMPK). Studies have shown that SREBPs play roles in regulating lipid metabolism, cytokine production, inflammation, and the proliferation of germinal center B (GCB) cells. These functions are significant in the pathogenesis of rheumatic and immune diseases (Graphical abstract). Therefore, this paper reviews the potential mechanisms of SREBPs in the development of SLE, RA, and gout, based on an exploration of their functions.
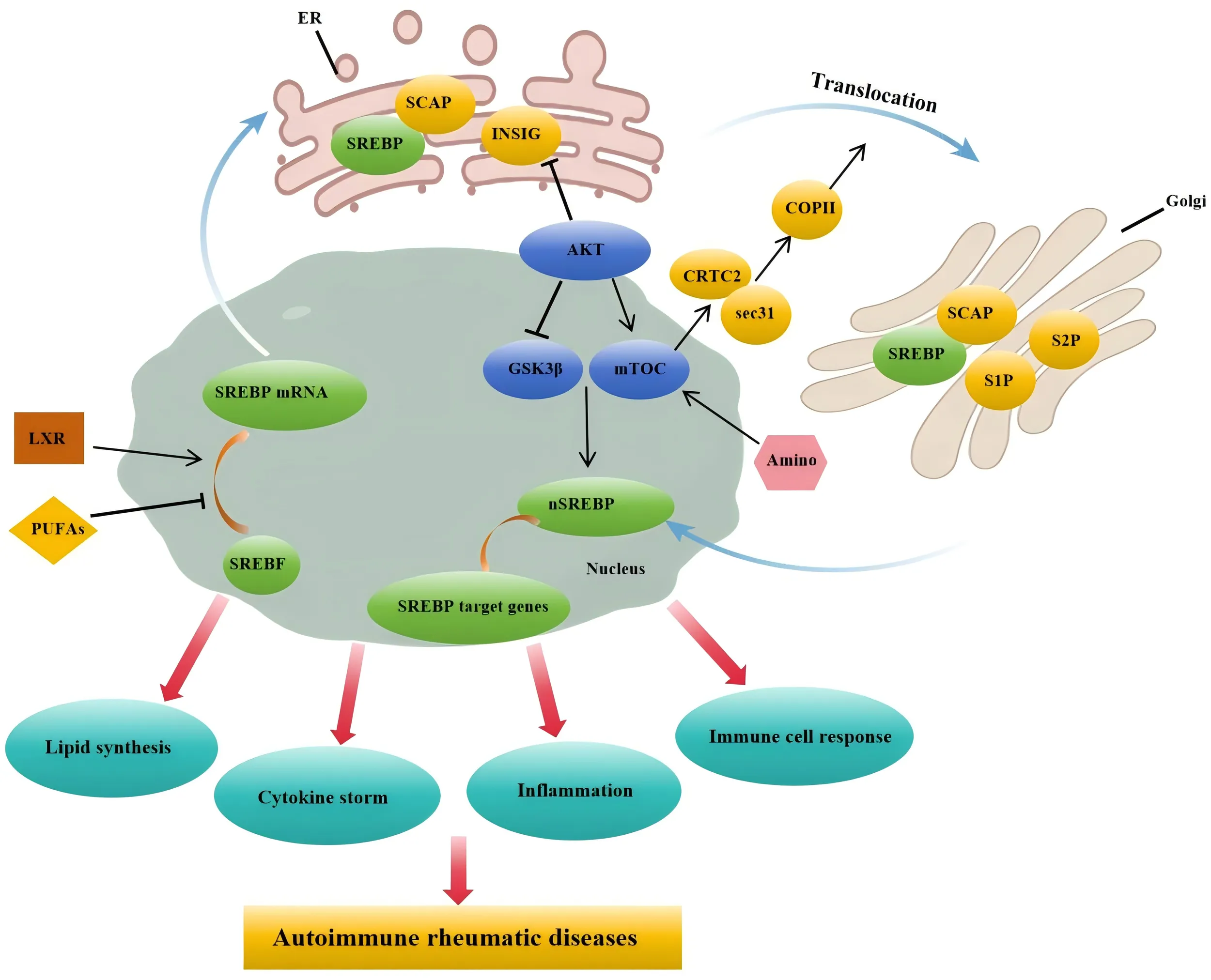
Under the influence of factors such as PUFAs and LXR, SREBP mRNA transcription is promoted, allowing the SCAP-SREBP complex to move from the endoplasmic reticulum to the Golgi apparatus. After cleavage by S1P and S2P in the Golgi, the complex enters the nucleus. Furthermore, under the influence of amino acids and the PI3K/AKT/mTOR signaling pathway, nuclear SREBP promotes the expression of target genes, thereby facilitating lipid synthesis, cytokine production, and accelerating inflammatory and immune cell responses. These functions are closely associated with the pathogenesis of rheumatic autoimmune diseases.
1 Introduction
Autoimmune rheumatic diseases represent a group of chronic disorders characterized by inflammation and autoimmunity, with the potential to affect any organ or system, leading to systemic damage. Current research indicates the involvement of various signaling pathways, including type I interferon pathways, immune cell pathways, immune metabolic pathways, complement, and coagulation, in the pathogenesis of these diseases. Adaptive immune cells, particularly B lymphocytes, play a significant role in the mechanisms underlying the development of autoimmune rheumatic diseases. B cells emerge as crucial contributors in many autoimmune disorders, such as RA, SLE, gout and multiple sclerosis (MS). In many of these conditions, the production of autoantibodies may constitute a primary pathogenic mechanism, highlighting the potential roles of B cell subsets and terminally differentiated antibody-producing plasma cells in autoimmunity (1). Innate immune cells mainly include macrophages, neutrophils, dendritic cells, natural killer cells, eosinophils, basophils, and other cell types, they circulate in the blood or reside in tissues, serving as the first line of defense against various pathogenic factors (2). Under the influence of relevant pathogenic factors, these cells produce cytokines or regulate the dynamic balance of tissue microenvironments through direct interactions with lymphocytes, participating in tissue damage and repair (3).
Inflammasomes, as crucial components of the innate immune response, play a vital role in the clearance of pathogens or damaged cells, providing a defense mechanism against pathogens and preventing pathological host damage. However, excessive activation of inflammasomes can also lead to autoimmune diseases, including RA, juvenile idiopathic arthritis (JIA), SLE and others (4–6). Inflammasomes and the interleukin-1 family of cytokines can promote the development of autoimmune diseases through the generation of adaptive immunity by T and B lymphocytes (4).
SREBPs were initially identified by Brown et al. as transcription factors that regulate the promoters of genes involved in cholesterol biosynthesis and the sterol regulatory pathway of the LDLR (7–9). Biological analysis underscores the significance of the SREBP pathway as a pivotal node in cell growth, metabolism, circadian rhythms, cellular stress, inflammation, and homeostasis (10). Recent research has indicated a correlation between SREBPs and various pathogenic processes, including endoplasmic reticulum (ER) stress, inflammation, cell apoptosis, and autophagy, with disease severity also being linked to SREBP levels (10–13). Although the SCAP-SREBP pathway plays a crucial role in cholesterol metabolism, its role in the immune system remains poorly understood. Recent studies have found that the activation of SREBP leads to inflammasome activation, induces macrophage inflammation, and triggers cytokine storms, thereby participating in related immune and inflammatory responses (14, 15). Additionally, research indicates a close association between the SREBP pathway-controlled cholesterol metabolism and adaptive immunity, including antibody and T follicular cell responses (16). Loss of SREBP in T cells severely impairs CD8 T cell activation (17). Moreover, the absence of SREBP signaling in B cells results in defective germinal center, memory B cell, and bone marrow plasma cell generation, preventing the generation of effective antibody responses (18).
Consequently, this article provides a comprehensive review of the SREBP’s upstream regulators, intracellular distribution, and identified biological functions. Furthermore, based on the established biological functions, we hypothesize that this protein may play a crucial role in the development of autoimmune rheumatic diseases such as SLE, RA and gout, and we present a series of prospects for future investigation in this field, to provide a novel target for the treatment of this type of disease.
2 Regulators of SREBPs
SREBPs are considered transcription factors that serve as primary regulators of a series of lipogenesis pathways, comprising three subtypes: SREBP-1a, SREBP-1c, and SREBP-2 (19). The activity of SREBPs relies on the SCAP (20). SCAP is a polytopic membrane protein residing in the endoplasmic reticulum. It binds to SREBP within the endoplasmic reticulum and transports it to the Golgi apparatus for proteolytic processing. Additionally, two other ER-resident membrane proteins, insulin-induced gene 1 protein (INSIG1) and INSIG2, interact with SCAP, causing the retention of the SREBP-SCAP complex in the ER membrane (21, 22). INSIG can enhance its own stability by binding to 25-hydroxycholesterol (produced in the ER). Once SREBPs leave the ER and enter the Golgi apparatus, they undergo a two-step proteolytic processing by two proteases: site-1 protease and site-2 protease (23). Cholesterol 25-hydroxylase, an enzyme responsive to type I interferons, increases the production of 25-hydroxycholesterol, which inactivates SREBPs and subsequently exerts anti-inflammatory effects by reducing the secretion of IL-1β (24, 25).
The liver X receptor (LXR) serves as a pivotal transcriptional regulator of cholesterol, fatty acid, and phospholipid metabolism (26). The promoter region of SREBP1c possesses an SRE element that facilitates its self-regulatory activation (27), LXRα and LXRβ are powerful activators of the SREBP1c promoter. When LXR agonists are administered, they stimulate the activation of both SREBP1c and fatty acid synthase (28). Consumption of polyunsaturated fatty acids (PUFAs) can reduce hepatic SREBP1c activity (29). The inhibition of SREBP1c by PUFAs occurs through several mechanisms: reduced transcription, increased mRNA decay, inhibition of proteolytic cleavage, and enhanced proteasomal degradation of nuclear SREBP1c (30).
ATF6 is a transcription factor anchored in the ER membrane, overexpression of exogenous, active ATF6 or its activation through glucose depletion has been observed to suppress the expression of SREBP target genes. Specifically, ATF6 is known to interact directly with SREBP2 through its leucine-zipper domain. This interaction recruits histone deacetylase 1, thereby inhibiting the transcriptional activity of SREBP2 (31). Amino acid levels play a crucial role in the activation and regulation of SREBP expression. Specifically, amino acids activate the mechanistic target of rapamycin complex 1 (mTORC1) within the lysosome. By modulating mTORC1 activity, amino acids ensure that cells can appropriately adjust to metabolic demands, impacting both anabolic and catabolic pathways (32, 33).
The PI3K-AKT-mTOR pathway has been identified as a major upstream signaling pathway regulating SREBP (34). AKT phosphorylates GSK3β, inhibiting its activity, which reduces SREBP degradation via the FBXW7-mediated ubiquitin proteasome system (35). AKT phosphorylates and inhibits INSIG2A, freeing the SREBP–SCAP complex for transport to the Golgi (36). AKT suppresses TSC, activating mTORC1, which phosphorylates and relocates lipin-1 to the nucleus, activating nuclear SREBP1 (37, 38). mTORC1 phosphorylates CREB-regulated transcription coactivator 2, promoting SREBP1 translocation from the ER to the Golgi by releasing inhibitory SEC31 and facilitating COPII vesicle formation (39).
3 Biological functions of SREBPs
3.1 The role of SREBPs in lipogenesis regulation
The SREBP1 gene gives rise to two isoforms through transcription from distinct promoters. SREBP1c is the predominant subtype expressed in most tissues, whereas SREBP1a exhibits high expression only in specific tissues and cells, such as intestinal epithelium, cardiac tissue, macrophages, and bone marrow dendritic cells (40). SREBP1c lacks 24 amino acid residues in the N-terminal transactivation domain of SREBP1a. This domain allows SREBP1a to bind tightly to CREB-binding protein, and as SREBP1c lacks these amino acids, its transcriptional activity is relatively lower (41, 42). In most cultured cells, SREBP1a (rather than SREBP1c) is the predominant isoform, possibly because SREBP1a can stimulate the expression of lipogenic and cholesterol-synthetic genes, thus providing components necessary for membrane lipid synthesis (43, 44).
A series of animal studies employing transgenic and knockout mice for each SREBP gene and subtype indicate that SREBP1c primarily regulates the expression of fat synthesis genes, while SREBP2 controls genes related to cholesterol metabolism (20, 41, 43). Physiologically, SREBP1a robustly activates total fat synthesis in rapidly growing cells, while SREBP1c plays a role in the nutritional regulation of fatty acids and triglycerides in fat-producing organs, such as the liver. In contrast, SREBP2 has a regulatory role in all tissues (20). This functional specificity is more evident in vivo than in vitro, but isoforms exhibit functional overlap when overexpressed. The partial specificity of SREBP subtypes for different target genes is explained by their unique binding dynamics with cholesterol synthesis genes, mainly SRE elements, as well as SREs and enhancer boxes in fat genes, and auxiliary factors such as SP1 and NFY (45). SREBP1a and SREBP2, but not SREBP1c, associate as coactivators with CBP and P300 (also known as EP300) to recruit the mediator complex (42). The SREBP family is also acetylated and stabilized by CBP and P300 (42, 46, 47).
3.2 SREBPs can elicit cytokine storms
Cytokine storms refer to the rapid and excessive release of pro-inflammatory cytokines by immune cells, which can lead to a hyperinflammatory response and contribute to various pathological conditions. The mechanisms through which SREBPs induce cytokine storms and their precise impact on immune regulation are areas of ongoing research. SREBP2 activation induces an inflammatory response and exacerbates inflammatory damage. Firstly, existing research suggests that SREBP2 can regulate the inflammatory phenotype by modulating cholesterol homeostasis. Increased cholesterol synthesis is involved in various immune pathways, such as interferon response, inflammasome activation, and innate immunity (14, 48, 49). Additionally, perturbation of cellular cholesterol may alter membrane dynamics and impact cell signal transduction (50). Secondly, SREBP2 can interact with several pro-inflammatory mediators and promote their transcription, such as IL1β, IL8, NLRP3, and NOX2 (51–54).
SREBP-2 serves as a transcription factor for lipid synthesis. However, it has been observed that COVID-19 patients maintain lower cholesterol levels, even though the expression levels of SREBP-2 in their plasma increase. A seminal discovery by Wonhwa (15) identified an elevated C-terminal segment of SREBP-2, termed SREBP-2C, in the blood of COVID-19 patients. This heightened level of SREBP-2C correlates closely with the excessive inflammation observed in the lung tissues of COVID-19 patients, inducing an upregulation of inflammatory responses that can lead to cytokine storms. Clinical studies have further revealed that ICU patients with elevated plasma levels of SREBP-2C exhibit more severe lung inflammation in CT images compared to non-ICU patients with lower SREBP-2C levels. Consequently, SREBP-2 can serve as a diagnostic marker for the severity of COVID-19 in critically ill patients and as a therapeutic target for preventing cytokine storms and lung damage. Additionally, in an infectious disease mouse model, the inhibition of SREBP-2 and NF-κB attenuated cytokine storms induced by viral infection and prevented lung injury. These findings underscore the potential clinical significance of SREBP-2 in assessing COVID-19 severity and its role in the prevention of cytokine storms and lung injury, offering novel prospects for the diagnosis and treatment of COVID-19.
3.3 SREBPs can promote macrophage inflammation
The inflammasome is a multiprotein complex formed in the cytoplasm following exposure to various stimuli from pathogenic sources. Its activation depends on sensor proteins recognizing ligands, subsequently recruiting adaptor protein ASC (55). This leads to ASC oligomerization and the recruitment and activation of caspase-1, an enzyme responsible for processing Pro-IL-1β into mature IL-1β. Studies indicate that conditioned media from BMDMs transduced with Hmgcr or Dhcr24 enhances their ability to stimulate T cells for IL-17A production (56). Furthermore, researchers have demonstrated that cholesterol-treated BMDM significantly promote IL-1β production. Inhibition of SREBP expression can suppress IL-1β-induced macrophage inflammation (14).
In macrophages, studies have revealed that the SCAP/SREBP2 shuttle complex directly interacts with the NLRP3 inflammasome, regulating inflammasome activation by translocating from the endoplasmic reticulum to the Golgi apparatus (57). Another set of research indicates that SREBP2 is highly activated in macrophages treated with TNFα, and nuclear SREBP2 binds to target genes involved in inflammation and interferon responses, promoting an M1-like inflammatory state (51). Moreover, several studies suggest that cellular cholesterol levels control immune phenotypes. The type I interferon (IFN) signal in macrophages reduces cholesterol synthesis, allowing the activation of STING on the ER to enhance IFN signaling (49). Furthermore, research has shown that restoring cholesterol biosynthesis in macrophages promotes inflammation (14, 50).
Additionally, inflammatory factors upregulate SCAP expression, facilitating the translocation of the SCAP/SREBP2 complex from the endoplasmic reticulum to the Golgi apparatus. This disrupts intracellular cholesterol homeostasis and contributes to atherosclerosis and non-alcoholic fatty liver disease (58, 59). We have also discovered that crosstalk between SCAP/SREBP and the TLR4-MyD88-NF-κB inflammation pathway mediates foam cell formation in atherosclerosis (60). Moreover, SCAP overexpression promotes the translocation of SCAP and NLRP3 inflammasomes to the Golgi apparatus, increasing the activation of the NLRP3 inflammasome pathway and thereby expediting atherosclerosis (61, 62). These findings highlight SCAP as a crucial molecular link between lipid metabolism and inflammation (63). The STING/TBK1 pathway, a classical innate immune signaling pathway, has recently been shown to play a critical role in the inflammatory response of metabolic diseases. SCAPs activation of the STING-NF-κB signaling pathway has been implicated in the pathogenesis of macrophage inflammation and lean non-alcoholic fatty liver disease (64).
3.4 SREBPs can regulate cellular energy metabolism to promote the proliferation of B cells and the production of autoantibodies
Recent research indicates that the SREBP pathway, governing sterol metabolism, is closely associated with adaptive immunity, including antibody and T follicular cell responses (16). Depletion of SREBP in T cells severely impedes CD8 T cell activation (17). In addition, recent research has revealed a connection between B-cell activation and lipid metabolism reprogramming (18). Activation of TLR4, CD40, and BCR signaling pathways significantly upregulates the expression of most genes involved in cholesterol biosynthesis in B cells. Notably, the lack of SCAP severely inhibits the regulation of these cholesterol biosynthesis-related genes, including those encoding key enzymes such as Hmgcr, Hmgcs1, Sqle, Dhcr24, and lipid synthesis-related genes Acsl3 and Acsl4. Pathway analysis further indicates that the lipid biosynthesis pathway is one of the highly impacted pathways associated with SCAP deficiency in B cells.
Studies have demonstrated that B cells undergo 2-5 rounds of division after stimulation with different TLRs or CD40, while SCAP-deficient B cells not only fail to undergo division but are also blocked from entering the S phase. This suggests that the SREBP signal is a key factor in mitogen-induced B-cell division and promotes the B-cell cycle process. Furthermore, SCAP deficiency leads to a sharp reduction in CD138 and YFP-positive GC-derived plasma cells, resulting in a significant decrease in antigen-specific IgG titers and reduced affinity maturation. These findings highlight the critical role of cholesterol synthesis-mediated SREBP signaling in maintaining lipid homeostasis, cell cycle progression, and plasma cell differentiation. Loss of SREBP signaling in B cells impairs the generation of germinal center, memory B cells, and bone marrow plasma cells, preventing the effective production of antibody responses (18).
3.5 SREBPs can facilitates cancer development
Dysregulated cellular lipid metabolism, driven by the SREBP pathway, is a prominent feature of cancer cells (65). Oncogenic growth signals render cells reliant on de novo lipogenesis, which encompasses extensive fatty acid synthesis from glucose and glutamine to meet the bioenergetic and biosynthetic demands of rapidly proliferating tumor cells (66). Insulin-mediated signals through the PI3K-AKT-mTORC1-SREBP axis play a pivotal role in regulating lipid synthesis in response to nutritional fluctuations. This signaling pathway also serves as an established survival route, activated structurally in many cancer types, exerting pronounced roles in growth, malignancy, anti-apoptosis, drug resistance, and metastasis (67). Numerous oncogenic signaling molecules, including P53, PTEN, PI3K, and KRAS, converge upon the PI3K-AKT-mTOR pathway to activate protein and lipid biosynthesis in cancer cells, satisfying the demand for lipids in cell growth. Even in conditions of low oxygen and high acidity, SREBP1a mediates the metabolic flux from enhanced glycolysis to lipid synthesis via PI3K-AKT signaling, upregulating the LDLR to facilitate cholesterol uptake. SREBP1a is highly expressed in cancer cells and exhibits robust promoter activity in actively proliferating cancer cells (68–71).
The mevalonate pathway is upregulated in many cancers, including liver cancer, possibly due to mutations in sterol-related genes, such as SREBP2 and SCAP, resulting from p53 mutations (71). Additionally, genome-wide expression analysis identified mutations in sterol gene promoters for p53 and SREBP in human breast tumors. These mutations disrupted breast tissue architecture through the mevalonate pathway (69–71). The tumor suppressor retinoblastoma protein and GTPase NRAS interact in an anticancer senescence pathway. retinoblastoma protein loss activates SREBPs and enhances geranylgeranylation in a reverse E2F-dependent manner, leading to NRAS activation, subsequent induction of DNA damage response, and p130-dependent cell senescence (72), thus promoting retinoblastoma development.
4 Prospects of SREBPs in rheumatic immune diseases
4.1 SREBP and systemic lupus erythematosus
SLE is a chronic autoimmune disease characterized by genetic, endocrine, environmental, and their interaction-induced autoantibody production, immune complex deposition, abnormal activation of various immune cells (such as T and B lymphocytes and granulocytes), and tissue damage in organs like the kidneys, skin, heart, and lungs (73–75). The exact etiology and pathogenesis of SLE remain incompletely understood (76). In China, the incidence of SLE is approximately 70 per 100,000, and it shows an increasing trend year by year (75). SLE has a complex pathogenesis closely related to genetics, immune dysregulation, viral infections, and environmental factors (77, 78). Recent research on the pathogenesis of SLE has provided a wealth of information (79–90). Among the multitude of mechanisms contributing to SLE, immune dysregulation stands out as one of the primary drivers of disease. Immune cells play a pivotal role in the initiation and progression of SLE’s autoimmune response. In SLE, immune cells exhibit overexpression of autoimmunity, imbalanced cytokine production, and increased apoptosis, all of which have a significant impact on the pathogenesis of SLE (91).
Lipids are crucial constituents of cell membranes, and changes in their composition and content can influence the normal function of cells. For instance, alterations in the composition, distribution, and dynamics of lipid rafts (microdomains primarily composed of cholesterol and sphingolipids) on T cells accelerate their activation in SLE patients, exacerbating the condition (92). Anomalies in lipid raft expression on T cells in SLE may lead to abnormal activation and signaling pathways, resulting in the production of aberrantly expressed cytokines and assisting in the abnormal response of B lymphocytes, leading to the production of autoantibodies and the development of SLE (93). Additionally, the cellular microenvironment plays a significant role in influencing cellular lipid metabolism, further contributing to cellular dysfunction (94). Research indicates that cholesterol buildup is essential for T cell proliferation and their response to antigen interactions. When cholesterol synthesis is blocked due to SCAP deficiency, T cell proliferation is entirely halted (17, 95). On the other hand, if cholesterol cannot be esterified because of Acat1 deficiency, there is an increase in plasma membrane cholesterol accumulation, which enhances T cell proliferation (96). Likewise (97), the lack of Abcg1-mediated cholesterol efflux results in increased plasma membrane cholesterol and further promotes T cell proliferation (Figure 1).
Figure 1
Research (98) has indicated a close relationship between IFN-γ expression and the severity of SLE in both human and murine models of the disease, the increase in nuclear form of SREBP (nSREBP) leads to cholesterol accumulation, which promotes the expression of IFN-γ (99). Spontaneously developed germinal centers containing autoreactive B cells that produce pathogenic autoantibodies play a role in promoting autoimmune responses and driving the development of systemic lupus erythematosus. Studies have demonstrated that IFN-γ-STAT1 signaling controls the formation of GCs by promoting the expression of T-bet in B cells and the production of IFN-γ (100). As previously discussed, the loss of nSREBP signaling in cells can lead to defects in cell cycle progression and metabolic reprogramming, thereby impeding the generation of germinal centers, memory B cells, and plasma cells required for an effective antibody response (18). We speculate that increased expression of nSREBP in B cells accelerates B cell cycle progression, promotes B cell activation and proliferation, leading to the formation of germinal centers. Subsequently, B cells complete Ig affinity maturation and class switching in germinal centers, resulting in excessive production of pathogenic autoantibodies. The binding of these autoantibodies to antigens forms immune complexes, ultimately contributing to the pathogenesis of SLE (Figure 1).
Macrophages can be polarized into inflammatory (M1) and anti-inflammatory (M2) phenotypes through various stimuli, such as IFN-γ and LPS for M1 polarization, or IL-4 for M2 polarization, playing crucial roles in immune regulation (101). Studies have shown that M1 macrophages in patients with SLE can regulate the activation status of T and B lymphocytes, thereby influencing disease activity (102). In MRL/lpr mice, transient ischemic renal injury upregulates CSF-1 expression in renal tubular epithelial cells, leading to increased release of CSF-1 and subsequent expansion of M1 macrophages, accelerating the onset of lupus nephritis (103). Moreover, recent research has revealed that human and murine macrophages, when stimulated by IgG immune complexes, undergo metabolic reprogramming dependent on mTOR and HIF-1α, resulting in the production of pro-inflammatory cytokine IL-1β, thereby promoting lupus nephritis (104). In the presence of cholesterol and oxygenated sterols, including 25/27-HC, the SREBP-SCAP complex remains sequestered within the endoplasmic reticulum along with INSIGs. Studies have indicated (15) that overexpression of nSREBP leads to a cytokine storm, while inhibiting nSREBP expression can suppress the production of inflammatory cytokines such as IL-1β and TNF-α, dampening macrophage inflammatory responses. Consequently, it is hypothesized that when 25/27-HC expression is reduced, preventing INSIGs from inhibiting the translocation of the SREBP-SCAP complex to the Golgi apparatus (Figure 1), excessive SREBP expression ensues, resulting in the overproduction of IL-1β, IL-6, and CSF-1, which promotes the expansion of M1 macrophages and accelerates the onset of lupus nephritis (Figure 2).
Figure 2
All of these findings collectively suggest that SREBPs may play a crucial role in the pathogenesis of SLE. Investigating the mechanistic involvement of SREBPs in SLE could offer a novel therapeutic target for the treatment of SLE.
4.2 SREBP and Rheumatoid Arthritis
RA is an autoimmune disease characterized by progressive synovial inflammation in multiple joints, resulting in widespread symmetrical joint swelling, pain, bone destruction, joint deformities, and potential involvement of connective tissues such as the heart, lungs, and eyes. It is considered a severe and challenging-to-treat disease (105). Epidemiological studies have indicated a global distribution of RA, with an average prevalence ranging from 0.5% to 1.5%. In China, the prevalence of RA in adults is approximately 0.4% and shows an increasing trend (106). The etiology and pathogenesis of RA remain inconclusive, but it is generally accepted that RA is an inflammatory disease triggered by external factors on a genetic susceptibility background. RA patients exhibit innate immune and adaptive immune response abnormalities, characterized by an imbalance in Th1/Th2 responses, an increased number of Th17 cells, and compromised functions of T and B lymphocytes that have immunosuppressive effects. These abnormalities lead to the production of a multitude of pro-inflammatory cytokines, including IFN-γ, TNF-α, IL-1β, IL-6, and IL-17 (107–109), which participate in a positive feedback loop, promoting the continuous progression of inflammation. Ultimately, this results in the infiltration of inflammatory cells into the synovium, cartilage and bone destruction, joint deformities, and peripheral tissue damage in RA patients. SREBP-2 promotes targets of the methoxyerate pathway and interferon-responsive genes in TNF-activated macrophages (51). Recent studies have shown that endotoxin-mediated IL-1β production is partially dependent on SREBP-1a. Caspase-11 interacts with the S1P-SREBP pathway, activating SREBP-1a, which subsequently drives downstream inflammatory responses (40, 110). Given the characteristics of SREBP overexpression promoting the release of inflammatory factors, we speculate that SREBP exacerbates the continuous progression of inflammation, leading to synovial infiltration of inflammatory cells, cartilage and bone destruction, joint deformities, and peripheral tissue damage in RA patients.
The JAK-STAT signaling pathway plays a significant role in the pathogenesis of RA (111–114). JAK2 activates downstream genes by phosphorylating STAT3 or STAT5, leading to the release of pro-inflammatory signals, such as IL-6, IFN-γ, IL-12, and other inflammatory factors (115–118). Researches have shown that blocking the JAK2-STAT3 pathway by activating AMPK can exert an anti-inflammatory effect. AMPK phosphorylates JAK2 at Ser515 and Ser518, inhibiting JAK2-mediated STAT3 activation (119, 120). Furthermore (121), study has shown that the activation of AMPK can suppress the expression of SREBP, while AMPK expression is inhibited in RA. Therefore, we speculate that SREBP may also play a role in the pathogenesis of RA.
NF-κB plays an essential role in joint destruction. When cells are stimulated, IκB are phosphorylated by IκB kinase, leading to the release of NF-κB. NF-κB enters the cell nucleus and, through a series of reactions, initiates the transcription and expression of downstream inflammatory factors such as IL-1β, IL-6, IL-12, IL-17, TNF-α, and others (122). This, in turn, activates NF-κB, causing an amplification of the initial inflammatory signal, resulting in a cascade reaction that sustains the development of the inflammatory response and structural damage. Clinical studies have found significantly elevated expression levels and activity of NF-κB in the serum and lymphatic endothelial cells of RA patients. Animal experiments have also confirmed that inhibiting the NF-κB pathway can significantly improve the degree of toe swelling and inflammatory responses in a mouse model of RA (123). Studies have found that SCAP and SREBP1 form a super complex with IkBα, bringing NF-kB close to the endoplasmic reticulum. Upon endotoxin stimulation, SCAP escorts this complex to the Golgi apparatus, where SREBP1 is cleaved by S1P/S2P, releasing IkBα for phosphorylation and activating subsequent inflammatory responses (124, 125). Additionally, the SCAP/SREBP/STING/TBK1 pathway can activate NF-κB in metabolic diseases and promote the expression of related inflammatory factors (64). HSP90 is a new regulatory factor of SREBP and can bind to the SREBP-SCAP complex in the endoplasmic reticulum and Golgi apparatus, stabilizing it. The inhibition of HSP90 results in the dependence of the complex on proteasomal degradation (126). HSP90β activates SREBP2, increasing cholesterol biosynthesis and NF-κB signaling to promote osteoclastogenesis (127). Inhibiting SREBP and subsequently suppressing NF-κB-related inflammatory responses may alleviate inflammation, inhibit bone destruction, reduce disability, and achieve disease remission in RA patients (Figure 3). This hypothesis merits further investigation.
Figure 3
4.3 SREBP and gout
Gout is an inflammatory disease characterized by the massive deposition of urate crystals in joints and surrounding tissues, resulting from disturbances in purine metabolism and/or abnormal uric acid excretion (128). It has become the second most prevalent metabolic disease in China, following diabetes (129). Recurrent gout attacks not only cause severe pain in affected joints but can also lead to joint damage, dislocation, deformities, and even joint disability or amputation. Furthermore, the extensive deposition of urate crystals in the kidneys can lead to renal damage and even acute kidney failure, which can be fatal (130). Currently, commonly used urate-lowering drugs such as febuxostat, while effective to some extent, have the potential for severe toxic side effects with long-term use, low patient tolerance, and a high disease recurrence rate, significantly impacting patient compliance (131).
Modern medicine recognizes that MSU crystals can strongly stimulate Toll-like receptors and the NLRP3 inflammasome, leading to the activation of the innate immune response, primarily involving macrophages and neutrophils. This eventually results in the release of IL-1β and other pro-inflammatory factors, triggering gout attacks (132, 133). Macrophages play a frontline role in the immune response, with different subtypes classified based on the cytokines they secrete and the cell surface adhesion molecules. Among these subtypes, M1 macrophages, when activated, promote the assembly of the NLRP3 inflammasome, leading to the release of high pro-inflammatory cytokines such as TNF-α and IL-1β, along with promoting the recruitment of neutrophils from peripheral blood to the inflammatory site, subsequently releasing pro-inflammatory cytokines and causing sustained inflammation. In contrast, M2 macrophages can suppress MSU crystal-induced inflammation and inhibit caspase-1 activation and IL-1β production (134). In M1 pro-inflammatory macrophages, the TCA cycle is disrupted, whereas M2 macrophages possess a complete TCA cycle and mainly rely on OXPHOS (135). The overexpression of SREBP1a promotes the repositioning of the NLRP3 inflammasome to the Golgi apparatus, thereby enhancing the activation of the NLRP3 inflammasome pathway (61, 62). SREBP1a also directly activates the transcription of NLRP genes and caspase-1, mediating the secretion of IL-1β by macrophages. This indicates an important function of macrophage SREBP1a, associating lipid generation and/or lipid toxicity with innate immune responses (40). MIR-33 is a microRNA involved in SREBP signaling, and is also involved in the production of pro-inflammatory and anti-inflammatory genes in M1 and M2 macrophages, respectively (136). In addition, SREBP and miR-33 inhibit cholesterol efflux through the ATP-binding cassette transporter A1 in macrophages (137). Loss of SCAP caused changes in cholesterol metabolism and induced proinflammatory M1 polarization in adipose tissue macrophages (125, 138).
Macrophage polarization is a complex process influenced by the interaction of various intracellular signaling molecules and pathways. It holds crucial significance in inflammation, metabolic diseases, autoimmune disorders, and other conditions. Among these factors, the regulation of the PI3K/Akt/mTOR signaling pathway plays a central role in controlling macrophage polarization (139). Furthermore, SREBP1 mediates metabolic flux from enhanced glycolysis to lipid generation via the PI3K-AKT pathway and upregulates LDLR, promoting cholesterol uptake (68–71). These factors can lead to macrophage inflammation, glucose and lipid metabolic abnormalities, ultimately contributing to the pathogenesis of gout (Figure 4).
Figure 4
While macrophages and neutrophils are considered the primary immune cells involved in gout pathogenesis (140, 141), recent researches have highlighted the role of T cell subsets in gout (142, 143). Targeting pro-inflammatory T cell subsets or their associated cytokines can improve MSU crystal-induced arthritis in mice. The gut microbiota and its metabolites play a significant role in maintaining intestinal homeostasis (144, 145) and regulating T cell differentiation (146), which are crucial in the development of autoimmune disorders and inflammatory diseases. A previous study indicated that the loss of SCAP in T cells severely impairs CD8 T cell activation (17). Due to the limited research on SREBP in gout, future investigations targeting this pathway may provide new strategies for gout treatment.
5 Conclusions
SREBP has emerged as a significant discovery in medical research in recent years, research on SREBP has primarily focused on the field of obesity and cancer. However, the functions of SREBP in lipid metabolism, cell growth, and inflammation are also closely related to the pathogenesis of rheumatic and immune diseases. Therefore, investigating the expression characteristics and signaling pathways of SREBP in patients with rheumatic immune diseases can provide deeper insights into the pathogenesis of these conditions. This, in turn, holds substantial significance for the identification of early biomarkers and the development of precise personalized treatment approaches.
Statements
Author contributions
XD: Funding acquisition, Project administration, Writing – original draft, Writing – review & editing. XX: Investigation, Methodology, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing. WJ: Investigation, Resources, Writing – original draft, Writing – review & editing. RC: Investigation, Methodology, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The work was partially supported by National Natural Science Foundation of China (No. 81973778).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
HofmannKClauderAKManzRA. Targeting B cells and plasma cells in autoimmune diseases. Front Immunol. (2018) 9:835. doi: 10.3389/fimmu.2018.00835
2
StögererTStägerS. Innate immune sensing by cells of the adaptive immune system. Front Immunol. (2020) 11:1081. doi: 10.3389/fimmu.2020.01081
3
HatoTDagherPC. How the innate immune system senses trouble and causes trouble. Clin J Am Soc Nephrol. (2015) 10:1459–69. doi: 10.2215/CJN.04680514
4
SharmaDKannegantiTD. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol. (2016) 213:617–29. doi: 10.1083/jcb.201602089
5
CorderoMDAlcocer-GómezERyffelB. Gain of function mutation and inflammasome driven diseases in human and mouse models. J Autoimmun. (2018) 91:13–22. doi: 10.1016/j.jaut.2018.03.002
6
EvavoldCLKaganJC. How inflammasomes inform adaptive immunity. J Mol Biol. (2018) 430:217–37. doi: 10.1016/j.jmb.2017.09.019
7
YokoyamaCWangXBriggsMRAdmonAWuJHuaXet al. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell. (1993) 75:187–97. doi: 10.1016/S0092-8674(05)80095-9
8
BrownMSGoldsteinJL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. (1997) 89:331–40. doi: 10.1016/S0092-8674(00)80213-5
9
GoldsteinJLDeBose-BoydRABrownMS. Protein sensors for membrane sterols. Cell. (2006) 124:35–46. doi: 10.1016/j.cell.2005.12.022
10
ShimanoHSatoR. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol. (2017) 13:710–30. doi: 10.1038/nrendo.2017.91
11
SeoYKChongHKInfanteAMImSSXieXOsborneTF. Genome-wide analysis of SREBP-1 binding in mouse liver chromatin reveals a preference for promoter proximal binding to a new motif. Proc Natl Acad Sci U S A. (2009) 106:13765–9. doi: 10.1073/pnas.0904246106
12
SeoYKJeonTIChongHKBiesingerJXieXOsborneTF. Genome-wide localization of SREBP-2 in hepatic chromatin predicts a role in autophagy. Cell Metab. (2011) 13:367–75. doi: 10.1016/j.cmet.2011.03.005
13
RomaniPBrianISantinonGPocaterraAAudanoMPedrettiSet al. Extracellular matrix mechanical cues regulate lipid metabolism through Lipin-1 and SREBP. Nat Cell Biol. (2019) 21:338–47. doi: 10.1038/s41556-018-0270-5
14
DangEVMcDonaldJGRussellDWCysterJG. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell. (2017) 171:1057–71.e11. doi: 10.1016/j.cell.2017.09.029
15
LeeWAhnJHParkHHKimHNKimHYooYet al. COVID-19-activated SREBP2 disturbs cholesterol biosynthesis and leads to cytokine storm. Signal Transduct Target Ther. (2020) 5:186. doi: 10.1038/s41392-020-00292-7
16
LiSSullivanNLRouphaelNYuTBantonSMaddurMSet al. Metabolic phenotypes of response to vaccination in humans. Cell. (2017) 169:862–77.e17. doi: 10.1016/j.cell.2017.04.026
17
KidaniYElsaesserHHockMBVergnesLWilliamsKJArgusJPet al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol. (2013) 14:489–99. doi: 10.1038/ni.2570
18
LuoWAdamskaJZLiCVermaRLiuQHaganTet al. SREBP signaling is essential for effective B cell responses. Nat Immunol. (2023) 24:337–48. doi: 10.1038/s41590-022-01376-y
19
KawamuraSMatsushitaYKurosakiSTangeMFujiwaraNHayataYet al. Inhibiting SCAP/SREBP exacerbates liver injury and carcinogenesis in murine nonalcoholic steatohepatitis. J Clin Invest. (2022) 132:e151895. doi: 10.1172/JCI151895
20
HortonJDGoldsteinJLBrownMS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. (2002) 109:1125–31. doi: 10.1172/JCI0215593
21
YangTEspenshadePJWrightMEYabeDGongYAebersoldRet al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. (2002) 110:489–500. doi: 10.1016/S0092-8674(02)00872-3
22
YabeDBrownMSGoldsteinJL. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci U S A. (2002) 99:12753–8. doi: 10.1073/pnas.162488899
23
BrownMSRadhakrishnanAGoldsteinJL. Retrospective on cholesterol homeostasis: the central role of scap. Annu Rev Biochem. (2018) 87:783–807. doi: 10.1146/annurev-biochem-062917-011852
24
ReboldiADangEVMcDonaldJGLiangGRussellDWCysterJG. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science. (2014) 345:679–84. doi: 10.1126/science.1254790
25
CashikarAGToral-RiosDTimmDRomeroJStricklandMLongJMet al. Regulation of astrocyte lipid metabolism and ApoE secretionby the microglial oxysterol, 25-hydroxycholesterol. J Lipid Res. (2023) 64:100350. doi: 10.1016/j.jlr.2023.100350
26
WaddingtonKERobinsonGARubio-CuestaBChrifi-AlaouiEAndreoneSPoonKSet al. LXR directly regulates glycosphingolipid synthesis and affects human CD4+ T cell function. Proc Natl Acad Sci U.S.A. (2021) 118(21):e2017394118. doi: 10.1073/pnas.2017394118
27
Amemiya-KudoMShimanoHYoshikawaTYahagiNHastyAHOkazakiHet al. Promoter analysis of the mouse sterol regulatory element-binding protein-1c gene. J Biol Chem. (2000) 275:31078–85. doi: 10.1074/jbc.M005353200
28
RepaJJLiangGOuJBashmakovYLobaccaroJMShimomuraIet al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. (2000) 14:2819–30. doi: 10.1101/gad.844900
29
SuFKoeberleA. Regulation and targeting of SREBP-1 in hepatocellular carcinoma. Cancer Metastasis Rev. (2024) 43:673–708. doi: 10.1007/s10555-023-10156-5
30
JumpDB. Dietary polyunsaturated fatty acids and regulation of gene transcription. Curr Opin Lipidol. (2002) 13:155–64. doi: 10.1097/00041433-200204000-00007
31
ZengLLuMMoriKLuoSLeeASZhuYet al. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. (2004) 23:950–8. doi: 10.1038/sj.emboj.7600106
32
GuoFCavenerDR. The GCN2 eIF2alpha kinase regulates fatty-acid homeostasis in the liver during deprivation of an essential amino acid. Cell Metab. (2007) 5:103–14. doi: 10.1016/j.cmet.2007.01.001
33
PanwarVSinghABhattMTonkRKAzizovSRazaASet al. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct Target Ther. (2023) 8:375. doi: 10.1038/s41392-023-01608-z
34
HagiwaraACornuMCybulskiNPolakPBetzCTrapaniFet al. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. (2012) 15:725–38. doi: 10.1016/j.cmet.2012.03.015
35
SundqvistABengoechea-AlonsoMTYeXLukiyanchukVJinJHarperJWet al. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab. (2005) 1:379–91. doi: 10.1016/j.cmet.2005.04.010
36
YeciesJLZhangHHMenonSLiuSYeciesDLipovskyAIet al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. (2011) 14:21–32. doi: 10.1016/j.cmet.2011.06.002
37
PorstmannTSantosCRGriffithsBCullyMWuMLeeversSet al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. (2008) 8:224–36. doi: 10.1016/j.cmet.2008.07.007
38
PetersonTRSenguptaSSHarrisTECarmackAEKangSABalderasEet al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. (2011) 146:408–20. doi: 10.1016/j.cell.2011.06.034
39
HanJLiEChenLZhangYWeiFLiuJet al. The CREB coactivator CRTC2 controls hepatic lipid metabolism by regulating SREBP1. Nature. (2015) 524:243–6. doi: 10.1038/nature14557
40
ImSSYousefLBlaschitzCLiuJZEdwardsRAYoungSGet al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. (2011) 13:540–9. doi: 10.1016/j.cmet.2011.04.001
41
ShimanoHHortonJDShimomuraIHammerREBrownMSGoldsteinJL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest. (1997) 99:846–54. doi: 10.1172/JCI119248
42
TothJIDattaSAthanikarJNFreedmanLPOsborneTF. Selective coactivator interactions in gene activation by SREBP-1a and -1c. Mol Cell Biol. (2004) 24:8288–300. doi: 10.1128/MCB.24.18.8288-8300.2004
43
ShimanoHHortonJDHammerREShimomuraIBrownMSGoldsteinJL. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J Clin Invest. (1996) 98:1575–84. doi: 10.1172/JCI118951
44
SzántóMGupteRKrausWLPacherPBaiP. PARPs in lipid metabolism and related diseases. Prog Lipid Res. (2021) 84:101117. doi: 10.1016/j.plipres.2021.101117
45
Amemiya-KudoMShimanoHHastyAHYahagiNYoshikawaTMatsuzakaTet al. Transcriptional activities of nuclear SREBP-1a, -1c, and -2 to different target promoters of lipogenic and cholesterogenic genes. J Lipid Res. (2002) 43:1220–35. doi: 10.1194/jlr.M100417-JLR200
46
OlinerJDAndresenJMHansenSKZhouSTjianR. SREBP transcriptional activity is mediated through an interaction with the CREB-binding protein. Genes Dev. (1996) 10:2903–11. doi: 10.1101/gad.10.22.2903
47
YangFVoughtBWSatterleeJSWalkerAKJim SunZYWattsJLet al. An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature. (2006) 442:700–4. doi: 10.1038/nature04942
48
BekkeringSArtsRJWNovakovicBKourtzelisIvan der HeijdenCLiYet al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. (2018) 172:135–46.e9. doi: 10.1016/j.cell.2017.11.025
49
YorkAGWilliamsKJArgusJPZhouQDBrarGVergnesLet al. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell. (2015) 163:1716–29. doi: 10.1016/j.cell.2015.11.045
50
AraldiEFernández-FuertesMCanfrán-DuqueATangWClineGWMadrigal-MatuteJet al. Lanosterol modulates TLR4-mediated innate immune responses in macrophages. Cell Rep. (2017) 19:2743–55. doi: 10.1016/j.celrep.2017.05.093
51
KusnadiAParkSHYuanRPannelliniTGiannopoulouEOliverDet al. The cytokine TNF promotes transcription factor SREBP activity and binding to inflammatory genes to activate macrophages and limit tissue repair. Immunity. (2019) 51:241–57.e9. doi: 10.1016/j.immuni.2019.06.005
52
XiaoHLuMLinTYChenZChenGWangWCet al. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation. (2013) 128:632–42. doi: 10.1161/CIRCULATIONAHA.113.002714
53
YehMColeALChoiJLiuYTulchinskyDQiaoJHet al. Role for sterol regulatory element-binding protein in activation of endothelial cells by phospholipid oxidation products. Circ Res. (2004) 95:780–8. doi: 10.1161/01.RES.0000146030.53089.18
54
WillemsenLPrangeKHMNeeleAEvan RoomenCGijbelsMGriffithGRet al. DOT1L regulates lipid biosynthesis and inflammatory responses in macrophages and promotes atherosclerotic plaque stability. Cell Rep. (2022) 41:111703. doi: 10.1016/j.celrep.2022.111703
55
RathinamVAFitzgeraldKA. Inflammasome complexes: emerging mechanisms and effector functions. Cell. (2016) 165:792–800. doi: 10.1016/j.cell.2016.03.046
56
RobinsonLEShridarMSmithPMurrell-LagnadoRD. Plasma membrane cholesterol as a regulator of human and rodent P2X7 receptor activation and sensitization. J Biol Chem. (2014) 289:31983–94. doi: 10.1074/jbc.M114.574699
57
GuoCChiZJiangDXuTYuWWangZet al. Cholesterol homeostatic regulator SCAP-SREBP2 integrates NLRP3 inflammasome activation and cholesterol biosynthetic signaling in macrophages. Immunity. (2018) 49:842–56.e7. doi: 10.1016/j.immuni.2018.08.021
58
ChenYKuHZhaoLWheelerDCLiLCLiQet al. Inflammatory stress induces statin resistance by disrupting 3-hydroxy-3-methylglutaryl-CoA reductase feedback regulation. Arterioscler Thromb Vasc Biol. (2014) 34:365–76. doi: 10.1161/ATVBAHA.113.301301
59
YeQChenYLeiHLiuQMoorheadJFVargheseZet al. Inflammatory stress increases unmodified LDL uptake via LDL receptor: an alternative pathway for macrophage foam-cell formation. Inflammation Res. (2009) 58:809–18. doi: 10.1007/s00011-009-0052-4
60
MaKLRuanXZPowisSHChenYMoorheadJFVargheseZ. Inflammatory stress exacerbates lipid accumulation in hepatic cells and fatty livers of apolipoprotein E knockout mice. Hepatology. (2008) 48:770–81. doi: 10.1002/hep.v48:3
61
LiLCVargheseZMoorheadJFLeeCTChenJBRuanXZ. Cross-talk between TLR4-MyD88-NF-κB and SCAP-SREBP2 pathways mediates macrophage foam cell formation. Am J Physiol Heart Circ Physiol. (2013) 304:H874–84. doi: 10.1152/ajpheart.00096.2012
62
OuyangNGanHHeQLeiHWangSYLiuQet al. Dysfunction of cholesterol sensor SCAP promotes inflammation activation in THP-1 macrophages. Exp Cell Res. (2018) 367:162–9. doi: 10.1016/j.yexcr.2018.03.032
63
LiDLiuMLiZZhengGChenAZhaoLet al. Sterol-resistant SCAP overexpression in vascular smooth muscle cells accelerates atherosclerosis by increasing local vascular inflammation through activation of the NLRP3 inflammasome in mice. Aging Dis. (2021) 12:747–63. doi: 10.14336/AD.2020.1120
64
HuangXYaoYHouXWeiLRaoYSuYet al. Macrophage SCAP contributes to metaflammation and lean NAFLD by activating STING-NF-κB signaling pathway. Cell Mol Gastroenterol Hepatol. (2022) 14:1–26. doi: 10.1016/j.jcmgh.2022.03.006
65
PanYChenHZhangXLiuWDingYHuangDet al. METTL3 drives NAFLD-related hepatocellular carcinoma and is a therapeutic target for boosting immunotherapy. Cell Rep Med. (2023) 4:101144. doi: 10.1016/j.xcrm.2023.101144
66
MenendezJALupuR. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. (2007) 7:763–77. doi: 10.1038/nrc2222
67
GuoDBellEHMischelPChakravartiA. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharm Des. (2014) 20:2619–26. doi: 10.2174/13816128113199990486
68
RicoultSJYeciesJLBen-SahraIManningBD. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene. (2016) 35:1250–60. doi: 10.1038/onc.2015.179
69
Freed-PastorWAMizunoHZhaoXLangerødAMoonSHRodriguez-BarruecoRet al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. (2012) 148:244–58. doi: 10.1016/j.cell.2011.12.017
70
TeresiREPlanchonSMWaiteKAEngC. Regulation of the PTEN promoter by statins and SREBP. Hum Mol Genet. (2008) 17:919–28. doi: 10.1093/hmg/ddm364
71
BathaieSZAshrafiMAzizianMTamanoiF. Mevalonate pathway and human cancers. Curr Mol Pharmacol. (2017) 10:77–85. doi: 10.2174/1874467209666160112123205
72
ShammaATakegamiYMikiTKitajimaSNodaMObaraTet al. Rb Regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer Cell. (2009) 15:255–69. doi: 10.1016/j.ccr.2009.03.001
73
HerradaAAEscobedoNIruretagoyenaMValenzuelaRABurgosPICuitinoLet al. Innate immune cells' Contribution to systemic lupus erythematosus. Front Immunol. (2019) 10:772. doi: 10.3389/fimmu.2019.00772
74
GoulielmosGNZervouMIVazgiourakisVMGhodke-PuranikYGaryfallosANiewoldTB. The genetics and molecular pathogenesis of systemic lupus erythematosus (SLE) in populations of different ancestry. Gene. (2018) 668:59–72. doi: 10.1016/j.gene.2018.05.041
75
LiangYXieSBWuCHHuYZhangQLiSet al. Coagulation cascade and complement system in systemic lupus erythematosus. Oncotarget. (2018) 9:14862–81. doi: 10.18632/oncotarget.v9i19
76
TsokosGC. Systemic lupus erythematosus. N Engl J Med. (2011) 365:2110–21. doi: 10.1056/NEJMra1100359
77
GondretFFerréPDugailI. ADD-1/SREBP-1 is a major determinant of tissue differential lipogenic capacity in mammalian and avian species. J Lipid Res. (2001) 42:106–13. doi: 10.1016/S0022-2275(20)32341-5
78
AraiTKimHJChibaHMatsumotoA. Anti-obesity effect of fish oil and fish oil-fenofibrate combination in female KK mice. J Atheroscler Thromb. (2009) 16:674–83. doi: 10.5551/jat.1313
79
VanarsaKYeYHanJXieCMohanCWuT. Inflammation associated anemia and ferritin as disease markers in SLE. Arthritis Res Ther. (2012) 14:R182. doi: 10.1186/ar4012
80
UrregoTOrtiz-ReyesBVanegas-GarcíaALMuñozCHGonzálezLAVásquezGet al. Utility of urinary transferrin and ceruloplasmin in patients with systemic lupus erythematosus for differentiating patients with lupus nephritis. Reumatol Clin (Engl Ed). (2020) 16:17–23. doi: 10.1016/j.reuma.2018.02.002
81
BrownAC. Lupus erythematosus and nutrition: a review of the literature. J Ren Nutr. (2000) 10:170–83. doi: 10.1053/jren.2000.16323
82
NankivellBJBoadleRAHarrisDC. Iron accumulation in human chronic renal disease. Am J Kidney Dis. (1992) 20:580–4. doi: 10.1016/S0272-6386(12)70222-6
83
van RaaijSvan SwelmRBoumanKCliteurMvan den HeuvelMCPertijsJet al. Tubular iron deposition and iron handling proteins in human healthy kidney and chronic kidney disease. Sci Rep. (2018) 8:9353. doi: 10.1038/s41598-018-27107-8
84
LeiterLMReuhlKRRacisSPJr.ShermanAR. Iron status alters murine systemic lupus erythematosus. J Nutr. (1995) 125:474–84. doi: 10.1093/jn/125.3.474
85
MarksESBonnemaisonMLBrusnahanSKZhangWFanWGarrisonJCet al. Renal iron accumulation occurs in lupus nephritis and iron chelation delays the onset of albuminuria. Sci Rep. (2017) 7:12821. doi: 10.1038/s41598-017-13029-4
86
ScIndiaYWlazloEGhiasECechovaSLoiVLeedsJet al. Modulation of iron homeostasis with hepcidin ameliorates spontaneous murine lupus nephritis. Kidney Int. (2020) 98:100–15. doi: 10.1016/j.kint.2020.01.025
87
WlazloEMehradBMorelLScIndiaY. Iron metabolism: an under investigated driver of renal pathology in lupus nephritis. Front Med (Lausanne). (2021) 8:643686. doi: 10.3389/fmed.2021.643686
88
ChenHQiQWuNWangYFengQJinRet al. Aspirin promotes RSL3-induced ferroptosis by suppressing mTOR/SREBP-1/SCD1-mediated lipogenesis in PIK3CA-mutant colorectal cancer. Redox Biol. (2022) 55:102426. doi: 10.1016/j.redox.2022.102426
89
KimJGuptaRBlancoLPYangSShteinfer-KuzmineAWangKet al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science. (2019) 366:1531–6. doi: 10.1126/science.aav4011
90
ZhaoLHuXXiaoFZhangXZhaoLWangM. Mitochondrial impairment and repair in the pathogenesis of systemic lupus erythematosus. Front Immunol. (2022) 13:929520. doi: 10.3389/fimmu.2022.929520
91
CardielMHTumlinJAFurieRAWallaceDJJohTLinnikMD. Abetimus sodium for renal flare in systemic lupus erythematosus: results of a randomized, controlled phase III trial. Arthritis Rheumatol. (2008) 58:2470–80. doi: 10.1002/art.23673
92
DongJWangQXZhouCYMaXFZhangYC. Activation of the STAT1 signalling pathway in lupus nephritis in MRL/lpr mice. Lupus. (2007) 16:101–9. doi: 10.1177/0961203306075383
93
LarreaEAldabeRMolanoEFernandez-RodriguezCMAmetzazurraACiveiraMPet al. Altered expression and activation of signal transducers and activators of transcription (STATs) in hepatitis C virus infection: in vivo and in vitro studies. Gut. (2006) 55:1188–96. doi: 10.1136/gut.2005.070060
94
LetexierDPinteurCLargeVFréringVBeylotM. Comparison of the expression and activity of the lipogenic pathway in human and rat adipose tissue. J Lipid Res. (2003) 44:2127–34. doi: 10.1194/jlr.M300235-JLR200
95
BaziotiVHalmosBWesterterpM. T-cell cholesterol accumulation, aging, and atherosclerosis. Curr Atheroscler Rep. (2023) 25:527–34. doi: 10.1007/s11883-023-01125-y
96
YangWBaiYXiongYZhangJChenSZhengXet al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature. (2016) 531:651–5. doi: 10.1038/nature17412
97
ChengHYGaddisDEWuRMcSkimmingCHaynesLDTaylorAMet al. Loss of ABCG1 influences regulatory T cell differentiation and atherosclerosis. J Clin Invest. (2016) 126:3236–46. doi: 10.1172/JCI83136
98
PollardKMCauviDMToomeyCBMorrisKVKonoDH. Interferon-γ and systemic autoimmunity. Discovery Med. (2013) 16:123–31.
99
ZhangJRColemanTLangmadeSJScherrerDELaneLLanierMHet al. Niemann-Pick C1 protects against atherosclerosis in mice via regulation of macrophage intracellular cholesterol trafficking. J Clin Invest. (2008) 118:2281–90. doi: 10.1172/JCI32561
100
DomeierPPChodisettiSBSoniCSchellSLEliasMJWongEBet al. IFN-γ receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J Exp Med. (2016) 213:715–32. doi: 10.1084/jem.20151722
101
MantovaniASicaASozzaniSAllavenaPVecchiALocatiM. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
102
LabonteACKegerreisBGeraciNSBachaliPMadamanchiSRoblRet al. Identification of alterations in macrophage activation associated with disease activity in systemic lupus erythematosus. PloS One. (2018) 13:e0208132. doi: 10.1371/journal.pone.0208132
103
IwataYBoströmEAMenkeJRabacalWAMorelLWadaTet al. Aberrant macrophages mediate defective kidney repair that triggers nephritis in lupus-susceptible mice. J Immunol. (2012) 188:4568–80. doi: 10.4049/jimmunol.1102154
104
JingCCastro-DopicoTRichozNTuongZKFerdinandJRLokLSCet al. Macrophage metabolic reprogramming presents a therapeutic target in lupus nephritis. Proc Natl Acad Sci U S A. (2020) 117:15160–71. doi: 10.1073/pnas.2000943117
105
SparksJA. Rheumatoid arthritis. Ann Intern Med. (2019) 170:Itc1–itc16. doi: 10.7326/AITC201901010
106
FinckhAGilbertBHodkinsonBBaeSCThomasRDeaneKDet al. Global epidemiology of rheumatoid arthritis. Nat Rev Rheumatol. (2022) 18:591–602. doi: 10.1038/s41584-022-00827-y
107
MetawiSAAbbasDKamalMMIbrahimMK. Serum and synovial fluid levels of interleukin-17 in correlation with disease activity in patients with RA. Clin Rheumatol. (2011) 30:1201–7. doi: 10.1007/s10067-011-1737-y
108
KoendersMIMarijnissenRJDevesaILubbertsEJoostenLARothJet al. Tumor necrosis factor-interleukin-17 interplay induces S100A8, interleukin-1β, and matrix metalloproteinases, and drives irreversible cartilage destruction in murine arthritis: rationale for combination treatment during arthritis. Arthritis Rheumatol. (2011) 63:2329–39. doi: 10.1002/art.30418
109
GenoveseMCVan den BoschFRobersonSABojinSBiaginiIMRyanPet al. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheumatol. (2010) 62:929–39. doi: 10.1002/art.27334
110
ChengYManabeIHayakawaSEndoYOishiY. Caspase-11 contributes to site-1 protease cleavage and SREBP1 activation in the inflammatory response of macrophages. Front Immunol. (2023) 14:1009973. doi: 10.3389/fimmu.2023.1009973
111
Jadidi-NiaraghFMirshafieyA. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand J Immunol. (2011) 74:1–13. doi: 10.1111/j.1365-3083.2011.02536.x
112
LaBrancheTPJessonMIRadiZAStorerCEGuzovaJABonarSLet al. JAK inhibition with tofacitinib suppresses arthritic joint structural damage through decreased RANKL production. Arthritis Rheumatol. (2012) 64:3531–42. doi: 10.1002/art.34649
113
MadanBGohKCHartSWilliamADJayaramanREthirajuluKet al. SB1578, a novel inhibitor of JAK2, FLT3, and c-Fms for the treatment of rheumatoid arthritis. J Immunol. (2012) 189:4123–34. doi: 10.4049/jimmunol.1200675
114
MoonYMLeeJLeeSYHerYMRyuJGKimEKet al. Gene associated with retinoid-interferon-induced mortality 19 attenuates murine autoimmune arthritis by regulation of th17 and treg cells. Arthritis Rheumatol. (2014) 66:569–78. doi: 10.1002/art.38267
115
ParkJSLeeJLimMAKimEKKimSMRyuJGet al. JAK2-STAT3 blockade by AG490 suppresses autoimmune arthritis in mice via reciprocal regulation of regulatory T Cells and Th17 cells. J Immunol. (2014) 192:4417–24. doi: 10.4049/jimmunol.1300514
116
ChoSOLimJWKimH. Red ginseng extract inhibits the expression of MCP-1 and iNOS in Helicobacter pylori-infected gastric epithelial cells by suppressing the activation of NADPH oxidase and Jak2/Stat3. J Ethnopharmacol. (2013) 150:761–4. doi: 10.1016/j.jep.2013.09.013
117
SchwarzABonaterraGASchwarzbachHKinscherfR. Oxidized LDL-induced JAB1 influences NF-κB independent inflammatory signaling in human macrophages during foam cell formation. J BioMed Sci. (2017) 24:12. doi: 10.1186/s12929-017-0320-5
118
Aguirre-RuedaDGuerra-OjedaSAldasoroMIradiAObradorEOrtegaAet al. Astrocytes protect neurons from Aβ1-42 peptide-induced neurotoxicity increasing TFAM and PGC-1 and decreasing PPAR-γ and SIRT-1. Int J Med Sci. (2015) 12:48–56. doi: 10.7150/ijms.10035
119
RutherfordCSpeirsCWilliamsJJEwartMAManciniSJHawleySAet al. Phosphorylation of Janus kinase 1 (JAK1) by AMP-activated protein kinase (AMPK) links energy sensing to anti-inflammatory signaling. Sci Signal. (2016) 9:ra109. doi: 10.1126/scisignal.aaf8566
120
WangDTHeJWuMLiSMGaoQZengQP. Artemisinin mimics calorie restriction to trigger mitochondrial biogenesis and compromise telomere shortening in mice. PeerJ. (2015) 3:e822. doi: 10.7717/peerj.822
121
LiYXuSMihaylovaMMZhengBHouXJiangBet al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. (2011) 13:376–88. doi: 10.1016/j.cmet.2011.03.009
122
LiangJChangBHuangMHuangWMaWLiuYet al. Oxymatrine prevents synovial inflammation and migration via blocking NF-κB activation in rheumatoid fibroblast-like synoviocytes. Int Immunopharmacol. (2018) 55:105–11. doi: 10.1016/j.intimp.2017.12.006
123
PauleyKMSatohMChanALBubbMRReevesWHChanEK. Upregulated miR-146a expression in peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Res Ther. (2008) 10:R101. doi: 10.1186/ar2493
124
FeiXHuangJLiFWangYShaoZDongLet al. The Scap-SREBP1-S1P/S2P lipogenesis signal orchestrates the homeostasis and spatiotemporal activation of NF-κB. Cell Rep. (2023) 42:112586. doi: 10.1016/j.celrep.2023.112586
125
VassiliouEFarias-PereiraR. Impact of lipid metabolism on macrophage polarization: implications for inflammation and tumor immunity. Int J Mol Sci. (2023) 24(15):12032. doi: 10.3390/ijms241512032
126
KuanYCHashidumeTShibataTUchidaKShimizuMInoueJet al. Heat shock protein 90 modulates lipid homeostasis by regulating the stability and function of sterol regulatory element-binding protein (SREBP) and SREBP cleavage-activating protein. J Biol Chem. (2017) 292:3016–28. doi: 10.1074/jbc.M116.767277
127
ChengHMXingMZhouYPZhangWLiuZLiLet al. HSP90β promotes osteoclastogenesis by dual-activation of cholesterol synthesis and NF-κB signaling. Cell Death Differ. (2023) 30:673–86. doi: 10.1038/s41418-022-01071-3
128
SoAKMartinonF. Inflammation in gout: mechanisms and therapeutic targets. Nat Rev Rheumatol. (2017) 13:639–47. doi: 10.1038/nrrheum.2017.155
129
DalbethNChoiHKJoostenLABKhannaPPMatsuoHPerez-RuizFet al. Gout. Nat Rev Dis Primers. (2019) 5:69. doi: 10.1038/s41572-019-0115-y
130
DalbethNGoslingALGaffoAAbhishekA. Gout. Lancet. (2021) 397:1843–55. doi: 10.1016/S0140-6736(21)00569-9
131
StampLKDalbethN. Prevention and treatment of gout. Nat Rev Rheumatol. (2019) 15:68–70. doi: 10.1038/s41584-018-0149-7
132
FranchiLEigenbrodTMuñoz-PlanilloRNuñezG. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. (2009) 10:241–7. doi: 10.1038/ni.1703
133
DalbethNMerrimanTRStampLK. Gout. Lancet. (2016) 388:2039–52. doi: 10.1016/S0140-6736(16)00346-9
134
RuytinxPProostPVan DammeJStruyfS. Chemokine-induced macrophage polarization in inflammatory conditions. Front Immunol. (2018) 9:1930. doi: 10.3389/fimmu.2018.01930
135
MichaudelCSokolH. The gut microbiota at the service of immunometabolism. Cell Metab. (2020) 32:514–23. doi: 10.1016/j.cmet.2020.09.004
136
AhangariFPriceNLMalikSChioccioliMBärnthalerTAdamsTSet al. microRNA-33 deficiency in macrophages enhances autophagy, improves mitochondrial homeostasis, and protects against lung fibrosis. JCI Insight. (2023) 8(4):e158100. doi: 10.1172/jci.insight.158100
137
Najafi-ShoushtariSHKristoFLiYShiodaTCohenDEGersztenREet al. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. (2010) 328:1566–9. doi: 10.1126/science.1189123
138
LeeJHLeeSHLeeEHChoJYSongDKLeeYJet al. SCAP deficiency facilitates obesity and insulin resistance through shifting adipose tissue macrophage polarization. J Adv Res. (2023) 45:1–13. doi: 10.1016/j.jare.2022.05.013
139
BeharkaAACrowtherJEMcCormackFXDenningGMLeesJTibesarEet al. Pulmonary surfactant protein A activates a phosphatidylinositol 3-kinase/calcium signal transduction pathway in human macrophages: participation in the up-regulation of mannose receptor activity. J Immunol. (2005) 175:2227–36. doi: 10.4049/jimmunol.175.4.2227
140
ChoulakiCPapadakiGRepaAKampourakiEKambasKRitisKet al. Enhanced activity of NLRP3 inflammasome in peripheral blood cells of patients with active rheumatoid arthritis. Arthritis Res Ther. (2015) 17:257. doi: 10.1186/s13075-015-0775-2
141
DunneARossPJPospisilovaEMasinJMeaneyASuttonCEet al. Inflammasome activation by adenylate cyclase toxin directs Th17 responses and protection against Bordetella pertussis. J Immunol. (2010) 185:1711–9. doi: 10.4049/jimmunol.1000105
142
KoolMWillartMAvan NimwegenMBergenIPouliotPVirchowJCet al. An unexpected role for uric acid as an inducer of T helper 2 cell immunity to inhaled antigens and inflammatory mediator of allergic asthma. Immunity. (2011) 34:527–40. doi: 10.1016/j.immuni.2011.03.015
143
DaiXJTaoJHFangXXiaYLiXMWangYPet al. Changes of treg/th17 ratio in spleen of acute gouty arthritis rat induced by MSU crystals. Inflammation. (2018) 41:1955–64. doi: 10.1007/s10753-018-0839-y
144
MaynardCLWeaverCT. Intestinal effector T cells in health and disease. Immunity. (2009) 31:389–400. doi: 10.1016/j.immuni.2009.08.012
145
WenXLouYSongSHeZChenJXieZet al. Qu-zhuo-tong-bi decoction alleviates gouty arthritis by regulating butyrate-producing bacteria in mice. Front Pharmacol. (2020) 11:610556. doi: 10.3389/fphar.2020.610556
146
WangXSunGFengTZhangJHuangXWangTet al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer's disease progression. Cell Res. (2019) 29:787–803. doi: 10.1038/s41422-019-0216-x
Glossary
AMPKadenine mononucleotide activated protein kinaseAKTprotein kinase BBMDMbone marrow-derived macrophageCREB1cAMP responsive element binding protein1CSF-1colony-stimulating factor-1GCgerminal centerGTPguanosine triphosphateHIF-1αhypoxia-inducible factor-1αHSP90heat shock protein 90IFN-γinterferon-γIL-1βinterleukin-1βIL-4interleukin-4INSIGsinsulin -induced gene proteinIκBNF-κB inhibitor proteinsJAK-STATjanus kinase-signal transducer and activator of transcriptionKRASkirsten rat sarcoma viral oncogene homologLECslymphatic endothelial cellsLDLRlow density lipoprotein receptorLPSlipopolysaccharideM1macrophages can polarize to inflammatory phenotypemTORmammalian target of rapamycinMyD88myeloiddifferentiation factor88MSUmonosodiumuratecrystalsNF-κBnuclear factor-κBNLRP3nucleotide-binding oligomerization domain, leucine- rich repeat and pyrin domain-containing 3NRASneuroblastoma RAS viral oncogene homologOXPHOSoxidative phosphorylationPI3Kphosphatidylinositol 3-kinasePTENphosphatase and tensin homologRArheumatoid arthritisSCAPSREBP cleavage activating proteinSLEsystemic lupus erythematosusSREBPssterol regulatory element binding proteinsSTINGstimulator of interferon genesTACtricarboxylic acidTBK1TANK-binding kinase 1TLR4toll-like receptor 425/27-HC25-hydroxycholesterol and 27-hydroxycholesterol
Summary
Keywords
sterol regulatory element binding proteins, cytokine storm, autoantibodies, autoimmune rheumatic diseases, inflammation, immune cells
Citation
Xu X, Jin W, Chang R and Ding X (2024) Research progress of SREBP and its role in the pathogenesis of autoimmune rheumatic diseases. Front. Immunol. 15:1398921. doi: 10.3389/fimmu.2024.1398921
Received
11 March 2024
Accepted
31 July 2024
Published
19 August 2024
Volume
15 - 2024
Edited by
Monica Neagu, Victor Babes National Institute of Pathology (INCDVB), Romania
Reviewed by
Angela Bonura, National Research Council (CNR), Italy
Xuejun Gao, Yangtze University, China
Updates
Copyright
© 2024 Xu, Jin, Chang and Ding.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinghong Ding, dxh@zcmu.edu.cn
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.