 Zinat Sharmin1
Zinat Sharmin1 Kehua Jin1,2Ai-Yu Gong1
Kehua Jin1,2Ai-Yu Gong1 Silu Deng1Chansorena Pok1Marion L. Graham1Shuhong Wang1Nicholas W. Mathy3
Silu Deng1Chansorena Pok1Marion L. Graham1Shuhong Wang1Nicholas W. Mathy3 Annemarie Shibata4
Annemarie Shibata4 Xian-Ming Chen1*
Xian-Ming Chen1*- 1Department of Microbial Pathogens and Immunity, Rush University Medical Center, Chicago, IL, United States
- 2Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Hubei University of Science and Technology, Xianning, Hubei, China
- 3Department of Medical Microbiology and Immunology, Creighton University School of Medicine, Omaha, NE, United States
- 4Department of Biology, Creighton University College of Arts and Sciences, Creighton University, Omaha, NE, United States
Intestinal epithelial cells possess the requisite molecular machinery to initiate cell-intrinsic defensive responses against intracellular pathogens, including intracellular parasites. Interferons(IFNs) have been identified as cornerstones of epithelial cell-intrinsic defense against such pathogens in the gastrointestinal tract. Long non-coding RNAs (lncRNAs) are RNA transcripts (>200 nt) not translated into protein and represent a critical regulatory component of mucosal defense. We report here that lncRNA Nostrill facilitates IFN-γ-stimulated intestinal epithelial cell-intrinsic defense against infection by Cryptosporidium, an important opportunistic pathogen in AIDS patients and a common cause of diarrhea in young children. Nostrill promotes transcription of a panel of genes controlled by IFN-γ through facilitating Stat1 chromatin recruitment and thus, enhances expression of several genes associated with cell-intrinsic defense in intestinal epithelial cells in response to IFN-γ stimulation, including Igtp, iNos, and Gadd45g. Induction of Nostrill enhances IFN-γ-stimulated intestinal epithelial defense against Cryptosporidium infection, which is associated with an enhanced autophagy in intestinal epithelial cells. Our findings reveal that Nostrill enhances the transcription of a set of genes regulated by IFN-γ in intestinal epithelial cells. Moreover, induction of Nostrill facilitates the IFN-γ-mediated epithelial cell-intrinsic defense against cryptosporidial infections.
Introduction
Cell-intrinsic immunity denotes the capacity of a host cell to eliminate an invasive/intracellular infectious agent at the cellular level which is a first line of host defense against intracellular pathogens including intracellular parasites (1). Intestinal epithelial cells are equipped with the necessary molecular machinery to mount cell-intrinsic defensive responses by themselves. Activation of pattern recognition receptors in intestinal epithelial cells leads to the upregulation of antimicrobial factors, secretion of cytokines and chemokines, and priming of immune cells for direct antimicrobial action or for guiding adaptive immune responses. On the other hand, intestinal epithelial cells serve as targets of mucosal immune mediators released from immune cells residing in the mucosa (2). IFNs have been identified as key elements of epithelial cell-intrinsic defense against intracellular pathogens in both mice and humans (3, 4).
The IFN family can typically be categorized into three main types: type I (e.g., IFN-α and IFN-β), type II (IFN-γ), and type III (IFN-λ family) (5). Type I IFNs bind to the conserved receptor IFNAR1/2 to induce transcription of type I IFN-stimulated genes (6). Although type III IFNs bind to the IFNLR1/IL-10RB receptor, they induce highly similar transcriptional responses as type I IFNs (5). IFN-γ binds to the heterodimeric receptor of IFNGR1 and IFNGR2, initiating the transcription of IFN-γ-stimulated genes (ISGs) (5). Canonical IFN-γ signaling employs the JAK/STAT pathway, leading to the activation of STAT1 and subsequent formation of active STAT1 homodimers. These homodimers then bind to gamma-activated sites (GASs) located in the promoters or enhancers of ISGs (6, 7). While the activation of various types of IFN signaling pathways is not mutually exclusive, mounting evidence suggests that IFN-γ plays a crucial role in anti-bacterial and anti-parasite immunity, whereas type I and type III IFNs are primarily associated with anti-viral immunity (3–6). Nonetheless, dysregulation of IFN signaling pathways can have detrimental effects on host defense mechanisms (7, 8). Therefore, activation of the IFN signal pathway is finely controlled and crosstalk between various types of IFN signaling has been demonstrated (6–9). Intestinal epithelial cells have the capacity to generate various type I and III IFNs, exerting autocrine effects (9). Although intestinal epithelial cells themselves do not produce IFN-γ, it is well established that IFN-γ produced from immune cells residing at the intestinal epithelium is essential to intestinal epithelial antimicrobial defense (10). IFN-γ triggers a broad spectrum of cell intrinsic responses aimed at combating intracellular pathogens through mechanisms like nutrient deprivation and the production of potent defense molecules, including reactive oxygen and nitrogen species, immunity-related GTPases (IRGs), and guanylate-binding proteins (GBPs) (3, 4, 8–10). However, numerous intracellular pathogens have developed sophisticated strategies to escape the immune response (11, 12). The specific regulatory components that govern IFN-γ-mediated defense by intestinal epithelial cells against diverse pathogens remain elusive.
Enteric infections caused by a growing spectrum of bacterial, parasitic, and viral pathogens exert significant impacts on intestinal absorption, nutritional status, childhood development, and contribute substantially to global mortality rates (13). Cryptosporidium, a coccidian parasite and an NIAID Category B priority pathogen, infects the intestinal epithelium and is a leading cause of infectious diarrhea and diarrheal-related death in children worldwide (14–16). Moreover, this infection can precipitate life-threatening diarrheal illnesses in individuals with AIDS (16). The majority of human Cryptosporidium infections are caused by two species: C. parvum and C. hominis (14), through ingesting Cryptosporidium oocysts. After excystation in the gastrointestinal tract to release infective sporozoites, each sporozoite then attaches to the apical membrane of intestinal epithelial cells and forms a parasitophorous vacuole, preventing further infection of other cell types (14). Consequently, intestinal epithelial cells serve as the frontline defense, with epithelial cell-intrinsic defense mechanisms playing a pivotal role in initiating, modulating, and resolving both innate and adaptive immune responses to Cryptosporidium infection (17, 18). IFN-γ is a key regulator of cell-intrinsic defense to Cryptosporidium infection (17, 18).
Long non-coding RNAs (lncRNAs), RNA transcripts exceeding 200 nucleotides in length, are notable for their non-protein-coding nature, yet many exhibit functional roles (19, 20). Accumulating evidence underscores the critical regulatory contribution of lncRNAs to mucosal defense mechanisms (21, 22). In our previous studies, we identified a lncRNA originating from the 2500002B13Rik gene, which we termed Nostrill (iNOS transcriptional regulatory intergenic lncRNA locus) (23). We found that Nostrill may facilitate intestinal epithelial defense against C. parvum through regulation of the interferon response, albeit the underlying mechanisms remained elusive (24). In this study, we explore the role for Nostrill in IFN-γ-stimulated gene transcription and antimicrobial defense. Our findings indicate that Nostrill promotes the transcription of a subset of genes stimulated by IFN-γ and that the induction of Nostrill enhances the IFN-γ-mediated intestinal epithelial cell-intrinsic defense against Cryptosporidium infection.
Results
Nostrill knockdown alters gene expression profiles in intestinal epithelium following Cryptosporidium infection
Several signaling pathways associated with cell-intrinsic defense are activated in the intestinal epithelium against Cryptosporidium following its infection, including the TLR/NF-кB signaling and IFN signaling (17, 18). Whereas signaling of all IFN types are activated in the intestine during in vivo Cryptosporidium infection, only IFN-I and IFN-III are activated in vitro infection models using intestinal epithelial cells because these cells in culture themselves cannot produce IFN-γ (10, 17, 18). In our previous studies, we demonstrated that knockdown of the lncRNA Nostrill using siRNA leads to a higher parasite burden of Cryptosporidium in intestinal epithelial cells (24). This suggested that Nostrill might play a role in the cell-intrinsic anti-parasitic defense. Expanding on this, we carried out comprehensive study by examining the transcriptome of cultured mouse intestinal epithelial IEC4.1 cells cells following C. parvum infection at a genome-wide level. These cells were subjected to treatment with a pool of siRNAs to knockdown Nostrill (referred as siR_Nostrill), followed by exposure to C. parvum infection for 24h. Consistent with our previous observations (25), siRNA knockdown of Nostrill in IEC4.1 cells increased the parasite infection burden (Supplementary Figure 1). The infection also triggered substantial changes in the gene expression patterns in IEC4.1 cells. Specifically, while comparing the gene expression between cells treated with siR_Ctrl (scramble siRNA control) with or without C. parvum infection, we identified a significant alteration in the expression levels of 11,761 genes. This shift encompassed both downregulation (5,590 genes) and upregulation (6,171 genes), demonstrating statistical significance at p<0.05 and a fold change >1 (Figure 1A). Intriguingly, the extent of gene alterations escalated further in infected cells subjected to siR_Nostrill treatment, revealing a higher count of changes. This encompassed both downregulated (5,791 genes) and upregulated (7,174 genes) entities, all adhering to the same rigorous statistical criteria (Figure 1B).
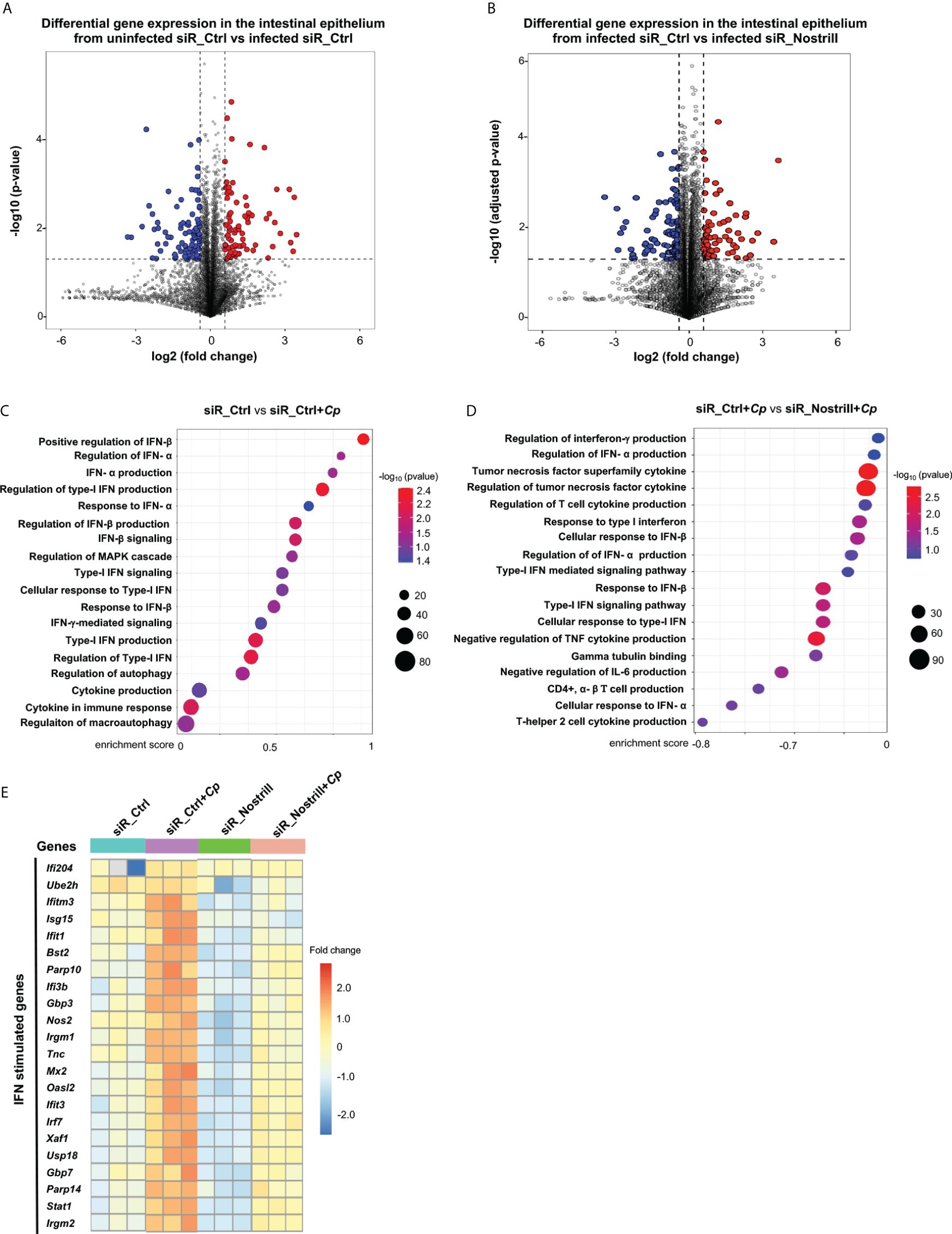
Figure 1 Transcriptomic responses in Cryptosporidium-infected IEC4.1 cells with Nostrill knockdown. (A, B) Volcano plots illustrating the differential gene expression between two groups. Statistical analysis utilized two-tailed Wald tests. The dashed line represents a false discovery rate cutoff of 0.05. Data were derived from three biological replicates for each group (p < 0.05 with a fold change > 1). Differentially expressed genes between infected siR_Ctrl (24h post-infection) versus uninfected siR_Ctrl (non-specific control siRNA) (A) and between infected siR_Nostrill versus infected siR_Ctrl (siRNA to Nostrill) (B) are shown. (C, D) Gene Set Enrichment Analysis (GSEA) depicting immune defense-related pathways in IEC4.1 cells from infected siR_Ctrl (24h post-infection) compared to uninfected siR_Ctrl (C) and in infected siR_Ctrl compared to infected siR_Nostrill (24h post-infection) (D). p-values were calculated using the Kolmogorov–Smirnov test and adjusted via the Benjamini–Hochberg method. Enrichment scores and -log10 p-values for signaling pathways are presented. Red dots indicate smaller p-values, while blue dots indicate larger p-values. (E) Heatmap displaying representative transcriptional expression patterns for interferon-stimulated genes (ISGs) across different groups.
Next, through Gene Set Enrichment Analysis (GSEA), we investigated the gene expression profiles in IEC4.1 cells treated with the siR_Ctrl upon infection. In consistent with results from previous studies (17, 18), our analysis revealed activation of several innate defense pathways at the cellular level, such as IFN-mediated signaling and other cytokine-mediated signaling, in the infected cells (Figure 1C). Nonetheless, it was observed that pathways related to cellular defense appeared to be suppressed in cells treated with siR_Nostrill following infection, compared with infected cells treated with siR_Ctrl (Figure 1D). Genes whose expression levels were significantly altered and differentially expressed between siR_Ctrl-treated IEC4.1 and siR_Nostrill-treated IEC4.1 cells following infection were listed in Supplementary Table 1. It’s noteworthy that many of the well-known ISGs exhibited significant changes unique to each sample type. To visualize these changes, we employed unsupervised hierarchical clustering analysis, which generated a heat map illustrating the expression patterns of these differentially expressed immune defense genes, including many ISGs (Figure 1E). Intriguingly, genes such as Ifi204, Irgm1/2, Nos2, Gbp3/7, Usp18, Irf7, Stat1, and Isg15, which were upregulated in response to infection in cells treated with siR_Ctrl, showed decreased upregulation in cells treated with siR_Nostrill following infection. These findings highlight distinct gene expression profiles between infected siR_Ctrl-treated and siR_Nostrill-treated IEC4.1 cells, indicating a compromised cell-intrinsic defense response in cells treated with siR_Nostrill.
Nostrill modulates the gene expression pattern of intestinal epithelial cells following IFN-γ stimulation
We then asked whether Nostrill induction acts as a host defense response to Cryptosporidium infection and can modulate IFN-mediated cell-intrinsic anti-parasitic defense in intestinal epithelial cells. Whereas signaling of all IFN types are activated in the intestine during in vivo Cryptosporidium infection, only IFN-I and IFN-III are activated in vitro infection models using intestinal epithelial cells because these cells in culture themselves cannot produce IFN-γ (10). Taken advantage on the absence of IFN-γ in the in vitro infection models, we investigated the impact of Nostrill induction on cell-intrinsic anti-parasitic defense in response to exogenous IFN-γ using our in vitro IEC4.1 cell infection model. We first performed a genome-wide transcriptome analysis of cultured IEC4.1 cells treated with the siR_Ctrl or siR_Nostrill in response to IFN-γ. Upon comparing the gene expression patterns between the siR_Ctrl-treated cells with and without IFN-γ stimulation, we have identified many genes that displayed significant differential expression (p<0.05, fold change > 0.5) (Figure 2A). Most of these genes are upregulated, confirming the impact of IFN-γ treatment on the cells in consistent with previous studies (26–28). When comparing the basal gene expression levels between the siR_Ctrl- and siR_Nostrill-treated cells, without IFN-γ treatment, we observed 186 genes that exhibited differential expression (Figure 2B). Nevertheless, when comparing expression levels of genes in the siR_Ctrl + IFN-γ-treated cells versus siR_Nostrill + IFN-γ-treated cells, a total of 1,559 genes showed significant differential expression, indicating a notable impact of Nostrill on the gene expression induced by IFN-γ treatment (Figure 2C). Out of these genes, 883 were genes whose expression levels were significantly lower in the siR_Nostrill + IFN-γ-treated cells than that in the siR_Ctrl + IFN-γ-treated cells (Figure 2C). The normalized transcript expression levels of selected genes out of these 883 genes were shown in Figure 2D, including Igtp, iNos, and Gadd45g. The complete dataset, containing a comprehensive list of genes showing differential expression between these groups, is available in the Supplementary Table 2.
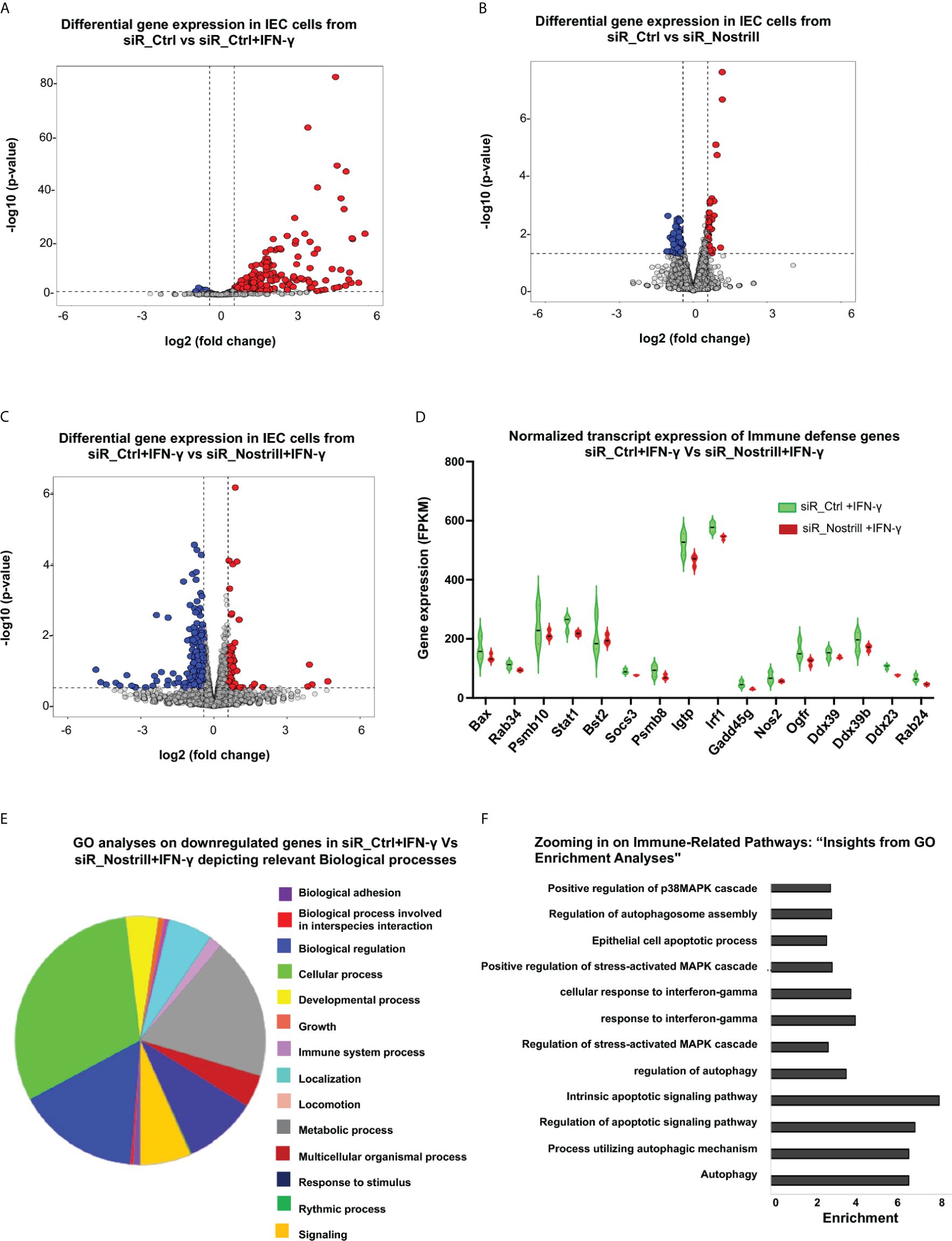
Figure 2 Impact of Nostrill on gene expression in intestinal epithelial cells upon IFN-γ stimulation. (A-C) Volcano plots illustrating the differential gene expression between two groups. Statistical analysis utilized two-tailed Wald tests. The dashed line represents a false discovery rate cutoff of 0.05. Data were derived from three biological replicates for each group (p < 0.05 with a fold change > 0.5). Differentially expressed genes between cells treated with siR_Ctrl + IFN‐γ (4 h post-treatment at a dose of 1 ng/ml) versus with siR_Ctrl (A, B) cells treated with siR_Nostrill versus siR_Ctrl (without IFN-γ treatment), and cells treated with siR_Nostrill + IFN‐γ (4 h post-treatment at a dose of 1 ng/mL) versus with siR_Ctrl + IFN‐γ (C) are shown. (D) Violin plot depicting the comparison of normalized transcript expression of host defense genes between siR_Ctrl- or siR_Nostrill-treated cells in response to IFN‐γ stimulation (4 h post-treatment at a dose of 1 ng/ml). (E) Gene ontology analyses using PANTHER GO-Slim tool revealed biological processes associated with downregulated genes in siR_Nostrill + IFN‐γ group when compared with that of siR_Ctrl + IFN‐γ group. (F) GO-KEGG analysis illustrating the enrichment of decreased Immune-related signaling pathways in response to siR_Nostrill + IFN‐γ treatment.
We then performed gene ontology (GO) analysis to understand the functional enrichment and biological pathways associated with these differentially expressed genes associated with Nostrill in IFN-γ-treated cells. The PANTHER GO-slim tool revealed that signaling pathways associated with cellular process, biological regulation, and immune system process were generally suppressed in siR_Nostrill-treated IEC4.1 cells upon IFN-γ stimulation, compared with that in siR_Ctrl- and IFN-γ-treated cells (Figure 2E). To get a deeper understanding of these immune system processes, we integrated GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases to perform functional analysis. The GO KEGG analysis revealed various immune system processes that were downregulated in siR_Nostrill-treated cells in response to IFN-γ, including IFN-γ-mediated cellular responses, regulation of MAPK cascade and autophagy mediators (Figure 2F). Expression levels of selected genes associated with cell-intrinsic defense were further validated at the RNA level in cells treated with siR_Nostrill or cells overexpressing Nostrill, using quantitative real-time reverse transcription PCR (qRT-PCR) (Supplementary Figure 1).
Nostrill facilitates chromatin recruitment of Stat1 to promote IFN-γ-mediated gene transcription in intestinal epithelial cells
The canonical IFN-γ signaling utilizes the JAK/STAT pathway to activate Stat1, resulting in the formation of active Stat1 homodimers (5). This complex then binds to gamma-activated sites in the promoters/enhancers of ISGs (5). To determine whether Nostrill and the Stat1 transcription factor have a direct physical relationship with one another, we performed formaldehyde crosslinking RNA immunoprecipitation (RIP) analysis. We observed that the anti-Stat1 immunoprecipitates from IFN-γ-stimulated cells contained a significant amount of Nostrill. In IFN-γ-stimulated cells, immunoprecipitation of Stat1 indicated a 1.8-fold increase in Nostrill enrichment as compared to the untreated control cells (Figure 3A).
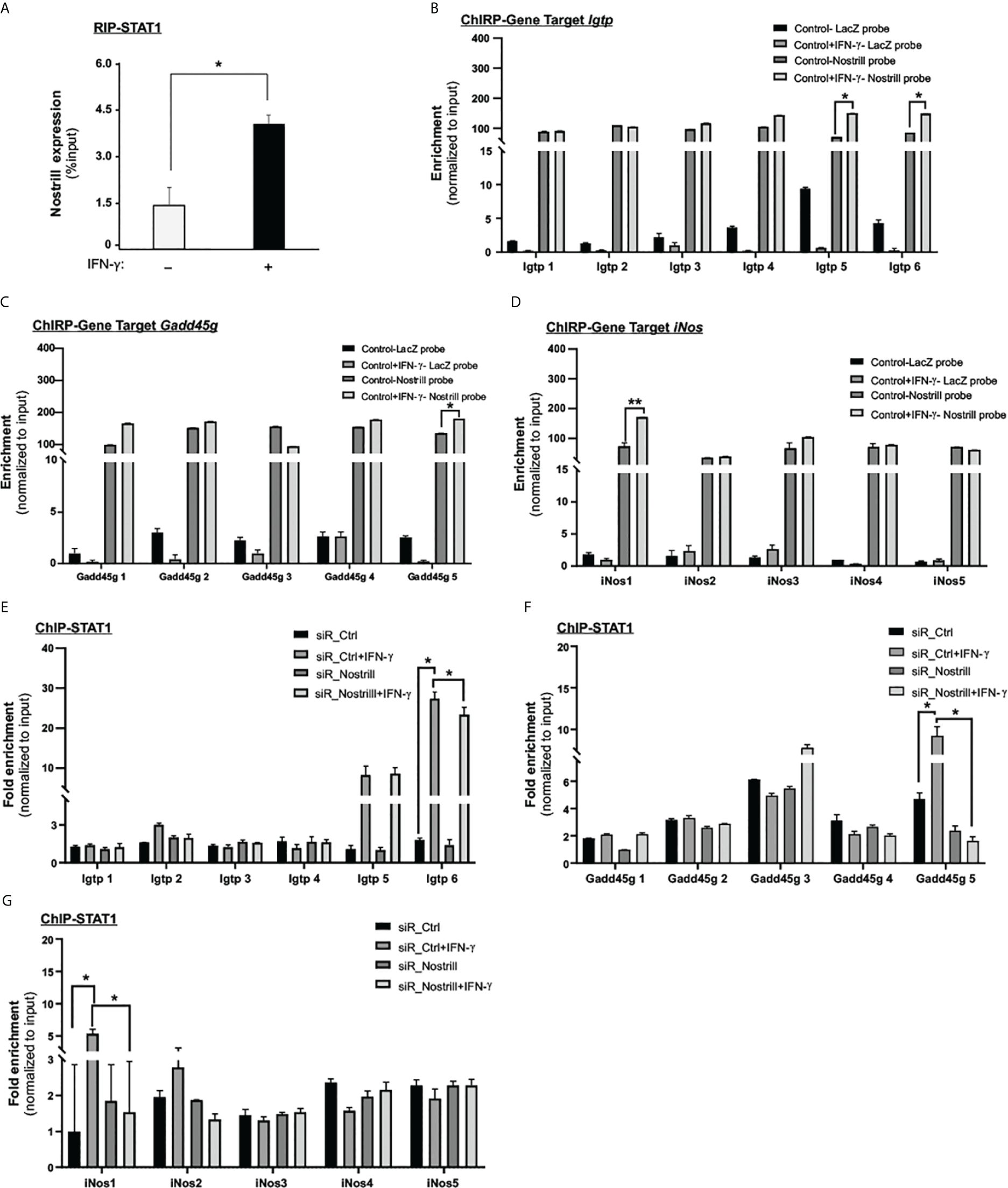
Figure 3 Molecular insights into Nostrill and STAT1 interaction in regulating ISG transcription. (A) Evidence of physical interaction between Nostrill and p65 in intestinal epithelial cells. IEC4.1 cells were exposed to a 5 ng/mL dose of IFN‐γ for 2h, followed by RNA immunoprecipitation (RIP) analysis utilizing an anti-Stat1 antibody. The presence of Nostrill was quantified through qRT-PCR and normalized against the percent input. Significance (*p < 0.05) was determined when compared to the control between the indicated groups. (B–D) Recruitment of Nostrill to ISG promoter sites following IFN‐γ treatment. IEC4.1 cells were exposed to a 5 ng/mL dose of IFN‐γ for 2h, and subsequent ChIRP analysis was carried out using pool of probes specific to Nostrill, along with designed PCR primer sets targeting Stat1 binding sites of Igtp, Gadd45g, and iNos. A pool of Lacz probes was used as negative control. Significance levels (*p < 0.05) were assessed in comparison to the control. (E–G) Influence of Nostrill siRNA knockdown on Stat1 recruitment to Igtp, Gadd45g, and iNos promoter sites upon IFN‐γ stimulation. IEC4.1 cells were transfected with siR_Nostrill or siR_Ctrl for 24h, then exposed to IFN‐γ for 2h, followed by ChIP analysis using anti-p65 antibody and designed PCR primer sets. Statistical significance (*p < 0.05) was determined in comparison to siR_Ctrl. Data are presented as means ± SEM with “n” of 3.
To determine if Nostrill binds to specific genomic sites in the promoters of ISGs, potentially facilitating the binding of Stat1, we employed the Chromatin Isolation by RNA Purification (ChIRP) technique. We used biotinylated probes specific to Nostrill to isolate chromatin fragments and then measured the enrichment of Nostrill to the promoter regions of selected ISGs, such as, Igtp, iNos, and Gadd45g. A pool of eight biotinylated tiling oligonucleotide probes unique to Nostrill were utilized to isolate the associated chromatin fragments and a pool of six LacZ probes were used as control. Following the Nostrill pull-down, we conducted qRT-PCR assays employing primers designed to target the Stat1 binding sites within the respective gene promoters. The findings revealed enhanced associations of Nostrill with the promoter regions of these genes in response to IFN-γ stimulation (Figures 3B–D). Notably, there was an amplified interaction between Nostrill and the Igtp promoter regions 5 and 6 following IFN-γ stimulation (Figure 3B). In the case of the Gadd45g promoter site 5, a noticeable increase in Nostrill abundance was observed (Figure 3C). Likewise, the iNos promoter region 1 exhibited an increased enrichment of Nostrill upon IFN-γ stimulation (Figure 3D).
To further investigate the potential involvement of Nostrill in docking Stat1 at the promoter regions of three genes: Igtp, iNos, and Gadd45g, we employed chromatin immunoprecipitation (ChIP) to assess Stat1 recruitment in IEC4.1 cells treated with siR_Nostrill (Figures 3E, F). The results showed that in siR_Ctrl-transfected cells stimulated with IFN-γ, there was a significant increase of Stat1 enrichment at the site 5 and site 6 regions of the Igtp promoter compared to the non-IFN-γ-treated control (p = 0.05) (Figure 3E). This suggests that Stat1 plays a role in the transcriptional regulation of Igtp in response to IFN-γ. Furthermore, when Nostrill expression was silenced in IEC4.1 cells and then exposed to IFN-γ stimulation, the enrichment of Stat1 at the site 6 region of the Igtp promoter was significantly reduced (p = 0.02). Of note, Nostrill was also recruited to the same site 6 region in the IFN-γ-treated IEC4.1 cells (Figure 3B). This indicates that Nostrill is involved in the docking or recruitment of Stat1 to the Igtp transcriptional region, potentially acting as a co-regulator. Similar observations were made for the Gadd45g and iNos promoter regions. Following IFN-γ stimulation, enrichment of Stat1 at the site 5 region of the Gadd45g promoter showed a significant increase (Figure 3F), while at the site 1 region of the iNos promoter, it increased significantly in the siR_Ctrl-transfected IEC4.1 cells (Figure 3G). However, silencing of Nostrill in cells significantly reduced the enrichment of Stat1 at these corresponding regions of the Gadd45g and iNos promoters (Figures 3F, G). Recruitment of Nostrill was observed to the same regions of the Gadd45g and iNos promoters in IFN-γ-treated IEC4.1 cells (Figures 3C, D). These findings suggest that Nostrill interacts with Stat1 and may function as a co-regulator for the chromatin enrichment of Stat1 to promote IFN-γ-stimulated gene transcription in intestinal epithelial cells.
Nostrill promotes IFN-γ-mediated intestinal epithelial cell-intrinsic anti-Cryptosporidium defense
Our lab has previously demonstrated the induction of Nostrill in intestinal epithelial cells in response to C. parvum infection (24). To this end, IEC4.1 cells were infected with C. parvum for up to 24h. At 24h following infection, the Nostrill’s induction was apparent, demonstrating a 72% increase in its expression (Figure 4A). To investigate the expression profile of Nostrill in a relevant in vivo model and gain insights into its potential role in the context of intestinal epithelium, neonatal mice (5 days old) were infected with C. parvum via oral gavage. Infection of the small intestinal epithelium by C. parvum was evident by immunostaining (Figure 4B). Consistent with previous reports (24), the expression of Nostrill was significantly increased at 24h after the infection in the intestinal epithelium (Figure 4C).
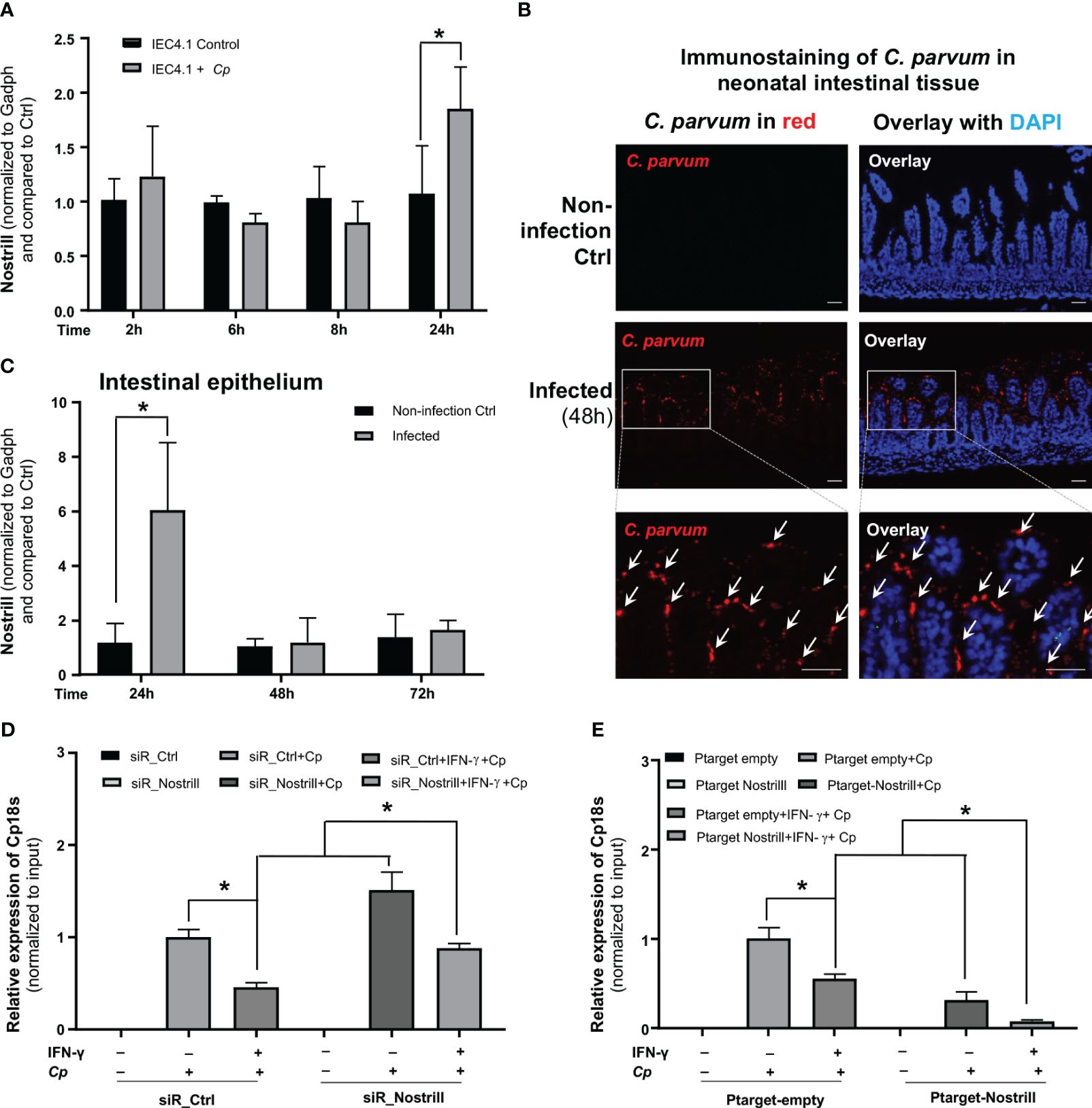
Figure 4 Impact of Nostrill on C. parvum infection burden in intestinal epithelial cells in response to IFN-γ. (A) Upregulation of Nostrill in cultured intestinal epithelial cells following exposure to C. parvum infection. IEC4.1 cells were infected with C. parvum for 2-24h and the level of Nostrill was measured via qRT-PCR. (B) C. parvum infection of the intestinal epithelium in neonatal mice. Neonatal mice, aged 5 days, were orally exposed to C. parvum, while control mice received PBS. C. parvum infection was confirmed by immunostaining of the parasite in the infected intestinal epithelium (48h post-infection). C. parvum was stained in red with the counterstaining of cell nuclei with DAPI in blue. Representative images are shown. Bar = 50 µm. (C) Nostrill induction in the intestinal epithelium from infected neonatal mice in response to C. parvum infection. Intestinal epithelium samples were collected at 24, 48, and 72h post-infection, and Nostrill expression was quantified using qRT-PCR. (D, E) Impact of Nostrill on C. parvum infection burden, (D) Effect of Nostrill knockdown on C. parvum infection burden in a cell culture. IEC4.1 cells were transfected with either siR_Ctrl or siR_Nostrill. Subsequently, they were exposed to IFN-γ 10 ng/ml for 24h and then infected with C. parvum for 8h. Quantification of C. parvum infection was performed using qRT-PCR. (E) The impact of Nostrill overexpression on C. parvum infection in vitro. IEC4.1 cells were transfected with a Nostrill overexpression plasmid (PTarget-Nostrill) or an empty vector control (PTarget-Empty). After 24h of treatment with IFN-γ 10 ng/ml, the cells were infected with C. parvum for 8h, and the extent of infection was quantified via qRT-PCR. Statistical significance was calculated using t-test. Significance levels (*p < 0.05). Data are presented as means ± SEM with “n” of 3.
Given the significant impact of Nostrill on IFN-γ-mediated gene transcription in intestinal epithelial cells, we sought to ask whether Nostrill induction may influence IFN-γ-mediated intestinal epithelial cell-intrinsic defense against C. parvum infection. To investigate this, we initially primed siR_Ctrl- and siR_Nostrill-treated IEC4.1 cells with IFN-γ and subsequently infected them with C. parvum, followed by an assessment of the resulting infection burden. Consistent with previous reports (25, 29), we detected a decreased infection burden in siR_Ctrl-treated IEC4.1 cells primed with IFN-γ (Figure 4D). Intriguingly, knockdown of Nostrill with the siRNA in IEC4.1 cells resulted in a significant suppression of IFN-γ-mediated epithelial defense, reflected by a higher infection burden in siR_Nostrill-treated and IFN-γ-primed cells compared with that in IFN-γ-primed cells treated with the siR_Ctrl (Figure 4D). Complementarily, overexpression of Nostrill resulted in enhanced IFN-γ-mediated inhibition of infection burden in IEC4.1 cells (Figure 4E). These data suggest that Nostrill promotes IFN-γ-mediated epithelial cell-intrinsic anti-Cryptosporidium defense.
Nostrill exerts anti-Cryptosporidium defense through IFN-γ-induced autophagy
Autophagy has been recognized as one of the immune responses elicited by IFN-γ against intracellular pathogens (30). Several genes, whose expression in response to IFN-γ in intestinal epithelial cells was associated with Nostrill, including Igtp, iNos, and Gadd45g, have been implicated in the regulation of cellular autophagy (30, 31). To examine the role of Nostrill in IFN-γ-induced autophagy, we first assessed the autophagy levels in IEC4.1 cells following exposure to IFN-γ at a dose of 10 ng/mL for 24h. We quantified autophagosome formation using LC3-II release as the marker as previously reported (32, 33). Our results clearly demonstrated the induction of autophagy in IEC4.1 cells following IFN-γ stimulation, as corroborated by both Western blot and qRT-PCR analyses of LC3-II release (Figures 5A, B, Supplementary Figure 2). Subsequently, we sought to determine the influence of Nostrill on this process. To do so, we utilized siR_Nostrill to knockdown its expression in the cells before exposure to IFN-γ. Our findings revealed a significant inhibition of IFN-γ-induced autophagy in cells treated with siR_Nostrill (Figures 5A, B).
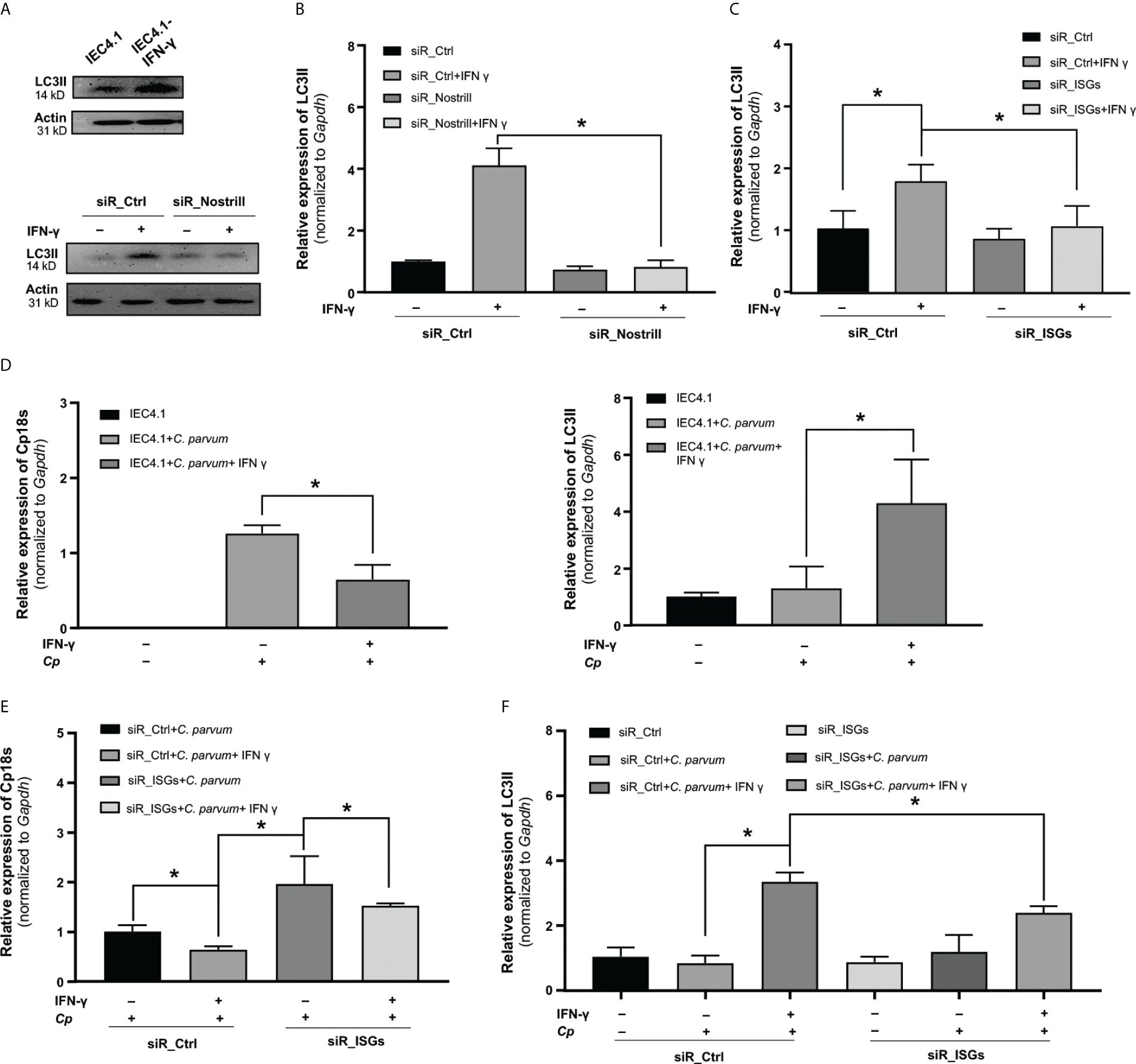
Figure 5 Evidence for Nostrill-mediated enhancement of anti-Cryptosporidium defense via IFN-γ-induced autophagy in intestinal epithelial cells. (A, B) Assessment of autophagy in Nostrill knockdown cells. (A) IEC4.1 cells were exposed to IFN-γ for 24h, followed by Western blot analysis to evaluate the autophagy marker LC3II (14kDa), with beta-actin (31kD) serving as a reference protein. A similar analysis was conducted on Nostrill-knocked down cells exposed to IFN-γ. (B) Quantification of LC3II levels in Nostrill knockdown cells was performed via qPCR. (C) Impact of Nostrill associated genes on autophagy. A combination of siRNAs was used to knockdown Igtp, iNos, and Gadd45g (referred to as siR_ISGs) in IEC4.1 cells, followed by treatment with 10 ng/ml of IFN-γ for 24h. The level of LC3II marker was quantified through qRT-PCR. (D-F) Impact of Nostrill associated ISGs in defending C. parvum through autophagy. (D) IEC cells were treated with 10 ng/ml of IFN γ for 24h and then infected with C. parvum for 8h. Level of Cp18s and LC3II were measured by qRT-PCR. (E, F) IEC4.1 cells were treated with siR_ISGs (260 nM), which consisted of a pool of siRNAs targeting Igtp (80 nM), iNos (60 nM), and Gadd45g (120 nM), or a scrambled siR_Ctrl at 260 nM. Subsequently, cells were treated with 10 ng/ml of IFN-γ for 24h and then infected with C. parvum for 8h. (E) Cp18s, representative of C. parvum burden, and (F) LC3II levels as an indicator of autophagy were quantified using qRT-PCR. Statistical significance was calculated using t-test. Significance levels (*p < 0.05). Data are presented as means ± SEM with “n” of 3.
To further investigate the contribution of Nostrill-mediated genes, including Igtp, iNos, and Gadd45g, to IFN-γ-induced autophagy, we employed a combination of siRNAs (siR_ISGs) to silence their expression in IEC4.1 cells (Supplementary Figure 3). We then examined the impact of siR_ISG knockdown on IFN-γ-induced autophagy. Silencing Igtp, iNos, and Gadd45g with the siR_ISGs led to a significant inhibition of IFN-γ-induced autophagy (Figure 5C).
The subsequent question we aimed to address was whether these Nostrill-associated ISGs within the IFN-γ pathway play a role in combating Cryptosporidium infection through autophagy. To investigate this, we first assessed the expression of LC3II in IEC4.1 cells treated with IFN-γ and subsequently infected with C. parvum. As shown in Figure 5D, IFN-γ treatment led to a reduction in C. parvum burden in the cells. Meanwhile, there was a significant increase in the LC3II levels in the IFN-γ-treated cells following C. parvum infection (Figure 5D). This suggests that IFN-γ may reduce C. parvum burden in the cells through induction of autophagy. To further substantiate the significance of IFN-γ-induced autophagy in the defense against Cryptosporidium, we assessed both the Cryptosporidium infection burden and the LC3II levels in cells with or without treatment with the siR_ISGs. IEC4.1 cells were first treated with siR_Ctrl or siR_ISGs for 24h, pre-treated with IFN-γ for additional 24h, and subsequently infected with Cryptosporidium for 24h. Similar to our observations in Nostrill knockdown cells, the knockdown of selected ISGs in IEC4.1 cells resulted in a significant suppression of IFN-γ-mediated epithelial defense, as evidenced by a higher infection burden in the siR_ISGs-treated and IFN-γ primed cells compared to those treated with the siR_Ctrl (Figure 5E). Furthermore, we observed a reduction in LC3II levels in cells transfected with siR_ISGs, specifically when treated with IFN-γ and subsequently infected with C. parvum (Figure 5F). Taken together, the above data suggest that Nostrill facilitates the transcription of Igtp, Gadd45g, and iNos induced by IFN-γ, which in turn positively regulates autophagy, ultimately contributing to the epithelial cell-intrinsic defense against Cryptosporidium infection.
Discussion
Intestinal epithelial cell-intrinsic immunity provides the first line of host defense against intracellular pathogens in the intestine (2). This defense mechanism operates through layered innate immune signaling networks within intestinal epithelial cells, which detect microbial pathogens and trigger downstream mechanisms for pathogen eradication. These mechanisms include the production of antimicrobial proteins, activation of specialized degradative compartments, and initiation of programmed host cell death. IFNs stand as one of the most potent signals derived from vertebrates, mobilizing antimicrobial effector functions against intracellular pathogens (2, 9) and serving as cornerstones of intestinal epithelial cell-intrinsic defense. Emerging evidence highlights the critical role of long lncRNAs as regulatory components in gastrointestinal defense against microbial infection (22, 24, 34, 35). In this study, we investigated the impact of Nostrill, a MyD88/NF-кB-associated lncRNA as we previously identified (23), on the regulation of IFN-γ-mediated antimicrobial defense in intestinal epithelial cells. Our findings demonstrate that Nostrill enhances the transcription of a set of genes regulated by IFN-γ in intestinal epithelial cells and induction of Nostrill facilitates IFN-γ-stimulated epithelial cell-intrinsic anti-cryptosporidial defense.
Several lncRNAs are induced in innate immune cells and are likely to play key roles in the regulation of innate defense. For instance, lncRNAs exhibit differential regulation in virus-infected cells (36) and in dendritic cells or macrophages upon stimulation by ligands for TLR4 and TLR2 (21). LncRNA-Cox2, among the most highly induced lncRNAs in macrophages, has been demonstrated to mediate both the activation and repression of distinct classes of immune genes (21, 37–39). Recent research has shown that various IFN responses are exquisitely regulated by lncRNAs and these IFN-related lncRNAs are also highly tissue- and cell-type-specific, rendering them as promising biomarkers or therapeutic candidates to modulate specific stages of the antiviral immune response with fewer adverse effects (40). LncRNAs are implicated in fundamental roles as inducers or repressors at nearly every stage of the IFN response. Our findings align with this notion, indicating that Nostrill can modulate gene transcription in intestinal epithelial cells in response to IFN-γ stimulation. Modes of action for lncRNAs encompass various mechanisms, such as regulation of gene transcription either in cis or in trans by recruiting proteins or molecular complexes to specific gene loci, acting as scaffolds for nuclear complexes, titrating RNA-binding proteins, and forming RNA-RNA pairs to induce posttranscriptional regulation (41). Our observations indicate that Nostrill facilitates the recruitment of Stat1 to the promoters of IFN-γ-stimulated genes. Similar mechanisms have been previously demonstrated with lncRNA XR_001779380 in regulating IFN-γ-stimulated gene transcription in intestinal epithelial cells (22). Intriguingly, Nostrill selectively promotes the transcription of a subset, rather than all, of IFN-γ-stimulated genes in IEC4.1 cells. In addition, not all the promoter regions with an increased enrihment of Stat1 are accompanied with an enhanced Nostrill recruitment. Obviously, besides Stat1 promoter recruitment, other determinants may be involved in Nostrill-mediated gene transcription in intestinal epithelial cells in response to IFN-γ stimulation. In our previous studies (24), we found that Nostrill can recruit IRF7 and NF-кB p65 to several defense gene loci in cells following cryptosporidial infection. It would be worthwhile to explore whether the recruitment of IRF7 and NF-кB p65 is also implicated in Nostrill-mediated gene transcription in intestinal epithelial cells in response to IFN-γ.
As a result, the induction of Nostrill may enhance IFN-γ-mediated cell-intrinsic defense against intracellular pathogens in intestinal epithelial cells by facilitating gene transcription in response to IFN-γ stimulation. Indeed, we observed a greater burden of Cryptosporidium infection (indicating diminished cellular defense) in intestinal epithelial cells treated with siRNA to knockdown Nostrill following IFN-γ stimulation, compared to control-siRNA-treated cells. Hence, the induction of Nostrill in intestinal epithelial cells following Cryptosporidium infection through activation of the MyD88/NF-кB pathway may prime host epithelial cells for a more efficient cell-intrinsic defense in response to IFN-γ released upon activation of immune cells residing at the epithelium. Induction of another lncRNA, XR_001779380, has been demonstrated to provide a similar promoting effect on cellular response to IFN-γ during Cryptosporidium infection in the intestine through promoting Swi/Snf-mediated gene transcription (22).
IFN-γ instigates a diverse array of cell-intrinsic responses aimed at inhibiting parasite growth through mechanisms such as nutrient deprivation and the production of potent defense molecules, including reactive oxygen and nitrogen species, IRGs, and GBPs (3, 4, 8–10). Notably, Nostrill can promote the transcription of many of these genes in intestinal epithelial cells following IFN-γ stimulation. Among them, certain genes such as Igtp, iNos, and Gadd45g are of particular interest due to their regulatory roles in autophagy. Their upregulation may contribute to enhanced cell-intrinsic defense in response to IFN-γ associated with Nostrill induction. Stimulation with IFN-γ induces autophagy in cultured intestinal epithelial cells, a process that is partially impeded by knockdown of Nostrill, Igtp, iNos, or Gadd45g. These findings suggest that Nostrill may augment IFN-γ-mediated cell-intrinsic defense against Cryptosporidium infection in intestinal epithelial cells by promoting the transcription of genes associated with autophagy. Autophagy serves as a critical defense mechanism against infections, facilitating the capture and elimination of intracellular parasites (42). Recent studies have reported the induction of autophagy in intestinal epithelial cells via mTOR inactivation during Cryptosporidium infection of Caco-2 cells (43). Diminished host cell autophagy can promote parasitic survival in host cells following Cryptosporidium infection (44, 45).
Given the fact that all three types of IFN signaling are activated in the intestinal epithelium following Cryptosporidium infection (25, 46–48), it would be intriguing to investigate whether Nostrill also regulates gene transcription in intestinal epithelial cells in response to stimulation by other type IFNs. While type II (IFN-γ) and III IFNs demonstrate a pronounced protective role (47–51), type I IFN signaling exhibits a pro-parasitic effect, as evidenced by the resistance of mice lacking Ifnar1 to C. parvum infection (25, 47). Furthermore, elucidating the role of Nostrill in regulating IFN-mediated intestinal epithelial anti-Cryptosporidium defense warrants further investigation using in vivo infection models employing conditional knockout strains of various IFN types.
In conclusion, our findings suggest that Nostrill may enhance intestinal epithelial defense against Cryptosporidium infection through its regulation of interferon responses. We investigated the role of Nostrill in IFN-γ-stimulated gene transcription and antimicrobial defense. Our results indicate that Nostrill promotes the transcription of a subset of genes controlled by IFN-γ, and its induction facilitates IFN-γ-mediated intestinal epithelial cell-intrinsic defense against Cryptosporidium. A deeper understanding of how lncRNAs like Nostrill modulate the transcription of host defense genes would offer new insights into mucosal immunity to Cryptosporidium and potentially other pathogens in general.
Materials and methods
Cell culture
Dr. Pingchang Yang of McMaster University in Hamilton, Canada, generously provided the newborn mouse intestinal epithelial cell line (IEC4.1) (52). Cultures of IEC4.1 cells were maintained in DMEM media with 10% fetal bovine serum (FBS; Gibco; Carlsbad, CA, USA). Streptomycin at 100 μg/ml and penicillin at 100 U/ml (both from Gibco) were added to the growth media. Tissue culture flasks were used to cultivate the cells in a 37°C, 5% CO2 incubator.
C. parvum oocyst
Cryptosporidium parvum oocysts, sourced from the Iowa strain provided by Harley Moon at the National Animal Disease Center (Ames, IA), were purchased commercially from the Bunch Grass Farms in Deary, ID. To purify the oocysts, a modified ether extraction technique was employed. Subsequently, the oocysts were suspended in phosphate-buffered saline (PBS) and stored at a temperature of 4°C. For both in vitro and in vivo infection experiments, the oocysts were subjected to a treatment with 1% sodium hypochlorite while being preserved on ice for 20 minutes. After this treatment, the oocysts were washed three times using Dulbecco’s modified Eagle medium (DMEM) culture media.
Infection models and assays
For in vitro infection studies using IEC4.1 cells, viable C. parvum oocysts were utilized. These oocysts were treated with 1% sodium hypochlorite and were added to a culture medium consisting of DMEM-F-12 supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin. The infection was established by combining oocysts and host cells in a 1:1 ratio. The cell cultures were then incubated at 37°C for 4h to facilitate parasite attachment and invasion. After this, the cells were thoroughly washed with DMEM-F-12 medium three times to remove any free parasites. The cells were further cultured for varying time periods based on the experimental requirements. For the in vivo experiments, a neonatal murine model of intestinal cryptosporidiosis was employed (25). Neonates, aged 5 days after birth, were orally administered C. parvum oocysts (10^5 oocysts per mouse) through gavage to induce intestinal cryptosporidiosis. Control mice were given PBS via the same method. Following 24, 48, and 72h from the administration of C. parvum oocysts or PBS, the animals were sacrificed, and samples of ileum intestine tissues were collected. Epithelial tissues from the ileum were extracted from at least five animals within each experimental group. The assessment of C. parvum was done using qRT-PCR both in vitro and in vivo context.
siRNA-mediated knockdown
To silence the Nostrill gene in murine cells, a pool of three small interfering RNA (siRNA) duplexes (siR_Nostrill) was synthesized using the WI siRNA Selection Program at the Massachusetts Institute of Technology. The specific siRNA sequences used were as follows: 25b_1: sense 5’ CCUGGGAAGAAGCAUUAAU 3’, 25b_2: sense 5’ CCACCAGGAACACACAAAU 3’, and 25b_3: sense 5’ GCCUAUAAGUCGUUCUAAU 3’. The IEC4.1 cells were cultured in 24-well plates at a density of 1.5 x 10^5 cells per milliliter. The next day, the cells were transfected with 120 nM of the siRNA pool (40 nM of each of the siRNA) using Lipofectamine RNAiMax from Life Technologies, Inc., following the manufacturer’s instructions. As a negative control, non-specific scrambled siRNA at a concentration of 120 nM was used as the control siRNA (siR_Ctrl). The transfection process was allowed to continue for 24h, after which the cells were treated with IFN-γ. RNA was then isolated from the cells using a previously described method.
Igtp, iNos, and Gadd45g were targeted for knockdown of ISGs in mouse IEC4.1 cells. The siRNA-mediated knockdown approach was employed to silence these genes. The siRNA to iNos was synthesized using the WI siRNA Selection Program at the Massachusetts Institute of Technology (MIT) and the sequence was sense 5’ GCCGCUCUAAUACUUAGCU 3’. For silencing Igtp and Gadd45g, siRNA duplexes were purchased from Santacruz Biotechnology with the catalogue numbers sc-41792 and sc-37419, respectively. The IEC4.1 cells were cultured in 24-well plates at a density of 1.5 x 10^5 cells/ml. After overnight incubation, the cells were transfected with a pool of the three sets of siRNAs to achieve knockdown of Igtp, Gadd45g, and iNos (as siR_ISGs). The concentrations of gene-specific siRNAs used for transfection were as follows: siR_Igtp at 80 nM, siR_Gadd45g at 120 nM, and siR_Nos at 60 nM. siR_Ctrl at a concentration of 260 nM was used for control. Lipofectamine RNAiMax from Life Technologies, Inc. was used as the transfection reagent, and the transfection process was carried out following the manufacturer’s instructions.
RNA isolation
Total RNA from the cells was extracted using the TRIzol reagent from Invitrogen as per the manufacturer’s instructions. The concentration and quality of the RNA were assessed using a spectrophotometer from Beckman in Brea, CA. For subsequent steps, 500 ng of the RNA sample was utilized as a template for cDNA synthesis, which was performed using the M-MLV Reverse Transcriptase kit from Invitrogen, located in Carlsbad, CA. The reaction was carried out in a total volume of 20 µl.
qRT-PCR
The qRT-PCR was conducted using the Invitrogen™ SYBR GreenER™ qPCR SuperMix Universal from Thermo Fisher Scientific in Waltham, MA. The qRT-PCR was performed on the Bio-Rad CFX96 Touch™ Real-Time PCR Detection System. The expression levels of the RNA samples were determined using the threshold cycle (ΔΔCT) method and normalized to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (Gapdh). The primer sequences are displayed in Supplementary Table 3.
RNA sequencing and bioinformatics analysis
RNA-Seq was performed in biological triplicates in IEC4.1 cells treated with siR_Ctrl or siR_Nostrill. Total RNA molecules were isolated using Trizol reagent. The BGI Americas Corporation in Cambridge, MA, carried out transcriptome sequencing and data processing using DNBSEQ™ sequencing technology platforms. To assess RNA quality, a 2100 Bioanalyzer from Agilent Technologies was employed. RNA samples meeting the criteria of consistent visual RNA patterns, an RNA Integrity Number (RIN) greater than 7, and a concentration exceeding 20 ng/μl were categorized as having good quality. The complete RNA was broken down into smaller fragments, and specific mRNA molecules were concentrated using magnetic beads with oligo (dT) sequences. Subsequently, complementary DNA (cDNA) was synthesized from the enriched mRNA. This double-stranded cDNA underwent purification and was amplified using PCR. The sequenced library products were processed on the DNBSEQ-500 platform with paired-end reads of 100 base pairs, generating an average of approximately 4.94 Gb per sample. To ensure data quality, subpar reads were excluded. This included reads with base qualities lower than 10 for over 20% of their bases, reads with adaptors, and those containing more than 5% bases marked as unknown (N bases). This filtration was achieved using internal software called SOAPnuke (version: v1.5.2) on the DNBSEQ-500 platform, with specific parameters (-I 15-q0.5-n0.1). The resultant high-quality reads were stored in the FASTQ format. Reads were aligned to the Mus_musculus genome (GCF_000001635.26_GRCm38.p6) from the NCBI database using two different software tools: HISAT2 (version: v2.0.4) and Bowtie2 (version: v2.2.5). The alignment was guided by parameters tailored to the specifics of the process (-phred64-sensitive-no-discordant-no-mixed-I 1-X 1000). Raw RNA-seq data were further refined by filtering out unreliable sites, accomplished through the application of the GATK program. This step targeted the removal of low-quality reads from each sample. Finally, the gene read counts were normalized to RPKM, which represents the expression level of genes in terms of reads per kilobase of transcript per Million mapped reads. This normalization allowed for meaningful comparison of relative gene expression levels across samples.
Differential gene-expression analyses were conducted using DESeq2. Differences in gene expression, whether increased or decreased, were quantified using logarithmically transformed fold change values. These values were represented as either log2FC (log base 2) or log10FC (log base 10), which were computed as the logarithmic difference between gene expression values under distinct treatment conditions (log2FC = log2[B] - log2[A] or log10FC = log10[B] - log10[A], where A and B denote the gene expression values). GO enrichment analysis was performed using PANTHER. To further analyze the data, Gene Set Enrichment Analysis (GSEA) was conducted using the “clusterProfiler” R package. This analysis was carried out in a pre-ranked manner, where all genes were ranked based on their log2FC values derived from the differential expression analysis. The weighted Kolmogorov-Smirnov test was applied, and p-values were adjusted using the Benjamini-Hochberg method. The findings from GSEA were depicted visually using the “ggplot2” R package, offering insights into patterns of gene set enrichment. Additionally, a volcano plot was generated with the “ggplot2” package, utilizing log2FC and adjusted p-values from the differential expression analysis to visually convey the significance and extent of gene expression alterations. For a comprehensive overview of gene expression trends, heatmaps were generated using the “ComplexHeatmap” R package. These heatmaps visually summarized gene expression patterns across different conditions. Furthermore, the expression z-score was computed based on relative gene expression levels. This score aids in standardizing and comparing gene expression values across diverse samples or conditions, facilitating more meaningful interpretation of expression patterns.
RIP assay
The formaldehyde crosslinking RNA immunoprecipitation (RIP) assay was conducted as described previously (22, 53). To summarize, the process involved the initial cleaning of lysates using 20 μl of Magna ChIP Protein A + G Magnetic Beads (Millipore, Massachusetts) that had been pre-washed with PBS. Subsequently, the pre-cleaned lysate (250 μl) was mixed with an equivalent volume of the whole cell extract buffer (250 μl). This mixture was then combined with beads coated with an antibody to Stat1 (#9172S, Cell Signaling, Danvers, MA). The entire assembly was then rotated at 4°C overnight. Following this, the mixture underwent four rounds of washing using the whole cell extract buffer supplemented with protease and RNase inhibitors. The immunoprecipitated ribonucleoprotein (RNP) complexes and the input material were subjected to digestion in RNA PK Buffer pH 7.0 (comprising 100 mM NaCl, 10 mM TrisCl pH 7.0, 1 mM EDTA, and 0.5% SDS) in the presence of 10 μg of Proteinase K. This mixture was then incubated at 50°C for 45 minutes with continuous shaking at 400 rpm. The formaldehyde cross-links were subsequently reversed through incubation at 65°C with rotation for 4 hours. RNA was isolated from the samples using Trizol, following the protocol provided by Invitrogen. The presence of RNA was evaluated using qRT-PCR, conducted with the CFX96 Touch™ Real-Time PCR Detection System from Bio-Rad. The specific primer pairs for PCR can be found in Supplementary Table 3.
ChIP assay
The detailed protocol is described elsewhere (22, 54). Briefly, cells were treated with 1% formaldehyde for 10 minutes to cross-link the proteins to the DNA, preserving the protein-DNA interactions at the time of fixation. The fixed cells were collected in 1x ice-cold PBS (Phosphate-Buffered Saline) and resuspended in an SDS (Sodium Dodecyl Sulfate) lysis buffer. The genomic DNA was then fragmented into smaller pieces through sonication resulting in fragments ranging from 200 to 500 base pairs in length. One percent of the cell extract was kept aside as input. The cell extracts containing the sheared DNA are incubated overnight at 4°C with anti-Stat1 and added protein G-agarose beads to precipitate the antibody-protein-DNA complexes. The immunoprecipitates (antibody-protein-DNA complexes bound to the beads) were washed sequentially to remove any non-specific interactions. The washing steps involve low-salt buffer, high-salt buffer, LiCl buffer, and Tris EDTA buffer. The DNA-protein complexes were eluted from the beads afterwards, separating the DNA from the proteins. The proteins in the eluted solution were then digested at 45°C for 1hr using proteinase K. To detect and quantify the DNA, qRT-PCR analysis was employed, which allowed for the measurement of the DNA content, enabling the quantification of protein binding levels at particular regions of the genome.
ChIRP assay
Chromatin isolation by RNA purification (ChIRP) analysis was conducted as described in a previous study (22, 55). In summary, chromatin was isolated after cross-linking the cells with glutaraldehyde. A set of tiling oligonucleotide probes were designed to specifically bind to the Nostrill sequence. The sequences for these probes are available in Supplementary Table 1. Verification of the DNA sequences in the chromatin immunoprecipitates was performed using qRT-PCR. The same primers that cover the gene promoter regions of interest, similar to the ChIP analysis, were utlized for the ChIRP analyses. As a control, a collection of scrambled oligonucleotide probes targeting LacZ (listed in Supplementary Table 3) was used.
Autophagy assay
Cell Culture and Lysate Preparation: Cells were plated in six-well plates with 2 ml of medium per well to achieve 80%–90% confluency within 48h. After 24h, the culture medium was replaced with treatment (such as, IFN-γ at a dose of 10 ng/ml) or control medium and incubated for an additional 24h. Two hours prior to collection, 1:100 dilutions of the stock solutions of leupeptin (10 mM) and NH4Cl (2 M) were added to the medium to yield final concentrations of 100 μM and 20 mM, respectively. The cell pellet was collected using EDTA/PBS, washed with ice-cold PBS, and lysed with pre-cooled lysis buffer. Lysates were sonicated for 10–15 sec or treated with syringe to complete cell lysis and shear DNA to reduce sample viscosity. The lysate was centrifuged at 12,000 rpm for 10 minutes at 4°C, and the supernatant was transferred to a new tube. Protein concentration was determined, samples were adjusted for equal concentrations, mixed with loading buffer, and boiled before storing at -80°C.
For SDS-PAGE and Western blotting, equal protein amounts (10–20 μg) were loaded onto the SDS-PAGE gel with a molecular weight marker. The gel was run at 95 V for 20 minutes and then at 150 V for approximately 1–2 hours until the dye reached 1/4 inch from the bottom of the gel. LC3-II migrated at ~14 kD. The separated proteins were transferred onto a PVDF membrane, chosen for its enhanced LC3-II binding. Following transfer, the membrane was blocked with 5% BSA blocking buffer for one hour at room temperature and probed with primary anti-LC3II antibody (1:1000, cat#2775S from Cell Signaling) overnight at 4°C. The membrane was washed three times for 15 minutes each with TBST at room temperature, probed with secondary HRP-conjugated anti-rabbit IgG antibody (1:5000) for one hour at room temperature, and washed again. An ECL solution mixture was applied to the membrane, and the emitted light was captured. The levels of LC3-II in each sample were normalized to β-actin (1:1000, cat#A1978 from Cell Signaling) for analysis.
Statistical analysis
Data are given as mean ± SEM. from at least three independent experiments or biological replicates. The two-tailed unpaired Student’s t test was used for compassion between two groups and one-way ANOVA were used for compassion among three or more groups. p values < 0.05 were considered statistically significant.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GSE245211 (GEO).
Ethics statement
The animal study was approved by IACUC, Rush University Medical Center. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
ZS: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Software, Validation, Visualization, Writing – original draft. KJ: Data curation, Investigation, Validation, Writing – review & editing. A-YG: Data curation, Formal Analysis, Investigation, Methodology, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing. SD: Methodology, Software, Writing – review & editing. CP: Methodology, Visualization, Writing – review & editing. MG: Methodology, Visualization, Writing – review & editing. SW: Methodology, Visualization, Writing – review & editing. NM: Formal Analysis, Investigation, Methodology, Writing – review & editing. AS: Formal Analysis, Resources, Writing – review & editing. X-MC: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by funding from the National Institutes of Health (AI116323, AI136877, AI156370, and AI177188 to X-MC).
Acknowledgments
We thank Dr. Pingchang Yang (McMaster University, Hamilton, Canada) for sharing IEC4.1 cells.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1397117/full#supplementary-material
Supplementary Table 1 | List of genes from RNA-Seq in IEC4.1 cells following Cryptosporidium infection.
Supplementary Table 2 | List of genes from RNA-Seq in IEC4.1 cells following IFN-γ stimulation.
Supplementary Table 3 | Primers for PCR, RIP, ChIP, ChIRP and construct generating.
References
1. Randow F, MacMicking JD, James LC. Cellular self-defense: how cell-autonomous immunity protects against pathogens. Science. (2013) 340:701–6. doi: 10.1126/science.1233028
2. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Publ Gr. (2014) 14(3):141–53. doi: 10.1038/nri3608
3. Meunier E, Broz P. Microreview Interferon-inducible GTPases in cell autonomous and innate immunity. Cell Microbiol. (2015) 18(2):168–80. doi: 10.1111/cmi.12546
4. MacMicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol. (2012) 12:367. doi: 10.1038/nri3210
5. Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. (2004) 202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x
6. González-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. (2012) 12:125–35. doi: 10.1038/nri3133
7. Lurie RH, Platanias LC. Mechanisms of type-I- and type-II interferon-mediated signaling. (2005). doi: 10.1038/nri1604
8. Hu X, Ivashkiv LB. Cross-regulation of signaling and immune responses by IFN-γ and STAT1. Immunity. (2009) 31(4):539–50. doi: 10.1016/j.immuni.2009.09.002
9. Stanifer ML, Guo C, Doldan P, Boulant S, Walter MR, Pierangeli A, et al. Importance of type I and III interferons at respiratory and intestinal barrier surfaces. Front Immunol. (2020) 11:1. doi: 10.3389/fimmu.2020.608645
10. Borger J, Wilhelm C, Colonna M, Panda SK. Innate lymphoid cells in mucosal immunity. Front Immunol. (2019) 1:861. doi: 10.3389/fimmu.2019.00861
11. Ouwendijk WJD, Van Veen S, Mahalingam R, Verjans GMGM. Simian varicella virus inhibits the interferon gamma signalling pathway. J Gen Virol. (2017) 98:2582–8. doi: 10.1099/jgv.0.000925
12. Krishnamurthy S, Maru P, Wang Y, Bitew MA, Mukhopadhyay D, Yamaryo-Botté Y, et al. CRISPR screens identify toxoplasma genes that determine parasite fitness in interferon gamma-stimulated human cells. mBio. (2023) 14(2):e0006023. doi: 10.1128/mbio.00060-23
13. Petri WA, Miller M, Binder HJ, Levine MM, Dillingham R, Guerrant RL. Enteric infections, diarrhea, and their impact on function and development. J Clin Invest. (2008) 118:1277–90. doi: 10.1172/JCI34005
14. Checkley W, White AC, Jaganath D, Arrowood MJ, Chalmers RM, Chen X-M, et al. Review A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for Cryptosporidium. Lancet Infect Dis. (2015) 15(1):85–94. doi: 10.1016/S1473-3099(14)70772-8
15. Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet. (2013) 382(9888):209–22. doi: 10.1016/S0140-6736(13)60844-2
16. Colford JM, Tager IB, Hirozawa AM, Lemp GF, Aragon T, Petersen C. Sponsored by the society for epidemiologic research ORIGINAL CONTRIBUTIONS cryptosporidiosis among patients infected with human immunodeficiency virus factors related to symptomatic infection and survival. Am J Epidemiol. (1996) 144:807–16. doi: 10.1093/oxfordjournals.aje.a009015
17. Pantenburg B, Dann SM, Wang HC, Robinson P, Castellanos-Gonzalez A, Lewis DE, et al. Intestinal immune response to human Cryptosporidium sp. infection. Infect Immun. (2008) 76:23–9. doi: 10.1128/IAI.00960-07
18. Ludington JG, Ward HD. Systemic and mucosal immune responses to cryptosporidium-vaccine development. Curr Trop Med Rep. (2015) 2(3):171–80. doi: doi: 10.1007/s40475-015-0054-y
19. Ulitsky I, Bartel DP. Leading edge review lincRNAs: genomics, evolution, and mechanisms. Cell. (2013) 154(1):26–46. doi: 10.1016/j.cell.2013.06.020
20. Bocchetti M, Scrima M, Melisi F, Luce A, Sperlongano R, Caraglia M, et al. Molecular sciences lncRNAs and immunity: coding the immune system with noncoding oligonucleotides. Int J Mol Sci. (2021) 22(4):1741. doi: 10.3390/ijms22041741
21. Carpenter S, Atianand M, Aiello D, Ricci E, Gandhi P, Hall LL, et al. A long noncoding RNA induced by TLRs mediates both activation and repression of immune response genes HHS Public Access. Sci (80-). (2013) 341:789–92. doi: 10.1126/science.1240925
22. Gong A-Y, Wang Y, Li M, Zhang X-T, Deng S, Chen JM, et al. LncRNA XR_001779380 primes epithelial cells for IFN-g-mediated gene transcription and facilitates age-dependent intestinal antimicrobial defense. mBio. (2021) 12(5):e0212721. doi: 10.1128/mBio.02127-21
23. Mathy NW, Burleigh O, Kochvar A, Whiteford ER, Behrens M, Marta P, et al. A novel long intergenic non-coding RNA, Nostrill, regulates iNOS gene transcription and neurotoxicity in microglia. J Neuroinflammation. (2021) 18(1):16. doi: 10.1186/s12974-020-02051-5
24. Mathy NW, Deng S, Gong A-Y, Li M, Wang Y, Burleigh O, et al. The Long Non-Coding RNA Nostrill Regulates Transcription of Irf7 Through Interaction With NF-kB p65 to Enhance Intestinal Epithelial Defense Against Cryptosporidium parvum. Front Immunol. (2022) 13:1. doi: 10.3389/fimmu.2022.863957
25. Deng S, He W, Gong A-Y, Li M, Wang Y, Xia Z, et al. Cryptosporidium uses CSpV1 to activate host type I interferon and attenuate antiparasitic defenses. Nat Commun. (2023) 14(1):1456. doi: 10.1038/s41467-023-37129-0
26. Fu Y, Zhu Z, Chang H, Liu Z, Liu J, Chen H. Comparative transcriptome analyses indicate enhanced cellular protection against FMDV in PK15 cells pretreated with IFN-γ. Gene. (2016) 586(2):206–15. doi: 10.1016/j.gene.2016.03.027
27. Burian K, Endresz V, Deak J, Kormanyos Z, Pal A, Nelson D, et al. Transcriptome analysis indicates an enhanced activation of adaptive and innate immunity by chlamydia-infected murine epithelial cells treated with interferon-gamma. J Infect Dis. (2010) 202(9):1405. doi: 10.1086/656526
28. Kota RS, Rutledge JC, Gohil K, Kumar A, Enelow RI, Ramana CV. Regulation of gene expression in RAW 264.7 macrophage cell line by interferon-c. Biochem Biophys Res Commun. (2006) 342(4):1137–46. doi: 10.1016/j.bbrc.2006.02.087
29. Pollok RCG, Farthing MJG, Bajaj-Elliott M, Sanderson IR, Mcdonald V. Interferon gamma induces enterocyte resistance against infection by the intracellular pathogen cryptosporidium parvum. Gastroenterology. (2001) 120(1):99–107. doi: 10.1053/gast.2001.20907
30. Goswami AB, Karadarević D, Castaño-Rodríguez N. Immunity-related GTPase IRGM at the intersection of autophagy, inflammation, and tumorigenesis. Inflammation Res. (2022) 71:785–95. doi: 10.1007/s00011-022-01595-x
31. McAllaster MR, Bhushan J, Balce DR, Orvedahl A, Park A, Hwang S, et al. Autophagy gene-dependent intracellular immunity triggered by interferon-γ. MBio. (2023) 14:02332–23. doi: 10.1128/mbio.02332-23
32. Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. (2004) 36:249. doi: 10.1016/j.biocel.2004.02.005
33. Kimura S, Fujita N, Noda T, Yoshimori T. Chapter 1 - monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Autophagy Mamm Syst Part B. (2009) 452:1–12. doi: 10.1016/S0076-6879(08)03601-X
34. Lashgarian HE, Karkhane M, Marzban A, Yazdi M, Shahzamani K. Emerging Involvement of long non-coding RNAs in gastrointestinal associated inflammatory disorders. Comp Immunol Microbiol Infect Dis. (2020) 69:101428. doi: 10.1016/j.cimid.2020.101428
35. Zhu H, Wang Q, Yao Y, Fang J, Sun F, Ni Y, et al. Microarray analysis of Long non-coding RNA expression profiles in human gastric cells and tissues with Helicobacter pylori Infection. BMC Med Genomics. (2015) 8:84. doi: 10.1186/s12920-015-0159-0
36. Peng X, Gralinski L, Armour CD, Ferris MT, Thomas MJ, Proll S, et al. Unique signatures of long noncoding RNA expression in response to virus infection and altered innate immune signaling. mBio. (2010) 1(5):e00206–10. doi: 10.1128/mBio.00206-10
37. Hu G, Gong A-Y, Wang Y, Ma S, Chen X, Chen J, et al. LincRNA-cox2 promotes late inflammatory gene transcription in macrophages through modulating SWI/SNF-mediated chromatin remodeling. J Immunol. (2016) 196:2799–808. doi: 10.4049/jimmunol.1502146
38. Tong Q, Gong AY, Zhang XT, Lin C, Ma S, Chen J, et al. LincRNA-Cox2 modulates TNF-α-induced transcription of Il12b gene in intestinal epithelial cells through regulation of Mi-2/NuRD-mediated epigenetic histone modifications. FASEB J. (2016) 30:1187–97. doi: 10.1096/fj.15-279166
39. Ma S, Ming Z, Gong AY, Wang Y, Chen X, Hu G, et al. A long noncoding RNA, lincRNA-Tnfaip3, acts as a coregulator of NF-κB to modulate inflammatory gene transcription in mouse macrophages. FASEB J. (2017) 31:1215–25. doi: 10.1096/fj.201601056R
40. Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. (2012) 81:145–66. doi: 10.1146/annurev-biochem-051410-092902
41. Mathy NW, Chen X-M, Carman GM. Long non-coding RNAs (lncRNAs) and their transcriptional control of inflammatory responses. J Biol Chem. (2017) 292(30):12375–82. doi: 10.1074/jbc.R116.760884
42. Xiao L, Subauste C, Carruthers VB, Chen Y, Du J, Wu M, et al. The host autophagy during Toxoplasma Infection. Host Autophagy Dur Toxoplasma Infect Front Microbiol. (2020) 11:589604. doi: 10.3389/fmicb.2020.589604
43. Priyamvada S, Jayawardena D, Bhalala J, Kumar A, Anbazhagan AN, Alrefai WA, et al. Cryptosporidium parvum infection induces autophagy in intestinal epithelial cells. Cell Microbiol. (2021) 23:13298. doi: 10.1111/cmi.13298
44. El-Refai SA, Helwa MA, Rakha EB, Atia AF. Autophagy-related 16-like 1 single nucleotide gene polymorphism increases the risk and severity of Cryptosporidium parvum infection. Mol Biochem Parasitol. (2021) 245:111407. doi: 10.1016/j.molbiopara.2021.111407
45. Yang H, Zhang M, Wang X, Gong P, Zhang N, Zhang X, et al. Cryptosporidium parvum maintains intracellular survival by activating the host cellular EGFR-PI3K/Akt signaling pathway. Mol Immunol. (2023) 154:69–79. doi: 10.1016/j.molimm.2023.01.002
46. Heo I, Dutta D, Schaefer DA, Iakobachvili N, Artegiani B, Sachs N, et al. Modelling Cryptosporidium infection in human small intestinal and lung organoids. Nat Microbiol. (2018) 3:814–23. doi: 10.1038/s41564-018-0177-8
47. Gibson AR, Sateriale A, Dumaine JE, Engiles JB, Pardy R, Gullicksrud JA, et al. A genetic screen identifies a protective type III interferon response to Cryptosporidium that requires TLR3 dependent recognition. PloS Pathog. (2022) 18:1010003. doi: 10.1371/journal.ppat.1010003
48. Greigert V, Saraav I, Son J, Zhu Y, Dayao D, Antia A, et al. Cryptosporidium infection of human small intestinal epithelial cells induces type III interferon and impairs infectivity of Rotavirus. Gut Microbes. (2024) 16:555581. doi: 10.1080/19490976.2023.2297897
49. Tessema TS, Schwamb B, Lochner M, Förster I, Jakobi V, Petry F. Dynamics of gut mucosal and systemic Th1/Th2 cytokine responses in interferon-gamma and interleukin-12p40 knock out mice during primary and challenge Cryptosporidium parvum infection. Immunobiology. (2009) 214:454–66. doi: 10.1016/j.imbio.2008.11.015
50. Chen W, Harp JA, Harmsen AG, Havell’ EA. Gamma interferon functions in resistance to cryptosporidium parvum infection in severe combined immunodeficient mice. INFECrION Immun. (1993) 61(8):3548–51. doi: 10.1128/iai.61.8.3548-3551.1993
51. Barakat FM, McDonald V, Di Santo JP, Korbel DS. Roles for NK cells and an NK cell-independent source of intestinal gamma interferon for innate immunity to Cryptosporidium parvum infection. Infect Immun. (2009) 77:5044–9. doi: 10.1128/IAI.00377-09
52. Li XC, Jevnikar AM, Grant DR. Expression of functional ICAM-1 and VCAM-1 adhesion molecules by an immortalized epithelial cell clone derived from the small intestine. Cell Immunol. (1997) 175:58–66. doi: 10.1006/cimm.1996.1050
53. Niranjanakumari S, Lasda E, Brazas R, Garcia-Blanco MA. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods. (2002) 26(2):182–90.
54. Subramaniam D, Ramalingam S, May R, Dieckgraefe BK, Berg DE, Pothoulakis C, et al. Gastrin-mediated interleukin-8 and cyclooxygenase-2 gene expression: differential transcriptional and posttranscriptional mechanisms. Gastroenterology. (2008) 134(4):1070–82. doi: 10.1053/j.gastro.2008.01.040
Keywords: cryptosporidium, interferon, lncRNAs, Nostrill, cell-intrinsic defense, intestinal epithelium, transcription
Citation: Sharmin Z, Jin K, Gong A-Y, Deng S, Pok C, Graham ML, Wang S, Mathy NW, Shibata A and Chen X-M (2024) LncRNA Nostrill promotes interferon-γ-stimulated gene transcription and facilitates intestinal epithelial cell-intrinsic anti-Cryptosporidium defense. Front. Immunol. 15:1397117. doi: 10.3389/fimmu.2024.1397117
Received: 06 March 2024; Accepted: 24 June 2024;
Published: 08 July 2024.
Edited by:
Reginaldo G. Bastos, Agricultural Research Service (USDA), United StatesReviewed by:
Celio Geraldo Freire-de-Lima, Federal University of Rio de Janeiro, BrazilSonali Das, National Heart, Lung, and Blood Institute (NIH), United States
Copyright © 2024 Sharmin, Jin, Gong, Deng, Pok, Graham, Wang, Mathy, Shibata and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xian-Ming Chen, eGlhbl9tX2NoZW5AcnVzaC5lZHU=