 Mengya Zhao
Mengya Zhao Yiming Guan
Yiming Guan Jin Lin
Jin Lin Yu Qiu1
Yu Qiu1 Shen Zhao
Shen Zhao Meili Duan
Meili Duan- 1Department of Critical Care Medicine, Beijing Friendship Hospital, Capital Medical University, Beijing, China
- 2Department of Nephrology, Beijing Friendship Hospital, Capital Medical University, Beijing, China
Hemophagocytic lymphohistiocytosis (HLH) is an immune dysfunction characterized by an exaggerated and pathological inflammatory response, potentially leading to systemic inflammatory reactions and multiple-organ failure, including renal involvement. HLH can be classified as primary or secondary, with primary HLH associated with genetic mutations affecting cell degranulation capacity, and secondary HLH often linked to infections, tumors, and autoimmune diseases. The pathogenesis of HLH is not fully understood, but primary HLH is typically driven by genetic defects, whereas secondary HLH involves the activation of CD8+ T cells and macrophages, leading to the release of inflammatory cytokines and systemic inflammatory response syndrome (SIRS). The clinical presentation of HLH includes non-specific manifestations, making it challenging to differentiate from severe sepsis, particularly secondary HLH due to infections. Shared features include prolonged fever, hepatosplenomegaly, hematopenia, hepatic dysfunction, hypertriglyceridemia, and hypofibrinogenemia, along with histiocytosis and hemophagocytosis. However, distinctive markers like dual hemocytopenia, hypertriglyceridemia, hypofibrinogenemia, and elevated sCD25 levels may aid in differentiating HLH from sepsis. Indeed, no singular biomarker effectively distinguishes between hemophagocytic lymphohistiocytosis and infection. However, research on combined biomarkers provides insights into the differential diagnosis. Renal impairment is frequently encountered in both HLH and sepsis. It can result from a systemic inflammatory response triggered by an influx of inflammatory mediators, from direct damage caused by these factors, or as a consequence of the primary disease process. For instance, macrophage infiltration of the kidney can lead to structural damage affecting various renal components, precipitating disease. Presently, tubular necrosis remains the predominant form of renal involvement in HLH-associated acute kidney injury (HLH-AKI). However, histopathological changes may also encompass interstitial inflammation, glomerular abnormalities, microscopic lesions, and thrombotic microangiopathy. Treatment approaches for HLH and sepsis diverge significantly. HLH is primarily managed with repeated chemotherapy to eliminate immune-activating stimuli and suppress hypercellularity. The treatment approach for sepsis primarily focuses on anti-infective therapy and intensive symptomatic supportive care. Renal function significantly influences clinical decision-making, particularly regarding the selection of chemotherapy and antibiotic dosages, which can profoundly impact patient prognosis. Conversely, renal function recovery is a complex process influenced by factors such as disease severity, timely diagnosis, and the intensity of treatment. A crucial aspect in managing HLH-AKI is the timely diagnosis, which plays a pivotal role in reversing renal impairment and creating a therapeutic window for intervention, may have opportunity to improve patient prognosis. Understanding the clinical characteristics, underlying causes, biomarkers, immunopathogenesis, and treatment options for hemophagocytic lymphohistiocytosis associated with acute kidney injury (HLH-AKI) is crucial for improving patient prognosis.
Introduction
Hemophagocytic syndromes (HPS), also known as hemophagocytic lymphohistiocytosis (HLH), represent an immune dysfunction characterized by an exaggerated and pathological inflammatory response, resulting in hypercytokinemia and potential multiorgan failure. This condition can lead to significant damage in target organs, with acute kidney injury (AKI) being particularly noteworthy as a strong predictor of poor prognosis. Morbidity in HLH patients, reaching up to 50%, underscores the severity of the disease (1–4). Given its rapid progression and high mortality rates (5, 6), early diagnosis and intervention are pivotal for improving patient outcomes.
Recognizing the critical role of renal involvement, especially AKI, it becomes imperative to comprehend the clinical presentation, diagnosis, and treatment of HLH. Identifying high-risk factors for AKI in HLH patients and promptly admitting them to intensive care units for intervention are essential components of patient care. Unfortunately, the early recognition of AKI in HLH patients is presently underestimated, with limited reported studies. A comprehensive understanding of the incidence, clinical features, and prognosis of AKI in HLH patients is still lacking. Addressing these gaps in knowledge is crucial for enhancing the overall management and prognosis of patients with HLH.
In summary, this article provides an overview of the clinical manifestations, underlying causes, biomarkers, immunopathogenesis, and management strategies for patients presenting with hemophagocytic syndrome complicated by acute kidney injury (AKI), intended to assist clinicians in their understanding and management of this condition.
Clinical features
Hemophagocytic lymphohistiocytosis (HLH) is a rare and inadequately diagnosed disease with an unknown true prevalence due to the non-specific nature of its clinical presentation. Consequently, the epidemiology of HLH-associated acute kidney injury (HLH-AKI) remains poorly characterized.
First documented by Scott and Robb-Smith in 1939, HLH is a syndrome characterized by fever, hepatosplenomegaly, peripheral hypoplasia, and macrophage proliferation in the bone marrow and other hematopoietic organs (7). The initial familial case was reported by Farquhar in 1952 (8), and infection-associated HLH was recognized as a distinct clinical and pathologic feature in 1979 (9). The term “Hemophagocytic lymphohistiocytosis” was officially adopted by the International Histiocyte Association in 1991 (10).
The clinical presentation of HLH is heterogeneous, constituting a group of diseases marked by prolonged fever, hepatosplenomegaly, hematopenia, hepatic dysfunction, hypertriglyceridemia, and hypofibrinogenemia. It is characterized by histiocyte proliferation and increased hemophagocytosis. The HLH-2004 criteria (11) remain internationally recognized for diagnosis (Table 1) but are more tailored to pediatric cases and have limitations when applied to adults. The 2023 HiHASC guidelines (12) propose rapid identification markers, such as fever, decreased blood counts, and elevated ferritin levels; ferritin thresholds are not the same in adults and children. The guidelines recommend that the quick screen should be available within 6 h–12 h. Confirmation of HLH diagnosis and investigation into potential triggers or underlying causes should be completed within the subsequent 24 h–48 h. Among the scoring systems utilized to assess HLH probability in patients, the H-score is presently deemed most suitable for adults (13). It incorporates three clinical variables (underlying immunosuppression, fever, organomegaly), five routine blood tests (ferritin, triglycerides, aspartate aminotransferase, fibrinogen, and cytopenias), and one histopathologic factor (tissue phagocytosis), resulting in a total score of 337 points. The probability of diagnosing HLH exceeds 99% for scores exceeding 250, with a cutoff value of 169. With ongoing improvements in clinical diagnosis leading to increased case identification, secondary HLH is more frequently diagnosed. It often occurs in conjunction with various underlying disease states, particularly in patients with malignant tumors, severe infections, and various critical illnesses causing organ dysfunction, including renal impairment. HLH secondary to rheumatic diseases, is designated as macrophage activation syndrome (MAS). MAS, a subtype of HLH, represents a severe complication of rheumatic diseases, arising from the dysregulated activation and proliferation of T lymphocytes and macrophages. MAS, particularly with heightened phagocytic activity, presents a grave and potentially lethal condition. Rheumatic disorders implicated in MAS encompass systemic juvenile idiopathic arthritis (SJIA), systemic lupus erythematosus (SLE), Kawasaki disease (KD), and dermatomyositis, each characterized by clinical features indicative of HLH. Secondary HLH manifests through diverse etiologies, as outlined in Table 2, detailing the underlying diseases contributing to HLH development (14).

Table 1 Diagnostic criteria for HLH-2004.
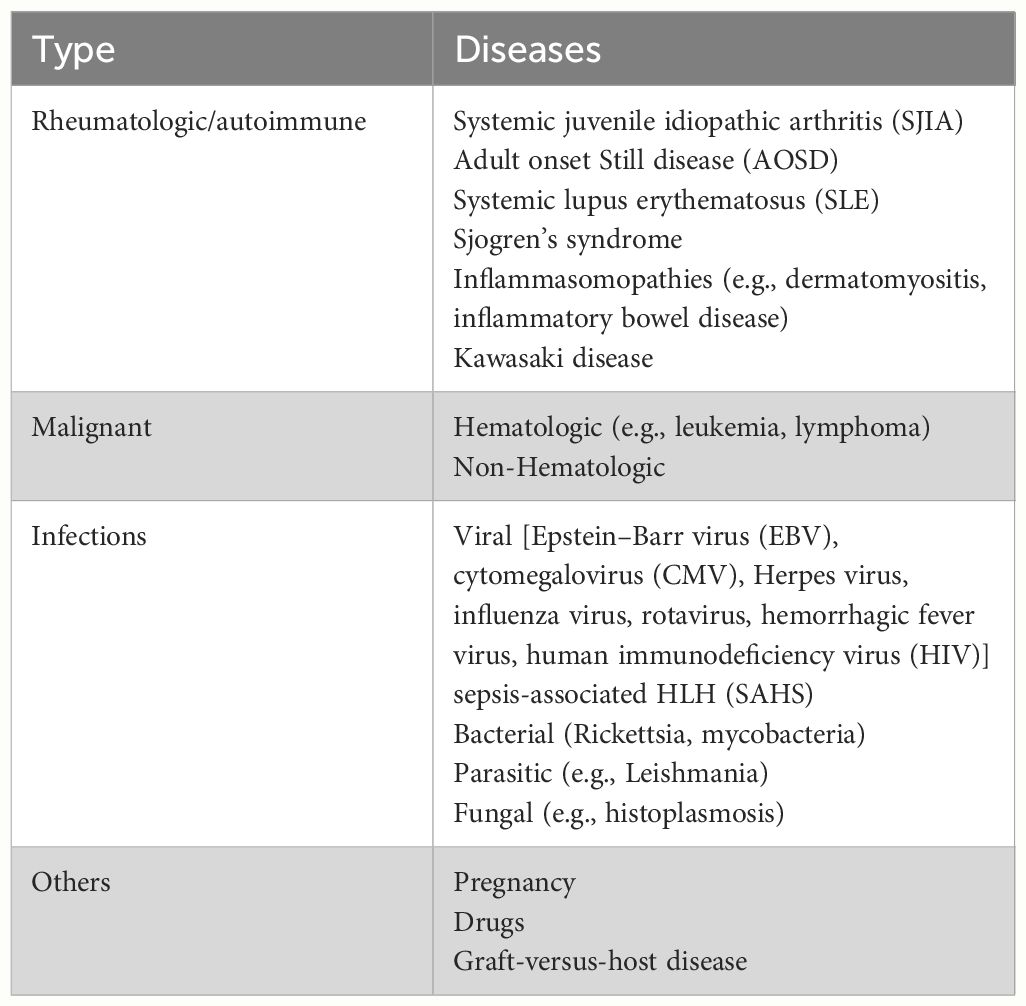
Table 2 HLH-related secondary diseases.
However, discerning patients with hemophagocytic lymphohistiocytosis (HLH) from those experiencing severe sepsis presents a formidable clinical challenge. While experienced hematologists typically conduct early screening for suspected HLH cases, routine assessments for the majority of critically ill patients with similar clinical presentations rarely include specialized tests such as sCD25 and NK cell activity. This limited utilization of advanced diagnostic tools may result in the oversight or delayed diagnosis of HLH in a subset of patients, contributing to the rapid progression of the syndrome and elevated rates of morbidity and mortality. Incorporating these specialized tests into routine evaluations for critically ill patients with risk factors could enhance diagnostic accuracy, facilitating more timely interventions and potentially improving outcomes in cases of HLH.
Secondary HLH, particularly when induced by infection, closely aligns with the definition of sepsis, manifesting as organ dysfunction triggered by a cytokine storm. Due to their overlapping features, approximately one-fifth of septic patients may exhibit characteristics resembling HLH (15). In cases of septic shock, misdiagnosis with HLH is not uncommon (16) and the diagnosis of HLH is not sufficient for patients admitted to the ICU (17). Routine diagnostic evaluations of patients with systemic inflammatory syndromes at the Mayo Clinic have revealed that the prevalence of HLH in sepsis patients can be as high as 1 in 2000 (18). Notably, the phenomenon of tissue phagocytosis lacks sensitivity and specificity, making it challenging to differentiate between secondary HLH and sepsis. Phagocytosis is also observed in a subset of sepsis cases, complicating the distinction between phagocytosis directly induced by sepsis versus that triggered by sepsis-associated secondary HLH (17, 19, 20). In a multinational study involving 362 patients with systemic juvenile idiopathic arthritis (JIA) complicated by macrophage activation syndrome (MAS), phagocytosis was identified in only 60.7% of patients upon bone marrow examination (21). This underscores the complexity of the relationship between infection and HLH. The intricate relationship between HLH and sepsis is compounded by HLH’s inherent immune dysregulation. Moreover, chemotherapy and immunosuppressive treatments exacerbate mucosal damage, intensify immune dysfunction, and induce cytopenias, rendering patients more vulnerable to secondary infections. A study noted that 56% of HLH patients exhibited concurrent infections, with 67% succumbing directly to infectious complications (22).
HLH can be further complicated by infections during treatment due to immunosuppression. Additionally, infections can give rise to reactive hemophagocytic syndrome (RHS) (23), which may contribute to multiple-organ dysfunction syndrome (MODS). Both HLH and RHS may occur simultaneously, potentially triggered by the same cytokines. However, there is currently insufficient evidence to support a direct causal relationship between the two.
Regrettably, there is no specific diagnostic test for HLH and diagnosis continues to rely on clinical presentation. Efforts to enhance diagnostic precision in critically ill patients, particularly those with overlapping symptoms of HLH and sepsis, remain a critical area for future research.
Therefore, we propose several features that may distinguish HLH from sepsis:
1. Hematologic and immunologic markers:
● Double hemocytopenia, hypertriglyceridemia, hypofibrinogenemia, and elevated sCD25 levels are less common in sepsis and may indicate true HLH (24).
● Methemoglobinemia is a distinctive feature of HLH, and nearly all cases exhibit markedly elevated serum ferritin levels. However, significant variations in ferritin levels exist among patients with HLH, sepsis, infectious shock, and other conditions. The highest ferritin levels, observed in patients with HLH especially in deceased patients (25, 26), serve as well-recognized markers of HLH activity and prognosis (27, 28). Nevertheless, defining a precise cutoff value for serum ferritin remains controversial (25, 29).
● Hypofibrinogenemia, particularly reported in children, may prove useful in identifying patients with sepsis/HLH overlap (30).
2. Renal manifestations in HLH-AKI:
● Patients with HLH-AKI typically present with a clear clinical picture, including oliguria, azotemia, and nephrotic syndrome. Approximately 24% exhibit proteinuria in the nephrotic range (31), which may or may not be accompanied by evidence of phagocytic activity.
● The Kidney Disease Improving Global Outcomes (KDIGO) (32) criteria are widely recognized as the standard for assessing renal impairment in critically ill patients (Table 3).
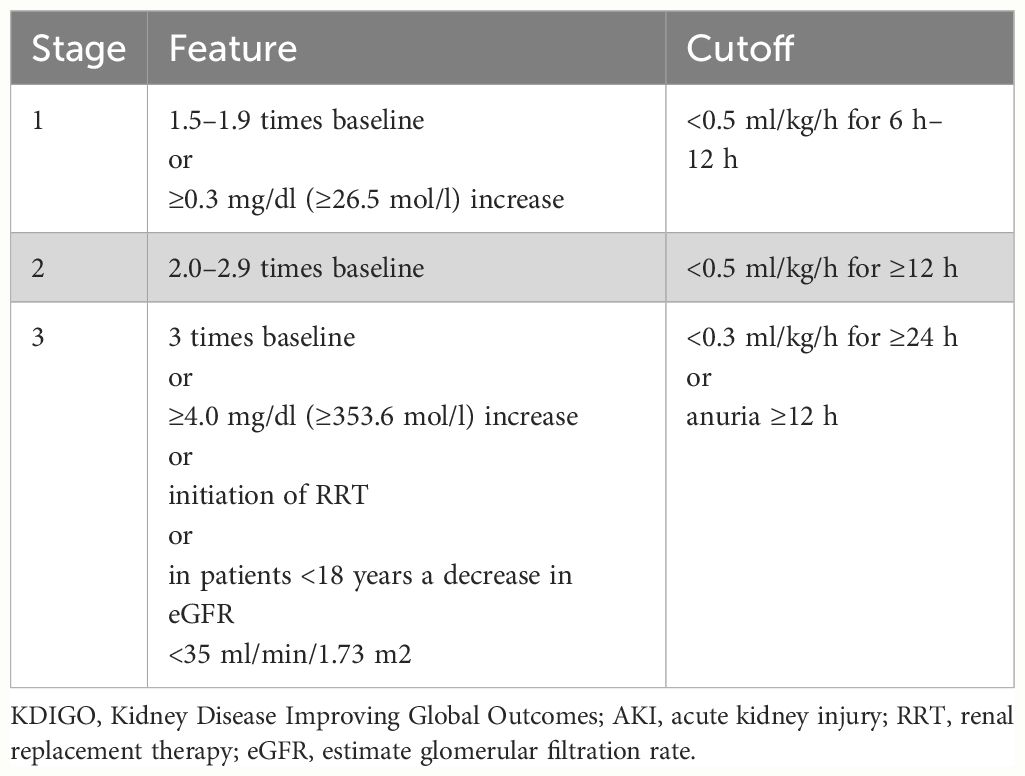
Table 3 Diagnostic criteria for KDIGO staging of AKI.
These proposed features aim to facilitate the differentiation between HLH and sepsis, providing clinicians with valuable clinical indicators to enhance diagnostic accuracy, especially in critically ill patients.
Causes
The etiology of HLH-AKI, characterized by acute kidney injury complicated by phagocytic syndrome, can be broadly categorized into direct injury by inflammatory mediators, hemodynamic instability, tumor-associated renal injury, and the application of nephrotoxic drugs. It is challenging to pinpoint the specific cause of AKI, as it may involve multiple factors simultaneously.
Familial cases of HLH reported in the literature highlight activated macrophage infiltration as a direct consequence of specific renal parenchymal involvement (33). These cases confirm that HLH induces direct inflammatory damage to the kidneys. Patients in these familial cases responded well to high-dose intravenous glucocorticoids and continuous blood purification, showing a reversible transition from renal injury.
Secondary HLH, frequently triggered by infectious diseases, constitutes 44.5% of the etiology (34), with viruses, particularly herpesviruses like EBV and CMV, being predominant pathogens. Severe infectious cases can precipitate septic shock, resulting in renal hypoperfusion. Concurrently, the amplified cascade of inflammatory responses, triggered by severe infections, prompts the release of diverse inflammatory factors directly affecting the kidneys. This cascade leads to renal dysfunction, suggesting that the primary cause of acute kidney injury (AKI) is attributable to both renal hypoperfusion and the direct impact of inflammatory mediators on kidney function. Aulagnon et al. reported that hypoperfusion was the second-highest contributor to AKI etiology in 95 cases of secondary HLH, with most patients experiencing renal dysfunction at KDIGO stages 2 or 3 (31).
In patients with hematologic malignancies, AKI accounts for 35% of the etiology (34), primarily associated with B-cell lymphoma. Tumor-induced monoclonal gammopathy, renal injury due to malignant cell infiltration, and paraneoplastic tumors are potential AKI causes (35). Prerenal and acute tubular necrosis are common, often stemming from the direct effects of tumors, tumor complications, and chemotherapy.
Examination-related drugs, antitumor drugs, and anti-infective drugs used in diagnosis and treatment are frequently nephrotoxic, contributing to AKI in HLH patients. Previous exposure to nephrotoxic drugs has been reported in 72.3% of AKI patients (2), involving agents such as contrast agents, vancomycin, aminoglycosides, and nonsteroidal anti-inflammatory drugs, as well as methotrexate, siderophores, cyclophosphamide, and oxaliplatin. Recommended therapy for HLH include chemotherapy such as etoposide, which is cleared by the kidney.
Pathophysiology
Primary HLH predominantly affects children, and immune dysfunction is typically irreversible. The primary cause is often autosomal recessive, resulting from defects in the release of cytoplasmic granules containing enzymes like perforin and granzyme B by cytotoxic T cells and NK cells. HLH leads to compromised cytotoxic T-cell and natural killer (NK) cell functionality. Pathogenic mutations in four genes (PRF1, UNC13D, STX11, and STXBP2) are identified (36–39), all impacting the perforin pathway crucial for lymphocyte cytotoxicity. These enzymes are crucial for cytotoxic activity, and their deficiency leads to uncontrolled proliferation of lymphocytes and macrophages. The subsequent phagocytosis is amplified by the excessive production of pro-inflammatory cytokines (Figures 1, 2).
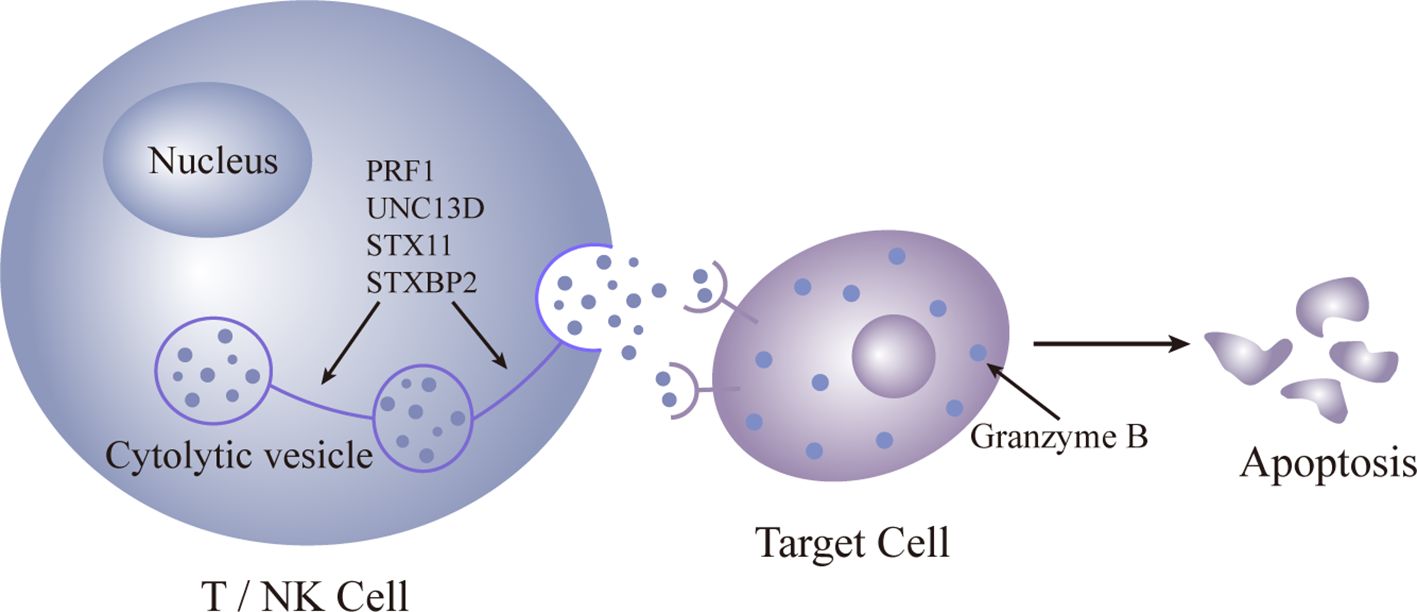
Figure 1 Pattern diagram of normal gene expression.
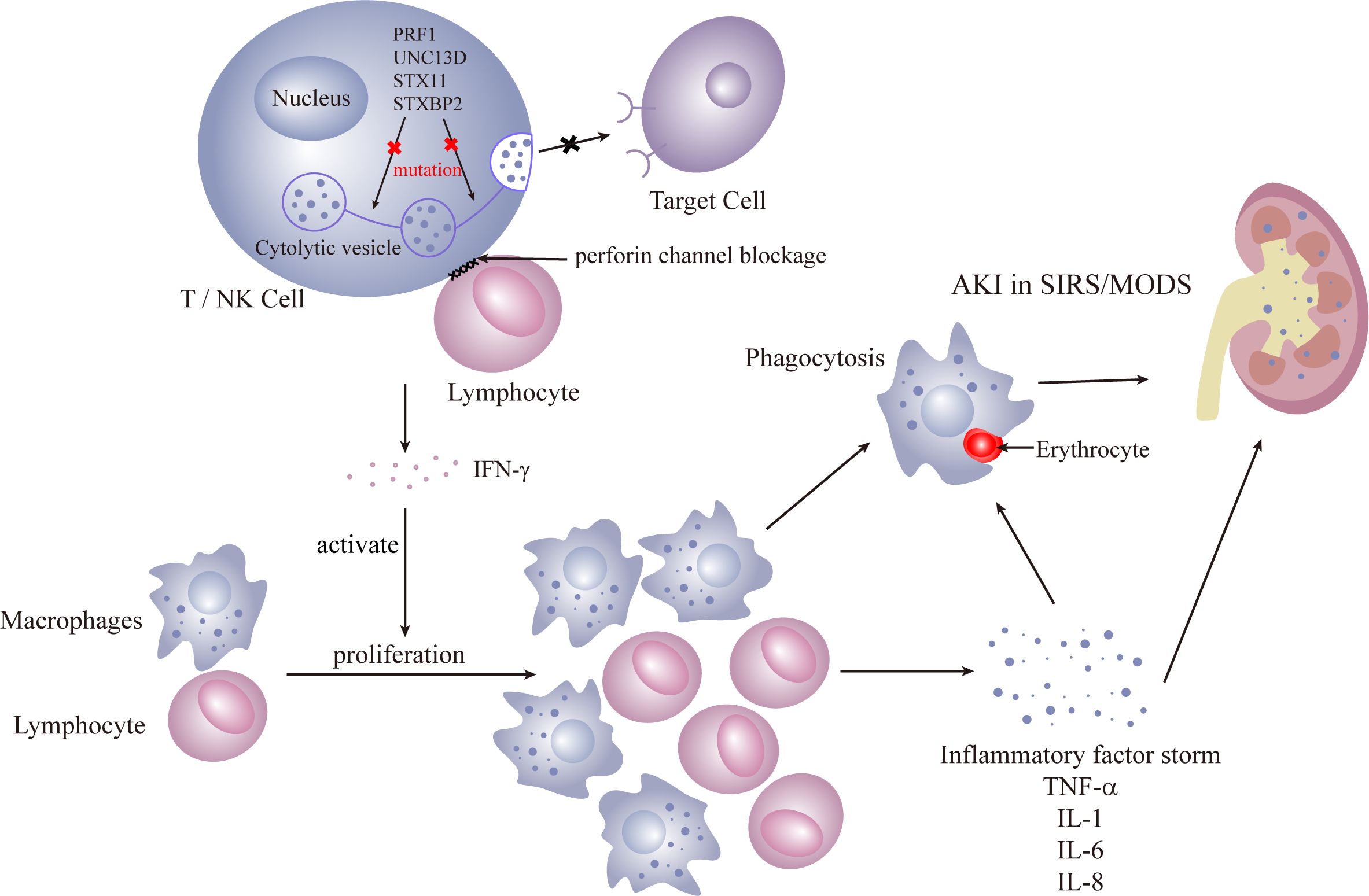
Figure 2 Primary HLH.
Secondary HLH, more prevalent in adults, is triggered by infections, malignancies, and autoinflammatory/immune disorders. Infections are the most common cause, with the initial response coming from activation of antigen-presenting cells (APC), initiating a TH-1 response that induces cytotoxic T cells and NK cells to proliferate. Reversible dysfunction of NK or CD8+ T cells characterizes immune dysfunction. The release of interferon (IFN) and granulocyte-macrophage colony-stimulating factor (GM-CSF) by cytotoxic T cells leads to sustained expansion and activation of cytotoxic CD8+ T cells and macrophages, histiocyte proliferation, and infiltration into organs, including the kidneys. Renal infiltration of activated cytotoxic T cells and macrophages is recognized as a potential mechanism of AKI (40). Inflammatory factors such as TNF, IL-1, and IL-6 contribute to a cytokine storm, resulting in tissue damage, severe systemic inflammatory response syndrome (SIRS), and multiorgan failure (41) (Figure 3).
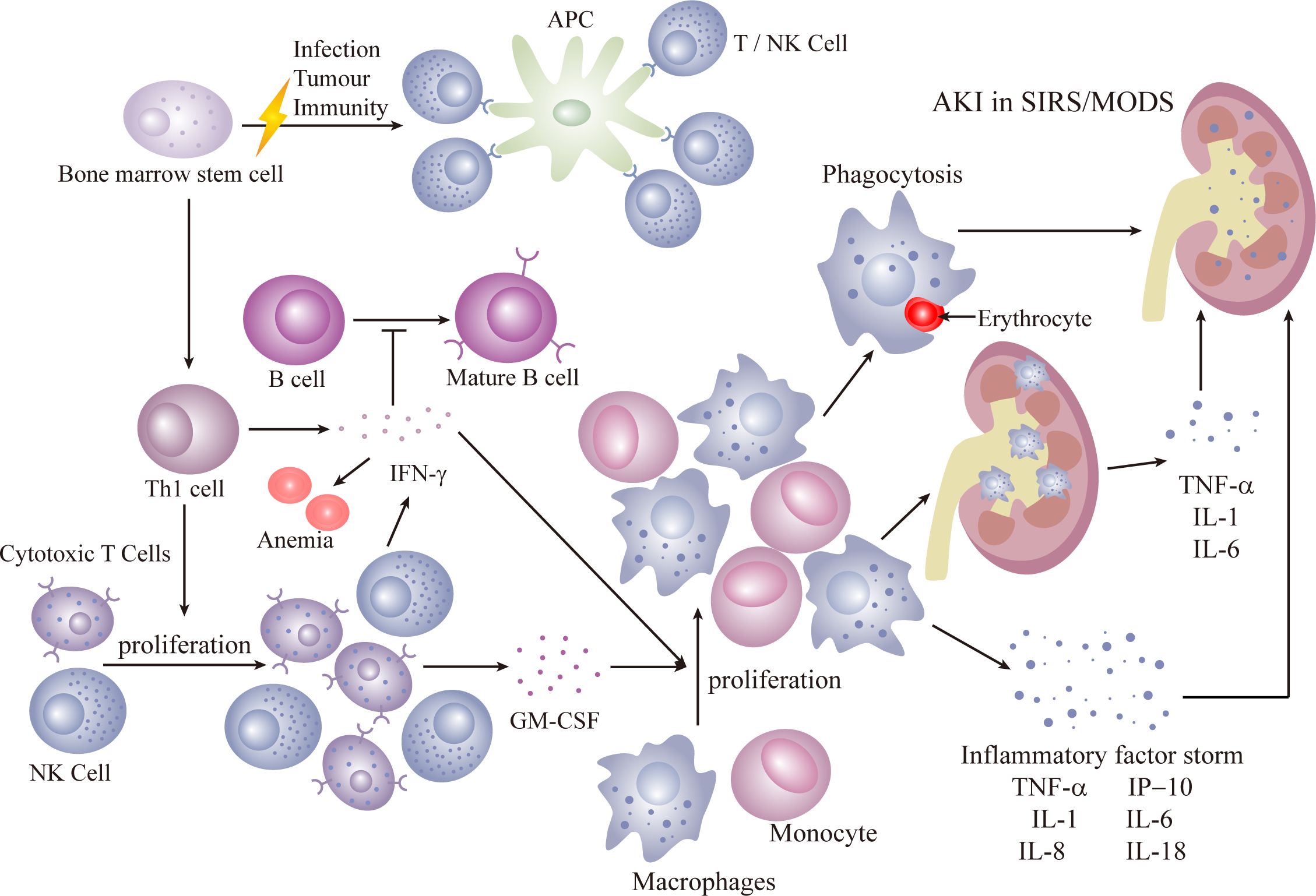
Figure 3 Secondary HLH.
Both primary and secondary HLH involve an overactivation of the immune response, causing a cytokine storm that can lead to multiple-organ insufficiencies, including renal complications. Hallmarks of HLH include bone marrow infiltration and the presence of non-malignant macrophages in organs. Non-malignant macrophages, associated with phagocytic activity, can affect various kidney structures. In HLH-AKI, tubular involvement is prevalent (31), often manifesting as acute tubular necrosis, with an incidence of up to 62%. The majority of AKI cases progress to KDIGO 2 or KDIGO 3. Histopathological alterations reveal acute tubular necrosis accompanied by interstitial inflammation, observed in 45% of HLH patients (1). Tubulointerstitial damage may arise from hemodynamic shifts and coagulation disorders during the acute phase of severe sepsis. While glomerular involvement is infrequent, it can lead to collapsed glomerulopathy, focal segmental glomerulosclerosis, microscopic lesions, and thrombotic microangiopathy (3, 42–44). A direct deleterious effect of pro-inflammatory cytokines on the kidney is evident. Observations suggest that the direct toxic impact of pro-inflammatory cytokines (IL-6 and TNF-α) on renal tubular cells may be a mechanism in the development of AKI in HLH (1). Glomerular hemophagocytic macrophages may locally release nephrotoxic cytokines (45), inducing dysfunction in the local glomerular capillary wall and resulting in protein leakage, leading to proteinuria and renal dysfunction. Infiltration of the renal parenchyma by activated macrophages can contribute to reversible AKI (33).
Biomarkers
Given the substantial morbidity and mortality associated with HLH-AKI and its non-specific clinical presentation, clinicians face a significant challenge in timely diagnosis and treatment. To enhance early identification of this patient group and improve clinical outcomes, various biomarkers have been reported. However, these biomarkers are often case-specific, lacking a standardized marker for accurate identification. Based on recent reports, we summarize the current understanding of these biomarkers as follows (Table 4).
1. Glycosylated ferritin:
● HLH patients exhibit impaired cytotoxic function characterized by diminished perforin expression, leading to heightened cell lysis. Additionally, there is rapid ferritin synthesis, which may surpass the glycosylation process, or active suppression of glycosylation, resulting in reduced levels of glycosylated ferritin (46, 48–51).
● Reduced percentage of glycosylated ferritin is considered a specific marker for confirming HLH diagnosis (46, 67).
● Studies suggest that the pattern of released inflammatory cytokines may aid in differentiating HLH from infections and other causes of methemoglobinemia (48, 52, 60, 64, 68).
● As a widely distributed globular protein, ferritin comprises 24 heavy chain (FtH) and light chain (FtL) subunits. Its expression escalates in both HLH and sepsis, with hyperferritinemia typically correlating with sepsis severity (69, 70). FtL may facilitate immunomodulation and mitigate septic acute kidney injury (AKI). Some studies indicate that ferritin levels ≥500 ng/ml aid HLH diagnosis (71), whereas others propose a threshold of >13,405 ng/ml for distinguishing infection from HLH (72). Zhang et al. put forth a diagnostic framework concerning ferritin (73).
● Ferritin mediates a positive feedback loop involving the activation of inflammatory vesicles, resulting in increased production of IL-1 and IL-18. Elevated extracellular ferritin levels subsequently induce activation of innate immune cells while concurrently suppressing adaptive immune cell function (47).
● Elevated plasma catalytic ferric ion levels were correlated with increased rates of death/requirement for renal replacement therapy, AKI, and in-hospital mortality. This association was observed in a study comprising 121 critically ill patients admitted to medical or surgical intensive care units (74).
3. IL-18 and new diagnostic parameters:
● IL-18, synthesized primarily by activated macrophages and prominently expressed in cytotoxic lymphocytes, stimulates the secretion of additional pro-inflammatory cytokines. It augments the impact of IFN-γ production by lymphocytes, thereby amplifying the inflammatory response (47, 53, 60).
● Ferritin, IL-18, and glycosylated ferritin are proposed as effective parameters for early HLH diagnosis.
● A new scale combining IL-18 with the H-Score is considered more specific. IL-18 levels are reported to be higher in HLH caused by malignant diseases compared with infections (47). Chronic elevation of IL-18 has shown a significant correlation with HLH (60).
● Furthermore, IL-18 has emerged as a potential novel biomarker for AKI. Studies indicate that IL-18-deficient mice exhibit protection against ischemia/reperfusion-induced AKI (75). Administration of IL-18-binding protein before ischemia/reperfusion injury has been shown to mitigate renal injury in rats (76).
4. Soluble interleukin 2 receptor (sCD25):
● Reactive T-lymphocyte activation and proliferation, along with the expansion of transplanted lymphocytes, can lead to immune dysfunction and interference with normal immune processes (48, 52).
● Serum sCD25 is another important marker reflecting immune activation in autoimmune diseases, tumors, and infections (55).
● O Soluble CD25 (sCD25) can be directly shed from the surface of tumor cells. Higher levels of sCD25 are observed in patients with malignant tumors; excessively low levels (≤2,400 U/ml) of sIL-2r help exclude HLH (52).
5. CD38high/HLA-DR+ cell expression:
● CD38high/HLA-DR+ cells are indicative of activated T cells. Increased CD38high/HLA-DR+ cell expression in T cells from HLH patients is proposed as a potential diagnostic marker. Expression in patients with sepsis is indistinguishable from that of healthy individuals (65).
● Further evidence from large prospective studies is needed to validate its diagnostic utility.
6. Procalcitonin (PCT):
● Procalcitonin (PCT), as a marker of inflammation, aids in distinguishing between bacterial and non-bacterial infections. It is frequently employed in the diagnosis of acute infections and for monitoring infection progression. PCT monitoring facilitates the evaluation of infection response and informs the development of anti-infective treatment strategies (73). The utilization of PCT for guiding antimicrobial therapy in severe infections and sepsis has been documented (77).
● Serum procalcitonin levels exhibit notable elevation in individuals with severe bacterial infections compared with those with viral infections or non-specific inflammatory conditions (78). Research in patients with critical infections demonstrates that utilizing PCT to guide therapy effectively reduces antibiotic utilization and correlates significantly with improved mortality rates (79). Furthermore, studies involving patients with systemic lupus erythematosus (SLE) indicate that PCT can facilitate early detection of bacterial or fungal infections during the febrile phase of SLE (80), enabling differentiation between infectious etiologies and active SLE. In individuals with concurrent renal insufficiency, PCT serves as a reliable marker for bacterial infections (81, 82).
● Several studies have noted a correlation between procalcitonin levels and ferritin, hinting at the possibility that PCT is involved in the cytokine pattern of HLH (83). Specific mechanisms underlying this relationship warrant further investigation in subsequent studies.
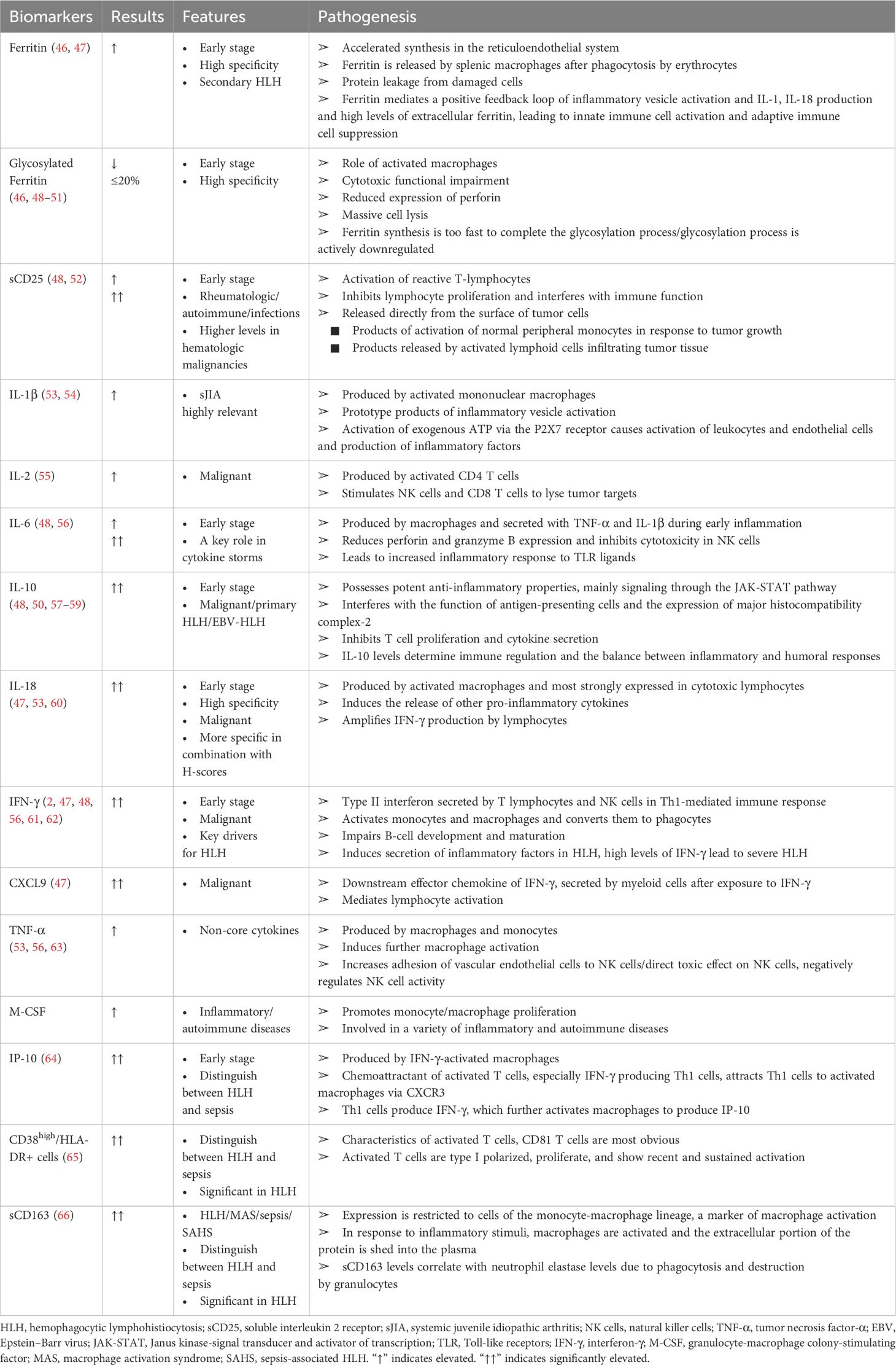
Table 4 HLH-related biomarkers.
No singular biomarker effectively distinguishes between HLH and sepsis; however, some studies explore combined biomarkers providing insights into the differential diagnosis. A distinctive cytokine profile characterized by markedly elevated levels of IFN-γ and IL-10, coupled with moderately elevated IL-6 levels, exhibits high diagnostic accuracy for HLH (48). This cytokine pattern aids in distinguishing HLH from infection. Additionally, the cytokine profile consisting of IL-18, IL-1β, and IFN-γ demonstrates good sensitivity and specificity in identifying severe infections (84).
In conclusion, whereas various biomarkers show promise in aiding HLH diagnosis, the lack of a standardized marker necessitates ongoing research and validation through large-scale prospective studies.
Treatment
Despite the similarities between HLH and sepsis, including multiorgan dysfunction and similar clinical features, their treatment approaches differ significantly. HLH treatment focuses on repetitive chemotherapy to eliminate immune activation stimuli and suppress hypercytokinemia, patients with rapid clinical deterioration are the target population for HLH chemotherapy, and potential triggers need to be identified and treated in patients whose clinical status is stable, whereas sepsis treatment primarily relies on anti-infective therapy and intensive symptomatic care (Table 5, Figure 4).
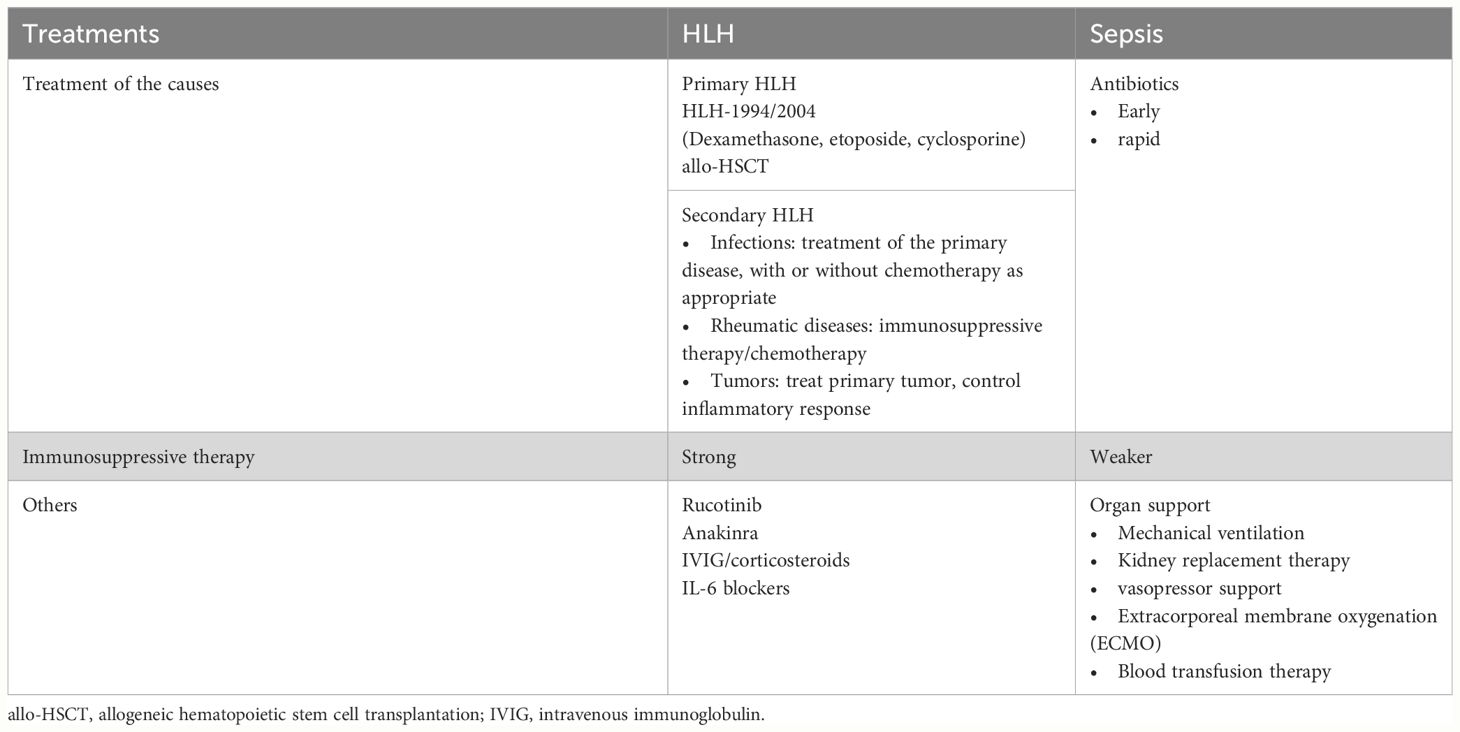
Table 5 Treatments of HLH and sepsis.
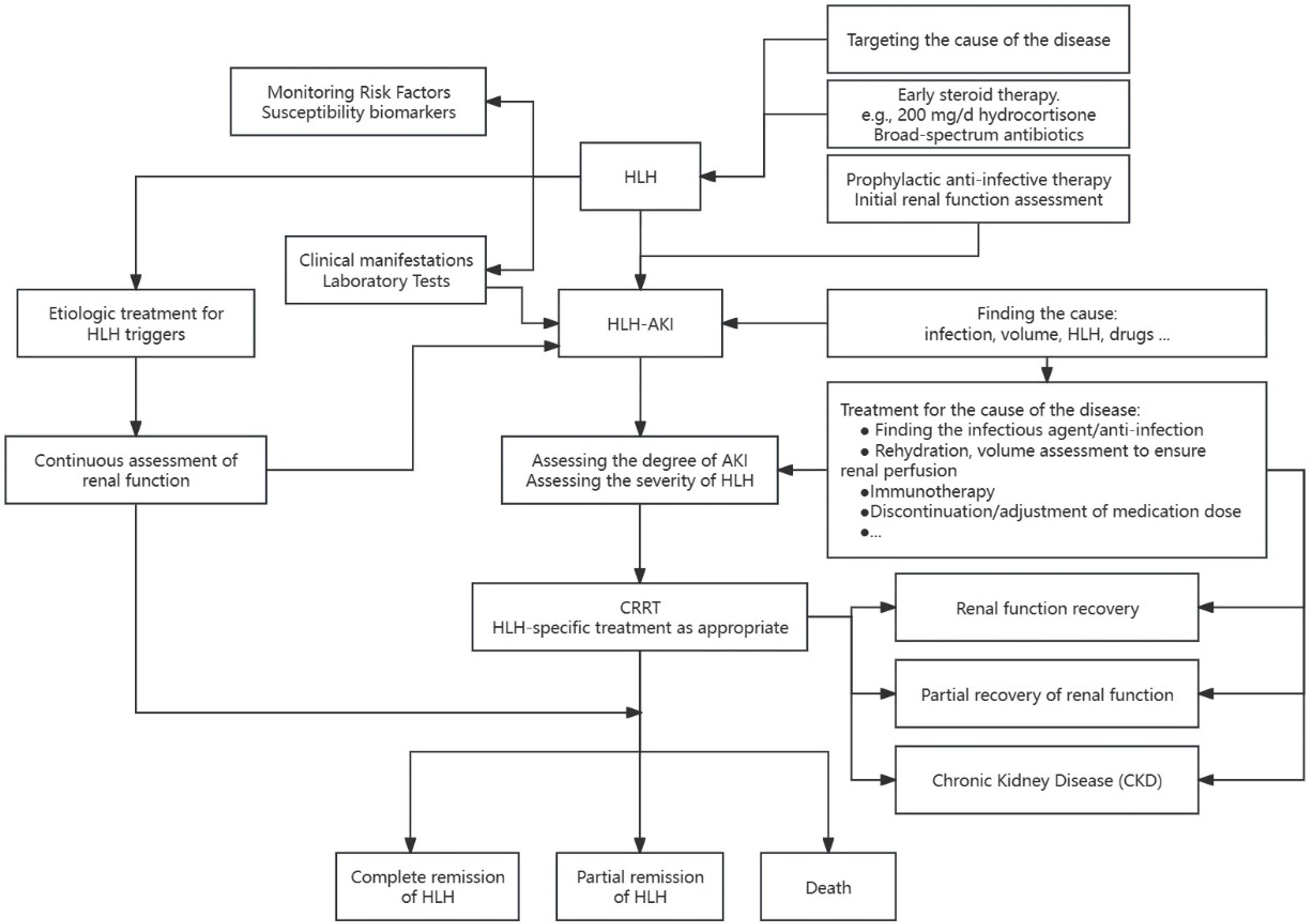
Figure 4 Management of patients with HLH-AK.
HLH treatment
● Cause-specific therapy involves induction therapy, following the HLH-1994/2004 regimen with dexamethasone, etoposide, with or without cyclosporine, and allogeneic hematopoietic stem cell transplantation (allo-HSCT) for familial, persistent, and recurrent HLH (11).
● Treatment protocols are largely based on pediatric studies, and their applicability to adults is uncertain. It has also been suggested that the intensity of immunosuppression in pediatric HLH94 protocols is neither appropriate nor necessary for most adults (21, 85). Weaker immunosuppressive and cytotoxic methods may be preferable for infection-related HLH (86).
● Ruxolitinib, a JAK1 and JAK2 inhibitor, and other treatments like interleukin-1 antagonist (anakinra), intravenous immunoglobulin (IVIG), and/or corticosteroids, have shown promise (87–93).
● No standardized protocols exist, requiring individualized treatment plans based on factors such as disease progression, inflammatory response, triggers, and concomitant diseases, with current experience relying primarily on observational studies and case reports.
Supportive interventions in critically ill patients
● Critically ill HLH patients, especially those with acute kidney injury, may require aggressive supportive interventions (94) such as broad-spectrum antibiotics, vasopressors, renal replacement therapy, mechanical ventilation, blood product transfusions and coagulopathy management, and even extracorporeal membrane oxygenation (ECMO) (5, 95, 96). Patients who fail to respond to initial therapy typically experience a poorer prognosis, often accompanied by a higher 30-day mortality rate (97).
● Early steroid therapy is valuable in stabilizing the disease, particularly in primary HLH and previously untreated secondary HLH (94), and that steroid therapy should not be delayed, especially in the presence of organ dysfunction. In difficult-to-diagnose cases, short-term treatment with corticosteroids is suggested (71), along with broad-spectrum antibiotics, as 200 mg/day of hydrocortisone has been shown to be beneficial in patients with sepsis shock (98),whereas if HLH is suspected, it is even more recommended that higher doses of corticosteroids be administered as early as possible, before the patient goes into sepsis shock. In acutely critically ill patients, the administration of full-dose chemotherapy regimens for treating HLH is suboptimal and can even lead to fatalities in some instances (99, 100). Consequently, guidelines advocate for early intervention directed at mitigating the cytokine storm through the use of reduced doses of etoposide (101).
● Mortality rates among critically ill patients with HLH have shown no recent decrease, and several factors may contribute to this trend. These include high-intensity treatment leading to severe immunosuppression, which is secondary to severe infections. The inappropriate application of drugs, such as in cases of combined hepatic and renal dysfunction, emphasizes the need to reduce the dose of etoposide rather than discontinuing the drug (18, 94, 102–104). Additionally, delayed diagnosis and treatment could also play a role in the sustained mortality rates.
Continuous renal replacement therapy
● Continuous renal replacement therapy (CRRT) serves as a crucial adjunctive support for renal function replacement in patients with HLH-AKI, contributing to internal environment stability and inflammatory factor removal.
● There are no standardized protocols for the treatment of HLH-AKI, either in case reports or in clinical studies, and the selection of the timing of treatment, the application of different filters, the formulation of the therapeutic dose, and the assessment of the end of treatment are all based on the experience of individual clinical hospitals, and the choices vary widely from patient to patient and from physician to physician (26, 66).
● While continuous renal replacement therapy (CRRT) has been reported to provide stabilization in patients, the persistently high morbidity and mortality rates may be associated with factors such as the severity of the disease, the timeliness of diagnosis, and the intensity of treatment. Moreover, the long-term prognosis of this patient cohort remains uncertain, with potential progression to chronic kidney disease (CKD) (40).
In conclusion, while advancements have been made in HLH treatment strategies, challenges persist, emphasizing the need for individualized approaches and ongoing research. The effectiveness of CRRT in HLH-AKI underscores its role as a supportive measure, but further studies are warranted to establish standardized protocols.
Conclusions
Timely diagnosis plays a pivotal role in the identification and management of patients with hemophagocytic lymphohistiocytosis (HLH), facilitating the reversal of renal impairment and creating a therapeutic window for HLH intervention. Collaborative efforts involving hematologists, intensivists, and nephrologists are essential for a comprehensive understanding of HLH pathogenesis, early identification of critically ill patients, prompt initiation of immunomodulatory therapy, and the optimization of organ supportive care. These measures hold the potential to enhance the prognosis and increase the complete remission rate among patients with HLH. Future research endeavors should focus on further unraveling the complexities of HLH and refining multidisciplinary approaches for improved patient outcomes.
Author contributions
MZ: Writing – original draft, Conceptualization. YG: Writing – original draft, Visualization. JL: Writing – review & editing, Supervision. YQ: Writing – review & editing, Validation. SZ: Writing – review & editing, Validation, Supervision. MD: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Karras A. What nephrologists need to know about hemophagocytic syndrome. Nat Rev Nephrol. (2009) 5:329–36. doi: 10.1038/nrneph.2009.73
2. Canet E, Zafrani L, Lambert J, Thieblemont C, Galicier L, Schnell D, et al. Acute kidney injury in patients with newly diagnosed high-grade hematological Malignancies: impact on remission and survival. PloS One. (2013) 8:e55870. doi: 10.1371/journal.pone.0055870
3. Thaunat O, Delahousse M, Fakhouri F, Martinez F, Stephan J-L, Noe¨l L-H, et al. Nephrotic syndrome associated with hemophagocytic syndrome. Kidney Int. (2006) 69:1892–8. doi: 10.1038/sj.ki.5000352
4. Karapinar B, Yilmaz D, Balkan C, Akin M, Ay Y, Kvakli K. An unusual cause of multiple organ dysfunction syndrome in the pediatric intensive care unit: hemophagocytic lymphohistiocytosis. Pediatr Crit Care Med. (2009) 10:285–90. doi: 10.1097/PCC.0b013e318198868b
5. Buyse S, Teixeira L, Galicier L, Mariotte E, Lemiale V, Seguin A, et al. Critical care management of patients with hemophagocytic lymphohistiocytosis. Intensive Care Med. (2010) 36:1695–702. doi: 10.1007/s00134-010-1936-z
6. Barba T, Maucort-Boulch D, Iwaz J, Bohe´ J, Ninet J, Hot A, et al. Hemophagocytic lymphohistiocytosis in intensive care unit: A 71-case strobe-compliant retrospective study. Med (Baltimore). (2015) 94:e2318. doi: 10.1097/MD.0000000000002318
7. Scott RB, et al. Histiocytic medullary reticulocytosis. Lancet. (1939) 2:194–8. doi: 10.1016/S0140-6736(00)61951-7
8. Farquhar J, Macgregor A, Richmond J, et al. Familial hemophagocytic reticulocytosis. Arch Dis Child. (1952) 27:519–25. doi: 10.1136/adc.27.136.519
9. Risdall RJ, McKenna RW, Nesbit ME, Krivit W, Balfour HH Jr, Simmons RL, et al. Virus-associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from Malignant histiocytosis[J]. Cancer. (1979) 44:993–1002. doi: 10.1002/1097–0142(197909)44:3<993::aid-cncr2820440329<3.0.co;2–5
10. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society[J]. Semin Oncol. (1991) 18:29–33.
11. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis[J]. Pediatr Blood Cancer. (2007) 48:124–31. doi: 10.1002/pbc.21039
12. Cox MF, Mackenzie S, Low R, Brown M, Sanchez E, Carr A, et al. Diagnosis and investigation of suspected haemophagocytic lymphohistiocytosis in adults: 2023 Hyperinflammation and HLH Across Speciality Collaboration (HiHASC) consensus guideline[J]. Lancet Rheumatol. (2024) 6:e51–62. doi: 10.1016/S2665–9913(23)00273–4
13. Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome[J]. Arthritis Rheumatol. (2014) 66:2613–20. doi: 10.1002/art.38690
14. Cron RQ, Goyal G, Chatham WW. Cytokine storm syndrome[J]. Annu Rev Med. (2023) 74:321–37. doi: 10.1146/annurev-med-042921–112837
15. Bursa D, Bednarska A, Pihowicz A, Paciorek M, Horban A, et al. Analysis of the occurrence of hemophagocytic lymphohistiocytosis (HLH) features in patients with sepsis: a prospective study[J]. Sci Rep. (2021) 11:10529. doi: 10.1038/s41598–021-90046–4
16. Stéphan F, Thiolière B, Verdy E, Tulliez M, et al. Role of hemophagocytic histiocytosis in the etiology of thrombocytopenia in patients with sepsis syndrome or septic shock[J]. Clin Infect Dis. (1997) 25:1159–64. doi: 10.1086/516086
17. Strauss R, Neureiter D, Westenburger B, Wehler M, Kirchner T, Hahn EG, et al. Multifactorial risk analysis of bone marrow histiocytic hyperplasia with hemophagocytosis in critically ill medical patients–a postmortem clinicopathologic analysis[J]. Crit Care Med. (2004) 32:1316–21. doi: 10.1097/01.ccm.0000127779.24232.15
18. Parikh SA, Kapoor P, Letendre L, Kumar S, Wolanskyj AP, et al. Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis[J]. Mayo Clin Proc. (2014) 89:484–92. doi: 10.1016/j.mayocp.2013.12.012
19. Gupta A, Weitzman S, Abdelhaleem M. The role of hemophagocytosis in bone marrow aspirates in the diagnosis of hemophagocytic lymphohistiocytosis[J]. Pediatr Blood Cancer. (2008) 50:192–4. doi: 10.1002/pbc.21441
20. Ho C, Yao X, Tian L, Li FY, Podoltsev N, Xu ML, et al. Marrow assessment for hemophagocytic lymphohistiocytosis demonstrates poor correlation with disease probability[J]. Am J Clin Pathol. (2014) 141:62–71. doi: 10.1309/AJCPMD5TJEFOOVBW
21. Minoia F, Davì S, Horne A, Demirkaya E, Bovis F, Li C, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients[J]. Arthritis Rheumatol. (2014) 66:3160–9. doi: 10.1002/art.38802
22. Sung L, Weitzman SS, Petric M, King SM, et al. The role of infections in primary hemophagocytic lymphohistiocytosis: a case series and review of the literature[J]. Clin Infect Dis. (2001) 33:1644–8. doi: 10.1086/323675
23. Gauvin F, Toledano B, Champagne J, Lacroix J, et al. Reactive hemophagocytic syndrome presenting as a component of multiple organ dysfunction syndrome[J]. Crit Care Med. (2000) 28:3341–5. doi: 10.1097/00003246–200009000–00038
24. El-Sayed ZA, El-Owaidy RH, Khamis MA, Rezk AR, et al. Screening of hemophagocytic lymphohistiocytosis in children with severe sepsis in pediatric intensive care[J]. Sci Prog. (2021) 104:368504211044042. doi: 10.1177/00368504211044042
25. Lachmann G, Knaak C, Vorderwülbecke G, La Rosée P, Balzer F, Schenk T, et al. Hyperferritinemia in critically ill patients[J]. Crit Care Med. (2020) 48:459–65. doi: 10.1097/CCM.0000000000004131
26. Gregory J, Greenberg J, Basu S. Outcomes analysis of children diagnosed with hemophagocytic lymphohistiocytosis in the PICU[J]. Pediatr Crit Care Med. (2019) 20:e185–90. doi: 10.1097/PCC.0000000000001827
27. Janka G, zur Stadt U. Familial and acquired hemophagocytic lymphohistiocytosis[J]. Hematol Am Soc Hematol Educ Program. (2005) 1:82–8. doi: 10.1182/asheducation-2005.1.82
28. Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL, et al. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality[J]. Pediatr Blood Cancer. (2011) 56:154–5. doi: 10.1002/pbc.22774
29. Karakike E, Giamarellos-Bourboulis EJ. Macrophage activation-like syndrome: A distinct entity leading to early death in sepsis[J]. Front Immunol. (2019) 10:55. doi: 10.3389/fimmu.2019.00055
30. Signoff JK, Fitzgerald JC, Teachey DT, Balamuth F, Weiss SL, et al. Hypofibrinogenemia is associated with poor outcome and secondary hemophagocytic lymphohistiocytosis/macrophage activation syndrome in pediatric severe sepsis[J]. Pediatr Crit Care Med. (2018) 19:397–405. doi: 10.1097/PCC.0000000000001507
31. Aulagnon F, Lapidus N, Canet E, Galicier L, Boutboul D, Peraldi MN, et al. Acute kidney injury in adults with hemophagocytic lymphohistiocytosis[J]. Am J Kidney Dis. (2015) 65:851–9. doi: 10.1053/j.ajkd.2014.10.012
32. Khwaja A. KDIGO clinical practice guidelines for acute kidney injury[J]. Nephron Clin Pract. (2012) 120:c179–184. doi: 10.1159/000339789
33. Malaga-Dieguez L, Ming W, Trachtman H. Direct reversible kidney injury in familial hemophagocytic lymphohistiocytosis type 3[J]. J Am Soc Nephrol. (2015) 26:1777–80. doi: 10.1681/ASN.2014111090
34. Birndt S, Schenk T, Heinevetter B, Brunkhorst FM, Maschmeyer G, Rothmann F, et al. Hemophagocytic lymphohistiocytosis in adults: collaborative analysis of 137 cases of a nationwide German registry[J]. J Cancer Res Clin Oncol. (2020) 146:1065–77. doi: 10.1007/s00432–020-03139–4
35. Bridoux F, Cockwell P, Glezerman I, Gutgarts V, Hogan JJ, Jhaveri KD, et al. Kidney injury and disease in patients with haematological Malignancies[J]. Nat Rev Nephrol. (2021) 17:386–401. doi: 10.1038/s41581–021-00405–7
36. Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13–4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3)[J]. Cell. (2003) 115:461–73. doi: 10.1016/s0092–8674(03)00855–9
37. zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter JI, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11[J]. Hum Mol Genet. (2005) 14:827–34. doi: 10.1093/hmg/ddi076
38. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis[J]. Science. (1999) 286:1957–9. doi: 10.1126/science.286.5446.1957
39. Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis[J]. Blood. (2020) 135:1332–43. doi: 10.1182/blood.2019000936
40. Nahum E, Ben-Ari J, Stain J, Schonfeld T, et al. Hemophagocytic lymphohistiocytic syndrome: Unrecognized cause of multiple organ failure[J]. Pediatr Crit Care Med. (2000) 1:51–4. doi: 10.1097/00130478–200007000–00010
41. Janka GE, Lehmberg K. Hemophagocytic syndromes–an update[J]. Blood Rev. (2014) 28:135–42. doi: 10.1016/j.blre.2014.03.002
42. Santoriello D, Hogan J, D'Agati VD. Hemophagocytic syndrome with histiocytic glomerulopathy and intraglomerular hemophagocytosis[J]. Am J Kidney Dis. (2016) 67:978–83. doi: 10.1053/j.ajkd.2015.11.017
43. Bae MN, Kwak DH, Park SJ, Choi BS, Park CW, Choi YJ, et al. Acute kidney injury induced by thrombotic microangiopathy in a patient with hemophagocytic lymphohistiocytosis[J]. BMC Nephrol. (2016) 17:4. doi: 10.1186/s12882–015-0217-z
44. Chidambaram AC, Maulik K, Ramamoorthy JG, Parameswaran N, et al. A novel mutation of adenosine deaminase causing SCID presenting as hemophagocytic lymphohistiocytosis with acute kidney injury[J]. Br J Haematol. (2020) 191:509–12. doi: 10.1111/bjh.17058
45. Cao L, Wallace WD, Eshaghian S, Linhares Y, Marder VJ, et al. Glomerular hemophagocytic macrophages in a patient with proteinuria and clinical and laboratory features of hemophagocytic lymphohistiocytosis (HLH)[J]. Int J Hematol. (2011) 94:483–7. doi: 10.1007/s12185–011-0936–2
46. Fardet L, Coppo P, Kettaneh A, Dehoux M, Cabane J, Lambotte O, et al. Low glycosylated ferritin, a good marker for the diagnosis of hemophagocytic syndrome[J]. Arthritis Rheum. (2008) 58:1521–7. doi: 10.1002/art.23415
47. Debaugnies F, Mahadeb B, Nagant C, Meuleman N, De Bels D, Wolff F, et al. Biomarkers for early diagnosis of hemophagocytic lymphohistiocytosis in critically ill patients[J]. J Clin Immunol. (2021) 41:658–65. doi: 10.1007/s10875–020-00950-z
48. Xu XJ, Tang YM, Song H, Yang SL, Xu WQ, Zhao N, et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children[J]. J Pediatr. (2012) 160:984–990.e1. doi: 10.1016/j.jpeds.2011.11.046
49. Vandenhaute J, Wouters CH, Matthys P. Natural killer cells in systemic autoinflammatory diseases: A focus on systemic juvenile idiopathic arthritis and macrophage activation syndrome[J]. Front Immunol. (2019) 10:3089. doi: 10.3389/fimmu.2019.03089
50. Ou W, Zhao Y, Wei A, Ma H, Zhang Q, Zhang L, et al. Serum cytokine pattern in children with hemophagocytic lymphohistiocytosis[J]. Ann Hematol. (2023) 102:729–39. doi: 10.1007/s00277–023-05132–6
51. Grom AA. Natural killer cell dysfunction: A common pathway in systemic-onset juvenile rheumatoid arthritis, macrophage activation syndrome, and hemophagocytic lymphohistiocytosis?[J]. Arthritis Rheum. (2004) 50:689–98. doi: 10.1002/art.20198
52. Hayden A, Lin M, Park S, Pudek M, Schneider M, Jordan MB, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH[J]. Blood Adv. (2017) 1:2529–34. doi: 10.1182/bloodadvances.2017012310
53. Schulert GS, Grom AA. Pathogenesis of macrophage activation syndrome and potential for cytokine- directed therapies[J]. Annu Rev Med. (2015) 66:145–59. doi: 10.1146/annurev-med-061813–012806
54. Gloria L, David B. Understanding the mechanism of IL-1β secretion[J]. Cytokine Growth FACTOR Rev. (2011) 22:189–95. doi: 10.1016/j.cytogfr.2011.10.001
55. Bien E, Balcerska A. Serum soluble interleukin 2 receptor alpha in human cancer of adults and children: a review[J]. Biomarkers. (2008) 13:1–26. doi: 10.1080/13547500701674063
56. Zhang HQ, Yang SW, Fu YC, Chen MC, Yang CH, Yang MH, et al. Cytokine storm and targeted therapy in hemophagocytic lymphohistiocytosis. Immunol Res. (2022) 70:566–77. doi: 10.1007/s12026-022-09285-w
57. Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG, et al. Regulation and functions of the IL-10 family of cytokines in inflammation and disease[J]. Annu Rev Immunol. (2011) 29:71–109. doi: 10.1146/annurev-immunol-031210–101312
58. de Waal Malefyt R, Haanen J, Spits H, Roncarolo MG, te Velde A, Figdor C, et al. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression[J]. J Exp Med. (1991) 174:915–24. doi: 10.1084/jem.174.4.915
59. An Q, Hu SY, Xuan CM, Jin MW, Ji Q, Wang Y, et al. Interferon gamma and interleukin 10 polymorphisms in Chinese children with hemophagocytic lymphohistiocytosis[J]. Pediatr Blood Cancer. (2017) 64:e26505. doi: 10.1002/pbc.26505
60. Weiss ES, Girard-Guyonvarc'h C, Holzinger D, de Jesus AA, Tariq Z, Picarsic J, et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome[J]. Blood. (2018) 131:1442–55. doi: 10.1182/blood-2017–12-820852
61. Bracaglia C, de Graaf K, Pires Marafon D, Guilhot F, Ferlin W, Prencipe G, et al. Elevated circulating levels of interferon-γ and interferon-γ-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis[J]. Ann Rheum Dis. (2017) 76:166–72. doi: 10.1136/annrheumdis-2015–209020
62. Gao Z, Wang Y, Wang J, Zhang J, Wang Z, et al. The inhibitory receptors on NK cells and CTLs are upregulated in adult and adolescent patients with secondary hemophagocytic lymphohistiocytosis[J]. Clin Immunol. (2019) 202:18–28. doi: 10.1016/j.clim.2019.03.006
63. Crayne CB, Albeituni S, Nichols KE, Cron RQ, et al. The immunology of macrophage activation syndrome[J]. Front Immunol. (2019) 10:119. doi: 10.3389/fimmu.2019.00119
64. Maruoka H, Inoue D, Takiuchi Y, Nagano S, Arima H, Tabata S, et al. IP-10/CXCL10 and MIG/CXCL9 as novel markers for the diagnosis of lymphoma-associated hemophagocytic syndrome[J]. Ann Hematol. (2014) 93:393–401. doi: 10.1007/s00277–013-1878-y
65. Chaturvedi V, Marsh RA, Zoref-Lorenz A, Owsley E, Chaturvedi V, Nguyen TC, et al. T-cell activation profiles distinguish hemophagocytic lymphohistiocytosis and early sepsis[J]. Blood. (2021) 137:2337–46. doi: 10.1182/blood.2020009499
66. Cui Y, Zhang YC, Kang YL, Ren YQ, Miao HJ, Wang F, et al. High-volume hemofiltration in critically ill patients with secondary hemophagocytic lymphohistiocytosis/macrophage activation syndrome: A prospective study in the PICU[J]. Pediatr Crit Care Med. (2016) 17:e437–43. doi: 10.1097/PCC.0000000000000896
67. Nabergoj M, Marinova M, Binotto G, Brugnaro L, Zaninotto M, Plebani M, et al. Diagnostic and prognostic value of low percentage of glycosylated ferritin in acquired hemophagocytic lymphohistiocytosis: A single-center study[J]. Int J Lab Hematol. (2017) 39:620–4. doi: 10.1111/ijlh.12713
68. Cui Y, Xiong X, Ren Y, Wang F, Wang C, Zhang Y, et al. CD163 as a valuable diagnostic and prognostic biomarker of sepsis-associated hemophagocytic lymphohistiocytosis in critically ill children[J]. Pediatr Blood Cancer. (2019) 66:e27909. doi: 10.1002/pbc.27909
69. Kernan KF, Carcillo JA. Hyperferritinemia and inflammation[J]. Int Immunol. (2017) 29:401–9. doi: 10.1093/intimm/dxx031
70. Balla J, Balla G, Zarjou A. Ferritin in kidney and vascular related diseases: novel roles for an old player[J]. Pharm (Basel). (2019) 12:96. doi: 10.3390/ph12020096
71. Machowicz R, Janka G, Wiktor-Jedrzejczak W. Similar but not the same: Differential diagnosis of HLH and sepsis[J]. Crit Rev Oncol Hematol. (2017) 114:1–12. doi: 10.1016/j.critrevonc.2017.03.023
72. Fauter M, Mainbourg S, El Jammal T, Guerber A, Zaepfel S, Henry T, et al. Extreme hyperferritinemia: causes and prognosis[J]. J Clin Med. (2022) 11:5438. doi: 10.3390/jcm11185438
73. Zhang Y, Cheng Z, Hu Y, Tang LV, et al. Management of complex infections in hemophagocytic lymphohistiocytosis in adults[J]. Microorganisms. (2023) 11:1694. doi: 10.3390/microorganisms11071694
74. Leaf DE, Rajapurkar M, Lele SS, Mukhopadhyay B, Waikar SS, et al. Plasma catalytic iron, AKI, and death among critically ill patients[J]. Clin J Am Soc Nephrol. (2014) 9:1849–56. doi: 10.2215/CJN.02840314
75. Wu H, Craft ML, Wang P, Wyburn KR, Chen G, Ma J, et al. IL-18 contributes to renal damage after ischemia-reperfusion[J]. J Am Soc Nephrol. (2008) 19:2331–41. doi: 10.1681/ASN.2008020170
76. Wang J, Long Q, Zhang W, Chen N, et al. Protective effects of exogenous interleukin 18-binding protein in a rat model of acute renal ischemia-reperfusion injury[J]. Shock. (2012) 37:333–40. doi: 10.1097/SHK.0b013e318240bdc8
77. Tan M, Lu Y, Jiang H, Zhang L, et al. The diagnostic accuracy of procalcitonin and C-reactive protein for sepsis: A systematic review and meta-analysis[J]. J Cell Biochem. (2019) 120:5852–9. doi: 10.1002/jcb.27870
78. Branche AR, Walsh EE, Vargas R, Hulbert B, Formica MA, Baran A, et al. Serum procalcitonin measurement and viral testing to guide antibiotic use for respiratory infections in hospitalized adults: A randomized controlled trial[J]. J Infect Dis. (2015) 212:1692–700. doi: 10.1093/infdis/jiv252
79. de Jong E, van Oers JA, Beishuizen A, Vos P, Vermeijden WJ, Haas LE, et al. Efficacy and safety of procalcitonin guidance in reducing the duration of antibiotic treatment in critically ill patients: a randomised, controlled, open-label trial[J]. Lancet Infect Dis. (2016) 16:819–27. doi: 10.1016/S1473–3099(16)00053–0
80. Serio I, Arnaud L, Mathian A, Hausfater P, Amoura Z, et al. Can procalcitonin be used to distinguish between disease flare and infection in patients with systemic lupus erythematosus: a systematic literature review[J]. Clin Rheumatol. (2014) 33:1209–15. doi: 10.1007/s10067–014-2738–4
81. Dumea R, Siriopol D, Hogas S, Mititiuc I, Covic A, et al. Procalcitonin: diagnostic value in systemic infections in chronic kidney disease or renal transplant patients[J]. Int Urol Nephrol. (2014) 46:461–8. doi: 10.1007/s11255–013-0542–8
82. Lu XL, Xiao ZH, Yang MY, Zhu YM, et al. Diagnostic value of serum procalcitonin in patients with chronic renal insufficiency: a systematic review and meta-analysis[J]. Nephrol Dial Transplant. (2013) 28:122–9. doi: 10.1093/ndt/gfs339
83. Strenger V, Merth G, Lackner H, Aberle SW, Kessler HH, Seidel MG, et al. Malignancy and chemotherapy induced haemophagocytic lymphohistiocytosis in children and adolescents-a single centre experience of 20 years[J]. Ann Hematol. (2018) 97:989–98. doi: 10.1007/s00277–018-3254–4
84. Luo H, Wang N, Huang L, Zhou X, Jin J, Li C, et al. Inflammatory signatures for quick diagnosis of life-threatening infection during the CAR T-cell therapy[J]. J Immunother Cancer. (2019) 7:271. doi: 10.1186/s40425–019-0767-x
85. Bergsten E, Horne A, Aricó M, Astigarraga I, Egeler RM, Filipovich AH, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study[J]. Blood. (2017) 130:2728–38. doi: 10.1182/blood-2017–06-788349
86. Rajajee S, Ashok I, Manwani N, Rajkumar J, Gowrishankar K, Subbiah E, et al. Profile of hemophagocytic lymphohistiocytosis; efficacy of intravenous immunoglobulin therapy[J]. Indian J Pediatr. (2014) 81:1337–41. doi: 10.1007/s12098–014-1461–0
87. Parajuli B, Angurana SK, Awasthi P, Nallasamy K, Baranwal A, Bansal A, et al. Hemophagocytic lymphohistiocytosis in a PICU of a developing economy: clinical profile, intensive care needs, outcome, and predictors of mortality[J]. Pediatr Crit Care Med. (2021) 22:e44–57. doi: 10.1097/PCC.0000000000002539
88. Yan WL, Yang SL, Zhao FY, Xu XJ, et al. Ruxolitinib is an alternative to etoposide for patient with hemophagocytic lymphohistiocytosis complicated by acute renal injury: A case report[J]. J Oncol Pharm Pract. (2022) 28:222–7. doi: 10.1177/10781552211020821
89. Wang J, Zhang R, Wu X, Li F, Yang H, Liu L, et al. Ruxolitinib-combined doxorubicin-etoposide-methylprednisolone regimen as a salvage therapy for refractory/relapsed haemophagocytic lymphohistiocytosis: a single-arm, multicentre, phase 2 trial[J]. Br J Haematol. (2021) 193:761–8. doi: 10.1111/bjh.17331
90. Wohlfarth P, Agis H, Gualdoni GA, Weber J, Staudinger T, Schellongowski P, et al. Interleukin 1 receptor antagonist anakinra, intravenous immunoglobulin, and corticosteroids in the management of critically ill adult patients with hemophagocytic lymphohistiocytosis[J]. J Intensive Care Med. (2019) 34:723–31. doi: 10.1177/0885066617711386
91. Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial[J]. Crit Care Med. (2016) 44:275–81. doi: 10.1097/CCM.0000000000001402
92. Eloseily EM, Weiser P, Maruoka H, Haines H, Mannion ML, Stoll ML, et al. Benefit of anakinra in treating pediatric secondary hemophagocytic lymphohistiocytosis[J]. Arthritis Rheumatol. (2020) 72:326–34. doi: 10.1002/art.41103
93. Miettunen PM, Narendran A, Jayanthan A, Behrens EM, Cron RQ, et al. Successful treatment of severe paediatric rheumatic disease-associated macrophage activation syndrome with interleukin-1 inhibition following conventional immunosuppressive therapy: case series with 12 patients[J]. Rheumatol (Oxford). (2011) 50:417–9. doi: 10.1093/rheumatology/keq218
94. Hines MR, von Bahr Greenwood T, Beutel G, Beutel K, Hays JA, Horne A, et al. Consensus-based guidelines for the recognition, diagnosis, and management of hemophagocytic lymphohistiocytosis in critically ill children and adults[J]. Crit Care Med. (2022) 50:860–72. doi: 10.1097/CCM.0000000000005361
95. Knaak C, Schuster FS, Spies C, Vorderwülbecke G, Nyvlt P, Schenk T, et al. Hemophagocytic lymphohistiocytosis in critically ill patients[J]. Shock. (2020) 53:701–9. doi: 10.1097/SHK.0000000000001454
96. Leow EH, Soh SY, Tan AM, Mok YH, Chan MY, Lee JH, et al. Critically ill children with hemophagocytic lymphohistiocytosis: A case series of 14 patients[J]. J Pediatr Hematol Oncol. (2017) 39:e303–6. doi: 10.1097/MPH.0000000000000916
97. Wang Y, Huang W, Hu L, Cen X, Li L, Wang J, et al. Multicenter study of combination DEP regimen as a salvage therapy for adult refractory hemophagocytic lymphohistiocytosis[J]. Blood. (2015) 126:2186–92. doi: 10.1182/blood-2015–05-644914
98. Annane D. Corticosteroids for severe sepsis: an evidence-based guide for physicians[J]. Ann Intensive Care. (2011) 1:7. doi: 10.1186/2110–5820-1–7
99. Jin Z, Wang Y, Wei N, Wang Z, et al. Hodgkin lymphoma-associated hemophagocytic lymphohistiocytosis-a dangerous disease[J]. Ann Hematol. (2020) 99:1575–81. doi: 10.1007/s00277–020-04093–4
100. Nicholson MC, Naeije L, Hayden AR, Mattman A, Dix D, Chen LYC, et al. Etoposide-based treatment of adult HLH is associated with high biochemical response but poor survival outcomes[J]. EJHaem. (2020) 1:277–80. doi: 10.1002/jha2.57
101. La Rosée P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults[J]. Blood. (2019) 133:2465–77. doi: 10.1182/blood.2018894618
102. Janus N, Launay-Vacher V, Byloos E, Machiels JP, Duck L, Kerger J, et al. Cancer and renal insufficiency results of the BIRMA study[J]. Br J Cancer. (2010) 103:1815–21. doi: 10.1038/sj.bjc.6605979
103. Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL, et al. How I treat hemophagocytic lymphohistiocytosis[J]. Blood: J Am Soc Hematol. (2011) 118:4041–52. doi: 10.1182/blood-2011-03-278127
Keywords: hemophagocytic lymphohistiocytosis, acute kidney injury, sepsis, critical care, intensive care unit
Citation: Zhao M, Guan Y, Lin J, Qiu Y, Zhao S and Duan M (2024) Acute kidney injury in critical care: complications of hemophagocytic lymphohistiocytosis. Front. Immunol. 15:1396124. doi: 10.3389/fimmu.2024.1396124
Received: 05 March 2024; Accepted: 29 May 2024;
Published: 18 June 2024.
Edited by:
Andrzej Lange, Polish Academy of Sciences, PolandReviewed by:
Yasuhiro Shimojima, Shinshu University, JapanKatharina L. Willmann, Gulbenkian Institute of Science (IGC), Portugal
Copyright © 2024 Zhao, Guan, Lin, Qiu, Zhao and Duan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shen Zhao, anpremhhb3NoZW5AMTYzLmNvbQ==; Meili Duan, ZG1laWxpQGNjbXUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work