 Meng-Yao Xu†
Meng-Yao Xu† Zhi-Yu Xia†
Zhi-Yu Xia† Jian-Xuan Sun
Jian-Xuan Sun Chen-Qian Liu
Chen-Qian Liu Ye An
Ye An Jin-Zhou Xu
Jin-Zhou Xu Si-Han Zhang
Si-Han Zhang Xing-Yu Zhong
Xing-Yu Zhong Na Zeng
Na Zeng Si-Yang Ma
Si-Yang Ma Hao-Dong He
Hao-Dong He Shao-Gang Wang*‡
Shao-Gang Wang*‡ Qi-Dong Xia*‡
Qi-Dong Xia*‡- Department of Urology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
The emergence of resistance to prostate cancer (PCa) treatment, particularly to androgen deprivation therapy (ADT), has posed a significant challenge in the field of PCa management. Among the therapeutic options for PCa, radiotherapy, chemotherapy, and hormone therapy are commonly used modalities. However, these therapeutic approaches, while inducing apoptosis in tumor cells, may also trigger stress-induced premature senescence (SIPS). Cellular senescence, an entropy-driven transition from an ordered to a disordered state, ultimately leading to cell growth arrest, exhibits a dual role in PCa treatment. On one hand, senescent tumor cells may withdraw from the cell cycle, thereby reducing tumor growth rate and exerting a positive effect on treatment. On the other hand, senescent tumor cells may secrete a plethora of cytokines, growth factors and proteases that can affect neighboring tumor cells, thereby exerting a negative impact on treatment. This review explores how radiotherapy, chemotherapy, and hormone therapy trigger SIPS and the nuanced impact of senescent tumor cells on PCa treatment. Additionally, we aim to identify novel therapeutic strategies to overcome resistance in PCa treatment, thereby enhancing patient outcomes.
1 Introduction
Prostate cancer (PCa) is a significant health concern for men globally, and in the United States, PCa is the most common malignancy and the second leading cause of death among men (1), with over 1.2 million new cases diagnosed worldwide annually (2–4).
The European Association of Urology (EAU) suggests stratifying PCa based on risk factors, such as prostate-specific antigen (PSA) and Gleason score (GS), to assist medical professionals in the evidence-based management of different risk PCa and predict recurrence risk after definitive treatment (5). Among newly diagnosed cases of localized low-risk PCa, active surveillance (AS) or radical prostatectomy is advocated as the primary treatment options, while the remaining 15-20% of high-risk PCa patients necessitate a comprehensive treatment regimen comprising chemotherapy, radiation therapy, and hormone therapy (4, 6).
The treatment landscape for PCa has come a long way in recent decades, but the challenge of treatment resistance persists, particularly in managing intermediate- and high-risk patients. Treatment resistance, classified as either primary (intrinsic) or secondary (acquired), continues to hinder effective treatment (7, 8). Primary resistance occurs when the cancer does not respond to treatment at the outset, while secondary resistance represents the local or distant recurrence of malignancy, following an initial clinical response (9–11). Hormone therapy remains a cornerstone for managing advanced PCa, but after a median treatment time of 18~24 months, the majority of these patients progress to castration-resistant prostate cancer (CRPC) (12, 13), underscoring the limitations of current treatments including first-line anti-androgen therapies like enzalutamide, which are not immune to the emergence of primary or acquired drug resistance (12, 14). Chemotherapy, particularly with docetaxel as the first-line regimen, plays a critical role in treating high-risk PCa (15, 16), either as a standalone treatment or in combination with other agents like abiraterone and prednisone for metastatic CRPC or as adjunct therapy post-surgery (16, 17). Docetaxel used as chemotherapy mainly interferes with the microtube network essential for cell mitosis and interphase cell function, leading to cell cycle arrest (18). However, chemotherapy often initially shows the desired effect, but they are prone to develop resistance over time, leading to treatment failure. Similarly, radiation therapy, which targets tumor cells through direct DNA physical damage or indirect damage from active oxygen free radicals (19–21). Cancer cells can adapt to radiation-induced apoptosis through dynamic interactions and regulation of multiple survival factors, ultimately leading to treatment failure (17, 22).
Previously, it was believed that the emergence of treatment resistance in tumor cells was due to genomic instability (23). This instability arises from genetic alterations such as gene mutations, amplifications, deletions, or chromosome translocations. These changes can lead to target proteins mutating and unable to bind to drugs or activate downstream signaling molecules or alternative survival pathways. As a result, tumor cells evade treatment, continuing to survive and proliferate (24, 25). These genetic changes might originate from cancer stem cells (CSCs), contributing to primary (intrinsic) resistance, or develop during treatment, leading to secondary (acquired) resistance.
With the advancements in technologies that capture the magnitude and dynamics of both genetic and non-genetic intratumor heterogeneity in the 4D (spatial and temporal) space and at single-cell resolution (8), it is now understood that tumor cells may develop treatment resistance through non-genetic mechanisms. These on-genetic mechanisms mainly involve alterations in tumor cell phenotype, such as tumor cell plasticity (TCP) (9, 10), and adaptive changes in the tumor microenvironment (TME). TCP, through processes like epithelial-to-mesenchymal transition (EMT) (26, 27), transdifferentiation, and cancer stem cell formation (28), enables tumor cells to undergo irreversible phenotypic transitions. This versatility allows tumor cells to switch between proliferative, metastatic tumor cells, slow-cycling cells, and drug-tolerant persister (DTP) (29, 30). Currently, this adaptive non-genetic change is recognized as a common mechanism for treatment resistance in tumor cells.
Cellular senescence and the emergence of tumor treatment resistance are processes that transition from order to disorder. López-Otín et al. highlighted similarities between cellular senescence and cancer, identifying four meta-hallmarks of cellular senescence (genomic instability, epigenetic alteration, chronic inflammation, and dysbiosis) and four hallmarks of cancer (genomic instability & mutation, epigenetic reprogramming, tumor promoting inflammation, and polymorphic microbiomes) (31, 32). This similarity underscores the critical interaction between cellular senescence and tumor progression. In 1965, Hayflick’s discovery that normal human cells in culture have a limited capacity to divide, known as “Hayflick limit”, serves as a marker of cellular senescence, and the process is referred to as “replicative senescence” (33). Additionally, exposure to external stimuli such as reactive oxygen species (ROS) (34), DNA-damaging agents (35), or activated proto-oncogenes can induce stress-induced premature senescence (SIPS), resembling the senescent cell phenotype (36, 37). Similarly, local ionizing radiation inflicts DNA damage in PCa cells, both directly and indirectly through increasing ROS, akin to chemotherapy effects (38). High-dose chemoradiation triggers tumor cell apoptosis, while low-dose promotes SIPS in tumor cells. These senescent tumor cells secrete various chemotactic factors, cytokines, and extracellular matrix (ECM), which induces phenotypic transition (EMT, transdifferentiation, and CSCs formation) in other tumor cells, leading to treatment resistance (39).
This review aims to examine the current status of treatment resistance in PCa and explore its interaction with cellular senescence. By providing a comprehensive overview of treatment resistance in PCa, it assists clinician in better understanding and addressing this challenge in practice.
2 Cellular senescence
Cellular senescence is accompanied by changes in molecular markers, in addition to the degenerative changes in cellular morphological structures. Senescence-associated β-galactosidase (SA-β-Gal) activity increase is one of the important biomarkers of cellular senescence (40, 41). This enzyme hydrolyzes X-Gal substrate to produce insoluble blue crystals in cytoplasm (42), which can be observed under optical microscope. In senescent cells, lysosomes swell and increase, leading to a large accumulation of SA-β-Gal in lysosomes and a shift in pH from the normal range of 4.5 to 6.0 (43, 44). Another typical feature of senescent cells is the senescence-associated heterochromatin foci (SAHF), which are highly folded and tightly bound DNA double-strand structures that prevent DNA expression (45, 46). After DAPI staining, punctate heterochromatin structures can be observed in the cell nucleus under fluorescence microscope, indicating SAHF’s role in inhibiting the expression of genes regulated by transcription factors such as MCM3, PCNA, or cyclin A (47). Additionally, senescent cells develop a senescence-associated secretory phenotype (SASP) (48–50), consisting of a group of pro-inflammatory (IL-1, IL-6, TNF-α), pro-angiogenic (VEGF, FGF), and growth stimulating factors (amphiregulin, epiregulin, angiogenin) (51, 52). The NF-κB signaling pathway plays a key role in regulating SASP expression (52, 53), while other signaling pathways such as cGAS-STING, p38 MAPK, and mTOR are also closely related to SASP expression (49, 50, 54).
The molecular mechanism of cellular senescence is a complex and multifaceted process that involves the interaction of multiple pathways and factors. The P53-P21 pathway is crucial for cellular senescence, primarily causing telomere shortening and cellular senescence caused by DNA damage (55). Upon DNA damage, P53 initiates the expression of its downstream gene, p21, which encodes a Cyclin-dependent protein kinase (Cdk) inhibitor to inhibit the Cyclin E-CDK2 activity. This prevention stops Cyclin-Cdk from phosphorylating Rb, keeping Rb unphosphorylated. The unphosphorylated Rb maintains binding with E2F, preventing the activation of the transcription regulator E2F, which ultimately results in cell cycle arrest at the G1 phase (56, 57). The p16-pRb pathway also matters for cellular senescence (58). p16 impedes the phosphorylation of the Cyclin D-CDK4/6-PBRB complex (59), keeping PRB in an active, non-phosphorylated state. This state allows pRb to attach to E2F proteins, obstructing the transcriptional activation of effectors of the cell cycle progression target proteins (57). The efficacy of these mechanisms relies on p16 keeping pRb active and non-phosphorylated. Furthermore, the significance of the PTEN-p27 pathway in cellular senescence is undeniable. PTEN, known for its tumor-suppressing properties, possesses both lipid and protein phosphatase activities that counteract the PI3K/AKT pathway (60). Mutation or deletion in PTEN prevent the dephosphorylation of Phosphatidylinositol-3,4,5-triphosphate (PIP3), leading to the activation of the PI3K/AKT signaling pathway (61, 62). p27kip1, a Cyclin-dependent kinase inhibitor, attaches to CDK2, blocking its kinase activity and halting cell cycle progression. p27 has a high expression level in the G1 phase and reduces to a minimum level in the S phase (63). PTEN upregulates p27 expression and decrease cyclin D1 activity to regulates the G1-S transition (Figure 1).
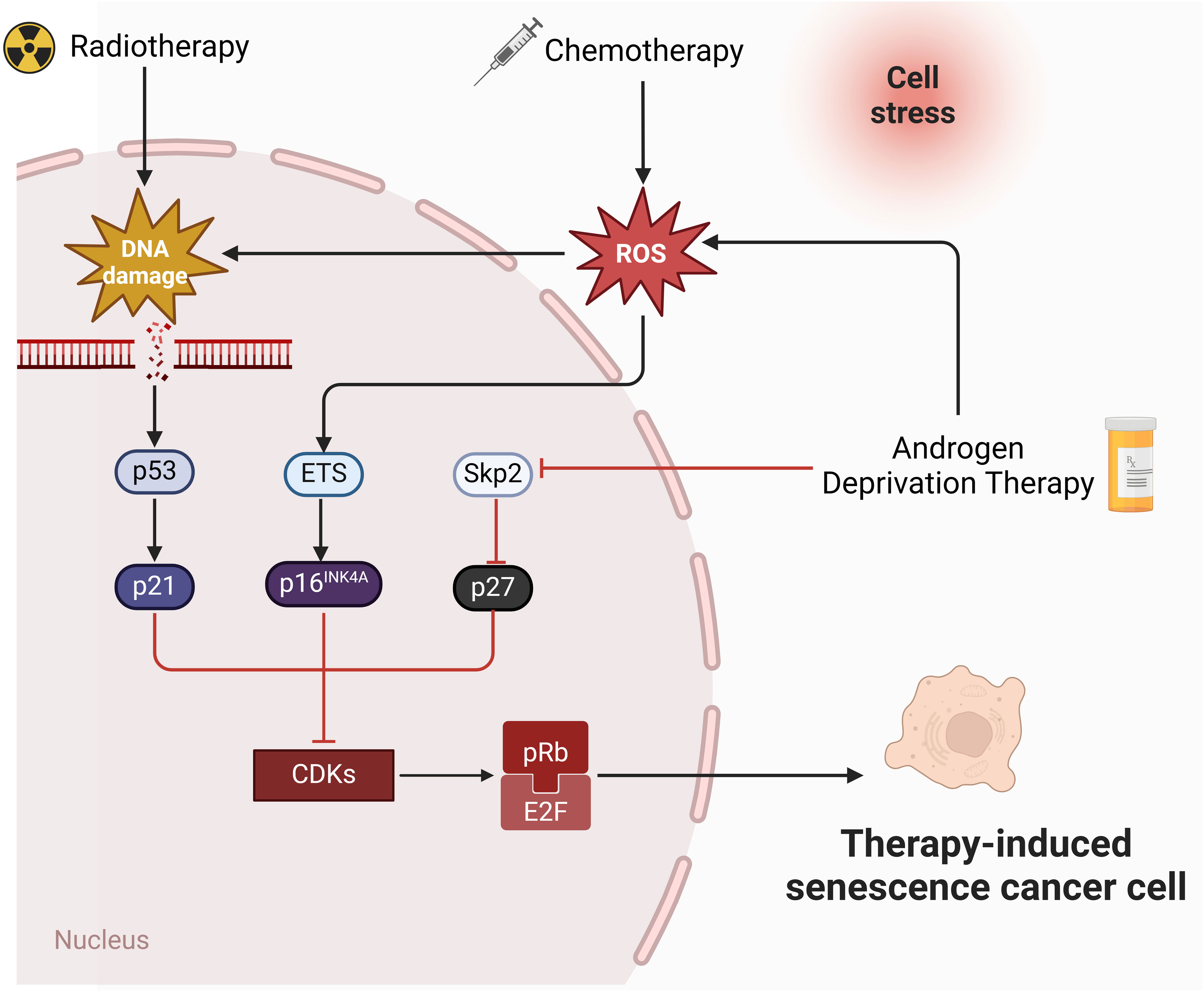
Figure 1 Molecular pathways of therapies-induced senescence in PCa. Radiotherapy and chemotherapy lead to irreversible DNA damage, ultimately triggering the activation of p53 and p21. p21 inhibits CDKs and mediates senescence by preventing the phosphorylation of Rb. In addition, chemotherapy induces senescence through ROS-ETS-p16 pathway. Androgen deprivation therapy induce senescence through Skp2-p27 pathway (created in Biorender.com).
3 Resistance to androgen deprivation therapy (ADT) and cell senescence in PCa
Currently, the main treatment methods for PCa include surgery (radical prostatectomy and focal ablation therapy) (64), radiotherapy, chemotherapy, and endocrine therapy, among which endocrine therapy is the first-line treatment for advanced metastatic PCa (5). Exposure of PCa to therapies targeting AR, different anticancer compounds, and ionizing radiation can induce a senescent phenotype, termed therapy-induced senescence (TIS) (65). The shared mechanisms of senescence and PCa encompass disruptions in proteostasis, dysbiosis of the microbiota, persistent chronic inflammation, and widespread immunosenescence (66). TIS may benefit patients in the treatment of PCa (67). A reason for this is the activation of inflammatory cytokines targeting tumor cells, which enhances the innate immune response (68). Nonetheless, senescent cells could also facilitate tumor growth (69). SASP possesses tumor-promoting properties, such as chronic inflammation, angiogenesis, stemness, migration, and invasion (70). Therefore, TIS is a double-edged sword that can lead to reduced or enhanced tumor growth. Investigating its role in different treatments can help address resistance in PCa treatment and provide new clinical solutions.
3.1 Development of ADT resistance
Therapies that suppress the secretion of testicular androgens or the activity of androgens are collectively known as ADT, serving as the hormonal treatment for PCa (71). ADT encompasses both castration treatments and anti-androgen therapies. Castration therapies are further classified into surgical castration, which is the removal of both testicles through surgery, and medical castration, employing luteinizing hormone-releasing hormone (LHRH) analogs to inhibit the production and secretion of androgens. Anti-androgen therapies involve the use of steroidal or non-steroidal anti-androgens that competitively bind to androgen receptors (ARs), thus suppressing the action of androgens (72). As advanced PCa evolves from being androgen-dependent to androgen-independent, it transitions into castration-resistant CRPC, in which cancer progression can be driven by even low levels of androgens (73, 74). In early-stage CRPC, there’s still dependence on the androgen receptor (AR) signaling pathway, with drugs like steroidogenesis inhibitors (abiraterone) and AR antagonists (enzalutamide), which are AR pathway inhibitors (ARPIs), emerging as frontline treatments for CRPC (13). Under the continuous action of ARPIs, resistance quickly becomes a new challenge, with CRPC gradually evolving into a tumor that does not depend on the AR signaling pathway, resulting in neuroendocrine prostate cancer (NEPC) (75). The development of ADT resistance depends on adaptive changes and reactivation of the AR signaling pathway, including intratumoral production of androgens in PCa tissues, AR gene amplification (76), point mutations in AR (77), and constitutively active AR splice variants (78).
3.2 ADT can induce cellular senescence
While contributing to the development of resistance in PCa, ADT can also induce cell senescence. Studies in vitro indicate that various PCa cell lines, such as LNCaP and LAPC-4, exhibit senescent phenotypes after being cultured in charcoal-stripped serum (CSS), which is a medium that depletes androgens along with other steroids, thyroid, and vitamin D3 hormones (79). Signs of this cellular senescence are characterized by SA-β-gal positive staining, formation of heterochromatin foci (signaling epigenetic alterations), and an upregulation in SASP components expression, for instance, insulin-like growth factor-binding protein 3 (IGFBP3) and tissue plasminogen activator B (80). However, AR-negative and androgen-independent PCa cell line PC3 does not undergo cellular senescence under ADT, highlighting the crucial role of AR signaling in the proliferation arrest induced by ADT, with AR signals facilitating the transition of cells from G1 to S phase (81). The senescence induced by ADT is partly mediated by Skp2-dependent cyclin-dependent kinase inhibitor p27Kip1 (80). Some studies also suggest that CSS and bicalutamide induce p27Kip1-independent cell senescence, a process that may be executed through ROS-induced DNA damage and the p16INK4a pathway (82). Additionally, the upregulation of the transcription factor CCAAT/Enhancer Binding Protein (C/EBP β) could also propel the aging response (83).
Clinical research shows that aging in PCa cells can be induced by non-steroidal AR antagonists such as bicalutamide, enzalutamide, darolutamide, and ataric acid treatments. Bicalutamide, a first-generation AR antagonist, triggers senescence in PCa cells by elevating the levels of CDK inhibitors p16INK4a and p27Kip1 (84). Enzalutamide and darolutamide are second-generation AR antagonists that inhibit cell proliferation and induce PCa cell aging through a mechanism involving p16INK4a induction (85, 86). Atraric acid, a natural AR antagonist, suppresses proliferation and induces senescence in PCa cells—both androgen-dependent (LNCaP) and castration-resistant (C4-2), as well as in PCa-like tumors from prostatectomies via the p16INK4a-pRb-E2F1 CyclinD1signaling pathway (87).
3.3 Cellular senescence benefits ADT therapy
Cellular senescence was initially described as a physiological suppression mechanism of tumor cells, as the development of cancer requires cell proliferation (88). Senescent cells cannot respond to mitotic signals nor re-enter the cell cycle, preventing damaged or stressed cells from dividing and forming tumors (89). There is evidence that TIS can yield beneficial effects for ADT, including the activation of the immune system and upregulation of inflammatory cytokines targeting tumor cells (90). Senescent cells secrete various chemokines, cytokines, and small molecules as components of SASP (91). A component of SASP, IL-1, is capable of inducing or amplifying Senescence-associated growth arrest and generating a pro-inflammatory environment, crucial in recruiting immune cells, thereby hindering cancer progression (92, 93). Moreover, lysosomal β-galactosidase (GLB1) is typically elevated in senescent cells (79), with a rise in GLB1 mRNA signifying better PCa outcomes (94). Studies indicate an increase in GLB1 protein levels in PCa patient samples within a month after initiating ADT (95), suggesting TIS aids ADT in PCa.
3.4 Cellular senescence promotes drug resistance in PCa
ADT-induced senescence can develop phenotypes favorable for cell survival, potentially evolving into clinically observed castration-resistant PCa through senescence evasion, cell-autonomous reprogramming, and the promotion of tumorigenic SASP, thereby countering the effectiveness of ADT (96).
Cellular senescence is not a stable state but rather a transitional phase, where, amidst complex epigenetic reprogramming, some PCa cells can regain the ability to proliferate after androgen deprivation ceases, known as TIS escape (97). Studies show that anti-androgen enzalutamide can trigger a reversible state akin to senescence, with no evidence of cell death or DNA damage (98).
During PCa progression, cellular senescence is tightly associated with telomere reduction (99, 100). Studies reveal that cancer cells typically undergo telomere shortening with anti-androgen treatment, likely due to treatment-induced stress and proliferation demands (101). The senescence induced by telomere shortening may drive CRPC development, allowing cancer cells to escape treatment suppression (102). Anti-androgen treatments can provoke DNA damage, with senescent cells’ reduced ability to repair DNA potentially diminishing treatment outcomes (79). Additionally, cellular senescence might activate DNA damage response pathways, prompting cancer cells to employ escape mechanisms for ongoing survival and proliferation (82).
Under the influence of ADT, various apoptotic regulatory factors become dysregulated, including the upregulation of BCL-2 (103), altering the activity of transcription factors during cell senescence, leading to transcriptomic-level control and gene expression reprogramming (97), a process previously identified as crucial for developing castration resistance (104). Following ADT, senescent PCa cells exhibit decreased sensitivity to various chemotherapies, including docetaxel (82, 83).
Cell senescence is also related to the regulation of inflammation and immune responses, with ADT indeed promoting the expression of SASP (105), which can alter the tissue microenvironment, with certain SASP paracrine components exhibiting tumor-promoting characteristics (106). IL-6 and IL-8, as components of SASP, can stimulate inflammation, epithelial-mesenchymal transition (EMT), and invasiveness (107), in addition to directly interacting with and activating AR (108, 109). Senescent fibroblasts and tumor cells can encourage the proliferation of nearby cells through paracrine activation of mechanisms, including the ERK1/2 signaling pathway, in both in vitro and in vivo settings (110, 111).
4 Resistance to chemotherapy and cellular senescence in PCa
4.1 Overview of chemotherapy resistance in PCa
Chemotherapy, a prevalent treatment for various malignancies, but PCa was once considered to be insensitive to chemotherapy. Since its introduction in 2004, docetaxel has become increasingly pivotal in treating metastatic castration-resistant PCa (112). Combining docetaxel with ADT and radiation therapy has been reported to enhance the recurrence-free survival in non-metastatic, locally advanced PCa (113, 114). While docetaxel remains a cornerstone for treating advanced stages of PCa, including castration-resistant variants, resistance to it markedly narrows treatment options (115). Chemotherapy resistance in PCa arises from various factors, including alterations in drug targets, epigenetic modifications, DNA repair mechanisms, cell death inhibition, and epithelial-mesenchymal transition (EMT) (116). Notably, research indicates that circARHGAP29 overexpression instigates docetaxel resistance and aerobic glycolysis within PCa cells (117). The cholinergic muscarinic M1 receptor (CHRM1) directly contributes to PCa cells’ resistance against docetaxel (118).
4.2 Chemotherapy triggers cellular senescence in PCa
Chemotherapy also leads to cellular senescence in cancer cells and TME components (119). It’s widely recognized that most cancer cells undergo growth arrest or death following chemotherapy (120). However, a minority of cancer cells enter prolonged growth arrest, exhibiting signs of cellular senescence (121). Studies indicate that DTX induces cellular senescence in the TC-1 and B16 tumor cell lines, marked by growth arrest, positive β-galactosidase staining, and elevated p21Waf1 (p21) expression (122). Clinical research on cancer survivors treated with chemotherapy shows increased levels of various cellular senescence markers post-treatment (123). Another clinical study analyzed paraffin-embedded tissue sections from PCa patients treated with neoadjuvant paclitaxel chemotherapy before radical prostatectomy, revealing specific detection of lipofuscin staining in the stroma of paclitaxel-treated patients (124).
Various chemotherapy drugs trigger cellular senescence through different mechanisms, including DNA damage, oxidative stress, and DNA methylation changes (125). Moreover, cellular senescence can be induced by impacting their metabolism and function. Research treating the TC-1 tumor cell line with DTX has revealed DNA double-strand breaks before or during mitosis, leading to ongoing activation of cell cycle checkpoints and the progression of subcellular cellular senescence (122). Oxidative damage to mitochondria from chemotherapy drugs leads to a reduction in cellular energy and functional shutdown, resulting in cellular senescence (126). During chemotherapy, alterations in DNA methylation and enzymes related to DNA methylation significantly impair cell function and mediate tumor cell cellular senescence by activating SASP, promoting a chronic inflammatory state (127).
4.3 The dual impact of chemotherapy-induced cellular senescence on PCa treatment
Chemotherapy drugs induce cancer cell senescence via DNA damage, cell cycle arrest, and apoptosis—a strategy that curtails cancer progression by reducing the proliferative capacity of senescent cells (128). The therapeutic effects of chemotherapy-induced cellular senescence extend to altering the TME (129). Signaling molecules from senescent cells can trigger inflammation, activate the immune system, and bolster the assault on cancer cells (130). Immune-mediated anticancer effects contribute to the elimination of residual cancer cells, lowering recurrence risk.
However, the cellular senescence triggered by chemotherapy drugs is closely linked to PCa resistance, with cancer cells deploying multiple strategies to bypass or diminish the adverse impacts of cellular senescence.
Enhancement of stem-like characteristics: German researchers discovered that cells enduring extreme chemotherapy environments transition into senescence, with these senescent cells sharing distinctive traits with resistant cells, including enhanced stem cell gene expression and heightened self-renewal capabilities, unbound by maturity constraints (131).
Activation of the p53 signaling pathway: The p53 gene, crucial for tumor suppression (132), activates in response to DNA damage, guiding cell repair or apoptosis (133). Nevertheless, research has identified p53 pathway anomalies in certain PCa cells, complicating their chemotherapy responses (134), thus facilitating their evasion of TIS and fostering resistance (135, 136).
Altered cell cycle regulation: Chemotherapy drugs may induce cell cycle arrest and modify the expression of cycle-regulating proteins, affording cells additional repair time for DNA damage and triggering senescence, culminating in treatment resistance (137, 138).
Alterations of TME: Chemotherapy drugs foster a senescent phenotype in stromal fibroblasts, triggering metabolic shifts and the release of paracrine factors, activating tumor cell survival pathways such as ERK1/2 signaling pathways (139), and enhancing PCa cells invasiveness and clonogenic potential. These alterations may render the TME more supportive of resistance development (Figure 2) (124).
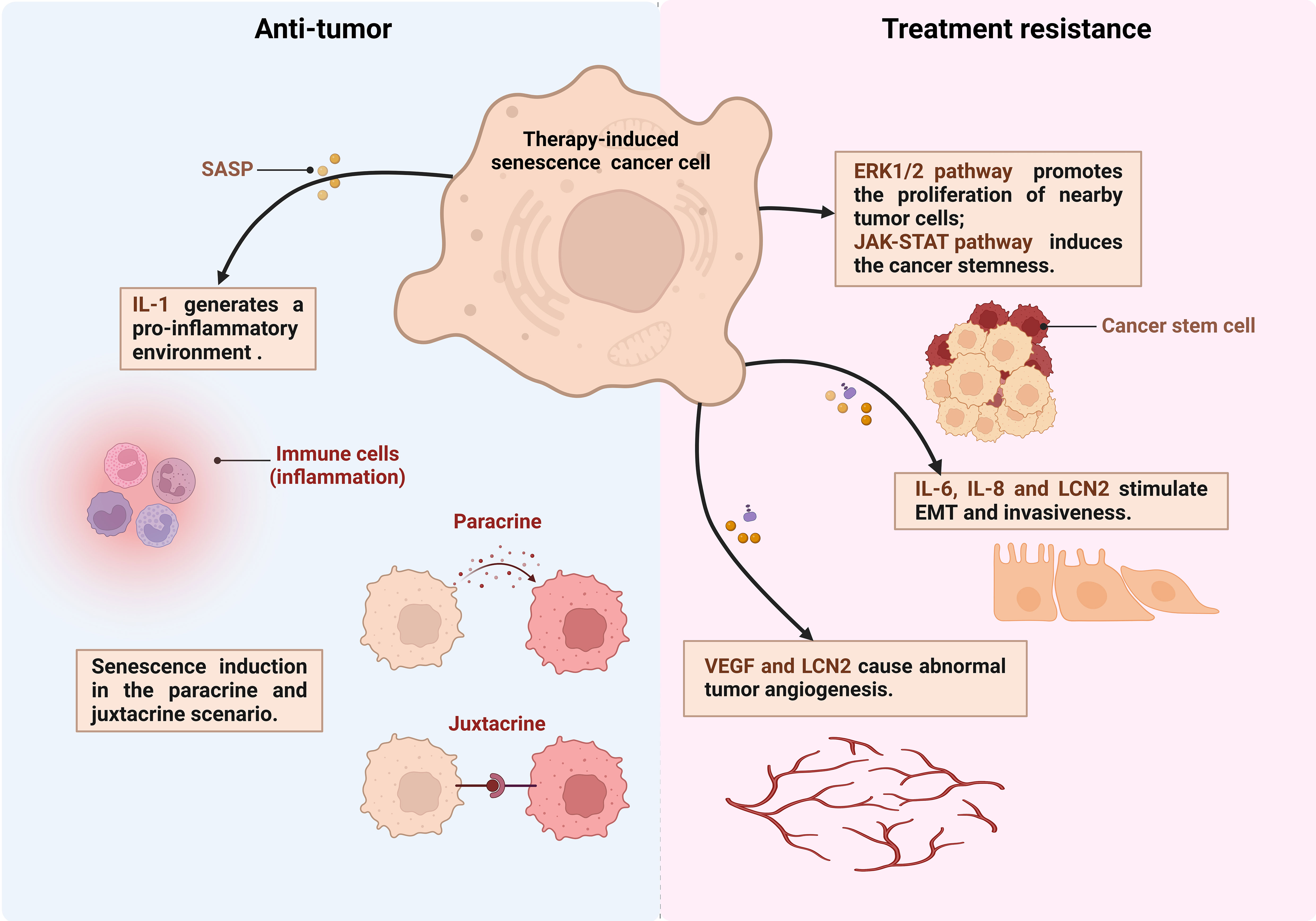
Figure 2 The dual effects of senescent cells in PCa. Therapy-induced senescent cancer cells secrete production of IL-6, IL-8, LCN2 and other senescence-associated secretory phenotype (SASP) factors. These factors exert juxtacrine and paracrine effects on the surrounding tumor microenvironment thereby anti-tumor or treatment resistance (created in Biorender.com). EMT, Epithelial-to-mesenchymal transition; VEGF, Vascular endothelial growth factor; LCN2, lipocalin 2.
5 Cellular senescence and resistance to radiation therapy for PCa
Radiation therapy, recognized as the most widely used cytotoxic therapy, causes irreversible DNA damage, including double-strand breaks, single-strand breaks, DNA interstrand crosslinks, through γ rays or X-rays (140), This process initiates tumor cell apoptosis. However, the emergence of radiation therapy resistance poses significant challenges, particularly for patients with high-risk PCa (141–143).
Larsen et al. discovered that radiation-induced exogenous DNA damage activates Caspase-activated Dnase (CAD) (144), further triggering endogenous DNA breaks. This dual effect accelerates the aging of tumor cells through DNA damage-induced senescence (145). The resulting genomic instability activates the G2 cell cycle checkpoint, preventing tumor cells from entering the highest radiation sensitivity G2/M phase (146, 147), thereby acquiring radiotherapy resistance (144, 148). Additionally, senescent tumor cells secrete vascular endothelial growth factor (VEGF), leading to abnormal tumor angiogenesis and contributing to both chronic, diffuse hypoxia and acute, transient perfusion-related hypoxia. Pan-cancer cells often exhibit elevated levels of ANGPTL4, a protein that interacts with integrins to generate O2−, significantly contributing to tumor growth and survival (149). Additionally, ANGPTL4 is crucial in endothelial cells, regulating metabolism and angiogenesis. Specifically, endothelial-specific deletion of ANGPTL4 decreases pathological neovascularization and reduces permeability, emphasizing its crucial role in endothelial cell metabolism and angiogenic functions (150). Hence, ANGPTL4 represents a potential therapeutic target in the treatment of prostate cancer, it may be possible to disrupt tumor growth and survival mechanisms, offering a novel approach for the treatment of prostate cancer. In another study, Zhang et al. discovered that hypoxia-induced ANGPTL4 protein promotes radiation resistance in lung cancer through two mechanisms. Hypoxia not only increases the expression and secretion of ANGPTL4 in lung cancer cells, inhibiting ferroptosis and mediating radiation resistance, but also allows ANGPTL4 protein to be loaded into exosomes derived from hypoxic tumor cells. These exosomes then transfer to surrounding normoxic tumor cells, inducing radiation resistance by inhibiting ferroptosis through GPX4 (151). LCN2, a 25 kDa secreted glycoprotein belonging to the lipocalin family of lipid-carrying proteins, is highly expressed in PCa cells and can be induced by cellular senescence (152, 153). Studies have demonstrated that LCN2 interacts with MMP9 to form complexes, exerting pro-angiogenic and pro-tumor effects (154, 155). Furthermore, overexpression of LCN2 induces epithelial-mesenchymal transition in PCa, promoting tumor metastasis (156).
In addition, DNA damage-induced senescent tumor cells are closely linked to cancer stemness (157). Characterized by their tumorigenic nature, rapid multiplication, and multi-directional differentiation, CSCs play a pivotal role in determining tumor radio-sensitivity (158, 159). Tumor recurrence is also closely associated with CSCs, and these newly formed tumor cells, having been previously exposed to irradiation, exhibit reduced sensitivity to the initial radiation dose and gradually develop radiotherapy resistance. Evidence suggests that activating the JAK-STAT pathway can transform ordinary tumor cells into CSCs (160, 161). Studies have demonstrated that the JAK-STAT pathway is more highly activate in senescent than in non-senescent cancer cells (162). Meanwhile, certain signaling pathway that induce cellular senescence, such as the p53 and MAPK pathways, are known to activate the JAK-STAT pathway (163, 164). Furthermore, Karabicici et al. showed that the senescent tumor cells increase the mRNA expression of stem cell-related molecules, such as CD34 and CD133 (165).
For a long time, the radiotherapy resistance of PCa has been overlooked (166). With the increase of tumor resistance to radiation, it is necessary to gradually increase the radiation dose, which may be a dilemma for patients. On one hand, tumors can develop acquired resistance to radiation therapy after multiple exposures. On the other hand, bladder and rectal toxicities increase during radiation therapy. Hence, it is necessary to conduct research in two aspects: firstly, to explore strategies that can enhance the sensitivity of tumor radiotherapy, such as the application of nanoparticles (167) and the establishment of cancer radiosensitivity regulation factors database (dbCRSR) (168), to achieve effective local tumor control at acceptable and safe radiation dose; secondly, to investigate the mechanisms of tumor radiotherapy resistance, providing theoretical basis for improving the efficacy of radiotherapy (Figure 3).
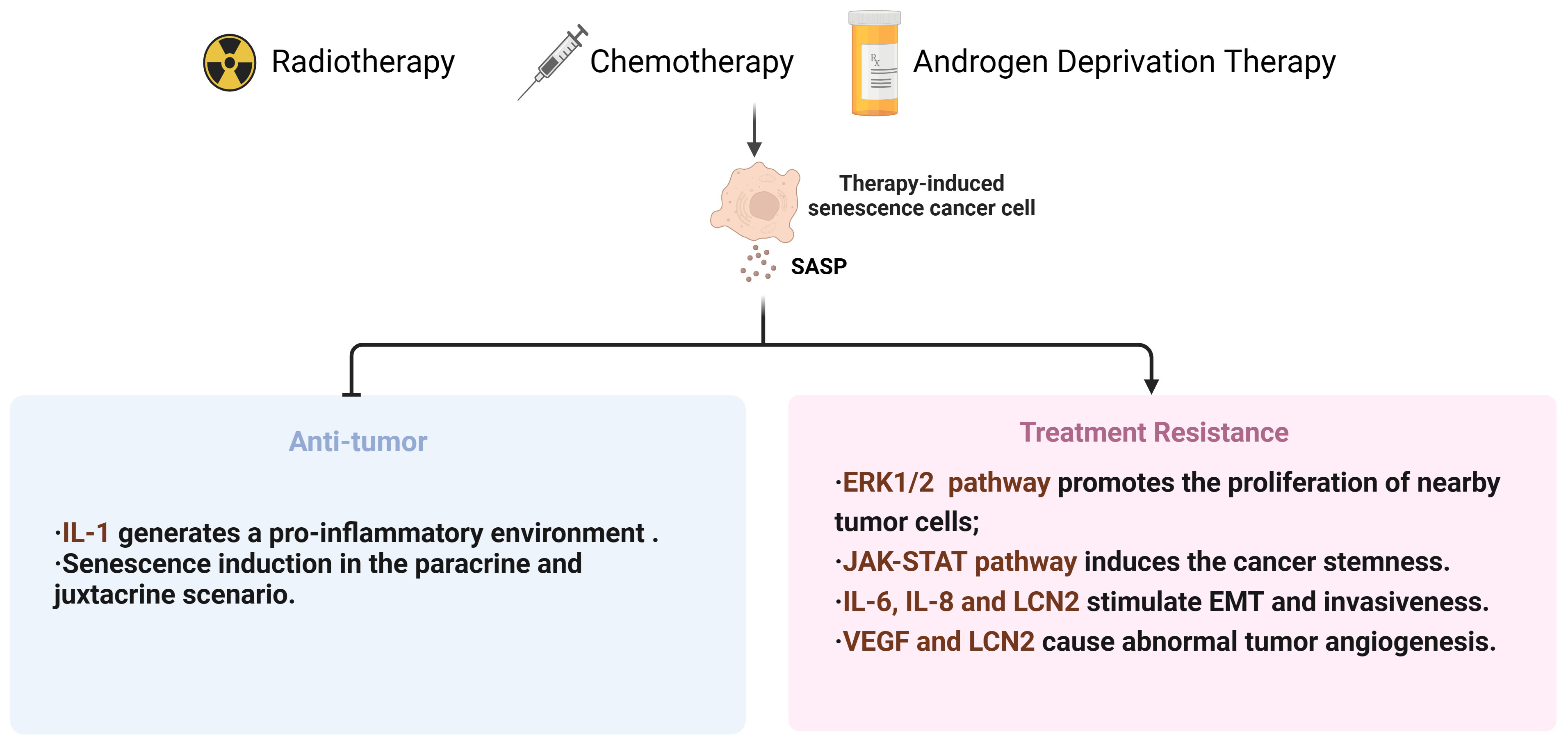
Figure 3 Summary diagram for therapies-induced senescence and dual effects in PCa. SASP, senescence-associated secretory phenotype; EMT, Epithelial-to-mesenchymal transition; VEGF, Vascular endothelial growth factor; LCN2, lipocalin 2.
6 The application and prospects of cellular senescence in PCa treatment resistance
6.1 The potential application of cellular senescence related markers in monitoring and prognostic evaluation of PCa therapy
As research into cellular senescence progresses, scientists have identified that biomarkers associated with cellular senescence offer promising applications in monitoring tumor treatment and assessing prognosis. In the context of PCa treatment, employing these biomarkers introduces innovative approaches for disease management and therapeutic intervention.
Biomarkers related to cellular senescence encompass an array of proteins, enzymes, and molecular signals, notably increased β-galactosidase activity, p16^INK4a, p21^CIP1/WAF1, and cyclin D1 (169). Significant changes in the expression levels of these markers during cellular senescence (170) make them valuable tools for monitoring and assessing the state of senescence.
Monitoring changes in senescence-associated biomarkers during PCa treatment offers a method to assess therapeutic effectiveness. Treatments like chemotherapy and radiation therapy can induce a senescent state in tumor cells, marked by a significant increase in the expression of senescence-related biomarkers (171). Regular monitoring of these biomarkers enables physicians to track treatment efficacy in real-time and promptly adjust therapeutic strategies as needed. Moreover, for innovative therapies that act by inducing tumor cell senescence, monitoring senescence-associated biomarkers directly indicates the efficacy of these drugs.
While biomarkers related to cellular senescence hold immense potential for therapy monitoring and prognostic evaluations, practical applications encounter significant challenges. Initially, cellular senescence, being a process influenced by numerous factors and stages, means a single biomarker might not adequately capture the complexity of the senescent state. Consequently, there is a need to devise methods for the combined detection of multiple biomarkers, aiming to boost the precision and dependability of monitoring efforts. Additionally, the expression patterns of senescence biomarkers may vary across different tumor types and among individuals, necessitating extensive validation and personalized assessment prior to their application.
6.2 Strategies for combating drug resistance in the treatment of PCa through cellular senescence
Cellular senescence, as a multifaceted biological phenomenon, has garnered significant interest in the realm of PCa treatment research in recent years. This process curtails tumor growth by inhibiting the proliferation of damaged or aberrant cells. In the context of tumor therapy, cellular senescence emerges as a potential double-edged sword. The accumulation of senescent cells can instigate inflammatory responses and alterations in the TME, potentially facilitating tumor progression. Tumor recurrence and severe, long-term adverse effects continue to pose significant challenges in the treatment of PCa patients. Consequently, the precise modulation of cellular senescence, harnessing its potential to combat treatment resistance in PCa, represents an emerging avenue of research (Table 1).
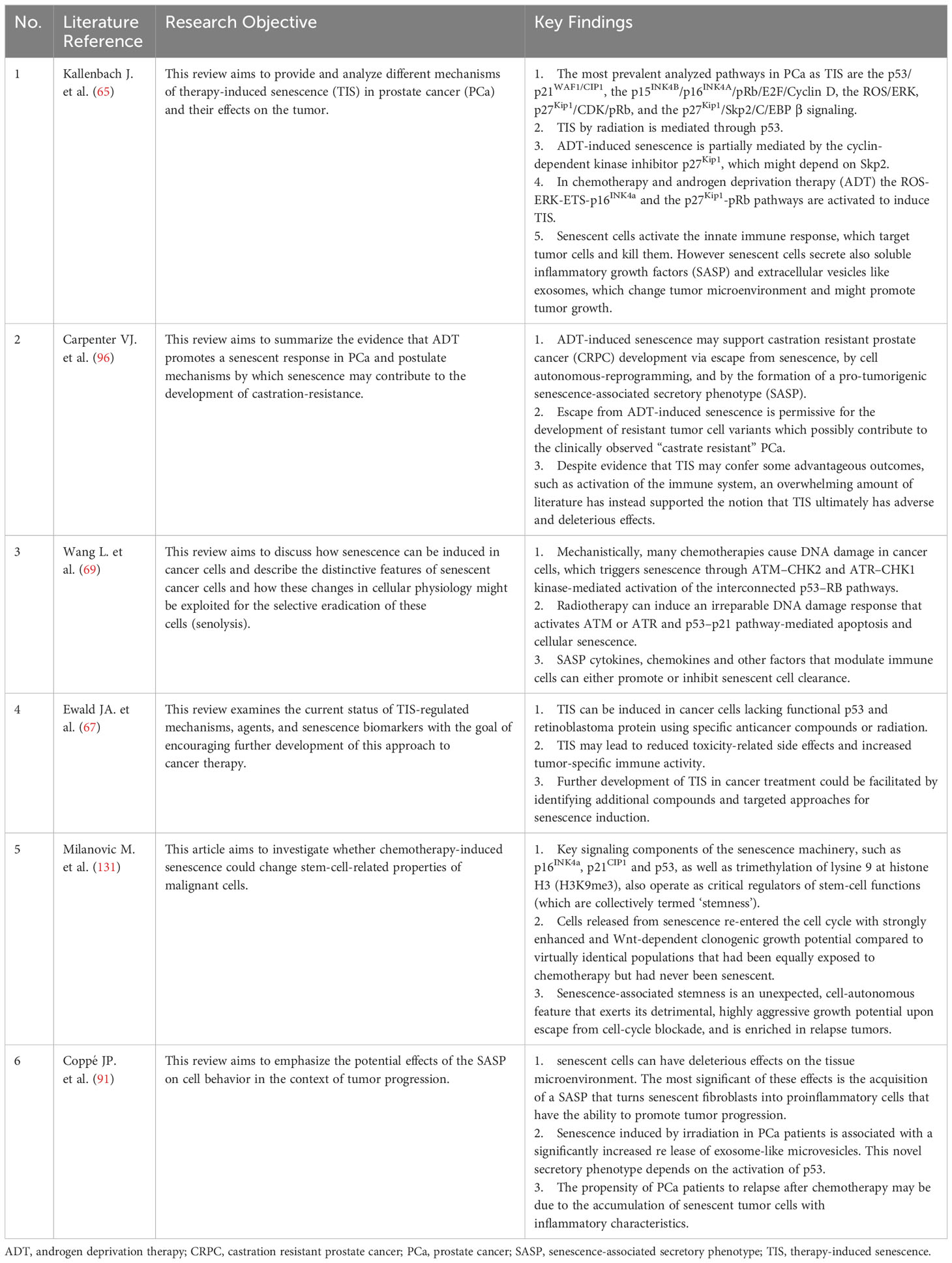
Table 1 Core literature summary table.
Strategy 1: Targeted induction of senescence in tumor cells. Specifically inducing senescence in tumor cells can effectively inhibit their proliferation, thus decelerating tumor growth (169, 172). The challenge lies in accurately targeting and inducing senescence in tumor cells while sparing normal cells from adverse effects. Research indicates that the NF-κB signaling pathway plays a crucial role in triggering SASP, with targeted intervention in this pathway effectively inducing tumor cell senescence (173). Additionally, activating the p53 pathway emerges as an effective strategy for promoting cellular senescence (174). Inhibiting the interaction between p53 and its negative regulator MDM4 can rejuvenate p53 activity in melanoma cells, enhancing tumor cell suppression and chemotherapy sensitivity (175). Thus, employing small-molecule drugs to activate p53 allows for the targeted induction of tumor cell senescence without harming normal cells. Despite the potential of activating the p53 pathway to promote cellular senescence in cancer therapy, current research faces multiple challenges. One possible explanation for the failure of MDM4 small-molecule inhibitors is that they may be too specific and unable to fully reactivate p53, as MDM2, which is structurally related to MDM4, can also bind and block the activity of p53. Therefore, a dual-targeting approach is considered crucial for success (176). Additionally, protein degraders offer a novel approach to reactivation of wild-type p53 by tagging and degrading target proteins through the combination of interest proteins and E3 ubiquitin ligases (177). Furthermore, mRNA-based therapies have demonstrated the potential to treat cancers with p53 mutations (178).
Strategy 2: Eliminating senescent cells. Inducing senescence in tumor cells can inhibit their proliferation, yet the accumulation of senescent cells might entail adverse outcomes, including the promotion of inflammation and alterations in the TME (179). Consequently, devising strategies to eliminate senescent cells represents a critical approach to surmounting treatment resistance. “Senolytics” refers to a category of drugs designed to specifically target and eliminate senescent cells (180). By targeting survival signaling pathways unique to senescent cells, such as navitoclax which targets the Bcl-2 protein family, these drugs mitigate the influence of senescent cells on the surrounding microenvironment (181), thus diminishing the risk of tumor progression. Targeting senescent cells shows promise for disease alleviation, yet its clinical translation remains intricate. The nonspecific nature of senescent cells in both tumor and healthy tissues poses a challenge for “Senolytics” agents, requiring discrimination to minimize collateral damage. Heterogeneity of the senescence phenotype complicates universal agent development, necessitating personalized approaches. Long-term impacts and safety profiles of “Senolytics” therapy remain uncertain, necessitating rigorous preclinical and clinical testing.
Strategy 3: Modulating the TME. Besides acting directly on tumor cells, modulating the TME is also an effective strategy for overcoming treatment resistance in PCa (182–184). Through the secretion of SASP, senescent cells influence neighboring cells and tissues, thereby fostering alterations in the TME (185). Inhibiting SASP secretion or blocking its effects can mitigate the adverse influence of senescent cells on the TME, thus diminishing tumor cell resistance (186). Utilizing anti-inflammatory drugs to curb inflammatory responses, or antibodies to obstruct critical SASP factors like IL-6 and IL-8, can significantly enhance the TME and curb tumor growth (187). However, current challenges lie in identifying specific targets within the heterogeneous TME, developing agents with high selectivity and low toxicity, and understanding the long-term impact of modulating the TME on PCa progression and patient outcomes.
7 Conclusion
In conclusion, cellular senescence in PCa therapy reveals both significant potential and formidable challenges. A profound comprehension of the interplay between cellular senescence and treatment resistance in PCa not only aids in uncovering mechanisms of resistance but also opens avenues for devising novel therapeutic strategies. In the future, research into cellular senescence will be pivotal in formulating innovative approaches to circumvent or reverse resistance and enhance therapeutic efficacy. Strategies focused on eliminating senescent cells or adjusting their secretions promise to yield more efficacious treatment alternatives for PCa patients.
Author contributions
M-YX: Writing – original draft, Writing – review & editing. Z-YX: Visualization, Writing – original draft. J-XS: Writing – review & editing. C-QL: Writing – review & editing. YA: Writing – review & editing. J-ZX: Writing – review & editing. S-HZ: Writing – review & editing. X-YZ: Writing – review & editing. NZ: Writing – review & editing. S-YM: Writing – review & editing. H-DH: Writing – review & editing. S-GW: Writing – review & editing. Q-DX: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
PCa, Prostate cancer; EAU, European Association of Urology; PSA, Prostate-specific antigen; GS, Gleason score; AS, Active surveillance; CRPC, Castration-resistant prostate cancer; CSCs, Cancer stem cells; TCP, Tumor cell plasticity; TME, Tumor microenvironment; EMT, Epithelial-to-mesenchymal transition; DTP, Drug-tolerant persister; ROS, Reactive oxygen species; SIPS, Stress-induced premature senescence; ECM, Extracellular matrix; SA-β-Gal, Senescence-associated β-galactosidase; SAHF, Senescence-associated heterochromatin foci; SASP, Senescence-associated secretory phenotype; Cdk, Cyclin-dependent protein kinase; ADT, Androgen deprivation therapy; TIS, Therapy-induced senescence; LHRH, Luteinizing hormone-releasing hormone; ARs, Androgen receptors; ARPIs, AR pathway inhibitors; NEPC, Neuroendocrine prostate cancer; CSS, Charcoal-stripped serum; IGFBP3, Insulin-like growth factor-binding protein 3; C/EBP β, CCAAT/Enhancer Binding Protein; GLB1, Lysosomal β-galactosidase; CHRM1, Cholinergic muscarinic M1 receptor; VEGF, Vascular endothelial growth factor; LCN2, lipocalin 2.
References
1. Bergengren O, Pekala KR, Matsoukas K, Fainberg J, Mungovan SF, Bratt O, et al. 2022 Update on prostate cancer epidemiology and risk factors-a systematic review. Eur Urol. (2023) 84:191–206. doi: 10.1016/j.eururo.2023.04.021
2. Gandaglia G, Leni R, Bray F, Fleshner N, Freedland SJ, Kibel A, et al. Epidemiology and prevention of prostate cancer. Eur Urol Oncol. (2021) 4:877–92. doi: 10.1016/j.euo.2021.09.006
3. Liu Y, Zhang PP, Zhang YH, Zheng LC, Xu WB, Hou DT, et al. Clinical characteristics and overall survival nomogram of second primary Malignancies after prostate cancer, a seer population-based study. Sci Rep. (2021) 11(1). doi: 10.1038/s41598-020-80534-4
4. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. Ca-a Cancer J Clin. (2020) 70:7–30. doi: 10.3322/caac.21590
5. Mottet N, van den Bergh RCN, Briers E, Van den Broeck T, Cumberbatch MG, De Santis M, et al. Eau-eanm-estro-esur-siog guidelines on prostate cancer-2020 update. Part 1: screening, diagnosis, and local treatment with curative intent. Eur Urol. (2021) 79:243–62. doi: 10.1016/j.eururo.2020.09.042
6. Moschini M, Carroll PR, Eggener SE, Epstein JI, Graefen M, Montironi R, et al. Low-risk prostate cancer: identification, management, and outcomes. Eur Urol. (2017) 72:238–49. doi: 10.1016/j.eururo.2017.03.009
7. Jin HJ, Wang LQ, Bernards R. Rational combinations of targeted cancer therapies: background, advances and challenges. Nat Rev Drug Discovery. (2023) 22:213–34. doi: 10.1038/s41573-022-00615-z
8. Marine JC, Dawson SJ, Dawson MA. Non-genetic mechanisms of therapeutic resistance in cancer. Nat Rev Cancer. (2020) 20:743–56. doi: 10.1038/s41568-020-00302-4
9. Arozarena I, Wellbrock C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat Rev Cancer. (2019) 19:377–91. doi: 10.1038/s41568-019-0154-4
10. Boumahdi S, de Sauvage FJ. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discovery. (2020) 19:39–56. doi: 10.1038/s41573-019-0044-1
11. Chatterjee N, Bivona TG. Polytherapy and targeted cancer drug resistance. Trends Cancer. (2019) 5:170–82. doi: 10.1016/j.trecan.2019.02.003
12. Desai K, McManus JM, Sharifi N. Hormonal therapy for prostate cancer. Endocrine Rev. (2021) 42:354–73. doi: 10.1210/endrev/bnab002
13. Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. (2015) 15:701–11. doi: 10.1038/nrc4016
14. Westaby D, Fenor de la Maza M, Paschalis A, Jimenez-Vacas JM, Welti J, de Bono J, et al. A new old target: androgen receptor signaling and advanced prostate cancer. Annu Rev Pharmacol Toxicol. (2022) 62:131–53. doi: 10.1146/annurev-pharmtox-052220-015912
15. Chi KN, Agarwal N, Bjartell A, Chung BH, Gomes A, Given R, et al. Apalutamide for metastatic, castration-sensitive prostate cancer. New Engl J Med. (2019) 381:13–24. doi: 10.1056/NEJMoa1903307
16. Davis ID, Martin AJ, Stockler MR, Begbie S, Chi KN, Chowdhury S, et al. Enzalutamide with standard first-line therapy in metastatic prostate cancer. New Engl J Med. (2019) 381:121–31. doi: 10.1056/NEJMoa1903835
17. Miller KD, Nogueira L, Devasia T, Mariotto AB, Yabroff KR, Jemal A, et al. Cancer treatment and survivorship statistics, 2022. Ca-a Cancer J Clin. (2022) 72:409–36. doi: 10.3322/caac.21731
18. Imran M, Saleem S, Chaudhuri A, Ali J, Baboota S. Docetaxel: an update on its molecular mechanisms, therapeutic trajectory and nanotechnology in the treatment of breast, lung and prostate cancer. J Drug Delivery Sci Technol. (2020) 60. doi: 10.1016/j.jddst.2020.101959
19. Khalaf K, Hana D, Chou JTT, Singh C, Mackiewicz A, Kaczmarek M. Aspects of the tumor microenvironment involved in immune resistance and drug resistance. Front Immunol. (2021) 12:656364. doi: 10.3389/fimmu.2021.656364
20. Liu Y, Dong YP, Kong L, Shi F, Zhu H, Yu JM. Abscopal effect of radiotherapy combined with immune checkpoint inhibitors. J Hematol Oncol. (2018) 11. doi: 10.1186/s13045-018-0647-8
21. Ma G-L, Lin W-F. Immune checkpoint inhibition mediated with liposomal nanomedicine for cancer therapy. Military Med Res. (2023) 10(1). doi: 10.1186/s40779-023-00455-x
22. Kumar R, Sena LA, Denmeade SR, Kachhap S. The testosterone paradox of advanced prostate cancer: mechanistic insights and clinical implications. Nat Rev Urol. (2023) 20:265–78. doi: 10.1038/s41585-022-00686-y
23. Sarmento-Ribeiro AB, Scorilas A, Goncalves AC, Efferth T, Trougakos IP. The emergence of drug resistance to targeted cancer therapies: clinical evidence. Drug Resistance Updates. (2019) 47. doi: 10.1016/j.drup.2019.100646
24. Ward RA, Fawell S, Floc'h N, Flemington V, McKerrecher D, Smith PD. Challenges and opportunities in cancer drug resistance. Chem Rev. (2021) 121:3297–351. doi: 10.1021/acs.chemrev.0c00383
25. Zhong L, Li YS, Xiong L, Wang WJ, Wu M, Yuan T, et al. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal Transduction Targeted Ther. (2021) 6(1). doi: 10.1038/s41392-021-00572-w
26. Brabletz S, Schuhwerk H, Brabletz T, Stemmler MP. Dynamic emt: A multi-tool for tumor progression. EMBO J. (2021) 40(18). doi: 10.15252/embj.2021108647
27. De las Rivas J, Brozovic A, Izraely S, Casas-Pais A, Witz IP, Figueroa A. Cancer drug resistance induced by emt: novel therapeutic strategies. Arch Toxicol. (2021) 95:2279–97. doi: 10.1007/s00204-021-03063-7
28. Mavs antagonizes human stem cell senescence as a mitochondrial stabilizer. Research. (2023) 2023(7).
29. Lambert AW, Weinberg RA. Linking emt programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer. (2021) 21:325–38. doi: 10.1038/s41568-021-00332-6
30. Shi ZD, Pang K, Wu ZX, Dong Y, Hao L, Qin JX, et al. Tumor cell plasticity in targeted therapy-induced resistance: mechanisms and new strategies. Signal Transduction Targeted Ther. (2023) 8(1). doi: 10.1038/s41392-023-01383-x
31. López-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. (2023) 186:243–78. doi: 10.1016/j.cell.2022.11.001
32. López-Otín C, Pietrocola F, Roiz-Valle D, Galluzzi L, Kroemer G. Meta-hallmarks of aging and cancer. Cell Metab. (2023) 35:12–35. doi: 10.1016/j.cmet.2022.11.001
33. Shay JW, Wright WE. Hayflick, his limit, and cellular ageing. Nat Rev Mol Cell Biol. (2000) 1:72–6. doi: 10.1038/35036093
34. Marazita MC, Duguor A, Marquioni-Ramella MD, Figueroa JM, Suburo AM. Oxidative stress-induced premature senescence dysregulates Vegf and Cfh expression in retinal pigment epithelial cells: implications for age-related macular degeneration. Redox Biol. (2016) 7:78–87. doi: 10.1016/j.redox.2015.11.011
35. Lazzarini E, Lodrini AM, Arici M, Bolis S, Vagni S, Panella S, et al. Stress-induced premature senescence is associated with a prolonged Qt interval and recapitulates features of cardiac aging. Theranostics. (2022) 12:5237–57. doi: 10.7150/thno.70884
36. Parker SM, Ward C, Johnson G, Black F, Lordan JL, Corris PA, et al. Stress-induced premature senescence in airway epithelium and accelerated lung allograft aging. J Heart Lung Transplant. (2007) 26:S84–S. doi: 10.1016/j.healun.2006.11.083
37. Toussaint O, Royer V, Salmon M, Remacle J. Stress-induced premature senescence and tissue ageing. Biochem Pharmacol. (2002) 64:1007–9. doi: 10.1016/S0006-2952(02)01170-X
38. Seiwert TY, Salama JK, Vokes EE. The concurrent chemoradiation paradigm - general principles. Nat Clin Pract Oncol. (2007) 4:86–100. doi: 10.1038/ncponc0714
39. Jeon H-M, Kim J-Y, Cho HJ, Lee WJ, Nguyen D, Kim SS, et al. Tissue factor is a critical regulator of radiation therapy-induced glioblastoma remodeling. Cancer Cell. (2023) 41:1480–+. doi: 10.1016/j.ccell.2023.06.007
40. Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (Sa-Bgal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. (2009) 4:1798–806. doi: 10.1038/nprot.2009.191
41. Zhang PS, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang SL, et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an alzheimer's disease model. Nat Neurosci. (2019) 22:719–+. doi: 10.1038/s41593-019-0372-9
42. Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest. (2018) 128:1238–46. doi: 10.1172/jci95148
43. Huang WJ, Hickson LJ, Eirin A, Kirkland JL, Lerman LO. Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol. (2022) 18:611–27. doi: 10.1038/s41581-022-00601-z
44. Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. (2015) 15:397–408. doi: 10.1038/nrc3960
45. Narita M, Narita M, Krizhanovsky V, Nuñez S, Chicas A, Hearn SA, et al. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell. (2006) 126:503–14. doi: 10.1016/j.cell.2006.05.052
46. Zhang RG, Chen W, Adams PD. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol. (2007) 27:2343–58. doi: 10.1128/mcb.02019-06
47. Di Micco R, Sulli G, Dobreva M, Liontos M, Botrugno OA, Gargiulo G, et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol. (2011) 13:292–U44. doi: 10.1038/ncb2170
48. Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol. (2016) 12:412–20. doi: 10.1038/nrrheum.2016.65
49. Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. (2013) 155:1119–30. doi: 10.1016/j.cell.2013.10.041
50. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. (2013) 123:966–72. doi: 10.1172/jci64098
51. Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. (2017) 23:775–+. doi: 10.1038/nm.4324
52. Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. (2016) 23:303–14. doi: 10.1016/j.cmet.2015.11.011
53. Dou ZX, Ghosh K, Vizioli MG, Zhu JJ, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. (2017) 550:402–6. doi: 10.1038/nature24050
54. Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou LL, et al. Mtor regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting il1a translation. Nat Cell Biol. (2015) 17:1049–U416. doi: 10.1038/ncb3195
55. Miyauchi H, Minamino T, Tateno K, Kunieda T, Toko H, Komuro I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a P53/P21-dependent pathway. EMBO J. (2004) 23:212–20. doi: 10.1038/sj.emboj.7600045
56. Gevry N, Chan HM, Laflamme L, Livingston DM, Gaudreau L. <I>P21</I> transcription is regulated by differential localization of histone H2a.Z. Genes Dev. (2007) 21:1869–81. doi: 10.1101/gad.1545707
57. Kumari R, Jat P. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol. (2021) 09:645593. doi: 10.3389/fcell.2021.645593
58. Jarrard DF, Sarkar S, Shi Y, Yeager TR, Magrane G, Kinoshita H, et al. P16/prb pathway alterations are required for bypassing senescence in human prostate epithelial cells. Cancer Res. (1999) 59:2957–64.
59. Larsson LG. Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin Cancer Biol. (2011) 21:367–76. doi: 10.1016/j.semcancer.2011.10.005
60. Steelman LS, Chappell WH, Abrams SL, Kempf CR, Long J, Laidler P, et al. Roles of the raf/mek/erk and pi3k/pten/akt/mtor pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging-Us. (2011) 3:192–222. doi: 10.18632/aging.100296
61. Chen ZB, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of P53-dependent cellular senescence in suppression of pten-deficient tumorigenesis. Nature. (2005) 436:725–30. doi: 10.1038/nature03918
62. Collado M, Serrano M. Senescence senescence in tumours: evidence from mice and humans. Nat Rev Cancer. (2010) 10:51–7. doi: 10.1038/nrc2772
63. Currier AW, Kolb EA, Gorlick RG, Roth ME, Gopalakrishnan V, Sampson VB. P27/kip1 functions as a tumor suppressor and oncoprotein in osteosarcoma. Sci Rep. (2019) 9. doi: 10.1038/s41598-019-42450-0
64. Feng D, Li D, Xiao Y, Wu R, Wang J, Zhang C. Focal ablation therapy presents promising results for selectively localized prostate cancer patients. Chin J Cancer Res. (2023) 35:424–30. doi: 10.21147/j.issn.1000-9604.2023.04.08
65. Kallenbach J, Atri Roozbahani G, Heidari Horestani M, Baniahmad A. Distinct mechanisms mediating therapy-induced cellular senescence in prostate cancer. Cell Biosci. (2022) 12:200. doi: 10.1186/s13578-022-00941-0
66. Shen W, He J, Hou T, Si J, Chen S. Common pathogenetic mechanisms underlying aging and tumor and means of interventions. Aging Dis. (2022) 13:1063–91. doi: 10.14336/ad.2021.1208
67. Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-induced senescence in cancer. J Natl Cancer Inst. (2010) 102:1536–46. doi: 10.1093/jnci/djq364
69. Wang L, Lankhorst L, Bernards R. Exploiting senescence for the treatment of cancer. Nat Rev Cancer. (2022) 22:340–55. doi: 10.1038/s41568-022-00450-9
70. Schmitt CA, Wang B, Demaria M. Senescence and cancer - role and therapeutic opportunities. Nat Rev Clin Oncol. (2022) 19:619–36. doi: 10.1038/s41571-022-00668-4
71. Yang Y. Comments on national guidelines for diagnosis and treatment of prostate cancer 2022 in China (English version). Chin J Cancer Res. (2022) 34:456–7. doi: 10.21147/j.issn.1000-9604.2022.05.05
72. Wala J, Nguyen P, Pomerantz M. Early treatment intensification in metastatic hormone-sensitive prostate cancer. J Clin Oncol. (2023) 41:3584–90. doi: 10.1200/jco.23.00723
73. Crona DJ, Milowsky MI, Whang YE. Androgen receptor targeting drugs in castration-resistant prostate cancer and mechanisms of resistance. Clin Pharmacol Ther. (2015) 98:582–9. doi: 10.1002/cpt.256
74. Karantanos T, Evans CP, Tombal B, Thompson TC, Montironi R, Isaacs WB. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur Urol. (2015) 67:470–9. doi: 10.1016/j.eururo.2014.09.049
75. de Kouchkovsky I, Chan E, Schloss C, Poehlein C, Aggarwal R. Diagnosis and management of neuroendocrine prostate cancer. Prostate. (2024). doi: 10.1002/pros.24664
76. Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, et al. Androgen receptor gene amplification: A possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. (1997) 57:314–9.
77. Tilley WD, Buchanan G, Hickey TE, Bentel JM. Mutations in the androgen receptor gene are associated with progression of human prostate cancer to androgen independence. Clin Cancer Res. (1996) 2:277–85.
78. Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. (2008) 68:5469–77. doi: 10.1158/0008-5472.Can-08-0594
79. Ewald JA, Desotelle JA, Church DR, Yang B, Huang W, Laurila TA, et al. Androgen deprivation induces senescence characteristics in prostate cancer cells in vitro and in vivo. Prostate. (2013) 73:337–45. doi: 10.1002/pros.22571
80. Pernicová Z, Slabáková E, Kharaishvili G, Bouchal J, Král M, Kunická Z, et al. Androgen depletion induces senescence in prostate cancer cells through down-regulation of skp2. Neoplasia. (2011) 13:526–36. doi: 10.1593/neo.11182
81. Schiewer MJ, Augello MA, Knudsen KE. The Ar dependent cell cycle: mechanisms and cancer relevance. Mol Cell Endocrinol. (2012) 352:34–45. doi: 10.1016/j.mce.2011.06.033
82. Burton DG, Giribaldi MG, Munoz A, Halvorsen K, Patel A, Jorda M, et al. Androgen deprivation-induced senescence promotes outgrowth of androgen-refractory prostate cancer cells. PloS One. (2013) 8:e68003. doi: 10.1371/journal.pone.0068003
83. Barakat DJ, Zhang J, Barberi T, Denmeade SR, Friedman AD, Paz-Priel I. Ccaat/enhancer binding protein B Controls androgen-deprivation-induced senescence in prostate cancer cells. Oncogene. (2015) 34:5912–22. doi: 10.1038/onc.2015.41
84. Kokal M, Mirzakhani K, Pungsrinont T, Baniahmad A. Mechanisms of androgen receptor agonist- and antagonist-mediated cellular senescence in prostate cancer. Cancers (Basel). (2020) 12(7). doi: 10.3390/cancers12071833
85. Gupta S, Pungsrinont T, Ženata O, Neubert L, Vrzal R, Baniahmad A. Interleukin-23 represses the level of cell senescence induced by the androgen receptor antagonists enzalutamide and darolutamide in castration-resistant prostate cancer cells. Horm Cancer. (2020) 11:182–90. doi: 10.1007/s12672-020-00391-5
86. Fizazi K, Shore N, Tammela TL, Ulys A, Vjaters E, Polyakov S, et al. Darolutamide in nonmetastatic, castration-resistant prostate cancer. N Engl J Med. (2019) 380:1235–46. doi: 10.1056/NEJMoa1815671
87. Hessenkemper W, Roediger J, Bartsch S, Houtsmuller AB, van Royen ME, Petersen I, et al. A natural androgen receptor antagonist induces cellular senescence in prostate cancer cells. Mol Endocrinol. (2014) 28:1831–40. doi: 10.1210/me.2014-1170
88. Yang J, Liu M, Hong D, Zeng M, Zhang X. The paradoxical role of cellular senescence in cancer. Front Cell Dev Biol. (2021) 9:722205. doi: 10.3389/fcell.2021.722205
89. Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. (2001) 11:S27–31. doi: 10.1016/s0962-8924(01)02151-1
90. Huang SB, Rivas P, Yang X, Lai Z, Chen Y, SChadler KL, et al. Sirt1 inhibition-induced senescence as a strategy to prevent prostate cancer progression. Mol Carcinog. (2022) 61:702–16. doi: 10.1002/mc.23412
91. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
92. Capece D, Verzella D, Tessitore A, Alesse E, Capalbo C, Zazzeroni F. Cancer secretome and inflammation: the bright and the dark sides of nf-Kb. Semin Cell Dev Biol. (2018) 78:51–61. doi: 10.1016/j.semcdb.2017.08.004
93. Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic Ras and the P53 tumor suppressor. PloS Biol. (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
94. Wagner J, Damaschke N, Yang B, Truong M, Guenther C, McCormick J, et al. Overexpression of the novel senescence marker B-galactosidase (Glb1) in prostate cancer predicts reduced Psa recurrence. PloS One. (2015) 10:e0124366. doi: 10.1371/journal.pone.0124366
95. Blute ML Jr., Damaschke N, Wagner J, Yang B, Gleave M, Fazli L, et al. Persistence of senescent prostate cancer cells following prolonged neoadjuvant androgen deprivation therapy. PloS One. (2017) 12:e0172048. doi: 10.1371/journal.pone.0172048
96. Carpenter VJ, Patel BB, Autorino R, Smith SC, Gewirtz DA, Saleh T. Senescence and castration resistance in prostate cancer: A review of experimental evidence and clinical implications. Biochim Biophys Acta Rev Cancer. (2020) 1874:188424. doi: 10.1016/j.bbcan.2020.188424
97. Yang L, Fang J, Chen J. Tumor cell senescence response produces aggressive variants. Cell Death Discovery. (2017) 3:17049. doi: 10.1038/cddiscovery.2017.49
98. Malaquin N, Vancayseele A, Gilbert S, Antenor-Habazac L, Olivier MA, Ait Ali Brahem Z, et al. DNA damage- but not enzalutamide-induced senescence in prostate cancer promotes senolytic Bcl-xl inhibitor sensitivity. Cells. (2020) 9(7). doi: 10.3390/cells9071593
99. Liao P, Yan B, Wang C, Lei P. Telomeres: dysfunction, maintenance, aging and cancer. Aging Dis. (2024). doi: 10.14336/ad.2023.1128
100. Bernadotte A, Mikhelson VM, Spivak IM. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging. (2016) 8:3–11. doi: 10.18632/aging.100871
101. Cheung AS, Yeap BB, Hoermann R, Hui J, Beilby JP, Grossmann M. Effects of androgen deprivation therapy on telomere length. Clin Endocrinol (Oxf). (2017) 87:381–5. doi: 10.1111/cen.13382
102. Tatar C, Avci CB, Acikgoz E, Oktem G. Doxorubicin-induced senescence promotes resistance to cell death by modulating genes associated with apoptotic and necrotic pathways in prostate cancer Du145 Cd133(+)/Cd44(+) cells. Biochem Biophys Res Commun. (2023) 680:194–210. doi: 10.1016/j.bbrc.2023.09.032
103. O'Hara SP, Splinter PL, Trussoni CE, Guicciardi ME, Splinter NP, Al Suraih MS, et al. The transcription factor Ets1 promotes apoptosis resistance of senescent cholangiocytes by epigenetically up-regulating the apoptosis suppressor Bcl2l1. J Biol Chem. (2019) 294:18698–713. doi: 10.1074/jbc.RA119.010176
104. Lin Y, Fukuchi J, Hiipakka RA, Kokontis JM, Xiang J. Up-regulation of bcl-2 is required for the progression of prostate cancer cells from an androgen-dependent to an androgen-independent growth stage. Cell Res. (2007) 17:531–6. doi: 10.1038/cr.2007.12
105. Kawata H, Kamiakito T, Nakaya T, Komatsubara M, Komatsu K, Morita T, et al. Stimulation of cellular senescent processes, including secretory phenotypes and anti-oxidant responses, after androgen deprivation therapy in human prostate cancer. J Steroid Biochem Mol Biol. (2017) 165:219–27. doi: 10.1016/j.jsbmb.2016.06.007
106. Bavik C, Coleman I, Dean JP, Knudsen B, Plymate S, Nelson PS. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. (2006) 66:794–802. doi: 10.1158/0008-5472.Can-05-1716
107. Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. (2008) 133:1019–31. doi: 10.1016/j.cell.2008.03.039
108. Malinowska K, Neuwirt H, Cavarretta IT, Bektic J, Steiner H, Dietrich H, et al. Interleukin-6 stimulation of growth of prostate cancer in vitro and in vivo through activation of the androgen receptor. Endocr Relat Cancer. (2009) 16:155–69. doi: 10.1677/erc-08-0174
109. Seaton A, Scullin P, Maxwell PJ, Wilson C, Pettigrew J, Gallagher R, et al. Interleukin-8 signaling promotes androgen-independent proliferation of prostate cancer cells via induction of androgen receptor expression and activation. Carcinogenesis. (2008) 29:1148–56. doi: 10.1093/carcin/bgn109
110. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc Natl Acad Sci U.S.A. (2001) 98:12072–7. doi: 10.1073/pnas.211053698
111. Li Y, Xu X, Wang L, Liu G, Li Y, Wu X, et al. Senescent mesenchymal stem cells promote colorectal cancer cells growth via galectin-3 expression. Cell Biosci. (2015) 5:21. doi: 10.1186/s13578-015-0012-3
112. Scott E. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. Sweeney cj, chen yh, carducci M, liu G, jarrard df, eisenberger M, wong yn, hahn N, kohli M, cooney mm, dreicer R, vogelzang nj, picus J, shevrin D, hussain M, garcia ja, dipaola rs. Department of medicine; department of biostatistics and computational biology; dana-farber cancer institute, boston; harvard medical school, boston; johns hopkins university, baltimore; university of wisconsin carbone cancer center; school of medicine and public health; madison; fox chase cancer center, temple university health system, philadelphia; Indiana university melvin and bren simon cancer center, Indianapolis; mayo clinic, rochester, mn; university hospitals case medical center, seidman cancer center; cleveland clinic taussig cancer institute; both in cleveland; university of virginia cancer center, charlottesville; comprehensive cancer centers of nevada, las vegas; siteman cancer center, washington university school of medicine, st. Louis; northshore university health system, evanston, il; university of michigan comprehensive cancer center, ann arbor; rutgers cancer institute of new Jersey, new brunswick.N engl J med. 2015 aug 20;373(8):737-46. [Epub 2015 aug 5]. Doi: 10.1056/nejmoa1503747. Urol Oncol. (2017) 35:123. doi: 10.1016/j.urolonc.2016.12.021
113. James ND, Sydes MR, Clarke NW, Mason MD, Dearnaley DP, Spears MR, et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (Stampede): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet. (2016) 387:1163–77. doi: 10.1016/s0140-6736(15)01037-5
114. Pan J, Chi C, Qian H, Zhu Y, Shao X, Sha J, et al. Neoadjuvant chemohormonal therapy combined with radical prostatectomy and extended plnd for very high risk locally advanced prostate cancer: A retrospective comparative study. Urol Oncol. (2019) 37:991–8. doi: 10.1016/j.urolonc.2019.07.009
115. Tucci M, Bertaglia V, Vignani F, Buttigliero C, Fiori C, Porpiglia F, et al. Addition of docetaxel to androgen deprivation therapy for patients with hormone-sensitive metastatic prostate cancer: A systematic review and meta-analysis. Eur Urol. (2016) 69:563–73. doi: 10.1016/j.eururo.2015.09.013
116. Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, et al. Drug resistance in cancer: an overview. Cancers (Basel). (2014) 6:1769–92. doi: 10.3390/cancers6031769
117. Jiang X, Guo S, Wang S, Zhang Y, Chen H, Wang Y, et al. Eif4a3-induced circarhgap29 promotes aerobic glycolysis in docetaxel-resistant prostate cancer through Igf2bp2/C-Myc/Ldha signaling. Cancer Res. (2022) 82:831–45. doi: 10.1158/0008-5472.Can-21-2988
118. Wang J, Wei J, Pu T, Zeng A, Karthikeyan V, Bechtold B, et al. Cholinergic signaling via muscarinic M1 receptor confers resistance to docetaxel in prostate cancer. Cell Rep Med. (2024). doi: 10.1016/j.xcrm.2023.101388
119. Zhao W, Lin ZX, Zhang ZQ. Cisplatin-induced premature senescence with concomitant reduction of gap junctions in human fibroblasts. Cell Res. (2004) 14:60–6. doi: 10.1038/sj.cr.7290203
120. Hickman JA. Apoptosis induced by anticancer drugs. Cancer Metastasis Rev. (1992) 11:121–39. doi: 10.1007/bf00048059
121. Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, et al. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. (1999) 59:3761–7.
122. Sapega O, Mikyšková R, Bieblová J, Mrázková B, Hodný Z, Reiniš M. Distinct phenotypes and 'Bystander' Effects of senescent tumour cells induced by docetaxel or immunomodulatory cytokines. Int J Oncol. (2018) 53:1997–2009. doi: 10.3892/ijo.2018.4553
123. Scuric Z, Carroll JE, Bower JE, Ramos-Perlberg S, Petersen L, Esquivel S, et al. Biomarkers of aging associated with past treatments in breast cancer survivors. NPJ Breast Cancer. (2017) 3:50. doi: 10.1038/s41523-017-0050-6
124. Pardella E, Pranzini E, Nesi I, Parri M, Spatafora P, Torre E, et al. Therapy-induced stromal senescence promoting aggressiveness of prostate and ovarian cancer. Cells. (2022) 11:4026. doi: 10.3390/cells11244026
125. Chang BD, Swift ME, Shen M, Fang J, Broude EV, Roninson IB. Molecular determinants of terminal growth arrest induced in tumor cells by a chemotherapeutic agent. Proc Natl Acad Sci U.S.A. (2002) 99:389–94. doi: 10.1073/pnas.012602599
126. Wu Z, Qu J, Zhang W, Liu GH. Stress, epigenetics, and aging: unraveling the intricate crosstalk. Mol Cell. (2024) 84:34–54. doi: 10.1016/j.molcel.2023.10.006
127. Crouch J, Shvedova M, Thanapaul R, Botchkarev V, Roh D. Epigenetic regulation of cellular senescence. Cells. (2022) 11(4). doi: 10.3390/cells11040672
128. Costantino VV, Lobos-Gonzalez L, Ibañez J, Fernandez D, Cuello-Carrión FD, Valenzuela MA, et al. Dehydroleucodine inhibits tumor growth in a preclinical melanoma model by inducing cell cycle arrest, senescence and apoptosis. Cancer Lett. (2016) 372:10–23. doi: 10.1016/j.canlet.2015.12.004
129. Jiang MJ, Gu DN, Dai JJ, Huang Q, Tian L. Dark side of cytotoxic therapy: chemoradiation-induced cell death and tumor repopulation. Trends Cancer. (2020) 6:419–31. doi: 10.1016/j.trecan.2020.01.018
130. Sun Y, Nelson PS. Molecular pathways: involving microenvironment damage responses in cancer therapy resistance. Clin Cancer Res. (2012) 18:4019–25. doi: 10.1158/1078-0432.Ccr-11-0768
131. Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. (2018) 553:96–100. doi: 10.1038/nature25167
132. Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of P53. Oncogene. (2013) 32:5129–43. doi: 10.1038/onc.2012.640
133. Roake CM, Artandi SE. Control of cellular aging, tissue function, and cancer by P53 downstream of telomeres. Cold Spring Harb Perspect Med. (2017) 7(5). doi: 10.1101/cshperspect.a026088
134. Tamura RE, Lana MG, Costanzi-Strauss E, Strauss BE. Combination of cabazitaxel and P53 gene therapy abolishes prostate carcinoma tumor growth. Gene Ther. (2020) 27:15–26. doi: 10.1038/s41434-019-0071-x
135. Li H, Zhang W, Zhao K, Zhao D, Zheng S, Hu Y. A previously identified apoptosis inhibitor Iaspp confers resistance to chemotherapeutic drugs by suppressing senescence in cancer cells. J Biol Chem. (2020) 295:4049–63. doi: 10.1074/jbc.RA119.011411
136. Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A senescence program controlled by P53 and P16ink4a contributes to the outcome of cancer therapy. Cell. (2002) 109:335–46. doi: 10.1016/s0092-8674(02)00734-1
137. Netterfield TS, Ostheimer GJ, Tentner AR, Joughin BA, Dakoyannis AM, Sharma CD, et al. Biphasic jnk-erk signaling separates the induction and maintenance of cell senescence after DNA damage induced by topoisomerase ii inhibition. Cell Syst. (2023) 14:582–604.e10. doi: 10.1016/j.cels.2023.06.005
138. Hashemi M, Zandieh MA, Talebi Y, Rahmanian P, Shafiee SS, Nejad MM, et al. Paclitaxel and docetaxel resistance in prostate cancer: molecular mechanisms and possible therapeutic strategies. BioMed Pharmacother. (2023) 160:114392. doi: 10.1016/j.biopha.2023.114392
139. Sun X, Shi B, Zheng H, Min L, Yang J, Li X, et al. Senescence-associated secretory factors induced by cisplatin in melanoma cells promote non-senescent melanoma cell growth through activation of the Erk1/2-Rsk1 pathway. Cell Death Dis. (2018) 9:260. doi: 10.1038/s41419-018-0303-9
140. Yazbeck V, Alesi E, Myers J, Hackney MH, Cuttino L, Gewirtz DA. An overview of chemotoxicity and radiation toxicity in cancer therapy. In: Gewirtz DA, Fisher PB, editors. Strategies to mitigate the toxicity of cancer therapeutics. Advances in cancer research (2022). p. 1–27.
141. Deek MP, Yu C, Phillips R, Song DY, Deville C, Greco S, et al. Radiation therapy in the definitive management of oligometastatic prostate cancer: the johns hopkins experience. Int J Radiat Oncol Biol Phys. (2019) 105:948–56. doi: 10.1016/j.ijrobp.2019.08.008
142. Aminsharifi A, Polascik TJ. Radical prostatectomy, external beam radiotherapy, or external beam radiotherapy with brachytherapy boost and disease progression and mortality in patients with gleason score 9-10 prostate cancer. Eur Urol. (2018) 74:526–. doi: 10.1016/j.eururo.2018.05.031
143. Lyu F, Gao X, Shang S, Li S, Ren X, Chen J. Radiotherapy resistance in prostate cancer cells: Akr1c3 inhibition of ubiquitinated degradation of Nrf2 through interaction with keap1. Int J Radiat Oncol Biol Phys. (2023) 117:E248–E. doi: 10.1016/j.ijrobp.2023.06.1187
144. Larsen BD, Benada J, Yung PYK, Bell RAV, Pappas G, Urban V, et al. Cancer cells use self-inflicted DNA breaks to evade growth limits imposed by genotoxic stress. Science. (2022) 376:476–+. doi: 10.1126/science.abi6378
145. Schumacher B, Pothof J, Vijg J, Hoeijmakers JHJ. The central role of DNA damage in the ageing process. Nature. (2021) 592:695–703. doi: 10.1038/s41586-021-03307-7
146. Shimono H, Kaida A, Homma H, Nojima H, Onozato Y, Harada H, et al. Fluctuation in radioresponse of hela cells during the cell cycle evaluated based on micronucleus frequency. Sci Rep. (2020) 10(1). doi: 10.1038/s41598-020-77969-0
147. Basu S, Dong Y, Kumar R, Jeter C, Tang DG. Slow-cycling (Dormant) cancer cells in therapy resistance, cancer relapse and metastasis. Semin Cancer Biol. (2022) 78:90–103. doi: 10.1016/j.semcancer.2021.04.021
148. Larsen BD, Rampalli S, Burns LE, Brunette S, Dilworth FJ, Megeney LA. Caspase 3/caspase-activated dnase promote cell differentiation by inducing DNA strand breaks. Proc Natl Acad Sci United States America. (2010) 107:4230–5. doi: 10.1073/pnas.0913089107
149. Zhu P, Tan MJ, Huang R-L, Tan CK, Chong HC, Pal M, et al. Angiopoietin-like 4 protein elevates the prosurvival intracellular O2(-):H2O2 ratio and confers anoikis resistance to tumors. Cancer Cell. (2011) 19:401–15. doi: 10.1016/j.ccr.2011.01.018
150. Chaube B, Citrin KM, Sahraei M, Singh AK, de Urturi DS, Ding W, et al. Suppression of angiopoietin-like 4 reprograms endothelial cell metabolism and inhibits angiogenesis. Nat Commun. (2023) 14(1). doi: 10.1038/s41467-023-43900-0
151. Zhang YH, Liu XL, Zeng L, Zhao XR, Chen QP, Pan Y, et al. Exosomal protein angiopoietin-like 4 mediated radioresistance of lung cancer by inhibiting ferroptosis under hypoxic microenvironment. Br J Cancer. (2022) 127:1760–72. doi: 10.1038/s41416-022-01956-7
152. Barer L, Schroeder SK, Weiskirchen R, Bacharach E, Ehrlich M. Lipocalin-2 regulates the expression of interferon-stimulated genes and the susceptibility of prostate cancer cells to oncolytic virus infection. Eur J Cell Biol. (2023) 102(2). doi: 10.1016/j.ejcb.2023.151328
153. Al Jaberi S, Cohen A, Abdulrazzaq YM, Ojha S, Bastaki S, Adeghate EA. Lipocalin-2: structure, function, distribution and role in metabolic disorders. Biomedicine Pharmacotherapy. (2021) 142. doi: 10.1016/j.biopha.2021.112002
154. Huang H. Matrix metalloproteinase-9 (Mmp-9) as a cancer biomarker and mmp-9 biosensors: recent advances. Sensors. (2018) 18(10). doi: 10.3390/s18103249
155. Augoff K, Hryniewicz-Jankowska A, Tabola R, Stach K. Mmp9: A tough target for targeted therapy for cancer. Cancers. (2022) 14(7). doi: 10.3390/cancers14071847
156. Mongre R, Sodhi SS, Sharma N, Ghosh M, Hyunkim J, Kim N, et al. Epigenetic induction of epithelial to mesenchymal transition by LCN2 mediates metastasis and tumorigenesis, which is abrogated by nf-Kb inhibitor brm270 in a xenograft model of lung adenocarcinoma. Int J Oncol. (2016) 48:84–98. doi: 10.3892/ijo.2015.3245
157. Park SS, Choi YW, Kim J-H, Kim HS, Park TJ. Senescent tumor cells: an overlooked adversary in the battle against cancer. Exp Mol Med. (2021) 53:1834–41. doi: 10.1038/s12276-021-00717-5
158. Najafi M, Mortezaee K, Majidpoor J. Cancer stem cell (Csc) resistance drivers. Life Sci. (2019) 234. doi: 10.1016/j.lfs.2019.116781
159. Walcher L, Kistenmacher AK, Suo HZ, Kitte R, Dluczek S, Strauss A, et al. Cancer stem cells-origins and biomarkers: perspectives for targeted personalized therapies. Front Immunol. (2020) 11:1280. doi: 10.3389/fimmu.2020.01280
160. Jin W. Role of jak/stat3 signaling in the regulation of metastasis, the transition of cancer stem cells, and chemoresistance of cancer by epithelial-mesenchymal transition. Cells. (2020) 9(1). doi: 10.3390/cells9010217
161. Yang LQ, Shi PF, Zhao GC, Xu J, Peng W, Zhang JY, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduction Targeted Ther. (2020) 5(1). doi: 10.1038/s41392-020-0110-5
162. Xu M, Tchkonia T, Kirkland JL. Perspective: targeting the jak/stat pathway to fight age-related dysfunction. Pharmacol Res. (2016) 111:152–4. doi: 10.1016/j.phrs.2016.05.015
163. Birnie R, Bryce SD, Roome C, Dussupt V, Droop A, Lang SH, et al. Gene expression profiling of human prostate cancer stem cells reveals a pro-inflammatory phenotype and the importance of extracellular matrix interactions. Genome Biol. (2008) 9(5). doi: 10.1186/gb-2008-9-5-r83
164. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting notch, hedgehog, and wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. (2015) 12:445–64. doi: 10.1038/nrclinonc.2015.61
165. Karabicici M, Alptekin S, Karagonlar ZF, Erdal E. Doxorubicin-induced senescence promotes stemness and tumorigenicity in Epcam-/Cd133-nonstem cell population in hepatocellular carcinoma cell line, huh-7. Mol Oncol. (2021) 15:2185–202. doi: 10.1002/1878-0261.12916
166. Lyu F, Shang SY, Gao XS, Ma MW, Xie M, Ren XY, et al. Uncovering the secrets of prostate cancer's radiotherapy resistance: advances in mechanism research. Biomedicines. (2023) 11(6). doi: 10.3390/biomedicines11061628
167. Gong LY, Zhang YJ, Zhao J, Zhang YL, Tu KS, Jiao LY, et al. All-in-one biomimetic nanoplatform based on hollow polydopamine nanoparticles for synergistically enhanced radiotherapy of colon cancer. Small. (2022) 18(14). doi: 10.1002/smll.202107656
168. Wen PB, Xia JF, Cao XB, Chen B, Tao YP, Wu LJ, et al. Dbcrsr: A manually curated database for regulation of cancer radiosensitivity. Database-the J Biol Database Curation. (2018). doi: 10.1093/database/bay049
169. Shay JW, Roninson IB. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene. (2004) 23:2919–33. doi: 10.1038/sj.onc.1207518
170. Gewirtz DA, Holt SE, Elmore LW. Accelerated senescence: an emerging role in tumor cell response to chemotherapy and radiation. Biochem Pharmacol. (2008) 76:947–57. doi: 10.1016/j.bcp.2008.06.024
171. Schoetz U, Klein D, Hess J, Shnayien S, Spoerl S, Orth M, et al. Early senescence and production of senescence-associated cytokines are major determinants of radioresistance in head-and-neck squamous cell carcinoma. Cell Death Dis. (2021) 12:1162. doi: 10.1038/s41419-021-04454-5
173. Salminen A, Kauppinen A, Kaarniranta K. Emerging role of Nf-Kb signaling in the induction of senescence-associated secretory phenotype (Sasp). Cell Signal. (2012) 24:835–45. doi: 10.1016/j.cellsig.2011.12.006
174. Stegh AH. Targeting the P53 signaling pathway in cancer therapy – the promises, challenges and perils. Expert Opin Ther Targets. (2012) 16:67–83. doi: 10.1517/14728222.2011.643299
175. Mijit M, Caracciolo V, Melillo A, Amicarelli F, Giordano A. Role of P53 in the regulation of cellular senescence. Biomolecules. (2020) 10:420. doi: 10.3390/biom10030420
176. Wade M, Li YC, Wahl GM. Mdm2, mdmx and P53 in oncogenesis and cancer therapy. Nat Rev Cancer. (2013) 13:83–96. doi: 10.1038/nrc3430
177. Hines J, Lartigue S, Dong HQ, Qian YM, Crews CM. Mdm2-recruiting protac offers superior, synergistic antiproliferative activity via simultaneous degradation of brd4 and stabilization of P53. Cancer Res. (2019) 79:251–62. doi: 10.1158/0008-5472.Can-18-2918
178. Kong N, Tao W, Ling X, Wang JQ, Xiao YL, Shi SJ, et al. Synthetic mrna nanoparticle-mediated restoration of P53 tumor suppressor sensitizes p53-deficient cancers to Mtor inhibition. Sci Trans Med. (2019) 11(523). doi: 10.1126/scitranslmed.aaw1565
179. Davalos AR, Coppe JP, Campisi J, Desprez PY. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. (2010) 29:273–83. doi: 10.1007/s10555-010-9220-9
180. Chaib S, Tchkonia T, Kirkland JL. Cellular senescence and senolytics: the path to the clinic. Nat Med. (2022) 28:1556–68. doi: 10.1038/s41591-022-01923-y
181. Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. (2016) 15:428–35. doi: 10.1111/acel.12445
182. Ge R, Wang Z, Cheng L. Tumor microenvironment heterogeneity an important mediator of prostate cancer progression and therapeutic resistance. NPJ Precis Oncol. (2022) 6:31. doi: 10.1038/s41698-022-00272-w
183. Zetrini AE, Lip H, Abbasi AZ, Alradwan I, Ahmed T, He CS, et al. Remodeling tumor immune microenvironment by using polymer-lipid-manganese dioxide nanoparticles with radiation therapy to boost immune response of castration-resistant prostate cancer. Research. (2023) 2023(10).
184. Li P, Kong X, He Y, Liu Y, Peng X, Li Z, et al. Recent developments in application of single-cell Rna sequencing in the tumour immune microenvironment and cancer therapy. Military Med Res. (2023) 10:383–402.
185. Takasugi M, Yoshida Y, Hara E, Ohtani N. The role of cellular senescence and sasp in tumour microenvironment. FEBS J. (2023) 290:1348–61. doi: 10.1111/febs.16381
186. Özdemir A, Şimay Demir YD, Yeşilyurt ZE, Ark M. Senescent Cells and Sasp in Cancer Microenvironment: New Approaches in Cancer Therapy. Adv. Protein Chem. Struct. Biol. (2023) 133:115–58. doi: 10.1016/bs.apcsb.2022.10.002
Keywords: cellular senescence, drug resistance, treatment resistance, prostate cancer, SIPS
Citation: Xu M-Y, Xia Z-Y, Sun J-X, Liu C-Q, An Y, Xu J-Z, Zhang S-H, Zhong X-Y, Zeng N, Ma S-Y, He H-D, Wang S-G and Xia Q-D (2024) A new perspective on prostate cancer treatment: the interplay between cellular senescence and treatment resistance. Front. Immunol. 15:1395047. doi: 10.3389/fimmu.2024.1395047
Received: 03 March 2024; Accepted: 01 April 2024;
Published: 17 April 2024.
Edited by:
Dechao Feng, University College London, United KingdomReviewed by:
Bin Xu, Renmin Hospital of Wuhan University, ChinaYingkun Xu, Chongqing Medical University, China
Copyright © 2024 Xu, Xia, Sun, Liu, An, Xu, Zhang, Zhong, Zeng, Ma, He, Wang and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shao-Gang Wang, c2d3YW5ndGptQDE2My5jb20=; Qi-Dong Xia, cWlkb25neGlhX21kQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work