 Xing Yu
Xing Yu Jin Yuan2
Jin Yuan2 Lei Yue
Lei Yue- 1Department of Pathogen Biology and Immunology, Faculty of Basic Medical Science, Kunming Medical University, Kunming, China
- 2Clinical Laboratory, Puer Hospital of Traditional Chinese Medicine, Puer, China
- 3Institute of Medical Biology, Chinese Academy of Medical Sciences and Peking Union Medical College, Kunming, China
Necroptosis, a recently discovered form of cell-programmed death that is distinct from apoptosis, has been confirmed to play a significant role in the pathogenesis of bacterial infections in various animal models. Necroptosis is advantageous to the host, but in some cases, it can be detrimental. To understand the impact of necroptosis on the pathogenesis of bacterial infections, we described the roles and molecular mechanisms of necroptosis caused by different bacterial infections in this review.
1 Introduction
Initially, researchers thought there were only two ways of cell death: apoptosis and necrosis. Apoptosis is active and program-controlled, and its key regulator is caspase. The morphological changes associated with apoptosis mainly include cell shrinkage and chromatin condensation, the formation of apoptotic bodies and cytoskeleton disintegration. Necrosis is passive and unprogrammed and can be activated in various ways, e.g., by bacterial infection (1), toxins (2), and ischemia (3); the morphological changes characteristic of necrosis include cellular swelling, plasma membrane rupture, and the release of cellular content (4, 5). In 2005, Professor Junying Yuan first demonstrated that nonapoptotic cell death induced by death receptor signaling is programmed (6), and coined the term “programmed necrosis” (also known as necroptosis). Necroptosis was originally named receptor-interacting protein 1 (RIP1)-dependent necrosis, referring to a caspase-independent form of programmed cell death that was characterized by both necrosis and apoptosis. During necroptosis, cells undergo changes such as membrane rupture, organelle swelling, and nuclear and cytoplasmic disintegration (7, 8).
Necroptosis depends on the activity of receptor interacting protein kinase-1 (RIPK1), receptor interacting protein kinase-3 (RIPK3), and mixed lineage kinase domain-like protein (MLKL) (9–11). Necroptosis can be initiated by tumor necrosis factor receptor (TNFR) (12), pattern recognition receptors (PRRs), including Toll-like receptor (TLR3/4), Nod-like receptors (NLRs), and RIG-I-like receptors (RLRs) (4, 13–16), INF-α receptors (17, 18), adhesion receptors (19), and DNA-dependent activator of IFN (DAI) (also known as ZBP-1) (20, 21). The most classical pathway is TNF-induced necroptosis, which is described in Figure 1. After TNF binds to TNFR1 on the cell membrane, it recruits TNFR1-associated death domain protein (TRADD) and RIPK1 to form an early complex, which is subsequently detached from TNFR and recruits TNF receptor-associated factor 2 (TRAF2), cellular inhibitors of apoptosis (cIAPs) (including cIAP1/2) and linear ubiquitin chain assembly complex (LUBAC) to form complex I. At this time, RIPK1 is ubiquitinated by cIAPs and LUBAC, and the result of ubiquitination is the stabilization of complex I, which then continues to recruit downstream proteins, such as TGF activated kinase 1 (TAK1), TAK1 binding protein 2/3 (TAB2/3), and an IκB kinase complex (IKK) composed of IKKα, IKKβ, and NF-κB essential modulator (NEMO) (4, 22). The recruited downstream proteins activate the NF-κB and mitogen-activated protein kinase (MAPK) pathways, resulting in increased expression of proinflammatory genes, which contribute to the production of proinflammatory factors (4, 22). Blocking cIAPs or cylindromatosis protein (CYLD) to remove the ubiquitin chain on RIPK1 (23) can inhibit the ubiquitination of RIPK1 and induce the formation of complex IIa/b, which is composed of TRADD, RIPK1, and FAS-associated death domain protein (FADD). Complex IIa/b activates caspase-8 and then induces apoptosis (5, 24). RIPK1 contains an N-terminal kinase domain, a C-terminal death domain (DD), and an RIP homotypic interaction motif (RHIM). When the activity of caspase-8 is blocked and apoptosis is inhibited, RIPK1 recruits RIPK3 through interactions between the RHIM domains, and RIPK3 continues to recruit downstream MLKL and forms a complex, called a necrosome. RIPK3 recruits MLKL and phosphorylates MLKL, which subsequently translocates to the plasma membrane, triggers necroptosis, and releases damage-associated molecular patterns (DAMPs) from the cell (25–27), leading to excessive inflammation (28). However, the immune system has evolved RIPK1-independent necroptosis, and RHIM domain-containing protein molecules such as ZBP1 and TIR domain-containing adapter-inducing interferon-β (TRIF) can both induce necroptosis through the binding of their own RHIM domain to RIPK3 (20).
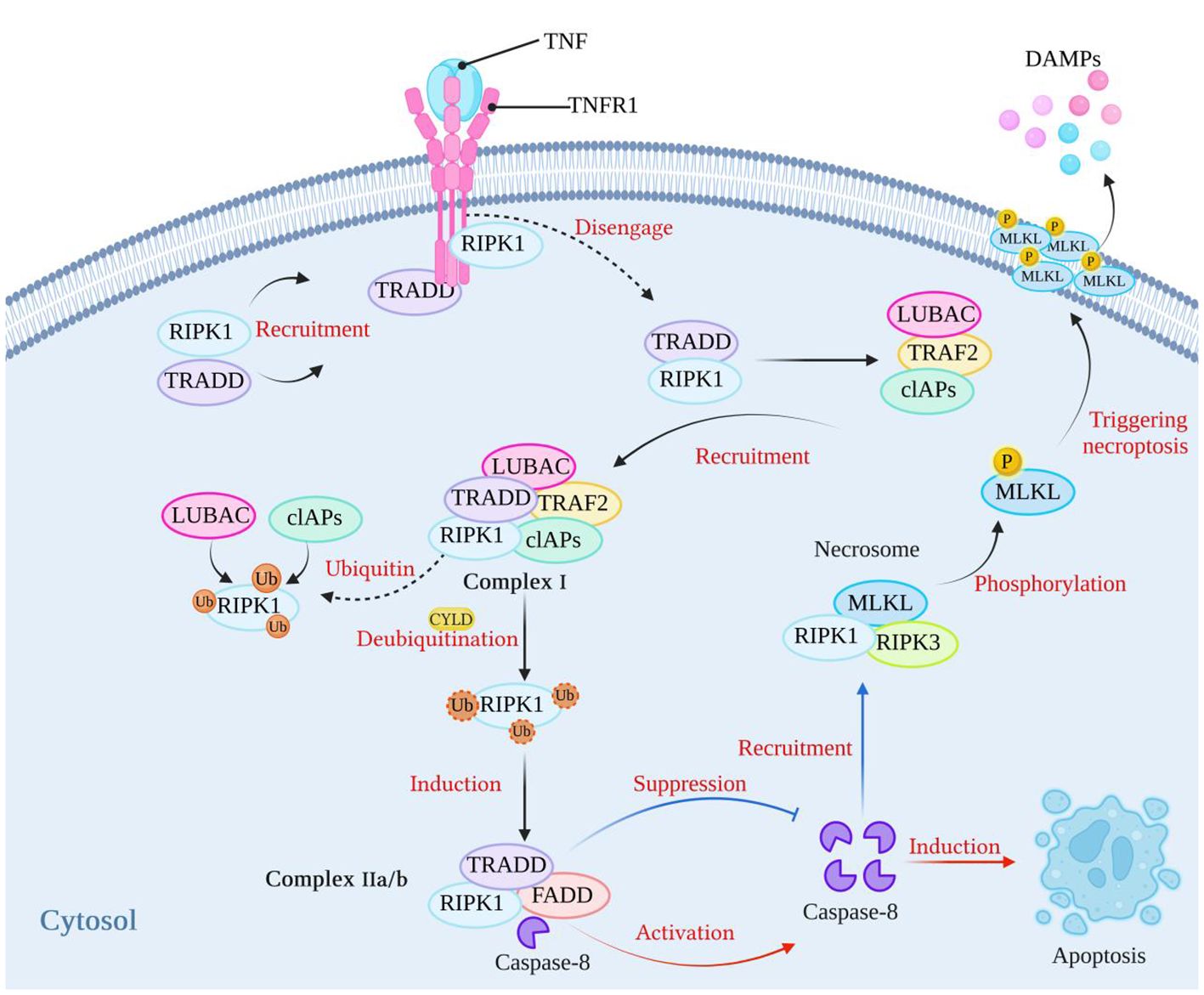
Figure 1 The classical mechanism of necroptosis. After TNF binds to its receptor, it can recruit RIPK1 and TRADD to form a complex. Subsequently, RIPK1 and TRADD dissociate from the TNF receptor and recruit LUBAC, TRAF2, and cIAPs to form complex I. At this stage, RIPK1 undergoes ubiquitination by LUBAC and cIAPs, which stabilizes the complex. When cIAPs are inhibited or when the ubiquitin chains on RIPK1 are removed using CYLD, a complex called complex IIa/b, consisting of TRADD, RIPK1, and FADD, is formed. Complex IIa/b activates caspase-8, which then induces cell apoptosis. When caspase-8 activity is blocked, cell apoptosis is inhibited. Subsequently, RIPK1 recruits RIPK3 through interactions between their RHIM domains. RIPK3 then recruits MLKL, forming necrosomes. MLKL is phosphorylated, and phosphorylated MLKL molecules aggregate and translocate to the plasma membrane, thereby triggering necroptosis. (Created with BioRender.com).
Dysregulated necroptosis can be involved in the occurrence of neurological diseases, such as Parkinson’s disease (29) and Alzheimer’s disease (30). In addition, necroptosis can play a different role in tumor diseases. For example, in gastric adenocarcinoma (31) and non-small cell lung cancer (32), the body can provide a favorable environment for the growth of cancer cells by downregulating the expression of proteins related to the necroptosis pathway. Therefore, activating necroptosis may be an effective anticancer strategy. However, some studies have also demonstrated that the upregulation of RIPK3 expression can also promote the occurrence of some tumor diseases, such as aggressive and recurrent breast cancer, which can promote the strong proliferation of cancer cells (33). This dual role of necroptosis in the body is also reflected in pathogen infection. Necroptosis is a growing concern in the pathogenesis of bacterial infections (34). Cell death is a common result of interactions between bacteria and hosts (35). Necroptosis is the lytic death of cells, is generally considered highly proinflammatory, and plays an important role in the pathogenesis of bacterial infections (4, 36, 37). Necroptosis may protect the host. Conversely, in some cases, necroptosis has an adverse effect on the host. Therefore, understanding the role of necroptosis in the process of bacterial infection and its mechanism of action in the defense against bacterial infection is important. This paper focuses on whether necroptosis during bacterial infection is beneficial or harmful and discusses the mechanism of action (Figure 2, Table 1).
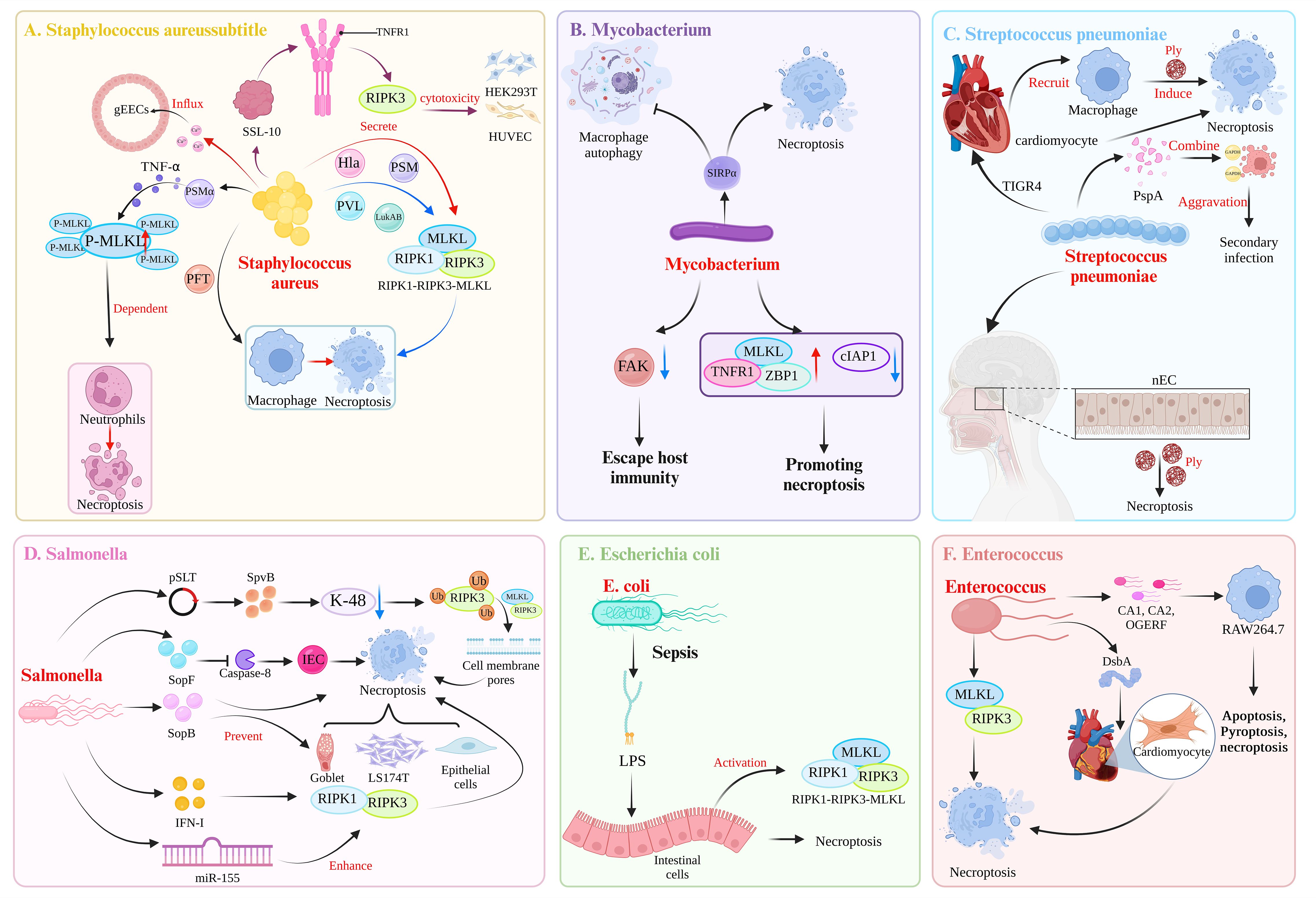
Figure 2 Mechanisms of bacterial induction of necroptosis. (A) Toxic virulence factors secreted by Staphylococcus aureus, such as Hla, PAM, PVL, and LukAB, can induce necroptosis in macrophages through the RIPK1-RIPK3-MLKL pathway. Among them, PSMα can induce TNFα secretion, leading to MLKL-dependent necroptosis in neutrophils. In addition, the virulence factor PFT can induce necroptosis in macrophages. The S. aureus-secreted SSL-10 binds to TNFR on the cell membrane to induce RIPK3-dependent necroptosis in HEK293T cell and HUVECs. S. aureus can induce necroptosis in gEECs, and the trigger is Ca2+ influx. (B) Mycobacterium tuberculosis triggers SIRPα in macrophages, leading to the inhibition of autophagy and the promotion of necroptosis. Additionally, M. tuberculosis can downregulate the expression of FAK in macrophages to evade host immunity. M. tuberculosis also shapes an environment that promotes necroptosis in macrophages by upregulating MLKL, TNFR1, ZBP1 expression and downregulating cIAP1 expression. (C) Following invasion of the heart by the TIGR4 strain of Streptococcus pneumoniae, the secretion of Ply can induce necroptosis in both cardiac myocytes and recruited macrophages. During the asymptomatic colonization of S. pneumoniae in the nasopharynx, Ply can induce necroptosis in nasopharyngeal epithelial cells (nECs). In the case of coinfection with influenza A virus and S. pneumoniae, the surface protein A (PspA) of S. pneumoniae acts as a cell adhesin and binds to GAPDH in dying cells, thereby increasing the localization of S. pneumoniae in the lower airways and exacerbating secondary infection following influenza. (D) The pSLT-encoded SpvB effector factor inhibits K-48-mediated ubiquitination of RIPK3, thereby mediating the formation of cell membrane pores through the RIPK3-MLKL pathway, resulting in necroptosis of intestinal epithelial cells (IECs). The effector factor SopF of the T3SS can induce necroptosis of IECs by blocking the activity of caspase-8, thereby enabling Salmonella enterica to spread to the intestinal lamina propria. SopB, encoded by SPI-1, can prevent the necroptosis of goblet cells, LS174T cells, and epithelial cells. S. typhimurium utilizes the host’s IFN-I response to induce necroptosis in macrophages mediated by RIPK1-RIPK3. Additionally, S. typhimurium induces the expression of miR-155, which promotes macrophages necroptosis. This effect is a result of miR-155 targeting of the RIPK1-RIPK3 pathway, which further promote apoptosis. (E) During sepsis, the lipopolysaccharide (LPS) by Escherichia coli can upregulate the expression of the necrosis-related proteins RIPK1, RIPK3, and MLKL, leading to intestinal epithelial cells necroptosis. (F) After infection with Enterococcus faecalis, DsbA can induce microinjury in the heart of Caenorhabditis elegans. Subsequently, at the site of microinjury in the heart, E. faecalis can induce the apoptosis and necroptosis of cardiomyocytes. In refractory apical periodontitis, E. faecalis can induce necroptosis in macrophages mediated by RIPK3-MLKL. Strains of E. faecalis isolated from root canals (CA1, CA2) and the OGERF strain can induce apoptosis, pyroptosis, and necroptosis in RAW264.7 macrophages. (Created with BioRender.com).
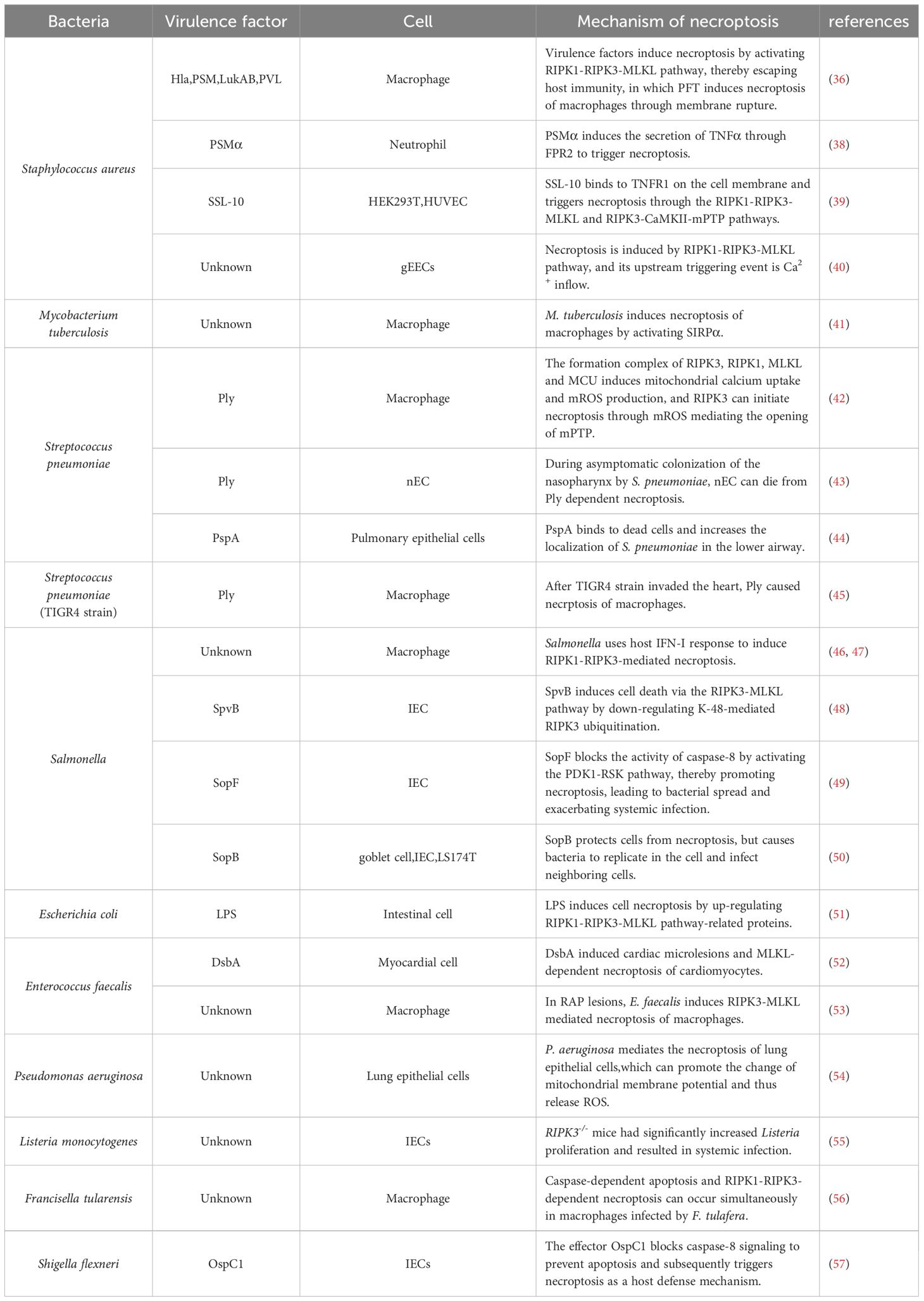
Table 1 Mechanism of necroptosis in bacteria.
2 The role of necroptosis in bacterial infection
2.1 Staphylococcus aureus
Staphylococcus aureus is the main pathogen of nosocomial infections and can cause a variety of diseases, such as pneumonia, endocarditis, sepsis, and osteomyelitis, which seriously threaten human health (58). In the process of infection, in addition to being able to induce inflammatory cells to release inflammatory factors and accumulate at the site of infection to trigger an inflammatory response (59–63), S. aureus can also induce necroptosis in host cells, such as macrophages (36) and neutrophils (38).
A typical feature of S. aureus pneumonia is toxin-induced necroptosis of immune and resident cells. Virulence factors secreted by S. aureus, such as Hla, PSM, LukAB and PVL, can induce necroptosis in macrophages through the RIPK1-RIPK3-MLKL pathway (36). PSMα reportedly triggers neutrophil necroptosis via FPR2-induced TNFα secretion (38). Some S. aureus phagocytosed by polymorphonuclear neutrophils (PMN) can survive in phagosomes, thereby inducing PMN necroptosis, and this process is dependent on RIPK3 and independent of RIPK1 and MLKL (64, 65). Examples of necroptosis independent of active RIPK1 or MLKL have been reported. TLR3 or TLR4 can activate RIPK3 through TRIF, thereby directly initiating necroptosis through TRIF-RIPK3-MLKL (16). RIPK3 can also induce necroptosis of cardiomyocytes through calmodulin-dependent protein kinase II (CaMKII) and mitochondrial pathways (66). Thus, delineating the molecular pathways that ultimately lead to PMN death will provide new targets for the treatment of S. aureus infections. After S. aureus is phagocytosed by macrophages, approximately 10% of the S. aureus population will persist in macrophages, with the remaining bacteria eliminated by bacteriolysis. However, excessive bacteriolysis can cause cell death (67). Further studies confirmed that this type of cell death is not apoptosis or pyroptosis (It is characterized by dependence on inflammatory caspase enzymes, mainly caspase-1, 4, 5, 11, accompanied by the release of a large number of pro-inflammatory cytokines) but rather AIM2-mediated necroptosis (67). Apoptosis is known to be important for clearing pathogens (68), and the authors propose a potential immune manipulation strategy by which S. aureus sacrifices the minority to trigger a limited necroptosis, thereby releasing signals from dead cells to inhibit apoptosis and other anti-inflammatory cascades of live cells, eventually surviving within host cells and establishing infection (67). In addition to AIM2, there are other pattern recognition receptors that mediate necroptosis, such as NLRC4, which inhibits the IL-17A-dependent recruitment of neutrophils by upregulating IL-18 expression and inducing necroptosis during S. aureus pneumonia. Loss of NLRC4 signaling contributes to host protection against S. aureus pneumonia, and treatment with necroptosis inhibitors or IL-18 gene ablation has been shown to enhance defenses against S. aureus in mice (69). Therefore, modulating the function of NLRC4 may be a potential approach for the treatment of S. aureus infection. However, stimulator of interferon genes (STING), which is an intracellular pattern recognition receptor (70), enhances the host response to S. aureus infection by inhibiting the necroptosis of macrophages.
In addition to immune cells, S. aureus-induced necroptosis is also observed in some nonimmune cells. When the human lung epithelial cell line A549 was infected with S. aureus, TNFα enhanced pulmonary epithelial cell injury by S. aureus, and the mechanism is related to RIPK3-mediated necroptosis (71). S. aureus secretes the toxic protein staphylococcal superantigen-like protein-10 (SSL-10), which has been shown to interfere with host cell inflammatory responses by binding to ERK2 (72), but whether SSL family proteins can induce cytotoxicity remains unknown. However, a recent study showed that SSL-10 can bind to the receptor TNFR1 on the cell membrane and exert strong cytotoxic effects in two types of nonimmune cells, HEK293T cells and HUVECs, by inducing RIPK3-dependent necroptosis; this study also showed that necroptosis is activated by two distinct signaling pathways, the RIPK1-RIPK3-MLKL and RIPK3-CaMKII mitochondrial permeability transport pore (mPTP) pathways (39). This study described the cytotoxicity of the SSL-10 protein, an inducer of necroptosis, and provided a potential target for the clinical treatment of S. aureus-related diseases (39). However, whether other proteins in the SSL family have the same effect remains to be further investigated. Furthermore, in another nonimmune cell type, S. aureus-induced necroptosis played a pathological role, and S. aureus-induced goat endometrial epithelial cells (gEECs), by increasing the expression of key proteins in the RIPK1-RIPK3-MLKL pathway, induced significant necroptosis. The authors found that the inducers of necroptosis in gEECs were not traditional TLRs or TNFRs but membrane disruption and ion imbalances (40), that is, Ca2+ influx; furthermore, blocking membrane permeability with glycine protected gEECs from S. aureus-induced cell death. Necroptosis initiated during S. aureus infection is mostly detrimental to the host; therefore, inhibitors targeting necroptosis may be an effective strategy for the treatment of S. aureus infection. In a recent report, a salt-inducible kinase (SIK) inhibitor, HG-9–91-01, was found to block necroptosis by inhibiting RIPK3 activity, thereby attenuating necroptosis-mediated inflammatory damage (73). However, the consequences of necroptosis are not necessarily proinflammatory. Necroptosis results in the elimination of cells that produce cytokines and inflammatory products, and therefore the bacteria inside the cells are released. These bacteria to be eliminated by neutrophils, leading to an overall reduction in inflammation (74, 75). In a previous study, necroptosis was found to play an immunoprotective role in a skin model of S. aureus infection, not by participating in cell death but by limiting damage caused by excessive inflammation (76). In the authors’ study, failure to activate necroptosis was associated with excessive local pathology and impaired S. aureus clearance. Thus, necroptosis contributes to host recovery after S. aureus infection.
2.2 Mycobacterium tuberculosis
Over a quarter of the world’s population is infected with Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB). TB remains a major burden on global public health (77). Understanding the host response to M. tuberculosis infection is a key aspect of eradicating TB through the development of effective vaccines and immunotherapies. During infection, M. tuberculosis can trigger substances in macrophages to damage the body. Signal regulatory protein alpha (SIRPα), which is mainly expressed in myeloid cells, such as monocytes/macrophages and dendritic cells (78), can participate in the pathogenesis of M. tuberculosis. The cytoplasmic region of SIRPα contains four immunoreceptor tyrosine-based inhibitory motifs (ITIMs) that, upon ligand binding, become phosphorylated and interact with the SH2-domain-containing tyrosine phosphatases (PTPase) SHP-1 and SHP-2 to mediate various biological functions (79). In the process of M. tuberculosis infection, SIRPα can inhibit the autophagy of macrophages and promote the necroptosis of macrophages. The occurrence of necroptosis is the main way that affects the weakening of the killing ability of macrophages against M. tuberculosis. The tail structure of SIRPα in the cell can directly bind to protein tyrosine kinase 2B (PTK2B) to affect the activity of PTK2B. When SIRPα is knocked out, it can promote the binding of SHP-1 to PTK2B, resulting in the activation of PTK2B. Therefore, PTK2B can bind to the death domain of RIPK1 to inhibit necroptosis of macrophages (41). The bactericidal capacity of macrophages was enhanced when necroptosis was blocked with the RIPK1 inhibitor NEC-1 (41). The results of this study suggest that targeting SIRPα may become a new therapy for the treatment of M. tuberculosis. However, studies have shown that SIRPα mutant mice have increased susceptibility to Salmonella typhimurium infection, suggesting that SIRPα may help the host defend against infection by the pathogen (80). Therefore, the role of SIRPα in host defense against pathogen infection is complex and requires further exploration. In addition, focal adhesion kinase (FAK) in human macrophages can block the occurrence of M. tuberculosis-induced macrophage necroptosis and M. tuberculosis can downregulate FAK expression, thereby evading host immunity (81). M. tuberculosis can also shape the environment to promote macrophage necroptosis by upregulating MLKL, TNFR1, and ZBP1 expression and downregulating cIAP1 expression (82). However, blocking necroptosis by knocking out the mlkl gene or inhibiting RIPK1 had no effect on the survival of infected human or mouse macrophages (82). Taken together, these results indicate that the inhibition of macrophage necroptosis may alleviate disease pathogenesis after M. tuberculosis infection, but not change the outcome of the disease.
2.3 Streptococcus pneumoniae
Streptococcus pneumoniae is a gram-positive bacterium that often colonizes the nasopharynx (83), and asymptomatic bacterial colonization can cause invasive diseases, such as pneumonia and meningitis (84), under certain conditions. S. pneumoniae carries two key virulence proteins, including pneumococcal surface protein A (PspA) and pulmonary hemolysin (Ply) (85, 86). RIPK3 is considered a key regulator of inflammation and cell death, and significantly elevated RIPK3 protein concentrations have been detected in patients with S. pneumoniae pneumonia (42). Further studies showed that RIPK3, RIPK1, MLKL and the mitochondrial calcium uniporter (MCU) combine to form complexes during S. pneumoniae infection, inducing mitochondrial calcium uptake and mROS production (42). In macrophages, RIPK3 can mediate mitochondrial permeability transport pore opening through mROS to initiate necroptosis and activate the NLRP3 inflammasome (inflammasome is a multiprotein complex assembled by cytoplasmic PRRs, inflammasome can recognize PAMPs or DAMPs, recruit and activate the proinflammatory protease caspase-1.) through the mROS-AKT pathway to protect against S. pneumoniae invasion (42). Furthermore, during asymptomatic colonization of the nasopharynx by S. pneumoniae, nasopharyngeal epithelial cells (nECs) die via Ply dependent necroptosis. When the mlkl gene was knocked out and bacteria were colonized, or wild-type mice were colonized with ply deficient strains, mice had reduced production of the antibody against the bacterial surface protein PspA, delayed bacterial clearance, and increased vulnerability to secondary attack by S. pneumoniae (43). Therefore, Ply induced necroptosis contributes to the protective immunity of the host. S. pneumoniae can invade the heart after causing bacteremia, both the TIGR4 and D39 strains can effectively invade the myocardium, and macrophages recruited after TIGR4 invades the heart die from pneumolysin-induced necroptosis (45). However, whether neutralizing pulmonary hemolysin or blocking necroptosis is beneficial to the host remains unclear. In a recent study, the TIGR4 strain induced necroptosis in cardiomyocytes after invading the heart, a process mediated by Ply, and treatment with a necroptosis inhibitor reduced pneumonia in heart streptococcal lesions and decreased serum troponin levels (87). Therefore, the cardiac injury that occurs during invasive pneumococcal disease is due in part to cardiomyocyte necroptosis, and necroptosis inhibitors may be an effective treatment. Coinfection with influenza A virus (IAV) and S. pneumoniae leads to high mortality (88). IAV infection can promote the translocation of S. pneumoniae to the heart, and IAV can promote the adhesion of S. pneumoniae to cardiomyocytes by upregulating the expression of cardiomyocyte adhesion factors. In an in vitro model of cardiomyocyte infection, IAV enhanced Ply induced necrotic cell death by promoting oxidative stress, thereby increasing S. pneumoniae cytotoxicity (89). IAV infection can cause the death of lung epithelial cells through apoptosis, pyroptosis and necroptosis. Recently, PspA was reported to act as a cytoadhesin and bind to GAPDH in dead cells, thereby increasing the localization of S. pneumoniae in the lower airways and exacerbating secondary infection after influenza infection (44). This finding also helps explain why IAV patients have an increased susceptibility to pneumococcal infection.
2.4 Salmonella
Salmonella enterica is one of the leading causes of bacterial gastrointestinal infections in humans and animals (90); Salmonella can be transmitted to humans and animals via the fecal-oral route, and the gastrointestinal tract is the first site of host–pathogen interaction after the ingestion of Salmonella. Salmonella virulence factors mainly include pathogenicity islands, virulence plasmids, enterotoxins and endotoxins (91, 92). Pathogenicity islands are directly related to bacterial invasiveness, especially for SPI-1 and SPI-2. SPI-1 is essential for nonphagocytic invasion and is responsible for Salmonella-induced inflammation in colitis, whereas SPI-2 is essential for bacterial survival and proliferation in phagocytes and plays an important role in systemic infection (93, 94). The type III secretion system (T3SS) encoded by SPI-2 is the most widely studied and important virulence factor in Salmonella. After Salmonella enters the host, it uses the T3SS to secrete effectors into host cells. These effectors can promote bacterial colonization and intracellular survival (95). Therefore, Salmonella can successfully localize to the gut and infect a variety of cell types, such as intestinal epithelial cells and macrophages (95).
A key virulence strategy of Salmonella typhimurium is the induction of macrophage death. S. typhimurium can exploit the host’s type I IFN (IFN-I) response to eliminate macrophages by inducing RIPK1-RIPK3-mediated necroptosis (46). In this process, IFN-I activates RIPK3, which in turn impairs the Nrf2-dependent antioxidant stress response, thereby enhancing necroptosis in macrophages (47). In addition, S. typhimurium-induced miR-155 also enhances necroptosis in macrophages by targeting RIPK1-RIPK3 (96). Intestinal epithelial cells (IECs) and the microbiota in the intestinal mucosa form a barrier to protect against invasion by foreign pathogens (97), and when intestinal epithelial cells are damaged, pathological damage to the intestine can be induced. Recently reports show that Salmonella can disrupt the integrity of the intestinal epithelial barrier by inducing IECs necroptosis, thereby promoting the invasion of Salmonella into the intestine. In this process, the virulence plasmid of S. typhimurium (pSLT) that encodes the SpvB effector plays a crucial role. SpvB mediates the formation of cell membrane pores via the RIPK3-MLKL pathway by inhibiting K-48-mediated RIPK3 ubiquitination to achieve cell death (48). This process is considered RIPK1-independent necroptosis because cell death still occurred after the use of an RIPK1 inhibitor (48). SopF is a newly discovered T3SS effector that promotes bacterial dissemination in mice (98). During S. typhimurium infection, SopF blocks the activity of caspase-8, which is defined as the molecular switch for PANoptosis. When the activity of caspase-8 is blocked, it can inhibit apoptosis and pyrodeath of IEC cells, and promote IEC necroptosis of IEC (49). Therefore, necroptosis may related to the S.typhimurium to spread the lamina propria and cause systemic infection. Further studies revealed that SopF, as a phosphoinositide (PIP)-binding effector, can block caspase-8 by activating the 3-phosphoinositide-dependent protein kinase 1 (PDK1)-ribosomal S6 kinase (RSK) signaling pathway and, after treatment with AR-12 (PDK1 inhibitor) and BI-D1870 (RSK inhibitor), can reverse abnormal apoptosis, pyroptosis and necroptosis (49). In addition, Salmonella exoprotein B (SopB), encoded by SPI-1, plays an important role in the Salmonella infection. Studies have shown that SopB is responsible for cell invasion after Salmonella infection (99). In contrast to SpvB-induced necroptosis, SopB plays a role in preventing necroptosis; for example, SopB can prevent necroptosis in nonimmune cells such as goblet cells, LS174T cells, and epithelial cells. Necroptosis in cecal goblet cells, LS174T cells, and epithelial cells can be promoted by increasing MLKL phosphorylation after infection with SopB-deficient strains (50). However, even though the presence of SopB protects epithelial cells from necroptosis, it also allows Salmonella to replicate in epithelial cells, which subsequently promotes bacterial escape from epithelial cells and increases their ability to infect neighboring cells, which may play a role in bacterial dissemination (50). In addition, SseK1 and SseK3 inhibited NF-κB activation and necrotizing apoptosis during Salmonella infection of macrophages in a study of the T3SS effector protein SseK (100). Inhibition of both proinflammatory signaling and host cell death by SseK1 and SseK3 may be a strategy that Salmonella plays to reproduce in host cells, thus providing Salmonella with robustness and flexibility in counteracting host immune responses.
However, many studies have also shown that Salmonella does not activate necroptosis. Non-invasive S. typhimurium does not naturally induce RIPK3-dependent macrophage death, whereas macrophage necroptosis can only be induced when caspase is inhibited using Z-VAD-FMK, and RIPK3 induction (after caspase inhibition) does not affect host survival after systemic Salmonella infection (101). Furthermore, the induction of RIPK3 leads to the recruitment of hypoinflammatory myeloid cells, contrary to the usual characterization of necroptosis as highly proinflammatory. Similarly, the synergistic role of RIPK3 and caspase-3/11 in regulating Salmonella burden in vivo was described in another study (102). Furthermore, mice with previously known caspase-3/11 deletions were shown to have an impaired ability to control the Salmonella burden, whereas RIPK3 deletion alone did not affect the innate immune response to Salmonella infection (102). In addition, studies in recent years have shown that single deletion of cell death, apoptosis, or necroptosis has little effect on Salmonella control, and that combination of these cell death pathways leads to loss of bacterial control in mice and their macrophages (103).
2.5 Escherichia coli
Escherichia coli is a common gram-negative bacterium with many virulence factors, including endotoxin, a capsule, a type III secretion system, adhesin and exotoxins; exotoxins include Shiga toxin, thermostable enterotoxin and heat labile enterotoxin (104). When these virulence factors are present in different types of E. coli, they have different pathogeneses and disease outcomes. Lipopolysaccharide (LPS) is a component of the outer cell wall of gram-negative bacteria, and LPS is an endotoxin that has toxic effects on the host. LPS induces necroptosis in host cells to promote disease (105). During sepsis, E. coli LPS can induce the intestinal cell necroptosis, activate the necroptosis signaling pathway, and upregulate the expression of the necrosis-related proteins RIPK1, RIPK3, and MLKL; furthermore, LPS is associated with intestinal morphology and functional damage (51). Pretreatment with the RIPK1 inhibitor NEC-1 reduces the extent of ultrastructural changes caused by necroptosis (51). Therefore, NEC-1 may prevent some intestinal damage during sepsis. Intestinal epithelial cell injury and inflammation can also be induced by enterotoxigenic E. coli (ETEC), but unsaturated fatty acids (EPA) and arachidonic acid (ARA) can alleviate enterotoxigenic E. coli induced intestinal damage by modulating necroptosis signals (106) because EPA and ARA inhibit the expression of the RIPK1, RIPK3, and MLKL proteins. The T3SS effectors NleB and EspL of EPEC block cell necroptosis; NleB works by inactivating the death domains of proteins, including TRADD, FADD, RIPK1, and TNFR1, to block TNFα-induced necroptosis (107). EspL inhibits TNF-induced necroptosis by cleaving the RHIM domains of RIPK1, RIPK3, TRIF and ZBP1/DAI (108). Therefore, the inhibition of necroptosis contributes to the continued colonization of EPEC in vivo, thereby contributing to disease progression.
2.6 Enterococcus faecalis
Enterococcus faecalis is a common gram-positive bacterium and part of the normal flora in the intestinal tract of animals. Generally, E. faecalis is harmless to humans and animals. However, recent studies have shown that some E. faecalis strains have evolved more virulence genes that allow them to infect the human body and cause a variety of diseases, such as endocarditis and peritonitis (109). Disulfide bonding protein A (DsbA) was found to be essential for fecal enterococcal virulence in a model of E. faecalis infection of the Shirley Cryptococcus nematode. DsbA can cause microdamage during heart formation. Subsequently, at the site of cardiac microinjury, E. faecalis can induce cardiomyocyte apoptosis and necroptosis, which in turn contribute to cardiac microinjury (52); however, although the EntV protein is a substrate of DsbA, in this study, the absence of EntV did not alleviate the symptoms of the disease, and therefore, future studies should explore and identify other substrates of DsbA to determine whether they contribute to cardiac micropathology. In addition, in immune cell studies, root canal isolates (CA1 and CA2) and OGERF induced upregulated expression of RAW264.7 macrophage apoptosis-related proteins associated with pyroptosis and necroptosis (110). In refractory apical periodontitis, E. faecalis can induce RIPK3-MLKL-mediated necroptosis in macrophages. This study suggested that inhibitors or treatments targeting necroptosis are a viable strategy for the treatment of refractory apical periodontitis (53). Notably, the increase in MLKL phosphorylation does not necessarily indicate necroptosis (111).
2.7 Other bacteria
2.7.1 Pseudomonas aeruginosa
Acute lung injury caused by Pseudomonas aeruginosa is a disease that seriously endangers public health. Recent reports indicated that P. aeruginosa-mediated necroptosis of epithelial cells plays an important role in this pathological process. P. aeruginosa mediated acute lung injury and lung inflammation can be alleviated by inhibiting the necroptosis pathway. Moreover, the NLRP3 inflammasome is involved in this pathological process, and MLKL-dependent necroptosis signaling can promote changes in mitochondrial membrane potential, thereby releasing reactive oxygen species (ROS), which are important triggers for inflammasome activation (54). In addition, in a recent study showing that the RIPK3 scaffold plays a regulatory role in lung inflammation during P. aeruginosa infection, blocking the RHIM domain in RIPK3 with M45 reduced the inflammatory response to infection in vitro (112). Therefore, the inhibition of RHIM signaling is a potential strategy for reducing lung inflammation during infection.
2.7.2 Listeria monocytogenes
As an important mediator of necroptosis, RIPK3 can be abundantly expressed in the gastrointestinal; and after oral infection with Listeria monocytogenes, compared with wild-type mice, RIPK3-/- mice exhibited significantly increased Listeria proliferation, resulting in systemic infection (55). Moreover, Studies have shown that the activation of MLKL induced by Listeria infection does not cause intestinal epithelial cell necroptosis, on the contrary, MLKL can directly bind to Listeria, thereby inhibiting pathogen replication (55). This finding illustrates the importance of necroptosis in the defense against bacterial infection. However, in Listeria-induced acute liver injury, the inhibition of necroptosis significantly ameliorated mitochondrial dysfunction in mouse livers (113). In addition, Listeria infection can also rapidly induce necroptosis of macrophages (114, 115).
2.7.3 Francisella tularensis
Francisella tularensis is the pathogen responsible for tularemia, and the infection of host cells induces host cell death (116). Caspase-dependent apoptosis and RIPK1-RIPK3-dependent necroptosis can occur simultaneously in macrophages infected by F. tularensis, and the presence of z-VAD-FMK (caspase inhibitor) and NEC-1 significantly reduces the level of cell death (56). What has not been fully explained, however, is how cell necroptosis is initiated in the early (< 72h) period of tularemia without TNF production (56). We believe that this understanding of TNF independent necroptosis mechanisms could help identify drug targets.
2.7.4 Shigella flexneri
Shigella flexneri is a pathogen responsible for bacillary dysentery that can invade and colonize the intestinal epithelial cells of the host, eventually leading to severe inflammatory colitis. Cell death is thought to be a key aspect of host resistance to bacterial invasion (117, 118). However, in Shigella-infected intestinal epithelial cells, no cell death was observed (119). A recent study demonstrated that effectors of Shigella’s T3SS play a crucial role in blocking host cell death. After Shigella infects intestinal epithelial cells, the effector OspC1 blocks caspase-8 signaling to prevent apoptosis and subsequently triggers necroptosis as a host defense mechanism (57). However, to counteract the host response to bacterial infection, Shigella employs OspD3 to target and cleave the RHIM domains of RIPK1 and RIPK3, thereby degrading RIPK1 and RIPK3 and inhibiting necroptosis in host cells (57). Therefore, this phenomenon of “cell death cross-talk” promotes the survival and proliferation of Shigella within host cells.
3 Conclusion and opinions
Currently, the emergence of multidrug-resistant bacteria, such as methicillin-resistant Staphylococcus aureus (120), has become a focus of concern; therefore, finding more effective targets to fight infection by pathogens is critical. In this article, we describe the roles and molecular mechanisms of necroptosis caused by different bacterial infections. In recent years, a wealth of evidence has indicated that necroptosis plays a significant role in various types of bacteria. Bacteria can utilize their own virulence factors or alter the composition of host cells to activate or inhibit necroptosis. These effects can benefit the host (42) or harm the host (105). Therefore, unraveling the role of necroptosis in different bacterial infections is crucial. Here, we review the current understanding of necroptosis in bacterial infection. More detailed information is provided for S. aureus, M. tuberculosis, S. pneumoniae, Salmonella, E. coli, and E. faecalis in this review because of the comprehensive or controversial functions of necroptosis reported in these bacteria compared with other bacteria.
After bacteria invade the host, necroptosis mainly occurs in two major types of cells. One is immune cells that play a phagocytic role after bacteria enter the body. In this article, we mainly focus on neutrophils and macrophages because they are the main cells that play a role in the early stages of bacterial infection. The other is resident cell at the site of bacterial invasion or colonization.
Neutrophils and macrophages are important components of the natural immune system. Neutrophils are the most abundant white blood cells in the systemic circulation, that can exert bactericidal effects through both oxygen-dependent pathways (myeloperoxidase, MPO) and oxygen-independent pathways (antimicrobial peptides and proteins) (121). Myeloperoxidase (MPO) is released from primary particles, and produces reactive oxygen species (ROS). Primary particles can also release antimicrobial peptides, such as defensins. Secondary particles can release antimicrobial proteins, such as lysozyme. Bacteria have evolved multiple strategies to interfere with the bactericidal mechanism of neutrophils. Inducing the death of neutrophils is one of these strategies, and in this article, we mainly focus on necroptosis as a programmed cell death pathway. For example, S. aureus induces necroptosis in neutrophils by releasing its virulence factor, PSMα (38). Macrophages have a range of mechanisms to clear pathogens, including the release of reactive oxygen species (ROS), active nitrogen (RNS), enzymes and antimicrobial peptides, as well as the acidification of phagolysosomes, nutrient restriction and autophagy (62). Similarly, bacteria can evade macrophage killing through a variety of strategies. S. aureus induces necroptosis in macrophages through its virulence factors Hla, PSM, LukAB and PVL (36). In addition, Salmonella (46) and E. faecalis (53) also induce necroptosis of macrophages to interfere with bacterial clearance by macrophages. Taken together, these examples suggest that immune cell death is detrimental to the host and that inhibiting key proteins associated with necroptosis to prevent immune cell death may be an effective strategy for treating bacterial infection. In addition, signaling regulatory protein alpha (SIRPα), which is expressed mainly on the surface of myeloid cells, may be an important target for the treatment of M. tuberculosis infection. SIRPα promotes the necroptosis of macrophages to reduce their ability to kill M. tuberculosis (41). However, mice with SIRPα deficiency showed increased susceptibility to S. typhimurium infection, indicating that SIRPα may contribute to host defense against Salmonella infection (80). We speculate that the different effects of SIRPα on the host may be related to the different gram-negative nature of the bacteria.
Research has shown that the occurrence of necroptosis depends on the activity of RIPK1, RIPK3, and MLKL. RIPK3, a key protein in the necroptotic pathway, induces necroptosis in macrophages during S. pneumoniae infection and activates the NLRP3 inflammasome in response to S. pneumoniae infection (42). RIPK3 regulates the balance between inflammatory signaling, which promotes bacterial clearance, and the lethal consequences of excessive inflammation. RIPK3 activates the NLRP3 inflammasome to secrete proinflammatory cytokines, thereby inducing immune cell recruitment and bacterial clearance while also regulating necroptosis by clearing dead bacteria and cell debris to prevent excessive inflammation and maintain immune homeostasis (42). In recent years, there have been many reports of macrophages necroptosis induced by bacterial infection. However, neutrophils, as the most common circulating white blood cells in the body, are rarely reported to undergo induce necroptosis after bacterial infection. This distinction may be due to macrophages expressing a wide range of pattern recognition receptors, which can trigger a variety of bacterial sensing systems that induce necroptosis.
Epithelial cells are cells on the surface of the skin or lumen that maintain tissue function by forming a barrier while also participating in the immune response. Epithelial cell death caused by bacterial invasion of the host may lead to barrier destruction, thus facilitating bacterial invasion. For example, S. aureus can induce necroptosis in gEECs (40). Salmonella and E. coli can cause IEC death after intestinal invasion (48, 51). The process of Salmonella infection in epithelial cells is complex. To establish infection, Salmonella enters and replicates in epithelial cells and subsequently escapes from them with the help of effectors (122). Here, the authors propose that Salmonella has a time-regulating ability. In the initial stages of infection, bacteria must prevent cell death; for example, SopB, encoded by Salmonella SPI-1, protects epithelial cells from necroptosis (50), thus providing a favorable environment for bacteria to replicate. In the final stage of infection, cell death is needed to promote Salmonella escape from epithelial cells, a role reversal that requires the time regulation of effector proteins (50). The ability of bacteria to regulate time may provide a theoretical basis for understanding the pathogenesis of bacteria.
Successful control of S. aureus infection requires two major host responses: rapid suppression of S. aureus replication and rapid regulation of the subsequent excessive inflammatory response (75, 123). Therefore, we speculate that the dual role of necroptosis after bacterial infection is related to the two factors. On the one hand, cells can provide a favorable environment for bacteria to replicate, and properly triggering necroptosis can inhibit bacterial replication. However, excessive cell death may cause a breakdown of the host barrier and allow for the release of bacteria, which are then internalized by macrophages and spread throughout the body. Alternatively, excessive immune cell death allows the bacteria to escape host immunity. On the other hand, necroptosis may be involved in regulating the balance between proinflammatory signals and excessive inflammation, which can lead to the death of cells that produce cytokines and inflammatory products, thus limiting excessive inflammation. For example, necroptosis played a protective role in a model of S. aureus skin infection by limiting excessive inflammation (76). This imbalance may be related to the type of bacteria and the severity of the infection. Overall, exploring the beneficial and detrimental effects of necroptosis on the host is helpful for identifying effective strategies for the treatment of bacterial infections. Furthermore, research into the mechanisms and physiological effects of necrotic apoptosis is needed to understand the effectors that target cell death, which may trigger cancer cell death or an anticancer immune response.
Author contributions
XY: Writing – original draft. JY: Writing – original draft. LS: Writing – original draft. SD: Writing – original draft. LY: Writing – review & editing. MY: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (grant number 81800012 and 31560271), the Science and Technology Project of Yunnan Province (grant number 202201AS070060), the Foundation for High-Level Talents in Health and Health Technical of Yunnan (grant number H-2018061), the Joint Special Project of Yunnan Science and Technology Department and Kunming Medical University (grant number 202201AY070001-015), the Foundation for High-Level Scientific and Technological Talents Selection Special Project of Yunnan (grant number 202105AC160025), and Major science and technology special programs of Yunnan (grant number 202302AA310004). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ricucci D, Siqueira JF Jr., Abdelsayed RA, Lio SG, Rôças IN. Bacterial invasion of pulp blood vessels in teeth with symptomatic irreversible pulpitis. J Endod. (2021) 47:1854–64. doi: 10.1016/j.joen.2021.09.010
2. Awad MM, Bryant AE, Stevens DL, Rood JI. Virulence studies on chromosomal alpha-toxin and theta-toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of alpha-toxin in Clostridium perfringens-mediated gas gangrene. Mol Microbiol. (1995) 15:191–202. doi: 10.1111/j.1365-2958.1995.tb02234.x
3. Zhu W, Zhang F, Lu J, Ma C, Shen L, Hu D, et al. The analysis of Modified Qing' E Formula on the differential expression of exosomal miRNAs in the femoral head bone tissue of mice with steroid-induced ischemic necrosis of femoral head. Front Endocrinol (Lausanne). (2022) 13:954778. doi: 10.3389/fendo.2022.954778
4. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. (2015) 517:311–20. doi: 10.1038/nature14191
5. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. (2017) 18:127–36. doi: 10.1038/nrm.2016.149
6. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. (2005) 1:112–9. doi: 10.1038/nchembio711
7. Proskuryakov SY, Konoplyannikov AG, Gabai VL. Necrosis: a specific form of programmed cell death? Exp Cell Res. (2003) 283:1–16. doi: 10.1016/S0014-4827(02)00027-7
8. Nirmala JG, Lopus M. Cell death mechanisms in eukaryotes. Cell Biol Toxicol. (2020) 36:145–64. doi: 10.1007/s10565-019-09496-2
9. Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S. (2012) 109:5322–7. doi: 10.1073/pnas.1200012109
10. Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. (2016) 23:76–88. doi: 10.1038/cdd.2015.70
11. Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. (2013) 23:994–1006. doi: 10.1038/cr.2013.91
12. Santos LD, Antunes KH, Muraro SP, de Souza GF, da Silva AG, Felipe JS, et al. TNF-mediated alveolar macrophage necroptosis drives disease pathogenesis during respiratory syncytial virus infection. Eur Respir J. (2021) 57:2003764. doi: 10.1183/13993003.03764-2020
13. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. (2014) 15:135–47. doi: 10.1038/nrm3737
14. He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S. (2011) 108:20054–9. doi: 10.1073/pnas.1116302108
15. Humphries F, Yang S, Wang B, Moynagh PN. RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ. (2015) 22:225–36. doi: 10.1038/cdd.2014.126
16. Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. (2013) 288:31268–79. doi: 10.1074/jbc.M113.462341
17. McComb S, Cessford E, Alturki NA, Joseph J, Shutinoski B, Startek JB, et al. Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc Natl Acad Sci U S. (2014) 111:E3206–13. doi: 10.1073/pnas.1407068111
18. Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S. (2013) 110:E3109–18. doi: 10.1073/pnas.1301218110
19. Wang X, He Z, Liu H, Yousefi S, Simon HU. Neutrophil necroptosis is triggered by ligation of adhesion molecules following GM-CSF priming. J Immunol (Baltimore Md: 1950). (2016) 197:4090–100. doi: 10.4049/jimmunol.1600051
20. Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. (2012) 11:290–7. doi: 10.1016/j.chom.2012.01.016
21. Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, et al. DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe. (2016) 20:674–81. doi: 10.1016/j.chom.2016.09.014
22. Annibaldi A, Meier P. Checkpoints in TNF-induced cell death: implications in inflammation and cancer. Trends Mol Med. (2018) 24:49–65. doi: 10.1016/j.molmed.2017.11.002
23. Legarda D, Justus SJ, Ang RL, Rikhi N, Li W, Moran TM, et al. CYLD proteolysis protects macrophages from TNF-mediated auto-necroptosis induced by LPS and licensed by type I IFN. Cell Rep. (2016) 15:2449–61. doi: 10.1016/j.celrep.2016.05.032
24. He S, Wang X. RIP kinases as modulators of inflammation and immunity. Nat Immunol. (2018) 19:912–22. doi: 10.1038/s41590-018-0188-x
25. Samson AL, Garnish SE, Hildebrand JM, Murphy JM. Location, location, location: A compartmentalized view of TNF-induced necroptotic signaling. Sci Signal. (2021) 14:eabc6178. doi: 10.1126/scisignal.abc6178
26. Kolbrink B, Riebeling T, Kunzendorf U, Krautwald S. Plasma membrane pores drive inflammatory cell death. Front Cell Dev Biol. (2020) 8:817. doi: 10.3389/fcell.2020.00817
27. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. (2012) 148:213–27. doi: 10.1016/j.cell.2011.11.031
28. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. (2010) 11:700–14. doi: 10.1038/nrm2970
29. Hu YB, Zhang YF, Wang H, Ren RJ, Cui HL, Huang WY, et al. miR-425 deficiency promotes necroptosis and dopaminergic neurodegeneration in Parkinson's disease. Cell Death Dis. (2019) 10:589. doi: 10.1038/s41419-019-1809-5
30. Koper MJ, Tomé SO, Gawor K, Belet A, Van Schoor E, Schaeverbeke J, et al. LATE-NC aggravates GVD-mediated necroptosis in Alzheimer's disease. Acta Neuropathol Commun. (2022) 10:128. doi: 10.1186/s40478-022-01432-6
31. Khan M, Lin J, Wang B, Chen C, Huang Z, Tian Y, et al. A novel necroptosis-related gene index for predicting prognosis and a cold tumor immune microenvironment in stomach adenocarcinoma. Front Immunol. (2022) 13:968165. doi: 10.3389/fimmu.2022.968165
32. Long K, Gu L, Li L, Zhang Z, Li E, Zhang Y, et al. Small-molecule inhibition of APE1 induces apoptosis, pyroptosis, and necroptosis in non-small cell lung cancer. Cell Death Dis. (2021) 12:503. doi: 10.1038/s41419-021-03804-7
33. Lin CC, Mabe NW, Lin YT, Yang WH, Tang X, Hong L, et al. RIPK3 upregulation confers robust proliferation and collateral cystine-dependence on breast cancer recurrence. Cell Death Differ. (2020) 27:2234–47. doi: 10.1038/s41418-020-0499-y
34. Place DE, Lee S, Kanneganti TD. PANoptosis in microbial infection. Curr Opin Microbiol. (2021) 59:42–9. doi: 10.1016/j.mib.2020.07.012
35. Moraco AH, Kornfeld H. Cell death and autophagy in tuberculosis. Semin Immunol. (2014) 26:497–511. doi: 10.1016/j.smim.2014.10.001
36. Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, et al. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PloS Pathog. (2015) 11:e1004820. doi: 10.1371/journal.ppat.1004820
37. Parker D, Prince A. Immunoregulatory effects of necroptosis in bacterial infections. Cytokine. (2016) 88:274–5. doi: 10.1016/j.cyto.2016.09.024
38. Zhou Y, Niu C, Ma B, Xue X, Li Z, Chen Z, et al. Inhibiting PSMα-induced neutrophil necroptosis protects mice with MRSA pneumonia by blocking the agr system. Cell Death Dis. (2018) 9:362. doi: 10.1038/s41419-018-0398-z
39. Jia N, Li G, Wang X, Cao Q, Chen W, Wang C, et al. Staphylococcal superantigen-like protein 10 induces necroptosis through TNFR1 activation of RIPK3-dependent signal pathways. Commun Biol. (2022) 5:813.
40. Yi Y, Gao K, Lin P, Chen H, Zhou D, Tang K, et al. Staphylococcus aureus-induced necroptosis promotes mitochondrial damage in goat endometrial epithelial cells. Anim (Basel). (2022) 12:2218. doi: 10.3390/ani12172218
41. Wang D, Lin Y, Xu F, Zhang H, Zhu X, Liu Z, et al. SIRPα maintains macrophage homeostasis by interacting with PTK2B kinase in Mycobacterium tuberculosis infection and through autophagy and necroptosis. EBioMedicine. (2022) 85:104278. doi: 10.1016/j.ebiom.2022.104278
42. Huang HR, Cho SJ, Harris RM, Yang J, Bermejo S, Sharma L, et al. RIPK3 activates MLKL-mediated necroptosis and inflammasome signaling during streptococcus infection. Am J Respir Cell Mol Biol. (2021) 64:579–91. doi: 10.1165/rcmb.2020-0312OC
43. Riegler AN, Brissac T, Gonzalez-Juarbe N, Orihuela CJ. Necroptotic cell death promotes adaptive immunity against colonizing pneumococci. Front Immunol. (2019) 10:615. doi: 10.3389/fimmu.2019.00615
44. Park SS, Gonzalez-Juarbe N, Riegler AN, Im H, Hale Y, Platt MP, et al. Streptococcus pneumoniae binds to host GAPDH on dying lung epithelial cells worsening secondary infection following influenza. Cell Rep. (2021) 35:109267. doi: 10.1016/j.celrep.2021.109267
45. Gilley RP, González-Juarbe N, Shenoy AT, Reyes LF, Dube PH, Restrepo MI, et al. Infiltrated macrophages die of pneumolysin-mediated necroptosis following pneumococcal myocardial invasion. Infect Immun. (2016) 84:1457–69. doi: 10.1128/IAI.00007-16
46. Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. (2012) 13:954–62. doi: 10.1038/ni.2397
47. Hos NJ, Ganesan R, Gutiérrez S, Hos D, Klimek J, Abdullah Z, et al. Type I interferon enhances necroptosis of Salmonella Typhimurium-infected macrophages by impairing antioxidative stress responses. J Cell Biol. (2017) 216:4107–21. doi: 10.1083/jcb.201701107
48. Dong K, Zhu Y, Deng Q, Sun L, Yang S, Huang K, et al. Salmonella pSLT-encoded effector SpvB promotes RIPK3-dependent necroptosis in intestinal epithelial cells. Cell Death Discovery. (2022) 8:44. doi: 10.1038/s41420-022-00841-9
49. Yuan H, Zhou L, Chen Y, You J, Hu H, Li Y, et al. Salmonella effector SopF regulates PANoptosis of intestinal epithelial cells to aggravate systemic infection. Gut Microbes. (2023) 15:2180315. doi: 10.1080/19490976.2023.2180315
50. Hu GQ, Yang YJ, Qin XX, Qi S, Zhang J, Yu SX, et al. Salmonella outer protein B suppresses colitis development via protecting cell from necroptosis. Front Cell Infect Microbiol. (2019) 9:87. doi: 10.3389/fcimb.2019.00087
51. Liu Y, Xu Q, Wang Y, Liang T, Li X, Wang D, et al. Necroptosis is active and contributes to intestinal injury in a piglet model with lipopolysaccharide challenge. Cell Death Dis. (2021) 12:62. doi: 10.1038/s41419-020-03365-1
52. Brown AO, Singh KV, Cruz MR, Kaval KG, Francisco LE, Murray BE, et al. Cardiac microlesions form during severe bacteremic Enterococcus faecalis infection. J Infect Dis. (2021) 223:508–16. doi: 10.1093/infdis/jiaa371
53. Dai X, Ma R, Jiang W, Deng Z, Chen L, Liang Y, et al. Enterococcus faecalis-induced macrophage necroptosis promotes refractory apical periodontitis. Microbiol Spectr. (2022) 10:e0104522. doi: 10.1128/spectrum.01045-22
54. Li H, Guan J, Chen J, Sun W, Chen H, Wen Y, et al. Necroptosis signaling and NLRP3 inflammasome cross-talking in epithelium facilitate Pseudomonas aeruginosa mediated lung injury. Biochim Biophys Acta Mol Basis Dis. (2023) 1869:166613. doi: 10.1016/j.bbadis.2022.166613
55. Sai K, Parsons C, House JS, Kathariou S, Ninomiya-Tsuji J. Necroptosis mediators RIPK3 and MLKL suppress intracellular Listeria replication independently of host cell killing. J Cell Biol. (2019) 218:1994–2005. doi: 10.1083/jcb.201810014
56. Singh A, Periasamy S, Malik M, Bakshi CS, Stephen L, Ault JG, et al. Necroptotic debris including damaged mitochondria elicits sepsis-like syndrome during late-phase tularemia. Cell Death Discovery. (2017) 3:17056. doi: 10.1038/cddiscovery.2017.56
57. Ashida H, Sasakawa C, Suzuki T. A unique bacterial tactic to circumvent the cell death crosstalk induced by blockade of caspase-8. EMBO J. (2020) 39:e104469. doi: 10.15252/embj.2020104469
58. Murray RJ. Recognition and management of Staphylococcus aureus toxin-mediated disease. Intern Med J. (2005) 35 Suppl 2:S106–19. doi: 10.1111/j.1444-0903.2005.00984.x
59. Nakagawa S, Matsumoto M, Katayama Y, Oguma R, Wakabayashi S, Nygaard T, et al. Staphylococcus aureus virulent PSMα Peptides induce keratinocyte alarmin release to orchestrate IL-17-dependent skin inflammation. Cell Host Microbe. (2017) 22:667–77.e5. doi: 10.1016/j.chom.2017.10.008
60. Peters N, Peters AT. Atopic dermatitis. Allergy Asthma Proc. (2019) 40:433–6. doi: 10.2500/aap.2019.40.4265
61. Fujieda S, Imoto Y, Kato Y, Ninomiya T, Tokunaga T, Tsutsumiuchi T, et al. Eosinophilic chronic rhinosinusitis. Allergol Int. (2019) 68:403–12. doi: 10.1016/j.alit.2019.07.002
62. Pidwill GR, Gibson JF, Cole J, Renshaw SA, Foster SJ. The role of macrophages in Staphylococcus aureus infection. Front Immunol. (2020) 11:620339. doi: 10.3389/fimmu.2020.620339
63. Tatsuno K, Fujiyama T, Yamaguchi H, Waki M, Tokura Y. TSLP directly interacts with skin-homing Th2 cells highly expressing its receptor to enhance IL-4 production in atopic dermatitis. J Invest Dermatol. (2015) 135:3017–24. doi: 10.1038/jid.2015.318
64. Greenlee-Wacker MC, Rigby KM, Kobayashi SD, Porter AR, DeLeo FR, Nauseef WM. Phagocytosis of Staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J Immunol (Baltimore Md: 1950). (2014) 192:4709–17. doi: 10.4049/jimmunol.1302692
65. Greenlee-Wacker MC, Kremserová S, Nauseef WM. Lysis of human neutrophils by community-associated methicillin-resistant Staphylococcus aureus. Blood. (2017) 129:3237–44. doi: 10.1182/blood-2017-02-766253
66. Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. (2016) 22:175–82. doi: 10.1038/nm.4017
67. Feng S, Yang Y, Liu Z, Chen W, Du C, Hu G, et al. Intracellular bacteriolysis contributes to pathogenicity of Staphylococcus aureus by exacerbating AIM2-mediated inflammation and necroptosis. Virulence. (2022) 13:1684–96. doi: 10.1080/21505594.2022.2127209
68. Preston JA, Bewley MA, Marriott HM, McGarry Houghton A, Mohasin M, Jubrail J, et al. Alveolar macrophage apoptosis-associated bacterial killing helps prevent murine pneumonia. Am J Respir Crit Care Med. (2019) 200:84–97. doi: 10.1164/rccm.201804-0646OC
69. Paudel S, Ghimire L, Jin L, Baral P, Cai S, Jeyaseelan S. NLRC4 suppresses IL-17A-mediated neutrophil-dependent host defense through upregulation of IL-18 and induction of necroptosis during Gram-positive pneumonia. Mucosal Immunol. (2019) 12:247–57. doi: 10.1038/s41385-018-0088-2
70. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. (2008) 455:674–8
71. Wen SH, Lin LN, Wu HJ, Yu L, Lin L, Zhu LL, et al. TNF-α increases Staphylococcus aureus-induced death of human alveolar epithelial cell line A549 associated with RIP3-mediated necroptosis. Life Sci. (2018) 195:81–6.
72. Dutta D, Mukherjee D, Mukherjee IA, Maiti TK, Basak A, Das AK. Staphylococcal superantigen-like proteins interact with human MAP kinase signaling protein ERK2. FEBS Lett. (2020) 594:266–77
73. Huang D, Chen P, Huang G, Sun H, Luo X, He C, et al. Salt-inducible kinases inhibitor HG-9–91-01 targets RIPK3 kinase activity to alleviate necroptosis-mediated inflammatory injury. Cell Death Dis. (2022) 13:188. doi: 10.1038/s41419-022-04633-y
74. Kearney CJ, Cullen SP, Tynan GA, Henry CM, Clancy D, Lavelle EC, et al. Necroptosis suppresses inflammation via termination of TNF- or LPS-induced cytokine and chemokine production. Cell Death Differ. (2015) 22:1313–27. doi: 10.1038/cdd.2014.222
75. Stephenson HN, Herzig A, Zychlinsky A. Beyond the grave: When is cell death critical for immunity to infection? Curr Opin Immunol. (2016) 38:59–66. doi: 10.1016/j.coi.2015.11.004
76. Kitur K, Wachtel S, Brown A, Wickersham M, Paulino F, Peñaloza HF, et al. Necroptosis promotes Staphylococcus aureus clearance by inhibiting excessive inflammatory signaling. Cell Rep. (2016) 16:2219–30. doi: 10.1016/j.celrep.2016.07.039
77. Glaziou P, Floyd K, Raviglione MC. Global epidemiology of tuberculosis. Semin Respir Crit Care Med. (2018) 39:271–85. doi: 10.1055/s-0038-1651492
78. Barclay AN. Signal regulatory protein alpha (SIRPalpha)/CD47 interaction and function. Curr Opin Immunol. (2009) 21:47–52. doi: 10.1016/j.coi.2009.01.008
79. Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPα) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. (2014) 32:25–50. doi: 10.1146/annurev-immunol-032713-120142
80. Li LX, Atif SM, Schmiel SE, Lee SJ, McSorley SJ. Increased susceptibility to Salmonella infection in signal regulatory protein α-deficient mice. J Immunol (Baltimore Md: 1950). (2012) 189:2537–44. doi: 10.4049/jimmunol.1200429
81. Afriyie-Asante A, Dabla A, Dagenais A, Berton S, Smyth R, Sun J. Mycobacterium tuberculosis exploits focal adhesion kinase to induce necrotic cell death and inhibit reactive oxygen species production. Front Immunol. (2021) 12:742370. doi: 10.3389/fimmu.2021.742370
82. Stutz MD, Ojaimi S, Allison C, Preston S, Arandjelovic P, Hildebrand JM, et al. Necroptotic signaling is primed in Mycobacterium tuberculosis-infected macrophages, but its pathophysiological consequence in disease is restricted. Cell Death Differ. (2018) 25:951–65. doi: 10.1038/s41418-017-0031-1
83. Chao Y, Marks LR, Pettigrew MM, Hakansson AP. Streptococcus pneumoniae biofilm formation and dispersion during colonization and disease. Front Cell Infect Microbiol. (2014) 4:194. doi: 10.3389/fcimb.2014.00194
84. Loughran AJ, Orihuela CJ, Tuomanen EI. Streptococcus pneumoniae: invasion and inflammation. Microbiol Spectr. (2019) 7:10.1128/microbiolspec.gpp3-0004-2018. doi: 10.1128/microbiolspec.GPP3-0004-2018
85. Crain MJ, Waltman WD 2nd, Turner JS, Yother J, Talkington DF, McDaniel LS, et al. Pneumococcal surface protein A (PspA) is serologically highly variable and is expressed by all clinically important capsular serotypes of Streptococcus pneumoniae. Infect Immun. (1990) 58:3293–9. doi: 10.1128/iai.58.10.3293-3299.1990
86. Kanclerski K, Möllby R. Production and purification of Streptococcus pneumoniae hemolysin (pneumolysin). J Clin Microbiol. (1987) 25:222–5. doi: 10.1128/jcm.25.2.222-225.1987
87. Beno SM, Riegler AN, Gilley RP, Brissac T, Wang Y, Kruckow KL, et al. Inhibition of necroptosis to prevent long-term cardiac damage during pneumococcal pneumonia and invasive disease. J Infect Dis. (2020) 222:1882–93. doi: 10.1093/infdis/jiaa295
88. Zhang YY, Tang XF, Du CH, Wang BB, Bi ZW, Dong BR. Comparison of dual influenza and pneumococcal polysaccharide vaccination with influenza vaccination alone for preventing pneumonia and reducing mortality among the elderly: A meta-analysis. Hum Vaccin Immunother. (2016) 12:3056–64. doi: 10.1080/21645515.2016.1221552
89. Platt MP, Lin YH, Wiscovitch-Russo R, Yu Y, Gonzalez-Juarbe N. Pandemic influenza infection promotes streptococcus pneumoniae infiltration, necrotic damage, and proteomic remodeling in the heart. mBio. (2022) 13:e0325721. doi: 10.1128/mbio.03257-21
90. Boyle EC, Bishop JL, Grassl GA, Finlay BB. Salmonella: from pathogenesis to therapeutics. J Bacteriol. (2007) 189:1489–95. doi: 10.1128/JB.01730-06
91. Jennings E, Thurston TLM, Holden DW. Salmonella SPI-2 type III secretion system effectors: molecular mechanisms and physiological consequences. Cell Host Microbe. (2017) 22:217–31. doi: 10.1016/j.chom.2017.07.009
92. Abraham A, Ifeanyi SS, Muinah F, Ibidunni Oreoluwa BS, Coulibaly KJ, Adeyemi AI. Plasmid profile and role in virulence of salmonella enterica serovars isolated from food animals and humans in Lagos Nigeria. Pathog Glob Health. (2019) 113:282–7. doi: 10.1080/20477724.2019.1691364
93. Barthel M, Hapfelmeier S, Quintanilla-Martínez L, Kremer M, Rohde M, Hogardt M, et al. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun. (2003) 71:2839–58. doi: 10.1128/IAI.71.5.2839-2858.2003
94. Abrahams GL, Hensel M. Manipulating cellular transport and immune responses: dynamic interactions between intracellular Salmonella enterica and its host cells. Cell Microbiol. (2006) 8:728–37. doi: 10.1111/j.1462-5822.2006.00706.x
95. Crowley SM, Knodler LA, Vallance BA. Salmonella and the inflammasome: battle for intracellular dominance. Curr Top Microbiol Immunol. (2016) 397:43–67. doi: 10.1007/978-3-319-41171-2_3
96. Ro YT, Jo GH, Jung SA, Lee EH, Shin J, Lee JH. Salmonella−induced miR−155 enhances necroptotic death in macrophage cells via targeting RIP1/3. Mol Med Rep. (2018) 18:5133–40. doi: 10.3892/mmr.2018.9525
97. Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. (2009) 1:a002584. doi: 10.1101/cshperspect.a002584
98. Lau N, Haeberle AL, O'Keeffe BJ, Latomanski EA, Celli J, Newton HJ, et al. SopF, a phosphoinositide binding effector, promotes the stability of the nascent Salmonella-containing vacuole. PloS Pathog. (2019) 15:e1007959. doi: 10.1371/journal.ppat.1007959
99. Piscatelli HL, Li M, Zhou D. Dual 4- and 5-phosphatase activities regulate SopB-dependent phosphoinositide dynamics to promote bacterial entry. Cell Microbiol. (2016) 18:705–19. doi: 10.1111/cmi.12542
100. Günster RA, Matthews SA, Holden DW, Thurston TLM. SseK1 and SseK3 type III secretion system effectors inhibit NF-κB signaling and necroptotic cell death in salmonella-infected macrophages. Infect Immun. (2017) 85:e00010-17. doi: 10.1128/IAI.00010-17
101. Satkovich J, Anderson CJ, Medina CB, Ottolini M, Lukens JR, Kendall MM. RIPK3-dependent recruitment of low-inflammatory myeloid cells does not protect from systemic salmonella infection. mBio. (2020) 11:e02588-20. doi: 10.1128/mBio.02588-20
102. Shutinoski B, Patel R, Tomlinson JJ, Schlossmacher MG, Sad S. Ripk3 licenced protection against microbial infection in the absence of Caspase1–11 inflammasome. Microbes Infect. (2020) 22:40–5. doi: 10.1016/j.micinf.2019.08.002
103. Doerflinger M, Deng Y, Whitney P, Salvamoser R, Engel S, Kueh AJ, et al. Flexible usage and interconnectivity of diverse cell death pathways protect against intracellular infection. Immunity. (2020) 53:533–47.e7. doi: 10.1016/j.immuni.2020.07.004
104. Pakbin B, Brück WM, Rossen JWA. Virulence factors of enteric pathogenic Escherichia coli: A review. Int J Mol Sci. (2021) 22:9922. doi: 10.3390/ijms22189922
105. Wang J, Luan Y, Fan EK, Scott MJ, Li Y, Billiar TR, et al. TBK1/IKKϵ Negatively regulate LPS-induced neutrophil necroptosis and lung inflammation. Shock. (2021) 55:338–48. doi: 10.1097/SHK.0000000000001632
106. Xiao K, Yang Y, Zhang Y, Lv Q, Huang F, Wang D, et al. Long-chain PUFA ameliorate enterotoxigenic Escherichia coli-induced intestinal inflammation and cell injury by modulating pyroptosis and necroptosis signaling pathways in porcine intestinal epithelial cells. Br J Nutr. (2022) 128:835–50. doi: 10.1017/S0007114521005092
107. Li S, Zhang L, Yao Q, Li L, Dong N, Rong J, et al. Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature. (2013) 501:242–6. doi: 10.1038/nature12436
108. Pearson JS, Giogha C, Mühlen S, Nachbur U, Pham CL, Zhang Y, et al. EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat Microbiol. (2017) 2:16258. doi: 10.1038/nmicrobiol.2016.258
109. Chirouze C, Athan E, Alla F, Chu VH, Ralph Corey G, Selton-Suty C, et al. Enterococcal endocarditis in the beginning of the 21st century: analysis from the International Collaboration on Endocarditis-Prospective Cohort Study. Clin Microbiol Infect. (2013) 19:1140–7. doi: 10.1111/1469-0691.12166
110. Chi D, Lin X, Meng Q, Tan J, Gong Q, Tong Z. Real-time induction of macrophage apoptosis, pyroptosis, and necroptosis by Enterococcus faecalis OG1RF and two root canal isolated strains. Front Cell Infect Microbiol. (2021) 11:720147. doi: 10.3389/fcimb.2021.720147
111. Mishra PK, Adameova A, Hill JA, Baines CP, Kang PM, Downey JM, et al. Guidelines for evaluating myocardial cell death. Am J Physiol Heart Circ Physiol. (2019) 317:H891–h922. doi: 10.1152/ajpheart.00259.2019
112. Lyons JD, Mandal P, Otani S, Chihade DB, Easley KF, Swift DA, et al. The RIPK3 scaffold regulates lung inflammation during Pseudomonas aeruginosa pneumonia. Am J Respir Cell Mol Biol. (2023) 68:150–60. doi: 10.1165/rcmb.2021-0474OC
113. Qian Z, Shuying W, Ranran D. Inhibitory effects of JQ1 on listeria monocytogenes-induced acute liver injury by blocking BRD4/RIPK1 axis. BioMed Pharmacother. (2020) 125:109818. doi: 10.1016/j.biopha.2020.109818
114. Blériot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity. (2015) 42:145–58. doi: 10.1016/j.immuni.2014.12.020
115. González-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, et al. Pore-forming toxins induce macrophage necroptosis during acute bacterial pneumonia. PloS Pathog. (2015) 11:e1005337. doi: 10.1371/journal.ppat.1005337
116. Parmely MJ, Fischer JL, Pinson DM. Programmed cell death and the pathogenesis of tissue injury induced by type A Francisella tularensis. FEMS Microbiol Lett. (2009) 301:1–11. doi: 10.1111/j.1574-6968.2009.01791.x
117. Rudel T, Kepp O, Kozjak-Pavlovic V. Interactions between bacterial pathogens and mitochondrial cell death pathways. Nat Rev Microbiol. (2010) 8:693–705. doi: 10.1038/nrmicro2421
118. Lamkanfi M, Dixit VM. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. (2010) 8:44–54. doi: 10.1016/j.chom.2010.06.007
119. Kobayashi T, Ogawa M, Sanada T, Mimuro H, Kim M, Ashida H, et al. The Shigella OspC3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe. (2013) 13:570–83. doi: 10.1016/j.chom.2013.04.012
120. Sakoulas G, Moellering RC Jr. Increasing antibiotic resistance among methicillin-resistant Staphylococcus aureus strains. Clin Infect Dis. (2008) 46 Suppl 5:S360–7. doi: 10.1086/533592
121. Kobayashi SD, Malachowa N, DeLeo FR. Neutrophils and bacterial immune evasion. J Innate Immun. (2018) 10:432–41. doi: 10.1159/000487756
122. Fàbrega A, Vila J. Salmonella enterica serovar Typhimurium skills to succeed in the host: virulence and regulation. Clin Microbiol Rev. (2013) 26:308–41. doi: 10.1128/CMR.00066-12
Keywords: necroptosis, bacterial infection, inflammatory cells, ripk1, ripk3, mlkl
Citation: Yu X, Yuan J, Shi L, Dai S, Yue L and Yan M (2024) Necroptosis in bacterial infections. Front. Immunol. 15:1394857. doi: 10.3389/fimmu.2024.1394857
Received: 02 March 2024; Accepted: 29 May 2024;
Published: 12 June 2024.
Edited by:
Felix Ngosa Toka, Ross University School of Veterinary Medicine, Saint Kitts and NevisReviewed by:
Norberto Gonzalez-Juarbe, J. Craig Venter Institute (La Jolla), United StatesDanielle Oliveira Nascimento, Federal Rural University of Rio de Janeiro, Brazil
Copyright © 2024 Yu, Yuan, Shi, Dai, Yue and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Yan, eWFubWluMDMxMDYwNkAxMjYuY29t; Lei Yue, bGVpX3l1ZUBvdXRsb29rLmNvbQ==