
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 27 June 2024
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1394690
This article is part of the Research TopicAutoimmune Diseases in ChildhoodView all 24 articles
 Ancuta Lupu1†
Ancuta Lupu1† Maria Oana Sasaran2*†
Maria Oana Sasaran2*† Elena Jechel1*†
Elena Jechel1*† Alice Azoicai1†
Alice Azoicai1† Monica Mihaela Alexoae1†
Monica Mihaela Alexoae1† Iuliana Magdalena Starcea1†
Iuliana Magdalena Starcea1† Adriana Mocanu1†
Adriana Mocanu1† Alin Horatiu Nedelcu3†Anton Knieling3†
Alin Horatiu Nedelcu3†Anton Knieling3† Delia Lidia Salaru3†Stefan Lucian Burlea4†
Delia Lidia Salaru3†Stefan Lucian Burlea4† Vasile Valeriu Lupu1*†
Vasile Valeriu Lupu1*† Ileana Ioniuc1†
Ileana Ioniuc1†Connective tissue represents the support matrix and the connection between tissues and organs. In its composition, collagen, the major structural protein, is the main component of the skin, bones, tendons and ligaments. Especially at the pediatric age, its damage in the context of pathologies such as systemic lupus erythematosus, scleroderma or dermatomyositis can have a significant negative impact on the development and optimal functioning of the body. The consequences can extend to various structures (e.g., joints, skin, eyes, lungs, heart, kidneys). Of these, we retain and reveal later in our manuscript, mainly the respiratory involvement. Manifested in various forms that can damage the chest wall, pleura, interstitium or vascularization, lung damage in pediatric systemic inflammatory diseases is underdeveloped in the literature compared to that described in adults. Under the threat of severe evolution, sometimes rapidly progressive and leading to death, it is necessary to increase the popularization of information aimed at physiopathological triggering and maintenance mechanisms, diagnostic means, and therapeutic directions among medical specialists. In addition, we emphasize the need for interdisciplinary collaboration, especially between pediatricians, rheumatologists, infectious disease specialists, pulmonologists, and immunologists. Through our narrative review we aimed to bring up to date, in a concise and easy to assimilate, general principles regarding the pulmonary impact of collagenoses using the most recent articles published in international libraries, duplicated by previous articles, of reference for the targeted pathologies.
Connective tissue diseases (CTDs) that frequently affect pediatric patients are systemic lupus erythematosus (SLE), scleroderma (Sc) and dermatomyositis (DM). In more particular cases, we can find antiphospholipid antibody syndrome, Sjögren's syndrome or associations of pathologies (overlap syndrome). The difficulty of approaching the cases lies both in the variety of clinical manifestations of the underlying disease, as well as in the multiple systemic damages that can occur (e.g., cerebral, pulmonary, cardiac, renal) (1). Regarding pulmonary damage, the pathogenic mechanisms can be diverse starting from granulomatous reaction, interstitial inflammation, primary vasculitis and ending with disease mediated by immune complexes. The functional imbalance can be objectified equally in patients who do not present pulmonary imaging changes (2). Consequently, given the impact played by pulmonary pathology on the patient's quality of life, understanding the pathological mechanisms underlying them, early detection and countering/slowing down the decline represent the goals of modern, preventive medicine.
The current narrative review aims to outline the most current medical information regarding the pathophysiological cascade underlying lung degradation in CTD. In this sense, we will consider a concise presentation regarding the main respiratory damage that can occur in the evolution of the pediatric patient with collagenoses. Additionally, we will not omit from the discussion the current standards of their diagnosis and management. Finally, we will combine the results certified in the literature with those currently being researched, thus broadening the horizons of approaching pediatric children with collagenoses from the perspective of respiratory system disorders. The desired practical purpose is to obtain an increase in the quality of life by drawing attention to the chameleon-like ways of presentation, encouraging early detection and appropriate, individualized management. In order to achieve our objectives, we performed a screening of the most recent publications in the field of interest. We have added reference articles to these. The databases entered were PubMed, Google Scholar, Web of Science, Scopus and Embase. No linguistic criteria were imposed, in order to limit the risk of bias. The terms used for the search were "pulmonary disease", "collagenosis", "collagenoses", "systemic lupus erythematosus", "scleroderma", "dermatomyositis", "children" and various combinations thereof.
CTDs are thus broadly represented by the triad SLE – Sc – DM. Although the characteristics at the pediatric age are slightly different from those of adults, their importance cannot be neglected from the perspective of the increased morbidity and mortality that they print.
SLE is a multisystemic autoimmune condition, with a pathophysiology based on genetic and epigenetic factors and a disabling evolution. The genetic involvement is demonstrated by recent studies regarding the increased susceptibility to develop the juvenile form of SLE (jSLE) among children with positive mothers for SLE or Sjögren's syndrome. Compared to the adult form, the juvenile onset is marked by a higher activity of the disease, important organic lesions, and significant therapeutic burden. The aggressiveness of the disease is inversely proportional to the age at diagnosis, under 5 years of age atypical manifestations of the disease can also be observed (e.g., lack of autoantibodies). Therefore, an overall mortality rate of about three times the usual rate is estimated (3–5). Due to these considerations, the medical world is focusing its efforts on the discovery of new biomarkers (blood/urine) useful in the diagnosis, monitoring of jSLE, and predicting the response to treatment (6, 7). It is also worth mentioning the existence of a neonatal form of SLE that occurs in the case of transplacental transfer of the maternal antigen. The clinical manifestations are violent, including skin, cardiac, hepatobiliary, and hematological damage and regressing (with the exception of cardiac symptoms) once the antibodies are cleared (8). Representing up to 15% of the total number of cases, jSLE notes an incidence that varies between 0.36–2.5/100,000 children, with a prevalence of 1.89–34.1/100,000 children. It is noted that most patients have an onset of jSLE in peri-puberty/adolescence. Regarding the racial distribution, jSLE appears to present more classic clinico-biological characteristics among African/Caribbean patients compared to Caucasian patients. On the other hand, the renal damage was more pronounced in the first category, reaching more frequent therapies with Cyclophosphamide and Rituximab. The gender distribution is in favor of girls, who are affected up to ten times more often than boys (3, 9). Organic damage is an invariable and apparently multifocal phenomenon in the evolution of SLE. Factors such as demographic characteristics, duration of the disease and age at onset, genetic predisposition, the presence of antiphospholipid antibodies or pharmacotherapy appear to play a role in its initiation and maintenance (10). Recent theories also place in the area of interest the impact played by sensitization due to infections (e.g., Covid 19, Epstein-Barr virus) or the imbalance of the microenvironment (e.g., dysbiosis, nutritional/immune deficiency, atopic terrain, hormonal or neuro-psychic disturbances) (11–14). The therapeutic protocol is largely based on the adaptation of the one used among adult patients (7). Thus, considering the stated conditions, it is seen necessary to undertake global efforts aimed at preventing and reducing the negative impact of jSLE on the quality of life.
Juvenile scleroderma (jSc) is considered the third most common rheumatic disease in childhood. Clinically, it gathers under its umbrella manifestations such as inflammation, vasculopathy and fibrosis. According to the degree of its damage, it can be localized (jLSc) or systemic (jSSc). Although jSc is primarily considered a localized disease, the results obtained by Zulian F. et al. contradict the paradigm. They expose the possibility of single or multi-organ damage within jSc, without increasing the risk of progression to jSSc. Consequently, a thorough and extensive evaluation of the patients is required. In turn, the systemic form is divided into diffuse cutaneous (diffuse and rapidly progressive thickening of the skin that associates early pulmonary, cardiac or renal damage), limited cutaneous (restricted and non-progressive thickening of the skin, limited to the distal extremities, which associates late pulmonary arterial hypertension or malabsorption) and overlap syndrome that may associate features of another connective tissue disease (e.g., dermatomyositis, SLE). Respiratory, the difference between the 3 forms is dictated by the more frequent cardiac damage in the skin-limited one and pulmonary in the others. Regardless of the form we are dealing with, it should be known that up to 1/3 of scleroderma cases begin in childhood. The average age of diagnosis is 6–8 years (jLSc), and respectively 8–11 years (jSSc). The incidence varies between forms from 0.34–2.7/100,000 children/year in the localized form, to 0.27/1,000,000 children/year in the systemic one. The gender ratio is against girls in both forms, while the racial predominance of jLSc is higher among Caucasians (15–19). The organic damage seems to be similar between children and adults. However, compared to the adult form, the prognosis in the medium and long term (5 to 15 years) is more favorable in the juvenile form. However, the data must be interpreted with caution due to the disproportionality of the compared cohorts. Martini G. et al. also mentions the reverse side of the coin, represented by the existence of a form with very rapid progression and early signs of internal organ involvement. There are also rare reports of apparently jLSc forms that have progressed to jSSc. The main factors influencing mortality are damage to vital organs such as the heart (pericarditis), kidneys (increase in creatinine) and lungs (pulmonary fibrosis) (17, 20–23).
Juvenile dermatomyositis (jDM) is the last collagenoses disease on which we focus our attention. The onset of vasculopathy is usually between 4 and 10 years, with an incidence of approximately 3/1 million children and a double frequency in girls. Although of unknown etiology, jDM affects the skin (e.g., heliotrope eruptions, Gottron papules) and the proximal muscles progressively, often with systemic involvement (pulmonary, cardiac, gastrointestinal, endocrine). In addition, we cannot overlook the citation in the literature of SARS-CoV-2 as a trigger mechanism of jDM. Excluding the clinical-biological involvement specific to the pathology (also useful for confirming the diagnosis), Livermore P. et al. completes the symptomatic profile by highlighting the psychosocial manifestations identified among the patients. Their awareness is vital in the optimal, multifactorial approach to the consequences of the disease. It should be mentioned that the gold standard in diagnosis (muscle biopsy) is variably performed in children. The basic treatment used is an aggressive regimen of corticosteroid therapy, associated with immunosuppressants (Methotrexate, Ciclosporin, Cyclophosphamide, Azathioprine) or intravenous immunoglobulins. However, it is recommended to customize the therapy according to the patient's profile, following a "treat to target" approach. Biological agents are being studied (24–30).
Pulmonary function evaluation must be used both to refine the differential diagnosis and to dynamically monitor the severity of the disease. Early diagnosis allows targeted therapies to be instituted, thus maximizing results (31).
In addition to cardiac, renal or neurological damage, the lung damage identified in the case of jSLE is among the most diverse, bringing together both pleuropulmonary damage (pleurisy, pleurisy, acute lupus pneumonia, chronic interstitial lung disease, shrinking lung syndrome, pulmonary hemorrhages) and vascular (hypertension) pulmonary). Veiga CS. et col. note that approximately half of jSLE patients may present subclinical pulmonary abnormalities. The most frequent anomalies of the respiratory functional tests are the alteration of the carbon monoxide diffusion capacity and the total lung capacity. Its frequency is variable, being between 7% and 75%. On the other hand, the prevalence can register an upward curve, depending on the duration of the disease manifestation. However, Trapani S. et al. fail to identify a significant correlation between the changes in pulmonary function tests and the duration/activity of the disease or its immunological profile. Against them, recently, Dai G. et al. demonstrated the impact of both the duration of the disease and the immunological profile (positive anti-RNP and ANCA antibody) in dictating the risk of lung damage. After the research that involved 120 patients, Haupt HM. et al. attribute some of these changes to intercurrent infections, oxygen toxicity, or associated cardiac/renal damage (32–37). Consequently, pulmonary damage in jSLE is a vast field that lends itself to in-depth research due to its controversies.
Pulmonary involvement in jSSc is described by Murray KJ. et al. as being "a frequent, almost universal characteristic, if it is looked for carefully". Reports in the literature vary between 30% to 70%, noting an early involvement of the lungs in jSSc. Manifestations include interstitial lung disease, restrictive/obstructive pulmonary syndrome, pulmonary arterial hypertension (primary/secondary), and alteration of pulmonary function tests such as forced vital capacity or carbon monoxide diffusing capacity (17, 38, 39). Additionally, the medical world recognizes the association between SSc and malignant pathologies, including lung (e.g., adenocarcinoma), attributed in part to the genetic susceptibility of patients (40–42). In a practical way, since the specialized reports aimed at children are low in frequency, we emphasize the comparison obtained by Adrovic A. et al. between adult and juvenile disease phenotypes. Although the juvenile form seems to mainly affect the skin and joints, the authors objectify the presence of interstitial lung disease in a significant percentage of children (30%), compared to that of adults (50.9%). Thus, although rarely found in the clinical profile, possible lung damage in children with jSSc must be considered when diagnosing the pathology or in its evolution (21). To reinforce what was stated previously, the medical literature exposes cases of jSSc that are complicated by cardio-pulmonary damage (43, 44). Next, Adrovic A. et al. argue that it is the cardiovascular and pulmonary damage that largely dictates the prognosis of juvenile scleroderma (45).
Pulmonary involvement in jDM is a serious complication, but with a low prevalence. Manifestations include interstitial lung disease, aspiration pneumonia secondary to pharyngeal/esophageal dysmotility, and alveolar hypoventilation secondary to respiratory muscle weakness. The prevalence of DM symptoms depends on the spectrum of autoantibodies. Although lung damage is not correlated with the severity of the underlying condition, it negatively interferes with mortality. The importance of early diagnosis and treatment is therefore emphasized. Not to be neglected, despite its rarity, is the existence in the literature of cases with respiratory impairment due to CTD overlap syndrome. Popescu NA. et al. describes such a case of jDM-jSSc overlap. The importance of popularization lies in the need to add anti-PM/Scl antibodies in the diagnostic protocol (27, 46–49).
The heterogeneity of the pleuro-pulmonary damage in CTDs suggests divergent directions of the main pathophysiological mechanisms incriminated in the structural and functional decline of the respiratory system. Consequently, in what follows, we detail the general molecular lines of the degenerative process by correlation with the three entities that are the subject of the manuscript.
Although the etiology of jSLE is still open to research, it is certain that among the mechanisms involved in organic damage we find circulating immune complexes (CIC). The findings are supported by animal and human studies that reveal the involvement of autoantibodies in the pathogenesis of pulmonary complications. Quantitative research demonstrates an increased level of antibodies in lung tissue compared to circulating blood levels. Thus, CICs were isolated in alveolar septa, alveolar basement membrane, capillaries, large vessels, bronchi, and pleural effusion. Additional evidence is provided by the therapeutic efficacy of plasmapheresis (50–53). The latter represents an extracorporeal therapy that allows the removal of pathogens from the plasma. In the past, it was frequently used for complications such as renal or cerebral damage, thrombocytopenia, recurrent pulmonary hemorrhages, or antiphospholipid syndrome. Currently, use has been restricted to severe cerebral/pulmonary complications. Adverse effects were divided into mild in 2.4% of cases (related to the vascular approach in 54% and to the device in 7% of cases, in addition to hypotension and tingling), moderate in 3% (tingling, urticaria, hypotension and nausea) and severe in 0.4% of treatments (hypotension and syncope, urticaria, chills or fever, arrhythmia or asystole, nausea or vomiting). In addition to these, we note the risk of disturbing the coagulant and/or acid-base balance, infections, transfusion incompatibility, acute lung injuries related to transfusion and, respectively, the risk of diseases transmitted through the use of contaminated donor plasma. A report by Lu J. et al. which included 120 children with jSLE also noted a 2.5% incidence of toxic epidermal necrolysis after plasmapheresis. Consequently, although a well-tolerated procedure with excellent results in improving the general prognosis, among children prone to such complications, the risk-benefit ratio should be weighed judiciously. To increase the efficiency of the procedure, the clinician must consider the plasma volume (PV) that needs to be purified. It is calculated individually, according to the formula: PV = body weight (kg) × 0.065 × (1-hematocrit). It is estimated that to remove 75%-95% of a noxious substance, 1.4–3 PV must be changed during a procedure. To avoid the return of the pathogen in the blood circulation, the procedure must be repeated 4–5 times, at an interval of 24–48 hours (54–58).
At the molecular level, increased expression of genes regulated by type I interferon (IFN-1) is known. These are crucial in the process of promoting the synthesis of autoantibodies and deregulation of immune tolerance. Thus, pulmonary involvement may coexist with high levels of antibodies such as anti-double-stranded DNA (dsDNA), anti-La, anti-Scl-70, anti-SM, and anti-U1RNP, in contrast to decreased complement. Proinflammatory cytokines such as IFN-γ, tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-8, IL-12, IL-17 also recorded high levels in patients with SLE that presented lung damage. At the opposite pole, IL-10 can be considered a protective factor for pulmonary manifestations in SLE. Other molecules involved in the pathogenesis of SLE are CXC chemokines. Among these, CXCL10 and CXCL11 are associated with the accumulation of neutrophils in the alveolar space, being therefore considered potentiating factors of interstitial fibrosis. Also, findings in cases with pulmonary fibrosis reveal overexpression of CX3C motif chemokine receptor 1 (CX3CR1) and CX3C chemokine ligand 1 (CX3CL1) (59–63). With regard to the pathogenesis of diffuse alveolar hemorrhages, during the research it was incriminated about its possible overlap with the COPA syndrome (mutations that damage the endoplasmic reticulum) (64). Despite the intense research in the field of molecular pathophysiology of jSLE, studies could not associate the changes of biomarkers with a pattern of clinical-imaging manifestations characteristic of lung damage from jSLE. Part of this limitation is attributed to the polymorphic manifestations. They could not allow the formation of sufficient study groups to be able to issue findings. Next, Ding H. et al. note the potential of using biomarkers in the development of precision medicine in SLE (65, 66). We encourage the concentration of future efforts towards the deepening of research in the field. We strongly believe that they could bring a new perspective in the anticipation of the evolutionary pattern and the personalized management of children with jSLE.
In SSc, lung damage mainly follows two directions similar to the basic pathology, represented by fibrotic and immuno-inflammatory pathways. In addition, we consider the endothelial dysfunction that is considered as a bridge between the immune and fibrotic damage. This is triggered by the autoimmune attack doubled by the influence of genetic and environmental factors. Although apparently common with those of idiopathic interstitial lung disease, the pathogenic pathways followed in autoimmune diseases are delimited by the dynamics of their involvement (e.g., divergent IFN signaling patterns). Summarizing, alveolar, and microcirculatory injury can be considered the trigger factors both in the activation and proliferation of lung-resident immune cells, as well as in the recruitment of inflammatory cells (e.g., monocytes, neutrophils, mast cells and natural killer cells). To these is added as an aggravating factor the dysregulation of microvessels and the regeneration capacity of lung progenitor cells (type 2 alveolar cells). Subsequently, pro-fibrotic mediators modulate the activation of the Toll Like 4 receptor (TLR4), promoting the uncontrolled, excessive deposition of extracellular matrix and potentiating transforming growth factor-β (TGF-β) signaling. The finality of the process is illustrated by epithelial and endotheliosis mesenchymal transition, promoting the transition of fibroblasts to activated myofibroblasts and extensive lung remodeling, culminating in the irreversible disorganization of the lung structure, increased stiffness and functional impairment. Consequently, the beneficial effects of modulating the TLR4 pathway in the management of pulmonary fibrosis are suggested. Autoreactive B lymphocytes also participate in this cascade, by favoring the secretion of pro-fibrotic mediators and autoantibodies. The pro-inflammatory balance is particularly supported by neutrophil-derived alpha-defensins (human neutrophil peptides), cytokines and chemokines (IL-8, IL-1α, IL-10, CCL2, CCL7, CCL18, macrophage inflammatory protein-1α and MCP- 1). An inverse correlation was also observed between lung function and myeloid-derived suppressor cell (MDSC) levels, thus attesting to their potential involvement in the pathogenesis of pulmonary fibrosis (67–74). Other pathways and molecules recently described as being involved in the balance of pulmonary fibrosis associated with SSc are: Wnt/β-catenin signaling, Yes-associated protein (YAP) and transcription coactivator with PDZ-binding motif (TAZ), nuclear receptor subfamily 4A (NR4A), CXCL4, Sirtuin1, matrix metalloproteinases, cathepsins or endostatins (73, 75–77).
The causative factors of jDM appear to be an entanglement of genetic susceptibility (e.g., HLA-DQA1 * 0501, HLA-DQA * 0301, HLA-DRB1 * 0301) with disturbing environmental factors. The major pathogenic event is complement-mediated damage to vessels, triggering additional cytokine release via the membrane attack complex. From this point, an inflammatory cascade centered on IFN-1 leads to major histocompatibility complex class I (MHC I) overexpression and dendritic cell (DC) maturation. Downstream appears the regulation of adhesion molecules, the modulation of lymphocyte migration and the inflammatory infiltration of the muscles. Further, the extensive studies on IFN-1 suggest the involvement of the cytokine in the relation of anti-melanoma differentiation associated gene 5 (MDA5) autoantibodies - interstitial lung disease (ILD) within the JDM subtype characteristic of them. The importance of knowing and recognizing the amyopathic form of DM to which we refer (DM5) resides in its high prevalence in pediatrics (10–40% of jDM cases) and the rapidly progressive evolution, with high early mortality, of ILD. In this case, the indications for therapy with aggressive corticosteroids are controversial, the only one noted being the decrease in the possibility of progression to classic jDM. After associative therapy, anti-MDA5 antibodies may become negative. The genetic predisposition is equally felt. Thus, the genotype-phenotype correlation indicates HLA-DRB1*03 as a predictive factor of the risk of ILD, independent of the presence of autoantibodies. To all this is added the possible trigger effect of RNA virus infections (e.g., coxsackie, parvovirus B19, SARS-CoV-2) (27, 78–81).
The pathological mechanism of pulmonary murmur in dermatomyositis is complex and still open to research. Regarding ILD, CD8 T lymphocytes are thought to exert a diffuse cytotoxic effect on muscle cells, while B lymphocytes and CD4 T lymphocytes (predominant in perivascular areas) may be responsible for muscle vasculitis. B cells also seem to be correlated with disease activity, being able to be used both as a biomarker and as a therapeutic target. This theory was supported by the lung biopsy results that revealed numerous CD8 lymphocytes both in the affected tissue and in the partially preserved tissue. Also, their percentage in the bronchoalveolar lavage fluid could be correlated with the response to corticosteroid treatment. Pulmonary arterial hypertension (PAH) occurs rather as a consequence of ILD, being partly precipitated by vasoconstriction secondary to chronic hypoxia and vascular remodeling (78, 82).
Summarizing the previously developed directions, Figure 1 shows the main pathological mechanisms underlying the pulmonary damage in collagenoses, respectively jSLE, jSSc and jDM.
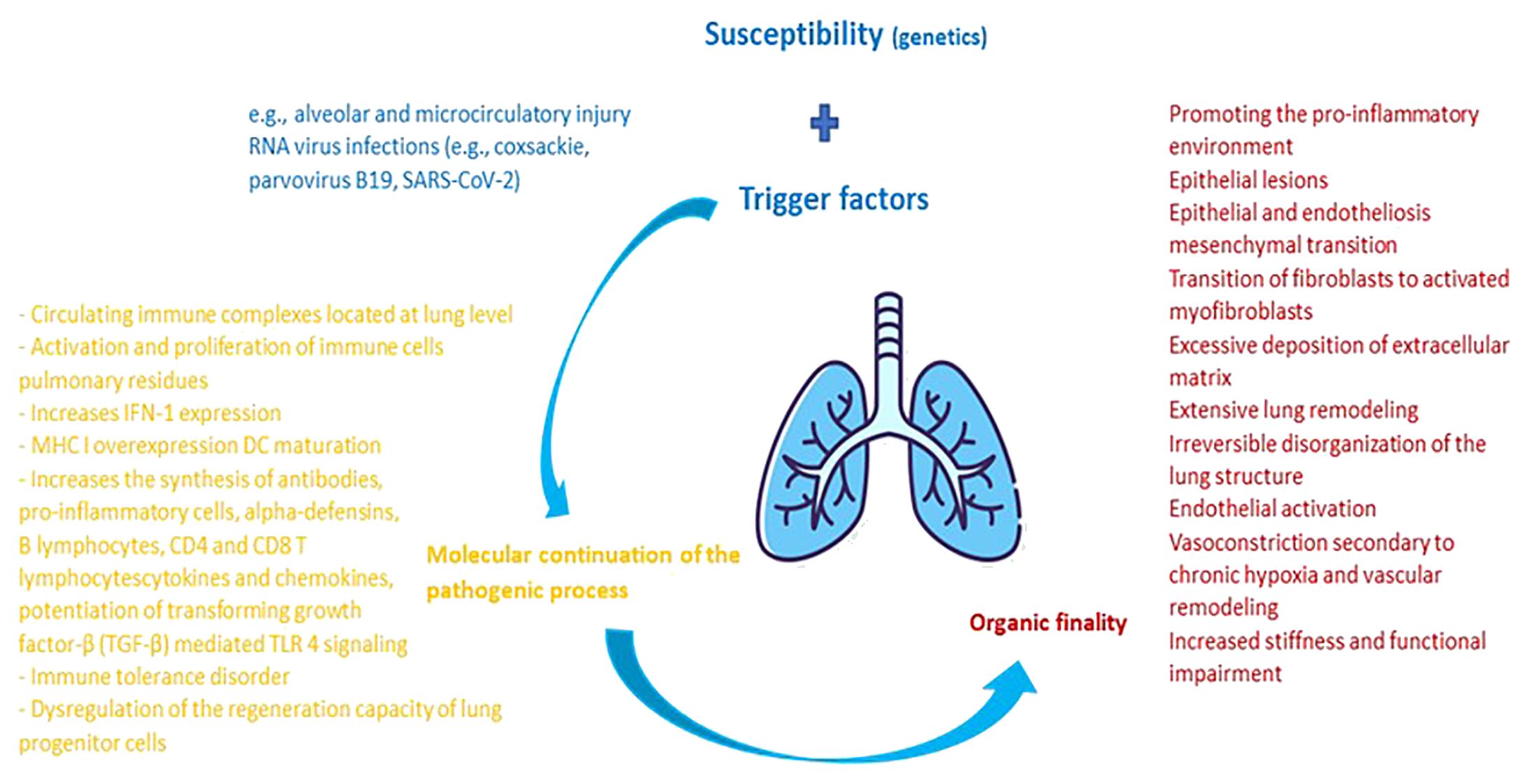
Figure 1 Pathophysiological cascade characteristic of pulmonary damage from collagenoses. The main factor of pulmonary damage from collagenoses is presented by genetic susceptibility. External triggers are additionally added to it. Upon contact of an organism genetically predisposed to the development of a collagenoses with external factors that potentiate the pulmonary injury, an activation of the host's immune system takes place. The molecular balance at this level maintains the pathological process, having the practical purpose of inflammation and damage to the adjacent pulmonary and vascular parenchyma. In dynamics, the lesions heal through remodeling and fibrosis, thus imprinting pulmonary elasticity and organic functionality.
Pulmonary damage in CTDs is diverse, gathering under its umbrella pathologies such as pleurisy, pleural effusion, acute pneumonia, emaciated lung syndrome, interstitial lung disease, diffuse alveolar hemorrhage, pulmonary arterial hypertension, and pulmonary embolism. Although the clinical profile and the diagnostic method are broadly similar, the division of the diseases in terms of frequency is dependent on the collagenoses pattern.
Pleural involvement in lupus is one of the most frequent consequences of the disease (approximately 80% of jSLE cases), being included in the classification criteria. Further, pleurisy notes a frequency of 12.5–32%, being able to be complicated by the appearance of pleural effusion. The localization can be unilateral or bilateral, of small to moderate amount and with the possibility of recurrence. We can rarely find the existence of the "dry" form of pleurisy. Clinically, it can evolve from an asymptomatic phase to one manifested by chest pain accentuated by inspiration, dry cough, fever and dyspnea. The diagnosis must be confirmed by chest X-ray or high-resolution computed tomography (HRCT). The balance is later completed by measuring the C-reactive protein (CRP), the erythrocyte sedimentation rate (ESR), lactate dehydrogenase, thrombomodulin and the pleural puncture which reveals a sterile, yellow serous liquid, with a predominance of polymorphonuclear neutrophils or lymphocytes. Immunologically, the high level of antinuclear antibodies (>1/160) is a potent indicator of the involvement of SLE in the pleural etiology. In the absence of SLE as etiology, the high level of antibodies frequently indicates paraneoplastic causes. Finally, susceptibility to pleuropulmonary lesions is suggested by leukopenia, low complement C3 fraction or dsDNA antibodies (+) (32, 50, 83–85). Pleural effusion in scleroderma is rarely reported in the literature, both in the pediatric and adult populations. However, its occurrence has recently been correlated with PAH within CTDs or, more rarely, with the coexistence of Meigs syndrome (recurrent pleural effusion, ascites and often benign ovarian tumor formation) in adults. This aspect can partly explain the interest in using elevated serum CA125 values as a marker of serositis in CTDs (86–89). In jDM, pleural damage is rarely found, the exudative effusion can appear isolated or in association with the pericardial effusion (78).
It is relatively rare in the evolution of jSLE, and may also be the initial manifestation of the disease, as presented by Şişmanlar Eyüboğlu T. et al. The frequency noted in its case is approximately 11% of patients. Histologically, diffuse alveolar lesions, interstitial edema, formation of hyaline membranes and even hemorrhagic areas can be observed. Clinically, it is defined by non-specific signs such as fever, cough, dyspnea and cyanosis due to hypoxia. Consequently, the factors that induce a correct diagnosis are the onset of ALP in correlation with the exacerbation of the underlying disease involving other organs, doubled by the positivity of anti-SSA antibodies in most cases. In dynamics, it may appear necessary for fan support. Imaging we observe pulmonary infiltrates in the presence/absence of pleural effusion. Important in the management of ALP are the diagnosis and countermeasures of associated infections and pulmonary hemorrhage, entities that can mimic the clinical profile. Because infections are the leading cause of death in patients with SLE, the diagnostic workup must include appropriate cultures, doubled as needed by fiberoptic bronchoscopy, transbronchial biopsy, and open lung biopsy. Therapeutically, the administration of immunoglobulins must be carried out with caution due to the risk of precipitation of renal lesions (32, 50, 90–92).
ILD within CTDs can currently be divided according to the histological characteristics into usual interstitial pneumonia, non-specific interstitial pneumonia, desquamative interstitial pneumonia and/or respiratory bronchiolitis-associated interstitial lung disease, organizing pneumonia, lymphocytic interstitial pneumonia, pleuroparenchymal fibroelastosis and alveolar lesions loudspeaker (93). Studies on ILD in jSLE are few. However, studies that included adults demonstrated a weak ILD-SLE association in the absence of overlap of other autoimmunity (94). The characteristic damage of jSSc begins with the accumulation of a mixed cellular infiltrate (bronchoalveolar lavage positive for neutrophils and eosinophils) in the pulmonary interstitium that will overflow into the alveolar spaces, constituting the first, inflammatory phase. The image of ground-glass opacities on the HRCT of the lungs is suggestive at this stage. Progressively, the inflammation decreases, being replaced by a transition towards thickening of the alveolar walls, fibrosis and pulmonary remodeling. Moderate restrictive lung disease/severe decrease in lung volumes develops consecutively. Clinically, it is mainly objectified slowly progressive dyspnea on exertion and productive cough. Other suggestive aspects in evolution on HRCT are reticular linear opacities, honeycombing, nodules, cylindrical bronchiectasis, and parenchymal bands. Associations between esophageal damage and respiratory dysfunction in children with jSSc have also been described. An accompanying complication can be aspiration pneumonia following esophageal dysmotility associated with jSSc (17, 32, 95–97). It should be mentioned that chest x-rays do not show pathological aspects except in the late stages of ILD, thus being included in the category of insensitive diagnostic methods. Likewise, repeating the bronchoalveolar lavage is not justified, since no post-therapeutic changes were objective (95). In conclusion, we emphasize the strong correlation between ILD, and mortality encountered in SSc. Castelino FV. et al. expose, with the help of a cohort of adults, the precocity of the development of restrictive lung disease independent of individual characteristics (age, gender, duration of the disease) and the importance of screening in the population at risk (98). In agreement, Hoffmann-Vold AM. et al. underlines the need to create some classifications of ILD from the perspective of severity and the risk of progression, but also the importance of early screening. The desired purpose is the optimization and individualization of therapeutic schemes to increase the quality of life (99).
ILD in jDM has been reported to affect approximately 8% of patients. The most common forms described are nonspecific interstitial pneumonia and cryptogenic organizing pneumonia. Among the early markers of interstitial disease in CTDs, we note Krebs von den Lungen-6 (KL6), expressed by damaged type II alveolar cells. IL-19 and ferritin (>1000 ng/ml) are added to this. The correlation between KL-6 and IL-18 suggests the possible association of alveolar macrophages in the pathogenesis of ILD. At the same time, anti-aminoacyl transfer ribonucleic acid synthetase (ARS) antibodies, rare in JDM patients, are associated in a proportion of 63% with ILD in juvenile idiopathic inflammatory myopathies. Clinically, the symptomatology is non-specific, dry cough and respiratory difficulties being described. Spontaneous pneumothorax and pneumomediastinum are not frequently encountered, probably due to vasculopathy. However, respiratory function tests, including carbon monoxide diffusing capacity, should be evaluated at diagnosis and in dynamics (79, 84, 100–103). ILD has also been reported in anti-MDA5 jDM. The described pattern includes skin, mucous and joint damage, in addition to the pulmonary component. Contrary to adults, the rapidly progressive evolution was variable, also describing a favorable therapeutic response compared to the anti-MD5 negative form. However, there are also citations in the literature of fatal cases of IDL with negative anti-MDA5. Compared to the general form, the clinical profile of jDM5 can be complicated by pneumomediastinum in 15% of cases, a condition clinically characterized by increased dyspnea, cervical or facial swelling, subcutaneous emphysema, cervical pain, and cough. It has a negative impact on mortality, especially in cases with non-invasive ventilation in the therapeutic scheme. Early recognition, the establishment of optimal treatment, as well as screening in risk populations are measures that lead to improving the prognosis dictated by lung function (78, 79, 104–106). For this purpose, Hu M. et al. defined, with the help of a cohort of 93 children, a nomogram based on variables such as ESR, IL-10 and MDA-5 antibodies. This has proven effective in the clinical evaluation and prediction of long-term prognosis of ILD associated with jDM (107).
CILD in patients with SLE is characterized by inflammatory cell infiltrate in various degrees, peribronchial lymphoid hyperplasia, homogeneous interstitial fibrosis and pneumocyte hyperplasia. In particular, the medical literature describes the existence of lymphoid interstitial pneumonia, characterized by infiltration of the interstitium with polyclonal lymphocytes and pneumocyte hyperplasia, along with moderate macrophage and lymphocytic alveolitis. Clinico-biologically, pulmonary function is affected similarly to diffuse infiltrative pneumonia. Radiologically, fibrosis is rare (50). Furthermore, it is estimated to affect approximately 14% of children with jSLE. The clinical-imaging presentation is marked by signs and symptoms such as chronic cough, dyspnea, exercise intolerance, fatigue, or pleural effusion. The diagnostic gold standard is HRCT (ground glass appearance or evidence of fibrosis) due to the reduced sensitivity of chest X-ray. Given the chronic and minimally symptomatic nature of the condition, the negative imprint of pulmonary function is less often objective and more difficult to interpret (32).
It is a rare manifestation but cited in the literature of jSLE. From an etiological point of view, the means of its occurrence is unclear, being dysfunctions or hypo functions of the diaphragm (unilateral or bilateral) due to muscle or nerve damage (phrenic nerve). The clinical pattern of SLS includes progressive, episodic dyspnea with orthopnea, hypoxia, pleural pain aggravated by inspiration, pleurisy, a restrictive pattern of lung volumes, with/without parenchymal imaging abnormalities (frequently, however, atelectasis may be present). Percussion reveals dullness in the lower lung lobes, characteristic of reduced expansion. SLS usually appears months to years after the diagnosis of SLE. Chest X-ray shows small lungs, without pleuropulmonary damage (except for atelectasis). Additionally, for diagnostic purposes, the use of diaphragmatic ultrasound in M mode or fluoroscopy is recommended. The response to early optimal therapy can be favorable. Consequently, we emphasize the importance of including SLS in the category of differential diagnoses of dyspnea in children with SLE (32, 50, 108–111).
PAH is defined by the progressive increase in vascular resistance and obstructive vascular remodeling, which over time leads to increased mortality. PAH is currently considered a pulmonary vascular disease with systemic noise. Recently, the possibility of distinguishing between the idiopathic form of PAH and that associated with scleroderma has been postulated based on the changes evident in the level of circulating bioactive metabolites (e.g., acid metabolites, eicosanoids/oxylipins, sex hormone metabolites). The pathogenesis is multifactorial bringing together, among others, inadequate angiogenesis, inflammation/vascular obstruction, metabolic/immune disorders, DNA damage, genetic mutations, and impairment of vasoreactivity. Initial evaluation can be performed with the help of echocardiogram. The condition often appears 1.5 years after the initial manifestations of CTDs. The gold standard in diagnosis is represented by right heart catheterization. The technique can measure elevated mean pulmonary arterial pressure (>25 mmHg at rest) by reference to normal pulmonary capillary pressure (<15 mmHg). The prevalence in the pediatric population with SSc is lower compared to adults (prevalence estimates <10%). Accordingly, children with PAH showed a lower risk of heart failure compared to adults, although the risk of syncope is higher. Current medical literature ranks SLE as the second cause of PAH precipitated by CTDs, after SSc. The risk factors for PAH in jSLE are Raynaud's phenomenon and the presence of antiphospholipid antibodies, although Anuardo P. et al. did not objectify a PAH pathogenic correlation in jSLE and the antiphospholipid antibody syndrome. At the opposite pole, the prognostic factor considered important is PaO2. Optimal and early management has improved life expectancy. In pediatrics, it mainly addresses congenital heart defects, as there are no specific recommendations specifically addressed to cases associated with scleroderma. Therapeutic combinations (e.g., Bosentan and Iloprost/Sildenafil) require further studies. A viable alternative seems to be lung transplantation (17, 32, 50, 84, 95, 112–115). PAH in DM is rare in the absence of ILD (78).
AH occurs most frequently as a consequence of pulmonary microcirculation damage, being categorized as one of the emergencies in jSLE. It occurs most frequently in girls, in cases with previously diagnosed SLE, being often associated with lupus nephritis. In this context, the suspicion arises that corticosteroid therapy for kidney damage is among the precipitating factors of AH. Of all the pulmonary manifestations, AH is marked by an acute onset with hemoptysis, anemia, and diffuse pulmonary infiltrates on chest X-ray. To these can be added cough, fever, dyspnea, hypoxia, asthenia, tachycardia, increased inflammatory markers, positive direct Coombs test, thrombocytopenia, and consumption of complement factors. Radiologically, it is presented by diffuse infiltrates. The pulmonary diffusing capacity of carbon monoxide is increased, the bronchoalveolar lavage (the key investigation in the examination) demonstrating the presence of blood or hemosiderin-laden macrophages. The biological consequence is a significant decrease in hemoglobin. Simultaneously, immunology described the presence of immune complexes and IgG and C3 deposits in alveolar, capillary, and interstitial cells. The incidence and mortality are high in the pediatric disease, being clearly higher than that reported in adults. Management requires an aggressive approach, centered on mechanical ventilation, therapy with corticosteroids, immunosuppressants, intravenous immunoglobulin or plasmapheresis depending on the needs (32, 50, 84, 116–119). Recently, such episodes have been correlated in the medical literature with acute respiratory infection caused by the SARS-Cov-2 virus (120).
It can be found especially among adolescents with jSLE, being precipitated by smoking, estrogen-based contraceptives and proteinuria resulting from nephrotic disease. There are described in the literature cases of pulmonary thromboembolism as the first symptom in jSLE, from the investigation of which the diagnosis of the underlying disease was started. It is mainly a complication of the coexistence of antiphospholipid antibodies, especially the lupus anticoagulant. Pulmonary thromboembolism can be found especially among adolescents with jSLE, being precipitated by smoking, estrogen-based contraceptives and proteinuria resulting from nephrotic disease. There are described in the literature cases of pulmonary thromboembolism as the first symptom in jSLE, from the investigation of which the diagnosis of the underlying disease was started. It is mainly a complication of the coexistence of antiphospholipid antibodies, especially the lupus anticoagulant (50, 84, 121–126).
In closing the manuscript, we would like to emphasize the importance of multidisciplinary monitoring in the dynamics of patients with CTDs. This must be carried out optimally, including clinical examination and paraclinical investigations (biological/imaging), mainly due to the risk of overlap in the evolution of pathologies. Argumentatively, we bring to attention the report submitted by Lin HK. et al. They describe the case of a 15-year-old child (known to have SSc) in whom, following the investigations initiated by the systemic decline of the underlying condition, the coexistence of the diagnostic criteria for SLE and pulmonary involvement (serositis, fibrosis and PAH) is objectified (127). The literature in this regard is vast, also recognizing cases of triple association of pathologies in dynamics (SLE, DM and later SSc) (128). In such situations, we should not lose sight of the possible involvement of microbiome disturbances in the imprinting of the risk of development, association or aggravation of pathologies, as described in the literature and in case of other diseases (e.g., atopies, autoimmunities, gastrointestinal or renal pathologies) (129–136).
Also, taking JSLE as an exemplary model, patient monitoring must be done gradually, starting with primary care doctors who must recognize the diagnostic/alarm signs of the pathology and continuing with rheumatologists, experts in the field of pathogenesis, pediatricians, cardiologists, orthopedists, and other medical categories that can be skilled in the management of complications. In the work team, an essential role is occupied both by clinical pharmacologists, competent to anticipate and prevent possible drug interactions, and psychotherapists. Good staffing leads to optimized therapeutics by early recognition of disease complications, minimizing drug toxicity, educating families about prevention, promoting school performance, addressing reproductive health concerns, and easing the transition to health care of the young adult (137). Chang JC. et al. notes that children who benefited from a multidisciplinary approach recorded better levels of the pediatric index of care, compared to those who benefited from monodisciplinary follow-up (138). Being a chronic pathology, current studies have also focused their attention on the barriers regarding good therapeutic practices. Among these, we mainly note the patient's lack of compliance due to psychological, systemic degradation or the deficit in primary education, the brutal transition between the team of pediatric specialists and that of adults, non-involvement/non-information of the patients regarding the natural course of the pathology and management methods, material shortages, biases of the medical system regarding financing, communication and the collaboration of specialists and social stigmatization. Looking at the pediatric-adult transition, a recent study showed that this period is characterized by moderate disease activity, partially precipitated by the emotional attachment to the previous medical team, doubled by a prolonged time until being noticed by the new medical team (139, 140).
Summarizing the previously presented data and supplementing them with other references from the literature, Table 1 shows the main lines in the recognition and management of pulmonary disorders in CTDs in children. We thus outline a clinical guide that summarizes the correlation between the type of collagenoses and the frequency of pulmonary injury, the paraclinical investigations (biological and imaging) that must be undertaken to certify a correct diagnosis and effective management means, currently certified. Additionally, we review the particularities of the conditions and the pharmacological substances used, where appropriate, but also the biases and future directions in the individualized therapy of the affected child.
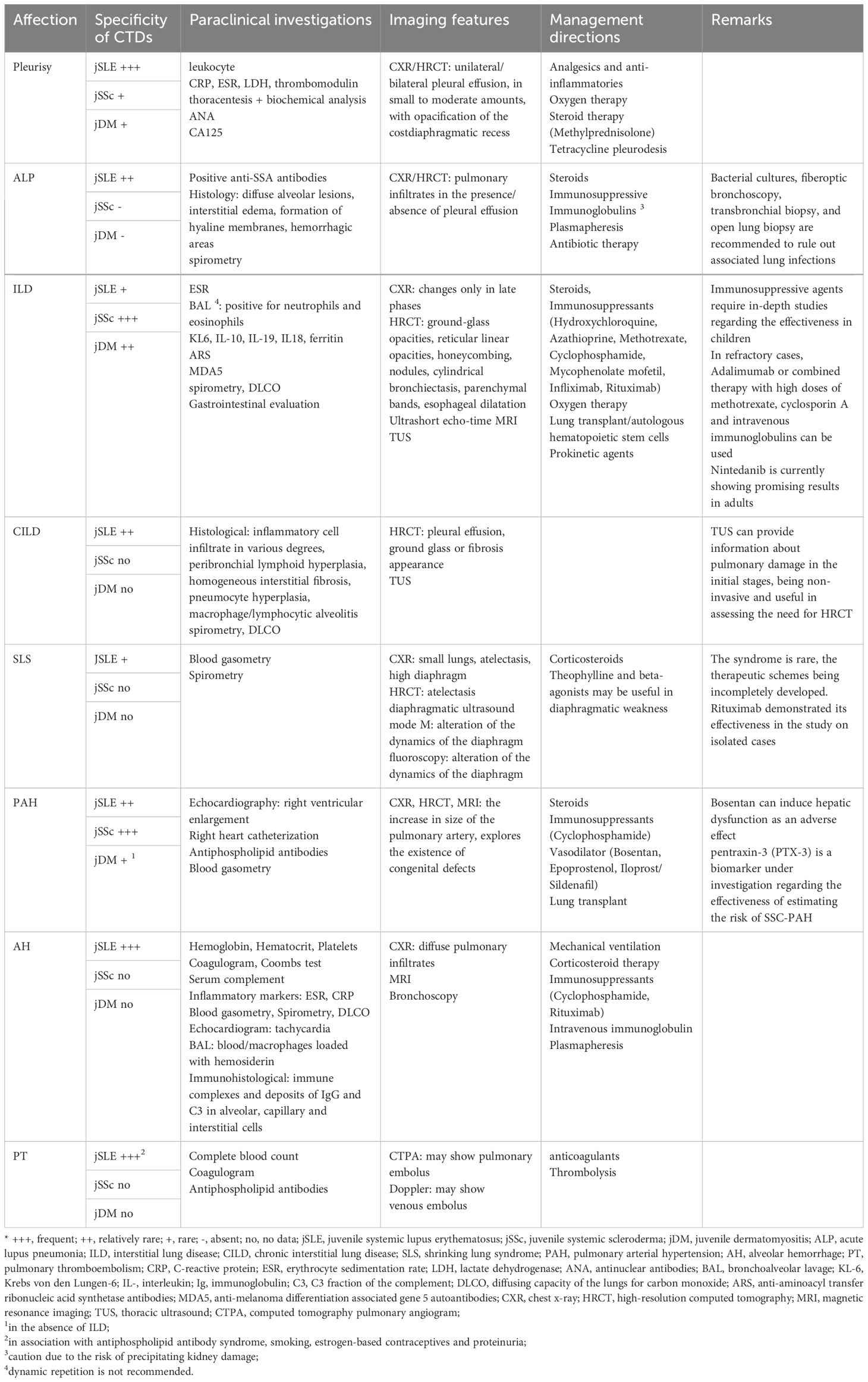
Table 1 Practical clinical guideline in the recognition and management of pulmonary lesions in CTD in children - current and future trends (adapted from Ramphul M. et al., García-Peña P. et al., Lammi MR. et al., Landini N. et al., Buda N. et al., Sperandeo M. et al., DeCoste C. et al., Shimizu M. et al., Eyraud A. et al. and Shin JI. et al.) (84, 141–149).
In conclusion, CTDs represent a topic of interest in pediatrics, the approach of which requires the formation of multidisciplinary teams composed, among others, of rheumatologists, pulmonologists, cardiologists, pediatricians, physio/kinetotherapists and psychologists. Although they differ in some respects from the clinical pattern found in adults, they are not without damage at a systemic level. Treatment can broadly include immunosuppressive drugs, corticosteroids, and other therapies designed to control inflammation and relieve symptoms. Given the burden on the quality of life, psychotherapy sessions can be necessary and useful. Focusing on the pulmonary damage, we reiterate that the damage to the respiratory system can be multiple, both at the pleural, interstitial, and vascular levels. At the same time, the functional impairment can also be found in children without suggestive pulmonary imaging changes. Therefore, we consider that the current work has achieved its defined objective by promoting the increase of information, diagnosis, and optimal management among children with collagenoses. The value of the work is given by the mirror approach to respiratory damage in the three frequent pathological entities (SLE, Sc and DM), both from a physiopathological and a management point of view. Also, through the final subchapter we bring to light both fundamental knowledge, already postulated in the literature and current research directions in the field. In the end, we reiterate the importance of screening in at-risk populations to reduce the burden of subsequent comorbidities and increase the patient's quality of life and his capacity for social integration.
AL: Conceptualization, Investigation, Writing – original draft. MS: Project administration, Validation, Writing – review & editing. EJ: Investigation, Methodology, Writing – original draft. AA: Investigation, Software, Writing – original draft. MA: Investigation, Software, Writing – original draft. IS: Validation, Visualization, Writing – review & editing. AM: Investigation, Software, Writing – original draft. AN: Validation, Visualization, Writing – review & editing. AK: Software, Validation, Writing – review & editing. DS: Software, Validation, Writing – review & editing. SB: Validation, Writing – review & editing. VL: Methodology, Supervision, Writing – review & editing. II: Investigation, Methodology, Writing – original draft.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Navallas M, Inarejos Clemente EJ, Iglesias E, Rebollo-Polo M, Antón J, Navarro OM. Connective tissue disorders in childhood: are they all the same? Radiographics. (2019) 39:229–50. doi: 10.1148/rg.2019180078
2. Dinwiddie R, Sonnappa S. Systemic diseases and the lung. Paediatr Respir Rev. (2005) 6:181–9. doi: 10.1016/j.prrv.2005.06.007
3. Charras A, Smith E, Hedrich CM. Systemic lupus erythematosus in children and young people. Curr Rheumatol Rep. (2021) 23:20. doi: 10.1007/s11926-021-00985-0
4. Harley ITW, Sawalha AH. Systemic lupus erythematosus as a genetic disease. Clin Immunol. (2022) 236:108953. doi: 10.1016/j.clim.2022.108953
5. Cody EM, Brunner HI. Biomarkers in childhood-onset systemic lupus erythematosus. Rheum Dis Clin North Am. (2022) 48:271–85. doi: 10.1016/j.rdc.2021.09.003
6. Wu CH, Chen CA, Lin SH, Weng CT, Kuo PL, Shieh CC. Increased risk of early-onset childhood systemic lupus erythematosus for children born to affected parents: a nationwide child-parent cohort study. Front Immunol. (2022) 13:966809. doi: 10.3389/fimmu.2022.966809
7. Smith EMD, Lythgoe H, Hedrich CM. Current views on lupus in children. Curr Opin Rheumatol. (2023) 35:68–81. doi: 10.1097/BOR.0000000000000913
8. Derdulska JM, Rudnicka L, Szykut-Badaczewska A, Mehrholz D, Nowicki RJ, Barańska-Rybak W, et al. Neonatal lupus erythematosus - practical guidelines. J Perinat Med. (2021) 49:529–38. doi: 10.1515/jpm-2020-0543
9. Lythgoe H, Lj M, Hedrich CM, Aringer M. Classification of systemic lupus erythematosus in children and adults. Clin Immunol. (2022) 234:108898. doi: 10.1016/j.clim.2021.108898
10. Ceccarelli F, Perricone C, Natalucci F, Picciariello L, Olivieri G, Cafaro G, et al. Organ damage in Systemic Lupus Erythematosus patients: A multifactorial phenomenon. Autoimmun Rev. (2023) 22:103374. doi: 10.1016/j.autrev.2023.103374
11. Lupu A, Miron IC, Gavrilovici C, Raileanu AA, Starcea IM, Ioniuc I, et al. Pediatric systemic lupus erythematous in COVID-19 era. Viruses. (2023) 15:272. doi: 10.3390/v15020272
12. Lupu VV, Jechel E, Mihai CM, Mitrofan EC, Lupu A, Starcea IM, et al. Connection between celiac disease and systemic lupus erythematosus in children-A development model of autoimmune diseases starting from what we inherit to what we eat. Nutrients. (2023) 15:2535. doi: 10.3390/nu15112535
13. Lupu VV, Butnariu LI, Fotea S, Morariu ID, Badescu MC, Starcea IM, et al. The disease with a thousand faces and the human microbiome-A physiopathogenic intercorrelation in pediatric practice. Nutrients. (2023) 15:3359. doi: 10.3390/nu15153359
14. Lupu VV, Lupu A, Jechel E, Starcea IM, Stoleriu G, Ioniuc I, et al. The role of vitamin D in pediatric systemic lupus erythematosus - a double pawn in the immune and microbial balance. Front Immunol. (2024) 15:1373904. doi: 10.3389/fimmu.2024.1373904
15. Li SC. Scleroderma in children and adolescents: localized scleroderma and systemic sclerosis. Pediatr Clin North Am. (2018) 65:757–81. doi: 10.1016/j.pcl.2018.04.002
16. Zulian F. Scleroderma in children. Best Pract Res Clin Rheumatol. (2017) 31:576–95. doi: 10.1016/j.berh.2018.02.004
17. Torok KS. Pediatric scleroderma: systemic or localized forms. Pediatr Clin North Am. (2012) 59:381–405. doi: 10.1016/j.pcl.2012.03.011
18. Zulian F, Vallongo C, Woo P, Russo R, Ruperto N, Harper J, et al. Juvenile Scleroderma Working Group of the Pediatric Rheumatology European Society (PRES). Localized scleroderma in childhood is not just a skin disease. Arthritis Rheum. (2005) 52:2873–81. doi: 10.1002/art.21264
19. Foeldvari I, Klotsche J, Kasapcopur O, Adrovic A, Terreri MT, Sakamoto AP, et al. Differences sustained between diffuse and limited forms of juvenile systemic sclerosis in an expanded international cohort. Arthritis Care Res (Hoboken). (2022) 74:1575–84. doi: 10.1002/acr.24609
20. Foeldvari I, Tyndall A, Zulian F, Müller-Ladner U, Czirjak L, Denton C, et al. Juvenile and young adult-onset systemic sclerosis share the same organ involvement in adulthood: data from the EUSTAR database. Rheumatol (Oxford). (2012) 51:1832–7. doi: 10.1093/rheumatology/kes144
21. Adrovic A, Karatemiz G, Esatoglu SN, Yildiz M, Sahin S, Barut K, et al. Juvenile and adult-onset scleroderma: different clinical phenotypes. Semin Arthritis Rheum. (2023) 60:152197. doi: 10.1016/j.semarthrit.2023.152197
22. Martini G, Vittadello F, Kasapçopur O, Magni Manzoni S, Corona F, Duarte-Salazar C, et al. Juvenile Scleroderma Working Group of Paediatric Rheumatology European Society (PRES). Factors affecting survival in juvenile systemic sclerosis. Rheumatol (Oxford). (2009) 48:119–22. doi: 10.1093/rheumatology/ken388
23. Mayorquin FJ, McCurley TL, Levernier JE, Myers LK, Becker JA, Graham TP, et al. Progression of childhood linear scleroderma to fatal systemic sclerosis. J Rheumatol. (1994) 21:1955–7.
24. McCann LJ, Livermore P, Wilkinson MGL, Wedderburn LR. Juvenile dermatomyositis. Where are we now? Clin Exp Rheumatol. (2022) 40:394–403. doi: 10.55563/clinexprheumatol/56ilob
25. Livermore P, Gray S, Mulligan K, Stinson JN, Wedderburn LR, Gibson F. Being on the juvenile dermatomyositis rollercoaster: a qualitative study. Pediatr Rheumatol Online J. (2019) 17:30. doi: 10.1186/s12969-019-0332-7
26. Leung AKC, Lam JM, Alobaida S, Leong KF, Wong AHC. Juvenile dermatomyositis: advances in pathogenesis, assessment, and management. Curr Pediatr Rev. (2021) 17:273–87. doi: 10.2174/1573396317666210426105045
28. Hinze C. Juvenile dermatomyositis-what's new? Z Rheumatol. (2019) 78:627–35. doi: 10.1007/s00393-019-0643-6
29. Wu Q, Wedderburn LR, McCann LJ. Juvenile dermatomyositis: Latest advances. Best Pract Res Clin Rheumatol. (2017) 31:535–57. doi: 10.1016/j.berh.2017.12.003
30. Liquidano-Perez E, García-Romero MT, Yamazaki-Nakashimada M, Maza-Morales M, Rivas-Calderón MK, Bayardo-Gutierrez B, et al. Juvenile dermatomyositis triggered by SARS-CoV-2. Pediatr Neurol. (2021) 121:26–7. doi: 10.1016/j.pediatrneurol.2021.05.011
31. Richardson AE, Warrier K, Vyas H. Respiratory complications of the rheumatological diseases in childhood. Arch Dis Child. (2016) 101:752–8. doi: 10.1136/archdischild-2014-306049
32. Huggins JL, Holland MJ, Brunner HI. Organ involvement other than lupus nephritis in childhood-onset systemic lupus erythematosus. Lupus. (2016) 25:857–63. doi: 10.1177/0961203316644339
33. Haupt HM, Moore GW, Hutchins GM. The lung in systemic lupus erythematosus. Analysis of the pathologic changes in 120 patients. Am J Med. (1981) 71:791–8. doi: 10.1016/0002-9343(81)90366-1
34. Trapani S, Camiciottoli G, Ermini M, Castellani W, Falcini F. Pulmonary involvement in juvenile systemic lupus erythematosus: a study on lung function in patients asymptomatic for respiratory disease. Lupus. (1998) 7:545–50. doi: 10.1191/096120398678920631
35. Dai G, Li L, Wang T, Jiang W, Ma J, Yan Y, et al. Pulmonary involvement in children with systemic lupus erythematosus. Front Pediatr. (2021) 8:617137. doi: 10.3389/fped.2020.617137
36. Altchek AJ, Moorthy LN, Ramagopal M, Salvant C, Uppaluri LP. A 5-year follow-up of pulmonary function tests in childhood-onset systemic lupus erythematosus: a single-center retrospective study. Lupus. (2023) 32:688–93. doi: 10.1177/09612033231163831
37. Veiga CS, Coutinho DS, Nakaie CM, Campos LM, Suzuki L, Cunha MT, et al. Subclinical pulmonary abnormalities in childhood-onset systemic lupus erythematosus patients. Lupus. (2016) 25:645–51. doi: 10.1177/0961203316629554
38. Murray KJ, Laxer RM. Scleroderma in children and adolescents. Rheum Dis Clin North Am. (2002) 28:603–24. doi: 10.1016/s0889-857x(02)00010-8
39. Garty BZ, Athreya BH, Wilmott R, Scarpa N, Doughty R, Douglas SD. Pulmonary functions in children with progressive systemic sclerosis. Pediatrics. (1991) 88:1161–7. doi: 10.1542/peds.88.6.1161
40. Dolcino M, Pelosi A, Fiore PF, Patuzzo G, Tinazzi E, Lunardi C, et al. Gene profiling in patients with systemic sclerosis reveals the presence of oncogenic gene signatures. Front Immunol. (2018) 9:449. doi: 10.3389/fimmu.2018.00449
41. Bento J, Fernandes G, Barbosa MA, Magalhães A, Santos AR, Hespanhol V. Lung adenocarcinoma associated with systemic sclerosis: a case report. Rev Port Pneumol. (2009) 15:93–9. doi: 10.1016/s0873-2159(15)30113-6
42. Yoshimi R, Takeno M, Yamanaka S, Shiina M, Kirino Y, Takeda Y, et al. Systemic sclerosis and pseudomesotheliomatous adenocarcinoma of the lung. Mod Rheumatol. (2006) 16:165–8. doi: 10.1007/s10165-006-0472-8
43. Liang CD, Ko SF, Huang CB, Niu CK. Systemic sclerosis with pulmonary involvement and right ventricular failure in a child. J Formos Med Assoc. (1997) 96:917–20.
44. Kurosawa R, Umebayashi H, Imagawa T, Katakura S, Mori M, Aihara Y, et al. A case of systemic scleroderma complicating pulmonary hypertension. Nihon Rinsho Meneki Gakkai Kaishi. (2006) 29:378–83. doi: 10.2177/jsci.29.378
45. Adrovic A, Oztunc F, Barut K, Koka A, Gojak R, Sahin S, et al. The frequency of pulmonary hypertension in patients with juvenile scleroderma. Bosn J Basic Med Sci. (2015) 15:30–5. doi: 10.17305/bjbms.2015.596
46. Popescu NA, Manea D, Capitanescu G, Cinteza E, Ionescu MD, Balgradean M. Respiratory failure in a rare case of juvenile dermatomyositis - systemic scleroderma overlap syndrome. Maedica (Bucur). (2020) 15:394–400. doi: 10.26574/maedica.2020.15.3.394
47. Pouessel G, Deschildre A, Le Bourgeois M, Cuisset JM, Catteau B, Karila C, et al. The lung is involved in juvenile dermatomyositis. Pediatr Pulmonol. (2013) 48:1016–25. doi: 10.1002/ppul.22742
48. Pouessel G, Thumerelle C, Nève V, Santangelo T, Flammarion S, Pruvot I, et al. Lung is also involved in juvenile dermatomyositis. Rev Med Interne. (2014) 35:461–5. doi: 10.1016/j.revmed.2014.04.005
49. He L. Recent research on myositis-specific autoantibodies in juvenile dermatomyositis. Zhongguo Dang Dai Er Ke Za Zhi. (2021) 23:1064–8. doi: 10.7499/j.issn.1008-8830.2106011
50. Carmier D, Marchand-Adam S, Diot P, Diot E. Respiratory involvement in systemic lupus erythematosus. Rev Mal Respir. (2010) 27:e66–78. doi: 10.1016/j.rmr.2010.01.003
51. Pan L, Lu MP, Wang JH, Xu M, Yang SR. Immunological pathogenesis and treatment of systemic lupus erythematosus. World J Pediatr. (2020) 16:19–30. doi: 10.1007/s12519-019-00229-3
52. Canny SP, Jackson SW. B cells in systemic lupus erythematosus: from disease mechanisms to targeted therapies. Rheum Dis Clin North Am. (2021) 47:395–413. doi: 10.1016/j.rdc.2021.04.006
53. Ahamada MM, Jia Y, Wu X. Macrophage polarization and plasticity in systemic lupus erythematosus. Front Immunol. (2021) 12:734008. doi: 10.3389/fimmu.2021.734008
54. Altobelli C, Anastasio P, Cerrone A, Signoriello E, Lus G, Pluvio C, et al. Therapeutic plasmapheresis: A revision of literature. Kidney Blood Press Res. (2023) 48:66–78. doi: 10.1159/000528556
55. Lu J, Dong L, Zhang L, Zhang H, Wang L, Zhang J, et al. Toxic epidermal necrolysis after therapeutic plasma exchange in pediatric lupus patients and associated risk factors analysis. Lupus. (2021) 30:465–72. doi: 10.1177/0961203320981128
56. Khair AM. Utility of plasmapheresis in autoimmune-mediated encephalopathy in children: potentials and challenges. Neurol Res Int. (2016) 2016:7685807. doi: 10.1155/2016/7685807
57. Misanovic V, Pokrajac D, Zubcevic S, Hadzimuratovic A, Rahmanovic S, Dizdar S, et al. Plasmapheresis in pediatric intensive care unit. Med Arch. (2016) 70:332–5. doi: 10.5455/medarh.2016.70.332-335
58. Eyre M, Hacohen Y, Barton C, Hemingway C, Lim M. Therapeutic plasma exchange in paediatric neurology: a critical review and proposed treatment algorithm. Dev Med Child Neurol. (2018) 60:765–79. doi: 10.1111/dmcn.13925
59. Richter P, Cardoneanu A, Dima N, Bratoiu I, Rezus C, Burlui AM, et al. Interstitial lung disease in systemic lupus erythematosus and systemic sclerosis: how can we manage the challenge? Int J Mol Sci. (2023) 24:9388. doi: 10.3390/ijms24119388
60. Nielepkowicz-Goździńska A, Fendler W, Robak E, Kulczycka-Siennicka L, Górski P, Pietras T, et al. Exhaled cytokines in systemic lupus erythematosus with lung involvement. Pol Arch Med Wewn. (2013) 123:141–8. doi: 10.20452/pamw.1676
61. Hammad A, Osman E, Mosaad Y, Wahba M. Serum interleukin-17 in Egyptian children with systemic lupus erythematosus: is it related to pulmonary affection? Lupus. (2017) 26:388–95. doi: 10.1177/0961203316665709
62. Nielepkowicz-Goździńska A, Fendler W, Robak E, Kulczycka-Siennicka L, Górski P, Pietras T, et al. The role of CXC chemokines in pulmonary fibrosis of systemic lupus erythematosus patients. Arch Immunol Ther Exp (Warsz). (2015) 63:465–73. doi: 10.1007/s00005-015-0356-8
63. Nielepkowicz-Goździńska A, Fendler W, Robak E, Kulczycka-Siennicka L, Górski P, Pietras T, et al. Exhaled IL-8 in systemic lupus erythematosus with and without pulmonary fibrosis. Arch Immunol Ther Exp (Warsz). (2014) 62:231–8. doi: 10.1007/s00005-014-0270-5
64. Zhuang H, Hudson E, Han S, Arja RD, Hui W, Lu L, et al. Microvascular lung injury and endoplasmic reticulum stress in systemic lupus erythematosus-associated alveolar hemorrhage and pulmonary vasculitis. Am J Physiol Lung Cell Mol Physiol. (2022) 323:L715–29. doi: 10.1152/ajplung.00051.2022
65. Aguilera-Pickens G, Abud-Mendoza C. Pulmonary manifestations in systemic lupus erythematosus: pleural involvement, acute pneumonitis, chronic interstitial lung disease and diffuse alveolar hemorrhage. Reumatol Clin (Engl Ed). (2018) 14:294–300. doi: 10.1016/j.reuma.2018.03.012
66. Ding H, Shen Y, Hong SM, Xiang C, Shen N. Biomarkers for systemic lupus erythematosus - a focus on organ damage. Expert Rev Clin Immunol. (2024) 20:39–58. doi: 10.1080/1744666X.2023.2260098
67. Khedoe P, Marges E, Hiemstra P, Ninaber M, Geelhoed M. Interstitial lung disease in patients with systemic sclerosis: toward personalized-medicine-based prediction and drug screening models of systemic sclerosis-related interstitial lung disease (SSc-ILD). Front Immunol. (2020) 11:1990. doi: 10.3389/fimmu.2020.01990
68. Asano Y. The pathogenesis of systemic sclerosis: an understanding based on a common pathologic cascade across multiple organs and additional organ-specific pathologies. J Clin Med. (2020) 9:2687. doi: 10.3390/jcm9092687
69. Codes H, Avanoglu Guler A, Campochiaro C, Matucci Cerinic M, Castelliv I. Systemic sclerosis and interstitial lung disease: From pathogenesis, to screening, diagnosis, and classification. Rev Col Rheumato. (2023) 31(1):54–66. doi: 10.1016/j.rcreu.2023.09.001
70. Bagnato G, Harari S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur Respir Rev. (2015) 24:102–14. doi: 10.1183/09059180.00003214
71. Nihtyanova SI, Denton CP. Pathogenesis of systemic sclerosis associated interstitial lung disease. J Scleroderma Relat Disord. (2020) 5:6–16. doi: 10.1177/2397198320903867
72. Valenzi E, Tabib T, Papazoglou A, Sembrat J, Trejo Bittar HE, Rojas M, et al. Disparate interferon signaling and shared aberrant basaloid cells in single-cell profiling of idiopathic pulmonary fibrosis and systemic sclerosis-associated interstitial lung disease. Front Immunol. (2021) 12:595811. doi: 10.3389/fimmu.2021.595811
73. Mouawad JE, Feghali-Bostwick C. The molecular mechanisms of systemic sclerosis-associated lung fibrosis. Int J Mol Sci. (2023) 24:2963. doi: 10.3390/ijms24032963
74. van Bon L, Cossu M, Loof A, Gohar F, Wittkowski H, Vonk M, et al. Proteomic analysis of plasma identifies the Toll-like receptor agonists S100A8/A9 as a novel possible marker for systemic sclerosis phenotype. Ann Rheum Dis. (2014) 73:1585–9. doi: 10.1136/annrheumdis-2013-205013
75. Servaas NH, Hiddingh S, Chouri E, Wichers CGK, Affandi AJ, Ottria A, et al. Nuclear receptor subfamily 4A signaling as a key disease pathway of CD1c+ Dendritic cell dysregulation in systemic sclerosis. Arthritis Rheumatol. (2023) 75:279–29. doi: 10.1002/art.42319
76. Affandi AJ, Carvalheiro T, Ottria A, de Haan JJ, Brans MAD, Brandt MM, et al. CXCL4 drives fibrosis by promoting several key cellular and molecular processes. Cell Rep. (2022) 38:110189. doi: 10.1016/j.celrep.2021.110189
77. Chu H, Jiang S, Liu Q, Ma Y, Zhu X, Liang M, et al. Sirtuin1 protects against systemic sclerosis-related pulmonary fibrosis by decreasing proinflammatory and profibrotic processes. Am J Respir Cell Mol Biol. (2018) 58:28–39. doi: 10.1165/rcmb.2016-0192OC
78. Lega JC, Reynaud Q, Belot A, Fabien N, Durieu I, Cottin V. Idiopathic inflammatory myopathies and the lung. Eur Respir Rev. (2015) 24:216–38. doi: 10.1183/16000617.00002015
79. Mehta P, MaChado PM, Gupta L. Understanding and managing anti-MDA 5 dermatomyositis, including potential COVID-19 mimicry. Rheumatol Int. (2021) 41:1021–36. doi: 10.1007/s00296-021-04819-1
80. Walling HW, Gerami P, Sontheimer RD. Juvenile-onset clinically amyopathic dermatomyositis: an overview of recent progress in diagnosis and management. Paediatr Drugs. (2010) 12:23–34. doi: 10.2165/10899380-000000000-00000
81. Hou J, Zhou ZX, Li JG, Xu YJ, Ding YC. Three cases report of juvenile dermatomyositis with positive anti-melanoma differentiation associated gene 5 (MDA5) antibody and severe interstitial lung disease and literature review. Zhonghua Er Ke Za Zhi. (2019) 57:928–33. doi: 10.3760/cma.j.issn.0578-1310.2019.12.007
82. Costin C, Khojah A, Ochfeld E, Morgan G, Subramanian S, Klein-Gitelman M, et al. B cell lymphocytosis in juvenile dermatomyositis. Diagnost (Basel). (2023) 13:2626. doi: 10.3390/diagnostics13162626
83. Zhao YF, Lu MP, Chen ZM. Clinical features of systemic lupus erythematosus with pulmonary pleural lesion in children. Zhejiang Da Xue Bao Yi Xue Ban. (2012) 41:327–31. doi: 10.3785/j.issn.1008-9292.2012.03.016
84. Ramphul M, Gallagher K, Warrier K, Jagani S, Bhatt JM. Why is a paediatric respiratory specialist integral to the paediatric rheumatology clinic? Breathe (Sheff). (2020) 16:200212. doi: 10.1183/20734735.0212-2020
85. Mochizuki T, Aotsuka S, Satoh T. Clinical and laboratory features of lupus patients with complicating pulmonary disease. Respir Med. (1999) 93:95–101. doi: 10.1016/s0954-6111(99)90297-4
86. Luo YF, Robbins IM, Karatas M, Brixey AG, Rice TW, Light RW. Frequency of pleural effusions in patients with pulmonary arterial hypertension associated with connective tissue diseases. Chest. (2011) 140:42–7. doi: 10.1378/chest.10-0227
87. Su F, Cummings KW, Krigman H, Ranganathan P. Meigs' syndrome: a rare cause of recurrent pleural effusion in scleroderma. Rheumatol Int. (2013) 33:2647–51. doi: 10.1007/s00296-012-2437-x
88. Kimura K, Ezoe K, Yokozeki H, Katayama I, Nishioka K. Elevated serum CA125 in progressive systemic sclerosis with pleural effusion. J Dermatol. (1995) 22:28–31. doi: 10.1111/j.1346-8138.1995.tb03336.x
89. Funauchi M, Ikoma S, Yu H, Sugiyama M, Ohno M, Kinoshita K, et al. A case of progressive systemic sclerosis complicated by massive pleural effusion with elevated CA125. Lupus. (2000) 9:382–5. doi: 10.1191/096120300678828334
90. Şişmanlar Eyüboğlu T, Aslan AT, Özdemir Y, Gezgin Yıldırım D, Buyan N, Boyunağa Ö. Isolated acute lupus pneumonitis as the initial presentation of systemic lupus erythematosus in an 8-year-old girl. Auto Immun Highlights. (2018) 9:4. doi: 10.1007/s13317-018-0104-2
91. Ciftçi E, Yalçinkaya F, Ince E, Ekim M, Ileri M, Orgerin Z, et al. Pulmonary involvement in childhood-onset systemic lupus erythematosus: a report of five cases. Rheumatol (Oxford). (2004) 43:587–91. doi: 10.1093/rheumatology/keh120
92. Ion DA, Chivu RD, Chivu LI. Aspects of pleural/pulmonary involvement in systemic lupus erythematosus. Pneumologia. (2006) 55:151–5.
93. Wells AU, Denton CP. Interstitial lung disease in connective tissue disease–mechanisms and management. Nat Rev Rheumatol. (2014) 10:728–39. doi: 10.1038/nrrheum.2014.149
94. Mageau A, Borie R, Crestani B, Timsit JF, Papo T, Sacre K. Epidemiology of interstitial lung disease in systemic lupus erythematosus in France: A nation-wide population-based study over 10 years. Respirology. (2022) 27:630–4. doi: 10.1111/resp.14268
95. Rabinovich CE. Challenges in the diagnosis and treatment of juvenile systemic sclerosis. Nat Rev Rheumatol. (2011) 7:676–80. doi: 10.1038/nrrheum.2011.148
96. Ambartsumyan L, Zheng HB, Iyer RS, Soares J, Henstorf G, Stevens AM. Relationship between esophageal abnormalities on fluoroscopic esophagram and pulmonary function testing in juvenile systemic sclerosis. Arthritis Care Res (Hoboken). (2019) 71:1444–9. doi: 10.1002/acr.23778
97. Li JC, Tadros S, Rosser F, Torok KS. Pulmonary nodules in juvenile systemic sclerosis: A case-series from the national registry for childhood onset scleroderma (NRCOS). Diagnost (Basel). (2023) 13:2103. doi: 10.3390/diagnostics13122103
98. Castelino FV, VanBuren JM, Startup E, Assassi S, Bernstein EJ, Chung L, et al. Baseline characteristics of systemic sclerosis patients with restrictive lung disease in a multi-center US-based longitudinal registry. Int J Rheum Dis. (2022) 25:163–74. doi: 10.1111/1756-185X.14253
99. Hoffmann-Vold AM, Molberg Ø. Detection, screening, and classification of interstitial lung disease in patients with systemic sclerosis. Curr Opin Rheumatol. (2020) 32:497–504. doi: 10.1097/BOR.0000000000000741
100. Wakiguchi H. Multispecialty approach for improving outcomes in juvenile dermatomyositis. J Multidiscip Healthc. (2019) 12:387–94. doi: 10.2147/JMDH.S171095
101. Kilinc AA, Arslan A, Yildiz M, Kucur M, Adrovic A, Barut K, et al. Serum KL-6 level as a biomarker of interstitial lung disease in childhood connective tissue diseases: a pilot study. Rheumatol Int. (2020) 40:1701–6. doi: 10.1007/s00296-019-04485-4
102. El-Beheidy R, Domouky AM, Zidan H, Amer YA. Serum KL-6 as predictive and prognostic marker of interstitial lung disease in childhood connective tissue diseases: a pilot study. Reumatismo. (2021) 73. doi: 10.4081/reumatismo.2021.1399
103. Benyamine A, Heim X, Resseguier N, Bertin D, Gomez C, Ebbo M, et al. Elevated serum Krebs von den Lungen-6 in systemic sclerosis: a marker of lung fibrosis and severity of the disease. Rheumatol Int. (2018) 38:813–9. doi: 10.1007/s00296-018-3987-3
104. Yeşilbaş O, Yıldız M, Yozgat CY, Tahaoğlu I, Yazan H, Çakır E, et al. A fatal interstitial lung disease in an anti-melanoma differentiation-associated gene 5 (anti-MDA5) antibody negative patient with juvenile dermatomyositis. Turk J Pediatr. (2021) 63:903–8. doi: 10.24953/turkjped.2021.05.018
105. Yeung TW, Cheong KN, Lau YL, Tse KN. Adolescent-onset anti-MDA5 antibody-positive juvenile dermatomyositis with rapidly progressive interstitial lung disease and spontaneous pneumomediastinum: a case report and literature review. Pediatr Rheumatol Online J. (2021) 19:103. doi: 10.1186/s12969-021-00595-1
106. Kobayashi I, Okura Y, Yamada M, Kawamura N, Kuwana M, Ariga T. Anti-melanoma differentiation-associated gene 5 antibody is a diagnostic and predictive marker for interstitial lung diseases associated with juvenile dermatomyositis. J Pediatr. (2011) 158:675–7. doi: 10.1016/j.jpeds.2010.11.033
107. Hu M, Shen C, Zheng F, Zhou Y, Teng L, Zheng R, et al. Clinical nomogram assisting in discrimination of juvenile dermatomyositis-associated interstitial lung disease. Respir Res. (2023) 24:286. doi: 10.1186/s12931-023-02599-9
108. Torres Jimenez AR, Ruiz Vela N, Cespedes Cruz AI, Velazquez Cruz A, Bernardino Gonzalez AK. Shrinking lung syndrome in pediatric systemic lupus erythematosus. Lupus. (2021) 30:1175–9. doi: 10.1177/09612033211010331
109. Ferguson PJ, Weinberger M. Shrinking lung syndrome in a 14-year-old boy with systemic lupus erythematosus. Pediatr Pulmonol. (2006) 41:194–7. doi: 10.1002/ppul.20357
110. Burns NS, Stevens AM, Iyer RS. Shrinking lung syndrome complicating pediatric systemic lupus erythematosus. Pediatr Radiol. (2014) 44:1318–22. doi: 10.1007/s00247-014-2979-z
111. Meinicke H, Heinzmann A, Geiger J, Berner R, Hufnagel M. Symptoms of shrinking lung syndrome reveal systemic lupus erythematosus in a 12-year-old girl. Pediatr Pulmonol. (2013) 48:1246–9. doi: 10.1002/ppul.22704
112. Alotaibi M, Shao J, Pauciulo MW, Nichols WC, Hemnes AR, Malhotra A, et al. Metabolomic profiles differentiate scleroderma-PAH from idiopathic PAH and correspond with worsened functional capacity. Chest. (2023) 163:204–15. doi: 10.1016/j.chest.2022.08.2230
113. Nickel NP, Yuan K, Dorfmuller P, Provencher S, Lai YC, Bonnet S, et al. Beyond the lungs: systemic manifestations of pulmonary arterial hypertension. Am J Respir Crit Care Med. (2020) 201:148–57. doi: 10.1164/rccm.201903-0656CI
114. Xing Y, Song HM, Wu XY, He YY, Wei M. Clinical analysis of pulmonary arterial hypertension secondary to connective tissue disease in children. Zhonghua Er Ke Za Zhi. (2008) 46:822–6.
115. Anuardo P, Verdier M, Gormezano NW, Ferreira GR, Leal GN, Lianza A, et al. Subclinical pulmonary hypertension in childhood systemic lupus erythematosus associated with minor disease manifestations. Pediatr Cardiol. (2017) 38:234–9. doi: 10.1007/s00246-016-1504-6
116. Jiang T, Li QB. Diffuse alveolar hemorrhage in children. Zhongguo Dang Dai Er Ke Za Zhi. (2019) 21:949–54. doi: 10.7499/j.issn.1008-8830.2019.09.020
117. Cucuzza ME, Marino SD, Schiavone L, Smilari P, Filosco F, Barone P. Diffuse alveolar haemorrage as initial presentation of systemic lupus erythematosus: a case report. Lupus. (2018) 27:507–10. doi: 10.1177/0961203317713144
118. Blay G, Rodrigues JC, Ferreira JCO, Leal GN, Gormezano NW, Novak GV, et al. Brazilian Childhood-onset Systemic Lupus Erythematosus Group. Diffuse alveolar hemorrhage in childhood-onset systemic lupus erythematosus: a severe disease flare with serious outcome. Adv Rheumatol. (2018) 58:39. doi: 10.1186/s42358-018-0038-4
119. Shen M, Zeng X, Tian X, Zhang F, Zeng X, Zhang X, et al. Diffuse alveolar hemorrhage in systemic lupus erythematosus: a retrospective study in China. Lupus. (2010) 19:1326–30. doi: 10.1177/0961203310373106
120. Asseri AA, Al-Murayeh R, Abudiah AM, Elgebally EI, Aljaser AM. A case report of pediatric systemic lupus erythematosus with diffuse alveolar hemorrhage following COVID-19 infection: Causation, association, or chance? Med (Baltimore). (2022) 101:e30071. doi: 10.1097/MD.0000000000030071
121. Harroche A, Remus N, Gaubicher S, Dunand O, Colombat M, Delacourt C, et al. Pulmonary thrombosis as the first manifestation of systemic lupus erythematosus in a 14-year-old boy. Pediatr Nephrol. (2009) 24:857–61. doi: 10.1007/s00467-008-1037-1
122. Bhat MA, Qureshi UA, Ali SW, Bhat JI, Din N, Robbani I. Pulmonary thromboembolism as the initial manifestation in a child with antiphospholipid syndrome in the emergency department. Pediatr Emerg Care. (2011) 27:205–7. doi: 10.1097/PEC.0b013e31820d8dd4
123. Dell'Era L, Corona F, Defilippi AC, Esposito A, Principi N, Esposito S. Systemic lupus erythematosus presenting with pulmonary thromboembolism in a 15-year-old girl. Rheumatol Int. (2012) 32:2925–8. doi: 10.1007/s00296-010-1483-5
124. Wincup C, Ioannou Y. The differences between childhood and adult onset antiphospholipid syndrome. Front Pediatr. (2018) 6:362. doi: 10.3389/fped.2018.00362
125. Montes de Oca MA, Babron MC, Blétry O, Broyer M, Courtecuisse V, Fontaine JL, et al. Thrombosis in systemic lupus erythematosus: a French collaborative study. Arch Dis Child. (1991) 66:713–7. doi: 10.1136/adc.66.6.713
126. Levy DM, Massicotte MP, Harvey E, Hebert D, Silverman ED. Thromboembolism in paediatric lupus patients. Lupus. (2003) 12:741–6. doi: 10.1191/0961203303lu458oa
127. Lin HK, Wang JD, Fu LS. Juvenile diffuse systemic sclerosis/systemic lupus erythematosus overlap syndrome–a case report. Rheumatol Int. (2012) 32:1809–11. doi: 10.1007/s00296-011-1937-4
128. Nitta Y, Muramatsu M. A juvenile case of overlap syndrome of systemic lupus erythematosus and polymyositis, later accompanied by systemic sclerosis with the development of anti-Scl 70 and anti-Ku antibodies. Pediatr Dermatol. (2000) 17:381–3. doi: 10.1046/j.1525-1470.2000.017005381.x
129. Pantazi AC, Balasa AL, Mihai CM, Chisnoiu T, Lupu VV, Kassim MAK, et al. Development of gut microbiota in the first 1000 days after birth and potential interventions. Nutrients. (2023) 15:3647. doi: 10.3390/nu15163647
130. Lupu A, Jechel E, Mihai CM, Mitrofan EC, Fotea S, Starcea IM, et al. The footprint of microbiome in pediatric asthma—A complex puzzle for a balanced development. Nutrients. (2023) 15:3278. doi: 10.3390/nu15143278
131. Pantazi AC, Mihai CM, Balasa AL, Chisnoiu T, Lupu A, Frecus CE, et al. Relationship between gut microbiota and allergies in children: A literature review. Nutrients. (2023) 15:2529. doi: 10.3390/nu15112529
132. Lupu VV, Bratu RM, Trandafir LM, Bozomitu L, Paduraru G, Gimiga N, et al. Exploring the microbial landscape: gut dysbiosis and therapeutic strategies in pancreatitis—A narrative review. Biomedicines. (2024) 12:645. doi: 10.3390/biomedicines12030645
133. Lupu VV, Trandafir LM, Raileanu AA, Mihai CM, Morariu ID, Starcea IM, et al. Advances in understanding the human gut microbiota and its implication in pediatric celiac disease—A narrative review. Nutrients. (2023) 15:2499. doi: 10.3390/nu15112499
134. Lupu VV, Ghiciuc CM, Stefanescu G, Mihai CM, Popp A, Sasaran MO, et al. Emerging role of the gut microbiome in post-infectious irritable bowel syndrome: A literature review. World J Gastroenterol. (2023) 29:3241–56. doi: 10.3748/wjg.v29.i21.3241
135. Pantazi AC, Kassim MAK, Nori W, Tuta LA, Mihai CM, Chisnoiu T, et al. Clinical perspectives of gut microbiota in patients with chronic kidney disease and end-stage kidney disease: where do we stand? Biomedicines. (2023) 11:2480. doi: 10.3390/biomedicines11092480
136. Mocanu A, Bogos RA, Lazaruc TI, Trandafir LM, Lupu VV, Ioniuc I, et al. Exploring a complex interplay: kidney–gut axis in pediatric chronic kidney disease. Nutrients. (2023) 15:3609. doi: 10.3390/nu15163609
137. Ardoin S, Schanberg L. The management of pediatric systemic lupus erythematosus. Nat Rev Rheumatol. (2005) 1:82–92. doi: 10.1038/ncprheum0046
138. Chang JC, Varghese SA, Behrens EM, Gmuca S, Kennedy JS, Liebling EJ, et al. Improving outcomes of pediatric lupus care delivery with provider goal-setting activities and multidisciplinary care models. Arthritis Care Res (Hoboken). (2023) 75:2267–76. doi: 10.1002/acr.25134
139. Thakral A, Klein-Gitelman MS. An update on treatment and management of pediatric systemic lupus erythematosus. Rheumatol Ther. (2016) 3:209–19. doi: 10.1007/s40744-016-0044-0
140. Ikram N, Lewandowski LB, Watt MH, Scott C. Barriers and facilitators to medical care retention for pediatric systemic lupus erythematosus in South Africa: a qualitative study. Res Sq. (2024) 22:59. doi: 10.21203/rs.3.rs-3919073/v1
141. García-Peña P, Boixadera H, Barber I, Toran N, Lucaya J, Enríquez G. Thoracic findings of systemic diseases at high-resolution CT in children. Radiographics. (2011) 31:465–82. doi: 10.1148/rg.312095160
142. Lammi MR, Kolstad KD, Saketkoo LA, Khatri A, Utz PJ, Steen VD, et al. Endothelial biomarkers of systemic sclerosis-associated pulmonary hypertension. Arthritis Care Res (Hoboken). (2023). doi: 10.1002/acr.25180
143. Landini N, Orlandi M, Occhipinti M, Nardi C, Tofani L, Bellando-Randone S, et al. Ultrashort echo-time magnetic resonance imaging sequence in the assessment of systemic sclerosis-interstitial lung disease. J Thorac Imaging. (2023) 38:97–103. doi: 10.1097/RTI.0000000000000637
144. Buda N, Piskunowicz M, Porzezińska M, Kosiak W, Zdrojewski Z. Lung ultrasonography in the evaluation of interstitial lung disease in systemic connective tissue diseases: criteria and severity of pulmonary fibrosis - analysis of 52 patients. Ultraschall Med. (2016) 37:379–85. doi: 10.1055/s-0041-110590
145. Sperandeo M, De Cata A, Molinaro F, Trovato FM, Catalano D, Simeone A, et al. Ultrasound signs of pulmonary fibrosis in systemic sclerosis as timely indicators for chest computed tomography. Scand J Rheumatol. (2015) 44:389–98. doi: 10.3109/03009742.2015.1011228
146. DeCoste C, Mateos-Corral D, Lang B. Shrinking lung syndrome treated with rituximab in pediatric systemic lupus erythematosus: a case report and review of the literature. Pediatr Rheumatol Online J. (2021) 19:7. doi: 10.1186/s12969-020-00491-0
147. Shimizu M, Hashida Y, Ueno K, Yokoyama T, Nakayama Y, Saito T, et al. Successful treatment with bosentan for pulmonary hypertension and reduced peripheral circulation in juvenile systemic sclerosis. Pediatr Cardiol. (2011) 32:1040–2. doi: 10.1007/s00246-011-0056-z
148. Eyraud A, Scouppe L, Barnetche T, Forcade E, Lazaro E, Duffau P, et al. Efficacy and safety of autologous haematopoietic stem cell transplantation in systemic sclerosis: a systematic review of the literature. Br J Dermatol. (2018) 178:650–8. doi: 10.1111/bjd.15993
Keywords: systemic lupus erythematosus, scleroderma, juvenile dermatomyositis, pulmonary damage, physiopathological mechanisms, overlap syndrome, child
Citation: Lupu A, Sasaran MO, Jechel E, Azoicai A, Alexoae MM, Starcea IM, Mocanu A, Nedelcu AH, Knieling A, Salaru DL, Burlea SL, Lupu VV and Ioniuc I (2024) Undercover lung damage in pediatrics - a hot spot in morbidity caused by collagenoses. Front. Immunol. 15:1394690. doi: 10.3389/fimmu.2024.1394690
Received: 01 March 2024; Accepted: 11 June 2024;
Published: 27 June 2024.
Edited by:
Bernadete Liphaus, University of São Paulo, BrazilReviewed by:
Tusharkanti Ghosh, University of Colorado Anschutz Medical Campus, United StatesCopyright © 2024 Lupu, Sasaran, Jechel, Azoicai, Alexoae, Starcea, Mocanu, Nedelcu, Knieling, Salaru, Burlea, Lupu and Ioniuc. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Oana Sasaran, b2FuYW05M0B5YWhvby5jb20=; Elena Jechel, ZWxlbmEuamVjaGVsQHlhaG9vLmNvbQ==; Vasile Valeriu Lupu, dmFsZXJpdWx1cHVAeWFob28uY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.