 Martina Morelli
Martina Morelli Stefania Madonna
Stefania Madonna Cristina Albanesi
Cristina Albanesi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 21 June 2024
Sec. Cytokines and Soluble Mediators in Immunity
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1393799
This article is part of the Research Topic SOCS: Regulation of the Immune System at a Whole New Level View all 10 articles
SOCS are a family of negative inhibitors of the molecular cascades induced by cytokines, growth factors and hormones. At molecular level, SOCS proteins inhibit the kinase activity of specific sets of receptor-associated Janus Activated Kinases (JAKs), thereby suppressing the propagation of intracellular signals. Of the eight known members, SOCS1 and SOCS3 inhibit activity of JAKs mainly induced by cytokines and can play key roles in regulation of inflammatory and immune responses. SOCS1 and SOCS3 are the most well-characterized SOCS members in skin inflammatory diseases, where their inhibitory activity on cytokine activated JAKs and consequent anti-inflammatory action has been widely investigated in epidermal keratinocytes. Structurally, SOCS1 and SOCS3 share the presence of a N-terminal domain containing a kinase inhibitory region (KIR) motif able to act as a pseudo-substrate for JAK and to inhibit its activity. During the last decades, the design and employment of SOCS1 and SOCS3-derived peptides mimicking KIR domains in experimental models of dermatoses definitively established a strong anti-inflammatory and ameliorative impact of JAK inhibition on skin inflammatory responses. Herein, we discuss the importance of the findings collected in the past on SOCS1 and SOCS3 function in the inflammatory responses associated to skin immune-mediated diseases and malignancies, for the development of the JAK inhibitor drugs. Among them, different JAK inhibitors have been introduced in the clinical practice for treatment of atopic dermatitis and psoriasis, and others are being investigated for skin diseases like alopecia areata and vitiligo.
SOCS1 and SOCS3 are proteins that play crucial roles in the regulation of JAK/STAT signaling pathways (1). SOCS1 and SOCS3 belong to SOCS protein family, comprising eight family members: SOCS1–SOCS7 and the cytokine-inducible SH2-domain-containing protein (CIS). All SOCS proteins show conserved structural similarities and mechanisms of action but with unique aspects for different family members (1, 2). Both SOCS1 and SOCS3 control the intensity and duration of immune responses and prevent uncontrolled inflammation. In particular, SOCS1 and SOCS3 are most closely involved in the regulation of the effects induced by IL-4, IL-6, and IFN-γ, which influence the polarization of lymphocytes and the activation of myeloid cells (2).
SOCS1 and SOCS3 dysregulation has been linked to various pathological conditions, making them potential targets for therapeutic interventions in diseases characterized by excessive immune responses and inflammatory cytokine stimulation (2). For example, reduced expression of SOCS1 has been associated with autoimmune diseases such as rheumatoid arthritis and systemic lupus erithematosus. SOCS1 has also implications in cancer, where its dysregulation can contribute to abnormal immune responses (3). Dysregulation of SOCS3 has been associated with various inflammatory conditions, including rheumatoid arthritis, inflammatory bowel disease, and psoriasis (4). As for SOCS1, aberrant expression of SOCS3 has been observed in certain cancers, influencing the tumor microenvironment and immune responses (5).
There is considerable evidence that SOCS1 and SOCS3 play important roles in viral immune evasion involving a broad range of viruses. In fact, SOCS1 and SOCS3 are broadly hijacked by viruses to function effectively as viral virulence factors (6).
In this review, we will retrace the numerous studies obtained during the last two decades and aimed at deciphering the structure and function of SOCS1 and SOCS3 in the inflammatory contexts associated to skin immune-mediated diseases and malignancies. We discussed on how this investigative studies have led to the design and development of peptides mimicking SOCS1 and SOCS3 inhibitory activity, and to the formulation of small molecules inhibiting JAKs as novel therapeutics for skin dermatoses.
SOCS1 and SOCS3 proteins are characterized by distinct functional domains contributing to their biological activities.
Structurally, SOCS1 and SOCS3 share similarities in their overall architecture, both containing a SH2 domain flanked by a similarly sized N-terminal domain, a kinase inhibitory region (KIR) located upstream of the central SH2 domain and a 40-amino acid SOCS box domain in the C-terminal region (7–10) (Figure 1).
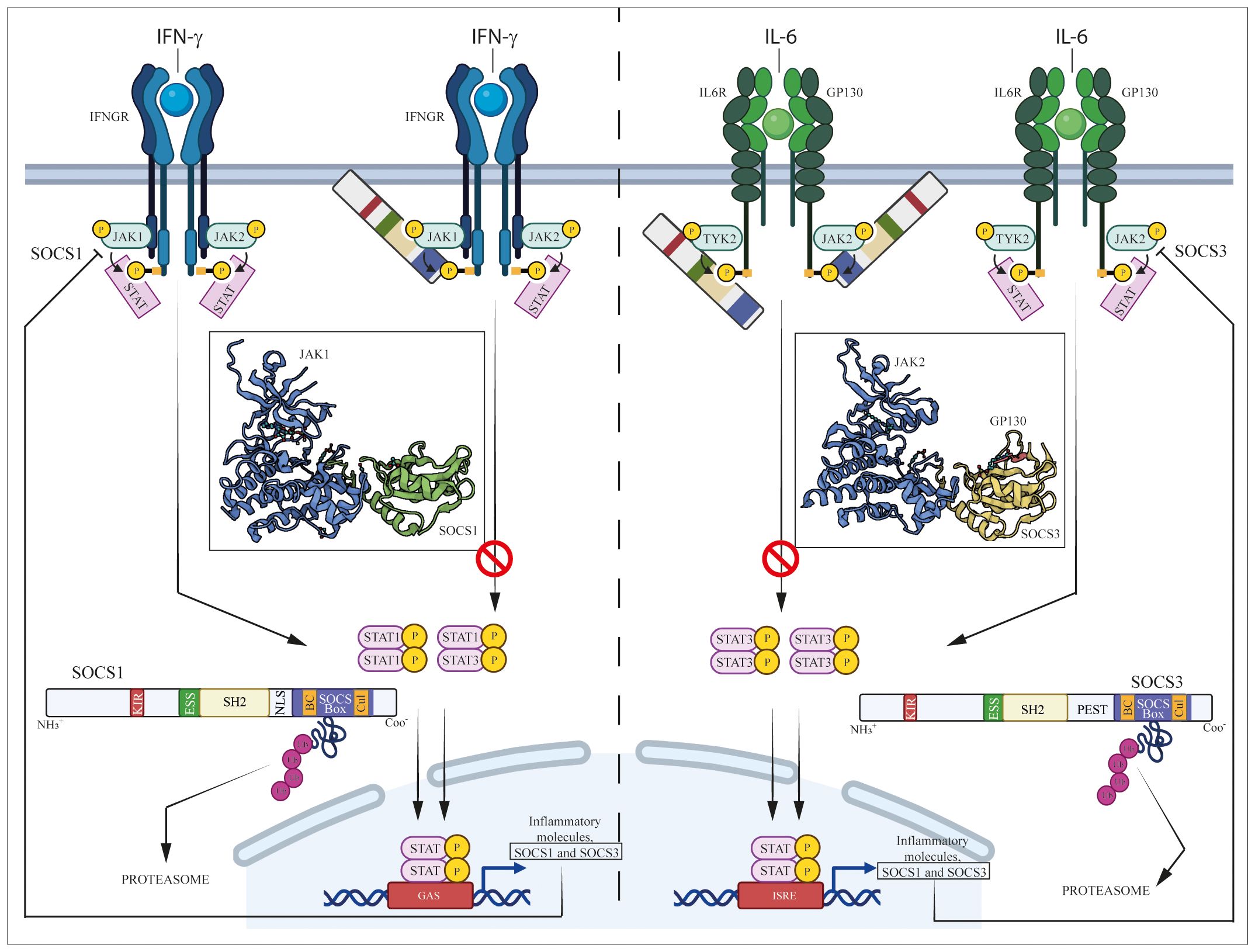
Figure 1 Regulation of JAK/STAT pathways induced by IFN-γ and IL-6 cytokine by SOCS1 and SOCS3. IFN-γ and IL-6 bind their specific IFN-γ and IL-6 receptors (IFNGR and IL-6R/GP130, respectively), causing the activation of associated intracellular JAK complexes (JAK1/JAK2 for IFN-γ and TYK2/JAK2 for IL-6 signaling), which mediate phosphorylation of tyrosine residues in components of receptor complexes. Docking site formation permit binding to receptors of STAT1 or STAT3, which form STAT dimers (STAT1/STAT1 or STAT1/STAT3 dimers for IFN-γ signaling, STAT3/STAT3 dimer for IL-6 signaling) following their phosphorylation. STAT homo- or heterodimers translocate into the nucleus where they activate expression of inflammatory molecules, as well as SOCS1 and SOCS3 themselves that provide negative feedback regulation.
SH2 domain plays a pivotal role in interacting with phosphotyrosines present in target proteins, allowing SOCS1 and SOCS3 to bind to cytokine receptors and other proteins involved in signaling pathways (11). However, unlike the canonical SH2 domain found in many proteins, SH2 domains of SOCS proteins contain a unique N-terminal α-helical region, known as the extended SH2 (ESS) domain, that directly contacts residues crucial for phosphotyrosine binding (7). Immediately upstream of the ESS, both SOCS1 and SOCS3 contain KIR domain, a short amino acid stretch of 12 residues that acts as a pseudosubstrate, blocking the substrate binding groove of JAK to prevent detrimental kinase activity (12). Similar to SOCS1, SOCS3 also contains a C-terminal SOCS box domain able to assemble components of an E3 ubiquitin ligase complex through two motifs, the Elongin B/C (BC) box and the Cullin (Cul) box. This interaction with protein complexes mediates ubiquitination, leading to receptor degradation and thereby regulating the duration and intensity of signaling responses (13, 14). However, the main structural differences between SOCS1 and SOCS3 resides in the central region. Specifically, SOCS1 only contains a nuclear localization sequence (NLS) positioned between the SH2 domain and SOCS box, that mediates transport of SOCS1 into the cell nucleus and supports its role in terminating NF-kB signaling (15). On the other hand, SOCS3 features a 35-residue PEST motif between the SH2 domain and SOCS box that enhances SOCS3 turnover, influencing not only its degradation pathway but also its intracellular stability (7, 16). Both SOCS1 and SOCS3 play a key role in regulating immune responses, and their modular structure reflects the adaptability and specificity of cellular responses modulated by these proteins (Figure 1).
The activation of the JAK-STAT pathway is integral to function of the largest group of immunomodulatory and inflammatory cytokines, including IL-2, IL-4, IL-13, IFN-α and IFN-γ. The activation of cytokine receptors results in the phosphorylation of intracellular receptor-associated JAK, which serves as docking sites for downstream transcription factors, such as members of the STAT family. Activated STAT dimers then translocate to the nucleus, where they bind to target elements, leading the transcriptional activation of multiple genes.
SOCS1 and SOCS3 are endogenous negative inhibitors of the activity of JAK and their related intracellular signaling pathways, which control the excessive and detrimental effects of cytokines trough different mechanisms. These mechanisms include the inhibition of JAK kinase activity, competitive binding with activated cytokine receptors and the targeting of proteins for degradation or rerouting (2, 17) (Figure 1). Aimed at inhibiting the tyrosine kinase activity, the N-terminal region containing KIR and the SH2 domains of SOCS3 is essential for impeding the catalytic activity of JAK1, JAK2, and TYK2. This inhibition is achieved through interaction with a GQM motif in JAK insertion loop. In particular, the first and third of these residues, G1071 and M1073, are indispensable for SOCS3 inhibitory function (18) (Figure 1).
The SH2 domain typically permits to SOCS proteins to directly interact with the exposed phosphotyrosines present on activated cytokine receptor. SOCS1 has been demonstrated to bind to a tyrosine residue (Y1007) on the JH2 domain of JAK2 proteins directly through its SH2 domain, leading to the inhibition of JAK2 catalytic activity (19). Furthermore, SOCS1 is able to bind JAK1, JAK3 and TYK2, inhibiting their activity (20). In contrast, SOCS3 binds JAK2 with a lower affinity and requires a higher expression than SOCS1 to achieve equivalent inhibition of kinase activity (21, 22).
Both SOCS1 and SOCS3 contain a KIR motif, essential for suppressing JAK tyrosine kinase activity. KIR domain acts as a pseudosubstrate blocking the substrate-binding groove of the JAK kinase domain and thereby prevents JAK from phosphorylating downstream substrates (13, 19). Babon et al. demonstrated that KIR binds to the surface of JH1 domain of JAK protein, rather than to the catalytic pocket, changing JH1 3D-conformation that inhibits phosphate transfer from ATP to the substrate peptide (23) (Figure 1).
SH2 domain present in both SOCS1 and SOCS3 is structurally similar to that of JAKs, and enables SOCS to compete with JAKs for binding to phosphotyrosines on cytokine receptors and sterically block binding of other molecules. Once bound to the activated receptor, SOCS1 and SOCS3 impede binding of other SH2 domain-containing proteins, such as STATs, thereby block STATs recruitment (17).
Both SH2 and KIR domains confer to SOCS proteins the specificity of inhibition of kinase activity or competitive binding with cytokine receptors. Indeed, although induced by many cytokines, each SOCS protein is specific for only a subset of cytokines. For example, SOCS1 is up-regulated by IL-2, IL-3, IL-6, IL-13, LIF, GM-CSF, IFN-γ, GH and prolactin but it regulates the intracellular signaling induced by the only IL-2, IL-6, IL-13, and IFN-γ cytokines (21, 24–30) (Figure 1).
SOCS1 appears to play an important role also in the regulation of other cytokines, such as IL-2, IL-4/13, IL-6, IL-12/23, IL-15 and the type-I IFNs (21, 31–36). Moreover, other than controlling the intracellular transduction of erythropoietin receptor by inhibiting JAK2/STAT5 signaling (37, 38), SOCS3 is involved in insulin and IGF-I signaling. Emmanuelli et al. (39) found that SOCS3 interacts directly with the insulin receptor on Tyr960, and inhibits insulin signaling by preventing STAT5β activation.
Considerable evidence showed SOCS3 exhibiting a high affinity for glycoprotein130 (gp130)-related receptors, especially IL-6 receptor (19, 40). The negative regulation of gp130 receptor subunit-mediated signaling by SOCS3 involves SH2 domain mediating interactions with a phosphorylated tyrosine residue (pY757) (41). These interactions induces a conformational change of SOCS3 that can bind to the surface of the JH1 kinase domain, including the GQM motif (42). Prolonged activation of STAT1 and STAT3 and induction of IFN-γ-inducible genes in response to IL-6 has been described in macrophages lacking SOCS3, suggesting an important role for preventing an IFN-γ-like response to IL-6 signaling (43).
Furthermore, SOCS3 is a leptin-inducible inhibitor of leptin signaling through specific bind to pTyr985 of leptin receptor. Leptin, secreted by adipocytes, activates the JAK/STAT pathway by binding to the long-form leptin receptor and SOCS3 represents a potential mechanism for leptin-resistant obesity (44, 45).
A third line of evidence shows that SOCS proteins act by promoting the degradation of specific signaling proteins. Indeed, Zhang et al. suggested that SOCS box-containing proteins act as adapter molecules that address activated signaling proteins to the proteasome machinery (46). Once bound to JAKs or receptors, SOCS box domain of SOCS1 and SOCS3 are able to interacts whit Elongin BC–Cullin5–RING-finger-domain that function as E3 ubiquitin ligases. E3 ligases define substrate specificity and covalently attach ubiquitin to lysine side chains in the substrate. Specifically, since SOCS proteins contain SH2 domains that bind tyrosine-phosphorylated signaling proteins, they can thus act as adapters that bring the E3 ubiquitin ligase complex in proximity of activated proteins, leading to their proteasomal degradation. SOCS proteins could themselves be ubiquitinated and degraded in this process (47) (Figure 1).
In conclusion, both SOCS1 and SOCS3 share a general strategy for JAK inhibition involving receptor interaction, competition for phosphotyrosines, blockade of catalytic activity, and ubiquitination followed by proteasomal degradation. However, they also exhibit specificity towards different JAKs and signaling pathways.
SOCS mRNAs are triggered by cytokines, leading to the activation of SOCS proteins, which in turn inhibit cytokine-induced signaling pathways via a conventional negative feedback loop that influence its gene expression. The expression of SOCS1 and SOCS3 is not only influenced by several cytokines but also by hormones and growth factors (21, 48–50). Indeed, it has been found that that SOCS1 and SOCS3 are induced in fibroblasts by GH (51), whereas SOCS1 is up-regulated by insulin in liver and adipose tissue. SOCS1, in turn, negatively regulates insulin signaling by inhibiting insulin receptor substrate (IRS) proteins (52). Instead, leptin induces SOCS3 expression in keratinocytes, resulting in the inhibition of leptin signaling through negative feedback and insulin resistance, which interfered with their differentiation (53). However, the downstream transcriptional factors regulating SOCS1 and SOCS3 induction by growth factors and hormones remain unexplored.
This regulatory process occurs at both transcriptional and post-transcriptional levels, thereby contributing to maintaining a balance in immune and inflammatory responses (54, 55).
At the transcriptional level, SOCS1 and SOCS3 are regulated by transcription factors that can act as activators or repressors of transcription. STATs and NF-kB, for instance, can bind to regulatory elements within the promoters of the socs1 and socs3 genes, strongly inducing their expression (56–58). In particular, the transcriptional induction of SOCS1 by IFN-γ is mediated by STAT1 through IRF-1 binding to multiple GAAA sequences in the promoter region, rather than the gamma activated sequence (GAS) binding site typically used by the STAT1 homodimer (59). However, binding sites for STAT3 and STAT6 have also been identified within SOCS1 promoter (56).
The SOCS1 promoter contains putative STAT1-, STAT3- and STAT6-binding sites, with evidence suggesting that a dominant negative form of STAT3 can inhibit the IL-6-induced expression of SOCS1 mRNA (60). A study demonstrated that STAT1 acts indirectly on socs1 transcriptional regulation by inducing the expression of IRF-1 transcription factor, which in turn stimulates transcription of the SOCS1gene (60). Moreover, it was found that Sp1 and IFN regulatory factor-1 transcription factors are responsible for the basal and IFN-γ–induced activity of socs1 promoter, respectively. The activity of the SOCS1 promoter dependent on IFN-γ is also negatively regulated by two transcriptional repressors, namely, growth factor independence-1b (GFI-1b) and Kruppel-like factor 4 (KLF4), which tightly control SOCS1 transcription (61). Also SOCS3 expression is regulated by STATs. In fact, SOCS3 promoter contains a STAT1/STAT3 binding element which is necessary and sufficient for its activation by LIF (62).
Of note, SOCS genes can be epigenetically silenced through the methylation of CpG islands or histone deacetylation (63, 64). SOCS1 and SOCS3 genes are often silenced by hypermethylation in several solid tumors, suggesting that hypomethylation plays a crucial role in disease onset and progression (65–67). For instance, epigenetic silencing of SOCS3 has been observed in malignant human melanoma. The repression of SOCS3 in malignant melanoma cells contributes to anti-apoptotic mechanisms and accelerates cell proliferation, suggesting that constitutive SOCS3 expression confers a proliferative advantage to human melanoma cells (68).
SOCS1 and SOCS3 mRNA can be subjected to post-transcriptional regulation through processes that can influence mRNA stability, subcellular localization, and translation of SOCS3 mRNA.
Specific elements within the 3’ UTR of SOCS3 mRNA can influence its stability. RNA interference or mRNA-binding proteins can regulate its degradation or stabilization.
Both genes can be subjected to regulation via RNA interference mechanisms, including the action of microRNAs that typically bind to sites located in the 3′UTR. This interference can disrupt the translation process or promote mRNA degradation (69–71).
Evidence indicated that miR-155 drives Treg/Th17 cells differentiation and enhances Th17 cell function by directly inhibiting SOCS1 protein expression (72). Interestingly, several studies showed that miR-155 promotes inflammatory cytokine expression and IFNs response in primary macrophages and dendritic cells (73).
Based on bioinformatic analysis, SOCS1 was identified as a putative target gene for miR-19b-3p and was also a negative regulator of NF-κB signaling pathway. In vitro studies showed that both mRNA and protein expressions of SOCS1 were inhibited and miR-19b-3p was internalized by macrophages, leading to M1 phenotype polarization (74).
With the aim of discovering new treatment targets, it was observed that miR-30a negatively regulated SOCS1. High miR-30a levels and reduced SOCS1 expression led to increased levels of JAK2, STAT3, Bax, TLR4, and HMGB1 as well as to the extent of JAK2 and STAT3 phosphorylation. By targeting and negatively regulating SOCS1 via the JAK/STAT signaling pathway, miR-30a inhibited the liver cell proliferation and promoted cell apoptosis in rats with sepsis (75).
Furthermore, miR-122 is involved in enhancing IFN-α signaling in the liver by regulating SOCS3 promoter methylation (76). miR-19a can also control SOCS3 expression by binding the 3’-UTR and functionally contributes to the regulation of anti-viral and pro-inflammatory responses induced by IFNs (70).
According to Sonkoly et al., the downregulation of SOCS3 in psoriatic lesions is associated with the upregulation of miR-203 (77). Xu et al. identified an opposite link between miR-203 and SOCS3 expression in IL-17-activated HaCaT cells (78). Taken together, these findings suggest that SOCS3 is a direct target of miR-203 and may influence the IL-17-induced VEGF production. The suppression of SOCS3 by miR-203 adds complexity to SOCS3 regulation, potentially impacting keratinocyte functions in both developing and adult skin (79).
Post-translational modifications, such as phosphorylation of the SOCS box, can regulate the activity and stability of SOCS proteins, influencing their ability to suppress signaling pathways (80, 81). Finally, SOCS3 features a PEST motif adjacent to its SH2 domain, providing an additional mechanism to influence protein stability (7).
The skin serves as a crucial barrier between the body and the external environment, and is essential for homeostasis maintaining a delicate balance in immune responses. The control of cell proliferation is also fundamental in the skin context, as aberrations in cell-growth regulatory mechanisms contribute to skin tumorigenesis.
STAT proteins play a pivotal role in mediating intracellular responses to cytokines, and SOCS, in particular SOCS1 and SOCS3, finely regulate these signaling cascades by preventing excessive or prolonged proinflammatory and pro-proliferative responses. Aberrant STAT hyperactivation and consequent SOCS1 or SOCS3 intervention is observed in immune-mediated inflammatory diseases and malignancies of the skin.
SOCS1 and SOCS3 are expressed in various cell types within the skin, including keratinocytes, fibroblasts, and immune cells. During development of skin inflammatory diseases, keratinocytes, the predominant cell type in the epidermis, express both SOCS1 and SOCS3 molecules in response to a plethora of IFNs and ILs, mostly derived from T lymphocytes, which inhibit in a negative feedback loop the JAK/STAT pathways activated by these inducing stimuli (82). Among them, type-1 (IFN-γ), type-2 (IL-4, IL-13) and IL-10 superfamily (IL-10, IL-20, IL-22) cytokines are potent inducers of SOCS1 and SOCS3 in keratinocytes (82–86), with different consequent effects.
Concerning IFN-γ, it aberrantly influences the inflammation and apoptotic/growth rate in keratinocytes, and in parallel induces SOCS1 and SOCS3 which in turn downregulate the expression of inflammatory genes, such as major histocompatibility complex class (MHC)-II, ICAM-1, and CXCL10, CXCL9, and CCL2 chemokines (82). Moreover, keratinocytes overexpressing SOCS1 show low susceptibility to IFN-γ-mediated growth inhibition, and constitutively show higher levels of the mitogenic cytokine CXCL8, and of RAS/ERK1/2 activities (82, 87).
Interestingly, keratinocytes of psoriasis skin express increased levels of SOCS1, as compared to healthy cells, as consequence of an unbalanced binding of transcriptional activators and repressors to SOCS1 promoter after IFN-γ stimulation (61). In fact, while Sp1 and IRF-1 transcription activators of SOCS1 are expressed at similar levels in keratinocytes of healthy and psoriasis donors, the transcriptional repressors of SOCS1, the GFI-1b and KLF4, are downregulated in psoriatic cells. SOCS1 upregulation could represent a mechanism in psoriatic keratinocytes through which these cells attempt to protect themselves from IFN-γ-induced inflammatory effects (88).
Similarly to SOCS1, SOCS3 is also upregulated in psoriasis epidermis, especially in areas close to CD3+ dermal infiltrate likely producing inflammatory cytokines (89). IFN-γ/TNF-α-treated keratinocytes of psoriasis patients strongly express SOCS3 and SOCS1, at higher levels as compared to healthy cells. In psoriasis, SOCS3 and SOCS1 suppress cytokine-induced apoptosis by sustaining the activation of the PI3K/AKT pathway in keratinocytes. The latter determines the activation of the anti-apoptotic NF-κB cascade and, in parallel, the inhibition of the pro-apoptotic BAD function in keratinocytes (89). Both AKT and BAD are strongly expressed in lesional skin and contribute to the increase of dysfunctional apoptosis and, thus, the peculiar thickening of psoriatic skin (90). In addition, SOCS3 has been shown to be involved in controlling keratinocyte proliferation, as demonstrated in transgenic mice overexpressing SOCS3 in keratinocytes (tsgn-K5/SOCS3) of the basal layer of epidermis (86). Tsgn-K5/SOCS3 mice show full inhibition of STAT3 phosphorylation and atrophied epithelia as result of a marked reduction of proliferating cells and total keratinocyte numbers. Therefore, experimentally mimicked SOCS3 overexpression might turn out to be beneficial in psoriasis by reducing STAT3-dependent inflammation and proliferation. In contrast, SOCS3 overexpression could be deleterious in a hampered cutaneous wound healing context, such as diabetic ulcer, where keratinocyte proliferation and inflammatory responses are fundamental to restore skin integrity after damage. The crucial role of SOCS3 in maintaining homeostasis and preventing skin inflammation has also been demonstrated in SOCS3 gene deficient mice (86). In fact, Socs3 cKO causes severe skin inflammation with epidermal hyperplasia and enhanced inflammatory gene expression. The inflamed skin showed constitutive STAT3 activation and up-regulation of IL-6 and IL-20 receptor (IL-20R) related cytokines (i.e. IL-19, IL-20 and IL-24). Disease symptoms can be counteracted by deletion of Il6 gene, but not by the deletion of Il23, Il4r, or Rag1 genes. The expression of IL-6 in socs3 cKO keratinocytes increases expression of IL-20R-related cytokines that further facilitates STAT3 hyperactivation, epidermal hyperplasia and neutrophilia. These results demonstrate that the IL-6-STAT3-SOCS3 axis strictly regulates skin homeostasis. Moreover, the SOCS3-mediated negative feedback loop in keratinocytes has a critical mechanistic role in the prevention of skin inflammation caused by hyperactivation of STAT3 (86).
Under the influence of proinflammatory cytokines released by leukocytes infiltrating the dermis, also psoriatic dermal fibroblasts upregulate SOCS3 (91). This increased SOCS3 expression correlates with the enhanced activation of TGF-β signaling pathway in psoriasis microenvironment, and elevated production of extracellular matrix (ECM) components, such as collagen and fibronectin, thus indirectly impacting inflammation and excessive proliferation of keratinocytes (92). Indeed, TGF-β can deregulate SOCS3 expression in fibroblasts by inducing the hypermethylation of its promoter and mRNA silencing. As a consequence of SOCS3 silencing, STAT3 is hyperactivated and fibroblasts highly release collagen and promote fibrosis in vitro and in vivo (92). Furthermore, SOCS3 can be responsible for fibroblast proliferation and migration by activating p120 Ras-Gap and ERK1/2 signaling in response to IL-6 and other mitogenic cytokines released in psoriatic skin (93, 94).
Central to psoriasis pathogenesis is the dysregulated signaling of cytokines influencing pathogenic immune responses, particularly IL-23 and IL-17A (88). The pro-inflammatory cytokine IL-23 is critical for the differentiation and maintenance of Th17 cells, and SOCS1 greatly influences IL-23-dependent STAT3 signaling downstream of IL-23 receptor in Th17 cells (94). In psoriasis, the diminished expression of SOCS1 in T cells would contribute to the sustained activation of IL-23-driven pathways and differentiation of Th17 lymphocytes, thus fostering an environment conducive to chronic inflammation and aberrant keratinocyte proliferation. In parallel, dysregulation of SOCS1 in psoriasis has been associated with enhanced dendritic cell activation, resulting in an increase of autoreactive T-cell priming s and perpetuation of the immune cascades (95).
AD, characterized by pruritic and inflamed skin lesions, represents another immune-mediated skin disease where SOCS1 and SOCS3 involvement is evident. The pathogenesis of AD involves a complex interplay of immune dysregulation, barrier dysfunction, and a predisposition to allergic reactions (96).
IL-4 and IL-13, cytokines associated with allergic inflammation, play a pivotal role in AD pathogenesis. SOCS1 and SOCS3, as negative regulators of JAK1/JAK3/STAT6 and JAK1/TYK2/STAT3/STAT6 signaling downstream of IL-4 and IL-13 receptors (97), influences the intensity and duration of these inflammatory responses. SOCS3 has been shown to have an important role in regulating the onset and maintenance of Th2-mediated allergic and atopic diseases, such as AD and asthma (98). In fact, SOCS3 transgenic mice overexpressing SOCS3 in T cells show increased Th2 responses and multiple pathological features typical of asthma in an airway hypersensitivity model system. In contrast, Th2 responses are deficient in both homozygous and heterozygous SOCS3 −/− knockout transgenic mice (98). Consistently, SOCS3 in Th2 cells evokes type-2 cytokine production, IgE production and eosinophilia by inhibiting Th1 differentiation process and thus controlling the balance between Th1 and Th2 cells (99). On the other hand, SOCS3 and SOCS1 in resident AD fibroblasts can impact the recruitment of Th2 cells and eosinophils by deregulating the IL-4-induced expression of the chemokine eotaxin, achieved through the direct inhibition of STAT6 (91).
Beyond its role in cytokine signaling, SOCS3 can influence the maintenance of the epidermal barrier. AD is characterized by immune cell infiltrates and impaired barrier function. SOCS3, by modulating IL-4 and IL-13-dependent signaling pathways regulating the expression of filaggrin, may indirectly impact the progression and severity of AD. Finally, AD is also complicated by recurrent infections, which have been associated with decreased or impaired production of the antimicrobial peptide defensins. Among them, the β-defensins HBD-2 and HBD-3 are downregulated in AD, as consequence of STAT1 inhibition by SOCS1 and SOCS3, transcriptionally activated by IL-4 and IL-13 via STAT6 (83). Due to their role in modulating fibrosis and fibroblast collagen deposition, SOCS1 and SOCS3 may regulate the aberrant responses and excessive ECM production induced by IL-4 and IL-13 in fibroblasts, leading to dermal fibrosis and impaired wound healing characterizing AD skin (92).
SOCS1 and SOCS3 control on immune-mediated skin diseases also depends on the effects that they exert on T-cell polarization. SOCS1 has been shown to downregulate Th1 differentiation by inhibiting IFN-γ-mediated STAT1 activation (100). Accordingly, deficiency of SOCS1 in CD4+ T cells determines increased Th1 responses due to the upregulated IFN-γ signaling, but also indirectly through reduced IL-6/STAT3-mediated Th1 suppression mediated by SOCS3 (101). SOCS1 can also inhibit IL-4-mediated STAT6 activation that drives Th2 polarization, and IL-12-mediated Th1 polarization (102). SOCS1−/− IFN-γ−/− knockout mice were skewed toward Th2 responses, unveiling the role of SOCS1 in the inhibition of Th2 polarization (103). In addition, loss of SOCS1 in CD4+ T cells suppresses Th17 cell development (101).
Genetic factors play a significant role in predisposing individuals to psoriasis or AD. Research has identified specific genetic variations, or polymorphisms, in both socs1 and socs3 genes that may predispose to psoriasis and atopy conditions. Among them, rs4780355 and rs33989964 single-nucleotide polymorphism (SNP) in SOCS1 associate with psoriasis and asthma conditions, respectively (104, 105). On the other hand, rs8074003 SNP in SOCS3 predisposes to atopic disease development, maybe leading to increased SOCS3 expression and IL-4 levels in AD patients (106). These SNPs could influence the expression levels of SOCS and/or their function on JAK/STAT signaling and specific signaling molecules. As a consequence of genetic variations presence in psoriasis or AD patients, the fine-tuning of immune responses could be compromised and potentially tip the balance towards aberrant inflammation.
SOCS1 and SOCS3 proteins were initially described as onco-suppressors, being silenced in many tumors (42) as consequence of hypermethylation of their promoters or mutations influencing proliferation, differentiation and survival of immune cells controlling tumor expansion (107). SOCS1 and SOCS3 expression and anti-tumor function have been also elucidated in skin cancer contexts, such as non-melanoma skin cancer (NMSC) and melanoma.
NMSC constitutes a prevalent group of malignancies, with basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) as its primary subtypes (108). These cancers arise from the neoplastic transformation of keratinocytes of the basal or suprabasal layers of epidermis, driven by cumulative sun exposure, genetic factors, and immune dysregulation. SOCS1 and SOCS3 have garnered increasing attention for their intricate involvement and multifaceted roles in the pathogenesis of NMSC. In BCC and SCC skin lesions, SOCS1 and SOCS3 are moderately detected in vivo (85). Their decrease commonly associates with a transcriptional silencing or selective unresponsiveness to the inducing cytokines. Consistently, hyper-methylation of the SOCS3 promoter is found in 90% of head and neck cancer (109), suggesting that SOCS3 inactivation induced by methylation gene may be an early event in these cancers. DNA hypermethylation of SOCS1 is also frequently found in certain types of lymphomas, with a consequent enhancement of STAT1 and JAK2 activity and hence cell proliferation (110). SOCS1 and SOCS3 expression is reduced in tumor lesions of BCC and SCC, as compared to other skin inflammatory conditions such as psoriasis, despite the high number of IL-22-secreting tumor-infiltrating lymphocytes (85). The latter cells, together with IL-17-producing T cells, promote tumor progression in NMSC by upregulating proliferation and pro-survival pathways dependent on STAT3, NF-κB, and AKT signaling (111). Interestingly, IL-22 is not able to induce SOCS3 transcriptional expression in BCC-or SCC-derived keratinocytes, contrarily to healthy cells. SOCS3 deficiency in NMSC indirectly sustains tumorigenesis by enabling aberrant activation of IL-22-induced STAT3, which promotes cell-cycle progress and suppresses anti-apoptotic events. Potential target genes of STAT3 are involved in cell survival, including Bcl-2 and Bcl-xL, and cell cycle regulators, such as cyclin D1 and cyclin E1, and p21 (112). Vascular endothelial growth factor (VEGF), which is among the target of STAT3, also contributes to tumor angiogenesis (113). Of note, in vitro and in vivo studies conducted on SCC models showed that SOCS3 (and SOCS1) forced expression by SOCS3-derived peptide (KIR-ESS, see paragraph 4.1) efficiently suppresses IL-22-induced tumor growth (85).
SOCS1 and SOCS3 are key players also implicated in melanoma cell growth and tumor development, even if controversial effects have been described. In early studies, SOCS1 was considered as a progression marker of human melanoma, being its expression related to tumor invasion, tumor thickness and stage of the disease (114). Consistently, SOCS1 inhibition in murine B16F10 melanoma cells has been shown to revert the tumorigenic phenotype and inhibit cell metastasis, by inducing cell-cycle arrest at S-phase and decreasing melanoma cell invasion (115). In addition, down-regulation of SOCS1 was capable to decrease the activation of different pro-tumorigenic signals in melanoma cells (115). On the other hand, SOCS1 has been found to block mitosis in melanoma cell lines, by reducing levels of cell-cycle G1 phase regulators, such as cyclin D and cyclin E levels and altering M-phase protein levels (116). SOCS1 also significantly diminished the metastatic capability of melanoma cells, as consequence of its negative control of STAT3 signaling and matrix metalloproteinase-2, basic fibroblast growth factor, and VEGF expression (117).
Similarly to SOCS1, the role of SOCS3 in melanoma appears to be context-dependent, exhibiting both tumor-suppressive and pro-tumorigenic functions (118). Constitutive expression of SOCS3 has not reported for all melanoma cell lines, and high SOCS3 expression has been correlated with insensitivity to the growth-inhibitory effect of IL-6 (119), IFN-γ and IFN-α (120). Suppression of SOCS3 re-established the growth-inhibitory role of IL-6. Consistently, the targeted inhibition of SOCS3 activity in macrophages determined the suppression of melanoma metastasis and prolonged survival in mice xenografted with B16F10 cells (121). On the other hand, CCR5 blockade suppresses melanoma development through inhibition of IL-6-STAT pathway via upregulation of SOCS3 (122). Finally, evidence of a direct role of SOCS3 in tumor progression in terms of disruption of cell cycle does not exist, likely due to the fact that SOCS3, differently from SOCS1, cannot localize in the cell nucleus, since it lacks a nuclear translocation domain (15).
The development of SOCS1 and SOCS3 mimetic peptides started from the detailed characterization of the activation/autophosphorylation loop of JAK2, with the aim at inhibiting its activity independent of the regulation of JAKs by SOCS (123). This study permitted to identifying the amino acid region LPQDKEYYKVKEP of human JAK2, depicted symbolically as pJAK2(1001–1013) (with the p of pJAK2 indicative of phosphorylation of tyrosine 1007), as a focal starting point for the design of SOCS1 mimetic peptides (124, 125).
A first 12-amino acid SOCS1 mimetic peptide, named Tkip, was developed by explorating hydropathic complementarity to the JAK2 activation loop and was characterized by the sequence WLVFFVIFYFFR (126, 127). This predicted peptide showed homology with the F56, F59, and R60 residues of KIR sequence and bound the activation loop of JAK2, thus inhibiting IFN-γ-induced activation of JAK2 and STAT1α, as well as phosphorylation of IFN-γ receptor subunit IFNGR1 (128). Functionally, Tkip inhibited antiviral activity of IFN-γ, as well as expressipon of MHC class I molecules in fibroblasts (128, 129). Furthermore, Tkip reduced inflammatory symptoms in murine encephalomyelitis model, by inhibiting IFN-γ signaling and suppressing the effector functions of T-cells (130, 131) (Table 1).

Table 1 List of SOCS1 and SOCS3 mimetic peptides used in preclinical studies.
As previously described, SOCS1 and SOCS3 KIR region of are located in the N-terminus of the proteins and are adjacent to a similarly short ESS sequence. Peptide corresponding to the ESS sequence was not able to bind the pJAK2(1001–1013) peptide. Thus, Tkip fortuitously led to the KIR region of SOCS1 as a potential SOCS1 mimetics. Analysis of dose–response competitive binding suggested that Tkip and SOCS1-KIR recognized the activation loop of the pJAK2(1001–1013) peptide with similar affinity (127). Differently from Tkip, SOCS1-KIR did not inhibit JAK2 autophosphorylation. However, both peptides inhibited STAT1α activation, IFN-γ-induced activation of macrophages, and antigen-specific lymphocyte proliferation. Of note, topical administration of SOCS1-KIR peptide successfully prevented uveitis and ocular damage (132). Furthermore, a modified version of SOCS1-KIR, named R9-SOCS1-KIR, containing a N-terminal poly-arginine sequence for penetration into plasma membrane, ameliorated autoimmune uveitis in mice (133) (Table 1).
In 2012, a new mimetic peptide of the kinase-inhibitory region of SOCS1, named PS-5, was designed (134). In detail, staring from the 52–67 KIR sequence of SOCS1, through an alanine scanning approach, a truncated linear peptide (62–61), named new KIR, was designed, showing ability to bind to JAK2 (Table 1). Subsequently, new KIR was transformed in a new lead compound, named PS5, carrying the mutations His54/Cys(Acm), Phe55/Arg, and Arg56/Gln, by using a screening of “combinatorial focused libraries” in positional scanning (PS) format (141–143). The new unnatural sequence DTC(Acm)RQTFRSH of PS5 improved JAK2 binding by establishing enhanced electrostatic interactions with the negative phosphate moiety on Y1007 of JAK2. In addition, due to the presence of non-natural residue (Cys (Acm), PS-5 exhibited greater protease stability and effectively inhibited STAT1 phosphorylation and expression levels of IRF-1 expression in human keratinocyte cultures (134). Ps-5 peptide showed also antioxidant and athero-protective effects in in vitro and in vivo model of atherosclerosis (144) (Table 1). Un-natural residues and a lactam internal bridge were further introduced within SOCS1-KIR motif of PS-5 mimetic, generating a set of internal cyclic PS5 analogues These new peptides were able to reduce cytokine-induced proinflammatory gene expression, oxidative stress generation and cell migration, by inhibiting JAK-mediated tyrosine phosphorylation of STAT1 and to (135). Starting from the cyclic peptidomimetic of KIR-SOCS1, icPS5(Nal1), identified in the previous study, La Manna et al. designed novel derivatives, containing crucial amino acids substitutions and/or modifications affecting the ring size that ensured the maintenance of low-micromolar affinity toward JAK2, and a great serum stability (136).
In 2013, the crystal structure of the ternary complex among murine SOCS3/JAK2 kinase domain/gp130 phosphotyrosine-peptide was resolved and allowed to design mimetics of SOCS3 (145). From its inspection, Leu22, Lys23, and Thr24 residues of KIR domain and Val34, Val35, and Val38 of ESS domain of SOCS3 appeared to interact with the GQM” and helix G motifs of JAK2 through hydrophobic interactions (40). Additional contacts involved a “hinge” region between ESS and helix A (HA) of SH2, where the stretch 46–52, called CONG, bears three adjacent aromatic residues, FYW. With the aim to obtain SOCS3 proteomimetics, the ability of different linear peptides, overlapping KIR domain, was analyzed in their ability to recognize JAK2 and mimic SOCS3 cellular effects. Initially, KIR-SOCS3 and ESS-SOCS3 peptides were analyzed isolated and in combination: the entire KIR-ESS-SOCS3 region exhibited good affinity for JAK2 and an efficient suppression of both IL-22 signaling in keratinocytes (85). Aimed at exploring the contribution of other SOCS3 regions, the CONG region was transformed in a chimeric peptide, named KIRCONG chim, that contained non-contiguous fragments: a reduced region of KIR (25–33) and CONG (46–52) connected by β-alanines as spacers (144). This proteomimetic showed anti-inflammatory properties in vascular smooth muscle cells and macrophages, by suppressing STAT3 phosphorylation, as well as the expression of genes as CXCL10 and CCL5 chemokines and the superoxide-generating enzyme NOX2 (138, 139). The observed anti-inflammatory and antioxidant properties of KIRCONG chim corroborated the potential application of SOCS3 mimetics in inflammatory diseases. However, their direct use as drugs are hampered by low aqueous solubility and high molecular weights (Table 1).
Pathogenic mechanisms leading to the manifestation of immune-mediated skin disorders, such as psoriasis and allergic contact dermatitis, are mostly driven by Th1 and Th17 lymphocytes (146, 147). Other than IFN-γ and IL-17, Th1 and Th17 cells can release high amounts of TNF-α, which act in synergy with IFN-γ and IL-17 in reinforcing the inflammatory responses of target cells, among which epidermal keratinocytes (148). Th22 cells also contribute to disease pathogenesis, stimulating defined signaling pathways in epidermal keratinocytes, in several immune-mediated skin diseases (146). Despite recent studies demonstrating that IL-23/IL-17 axis has a pathogenic role in the development of psoriasis, IFN-γ remains a crucial cytokine trigger of resident skin cells, as it potently enhances the expression of pro-inflammatory genes in epidermal keratinocytes activated by the transcription factor STAT1 and alters their apoptotic/growth rate. These pro-inflammatory genes include IRF-1 transcription factor, as well as chemokines and adhesion molecules that contribute to recruitment of leukocytes into the inflammatory sites (149, 150).
We definitively assessed that SOCS1 negatively regulates the molecular cascades triggered by IFN-γ by disabling JAK2 phosphorylation in epidermal keratinocytes (82, 87). Based on this knowledge, the therapeutic potential of PS-5 has been explored in managing pathogenesis of inflammatory skin diseases mediated by IFN-γ (84, 137). In particular, the ability of the PS-5 SOCS1 mimetic peptide, compared to that of SOCS1-KIR peptide, in suppressing the inflammatory responses was evaluated in IFN-γ-activated epidermal keratinocytes by using in vitro cultures and whole-skin explants exposed to IFN-γ. We found that PS-5 efficiently suppressed the IFN-γ molecular signaling in keratinocytes, for instance the JAK2-STAT1-IRF-1 cascade, as well as the downstream expression of STAT1-IRF-1-dependent genes (84, 137). As a direct consequence of the inhibition of CXCL10 and CCL2 chemokine release and partial reduction of ICAM-1 induction, PS-5-treated keratinocytes could no longer retain and induce migration of T lymphocytes in response to IFN-γ. In addition, human skin explants treated with PS-5 did not exhibit the inflammatory signature typically induced by IFN-γ, confirming the ability of this peptide in reducing the inflammatory responses of IFN-γ-activated keratinocytes (84, 137) (Table 1; Figure 2).
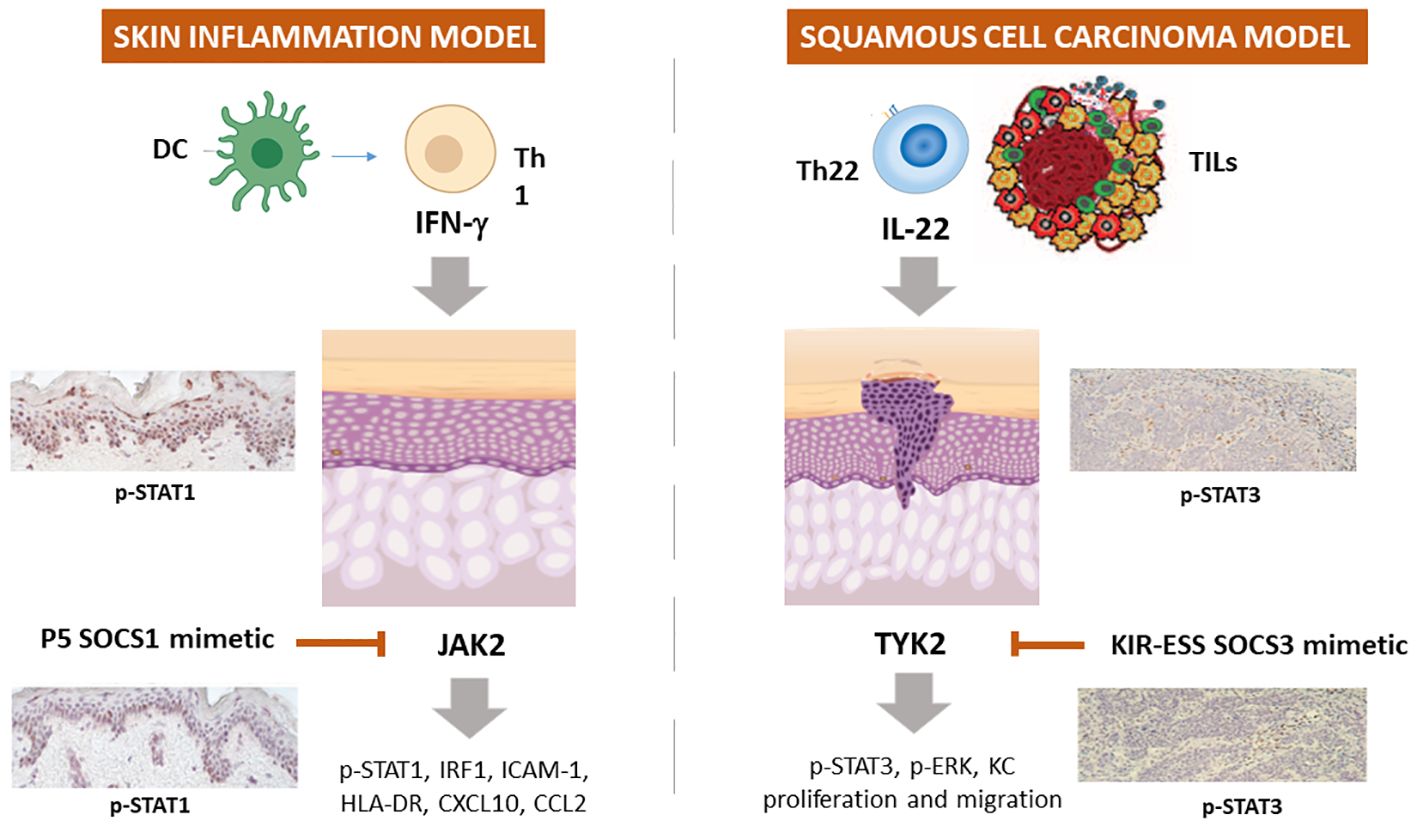
Figure 2 Schematic representation of the employment of SOCS1 and SOCS3 mimetic peptides in experimental models of skin inflammation and non-melanoma skin cancer. In IFN-γ-activated keratinocytes or skin explants, SOCS1 and its mimetic peptide PS-5 inhibit the expression of ICAM-1, HLA-DR, CXCL10, and CCL2 through the blockade of JAK2 tyrosine kinase activity, that leads to STAT1 and IRF-1 activation. In IL-22 activated keratinocytes and skin biopsies of squamous cell carcinoma murine model, KIR-ESS SOCS3 peptide inhibits the expression of p-STAT3 and p-ERK1/2, as well as keratinocyte proliferation and migration.
Epidermal keratinocytes are also involved in BCC and SCC and IL-22-releasing TILs participates to BCC and SCC growth by inducing keratinocyte proliferation and migration, as well as the expression of inflammatory, anti-apoptotic and pro-angiogenic genes. In 2017, we performed a study aimed at rescuing the decreased SOCS3 activity in these tumor contexts by using a SOCS3-derived KIR-ESS peptide (85). We found that SOCS3-KIR-ESS peptide suppressed the IL-22 molecular signaling in keratinocytes, by acting on IL-22R1 and JAK1 phosphorylation and STAT3 and Erk1/2 activation, as well as on the expression of STAT3-dependent downstream genes. As a direct consequence, KIR-ESS peptide counteracted in vitro proliferative and migratory potential of NMSC-derived keratinocytes induced by IL-22. Consistently with in vitro results, KIR-ESS peptide reduced tumor growth in athymic nude mice bearing SCC xenografts, especially when xenografts were subjected to IL-22 administration (85). Inhibition of tumor growth by KIR-ESS peptide was associated to its efficacy in reducing phosphorylation of STAT3 within tumor where keratinocytes are responsible for maintaining tumor growth (151). In line, KIR-ESS peptide showed a significant inhibition of primary tumor growth and pulmonary metastasis in mouse xenografts of MDA-MB-231-luci tumors usually used as models of human triple negative breast cancer (140).
As a whole, these data provided a rationale for the use of peptides mimicking the action of SOCS1 and SOCS3 in the amelioration of skin inflammation, as well as of skin BCC and SCC tumors (Table 1; Figure 2).
Despite various strategies are currently being employed to improve peptide properties, mimetic peptide drugs present limited permeability, proteolytic vulnerability, short half-life, swift in vivo clearance, and limited oral bioavailability (152). However, the design of peptides mimicking the JAKs inhibitory region of SOCS1 and SOCS3 proteins and their employment in managing pathogenesis of inflammatory immune-mediated skin disease led the way towards the development of a class of small molecules inhibiting specific JAKs, known as JAK inhibitors. JAK inhibitors are designed to modulate aberrant JAK activity and downstream signaling pathways, providing a targeted approach for treating diseases driven by dysregulated immune responses. Similarly to SOCS1 and SOCS3 KIR regions, these inhibitors selectively target JAK enzyme family, which plays a crucial role in the JAK/STAT signaling pathway involved in immune response and inflammation. In detail, JAK inhibitors exhibit anti-inflammatory effects suppressing cytokine production involved in Th1, Th2, Th17, and Th22 immune pathways (153). This mechanism contrasts with that of biological disease modifying drugs, which are monoclonal antibodies targeted against only one or two specific cytokines, such as TNF-α, IL-23 or IL-17 blockers for psoriasis or IL-4 and IL-13 inhibitors for atopic dermatitis.
The development of JAK inhibitors has undergone several phases, from early preclinical research to extensive clinical trials. To date, several JAK inhibitors have received the US Food and Drug Administration (FDA) approval for specific autoimmune/inflammatory disorders and are currently being evaluated for additional conditions. Depending on selectivity, JAK inhibitors can be classified as first-generation or poor-selective (e.g., tofacitinib, baricitinib, ruxolitinib) or second generation (e.g., upadacitinib, abrocitinib) inhibitors, with the latter being considered the most selective for each JAK subtypes (Figure 3).
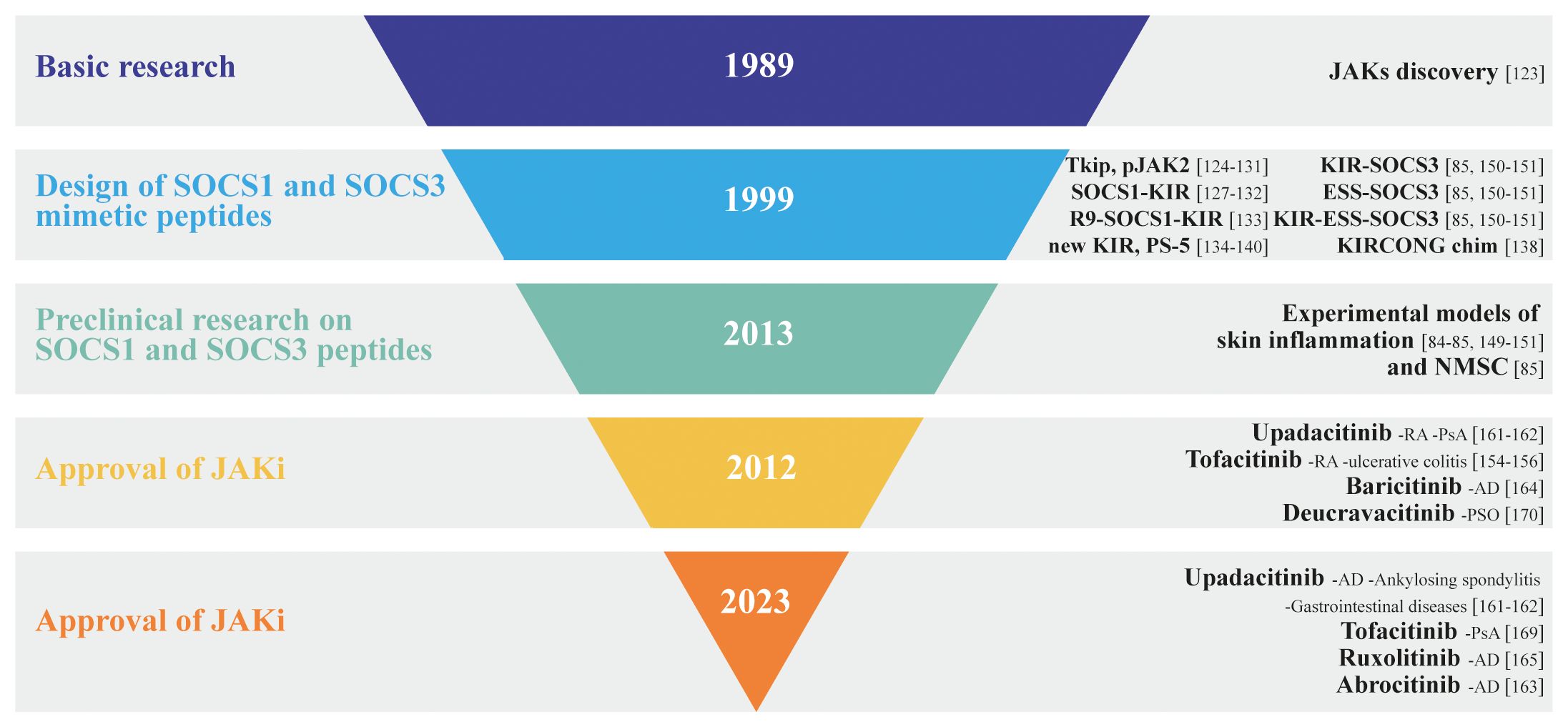
Figure 3 Inverted pyramid model illustrating the design and employment of SOCS1 and SOCS3 mimetic peptides in skin disorders, as well as the discovery and approval of JAK inhibitors, highlighting the key milestones and stages in the process.
Tofacitinib was the first JAK inhibitor approved by the FDA for treating moderate-to-severe rheumatoid arthritis, psoriatic arthritis (PsA), and ulcerative colitis in adult patients. Being a first-generation JAK inhibitor, it is non-selective by acting against JAK1 and JAK3 isoforms along with some JAK2 inhibition. Since all JAK isoforms are inhibited, it effectively blocks several gamma chain cytokines including IL-2, IL-4, IL-15 and IL-21 (154). Tofacitinib is capable of reducing TNF-α, IL-1β, and type I IFN production in dendritic cells stimulated with antigenic lipopolysaccharide (155). In a 2018 study, our group contributed to test the anti-inflammatory effects of tofacitinib in preclinical models of psoriasis. In detail, we evaluated the immunomodulatory effects of tofacitinib on epidermal keratinocytes activated by IFN-γ, IL-22, TNF-α or IL-17. We demonstrated that, similarly to full-length SOCS1 or SOCS3 in keratinocyte clones (82), tofacitinib suppressed expression of IFN-γ-dependent inflammatory genes, and restored normal differentiative programs altered by IL-22 in psoriatic keratinocytes, whereas it did not influence TNF-α- or IL-17-induced molecular cascades. Of note, tofacitinib strongly ameliorated the psoriasiform phenotype of an imiquimod-induced murine model of psoriasis by breaking the inflammatory cross-talks between stromal resident cells and infiltrating immune cells (156) (Figure 3).
Currently, several JAK inhibitors are employed in dermatology, and others are being investigated for skin diseases like alopecia areata, psoriasis, and vitiligo.
The most of FDA-approved JAK inhibitors are currently employed for AD management. AD is an immune-mediated skin disease, characterized by an acute phase occurring within the first 72 h after lesion onset, determined by Th2-driven immune responses with a consequent upregulation of the Th22 axis (157). Thereafter, a chronic phase manifests with continued increase in Th2 and Th22 infiltration and new upregulation of Th1 responses (158). A variety of Th2-related cytokines, in particular IL-4 and IL-13, are involved in the pathogenesis of AD. Other than inhibiting the expression of anti-microbial peptides, IL-4 and IL-13 suppress the expression of filaggrin (FLG), resulting in skin barrier dysfunction. Furthermore, IL-4 and IL-13, together with IL-31 and thymic stromal lymphopoietin (TSLP) are involved in pruritus, whereas Th2-released IL-5 activates eosinophils. Finally, Th22-released IL-22 drives keratinocyte proliferation in the chronic phase (96) (Figure 3).
All these cytokines bind to specific receptors and activate specific arrays of JAKs and STATs pathways. Since JAK1 is involved in most of the signaling pathways of those key cytokines for AD (159), inhibition of JAK1 leads to improvement of AD. JAK inhibitors inhibit a wider range of intracellular molecular cascades activated by AD-related cytokines. However, JAK1 associates to receptors of AD-unrelated cytokines or growth factors that contribute to homeostasis and the immune response (160). For instance, IFN-α plays an important role in the innate immune response, and its receptor harbors JAK1 and TYK2, whereas IFN-γ receptor harbors JAK1 and JAK2. Of note, GM-CSF and erythropoietin are involved in erythropoiesis, myelopoiesis, and platelet production, and their receptors harbor JAK2. Therefore, strong inhibition of JAK1 and JAK2 could cause adverse events in addition to amelioration of AD (Figure 3). Upadacitinib, a selective oral JAK1 inhibitor, was approved by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) for the treatment of rheumatoid arthritis in 2019. Currently, upadacitinib is also approved for atopic dermatitis, psoriatic arthritis, ankylosing spondylitis, and gastrointestinal diseases (161). Indeed, upadacitinib exhibited > 40-, 130-, and 190-fold greater selectivity for JAK1 versus JAK2, JAK3, and TYK2, respectively, in cellular assays (162).
As for upadacitinib, abrocitinib is a selective oral JAK1 inhibitor recently FDA-approved for patients ≥ 18 years old with moderate-to-severe AD (163), whereas baricitinib, recently approved in Europe and Japan for the treatment of AD in adult patients (164), is an oral inhibitor of JAK1 and JAK2 with modest activity against JAK3 and TYK2. Finally, ruxolitinib, a JAK1/JAK2 inhibitor, has been recently approved for the topical treatment of AD (165) (Figure 3).
Of note, although these three JAK inhibitors have different selectivity, increased dose of their administration can inhibit other JAKs. For instance, although upadacitinib is a JAK1-selective inhibitor, the incidence of anemia, one of the adverse effects of JAK2 inhibition, was higher in patients treated with 30 mg of upadacitinib than in those treated with 15 mg of upadacitinib or placebo during 16 weeks (166). Furthermore, the degree of inhibition is also important in understanding the differences in efficacy and side effects among JAK inhibitors. For example, although baricitinib inhibits both JAK2 and JAK1, its degree of inhibition is moderate, thus having mild efficacy with tolerable safety. In particular, severe anemia, one of the possible adverse effects caused by JAK2 inhibition, was not observed in clinical trials of baricitinib (167) (Figure 3).
Psoriasis is a chronic inflammatory skin disease, affecting up to 3% of the worldwide population (88). Disease pathogenesis results from sustained innate and adaptive immune responses that lead to uncontrolled keratinocyte proliferation and dysfunctional differentiation. The activation of plasmacytoid dendritic cells (pDC) by LL37/cathelicidin released by damaged keratinocytes and complexed with self-genetic material is considered as the triggering event for the development of the psoriatic plaque. pDC thus produce type I IFN promoting myeloid dendritic cell phenotypic maturation, thus leading to Th1 and Th17 cell activation.
Several cytokines have been found to play a key role in the development and maintenance of the inflammatory processes behind psoriatic lesions. In particular, the IL-23/Th17 axis is believed to play a central role (88). Indeed, IL-23 drives the differentiation and proliferation of Th17 cells, which produce high levels of IL-17 and IL-22, causing proliferation of epidermal keratinocytes and impairing their terminal differentiation. Psoriasis has several extracutaneous manifestations, such as inflammatory articular and entheseal involvement, leading to PsA. Recently, tofacitinib and upadacitinib were added to the therapeutic armamentarium for treating PsA (168). In September 2022, deucravacitinib, a selective TYK2 inhibitor, has been approved for the treatment of moderate-to-severe psoriasis (169). Its unique mechanism determines greater selectivity and a reduced risk of adverse events (Figure 3).
Alopecia areata (AA) is a CD8+ T cell-mediated autoimmune disease, characterized by inflammatory cell infiltration around lesion follicles, that affects hair follicles (170, 171). In normal conditions, the anagen hair bulb is preserved by complex immune mechanisms. During the anagen phase, the follicle is protected by its low levels of MHC protein and β2 microglobulin expression, which reduce self-antigen presentation and prevent T-cell activation and proliferation. In AA, cytokines such as IFN-γ and common gamma chain cytokines (IL-2, IL-7 and IL-15), as well as inflammatory NKG2D+ CD8+ T cells and CD4+ T cells predominate. These T cells activate IFN-γ, creating a positive feedback loop, which leads to the premature end of the anagen phase, with entry in catagen phase, which is characterized by apoptosis of the hair cells (172). Tofacitinib, a pan-JAK inhibitor, has been used in the off-label management of AA, resulting to be effective in the treatment of adults and adolescents with AA (173). However, being a pan-JAK inhibitor, tofacitinib may be associated with adverse events (174) (Figure 3).
In June 2022, baricitinib has been approved from FDA for AA (175). Numerous other JAK inhibitors are now being studied in AA management. Oral JAK inhibitors seem to be superior to topical JAK inhibitors but a role for topical JAK inhibitors in the management of some aspects of AA treatment (eyebrows, eyelashes, pediatric AA) cannot be excluded.
Although JAK inhibitors are among the most promising therapeutics for AA, the long-term safety and efficacy of JAK inhibitors need to be deepened (Figure 3).
Understanding the functions and roles of SOCS1 and SOCS3 is essential for appreciating their contributions to immune regulation and their implications in various diseases. Their study has opened avenues for important therapeutic interventions, culminating in the development of the current JAK inhibitors, aimed at modulating immune responses in specific conditions, including inflammatory immune-mediated skin diseases. The field of JAK inhibitors is dynamic, with ongoing research contributing to a deeper understanding of their mechanisms and potential applications.
For example, JAK/STAT pathway inhibition can be considered as a promising target for the treatment of vitiligo, a complex skin depigmentation disease that involves multiple pathogenetic mechanisms, including genetic predisposition, oxidative mechanisms and environmental factors, which culminate in the destruction of melanocytes by CD8+ cytotoxic T cells (176). IFN-γ-induced signature has been observed in human skin with vitiligo, with the upregulation of cytokines CXCL9 and CXCL10 via JAK1/2. These cytokines contribute to the recruitment of cytotoxic T lymphocytes responsible for melanocyte destruction (177). This is the reason why inhibition of JAK proteins can be considered an effective therapeutic strategy for the treatment of vitiligo (178). Consistently, topical ruxolitinib exhibited efficacy for re-pigmentation in face vitiligo in a preliminary open-label study (179). Currently, there is an ongoing phase-2 study in which patients received a combination therapy of baricitinib and phototherapy (180).
In contrast, more studies are highly needed to test the validity of JAK inhibitors for hidradenitis suppurativa, a chronic, inflammatory, recurrent, and debilitating skin disease of the hair follicles characterized by inflammatory, painful, deep-rooted lesions. There is only one published clinical trial in the literature (JAK1 inhibitor INCB054707), a real-life study with 15 patients up to week 24 with upadacitinib and a case report showing the successful use of tofacitinib (181). Conversely, there are several ongoing clinical trials.
In conclusion, more efforts aimed at deepening the role of SOCS1 and SOCS3 in “treatment orphan” inflammatory or autoimmune skin diseases and improving efficiency and selectivity of JAK inhibitors will permit to advance treatment options for individuals suffering from debilitating skin conditions.
MM: Data curation, Formal analysis, Visualization, Writing – original draft. SM: Conceptualization, Supervision, Visualization, Writing – original draft. CA: Conceptualization, Supervision, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by grants obtained by IDI-IRCCS from the Italian Ministry of Health (Ricerca Corrente 2022 and 2023).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
SOCS, Suppressors of cytokine signaling; LIF, Leukemia inhibitory factor; JAK/STAT, Janus kinase/signal transducer and activator of transcription; IL, interleukin; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN-γ, Interferon-gamma; SH2, Src Homology 2; KIR, kinase inhibitory region; IGF, Insulin growth factor; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; IRF1, Interferon regulatory factor 1; TNF-α, Tumor necrosis factor-alpha; PI3K, Phosphatidylinositol 3 kinase; TGF-b, Transforming growth factor B; HBD-2/3, Human beta defensin-2/3; TYK2, Tyrosine kinase 2.
1. Durham GA, Williams JJL, Nasim MT, Palmer TM. Targeting SOCS proteins to control JAK-STAT signalling in disease. Trends Pharmacol Sci. (2019) 40:298–308. doi: 10.1016/j.tips.2019.03.001
2. Sobah ML, Liongue C, Ward AC. SOCS proteins in immunity, inflammatory diseases, and immune-related cancer. Front Med. (2021) 8:727987. doi: 10.3389/fmed.2021.727987
3. Ying J, Qiu X, Lu Y, Zhang M. SOCS1 and its potential clinical role in tumor. Pathol Oncol Res. (2019) 25:1295–301. doi: 10.1007/s12253-019-00612-5
4. Yin Y, Liu W, Dai Y. SOCS3 and its role in associated diseases. Hum Immunol. (2015) 76:775–80. doi: 10.1016/j.humimm.2015.09.037
5. Dai L, Li Z, Tao Y, Liang W, Hu W, Zhou S, et al. Emerging roles of suppressor of cytokine signaling 3 in human cancers. Biomed Pharmacother. (2021) 144:112262. doi: 10.1016/j.biopha.2021.112262
6. Johnson HM, Lewin AS, Ahmed CM. SOCS, intrinsic virulence factors, and treatment of COVID-19. Front Immunol. (2020) 11:582102. doi: 10.3389/fimmu.2020.582102
7. Babon JJ, McManus EJ, Yao S, DeSouza DP, Mielke LA, Sprigg NS, et al. The structure of SOCS3 reveals the basis of the extended SH2 domain function and identifies an unstructured insertion that regulates stability. Mol Cell. (2006) 22:205–16. doi: 10.1016/j.molcel.2006.03.024
8. Starr R, Hilton DJ. SOCS: suppressors of cytokine signalling. Int J Biochem Cell Biol. (1998) 30:1081–5. doi: 10.1016/S1357-2725(98)00067-3
9. Kile BT, Schulman BA, Alexander WS, Nicola NA, Martin HME, Hilton DJ. The SOCS box: a tale of destruction and degradation. Trends Biochem Sci. (2002) 27:235–41. doi: 10.1016/S0968-0004(02)02085-6
10. Linossi EM, Nicholson SE. The SOCS box-adapting proteins for ubiquitination and proteasomal degradation. IUBMB Life. (2012) 64:316–23. doi: 10.1002/iub.1011
11. Kükenshöner T, Schmit NE, Bouda E, Sha F, Pojer F, Koide A, et al. Selective targeting of SH2 domain-phosphotyrosine interactions of src family tyrosine kinases with monobodies. J Mol Biol. (2017) 429:1364–80. doi: 10.1016/j.jmb.2017.03.023
12. Ushiki T, Huntington ND, Glaser SP, Kiu H, Georgiou A, Zhang J-G, et al. Rapid inflammation in mice lacking both SOCS1 and SOCS3 in hematopoietic cells. PloS One. (2016) 11:e0162111. doi: 10.1371/journal.pone.0162111
13. Kamura T, Maenaka K, Kotoshiba S, Matsumoto M, Kohda D, Conaway RC, et al. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. (2004) 18:3055–65. doi: 10.1101/gad.1252404
14. Babon JJ, Sabo JK, Zhang J-G, Nicola NA, Norton RS. The SOCS box encodes a hierarchy of affinities for Cullin5: implications for ubiquitin ligase formation and cytokine signalling suppression. J Mol Biol. (2009) 387:162–74. doi: 10.1016/j.jmb.2009.01.024
15. Baetz A, Koelsche C, Strebovsky J, Heeg K, Dalpke AH. Identification of a nuclear localization signal in suppressor of cytokine signaling 1. FASEB J Off Publ Fed Am Soc Exp Biol. (2008) 22:4296–305. doi: 10.1096/fj.08-116079
16. Yoshimura A, Ohkubo T, Kiguchi T, Jenkins NA, Gilbert DJ, Copeland NG, et al. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. (1995) 14:2816–26. doi: 10.1002/embj.1995.14.issue-12
17. Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. (2018) 27:1984–2009. doi: 10.1002/pro.3519
18. Chikuma S, Kanamori M, Mise-Omata S, Yoshimura A. Suppressors of cytokine signaling: Potential immune checkpoint molecules for cancer immunotherapy. Cancer Sci. (2017) 108:574–80. doi: 10.1111/cas.13194
19. Liau NPD, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E, et al. The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun. (2018) 9:1558. doi: 10.1038/s41467-018-04013-1
20. Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, et al. Structure and function of a new STAT-induced STAT inhibitor. Nature. (1997) 387:924–9. doi: 10.1038/43219
21. Pezet A, Favre H, Kelly PA, Edery M. Inhibition and restoration of prolactin signal transduction by suppressors of cytokine signaling. J Biol Chem. (1999) 274:24497–502. doi: 10.1074/jbc.274.35.24497
22. Nicholson SE, Willson TA, Farley A, Starr R, Zhang JG, Baca M, et al. Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. EMBO J. (1999) 18:375–85. doi: 10.1093/emboj/18.2.375
23. Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, et al. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity. (2012) 36:239–50. doi: 10.1016/j.immuni.2011.12.015
24. Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, et al. A family of cytokine-inducible inhibitors of signalling. Nature. (1997) 387:917–21. doi: 10.1038/43206
25. Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. (1997) 387:921–4. doi: 10.1038/43213
26. Losman JA, Chen XP, Hilton D, Rothman P. Cutting edge: SOCS-1 is a potent inhibitor of IL-4 signal transduction. J Immunol. (1999) 162:3770–4. doi: 10.4049/jimmunol.162.7.3770
27. Ram PA, Waxman DJ. SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. J Biol Chem. (1999) 274:35553–61. doi: 10.1074/jbc.274.50.35553
28. Hansen JA, Lindberg K, Hilton DJ, Nielsen JH, Billestrup N. Mechanism of inhibition of growth hormone receptor signaling by suppressor of cytokine signaling proteins. Mol Endocrinol. (1999) 13:1832–43. doi: 10.1210/mend.13.11.0368
29. Song MM, Shuai K. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon-mediated antiviral and antiproliferative activities. J Biol Chem. (1998) 273:35056–62. doi: 10.1074/jbc.273.52.35056
30. Zong CS, Chan J, Levy DE, Horvath C, Sadowski HB, Wang LH. Mechanism of STAT3 activation by insulin-like growth factor I receptor. J Biol Chem. (2000) 275:15099–105. doi: 10.1074/jbc.M000089200
31. Gingras S, Parganas E, de Pauw A, Ihle JN, Murray PJ. Re-examination of the role of suppressor of cytokine signaling 1 (SOCS1) in the regulation of toll-like receptor signaling. J Biol Chem. (2004) 279:54702–7. doi: 10.1074/jbc.M411043200
32. Eyles JL, Metcalf D, Grusby MJ, Hilton DJ, Starr R. Negative regulation of interleukin-12 signaling by suppressor of cytokine signaling-1. J Biol Chem. (2002) 277:43735–40. doi: 10.1074/jbc.M208586200
33. Starr R, Metcalf D, Elefanty AG, Brysha M, Willson TA, Nicola NA, et al. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci USA. (1998) 95:14395–9. doi: 10.1073/pnas.95.24.14395
34. Naka T, Tsutsui H, Fujimoto M, Kawazoe Y, Kohzaki H, Morita Y, et al. SOCS-1/SSI-1-deficient NKT cells participate in severe hepatitis through dysregulated cross-talk inhibition of IFN-gamma and IL-4 signaling in vivo. Immunity. (2001) 14:535–45. doi: 10.1016/S1074-7613(01)00132-7
35. Davey GM, Starr R, Cornish AL, Burghardt JT, Alexander WS, Carbone FR, et al. SOCS-1 regulates IL-15-driven homeostatic proliferation of antigen-naive CD8 T cells, limiting their autoimmune potential. J Exp Med. (2005) 202:1099–108. doi: 10.1084/jem.20050003
36. Sporri B, Kovanen PE, Sasaki A, Yoshimura A, Leonard WJ. JAB/SOCS1/SSI-1 is an interleukin-2-induced inhibitor of IL-2 signaling. Blood. (2001) 97:221–6. doi: 10.1182/blood.V97.1.221
37. Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D, et al. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell. (1999) 98:609–16. doi: 10.1016/S0092-8674(00)80048-3
38. Sasaki A, Yasukawa H, Shouda T, Kitamura T, Dikic I, Yoshimura A. CIS3/SOCS-3 suppresses erythropoietin (EPO) signaling by binding the EPO receptor and JAK2. J Biol Chem. (2000) 275:29338–47. doi: 10.1074/jbc.M003456200
39. Emanuelli B, Peraldi P, Filloux C, Sawka-Verhelle D, Hilton D, Van Obberghen E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem. (2000) 275:15985–91. doi: 10.1074/jbc.275.21.15985
40. Babon JJ, Varghese LN, Nicola NA. Inhibition of IL-6 family cytokines by SOCS3. Semin Immunol. (2014) 26:13–9. doi: 10.1016/j.smim.2013.12.004
41. Nicholson SE, De Souza D, Fabri LJ, Corbin J, Willson TA, Zhang JG, et al. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc Natl Acad Sci USA. (2000) 97:6493–8. doi: 10.1073/pnas.100135197
42. Inagaki-Ohara K, Kondo T, Ito M, Yoshimura A. SOCS, inflammation, and cancer. JAK-STAT. (2013) 2:e24053. doi: 10.4161/jkst.24053
43. Lang R, Pauleau A-L, Parganas E, Takahashi Y, Mages J, Ihle JN, et al. SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol. (2003) 4:546–50. doi: 10.1038/ni932
44. Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, et al. SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem. (2000) 275:40649–57. doi: 10.1074/jbc.M007577200
45. Wang Z, Zhou YT, Kakuma T, Lee Y, Kalra SP, Kalra PS, et al. Leptin resistance of adipocytes in obesity: role of suppressors of cytokine signaling. Biochem Biophys Res Commun. (2000) 277:20–6. doi: 10.1006/bbrc.2000.3615
46. Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci USA. (1999) 96:2071–6. doi: 10.1073/pnas.96.5.2071
47. Piessevaux J, Lavens D, Peelman F, Tavernier J. The many faces of the SOCS box. Cytokine Growth Factor Rev. (2008) 19:371–81. doi: 10.1016/j.cytogfr.2008.08.006
48. Tam SP, Lau P, Djiane J, Hilton DJ, Waters MJ. Tissue-specific induction of SOCS gene expression by PRL. Endocrinology. (2001) 142:5015–26. doi: 10.1210/endo.142.11.8466
49. Peraldi P, Filloux C, Emanuelli B, Hilton DJ, Van Obberghen E. Insulin induces suppressor of cytokine signaling-3 tyrosine phosphorylation through janus-activated kinase. J Biol Chem. (2001) 276:24614–20. doi: 10.1074/jbc.M102209200
50. Emilsson V, Arch JR, de Groot RP, Lister CA, Cawthorne MA. Leptin treatment increases suppressors of cytokine signaling in central and peripheral tissues. FEBS Lett. (1999) 455:170–4. doi: 10.1016/S0014-5793(99)00874-1
51. Adams TE, Hansen JA, Starr R, Nicola NA, Hilton DJ, Billestrup N. Growth hormone preferentially induces the rapid, transient expression of SOCS-3, a novel inhibitor of cytokine receptor signaling. J Biol Chem. (1998) 273:1285–7. doi: 10.1074/jbc.273.3.1285
52. Davey GM, Heath WR, Starr R. SOCS1: a potent and multifaceted regulator of cytokines and cell-mediated inflammation. Tissue Antigens. (2006) 67:1–9. doi: 10.1111/j.1399-0039.2005.00532.x
53. Wang R, Yu C, Tang Z, Sun J, Wang Y, Zhao Z, et al. Leptin induces altered differentiation of keratinocytes by inducing insulin resistance: implications for metabolic syndrome-induced resistance of psoriatic therapy. J Dermatolog Treat. (2024) 35:2309305. doi: 10.1080/09546634.2024.2309305
54. O’Sullivan LA, Liongue C, Lewis RS, Stephenson SEM, Ward AC. Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol Immunol. (2007) 44:2497–506. doi: 10.1016/j.molimm.2006.11.02
55. Kazi JU, Kabir NN, Flores-Morales A, Rönnstrand L. SOCS proteins in regulation of receptor tyrosine kinase signaling. Cell Mol Life Sci. (2014) 71:3297–310. doi: 10.1007/s00018-014-1619-y
56. Krebs DL, Hilton DJ. SOCS: physiological suppressors of cytokine signaling. J Cell Sci. (2000) 113:2813–9. doi: 10.1242/jcs.113.16.2813
57. Chen XP, Losman JA, Rothman P. SOCS proteins, regulators of intracellular signaling. Immunity. (2000) 13:287–90. doi: 10.1016/S1074-7613(00)00028-5
58. Yasukawa H, Sasaki A, Yoshimura A. Negative regulation of cytokine signaling pathways. Annu Rev Immunol. (2000) 18:143–64. doi: 10.1146/annurev.immunol.18.1.143
59. O’Keefe GM, Nguyen VT, Ping Tang LL, Benveniste EN. IFN-gamma regulation of class II transactivator promoter IV in macrophages and microglia: involvement of the suppressors of cytokine signaling-1 protein. J Immunol. (2001) 166:2260–9. doi: 10.4049/jimmunol.166.4.2260
60. Saito H, Morita Y, Fujimoto M, Narazaki M, Naka T, Kishimoto T. IFN regulatory factor-1-mediated transcriptional activation of mouse STAT-induced STAT inhibitor-1 gene promoter by IFN-gamma. J Immunol. (2000) 164:5833–43. doi: 10.4049/jimmunol.164.11.5833
61. Madonna S, Scarponi C, Sestito R, Pallotta S, Cavani A, Albanesi C. The IFN-gamma-dependent suppressor of cytokine signaling 1 promoter activity is positively regulated by IFN regulatory factor-1 and Sp1 but repressed by growth factor independence-1b and Krüppel-like factor-4, and it is dysregulated in psoriatic keratino. J Immunol. (2010) 185:2467–81. doi: 10.4049/jimmunol.1001426
62. Auernhammer CJ, Bousquet C, Melmed S. Autoregulation of pituitary corticotroph SOCS-3 expression: characterization of the murine SOCS-3 promoter. Proc Natl Acad Sci USA. (1999) 96:6964–9. doi: 10.1073/pnas.96.12.6964
63. Kim M-H, Kim M-S, Kim W, Kang MA, Cacalano NA, Kang S-B, et al. Suppressor of cytokine signaling (SOCS) genes are silenced by DNA hypermethylation and histone deacetylation and regulate response to radiotherapy in cervical cancer cells. PloS One. (2015) 10:e0123133. doi: 10.1371/journal.pone.0123133
64. Boosani CS, Agrawal DK. Methylation and microRNA-mediated epigenetic regulation of SOCS3. Mol Biol Rep. (2015) 42:853–72. doi: 10.1007/s11033-015-3860-3
65. Liu T-C, Lin S-F, Chang J-G, Yang M-Y, Hung S-Y, Chang C-S. Epigenetic alteration of the SOCS1 gene in chronic myeloid leukaemia. Br J Haematol. (2003) 123:654–61. doi: 10.1046/j.1365-2141.2003.04660.x
66. Sutherland KD, Lindeman GJ, Choong DYH, Wittlin S, Brentzell L, Phillips W, et al. Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene. (2004) 23:7726–33. doi: 10.1038/sj.onc.1207787
67. Sobti RC, Singh N, Hussain S, Suri V, Nijhawan R, Bharti AC, et al. Aberrant promoter methylation and loss of suppressor of cytokine signalling-1 gene expression in the development of uterine cervical carcinogenesis. Cell Oncol (Dordr). (2011) 34:533–43. doi: 10.1007/s13402-011-0056-2
68. Tokita T, Maesawa C, Kimura T, Kotani K, Takahashi K, Akasaka T, et al. Methylation status of the SOCS3 gene in human Malignant melanomas. Int J Oncol. (2007) 30:689–94. doi: 10.3892/ijo
69. Wu T, Xie M, Wang X, Jiang X, Li J, Huang H. miR-155 modulates TNF-α-inhibited osteogenic differentiation by targeting SOCS1 expression. Bone. (2012) 51:498–505. doi: 10.1016/j.bone.2012.05.013
70. Collins AS, McCoy CE, Lloyd AT, O’Farrelly C, Stevenson NJ. miR-19a: an effective regulator of SOCS3 and enhancer of JAK-STAT signalling. PloS One. (2013) 8:e69090. doi: 10.1371/journal.pone.0069090
71. Lin X-M, Chen H, Zhan X-L. MiR-203 regulates JAK-STAT pathway in affecting pancreatic cancer cells proliferation and apoptosis by targeting SOCS3. Eur Rev Med Pharmacol Sci. (2019) 23:6906–13. doi: 10.26355/eurrev_201908_18730
72. Yao R, Ma Y-L, Liang W, Li H-H, Ma Z-J, Yu X, et al. MicroRNA-155 modulates Treg and Th17 cells differentiation and Th17 cell function by targeting SOCS1. PloS One. (2012) 7:e46082. doi: 10.1371/journal.pone.0046082
73. Contreras J, Rao DS. MicroRNAs in inflammation and immune responses. Leukemia. (2012) 26:404–13. doi: 10.1038/leu.2011.356
74. Lv L-L, Feng Y, Wu M, Wang B, Li Z-L, Zhong X, et al. Exosomal miRNA-19b-3p of tubular epithelial cells promotes M1 macrophage activation in kidney injury. Cell Death Differ. (2020) 27:210–26. doi: 10.1038/s41418-019-0349-y
75. Yuan F-H, Chen Y-L, Zhao Y, Liu Z-M, Nan C-C, Zheng B-L, et al. microRNA-30a inhibits the liver cell proliferation and promotes cell apoptosis through the JAK/STAT signaling pathway by targeting SOCS-1 in rats with sepsis. J Cell Physiol. (2019) 234:17839–53. doi: 10.1002/jcp.28410
76. Yoshikawa T, Takata A, Otsuka M, Kishikawa T, Kojima K, Yoshida H, et al. Silencing of microRNA-122 enhances interferon-α signaling in the liver through regulating SOCS3 promoter methylation. Sci Rep. (2012) 2:637. doi: 10.1038/srep00637
77. Sonkoly E, Wei T, Janson PCJ, Sääf A, Lundeberg L, Tengvall-Linder M, et al. MicroRNAs: novel regulators involved in the pathogenesis of psoriasis? PloS One. (2007) 2:e610. doi: 10.1371/journal.pone.0000610
78. Xu Y, Ji Y, Lan X, Gao X, Chen H-D, Geng L. miR−203 contributes to IL−17−induced VEGF secretion by targeting SOCS3 in keratinocytes. Mol Med Rep. (2017) 16:8989–96. doi: 10.3892/mmr.2017.7759
79. Mostafa SA, Mohammad MHS, Negm WA, Batiha GES, Alotaibi SS, Albogami SM, et al. Circulating microRNA203 and its target genes’ role in psoriasis pathogenesis. Front Med. (2022) 9:988962. doi: 10.3389/fmed.2022.988962
80. Chen XP, Losman JA, Cowan S, Donahue E, Fay S, Vuong BQ, et al. Pim serine/threonine kinases regulate the stability of Socs-1 protein. Proc Natl Acad Sci USA. (2002) 99:2175–80. doi: 10.1073/pnas.042035699
81. Haan S, Ferguson P, Sommer U, Hiremath M, McVicar DW, Heinrich PC, et al. Tyrosine phosphorylation disrupts elongin interaction and accelerates SOCS3 degradation. J Biol Chem. (2003) 278:31972–9. doi: 10.1074/jbc.M303170200
82. Federici M, Giustizieri ML, Scarponi C, Girolomoni G, Albanesi C. Impaired IFN-gamma-dependent inflammatory responses in human keratinocytes overexpressing the suppressor of cytokine signaling 1. J Immunol. (2002) 169:434–42. doi: 10.4049/jimmunol.169.1.434
83. Albanesi C, Fairchild HR, Madonna S, Scarponi C, De Pità O, Leung DYM, et al. IL-4 and IL-13 negatively regulate TNF-alpha- and IFN-gamma-induced beta-defensin expression through STAT-6, suppressor of cytokine signaling (SOCS)-1, and SOCS-3. J Immunol. (2007) 179:984–92. doi: 10.4049/jimmunol.179.2.984
84. Madonna S, Scarponi C, Doti N, Carbone T, Cavani A, Scognamiglio PL, et al. Therapeutical potential of a peptide mimicking the SOCS1 kinase inhibitory region in skin immune responses. Eur J Immunol. (2013) 43:1883–95. doi: 10.1002/eji.201343370
85. Madonna S, Scarponi C, Morelli M, Sestito R, Scognamiglio PL, Marasco D, et al. SOCS3 inhibits the pathological effects of IL-22 in non-melanoma skin tumor-derived keratinocytes. Oncotarget. (2017) 8:24652–67. doi: 10.18632/oncotarget.v8i15
86. Uto-Konomi A, Miyauchi K, Ozaki N, Motomura Y, Suzuki Y, Yoshimura A, et al. Dysregulation of suppressor of cytokine signaling 3 in keratinocytes causes skin inflammation mediated by interleukin-20 receptor-related cytokines. PloS One. (2012) 7:e40343. doi: 10.1371/journal.pone.0040343
87. Madonna S, Scarponi C, De Pità O, Albanesi C. Suppressor of cytokine signaling 1 inhibits IFN-gamma inflammatory signaling in human keratinocytes by sustaining ERK1/2 activation. FASEB J Off Publ Fed Am Soc Exp Biol. (2008) 22:3287–97. doi: 10.1096/fj.08-106831
88. Albanesi C, Madonna S, Gisondi P, Girolomoni G. The interplay between keratinocytes and immune cells in the pathogenesis of psoriasis. Front Immunol. (2018) 9:1–7. doi: 10.3389/fimmu.2018.01549
89. Madonna S, Scarponi C, Pallotta S, Cavani A, Albanesi C. Anti-apoptotic effects of suppressor of cytokine signaling 3 and 1 in psoriasis. Cell Death Dis. (2012). 3:e334. doi: 10.1038/cddis.2012.69
90. Raj D, Brash DE, Grossman D. Keratinocyte apoptosis in epidermal development and disease. J Invest Dermatol. (2006) 126:243–57. doi: 10.1038/sj.jid.5700008
91. Horiuchi Y, Bae S-J, Katayama I. Overexpression of the suppressor of cytokine signalling 3 (SOCS3) in severe atopic dermatitis. Clin Exp Dermatol. (2006) 31:100–4. doi: 10.1111/j.1365-2230.2005.01979.x
92. Dees C, Pötter S, Zhang Y, Bergmann C, Zhou X, Luber M, et al. TGF-β-induced epigenetic deregulation of SOCS3 facilitates STAT3 signaling to promote fibrosis. J Clin Invest. (2020) 130:2347–63. doi: 10.1172/JCI122462
93. Luckett-Chastain LR, Ihnat MA, Mickle-Kawar BM, Gallucci RM. SOCS3 modulates interleukin-6R signaling preference in dermal fibroblasts. J Interf Cytokine Res Off J Int Soc Interf Cytokine Res. (2012) 32:207–15. doi: 10.1089/jir.2011.0086
94. Linke A, Goren I, Bösl MR, Pfeilschifter J, Frank S. The suppressor of cytokine signaling (SOCS)-3 determines keratinocyte proliferative and migratory potential during skin repair. J Invest Dermatol. (2010) 130:876–85. doi: 10.1038/jid.2009.344
95. Hong B, Ren W, Song X-T, Evel-Kabler K, Chen S-Y, Huang XF. Human suppressor of cytokine signaling 1 controls immunostimulatory activity of monocyte-derived dendritic cells. Cancer Res. (2009) 69:8076–84. doi: 10.1158/0008-5472.CAN-09-1507
96. Fania L, Moretta G, Antonelli F, Scala E, Abeni D, Albanesi C, et al. Multiple roles for cytokines in atopic dermatitis: from pathogenic mediators to endotype-specific biomarkers to therapeutic targets. Int J Mol Sci. (2022) 23:2684. doi: 10.3390/ijms23052684
97. Huang I-H, Chung W-H, Wu P-C, Chen C-B. JAK-STAT signaling pathway in the pathogenesis of atopic dermatitis: An updated review. Front Immunol. (2022) 13:1068260. doi: 10.3389/fimmu.2022.1068260
98. Seki Y, Inoue H, Nagata N, Hayashi K, Fukuyama S, Matsumoto K, et al. SOCS-3 regulates onset and maintenance of T(H)2-mediated allergic responses. Nat Med. (2003) 9:1047–54. doi: 10.1038/nm896
99. Kubo M, Inoue H. Suppressor of cytokine signaling 3 (SOCS3) in Th2 cells evokes Th2 cytokines, IgE, and eosinophilia. Curr Allergy Asthma Rep. (2006) 6:32–9. doi: 10.1007/s11882-006-0007-6
100. Takahashi R, Nakatsukasa H, Shiozawa S, Yoshimura A. SOCS1 is a key molecule that prevents regulatory T cell plasticity under inflammatory conditions. J Immunol. (2017) 199:149–58. doi: 10.4049/jimmunol.1600441
101. Tanaka K, Ichiyama K, Hashimoto M, Yoshida H, Takimoto T, Takaesu G, et al. Loss of suppressor of cytokine signaling 1 in helper T cells leads to defective Th17 differentiation by enhancing antagonistic effects of IFN-gamma on STAT3 and Smads. J Immunol. (2008) 180:3746–56. doi: 10.4049/jimmunol.180.6.3746
102. Fujimoto M, Tsutsui H, Yumikura-Futatsugi S, Ueda H, Xingshou O, Abe T, et al. A regulatory role for suppressor of cytokine signaling-1 in T(h) polarization in vivo. Int Immunol. (2002) 14:1343–50. doi: 10.1093/intimm/dxf094
103. Lee C, Kolesnik TB, Caminschi I, Chakravorty A, Carter W, Alexander WS, et al. Suppressor of cytokine signalling 1 (SOCS1) is a physiological regulator of the asthma response. Clin Exp Allergy J Br Soc Allergy Clin Immunol. (2009) 39:897–907. doi: 10.1111/j.1365-2222.2009.03217.x
104. Ellinghaus D, Ellinghaus E, Nair RP, Stuart PE, Esko T, Metspalu A, et al. Combined analysis of genome-wide association studies for Crohn disease and psoriasis identifies seven shared susceptibility loci. Am J Hum Genet. (2012) 90:636–47. doi: 10.1016/j.ajhg.2012.02.020
105. Harada M, Nakashima K, Hirota T, Shimizu M, Doi S, Fujita K, et al. Functional polymorphism in the suppressor of cytokine signaling 1 gene associated with adult asthma. Am J Respir Cell Mol Biol. (2007) 36:491–6. doi: 10.1165/rcmb.2006-0090OC
106. Hussain S, Rasool R, Shafi T, Gull A, Jan R, Bhat IA, et al. Gene variants and mRNA expression analysis of SOCS3 and its association with serum IL-4 levels in atopic diseases. Immunobiology. (2023) 228:152387. doi: 10.1016/j.imbio.2023.152387
107. Ilangumaran S, Bobbala D, Ramanathan S. SOCS1: regulator of T cells in autoimmunity and cancer. Curr Top Microbiol Immunol. (2017) 410:159–89. doi: 10.1007/82_2017_63
108. Zambrano-Román M, Padilla-Gutiérrez JR, Valle Y, Muñoz-Valle JF, Valdés-Alvarado E. Non-melanoma skin cancer: A genetic update and future perspectives. Cancers (Basel). (2022) 14:2371. doi: 10.3390/cancers14102371
109. Rossa CJ, Sommer G, Spolidorio LC, Rosenzweig SA, Watson DK, Kirkwood KL. Loss of expression and function of SOCS3 is an early event in HNSCC: altered subcellular localization as a possible mechanism involved in proliferation, migration and invasion. PloS One. (2012) 7:e45197. doi: 10.1371/journal.pone.0045197
110. Chim CS, Wong KY, Loong F, Srivastava G. SOCS1 and SHP1 hypermethylation in mantle cell lymphoma and follicular lymphoma: implications for epigenetic activation of the Jak/STAT pathway. Leukemia. (2004) 18:356–8. doi: 10.1038/sj.leu.2403216
111. Nardinocchi L, Sonego G, Passarelli F, Avitabile S, Scarponi C, Failla CM, et al. Interleukin-17 and interleukin-22 promote tumor progression in human nonmelanoma skin cancer. Eur J Immunol. (2015) 45:922–31. doi: 10.1002/eji.201445052
112. Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. (2000) 19:2548–56. doi: 10.1038/sj.onc.1203551
113. Xu Q, Briggs J, Park S, Niu G, Kortylewski M, Zhang S, et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene. (2005) 24:5552–60. doi: 10.1038/sj.onc.1208719
114. Li Z, Metze D, Nashan D, Müller-Tidow C, Serve HL, Poremba C, et al. Expression of SOCS-1, suppressor of cytokine signalling-1, in human melanoma. J Invest Dermatol. (2004) 123:737–45. doi: 10.1111/j.0022-202X.2004.23408.x
115. Scutti JAB, Matsuo AL, Pereira FV, Massaoka MH, Figueiredo CR, Moreira DF, et al. Role of SOCS-1 gene on melanoma cell growth and tumor development. Transl Oncol. (2011) 4:101–9. doi: 10.1593/tlo.10250
116. Parrillas V, Martínez-Muñoz L, Holgado BL, Kumar A, Cascio G, Lucas P, et al. Suppressor of cytokine signaling 1 blocks mitosis in human melanoma cells. Cell Mol Life Sci. (2013) 70:545–58. doi: 10.1007/s00018-012-1145-8
117. Huang F-J, Steeg PS, Price JE, Chiu W-T, Chou P-C, Xie K, et al. Molecular basis for the critical role of suppressor of cytokine signaling-1 in melanoma brain metastasis. Cancer Res. (2008) 68:9634–42. doi: 10.1158/0008-5472.CAN-08-1429
118. Culig Z. Suppressors of cytokine signalling-3 and -1 in human carcinogenesis. Front Biosci (Schol Ed). (2013) 5:277–83. doi: 10.2741/S372
119. Komyod W, Böhm M, Metze D, Heinrich PC, Behrmann I. Constitutive suppressor of cytokine signaling 3 expression confers a growth advantage to a human melanoma cell line. Mol Cancer Res. (2007) 5:271–81. doi: 10.1158/1541-7786.MCR-06-0274
120. Lesinski GB, Zimmerer JM, Kreiner M, Trefry J, Bill MA, Young GS, et al. Modulation of SOCS protein expression influences the interferon responsiveness of human melanoma cells. BMC Cancer. (2010) 10:142. doi: 10.1186/1471-2407-10-142
121. Hiwatashi K, Tamiya T, Hasegawa E, Fukaya T, Hashimoto M, Kakoi K, et al. Suppression of SOCS3 in macrophages prevents cancer metastasis by modifying macrophage phase and MCP2/CCL8 induction. Cancer Lett. (2011) 308:172–80. doi: 10.1016/j.canlet.2011.04.024
122. Tang Q, Jiang J, Liu J. CCR5 blockade suppresses melanoma development through inhibition of IL-6-stat3 pathway via upregulation of SOCS3. Inflammation. (2015) 38:2049–56. doi: 10.1007/s10753-015-0186-1
123. Ahmed CMI, Larkin J 3rd, Johnson HM. SOCS1 mimetics and antagonists: A complementary approach to positive and negative regulation of immune function. Front Immunol. (2015) 6:183. doi: 10.3389/fimmu.2015.00183
124. Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, et al. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. (1999) 18:1309–20. doi: 10.1093/emboj/18.5.1309
125. Ahmed CM, Grams TR, Bloom DC, Johnson HM, Lewin AS. Individual and synergistic anti-coronavirus activities of SOCS1/3 antagonist and interferon α1 peptides. Front Immunol. (2022) 13:902956. doi: 10.3389/fimmu.2022.902956
126. Flowers LO, Johnson HM, Mujtaba MG, Ellis MR, Haider SMI, Subramaniam PS. Characterization of a peptide inhibitor of Janus kinase 2 that mimics suppressor of cytokine signaling 1 function. J Immunol. (2004) 172:7510–8. doi: 10.4049/jimmunol.172.12.7510
127. Waiboci LW, Ahmed CM, Mujtaba MG, Flowers LO, Martin JP, Haider MI, et al. Both the suppressor of cytokine signaling 1 (SOCS-1) kinase inhibitory region and SOCS-1 mimetic bind to JAK2 autophosphorylation site: implications for the development of a SOCS-1 antagonist. J Immunol. (2007) 178:5058–68. doi: 10.4049/jimmunol.178.8.5058
128. Frey KG, Ahmed CMI, Dabelic R, Jager LD, Noon-Song EN, Haider SM, et al. HSV-1-induced SOCS-1 expression in keratinocytes: use of a SOCS-1 antagonist to block a novel mechanism of viral immune evasion. J Immunol. (2009) 183:1253–62. doi: 10.4049/jimmunol.0900570
129. Ahmed CM, Dabelic R, Waiboci LW, Jager LD, Heron LL, Johnson HM. SOCS-1 mimetics protect mice against lethal poxvirus infection: identification of a novel endogenous antiviral system. J Virol. (2009) 83:1402–15. doi: 10.1128/JVI.01138-08
130. Mujtaba MG, Flowers LO, Patel CB, Patel RA, Haider MI, Johnson HM. Treatment of mice with the suppressor of cytokine signaling-1 mimetic peptide, tyrosine kinase inhibitor peptide, prevents development of the acute form of experimental allergic encephalomyelitis and induces stable remission in the chronic relapsing/remi. J Immunol. (2005) 175:5077–86. doi: 10.4049/jimmunol.175.8.5077
131. Jager LD, Dabelic R, Waiboci LW, Lau K, Haider MS, Ahmed CMI, et al. The kinase inhibitory region of SOCS-1 is sufficient to inhibit T-helper 17 and other immune functions in experimental allergic encephalomyelitis. J Neuroimmunol. (2011) 232:108–18. doi: 10.1016/j.jneuroim.2010.10.018
132. He C, Yu C-R, Mattapallil MJ, Sun L, Larkin Iii J, Egwuagu CE. SOCS1 mimetic peptide suppresses chronic intraocular inflammatory disease (Uveitis). Mediators Inflamm. (2016) 2016:2939370. doi: 10.1155/2016/2939370
133. Ahmed CM, Patel AP, Ildefonso CJ, Johnson HM, Lewin AS. Corneal application of R9-SOCS1-KIR peptide alleviates endotoxin-induced uveitis. Transl Vis Sci Technol. (2021) 10:25. doi: 10.1167/tvst.10.3.25
134. Doti N, Scognamiglio PL, Madonna S, Scarponi C, Ruvo M, Perretta G, et al. New mimetic peptides of the kinase-inhibitory region (KIR) of SOCS1 through focused peptide libraries. Biochem J. (2012) 443:231–40. doi: 10.1042/BJ20111647
135. La Manna S, Lopez-Sanz L, Bernal S, Fortuna S, Mercurio FA, Leone M, et al. Cyclic mimetics of kinase-inhibitory region of Suppressors of Cytokine Signaling 1: Progress toward novel anti-inflammatory therapeutics. Eur J Med Chem. (2021) 221:113547. doi: 10.1016/j.ejmech.2021.113547
136. La Manna S, Fortuna S, Leone M, Mercurio FA, Di Donato I, Bellavita R, et al. Ad-hoc modifications of cyclic mimetics of SOCS1 protein: Structural and functional insights. Eur J Med Chem. (2022) 243:114781. doi: 10.1016/j.ejmech.2022.114781
137. Kubo M. Therapeutic hope for psoriasis offered by SOCS (suppressor of cytokine signaling) mimetic peptide. Eur J Immunol. (2013) 43:1702–5. doi: 10.1002/eji.201343748
138. La Manna S, Leone M, Mercurio FA, Florio D, Marasco D. Structure-activity relationship investigations of novel constrained chimeric peptidomimetics of SOCS3 protein targeting JAK2. Pharm (Basel). (2022) 15:458. doi: 10.3390/ph15040458
139. La Manna S, Lopez-Sanz L, Mercurio FA, Fortuna S, Leone M, Gomez-Guerrero C, et al. Chimeric peptidomimetics of SOCS 3 able to interact with JAK2 as anti-inflammatory compounds. ACS Med Chem Lett. (2020) 11:615–23. doi: 10.1021/acsmedchemlett.9b00664
140. La Manna S, Lee E, Ouzounova M, Di Natale C, Novellino E, Merlino A, et al. Mimetics of suppressor of cytokine signaling 3: Novel potential therapeutics in triple breast cancer. Int J Cancer. (2018) 143:2177–86. doi: 10.1002/ijc.31594
141. Marasco D, Perretta G, Sabatella M, Ruvo M. Past and future perspectives of synthetic peptide libraries. Curr Protein Pept Sci. (2008) 9:447–67. doi: 10.2174/138920308785915209
142. Lonardo E, Parish CL, Ponticelli S, Marasco D, Ribeiro D, Ruvo M, et al. A small synthetic cripto blocking Peptide improves neural induction, dopaminergic differentiation, and functional integration of mouse embryonic stem cells in a rat model of Parkinson’s disease. Stem Cells. (2010) 28:1326–37. doi: 10.1002/stem.458
143. Ponticelli S, Marasco D, Tarallo V, Albuquerque RJC, Mitola S, Takeda A, et al. Modulation of angiogenesis by a tetrameric tripeptide that antagonizes vascular endothelial growth factor receptor 1. J Biol Chem. (2008) 283:34250–9. doi: 10.1074/jbc.M806607200
144. La Manna S, Lopez-Sanz L, Bernal S, Jimenez-Castilla L, Prieto I, Morelli G, et al. Antioxidant effects of PS5, a peptidomimetic of suppressor of cytokine signaling 1, in experimental atherosclerosis. Antioxidants (Basel Switzerland). (2020) 9:754. doi: 10.3390/antiox9080754
145. Kershaw NJ, Murphy JM, Liau NPD, Varghese LN, Laktyushin A, Whitlock EL, et al. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol. (2013) 20:469–76. doi: 10.1038/nsmb.2519
146. Eyerich S, Eyerich K, Cavani A, Schmidt-Weber C. IL-17 and IL-22: siblings, not twins. Trends Immunol. (2010) 31:354–61. doi: 10.1016/j.it.2010.06.004
147. Di Meglio P, Perera GK, Nestle FO. The multitasking organ: recent insights into skin immune function. Immunity. (2011) 35:857–69. doi: 10.1016/j.immuni.2011.12.003
148. Albanesi C, Scarponi C, Cavani A, Federici M, Nasorri F, Girolomoni G. Interleukin-17 is produced by both Th1 and Th2 lymphocytes, and modulates interferon-gamma- and interleukin-4-induced activation of human keratinocytes. J Invest Dermatol. (2000) 115:81–7. doi: 10.1046/j.1523-1747.2000.00041.x
149. Lew W, Bowcock AM, Krueger JG. Psoriasis vulgaris: cutaneous lymphoid tissue supports T-cell activation and “Type 1” inflammatory gene expression. Trends Immunol. (2004) 25:295–305. doi: 10.1016/j.it.2004.03.006
150. Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. (2007) 445:866–73. doi: 10.1038/nature05663
151. Patel GK, Yee CL, Terunuma A, Telford WG, Voong N, Yuspa SH, et al. Identification and characterization of tumor-initiating cells in human primary cutaneous squamous cell carcinoma. J Invest Dermatol. (2012) 132:401–9. doi: 10.1038/jid.2011.317
152. Lee MF, Poh CL. Strategies to improve the physicochemical properties of peptide-based drugs. Pharm Res. (2023) 40:617–32. doi: 10.1007/s11095-023-03486-0
153. Cartron AM, Nguyen TH, Roh YS, Kwatra MM, Kwatra SG. Janus kinase inhibitors for atopic dermatitis: a promising treatment modality. Clin Exp Dermatol. (2021) 46:820–4. doi: 10.1111/ced.14567
154. Virtanen A T, Haikarainen T, Raivola J, Silvennoinen O. Selective JAKinibs: prospects in inflammatory and autoimmune diseases. BioDrugs. (2019) 33:15–32. doi: 10.1007/s40259-019-00333-w
155. Kubo S, Yamaoka K, Kondo M, Yamagata K, Zhao J, Iwata S, et al. The JAK inhibitor, tofacitinib, reduces the T cell stimulatory capacity of human monocyte-derived dendritic cells. Ann Rheumatol Dis. (2014) 73:2192–8. doi: 10.1136/annrheumdis-2013-203756
156. Morelli M, Scarponi C, Mercurio L, Facchiano F, Pallotta S, Madonna S, et al. Selective immunomodulation of inflammatory pathways in keratinocytes by the janus kinase (JAK) inhibitor tofacitinib: implications for the employment of JAK-targeting drugs in psoriasis. J Immunol Res. (2018) 2018:7897263. doi: 10.1155/2018/7897263
157. Gittler JK, Shemer A, Suárez-Fariñas M, Fuentes-Duculan J, Gulewicz KJ, Wang CQF, et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. (2012) 130:1344–54. doi: 10.1016/j.jaci.2012.07.012
158. Guttman-Yassky E, Nograles KE, Krueger JG. Contrasting pathogenesis of atopic dermatitis and psoriasis–part I: clinical and pathologic concepts. J Allergy Clin Immunol. (2011) 127:1110–8. doi: 10.1016/j.jaci.2011.01.053
159. Chovatiya R, Paller AS. JAK inhibitors in the treatment of atopic dermatitis. J Allergy Clin Immunol. (2021) 148:927–40. doi: 10.1016/j.jaci.2021.08.009
160. Traves PG, Murray B, Campigotto F, Galien R, Meng A, Di Paolo JA. JAK selectivity and the implications for clinical inhibition of pharmacodynamic cytokine signalling by filgotinib, upadacitinib, tofacitinib and baricitinib. Ann Rheumatol Dis. (2021) 80:865–75. doi: 10.1136/annrheumdis-2020-219012
161. Mohamed M-EF, Bhatnagar S, Parmentier JM, Nakasato P, Wung P. Upadacitinib: Mechanism of action, clinical, and translational science. Clin Transl Sci. (2024) 17:e13688. doi: 10.1111/cts.13688
162. Parmentier JM, Voss J, Graff C, Schwartz A, Argiriadi M, Friedman M, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. (2018) 2:23. doi: 10.1186/s41927-018-0031-x
163. Lé AM, Gooderham M, Torres T. Abrocitinib for the treatment of atopic dermatitis. Immunotherapy. (2023) 15:1351–62. doi: 10.2217/imt-2023-0057
164. Radi G, Simonetti O, Rizzetto G, Diotallevi F, Molinelli E, Offidani A. Baricitinib: the first jak inhibitor approved in europe for the treatment of moderate to severe atopic dermatitis in adult patients. Healthc (Basel Switzerland). (2021) 9:1575. doi: 10.3390/healthcare9111575
165. Papp K, Szepietowski JC, Kircik L, Toth D, Eichenfield LF, Leung DYM, et al. Efficacy and safety of ruxolitinib cream for the treatment of atopic dermatitis: Results from 2 phase 3, randomized, double-blind studies. J Am Acad Dermatol. (2021) 85:863–72. doi: 10.1016/j.jaad.2021.04.085
166. Guttman-Yassky E, Teixeira HD, Simpson EL, Papp KA, Pangan AL, Blauvelt A, et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet (London England). (2021) 397:2151–68. doi: 10.1016/S0140-6736(21)00588-2
167. Bieber T, Thyssen JP, Reich K, Simpson EL, Katoh N, Torrelo A, et al. Pooled safety analysis of baricitinib in adult patients with atopic dermatitis from 8 randomized clinical trials. J Eur Acad Dermatol Venereol. (2021) 35:476–85. doi: 10.1111/jdv.16948
168. Nash P, Coates LC, Kivitz AJ, Mease PJ, Gladman DD, Covarrubias-Cobos JA, et al. Safety and efficacy of tofacitinib in patients with active psoriatic arthritis: interim analysis of OPAL balance, an open-label, long-term extension study. Rheumatol Ther. (2020) 7:553–80. doi: 10.1007/s40744-020-00209-4
169. Roskoski RJ. Deucravacitinib is an allosteric TYK2 protein kinase inhibitor FDA-approved for the treatment of psoriasis. Pharmacol Res. (2023) 189:106642. doi: 10.1016/j.phrs.2022.106642
170. Petukhova L, Duvic M, Hordinsky M, Norris D, Price V, Shimomura Y, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. (2010) 466:113–7. doi: 10.1038/nature09114
171. Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med. (2014) 20:1043–9. doi: 10.1038/nm.3645
172. de Oliveira AB, Alpalhão M, Filipe P, Maia-Silva J. The role of Janus kinase inhibitors in the treatment of alopecia areata: A systematic review. Dermatol Ther. (2019) 32:e13053. doi: 10.1111/dth.13053
173. Craiglow BG, Liu LY, King BA. Tofacitinib for the treatment of alopecia areata and variants in adolescents. J Am Acad Dermatol. (2017) 76:29–32. doi: 10.1016/j.jaad.2016.09.006
174. Qian J, Xue X, Shannon J. Characteristics of adverse event reporting of Xeljanz/Xeljanz XR, Olumiant, and Rinvoq to the US Food and Drug Administration. J Manage Care Spec Pharm. (2022) 28:1046–52. doi: 10.18553/jmcp.2022.28.9.1046
175. King B, Ohyama M, Kwon O, Zlotogorski A, Ko J, Mesinkovska NA, et al. Two phase 3 trials of baricitinib for alopecia areata. N Engl J Med. (2022) 386:1687–99. doi: 10.1056/NEJMoa2110343
176. Garcia-Melendo C, Cubiró X, Puig L. Janus kinase inhibitors in dermatology: part 2: applications in psoriasis, atopic dermatitis, and other dermatoses. Actas Dermosifiliogr. (2021) 112:586–600. doi: 10.1016/j.adengl.2021.05.008
177. Rosmarin D, Pandya AG, Lebwohl M, Grimes P, Hamzavi I, Gottlieb AB, et al. Ruxolitinib cream for treatment of vitiligo: a randomised, controlled, phase 2 trial. Lancet (London England). (2020) 396:110–20. doi: 10.1016/S0140-6736(20)30609-7
178. Perez-Bootello J, Cova-Martin R, Naharro-Rodriguez J, Segurado-Miravalles G. Vitiligo: pathogenesis and new and emerging treatments. Int J Mol Sci. (2023) 24:17306. doi: 10.3390/ijms242417306
179. Rothstein B, Joshipura D, Saraiya A, Abdat R, Ashkar H, Turkowski Y, et al. Treatment of vitiligo with the topical Janus kinase inhibitor ruxolitinib. J Am Acad Dermatol. (2017) 76:1054–1060.e1. doi: 10.1016/j.jaad.2017.02.049
180. Dong J, Huang X, Ma L-P, Qi F, Wang S-N, Zhang Z-Q, et al. Baricitinib is Effective in Treating Progressing Vitiligo in vivo and in vitro. Dose Response. (2022) 20:15593258221105370. doi: 10.1177/15593258221105370
Keywords: suppressors of cytokine signaling (SOCS), Janus Activated Kinases (JAK), skin inflammation, immunemediated skin diseases, skin cancer, JAK inhibitors
Citation: Morelli M, Madonna S and Albanesi C (2024) SOCS1 and SOCS3 as key checkpoint molecules in the immune responses associated to skin inflammation and malignant transformation. Front. Immunol. 15:1393799. doi: 10.3389/fimmu.2024.1393799
Received: 29 February 2024; Accepted: 07 June 2024;
Published: 21 June 2024.
Edited by:
Daniela Marasco, University of Naples Federico II, ItalyReviewed by:
Xiaohui Mo, Shanghai Jiao Tong University, ChinaCopyright © 2024 Morelli, Madonna and Albanesi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefania Madonna, cy5tYWRvbm5hQGlkaS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.