 Shan Zhang1†
Shan Zhang1† Xueying Zhu
Xueying Zhu Peiyu Shi
Peiyu Shi Qing Ni
Qing Ni
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 07 May 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1393392
This article is part of the Research Topic Differential Activation of Cell Death Pathways in Macrophages as a Result of Adaptation to Divergent Microenvironment View all 7 articles
This review provides a comprehensive analysis of the critical role played by macrophages and their underlying mechanisms in the progression of diabetic cardiomyopathy (DCM). It begins by discussing the origins and diverse subtypes of macrophages, elucidating their spatial distribution and modes of intercellular communication, thereby emphasizing their significance in the pathogenesis of DCM. The review then delves into the intricate relationship between macrophages and the onset of DCM, particularly focusing on the epigenetic regulatory mechanisms employed by macrophages in the context of DCM condition. Additionally, the review discusses various therapeutic strategies aimed at targeting macrophages to manage DCM. It specifically highlights the potential of natural food components in alleviating diabetic microvascular complications and examines the modulatory effects of existing hypoglycemic drugs on macrophage activity. These findings, summarized in this review, not only provide fresh insights into the role of macrophages in diabetic microvascular complications but also offer valuable guidance for future therapeutic research and interventions in this field.
Diabetes Mellitus (DM) is a chronic metabolic disorder characterized by persistently elevated blood glucose levels (1). Reports from 2019 estimate the global prevalence of diabetes to be 9.3% (463 million individuals), with projections suggesting an increase to 10.2% (578 million individuals) by 2030 (2). Among the cardiovascular complications induced by diabetes, microvascular disease, primarily in the form of DCM, is often diagnosed in its later stages and is considered a leading cause of heart failure in diabetic patients (3, 4). Macrovascular complications primarily refer to accelerated atherosclerosis, including coronary artery disease (CAD) that can lead to acute myocardial infarction (MI) (5). A low-grade inflammatory state (metabolic inflammation) is one of the hallmarks of DCM and the accelerated atherosclerosis associated with diabetes (6, 7). In sites of chronic inflammation in diabetic patients, monocytes/macrophages are excessively recruited, playing an indispensable role in DCM (8).
This article delves into the mechanisms by which macrophages contribute to the development and progression of DCM, as well as the impact of existing therapeutic strategies on these cells. Through a comprehensive analysis of these key mechanisms and cell types, the article aims to offer new perspectives and strategies for the prevention and treatment of diabetic cardiovascular complications, thereby aiming to enhance the quality of life and prognosis for individuals with diabetes.
In 1968, Van Furth and Cohen first defined macrophages in mice as being derived entirely from blood monocytes (9). It was not until 1984 that they discovered macrophage populations in the spleen to be independent of monocyte influx, thus bringing tissue-resident macrophages into focus (10). The mammalian heart contains a significant number of cardiac tissue-resident macrophages (cMacs). The epigenetic mechanisms dependent on the environment are one of the bases for regulating the transcriptional differences among heterogeneous subgroups of cMacs (11). Previous studies have identified three distinct subgroups of human cardiac macrophages based on markers such as C-C chemokine receptor type 2 (CCR2) and HLA-DR (a human major histocompatibility complex II [MHC-II] molecule), namely CCR2+ HLA-DRlow, CCR2+ HLA-DRhigh, and CCR2− HLA-DRhigh (12). Furthermore, based on markers like lymphocyte antigen 6 complex, locus C (Ly6C), and MHC-II, four distinct subgroups of mouse macrophages can be effectively distinguished (13).
With the advent of new technologies such as genetic fate mapping and lineage tracing, we are now able to label, track, and monitor the origins and developmental processes of macrophages in tissues (14). The earliest macrophage precursor cells originate from the extraembryonic yolk sac between embryonic day 7.0 (E7.0) and E9.0. As embryonic vasculature and the aorta-gonad-mesonephros (AGM) region develop, hematopoietic stem cells (HSCs) are produced and eventually colonize the fetal liver and bone marrow. Thus, at least two mechanisms (yolk sac and fetal liver) are responsible for establishing cMacs before birth (15). Monocyte-derived M1 macrophages primarily differentiate from blood monocytes produced by bone marrow HSCs, under the stimulation of colony-stimulating factor (CSF) and granulocyte-macrophage CSF, developing into monocytes, pre-monocytes, and mature monocytes. Subsequently, they proliferate into the body’s epithelial and submucosal tissues, which are essential for immune surveillance, self-stability, and resistance to infection (16). The earliest yolk sac-derived M2 macrophages are mostly attributed to cMacs, appearing in cardiac tissue between E9.5 and E10.5, followed by CCR2+ cMacs from fetal monocyte precursor cells colonizing the heart by E14.5 (17) (Figure 1).
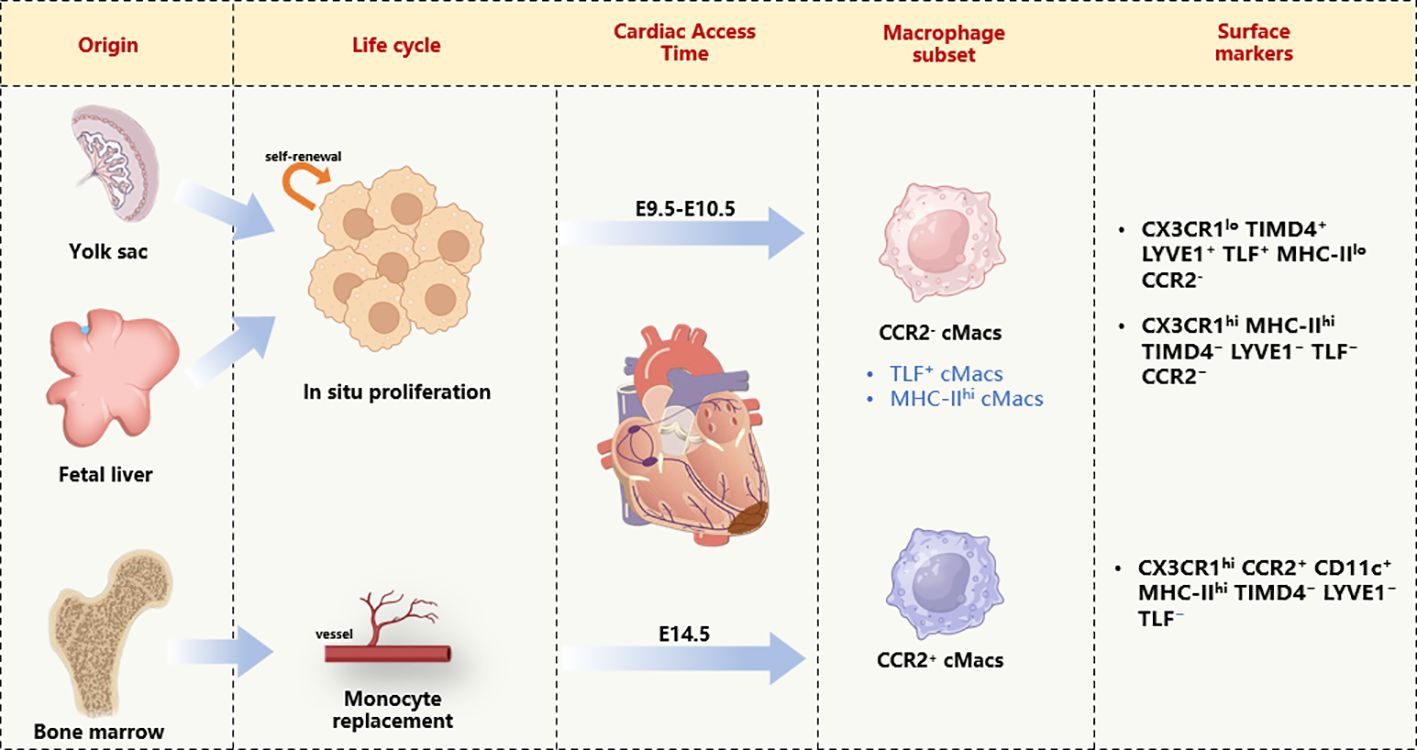
Figure 1 Cardiac macrophage diversity on the basis of developmental origin, life cycle, and transcriptional signature. cMacs, Cardiac resident macrophages; CD11c, Integrin alpha X; CCR2, CC- chemokine receptor 2; CX3CR, Chemokine (C-X3-C motif) receptor; LYVE1, lymphatic vessel endothelial hyaluronan receptor 1; MHC-II, major histocompatibility complex class II; TLF, folate receptor beta; TIMD4, T cell immunoglobulin and mucin domain containing 4.
The M1/M2 binary classification of macrophages, as initially described by Mills CD and colleagues, is based on their responses to different stimuli In vitro (18). M1 macrophages are typically associated with pro-inflammatory and antimicrobial activities, while M2 macrophages are generally linked to anti-inflammatory actions, tissue repair, and tumor-promoting effects. Although this classification has its limitations and does not fully capture the complexity and diversity of macrophage functions in vivo, it remains a useful framework for researching and understanding macrophage roles under various physiological and pathological conditions. Recent studies, including the work of Xue J et al., have proposed a ‘spectrum model’ of macrophage activation, which offers a more nuanced view of macrophage function across a continuum of activation states (19). This model underscores the importance of considering the full range of macrophage activation in understanding their roles in health and disease.
M1 macrophages: Their surface markers include HLA-DR, CD80, CD86, and CD197, and they produce pro-inflammatory cytokines (20). Typically, Th1 (interferon [IFN]-γ, tumor necrosis factor-[TNF]-α) or bacterial lipopolysaccharide (LPS) can induce and activate M1 polarization, leading to the release of a large amount of pro-inflammatory cytokines, including TNF-α, interleukins (IL)-1, IL-6, IL-12, IL-18, IL-23, and chemokine ligands CXCL-9 and CXCL-10. In T2DM, the islets undergo inflammation, with a significant accumulation of M1-like macrophages, although it remains unclear what triggers this inflammatory cascade. Recent studies report that islet macrophages and β-cells are frontline responders to systemic metabolic changes. Furthermore, M1-like macrophages produce pro-inflammatory cytokines that promote β-cell dysfunction, creating a vicious cycle with β-cell secretion of chemokines and M1-like macrophage-secreted cytokines, accelerating islet inflammation (21).
M2 macrophages: Based on multiple activation signals received by M2 macrophages, four different M2 macrophage groups can be identified: M2a, M2b, M2c, and M2d. M2-like macrophages are maintained through the local proliferation of resident macrophages, playing an indispensable role in the proliferation of β-cells and participating in maintaining a healthy islet microenvironment (22). During the metabolic inflammation process in the islets, including macrophages, endocrine cells, and endothelial cells (ECs), jointly secrete a variety of cytokines, such as CSF-1, TGF-β1, and vascular endothelial growth factor-A (VEGFA), promoting the polarization of M2-type islet macrophages (23, 24). Tissue-resident macrophages belong to the M2 class, in addition, activated M2-type macrophages include anti-inflammatory macrophages, pro-fibrotic macrophages, and pro-angiogenic macrophages, which are involved in the proliferation of β-cells during development and in adult mice (25, 26) (Table 1).
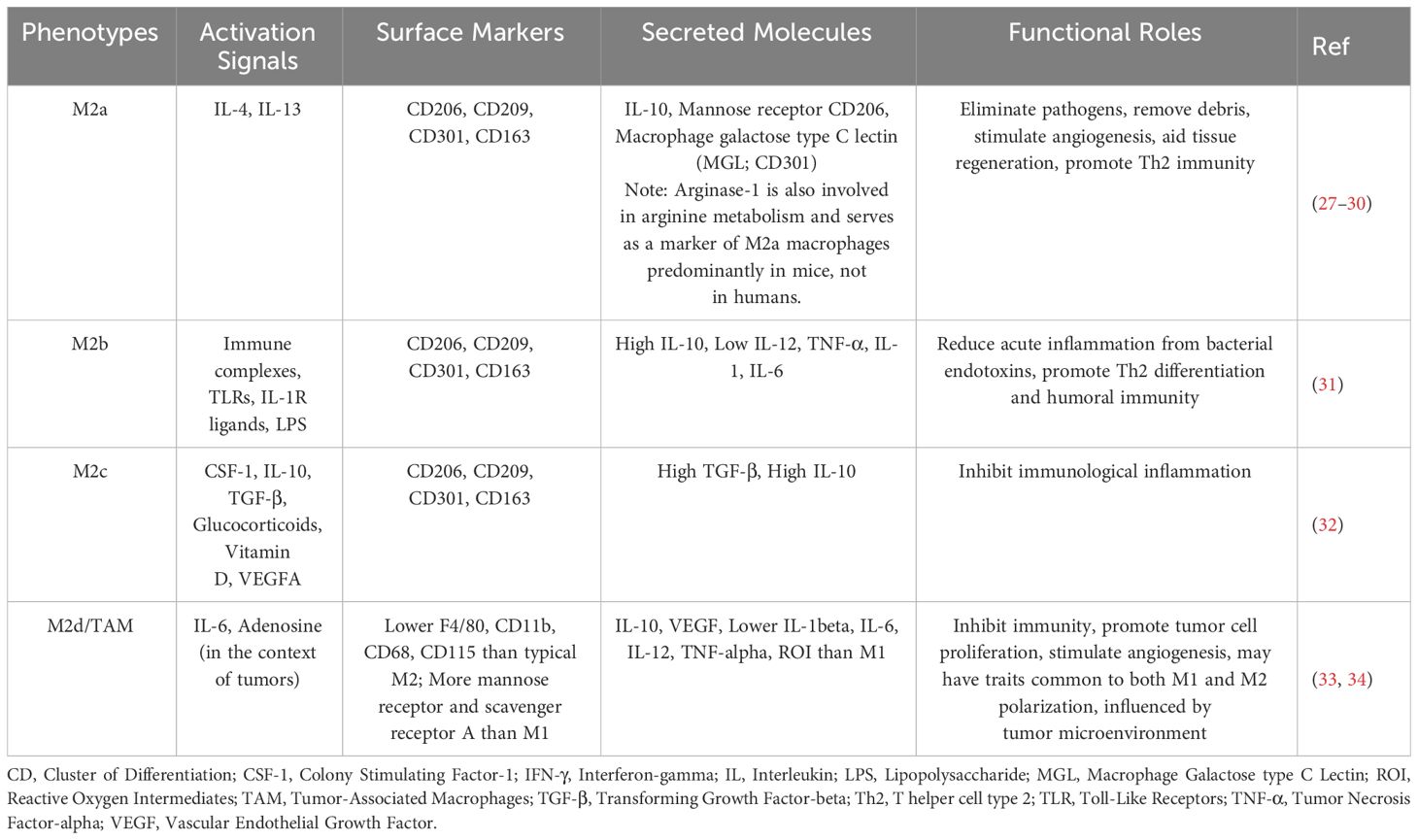
Table 1 Phenotypes, stimulations, secretions, markers, and functional roles of M2 macrophages.
The heart’s structural complexity and its physiological and pathological environments dictate the distinct characteristics displayed by cMacs in their respective niches. The heart, being a highly vascularized organ, sees a significant accumulation of CCR2- cMacs around blood vessels (17). These cMacs regulate vascular homeostasis through their interactions with smooth muscle cells and collagen by expressing lymphatic vessel endothelial hyaluronan receptor 1 (LYVE-1) (35, 36). Communication between macrophages and ECs via extracellular vesicles (EVs) leads to changes in macrophage phenotypes, resulting in reduced microvascular repair and compromised myocardial perfusion (37). This process, in turn, promotes atherosclerosis, chronic inflammation, and fibrosis (38). Furthermore, interactions between macrophages and ECs through small extracellular vesicles (sEVs) also play a role in diabetic complications. High glucose levels in pro-inflammatory macrophages induce the production of sEVs carrying elevated levels of IL-1β, inducible nitric oxide synthase, human antigen R (HuR), miR-21-5p, miR-486-5p, and TGF-β mRNA (39–43). These sEVs can effectively transfer pathogenic miRNAs to the subendothelium, leading to vasoconstriction, dysfunction, and inflammation, which further promote atherosclerosis and thrombosis (44). Conversely, sEVs derived from ECs containing MALAT-1 promote the polarization of anti-inflammatory macrophages (45, 46).
In the context of cardiac electrophysiology, confocal fluorescence microscopy has shown that CCR2- cMacs are abundantly present at the atrioventricular node (47). These cMacs connect with conducting cardiomyocytes through gap junctions involving connexin 43 (Cx43) and regulate the electrical activity of cardiomyocytes through periodic depolarization (47). cMacs can also indirectly prevent cardiac conduction block and ventricular arrhythmias. In models of right heart pressure overload (pulmonary hypertension), macrophage-derived amphiregulin (Areg) has been shown to prevent lethal arrhythmias and sudden death (48). Areg plays a crucial role in controlling the phosphorylation and translocation of cardiomyocyte Cx43; its deletion leads to the disassembly of gap junctions (48). Research by Simon-Chica et al. revealed a mirroring effect in the expression of ion channel subunits between cardiomyocytes and cMacs, including Kir2.1, Kv1.5, Kv1.3, Cx43, and Nav1.5, thereby accelerating the repolarization of cardiomyocytes (49). This suggests that changes in macrophage phenotypes in DCM could be a potential cause of the cardiac electrical remodeling often observed in patients with type 1 and type 2 diabetes (50).
DCM is a myocardial-specific microvascular complication that occurs independently of CAD, valvular disease, and hypertension (51). It increases the risk of heart failure in diabetic patients (52). The overproduction of free radicals in a hyperglycemic environment has been identified as a key pathogenic factor in the development of DCM. This process induces oxidative stress, mitochondrial dysfunction, apoptosis, and myocardial fibrosis, thereby impairing myocardial contractility and normal cardiac function. Abnormal cellular metabolism and the accumulation of damaged or defective organelles are associated with the pathogenesis of DCM (53) (Figure 2).
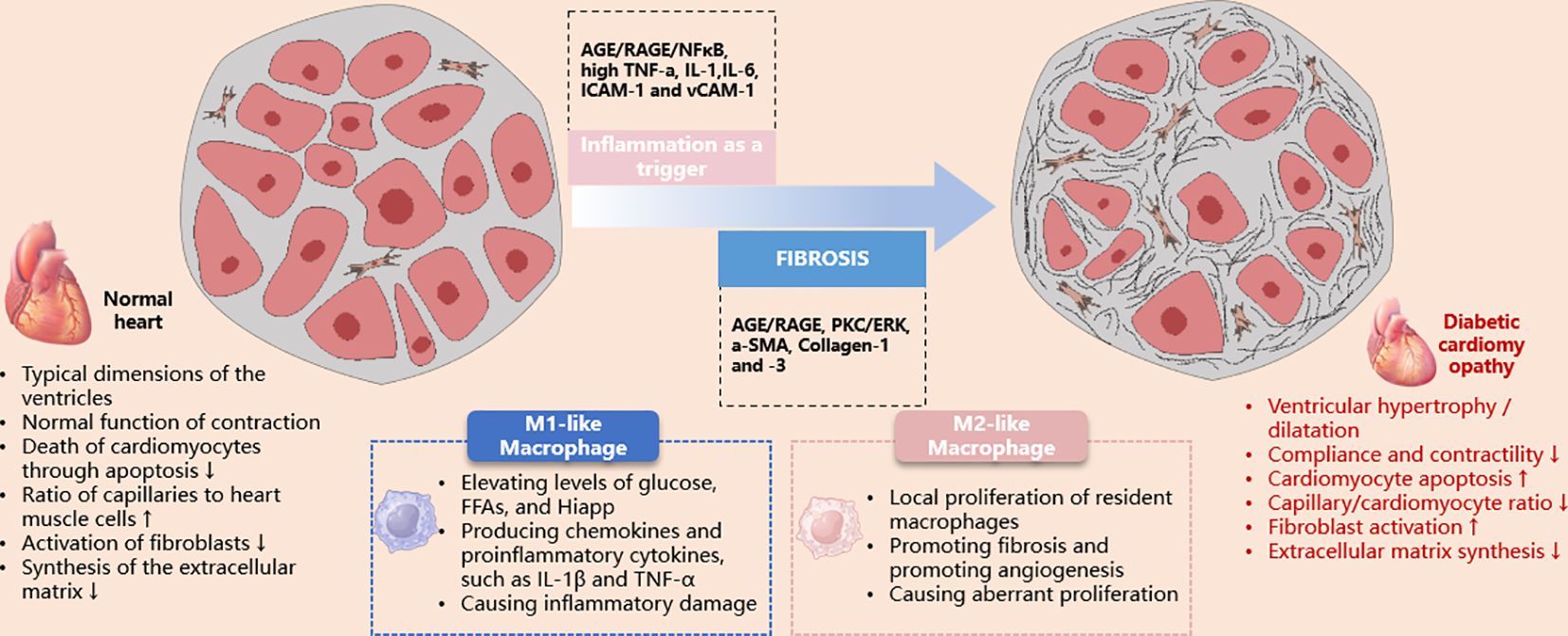
Figure 2 Schematic illustration of the role of macrophages in the pathological damage of diabetic cardiomyopathy. AGE, advanced glycation end products; ERK, extracellular signal-regulated kinase; ICAM-1, intercellular adhesion molecule-1; IL, interleukin; iNOS, inducible nitric oxide synthase; PKC, protein kinase C; RAGE, receptor for advanced glycation end products; TNF, tumor necrosis factor; VCAM-1, vascular cell adhesion molecule-1.
The involvement of the innate immune system and pro-inflammatory responses is associated with the development of DCM. The activation and expression of pro-inflammatory cytokines, such as TNFα, IL-6 and 8, monocyte chemoattractant protein 1 (MCP-1), intercellular adhesion molecule 1, and vascular cell adhesion molecule 1, contribute to cardiac oxidative stress, remodeling, fibrosis, and diastolic dysfunction (54). The expression of these cytokines is regulated by the nuclear transcription factor NF-κB (54). Additionally, Toll-like receptor 4 plays a significant role in triggering NF-κB activation, enhancing pro-inflammatory and innate immune responses (55). The increased migration of monocytes/macrophages into the coronary endothelium elevates resident cardiac macrophages. Under conditions of increased ROS and reduced bioavailable NO, these macrophages can polarize into a pro-inflammatory M1 phenotype (54–56). Recent studies have observed an upregulation of pro-inflammatory M1 polarization in diabetic cardiac tissue, while the M2 anti-inflammatory response is suppressed (57–61).
Beyond the apparent pro-inflammatory state in diabetes, the body’s attempt to recover from this persistent damage may also be impaired. Infiltrating macrophages exhibit defective efferocytosis of apoptotic neutrophils at sites of vascular inflammatory damage (62). Thus, defective resolution of inflammation may represent another mechanism for cardiac injury induced by diabetes, secondary to macrophage infiltration, as is evident in other tissues (63). Approaches including targeting the chemokine receptor CCR2 or enhancing heme oxygenase-1 (downstream of Nrf2) favor macrophage polarization towards an M2-like state, mitigating oxidative stress, remodeling, and dysfunction induced by diabetes (64–66).
A significant population of resident macrophages exists within the heart, which is thought to be related to cardiac electrophysiology and myocardial mitochondrial homeostasis in mouse models (47, 67). When myocardial injury occurs, monocytes are recruited to replace these resident macrophages, performing functions such as phagocytosing dead cells, regulating the inflammatory response, and stimulating the growth of fibroblasts and vascular cells (68). There is ample evidence that macrophages can activate fibroblasts in post-infarction and pressure-overloaded hearts, and in a hyperglycemic environment, induce the expression of adhesion molecules and CC chemokines, which are factors that promote inflammatory responses (69). Therefore, it is reasonable to assume that the recruitment and activation of macrophages may be involved in the process of cardiac fibrosis in diabetes. This hypothesis is supported by several pieces of evidence: infiltration of monocytes and macrophages in the hearts of models of both type 1 and type 2 diabetes (70, 71); cytokines and chemokines produced by macrophages under high glucose conditions can activate fibroblasts, promoting the secretion of fibrotic mediators and proteases (72); exosomes released by activated macrophages are considered to stimulate fibroblasts in diabetic hearts (73); genetic 3 deletion or pharmacological inhibition of the chemokine receptor CCR2 slows cardiac fibrosis in STZ-induced diabetes models (74).
The relationship between macrophage epigenetic regulation and diabetic cardiomyopathy (DCM) is complex, engaging a variety of mechanisms. Chronic inflammation and macrophage infiltration play critical roles in the etiology of cardiovascular disorders, including DCM (75). Key epigenetic modifications—DNA methylation, histone adjustments, and RNA alterations—are crucial for macrophage polarization and function. In DCM, inflammation and fibrosis are central to disease progression, with epigenetic mechanisms in macrophages influencing their activation, inflammatory cytokine production, and tissue repair capabilities, thus intensifying myocardial damage and fibrosis (76). Additionally, microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) significantly impact cardiac remodeling in DCM, affecting essential processes like fibrosis and hypertrophy.
lncRNA encompasses all non-coding RNAs longer than 200 nucleotides, roughly classified into linear lncRNAs and circular RNAs (circRNAs), the latter produced through back-splicing of precursor coding and lncRNA transcripts (77).
DRAIR, or Diabetes Regulated anti-inflammatory RNA, is a long non-coding RNA that plays a significant anti-inflammatory role in inflammation regulation related to diabetes (78), often downregulated in monocytes of diabetic patients (78). Silencing DRAIR inhibits the expression of anti-inflammatory genes in THP1 monocytes and enhances the adhesion of monocytes to ECs and the expression of pro-inflammatory genes such as IL-1β (78). Conversely, overexpression of DRAIR in THP-1 cells increases differentiation of monocytes/macrophages and the expression of anti-inflammatory target genes, while inhibiting pro-inflammatory genes like TNF-α and Fcγ receptor IIIb, suggesting DRAIR regulates the inflammatory phenotype of human monocytes/macrophages through epigenetic mechanisms (78).
Dnm3os, or dynamin 3 opposite strand, is another lncRNA associated with inflammation responses that promote diabetes and accelerate atherosclerosis. Dnm3os is significantly upregulated in CD14+ monocytes of diabetic patients and observed in various diabetic and atherosclerosis-accelerated mouse models (78, 79). Overexpression of Dnm3os increases the expression of pro-inflammatory genes and phagocytic activity in macrophages, while its silencing produces the opposite effect. Dnm3os interacts with nucleolar proteins, which have protective roles in macrophages. In diabetic conditions, the expression levels of nucleolar proteins decrease, leading to an increase in Dnm3os and promoting the expression of inflammatory genes, indicating Dnm3os plays a role in diabetes and accelerated atherosclerosis (79).
miRNAs are non-coding small RNA molecules, about 22 nucleotides in length, playing significant roles in regulating gene expression in eukaryotes by binding to complementary sequences in the 3’UTRs of target mRNAs, causing mRNA degradation or translational repression, thus regulating protein production (80) (81). This regulatory mechanism affects various biological processes including cell differentiation, proliferation, apoptosis, and metabolism, essential for maintaining cellular homeostasis (82). Particularly in the context of diabetic cardiomyopathy, miRNAs such as miR-471 and miR-155 are involved in regulating cell death pathways in macrophages, including apoptosis, necroptosis, and pyroptosis, which are crucial for modulating inflammatory responses and tissue remodeling in the diseased heart.
miR-471 is associated with cardiac hypertrophy, identified as a critical negative regulator in the process of mitigating pathological cardiac hypertrophy through preconditioning (83). In bone marrow-derived macrophages (BMDM) of db/db mice and RAW264.7 cells treated with advanced glycation end-products (AGE), miR-471-3p is significantly upregulated, and inhibition of miR-471-3p reduces inflammatory polarization of macrophages. Beyond inflammation, miR-471-3p also influences cell death pathways in macrophages, such as apoptosis and necroptosis, which could have implications for the progression and resolution of cardiac injury in diabetic cardiomyopathy. Bioinformatics analysis identified SIRT1 as a target of miR-471-3p, confirmed by dual-luciferase reporter assay and Western blot, indicating miR-471-3p negatively regulates SIRT1 expression. The SIRT1 agonist resveratrol can downregulate the increased ratio of M1 macrophages induced by AG (83).
miR-155 is involved in NLRP3 activation in macrophages by regulating the MEK/ERK/NFκB pathway induced by oxidized low-density lipoprotein (ox-LDL). Its role extends to influencing cell death processes, specifically pyroptosis, in macrophages. Overexpression of miR-155 in apoE-deficient mice via lentivirus accelerates atherosclerosis, indicating that miR-155 may promote inflammatory cell death and contribute to the exacerbation of cardiac damage in diabetic cardiomyopathy (84).
The role of miRNAs in macrophage polarization is complex and multifaceted. MiR-21, for instance, exhibits dual roles in macrophage polarization; some studies suggest it promotes M2 polarization, while others indicate a preference for M1 polarization (85). This discrepancy underscores the context-dependent nature of miRNA-mediated macrophage polarization. Other miRNAs like miR-9, miR-127, and miR-155 are noted for their roles in promoting M1 polarization, affecting various inflammatory and apoptotic pathways. Conversely, miRNAs such as miR-124, miR-223, and let-7c have been found to favor M2 polarization, highlighting their potential in modulating anti-inflammatory responses (85).
miRNAs regulate macrophage phenotypes in various disease processes. For example, miR155 directly blocks IL-13-induced M2 macrophage phenotype by inhibiting IL-13Rα1 expression (86). Imbalance of M1/M2 exacerbates cardiomyopathy in orchiectomized diabetic mice. Intravenous injection of miR155-AuNP, where thiol-modified antago-miR155 covalently binds to gold nanoparticles (AuNP), preferentially delivers nucleic acids to macrophages through phagocytosis. Delivery of antago-miR155 in vivo reduces apoptosis and restores cardiac function by increasing the M2 ratio and reducing inflammation (87).
In diabetic rat hearts, levels of substance P (SP), a sensory neuropeptide, are decreased, resulting in the loss of cardioprotective effects post-ischemic treatment. SP promotes the transition to a reparative M2 macrophage phenotype, which in mice is characterized by markers such as arginase 1 and IL-10. It is important to note that arginase 1 serves as a marker for the M2a phenotype predominantly in murine models, and its expression and functional role may differ in human macrophages (29, 30). Leprdb/db mice show increased LV M1 phenotype macrophages and an elevated M1/M2 ratio. Substitution of SP in Leprdb/db mice restores a favorable M1 to M2 balance, suggesting SP deficiency makes diabetic hearts prone to fibrosis. The anti-fibrotic effects of SP substitution involve direct effects on cardiac fibroblasts and macrophages to counter adverse phenotypic changes (88).
Bone morphogenetic protein (BMP)-7, a novel mediator of monocyte polarization, activates infiltrating monocytes into anti-inflammatory M2 macrophages, inhibiting apoptosis and fibrosis and improving cardiac function. Additionally, considering the nonresolving nature of inflammation in Type I diabetes, efferocytic macrophages could play a crucial role in mitigating the persistent inflammation associated with DCM. Efferocytic macrophages, adept at clearing apoptotic cells, might help to promote the resolution of inflammation, thus offering a potential therapeutic strategy for DCM. Employing pro-resolving factors, as highlighted by Nathan C et al. and Saas P et al., could further facilitate this process by enhancing the efferocytosis and restoring homeostasis (89–92).
Triptolide, derived from the root of the Tripterygium wilfordii plant, harbors triptolide as its most pivotal bioactive constituent. This herb, entrenched in traditional Chinese medicine, has been employed for over three decades in the management of glomerulonephritis and immune-mediated disorders, such as complex nephritis and systemic lupus erythematosus, attributed to its immunosuppressive, antiproliferative, and anti-inflammatory properties (93). Triptolide effectively protects against DCM, preventing immune dysregulation, myocardial inflammation, and cardiac fibrosis in diabetic rats, thereby alleviating left ventricular dysfunction. Triptolide’s protective effects on cardiac tissue may be attributed to inhibiting the TLR4-induced NF-κB/IL-1β immune pathway, the NF-κB/TNF-α/VCAM-1-mediated inflammatory pathway, and downregulating TGF-β1/α-SMA/Vimentin involved in cardiac fibrosis, indicating potential beneficial effects of triptolide on DCM (93).
Ginsenoside Rg1, one of the paramount components found in Panax ginseng (94), is acclaimed for its cardiovascular protective effects, primarily through the inhibition of cellular apoptosis and mitigation of oxidative stress (95). Research focusing on the cardioprotective mechanisms of ginsenoside Rg1 predominantly targets conditions such as hypertrophic cardiomyopathy, acute myocardial infarction, and myocardial ischemia/reperfusion injury (96–98). Ginsenoside RG1 improves DCM in diabetic rats by inhibiting endoplasmic reticulum stress-induced apoptosis and alleviating oxidative stress and myocardial cell apoptosis induced by streptozotocin in diabetic rats. Ginsenoside RG1 has been shown to be an effective regulator of MSCs. RG1-induced MSC-secreted exosomal circNOTCH1 alleviates DCM by activating NOTCH signaling, promoting macrophage M2 polarization pathways (99).
Curcumin, the principal bioactive constituent of turmeric, derived from the rhizomes of the Curcuma longa plant (100), exhibits multifaceted pharmacological activities, including antioxidative, free radical scavenging, anti-inflammatory, antineoplastic, and antimicrobial properties (101). Previous investigations have elucidated that curcumin treatment ameliorates diabetic cardiomyopathy (DCM) through attenuation of reactive oxygen species (ROS), apoptosis, and inflammation, thereby significantly enhancing cardiac function and mitigating fibrosis (101). JM-2, a novel curcumin analog, has been demonstrated to effectively prevent cardiac functional and structural aberrations, reducing cardiac inflammation and fibrosis. The therapeutic administration of JM-2 mitigates DCM by inhibiting the activation of NF-κB in cardiac tissue. Moreover, JM-2 treatment completely abrogated the elevation of pro-inflammatory cytokines and macrophage infiltration in models of type 1 and type 2 diabetes mellitus. RNA sequencing analyses have revealed that the anti-inflammatory activity of JM-2 correlates with the suppression of NF-κB activation. In vitro, JM-2 attenuates high glucose-induced cardiomyocyte hypertrophy and fibrosis, concurrently inhibiting the activation of NF-κB (58).
Puerarin, also known as dihydroxyisoflavone, isolated from the roots of the traditional Chinese medicinal herb Pueraria lobata, possesses a spectrum of beneficial activities against cardiovascular disorders, including myocardial infarction (102). Puerarin has been reported to prevent diabetic cardiomyopathy both In vitro and in vivo through the inhibition of inflammatory pathways (103). Studies have shown that puerarin mitigates cardiomyocyte and macrophage necroptosis via the inhibition of the NLRP3-Caspase-1-GSDMD pathway mediated by the P2X7 receptor (104). In vitro, puerarin treatment significantly reduces D-glucose-induced cell death in H9C2 cardiomyocytes and exerts anti-inflammatory effects on RAW264.7 macrophages. The P2X7 receptor-mediated necroptosis is posited as a pivotal mechanism in the therapeutic action of puerarin against diabetic cardiomyopathy (104).
Syringaresinol (SYR), a phenolic compound prevalent in various cereals and medicinal plants, is noted for its anti-inflammatory and antioxidative pharmacological properties (105). Oral administration of SYR bi-daily for eight weeks in STZ-induced type 1 diabetic mice significantly ameliorated cardiac dysfunction and prevented cardiac hypertrophy and fibrosis. SYR also inhibited macrophage infiltration and oxidative stress biomarkers without impacting hyperglycemia and body weight. In neonatal cardiomyocytes, SYR effectively reduced high glucose-induced apoptosis and fibrosis, and incubation with SYR alleviated inflammatory response and oxidative stress. Mechanistically, SYR may exert protective effects by restoring suppression of antioxidant kelch-like ECH-associated protein 1 (Keap1)/nuclear factor-E2-related factor 2 (Nrf2) system and abnormal activation of transforming growth factor-β (TGF-β)/mothers against decapentaplegic homolog (Smad) signaling pathway In vitro and in vivo (106).
Aza-resveratrol-chalcone compounds, considered precursors to resveratrol and chalcone, are ubiquitously present in edible plants and have been reported to exhibit diverse pharmacological effects on vascular diseases, cancer, viral infections, and inflammation In vitro and in vivo (107, 108). A synthesized derivative, 6b, effectively inhibited LPS-induced macrophage inflammatory response. Inflammation and oxidative stress are implicated in the pathogenesis of DCM, and studies have demonstrated that 6b prevents inflammation, oxidative stress, hypertrophy, fibrosis, and apoptosis in DCM through blocking NF-κB nuclear translocation and activating the Nrf2 pathway both In vitro and in vivo (109).
Isoliquiritigenin (4,2’,4’-trihydroxychalcone) (ISL), a widespread chalcone present in Glycyrrhiza uralensis Fisch., Dianthus chinensis L., and Astragalus membranaceus Moench, is renowned for its anti-inflammatory, antioxidant, and antitumor properties (110, 111). It was generally recognized than ISL possess pharmacological properties, such as anti-inflammatory, antioxidant and antitumor activities (112). Research has indicated that ISL mitigates diabetic cardiomyopathy by suppressing high glucose-induced inflammatory responses and oxidative stress (113). ISL also possesses anti-inflammatory effects on LPS-stimulated macrophages and inhibits macrophage infiltration in cardiac tissue (113, Figure 3).
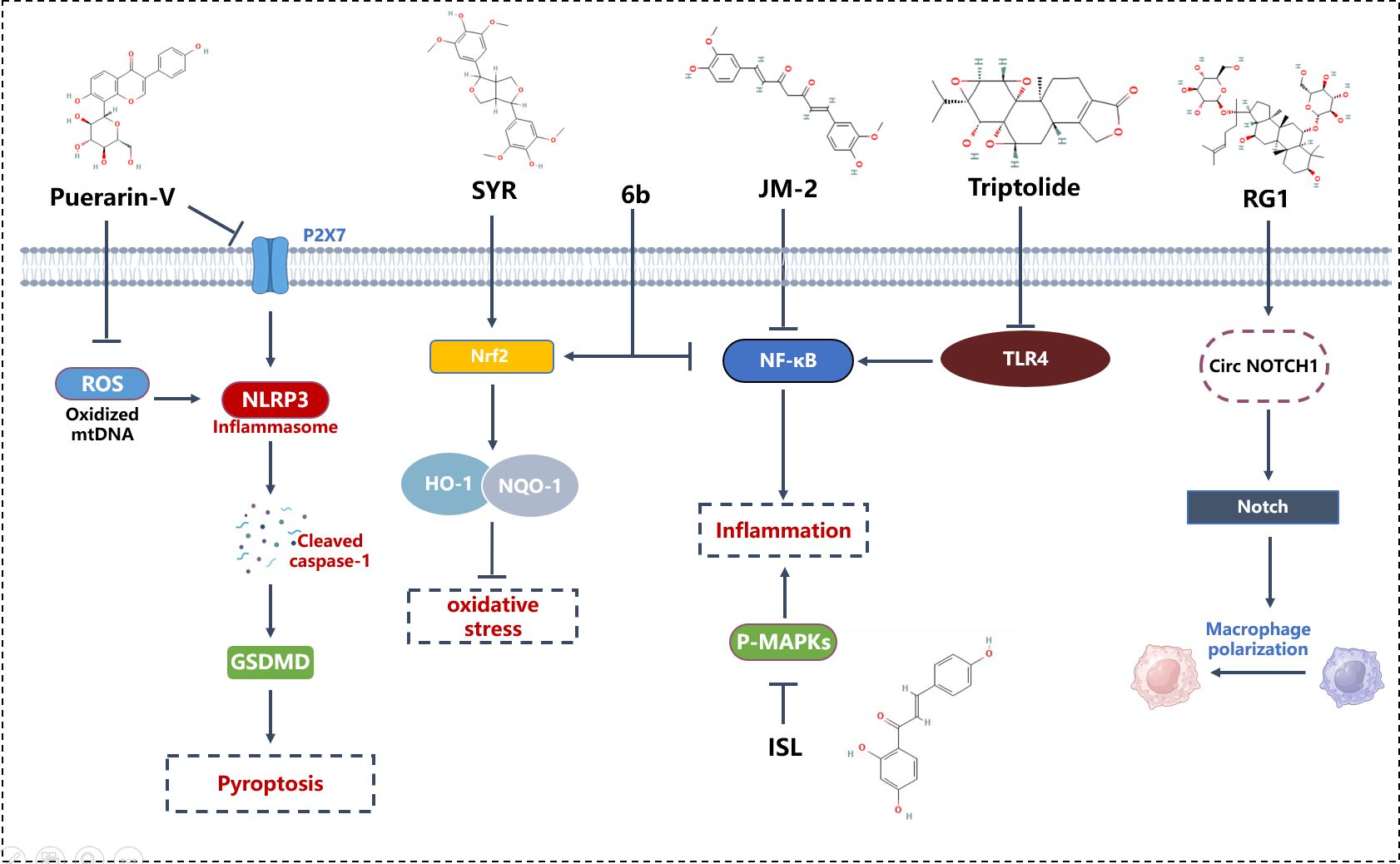
Figure 3 Mechanism of phytomedicine in improving DCM through macrophage modulation. 6b, Aza resveratrol–chalcone compounds; GSDMD, Gasdermin D; HO-1, Heme Oxygenase-1; ISL, Isoliquiritigenin; JM-2, Curcumin analog; NF-κB, Nuclear Factor kappa-light-chain-enhancer of activated B cells; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; NQO-1, NAD(P)H Quinone Dehydrogenase 1; Nrf2, Nuclear Factor Erythroid 2–Related Factor 2; P-MAPKs, Phosphorylated Mitogen-Activated Protein Kinases; RG1, Ginsenoside RG1; ROS, Reactive Oxygen Species; SYR, Syringaresinol; TLR4, Toll-Like Receptor 4.
Sodium-glucose cotransporter-2 (SGLT2) is primarily expressed in the S1 and S2 segments of the renal proximal tubules, the region responsible for reabsorbing 90% of the filtered glucose (114). Recently, SGLT-2 inhibitors have been developed as novel therapeutic drugs for T2DM patients. Clinical trials suggest that SGLT2 inhibitors may offer benefits beyond diabetes treatment, including reduced risk of hospitalization for heart failure or cardiovascular death. These benefits are now available to individuals with and without T2DM (115). In a randomized controlled trial, dapagliflozin reduced heart failure by 27% in T2DM (type 2 diabetes mellitus) patients with cardiovascular disease. The effectiveness was 35% for empagliflozin and 33% for canagliflozin (116). SGLT2 inhibitors might benefit the cardiovascular system by reducing adipose tissue-mediated inflammation and pro-inflammatory cytokine production, promoting ketone bodies as metabolic substrates for the heart and kidneys, decreasing oxidative stress, lowering serum uric acid levels, and reducing glomerular hyperfiltration and albuminuria (117).
A prospective, randomized, open-label, active-controlled, two-arm parallel intervention study in T2DM patients with high cardiovascular risk treated with SGLT2 inhibitors or sulfonylureas for 30 days analyzed the activation of NLRP3 inflammasomes in macrophages. Although the glucose-lowering ability of SGLT2 inhibitors was similar to sulfonylureas, they showed greater reduction in IL-1β secretion, accompanied by increased serum β-hydroxybutyrate (BHB) and decreased serum insulin. Ex-vivo experiments with macrophages confirmed the inhibitory effect of high BHB and low insulin levels on NLRP3 inflammasome activation (118).
Studies show that Empagliflozin reduces the accumulation of M1 polarized macrophages while inducing an anti-inflammatory M2 phenotype in adipose and liver macrophages, alleviating obesity-induced inflammation and insulin resistance. Empagliflozin increases energy expenditure, thermogenesis, and expression of uncoupling protein 1 in brown adipose tissue, inguinal and epididymal white adipose tissue (WAT). Furthermore, Empagliflozin reduces the accumulation of M1 polarized macrophages while inducing an anti-inflammatory M2 phenotype in WAT and liver, lowering plasma TNFα levels and alleviating obesity-related chronic inflammation (119).
In a 5/6 nephrectomy rat model, the beneficial effects of Empagliflozin on renal function and morphology are partly attributed to inhibition of CD206+ CD68+ M2 macrophage polarization by targeting the mTOR and mitochondrial autophagy pathways and attenuating inflammation signals from CD8+ effector T cells (120).
Research indicates that Canagliflozin can also inhibit atherosclerosis (AS). In macrophages, Canagliflozin enhances autophagy, promoting cholesterol efflux and reducing intracellular lipid droplet concentration. More importantly, Canagliflozin increases macrophage autophagy in atherosclerotic plaques of ApoE-/- mice, suggesting it could help inhibit the development of AS. Overall, these actions of Canagliflozin reduce the formation of atherosclerotic lesions, providing new insights into the potential mechanisms by which SGLT2 inhibitors slow the progression of this disease (121).
Liraglutide (LRG), a glucagon-like peptide 1 analog (GLP1A), can reduce weight in T2DM. In a randomized, placebo-controlled trial, patients with type 2 diabetes mellitus, devoid of prior cardiovascular disease, demonstrated a reduction in early diastolic left ventricular filling and left ventricular filling pressure upon treatment with liraglutide, as opposed to the addition of a placebo to the standard therapy. This effect ameliorated left ventricular load. Concurrently, left ventricular systolic function diminished yet remained within normal limits (122).
Lixisenatide has been reported to prevent acute cardiovascular events in Type 2 Diabetes (123). Treatment with Lixisenatide in Apoe-/- Irs2+/- mice (a mouse model of insulin resistance, metabolic syndrome, and AS resulted in more stable atherosclerotic plaques with less inflammatory infiltration, reduced necrotic core, and a thicker fibrous cap. Lixisenatide reduced the secretion of the pro-inflammatory cytokine IL-6 while enhancing the activation of Signal Transducer and Activator of Transcription (STAT)3, a determinant for M2 macrophage differentiation. The activation of STAT1, necessary for M1 phenotype, was also diminished. The study suggests that Lixisenatide reduces the size and instability of atherosclerotic plaques in Apoe-/- Irs2+/- mice by reprogramming macrophages to the M2 phenotype, thereby reducing inflammation (124).
Exendin-4 has restorative effects on tubular damage in a STZ-induced diabetic model. Exendin-4 improved proteinuria, glomerular hyperfiltration, glomerular hypertrophy, and mesangial matrix expansion in diabetic rats without altering blood pressure or body weight. Exendin-4 also prevented macrophage infiltration, reduced the protein levels of ICAM-1, and type IV collagen, and decreased oxidative stress and nuclear factor-κB activation in renal tissue (125). Exendin-4 acts directly on GLP-1 receptors and attenuates the release of pro-inflammatory cytokines in macrophages and the production of ICAM-1 on glomerular ECs (126).
Additionally, Exendin-4 has acute cardioprotective effects and benefits experimental/clinical heart failure. Exendin-4 prevents post-MI remodeling through preferential action on inflammation and the extracellular matrix (127). Exendin-4 and insulin improved metabolic parameters in diabetic mice after 12 weeks, but only Exendin-4 reduced diastolic dysfunction and interstitial fibrosis while altering ECM gene expression. Exendin-4 prevents ECM remodeling and diastolic dysfunction in experimental diabetes by preferentially preventing paracrine communication between infiltrating macrophages and resident fibroblasts, suggesting that cell-specific targeting of GLP-1 signaling might be a viable treatment strategy in this context (128).
Dipeptidyl peptidase-4 (DPP-4) inhibitors, employed primarily to manage type 2 diabetes, function by inhibiting the degradation of glucagon-like peptide-1 (GLP-1), thus facilitating glycemic control. Beyond their role in glucose regulation, DPP-4 inhibitors have been substantiated to exert cardioprotective effects in ischemic heart models (129, 130). Alogliptin inhibits monocyte activation/chemotaxis, playing an anti-atherosclerotic role and reducing inflammation. In a male LDLR-/- mouse model fed a high-fat diet, Alogliptin reduced visceral adipose tissue macrophage content (adipose tissue macrophages; CD11b+, CD11c+, Ly6Chi) while upregulating CD163. DPP-4 is highly expressed in bone marrow-derived CD11b+ cells, and Alogliptin downregulates pro-inflammatory genes in these cells. DPP-4i reduced aortic plaques and significantly decreased plaque macrophages. In vitro, Alogliptin blocked monocyte migration and actin polymerization through a Rac-dependent mechanism and prevented the migration of labeled monocytes responsive to exogenous TNF-α and DPP-4 to the aorta (131).
Thiazolidinediones (TZDs) represent a class of drugs that directly target insulin resistance through activation of PPARγ agonists. These agents are efficacious antihyperglycemic medications with numerous potential cardiovascular advantages (132, 133). The prospective pioglitazone clinical trial in major vascular events, known as the PROactive study, indicated that TZDs, specifically pioglitazone, reduced the risk of the primary endpoint—a composite of disease and surgery-related outcomes—by 10% (134). TZDs are ligands for Peroxisome Proliferator-Activated Receptor-γ (PPAR-γ) and possess anti-inflammatory effects independent of their insulin-sensitizing actions. Pioglitazone (PIO) treatment did not change blood glucose and HbA1c. PIO reduced urinary albumin excretion and glomerular hypertrophy, inhibiting expression of transforming growth factor (TGF)-β, type IV collagen, ICAM-1, and macrophage infiltration in diabetic rat kidneys. PPAR-γ is expressed in glomerular ECs of diabetic kidneys and cultured glomerular ECs. HG conditions increased ICAM-1 expression and NF-κB activation in cultured glomerular ECs. PIO, ciglitazone, and pyrrolidine dithiocarbamate (a NF-κB inhibitor) reduced these changes. PIO’s preventive effects are likely mediated through its anti-inflammatory actions, including inhibition of NF-κB activation, ICAM-1 expression, and macrophage infiltration in diabetic kidneys (135). Additionally, PIO reduces inflammation in high-fat diet diabetic mice by inhibiting AGE-induced classical macrophage activation (136).
Rositiglitazone (ROSI), as a modulator of macrophage polarization, can mitigate renal calcium oxalate (CaOx) deposition by alleviating oxidative stress and inflammatory responses. CaOx crystals upregulate NADPH oxidase p47phox, leading to excessive production of ROS in RTECs. The pro-inflammatory action of macrophages further intensifies this effect (137). ROSI significantly reduces renal crystal deposition, tubular damage, crystal adherence, cellular apoptosis, oxidative stress, and inflammatory responses. In vitro, ROSI markedly inhibits renal injury, cellular apoptosis, and crystal adhesion in HK-2 cells, and significantly transforms COM-stimulated M1 macrophages into M2 macrophages, exhibiting anti-inflammatory effects. Moreover, ROSI significantly suppresses oxidative stress by promoting the Nrf2/HO-1 pathway in HK-2 cells. These findings suggest that ROSI can ameliorate tubular damage caused by oxidative stress and inflammatory responses by inhibiting M1 macrophage polarization and promoting M2 macrophage polarization (138).
Metformin is among the most frequently employed antidiabetic agents in clinical practice (139). Research has substantiated its efficacy in ameliorating cardiac fibrosis and oxidative stress, as well as in augmenting the levels of mitochondrial antioxidant enzymes (140). Furthermore, metformin has been reported to enhance glycemic control and mitigate the effects of diabetic (140). The therapeutic potential of Metformin in treating obese/diabetic patients is linked to its capacity to counteract insulin resistance. LPS activate M1 macrophages, promoting paracrine activation of HIF-1α in brown adipocytes, subsequently diminishing insulin signaling and glucose uptake, as well as β-adrenergic sensitivity. The addition of Metformin to M1 polarized macrophages attenuates these indications of brown adipocyte functional impairment. At the molecular level, Metformin induces the degradation of HIF1α by inhibiting mitochondrial complex I activity, thereby curtailing the HIF1α-mediated inflammatory program in macrophages and reducing oxygen consumption in a ROS-independent manner. In summary, Metformin’s inhibition of HIF1α-dependent pro-inflammatory programs in macrophages may indirectly elicit beneficial effects on insulin-mediated glucose uptake and β-adrenergic responses in brown adipocytes (141).
This article contributes to the evolving understanding of the complex role of macrophages in diabetic cardiomyopathy (DCM), indicating their potential as therapeutic targets. It has highlighted intricate mechanisms through which macrophages influence DCM, particularly focusing on epigenetic modulation and their interactions within the cardiac microenvironment. Therapeutically, modulating macrophage activity appears to hold promise in addressing DCM. Strategies such as epigenetic adjustments, the targeted manipulation of macrophage functions, and the exploration of natural substances with anti-inflammatory effects are being considered. These methods aim to subtly shift macrophage behavior towards a more reparative phenotype, potentially easing myocardial impairment in DCM.
Moreover, the review suggests that a deeper insight into macrophage dynamics in DCM could lead to more balanced therapeutic strategies. These would ideally leverage the protective roles of macrophages while minimizing their harmful aspects. Incorporating macrophage-focused treatments into the broader management strategy for DCM could offer a valuable pathway for research and might improve clinical outcomes. In closing, while targeting macrophages in DCM shows potential, it is presented here as a promising but exploratory approach. The common mechanisms identified, including the regulation of inflammation, epigenetic changes, and improved cell signaling, warrant further investigation. Continuing research in this direction may provide a foundation for developing nuanced and effective macrophage-targeted therapies, aiming to alleviate the impact of diabetic cardiomyopathy.
SZ: Writing – original draft, Writing – review & editing. XZ: Writing – review & editing. YC: Conceptualization, Writing – review & editing. ZW: Formal analysis, Resources, Writing – review & editing. PS: Conceptualization, Writing – review & editing. QN: Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82174354); Construction Special funding project of the Centers for Clinical Medicine Research Center of Guang’anmen Hospital, China Academy of Chinese Medical Sciences (2022LYJSZX02); the China Postdoctoral Science Foundation (2023M733912); Natural Science Foundation of Beijing, China (7244496); Clinical research business expenses of the Central High-level Hospital of Traditional Chinese Medicine (HLCHPP2023084); Natural Science Foundation of Shandong, China (ZR202111230184, ZR2022LZY010).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Arora A, Behl T, Sehgal A, Singh S, Sharma N, Bhatia S, et al. Unravelling the involvement of gut microbiota in type 2 diabetes mellitus. Life Sci. (2021) 273:119311. doi: 10.1016/j.lfs.2021.119311
2. Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res Clin Pract. (2019) 157:107843. doi: 10.1016/j.diabres.2019.107843
3. Li W, Lou X, Zha Y, Qin Y, Zha J, Hong L, et al. Single-cell RNA-seq of heart reveals intercellular communication drivers of myocardial fibrosis in diabetic cardiomyopathy. eLife. (2023) 12:e80479. doi: 10.7554/eLife.80479
4. Januzzi JL, Del Prato S, Rosenstock J, Butler J, Ezekowitz J, Ibrahim NE, et al. Characterizing diabetic cardiomyopathy: baseline results from the ARISE-HF trial. Cardiovasc Diabetol. (2024) 23(1):49. doi: 10.1186/s12933-024-02135-z
5. Negre-Salvayre A, Salvayre R, Augé N, Pamplona R, Portero-Otín M. Hyperglycemia and glycation in diabetic complications. Antioxid Redox Signaling. (2009) 11(12):3071–109. doi: 10.1089/ars.2009.2484
6. Wolf D, Ley K. Immunity and Inflammation in Atherosclerosis. Circ Res. (2019) 124(2):315–27. doi: 10.1161/circresaha.118.313591
7. Spinetti G, Sangalli E, Tagliabue E, Maselli D, Colpani O, Ferland-McCollough D, et al. MicroRNA-21/PDCD4 proapoptotic signaling from circulating CD34(+) cells to vascular endothelial cells: a potential contributor to adverse cardiovascular outcomes in patients with critical limb ischemia. Diabetes Care. (2020) 43(7):1520–9. doi: 10.2337/dc19-2227
8. Prame Kumar K, Nicholls AJ, Wong CHY. Partners in crime: neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. (2018) 371(3):551–65. doi: 10.1007/s00441-017-2753-2
9. van Furth R, Cohn ZA. The origin and kinetics of mononuclear phagocytes. J Exp Med. (1968) 128(3):415–35. doi: 10.1084/jem.128.3.415
10. van Furth R, Diesselhoff-den Dulk MM. Dual origin of mouse spleen macrophages. J Exp Med. (1984) 160(5):1273–83. doi: 10.1084/jem.160.5.1273
11. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. (2014) D159(6):1312–26. doi: 10.1016/j.cell.2014.11.018
12. Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med. (2018) 24(8):1234–45. doi: 10.1038/s41591-018-0059-x
13. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38(1):79–91. doi: 10.1016/j.immuni.2012.12.001
14. McGrath KE, Frame JM, Palis J. Early hematopoiesis and macrophage development. Semin Immunol. (2015) 27(6):379–87. doi: 10.1016/j.smim.2016.03.013
15. Cohen HB, Mosser DM. Cardiac macrophages: how to mend a broken heart. Immunity. (2014) 40(1):3–5. doi: 10.1016/j.immuni.2013.12.005
16. Perdiguero EG, Geissmann F. The development and maintenance of resident macrophages. Nat Immunol. (2016) 17(1):2–8. doi: 10.1038/ni.3341
17. Leid J, Carrelha J, Boukarabila H, Epelman S, Jacobsen SE, Lavine KJ. Primitive Embryonic Macrophages are Required for Coronary Development and Maturation. Circ Res. (2016) 118(10):1498–511. doi: 10.1161/circresaha.115.308270
18. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol (Baltimore Md: 1950).. (2000) 164(12):6166–73. doi: 10.4049/jimmunol.164.12.6166
19. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. (2014) 40(2):274–88. doi: 10.1016/j.immuni.2014.01.006
20. Costantini A, Viola N, Berretta A, Galeazzi R, Matacchione G, Sabbatinelli J, et al. Age-related M1/M2 phenotype changes in circulating monocytes from healthy/unhealthy individuals. Aging. (2018) 10(6):1268–80. doi: 10.18632/aging.101465
21. Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, et al. Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab. (2012) 15(4):518–33. doi: 10.1016/j.cmet.2012.01.023
22. Ying W, Lee YS, Dong Y, Seidman JS, Yang M, Isaac R, et al. Expansion of islet-resident macrophages leads to inflammation affecting β cell proliferation and function in obesity. Cell Metab. (2019) 29(2):457–474.e5. doi: 10.1016/j.cmet.2018.12.003
23. Brissova M, Shostak A, Shiota M, Wiebe PO, Poffenberger G, Kantz J, et al. Pancreatic islet production of vascular endothelial growth factor–a is essential for islet vascularization, revascularization, and function. Diabetes. (2006) 55(11):2974–85. doi: 10.2337/db06-0690
24. Tessem JS, Jensen JN, Pelli H, Dai XM, Zong XH, Stanley ER, et al. Critical roles for macrophages in islet angiogenesis and maintenance during pancreatic degeneration. Diabetes. (2008) 57(6):1605–17. doi: 10.2337/db07-1577
25. Criscimanna A, Coudriet GM, Gittes GK, Piganelli JD, Esni F. Activated macrophages create lineage-specific microenvironments for pancreatic acinar- and β-cell regeneration in mice. Gastroenterology. (2014) 147(5):1106–1118.e11. doi: 10.1053/j.gastro.2014.08.008
26. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. (2011) 11(11):723–37. doi: 10.1038/nri3073
27. Zhou L, Wang D, Qiu X, Zhang W, Gong Z, Wang Y, et al. DHZCP modulates microglial M1/M2 polarization via the p38 and TLR4/NF-κB signaling pathways in LPS-stimulated microglial cells. Front Pharmacol. (2020) 11:1126. doi: 10.3389/fphar.2020.01126
28. Topf MC, Tuluc M, Harshyne LA, Luginbuhl A. Macrophage type 2 differentiation in a patient with laryngeal squamous cell carcinoma and metastatic prostate adenocarcinoma to the cervical lymph nodes. J Immunother Cancer.. (2017) 5(1):60. doi: 10.1186/s40425-017-0264-z
29. Raes G, Van den Bergh R, De Baetselier P, Ghassabeh GH, Scotton C, Locati M, et al. Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. J Immunol (Baltimore Md: 1950).. (2005) 174(11):6561. doi: 10.4049/jimmunol.174.11.6561
30. Saas P, Chagué C, Maraux M, Cherrier T. Toward the Characterization of Human Pro-Resolving Macrophages? Front Immunol. (2020) 11:593300. doi: 10.3389/fimmu.2020.593300
31. Fan W, Huang W, Chen J, Li N, Mao L, Hou S. Retinal microglia: Functions and diseases. Immunology. (2022) 166(3):268–86. doi: 10.1111/imm.13479
32. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23(11):549–55. doi: 10.1016/s1471-4906(02)02302-5
33. Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front Immunol. (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
34. Zhu C, Kros JM, Cheng C, Mustafa D. The contribution of tumor-associated macrophages in glioma neo-angiogenesis and implications for anti-angiogenic strategies. Neuro-oncology. (2017) 19(11):1435–46. doi: 10.1093/neuonc/nox081
35. Lim HY, Lim SY, Tan CK, Thiam CH, Goh CC, Carbajo D, et al. Hyaluronan Receptor LYVE-1-Expressing Macrophages Maintain Arterial Tone through Hyaluronan-Mediated Regulation of Smooth Muscle Cell Collagen. Immunity. (2018) 49(2):326–341.e7. doi: 10.1016/j.immuni.2018.06.008
36. Chen C, Wang J, Liu C, Hu J. Cardiac resident macrophages: key regulatory mediators in the aftermath of myocardial infarction. Front Immunol. (2023) 14:1207100. doi: 10.3389/fimmu.2023.1207100
37. Lin P, Ji HH, Li YJ, Guo SD. Macrophage Plasticity and Atherosclerosis Therapy. Front Mol Biosci. (2021) 8:679797. doi: 10.3389/fmolb.2021.679797
38. Stratmann B, Tschoepe D. The diabetic heart: sweet, fatty and stressed. Expert Rev Cardiovasc Ther. (2011) 9(9):1093–6. doi: 10.1586/erc.11.109
39. Bouchareychas L, Duong P, Phu TA, Alsop E, Meechoovet B, Reiman R, et al. High glucose macrophage exosomes enhance atherosclerosis by driving cellular proliferation & hematopoiesis. iScience. (2021) 24(8):102847. doi: 10.1016/j.isci.2021.102847
40. Ding X, Jing N, Shen A, Guo F, Song Y, Pan M, et al. MiR-21-5p in macrophage-derived extracellular vesicles affects podocyte pyroptosis in diabetic nephropathy by regulating A20. J Endocrinol Invest.. (2021) 44(6):1175–84. doi: 10.1007/s40618-020-01401-7
41. Kawakami R, Katsuki S, Travers R, Romero DC, Becker-Greene D, Passos LSA, et al. S100A9-RAGE axis accelerates formation of macrophage-mediated extracellular vesicle microcalcification in diabetes mellitus. Arteriosclerosis Thromb Vasc Biol. (2020) 40(8):1838–53. doi: 10.1161/atvbaha.118.314087
42. Zhu M, Sun X, Qi X, Xia L, Wu Y. Exosomes from high glucose-treated macrophages activate macrophages andinduce inflammatory responses via NF-κB signaling pathway in vitro and in vivo. Int Immunopharmacol. (2020) 84:106551. doi: 10.1016/j.intimp.2020.106551
43. Zhu QJ, Zhu M, Xu XX, Meng XM, Wu YG. Exosomes from high glucose-treated macrophages activate glomerular mesangial cells via TGF-β1/Smad3 pathway in vivo and in vitro. FASEB J. (2019) 33(8):9279–90. doi: 10.1096/fj.201802427RRR
44. Li K, Cui M, Zhang K, Wang G, Zhai S. M1 macrophages-derived extracellular vesicles elevate microRNA-185-3p to aggravate the development of atherosclerosis in ApoE(-/-) mice by inhibiting small mothers against decapentaplegic 7. Int Immunopharmacol.. (2021) 90:107138. doi: 10.1016/j.intimp.2020.107138
45. Wang L, Qi Y, Wang Y, Tang H, Li Z, Wang Y, et al. LncRNA MALAT1 suppression protects endothelium against oxLDL-Induced inflammation via inhibiting expression of MiR-181b target gene TOX. Oxid Med Cell Longevity.. (2019) 2019:8245810. doi: 10.1155/2019/8245810
46. Huang C, Han J, Wu Y, Li S, Wang Q, Lin W, et al. Exosomal MALAT1 derived from oxidized low-density lipoprotein-treated endothelial cells promotes M2 macrophage polarization. Mol Med Rep. (2018) 18(1):509–15. doi: 10.3892/mmr.2018.8982
47. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, et al. Macrophages facilitate electrical conduction in the heart. Cell. (2017) 169(3):510–522.e20. doi: 10.1016/j.cell.2017.03.050
48. Sugita J, Fujiu K, Nakayama Y, Matsubara T, Matsuda J, Oshima T, et al. Cardiac macrophages prevent sudden death during heart stress. Nat Commun. (2021) 12(1):1910. doi: 10.1038/s41467-021-22178-0
49. Simon-Chica A, Fernández MC, Wülfers EM, Lother A, Hilgendorf I, Seemann G, et al. Novel insights into the electrophysiology of murine cardiac macrophages: relevance of voltage-gated potassium channels. Cardiovasc Res. (2022) 118(3):798–813. doi: 10.1093/cvr/cvab126
50. Gallego M, Zayas-Arrabal J, Alquiza A, Apellaniz B, Casis O. Electrical features of the diabetic myocardium. arrhythmic and cardiovascular safety considerations in diabetes. Front Pharmacol. (2021) 12:687256. doi: 10.3389/fphar.2021.687256
51. Arnold SV, Echouffo-Tcheugui JB, Lam CSP, Inzucchi SE, Tang F, McGuire DK, et al. Patterns of glucose-lowering medication use in patients with type 2 diabetes and heart failure. Insights from the Diabetes Collaborative Registry (DCR). Am Heart J. (2018) 203:25–9. doi: 10.1016/j.ahj.2018.05.016
52. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. (2018) 122(4):624–38. doi: 10.1161/circresaha.117.311586
53. Dewanjee S, Vallamkondu J, Kalra RS, John A, Reddy PH, Kandimalla R. Autophagy in the diabetic heart: A potential pharmacotherapeutic target in diabetic cardiomyopathy. Ageing Res Rev. (2021) 68:101338. doi: 10.1016/j.arr.2021.101338
54. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. (2016) 12(3):144–53. doi: 10.1038/nrendo.2015.216
55. Uchimura K, Hayata M, Mizumoto T, Miyasato Y, Kakizoe Y, Morinaga J, et al. The serine protease prostasin regulates hepatic insulin sensitivity by modulating TLR4 signalling. Nat Commun. (2014) 5:3428. doi: 10.1038/ncomms4428
56. Jia G, Habibi J, DeMarco VG, Martinez-Lemus LA, Ma L, Whaley-Connell AT, et al. Endothelial Mineralocorticoid Receptor Deletion Prevents Diet-Induced Cardiac Diastolic Dysfunction in Females. Hypertension (Dallas Tex 1979).. (2015) 66(6):1159–67. doi: 10.1161/hypertensionaha.115.06015
57. Li Q, Li Y, Huang W, Wang X, Liu Z, Chen J, et al. Loss of lipocalin 10 exacerbates diabetes-induced cardiomyopathy via disruption of nr4a1-mediated anti-inflammatory response in macrophages. Front Immunol. (2022) 13:930397. doi: 10.3389/fimmu.2022.930397
58. Wang M, Jin L, Zhang Q, Zhu W, He H, Lou S, et al. Curcumin analog JM-2 alleviates diabetic cardiomyopathy inflammation and remodeling by inhibiting the NF-κB pathway. BioMed Pharmacother = BioMed pharmacother.. (2022) 154:113590. doi: 10.1016/j.biopha.2022.113590
59. Miao H, Li X, Zhou C, Liang Y, Li D, Ji Q. NR4A2 alleviates cardiomyocyte loss and myocardial injury in rats by transcriptionally suppressing CCR5 and inducing M2 polarization of macrophages. Microvas Res. (2022) 140:104279. doi: 10.1016/j.mvr.2021.104279
60. Baumeier C, Escher F, Aleshcheva G, Pietsch H, Schultheiss HP. Plasminogen activator inhibitor-1 reduces cardiac fibrosis and promotes M2 macrophage polarization in inflammatory cardiomyopathy. Basic Res Cardiol. (2021) 116(1):1. doi: 10.1007/s00395-020-00840-w
61. Li Y, Deng S, Wang X, Huang W, Chen J, Robbins N, et al. Sectm1a deficiency aggravates inflammation-triggered cardiac dysfunction through disruption of LXRα signalling in macrophages. Cardiovasc Res. (2021) 117(3):890–902. doi: 10.1093/cvr/cvaa067
62. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Sci (New York NY).. (2013) 339(6116):161–6. doi: 10.1126/science.1230719
63. de Gaetano M, McEvoy C, Andrews D, Cacace A, Hunter J, Brennan E, et al. Specialized Pro-resolving Lipid Mediators: Modulation of Diabetes-Associated Cardio-, Reno-, and Retino-Vascular Complications. Front Pharmacol. (2018) 9:1488. doi: 10.3389/fphar.2018.01488
64. Rossi M, Korpak K, Doerfler A, Zouaoui Boudjeltia K. Deciphering the role of heme oxygenase-1 (HO-1) expressing macrophages in renal ischemia-reperfusion injury. Biomedicines. (2021) 9(3):306. doi: 10.3390/biomedicines9030306
65. Araujo JA, Zhang M, Yin F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front Pharmacol. (2012) 3:119. doi: 10.3389/fphar.2012.00119
66. Ruytinx P, Proost P, Van Damme J, Struyf S. Chemokine-induced macrophage polarization in inflammatory conditions. Front Immunol. (2018) 9:1930. doi: 10.3389/fimmu.2018.01930
67. Nicolás-Ávila JA, Lechuga-Vieco AV, Esteban-Martínez L, Sánchez-Díaz M, Díaz-García E, Santiago DJ, et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell. (2020) 183(1):94–109.e23. doi: 10.1016/j.cell.2020.08.031
68. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. (2007) 204(12):3037–47. doi: 10.1084/jem.20070885
69. Chow FY, Nikolic-Paterson DJ, Ma FY, Ozols E, Rollins BJ, Tesch GH. Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia. (2007) 50(2):471–80. doi: 10.1007/s00125-006-0497-8
70. Fukuda M, Nakamura T, Kataoka K, Nako H, Tokutomi Y, Dong YF, et al. Potentiation by candesartan of protective effects of pioglitazone against type 2 diabetic cardiovascular and renal complications in obese mice. J hypertension.. (2010) 28(2):340–52. doi: 10.1097/HJH.0b013e32833366cd
71. De Blasio MJ, Huynh N, Deo M, Dubrana LE, Walsh J, Willis A, et al. Defining the progression of diabetic cardiomyopathy in a mouse model of type 1 diabetes. . Front Physiol. (2020) 11:124. doi: 10.3389/fphys.2020.00124
72. Morey M, O'Gaora P, Pandit A, Hélary C. Hyperglycemia acts in synergy with hypoxia to maintain the pro-inflammatory phenotype of macrophages. PloS One. (2019) 14(8):e0220577. doi: 10.1371/journal.pone.0220577
73. Govindappa PK, Patil M, Garikipati VNS, Verma SK, Saheera S, Narasimhan G, et al. Targeting exosome-associated human antigen R attenuates fibrosis and inflammation in diabetic heart. FASEB J. (2020) 34(2):2238–51. doi: 10.1096/fj.201901995R
74. Tan X, Hu L, Shu Z, Chen L, Li X, Du M, et al. Role of CCR2 in the development of streptozotocin-treated diabetic cardiomyopathy. Diabetes. (2019) 68(11):2063–73. doi: 10.2337/db18-1231
75. Komal S, Han SN, Cui LG, Zhai MM, Zhou YJ, Wang P, et al. Epigenetic regulation of macrophage polarization in cardiovascular diseases. Pharm (Basel Switzerland).. (2023) 16(2):141. doi: 10.3390/ph16020141
76. Mittal A, Garg R, Bahl A, Khullar M. Molecular mechanisms and epigenetic regulation in diabetic cardiomyopathy. Front Cardiovasc Med. (2021) 8:725532. doi: 10.3389/fcvm.2021.725532
77. Braun S, Domdey H, Wiebauer K. Inverse splicing of a discontinuous pre-mRNA intron generates a circular exon in a HeLa cell nuclear extract. Nucleic Acids Res. (1996) 24(21):4152–7. doi: 10.1093/nar/24.21.4152
78. Reddy MA, Amaram V, Das S, Tanwar VS, Ganguly R, Wang M, et al. lncRNA DRAIR is downregulated in diabetic monocytes and modulates the inflammatory phenotype via epigenetic mechanisms. JCI Insight. (2021) 6(11):e143289. doi: 10.1172/jci.insight.143289
79. Das S, Reddy MA, Senapati P, Stapleton K, Lanting L, Wang M, et al. Diabetes Mellitus-Induced Long Noncoding RNA Dnm3os Regulates Macrophage Functions and Inflammation via Nuclear Mechanisms. Arterioscler Thromb Vasc Biol... (2018) 38(8):1806–20. doi: 10.1161/atvbaha.117.310663
80. van Rooij E, Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discovery.. (2012) 11(11):860–72. doi: 10.1038/nrd3864
82. Ni WJ, Leng XM. Dynamic miRNA-mRNA paradigms: New faces of miRNAs. Biochem Biophys Rep. (2015) 4:337–41. doi: 10.1016/j.bbrep.2015.10.011
83. Liu G, Yan D, Yang L, Sun Y, Zhan L, Lu L, et al. The effect of miR-471-3p on macrophage polarization in the development of diabetic cardiomyopathy. Life Sci. (2021) 268:118989. doi: 10.1016/j.lfs.2020.118989
84. Yin R, Zhu X, Wang J, Yang S, Ma A, Xiao Q, et al. MicroRNA-155 promotes the ox-LDL-induced activation of NLRP3 inflammasomes via the ERK1/2 pathway in THP-1 macrophages and aggravates atherosclerosis in ApoE-/- mice. Ann palliative Med. (2019) 8(5):676–89. doi: 10.21037/apm.2019.10.11
85. Essandoh K, Li Y, Huo J, Fan GC. MiRNA-mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock (Augusta Ga).. (2016) 46(2):122–31. doi: 10.1097/shk.0000000000000604
86. Martinez-Nunez RT, Louafi F, Sanchez-Elsner T. The interleukin 13 (IL-13) pathway in human macrophages is modulated by microRNA-155 via direct targeting of interleukin 13 receptor alpha1 (IL13Ralpha1). J Biol Chem. (2011) 286(3):1786–94. doi: 10.1074/jbc.M110.169367
87. Jia C, Chen H, Wei M, Chen X, Zhang Y, Cao L, et al. Gold nanoparticle-based miR155 antagonist macrophage delivery restores the cardiac function in ovariectomized diabetic mouse model. Int J nanomed.. (2017) 12:4963–79. doi: 10.2147/ijn.S138400
88. Widiapradja A, Kasparian AO, McCaffrey SL, Kolb LL, Imig JD, Lacey JL, et al. Replacement of Lost Substance P Reduces Fibrosis in the Diabetic Heart by Preventing Adverse Fibroblast and Macrophage Phenotype Changes. Cells. (2021) 10(10):2659. doi: 10.3390/cells10102659
89. Nathan C, Ding A. Nonresolving inflammation. Cell. (2010) 140(6):871–82. doi: 10.1016/j.cell.2010.02.029
90. Urbina P, Singla DK. BMP-7 attenuates adverse cardiac remodeling mediated through M2 macrophages in prediabetic cardiomyopathy. Am J Physiol HeartCirculatory Physiol. (2014). doi: 10.1152/ajpheart.00367.2014
91. Saas P, Vetter M, Maraux M, Bonnefoy F, Perruche S. Resolution therapy: Harnessing efferocytic macrophages to trigger the resolution of inflammation. Front Immunol. (2022) 13:1021413. doi: 10.3389/fimmu.2022.1021413
92. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. (2014) 510(7503):92–101. doi: 10.1038/nature13479
93. Guo X, Xue M, Li CJ, Yang W, Wang SS, Ma ZJ, et al. Protective effects of triptolide on TLR4 mediated autoimmune and inflammatory response induced myocardial fibrosis in diabetic cardiomyopathy. J Ethnopharmacol. (2016) 193:333–44. doi: 10.1016/j.jep.2016.08.029
94. Nah JJ, Hahn JH, Chung S, Choi S, Kim YI, Nah SY. Effect of ginsenosides, active components of ginseng, on capsaicin-induced pain-related behavior. Neuropharmacology. (2000) 39(11):2180–4. doi: 10.1016/s0028-3908(00)00048-4
95. Lee CH, Kim JH. A review on the medicinal potentials of ginseng and ginsenosides on cardiovascular diseases. J ginseng Res. (2014) 38(3):161–6. doi: 10.1016/j.jgr.2014.03.001
96. Li X, Chen JX, Sun JJ. Protective effects of Panax notoginseng saponins on experimental myocardial injury induced by ischemia and reperfusion in rat. Zhongguo yao li xue bao.. (1990) 11(1):26–9.
97. Wang XD, Gu TX, Shi EY, Lu CM, Wang C. Effect and mechanism of panaxoside Rg1 on neovascularization in myocardial infarction rats. Chin J Integr Med. (2010) 16(2):162–6. doi: 10.1007/s11655-010-0162-4
98. Zhang YJ, Zhang XL, Li MH, Iqbal J, Bourantas CV, Li JJ, et al. The ginsenoside Rg1 prevents transverse aortic constriction-induced left ventricular hypertrophy and cardiac dysfunction by inhibiting fibrosis and enhancing angiogenesis. J Cardiovasc Pharmacol. (2013) J62(1):50–7. doi: 10.1097/FJC.0b013e31828f8d45
99. Zhen J, Bai J, Liu J, Men H, Yu H. Ginsenoside RG1-induced mesenchymal stem cells alleviate diabetic cardiomyopathy through secreting exosomal circNOTCH1 to promote macrophage M2 polarization. Phytother research: PTR.. (2023) 38(4):1745–60. doi: 10.1002/ptr.8018
100. Kunnumakkara AB, Bordoloi D, Padmavathi G, Monisha J, Roy NK, Prasad S, et al. Curcumin, the golden nutraceutical: multitargeting for multiple chronic diseases. Br J Pharmacol. (2017) 174(11):1325–48. doi: 10.1111/bph.13621
101. Yu W, Wu J, Cai F, Xiang J, Zha W, Fan D, et al. Curcumin alleviates diabetic cardiomyopathy in experimental diabetic rats. PloS One. (2012) 7(12):e52013. doi: 10.1371/journal.pone.0052013
102. Cheng W, Wu P, Du Y, Wang Y, Zhou N, Ge Y, et al. Puerarin improves cardiac function through regulation of energy metabolism in Streptozotocin-Nicotinamide induced diabetic mice after myocardial infarction. Biochem Biophys Res Commun... (2015) 463(4):1108–14. doi: 10.1016/j.bbrc.2015.06.067
103. Yin MS, Zhang YC, Xu SH, Liu JJ, Sun XH, Liang C, et al. Puerarin prevents diabetic cardiomyopathy in vivo and in vitro by inhibition of inflammation. J Asian Natural prod Res. (2019) 21(5):476–93. doi: 10.1080/10286020.2017.1405941
104. Sun S, Gong D, Liu R, Wang R, Chen D, Yuan T, et al. Puerarin inhibits NLRP3-Caspase-1-GSDMD-mediated pyroptosis via P2X7 receptor in cardiomyocytes and macrophages. Int J Mol Sci. (2023) 24(17):13169. doi: 10.3390/ijms241713169
105. Liu C, Cheng T, Wang Y, Li G, Wang Y, Tian W, et al. Syringaresinol alleviates early diabetic retinopathy by downregulating HIF-1α/VEGF via activating Nrf2 antioxidant pathway. Mol Nutr Food Res. (2024) 68(4):e2200771. doi: 10.1002/mnfr.202200771
106. Li G, Yang L, Feng L, Yang J, Li Y, An J, et al. Syringaresinol protects against type 1 diabetic cardiomyopathy by alleviating inflammation responses, cardiac fibrosis, and oxidative stress. Mol Nutr Food Res. (2020) 64(18):e2000231. doi: 10.1002/mnfr.202000231
107. Wu H, Li GN, Xie J, Li R, Chen QH, Chen JZ, et al. Resveratrol ameliorates myocardial fibrosis by inhibiting ROS/ERK/TGF-β/periostin pathway in STZ-induced diabetic mice. BMC Cardiovasc Disord. (2016) 16:5. doi: 10.1186/s12872-015-0169-z
108. Pan Y, Wang Y, Zhao Y, Peng K, Li W, Wang Y, et al. Inhibition of JNK phosphorylation by a novel curcumin analog prevents high glucose-induced inflammation and apoptosis in cardiomyocytes and the development of diabetic cardiomyopathy. Diabetes. (2014) 63(10):3497–511. doi: 10.2337/db13-1577
109. You S, Qian J, Sun C, Zhang H, Ye S, Chen T, et al. An Aza resveratrol-chalcone derivative 6b protects mice against diabetic cardiomyopathy by alleviating inflammation and oxidative stress. J Cell Mol Med. (2018) 22(3):1931–43. doi: 10.1111/jcmm.13477
110. Kumar V, Kumar S, Hassan M, Wu H, Thimmulappa RK, Kumar A, et al. Novel chalcone derivatives as potent Nrf2 activators in mice and human lung epithelial cells. J Med Chem. (2011) 54(12):4147–59. doi: 10.1021/jm2002348
111. Liu Y, Liu J, Wu KX, Guo XR, Tang ZH. A rapid method for sensitive profiling of bioactive triterpene and flavonoid from Astragalus mongholicus and Astragalus membranaceus by ultra-pressure liquid chromatography with tandem mass spectrometry. J Chromatogr B Anal Technol BioMed Life Sci. (2018) 1085:110–8. doi: 10.1016/j.jchromb.2018.03.044
112. Ramalingam M, Kim H, Lee Y, Lee YI. Phytochemical and Pharmacological Role of Liquiritigenin and Isoliquiritigenin From Radix Glycyrrhizae in Human Health and Disease Models. Front Aging Neurosci. (2018) 10:348. doi: 10.3389/fnagi.2018.00348
113. Gu X, Shi Y, Chen X, Sun Z, Luo W, Hu X, et al. Isoliquiritigenin attenuates diabetic cardiomyopathy via inhibition of hyperglycemia-induced inflammatory response and oxidative stress. Phytomedicine. (2020) 78:153319. doi: 10.1016/j.phymed.2020.153319
114. Rieg T, Masuda T, Gerasimova M, Mayoux E, Platt K, Powell DR, et al. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am J Physiol Renal Physiol. (2014) 306(2):F188–193. doi: 10.1152/ajprenal.00518.2013
115. Kang A, Jardine MJ. SGLT2 inhibitors may offer benefit beyond diabetes. Nat Rev Nephrol. (2021) 17(2):83–4. doi: 10.1038/s41581-020-00391-2
116. Reifsnider OS, Kansal AR, Gandhi PK, Cragin L, Brand SB, Pfarr E, et al. Cost-effectiveness of empagliflozin versus canagliflozin, dapagliflozin, or standard of care in patients with type 2 diabetes and established cardiovascular disease. BMJ Open Diabetes Res Care. (2021) 9(1):e001313. doi: 10.1136/bmjdrc-2020-001313
117. Cowie MR, Fisher M. SGLT2 inhibitors: mechanisms of cardiovascular benefit beyond glycaemic control. Nat Rev Cardiol. (2020) 17(12):761–72. doi: 10.1038/s41569-020-0406-8
118. Kim SR, Lee SG, Kim SH, Kim JH, Choi E, Cho W, et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun. (2020) 11(1):2127. doi: 10.1038/s41467-020-15983-6
119. Xu L, Nagata N, Nagashimada M, Zhuge F, Ni Y, Chen G, et al. SGLT2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. EBioMedicine. (2017) 20:137–49. doi: 10.1016/j.ebiom.2017.05.028
120. Lu YP, Wu HW, Zhu T, Li XT, Zuo J, et al. Empagliflozin reduces kidney fibrosis and improves kidney function by alternative macrophage activation in rats with 5/6-nephrectomy. BioMed Pharmacother = BioMed pharmacother.. (2022) 156:113947. doi: 10.1016/j.biopha.2022.113947
121. Chen H, Teng D, Xu B, Wang C, Wang H, Jia W, et al. The SGLT2 inhibitor canagliflozin reduces atherosclerosis by enhancing macrophage autophagy. J Cardiovasc Trans Res. (2023) 16(5):999–1009. doi: 10.1007/s12265-023-10390-w
122. Bizino MB, Jazet IM, Westenberg JJM, van Eyk HJ, Paiman EHM, Smit JWA, et al. Effect of liraglutide on cardiac function in patients with type 2 diabetes mellitus: randomized placebo-controlled trial. Cardiovasc Diabetol. (2019) 18(1):55. doi: 10.1186/s12933-019-0857-6
123. Tashiro Y, Sato K, Watanabe T, Nohtomi K, Terasaki M, Nagashima M, et al. A glucagon-like peptide-1 analog liraglutide suppresses macrophage foam cell formation and atherosclerosis. Peptides. (2014) 54:19–26. doi: 10.1016/j.peptides.2013.12.015
124. Vinué Á, Navarro J, Herrero-Cervera A, García-Cubas M, Andrés-Blasco I, Martínez-Hervás S, et al. The GLP-1 analogue lixisenatide decreases atherosclerosis in insulin-resistant mice by modulating macrophage phenotype. Diabetologia. (2017) 60(9):1801–12. doi: 10.1007/s00125-017-4330-3
125. Kodera R, Shikata K, Kataoka HU, Takatsuka T, Miyamoto S, Sasaki M, et al. Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia. (2011) 54(4):965–78. doi: 10.1007/s00125-010-2028-x
126. Sancar-Bas S, Gezginci-Oktayoglu S, Bolkent S. Exendin-4 attenuates renal tubular injury by decreasing oxidative stress and inflammation in streptozotocin-induced diabetic mice. Growth factors (Chur Switzerland. 2015) 33(5-6):419–29. doi: 10.3109/08977194.2015.1125349
127. Robinson E, Cassidy RS, Tate M, Zhao Y, Lockhart S, Calderwood D, et al. Exendin-4 protects against post-myocardial infarction remodelling via specific actions on inflammation and the extracellular matrix. Basic Res Cardiol. (2015) 110(2):20. doi: 10.1007/s00395-015-0476-7
128. Tate M, Robinson E, Green BD, McDermott BJ, Grieve DJ. Exendin-4 attenuates adverse cardiac remodelling in streptozocin-induced diabetes via specific actions on infiltrating macrophages. Basic Res Cardiol. (2016) 111(1):1. doi: 10.1007/s00395-015-0518-1
129. Zhang X, Zhang Z, Zhao Y, Jiang N, Qiu J, Yang Y, et al. Alogliptin, a Dipeptidyl Peptidase-4 inhibitor, alleviates atrial remodeling and improves mitochondrial function and biogenesis in diabetic rabbits. J Am Heart Assoc. (2017) 6(5):e005945. doi: 10.1161/jaha.117.005945
130. Zhang X, Zhang Z, Yang Y, Suo Y, Liu R, Qiu J, et al. Alogliptin prevents diastolic dysfunction and preserves left ventricular mitochondrial function in diabetic rabbits. Cardiovasc Diabetol. (2018) 17(1):160. doi: 10.1186/s12933-018-0803-z
131. Shah Z, Kampfrath T, Deiuliis JA, Zhong J, Pineda C, Ying Z, et al. Long-term dipeptidyl-peptidase 4 inhibition reduces atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis. Circulation. (2011) 124(21):2338–49. doi: 10.1161/circulationaha.111.041418
132. Irons BK, Greene RS, Mazzolini TA, Edwards KL, Sleeper RB. Implications of rosiglitazone and pioglitazone on cardiovascular risk in patients with type 2 diabetes mellitus. Pharmacotherapy. (2006) 26(2):168–81. doi: 10.1592/phco.26.2.168
133. Qayyum R, Schulman P. Cardiovascular effects of the thiazolidinediones. Diabetes/metabol Res Rev. (2006) 22(2):88–97. doi: 10.1002/dmrr.596
134. Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet (London England).. (2005) 366(9493):1279–89. doi: 10.1016/s0140-6736(05)67528-9
135. Ohga S, Shikata K, Yozai K, Okada S, Ogawa D, Usui H, et al. Thiazolidinedione ameliorates renal injury in experimental diabetic rats through anti-inflammatory effects mediated by inhibition of NF-kappaB activation. Am J Physiol Renal Physiol. (2007) 292(4):F1141–1150. doi: 10.1152/ajprenal.00288.2005
136. Jin X, Liu L, Zhou Z, Ge J, Yao T, Shen C. Pioglitazone alleviates inflammation in diabetic mice fed a high-fat diet via inhibiting advanced glycation end-product-induced classical macrophage activation. FEBS J. (2016) 283(12):2295–308. doi: 10.1111/febs.13735
137. Umekawa T, Tsuji H, Uemura H, Khan SR. Superoxide from NADPH oxidase as second messenger for the expression of osteopontin and monocyte chemoattractant protein-1 in renal epithelial cells exposed to calcium oxalate crystals. BJU Int. (2009) 104(1):115–20. doi: 10.1111/j.1464-410X.2009.08374.x
138. Lu H, Sun X, Jia M, Sun F, Zhu J, Chen X, et al. Rosiglitazone suppresses renal crystal deposition by ameliorating tubular injury resulted from oxidative stress and inflammatory response via promoting the Nrf2/HO-1 pathway and shifting macrophage polarization. Oxid Med Cell Longevity.. (2021) 2021:5527137. doi: 10.1155/2021/5527137
139. Agvall B, Jonasson JM, Galozy A, Halling A. Factors influencing hospitalization or emergency department visits and mortality in type 2 diabetes following the onset of new cardiovascular diagnoses in a population-based study. Cardiovasc Diabetol. (2024) 23(1):124. doi: 10.1186/s12933-024-02211-4
140. Li Y, Liu X, Wan L, Han B, Ma S, Pan H, et al. Metformin suppresses cardiac fibroblast proliferation under high-glucose conditions via regulating the mitochondrial complex I protein Grim-19 involved in the Sirt1/Stat3 signaling pathway. Free Radical Biol Med. (2023) 206:1–12. doi: 10.1016/j.freeradbiomed.2023.06.013
Keywords: macrophages, diabetic cardiomyopathy, diabetes, inflammation, cardiovascular diseases
Citation: Zhang S, Zhu X, Chen Y, Wen Z, Shi P and Ni Q (2024) The role and therapeutic potential of macrophages in the pathogenesis of diabetic cardiomyopathy. Front. Immunol. 15:1393392. doi: 10.3389/fimmu.2024.1393392
Received: 29 February 2024; Accepted: 16 April 2024;
Published: 07 May 2024.
Edited by:
Philippe Saas, Etablissement Français du Sang AuRA, FranceReviewed by:
Aleksandr E. Vendrov, University of Michigan, United StatesCopyright © 2024 Zhang, Zhu, Chen, Wen, Shi and Ni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Ni, bmlxaW5nQGdhbXl5LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.