 Xiaohui Ren1†
Xiaohui Ren1† Juan Liu
Juan Liu- 1Department of Respiratory Medicine, Shanxi Province Cancer Hospital/Shanxi Hospital Affiliated to Cancer Hospital, Chinese Academy of Medical Sciences/Cancer Hospital Affiliated to Shanxi Medical University, Taiyuan, Shanxi, China
- 2Department of Hospice Care, Shanxi Province Cancer Hospital/Shanxi Hospital Affiliated to Cancer Hospital, Chinese Academy of Medical Sciences/Cancer Hospital Affiliated to Shanxi Medical University, Taiyuan, Shanxi, China
- 3Shanxi Province Cancer Hospital/Shanxi Hospital Affiliated to Cancer Hospital, Chinese Academy of Medical Sciences/Cancer Hospital Affiliated to Shanxi Medical University, Taiyuan, Shanxi, China
- 4Department of Special Needs Medicine, Shanxi Province Cancer Hospital/Shanxi Hospital Affiliated to Cancer Hospital, Chinese Academy of Medical Sciences/Cancer Hospital Affiliated to Shanxi Medical University, Taiyuan, Shanxi, China
Immunotherapy has been developed, which harnesses and enhances the innate powers of the immune system to fight disease, particularly cancer. PD-1 (programmed death-1) and PD-L1 (programmed death ligand-1) are key components in the regulation of the immune system, particularly in the context of cancer immunotherapy. PD-1 and PD-L1 are regulated by PTMs, including phosphorylation, ubiquitination, deubiquitination, acetylation, palmitoylation and glycosylation. PROTACs (Proteolysis Targeting Chimeras) are a type of new drug design technology. They are specifically engineered molecules that target specific proteins within a cell for degradation. PROTACs have been designed and demonstrated their inhibitory activity against the PD-1/PD-L1 pathway, and showed their ability to degrade PD-1/PD-L1 proteins. In this review, we describe how PROTACs target PD-1 and PD-L1 proteins to improve the efficacy of immunotherapy. PROTACs could be a novel strategy to combine with radiotherapy, chemotherapy and immunotherapy for cancer patients.
Introduction
Post-translational modification (PTM) is one kind of the chemical modification of a protein after its synthesis (1). PTM often occurs after the translation process in which ribosomes create proteins from mRNA templates. It has been known that PTMs have profound effects on protein stability, activity, localization, and interaction with other cellular molecules, leading to regulation of the cellular function that was governed by proteins (2). PTMs play a central role in a wide range of biological processes, including cell signaling pathway, immune response, DNA damage response, and metabolism (3, 4). Dysregulation of PTMs is associated with various diseases, including cancer (5–7), neurodegenerative disorders (8, 9), cardiovascular diseases (10), and metabolic syndromes (11, 12). It is critical to explore the regulatory mechanism of PTMs in order to discover the new targets for disease treatments.
The common types of PTMs include ubiquitination (13), acetylation (14, 15), phosphorylation (16), methylation (17), glycosylation (18, 19), SUMOylation (20), prenylation (21), GlcNAcylation (18), succinylation (22), palmitoylation (23), and neddylation (24, 25). Ubiquitination is one common type of PTMs, in which a target protein is added a small protein called ubiquitin or multiple ubiquitins, leading to protein degradation by the proteasome (26). This process involves three key enzymes: E1 (Ubiquitin-activating enzyme), E2 (Ubiquitin-conjugating enzyme) and E3 (Ubiquitin ligase). The E1 enzyme activates ubiquitin in an ATP-dependent manner, forming a high-energy thioester bond with ubiquitin. The activated ubiquitin is transferred to E2, maintaining the high-energy bond. The E3 enzyme facilitates the transfer of ubiquitin from E2 to the substrate protein (27). E3 ligases are responsible for the specificity of the ubiquitination process, as they recognize specific target proteins to be ubiquitinated (28). There are different outcomes of ubiquitination, which are dependent on the number and linkage type of ubiquitin molecules. Monoubiquitination means only single ubiquitin is added to the substrate protein (29). Polyubiquitination means multiple ubiquitin molecules are attached and forma chain (30). The common and well-studied forms are linked through the lysine 48 (K48) residue and lysine 63 (K63) residue of ubiquitin. K48-linked polyubiquitination often tags the substrate protein for degradation by the proteasome, a cellular complex that breaks down proteins (31). K63-linked polyubiquitination often has different roles, such as in signaling pathways, DNA repair, or trafficking of membrane proteins (32). Beyond K48 and K63, non-canonical protein ubiquitination through K6, K11, K27, K29 and K33 residues have been reported (33). K6-linked ubiquitination is involved in autophagy and DNA damage response, while K27-linked ubiquitination participates into innate immunity. K29-linked ubiquitination plays a role in neurodegenerative disorders, whereas K11- and K33-linked ubiquitination should be further investigated (33).
E3 ubiquitin ligases are crucial enzymes in the ubiquitin-proteasome system (UPS). E3 ligases play a pivotal role in the process of ubiquitination, which involves attaching ubiquitin molecules to specific substrate proteins (34). They act as a bridge between the E2 enzyme and the substrate, ensuring accurate ubiquitin transfer. The E3 ligases are primarily responsible for the specificity of ubiquitination, determining which proteins will be tagged for various fates, including degradation, localization, or involvement in various cellular processes (35). There are several types of E3 ligases, categorized mainly based on their mechanism of action and structural features. The common types are RING (Really interesting new gene) finger ligases and HECT (Homologous to E6-AP carboxyl terminus) ligases. RING ligases directly transfer ubiquitin from the E2 enzyme to the substrate. HECT ligases form a covalent bond with ubiquitin before transferring it to the substrate (36).
RING E3 ligases are a diverse group with several types and subtypes, each having unique structural features and mechanisms of action (37). The notable types of RING E3 ligases have single-subunit RING E3 ligases and multiple-subunit E3 ligases. The former type is composed of a single protein that contains both the RING domain necessary for E2 binding and a substrate-recognition domain, such as MDM2. MDM2 targets the tumor suppressors p53, and c-Cbl. The latter type are complexes made up of multiple protein subunits. One of the subunits contains the RING domain, while others are responsible for substrate recognition and regulation. Multiple-subunit E3 ligases mainly have cullin-RING ligases (CRLs) and RBR (RING-between-RING) E3 Ligases (38). CRLs are the largest family of multi-subunit RING E3 ligases, which include several subfamilies like SCF (Skp1-Cullin1-F-box protein) complexes. CRLs are involved in various cellular processes including cell cycle control and signal transduction (39). RBR are a unique class of E3 ligases that contain two RING domains separated by a non-RING domain. They function through a hybrid mechanism that has characteristics of both RING and HECT type E3 ligases (40).
Unlike RING E3 ligases, HECT E3 ligases form a covalent bond with ubiquitin before transferring it to the substrate protein. This family is characterized by the HECT domain, which is responsible for this unique enzymatic activity (41). HECT ligases are classified into different types based on structural features and functional domains. HECT ligases have several notable types, including NEDD4 family and HERC family (42). Members of this family typically have a C2 domain that binds to phospholipids, two to four WW domains that recognize proline-rich motifs in substrates, and the HECT domain for ubiquitin transfer (43). NEDD4 family is the largest group of HECT E3 ligases and is composed of nine members, each with distinct but sometimes overlapping substrate specificities and functions (44). These members typically include NEDD4 (Neural Precursor Cell Expressed Developmentally Downregulated 4), NEDD4L (NEDD4-Like), ITCH (Itchy E3 Ubiquitin Protein Ligase), WWP1 (WW Domain Containing E3 Ubiquitin Protein Ligase 1), WWP2, SMURF1 (SMAD Specific E3 Ubiquitin Protein Ligase 1), SMURF2, NEDL1 (NEDD4-Like E3 Ubiquitin Protein Ligase 1) and NEDL2 (45–48). HERC E3 ligases are characterized by their large size and the presence of one or more RCC1-like domains (RLDs) in addition to the HECT domain. The HERC family is involved in processes such as protein trafficking and cellular growth control (49, 50).
Ubiquitination is a dynamic and reversible process, with deubiquitinating enzymes (DUBs) able to reverse the ubiquitination of proteins (51). This process, known as deubiquitination, is essential for maintaining the balance and regulation of protein ubiquitination within the cell, impacting various cellular processes and homeostasis (52, 53). DUBs are a group of proteases that play a critical role in the UPS by removing ubiquitin molecules from substrate proteins. Different DUBs have specificity for different types of ubiquitin linkages. Some are specialized in trimming ubiquitin chains from the distal end, while others can cleave ubiquitin at specific linkage points within a chain. There are several classes of DUBs, based on their structural and functional characteristics (54). The two main classes are cysteine proteases and metalloproteases. These classes are further divided into families such as USP (ubiquitin-specific protease), UCH (ubiquitin C-terminal hydrolase), OTU (ovarian tumor protease), and JAMM/MPN+ (Jab1/MPN domain-containing metalloenzymes) (55–58).
PROTACs
PROTACs (Proteolysis Targeting Chimeras) are a type of new drug design technology. They are specifically engineered molecules that target specific proteins within a cell for degradation (59). PROTACs consist of three key components: a target protein ligand, a E3 ubiquitin ligand, and a linker. Target protein ligand is the part of the PROTAC molecule that specifically binds to the protein targeted for degradation (60). The selection of this ligand is critical as it ensures the specificity of the PROTAC for its intended protein target (61). The PROTAC recruits an E3 ubiquitin ligase, which is responsible for transferring ubiquitin molecules to the target protein, tagging it for degradation. The linker connects the target protein ligand and the E3 ligase ligand (Figure 1). The length and composition of the linker are crucial for the effective proximity of the target protein and the E3 ligase, facilitating the transfer of ubiquitin from the E3 ligase to the target protein (62, 63).
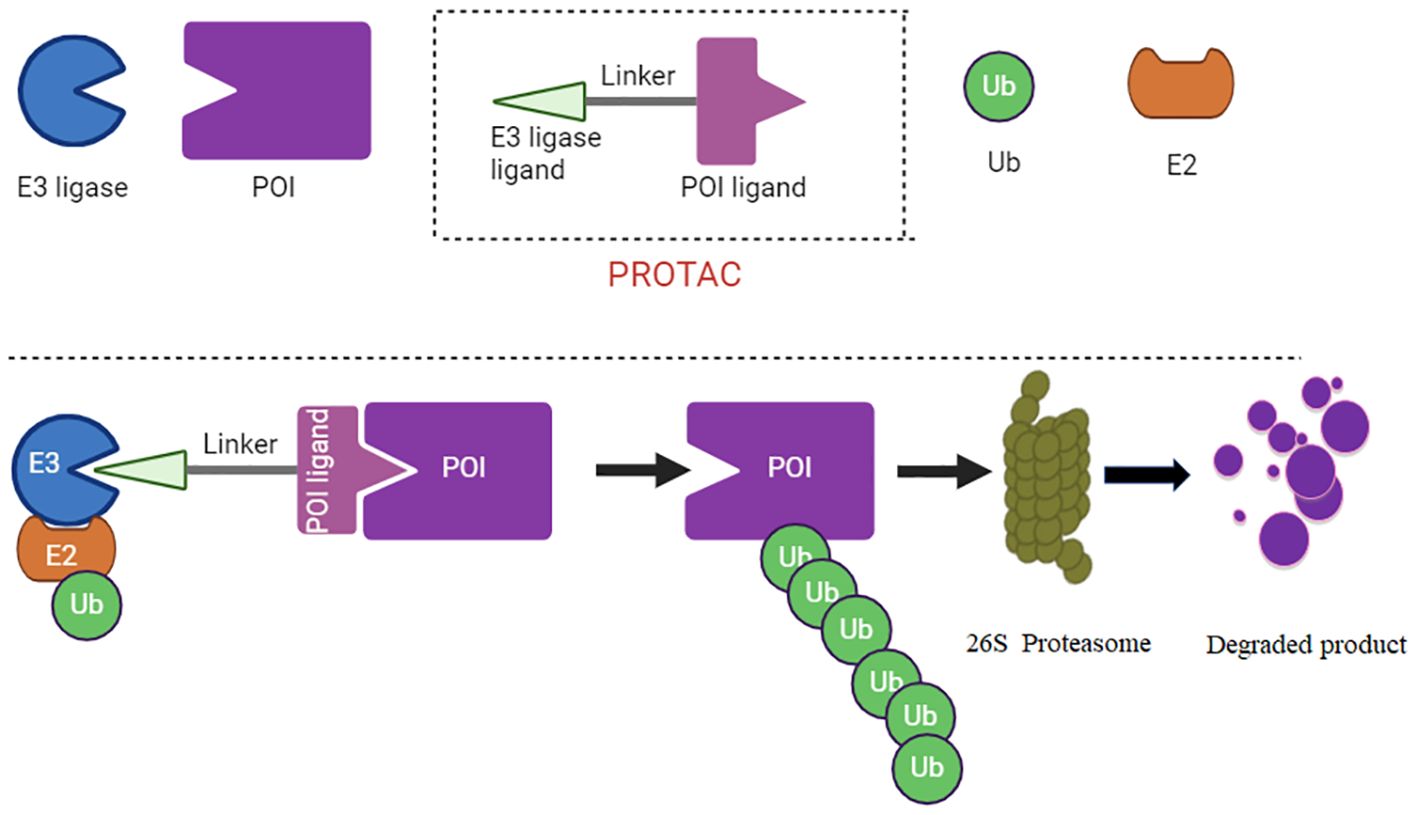
Figure 1 PROTAC diagram is illustrate. The PROTAC recruits an E3 ubiquitin ligase, which is responsible for transferring ubiquitin molecules to the target protein, tagging it for degradation. The linker connects the target protein ligand and the E3 ligase ligand.
Unlike traditional inhibitors that simply block protein activity, PROTACs work by recruiting an E3 ubiquitin ligase to tag the target protein with ubiquitin. This tag marks the protein for destruction by the cell’s proteasome, a protein complex responsible for degrading and recycling damaged or unneeded proteins (64). PROTAC technology allows for more precise control targeting protein levels within cells, which can be used to target proteins that are traditionally considered “undruggable” by conventional methods (65). AbTAC, molecular glue, LYTAC and Nano-PROTAC have been emerged to target proteins (66). PROTACs have been gaining attention in the field of drug development, particularly for their potential in treating diseases such as cancer, where certain proteins are overexpressed or mutated (67, 68).
Immunotherapy
Immunotherapy has been developed, which harnesses and enhances the innate powers of the immune system to fight disease, particularly cancer (69). Unlike chemotherapy and radiation, which directly target and kill cancer cells, immunotherapy performs its function via stimulating or restoring the immune system’s ability to detect and destroy tumor cells (70, 71). There are several types of immunotherapy to boost the immune response via employing a different strategy (72). Checkpoint inhibitors were discovered, which block proteins that prevent immune cells from attacking cancer cells (73). By inhibiting these “checkpoints”, checkpoint inhibitors enable the immune cells to recognize and destroy cancer cells, including inhibitors targeting PD-1, PD-L1, and CTLA-4 (74).
CAR T-cell therapy (Chimeric Antigen Receptor T-cell Therapy) involves genetically engineering a patient’s own T cells to produce special receptors on their surface (75, 76). These receptors will make T cells to better recognize and attack cancer cells. Cancer vaccines are designed to treat existing cancers by stimulating the immune system to attack cancer cells, such as targeting specific antigens found on cancer cells (77). Cytokine therapy boosts the immune system’s ability to fight cancer via using cytokines, such as interferons and interleukins (78, 79). Immunotherapy has shown remarkable overcomes in treating certain types of cancer, including melanoma, lung cancer, and leukemia. However, its effectiveness can vary widely among various cancer patients. In addition, immunotherapy could have side effects due to an overactive immune response and resistance (80). Therefore, immunotherapy represents a significant shift in cancer treatment, moving toward more personalized and targeted approaches that leverage the body’s natural defenses.
PD-1 and PD-L1
PD-1 (programmed death-1) and PD-L1 (programmed death ligand-1) are key components in the regulation of the immune system, particularly in the context of cancer immunotherapy (81). Their interaction plays a significant role in preventing the immune system from attacking normal cells, but it can also enable cancer cells to evade immune surveillance (82, 83). PD-1 is a protein receptor expressed on the surface of certain immune cells, such as T cells, B cells, and natural killer cells (84). PD-L1 is a ligand that is expressed on the surface of many cell types, including some cancer cells and immune cells (85). Under normal conditions, PD-L1 binding to PD-1 sends an inhibitory signal to T cells, telling them not to attack the cells presenting PD-L1. This is a natural mechanism to prevent autoimmunity and control inflammation (86). Many types of cancer cells exploit this pathway by overexpressing PD-L1. When cancer cells present high levels of PD-L1, they can bind to PD-1 receptors on T cells, effectively “turning off” these immune cells and preventing them from attacking the cancer (87). Given the role of the PD-1/PD-L1 pathway in allowing cancer cells to evade the immune system, blocking this interaction has become a key strategy in cancer immunotherapy (88). Drugs known as checkpoint inhibitors have been developed to target PD-1, PD-L1 and CTLA-4 (89, 90).
PD-1 inhibitors bind to PD-1 on immune cells, and block its interaction with PD-L1, such as nivolumab and pembrolizumab (91). PD-1 inhibitors allow the T cells to remain active and able to attack cancer cells. PD-L1 Inhibitors bind to PD-L1 on cancer cells or other cells in the tumor environment, which prevent it from binding to and inhibiting PD-1 on T cells. PD-L1 inhibitors include atezolizumab and durvalumab (92). PD-1 and PD-L1 inhibitors have shown significant success in treating various types of cancer, including melanoma, non-small cell lung cancer, kidney cancer, bladder cancer, and head and neck cancers. Although the development and use of PD-1 and PD-L1 inhibitors represent a major advancement in cancer therapy, offering hope for many patients with advanced or hard-to-treat cancers, side effects and resistance reduced the efficacy of PD-1/PD-L1 inhibitors in immunotherapy (93, 94).
PD-1 and PD-L1 are regulated by PTMs
It has been reported that PD-1 and PD-L1 are regulated by PTMs, including phosphorylation, ubiquitination, deubiquitination, acetylation, palmitoylation and glycosylation (95–97). PD-1/PD-L1 modification participated in governing immune escape and affecting cancer immunotherapy (98). Moreover, the E3 ubiquitin ligases improved tumor immunotherapy via controlling PD-1/PD-L1 protein accumulation in tumor microenvironment (99). In the following paragraphs, we will describe the role of PTMs in regulation of PD-1 and PD-L1 (Tables 1, 2).

Table 1 Ubiquitination and deubiquitination of PD-1/PD-L1 are summarized.
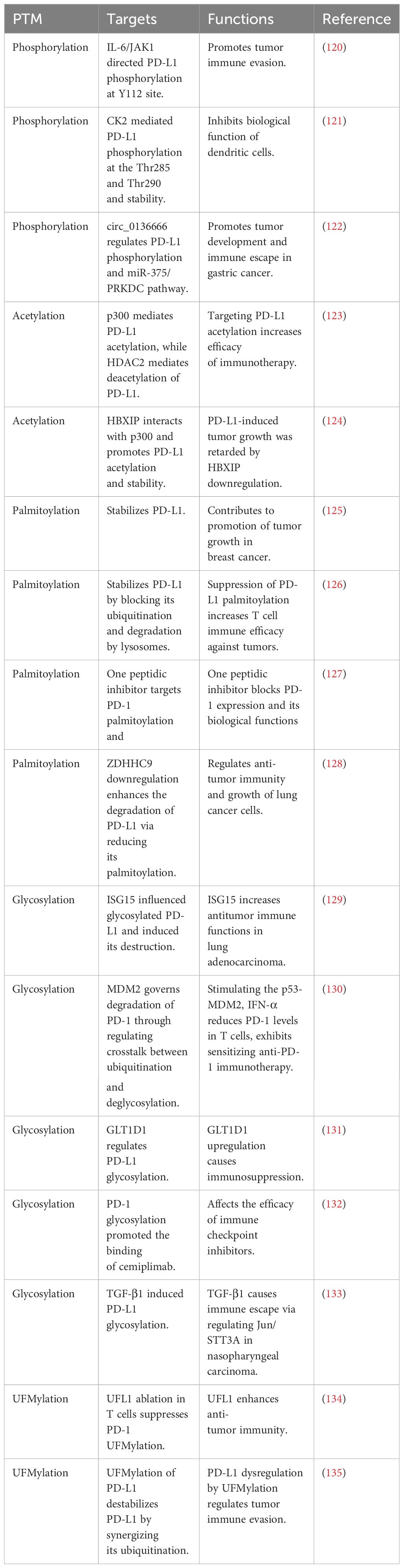
Table 2 PTMs of PD-1/PD-L1 are summarized.
Phosphorylation
PD-L1 phosphorylation has been found to regulate cancer immune evasion. For example, IL-6/JAK1 signaling pathway directed PD-L1 phosphorylation at Tyr112 site and promoted tumor immune evasion (120). When IL-6 activated JAK1, it led to the phosphorylation of PD-L1 at Tyr112. This phosphorylation attracted the N-glycosyltransferase STT3A from the endoplasmic reticulum, which initiated the glycosylation of PD-L1, thereby stabilizing it. By using an IL-6 antibody to inhibit IL-6, enhanced T cell killing effects in animal models were reported, especially when used in combination with anti-Tim-3 therapy. Furthermore, a direct relationship between the levels of IL-6 and PD-L1 was found in the tumor tissues of patients with hepatocellular carcinoma (120).
Another study revealed that in both cancer and dendritic cells (DC), PD-L1 underwent phosphorylation and subsequent stabilization by CK2 enzyme. This phosphorylation at the Thr285 and Thr290 sites on PD-L1 interfered with its binding to the adaptor protein, SPOP (speckle-type POZ protein). As a result, PD-L1 was shielded from CUL3/SPOP-mediated proteasomal degradation. When CK2 activity was hindered, there was a noticeable reduction in PD-L1 protein levels, facilitating the mobilization of CD80 from DC, which subsequently revitalized T-cell activities. Using a treatment combination of a CK2 inhibitor and a Tim-3 antibody in a mouse model, marked suppression of tumor growth and a signification prolongation of survival were reported. Hence, CK2-mediated PD-L1 phosphorylation and stability inhibited biological function of dendritic cells (121).
Recently, Miao et al. reported that circ_0136666 regulated PD-L1 phosphorylation and miR-375/PRKDC pathway, leading to promoting tumor development and immune escape in gastric cancer (122). Specifically, the expression of hsa_circ_0136666 was prominent in gastric cancer tissues and cell lines. Functionally, hsa_circ_0136666 enhanced the proliferation of gastric cancer and contributed to the development of the tumor microenvironment, enabling the tumor to evade immune surveillance through the modulatory activities on CD8+ T cells. At the mechanistic level, hsa_circ_0136666 induced the increase of PRKDC expression by competitively binding to miR-375-3p, affecting the phosphorylation and subsequent stabilization of PD-L1. Moreover, hsa_circ_0136666 suppressed the body’s immune vigilance and countering anti-cancer efficacy. Furthermore, lipid nanoparticle (LNP)-delivered siRNA strategies markedly improved anti-PD-L1 efficacy and inhibited immune escape (122).
Ubiquitination
SPOP targets PD-L1
Zhang et al. found that Cyclin D/CDK4 kinase leads to PD-L1 destabilization through SPOP-mediated degradation of PD-L1 and controls cancer immune surveillance (100). The levels of the PD-L1 protein are controlled through the actions of cyclin D-CDK4 and the CUL3-SPOP E3 ligase, which facilitate its degradation via the proteasome. Blocking the activity of CDK4 and CDK6 in vivo leads to an elevation in PD-L1 protein. This increase is due to the inhibition of the phosphorylation by cyclin D-CDK4 on the SPOP, which in turn enhances the breakdown of SPOP by the FZR1. Furthermore, mutations that result in a loss of SPOP function disrupt the PD-L1 degradation, resulting in heightened levels of PD-L1 and a decrease in the number of tumor-infiltrating lymphocytes in both mouse tumors and prostate cancer tissues. Importantly, CDK4/6 inhibitor therapy plus anti-PD-1 immunotherapy significantly boosts tumor shrinkage and substantially enhances survival rates in mouse models of cancer (100).
RIG-I modulated SPOP-mediated degradation of PD-L1 and elevated immune evasion in colon cancer (101). Suppressing RIG-I led to a decrease in the ability of T cells to eliminate tumor cells and reduced the growth of colon tumors in mice with a fully functional immune system. Conversely, increasing RIG-I expression accelerated tumor growth, and high levels of RIG-I made cells more responsive to anti-PD-1 treatment in vivo. Intriguingly, RIG-I modulates the expression of PD-L1, facilitating immune escape in colon cancer independently of type I interferon activation. RIG-I prevents PD-L1 destruction by SPOP (101).
FBXO38 targets PD-1
Meng et al. found that FBXO38 induced ubiquitination of PD-1 and controlled anti-tumor immunity of T cells (102). PD-1 on the surface of activated T cells was internalized, then underwent ubiquitination and proteasome-mediated degradation. FBXO38 targeted PD-1 for Lys48-linked polyubiquitination, leading to its degradation. Eliminating Fbxo38 in T cells accelerated tumor growth in mice due to increased PD-1 levels in the tumor-infiltrating T cells. Applying anti-PD-1 therapy counteracted the tumor growth observed with FBXO38 deficiency, indicating that PD-1 is a crucial target of FBXO38 in T cells. In both human tumor samples and a mouse cancer model, the expression levels of FBXO38 and Fbxo38 were reduced in tumor-infiltrating T cells. Nonetheless, IL-2 treatment was able to boost Fbxo38 expression, thus lowering PD-1 levels in PD-1-positive T cells in mice. FBXO38 controlled PD-1 levels, suggesting a novel strategy for inhibiting the PD-1 pathway by regulation of FBXO38 (102).
NEDD4 targets PD-L1
NEDD4 mediated PD-L1 ubiquitination and degradation and regulated T cell-induced immune surveillance in bladder cancer, which was regulated by fibroblast growth factor receptor 3 (FGFR3) (103). Blocking FGFR3 in bladder cancer with FGFR3 activation increased PD-L1 protein levels by altering its ubiquitination process, which in turn, hampered the anticancer activity of CD8+ T cells. An analysis of tissue microarrays from human urothelial carcinoma (UC) revealed a negative relationship between FGFR3 expression and PD-L1 levels. Additionally, NEDD4 became phosphorylated upon FGFR3 activation, playing a pivotal role in controlling PD-L1 ubiquitination. NEDD4 directly interacted with PD-L1, leading to the polyubiquitination of PD-L1 at the Lys48 (K48) linkage. In mice models with NEDD4-deficient bladder cancer, an increase in PD-L1 levels within the cancer cells resulted in reduced infiltration and antitumor activity of CD8+ T cells. In various FGFR3-activated tumor models, the diminished antitumor effectiveness due to targeted FGFR3 therapy could be counterbalanced by combining it with anti-PD-1 immunotherapy, thus significantly reducing tumor growth. NEDD4 could be a key E3 ubiquitin ligase that regulates PD-L1 for destruction in FGFR3-driven bladder cancer (103).
RNF125 targets PD-L1
Ubiquitin ligase RNF125 was reported by Wei et al. to promote the ubiquitination and degradation of PD-L1 in K48-linked manner (104). RNF125 binds to PD-L1, influencing its protein levels by facilitating K48-linked polyubiquitination, leading to its degradation. When RNF125 was knocked out in MC-38 and H22 cell lines, which were then implanted into C57BL/6 mice, an increase in PD-L1 levels and accelerated tumor growth were observed. Conversely, when RNF125 was overexpressed in these cell lines, PD-L1 levels decreased, and tumor growth slowed. Furthermore, MC-38 tumors with RNF125 overexpression had an increased in the infiltration of CD4+, CD8+ T cells, and macrophages. A positive association between RNF125 expression and the infiltration of CD4+, CD8+ T cells, and macrophages was observed in tumor tissues from TCGA public database. Additionally, RNF125 expression was found to be notably reduced in various human cancer tissues, inversely related to the clinical stage of the cancers, and tumor patients with elevated RNF125 levels displayed more favorable clinical outcomes (104).
In head and neck squamous cell carcinoma (HNSCC) cells, RNF125 expression was low. When RNF125 was overexpressed, it curtailed the immune evasion of HNSCC cells, as demonstrated by reduced proliferation, migration, and invasion of TSCCA cells, alongside enhanced proliferation of CD8+ T cells and increased levels of IL-2 and TNF-α. RNF125 also reduced PD-L1 expression in TSCCA cells and promoted its degradation. Overexpression of PD-L1 partially reversed the effects of RNF125 on the immune evasion of TSCCA cells. Additionally, RNF125 suppressed tumor formation and growth in mice. Collectively, RNF125 facilitates the ubiquitin-mediated degradation of PD-L1, thereby impeding immune escape in HNSCC (105).
TRIM21 targets PD-L1
Sun and coworkers reported that TRIM21 mediated PD-L1 ubiquitination and involved in regulation of anti-PD-L1 immunotherapeutic efficacy. LINC02418 acted as a suppressor of PD-L1 expression and was associated with increased CD8+ T cell infiltration, indicating better clinical outcomes for NSCLC patients. It reduced PD-L1 levels by promoting its ubiquitination through TRIM21. Both human LINC02418 and its mouse equivalent, mmu-4930573I07Rik, influenced the effectiveness of PD-L1-targeted therapies in NSCLC by facilitating T cell-mediated apoptosis. Additionally, the suppression of METTL3 by YTHDF2 led to the upregulation of hsa-LINC02418 and mmu-4930573I07Rik. In NSCLC patients, high levels of LINC02418 are linked to lower PD-L1 expression and a higher presence of CD8+ T cells (106). Inhibition of CDK5 reduced PD-L1 levels via the ubiquitination-proteasome pathway by TRIM21 and improved antitumor immunity in lung adenocarcinoma (107). Silencing Trim21 in mice increased HCC oncogenesis in a non-alcoholic steatohepatitis (NASH) context, which was due to overexpression of PD-1 in lymphocytes and PD-L1 in tumors (108).
c-Cbl targets PD-1
One group showed that casitas B lymphoma (c-Cbl) induced proteasomal degradation of PD-1 in immune cells and regulated colorectal cancer (CRC) growth (109). A significantly increased growth of xenografts and enhanced infiltration of immune cells in c-Cbl heterozygous (c-Cbl+/-) mice was observed compared to c-Cbl+/+. Tumor-associated CD8+ T-lymphocytes and macrophages in c-Cbl+/- mice exhibited upregulation of PD-1 levels. Macrophages from c-Cbl+/- mice displayed a 4-5 times decrease in their ability to phagocytize tumor cells, which was recovered by the application of an anti-PD-1 neutralizing antibody, indicating a regulatory role of c-Cbl on PD-1. The C-terminus of c-Cbl interacted with the cytoplasmic tail of PD-1, leading to PD-1 destabilization. Hence, c-Cbl could be as an E3 ligase for PD-1 and a modulator of the tumor microenvironment (109).
FBW7 targets PD-L1
Liu et al. reported that FBW7 enhanced sensitivity of anti-PD-1 immunotherapy via promotion of PD-1 ubiquitination and destruction in non-small cell lung cancer (110). FBW7 was identified to act as an E3 ubiquitin ligase targeting the PD-1 protein, specifically promoting K48-linked polyubiquitination at the Lys233 residue of PD-1. Targeting FBW7 caused faster degradation of the PD-1 protein, thereby boosting antitumor immunity effectively in vivo. Additionally, phosphorylation of the Ser261 residue by CDK1 enhanced the PD-1 protein for nuclear translocation and interaction with FBW7. A higher level of FBW7 was found in immunologically active TME, resulting in improved responses to PD-1 blockade therapy (110). Hence, ubiquitination plays a critical role in the regulation of PD-1/PD-L1 in tumor immunotherapy (136).
Deubiquitination
USP22 targets PD-L1
Huang et al. reported that USP22 deubiquitinated PD-L1 and maintained its stabilization, leading to inhibition of anticancer immunity. USP22 was identified as a deubiquitinase for PD-L1, where it binds directly to PD-L1’s C-terminus, leading to its deubiquitination and stabilization. USP22 showed high expression levels and frequent alterations in liver cancer, which was strongly linked to a dismal prognosis for these cancer patients. The genetic removal of USP22 led to the suppression of liver cancer growth, boosted tumor immunogenicity and the presence of tumor-infiltrating lymphocytes, and heightened the effectiveness of therapies targeting PD-L1 and CDDP-based chemotherapy in mouse models (111). Another study also showed that USP22 interacted with PD-L1, enhancing its stability by removing ubiquitin and preventing its breakdown by the proteasome. Additionally, USP22 forms a complex with CSN5, maintaining its stability via deubiquitination. USP22 or CSN5 can enhance the binding between PD-L1 and the other molecule. The reduction of USP22 levels was found to suppress tumor growth and enhance the cytotoxicity of T cells. Moreover, in samples from patients with non-small cell lung cancer, there was a notable positive relationship between the levels of USP22 and PD-L1 expression (112). Hence, USP22 plays a critical role in immune evasion in human cancer cells (137).
USP7 targets PD-L1
USP7 was reported to regulate PD-L1 stability and modulate sensitization of gastric cancer cells to T cells killing. Analyzing data from TCGA and tissues, a direct correlation between PD-L1 and USP7 expressions was found in gastric cancer. USP7 interacted with PD-L1, enhancing its stability, whereas inhibiting USP7 reduced the PD-L1/PD-1 interaction and made cancer cells more susceptible to T cell-mediated destruction. Additionally, USP7 inhibitors reduced cell growth by stabilizing p53 in gastric cancer. USP7 inhibitors not only hinder the cell proliferation but also reduce PD-L1 levels, thereby boosting the anti-tumor immune response (113). USP7 reprogrammed tumor-associated macrophages and modulates anti-tumor immune response in lung cancer. USP7 inhibitors upregulated the expression of PD-L1 in tumors, while inhibiting PD-1 had an effective anti-tumor activity (114). In glioma cells, USP7 showed high expression levels, but PD-L1 protein levels increased. Knocking down USP7 in glioma cells reduced their growth, increased apoptosis, and enhanced CD8+ T cell proliferation, thereby preventing immune evasion. USP7 contributed to the stabilization of PD-L1. The overexpression of PD-L1 counteracted the effects of USP7 silencing on the immune escape of glioma cells (115).
USP8 targets PD-L1
Blocking the USP8 significantly boosts the effectiveness of anti-PD-1/PD-L1 immunotherapy by altering the TME. Inhibiting USP8 elevated PD-L1 protein levels by promoting TRAF6-mediated K63-linked ubiquitination of PD-L1. USP8 blockade enhances the innate immune response and MHC-I expression via the activation of NF-κB signaling. Combining a USP8 inhibitor with PD-1/PD-L1 blockade markedly stimulates CD8+ T cell infiltration, leading to tumor suppression and improved survival in various mouse tumor models (116). In pancreatic cancer, USP8 levels were significantly higher in tumor tissues compared to normal tissues. The expression of USP8 was notably linked to the TNM stage in various pancreatic cancer patient cohorts. Inhibition of USP8 led to diminished tumor invasion, migration, and overall tumor size, enhancing anti-tumor immunity. USP8 inhibitor resulted in decreased tumor formation, and mice with Usp8 deficiency showed improved survival rates. Additionally, USP8 positively influenced PD-L1 expression by blocking its degradation in pancreatic cancer. A USP8 inhibitor and anti-PD-L1 together significantly halted pancreatic tumor growth through the activation of cytotoxic T-cells. The effectiveness of this anti-tumor immunity was primarily reliant on the PD-L1 pathway and CD8+ T cells. Hence, targeting USP8 sensitized anti-PD-L1 immunotherapy via regulation of PD-L1 degradation in pancreatic cancer (117).
OTUB1 targets PD-L1
CircRNA circIGF2BP3 stabilized OTUB1 mRNA and inhibited the ubiquitination and degradation of PD-L1 in a PKP3-dependent way, leading to tumor immune escape (118). circIGF2BP3 increases PKP3 expression by sequestering miR-328-3p and miR-3173-5p, thus impairing the cancer immune response. PKP3 then interacts with the RNA-binding protein FXR1 to enhance the stability of OTUB1 mRNA, which in turn boosts PD-L1 levels by promoting its deubiquitination. The elimination of tumor PD-L1 effectively nullified the influence of the circIGF2BP3/PKP3 axis on the CD8+ T cell response. Blocking the circIGF2BP3/PKP3 pathway improved the efficacy of anti-PD-1 treatment in a Lewis lung carcinoma mouse model (118).
OTUB2 targets PD-L1
Recently, targeting OTUB2 increased cytotoxic T cells efficacy via regulating PD-L1 degradation. OTUB2 directly bound to PD-L1, hindering its ubiquitination and subsequent degradation within the endoplasmic reticulum. Eliminating OTUB2 significantly reduced PD-L1 protein levels on tumor cells, enhancing their vulnerability to the cytotoxic effects of CD8+ T cells. A notable association between OTUB2 levels and PD-L1 was found in human non-small cell lung cancer samples. An OTUB2 inhibitor effectively diminished PD-L1 levels and inhibited tumor growth. OTUB2 plays a key role in regulating PD-L1 expression and tumor immune evasion (119).
Acetylation
PD-L1 acetylation has been uncovered by Gao and coworkers in 2020. This study showed that p300 mediated PD-L1 acetylation, while HDAC2 mediated deacetylation of PD-L1. Genetically or pharmacologically modulating acetylation of PD-L1 reduced its nuclear translocation, which promoted the anti-PD-1 efficacy via regulation of the expression of several immune-response-related genes in cancer cells. Targeting PD-L1 acetylation could increase the efficacy of tumor immunotherapy (123). Another study showed that HBXIP oncoprotein and PD-L1 expressions were upregulated in clinical breast tumor samples. Moreover, HBXIP expression was associated with PD-L1 expression in breast cancer tissues. Mechanistically, HBXIP elevated the PD-L1 transcription via activation of ETS2. Furthermore, HBXIP interacted with p300 and promoted PD-L1 acetylation and resulted in promotion of PD-L1 stability. PD-L1-induced tumor growth was retarded by downregulation of HBXIP in breast cancer (124).
Palmitoylation
Palmitoylation has been discovered to stabilize PD-L1 and contribute to promotion of tumor growth in breast cancer (125). Yao et al. reported that suppression of PD-L1 palmitoylation increased T cell immune efficacy against tumors (126). One peptidic inhibitor has been developed for targeting PD-1 palmitoylation and blocking PD-1 expression and its biological functions (127). Evidence showed that downregulation of ZDHHC9 enhanced the degradation of PD-L1 protein via reducing its palmitoylation levels in lung adenocarcinoma (128). Hence, PD-1/PD-L1 palmitoylation is involved in immunotherapy in human cancers.
Glycosylation
N-glycosylation and stabilization of PD-L1 were reported to inhibit T-cell activity. Glycogen synthase kinase 3β (GSK3β) can bind to PD-L1 and promote PD-L1 degradation by beta-TrCP in phosphorylation-dependent manner. Moreover, epidermal growth factor (EGF) treatment made PD-L1 stabilization due to inactivation of GSK3β in breast cancer. Consistently, gefitinib destabilized PD-L1 due to suppression of EGF pathway, which increased T-cell immunity and promoted PD-1 blockade efficacy in mice (138). ISG15 influenced glycosylated PD-L1 and induced its destruction to increase antitumor immune functions in lung adenocarcinoma (129). MDM2 (murine double minute 2) governed degradation of PD-1 through regulating crosstalk between ubiquitination and deglycosylation in tumor cells (130). Removal of N-glycosylation increased the detection of PD-L1 and provided the prediction of anti-PD-1/PD-L1 treatment efficacy (139). GLT1D1 upregulation caused immunosuppression via regulation of PD-L1 glycosylation and directed poor prognosis in B-cell lymphoma (131). N-glycosylation of PD-1 enhanced the interaction with a PD-1-specific monoclonal antibody, camrelizumab (140). PD-1 glycosylation promoted the binding of cemiplimab to affect the efficacy of immune checkpoint inhibitors (132). TGF-beta-1 induced PD-L1 glycosylation and led to immune escape via regulation of Jun/STT3A pathway in nasopharyngeal carcinoma (133).
UFMylation
UFMylation, a post-translational modification akin to ubiquitination, plays a crucial role in various biological functions, with its dysregulation linked to several human diseases, including cancer (141, 142). Mice lacking the UFMylation E3 ligase UFL1 specifically in T cells demonstrated superior tumor suppression. Through single-cell RNA sequencing, an increase in tumor-infiltrating cytotoxic CD8+ T cells was observed in these UFL1 conditional knockout (cKO) mice. UFL1 was found to enhance PD-1 stability by promoting its UFMylation, which counters PD-1 ubiquitination and subsequent degradation (134). Moreover, AMPK activation led to the phosphorylation of UFL1 at Thr536, which inhibited PD-1 UFMylation and promoted its degradation. Notably, the removal of UFL1 in T cells led to reduced PD-1 UFMylation, destabilizing PD-1 and thereby boosting CD8+ T cell activity. Consequently, tumors in UFL1 cKO mice responded more favorably to anti-CTLA-4 immunotherapy (134). Another study showed that PD-L1 underwent UFMylation, leading to destabilizing PD-L1 by promoting its ubiquitination. Disrupting PD-L1 UFMylation through the silencing of UFL1 or UFM1 resulted in PD-L1 stabilization in a variety of human and mouse cancer cell lines, which weakened antitumor immunity both in vitro and in mouse models. Reduced UFL1 levels were observed across several cancers, and lower UFL1 expression was associated with a diminished response to anti-PD1 therapy in melanoma patients. An inhibitor of UFSP2 enhances UFMylation activity and displayed potential to augment PD-1 blockade therapy (135).
PROTACs target PD-1/PD-L1
Numerous PROTACs have been developed for targeting PD-L1 and PD-1 (Table 3). In the following sections, we will highlight the multiple PROTACs that target PD-1/PD-L1 in human cancers (Figure 2).
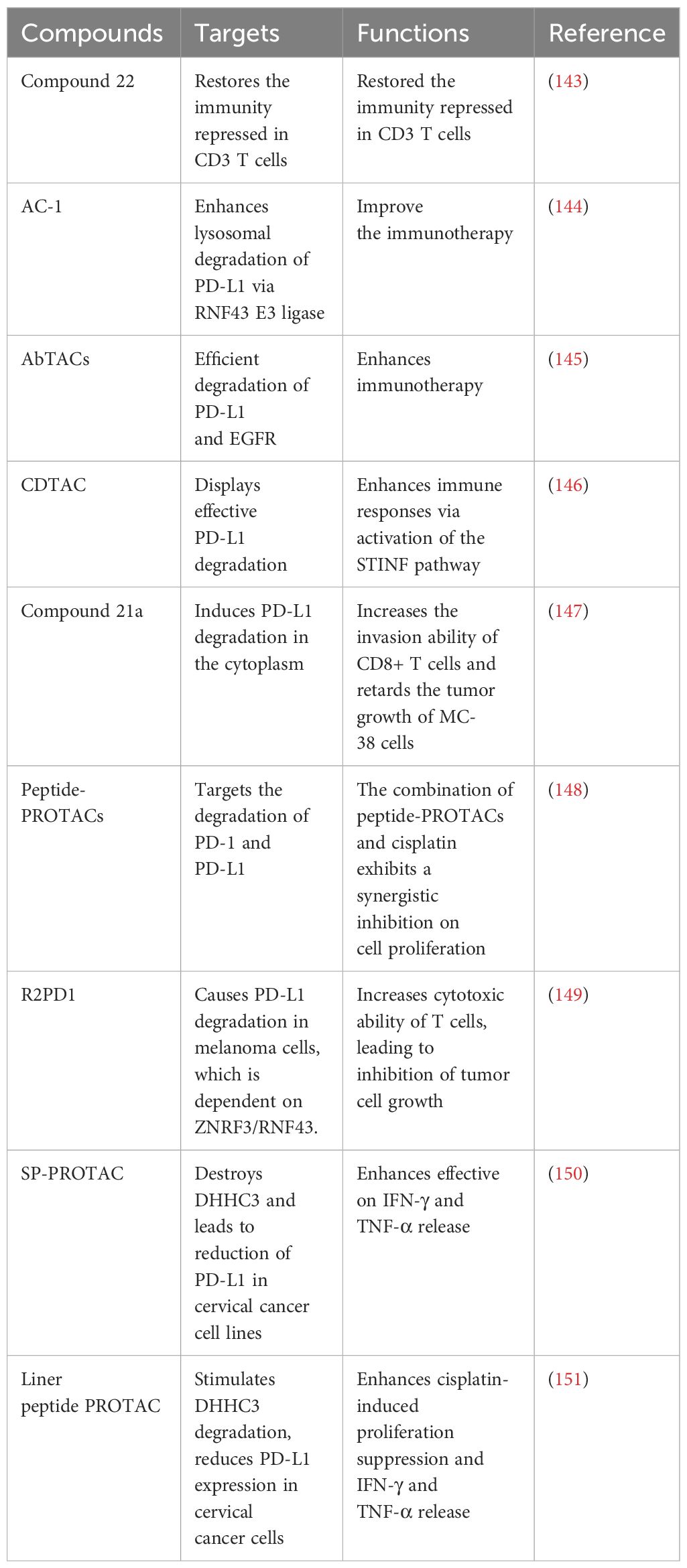
Table 3 PROTACs target PD-1/PD-L1 in cancer.
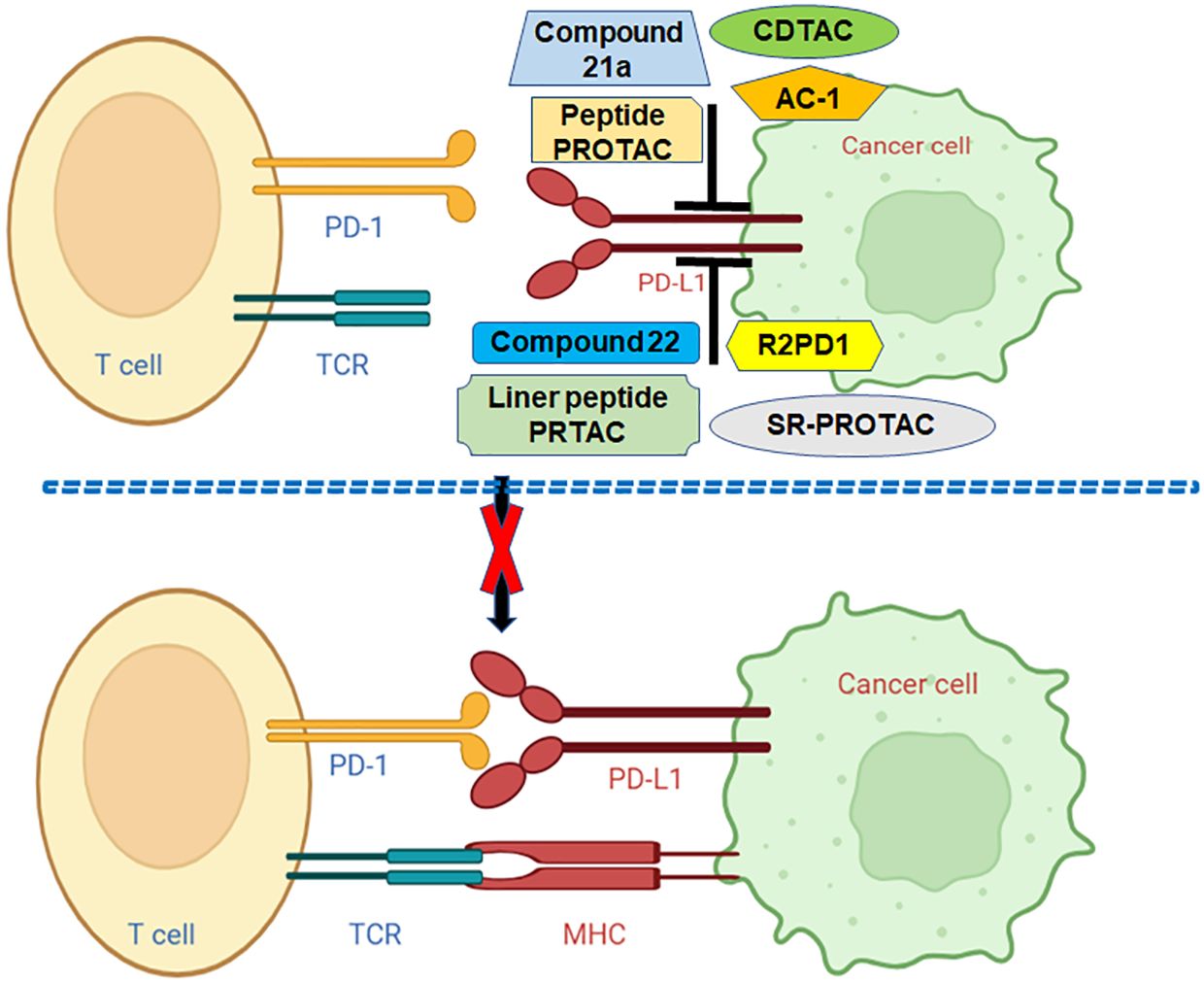
Figure 2 Various PROTACs target PD-L1 in human cancer. Linear peptide PROTAC, SP-PROTAC, R2PD1, Peptide-PROTACs, Compound 21a, CDTACs, AC-1, and Compound 22 target and decrease the PD-L1 and block the PD-1/PD-L1 pathway.
Compound 22 targets PD-L1
Novel resorcinol diphenyl ether-based PROTACs have been designed and demonstrated their inhibitory activity against the PD-1/PD-L1 pathway, and showed their ability to degrade PD-L1 protein. This study used HTRF binding assay to assess the inhibitory activities of PROTACs against PD-1/PD-L1. Most of PROTACs obtained excellent inhibition of PD-1/PD-L1. Compound 22 is one of the best PROTACs with low IC50 value. Moreover, except suppression of PD-1/PD-L1 interaction, compound 22 restored the immunity repressed in CD3 T cells and Hep3B/OS-8/hPD-L1 cells. Compound 22 decreased the expression of PD-L1 protein via lysosome-dependent pathway. Hence, compound 22 could work as a potential agent for the degradation of PD-L1 via PROTAC strategy (143).
AC-1 targets PD-L1
One group developed antibody-based PROTACs (AbTACs), which are fully recombinant bispecific antibodies. AbTACs degraded cell-surface proteins via recruitment of membrane-bound E3 ligases. AC-1, an AbTAC, enhanced the lysosomal degradation of PD-L1 via recruitment of the RNF43 E3 ligase. Moreover, AC-1 promoted the degradation of PD-L1 after one day treatment in three cancer cell lines, including MDA-MB-231, HCC827, and T24, indicating that AC-1 has wide cellular applicability. Furthermore, due to human parts of AbTAC, AC-1 makes it unlike to illicit an immune response. In summary, AC-1 could be a good AbTAC to target PD-L1 for degradation (144). New AbTACs were engineered for efficient degradation of two membrane proteins, PD-L1 and EGFR (145). In this research, they apply protein engineering techniques to study and enhance the degradation capabilities of PD-L1 and EGFR for therapeutic purposes. They created several antibodies targeting RNF43, discovering that the precise binding sites on RNF43 and the proteins of interest are crucial for degradation effectiveness, which have more binding strength than the AbTAC antibodies. Additionally, this group introduced ZNRF3, another E3 ligase, into our arsenal for degrading PD-L1 and EGFR. Notably, AbTACs targeting RNF43 and ZNRF3 do not trigger WNT signaling pathways. This work suggests that optimizing the development of AbTACs will enhance their utility for targeted protein degradation (145).
CDTACs target PD-L1
Carbon-dot (CD)-based PROTACs (CDTACs) have been established, which degrade membrane proteins via the UPS pathway. CDTACs were found to interact with PD-L1, and recruit cereblon (CRBN) to induce the ubiquitination and degradation of PD-L1 via proteasomes. Fasting-mimicking diet (FMD) was observed to promote the cellular uptake and proteasome activity. CDTACs treatment led to more than 90% of PD-L1 degradation in CT26 and B16-F10 cancer cells. Furthermore, CDTACs enhanced immune responses via activation of the STINF pathway. Consistently, the combination of CDTACs and FMD treatment suppressed the tumor growth of CT26 and B16-F10 cells. CDTACs displayed effective PD-L1 degradation and activation of immune system (146).
Compound 21a targets PD-L1
A new PROTAC, compound 21a, has been designed and demonstrated effective functions on the degradation of PD-L1 protein in multiple malignant cells via proteasome pathway, which was in time- and dose-dependent manners. These different cancer cells include MCF-7 breast cancer cell, SW-480 colon cancer cells, PC-3 prostate cancer cells, MB-49 murine bladder tumor cells, and hematological cancer cells (HL-60, Kasumi-1, Skno-1). Compound 21a was designed based on a BMS-37 derivates, which act as small molecule inhibitors against PD-L1. Compound 21a induced PD-L1 degradation in the cytoplasm. In vivo experiment data showed that compound 21a increased the invasion ability of CD8+ T cells and retarded the tumor growth of MC-38 cancer cells via reduction of PD-L1 protein levels. Altogether, compound 21a might be an alternative agent for cancer immunotherapy (147).
Peptide-PROTACs target PD-1/PD-L1
Dai et al. designed Peptide-PROTACs that target PD-1 and PD-L1 in human cancer cells (148). The peptide degraders include a cell-penetrating peptide (CPP) sequence, targeting protein recognition (TPR) peptide sequence, E3 recruitment peptide (ERP) sequence, and peptide linker. Peptides 1 and 2, consisting of VHL binder and CPR sequence, exhibited the highest efficiency for PD-1 and PD-L1 degradation, respectively. Based on TPR sequence, Peptides 1 and 6 targeted PD-1, and Peptide 11 targeted PD-L1. Peptide 1 displayed a higher potent for PD-1 degradation than peptide 6 in C33A cells (148). By AI-direct peptide design, Peptide 2 also targeted PD-L1 palmitoylation. Peptide 12 targeted PD-L1 palmitoylation without ERP sequence. Moreover, Peptide-PROTACs with CPP sequences were found to cross the cell membrane. Furthermore, Peptide-PROTACs displayed a low toxicity to C33A cells. Notably, a combination of Peptide-PROTACs and cisplatin exhibited a synergistic inhibition on cell proliferation via induction of cell apoptosis compared with cisplatin monotherapy (148). Peptide-PROTACs could provide better clinical benefit for cancer patients.
R2PD1 targets PD-L1
Sun and colleagues discovered ROTACs, which was bispecific WNT- and BMP-signaling-disabled R-spondin (RSPO) chimeras, to influence transmembrane protein degradation via leveraging the specificity of stem cell factors for ZNRF3/RNF43 ligases. R2PD1, a RSPO2 chimera, was uncovered to target the degradation of PD-L1. The R2PD1 protein interacted with PD-L1 and led to its lysosomal degradation. Surprisingly, R2PD1 caused 50% to 90% PD-L1 degradation in melanoma cell lines, which was dependent on ZNRF3/RNF43. Functionally, R2PD1 increased cytotoxic ability of T cells, leading to inhibition of tumor cell growth. Hence, ROTACs represent a new strategy for degrading cell surface proteins (149).
SP-PROTAC decreases PD-L1
A stapled peptide-based PROTAC (SP-PROTAC) was designed to specifically destroy palmitoyl-transferase (DHHC3), which led to the reduction of PD-L1 in cervical cancer cell lines. In C33A and HeLa cells, SP-PROTAC remarkably reduced PD-L1 protein levels at low concentration. The proteasome inhibitor MG132 attenuated the SP-PROTAC-induced PD-L1 degradation in cancer cells. When T cells and C33A cells were cocultured, SP-PROTAC enhanced IFN-γ and TNF-α release via promotion of PD-L1 degradation. Compared with BMS-8, an ICI, SP-PROTAC displayed more effective on IFN-γ and TNF-α release. Taken together, SP-PROTAC targeted DHHC3 and alleviated PD-L1 protein levels in human cervical cancer (150).
Liner peptide PROTAC decreases PD-L1
A cyclic peptide-based PROTAC was synthesized to stimulate the degradation of DHHC3 palmitoyltransferase, which resulted in reduce PD-L1 expression in cervical cancer cells (151). This cyclic peptide PROTAC consists of disulfide bonds to maintain their structure via keeping the stability of N- and C-termini of the peptide. MG132 can block cyclic peptide PROTAC-induced degradation of PD-L1. Moreover, cyclic peptide PROTAC enhanced cisplatin-induced proliferation suppression in C33A cells. Furthermore, after C33A and T cells were cocultured, cyclic peptide PROTAC blocked the PD-1/PD-L1 binding via enhancement of PD-L1 degradation, eventually contributing to increased secretion of IFN-γ and TNF-α. This cyclic peptide PROTAC promotes anti-PD-L1 activity in cancer cells (151).
Conclusion
In conclusion, PD-1 and PD-L1 are regulated by PTMs, including phosphorylation, ubiquitination, deubiquitination, acetylation, palmitoylation, glycosylation and UFMylation. Multiple PROTACs have been developed for targeting PD-L1 and PD-1, including compound 22, AC-1, AbTACs, CDTAC, compound 21a, peptide-PROTACs, R2PD1, SP-PROTAC, liner peptide PROTAC. PROTAC is rapidly evolving and could be a new approach for cancer therapies, including immunotherapy (Figure 2). Several issues need to be discussed regarding immunotherapy and PTMs of PD-1/PD-L1. Beyond the abovementioned PTMs, it is unclear whether other types of PTMs regulate PD-1 and PD-L1, which should be determined. Recent studies suggest that miRNAs, lncRNAs and circRNAs are involved in PD-1/PD-L1 expression and tumor immunotherapy (152). LncRNA MALAT1 modulated METTL3-mediated PD-L1 expression and affected immune infiltrates in pancreatic cancer (153). It is unclear whether noncoding RNAs regulate the PD-1/PD-L1 PTMs in human cancer, which needs to be clarified. Besides, PD-1 and PD-L1, whether other immune checkpoint factors are regulated by PTMs and involve in cancer immunotherapy. Although PROTACs have many advantages, such as modularity, targeting on undruggable targets, selectivity, specificity and compatibility, they have several disadvantages, including heavy-molecular weight, unclear pharmacokinetics (67). It is necessary to develop more general strategies for PROTAC-mediated protein degradation. Moreover, it is required to determine whether PROTACs could be used to combine with radiotherapy, chemotherapy and immunotherapy for a wider range of cancers and non-cancer diseases.
Author contributions
XR: Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Writing – original draft. LW: Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Writing – original draft. LL: Conceptualization, Formal analysis, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing. JL: Conceptualization, Project administration, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Kitamura N, Galligan JJ. A global view of the human post-translational modification landscape. Biochem J. (2023) 480:1241–65. doi: 10.1042/BCJ20220251
2. Wang S, Osgood AO, Chatterjee A. Uncovering post-translational modification-associated protein-protein interactions. Curr Opin Struct Biol. (2022) 74:102352. doi: 10.1016/j.sbi.2022.102352
3. Song H, Shen R, Liu X, Yang X, Xie K, Guo Z, et al. Histone post-translational modification and the DNA damage response. Genes Dis. (2023) 10:1429–44. doi: 10.1016/j.gendis.2022.04.002
4. Liu J, Qian C, Cao X. Post-translational modification control of innate immunity. Immunity. (2016) 45:15–30. doi: 10.1016/j.immuni.2016.06.020
5. Kumari S, Gupta R, Ambasta RK, Kumar P. Emerging trends in post-translational modification: Shedding light on Glioblastoma multiforme. Biochim Biophys Acta Rev Cancer. (2023) 1878:188999. doi: 10.1016/j.bbcan.2023.188999
6. Park MK, Lee H, Lee CH. Post-translational modification of ZEB family members in cancer progression. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms232315127
7. Li W, Li F, Zhang X, Lin HK, Xu C. Insights into the post-translational modification and its emerging role in shaping the tumor microenvironment. Signal Transduct Target Ther. (2021) 6:422. doi: 10.1038/s41392-021-00825-8
8. Yang J, Shen N, Shen J, Yang Y, Li HL. Complicated role of post-translational modification and protease-cleaved fragments of Tau in Alzheimer's disease and other tauopathies. Mol Neurobiol. (2023). doi: 10.1007/s12035-023-03867-x
9. Rani N, Sahu M, Ambasta RK, Kumar P. Triaging between post-translational modification of cell cycle regulators and their therapeutics in neurodegenerative diseases. Ageing Res Rev. (2023) 94:102174. doi: 10.1016/j.arr.2023.102174
10. Cheng X, Wang K, Zhao Y, Wang K. Research progress on post-translational modification of proteins and cardiovascular diseases. Cell Death Discov. (2023) 9:275. doi: 10.1038/s41420-023-01560-5
11. Buttari B, Profumo E, Capozzi A, Saso L, Sorice M, Rigano R. Post-translational modifications of proteins in antiphospholipid antibody syndrome. Crit Rev Clin Lab Sci. (2019) 56:511–25. doi: 10.1080/10408363.2019.1650714
12. Moiseenok AG, Kanunnikova NP. Brain CoA and acetyl CoA metabolism in mechanisms of neurodegeneration. Biochem (Mosc). (2023) 88:466–80. doi: 10.1134/S000629792304003X
13. Beck DB, Werner A, Kastner DL, Aksentijevich I. Disorders of ubiquitylation: unchained inflammation. Nat Rev Rheumatol. (2022) 18:435–47. doi: 10.1038/s41584-022-00778-4
14. Deng S, Marmorstein R. Protein N-terminal acetylation: structural basis, mechanism, versatility, and regulation. Trends Biochem Sci. (2021) 46:15–27. doi: 10.1016/j.tibs.2020.08.005
15. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. (2019) 20:156–74. doi: 10.1038/s41580-018-0081-3
16. Xu W, Huang Y. Regulation of inflammatory cell death by phosphorylation. Front Immunol. (2022) 13:851169. doi: 10.3389/fimmu.2022.851169
17. Biggar KK, Li SS. Non-histone protein methylation as a regulator of cellular signalling and function. Nat Rev Mol Cell Biol. (2015) 16:5–17. doi: 10.1038/nrm3915
18. He XF, Hu X, Wen GJ, Wang Z, Lin WJ. O-GlcNAcylation in cancer development and immunotherapy. Cancer Lett. (2023) 566:216258. doi: 10.1016/j.canlet.2023.216258
19. Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. (2017) 18:452–65. doi: 10.1038/nrm.2017.22
20. Vertegaal ACO. Signalling mechanisms and cellular functions of SUMO. Nat Rev Mol Cell Biol. (2022) 23:715–31. doi: 10.1038/s41580-022-00500-y
21. Wang M, Casey PJ. Protein prenylation: unique fats make their mark on biology. Nat Rev Mol Cell Biol. (2016) 17:110–22. doi: 10.1038/nrm.2015.11
22. Shen R, Ruan H, Lin S, Liu B, Song H, Li L, et al. Lysine succinylation, the metabolic bridge between cancer and immunity. Genes Dis. (2023) 10:2470–8. doi: 10.1016/j.gendis.2022.10.028
23. Villanueva CE, Hagenbuch B. Palmitoylation of solute carriers. Biochem Pharmacol. (2023) 215:115695. doi: 10.1016/j.bcp.2023.115695
24. He ZX, Yang WG, Zengyangzong D, Gao G, Zhang Q, Liu HM, et al. Targeting cullin neddylation for cancer and fibrotic diseases. Theranostics. (2023) 13:5017–56. doi: 10.7150/thno.78876
25. Yu Q, Jiang Y, Sun Y. Anticancer drug discovery by targeting cullin neddylation. Acta Pharm Sin B. (2020) 10:746–65. doi: 10.1016/j.apsb.2019.09.005
26. Rape M. Ubiquitylation at the crossroads of development and disease. Nat Rev Mol Cell Biol. (2018) 19:59–70. doi: 10.1038/nrm.2017.83
27. Toma-Fukai S, Shimizu T. Structural diversity of ubiquitin E3 ligase. Molecules. (2021) 26. doi: 10.3390/molecules26216682
28. Chen X, Ma J, Wang ZW, Wang Z. The E3 ubiquitin ligases regulate inflammation in cardiovascular diseases. Semin Cell Dev Biol. (2024) 154:167–74. doi: 10.1016/j.semcdb.2023.02.008
29. Chen Y, Zhou D, Yao Y, Sun Y, Yao F, Ma L. Monoubiquitination in homeostasis and cancer. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23115925
30. Livneh I, Kravtsova-Ivantsiv Y, Braten O, Kwon YT, Ciechanover A. Monoubiquitination joins polyubiquitination as an esteemed proteasomal targeting signal. Bioessays. (2017) 39. doi: 10.1002/bies.201700027
31. Verma R. Exploiting ubiquitin ligases for induced target degradation as an antiviral strategy. Adv Exp Med Biol. (2021) 1322:339–57. doi: 10.1007/978-981-16-0267-2_13
32. Martinez-Forero I, Rouzaut A, Palazon A, Dubrot J, Melero I. Lysine 63 polyubiquitination in immunotherapy and in cancer-promoting inflammation. Clin Cancer Res. (2009) 15:6751–7. doi: 10.1158/1078-0432.CCR-09-1225
33. Tracz M, Bialek W. Beyond K48 and K63: non-canonical protein ubiquitination. Cell Mol Biol Lett. (2021) 26:1. doi: 10.1186/s11658-020-00245-6
34. Liu Y, Duan C, Zhang C. E3 ubiquitin ligase in anticancer drugdsla resistance: recent advances and future potential. Front Pharmacol. (2021) 12:645864. doi: 10.3389/fphar.2021.645864
35. Wang W, Liu W, Chen Q, Yuan Y, Wang P. Targeting CSC-related transcription factors by E3 ubiquitin ligases for cancer therapy. Semin Cancer Biol. (2022) 87:84–97. doi: 10.1016/j.semcancer.2022.11.002
36. Yang L, Chen J, Huang X, Zhang E, He J, Cai Z. Novel insights into E3 ubiquitin ligase in cancer chemoresistance. Am J Med Sci. (2018) 355:368–76. doi: 10.1016/j.amjms.2017.12.012
37. Zhang X, Liu Y, Zhang T, Tan Y, Dai X, Yang YG, et al. Advances in the potential roles of Cullin-RING ligases in regulating autoimmune diseases. Front Immunol. (2023) 14:1125224. doi: 10.3389/fimmu.2023.1125224
38. Cai C, Tang YD, Zhai J, Zheng C. The RING finger protein family in health and disease. Signal Transduct Target Ther. (2022) 7:300. doi: 10.1038/s41392-022-01152-2
39. Uchida C, Kitagawa M. RING- HECT-. and RBR-type E3 ubiquitin ligases: involvement in human cancer. Curr Cancer Drug Targets. (2016) 16:157–74. doi: 10.2174/1568009616666151112122801
40. Wang P, Dai X, Jiang W, Li Y, Wei W. RBR E3 ubiquitin ligases in tumorigenesis. Semin Cancer Biol. (2020) 67:131–44. doi: 10.1016/j.semcancer.2020.05.002
41. Wang Y, Argiles-Castillo D, Kane EI, Zhou A, Spratt DE. HECT E3 ubiquitin ligases - emerging insights into their biological roles and disease relevance. J Cell Sci. (2020) 133:jcs228072. doi: 10.1242/jcs.228072
42. Zhang R, Shi S. The role of NEDD4 related HECT-type E3 ubiquitin ligases in defective autophagy in cancer cells: molecular mechanisms and therapeutic perspectives. Mol Med. (2023) 29:34. doi: 10.1186/s10020-023-00628-3
43. Bernassola F, Chillemi G, Melino G. HECT-type E3 ubiquitin ligases in cancer. Trends Biochem Sci. (2019) 44:1057–75. doi: 10.1016/j.tibs.2019.08.004
44. Wang ZW, Hu X, Ye M, Lin M, Chu M, Shen X. NEDD4 E3 ligase: Functions and mechanism in human cancer. Semin Cancer Biol. (2020) 67:92–101. doi: 10.1016/j.semcancer.2020.03.006
45. Xie S, Xia L, Song Y, Liu H, Wang ZW, Zhu X. Insights into the biological role of NEDD4L E3 ubiquitin ligase in human cancers. Front Oncol. (2021) 11:774648. doi: 10.3389/fonc.2021.774648
46. Hu X, Yu J, Lin Z, Feng R, Wang ZW, Chen G. The emerging role of WWP1 in cancer development and progression. Cell Death Discovery. (2021) 7:163. doi: 10.1038/s41420-021-00532-x
47. Behera A, Reddy ABM. WWP1 E3 ligase at the crossroads of health and disease. Cell Death Dis. (2023) 14:853. doi: 10.1038/s41419-023-06380-0
48. Yang L, Zhou W, Lin H. Posttranslational modifications of Smurfs: emerging regulation in cancer. Front Oncol. (2020) 10:610663. doi: 10.3389/fonc.2020.610663
49. Singh S, Ng J, Sivaraman J. Exploring the "Other" subfamily of HECT E3-ligases for therapeutic intervention. Pharmacol Ther. (2021) 224:107809. doi: 10.1016/j.pharmthera.2021.107809
50. Sala-Gaston J, Martinez-Martinez A, Pedrazza L, Lorenzo-Martin LF, Caloto R, Bustelo XR, et al. HERC ubiquitin ligases in cancer. Cancers (Basel). (2020) 12:1653. doi: 10.3390/cancers12061653
51. Ren J, Yu P, Liu S, Li R, Niu X, Chen Y, et al. Deubiquitylating enzymes in cancer and immunity. Adv Sci (Weinh). (2023) 10:e2303807. doi: 10.1002/advs.202303807
52. Dewson G, Eichhorn PJA, Komander D. Deubiquitinases in cancer. Nat Rev Cancer. (2023) 23:842–62. doi: 10.1038/s41568-023-00633-y
53. Ge F, Li Y, Yuan T, Wu Y, He Q, Yang B, et al. Deubiquitinating enzymes: Promising targets for drug resistance. Drug Discovery Today. (2022) 27:2603–13. doi: 10.1016/j.drudis.2022.06.009
54. Wang Y, Wang F. Post-translational modifications of deubiquitinating enzymes: expanding the ubiquitin code. Front Pharmacol. (2021) 12:685011. doi: 10.3389/fphar.2021.685011
55. Cruz L, Soares P, Correia M. Ubiquitin-specific proteases: players in cancer cellular processes. Pharm (Basel). (2021) 14:848. doi: 10.3390/ph14090848
56. Li Y, Reverter D. Molecular mechanisms of DUBs regulation in signaling and disease. Int J Mol Sci. (2021) 22:986. doi: 10.3390/ijms22030986
57. Wu L, Yu K, Chen K, Zhu X, Yang Z, Wang Q, et al. Fbxo45 facilitates pancreatic carcinoma progression by targeting USP49 for ubiquitination and degradation. Cell Death Dis. (2022) 13:231. doi: 10.1038/s41419-022-04675-2
58. Fang Y, Shen X. Ubiquitin carboxyl-terminal hydrolases: involvement in cancer progression and clinical implications. Cancer Metastasis Rev. (2017) 36:669–82. doi: 10.1007/s10555-017-9702-0
59. Li S, Chen T, Liu J, Zhang H, Li J, Wang Z, et al. PROTACs: Novel tools for improving immunotherapy in cancer. Cancer Lett. (2023) 560:216128. doi: 10.1016/j.canlet.2023.216128
60. Kelm JM, Pandey DS, Malin E, Kansou H, Arora S, Kumar R, et al. PROTAC'ing oncoproteins: targeted protein degradation for cancer therapy. Mol Cancer. (2023) 22:62. doi: 10.1186/s12943-022-01707-5
61. Kim Y, Kim EK, Chey Y, Song MJ, Jang HH. Targeted protein degradation: principles and applications of the proteasome. Cells. (2023) 12:1846. doi: 10.3390/cells12141846
62. Sincere NI, Anand K, Ashique S, Yang J, You C. PROTACs: emerging targeted protein degradation approaches for advanced druggable strategies. Molecules. (2023) 28:4014. doi: 10.3390/molecules28104014
63. Wang H, Chen M, Zhang X, Xie S, Qin J, Li J. Peptide-based PROTACs: current challenges and future perspectives. Curr Med Chem. (2024) 31:208–22. doi: 10.2174/0929867330666230130121822
64. He X, Weng Z, Zou Y. Progress in the controllability technology of PROTAC. Eur J Med Chem. (2024) 265:116096. doi: 10.1016/j.ejmech.2023.116096
65. Bekes M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discovery. (2022) 21:181–200. doi: 10.1038/s41573-021-00371-6
66. Chen S, Cui J, Chen H, Yu B, Long S. Recent progress in degradation of membrane proteins by PROTACs and alternative targeted protein degradation techniques. Eur J Med Chem. (2023) 262:115911. doi: 10.1016/j.ejmech.2023.115911
67. Choudhary D, Kaur A, Singh P, Chaudhary G, Kaur R, Bayan MF, et al. Target protein degradation by protacs: A budding cancer treatment strategy. Pharmacol Ther. (2023) 250:108525. doi: 10.1016/j.pharmthera.2023.108525
68. Schneider M, Radoux CJ, Hercules A, Ochoa D, Dunham I, Zalmas LP, et al. The PROTACtable genome. Nat Rev Drug Discovery. (2021) 20:789–97. doi: 10.1038/s41573-021-00245-x
69. Hamilton PT, Anholt BR, Nelson BH. Tumour immunotherapy: lessons from predator-prey theory. Nat Rev Immunol. (2022) 22:765–75. doi: 10.1038/s41577-022-00719-y
70. Cui JW, Li Y, Yang Y, Yang HK, Dong JM, Xiao ZH, et al. Tumor immunotherapy resistance: Revealing the mechanism of PD-1 / PD-L1-mediated tumor immune escape. BioMed Pharmacother. (2024) 171:116203. doi: 10.1016/j.biopha.2024.116203
71. Oliveira G, Wu CJ. Dynamics and specificities of T cells in cancer immunotherapy. Nat Rev Cancer. (2023) 23:295–316. doi: 10.1038/s41568-023-00560-y
72. Li YR, Halladay T, Yang L. Immune evasion in cell-based immunotherapy: unraveling challenges and novel strategies. J BioMed Sci. (2024) 31:5. doi: 10.1186/s12929-024-00998-8
73. Chen Z, Hu T, Zhou J, Gu X, Chen S, Qi Q, et al. Overview of tumor immunotherapy based on approved drugs. Life Sci. (2024) 340:122419. doi: 10.1016/j.lfs.2024.122419
74. Butterfield LH, Najjar YG. Immunotherapy combination approaches: mechanisms, biomarkers and clinical observations. Nat Rev Immunol. (2023). doi: 10.1038/s41577-023-00973-8
75. Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. (2016) 13:273–90. doi: 10.1038/nrclinonc.2016.25
76. Yang T, Xiong Y, Zeng Y, Wang Y, Zeng J, Liu J, et al. Current status of immunotherapy for non-small cell lung cancer. Front Pharmacol. (2022) 13:989461. doi: 10.3389/fphar.2022.989461
77. Lin MJ, Svensson-Arvelund J, Lubitz GS, Marabelle A, Melero I, Brown BD, et al. Cancer vaccines: the next immunotherapy frontier. Nat Cancer. (2022) 3:911–26. doi: 10.1038/s43018-022-00418-6
78. Pawlowski KD, Duffy JT, Gottschalk S, Balyasnikova IV. Cytokine modification of adoptive chimeric antigen receptor immunotherapy for glioblastoma. Cancers (Basel). (2023) 15:5852. doi: 10.3390/cancers15245852
79. Chen P, Paraiso WKD, Cabral H. Revitalizing cytokine-based cancer immunotherapy through advanced delivery systems. Macromol Biosci. (2023) 23:e2300275. doi: 10.1002/mabi.202300275
80. Liu L, Chen J. Therapeutic antibodies for precise cancer immunotherapy: current and future perspectives. Med Rev (2021). (2022) 2:555–69. doi: 10.1515/mr-2022-0033
81. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. (2016) 8:328rv4. doi: 10.1126/scitranslmed.aad7118
82. Mortezaee K. B7x in cancer immunity and immunotherapy. Int Immunopharmacol. (2023) 118:110133. doi: 10.1016/j.intimp.2023.110133
83. Sukari A, Nagasaka M, Al-Hadidi A, Lum LG. Cancer immunology and immunotherapy. Anticancer Res. (2016) 36:5593–606. doi: 10.21873/anticanres.11144
84. Chen M, Bie L, Ying J. Cancer cell-intrinsic PD-1: Its role in Malignant progression and immunotherapy. BioMed Pharmacother. (2023) 167:115514. doi: 10.1016/j.biopha.2023.115514
85. Zhang Y, Yang Y, Chen Y, Lin W, Chen X, Liu J, et al. PD-L1: Biological mechanism, function, and immunotherapy in gastric cancer. Front Immunol. (2022) 13:1060497. doi: 10.3389/fimmu.2022.1060497
86. Zhou YJ, Li G, Wang J, Liu M, Wang Z, Song Y, et al. PD-L1: expression regulation. Blood Sci. (2023) 5:77–91. doi: 10.1097/BS9.0000000000000149
87. Parvez A, Choudhary F, Mudgal P, Khan R, Qureshi KA, Farooqi H, et al. PD-1 and PD-L1: architects of immune symphony and immunotherapy breakthroughs in cancer treatment. Front Immunol. (2023) 14:1296341. doi: 10.3389/fimmu.2023.1296341
88. Jiao Z, Zhang J. Interplay between inflammasomes and PD-1/PD-L1 and their implications in cancer immunotherapy. Carcinogenesis. (2023) 44:795–808. doi: 10.1093/carcin/bgad072
89. Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol. (2016) 13:473–86. doi: 10.1038/nrclinonc.2016.58
90. Nishino M, Ramaiya NH, Hatabu H, Hodi FS. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol. (2017) 14:655–68. doi: 10.1038/nrclinonc.2017.88
91. Stark MC, Joubert AM, Visagie MH. Molecular farming of pembrolizumab and nivolumab. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms241210045
92. Tang Q, Chen Y, Li X, Long S, Shi Y, Yu Y, et al. The role of PD-1/PD-L1 and application of immune-checkpoint inhibitors in human cancers. Front Immunol. (2022) 13:964442. doi: 10.3389/fimmu.2022.964442
93. Li B, Jin J, Guo D, Tao Z, Hu X. Immune checkpoint inhibitors combined with targeted therapy: the recent advances and future potentials. Cancers (Basel). (2023) 15. doi: 10.3390/cancers15102858
94. Tian Y, Zhang C, Dang Q, Wang K, Liu Q, Liu H, et al. Risk of rash in PD-1 or PD-L1-related cancer clinical trials: A systematic review and meta-analysis. J Oncol. (2022) 2022:4976032. doi: 10.1155/2022/4976032
95. Feng C, Zhang L, Chang X, Qin D, Zhang T. Regulation of post-translational modification of PD-L1 and advances in tumor immunotherapy. Front Immunol. (2023) 14:1230135. doi: 10.3389/fimmu.2023.1230135
96. Nihira NT, Miki Y. Regulation of intrinsic functions of PD-L1 by post-translational modification in tumors. Front Oncol. (2022) 12:825284. doi: 10.3389/fonc.2022.825284
97. Dai X, Gao Y, Wei W. Post-translational regulations of PD-L1 and PD-1: Mechanisms and opportunities for combined immunotherapy. Semin Cancer Biol. (2022) 85:246–52. doi: 10.1016/j.semcancer.2021.04.002
98. Hu X, Lin Z, Wang Z, Zhou Q. Emerging role of PD-L1 modification in cancer immunotherapy. Am J Cancer Res. (2021) 11:3832–40.
99. Hou B, Chen T, Zhang H, Li J, Wang P, Shang G. The E3 ubiquitin ligases regulate PD-1/PD-L1 protein levels in tumor microenvironment to improve immunotherapy. Front Immunol. (2023) 14:1123244. doi: 10.3389/fimmu.2023.1123244
100. Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. (2018) 553:91–5. doi: 10.1038/nature25015
101. Zhang Y, Zeng L, Wang M, Yang Z, Zhang H, Gao L, et al. RIG-I promotes immune evasion of colon cancer by modulating PD-L1 ubiquitination. J Immunother Cancer. (2023) 11. doi: 10.1136/jitc-2023-007313
102. Meng X, Liu X, Guo X, Jiang S, Chen T, Hu Z, et al. FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature. (2018) 564:130–5. doi: 10.1038/s41586-018-0756-0
103. Jing W, Wang G, Cui Z, Xiong G, Jiang X, Li Y, et al. FGFR3 destabilizes PD-L1 via NEDD4 to control T-cell-mediated bladder cancer immune surveillance. Cancer Res. (2022) 82:114–29. doi: 10.1158/0008-5472.CAN-21-2362
104. Wei M, Mo Y, Liu J, Zhai J, Li H, Xu Y, et al. Ubiquitin ligase RNF125 targets PD-L1 for ubiquitination and degradation. Front Oncol. (2022) 12:835603. doi: 10.3389/fonc.2022.835603
105. Jiang C, He L, Xiao S, Wu W, Zhao Q, Liu F. E3 ubiquitin ligase RNF125 suppresses immune escape in head and neck squamous cell carcinoma by regulating PD-L1 expression. Mol Biotechnol. (2023) 65:891–903. doi: 10.1007/s12033-022-00587-w
106. Sun Z, Mai H, Xue C, Fan Z, Li J, Chen H, et al. Hsa-LINC02418/mmu-4930573I07Rik regulated by METTL3 dictates anti-PD-L1 immunotherapeutic efficacy via enhancement of Trim21-mediated PD-L1 ubiquitination. J Immunother Cancer. (2023) 11:e007415. doi: 10.1136/jitc-2023-007415
107. Gao L, Xia L, Ji W, Zhang Y, Xia W, Lu S. Knockdown of CDK5 down-regulates PD-L1 via the ubiquitination-proteasome pathway and improves antitumor immunity in lung adenocarcinoma. Transl Oncol. (2021) 14:101148. doi: 10.1016/j.tranon.2021.101148
108. Kara-Ali GH, Cano L, Dion S, Imerzoukene G, Hamon A, Simoes Eugenio M, et al. Trim21 deficiency in mice increases HCC carcinogenesis in a NASH context and is associated with immune checkpoint upregulation. Int J Cancer. (2024). doi: 10.1002/ijc.34869
109. Lyle C, Richards S, Yasuda K, Napoleon MA, Walker J, Arinze N, et al. c-Cbl targets PD-1 in immune cells for proteasomal degradation and modulates colorectal tumor growth. Sci Rep. (2019) 9:20257. doi: 10.1038/s41598-019-56208-1
110. Liu J, Wei L, Hu N, Wang D, Ni J, Zhang S, et al. FBW7-mediated ubiquitination and destruction of PD-1 protein primes sensitivity to anti-PD-1 immunotherapy in non-small cell lung cancer. J Immunother Cancer. (2022) 10:e005116. doi: 10.1136/jitc-2022-005116
111. Huang X, Zhang Q, Lou Y, Wang J, Zhao X, Wang L, et al. USP22 deubiquitinates CD274 to suppress anticancer immunity. Cancer Immunol Res. (2019) 7:1580–90. doi: 10.1158/2326-6066.CIR-18-0910
112. Wang Y, Sun Q, Mu N, Sun X, Wang Y, Fan S, et al. The deubiquitinase USP22 regulates PD-L1 degradation in human cancer cells. Cell Commun Signal. (2020) 18:112. doi: 10.1186/s12964-020-00612-y
113. Wang Z, Kang W, Li O, Qi F, Wang J, You Y, et al. Abrogation of USP7 is an alternative strategy to downregulate PD-L1 and sensitize gastric cancer cells to T cells killing. Acta Pharm Sin B. (2021) 11:694–707. doi: 10.1016/j.apsb.2020.11.005
114. Dai X, Lu L, Deng S, Meng J, Wan C, Huang J, et al. USP7 targeting modulates anti-tumor immune response by reprogramming Tumor-associated Macrophages in Lung Cancer. Theranostics. (2020) 10:9332–47. doi: 10.7150/thno.47137
115. Li B, Wang B. USP7 enables immune escape of glioma cells by regulating PD-L1 expression. Immunol Invest. (2022) 51:1921–37. doi: 10.1080/08820139.2022.2083972
116. Xiong W, Gao X, Zhang T, Jiang B, Hu MM, Bu X, et al. USP8 inhibition reshapes an inflamed tumor microenvironment that potentiates the immunotherapy. Nat Commun. (2022) 13:1700. doi: 10.1038/s41467-022-29401-6
117. Yang H, Zhang X, Lao M, Sun K, He L, Xu J, et al. Targeting ubiquitin-specific protease 8 sensitizes anti-programmed death-ligand 1 immunotherapy of pancreatic cancer. Cell Death Differ. (2023) 30:560–75. doi: 10.1038/s41418-022-01102-z
118. Liu Z, Wang T, She Y, Wu K, Gu S, Li L, et al. N(6)-methyladenosine-modified circIGF2BP3 inhibits CD8(+) T-cell responses to facilitate tumor immune evasion by promoting the deubiquitination of PD-L1 in non-small cell lung cancer. Mol Cancer. (2021) 20:105. doi: 10.1186/s12943-021-01398-4
119. Ren W, Xu Z, Chang Y, Ju F, Wu H, Liang Z, et al. Pharmaceutical targeting of OTUB2 sensitizes tumors to cytotoxic T cells via degradation of PD-L1. Nat Commun. (2024) 15:9. doi: 10.1038/s41467-023-44466-7
120. Chan LC, Li CW, Xia W, Hsu JM, Lee HH, Cha JH, et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J Clin Invest. (2019) 129:3324–38. doi: 10.1172/JCI126022
121. Zhao X, Wei Y, Chu YY, Li Y, Hsu JM, Jiang Z, et al. Phosphorylation and stabilization of PD-L1 by CK2 suppresses dendritic cell function. Cancer Res. (2022) 82:2185–95. doi: 10.1158/0008-5472.CAN-21-2300
122. Miao Z, Li J, Wang Y, Shi M, Gu X, Zhang X, et al. Hsa_circ_0136666 stimulates gastric cancer progression and tumor immune escape by regulating the miR-375/PRKDC Axis and PD-L1 phosphorylation. Mol Cancer. (2023) 22:205. doi: 10.1186/s12943-023-01883-y
123. Gao Y, Nihira NT, Bu X, Chu C, Zhang J, Kolodziejczyk A, et al. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat Cell Biol. (2020) 22:1064–75. doi: 10.1038/s41556-020-0562-4
124. Xu FF, Sun HM, Fang RP, Zhang L, Shi H, Wang X, et al. The modulation of PD-L1 induced by the oncogenic HBXIP for breast cancer growth. Acta Pharmacol Sin. (2022) 43:429–45. doi: 10.1038/s41401-021-00631-6
125. Yang Y, Hsu JM, Sun L, Chan LC, Li CW, Hsu JL, et al. Palmitoylation stabilizes PD-L1 to promote breast tumor growth. Cell Res. (2019) 29:83–6. doi: 10.1038/s41422-018-0124-5
126. Yao H, Lan J, Li C, Shi H, Brosseau JP, Wang H, et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat BioMed Eng. (2019) 3:306–17. doi: 10.1038/s41551-019-0375-6
127. Yao H, Li C, He F, Song T, Brosseau JP, Wang H, et al. A peptidic inhibitor for PD-1 palmitoylation targets its expression and functions. RSC Chem Biol. (2021) 2:192–205. doi: 10.1039/D0CB00157K
128. Li Z, Jiang D, Liu F, Li Y. Involvement of ZDHHC9 in lung adenocarcinoma: regulation of PD-L1 stability via palmitoylation. In Vitro Cell Dev Biol Anim. (2023) 59:193–203. doi: 10.1007/s11626-023-00755-5
129. Qu T, Zhang W, Yan C, Ren D, Wang Y, Guo Y, et al. ISG15 targets glycosylated PD-L1 and promotes its degradation to enhance antitumor immune effects in lung adenocarcinoma. J Transl Med. (2023) 21:341. doi: 10.1186/s12967-023-04135-1
130. Wu Z, Cao Z, Yao H, Yan X, Xu W, Zhang M, et al. Coupled deglycosylation-ubiquitination cascade in regulating PD-1 degradation by MDM2. Cell Rep. (2023) 42:112693. doi: 10.1016/j.celrep.2023.112693
131. Liu X, Zhang Y, Han Y, Lu W, Yang J, Tian J, et al. Overexpression of GLT1D1 induces immunosuppression through glycosylation of PD-L1 and predicts poor prognosis in B-cell lymphoma. Mol Oncol. (2020) 14:1028–44. doi: 10.1002/1878-0261.12664
132. Lu D, Xu Z, Zhang D, Jiang M, Liu K, He J, et al. PD-1 N58-glycosylation-dependent binding of monoclonal antibody cemiplimab for immune checkpoint therapy. Front Immunol. (2022) 13:826045. doi: 10.3389/fimmu.2022.826045
133. Ma XM, Luo YF, Zeng FF, Su C, Liu X, Li XP, et al. TGF-beta1-Mediated PD-L1 Glycosylation Contributes to Immune Escape via c-Jun/STT3A Pathway in Nasopharyngeal Carcinoma. Front Oncol. (2022) 12:815437. doi: 10.3389/fonc.2022.815437
134. He C, Xing X, Chen HY, Gao M, Shi J, Xiang B, et al. UFL1 ablation in T cells suppresses PD-1 UFMylation to enhance anti-tumor immunity. Mol Cell. (2024) 84(6):1120–1138.e8. doi: 10.1016/j.molcel.2024.01.024
135. Zhou J, Ma X, He X, Chen B, Yuan J, Jin Z, et al. Dysregulation of PD-L1 by UFMylation imparts tumor immune evasion and identified as a potential therapeutic target. Proc Natl Acad Sci U S A. (2023) 120:e2215732120. doi: 10.1073/pnas.2215732120
136. Hu X, Wang J, Chu M, Liu Y, Wang ZW, Zhu X. Emerging role of ubiquitination in the regulation of PD-1/PD-L1 in cancer immunotherapy. Mol Ther. (2021) 29:908–19. doi: 10.1016/j.ymthe.2020.12.032
137. Guo J, Zhao J, Fu W, Xu Q, Huang D. Immune evasion and drug resistance mediated by USP22 in cancer: novel targets and mechanisms. Front Immunol. (2022) 13:918314. doi: 10.3389/fimmu.2022.918314
138. Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. (2016) 7:12632. doi: 10.1038/ncomms12632
139. Lee HH, Wang YN, Xia W, Chen CH, Rau KM, Ye L, et al. Removal of N-linked glycosylation enhances PD-L1 detection and predicts anti-PD-1/PD-L1 therapeutic efficacy. Cancer Cell. (2019) 36:168–78.e4. doi: 10.1016/j.ccell.2019.06.008
140. Liu K, Tan S, Jin W, Guan J, Wang Q, Sun H, et al. N-glycosylation of PD-1 promotes binding of camrelizumab. EMBO Rep. (2020) 21:e51444. doi: 10.15252/embr.202051444
141. Komatsu M, Inada T, Noda NN. The UFM1 system: Working principles, cellular functions, and pathophysiology. Mol Cell. (2024) 84:156–69. doi: 10.1016/j.molcel.2023.11.034
142. Wang X, Xu X, Wang Z. The post-translational role of UFMylation in physiology and disease. Cells. (2023) 12:2543. doi: 10.3390/cells12212543
143. Cheng B, Ren Y, Cao H, Chen J. Discovery of novel resorcinol diphenyl ether-based PROTAC-like molecules as dual inhibitors and degraders of PD-L1. Eur J Med Chem. (2020) 199:112377. doi: 10.1016/j.ejmech.2020.112377
144. Cotton AD, Nguyen DP, Gramespacher JA, Seiple IB, Wells JA. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J Am Chem Soc. (2021) 143:593–8. doi: 10.1021/jacs.0c10008
145. Gramespacher JA, Cotton AD, Burroughs PWW, Seiple IB, Wells JA. Roadmap for optimizing and broadening antibody-based PROTACs for degradation of cell surface proteins. ACS Chem Biol. (2022) 17:1259–68. doi: 10.1021/acschembio.2c00185
146. Su W, Tan M, Wang Z, Zhang J, Huang W, Song H, et al. Targeted degradation of PD-L1 and activation of the STING pathway by carbon-dot-based PROTACs for cancer immunotherapy. Angew Chem Int Ed Engl. (2023) 62:e202218128. doi: 10.1002/anie.202218128
147. Wang Y, Zhou Y, Cao S, Sun Y, Dong Z, Li C, et al. In vitro and in vivo degradation of programmed cell death ligand 1 (PD-L1) by a proteolysis targeting chimera (PROTAC). Bioorg Chem. (2021) 111:104833. doi: 10.1016/j.bioorg.2021.104833
148. Dai MY, Shi YY, Wang AJ, Liu XL, Liu M, Cai HB. High-potency PD-1/PD-L1 degradation induced by Peptide-PROTAC in human cancer cells. Cell Death Dis. (2022) 13:924. doi: 10.1038/s41419-022-05375-7
149. Sun R, Meng Z, Lee H, Offringa R, Niehrs C. ROTACs leverage signaling-incompetent R-spondin for targeted protein degradation. Cell Chem Biol. (2023) 30:739–52.e8. doi: 10.1016/j.chembiol.2023.05.010
150. Shi YY, Wang AJ, Liu XL, Dai MY, Cai HB. Stapled peptide PROTAC induced significantly greater anti-PD-L1 effects than inhibitor in human cervical cancer cells. Front Immunol. (2023) 14:1193222. doi: 10.3389/fimmu.2023.1193222
151. Shi YY, Dong DR, Fan G, Dai MY, Liu M. A cyclic peptide-based PROTAC induces intracellular degradation of palmitoyltransferase and potently decreases PD-L1 expression in human cervical cancer cells. Front Immunol. (2023) 14:1237964. doi: 10.3389/fimmu.2023.1237964
152. Jiang W, Pan S, Chen X, Wang ZW, Zhu X. The role of lncRNAs and circRNAs in the PD-1/PD-L1 pathway in cancer immunotherapy. Mol Cancer. (2021) 20:116. doi: 10.1186/s12943-021-01406-7
Keywords: PROTAC, PD-1, PD-L1, immunotherapy, ubiquitination
Citation: Ren X, Wang L, Liu L and Liu J (2024) PTMs of PD-1/PD-L1 and PROTACs application for improving cancer immunotherapy. Front. Immunol. 15:1392546. doi: 10.3389/fimmu.2024.1392546
Received: 28 February 2024; Accepted: 22 March 2024;
Published: 04 April 2024.
Edited by:
Zong Sheng Guo, University at Buffalo, United StatesReviewed by:
Zhi-Wei Wang, Wenzhou Medical University, ChinaYizeng Fan, Xi’an Jiaotong University, China
Copyright © 2024 Ren, Wang, Liu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Liu, bGl1anVhbjgyOTFAMTYzLmNvbQ==; Likun Liu, ZHpxYnJzamRsbEAxNjMuY29t
†These authors have contributed equally to this work