 Hanming Yu
Hanming Yu Shi Liu
Shi Liu Shuo Wang
Shuo Wang- Department of Pulmonary and Critical Care Medicine, The Fourth Affiliated Hospital of China Medical University, Shenyang, China
Acute lung injury (ALI) and its severe counterpart, acute respiratory distress syndrome (ARDS), are critical respiratory conditions with high mortality rates due primarily to acute and intense pulmonary inflammation. Despite significant research advances, effective pharmacological treatments for ALI and ARDS remain unavailable, highlighting an urgent need for therapeutic innovation. Notably, idiopathic pulmonary fibrosis (IPF) is a chronic, progressive disease characterized by the irreversible progression of fibrosis, which is initiated by repeated damage to the alveolar epithelium and leads to excessive extracellular matrix deposition. This condition is further complicated by dysregulated tissue repair and fibroblast dysfunction, exacerbating tissue remodeling processes and promoting progression to terminal pulmonary fibrosis. Similar to that noted for ALI and ARDS, treatment options for IPF are currently limited, with no specific drug therapy providing a cure. Histone deacetylase 3 (HDAC3), a notable member of the HDAC family with four splice variants (HD3α, -β, -γ, and -δ), plays multiple roles. HDAC3 regulates gene transcription through histone acetylation and adjusts nonhistone proteins posttranslationally, affecting certain mitochondrial and cytoplasmic proteins. Given its unique structure, HDAC3 impacts various physiological processes, such as inflammation, apoptosis, mitochondrial homeostasis, and macrophage polarization. This article explores the intricate role of HDAC3 in ALI/ARDS and IPF and evaluates its therapeutic potential the treatment of these severe pulmonary conditions.
1 Introduction
Sepsis, a major global health threat, is predominantly characterized by high morbidity and mortality rates. An excessive inflammatory response induced by sepsis can precipitate multiorgan dysfunction and ultimately cause organ failure in severe cases (1). The lung is a particularly vulnerable organ to infection and the systemic inflammatory disorder sepsis (2). An immune and inflammatory response is evident within lung tissue during sepsis, impacting the integrity of the alveolar-capillary membrane barrier and contributing to edema formation (3). Due to the complexity of its underlying mechanisms, effective treatments for acute lung injury (ALI) remain elusive, with supportive care being the only available option to date.
ALI is an extreme clinicopathological entity characterized by unyielding hypoxemia and escalating dyspnea. In the absence of timely intervention, ALI has the potential to intensify to acute respiratory distress syndrome (ARDS) and multiple organ dysfunction syndrome (MODS). The latter two are important causes of death in critically ill patients (4–6). The central pathophysiological characteristics include enhanced pulmonary vascular permeability, interstitial edema, alveolar fibrin leakage, augmented intrapulmonary shunting, and a disruption in ventilation-perfusion equilibrium (7, 8). After acute inflammation and damage, the lung activates repair and remodeling processes to regain homeostasis. The resultant fibrosis and scarring potentially resolve over time or endure, accelerating persistent long-term pulmonary fibrotic damage (9–11). Signaling pathways act as crucial agents in the treatment of ALI and lung fibrosis. The initiation of these events by lipopolysaccharide (LPS) stimulation triggers several molecular intracellular signaling activities, including the classical activation of nuclear factor-kappa-B (NF-κB). This facilitates its translocation to the nucleus, consequently leading to the release of predominantly inflammatory cytokines, specifically interleukins (IL-1β, IL-2, and IL-6) and chemokines (macrophage inflammatory protein, MIP-1 α/β) (12–14). Within the scope of epigenetic mechanisms, strategic alterations in histone acetylation and deacetylation play a pivotal role in governing DNA accessibility (15). The deacetylation reaction is executed by histone deacetylases (HDACs), a series of enzymes partitioned into four classes (I-IV). Their primary function is to remove acetyl residues from histone tails, which facilitates DNA pairing with nucleosomes, prompts heterochromatin formulation, and hinders gene transcription (16). The activation and differentiation of fibroblasts into myofibroblasts, characterized by the expression of contractile proteins and an antiapoptotic phenotype, are critical factors involved in the pathogenesis of IPF (17, 18). The upregulation of HDAC in fibroblasts has been observed in IPF (19, 20). Furthermore, the use of HDAC inhibitors (HDACis) has demonstrated beneficial effects in preventing or reversing fibrogenesis (19–21).
IPF is an age-associated, progressively worsening, chronic, and ultimately lethal condition that manifests for several years and is characterized by inadequate remodeling of the lung parenchyma. The etiology of IPF is hypothesized to be centered on epithelial cell-mediated fibrosis. These cells are capable of activating fibroblasts that are responsible for extracellular matrix (ECM) components, leading to the fibrotic process (22, 23). IPF, a chronic and progressive fibrotic interstitial lung disease, remains elusive and is not entirely understood. Presumably, IPF is precipitated by repetitive harm to alveolar epithelial cells and consequent inadequate repair. The median survival duration postdiagnosis of IPF patients is projected to be between 2.5 and 3.5 years (22). Within the context of IPF, epigenetic reorientation occurs due to cell-microenvironment interactions, which establish a feedback loop that fosters a pro-fibrosis environment. This feedback loops subsequently bolsters the differentiation of cells into myofibroblasts and catalyzes the expression of genes implicated in the maladaptive restoration of the extracellular matrix (24–26). Presently, therapeutic options for IPF are notably limited, with lung transplantation remaining the only definitive treatment. The global prevalence of IPF is increasing, correlating with significant increases in morbidity and mortality. Moreover, this disease imposes a considerable economic burden on health care systems worldwide.
The roles of certain HDACs in ALI have been uncovered recently. For instance, in mice with pneumonia-triggered ALI, HDAC7 inhibition through trichostatin A significantly mitigates inflammatory damage in lung tissue, subsequently enhancing survival (27). The inhibition of histone deacetylase 3 (HDAC3) significantly curtails the mitochondrial pathway of apoptosis while preserving the mitochondrial membrane potential in cases of cold renal storage/transplantation injury (28). Furthermore, HDAC3 can regulate inflammatory reactions through its unconventional deacetylase-independent activity, thereby controlling the inflammatory response (29). HDAC3 has been shown to be a critical regulator necessary for the macrophage inflammatory response following stimulation by inflammatory factors (30). Additionally, HDAC3 affects phagocytosis in M2 phenotype macrophages by regulating the transforming growth factor-β (TGFβ) and HDAC3-associated Smad signaling pathways (31). Furthermore, HDAC3 plays a significant role in modulating the inflammatory response and endotoxin tolerance in human monocytes and macrophages (32). A recent study has demonstrated that the attenuation of HDAC3 significantly enhances survival in mice with LPS-induced endotoxemia (33).
Despite progress made in recent decades, an optimal therapeutic strategy for ALI has yet to be developed, and broadly targeted anti-inflammatory therapies have primarily been unsuccessful in improving mortality rates. This lack of progress is largely due to the underlying molecular mechanisms that drive ALI, which continue to be inadequately understood (8). On the other hand, the cause and pathogenesis of IPF are currently unclear. With patients having a grim prognosis and limited treatment options available, it has become fundamental to gain an in-depth understanding of the molecular mechanisms steering both ALI and IPF for the prospective management of these conditions. Epigenetic modifications, which influence gene expression and developmental pathways without altering the underlying DNA sequence, hold potential as targets for such novel interventions. Among the principal mechanisms of epigenetic regulation, modifications of histones, specifically through acetylation and deacetylation, have been implicated in an array of pathological and physiological processes within mammalian systems (34). Under various pathological conditions, epigenetic alterations of histones in eukaryotic cells are known to modulate gene expression by promoting chromatin structural stability, which can precipitate oxidative stress, inflammation, and cellular apoptosis (35, 36). HDAC3 exerts its regulatory role not only by deacetylating histones and thus modulating gene transcription but also by posttranslationally modifying a variety of nonhistone proteins, including select proteins within mitochondrial and cytoplasmic compartments (35). In this review, we outline the significance of HDAC3 and its inhibitors in the context of acute lung injury and pulmonary fibrosis, as observed in recent years, in an attempt to provide some clinical implications.
2 Biological properties of histone deacetylases
Histone deacetylases (HDACs) are a family of proteases that modify histone deacetylation, regulate chromatin state, and ultimately affect gene expression (37) and play an important role in epigenetic regulation. Eighteen different HDAC isoforms have been identified in mammals and are classified into four classes based on their homology and subcellular localization with yeast germline HDACs (38, 39). Class I HDACs include HDAC1, HDAC2, HDAC3 and HDAC8, the sequences of which are homologous to those of the yeast deacetylase reduced potassium dependency 3 (RPD3), which is mainly localized in the nucleus and involved in the regulation of cell proliferation (39, 40). Class II HDACs can be further divided into class IIa (HDAC4, HDAC5, HDAC7 and HDAC9) and class IIb (HDAC6 and HDAC10) (37, 41). Class III HDACs do not contain a zinc finger binding domain in their structure and are dependent on the metabolic cofactor nicotinamide adenine dinucleotide (NADD) (37, 42). The structure of class III HDACs does not contain a zinc finger binding domain, and these enzymes rely on the metabolic cofactor nicotinamide adenine dinucleotide (NAD+) for deacetylation. Class IV HDACs, localized in the nucleus and comprising solely HDAC11, possess a catalytic core region akin to that of Class I and Class II HDACs (43). HDAC11 has multiple functions, including the regulation of interleukin-10 (IL-10) expression (42) and the modulation of type I interferon signaling (44).
As a constituent of class I HDACs, HDAC3 is reported to be distinct within the HDAC chromatin modifier superfamily. In the nucleus, its activity differs with that of other deacetylases owing to its exclusive substrates. These include lysine 9 of histone 3 (H3K9ac) and lysine 5 (H3K5ac), along with lysine 9 of histone 4 (H4K9ac) (45, 46). HDAC3 plays a role in numerous inflammatory conditions, organ fibrosis, neurodegeneration, and ischemic injury. It accomplishes this by mediating histone deacetylation, subsequently fostering chromatin condensation and transcriptional repression (47). HDAC3 not only modulates gene transcription by deacetylating histones but also posttranslationally modifies nonhistone proteins, which include particular mitochondrial and cytoplasmic proteins (35, 36). Although HDAC3 is well-recognized for its deacetylation function, recent reports have demonstrated that HDAC3 can also act as an “eraser” of lactonylation (48, 49). Lactonylation, a novel epigenetic modification, plays a crucial role in macrophage activation and in the regulation of lung injury and fibrosis (50, 51). During the later stages of M1 macrophage polarization, increased histone lactylation induces the expression of homeostatic genes involved in wound healing, such as Arginase 1 (52). Recent studies have identified HDAC3 as a highly potent delactoylase, exhibiting activity levels thousands of times higher than SIRT2, previously considered the primary delactoylase in vivo (53). The de-L-lactamase activity of class I histone deacetylases (HDAC1-3) within cells has been further validated in a study by Moreno-Yruela et al. (49).HDAC3 either directly or indirectly interacts with various transcriptional regulatory proteins (Figure 1), suggesting that HDAC3 is involved in a diverse array of signaling pathways and cellular processes.

Figure 1. HDAC3 interacts directly or indirectly with a number of transcriptional regulatory proteins (from STRING.COM).
3 HDAC3 in acute lung injury
LPS stimulation significantly upregulated HDAC3 mRNA and protein levels in mouse lung tissues (33, 54, 55).
3.1 Impact of HDAC3 regulation on macrophage-driven lung inflammation
Macrophage cohorts in the lungs maintain homeostasis by phagocytizing inhaled particles and foreign pathogens, triggering cytokine production, and facilitating antigen presentation. These actions collectively contribute to the eradication of particulate antigens (56, 57). Although macrophages indeed play a pivotal role in fending off invading organisms, their overabundance may precipitate tissue damage (58). Within the context of sepsis-induced ALI, lung tissue experiences an influx of a significant number of peripheral macrophages. This infiltration results in the increased production of various proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin (IL)-1β. Additionally, increased release of high mobility group box 1 (HMGB1) is noted (12, 57). In subsequent processes, neutrophils in the circulatory system are mobilized toward the alveolar space and interstitial tissues, precipitating inflammatory exudation and tissue damage (57). When macrophages undergo LPS activation and in the absence of nuclear receptor corepressor 1/2 (NCoR1/2), HDAC3 encroaches upon the activating transcription factor 2 (ATF2) binding site, initiating the expression of inflammatory genes in a noncanonical fashion. Conversely, the deacetylase activity of HDAC3 selectively interacts with the ATF3 binding site, inhibiting Toll-like receptor (TLR) signaling. Interestingly, HDAC3 deletion in macrophages resulted in LPS resistance in mice (29).
A study led by Natoli and colleagues revealed that upon LPS stimulation, close to half of the inflammatory genes in macrophages were not expressed when HDAC3 was inhibited. This finding suggested that HDAC3 inhibition has a potential anti-inflammatory effect (59). HDAC3 is capable of initiating the cyclic GMP-AMP synthase (cGAS)–stimulator of interferon genes (STING) pathway by mitigating histone acetylation of the miR-4767 gene promoter, which is induced by LPS in macrophages. This trigger initiates an inflammatory response and thermokinetic-selectivity (33, 36). By modulating HDAC3 to suppress macrophage activation, the level of inflammation-related cytokines in the lung decreases, which subsequently improves the severity of LPS-induced septic shock. This process operates through the HMGB1/NF-κB axis (60). HDAC3 was identified as a critical modulator of IL-12b transcription. IL-10 reduces IL-12b synthesis by regulating HDAC3 activity on the IL-12b promoter. A notable reduction in the nuclear translocation of the p65 NF-κB subunit RelA in LPS-stimulated alveolar macrophages is observed with the disruption of the class I HDAC signaling axis by IL-10 (61).
3.2 Unraveling the role of HDAC3 in regulating the NF-κB pathway and inflammatory response
Several intracellular signaling events, including classic NF-κB activation, commence upon LPS stimulation. This action facilitates nuclear translocation, resulting in the release of primary inflammatory cytokines, chiefly interleukins (IL-1β, IL-2, and IL-6) and chemokines (macrophage inflammatory protein, MIP-1α/β) (12). In an extensive study, Elisabeth Ziesché and colleagues deciphered the pivotal role of HDAC-3, demonstrating its critical function as a coactivator in IL-1-induced inflammatory signaling, and this role is achieved through the removal of acetyls inhibited by p65NF-κB (62). Similarly, a study conducted by Leus and colleagues used RGFP966, an HDAC-3-selective inhibitor, in a lung inflammation model. They observed an increase in NF-κB transcription paired with a decrease in the expression of inflammatory cytokines, such as IL-1β, IL-6, and IL-12b, in macrophages. Concurrently, they noted an upregulation of the anti-inflammatory cytokine IL-10 (63). This notable study highlights the potential role of inhibiting NF-κB and HDAC-3 translocation. Such inhibition results in a reduction in proinflammatory cytokines and fosters redox homeostasis, thereby alleviating the inflammation typically associated with ARDS (55).
The significance of HDAC3 in promoting HMGB1 hyperacetylation and subsequent translocation is gaining recognition (60, 64, 65). Studies have shown that elevated HDAC3 levels inhibit LPS-induced HMGB1 translocation and secretion in RAW264.7 cells (65). Specifically, HDAC3 can enhance the transcriptional activity of the NF-κB p65 subunit and modulate the NF-κB signaling pathway through deacetylation, thereby reducing inflammatory responses (64). In in vitro experiments, HDAC3 reduces the acetylation level of HMGB1, inhibiting its translocation from the nucleus to the cytoplasm, a process associated with the intervention of NF-κB signaling (65). In vivo studies also show that the deletion or inhibition of HDAC3 leads to increased HMGB1 hyperacetylation, which can elevate the expression of inflammatory factors, indicating the crucial role of HDAC3 in regulating inflammatory responses (36). Moreover, in vivo research by Guo et al. corroborated the concept that HDAC3-dependent pathways may impair the nuclear factor erythroid 2-related factor 2 (Nrf2)/antioxidant response element (ARE) pathway through NF-κB activity, thereby exacerbating oxidative stress-induced necroptosis and ischemia/reperfusion injuries by facilitating the release of HMGB1 (66). Other studies have also demonstrated a key regulatory role for HDAC in HMGB1 translocation (55, 67, 68). Wen Q and colleagues demonstrated that HDAC3 modulation mitigated LPS-induced septic shock via the HMGB1/NF-κB pathway (60). On the other hand, HDAC3 suppression resulted in decreased TNF-α levels, which were accompanied by a correlated increase in acetylated p65. Notably, p65 is an NF-κB subunit that has been reported to play a significant role in inhibiting IκB-mediated NF-κB transcriptional activity (Figure 2) (69, 70).
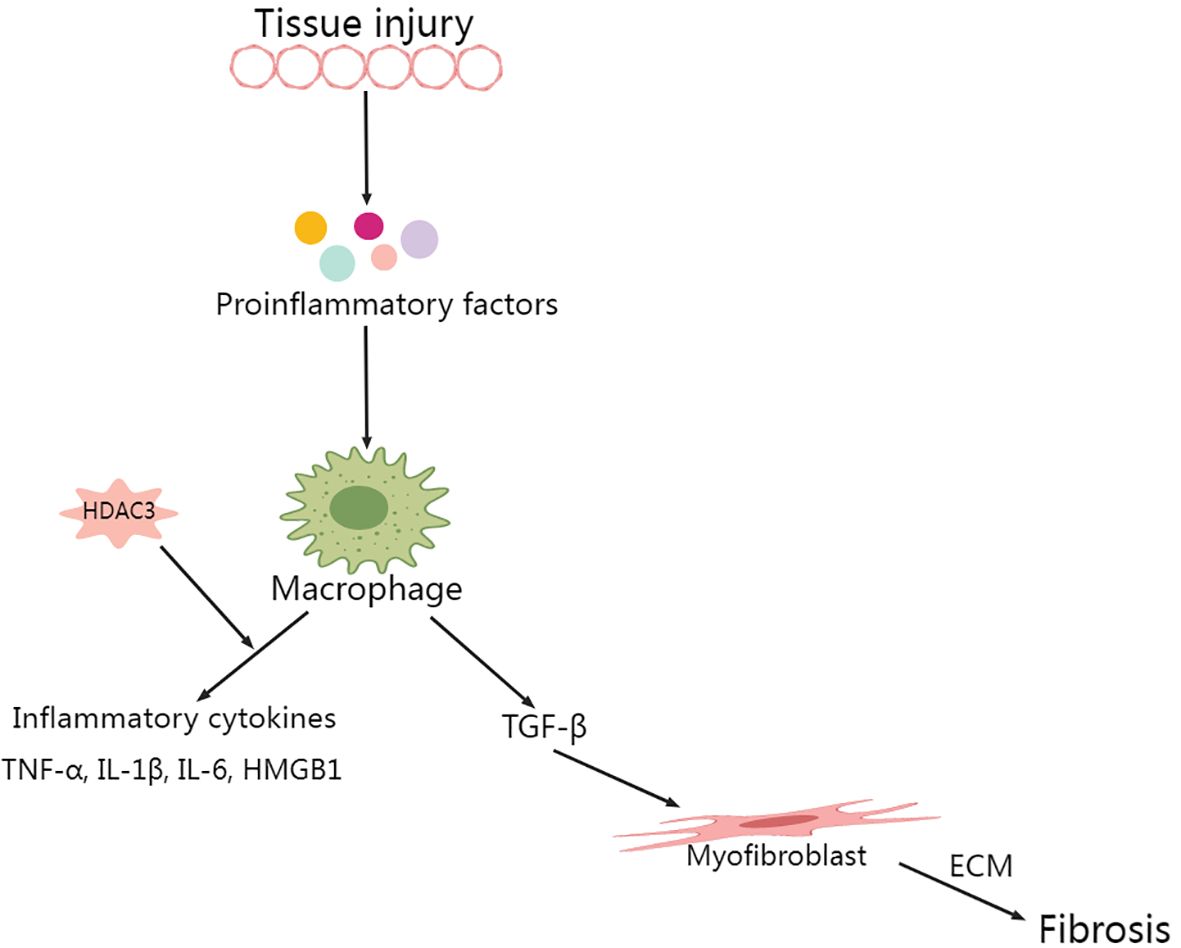
Figure 2. Role of HDAC3 in macrophage secretory spectrum during lung injury.
3.3 HDAC3 modulates mitochondrial balance, fatty acid metabolism, and epithelial disruption
Mitochondria, characterized by an elongated dual-membrane structure, reside in the cytoplasm of virtually all eukaryotic cells and possess a unique self-replicating genome (71). Wang XR and colleagues reported that mitoquinone (MitoQ) mitigated alveolar epithelial cell apoptosis and prevented barrier disintegration by controlling mitochondrial fission during sepsis-induced ALI (72).
HDAC3 facilitates the regulation of gene transcription through the deacetylation of histones but also posttranslationally modifies nonhistone proteins, notably certain cytoplasmic and mitochondrial proteins (35). A recent study highlighted the ability of HDAC3 to migrate to mitochondria, where its deacetylates an enzyme related to fatty acid oxidation (FAO), specifically the α-subunit of the mitochondrial trifunctional enzyme. This activity effectively impedes FAO within macrophages (73). RGFP966-mediated HDAC3 inhibition enhances mitochondrial membrane potential and diminish mitochondria-mediated apoptosis (28). Likewise, in scenarios involving diabetes and obesity, the pharmacological suppression of class I HDACs, accomplished through agents such as SAHA or MS275, augments mitochondrial function and oxidative capacity in adipose tissue and skeletal muscle (74). Indeed, HDAC3 plays a crucial role in energy metabolism and the maintenance of mitochondrial equilibrium. Remarkably, a significant decrease in the expression of genes related to mitochondrial oxidative phosphorylation was observed in brown adipose tissue deficient in HDAC3, resulting in diminished mitochondrial respiration. These findings suggest that HDAC3 plays an indispensable role in thermogenesis within brown adipose tissue (75). HDAC3 deficiency in brown adipose tissue resulted in considerable downregulation of mitochondrial oxidative phosphorylation genes, resulting in diminished mitochondrial respiration. This finding illustrates the crucial role of HDAC3 in thermogenesis within brown adipose tissue. Furthermore, HDAC3 facilitates Rho-associated, coiled-coil containing protein kinase 1 (ROCK1) transcription and activation via the deacetylation of Forkhead box O1 (FOXO1), which subsequently impairs mitochondrial quality control (MQC) in alveolar epithelial type II cell (AT2). This chain of events ultimately culminates in damage to the epithelial barrier and ALI (Figure 3) (54, 76).
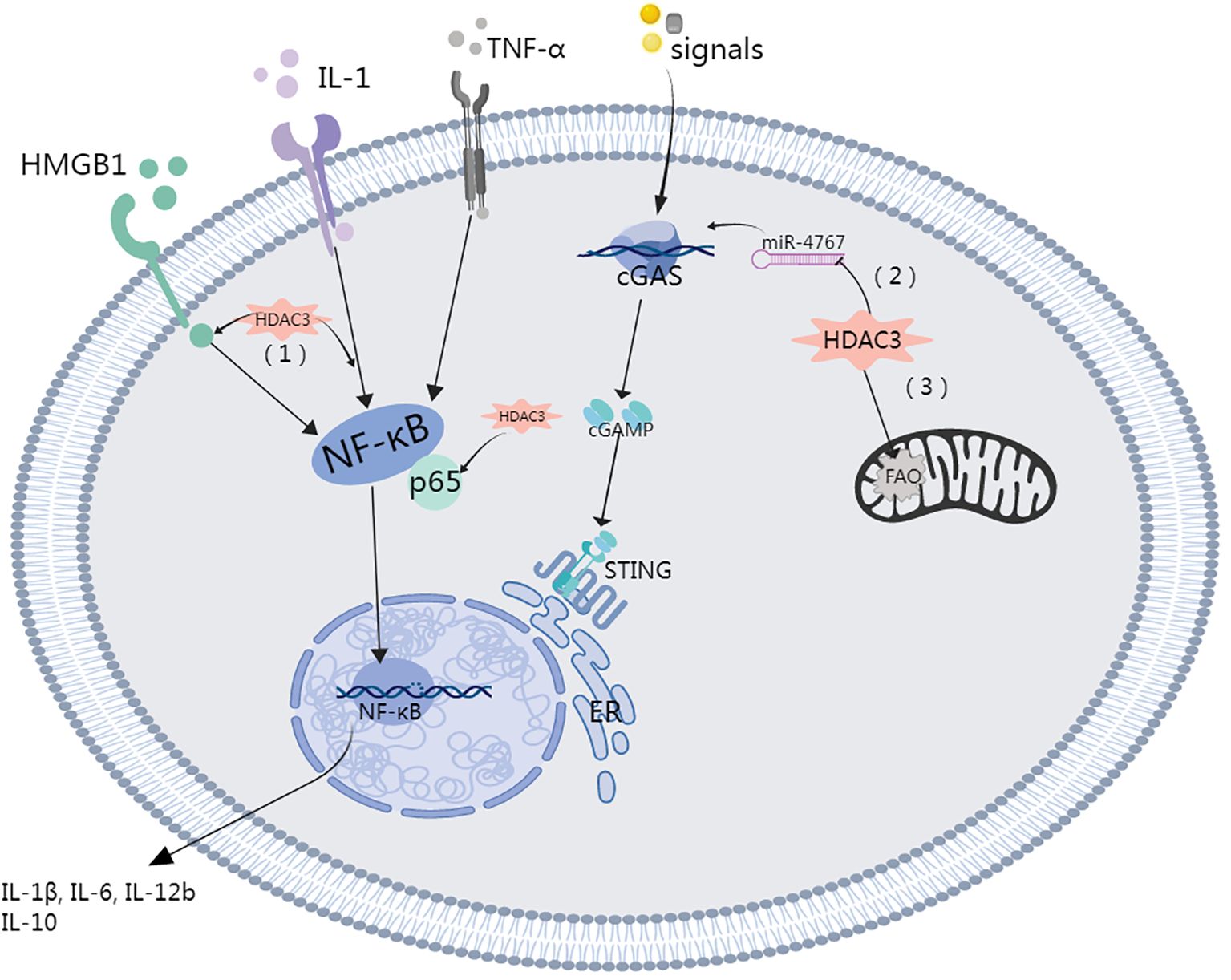
Figure 3. Role of HDAC3 in the lung injury.
4 HDAC3 in pulmonary fibrosis
The augmentation of HDAC3 is observed in diverse lung fibrosis models (77, 78), and HDAC3 is predominantly located within fibrotic lesions and pulmonary fibroblasts (79–81).
The formation of myofibroblasts from the epithelial–mesenchymal transition (EMT) of alveolar epithelial cells is a critical event in IPF (82, 83). Myofibroblasts not only are capable of synthesizing substantial amounts of ECM but also exhibit smooth muscle cell-like characteristics, such as the expression of α-smooth muscle actin (α-SMA) (84). These traits endow myofibroblasts with contractile abilities, further contributing to tissue stiffness and airway constriction (84, 85).
Lung tissue hypoxia is intrinsically linked to inflammation and fibrosis (86–88). In their seminal work, Zhou et al. elucidated how hypoxia triggers EMT under the influence of mitochondrial reactive oxygen species and hypoxia-inducible factor-1 (HIF-1) (89). Similarly, Higgins et al. revealed that hypoxia induces EMT via HIF-1 activation, ultimately promoting fibrogenesis (90). HDAC3 promotes the alveolar epithelial-mesenchymal transition and fibroblast migration under hypoxic conditions (91). This pathway directly enhances fibroblast migration and invasion. Intriguingly, a separate study implicated TGF-β1/SMAD3 signaling pathway activation as the direct trigger of the heightened HDAC3 expression typically observed in pulmonary fibrosis (92). Of paramount significance is the role of HDAC3 in mediating EMT in AT2 cells and pulmonary fibrosis. This is achieved through GATA3 deacetylation, which subsequently prevents its degradation (92). Additionally, miR-224 plays a crucial role in facilitating the HDAC3-induced EMT, primarily through the suppression of mRNA translation for recombinant forkhead box A1 (FOXA1) (91).
Pulmonary fibrosis is distinctively characterized by an increase in matrix stiffness, a consequence of the accumulation of ECM. This effectively established a self-propagating cycle that is favorable for fibroblast proliferation (93, 94). In this context, HDAC3 has been identified as a key player in the mechanotransduction response mechanism. Specifically, it contributes to regulating changes in cell morphology, chromatin accessibility, hyperacetylation, and gene expression that are typically driven by matrix stiffness or geometrical cellular constraints (95–97). HDAC3 is essential for preserving the acetylation-to-methylation ratio of H3K9 residues (98, 99). The absence of nuclear HDAC3 results in increased acetylation and decreased expression of both heterochromatin markers (H3K9me2) and fibronectin (100).
Recent research underscores the significant role of HDAC3 in the progression of fibrotic lung disease. This work indicated that RGFP966, a selective HDAC3 inhibitor, effectively diminished lung fibrosis in wild-type mice subjected to bleomycin treatment. Conversely, Hdac3 KO mice exhibit a high degree of resistance to bleomycin-induced lung fibrosis (79). Moreover, it was determined that IPF lungs exhibited HDAC3 overexpression on Days 14 and 21 after bleomycin treatment, which coincided with suppressed Nrf2 expression and expression of the pro-fibrosis forkhead box M1 (FOXM1) transcription factor during lung fibrosis progression. In contrast, RGFP966 therapy reduced HDAC3 and FOXM1 binding and increased the expression of acetylated histone H3 at the Nrf2 promoter site. These effects subsequently increased Nrf2 repression and concurrently increased the expression of Nrf2-induced antioxidants. In addition, this treatment restored epithelial E-calmodulin expression and decreased fibronectin expression (79). Similarly, Nrf2 expression was markedly absent in the fibroblast foci of IPF patients (101), whereas HDAC3 expression increased (19). It was determined that HDAC3 expedites the progression of pulmonary fibrosis by stimulating EMT and inflammation via the Notch1 or signal transducer and activator of transcription 1 (STAT1) signaling pathways (102). In summary, the atypical excessive expression of HDAC3 coupled with its inhibition of Nrf2 plays a pivotal role in IPF. Importantly, this can be moderated through the selective inhibition of HDAC3.
Moreover, HDAC3 increases interleukin 17 receptor A (IL17RA) expression in rheumatoid arthritis (RA)-associated interstitial lung disease (ILD) by down-regulating miR-19a-3p (81). Intriguingly, HDAC3 and IL17RA levels were elevated in the lung tissues of individuals with RA-ILD compared to healthy controls. Deeper examination revealed a positive correlation between the expression of HDAC3 and that of IL17RA in RA-ILD patients. Using a mouse model of RA-ILD, it was determined that HDAC3 downregulated the expression of miR-19a-3p in the lung fibroblasts of these mice, subsequently enhancing IL17/IL17RA signaling and ECM protein expression. However, this impact could be nullified through overexpression of miR-19a-3p or by silencing Il17ra or Hdac3 using siRNAs. Notably, the administration of Hdac3-targeted siRNA to RA-ILD mice noticeably ameliorated pulmonary fibrosis in vivo (Figure 4) (81).
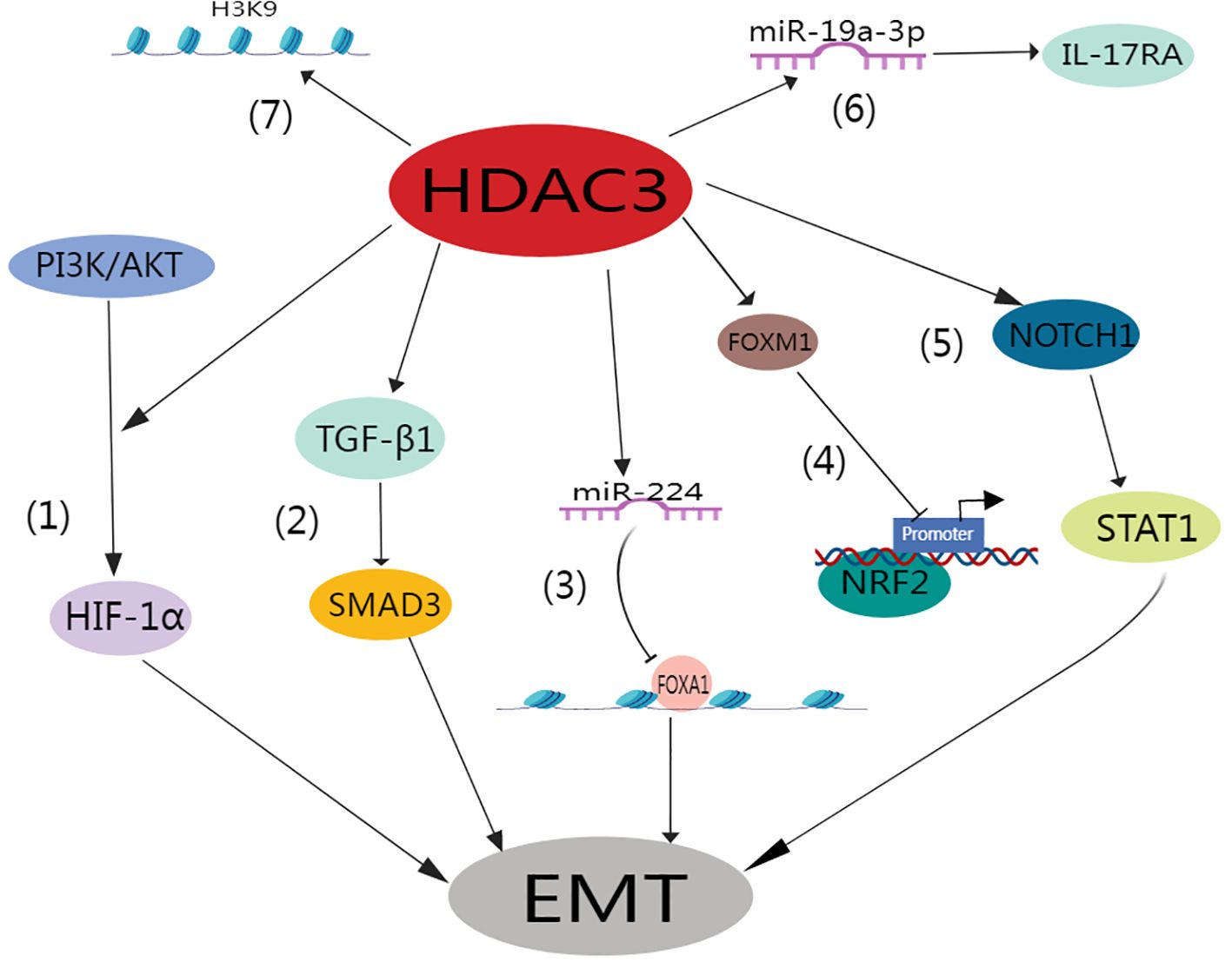
Figure 4. Role of HDAC3 in the pulmonary fibrosis.
5 Potential role of HDAC3 in ALI and IPF through NLRP3 activation
5.1 Role of HDAC3 in NLRP3 inflammasome initiation
The activation of NF-κB, which is promoted by primary signals, facilitates NLRP3 (NOD-, LRR-, and pyrin domain-containing (3)), pro-IL-1β, and pro-IL-18 transcription (103). The NF-κB transcription factor complex is at the epicenter of inflammatory gene regulation in response to the onset of immune and inflammatory stimuli. Notably, the acetylation status of p65, a subunit of NF-κB, has been implicated in its nuclear translocation (69, 104). Ziesché E et al. reported that HDAC3 is extensively coactivated via the deacetylation of NF-κB p65 and that the ablation of HDAC3 propels the acetylation of p65 (62). In LPS-stimulated mouse small glioma cells (BV2), HDAC3 inhibition correspondingly reduces the nuclear presence of p65, thereby curtailing NLRP3 protein expression and inflammation (105). These findings indicate that HDAC3 modulates p65 acetylation, thus amplifying the initiation of the NLRP3 inflammasome.
Furthermore, HDAC3 promotes NF-κB expression to increase pro-IL-1β transcription, IL-1β release, and inflammation via upstream NF-κB activators, including molecule myeloid differentiation factor 88 (Myd88), α-tubulin, and reactive oxygen species (ROS) (12, 62, 106–112). Documented interactions between HDAC3 and the TLR adapter molecule Myd88 suggest a modulatory influence (108, 111). A deficiency in HDAC3 seems to attenuate NF-κB induction through TLR4 stimulation and diminishes the secretion of inflammatory mediators, including IL-1β, IL-6, and TNF-α (108). HDAC3 inhibition increases the acetylation of α-tubulin, thus limiting microtubule depolymerization (109). ROS, primarily sourced from Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxidase (113, 114), comprises two membrane-bound subunits (p22phox and gp91phox), three cytosolic counterparts (p67phox, p47phox, and p40phox), and the small G protein Rac (Rac1 and Rac2) (115). HDAC3 upregulates the expression of Nox4-based NADPH oxidase subunits, augmenting ROS production (106, 110, 112). Hence, HDAC3 can promote NF-κB activation through Myd88, microtubules, or ROS, culminating in the engagement of the NLRP3 inflammasome; however, further elucidation of these mechanisms is imperative.
5.2 The role of HDAC3 in PPARγ/NLRP3 inflammasome activation
The peroxisome proliferator-activated receptor γ (PPARγ), a ligand-sensitive nuclear receptor, is regulated by posttranslational modifications, including phosphorylation, hemoylation, and ubiquitination (116). Recently, HDAC3 inhibition has been linked to increased acetylation and subsequent activation of PPARγ (117, 118) as well as elevated expression of PPARγ target genes (116). PPARγ activation can mitigate neuroinflammation by impeding the NLRP3 signaling cascade (119–122). Recent studies have shown that HDAC3 inhibition may attenuate neuroinflammation and enhance neurological outcomes by activating the PPARγ/NLRP3/GSDMD signaling pathway (120).
PPARγ can attenuate lung injury by inhibiting the activity of NLRP3 inflammatory vesicles through its anti-inflammatory effects (123, 124). Salvianolate Acid A (SA) efficaciously enhances the state of lipopolysaccharide-triggered acute lung injury in mice. This improvement is brought about by the diminution of M1 macrophage levels via the PPAR-γ/NLRP3 pathway (125). The activation of PPARγ, which targets the NLRP3/GSDMD/caspase-1 axis, by SYG has demonstrated potential for inhibiting macrophage pyroptosis (123). Another study showed that ACT001 (an anti-inflammatory agent) remarkably mitigates LPS-triggered acute lung injury. This occurs mainly through the modulation of the PPAR-γ/NLRP3/NF-κB signaling pathway embedded within macrophages (126). Vinpocetine activates PPAR-γ and down-regulates the NLRP3/NF-κB pathway, showing significant attenuation in lung fibrosis in vivo and in vitro (127). Moreover, in a murine model of acute liver failure, NLRP3 inflammasome-mediated inflammation and LPS-induced macrophage M1 polarization were ameliorated by modulating PPAR-γ methylation (128), which is consistent with the findings of Fu et al. (126).
The inhibitory effect of HDAC3 on NLRP3 inflammasome activation via PPARγ acetylation and activity enhancement is evident. Nevertheless, direct evidence in inflammation-afflicted macrophages remains elusive, and the current body of evidence suggests that the interplay between HDAC3 and PPARγ/NLRP3 may hold significance for treating inflammatory conditions by targeting macrophages.
5.3 Potential interplay of HDAC3 and NLRP3 in IPF pathogenesis
Recent studies have shown that in IPF patients, the inflammasome NLRP3 undergoes hyperactivation, contributing to an increase in the production of class I interleukins and collagen (129). The IL-1 superfamily is a group of 11 cytokines(IL-1α、IL-1β、IL-36α、IL-36β、IL-36γ、IL-36Ra、IL37、IL-38、IL-1Ra、IL-18和IL-33) that play a central role in the regulation of immune and inflammatory processes in response to a large group of stimuli (130). IL-18 and IL-18Ra expression is increased in patients with IPF, and experimental studies in mice have shown that administration of IL-18 exacerbates the progressive deposition of extracellular matrix components and the development of fibrosis (131). Within the lungs, IL-1β plays a crucial role in driving inflammation, promoting white blood cell migration, and contributing to the degradation of elastin fibers and the deposition of collagen (132). These pathological effects are, in part, mediated through the IL-17A signaling pathway (133, 134). These findings underscore the pivotal role of NLRP3 in fostering chronic lung injury and fibrosis (129). Furthermore, an investigation conducted by Giannarakis et al. (135) revealed a pronounced activation profile of the NLRP3 inflammasome in IPF, highlighting its significance in the pathological landscape of the disease.
Scutellaria (the principal active constituent in brevicaffin) has been demonstrated to mitigate inflammation and epithelial–mesenchymal transition (EMT) in bleomycin (BLM)-induced pulmonary fibrosis via the NF-κB/NLRP3 signaling axis (136). Concurrently, lycorine ameliorates BLM-induced pulmonary fibrosis by obstructing NLRP3 inflammasome activation and pyroptosis by targeting the pyrin domain (PYD) of the CARD (ASC) (137). These findings substantiate the integral role of the NLRP3 inflammasome in pulmonary fibrosis, indicating that certain anti-inflammatory therapeutics can modulate the disease course by inhibiting NLRP3 inflammasome activation, thereby attenuating pyroptosis.
Moreover, the activation of discoidin domain receptor 1 (DDR1) in macrophages has been identified as a proponent of IPF through the modulation of the NLRP3 inflammasome and macrophage responses (138). Similarly, SS-31 has been shown to ameliorate lung fibrosis and inflammation by inhibiting Nrf2-mediated NLRP3 inflammasome activation in macrophages (139). This insight provides a basis for future investigations into the interplay between macrophage-mediated HDAC3 and NLRP3 activation in the context of inflammation and fibrosis.
6 HDAC3 as a therapeutic target
HDAC3 is considered a potential therapeutic target for lung injury and pulmonary fibrosis. Studies have shown that HDAC3 is involved in the regulation of various biological processes, including inflammatory responses (102), immune cell function (140), cell survival and apoptosis (21, 79), and cell proliferation (54). All of these processes are associated with the onset and progression of lung injury and pulmonary fibrosis.
HDAC3 impacts various cellular signaling pathways, such as activating NOTCH1 and STAT1 signaling pathways, which influence macrophage activation and inflammatory responses from multiple angles (102). Additionally, research indicates that HDAC3 plays a crucial role in maintaining epithelial cell barrier integrity and mitochondrial quality control, particularly important in preventing acute lung injury (54). HDAC3 inhibitors have also shown potential in reducing pulmonary fibrosis by inducing fibroblast apoptosis and thus alleviating fibrosis (21, 92). Targeting HDAC3 specifically could provide a new treatment strategy for fibrosis, potentially improving therapeutic outcomes when combined with existing treatment methods (20, 91).
HDAC3 inhibitors may involve broad gene regulation, posing potential risks of side effects and toxicity, necessitating further research to confirm their safety (141). Additionally, current HDAC inhibitors lack target specificity, affecting other members of the HDAC family, which may lead to non-specific effects and side effects (79). Moreover, most research is still in the preclinical and animal experiment stage, lacking large-scale clinical trial data to confirm specific therapeutic effects and safety (140). Long-term use of HDAC3 inhibitors could also lead to drug resistance, which must be closely monitored in clinical applications and addressed through strategic planning (19).
Currently, the development of drugs that target HDAC3 involves a number of HDAC inhibitors; however, these agents tend to act on multiple HDAC isoforms rather than specifically targeting HDAC3. When developing therapeutics, establishing specific inhibitors is crucial for minimizing potential side effects and enhancing treatment efficacy.
7 Conclusion
Currently, the etiology and pathogenesis of ALI and IPF remain elusive, with patients often facing a dire prognosis and limited therapeutic options. HDAC3 inhibitors have emerged as potential pivotal agents for the treatment of acute lung injury and fibrotic diseases. HDAC3 influences the expression of inflammatory genes in macrophages, modulates the NF-κB signaling pathway, and affects the posttranslational modification of nonhistone proteins; these processes are crucial for the development and progression of lung injury and pulmonary fibrosis. Nonetheless, the precise mechanisms of action necessitate validation through extensive research in animal models of acute lung injury and pulmonary fibrosis as well as in vitro patient-derived studies. Therefore, targeting HDAC3 potentially represents an innovative therapeutic approach for acute lung injuries and fibrotic lung diseases. In summary, although research on the use of HDAC3 inhibitors for the treatment of lung injury and fibrotic diseases is in its infancy, with most studies being preclinical in nature, the preliminary evidence suggests that HDAC3 inhibitors have beneficial effects. However, translating these findings into clinical practice will require a substantial body of both basic and clinical research.
Author contributions
HY: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. SL: Investigation, Writing – review & editing. SW: Visualization, Writing – review & editing. XG: Conceptualization, Project administration, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Liaoning Provincial Department of Education, General project, LJKZ0768, Mechanism of alveolar epithelial CFTR chloride channel in acute lung injury.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. Bmj. (2016) 353:i1585. doi: 10.1136/bmj.i1585
2. Park I, Kim M, Choe K, Song E, Seo H, Hwang Y, et al. Neutrophils disturb pulmonary microcirculation in sepsis-induced acute lung injury. Eur Respir J. (2019) 53. doi: 10.1183/13993003.00786-2018
3. Guo RF, Ward PA. Role of oxidants in lung injury during sepsis. Antioxid Redox Signal. (2007) 9:1991–2002. doi: 10.1089/ars.2007.1785
4. Qian Y, Wang Z, Lin H, Lei T, Zhou Z, Huang W, et al. TRIM47 is a novel endothelial activation factor that aggravates lipopolysaccharide-induced acute lung injury in mice via K63-linked ubiquitination of TRAF2. Signal Transduct Target Ther. (2022) 7:148. doi: 10.1038/s41392-022-00953-9
5. Meyer NJ, Gattinoni L, Calfee CS. Acute respiratory distress syndrome. Lancet. (2021) 398:622–37. doi: 10.1016/S0140-6736(21)00439-6
6. Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol. (2011) 6:147–63. doi: 10.1146/annurev-pathol-011110-130158
7. Derwall M, Martin L, Rossaint R. The acute respiratory distress syndrome: pathophysiology, current clinical practice, and emerging therapies. Expert Rev Respir Med. (2018) 12:1021–9. doi: 10.1080/17476348.2018.1548280
8. Gill SE, Yamashita CM, Veldhuizen RA. Lung remodeling associated with recovery from acute lung injury. Cell Tissue Res. (2017) 367:495–509. doi: 10.1007/s00441-016-2521-8
9. Blackwell TS, Tager AM, Borok Z, Moore BB, Schwartz DA, Anstrom KJ, et al. Future directions in idiopathic pulmonary fibrosis research. An NHLBI workshop report. Am J Respir Crit Care Med. (2014) 189:214–22. doi: 10.1164/rccm.201306-1141WS
10. Thannickal VJ, Toews GB, White ES, Lynch JP 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med. (2004) 55:395–417. doi: 10.1146/annurev.med.55.091902.103810
11. Thannickal VJ, Zhou Y, Gaggar A, Duncan SR. Fibrosis: ultimate and proximate causes. J Clin Invest. (2014) 124:4673–7. doi: 10.1172/JCI74368
12. Ramana KV, Fadl AA, Tammali R, Reddy AB, Chopra AK, Srivastava SK. Aldose reductase mediates the lipopolysaccharide-induced release of inflammatory mediators in RAW264.7 murine macrophages. J Biol Chem. (2006) 281:33019–29. doi: 10.1074/jbc.M603819200
13. Takashima K, Matsushima M, Hashimoto K, Nose H, Sato M, Hashimoto N, et al. Protective effects of intratracheally administered quercetin on lipopolysaccharide-induced acute lung injury. Respir Res. (2014) 15:150. doi: 10.1186/s12931-014-0150-x
14. Deng G, He H, Chen Z, OuYang L, Xiao X, Ge J, et al. Lianqinjiedu decoction attenuates LPS-induced inflammation and acute lung injury in rats via TLR4/NF-κB pathway. BioMed Pharmacother. (2017) 96:148–52. doi: 10.1016/j.biopha.2017.09.094
15. Verdone L, Agricola E, Caserta M, Di Mauro E. Histone acetylation in gene regulation. Brief Funct Genomic Proteomic. (2006) 5:209–21. doi: 10.1093/bfgp/ell028
16. Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. (2007) 26:5310–8. doi: 10.1038/sj.onc.1210599
17. Vancheri C. Common pathways in idiopathic pulmonary fibrosis and cancer. Eur Respir Rev. (2013) 22:265–72. doi: 10.1183/09059180.00003613
18. Conte E, Gili E, Fruciano M, Korfei M, Fagone E, Iemmolo M, et al. PI3K p110γ overexpression in idiopathic pulmonary fibrosis lung tissue and fibroblast cells: in vitro effects of its inhibition. Lab Invest. (2013) 93:566–76. doi: 10.1038/labinvest.2013.6
19. Korfei M, Skwarna S, Henneke I, MacKenzie B, Klymenko O, Saito S, et al. Aberrant expression and activity of histone deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax. (2015) 70:1022–32. doi: 10.1136/thoraxjnl-2014-206411
20. Lyu X, Hu M, Peng J, Zhang X, Sanders YY. HDAC inhibitors as antifibrotic drugs in cardiac and pulmonary fibrosis. Ther Adv Chronic Dis. (2019) 10:2040622319862697. doi: 10.1177/2040622319862697
21. Sanders YY, Hagood JS, Liu H, Zhang W, Ambalavanan N, Thannickal VJ. Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in mice. Eur Respir J. (2014) 43:1448–58. doi: 10.1183/09031936.00095113
22. King TE Jr., Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. (9807) 2011:1949–61:378. doi: 10.1016/S0140-6736(11)60052-4
23. Pardo A, Selman M. The interplay of the genetic architecture, aging, and environmental factors in the pathogenesis of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. (2021) 64:163–72. doi: 10.1165/rcmb.2020-0373PS
24. Selman M, Pardo A. Fibroageing: An ageing pathological feature driven by dysregulated extracellular matrix-cell mechanobiology. Ageing Res Rev. (2021) 70:101393. doi: 10.1016/j.arr.2021.101393
25. Tsou PS, Varga J, O’Reilly S. Advances in epigenetics in systemic sclerosis: molecular mechanisms and therapeutic potential. Nat Rev Rheumatol. (2021) 17:596–607. doi: 10.1038/s41584-021-00683-2
26. Romero Y, Aquino-Gálvez A. Hypoxia in cancer and fibrosis: part of the problem and part of the solution. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22158335
27. Kasotakis G, Kintsurashvili E, Galvan MD, Graham C, Purves JT, Agarwal S, et al. Histone deacetylase 7 inhibition in a murine model of gram-negative pneumonia-induced acute lung injury. Shock. (2020) 53:344–51. doi: 10.1097/SHK.0000000000001372
28. Xiang X, Dong G, Zhu J, Zhang G, Dong Z. Inhibition of HDAC3 protects against kidney cold storage/transplantation injury and allograft dysfunction. Clin Sci (Lond). (2022) 136:45–60. doi: 10.1042/CS20210823
29. Nguyen HCB, Adlanmerini M, Hauck AK, Lazar MA. Dichotomous engagement of HDAC3 activity governs inflammatory responses. Nature. (2020) 584:286–90. doi: 10.1038/s41586-020-2576-2
30. Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, et al. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci U.S.A. (2012) 109:E2865–2874. doi: 10.1073/pnas.1121131109
31. Yang J, Yin S, Bi F, Liu L, Qin T, Wang H, et al. TIMAP repression by TGFβ and HDAC3-associated Smad signaling regulates macrophage M2 phenotypic phagocytosis. J Mol Med (Berl). (2017) 95:273–85. doi: 10.1007/s00109-016-1479-z
32. Ghiboub M, Zhao J, Li Yim AYF, Schilderink R, Verseijden C, van Hamersveld PHP, et al. HDAC3 mediates the inflammatory response and LPS tolerance in human monocytes and macrophages. Front Immunol. (2020) 11:550769. doi: 10.3389/fimmu.2020.550769
33. Li N, Liu B, He R, Li G, Xiong R, Fu T, et al. HDAC3 promotes macrophage pyroptosis via regulating histone deacetylation in acute lung injury. iScience. (2023) 26:107158. doi: 10.1016/j.isci.2023.107158
34. Du W, Shi G, Shan CM, Li Z, Zhu B, Jia S, et al. Mechanisms of chromatin-based epigenetic inheritance. Sci China Life Sci. (2022) 65:2162–90. doi: 10.1007/s11427-022-2120-1
35. Jiang LP, Yu XH, Chen JZ, Hu M, Zhang YK, Lin HL, et al. Histone deacetylase 3: A potential therapeutic target for atherosclerosis. Aging Dis. (2022) 13:773–86. doi: 10.14336/AD.2021.1116
36. Liao Y, Cheng J, Kong X, Li S, Li X, Zhang M, et al. HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics. (2020) 10:9644–62. doi: 10.7150/thno.47651
37. Lu X, Fong KW, Gritsina G, Wang F, Baca SC, Brea LT, et al. HOXB13 suppresses de novo lipogenesis through HDAC3-mediated epigenetic reprogramming in prostate cancer. Nat Genet. (2022) 54:670–83. doi: 10.1038/s41588-022-01045-8
38. Bassett SA, Barnett MP. The role of dietary histone deacetylases (HDACs) inhibitors in health and disease. Nutrients. (2014) 6:4273–301. doi: 10.3390/nu6104273
39. Batchu SN, Brijmohan AS, Advani A. The therapeutic hope for HDAC6 inhibitors in Malignancy and chronic disease. Clin Sci (Lond). (2016) 130:987–1003. doi: 10.1042/CS20160084
40. Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci U.S.A. (2004) 101:15064–9. doi: 10.1073/pnas.0404603101
41. Wang XX, Wan RZ, Liu ZP. Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur J Med Chem. (2018) 143:1406–18. doi: 10.1016/j.ejmech.2017.10.040
42. Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol. (2009) 10:92–100. doi: 10.1038/ni.1673
43. Gao L, Cueto MA, Asselbergs F, Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem. (2002) 277:25748–55. doi: 10.1074/jbc.M111871200
44. Cao J, Sun L, Aramsangtienchai P, Spiegelman NA, Zhang X, Huang W, et al. HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proc Natl Acad Sci U.S.A. (2019) 116:5487–92. doi: 10.1073/pnas.1815365116
45. Bossone KA, Ellis JA, Holaska JM. Histone acetyltransferase inhibition rescues differentiation of emerin-deficient myogenic progenitors. Muscle Nerve. (2020) 62:128–36. doi: 10.1002/mus.26892
46. Tao H, Li Q, Lin Y, Zuo H, Cui Y, Chen S, et al. Coordinated expression of p300 and HDAC3 upregulates histone acetylation during dentinogenesis. J Cell Biochem. (2020) 121:2478–88. doi: 10.1002/jcb.v121.3
47. Susetyo A, Ishii S, Fujiwara Y, Amano I, Koibuchi N. Histone deacetylase 3 inhibitor alleviates cerebellar defects in perinatal hypothyroid mice by stimulating histone acetylation and transcription at thyroid hormone-responsive gene loci. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23147869
48. Zhang Y, Zhang X. Virus-induced histone lactylation promotes virus infection in crustacean. Adv Sci (Weinh). (2024) 11:e2401017. doi: 10.1002/advs.202401017
49. Moreno-Yruela C, Zhang D, Wei W, Bæk M, Liu W, Gao J, et al. Class I histone deacetylases (HDAC1-3) are histone lysine delactylases. Sci Adv. (2022) 8:eabi6696. doi: 10.1126/sciadv.abi6696
50. Wu D, Spencer CB, Ortoga L, Zhang H, Miao C. Histone lactylation-regulated METTL3 promotes ferroptosis via m6A-modification on ACSL4 in sepsis-associated lung injury. Redox Biol. (2024) 74:103194. doi: 10.1016/j.redox.2024.103194
51. Cui H, Xie N, Banerjee S, Ge J, Jiang D, Dey T, et al. Lung myofibroblasts promote macrophage profibrotic activity through lactate-induced histone lactylation. Am J Respir Cell Mol Biol. (2021) 64:115–25. doi: 10.1165/rcmb.2020-0360OC
52. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1
53. Zessin M, Meleshin M, Praetorius L, Sippl W, Bařinka C, Schutkowski M. Uncovering robust delactoylase and depyruvoylase activities of HDAC isoforms. ACS Chem Biol. (2022) 17:1364–75. doi: 10.1021/acschembio.1c00863
54. Li N, Liu B, Xiong R, Li G, Wang B, Geng Q. HDAC3 deficiency protects against acute lung injury by maintaining epithelial barrier integrity through preserving mitochondrial quality control. Redox Biol. (2023) 63:102746. doi: 10.1016/j.redox.2023.102746
55. Pooladanda V, Thatikonda S, Bale S, Pattnaik B, Sigalapalli DK, Bathini NB, et al. Nimbolide protects against endotoxin-induced acute respiratory distress syndrome by inhibiting TNF-α mediated NF-κB and HDAC-3 nuclear translocation. Cell Death Dis. (2019) 10:81. doi: 10.1038/s41419-018-1247-9
56. Hesketh M, Sahin KB, West ZE, Murray RZ. Macrophage phenotypes regulate scar formation and chronic wound healing. Int J Mol Sci. (2017) 18. doi: 10.3390/ijms18071545
57. Puttur F, Gregory LG, Lloyd CM. Airway macrophages as the guardians of tissue repair in the lung. Immunol Cell Biol. (2019) 97:246–57. doi: 10.1111/imcb.2019.97.issue-3
58. Lu HL, Huang XY, Luo YF, Tan WP, Chen PF, Guo YB. Activation of M1 macrophages plays a critical role in the initiation of acute lung injury. Biosci Rep. (2018) 38. doi: 10.1042/BSR20171555
59. Hoeksema MA, Gijbels MJ, Van den Bossche J, van der Velden S, Sijm A, Neele AE, et al. Targeting macrophage Histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med. (2014) 6:1124–32. doi: 10.15252/emmm.201404170
60. Wen Q, Lau N, Weng H, Ye P, Du S, Li C, et al. Chrysophanol Exerts Anti-inflammatory Activity by Targeting Histone Deacetylase 3 Through the High Mobility Group Protein 1-Nuclear Transcription Factor-Kappa B Signaling Pathway in vivo and in vitro. Front Bioeng Biotechnol. (2020) 8:623866. doi: 10.3389/fbioe.2020.623866
61. Stanfield BA, Purves T, Palmer S, Sullenger B, Welty-Wolf K, Haines K, et al. IL-10 and class 1 histone deacetylases act synergistically and independently on the secretion of proinflammatory mediators in alveolar macrophages. PloS One. (2021) 16:e0245169. doi: 10.1371/journal.pone.0245169
62. Ziesché E, Kettner-Buhrow D, Weber A, Wittwer T, Jurida L, Soelch J, et al. The coactivator role of histone deacetylase 3 in IL-1-signaling involves deacetylation of p65 NF-κB. Nucleic Acids Res. (2013) 41:90–109. doi: 10.1093/nar/gks916
63. Leus NG, van der Wouden PE, van den Bosch T, Hooghiemstra WTR, Ourailidou ME, Kistemaker LE, et al. HDAC 3-selective inhibitor RGFP966 demonstrates anti-inflammatory properties in RAW 264.7 macrophages and mouse precision-cut lung slices by attenuating NF-κB p65 transcriptional activity. Biochem Pharmacol. (2016) 108:58–74. doi: 10.1016/j.bcp.2016.03.010
64. Chen S, Ye J, Chen X, Shi J, Wu W, Lin W, et al. Valproic acid attenuates traumatic spinal cord injury-induced inflammation via STAT1 and NF-κB pathway dependent of HDAC3. J Neuroinflamm. (2018) 15:150. doi: 10.1186/s12974-018-1193-6
65. Xu Q, Liu X, Mei L, Wen Q, Chen J, Miao J, et al. Paeonol reduces the nucleocytoplasmic transportation of HMGB1 by upregulating HDAC3 in LPS-Induced RAW264.7 cells. Inflammation. (2018) 41:1536–45. doi: 10.1007/s10753-018-0800-0
66. Guo X, Hong S, He H, Zeng Y, Chen Y, Mo X, et al. NFκB promotes oxidative stress-induced necrosis and ischemia/reperfusion injury by inhibiting Nrf2-ARE pathway. Free Radic Biol Med. (2020) 159:125–35. doi: 10.1016/j.freeradbiomed.2020.07.031
67. Banerjee S, Rakshit T, Sett S, Mukhopadhyay R. Interactions of histone acetyltransferase p300 with the nuclear proteins histone and HMGB1, as revealed by single molecule atomic force spectroscopy. J Phys Chem B. (2015) 119:13278–87. doi: 10.1021/acs.jpcb.5b07795
68. Zou JY, Crews FT. Release of neuronal HMGB1 by ethanol through decreased HDAC activity activates brain neuroimmune signaling. PloS One. (2014) 9:e87915. doi: 10.1371/journal.pone.0087915
69. Kiernan R, Brès V, Ng RW, Coudart MP, El Messaoudi S, Sardet C, et al. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J Biol Chem. (2003) 278:2758–66. doi: 10.1074/jbc.M209572200
70. Zhu H, Shan L, Schiller PW, Mai A, Peng T. Histone deacetylase-3 activation promotes tumor necrosis factor-alpha (TNF-alpha) expression in cardiomyocytes during lipopolysaccharide stimulation. J Biol Chem. (2010) 285:9429–36. doi: 10.1074/jbc.M109.071274
71. Zhang H, Feng YW, Yao YM. Potential therapy strategy: targeting mitochondrial dysfunction in sepsis. Mil Med Res. (2018) 5:41. doi: 10.1186/s40779-018-0187-0
72. Hou L, Zhang J, Liu Y, Fang H, Liao L, Wang Z, et al. MitoQ alleviates LPS-mediated acute lung injury through regulating Nrf2/Drp1 pathway. Free Radic Biol Med. (2021) 165:219–28. doi: 10.1016/j.freeradbiomed.2021.01.045
73. Chi Z, Chen S, Xu T, Zhen W, Yu W, Jiang D, et al. Histone deacetylase 3 couples mitochondria to drive IL-1β-Dependent inflammation by configuring fatty acid oxidation. Mol Cell. (2020) 80:43–58.e47. doi: 10.1016/j.molcel.2020.08.015
74. Galmozzi A, Mitro N, Ferrari A, Gers E, Gilardi F, Godio C, et al. Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes. (2013) 62:732–42. doi: 10.2337/db12-0548
75. Emmett MJ, Lim HW, Jager J, Richter HJ, Adlanmerini M, Peed LC, et al. Histone deacetylase 3 prepares brown adipose tissue for acute thermogenic challenge. Nature. (2017) 546:544–8. doi: 10.1038/nature22819
76. Ali I, Yang M, Wang Y, Yang C, Shafiq M, Wang G, et al. Sodium propionate protect the blood-milk barrier integrity, relieve lipopolysaccharide-induced inflammatory injury and cells apoptosis. Life Sci. (2021) 270:119138. doi: 10.1016/j.lfs.2021.119138
77. Li M, Zheng Y, Yuan H, Liu Y, Wen X. Effects of dynamic changes in histone acetylation and deacetylase activity on pulmonary fibrosis. Int Immunopharmacol. (2017) 52:272–80. doi: 10.1016/j.intimp.2017.09.020
78. Ye Q, Li Y, Jiang H, Xiong J, Xu J, Qin H, et al. Prevention of pulmonary fibrosis via trichostatin A (TSA) in bleomycin induced rats. Sarcoidosis Vasc Diffuse Lung Dis. (2014) 31:219–26.
79. Chen F, Gao Q, Zhang L, Ding Y, Wang H, Cao W. Inhibiting HDAC3 (Histone deacetylase 3) aberration and the resultant nrf2 (Nuclear factor erythroid-derived 2-related factor-2) repression mitigates pulmonary fibrosis. Hypertension. (2021) 78:e15–25. doi: 10.1161/HYPERTENSIONAHA.121.17471
80. Huang SK, Scruggs AM, Donaghy J, Horowitz JC, Zaslona Z, Przybranowski S, et al. Histone modifications are responsible for decreased Fas expression and apoptosis resistance in fibrotic lung fibroblasts. Cell Death Dis. (2013) 4:e621. doi: 10.1038/cddis.2013.146
81. Yuan H, Jiao L, Yu N, Duan H, Yu Y, Bai Y. Histone deacetylase 3-mediated inhibition of microRNA-19a-3p facilitates the development of rheumatoid arthritis-associated interstitial lung disease. Front Physiol. (2020) 11:549656. doi: 10.3389/fphys.2020.549656
82. Zhu L, Fu X, Chen X, Han X, Dong P. M2 macrophages induce EMT through the TGF-β/Smad2 signaling pathway. Cell Biol Int. (2017) 41:960–8. doi: 10.1002/cbin.10788
83. Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U.S.A. (2006) 103:13180–5. doi: 10.1073/pnas.0605669103
84. Zhou Y, Horowitz JC, Naba A, Ambalavanan N, Atabai K, Balestrini J, et al. Extracellular matrix in lung development, homeostasis and disease. Matrix Biol. (2018) 73:77–104. doi: 10.1016/j.matbio.2018.03.005
85. Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. (2007) 127:526–37. doi: 10.1038/sj.jid.5700613
86. Murdoch C, Muthana M, Lewis CE. Hypoxia regulates macrophage functions in inflammation. J Immunol. (2005) 175:6257–63. doi: 10.4049/jimmunol.175.10.6257
87. Scholz CC, Taylor CT. Targeting the HIF pathway in inflammation and immunity. Curr Opin Pharmacol. (2013) 13:646–53. doi: 10.1016/j.coph.2013.04.009
88. Tuder RM, Yun JH, Bhunia A, Fijalkowska I. Hypoxia and chronic lung disease. J Mol Med (Berl). (2007) 85:1317–24. doi: 10.1007/s00109-007-0280-4
89. Zhou G, Dada LA, Wu M, Kelly A, Trejo H, Zhou Q, et al. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol. (2009) 297:L1120–1130. doi: 10.1152/ajplung.00007.2009
90. Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. (2007) 117:3810–20. doi: 10.1172/JCI30487
91. Jeong SH, Son ES, Lee YE, Kyung SY, Park JW, Kim SH. Histone deacetylase 3 promotes alveolar epithelial-mesenchymal transition and fibroblast migration under hypoxic conditions. Exp Mol Med. (2022) 54:922–31. doi: 10.1038/s12276-022-00796-y
92. Xiong R, Geng B, Jiang W, Hu Y, Hu Z, Hao B, et al. Histone deacetylase 3 deletion in alveolar type 2 epithelial cells prevents bleomycin-induced pulmonary fibrosis. Clin Epigenet. (2023) 15:182. doi: 10.1186/s13148-023-01588-5
93. Huang X, Yang N, Fiore VF, Barker TH, Sun Y, Morris SW, et al. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol. (2012) 47:340–8. doi: 10.1165/rcmb.2012-0050OC
94. Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, et al. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. (2010) 190:693–706. doi: 10.1083/jcb.201004082
95. Stowers RS, Shcherbina A, Israeli J, Gruber JJ, Chang J, Nam S, et al. Matrix stiffness induces a tumorigenic phenotype in mammary epithelium through changes in chromatin accessibility. Nat BioMed Eng. (2019) 3:1009–19. doi: 10.1038/s41551-019-0420-5
96. Damodaran K, Venkatachalapathy S, Alisafaei F, Radhakrishnan AV, Sharma Jokhun D, Shenoy VB, et al. Compressive force induces reversible chromatin condensation and cell geometry-dependent transcriptional response. Mol Biol Cell. (2018) 29:3039–51. doi: 10.1091/mbc.E18-04-0256
97. Jain N, Iyer KV, Kumar A, Shivashankar GV. Cell geometric constraints induce modular gene-expression patterns via redistribution of HDAC3 regulated by actomyosin contractility. Proc Natl Acad Sci U.S.A. (2013) 110:11349–54. doi: 10.1073/pnas.1300801110
98. Zhang GF, Zhou ZQ, Guo J, Gu HW, Su MZ, Yu BC, et al. Histone deacetylase 3 in hippocampus contributes to memory impairment after chronic constriction injury of sciatic nerve in mice. Pain. (2021) 162:382–95. doi: 10.1097/j.pain.0000000000002056
99. Ji H, Zhou Y, Zhuang X, Zhu Y, Wu Z, Lu Y, et al. HDAC3 deficiency promotes liver cancer through a defect in H3K9ac/H3K9me3 transition. Cancer Res. (2019) 79:3676–88. doi: 10.1158/0008-5472.CAN-18-3767
100. Toscano-Marquez F, Romero Y, Espina-Ordoñez M, Cisneros J. Absence of HDAC3 by matrix stiffness promotes chromatin remodeling and fibroblast activation in idiopathic pulmonary fibrosis. Cells. (2023) 12. doi: 10.3390/cells12071020
101. Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med. (2014) 6:231ra247. doi: 10.1126/scitranslmed.3008182
102. Zheng Q, Lei Y, Hui S, Tong M, Liang L. HDAC3 promotes pulmonary fibrosis by activating NOTCH1 and STAT1 signaling and up-regulating inflammasome components AIM2 and ASC. Cytokine. (2022) 153:155842. doi: 10.1016/j.cyto.2022.155842
103. Pellegrini C, Antonioli L, Lopez-Castejon G, Blandizzi C, Fornai M. Canonical and non-canonical activation of NLRP3 inflammasome at the crossroad between immune tolerance and intestinal inflammation. Front Immunol. (2017) 8:36. doi: 10.3389/fimmu.2017.00036
104. Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. (2002) 21:6539–48. doi: 10.1093/emboj/cdf660
105. Sun W, Zhang N, Liu B, Yang J, Loers G, Siebert HC, et al. HDAC3 inhibitor RGFP966 ameliorated neuroinflammation in the cuprizone-induced demyelinating mouse model and LPS-stimulated BV2 cells by downregulating the P2X7R/STAT3/NF-κB65/NLRP3 activation. ACS Chem Neurosci. (2022) 13:2579–98. doi: 10.1021/acschemneuro.1c00826
106. Huang S, Chen G, Sun J, Chen Y, Wang N, Dong Y, et al. Histone deacetylase 3 inhibition alleviates type 2 diabetes mellitus-induced endothelial dysfunction via Nrf2. Cell Commun Signal. (2021) 19:35. doi: 10.1186/s12964-020-00681-z
107. Kobayashi T, Matsuoka K, Sheikh SZ, Russo SM, Mishima Y, Collins C, et al. IL-10 regulates Il12b expression via histone deacetylation: implications for intestinal macrophage homeostasis. J Immunol. (2012) 189:1792–9. doi: 10.4049/jimmunol.1200042
108. Li JM, Ge CX, Xu MX, Wang W, Yu R, Fan CY, et al. Betaine recovers hypothalamic neural injury by inhibiting astrogliosis and inflammation in fructose-fed rats. Mol Nutr Food Res. (2015) 59:189–202. doi: 10.1002/mnfr.201400307
109. Li X, Liu X, Gao M, Han L, Qiu D, Wang H, et al. HDAC3 promotes meiotic apparatus assembly in mouse oocytes by modulating tubulin acetylation. Development. (2017) 144:3789–97. doi: 10.1242/dev.153353
110. Lu Y, Chen Y, Xu S, Wei L, Zhang Y, Chen W, et al. HDAC inhibitor attenuates rat traumatic brain injury induced neurological impairments. Heliyon. (2023) 9:e18485. doi: 10.1016/j.heliyon.2023.e18485
111. Shen Y, Yang R, Zhao J, Chen M, Chen S, Ji B, et al. The histone deacetylase inhibitor belinostat ameliorates experimental autoimmune encephalomyelitis in mice by inhibiting TLR2/MyD88 and HDAC3/NF-κB p65-mediated neuroinflammation. Pharmacol Res. (2022) 176:105969. doi: 10.1016/j.phrs.2021.105969
112. Siuda D, Zechner U, El Hajj N, Prawitt D, Langer D, Xia N, et al. Transcriptional regulation of Nox4 by histone deacetylases in human endothelial cells. Basic Res Cardiol. (2012) 107:283. doi: 10.1007/s00395-012-0283-3
113. Buvelot H, Jaquet V, Krause KH. Mammalian NADPH oxidases. Methods Mol Biol. (2019) 1982:17–36. doi: 10.1007/978-1-4939-9424-3_2
114. Nauseef WM. The phagocyte NOX2 NADPH oxidase in microbial killing and cell signaling. Curr Opin Immunol. (2019) 60:130–40. doi: 10.1016/j.coi.2019.05.006
115. Kleniewska P, Piechota A, Skibska B, Gorąca A. The NADPH oxidase family and its inhibitors. Arch Immunol Ther Exp (Warsz). (2012) 60:277–94. doi: 10.1007/s00005-012-0176-z
116. Jiang X, Ye X, Guo W, Lu H, Gao Z. Inhibition of HDAC3 promotes ligand-independent PPARγ activation by protein acetylation. J Mol Endocrinol. (2014) 53:191–200. doi: 10.1530/JME-14-0066
117. Zhang MJ, Zhao QC, Xia MX, Chen J, Chen YT, Cao X, et al. The HDAC3 inhibitor RGFP966 ameliorated ischemic brain damage by downregulating the AIM2 inflammasome. FASEB J. (2020) 34:648–62. doi: 10.1096/fj.201900394RRR
118. Zhao Q, Yu Z, Zhang F, Huang L, Xing C, Liu N, et al. HDAC3 inhibition prevents oxygen glucose deprivation/reoxygenation-induced transendothelial permeability by elevating PPARγ activity in vitro. J Neurochem. (2019) 149:298–310. doi: 10.1111/jnc.2019.149.issue-2
119. Alvariño R, Alfonso A, Tabudravu JN, González-Jartín J, Al Maqbali KS, Elhariry M, et al. Psammaplin A and its analogs attenuate oxidative stress in neuronal cells through peroxisome proliferator-activated receptor γ Activation. J Nat Prod. (2024) 87:1187–96. doi: 10.1021/acs.jnatprod.4c00153
120. Li Y, Liu C, Wang G, Wang H, Liu X, Huang C, et al. HDAC3 inhibitor (BRD3308) modulates microglial pyroptosis and neuroinflammation through PPARγ/NLRP3/GSDMD to improve neurological function after intraventricular hemorrhage in mice. Neuropharmacology. (2023) 237:109633. doi: 10.1016/j.neuropharm.2023.109633
121. Meng QQ, Feng ZC, Zhang XL, Hu LQ, Wang M, Zhang HF, et al. PPAR-γ Activation exerts an anti-inflammatory effect by suppressing the NLRP3 inflammasome in spinal cord-derived neurons. Mediators Inflammation. (2019) 2019:6386729. doi: 10.1155/2019/6386729
122. Yang CC, Wu CH, Lin TC, Cheng YN, Chang CS, Lee KT, et al. Inhibitory effect of PPARγ on NLRP3 inflammasome activation. Theranostics. (2021) 11:2424–41. doi: 10.7150/thno.46873
123. Zhang S, Yang L, Hu D, He S, Cui L, Zhao J, et al. Syringaresinol alleviates IgG immune complex induced acute lung injury via activating PPARγ and suppressing pyroptosis. Int Immunopharmacol. (2023) 124:111071. doi: 10.1016/j.intimp.2023.111071
124. Raneros AB, Bernet CR, Flórez AB, Suarez-Alvarez B. An epigenetic insight into NLRP3 inflammasome activation in inflammation-related processes. Biomedicines. (2021) 9. doi: 10.3390/biomedicines9111614
125. Zhang Q, Zeng M, Zhang B, Ren Y, Li S, Wang R, et al. Salvianolactone acid A isolated from Salvia miltiorrhiza ameliorates lipopolysaccharide-induced acute lung injury in mice by regulating PPAR-γ. Phytomedicine. (2022) 105:154386. doi: 10.1016/j.phymed.2022.154386
126. Fu Q, Shen N, Fang T, Zhang H, Di Y, Liu X, et al. ACT001 alleviates inflammation and pyroptosis through the PPAR-γ/NF-κB signaling pathway in LPS-induced alveolar macrophages. Genes Genomics. (2024) 46:323–32. doi: 10.1007/s13258-023-01455-w
127. Hussein ZA, Abu-Raghif AR, Tahseen NJ, Rashed KA, Shaker NS, Fawzi HA. Vinpocetine alleviated alveolar epithelial cells injury in experimental pulmonary fibrosis by targeting PPAR-γ/NLRP3/NF-κB and TGF-β1/Smad2/3 pathways. Sci Rep. (2024) 14:11131. doi: 10.1038/s41598-024-61269-y
128. Ma J, Xu Y, Zhang M, Li Y. Geraniol ameliorates acute liver failure induced by lipopolysaccharide/D-galactosamine via regulating macrophage polarization and NLRP3 inflammasome activation by PPAR-γ methylation Geraniol alleviates acute liver failure. Biochem Pharmacol. (2023) 210:115467. doi: 10.1016/j.bcp.2023.115467
129. Colunga Biancatelli RML, Solopov PA, Catravas JD. The inflammasome NLR family pyrin domain-containing protein 3 (NLRP3) as a novel therapeutic target for idiopathic pulmonary fibrosis. Am J Pathol. (2022) 192:837–46. doi: 10.1016/j.ajpath.2022.03.003
130. Rivers-Auty J, Daniels MJD, Colliver I, Robertson DL, Brough D. Redefining the ancestral origins of the interleukin-1 superfamily. Nat Commun. (2018) 9:1156. doi: 10.1038/s41467-018-03362-1
131. Kitasato Y, Hoshino T, Okamoto M, Kato S, Koda Y, Nagata N, et al. Enhanced expression of interleukin-18 and its receptor in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. (2004) 31:619–25. doi: 10.1165/rcmb.2003-0306OC
132. Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. (2005) 32:311–8. doi: 10.1165/rcmb.2004-0309OC
133. Park MJ, Moon SJ, Lee EJ, Jung KA, Kim EK, Kim DS, et al. IL-1-IL-17 signaling axis contributes to fibrosis and inflammation in two different murine models of systemic sclerosis. Front Immunol. (2018) 9:1611. doi: 10.3389/fimmu.2018.01611
134. Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, et al. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med. (2010) 207:535–52. doi: 10.1084/jem.20092121
135. Lasithiotaki I, Giannarakis I, Tsitoura E, Samara KD, Margaritopoulos GA, Choulaki C, et al. NLRP3 inflammasome expression in idiopathic pulmonary fibrosis and rheumatoid lung. Eur Respir J. (2016) 47:910–8. doi: 10.1183/13993003.00564-2015
136. Peng L, Wen L, Shi QF, Gao F, Huang B, Meng J, et al. Scutellarin ameliorates pulmonary fibrosis through inhibiting NF-κB/NLRP3-mediated epithelial-mesenchymal transition and inflammation. Cell Death Dis. (2020) 11:978. doi: 10.1038/s41419-020-03178-2
137. Liang Q, Cai W, Zhao Y, Xu H, Tang H, Chen D, et al. Lycorine ameliorates bleomycin-induced pulmonary fibrosis via inhibiting NLRP3 inflammasome activation and pyroptosis. Pharmacol Res. (2020) 158:104884. doi: 10.1016/j.phrs.2020.104884
138. Wang H, Wen Y, Wang L, Wang J, Chen H, Chen J, et al. DDR1 activation in macrophage promotes IPF by regulating NLRP3 inflammasome and macrophage reaction. Int Immunopharmacol. (2022) 113:109294. doi: 10.1016/j.intimp.2022.109294
139. Nie Y, Li J, Zhai X, Wang Z, Wang J, Wu Y, et al. Elamipretide(SS-31) attenuates idiopathic pulmonary fibrosis by inhibiting the nrf2-dependent NLRP3 inflammasome in macrophages. Antioxidants (Basel). (2023) 12. doi: 10.3390/antiox12122022
140. Korfei M, Mahavadi P, Guenther A. Targeting histone deacetylases in idiopathic pulmonary fibrosis: A future therapeutic option. Cells. (2022) 11. doi: 10.3390/cells11101626
Keywords: histone deacetylase 3, acute lung injury, pulmonary fibrosis, inflammation, macrophage
Citation: Yu H, Liu S, Wang S and Gu X (2024) The involvement of HDAC3 in the pathogenesis of lung injury and pulmonary fibrosis. Front. Immunol. 15:1392145. doi: 10.3389/fimmu.2024.1392145
Received: 27 February 2024; Accepted: 05 September 2024;
Published: 26 September 2024.
Edited by:
Emilio Hirsch, University of Turin, ItalyReviewed by:
Heriberto Prado-Garcia, National Institute of Respiratory Diseases-Mexico (INER), MexicoAllan R. Brasier, University of Wisconsin-Madison, United States
Yungeng Wei, First Affiliated Hospital of Guangzhou Medical University, China
Copyright © 2024 Yu, Liu, Wang and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiu Gu, eGl1Z3VAY211LmVkdS5jbg==