 Jinyin Xiao
Jinyin Xiao Xiajun Guo3
Xiajun Guo3
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 28 March 2024
Sec. Microbial Immunology
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1385907
This article is part of the Research TopicCrosstalk Between the GI Redox Environment, Microbiome, and the Immune SystemView all 5 articles
The human intestinal tract constitutes a complex ecosystem, made up of countless gut microbiota, metabolites, and immune cells, with hypoxia being a fundamental environmental characteristic of this ecology. Under normal physiological conditions, a delicate balance exists among these complex “residents”, with disruptions potentially leading to inflammatory bowel disease (IBD). The core pathology of IBD features a disrupted intestinal epithelial barrier, alongside evident immune and microecological disturbances. Central to these interconnected networks is hypoxia-inducible factor-1α (HIF-1α), which is a key regulator in gut cells for adapting to hypoxic conditions and maintaining gut homeostasis. Short-chain fatty acids (SCFAs), as pivotal gut metabolites, serve as vital mediators between the host and microbiota, and significantly influence intestinal ecosystem. Recent years have seen a surge in research on the roles and therapeutic potential of HIF-1α and SCFAs in IBD independently, yet reviews on HIF-1α-mediated SCFAs regulation of IBD under hypoxic conditions are scarce. This article summarizes evidence of the interplay and regulatory relationship between SCFAs and HIF-1α in IBD, pivotal for elucidating the disease’s pathogenesis and offering promising therapeutic strategies.
Inflammatory Bowel Disease (IBD), encompassing Crohn’s Disease (CD) and Ulcerative Colitis (UC), represents chronic intestinal disorders characterized by recurrent inflammation. The annual surge in IBD prevalence marks it a major public health issue worldwide, across developed and developing countries alike (1). Factors contributing to IBD include genetic predisposition, diet, lifestyle, environmental factors, psychological stress, gut microbiota, and host immunity (2, 3). IBD pathology is primarily characterized by impaired intestinal epithelial barrier (IEB) function, significant immune dysregulation, and gut microbiome disturbances (4, 5). These pathologies are interrelated rather than isolated, with IEB impairment leading to heightened exposure of mucosal immune cells to luminal antigens and inflammatory mediators, as well as to gut microbiota and their metabolites. This exposure strains intestinal defenses, including the IEB and mucosal immunity, disrupting homeostasis and intensifying inflammation (6–9). Therefore, IEB integrity, immune function, and gut microbiome are critical in the pathogenesis and management of IBD and remain focal points in current and future research endeavors.
Currently, IBD is mostly incurable, focusing on symptom management and relapse prevention, sometimes requiring surgery. Conventional treatments are pharmacological, including various drugs like antibiotics, aminosalicylates, corticosteroids, immunomodulators, and biologics (10), but these have many limitations (11). Although many novel therapies, such as cell therapies and small-molecule drugs, are constantly being discovered, they are still in clinical trials with unclear risks (10, 12). Therefore, elucidating the mechanisms and links between the aforementioned pathological factors in IBD could enhance treatment strategies.
Hypoxia-inducible factor-1 (HIF-1), a key transcription factor during the hypoxic response, comprises two subunits, HIF-1α and HIF-1β (13). HIF-1α plays a broad physiological role, activating target genes to regulate body’s hypoxic and inflammatory responses, crucial for cell adaptation to hypoxia and inflammation (14, 15). Additionally, HIF-1α is central to regulating the integrity and permeability of IEB (16, 17). Hence, HIF-1α is vital for IEB function, gut immunity, and pathogen defense, marking it as a key target in treating inflammatory diseases like IBD. Short-chain fatty acids (SCFAs) are mainly fermented from dietary fiber by intestinal microorganisms like Firmicutes and Bacteroidetes, which mainly include acetate, propionate, and butyrate, which account for more than 95% (18–20). Recent studies have validated the significant physiological and pathological roles of SCFAs in maintaining IEB function, modulating gut immunity, and sustaining microbial homeostasis (21). Consequently, dysregulation of gut metabolites, especially SCFAs, is considered a key factor in causing IBD-related intestinal inflammation and IEB dysfunction (22). During IBD development, HIF-1α is also regulated by SCFAs, with complex pathways and effects, particularly on IEB function and gut immunity. Therefore, this article details the role and mechanism of SCFAs in regulating HIF-1α in IBD, aiming to provide new strategies and avenues for clinical treatment.
Normoxia and hypoxia represent two distinct oxygen concentration states in cellular environments (23). Normoxia denotes the normal oxygen levels in organs, tissues, and cells in healthy states, with an oxygen partial pressure of 10%-20% (23, 24). In this state, oxygen supply is sufficient for bodily functions without restriction. However, hypoxia arises when oxygen supply falls short of demand, resulting in subnormal levels, often under 5% oxygen partial pressure (23, 25). Nonetheless, hypoxia does not always indicate pathology. It occurs under normal physiological conditions as well, thus hypoxia is classified into physiological and pathological types (26, 27).
Physiological hypoxia is influenced by factors such as diet (28), exercise (29), and living environment (such as at high altitude) (30). The body triggers regulatory and adaptive responses to mitigate oxygen deficiency, restore hypoxic conditions for oxygen homeostasis, or boost cellular tolerance to hypoxia. For instance, the body may enhance cardiac output or elevate erythropoiesis to augment tissue oxygenation, processes intimately associated with HIF (30–32). Additionally, pathways such as HIF signaling are employed for tissue cell acclimatization to hypoxia (33). It’s critical to recognize that hypoxia is not inherently detrimental. In certain areas, such as the gastrointestinal tract, moderate and stable physiological hypoxia is essential for maintaining key physiological processes like immune homeostasis, cellular metabolism, and angiogenesis (34, 35). In contrast, pathological hypoxia, a common feature in many diseases, is more prevalent and severe, playing a vital role in the pathology of cardiac (36), pulmonary (37), autoimmune diseases (38), and cancer (39). Importantly, pathological hypoxia is significantly linked to physiological hypoxia. Disruption of physiological hypoxia can initiate a series of cellular alterations, precipitating pathological hypoxia (27). Both hypoxia types are modulated by HIF, with IBD research centering on gut physiological hypoxia. Therefore, understanding the role and mechanism of HIF-related signaling pathways is crucial for treating hypoxia-associated diseases, including IBD.
Relative hypoxia is a normal physiological condition for certain organs and tissues. The gastrointestinal tract, influenced by its anatomical structure, complex vascular system, and the distribution and metabolism of the microbiota, naturally forms steep oxygen gradients both horizontally and vertically, with extensive research conducted in the colon (40–42). Studies have confirmed a gradient decrease in oxygen tension from the stomach to the intestines, with gastric luminal oxygen tension at about 7.7 kPa, small intestinal luminal tension at 4.2 - 4.6 kPa, and colonic tension dropping to 0.4 - 1.5 kPa (43–45). Interestingly, a near-vertical oxygen tension drop is noted from various intestinal wall layers to the lumen, where muscularis mucosae holds much higher tensions than the lumen. Colonic muscle oxygen levels range from 5.6 to 9.5 kPa, the submucosa around 5.6 kPa, and the luminal mucosa surface between 0.13 to 0.8 kPa (34, 46–50). Thus, hypoxia and oxygen gradients are crucial physiological features of the gut, with disruption potentially leading to disease.
Notably, the established oxygen gradients are closely associated with HIF-1α and the spatial distribution of intestinal microbiota. Studies have indicated that in the oxygen-rich stomach and duodenum, aerobic and facultative anaerobic bacteria are dominant; in the small intestine, facultative anaerobes are most common; and in the oxygen-depleted large intestine, obligate anaerobes prevail (34, 51–53). These findings suggest that microbiota distribution critically influences intestinal oxygen gradients, as evidenced by germ-free mouse studies. Kelly et al. reported that antibiotics or germ-free conditions significantly reduce SCFAs levels, diminish HIF-1α in intestinal epithelial cells (IEC), and nearly eliminate physiological hypoxia and oxygen gradients (54). Wang et al. also discovered that gut microbiota and SCFAs, especially butyrate, are intimately linked to intestinal hypoxia; butyrate enhances oxygen consumption and stabilizes HIF via prolyl hydroxylase enzymes (PHD) inhibition (55). Additionally, the formation of intestinal oxygen gradients is also related to other metabolic products such as trimethylamine oxide and indole derivatives, with antibiotic exposure altering these processes (49). Research by Nicolas et al. validated these points (56). In summary, HIF-1α and/or the gut microbiota and their metabolites, SCFAs, are key factors in the formation of intestinal hypoxia.
HIF functions as a heterodimer composed of α and β subunits, primarily including HIF-1 (HIF-1α and HIF-1β), HIF-2 (HIF-2α and HIF-2β), and HIF-3 (HIF-3α and HIF-3β) (57, 58). Among these types, HIF-1 is the most thoroughly researched and is vital for hypoxic pathophysiology. HIF-1 is ubiquitous in tissues and cells, with HIF-1α expression regulated by oxygen levels and HIF-1β expressed continuously (44). HIF-1 exerts its extensive and potent biological functions only when HIF-1α and HIF-1β combine to form a heterodimeric structure. Under normoxia, HIF-1α, hydroxylated by oxygen-dependent PHD, binds to von Hippel-Lindau (VHL) protein, which recruits ubiquitin ligases to HIF-1α, leading to its proteasomal degradation and functional inactivity. However, in hypoxia, the absence of oxygen impedes the ubiquitination of HIF-1α, enabling its nuclear translocation to form a heterodimeric complex with HIF-1β. This complex then binds to specific DNA regions (hypoxia-response elements, HRE) of target genes, activating the transcription of numerous downstream genes. This activation plays a crucial role in processes such as cellular metabolism, intestinal barrier function, immune regulation, angiogenesis, erythropoiesis, tumorigenesis, cell apoptosis, and autophagy (33, 59, 60). In summary, HIF-1α plays a pivotal role in maintaining cellular oxygen homeostasis under hypoxic conditions and offers significant protection during pathological and physiological hypoxia by mediating adaptive responses to oxygen insufficiency (Figure 1).
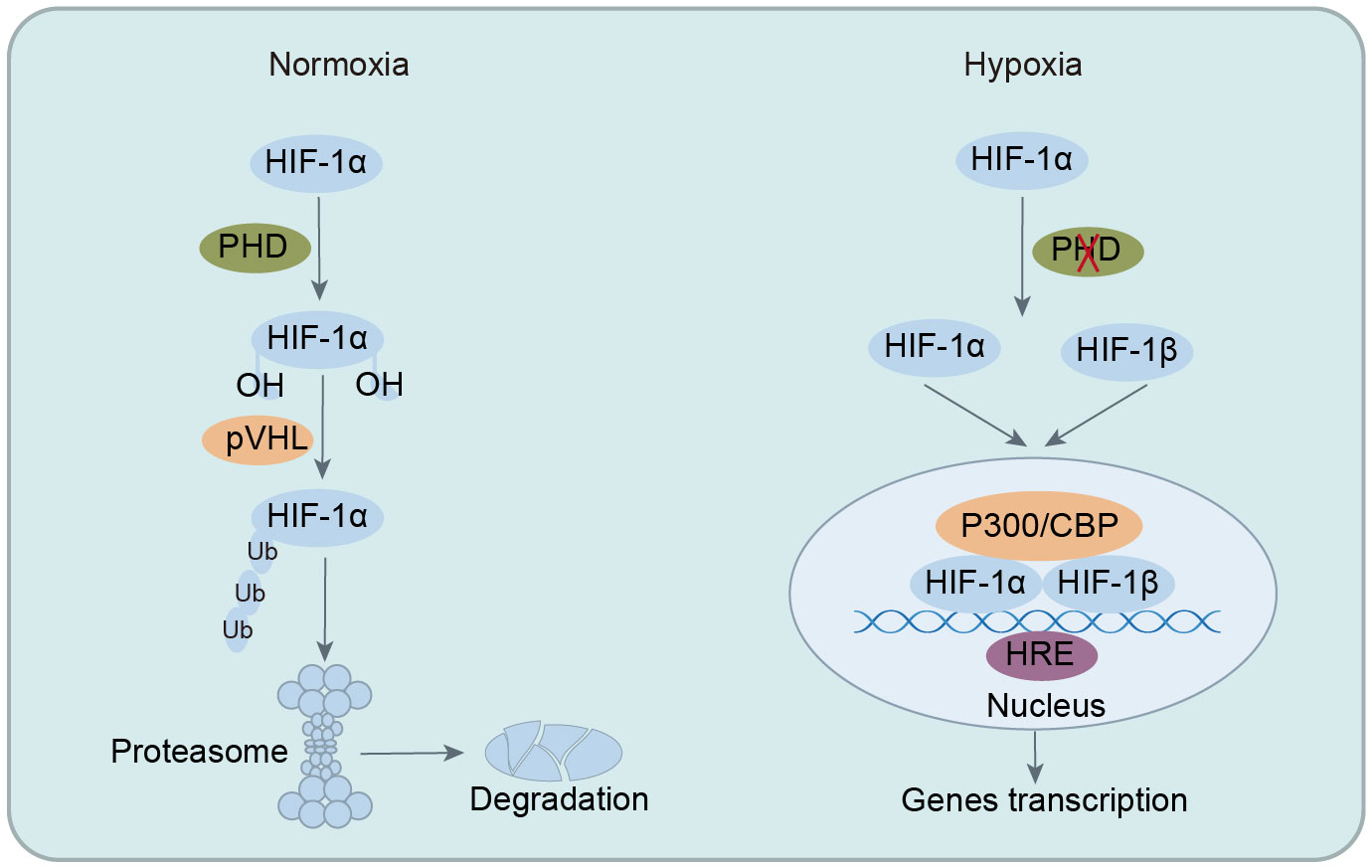
Figure 1 Regulation of HIF-1 stability under normoxia and hypoxic conditions. In normoxia, HIF-1α is hydroxylated by PHD, leading to ubiquitination and degradation, preventing its biological function. In hypoxia, ubiquitination of HIF-1α is inhibited, allowing its translocation into the nucleus to form heterodimeric complexes with HIF-1β. This complex then binds to specific DNA regions of target genes, activating the transcription of numerous downstream targets. HIF-1α, hypoxia-inducible factor-1α; PHD, prolyl hydroxylase enzymes; VHL, von Hippel-Lindau; CBP, CREB-binding protein; HRE, hypoxia-response elements.
Inflammatory states are considered a key pathological feature of IBD, while hypoxia is seen as a common characteristic of most inflammation. Research has confirmed significant hypoxia in IBD’s inflamed regions, which mutually affects each other, with HIF-1α being central to the connection between host hypoxia and inflammation (44). Current consensus in research posits physiological hypoxia as a crucial environmental characteristic of the gut, with IBD pathogenesis closely linked to disruptions in this hypoxic milieu. A stable hypoxic environment effectively controls and alleviates intestinal inflammation, closely linked to the activation of HIF-1α, which promotes adaptation to hypoxia, mitigates gut inflammation, and maintains intestinal barrier, serving a definitive protective role in IBD (48). For example, moderate hypoxia preserves the intestinal barrier in mouse dextran sulfate sodium (DSS)-induced colitis via HIF-1α modulation of Vitamin D Receptor (VDR) signaling (61). Additionally, hypoxia significantly reduces inflammatory gene expression in CD patients and colitis mice, linked to the inhibition of the NOD-like receptor thermal protein domain associated protein 3 (NLRP3)/mammalian target of rapamycin (mTOR) pathway and autophagy induction by hypoxia-driven HIF-1α (62). Furthermore, in colitis mouse models, butyrate upregulation activates IEC peroxisome proliferator-activated receptor γ (PPAR-γ), reducing cellular oxygenation in inflammation, boosting mitochondrial function and β-oxidation of SCFAs to sustain hypoxia and curb pathogenic bacterial growth in IBD (63–65). Conversely, deficiency or impairment of HIF-1α in the gut exacerbates intestinal inflammation. Myeloid HIF deficiency in DSS-induced colitis leads to increased infiltration of neutrophils and Ly6C monocytes, with a more marked impact from HIF-1α loss compared to HIF-2α (66).
Interestingly, research has indicated hypoxia induces or exacerbates inflammation, with HIF-1α activation contributing to IBD’s inflammatory processes. For instance, frequent high-altitude activities have been associated with gastrointestinal inflammation, increasing the risk of IBD flares (67). Hypoxia markers HIF-1α, macrophage inflammatory protein 3α (MIP-3α), and vascular endothelial growth factor (VEGF) were significantly elevated in colonic epithelial tissues and serum of both pediatric and adult patients with active IBD, correlating positively with histological severity (68). In DSS-induced colitis mice, myeloid HIF-1α deletion lessened inflammation, HIF-2α deletion worsened it, and the absence of both showed no significant changes (69); however, increased myeloid expression of HIF-1α accelerated colitis progression (70). Moreover, hypoxia was found to not only exacerbates DSS-induced colitis but also augments neuroinflammation in mice (71). However, the specific mechanisms and pathways by which hypoxia/HIF-1α induces or exacerbates intestinal inflammation have not been elucidated in these studies, necessitating further research for clarification. It is worth noting that inflammation also aggravates hypoxia. In the pathology of IBD, prolonged intestinal inflammation sharply increases the severity of mucosal hypoxia, potentially transitioning to a chronic pathological state (44). This exacerbates intestinal damage and perpetuates a vicious cycle. In IBD, the exacerbation of local hypoxia by intestinal inflammation may be associated with the following factors. On the one hand, inflammatory recurrence in IBD leads to vascular damage, endothelial dysfunction, and reduced perfusion (72, 73), while chronic inflammation increases oxygen consumption due to immune cell infiltration, exacerbating hypoxia (7, 74, 75). On the other hand, changes in the gut microbiota and their metabolites also consume oxygen, with obligate anaerobes switching to facultative anaerobes and less butyrate production, disrupting physiological hypoxia (76). Additionally, anaerobic bacteria metabolites enhance the consumption of oxygen in IEC, maintaining gut hypoxia (77–79). Hence, intestinal hypoxia is considered a common microenvironmental feature of IBD pathology, with inflammatory damage spreading hypoxia to deeper layers like the submucosa, exacerbating gut hypoxia.
Certainly, in the face of hypoxia, inflammation is not passive. In UC, it prompts a transcriptional reprogramming of the HIF pathway, downregulating PHD1 to favor HIF-1α’s protective role over HIF-2’s harmful effects, aiding mucosal repair (80). This highlights the close relationship between HIF-1α and the protective mechanisms against inflammatory hypoxia in IBD. Notably, different subtypes of HIF play distinct roles in hypoxia and the pathogenesis of IBD. HIF-1 is primarily involved in acute hypoxic response, whereas HIF-2 and HIF-3 are implicated in chronic hypoxia adaptation (33, 58). There is also cross-talk among these subtypes. A decrease in HIF-1 levels leads to an increase in HIF-2 and HIF-3, a shift reminiscent of cell polarization, which is necessary for the long-term hypoxic adaptation of cells (58). Moreover, in IBD, different HIF subtypes exert critical but opposing effects. As mentioned earlier, HIF-1α suppresses intestinal inflammation and promote resolution in IBD. Conversely, HIF-2α activation upregulates inflammatory expression, intensifying the inflammatory response, aggravating IBD, and is associated with colorectal cancer (CRC) progression (81–84).
Although the impact of hypoxia in IBD is contested, a stable hypoxic environment is pivotal for gastrointestinal wellness, with HIF-1α’s protective role acknowledged. HIF-1α orchestrates cellular adaptation to hypoxic and inflammatory conditions via downstream genes regulation, essential for cells coping with such stresses. Therefore, therapeutic approaches for IBD have diversified to include tactics like PHD inhibition to manage HIF-1α, enhancing treatment options (85, 86). The debate surrounding hypoxia’s pro-inflammatory or anti-inflammatory effects in IBD may hinge on varying experimental hypoxia parameters (the level, duration, and degree of hypoxia), HIF-1α’s cell-specific roles, the target genes or pathways affected by HIF-1α, and interactions between HIF isoforms. Thus, in exploring HIF’s role or therapeutic use in IBD, these considerations are essential.
The IEB, comprising IEC and the basement membrane, serves as a vital physiological barrier in the gut, essential for nutrient digestion and absorption, preserving gut hypoxia, and preventing luminal waste, microbiota, and allergens from entering (87, 88). Dysfunction of the IEB leads to intestinal damage, inflammation, and even sepsis and bacteremia (89). Therefore, the IEB plays a pivotal role in the pathology of IBD. IEC are primary targets of hypoxia, situated between the hypoxic lumen and the well-perfused lamina propria. Under physiological conditions, IEC are hypoxia-tolerant, aiding gut adaptation to low oxygen and maintaining IEB integrity (88, 90). The hypoxia-resistant of the IEB suggests a strong link between HIF-1α and IEB, with notable therapeutic implications for IBD management.
Studies have indicated HIF-1α improves IBD outcomes by augmenting IEB function through various pathways. HIF-1α fortifies the IEB first by regulating cellular junctions, especially tight junctions, and IEC permeability (91–93). It then directly controls barrier-protective genes for maintaining and repairing intestinal barrier, including intestinal trefoil factor (ITF), cluster of differentiation 39 (CD39), CD73, vascular endothelial growth factor (VASP), multidrug resistance protein 1 (MDR-1), and integrins α2, α6 (34, 94–96). Additionally, HIF-1α sustains IEB performance by managing mucosal immune homeostasis (97, 98), and by influencing the gut microenvironment and metabolic pathways (34, 44, 99). Finally, enteric neurons and enteric glia in the enteric nervous system (ENS) are important regulators of IEB function and play a key role in maintaining IEB and resisting inflammation (100). Research indicates that HIF-1α is closely linked to the protective and death regulation mechanisms of enteric neurons. In trinitro-benzene-sulfonic acid (TNBS)-induced colitis in rats, glial cell line-derived neurotrophic factor (GDNF) protects enteric neurons from cell death due to metabolic challenges by activating HIF-1α and the REarranged during Transfection (RET) pathway (101). Although the exact mechanism of HIF-1α within the ENS remains unclear, the critical role of HIF-1α in preserving intestinal barrier function is undeniable.
In the hypoxic environment of IBD, mucosal inflammation often involves extensive immune cells infiltration, which frequently determine the duration and intensity of the inflammatory response. The hypoxia signaling pathway centered on HIF-1 is present in nearly all cells, including immune cells, to help them adapt to hypoxic conditions (44, 57). Therefore, during the pathogenesis of IBD, HIF-1α inevitably interacts with immune cells, and under the conditions, the interactions between immune cells may also be mediated by HIF-1α. In IBD, HIF-1α plays a dual role in adaptive immunity. On the one hand, it modulates anti-inflammatory effects by regulating the functions and differentiation of T cells or B cells. HIF-1α is abundantly present in colonic T cells of IBD patients and colitis mice, restricting Th1 and Th17 cells activity and differentiation, while fostering regulatory T cells (Treg) differentiation and interleukin-10 (IL-10) release, thereby promoting T cells homeostasis and reducing gut inflammation (102–105). HIF-1α is also present in B cells, where it cooperates with phosphorylated signal transducer and activator of transcription 3 (p-STAT3) to regulate the transcription of CD11b, exerting an immunosuppressive effect to protect against IBD (106). It also enhances regulatory B cells (Breg) differentiation and IL-10 secretion to reduce intestinal inflammation (107, 108). On the other hand, HIF-1α may amplify inflammation by activating Th17 cells via ubiquitin-conjugating enzyme 9 (Ubc9) inhibition (109). The complex role of HIF-1α in adaptive immunity in IBD depends on its environment and regulatory mechanisms.
Recent research emphasizes the link between HIF-1α and innate immunity, especially its governance of macrophage functions crucial to IBD therapy (110). HIF-1α affects macrophage polarization, vital for immunological equilibrium in the gut. Macrophages exhibit a dual role, with M1 types inducing inflammation and M2 types exerting anti-inflammatory actions. These types are in a dynamic balance under physiological conditions, once disruption of this balance leads to inflammation. Mesenchymal stem cells, for example, encourage an anti-inflammatory state by shifting macrophages from M1 to M2 phenotypes through HIF-1α pathways, easing colitis (111–113). IBD medications like tiliroside, targeting the HIF-1α/glycolysis pathway (114), and spermidine, which boosts HIF-1α via reactive oxygen species (ROS)-activated Adenosine 5’-monophosphate (AMP)-activated protein kinase (AMPK) to favor M2 polarization (115), work by adjusting the M1/M2 balance. Dioscin’s anti-colitic effect also involves HIF-1α-mediated macrophage polarization (116). However, in IBD and murine colitis, hypoxia-driven HIF-1α in macrophages activates Wnt/mTOR pathways, disrupting autophagy in IEC (117). Since autophagy is key in IBD, regulating IEC metabolism, maintaining cell junctions, preventing IEC death, and aiding gut repair (118, 119), HIF-1α’s role in hypoxic immune cells may not uniformly benefit IBD pathology.
Dendritic cells (DC) are pivotal for intestinal immune balance, bridging innate and adaptive immunity. HIF-1α, abundant in DC, supports their function and adaption in hypoxia (57, 120). HIF-1α deletion in DC heightens pro-inflammatory factors and reduces Tregs, exacerbating intestinal inflammation (121). HIF-1α also interacts with neutrophils in IBD, where they serve ambivalent roles. Initially, they mitigate inflammation, but overactivation leads to barrier disruption and increased inflammation (122). Myeloid HIF-1α deficits encourage pro-inflammatory neutrophil and monocyte infiltration, hinder M2 polarization, and worsen colitis (66). Cyclosporine A eases severe UC by repressing sirtuin 6 (SIRT6)-driven neutrophil HIF-1α, enhancing glycolysis and tricarboxylic acid (TCA) cycle, restraining neutrophil overactivation, and preserving mucosal homeostasis (123). Additionally, advances in research on neutrophil extracellular traps (NET) in IBD suggests their dual role in inflammation (124), akin to HIF-1α, hinting at a potential hypoxia-driven HIF-1α-NET link in IBD pathogenesis worth further exploration. In summary, HIF-1α impacts intestinal immune equilibrium by modulating immune cells functions, thereby influencing IBD initiation and progression, though its precise role is modulated by variables like cell type, inflammation stage, and oxygen availability (Table 1).
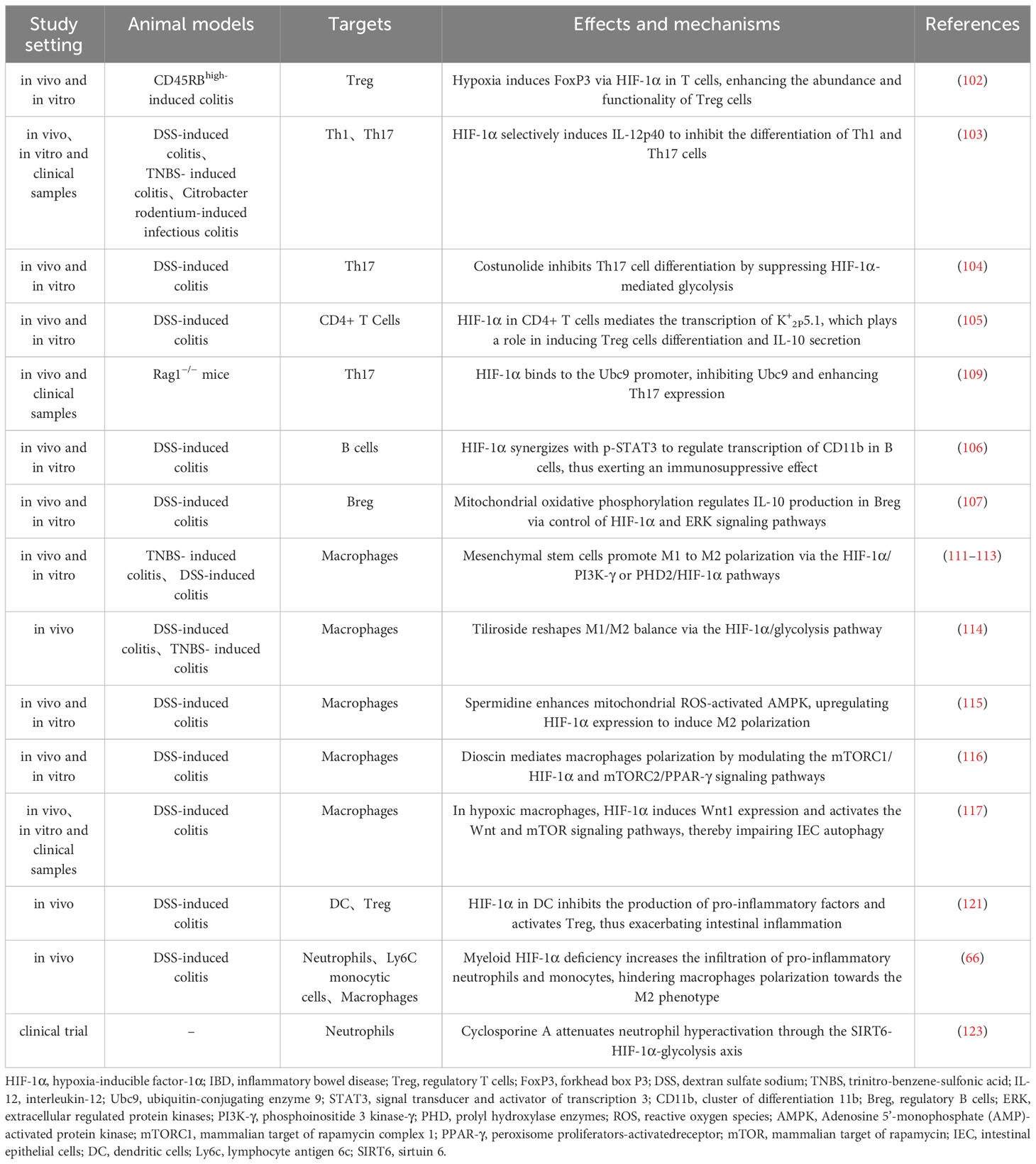
Table 1 Role and mechanisms of HIF-1α in regulating immune cells in IBD.
Intestinal fibrosis, a common complication of IBD, is primarily caused by chronic inflammation, with the pathogenesis mainly related to an excessive accumulation of extracellular matrix and an increase in the number of stromal-like cells in the submucosal layer (125). Previously considered irreversible and untreatable, this view of intestinal fibrosis has shifted due to recent studies. Extensive evidence has indicated that HIF-1α signaling plays a crucial role in various fibroses, including intestinal (17, 126–128). The autotaxin-lysophosphatidic acid (ATX-LPA) axis as a promising therapeutic target for CD-associated fibrosis, influencing fibroblast proliferation and differentiation, with HIF-1α modulating ATX expression (129). In IBD patients and colitis-induced mice, hypoxia alleviates fibrosis by inhibiting the transcription of pro-fibrotic genes (130). Betulinic acid hydroxamate, a PHD inhibitor targeting HIF, attenuates inflammation and fibrosis in colitis models, also aiding wound healing (131). Although evidence has supported HIF-1α’s antifibrotic effects, knowledge of its precise mechanisms is limited, and significant progress towards clinical application remains a lengthy process.
Growing research underscores the influence of gut microbiota and their metabolites, particularly SCFAs, on health and disease. SCFAs, as one of the gut microbiota’s main metabolite, are extensively confirmed to be a key contributor to the pathogenesis of IBD. IBD patients have fewer SCFAs-producing bacteria and lower fecal SCFAs levels than healthy individuals (132–134). Dietary fiber scarcity worsens colitis, while supplementation encourages SCFAs-producing bacteria and SCFAs levels in IBD, restoring gut homeostasis (135, 136). Notably, significant SCFAs concentration gradients exist along the gastrointestinal tract, with cecum and colon’s anaerobic conditions being prime for fermenting dietary fibers into SCFAs. Hence, the absence of the cecum decreases SCFAs-producing microbiota and SCFAs levels, intensifying inflammation (137). Understanding the role and related mechanisms of SCFAs in IBD is therefore crucial for the disease’s prevention and treatment.
The gut barrier, mainly composed of IEC, immune cells, and gut microbiota, encompasses mechanical, chemical, immune, and biological defenses, essential for protecting against harmful substances (138). Studies have indicated that SCFAs modulate IEB function through multiple pathways. SCFAs, particularly butyrate, serve as the main energy source for IEC, satisfying roughly 70% of colonic cells’ energy needs through mitochondrial β-oxidation, with the distal colon relying more heavily on butyrate than the proximal section (51, 139, 140). Besides, butyrate also enhances nutrient absorption in the gut by modulating NLRP3 pathway, providing further nourishment (141, 142). The energy from SCFAs is essential for regulating IEC activities such as proliferation, differentiation and apoptosis, which are critical for maintaining IEB integrity, intestinal energy metabolism, and immune regulation (22, 143). The “starving gut” hypothesis posits that IBD stems from mucosal malnutrition and energy deficiency associated with SCFAs scarcity, mainly manifesting as IEB defects and the persistence of the disease (139, 144, 145). Consequently, the energy provided by SCFAs is vital for the IEB function. Conversely, numerous perspectives critically refute the conventional view of butyrate as the first fuel of IEC, indicating that butyrate alone cannot fulfil the carbon needs for IEC synthesis, necessitating the involvement of DNA, RNA, proteins, and lipids. Hence, theoretically, butyrate may not be the sole carbon source for IEC proliferation (146). Moreover, several studies have shown that butyrate not only fail to promote IEC proliferation but may also inhibit it (147, 148), and even suppress the proliferation of colon cancer cells (149). The contradictions in the role of SCFAs in IEC proliferation might relate to the energy metabolism of different IEC types, such as undifferentiated versus differentiated cells, as not all IEC metabolize butyrate equally, with undifferentiated IEC preferring glucose over butyrate as a cellular fuel (150). Additionally, we believe that this might also be linked to the gut environment in various diseases and available energy sources for gut cells. Therefore, further research is necessary to elucidate the energy sources for IEC and the regulatory role of SCFAs in IEC proliferation.
SCFAs enhance gut barrier function by modulating microbiota composition and reducing luminal pH. The microbiota-driven bio-barrier is a crucial component of intestinal defenses. The increase of SCFAs significantly improve the structure of intestinal microbiota and accelerate the recovery of intestinal microflora, which is mainly manifested by increasing the number of beneficial bacteria while inhibiting harmful bacteria (151, 152). SCFAs reduce the colonic lumen pH and create a favorable acidic growth environment for probiotics such as Lactobacillus and bifidobacterium, thus maintaining the intestinal barrier function (153, 154). Besides, butyrate prevents colitis by modulating gut microbiota and reducing IgA-coated bacteria in the colon (155), and SCFAs inhibit the production of enterotoxin-producing bacteria, critical for protecting IEC and preventing intestinal diseases (156, 157). Additionally, due to their small molecular weight, SCFAs permeate IEB and exert antitoxin effects by modulating specific intracellular targets such as HIF-1, although the exact mechanisms remain unclear (156).
SCFAs also directly maintain IEB function by modulating tight junctions and mucous secretion. Tight junctions, vital for cellular polarity and permeability, consist of transmembrane proteins, cytoplasmic attachment proteins, and cytoskeletal proteins, with transmembrane proteins being most impactful (158). Their malfunction leads to compromised IEB (159). Occludin and claudins, especially claudins, are critical transmembrane proteins for tight junction (160). Studies have indicated SCFAs significantly upregulate tight junction proteins like zonula occludens-1 (ZO-1), occludin, and claudins, thus protecting IEB and attenuating inflammation (161, 162). The gut mucus layer, a physical barrier protecting IEC from harmful substances and the first line of defense for gut health, is mainly composed of mucins produced by goblet cells in IEC and nourishes the gut’s symbiotic flora (163–165). Therefore, mucus layer damage is linked to various diseases including IBD. SCFAs maintain the mucus barrier by regulating IEC mucin secretion. For instance, evidence has suggested that dietary fiber and SCFAs, particularly propionate and butyrate, promote the expression of mucins such as Mucin 2 (MUC2), enhancing the thickness of the intestinal mucus layer, thus alleviating gut inflammation and potentially reducing the risk of colitis associated colorectal cancer (CAC) (166–169). Moreover, mucins actively protect against gut infections by modulating antimicrobial peptide production, altering antigen-presenting cell activity, and enhancing bacterial clearance (170).
In IBD, the extensive presence of immune cells in inflamed gut tissue is a key indicator of disease. Likewise, SCFAs are abundant in the intestinal environment and regulate the immune system. They affect adaptive immune cells, particularly CD4+ T cells, by modulating their activity, proliferation, differentiation, and intercellular homeostasis. SCFAs represented by butyrate inhibit Th17 activity and differentiation, while promoting Treg activity and differentiation, thereby restoring Th17/Treg immune homeostasis to alleviate IBD (171–175). Additionally, pentanoate promotes IL-10 expression and reduces IL-17A secretion by reprogramming T and B cell metabolism and epigenetics (176). Notably, SCFAs immune modulation appears concentration-dependent, with normal gut concentrations (10-100 mM) key for mucosal immunity. Low butyrate levels encourage anti-inflammatory forkhead box P3 (Foxp3) Treg differentiation, while high levels drive pro-inflammatory gene expression in Treg and T cells via histone deacetylases (HDAC) inhibition (177).
SCFAs also modulate innate immune cells like neutrophils (178), macrophages (179), and dendritic cells (180) to preserve gut immunity. In mouse models, dietary acetate reduces colitis by controlling neutrophil recruitment (178). NET are vital defenses produced by neutrophils at inflammation sites that capture and destroy pathogens, effectively controlling infections (181). Although direct links between SCFAs and NET formation in IBD are not well-established, research on neuroendocrine tumors shows that SCFAs induce NET in a pH-dependent manner (182). Thus, the potential link between SCFAs and NET in the pathogenesis of IBD warrants further research for validation. Macrophages show notable adaptability to the gut microbiome, typically without inciting inflammation. However, IBD or antibiotic abuse disrupts the gut microbiome and SCFAs, unbalancing macrophages and T cells, triggering cytokine release, which butyrate supplementation improves above situation (179). Additionally, butyrate suppresses M1 macrophage polarization to reduce inflammation, and even to inhibit CAC progression (141, 183, 184). Moreover, butyrate works with chemokine cytokine receptor 9 (CCR9)-recruited Myeloid-derived suppressor cells (MDSC) to alleviate DSS-induced colitis (185). MDSC originate from bone marrow and accumulate in IBD-compromised intestines, serving as a key IBD treatment target (186). Studies show MDSC curb intestinal inflammation by restraining T cells proliferation, natural killer cells (NK) cytotoxicity, and promoting anti-inflammatory macrophages phenotype (187, 188). However, MDSC have also been found to enhance inflammation by stimulating T cells like Th17 reflecting their diverse roles in IBD (186). Besides, invariant natural killer T cells (iNKT), linking innate and adaptive immunity, are modulated by SCFAs to regulate gut balance and inflammation by secreting IL-10 and inhibiting Th1 and Th17 (189). While debates over SCFAs’ role in gut immunity, their overall positive influence on IBD is evident. It is imperative to emphasize that SCFAs primarily modulate gut immunity by influencing cytokine production, migration, and functionality. In summary, SCFAs are integral to intestinal immunity and profoundly influence IBD pathology. Therefore, SCFAs-mediated immune modulation is pivotal for advancing IBD therapy, but it’s indispensable to consider SCFAs type, concentration, intestinal environment (e.g., pH) and target cells (Table 2).
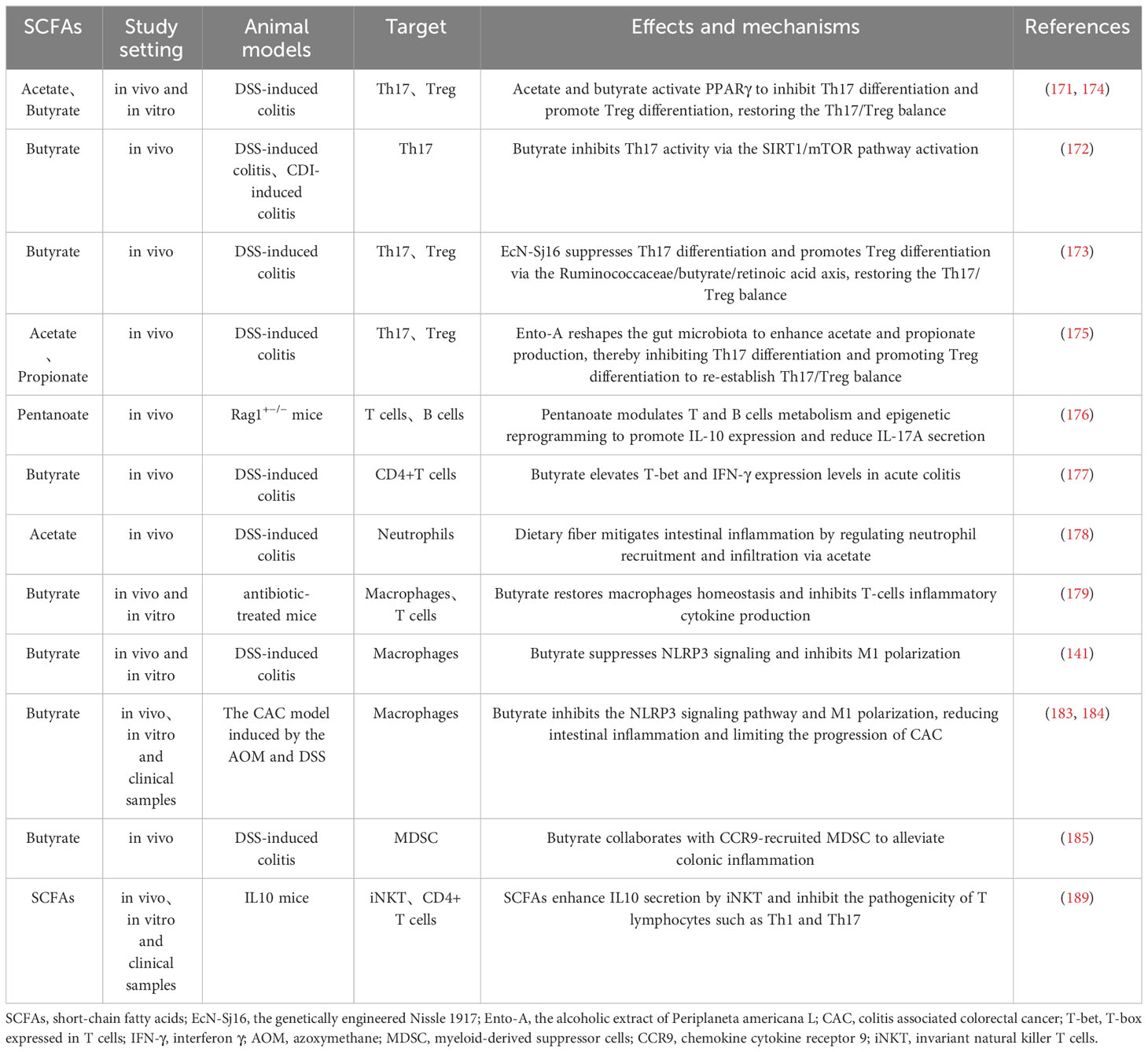
Table 2 Role and mechanisms of SCFAs in regulating immune cells in IBD.
It is important to note that the pathways of SCFAs in modulating gut barrier and immune functions are often closely linked with G protein-coupled receptor (GPCR) and/or HDAC signaling, regarded as bridges between the host and gut microbiota. On the one hand, SCFAs modulate IEC and immune cell expression and function directly through activating GPCR (primarily GPR41, GPR43, and GPR109a) (190–195) and/or inhibiting HDAC (191, 196–200). On the other hand, SCFAs intervene in IBD by targeting downstream signaling cascades via these mechanisms, for instance, GPCR mediate SCFA-induced activation of mitogen-activated protein kinase (MAPK) (201, 202), nuclear factor kappa-B (NF-κB) (201), phosphoinositide 3 kinase (PI3K)/mTOR (203), NLRP3 (204), and STAT (203, 205) pathways, essential for the expression of key immune and inflammatory mediators. HDAC mediate SCFAs regulation of Toll-like receptor 4 (TLR4) (206) and NF-κB (192) signaling pathways. Notably, in IBD, the potential mechanism of SCFAs to regulate intestinal function is not limited to the above two pathways. SCFAs also influence intestinal function by acting on HIF signaling pathway, which is another key mechanism for SCFAs in physiological regulation.
The gut microbiota and hypoxia are closely linked, engaging in crosstalk within the host’s intestines. Microbial-derived SCFAs elevate IEC oxygen consumption to maintain intestinal hypoxia which activates and stabilizes HIF, triggering the adaptive cellular responses to hypoxia essential for intestinal function and pathology (54). HIF-1α in the gut induces pyruvate dehydrogenase kinases (PDK or PDHK) expression, such as PDK1 and PDK3, inactivating the pyruvate dehydrogenase complex (PDC) and blocking pyruvate’s conversion to acetyl-CoA for the TCA cycle. Thus, colonocytes depend on SCFAs β-oxidation, especially of butyrate, for acetyl-CoA production, a highly oxygen-consuming process (207). Moreover, butyrate directly inhibits HDAC, inducing PDK1-4 expression (208). In short, SCFAs, particularly butyrate, directly or indirectly promote PDK activation to switch acetyl-CoA generation from glycolysis to SCFAs oxidation, key in SCFAs’ regulation of HIF-1α to preserve IEC energy supply and gut hypoxia. Thus understanding the physiological regulation of HIF-1α by SCFAs in IBD could inform prevention and treatment strategies for the condition.
SCFAs trigger HIF-1α in IEC, essential for maintaining IEB function and gut homeostasis. They enhance IEC survival, metabolism, and tight junction through HIF-1α activation, protecting the IEB from inflammatory and infectious damage (54, 209). For example, butyrate enhances IEB function by activating HIF-1α in IEC to upregulate tight junction protein expression, reducing barrier permeability and bacterial translocation, thereby mitigating colonic damage caused by colitis or Clostridioides difficile infection (CDI) (93, 210). However, butyrate’s precise mechanism in HIF-1α modulation remains unclear, suspected to involve physiological hypoxia. Additionally, butyrate stabilizes HIF-1α via HDAC inhibition, enhancing antimicrobial lysozyme activity (211). Research has revealed that the butyrate-producing bacterium Faecalibacterium prausnitzii can modulate HIF-1α to enhance IEC IL-18 expression, potentially aiding in mucosal healing in IBD, offering new insights into HIF-1α-mediated SCFA regulation of intestinal mucosal repair and IEB function maintenance (212). Antibiotic-induced experiment has suggested butyrate stabilizes colonic HIF-1α in mice by inhibiting PHD and increasing 2-oxoglutarate levels, beneficial for gut homeostasis (55). Moreover, SCFAs and HIF-1α play roles in VDR-mediated mucosal barrier protection under hypoxia. Moderate hypoxia induces HIF-1α-VDR promoter binding, enhancing barrier function, with VDR deficiency altering gut microbiota and reducing SCFAs-producing bacteria (61). The role of SCFAs in HIF-1α-regulated VDR signaling under hypoxia needs further investigation. Additionally, SCFAs stimulate mucin expression, critical for barrier function, with HIFs, particularly HIF-1α, being key mucin regulators (213). In asthma studies, butyrate affects HDAC1 through PI3K/Akt/HIF-1α/VEGF pathways, moderating mucus secretion and inflammation (214). However, current IBD research lacks understanding of the synergistic impact of SCFAs and HIF-1α on mucin. While HIF-2α is pivotal in propionate-induced mucin expression, promoting MUC2 secretion from goblet cells, no HIF-1α involvement has been observed, as propionate’s HIF-2α activation for MUC2 production may suppress HIF-1α influence (215). This aligns with findings that increased HIF-1α suppresses HIF-2α and HIF-3α, and vice versa (58). In conclusion, HIF-1α maintains stable expression in IEC under physiological hypoxia, with SCFAs, especially butyrate, being essential for this stability. Therefore, butyrate holds substantial promise as endogenous PHD inhibitors to stabilize HIF-1α and maintain IEB function (Figure 2).
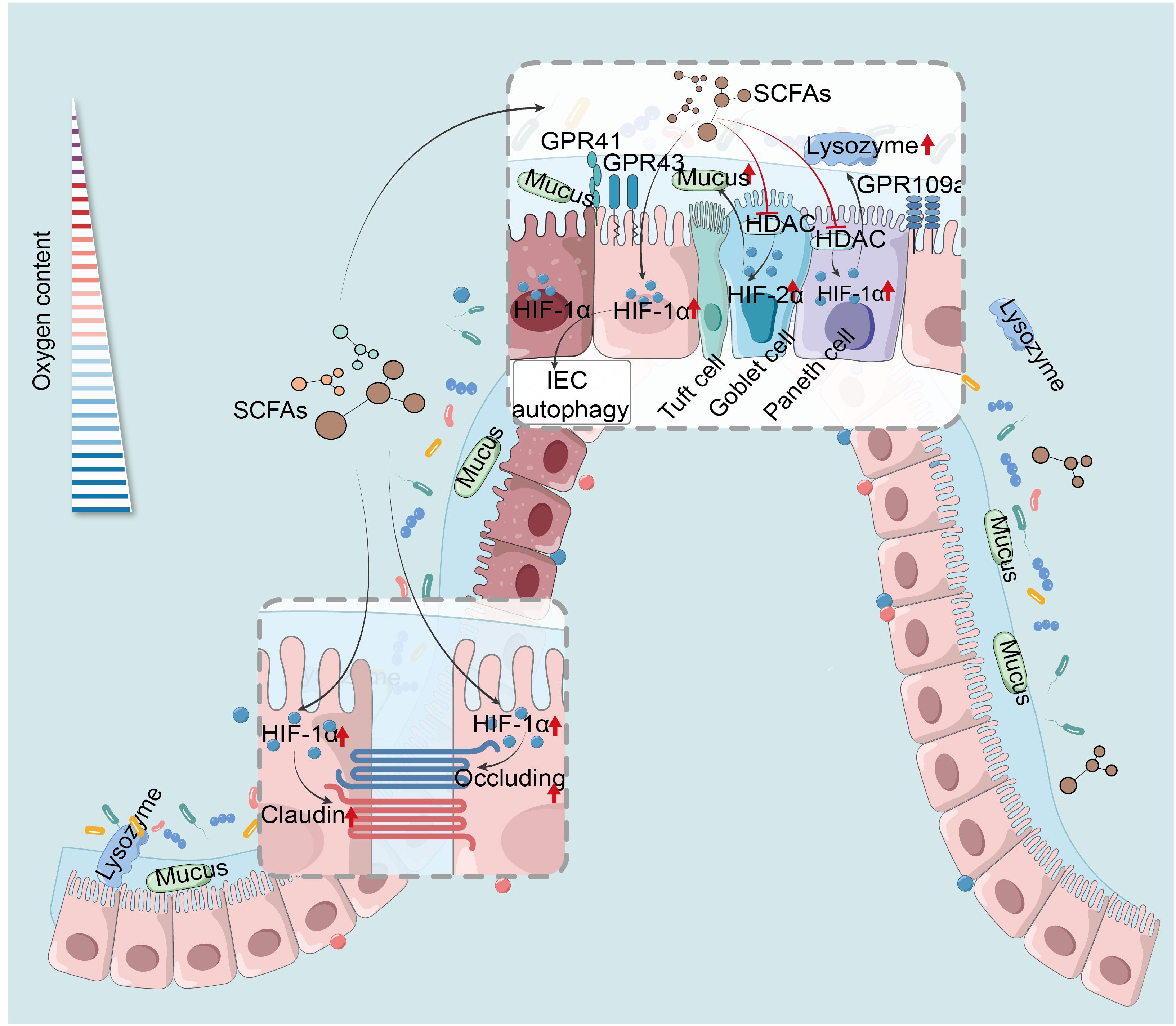
Figure 2 HIF mediates SCFAs regulation of the intestinal barrier and IEC autophagy. SCFAs reinforce IEB by activating HIF-1α in IEC, heightening tight junction proteins, and curtailing permeability and bacterial translocation (93, 210). Their anti-inflammatory action stabilizes HIF-1α via HDAC inhibition, boosting lysozyme activity (211). SCFAs activate HIF-2α, enhancing goblet cell MUC2 secretion and mucosal defense (215). SCFAs induce autophagy in IEC by up-regulation of HIF-1α (76). HIF-1α, hypoxia-inducible factor-1α; SCFAs, short-chain fatty acids; GPR41, G protein-coupled receptor 41; HDAC, histone deacetylases; IEC, intestinal epithelial cells.
In IBD, SCFAs stabilize HIF-1α, modulating immune cells function in the gut. José Luís’s team demonstrated that butyrate triggers the HIF-1α-IL-22 axis in ILC3s, reducing CDI-triggered gut inflammation and reinforcing IEB integrity (216). It also upregulates HIF-1α and Aryl hydrocarbon receptor (AhR) via GPR41 activation and HDAC inhibition, promoting IL-22 in CD4+ T cells and ILCs (217). This mechanism involves butyrate’s histone modifications, which increase HIF-1α binding to the IL-22 promoter, augmenting IL-22 expression (217). These studies provide new insights into SCFAs’ role in anti-inflammatory mediator production. Furthermore, butyrate enhances macrophage antibacterial activity by upregulating HIF-1α through HDAC inhibition without GPCR involvement, enhancing antimicrobial components’ expression like lysozyme and ROS, and raising oxygen consumption by about 50%. However, inhibition of HIF-1α could block the effects of butyrate (218). In research on Streptococcus pneumoniae, acetate boosts macrophage glycolysis, activating HIF-1α and NLRP3 inflammasome, triggering IL-1β release and enhancing nitric oxide (NO) synthesis, which in turn improves macrophage bactericidal function through NO reliance (219). Hence, the glycolysis-HIF-1α pathway is pivotal for SCFAs-mediated macrophages cytotoxicity regulation. However, focused research on HIF-1α in SCFAs-modulated colitis macrophages remains scarce, representing a promising research avenue, especially given its importance in conditions like pneumonia. Additionally, pentanoate facilitates iron uptake in intestinal cells, thereby augmenting HIF-2α expression and inducing c-Maf production, which is crucial for intestinal Treg differentiation and homeostasis (220). Notably, c-Maf typically acts as a positive regulator of IL-10 across various immune cells. However, whether pentanoate prompts Treg or other immune cells to secrete IL-10 was not clarified and warrants further study (Figure 3).
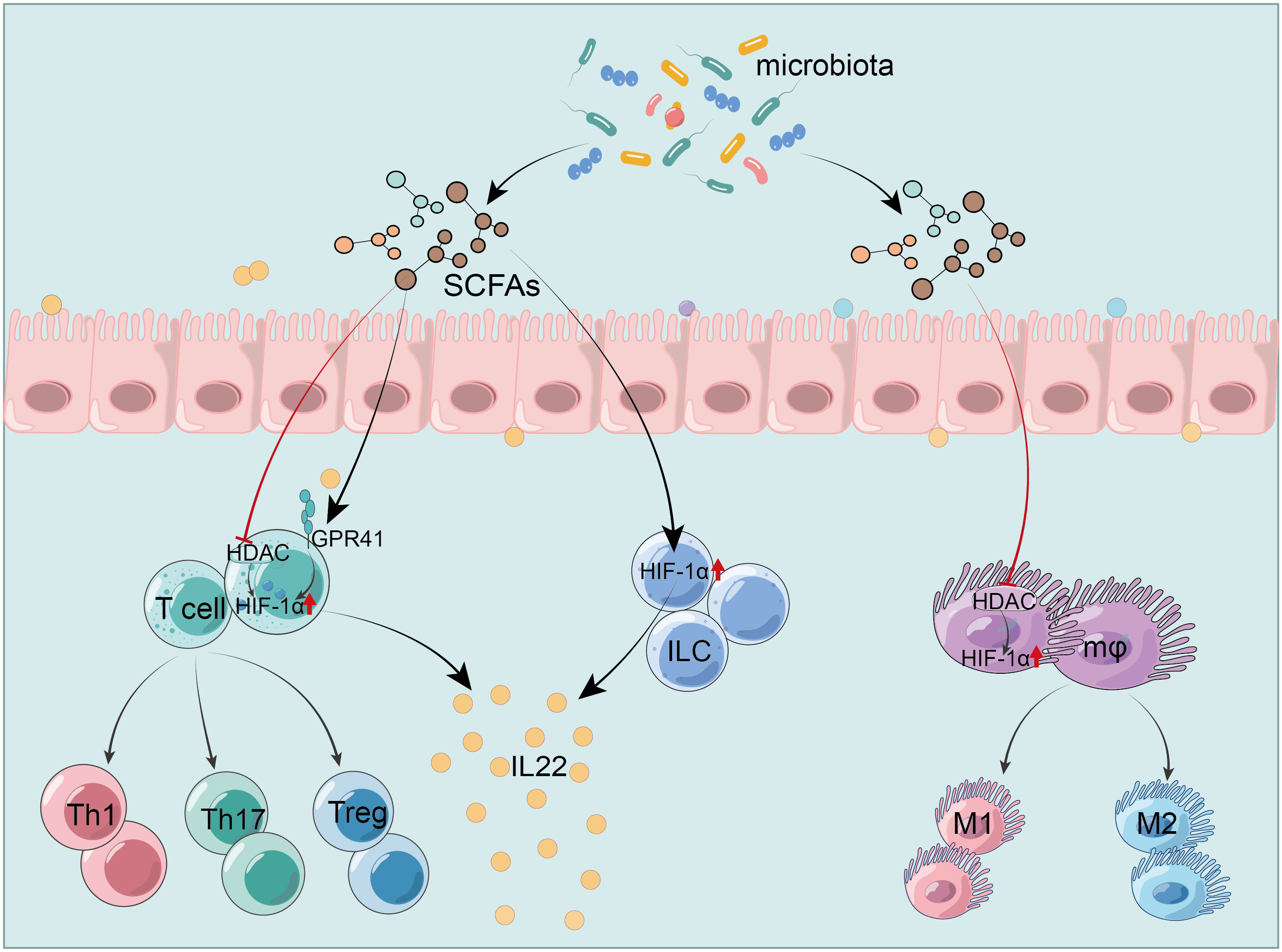
Figure 3 HIF-1α mediates SCFAs regulation of the intestinal immunity. SCFAs modulate HIF-1α to activate ILC and increase IL-22 production (216). They trigger IL-22 production in CD4+ T cells and ILC by activating GPR41 and inhibiting HDAC, which upregulates HIF-1α (217). SCFAs enhance the antibacterial activity of macrophages by inhibiting HDAC and up-regulating HIF-1α (218). HIF-1α, hypoxia-inducible factor-1α; SCFAs, short-chain fatty acids; GPR41, G protein-coupled receptor 41; HDAC, histone deacetylases; IEC, intestinal epithelial cells; ILC, innate lymphoid cells; Treg, regulatory T cells; IL-22, interleukin-22.
SCFAs not only modulate immune cells directly but also influence downstream inflammatory pathways through HIF-1α. Butyrate suppresses HDAC and activates the p-glycogen synthase kinase-3β (GSK-3β)/β-catenin/HIF-1α/NF-κB pathway, reducing inflammation in sleep deprivation-induced colitis mice (221). In human peripheral blood mononuclear cells, butyrate mitigates inflammation by blocking TLR4 signaling without affecting HIF-1α expression and the HIF-1α/HIF-2α ratio. In contrast, caprate, a medium-chain fatty acid (MCFA), activates inflammation via TLR2, reduces IL-10 and HIF-1α, and increases HIF-2α transcription, altering the HIF-1α/HIF-2α ratio (222). The differential immunomodulatory effects of fatty acids may relate to their origins or structures, with SCFAs being endogenous and MCFAs exogenous from diet. Structurally, SCFAs and MCFAs differ by a few carbon atoms. The variance in HIF-1α and HIF-2α expressions might be a compensatory response to inflammation induced by caprate. Besides, high concentrations of acetate in organoid-derived epithelial monolayers from UC patients exhibits anti-inflammatory effects and reinforces barrier function, concurrently elevating HIF-1α levels, though mechanisms are uncertain (223). Notably, acetate lowers IL-10 levels, which contrasts with the view that SCFAs promote IL-10 expression, suggesting that SCFAs type, concentration, or cell-type differences might play a role. Overall, SCFAs-targeted HIF-1α modulation offers a strategy to combat intestinal inflammation. The promotion of HIF-1α in IEC by SCFAs is largely linked to physiological hypoxia, and the HIF pathway is one of the most important pathways to communicate between the host and the intestinal microecology in addition to GPCR and HDAC signaling.
SCFAs activate HIF-1α to induce autophagy in IEC, beneficial for suppressing intestinal inflammation in IBD. In mouse IEC, butyrate eases DSS-induced colitis by upregulating HIF-1α to modulate autophagy and gut microbiota; however, HIF-1α deficiency lowers intestinal butyrate concentration and autophagy levels to increase susceptibility to colitis (Figure 2) (76). The reduced autophagy from HIF-1α deficiency may relate to the disruption of gut physiological hypoxia. Hypoxia or increased oxygen consumption also induces IEC autophagy (62), highlighting HIF-1α’s role in SCFAs-mediated autophagy regulation. Besides, SCFAs and HIF-1α are closely linked to PTEN induced putative kinase 1 (PINK1), an ubiquitin kinase essential for mitochondrial autophagy (224). In CAC models, PINK1 hampers tumor growth by activating p53 and HIF-1α/PDHK1/PDHE1α pathways, reducing acetyl-CoA production; contrastingly, acetate exposure increases acetyl-CoA in tumors, counteracting PINK1’s anti-tumorigenic effects (225). This indicates that the roles of SCFAs and HIF-1α in CAC and IBD are not constant and necessitate tailored analysis for future therapeutic applications.
In recent years, the intricate interplay among hypoxia, gut microbiota, and intestinal immunity has sparked significant interest among researchers. Substantial evidence has suggested that the dysregulation of gut microbiota and its metabolites, SCFAs, under hypoxic conditions, serves as a pivotal pathological feature of IBD and is a key contributor to the dysfunction of IEB and immune system. HIF-1α stands at the core of this complex ecological network, maintaining intestinal homeostasis. However, reviews on the role and mechanisms of HIF-1α-mediated SCFAs regulation in IBD under hypoxic conditions are scarce. Therefore, this review summarizes the roles and mechanisms of SCFAs in stabilizing HIF-1α to regulate IEB function and intestinal immunity under hypoxia, findings that are crucial for elucidating the pathophysiology of IBD and its prevention and treatment.
Hypoxia, a common physiological state in the host gut, provides a stable environment essential for maintaining cellular metabolism, immune homeostasis, and microbiota balance, with HIF-1α being the key regulatory factor under these conditions. HIF-1α and SCFAs preserve IEB function through regulating tight junctions, mucous secretion, and microbial homeostasis. Moreover, they modulate adaptive and innate immune cells homeostasis to suppress colonic inflammation, closely related to T-cells differentiation and macrophage polarization. Notably, SCFAs are closely linked to HIF-1α, playing a pivotal role in hypoxia-induced HIF-1α stabilization, and they synergistically modulate IBD pathogenesis. HIF-1α mediates SCFAs’ regulation of IEB function, intestinal immunity, and autophagy, a key mechanism in SCFAs’ therapeutic potential for IBD. Certainly, GPCR and HDAC are also significant pathways through which SCFAs regulate HIF-1α function. However, studies on the role of hypoxia/HIF-1α or SCFAs in IBD present contradictory conclusions, possibly related to factors such as colitis models, SCFA types and concentrations, gut environment (hypoxia levels, pH), and cell phenotype heterogeneity during experimentation. Considering the complexity of the gut environment and the significant impact of cellular phenotypic heterogeneity on IBD, current research challenges in elucidating the specific pathways and mechanisms by which HIF-1α and SCFAs regulate IBD. Therefore, future studies should focus on interdisciplinary collaboration, utilizing genomics, transcriptomics, and metabolomics, to comprehensively and multidimensionally explore the impact of these factors on the IEB and gut immunity, as well as their roles in different IBD stages. Moreover, single-cell RNA sequencing can reveal cellular phenotypic heterogeneity in the human gut, providing insights directly relevant to human diseases. Currently, animal models, mainly chemically induced such as DSS and TNBS, fail to fully replicate the intestinal characteristics and environment of human IBD. The development and adoption of animal models that closely mirror human physiological and pathological features, including gut microbiome composition, immune responses, and epithelial barrier functions, are essential. Genetic engineering can create models expressing or suppressing genes involved in human IBD, offering insights into underlying molecular pathways. If possible, future research should also more extensively incorporate data from human subjects, including using human intestinal organoids for a more accurate gut epithelial model and analyzing tissue samples from IBD patients.
Though the review presents SCFAs’ regulatory effects on HIF-1α in IBD and their proven therapeutic value in experimental studies, clinical treatments designed to boost SCFAs synthesis in the body, like dietary fiber, probiotics, and polyphenols, fall short of expectations due to human physiological complexities. Research shows that while widespread microbial dysregulation in the intestines of adult IBD patients during remission, their capacity to ferment fiber and produce SCFAs is comparable to healthy individuals (226). Merely supplementing dietary fibers or SCFAs-producing probiotics seems insufficient to correct the gut dysbiosis in IBD. Moreover, despite the regulatory importance of SCFAs in gut homeostasis and health, excessive SCFAs, particularly butyrate, in both the gut and blood disrupt gut ecology, hinder anti-cancer treatment sensitivity, and promote a tumor-friendly microenvironment, detrimental to cancer patients (227, 228). This negative impact could be due to SCFAs levels in the gut surpassing the host’s threshold, which potentially leading to pro-inflammatory and even carcinogenic effects, offering insight into inconsistent results in gut immunity research. Also, with the diverse and sometimes antagonistic interactions among SCFAs types, it’s unclear whether single or multiple SCFAs supplementation is beneficial. Therefore, it becomes necessary to develop clinical methods for the quantitative detection of SCFAs in intestinal cells and precise targeted drug delivery, which may become a focal point in future clinical therapeutic research on SCFAs. Additionally, HIF-1α plays a pivotal role in cellular adaptation to hypoxia, maintaining gut homeostasis and enhancing SCFAs functions, making it a vital target for IBD prevention and treatment. Acetate, a key component of SCFAs and an endogenous inhibitor of PHD stabilizing HIF-1α expression, holds significant implications as a therapeutic target for IBD.
JX: Conceptualization, Visualization, Writing – original draft. XG: Writing – review & editing. ZW: Conceptualization, Supervision, Validation, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Hunan Provincial Natural Science Foundation of China [2023JJ60040], Traditional Chinese Medicine Scientific Research Project of Hunan Province [2021020] and Changsha City Natural Science Foundation [kq2202482].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Agrawal M, Jess T. Implications of the changing epidemiology of inflammatory bowel disease in a changing world. United Eur Gastroenterol J. (2022) 10:1113–20. doi: 10.1002/ueg2.12317
2. Agrawal M, Allin KH, Petralia F, Colombel JF, Jess T. Multiomics to elucidate inflammatory bowel disease risk factors and pathways. Nat Rev Gastroenterol Hepatol. (2022) 19:399–409. doi: 10.1038/s41575-022-00593-y
3. Adolph TE, Zhang J. Diet fuelling inflammatory bowel diseases: preclinical and clinical concepts. Gut. (2022) 71:2574–86. doi: 10.1136/gutjnl-2021-326575
4. Campbell EL, Colgan SP. Control and dysregulation of redox signalling in the gastrointestinal tract. Nat Rev Gastroenterol Hepatol. (2019) 16:106–20. doi: 10.1038/s41575-018-0079-5
5. Cummins EP, Crean D. Hypoxia and inflammatory bowel disease. Microbes infection. (2017) 19:210–21. doi: 10.1016/j.micinf.2016.09.004
6. Saez A, Herrero-Fernandez B, Gomez-Bris R, Sánchez-Martinez H, Gonzalez-Granado JM. Pathophysiology of inflammatory bowel disease: innate immune system. Int J Mol Sci. (2023) 24:1526. doi: 10.3390/ijms24021526
7. Brown E, Taylor CT. Hypoxia-sensitive pathways in intestinal inflammation. J Physiol. (2018) 596:2985–9. doi: 10.1113/jp274350
8. Naito Y, Takagi T, Yoshikawa T. Molecular fingerprints of neutrophil-dependent oxidative stress in inflammatory bowel disease. J Gastroenterol. (2007) 42:787–98. doi: 10.1007/s00535-007-2096-y
9. Chen Y, Cui W, Li X, Yang H. Interaction between commensal bacteria, immune response and the intestinal barrier in inflammatory bowel disease. Front Immunol. (2021) 12:761981. doi: 10.3389/fimmu.2021.761981
10. Cai Z, Wang S, Li J. Treatment of inflammatory bowel disease: A comprehensive review. Front Med. (2021) 8:765474. doi: 10.3389/fmed.2021.765474
11. Beaugerie L, Rahier JF, Kirchgesner J. Predicting, preventing, and managing treatment-related complications in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. (2020) 18:1324–35.e2. doi: 10.1016/j.cgh.2020.02.009
12. Luo H, Cao G, Luo C, Tan D, Vong CT, Xu Y, et al. Emerging pharmacotherapy for inflammatory bowel diseases. Pharmacol Res. (2022) 178:106146. doi: 10.1016/j.phrs.2022.106146
13. Semenza GL. Hif-1 and mechanisms of hypoxia sensing. Curr Opin Cell Biol. (2001) 13:167–71. doi: 10.1016/s0955-0674(00)00194-0
14. Taylor CT, Colgan SP. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat Rev Immunol. (2017) 17:774–85. doi: 10.1038/nri.2017.103
15. Watts ER, Walmsley SR. Inflammation and hypoxia: Hif and Phd isoform selectivity. Trends Mol Med. (2019) 25:33–46. doi: 10.1016/j.molmed.2018.10.006
16. Glover LE, Colgan SP. Epithelial barrier regulation by hypoxia-inducible factor. Ann Am Thorac Soc. (2017) 14:S233–s6. doi: 10.1513/AnnalsATS.201608-610MG
17. Steiner CA, Cartwright IM, Taylor CT, Colgan SP. Hypoxia-inducible factor as a bridge between healthy barrier function, wound healing, and fibrosis. Am J Physiol Cell Physiol. (2022) 323:C866–c78. doi: 10.1152/ajpcell.00227.2022
18. Cronin P, Joyce SA, O'Toole PW, O'Connor EM. Dietary fibre modulates the gut microbiota. Nutrients. (2021) 13:1655. doi: 10.3390/nu13051655
19. Xia Y, Zhang L, Ocansey DKW, Tu Q, Mao F, Sheng X. Role of glycolysis in inflammatory bowel disease and its associated colorectal cancer. Front Endocrinol. (2023) 14:1242991. doi: 10.3389/fendo.2023.1242991
20. Krautkramer KA, Fan J, Bäckhed F. Gut microbial metabolites as multi-kingdom intermediates. Nat Rev Microbiol. (2021) 19:77–94. doi: 10.1038/s41579-020-0438-4
21. Ornelas A, Dowdell AS, Lee JS, Colgan SP. Microbial metabolite regulation of epithelial cell-cell interactions and barrier function. Cells. (2022) 11:944. doi: 10.3390/cells11060944
22. Shin Y, Han S, Kwon J, Ju S, Choi TG, Kang I, et al. Roles of short-chain fatty acids in inflammatory bowel disease. Nutrients. (2023) 15:4466. doi: 10.3390/nu15204466
23. Kumar H, Choi DK. Hypoxia inducible factor pathway and physiological adaptation: A cell survival pathway? Mediators Inflammation. (2015) 2015:584758. doi: 10.1155/2015/584758
24. Fagundes RR, Taylor CT. Determinants of hypoxia-inducible factor activity in the intestinal mucosa. J Appl Physiol (Bethesda Md 1985). (2017) 123:1328–34. doi: 10.1152/japplphysiol.00203.2017
26. Kierans SJ, Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (Hif): implications for cellular physiology. J Physiol. (2021) 599:23–37. doi: 10.1113/jp280572
27. Chen PS, Chiu WT, Hsu PL, Lin SC, Peng IC, Wang CY, et al. Pathophysiological implications of hypoxia in human diseases. J BioMed Sci. (2020) 27:63. doi: 10.1186/s12929-020-00658-7
28. Stellingwerff T, Peeling P, Garvican-Lewis LA, Hall R, Koivisto AE, Heikura IA, et al. Nutrition and altitude: strategies to enhance adaptation, improve performance and maintain health: A narrative review. Sports Med (Auckland NZ). (2019) 49:169–84. doi: 10.1007/s40279-019-01159-w
29. Wu D, Cao W, Xiang D, Hu YP, Luo B, Chen P. Exercise induces tissue hypoxia and hif-1α Redistribution in the small intestine. J sport Health Sci. (2020) 9:82–9. doi: 10.1016/j.jshs.2019.05.002
30. Villafuerte FC, Simonson TS, Bermudez D, León-Velarde F. High-altitude erythrocytosis: mechanisms of adaptive and maladaptive responses. Physiol (Bethesda Md). (2022) 37:0. doi: 10.1152/physiol.00029.2021
31. Rodriguez D, Watts D, Gaete D, Sormendi S, Wielockx B. Hypoxia pathway proteins and their impact on the blood vasculature. Int J Mol Sci. (2021) 22:9191. doi: 10.3390/ijms22179191
32. Santos CX, Anilkumar N, Zhang M, Brewer AC, Shah AM. Redox signaling in cardiac myocytes. Free Radical Biol Med. (2011) 50:777–93. doi: 10.1016/j.freeradbiomed.2011.01.003
33. Luo Z, Tian M, Yang G, Tan Q, Chen Y, Li G, et al. Hypoxia signaling in human health and diseases: implications and prospects for therapeutics. Signal transduction targeted Ther. (2022) 7:218. doi: 10.1038/s41392-022-01080-1
34. Singhal R, Shah YM. Oxygen battle in the gut: hypoxia and hypoxia-inducible factors in metabolic and inflammatory responses in the intestine. J Biol Chem. (2020) 295:10493–505. doi: 10.1074/jbc.REV120.011188
35. Krzywinska E, Sobecki M, Nagarajan S, Zacharjasz J, Tambuwala MM, Pelletier A, et al. The transcription factor Hif-1α Mediates plasticity of Nkp46+ Innate lymphoid cells in the gut. J Exp Med. (2022) 219:e20210909. doi: 10.1084/jem.20210909
36. Yu B, Wang X, Song Y, Xie G, Jiao S, Shi L, et al. The role of hypoxia-inducible factors in cardiovascular diseases. Pharmacol Ther. (2022) 238:108186. doi: 10.1016/j.pharmthera.2022.108186
37. Page LK, Staples KJ, Spalluto CM, Watson A, Wilkinson TMA. Influence of hypoxia on the epithelial-pathogen interactions in the lung: implications for respiratory disease. Front Immunol. (2021) 12:653969. doi: 10.3389/fimmu.2021.653969
38. Tang YY, Wang DC, Wang YQ, Huang AF, Xu WD. Emerging role of hypoxia-inducible factor-1α in inflammatory autoimmune diseases: A comprehensive review. Front Immunol. (2022) 13:1073971. doi: 10.3389/fimmu.2022.1073971
39. Wicks EE, Semenza GL. Hypoxia-inducible factors: cancer progression and clinical translation. J Clin Invest. (2022) 132:e159839. doi: 10.1172/jci159839
40. Kim R, Attayek PJ, Wang Y, Furtado KL, Tamayo R, Sims CE, et al. An in vitro intestinal platform with a self-sustaining oxygen gradient to study the human gut/microbiome interface. Biofabrication. (2019) 12:015006. doi: 10.1088/1758-5090/ab446e
41. Chen Y, Rudolph SE, Longo BN, Pace F, Roh TT, Condruti R, et al. Bioengineered 3d tissue model of intestine epithelium with oxygen gradients to sustain human gut microbiome. Advanced healthcare materials. (2022) 11:e2200447. doi: 10.1002/adhm.202200447
42. Glover LE, Lee JS, Colgan SP. Oxygen metabolism and barrier regulation in the intestinal mucosa. J Clin Invest. (2016) 126:3680–8. doi: 10.1172/jci84429
43. Keeley TP, Mann GE. Defining physiological normoxia for improved translation of cell physiology to animal models and humans. Physiol Rev. (2019) 99:161–234. doi: 10.1152/physrev.00041.2017
44. Dvornikova KA, Platonova ON, Bystrova EY. Hypoxia and intestinal inflammation: common molecular mechanisms and signaling pathways. Int J Mol Sci. (2023) 24:2425. doi: 10.3390/ijms24032425
45. Konjar Š, Pavšič M, Veldhoen M. Regulation of oxygen homeostasis at the intestinal epithelial barrier site. Int J Mol Sci. (2021) 22:9170. doi: 10.3390/ijms22179170
46. Knyazev E, Maltseva D, Raygorodskaya M, Shkurnikov M. Hif-dependent Nfatc1 activation upregulates Itga5 and plaur in intestinal epithelium in inflammatory bowel disease. Front Genet. (2021) 12:791640. doi: 10.3389/fgene.2021.791640
47. Zheng L, Kelly CJ, Colgan SP. Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A review in the theme: cellular responses to hypoxia. Am J Physiol Cell Physiol. (2015) 309:C350–60. doi: 10.1152/ajpcell.00191.2015
48. Colgan SP, Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol. (2010) 7:281–7. doi: 10.1038/nrgastro.2010.39
49. Schwerdtfeger LA, Nealon NJ, Ryan EP, Tobet SA. Human colon function ex vivo: dependence on oxygen and sensitivity to antibiotic. PloS One. (2019) 14:e0217170. doi: 10.1371/journal.pone.0217170
50. Dosh RH, Jordan-Mahy N, Sammon C, Le Maitre CL. Long-term in vitro 3d hydrogel co-culture model of inflammatory bowel disease. Sci Rep. (2019) 9:1812. doi: 10.1038/s41598-019-38524-8
51. de Vos WM, Tilg H, Van Hul M, Cani PD. Gut microbiome and health: mechanistic insights. Gut. (2022) 71:1020–32. doi: 10.1136/gutjnl-2021-326789
52. Donaldson GP, Lee SM, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol. (2016) 14:20–32. doi: 10.1038/nrmicro3552
53. McCallum G, Tropini C. The gut microbiota and its biogeography. Nat Rev Microbiol. (2023) 22:105–18. doi: 10.1038/s41579-023-00969-0
54. Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial Hif augments tissue barrier function. Cell Host Microbe. (2015) 17:662–71. doi: 10.1016/j.chom.2015.03.005
55. Wang RX, Henen MA, Lee JS, Vögeli B, Colgan SP. Microbiota-derived butyrate is an endogenous Hif prolyl hydroxylase inhibitor. Gut Microbes. (2021) 13:1938380. doi: 10.1080/19490976.2021.1938380
56. Kint N, Alves Feliciano C, Martins MC, Morvan C, Fernandes SF, Folgosa F, et al. How the anaerobic enteropathogen clostridioides difficile tolerates low O(2) tensions. mBio. (2020) 11:e01559–20. doi: 10.1128/mBio.01559-20
57. McGettrick AF, O'Neill LAJ. The role of hif in immunity and inflammation. Cell Metab. (2020) 32:524–36. doi: 10.1016/j.cmet.2020.08.002
58. Serocki M, Bartoszewska S, Janaszak-Jasiecka A, Ochocka RJ, Collawn JF, Bartoszewski R. Mirnas regulate the Hif switch during hypoxia: A novel therapeutic target. Angiogenesis. (2018) 21:183–202. doi: 10.1007/s10456-018-9600-2
59. Flück K, Fandrey J. Oxygen sensing in intestinal mucosal inflammation. Pflugers Archiv Eur J Physiol. (2016) 468:77–84. doi: 10.1007/s00424-015-1722-4
60. Shao Y, Wang K, Xiong X, Liu H, Zhou J, Zou L, et al. The landscape of interactions between hypoxia-inducible factors and reactive oxygen species in the gastrointestinal tract. Oxid Med Cell Longev. (2021) 2021:8893663. doi: 10.1155/2021/8893663
61. Wang Z, Yang H, Lv H, Huang C, Qian J. Vitamin D receptor-dependent protective effect of moderate hypoxia in a mouse colitis model. Front Physiol. (2022) 13:876890. doi: 10.3389/fphys.2022.876890
62. Cosin-Roger J, Simmen S, Melhem H, Atrott K, Frey-Wagner I, Hausmann M, et al. Hypoxia ameliorates intestinal inflammation through Nlrp3/Mtor downregulation and autophagy activation. Nat Commun. (2017) 8:98. doi: 10.1038/s41467-017-00213-3
63. Litvak Y, Byndloss MX, Bäumler AJ. Colonocyte metabolism shapes the gut microbiota. Sci (New York NY). (2018) 362:eaat9076. doi: 10.1126/science.aat9076
64. Byndloss MX, Olsan EE, Rivera-Chávez F, Tiffany CR, Cevallos SA, Lokken KL, et al. Microbiota-activated Ppar-Γ Signaling inhibits dysbiotic enterobacteriaceae expansion. Sci (New York NY). (2017) 357:570–5. doi: 10.1126/science.aam9949
65. Ge L, Qi J, Shao B, Ruan Z, Ren Y, Sui S, et al. Microbial hydrogen economy alleviates colitis by reprogramming colonocyte metabolism and reinforcing intestinal barrier. Gut Microbes. (2022) 14:2013764. doi: 10.1080/19490976.2021.2013764
66. Lin N, Shay JES, Xie H, Lee DSM, Skuli N, Tang Q, et al. Myeloid cell hypoxia-inducible factors promote resolution of inflammation in experimental colitis. Front Immunol. (2018) 9:2565. doi: 10.3389/fimmu.2018.02565
67. Vavricka SR, Rogler G, Maetzler S, Misselwitz B, Safroneeva E, Frei P, et al. High altitude journeys and flights are associated with an increased risk of flares in inflammatory bowel disease patients. J Crohn's colitis. (2014) 8:191–9. doi: 10.1016/j.crohns.2013.07.011
68. deZoeten EF, Battista KD, Colson SB, Lovell MA, Kessler BE, Isfort RW, et al. Markers of hypoxia correlate with histologic and endoscopic severity of colitis in inflammatory bowel disease. Hypoxia (Auckland NZ). (2020) 8:1–12. doi: 10.2147/hp.S219049
69. Kerber EL, Padberg C, Koll N, Schuetzhold V, Fandrey J, Winning S. The importance of hypoxia-inducible factors (Hif-1 and Hif-2) for the pathophysiology of inflammatory bowel disease. Int J Mol Sci. (2020) 21:8551. doi: 10.3390/ijms21228551
70. Kim YE, Lee M, Gu H, Kim J, Jeong S, Yeo S, et al. Hif-1α Activation in myeloid cells accelerates dextran sodium sulfate-induced colitis progression in mice. Dis Models Mech. (2018) 11:dmm033241. doi: 10.1242/dmm.033241
71. Han Y, Ding L, Cheng X, Zhao M, Zhao T, Guo L, et al. Hypoxia augments cerebral inflammation in a dextran sulfate sodium-induced colitis mouse model. Front Cell Neurosci. (2020) 14:611764. doi: 10.3389/fncel.2020.611764
72. Saijo H, Tatsumi N, Arihiro S, Kato T, Okabe M, Tajiri H, et al. Microangiopathy triggers, and inducible nitric oxide synthase exacerbates dextran sulfate sodium-induced colitis. Lab Invest. (2015) 95:728–48. doi: 10.1038/labinvest.2015.60
73. Cibor D, Domagala-Rodacka R, Rodacki T, Jurczyszyn A, Mach T, Owczarek D. Endothelial dysfunction in inflammatory bowel diseases: pathogenesis, assessment and implications. World J Gastroenterol. (2016) 22:1067–77. doi: 10.3748/wjg.v22.i3.1067
74. Zeitouni NE, Chotikatum S, von Köckritz-Blickwede M, Naim HY. The impact of hypoxia on intestinal epithelial cell functions: consequences for invasion by bacterial pathogens. Mol Cell Pediatr. (2016) 3:14. doi: 10.1186/s40348-016-0041-y
75. Cummins EP, Doherty GA, Taylor CT. Hydroxylases as therapeutic targets in inflammatory bowel disease. Lab Invest. (2013) 93:378–83. doi: 10.1038/labinvest.2013.9
76. Zhou C, Li L, Li T, Sun L, Yin J, Guan H, et al. Scfas induce autophagy in intestinal epithelial cells and relieve colitis by stabilizing Hif-1α. J Mol Med (Berlin Germany). (2020) 98:1189–202. doi: 10.1007/s00109-020-01947-2
77. Li Y, Wang Y, Shi F, Zhang X, Zhang Y, Bi K, et al. Phospholipid metabolites of the gut microbiota promote hypoxia-induced intestinal injury via Cd1d-dependent Γδ T cells. Gut Microbes. (2022) 14:2096994. doi: 10.1080/19490976.2022.2096994
78. Bossuet N, Guyonnet C, Chagneau CV, Tang-Fichaux M, Penary M, Loubet D, et al. Oxygen concentration modulates colibactin production. Gut Microbes. (2023) 15:2222437. doi: 10.1080/19490976.2023.2222437
79. Singh V, Lee G, Son H, Koh H, Kim ES, Unno T, et al. Butyrate producers, "the sentinel of gut": their intestinal significance with and beyond butyrate, and prospective use as microbial therapeutics. Front Microbiol. (2022) 13:1103836. doi: 10.3389/fmicb.2022.1103836
80. Brown E, Rowan C, Strowitzki MJ, Fagundes RR, Faber KN, Güntsch A, et al. Mucosal inflammation downregulates Phd1 expression promoting a barrier-protective Hif-1α Response in ulcerative colitis patients. FASEB J. (2020) 34:3732–42. doi: 10.1096/fj.201902103R
81. Singhal R, Mitta SR, Das NK, Kerk SA, Sajjakulnukit P, Solanki S, et al. Hif-2α Activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J Clin Invest. (2021) 131:e143691. doi: 10.1172/jci143691
82. Shah YM, Ito S, Morimura K, Chen C, Yim SH, Haase VH, et al. Hypoxia-inducible factor augments experimental colitis through an Mif-dependent inflammatory signaling cascade. Gastroenterology. (2008) 134:2036–48, 48.e1-3. doi: 10.1053/j.gastro.2008.03.009
83. Solanki S, Devenport SN, Ramakrishnan SK, Shah YM. Temporal induction of intestinal epithelial hypoxia-inducible factor-2α Is sufficient to drive colitis. Am J Physiol Gastrointestinal liver Physiol. (2019) 317:G98–g107. doi: 10.1152/ajpgi.00081.2019
84. Xue X, Ramakrishnan S, Anderson E, Taylor M, Zimmermann EM, Spence JR, et al. Endothelial pas domain protein 1 activates the inflammatory response in the intestinal epithelium to promote colitis in mice. Gastroenterology. (2013) 145:831–41. doi: 10.1053/j.gastro.2013.07.010
85. Bhat S, Rieder F. Hypoxia-inducible factor 1-alpha stabilizers in the treatment of inflammatory bowel diseases: oxygen as a novel Ibd therapy? J Crohn's colitis. (2022) 16:1924–32. doi: 10.1093/ecco-jcc/jjac092
86. Van Welden S, Selfridge AC, Hindryckx P. Intestinal hypoxia and hypoxia-induced signalling as therapeutic targets for Ibd. Nat Rev Gastroenterol Hepatol. (2017) 14:596–611. doi: 10.1038/nrgastro.2017.101
87. Martens EC, Neumann M, Desai MS. Interactions of commensal and pathogenic microorganisms with the intestinal mucosal barrier. Nat Rev Microbiol. (2018) 16:457–70. doi: 10.1038/s41579-018-0036-x
88. Manresa MC, Taylor CT. Hypoxia inducible factor (Hif) hydroxylases as regulators of intestinal epithelial barrier function. Cell Mol Gastroenterol Hepatol. (2017) 3:303–15. doi: 10.1016/j.jcmgh.2017.02.004
89. Chelakkot C, Ghim J, Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med. (2018) 50:1–9. doi: 10.1038/s12276-018-0126-x
90. Howe KL, Lorentz RJ, Assa A, Pinnell LJ, Johnson-Henry KC, Sherman PM. Transforming growth factor-B1 protects against intestinal epithelial barrier dysfunction caused by hypoxia-reoxygenation. Shock (Augusta Ga). (2015) 43:483–9. doi: 10.1097/shk.0000000000000333
91. Dowdell AS, Cartwright IM, Goldberg MS, Kostelecky R, Ross T, Welch N, et al. The Hif target Atg9a is essential for epithelial barrier function and tight junction biogenesis. Mol Biol Cell. (2020) 31:2249–58. doi: 10.1091/mbc.E20-05-0291
92. Lei Q, Qiang F, Chao D, Di W, Guoqian Z, Bo Y, et al. Amelioration of hypoxia and lps-induced intestinal epithelial barrier dysfunction by Emodin through the suppression of the Nf-Kb and Hif-1α Signaling pathways. Int J Mol Med. (2014) 34:1629–39. doi: 10.3892/ijmm.2014.1965
93. Yin J, Zhou C, Yang K, Ren Y, Qiu Y, Xu P, et al. Mutual regulation between butyrate and hypoxia-inducible factor-1α in epithelial cell promotes expression of tight junction proteins. Cell Biol Int. (2020) 44:1405–14. doi: 10.1002/cbin.11336
94. Rosenberger P, Khoury J, Kong T, Weissmüller T, Robinson AM, Colgan SP. Identification of vasodilator-stimulated phosphoprotein (Vasp) as an Hif-regulated tissue permeability factor during hypoxia. FASEB J. (2007) 21:2613–21. doi: 10.1096/fj.06-8004com
95. Xu LF, Teng X, Guo J, Sun M. Protective effect of intestinal trefoil factor on injury of intestinal epithelial tight junction induced by platelet activating factor. Inflammation. (2012) 35:308–15. doi: 10.1007/s10753-011-9320-x
96. Goggins BJ, Minahan K, Sherwin S, Soh WS, Pryor J, Bruce J, et al. Pharmacological Hif-1 stabilization promotes intestinal epithelial healing through regulation of A-integrin expression and function. Am J Physiol Gastrointestinal liver Physiol. (2021) 320:G420–g38. doi: 10.1152/ajpgi.00192.2020
97. Sun L, Li T, Tang H, Yu K, Ma Y, Yu M, et al. Intestinal epithelial cells-derived hypoxia-inducible factor-1α Is essential for the homeostasis of intestinal intraepithelial lymphocytes. Front Immunol. (2019) 10:806. doi: 10.3389/fimmu.2019.00806
98. Sun M, He C, Wu W, Zhou G, Liu F, Cong Y, et al. Hypoxia inducible factor-1α-induced interleukin-33 expression in intestinal epithelia contributes to mucosal homeostasis in inflammatory bowel disease. Clin Exp Immunol. (2017) 187:428–40. doi: 10.1111/cei.12896
99. Glover LE, Bowers BE, Saeedi B, Ehrentraut SF, Campbell EL, Bayless AJ, et al. Control of creatine metabolism by Hif is an endogenous mechanism of barrier regulation in colitis. Proc Natl Acad Sci U.S.A. (2013) 110:19820–5. doi: 10.1073/pnas.1302840110
100. Neunlist M, Van Landeghem L, Mahé MM, Derkinderen P, des Varannes SB, Rolli-Derkinderen M. The digestive neuronal-glial-epithelial unit: A new actor in gut health and disease. Nat Rev Gastroenterol Hepatol. (2013) 10:90–100. doi: 10.1038/nrgastro.2012.221
101. Kearon JE, Kocherry SC, Zoumboulakis D, Rivera D, Lourenssen SR, Blennerhassett MG. Gdnf requires Hif-1α and ret activation for suppression of programmed cell death of enteric neurons by metabolic challenge. Mol Cell Neurosci. (2021) 115:103655. doi: 10.1016/j.mcn.2021.103655
102. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-inducible factor-1 alpha-dependent induction of Foxp3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U.S.A. (2012) 109:E2784–93. doi: 10.1073/pnas.1202366109
103. Marks E, Naudin C, Nolan G, Goggins BJ, Burns G, Mateer SW, et al. Regulation of Il-12p40 by Hif controls Th1/Th17 responses to prevent mucosal inflammation. Mucosal Immunol. (2017) 10:1224–36. doi: 10.1038/mi.2016.135
104. Lv Q, Xing Y, Dong D, Hu Y, Chen Q, Zhai L, et al. Costunolide ameliorates colitis via specific inhibition of Hif1α/glycolysis-mediated Th17 differentiation. Int Immunopharmacol. (2021) 97:107688. doi: 10.1016/j.intimp.2021.107688
105. Endo K, Kito H, Tanaka R, Kajikuri J, Tanaka S, Elboray EE, et al. Possible contribution of inflammation-associated hypoxia to increased K(2p)5.1 K(+) channel expression in Cd4(+) T cells of the mouse model for inflammatory bowel disease. Int J Mol Sci. (2019) 21:38. doi: 10.3390/ijms21010038
106. Qian T, Hong J, Wang L, Wang Z, Lu Z, Li Y, et al. Regulation of Cd11b by Hif-1α and the Stat3 signaling pathway contributes to the immunosuppressive function of B cells in inflammatory bowel disease. Mol Immunol. (2019) 111:162–71. doi: 10.1016/j.molimm.2019.04.005
107. Zhu Y, Zhang X, Xie S, Bao W, Chen J, Wu Q, et al. Oxidative phosphorylation regulates interleukin-10 production in regulatory B cells via the extracellular signal-related kinase pathway. Immunology. (2022) 167:576–89. doi: 10.1111/imm.13554
108. Meng X, Grötsch B, Luo Y, Knaup KX, Wiesener MS, Chen XX, et al. Hypoxia-inducible factor-1α Is a critical transcription factor for Il-10-producing B cells in autoimmune disease. Nat Commun. (2018) 9:251. doi: 10.1038/s41467-017-02683-x
109. Kumar R, Singh AK, Starokadomskyy P, Luo W, Theiss AL, Burstein E, et al. Cutting edge: hypoxia-induced ubc9 promoter hypermethylation regulates Il-17 expression in ulcerative colitis. J Immunol (Baltimore Md 1950). (2021) 206:936–40. doi: 10.4049/jimmunol.2000015
110. Qiu B, Yuan P, Du X, Jin H, Du J, Huang Y. Hypoxia inducible factor-1α Is an important regulator of macrophage biology. Heliyon. (2023) 9:e17167. doi: 10.1016/j.heliyon.2023.e17167
111. Zhu W, Chen Q, Li Y, Wan J, Li J, Tang S. Hif-1α-overexpressing mesenchymal stem cells attenuate colitis by regulating M1-like macrophages polarization toward M2-like macrophages. Biomedicines. (2023) 11:825. doi: 10.3390/biomedicines11030825
112. Yuan Y, Ni S, Zhuge A, Li L, Li B. Adipose-derived mesenchymal stem cells reprogram M1 macrophage metabolism via Phd2/Hif-1α Pathway in colitis mice. Front Immunol. (2022) 13:859806. doi: 10.3389/fimmu.2022.859806
113. Gómez-Ferrer M, Amaro-Prellezo E, Dorronsoro A, Sánchez-Sánchez R, Vicente Á, Cosín-Roger J, et al. Hif-overexpression and pro-inflammatory priming in human mesenchymal stromal cells improves the healing properties of extracellular vesicles in experimental Crohn's disease. Int J Mol Sci. (2021) 22:11269. doi: 10.3390/ijms222011269
114. Zhuang H, Lv Q, Zhong C, Cui Y, He L, Zhang C, et al. Tiliroside ameliorates ulcerative colitis by restoring the M1/M2 macrophage balance via the Hif-1α/glycolysis pathway. Front Immunol. (2021) 12:649463. doi: 10.3389/fimmu.2021.649463
115. Liu R, Li X, Ma H, Yang Q, Shang Q, Song L, et al. Spermidine endows macrophages anti-inflammatory properties by inducing mitochondrial superoxide-dependent Ampk activation, Hif-1α Upregulation and autophagy. Free Radical Biol Med. (2020) 161:339–50. doi: 10.1016/j.freeradbiomed.2020.10.029
116. Wu MM, Wang QM, Huang BY, Mai CT, Wang CL, Wang TT, et al. Dioscin ameliorates murine ulcerative colitis by regulating macrophage polarization. Pharmacol Res. (2021) 172:105796. doi: 10.1016/j.phrs.2021.105796
117. Ortiz-Masiá D, Cosín-Roger J, Calatayud S, Hernández C, Alós R, Hinojosa J, et al. Hypoxic macrophages impair autophagy in epithelial cells through Wnt1: relevance in Ibd. Mucosal Immunol. (2014) 7:929–38. doi: 10.1038/mi.2013.108
118. Grizotte-Lake M, Vaishnava S. Autophagy: suicide prevention hotline for the gut epithelium. Cell Host Microbe. (2018) 23:147–8. doi: 10.1016/j.chom.2018.01.015
119. Foerster EG, Mukherjee T, Cabral-Fernandes L, Rocha JDB, Girardin SE, Philpott DJ. How autophagy controls the intestinal epithelial barrier. Autophagy. (2022) 18:86–103. doi: 10.1080/15548627.2021.1909406
120. Köhler T, Reizis B, Johnson RS, Weighardt H, Förster I. Influence of hypoxia-inducible factor 1α on dendritic cell differentiation and migration. Eur J Immunol. (2012) 42:1226–36. doi: 10.1002/eji.201142053
121. Flück K, Breves G, Fandrey J, Winning S. Hypoxia-inducible factor 1 in dendritic cells is crucial for the activation of protective regulatory T cells in murine colitis. Mucosal Immunol. (2016) 9:379–90. doi: 10.1038/mi.2015.67
122. Parkos CA. Neutrophil-epithelial interactions: A double-edged sword. Am J Pathol. (2016) 186:1404–16. doi: 10.1016/j.ajpath.2016.02.001
123. Lu H, Lin J, Xu C, Sun M, Zuo K, Zhang X, et al. Cyclosporine modulates neutrophil functions via the Sirt6-Hif-1α-glycolysis axis to alleviate severe ulcerative colitis. Clin Trans Med. (2021) 11:e334. doi: 10.1002/ctm2.334
124. Drury B, Hardisty G, Gray RD, Ho GT. Neutrophil extracellular traps in inflammatory bowel disease: pathogenic mechanisms and clinical translation. Cell Mol Gastroenterol Hepatol. (2021) 12:321–33. doi: 10.1016/j.jcmgh.2021.03.002
125. D'Alessio S, Ungaro F, Noviello D, Lovisa S, Peyrin-Biroulet L, Danese S. Revisiting fibrosis in inflammatory bowel disease: the gut thickens. Nat Rev Gastroenterol Hepatol. (2022) 19:169–84. doi: 10.1038/s41575-021-00543-0
126. Lv XM, Li MD, Cheng S, Liu BL, Liu K, Zhang CF, et al. Neotuberostemonine inhibits the differentiation of lung fibroblasts into myofibroblasts in mice by regulating hif-1α Signaling. Acta pharmacologica Sin. (2018) 39:1501–12. doi: 10.1038/aps.2017.202
127. Yuan S, Wei C, Liu G, Zhang L, Li J, Li L, et al. Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via Hif-1α/Slc7a11 pathway. Cell proliferation. (2022) 55:e13158. doi: 10.1111/cpr.13158
128. Zhao H, Han Y, Jiang N, Li C, Yang M, Xiao Y, et al. Effects of Hif-1α on renal fibrosis in cisplatin-induced chronic kidney disease. Clin Sci (Lond). (2021) 135:1273–88. doi: 10.1042/cs20210061
129. Huang L, Qian W, Xu Y, Guo Z, Yin Y, Guo F, et al. Mesenteric adipose tissue contributes to intestinal fibrosis in Crohn's disease through the Atx-Lpa axis. J Crohn's colitis. (2022) 16:1124–39. doi: 10.1093/ecco-jcc/jjac017
130. Simmen S, Maane M, Rogler S, Baebler K, Lang S, Cosin-Roger J, et al. Hypoxia reduces the transcription of fibrotic markers in the intestinal mucosa. Inflammatory intestinal Dis. (2021) 6:87–100. doi: 10.1159/000513061
131. Prados ME, García-Martín A, Unciti-Broceta JD, Palomares B, Collado JA, Minassi A, et al. Betulinic acid hydroxamate prevents colonic inflammation and fibrosis in murine models of inflammatory bowel disease. Acta pharmacologica Sin. (2021) 42:1124–38. doi: 10.1038/s41401-020-0497-0
132. Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. (2014) 63:1275–83. doi: 10.1136/gutjnl-2013-304833
133. Lo Sasso G, Khachatryan L, Kondylis A, Battey JND, Sierro N, Danilova NA, et al. Inflammatory bowel disease-associated changes in the gut: focus on Kazan patients. Inflammatory bowel Dis. (2021) 27:418–33. doi: 10.1093/ibd/izaa188
134. Kaczmarczyk O, Dąbek-Drobny A, Piątek-Guziewicz A, Woźniakiewicz M, Paśko P, Dobrowolska-Iwanek J, et al. The importance of nutritional aspects in the assessment of inflammation and intestinal barrier in patients with inflammatory bowel disease. Nutrients. (2022) 14:4622. doi: 10.3390/nu14214622
135. Facchin S, Vitulo N, Calgaro M, Buda A, Romualdi C, Pohl D, et al. Microbiota changes induced by microencapsulated sodium butyrate in patients with inflammatory bowel disease. Neurogastroenterol Motil. (2020) 32:e13914. doi: 10.1111/nmo.13914
136. Fritsch J, Garces L, Quintero MA, Pignac-Kobinger J, Santander AM, Fernández I, et al. Low-fat, high-fiber diet reduces markers of inflammation and dysbiosis and improves quality of life in patients with ulcerative colitis. Clin Gastroenterol Hepatol. (2021) 19:1189–99.e30. doi: 10.1016/j.cgh.2020.05.026
137. Brown K, Abbott DW, Uwiera RRE, Inglis GD. Removal of the cecum affects intestinal fermentation, enteric bacterial community structure, and acute colitis in mice. Gut Microbes. (2018) 9:218–35. doi: 10.1080/19490976.2017.1408763
138. Thoo L, Noti M, Krebs P. Keep calm: the intestinal barrier at the interface of peace and war. Cell Death Dis. (2019) 10:849. doi: 10.1038/s41419-019-2086-z
139. Roediger WE. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut. (1980) 21:793–8. doi: 10.1136/gut.21.9.793
140. Colgan SP, Wang RX, Hall CHT, Bhagavatula G, Lee JS. Revisiting the "Starved gut" Hypothesis in inflammatory bowel disease. Immunometabolism (Cobham Surrey). (2023) 5:e0016. doi: 10.1097/in9.0000000000000016
141. Yao Y, Sun S, Gu J, Ni H, Zhong K, Xu Q, et al. Roux-en-Y reconstruction alleviates radical gastrectomy-induced colitis via down-regulation of the butyrate/nlrp3 signaling pathway. EBioMedicine. (2022) 86:104347. doi: 10.1016/j.ebiom.2022.104347
142. Laffin M, Fedorak R, Zalasky A, Park H, Gill A, Agrawal A, et al. A high-sugar diet rapidly enhances susceptibility to colitis via depletion of luminal short-chain fatty acids in mice. Sci Rep. (2019) 9:12294. doi: 10.1038/s41598-019-48749-2
143. Martin-Gallausiaux C, Marinelli L, Blottière HM, Larraufie P, Lapaque N. Scfa: mechanisms and functional importance in the gut. Proc Nutr Soc. (2021) 80:37–49. doi: 10.1017/s0029665120006916
144. Roediger WE. The starved colon–diminished mucosal nutrition, diminished absorption, and colitis. Dis colon rectum. (1990) 33:858–62. doi: 10.1007/bf02051922
145. Roediger WE. The colonic epithelium in ulcerative colitis: an energy-deficiency disease? Lancet (London England). (1980) 2:712–5. doi: 10.1016/s0140-6736(80)91934-0
146. Fagundes RR, Belt SC, Bakker BM, Dijkstra G, Harmsen HJM, Faber KN. Beyond butyrate: microbial fiber metabolism supporting colonic epithelial homeostasis. Trends Microbiol. (2024) 32:178–89. doi: 10.1016/j.tim.2023.07.014
147. Wang Y, Kim R, Gunasekara DB, Reed MI, DiSalvo M, Nguyen DL, et al. Formation of human colonic crypt array by application of chemical gradients across a shaped epithelial monolayer. Cell Mol Gastroenterol Hepatol. (2018) 5:113–30. doi: 10.1016/j.jcmgh.2017.10.007
148. Ferrer-Picón E, Dotti I, Corraliza AM, Mayorgas A, Esteller M, Perales JC, et al. Intestinal inflammation modulates the epithelial response to butyrate in patients with inflammatory bowel disease. Inflammatory bowel Dis. (2020) 26:43–55. doi: 10.1093/ibd/izz119
149. Zeng H, Taussig DP, Cheng WH, Johnson LK, Hakkak R. Butyrate inhibits cancerous hct116 colon cell proliferation but to a lesser extent in noncancerous ncm460 colon cells. Nutrients. (2017) 9:25. doi: 10.3390/nu9010025
150. Salvi PS, Cowles RA. Butyrate and the intestinal epithelium: modulation of proliferation and inflammation in homeostasis and disease. Cells. (2021) 10:1775. doi: 10.3390/cells10071775
151. Lee JG, Lee J, Lee AR, Jo SV, Park CH, Han DS, et al. Impact of short-chain fatty acid supplementation on gut inflammation and microbiota composition in a murine colitis model. J Nutr Biochem. (2022) 101:108926. doi: 10.1016/j.jnutbio.2021.108926
152. Ren DD, Chen KC, Li SS, Zhang YT, Li ZM, Liu S, et al. Panax quinquefolius polysaccharides ameliorate ulcerative colitis in mice induced by dextran sulfate sodium. Front Immunol. (2023) 14:1161625. doi: 10.3389/fimmu.2023.1161625
153. Akhtar M, Chen Y, Ma Z, Zhang X, Shi D, Khan JA, et al. Gut microbiota-derived short chain fatty acids are potential mediators in gut inflammation. Anim Nutr (Zhongguo xu mu shou yi xue hui). (2022) 8:350–60. doi: 10.1016/j.aninu.2021.11.005
154. Godínez-Méndez LA, Gurrola-Díaz CM, Zepeda-Nuño JS, Vega-Magaña N, Lopez-Roa RI, Íñiguez-Gutiérrez L, et al. In vivo healthy benefits of galacto-oligosaccharides from lupinus albus (La-gos) in butyrate production through intestinal microbiota. Biomolecules. (2021) 11:1658. doi: 10.3390/biom11111658
155. Zhang T, Ding C, Zhao M, Dai X, Yang J, Li Y, et al. Sodium butyrate reduces colitogenic immunoglobulin a-coated bacteria and modifies the composition of microbiota in Il-10 deficient mice. Nutrients. (2016) 8:728. doi: 10.3390/nu8120728
156. Asadpoor M, Ithakisiou GN, Henricks PAJ, Pieters R, Folkerts G, Braber S. Non-digestible oligosaccharides and short chain fatty acids as therapeutic targets against enterotoxin-producing bacteria and their toxins. Toxins. (2021) 13:175. doi: 10.3390/toxins13030175
157. Saavedra PHV, Huang L, Ghazavi F, Kourula S, Vanden Berghe T, Takahashi N, et al. Apoptosis of intestinal epithelial cells restricts clostridium difficile infection in a model of pseudomembranous colitis. Nat Commun. (2018) 9:4846. doi: 10.1038/s41467-018-07386-5
158. Otani T, Furuse M. Tight junction structure and function revisited. Trends Cell Biol. (2020) 30:805–17. doi: 10.1016/j.tcb.2020.08.004
159. Horowitz A, Chanez-Paredes SD, Haest X, Turner JR. Paracellular permeability and tight junction regulation in gut health and disease. Nat Rev Gastroenterol Hepatol. (2023) 20:417–32. doi: 10.1038/s41575-023-00766-3
160. Fujiwara S, Nguyen TP, Furuse K, Fukazawa Y, Otani T, Furuse M. Tight junction formation by a Claudin mutant lacking the cooh-terminal Pdz domain-binding motif. Ann New York Acad Sci. (2022) 1516:85–94. doi: 10.1111/nyas.14881
161. Thomas S, Dilbarov N, Kelly J, Mercogliano G, Prendergast GC. Diet effects on colonic health influence the efficacy of Bin1 mab immunotherapy for ulcerative colitis. Sci Rep. (2023) 13:11802. doi: 10.1038/s41598-023-38830-2
162. Yang S, Shang J, Liu L, Tang Z, Meng X. Strains producing different short-chain fatty acids alleviate dss-induced ulcerative colitis by regulating intestinal microecology. Food Funct. (2022) 13:12156–69. doi: 10.1039/D2FO01577C
163. Reznik N, Gallo AD, Rush KW, Javitt G, Fridmann-Sirkis Y, Ilani T, et al. Intestinal mucin is a chaperone of multivalent copper. Cell. (2022) 185:4206–15.e11. doi: 10.1016/j.cell.2022.09.021
164. Ilani T, Reznik N, Yeshaya N, Feldman T, Vilela P, Lansky Z, et al. The disulfide catalyst Qsox1 maintains the colon mucosal barrier by regulating Golgi glycosyltransferases. EMBO J. (2023) 42:e111869. doi: 10.15252/embj.2022111869
165. Mahapatro M, Erkert L, Becker C. Cytokine-mediated crosstalk between immune cells and epithelial cells in the gut. Cells. (2021) 10:111. doi: 10.3390/cells10010111
166. Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell. (2016) 167:1339–53.e21. doi: 10.1016/j.cell.2016.10.043
167. Tajasuwan L, Kettawan A, Rungruang T, Wunjuntuk K, Prombutara P. Role of dietary defatted rice bran in the modulation of gut microbiota in Aom/Dss-induced colitis-associated colorectal cancer rat model. Nutrients. (2023) 15:1528. doi: 10.3390/nu15061528
168. Bilotta AJ, Cong Y. Gut microbiota metabolite regulation of host defenses at mucosal surfaces: implication in precision medicine. Precis Clin Med. (2019) 2:110–9. doi: 10.1093/pcmedi/pbz008
169. Burger-van Paassen N, Vincent A, Puiman PJ, van der Sluis M, Bouma J, Boehm G, et al. The regulation of intestinal mucin Muc2 expression by short-chain fatty acids: implications for epithelial protection. Biochem J. (2009) 420:211–9. doi: 10.1042/bj20082222
170. Breugelmans T, Oosterlinck B, Arras W, Ceuleers H, De Man J, Hold GL, et al. The role of mucins in gastrointestinal barrier function during health and disease. Lancet Gastroenterol Hepatol. (2022) 7:455–71. doi: 10.1016/s2468-1253(21)00431-3
171. Wen S, He L, Zhong Z, Zhao R, Weng S, Mi H, et al. Stigmasterol restores the balance of Treg/Th17 cells by activating the butyrate-pparγ Axis in colitis. Front Immunol. (2021) 12:741934. doi: 10.3389/fimmu.2021.741934
172. Wang S, Deng W, Li F, Xiang L, Lv P, Chen Y. Treatment with butyrate alleviates dextran sulfate sodium and clostridium difficile-induced colitis by preventing activity of Th17 cells via regulation of Sirt1/Mtor in mice. J Nutr Biochem. (2023) 111:109155. doi: 10.1016/j.jnutbio.2022.109155
173. Wang L, Liao Y, Yang R, Zhu Z, Zhang L, Wu Z, et al. An engineered probiotic secreting Sj16 ameliorates colitis via ruminococcaceae/butyrate/retinoic acid axis. Bioengineering Trans Med. (2021) 6:e10219. doi: 10.1002/btm2.10219
174. Zhang W, Cheng C, Han Q, Chen Y, Guo J, Wu Q, et al. Flos abelmoschus manihot extract attenuates dss-induced colitis by regulating gut microbiota and Th17/Treg balance. BioMed Pharmacother. (2019) 117:109162. doi: 10.1016/j.biopha.2019.109162
175. Zhao H, Wang Q, Zhao J, Wang D, Liu H, Gao P, et al. Ento-a alleviates Dss-induced experimental colitis in mice by remolding intestinal microbiota to regulate scfas metabolism and the Th17 signaling pathway. BioMed Pharmacother. (2024) 170:115985. doi: 10.1016/j.biopha.2023.115985
176. Luu M, Pautz S, Kohl V, Singh R, Romero R, Lucas S, et al. The short-chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic-epigenetic crosstalk in lymphocytes. Nat Commun. (2019) 10:760. doi: 10.1038/s41467-019-08711-2
177. Kespohl M, Vachharajani N, Luu M, Harb H, Pautz S, Wolff S, et al. The microbial metabolite butyrate induces expression of Th1-associated factors in Cd4(+) T cells. Front Immunol. (2017) 8:1036. doi: 10.3389/fimmu.2017.01036
178. Shen S, Prame Kumar K, Wen SW, Shim R, Wanrooy BJ, Stanley D, et al. Deficiency of dietary fiber modulates gut microbiota composition, neutrophil recruitment and worsens experimental colitis. Front Immunol. (2021) 12:619366. doi: 10.3389/fimmu.2021.619366
179. Scott NA, Andrusaite A, Andersen P, Lawson M, Alcon-Giner C, Leclaire C, et al. Antibiotics induce sustained dysregulation of intestinal T cell immunity by perturbing macrophage homeostasis. Sci Trans Med. (2018) 10:eaao4755. doi: 10.1126/scitranslmed.aao4755
180. Lavoie S, Chun E, Bae S, Brennan CA, Gallini Comeau CA, Lang JK, et al. Expression of free fatty acid receptor 2 by dendritic cells prevents their expression of interleukin 27 and is required for maintenance of mucosal barrier and immune response against colorectal tumors in mice. Gastroenterology. (2020) 158:1359–72.e9. doi: 10.1053/j.gastro.2019.12.027
181. Lee WL, Grinstein S. Immunology. The tangled webs that neutrophils weave. Sci (New York NY). (2004) 303:1477–8. doi: 10.1126/science.1095484
182. Íñiguez-Gutiérrez L, Godínez-Méndez LA, Fafutis-Morris M, Padilla-Arellano JR, Corona-Rivera A, Bueno-Topete MR, et al. Physiological concentrations of short-chain fatty acids induce the formation of neutrophil extracellular traps in vitro. Int J immunopathology Pharmacol. (2020) 34:2058738420958949. doi: 10.1177/2058738420958949
183. Shao X, Sun S, Zhou Y, Wang H, Yu Y, Hu T, et al. Bacteroides fragilis restricts colitis-associated cancer via negative regulation of the Nlrp3 axis. Cancer Lett. (2021) 523:170–81. doi: 10.1016/j.canlet.2021.10.002
184. Shao X, Liu L, Zhou Y, Zhong K, Gu J, Hu T, et al. High-fat diet promotes colitis-associated tumorigenesis by altering gut microbial butyrate metabolism. Int J Biol Sci. (2023) 19:5004–19. doi: 10.7150/ijbs.86717
185. Xiao T, Zhang P, Feng T, Lu K, Wang X, Zhou S, et al. Butyrate functions in concert with myeloid-derived suppressor cells recruited by Ccr9 to alleviate Dss-induced murine colitis. Int Immunopharmacol. (2021) 99:108034. doi: 10.1016/j.intimp.2021.108034
186. Wang Y, Ding Y, Deng Y, Zheng Y, Wang S. Role of myeloid-derived suppressor cells in the promotion and immunotherapy of colitis-associated cancer. J immunotherapy Cancer. (2020) 8:e000609. doi: 10.1136/jitc-2020-000609
187. Zheng W, Song H, Luo Z, Wu H, Chen L, Wang Y, et al. Acetylcholine ameliorates colitis by promoting Il-10 secretion of monocytic myeloid-derived suppressor cells through the Nachr/Erk pathway. Proc Natl Acad Sci U.S.A. (2021) 118:e2017762118. doi: 10.1073/pnas.2017762118
188. Zhao F, Gong W, Song J, Shen Z, Cui D. The paradoxical role of Mdscs in inflammatory bowel diseases: from bench to bedside. Front Immunol. (2022) 13:1021634. doi: 10.3389/fimmu.2022.1021634
189. Burrello C, Strati F, Lattanzi G, Diaz-Basabe A, Mileti E, Giuffrè MR, et al. Il10 secretion endows intestinal human Inkt cells with regulatory functions towards pathogenic T lymphocytes. J Crohn's colitis. (2022) 16:1461–74. doi: 10.1093/ecco-jcc/jjac049
190. Huang S, Hu S, Liu S, Tang B, Liu Y, Tang L, et al. Lithium carbonate alleviates colon inflammation through modulating gut microbiota and Treg cells in a Gpr43-dependent manner. Pharmacol Res. (2022) 175:105992. doi: 10.1016/j.phrs.2021.105992
191. Luu M, Visekruna A. Short-chain fatty acids: bacterial messengers modulating the immunometabolism of T cells. Eur J Immunol. (2019) 49:842–8. doi: 10.1002/eji.201848009
192. He XQ, Liu D, Liu HY, Wu DT, Li HB, Zhang XS, et al. Prevention of ulcerative colitis in mice by sweet tea (Lithocarpus litseifolius) via the regulation of gut microbiota and butyric-acid-mediated anti-inflammatory signaling. Nutrients. (2022) 14:2208. doi: 10.3390/nu14112208
193. Fachi JL, Sécca C, Rodrigues PB, Mato FCP, Di Luccia B, Felipe JS, et al. Acetate Coordinates Neutrophil and Ilc3 Responses against C. Difficile through Ffar2. J Exp Med. (2020) 217:e20190489. doi: 10.1084/jem.20190489
194. Bajic D, Niemann A, Hillmer AK, Mejias-Luque R, Bluemel S, Docampo M, et al. Gut microbiota-derived propionate regulates the expression of Reg3 mucosal lectins and ameliorates experimental colitis in mice. J Crohn's colitis. (2020) 14:1462–72. doi: 10.1093/ecco-jcc/jjaa065
195. Bilotta AJ, Ma C, Yang W, Yu Y, Yu Y, Zhao X, et al. Propionate enhances cell speed and persistence to promote intestinal epithelial turnover and repair. Cell Mol Gastroenterol Hepatol. (2021) 11:1023–44. doi: 10.1016/j.jcmgh.2020.11.011
196. He J, Zhang P, Shen L, Niu L, Tan Y, Chen L, et al. Short-chain fatty acids and their association with signalling pathways in inflammation, glucose and lipid metabolism. Int J Mol Sci. (2020) 21:6356. doi: 10.3390/ijms21176356
197. Chen L, Sun M, Wu W, Yang W, Huang X, Xiao Y, et al. Microbiota metabolite butyrate differentially regulates th1 and Th17 cells' Differentiation and function in induction of colitis. Inflammatory bowel Dis. (2019) 25:1450–61. doi: 10.1093/ibd/izz046
198. Alrafas HR, Busbee PB, Chitrala KN, Nagarkatti M, Nagarkatti P. Alterations in the gut microbiome and suppression of histone deacetylases by resveratrol are associated with attenuation of colonic inflammation and protection against colorectal cancer. J Clin Med. (2020) 9:1796. doi: 10.3390/jcm9061796
199. Dupraz L, Magniez A, Rolhion N, Richard ML, Da Costa G, Touch S, et al. Gut microbiota-derived short-chain fatty acids regulate Il-17 production by mouse and human intestinal Γδ T cells. Cell Rep. (2021) 36:109332. doi: 10.1016/j.celrep.2021.109332
200. Hao F, Tian M, Zhang X, Jin X, Jiang Y, Sun X, et al. Butyrate enhances cpt1a activity to promote fatty acid oxidation and itreg differentiation. Proc Natl Acad Sci U.S.A. (2021) 118:e2014681118. doi: 10.1073/pnas.2014681118
201. Chen YY, Chen SY, Chang HY, Liu YC, Chuang BF, Yen GC. Phyllanthus emblica L. Polysaccharides ameliorate colitis via microbiota modulation and dual inhibition of the rage/Nf-Kb and mapks signaling pathways in rats. Int J Biol macromolecules. (2023) 258:129043. doi: 10.1016/j.ijbiomac.2023.129043
202. Feng X, Du C, Wang C. Structural characterization of polysaccharide from yellow sweet potato and ameliorates Dss-induced mice colitis by active Gpr41/Mek/Erk 1/2 signaling pathway. Int J Biol macromolecules. (2021) 192:278–88. doi: 10.1016/j.ijbiomac.2021.09.175
203. Sepahi A, Liu Q, Friesen L, Kim CH. Dietary fiber metabolites regulate innate lymphoid cell responses. Mucosal Immunol. (2021) 14:317–30. doi: 10.1038/s41385-020-0312-8
204. Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite-sensing receptors Gpr43 and Gpr109a facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun. (2015) 6:6734. doi: 10.1038/ncomms7734
205. Jochum SB, Engen PA, Shaikh M, Naqib A, Wilber S, Raeisi S, et al. Colonic epithelial circadian disruption worsens dextran sulfate sodium-induced colitis. Inflammatory bowel Dis. (2023) 29:444–57. doi: 10.1093/ibd/izac219
206. Zhang Z, Zhang H, Chen T, Shi L, Wang D, Tang D. Regulatory role of short-chain fatty acids in inflammatory bowel disease. Cell communication Signaling CCS. (2022) 20:64. doi: 10.1186/s12964-022-00869-5
207. Kumar T, Pandey R, Chauhan NS. Hypoxia inducible factor-1α: the curator of gut homeostasis. Front Cell Infect Microbiol. (2020) 10:227. doi: 10.3389/fcimb.2020.00227
208. Blouin JM, Penot G, Collinet M, Nacfer M, Forest C, Laurent-Puig P, et al. Butyrate elicits a metabolic switch in human colon cancer cells by targeting the pyruvate dehydrogenase complex. Int J Cancer. (2011) 128:2591–601. doi: 10.1002/ijc.25599
209. Pral LP, Fachi JL, Corrêa RO, Colonna M, Vinolo MAR. Hypoxia and hif-1 as key regulators of gut microbiota and host interactions. Trends Immunol. (2021) 42:604–21. doi: 10.1016/j.it.2021.05.004
210. Fachi JL, Felipe JS, Pral LP, da Silva BK, Corrêa RO, de Andrade MCP, et al. Butyrate protects mice from clostridium difficile-induced colitis through an Hif-1-dependent mechanism. Cell Rep. (2019) 27:750–61.e7. doi: 10.1016/j.celrep.2019.03.054
211. Duan H, Wang L, Huangfu M, Li H. The impact of microbiota-derived short-chain fatty acids on macrophage activities in disease: mechanisms and therapeutic potentials. BioMed Pharmacother. (2023) 165:115276. doi: 10.1016/j.biopha.2023.115276
212. Fagundes RR, Bravo-Ruiseco G, Hu S, Kierans SJ, Weersma RK, Taylor CT, et al. Faecalibacterium prausnitzii promotes intestinal epithelial Il-18 production through activation of the Hhif1α Pathway. Front Microbiol. (2023) 14:1298304. doi: 10.3389/fmicb.2023.1298304
213. Dilly AK, Lee YJ, Zeh HJ, Guo ZS, Bartlett DL, Choudry HA. Targeting hypoxia-mediated Mucin 2 production as a therapeutic strategy for mucinous tumors. Trans Res. (2016) 169:19–30.e1. doi: 10.1016/j.trsl.2015.10.006
214. Islam R, Dash D, Singh R. An antioxidant ameliorates allergic airway inflammation by inhibiting Hdac 1 via Hif-1α/Vegf axis suppression in mice. Sci Rep. (2023) 13:9637. doi: 10.1038/s41598-023-36678-0
215. Ma S, Yeom J, Lim YH. Specific activation of hypoxia-inducible factor-2α by propionate metabolism via a B-oxidation-like pathway stimulates Muc2 production in intestinal goblet cells. BioMed Pharmacother. (2022) 155:113672. doi: 10.1016/j.biopha.2022.113672
216. Fachi JL, Pral LP, Assis HC, Oliveira S, Rodovalho VR, Dos Santos JAC, et al. Hyperbaric oxygen augments susceptibility to C. Difficile infection by impairing gut microbiota ability to stimulate the Hif-1α-Il-22 axis in Ilc3. Gut Microbes. (2024) 16:2297872. doi: 10.1080/19490976.2023.2297872
217. Yang W, Yu T, Huang X, Bilotta AJ, Xu L, Lu Y, et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell Il-22 production and gut immunity. Nat Commun. (2020) 11:4457. doi: 10.1038/s41467-020-18262-6
218. Zhang J, Zhang H, Liu M, Lan Y, Sun H, Mai K, et al. Short-chain fatty acids promote intracellular bactericidal activity in head kidney macrophages from turbot (Scophthalmus maximus L.) via hypoxia inducible factor-1α. Front Immunol. (2020) 11:615536. doi: 10.3389/fimmu.2020.615536
219. MaChado MG, Patente TA, Rouillé Y, Heumel S, Melo EM, Deruyter L, et al. Acetate improves the killing of streptococcus pneumoniae by alveolar macrophages via nlrp3 inflammasome and glycolysis-Hif-1α Axis. Front Immunol. (2022) 13:773261. doi: 10.3389/fimmu.2022.773261
220. Zhu L, Li G, Liang Z, Qi T, Deng K, Yu J, et al. Microbiota-assisted iron uptake promotes immune tolerance in the intestine. Nat Commun. (2023) 14:2790. doi: 10.1038/s41467-023-38444-2
221. Gao T, Wang Z, Dong Y, Cao J, Chen Y. Melatonin-mediated colonic microbiota metabolite butyrate prevents acute sleep deprivation-induced colitis in mice. Int J Mol Sci. (2021) 22:11894. doi: 10.3390/ijms222111894
222. Sam QH, Ling H, Yew WS, Tan Z, Ravikumar S, Chang MW, et al. The divergent immunomodulatory effects of short chain fatty acids and medium chain fatty acids. Int J Mol Sci. (2021) 22:6453. doi: 10.3390/ijms22126453
223. Deleu S, Arnauts K, Deprez L, Machiels K, Ferrante M, Huys GRB, et al. High acetate concentration protects intestinal barrier and exerts anti-inflammatory effects in organoid-derived epithelial monolayer cultures from patients with ulcerative colitis. Int J Mol Sci. (2023) 24:768. doi: 10.3390/ijms24010768
224. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase pink1 recruits autophagy receptors to induce mitophagy. Nature. (2015) 524:309–14. doi: 10.1038/nature14893
225. Yin K, Lee J, Liu Z, Kim H, Martin DR, Wu D, et al. Mitophagy protein pink1 suppresses colon tumor growth by metabolic reprogramming via P53 activation and reducing acetyl-coa production. Cell Death Differ. (2021) 28:2421–35. doi: 10.1038/s41418-021-00760-9
226. Gerasimidis K, Nichols B, McGowan M, Svolos V, Papadopoulou R, Kokkorou M, et al. The effects of commonly consumed dietary fibres on the gut microbiome and its fibre fermentative capacity in adults with inflammatory bowel disease in remission. Nutrients. (2022) 14:1053. doi: 10.3390/nu14051053
227. Behary J, Amorim N, Jiang XT, Raposo A, Gong L, McGovern E, et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat Commun. (2021) 12:187. doi: 10.1038/s41467-020-20422-7
Keywords: inflammatory bowel disease, hypoxia-inducible factor-1α, short-chain fatty acids, intestinal epithelial barrier, gut immunity
Citation: Xiao J, Guo X and Wang Z (2024) Crosstalk between hypoxia-inducible factor-1α and short-chain fatty acids in inflammatory bowel disease: key clues toward unraveling the mystery. Front. Immunol. 15:1385907. doi: 10.3389/fimmu.2024.1385907
Received: 14 February 2024; Accepted: 19 March 2024;
Published: 28 March 2024.
Edited by:
Guojun Wu, Rutgers, The State University of New Jersey, United StatesReviewed by:
Debora Decote-Ricardo, Federal Rural University of Rio de Janeiro, BrazilCopyright © 2024 Xiao, Guo and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenquan Wang, MzIwMDM1QGhudWNtLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.