 Lara Sophie Schlegel1†
Lara Sophie Schlegel1† Coralie Werbrouck
Coralie Werbrouck Michael Boettcher
Michael Boettcher Patrick Schlegel
Patrick Schlegel
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 June 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1383894
This article is part of the Research Topic Emerging Strategies to Overcome Current Roadblocks in CAR T Cell Immunotherapy View all 7 articles
Chimeric antigen receptor (CAR) T cell therapy has effectively complemented the treatment of advanced relapsed and refractory hematological cancers. The remarkable achievements of CD19- and BCMA-CAR T therapies have raised high expectations within the fields of hematology and oncology. These groundbreaking successes are propelling a collective aspiration to extend the reach of CAR therapies beyond B-lineage malignancies. Advanced CAR technologies have created a momentum to surmount the limitations of conventional CAR concepts. Most importantly, innovations that enable combinatorial targeting to address target antigen heterogeneity, using versatile adapter CAR concepts in conjunction with recent transformative next-generation CAR design, offer the promise to overcome both the bottleneck associated with CAR manufacturing and patient-individualized treatment regimens. In this comprehensive review, we delineate the fundamental prerequisites, navigate through pivotal challenges, and elucidate strategic approaches, all aimed at paving the way for the future establishment of multitargeted immunotherapies using universal CAR technologies.
Immune oncology has experienced a major breakthrough with the development of CAR expressing immune effector cells (1). CAR receptors are synthetic immune receptors comprised of cell-specific functional protein units strategically assembled to create dimeric receptors which facilitate a multifaceted response upon engagement of the CAR with its targeted molecular structure (2). Other than the recognition domain that can be derived from different species, the functional subunits of a CAR are generally derived from human proteins. Each module displays unique properties that mediate specifically defined functions. The CAR is comprised of an ectodomain, which consists of an antigen-recognition domain and a spacer, a transmembrane domain and the intracellular signaling unit that can be composed of one or multiple signaling domains (3).
In most instances, the extracellular signaling domain consists of a murine antibody-derived single chain variable fragment (scFv) (4) or a camelid heavy chain variable (VHH) (5) but may also be derived from fully human VHH (6), scFv or ligand (7) for the targeting of overexpressed cognate receptors in cancer (APRIL – BCMA & TACI) (8), other species’ antibody variable chains, or alternatively, artificial recognition domains, such as D-domains (9). To reduce the immunogenicity, humanized murine single chain variable fragment (scFv) (10) or deimmunized camelid VHH may be used (11).
The spacer is essential in the configuration of the CAR, as its length determines the binding proximity and prevents steric hindrances, hence facilitating access to the epitope. The transmembrane domain anchors and stabilizes the receptor in the cell membrane as well as connecting the extracellular components with the intracellular signaling compartment referred to as the endodomain that contains one or several costimulatory and/or signaling domains (3). The specific modules of the distinct CAR define the functional properties of the synthetic immune receptor (2). These are mainly the response and proliferation kinetics of the CAR expressing cell that are dependent on the signal transduction from the extracellular domain to the intracellular signaling domains (12). Therefore, the deliberate combination of functional protein units allows for optimized and requirement-adjusted functionality. The basic understanding and principles of the CAR introduced as a physicochemical receptor and its measurable effector functions are illustrated and explained in detailed in Figure 1.
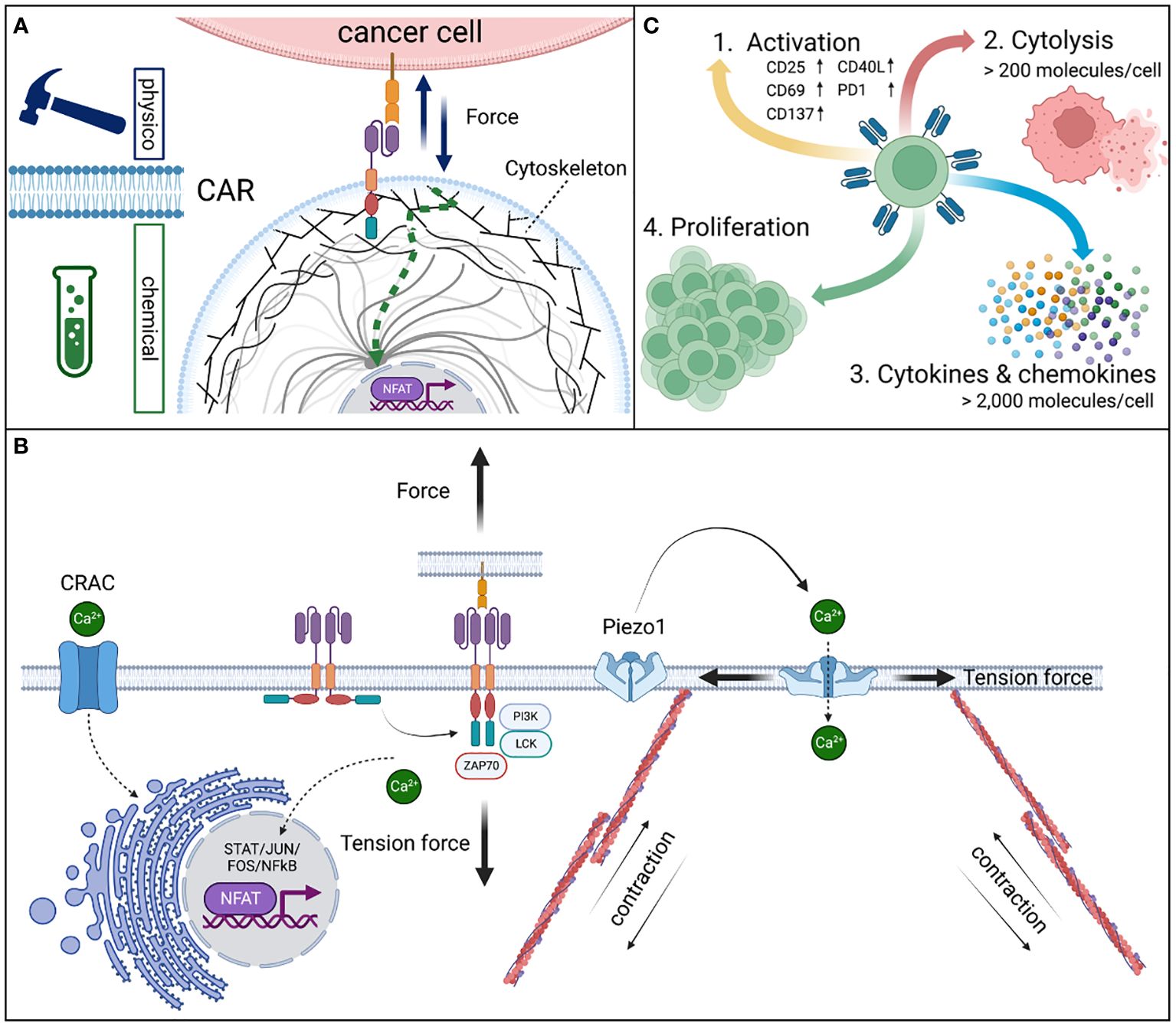
Figure 1 Physicochemical signal conversion of CAR receptors. CARs are (A) mechanoreceptors that allow for the transmission of physical tension force to convert into complex chemical signals from the outside of the cell into changing the activation state and downstream functions of the cell including immediate responses, such as the formation of a cytolytic synapse and migration or intermediate and long-term adaptions by changes in the gene expression. (B) The mechanical force on CAR expressing effector cells is mediated by the binding interaction of the CAR recognition domain and the antigen leading to a conformational change of the cytoplasmic signaling domains. These steric adaptions result in the accessibility of phosphorylation sites and the cognate alignment of signaling proteins that initiate a downstream signaling cascade which is based on enzymatic phosphorylation steps of subsequent signaling proteins in a defined order (13). Calcium serves as an important second messenger in the signaling process. Piezo1 calcium channels are opened by tension forces to the cell membrane by active deformation of the cytoskeleton via actin filaments, allowing calcium influx into the cytoplasm. Especially, during the formation of the cytolytic synapse Piezo1 dependent calcium influx is required (14). Further, cellular calcium metabolism is tightly regulated by the endoplasmic reticulum (ER). The ER serves as a large calcium storage filled with calcium by the calcium release activated channel (CRAC) (15). Upon calcium release from the ER and binding thereof to calcineurin, a phosphatase, is activated. Calcineurin serves as a key modulator of the transcription factor “nuclear factor of activated T cells” (NFAT) and thus serves as a key modulator of T cells in general (16). Dephosphorylation of the nucleus localization signal allows the translocation of NFAT into the nucleus and induce selective gene expression. Dysfunctional CRAC prevents the development of T cells and is the cause of the severe combined immunodeficiency (SCID), a life-threatening inborn immune deficiency (17). (C) The efficient transmission of the physicochemical signal leads to various effector functions in CAR expressing effector cells. In a resting CAR, 1st cell activation by the CAR receptor is detectable by the expression of activation markers. 2nd Cytolytic activity in high affinity CARs (KD < 1 nM) is induced at an antigen density of as low as >200 molecules per cell, whereas significant changes to the 3rd gene expression, cytokine and chemokine secretion require >2,000 molecules per cell (18). Thus, 4th a strong triple signal of i) CAR-mediated antigen recognition (CD3ζ), ii) co-stimulation (CD28), and iii) cytokine support (IL2) are required to induce proliferation.
Universality in CAR T cell therapy can refer to both 1st the universal applicability of a CAR T cell product with no recipient restrictions due to T cell receptor/HLA incompatibility (19) and 2nd the capability of targeting any antigen of interest facilitated by indirect CAR technologies via adapter molecules (20).
All US-FDA/EMA-approved CAR T products are manufactured from autologous patients’ T cells and have demonstrated high clinical efficacy (3). Per definition, autologous CAR T cell products cannot induce GvHD or is subject to HLA-based immune rejection. Autologous CD19- and BCMA-targeted CAR T cells robustly engraft in most patients (21). The CARs’ architecture define their biology, metabolism and persistence (4, 22, 23), which generally is not subject to primary immunological CAR rejection, however, the induction of humoral anti-CAR immunity may limit the engraftment and success of a second CAR T cell application in due course (24).
To date most CAR T cell therapies are based on autologous products which implies the donor and recipient to be the same individual (23). Posttransplant manufactured autologous CAR T cell products are considered autologous even if the genetic origin of the hematopoietic system is donor-derived (25) rather than self-derived. For example, a CAR T cell product created post-transplant can come from an HLA-matched donor or even a haploidentical donor with an HLA mismatch. The cell source can be either the recipient (patient) or the same donor used for the hematopoietic allogeneic stem cell transplantation (HSCT) (26). In general, HLA-matched and haploidentical allogeneic CAR T cells both require to be administered post transplantation. Otherwise, the CAR T cell product is rejected by the recipient’s immune system (27).
The major advantage of third-party allogeneic HLA-mismatched donor-derived immune effector cells, besides manufacturing costs, is the immediate availability of these products at any point in time (28). The manufacturing process of autologous CAR T is comprised of leukapheresis, T-cell isolation and stimulation, gene delivery, ex vivo expansion, cryopreservation and quality controls (29). The logistics in the complex multi-step preparation cells is prone to errors. Usually, time-consuming testing of T cells prior to manufacturing is required. Further, the therapeutic success of autologous CAR T cells may be constraint by the quantity and quality of the isolated peripheral blood cells of the patient (30).
The idea of third-party CAR T cells is driven by the success of third-party virus-specific T cells (ADV, CMV, EBV) used in the posttransplant setting to treat viral reactivation, as these allogeneic cells can transfer transient antiviral immunity to bridge the time until the patients’ T cells have reconstituted and provide viral immune protection (31, 32). In patients eligible for CAR T cell therapy, time is of the essence and one key critical determining factor of survival (33). Additionally, the risk for manufacturing failure in CAR T cell therapy varies significantly (>10%) depending on the pre-treatment history and the primary underlying cancer (34, 35). Further, the clinical efficiency of a bridging chemotherapy to enable the patient to reach the CAR T cell treatment a stable clinical state to receive the CAR T cell product (36). Thus, manufacturing failure can compromise the patients’ opportunity for a successful administration of CAR T cells (34, 37). As such, third-party allogeneic CAR T cell therapy provide a bridge to transplant for selected patients (38).
Partially HLA-matched third-party virus-specific T cells have been documented to contribute controlling virus reactivation posttransplant in immunocompromised patients (39). In order to guarantee anticancer activity by universal third-party CAR T cells, patients undergo a preparative regimen including the treatment with alemtuzumab (IgG1K, t1/2 = 6-21 days) targeted to CD52 which leads to a profound and enduring depletion of B- and T lymphocytes, NK cells and monocytes (40). Genetic modifications via CRISPR/Cas9 offer to disrupt the TRAC locus and CD52 genes, indicated in Figure 2. These genetic alterations prevent the immediate rejection of the HLA mismatched effector cells via the depletion of the recipients’ lymphocytic compartment by alemtuzumab and the inactivation of the TRAC leads to a collapse of the TcR preventing the induction of graft versus host disease (GvHD) by third-party T cells in the immunocompromised host (21, 46). A CD19-targeted allogeneic product was shown to be safe and efficacious in heavily pre-treated adult patients with relapsed and refractory B-ALL with allogeneic off-the-shelf UCART19 cells in the clinical trial [CALM] (NCT02746952) (21). Alternative cell sources for third-party off-the-shelf CAR products include NK cells isolated from cord blood (47), γδ T cells collected from healthy donors (48), or the leukemic cell line NK-92 (49, 50). NK cells and γδ T cells do not induce GvHD but are subject to rejection by the recipient’s immune system in the allogeneic setting and thus can only be used transiently post lymphodepleting chemotherapy (51).
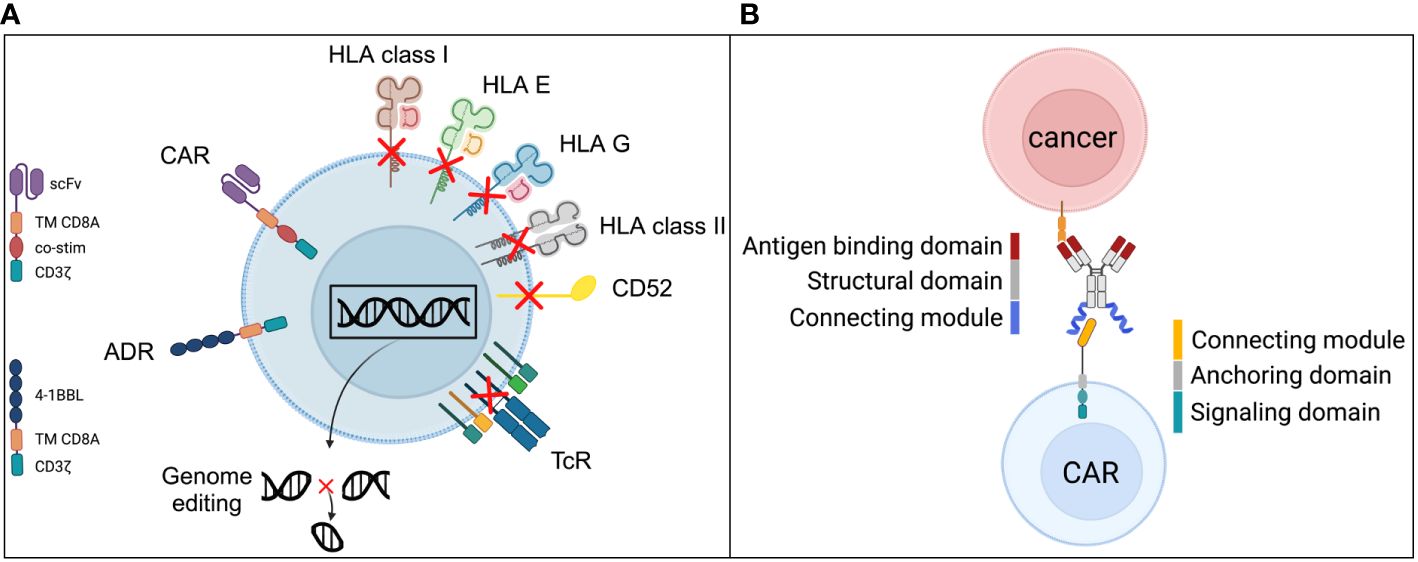
Figure 2 Universal CAR strategies have two meanings. Universal “allogeneic CARs” generated from iPSCs require a series of genetic alterations to enable their successful and safe clinical use. Current concepts (A) include modifications that reduce the likelihood of allorejection by gene disruption of ß2-microglobulin to abrogate the expression of HLA class I structures as well as by gene disruption of the class II MHC transactivator (CIITA) to abrogate HLA II expression (41, 42). To exclude iPSC-derived cells from harm through alemtuzumab, the CAMPATH-1 antigen CD52 is required to be knocked out as it has been done for third-party allogeneic CAR products (43). Alemtuzumab can then be used in the CAR preparative regimen and facilitate longer engraftments of the allogeneic cells. However, alemtuzumab induces long-lasting severe cellular and humoral immune deficiency which attracts serious infectious complications (44). In order to reduce the risk for graft-versus-host disease, the T cell receptor (TcR) expression has to be disrupted and is usually achieved by the genetic knockout of the constant alpha (TRAC) chain of the TcR. Since the TcR is the responsible receptor for alloreactivity in GvHD direction, the TcR-KO is the most effective strategy to silence the primary T cell function (42). Novel strategies to increase the resistance to potential allorejection was introduced by Mo et al in 2021 via an alloimmune defense receptor (ADR) (45). It’s a ligand based (4-1BBL) signal converting receptor by providing a CD3ζ signaling if the allogeneic iPSC derived CAR expressing cell gets in contact with activated immune cells, such as T cells and NK cells that express the co-stimulatory receptor 4-1BB (CD137). The CD3ζ signal increases the resistance to allorejection mechanisms induced by T cells and NK cells. (B) The basic principle of adapter CARs is the indirect targeting of the CAR expressing cells via advanced antibody-dependent cellular cytotoxicity (ADCC). Since the adapter molecule, e.g., an antibody is interchangeable the specificity of the targeting is theoretically unlimited. The structure of the adapter molecule is comprised of three domains with distinct functions. The antigen binding domain corresponds to the primary antibody binding capability to a structure expressed on target cells. The structural domain provides stability to the molecule and supports manufacturability, the connecting module interacts with the CAR receptor and facilitates the highly specific recognition of the adapter molecule only by the CAR expressing cell and no other immune cells. The counterpart is the CAR expressing cell and especially the CAR receptor itself comprised of the cognate connecting module to allow the highly specific recognition and interaction with the adapter molecule (20). Further, the anchoring domain stabilizes the receptor in the cell membrane and the signaling domain provides the cell with the downstream signaling to ignite cellular functions according to the design of the receptor.
Another approach that is proving to be viable involves using induced pluripotent stem cells (iPSCs) as the primary cell source for the cellular CAR product. iPSC-derived CAR T are at early stage of development. The main principles are illustrated and outlined in Figure 2A. iPSCs are used to create in silico designed, genetically tailored, highly defined and characterized cellular products according to unique functional and immunological requirements (52).
Genetic modifications are achieved using various gene engineering tools tailored for specific applications. The historical development, feasibility, scalability, advantages, and disadvantages of these tools are comprehensively discussed by Adli et al. in the review article “The CRISPR Toolkit for Genome Editing and Beyond” (53). Today, the key tools include CRISPR/Cas9, TALENs, ZFNs, homologous recombination, the PiggyBac transposon system, and the Sleeping Beauty transposon system, among others. Delivery systems for these genome editing tools include electroporation (54, 55), transfection with chemical compounds (56), AAV-based methods (57), and retroviral/lentiviral gene delivery (58). We reference primary literature and review articles that delve into the detailed aspects of these tools, such as “Genome Editing with CRISPR–Cas Nucleases, Base Editors, Transposases, and Prime Editors” by Anzalone et al (59).
Theoretically, iPSC-derived cellular products can grow endlessly and multiply countlessly in numbers, thus providing enough cells for third-party CAR T products from one single original donor source with no limits (28). Because the artificial iPSC-derived CAR T cells are HLA mismatched, they require at the minimum the same genetic modifications as “conventional” third-party CAR T cell products to reduce alloreactivity and graft rejection. However, counter to third-party allogeneic CAR T cells, there are no constraints in the genetic engineering as iPSC can be cultured infinitively (60). Of note, even though iPSC-derived cells can be well-characterized and extensively tested genetically and functionally, they bear a potential risk for mutations, genetic disruptions and rearrangements of functional genes (61) which could lead to secondary hard-to-treat iPSC-CAR-leukemia with resistance to treatment which has been observed in autologous CD19CAR T cells with piggyBac transposon-based gene delivery (62). In addition, decreased amplification and shorter persistence of iPSC-based CAR products in vivo in preclinical models required additional modifications to ensure robust anticancer activity (28). While iPSC-derived CAR T-cell therapy holds great promise for personalized cancer treatment, many challenges remain to ensure a safe and effective clinical translation (63).
The term “Universal CAR T” is also referred to technologies that enable CAR receptor expressing immune effector cells to engage with an unlimited variety of different antigens (64). This can be achieved with adapter CAR T platform technologies thoroughly discussed by Arndt (65) et al as well as Liu (66) et al. The basic principle of adapter CAR T cells is illustrated in Figure 2B. The antigen recognition and CAR signaling is decoupled and requires the correct assembly of three components 1st the adapter CAR expressing cell with their surface-expressed CAR receptors, 2nd the adapter molecule and 3rd the target antigen expressing cell (20). The recognition domain of adapter CARs is either targeted to a non-human “neo”-epitope (20, 67) non-existent in the human body or an inaccessible, e.g., intracellular (68) structure for the CAR recognition domain, but instead is only found as a targetable moiety on adapter molecules (65). In consequence, adapter CARs are functionally inert cells that only engage with target cells via the CAR receptor in the presence of the corresponding adapter molecule (65). Additionally, redirecting conventional CARs has also been employed to broaden the spectrum of targeting (69, 70). The specificity of the adapter molecule is interchangeable and thus the number of targetable antigens can be extended by introducing additional adapter molecules into the system (20, 71). Analogue to antibody-dependent cellular cytotoxicity (ADCC) with NK cells (72), adapter CAR technologies facilitate an artificially potentiated type of ADCC that allows for the tight regulation of effector functions (67), transient targeting, combinatorial synchronous multitargeting and sequential targeting (20).
An overview of adapter CAR systems is provided in Table 1 in alignment with Figure 2B. Contemporary translational aspects are summarized in the section - Clinical experience with adapter CAR technologies.
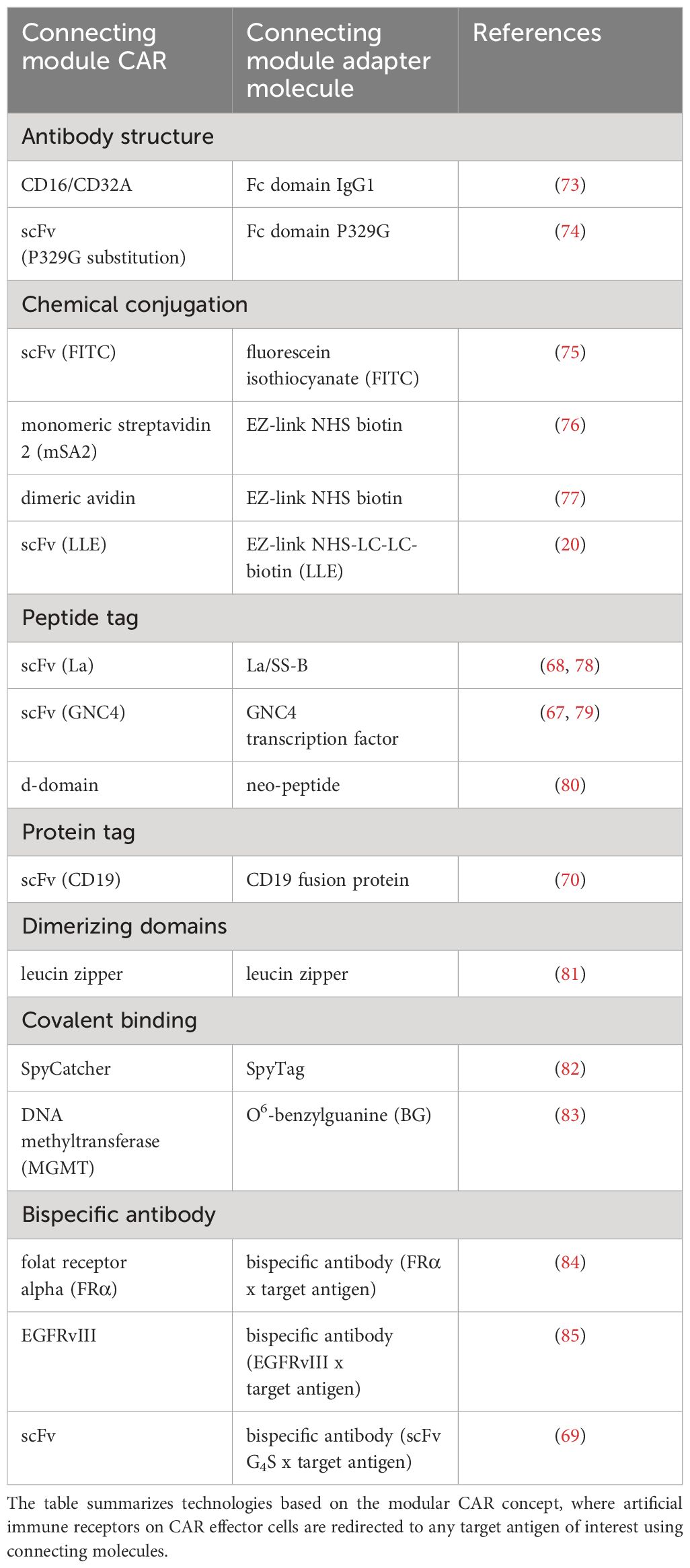
Table 1 Adapter CAR systems.
In a synergistic approach, iPSC derived third-party adapter CAR T cells may combine the features of iPSC-derived CAR immune effector cells (28) with the capabilities of versatile indirect CAR technologies – as adapter CAR 2.0 (20).
Patients who are eligible for CAR T cell therapy, are deadly sick and have no further treatment options. FDA/EMA-approved CD19- and BCMA-specific CAR T cell products are mainly last line therapies, however, ironically provide high complete remission induction rates and increase overall survival in B-lineage cancers including multiple myeloma substantially (86). With excellent response rates in CD19 positive cancers, a relevant proportion of patients may experience a sustained complete response (which can be considered a cure from the primary disease) in 40% of B-ALL (4, 87) and DLBCL patients (88, 89), whereas in CLL the sustained complete response rate is 20% (90).
Despite the great success of CAR T cells, in multiple myeloma, the cancer cells’ antigen heterogeneity and immunosuppressive factors in the tumor niche have highlighted future challenges to be overcome (91). BCMA-specific CAR T cells have demonstrated significant clinical benefits in multiple myeloma patients that otherwise have no further treatment available to date (92). Yet, multiple myeloma is continuously and largely considered incurable and new treatment strategies are required to improve outcomes (93). Obviously, potent therapies justify being considered earlier in the treatment algorithm and with less than 10% of newly diagnosed multiple myeloma patients to receive CAR T cell therapy (94) because they die before being eligible according to international guidelines. In due course adjustments are required to be made carefully based on clinical outcomes. It is noteworthy that multiple myeloma patients who received the BCMA CAR T cell product idecabtagene vicleucel (bb2121) in the clinical trial KarMMa (NCT03361748) showed a median overall survival of 19.4 months (92) which represents a significant survival benefit compared to untreated patients, non-responders and patients with partial response only.
Despite significant responses in patients with various solid cancers treated with CAR T cells, such as disialoganglioside GD2 in neuroblastoma (95) and H3K27M-mutated diffuse midline gliomas (96), mesothelin (MSLN) in mesothelioma (97), claudin 18.2 (CLDN18.2) in pancreatic cancer (98), and IL13Rα2 in combination with EGFR in glioblastoma multiforme (99), CAR-based therapies for solid cancers face major limitations and challenges. Although patients may experience objective tumor regression and even complete remissions, most cancers inevitably relapse due to target antigen loss or antigen heterogeneity, which hinders primary complete responses (95, 97). Additionally, reduced CAR T cell persistence (96), the inability of T cells to infiltrate tumors, and the inhibition of CAR T-cell function by the immunosuppressive microenvironment (97) are significant barriers to durable responses. These issues can potentially be addressed by combinatorial strategies, such as integrating CAR T cells with immune checkpoint inhibition (100).
Without immediate tumor control, patients may suffer from severe cancer-induced complications or die from cancer progression. Therefore, an immediate and effective anti-cancer response is essential for a successful CAR T cell therapy (4, 101). The containment of cancer growth is facilitated by the cellular cytotoxicity, the secretion of proinflammatory cytokines and the exponential proliferation, which are triggered by the antigen-specific CAR activation with rapid kinetics inducing effector functions in alignment with T cell receptor mediated immune responses (102). Especially, the trafficking and proliferation kinetics (103) to outnumber the cancer cells and the enduring anti-cancer response by the CAR T cells until the entirety of the cancer is cleared or reduced to a molecular residual disease are key requirements to prevent cancer recurrence (104). Additionally, the CAR must have a high sensitivity in terms of recognizing and eliminating low antigen density expressing tumor cells to ensure profound cancer control (12). Otherwise, the antigen low expressing cancer cells may escape the CAR-mediated recognition whereas the antigen negative cancer cell subsets cannot be recognized by the CAR T cells and in due course initiate the relapse (91, 105). Identifying the optimal CAR sensitivity and -kinetics depend on multiple factors, including the differential antigen expression in healthy versus cancerous tissue (106), the tumor microenvironment, the level of inflammation, and the responsiveness of the specific CAR design (12, 102). The activation state and exhaustion of CAR T cells are influenced by changing conditions during the anticancer response, including the cancer burden and the cytokine milieu (107, 108). Achieving a balance between initial and sustained response, tumor control, and managing both acute and chronic toxicity is crucial for effective therapy.
The basic primary effector functions of artificial immune cells like CAR T, CAR NK, chimeric TcR-like receptor and transgenic TcR expressing cells, are antigen-specific cellular cytotoxicity, cytokine production and proliferation (20, 57, 109, 110).
In reality, the immune effector cells are composed of a multitude of different cell subsets with various effector functions. The main αβT cell subsets are CD4+ T helper cells and CD8+ cytotoxic T cells. Yet, the various T cell subsets have been subclassified by the T cell receptor profile, the gene expression signature including decisive transcription factors, secretion of cytokines and chemokines as well as their cytokine- and chemokine receptor profiles which determine their responsiveness, adaptability and their specific functions (111–113).
While chemokine receptors and integrins determine the homing and preferential residence of T cells in certain tissues (114) the multifaceted effector functions including cytotoxicity and immunomodulation mediated by both CD4+ and CD8+ cells lead to their subclassification (115).
CD4+ αβT cells are classified as Th1, Th2, Tregs, TFH, TH22, TH17 and TH9. Cytokines range from proinflammatory IFNγ and TNFα released by Th1 cells to counterbalancing immunosuppressive cytokines IL10 and TGF-ß by Tregs (111). The interplay is important not only to prevent harm by hyperinflammation (116) but also to restore functions in order to maintain a potent immune response and circumvent terminal exhaustion of T cells (117).
CD8+ αβT cells are classified as Tc1, Tc2, Tc9, Tc17 and Tc22 and share a similar spectrum of cytokines like CD4+ cells ranging from proinflammatory to immunosuppressive functions as of IL12 and IL4, respectively (115). In relation to cancer entity the number and composition of tumor infiltrating lymphocytes (TILs), the frequency of CD8+ T cells and their subsets vary substantially (115). The prognostic value of TILs (CD4+ and CD8+) depends on the cancer type and the pathological TMN staging (118, 119).
Importantly, the maturation state of T cells defines their proliferative and regenerative capacity and is identified by the expression of cytokine- and chemokine receptors as well as activation and exhaustion markers (111).
Culturing conditions, including the cell culture media (e.g., glucose level) and supplements such as sera and cytokines (e.g., IL2, IL7, IL15, IL21, IL18), as well as cytokine dosing, initiation of T-cell activation and proliferation, and the duration of expansion, significantly impact the cellular composition of CAR products. These factors determine the maturation state and stemness of CAR effector cells, which in turn affect their proliferative capacity and clinical performance (120). The duration of CAR T cell expansion is crucial for supply, cost, and performance. CD19CAR-T tisagenlecleucel (CTL019), manufactured using the T-ChargeTM platform, is known as YTB323. This platform reduces manufacturing time from 9-10 days to less than 2 days. YTB323 has demonstrated that shortened manufacturing time preserves the stemness of CAR T cells, measured as the frequency of TN and TSCM, and achieves the same tumor control as tisagenlecleucel at a 25-fold lower dose (121). Other strategies to induce stemness and enhance CAR T cell functionality include supplementing the culture media with inosine which induced profound metabolic reprogramming, from glycolysis to mitochondrial oxidative metabolism and the epigenome toward greater stemness. The same effect was induced by genetically modifying CAR T cells to overexpress adenosine deaminase (ADA-OE), an enzyme that metabolizes adenosine to inosine, thereby preventing inhibitory effects through the ATP, ADP, and AMP CD39/CD73 pathway (122). Additionally, overexpressing FOXO1, a transcription factor involved in regulating gluconeogenesis and glycogenolysis mediated by insulin, has been shown to enhance stemness, metabolic fitness, and CAR T cell performance (123).
Based on their immunomodulatory functions CD4+ and CD8+ cell subsets harmonize the immune response. Severe immediate or chronic overactivation of T cells can lead to detrimental effects and death observed in viral infections (124), autoimmunity, serotherapy during conditioning for allogeneic stem cell transplantation and CAR T cell therapy (125).
In all of the US-FDA approved CAR T cell products the extracellular recognition domain is either based on a murine scFv or a llama-derived VHH, hence contain non-human sequences, which may be identified by the immune system as foreign and induce the generation of CAR-rejecting antibodies (24) or T cell mediated rejection (126, 127). Both allorejection mechanisms can lead to the loss of the CAR T cell function or to the complete elimination of the CAR T cells (126). Since immunogenicity is impacted by molecular size, sequence dissimilarity and conformational structure, camelid VHH are in general considered to be less immunogenic. VHH are superior in chemical and physical properties including higher solubility, stability, smaller size (15kDa), they have a higher resemblance in sequence and conformational structure to human VHH compared to murine scFv (30kDa), and hence exhibit lower immunogenicity (128). Immunogenicity of non-human proteins can be reduced by humanization of murine scFvs and deimmunization of camelid VHH (24, 129). However, CAR T cells targeted to B-lineage associated antigens, such as CD19, CD20, CD22 and BCMA protect themselves from rejection as they deplete or interfere with the immune compartment that is responsible for the generation of anti-CAR antibodies (129).
CAR T cell products are defined by their original cell source and cell number. Unstimulated peripheral blood leukaphereses contain a median of 9.8x109 total nucleated cells with 3.8x109 total CD3+ cells (130). Thus, one of the most compromising aspects of CAR T cell generation is the misrepresentation of their entire primary heterogeneity in the whole T cell pool of an estimated 4x1011 number of cells (131), 1st due to the low number of cells used for manufacturing [1x108] with a median transduction efficiency of 46% (132) which corresponds to 0.01% (one ten thousandth) of the T cell pool, and 2nd the collection from only one body compartment, the peripheral blood (130, 132). The heterogeneity of T cells in the peripheral blood and tissues are defined by differential functions dependent on the expression of certain integrins and chemokine receptors explaining the migration and residence tendency of the T cells in which they organize a complex interplay of the immune system (114).
Through the manufacturing process, various factors significantly impact CAR T characteristics. This process involves the drastic non-physiological antigen-independent activation of the T cell receptor complex (CD3) usually via the CD3 epsilon chain, often in combination with the co-activation of the CD28 costimulatory receptor (20). Additionally, continuous exposure to high concentrations of interleukin 7 (IL7), a T cell growth factor and regulator of Th1 and Th2 cytokine production (133) and/or the Th1-type cytokines, e.g., IL2 (120), IL15 (120, 134), IL21 (120) and/or the co-incubation with irradiated feeder cells (135), along with transgene delivery and the constitutive signaling and potential tonic signaling of the CAR receptor (136), collectively transform CAR expressing cells into artificial immune cells distinct from physiologic T and NK cells. Despite this distinction, CAR T cells share foundational properties with naturally matured and regrowing immune cells (137). Consequently, CAR expressing cells show similar behaviors and effector functions like natural immune cells but also exert additional functions and lack characteristics of their natural counterparts and have distinct metabolic signatures dependent on the CAR architecture (22). Signaling from T cell receptors and CAR receptors are fundamentally different even though CAR receptors mimic the TcR function and recruit the same downstream signaling proteins and engage with the same signaling pathways (138). Site-specific integration of CAR receptors into the TRAC locus facilitates the transgene expression according to the complex gene regulation of the TcR (57). The physiological CAR transgene expression has been shown to improve the CAR performance. Novel non-viral knock-in strategies may lower cost, complexity and time of CAR manufacturing. The technical challenge was to improve the delivery of homology-directed repair (HDR) templates of single-stranded DNA encoding the transgene instead of double-stranded DNA to reduce the toxicities in the electroporated cells (139).
These revolutionary gene editing tools have inspired the idea of site-specific integration of a novel immune receptor design mimicking the TcR. By exchanging the variable alpha and beta chains of the TcR with the antibody variable chains (VL and VH) of an antibody, the sensitivity of this advanced artificial immune receptor (HLA-independent TcR) was substantially increased to outperform classic CAR receptor design, demonstrated for antigen low expressing cancer cell line variants (NALM6CD19Low and MOLM13CD70Low). Yet, the constitutive co-expression of CD80 and 4-1BBL was required to enhance persistence (110). Interestingly, after three decades of CAR research, the advancements of synthetic biology and gene engineering have revived the original idea of Zelig Eshhar’s T-body, the forefather of the modern scFv-based CAR receptor (140).
The availability and suitability of target antigens are the greatest challenges in CAR T cell therapy. Due to the high requirements for target antigens, such as high and stable expression at relevant levels and preferably overexpression of the targeted antigen on cancerous cells compared to physiological tissues (141), the selection of potential target antigens are limited (142). Furthermore, the main mode of action of CAR T cells is the recognition and signaling of the CAR receptor upon engagement with the strictly defined epitope on a target antigen (143). Thus homogenous antigen expression is important for successful CAR T cell therapy, even though low frequency of antigen heterogeneity can be addressed by so called bystander effects whereas heterogenous cancers and antigen loss require multitargeted CAR approaches (144, 145).
Bystander effects through the recruitment of immune effector cells, such as T cells, NK cells and macrophages in the tumor niche have been shown to significantly contribute to tumor control for instance in the CD19 CAR T cell trial ZUMA-1 (NCT02348216) (146) and in preclinical models (147) and shall be exploited using next generation CAR designs, engineered to inducibly express cytokines impacting on the cytokine milieu (148). These effects are relevant but shall not be overinterpreted to enable CAR T cells to successfully control antigen negative tumor lesions.
In clinical practice there are three main strategies (i-iii) pursued to balance potency and on-target-off-tumor toxicities in targeted immunotherapies utilizing antibodies, bispecific T cell engagers (BiTEs), antibody-drug conjugates (ADCs) and CAR T cells.
All strategies are characterized by the inability to target cancer specifically and thus require to appreciate the potency limit set by the on-target-off-tumor toxicities comparable to dose-limiting toxicities (149) and cumulative dose-limiting toxicities in conventional chemotherapies (150). Despite the highly antigen-specific targeting mechanism, antibodies and medicines derived thereof are targeted to surface expressed structures that are co-expressed in physiological tissues (107, 151). As a consequence, all tissues that express the targeted antigen are subject to on-target-off-tumor toxicities. The impact of this effect is defined by biological factors on the target expressing cells as well as the CAR expressing effector cells. On the target cells, these include target antigen expression level and the primary resistance or general susceptibility of the target cells to CAR mediated effects for which a first indication of tumor control with acceptable toxicity was documented using GD2- (152) and HER2-specific CAR T cells (153). On the CAR T cell side, the affinity of the recognition domain and the signaling components both define the potency and the toxicity mediated on the antigen positive cells (154).
The most successful strategy i) in CAR T cell therapy is to accept the “complete” eradication of target antigen positive cells transiently and/or permanently. This has been observed in patients treated with CD19- and BCMA-targeted CAR T cells (23, 155, 156). Importantly, the level of the depletion of physiological antigen positive cells is a strong indicator of leukemia control in B-lineage acute lymphoblastic (107) and chronic lymphocytic leukemia (157). The function of the B-lineage compartment can be substituted by immunoglobulin replacement therapy and as such displays an exception in the human body since severe tissue damages or depletion of tissues in most organs are not compatible with life. However, the strategy to eliminate specific cell subsets in the body permanently is quite unique and is reserved for CAR T cell immunotherapy in B-lineage dependent oncology (4, 92) and it has been demonstrated to be beneficial in life-threatening autoimmune diseases, such as refractory systemic lupus erythematosus (SLE) (158).
The second strategy (ii) is to transiently accept severe toxicities and then terminate the CAR T cell function after a predefined period of time. This is especially important in AML because AML-associated immunotargets are co-expressed on vitally essential myeloid bone-marrow derived cells and progenitor cells (159). CAR T cell therapy including conditioning chemotherapy with fludarabine and cyclophosphamide but especially in AML is accompanied with severe myeloid toxicities leading to an immunocompromised state with an increased risk for life-threatening infectious complications (160).
The systematic preclinical evaluation of CD33-directed CAR T cell constructs identified the CD33-targeted lintuzumab-based 2nd generation CAR construct (LIN-CD28-CD3Z) to be most efficacious (161) and led to the multicenter CD33-specifc CAR T cell trial (NCT03971799) for pediatric patients with relapsed and refractory AML. In this context CAR T cells are used for remission induction prior to subsequent allogeneic stem cell transplantation. Using CD33-targeted CAR T cells requires the profound elimination of the CAR T cell function post treatment via the conditioning regimen to save patients from experiencing ongoing myelosuppression, unless patients are in parallel to the CAR T cell application, transplanted with genetically modified hematopoietic stem cells with a gene knockout for the CAR-targeted antigen, such as CD33 (162) or CD45 (163) to exclude the recovering autologous hematopoietic system from the CAR-targeting mechanism.
In the third strategy (iii) the on-target-off-tumor toxicity is tolerable and generally not life-threatening. This applies mainly to overexpressed target antigens like mesothelin (MSLN) (97), CEA cell adhesion molecule 5 (CEACAM5) (164), HER2 (153), GD2 (152), B7H3 (165), CD70 (166), IL13Ra2 (167) and others in solid cancers. The listed target antigens are expressed on a variety of different tissues or are upregulated in immune cells upon activation, such as CD70 (168, 169) and B7H3 (170). The toxicities experienced with CAR T cells for solid cancer treatment are generally tolerable at standard dose levels (152, 164) and even intracranially administered CAR T cells in brain cancers (glioblastoma multiforme) (167) are considered a legitimate approach. However, in some instances CAR T cells in solid cancer patients have been lethal due to on-target-off-tumor toxicities. In a patient with colorectal cancer, reportedly a high dose of HER2-specific CAR T cells (1x1010)CAR+ cells) induced a severe acute respiratory distress syndrome (ARDS) and subsequent cardiac arrest causing death within days (171).
One approach to overcome or limit CAR mediated on-target-off-tumor toxicities in physiological non-cancerous tissues is to reduce the affinity of the recognition domain (scFv, VHH) (172). In CD19 CAR T cells, the 40x lower recognition domain CAR induced significantly less toxicities including CRS and ICANS compared to FMC63-based CAR T cells in childhood BCP-ALL patients while demonstrating enhanced proliferative capacity and antitumor activity (108). However, reduced affinity of the recognition domain and reduced responsiveness based on the CAR architecture [4-1BB versus CD28 costimulatory domain] increases the risk for antigen low expressing tumors to evade the targeting mechanism whereas CAR persistence remains an independent discriminator of clinical success (4, 12, 108). Sparing toxicities by reducing the affinity has been demonstrated for ICAM-1 specific CAR T cells (micromolar affinity) in thyroid cancer (173), as well as in HER2- and EGFR-specific CAR T cells in various solid cancer models including ovarian and prostate (174). Genetic alteration of the CD3ζ chain signaling to reduce the number of immunoreceptor tyrosine-based activation motifs (ITAMs) alleviates exhaustion and can alter the antigen-density threshold of a CAR (175). Logic AND-gating circumvent on-target-off-tumor toxicity but in consequence lead to a sensitivity decrease of the CAR T cells since the CAR requires two independent signals for efficient CAR activation, e.g., the AND-gating of a dual-CAR construct [CEA-CD3ζ & MSLN-4-1BB] in which the CEA-CAR provides the CD3ζ signaling and the MSLN-CAR provides the 4-1BB costimulatory signaling (176).
Since allorejection limits the efficacy of CAR T cell therapies, strategies to reduce immunogenicity via de-immunization and humanization have become an integral part of CAR T design. Further, reduced immunogenicity is achieved by using fully human natural ligand- or receptor-based CAR T. By repurposing high affinity ligand-receptor interactions to target overexpressed ligands [NKG2DL via NKG2D] (177) and receptors [BCMA & TACI via APRIL] (8) on cancers have been explored in the preclinical setting and in clinical trials (178). However, the clinical success of ligand-based CAR T compared to conventional scFv-based CAR T design thus far has been disappointing in multiple myeloma (7).
Cytokine release syndrome (CRS), Immune effector cell associated neurotoxicity syndrome (ICANS) and Macrophage Activation Syndrome (MAS) are early onset toxicities, which can have fatal symptoms (179).
The acute CRS is a common systemic inflammatory response to the excessive secretion of cytokines in the CAR T cell activation and proliferation, which occurs in 77-93% of leukemia patients treated with CAR T cells. Upon CAR T cell activation, the increased cytokine concentrations, particularly of IL6, a pleiotropic and pro-inflammatory cytokine, co-activate macrophages and monocytes, which leads to a further secretion of cytokines, and can be diagnosed from the high levels of granulocyte-macrophage colony-stimulating factor (GMCSF) in the serum. CRS is symptomized by fever, rigors, hypoxia, nausea, and heart rhythm disorders, including tachycardia and arrhythmias. In addition to heart dysfunctions, multiorgan failure can cause life-threatening medical conditions. Detections at early stages can allow for specific anti-inflammatory therapy, for instance, using tocilizumab to block the IL6 receptors, which can minimize the adverse effects (3, 180).
In the beginning of CRS, the brain is protected from primary and secondary involvement of CRS by the blood-brain barrier. Hence, the migration of CAR T cells to the brain is slower compared to other compartments of the body. However, the secretion of IL1ß and IL6 triggers von Willebrand factors, a glycoprotein, which disrupts the highly selective semi permeability of the blood-brain barrier. This permits cytokines and cells to enter the central nervous system, which changes the cytokine concentration in the brain. The elevated cytokine levels in brain tissues have been linked to neurological complications (181).
20-70% of CAR T cell patients develop neurological symptoms, including initial manifestations of cognitive impairment, such as tremor, confusion, lethargy, mild expressive and receptive aphasia, stupor, apraxia and decreased attention, which can progress into severe ICANS, symptomized by agitation, status epilepticus, cerebral oedema and occasionally intracerebral hemorrhage (3, 182, 183).
In CD19-targeted CAR T cells, the primary on-target-off-tumor toxicity occurs in the B-lineage tissues due to the expression of CD19, a BCR co-receptor on B cells. However, due to low expressions of CD19 in neural cells, on-target-off-tumor toxicities may also occur in neural tissue in the hyperinflammatory state. Furthermore, there are associations between the serum elevations of cytokine IL15, IL3 and GM-CSF and the blood-brain barrier permeability and ICANS development and severity (181, 184). Nonetheless, the fundamental principles, responsible for the neural toxicities are not thoroughly comprehended and other factors are considered to influence the severity of ICANS symptoms.
Hemophagocytic lymphohistiocytosis (HLH) and macrophage activation syndrome (MAS) are severe hyperinflammatory syndromes, associated with the pathological dysregulation of macrophage proliferation, which are mostly triggered by viral infections, especially Epstein-Barr virus infection, genetic lymphoid immune cell disorder, rheumatic autoimmune diseases, for instance the systemic-onset juvenile idiopathic arthritis, systemic lupus erythematosus and genetic mutations (3, 185–187). In addition, HLH and MAS can occur in the context of BiTE and CAR T cell immunotherapy (185). The excessive release of proinflammatory cytokines IL1, IL6, IL18 and tumor necrosis factor, triggered by the uncontrolled activation and proliferation of macrophages, leads to dysregulated immune activity and induces systemic inflammation (186). MAS is characterized by hyperinflammatory clinical features, including persistent fever, cytopenia, coagulopathy, liver insufficiency, hepatosplenomegaly, lymphadenopathy and multi-organ failure (3, 186). Laboratory diagnostic characteristics are a decrease in blood cell count due to hemophagocytosis, hyperferritinemia, abnormal coagulation profile, elevated triglycerides and high cytokine levels. The treatment of MAS usually involves immunosuppressive medications, such as corticosteroid and immunomodulatory drugs, for instance IL1 and IL6 signaling inhibitors (179, 181).
A significant constraint in the effectiveness of CAR T therapies, particularly in treating solid tumors, is the immunosuppressive nature of the tumor microenvironment (TME) (188). The TME is composed of different cell types, matrix proteins and secreted factors (189). Immunosuppressive cell types like myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory T cells (Tregs) produce anti-inflammatory cytokines, such as interleukin-10 (IL10), IL4, transforming growth factor-beta (TGF-β) that inhibit the CAR T function. These cytokines hinder CAR T function, impede proliferation, induce T-cell exhaustion, reducing their efficacy against tumor cells by limiting their cytotoxic and cytokine-producing abilities. Additionally, dense fibrogenic extracellular matrix, dysregulated tumor vasculature, and the hypoxic TME, characterized by limited nutrients and oxygen, restrict CAR T infiltration and function (188).
Efforts to enhance CAR T efficacy involve engineering these cells to resist the TME-induced exhaustion or express additional effector functions to promote persistence and functionality within the immunosuppressive TME.
One strategy to overcome exhaustion is to target T cell intrinsic pathways, such as programmed death-1 (PD-1) or TGF-β (190). Activation of the TGF-β receptor upon binding of TGF-β present in the TME upregulates CD70 expression and induce T-cell exhaustion with increased expression of the inhibitory receptors PD-1 and TIM-3 (191). Sustained PD-1 expression positively correlates with an exhausted phenotype and dysfunction of the T cells (192, 193). Pre-clinical data indicates that knocking out of PD-1 and/or TGF-β receptor II genes in CAR T cells with CRISPR/Cas9 technology improve the CAR T function against tumors expressing PD-L1 and/or TGF-β release (194, 195). Current clinical trials, evaluate anti-MUC1 CAR T cells with PD-1 knockout in advanced esophageal cancer (NCT03706326) and anti-EGFR CAR T cells with TGFBR2 knockout in EGFR-positive solid tumors (NCT04976218). Expression of dominant-negative PD-1 or TGF-β receptors, receptors lacking intracellular domains necessary for downstream signaling, also show promising improvement of the CAR T persistence and have been shown to be safe in a phase I clinical trial (196, 197). Combining CAR T therapy with checkpoint inhibitors with already approved immunotherapies, such as anti-PD-L1 or CAR T cells engineered to secrete PD-1 scFv or nanobodies, promote the anti-tumor activity (97, 198). Other approaches such as chimeric switch receptor-expressing CAR T cells like PD-1/CD28 or TGF-β/IL7 also seems to improve the persistence of CAR T (199, 200).
Another strategy to enhance the anti-cancer functions of CAR T cells and recruit resident immune cells by modulating the TME is to use CAR T cells designed to produce pro-inflammatory cytokines (201). Many different cytokines have been used to potentiate CAR T cell function, such as interleukin (IL) IL7 (202), IL15 (134), IL21 (203), IL12 (204) and IL18 (86). Anticancer activity is substantially induced by the highly potent pro-inflammatory cytokine IL12, which is naturally secreted by monocytic cells, e.g., dendritic cells and macrophages. IL12 enhances the cytotoxic capacity of both NK cells and T cells by inducing granzyme B and perforin production and interferon (IFN)-γ secretion (205, 206) and reduce the immunosuppressive effects of regulatory T cells (207). However, despite the tempting and promising enhanced anticancer function of pro-inflammatory cytokine support for CAR T cells and accessory immune effector cells, severe life-threatening toxicities limit their use and require the incorporation of advanced safety measures (208). The use of inducible promoters incorporating responsive elements of NFAT, AP-1, NFκB, or other transcription factors and their combinations facilitates activation-related gene expression (209–211). Alternatively, small molecule-regulated gene expression systems, such as the tetracycline-dependent Tet-On system, can improve the control of transgenes, including CAR expression (212) and cytokine secretion. While gene regulation can be complex and prone to leakage, site-specific integration of transgenes offers new opportunities to harness physiological gene regulation mechanisms (57, 110). This approach can reverse immune functions, such as integrating activating transgenes (e.g., CAR) into inhibitory loci like immune checkpoint receptors (213). Functional control of transgenes at the protein level is more reliable (214).
Various adapter CAR T cell systems are being evaluated in clinical trials for their efficacy and safety in treating different types of cancers. Although these technologies utilize the fundamental principle of adapter CAR technology, each employs a unique adapter molecule format with differential size and pharmacokinetic and pharmacodynamic properties, including scFv (78, 215), monomeric Fab (79), silenced full-size antibodies (216), and d-domains (80). Additionally, the adapter molecule dosing can be significantly lower (10-1,000 times) than traditional antibody dosing used for therapeutic IgG1 antibodies like rituximab (217) at 375 mg/m2 and cetuximab (218) at 400 mg/m2 for most technologies, with exemption of the high-affinity CD16-polymorphism (158V) (73).
In the clinical trial ATTCK-20-03 (NCT03189836), the CD16-derived antibody-coupled T cell receptor (ACTR707) product was used in conjunction with rituximab at 375 mg/m² for the treatment of relapsed or refractory CD20+ B-cell lymphoma. Patients achieved a complete remission rate of 50% (3/6) with an acceptable toxicity profile (219). In the follow-up, 56% (14/25) of patients responded, with 40% (10/25) achieving complete remission. Although the trial was discontinued due to the adverse event of neutropenia, it serves as a proof-of-concept for ACTR-based CAR therapy (220).
The peptide-based sCAR system (derived from the yeast GCN4 transcription factor) is currently being evaluated in patients with relapsed/refractory B-cell Malignancies (NCT04450069). The first-in-human data involved two patients with follicular lymphoma and one patient with mantle cell lymphoma, who received 140x106 CAR+ cells in conjunction with 10 µg/kgBW/day SWI019, a CD19-targeted monomeric Fab fragment used as adapter molecule. Two patients achieved a complete response according to Lugano criteria. Acute toxicity, ICANS was successfully managed with dexamethasone and by reducing the SWI019 dose to 5 µg/kgBW (79). Another peptide-based adapter CAR system (UniCAR, derived from the nuclear autoantigen La/SS-B) is being clinically tested in patients with hematological malignancies (NCT04230265). The first patients treated with relapsed/refractory AML (rrAML) received 100 or 250x106 CAR+ cells with 0.5 or 1 mg/day TM123, a CD123-targeted scFv-based adapter molecule. Complete hematological remission with incomplete recovery was observed (215). In a follow-up report, 19 patients showed an overall response rate (ORR) of 53% (8/15) for rrAML and 75% (3/4) in patients with AML at minimal residual disease (MRD) level (78). The adapter CAR technology IBI345, utilizing silenced antibodies carrying the P329G substitution, is used for the treatment of claudin 18.2 (CLDN18.2) positive tumors, including esophagogastric junction-, gastric-, and pancreatic cancer (NCT05199519). Patients were administered 50-250x106 CAR+ cells plus 1 mg/kgBW/day IgG1, demonstrating limited clinical efficacy (216). Another promising technology in clinical evaluation is the d-domain-based adapter CAR system (SparX) for the treatment of multiple myeloma (NCT04155749) and AML (NCT05457010). The adapter molecules are d-domain-based and targeted to BCMA in multiple myeloma and CD123 in AML (80). Regarding increased safety compared to conventional CAR technologies, the sCAR and UniCAR trials have demonstrated clinical proof-of-concept for reducing toxicity by pausing the administration of adapter molecules (78, 79, 215).
The concept of universal CAR technologies is fascinating and tempting. Still in the early stages, adapter CAR technologies have shown first indications of clinical efficacy in humans, reported in 2021 in the clinical trial (NCT04230265) utilizing the UniCAR in conjunction with a CD123-targeted adapter molecule in relapsed/refractory AML (215). Furthermore, there are several clinical trials recruiting patients using an alternative peptide-tag based CAR system sCAR (switchable CAR) with CD19-targeted adapter molecules (NCT04450069) as well as a d-domain based BCMA-targeted trial in multiple myeloma (NCT04155749). As of today, no clinical outcomes of the sCAR and d-domain CAR trials have been published. Additionally, third-party CD19-CAR (21) and recently “first-ever” iPSC-derived CD19-CAR T cells have demonstrated preliminary clinical anticancer activity (221).
Nonetheless, since multitargeting is widely accepted the key challenge to address tumor antigen heterogeneity and antigen loss, adapter CAR technologies hold the promise to overcome this very limitation of contemporary CAR T design. Even though CD19-CD22 dual-targeted CAR constructs incorporated into one immune receptor (144) have not yet clearly proven to significantly increase long-term survival in patients, the future aspiration is that multitargeted approaches utilizing dual- or trispecific CAR products will prevent immune evasion (144, 222). Hence, versatile multitargeted adapter CAR technologies represent a pivotal stride towards the future of cancer immunotherapy. These innovative technologies have the potential to revolutionize treatment approaches by enabling the customization of patient-specific regimens, intricately tailored to the distinct antigen immune profiles of individual patients.
Besides finding the most suitable combination of targetable antigens, CAR T design will be most efficacious if genetically adjusted to the immune requirements of the specific tumor and combined with complementary treatment strategies. The complementary use of existing medicines can instantly contribute to improved outcomes. For instance, CAR T cell therapy in conjunction with immune checkpoint inhibitors, such as pembrolizumab has demonstrated to reverse CD19-CAR exhaustion in B-cell lymphoma patients (223) and enhance anticancer activity in malignant pleural disease using MSLN-targeted CAR T (97). Further, small molecules (224) and the application of cytokine or chemokine modulating chemotherapy (225) can improve the clinical efficacy of CAR T cell therapies.
As indicated in Figure 3 Universal CAR configurator, the comprehensive tumor profiling will facilitate to identify the best suitable complimentary therapies and help select the potential immune targets for a patient-individualized adapter molecule panel. Most importantly, it will provide the objective which additional effector functions may specifically increase the CAR performance in the corresponding subject.
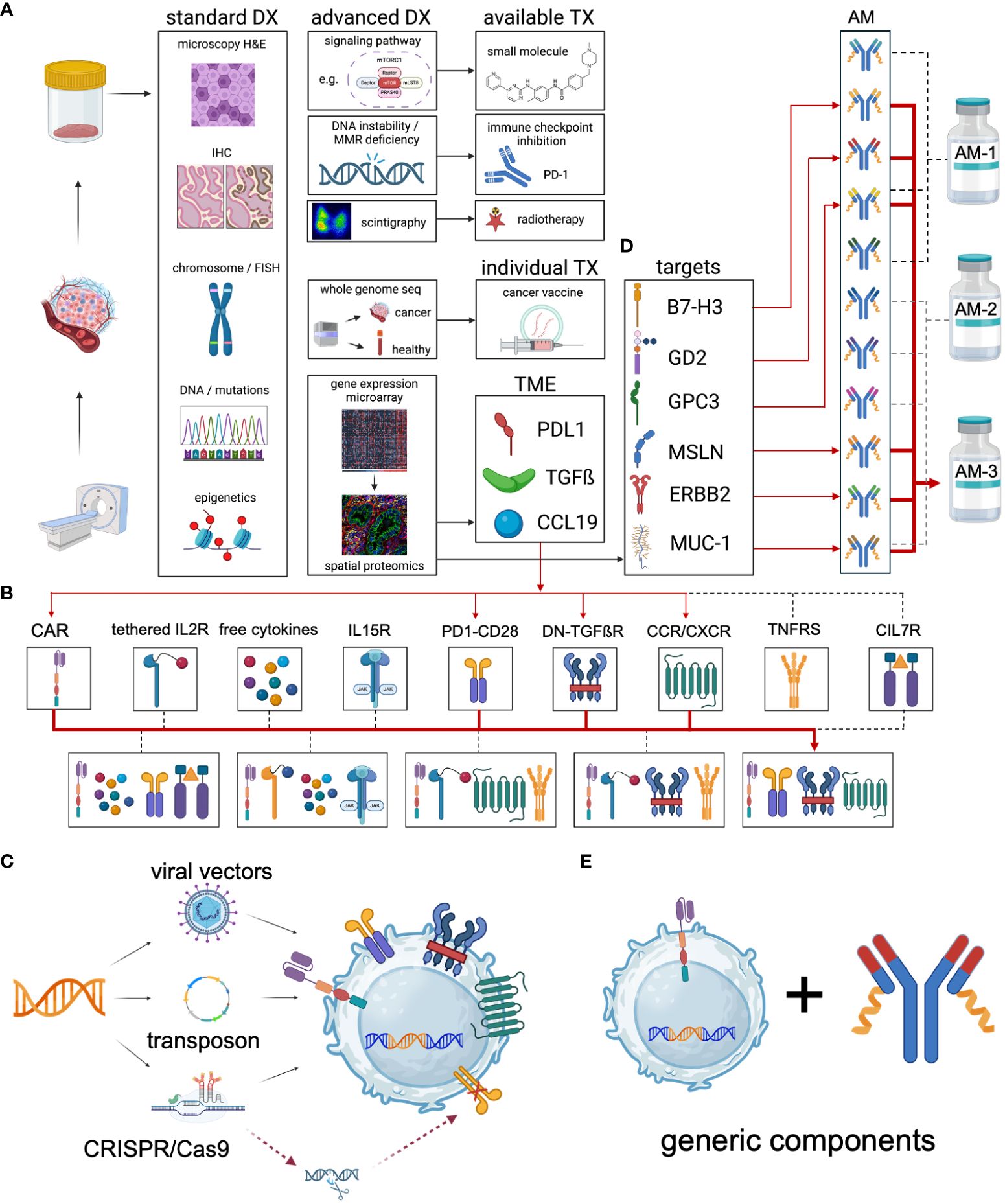
Figure 3 Universal CAR configurator. The concept of the Universal CAR Configurator is to design the most effective individual universal CAR for each specific cancer indication. It can be used with autologous immune effector cells or more advanced with iPSC-derived cell products to create “Universal CAR 2.0”. (A) The process begins with a comprehensive analysis of the cancer using state-of-the-art diagnostics. This includes basic microscopy, immunohistochemistry, and the detection of chromosomal aberrations (both structural and numerical) as well as utilizing fluorescence in situ hybridization (FISH). Additionally, targetable mutations and epigenetic changes are identified. Standard diagnostics confirm the diagnosis and guide the initial treatment regimen. Advanced diagnostics are employed to identify patients who could benefit from additional treatment options, either through established therapies or personalized treatments. Established therapies may involve the use of small molecules to inhibit upregulated signaling pathways such as kinases, mTOR, ABL, c-KIT, and FLT3. Moreover, patients with DNA instability due to mismatch repair deficiency, such as those with colorectal cancer, can significantly benefit from immune checkpoint inhibition (226). Further, theranostic approaches with antibody-guided radionuclides, such as Iodine-131 coupled to antibodies are used therapeutically (227). Whole genome sequencing is used to identify targetable mutations in the tumor with patient-individual cancer vaccines (228). Functionally tailored CAR-based therapies require a patient-individual two-step immune profiling. In the first step, the cancer is screened for the expression of relevant genes. Based on the results, in a second step a specific antibody panel is used for spatial proteomics to elucidate the patient’s tumor microenvironment (TME), exemplified as 1st PDL1 expression, 2nd TGFß secretion and 3rd CCL19 chemokine release by the tumor. (B) Therefore, the design of the TME-adjusted patient-individual CAR product requires the incorporation of 1st a PD1-CD28 signal converting receptor, 2nd a dominant negative TGFß receptor and 3rd the introduction of the chemokine receptor CCR7 to support the enrichment of CAR effector cells in and around the tumor. (C) The genetic payload of the relevant additional effector functions may be incorporated into the expression cassette and transferred via stable integration of the transgene using viral, transposon-based or CRISPR-based gene delivery. Additional genetic modifications can be utilized to inactivate immunosuppressive receptors such as PD-1 or CTLA-4 and others. (D) Spatial proteomics is used for the selection of patient-individual antibody panels that are specifically formulated for each patient. Combinatorial targeting is the key obstacle of targeted immunotherapies to overcome the antigen heterogeneity and immune escape evasion mechanisms. (E) Although patients receive individualized therapies, they built on generic key components - the CAR receptor and the adapter molecules. DX, diagnostics. TX, therapeutics. AM, adapter molecule. TGFß, TGF beta. IL15R, IL15 receptor. PD1-CD28, signal converting receptor. DN-TGFßR, dominant negative TGF beta receptor. CCR/CXCR, chemokine receptors. TNFRS, tumor necrosis factor receptor superfamily. CIL7, constitutive IL7 receptor.
These additional genetic modifications shall exploit pathways supportive of anticancer functions, such as cytokine signaling (IL7, IL12, IL15, IL18, IL21 and others) (86, 200, 204) and activating co-stimulatory receptor signaling (CD28, OX40 and others) (199) as well as the homing preference of cells via provision of chemokine receptors (229). Furthermore, they shall inactivate negative regulators of CAR T cell function, such as TGFßR signaling and immune checkpoints to overcome the immunosuppressive tumor microenvironment (TME) (195, 198, 200, 230). The disruption of immune homeostasis can lead to life-threatening, uncontrollable detrimental toxicities. Thus, in conventional CAR design it is more critical to employ a combination of boosting effector functions that may cause unmanageable hyperinflammatory complications if the targeted antigen is broadly expressed in physiological tissues. The decoupling of antigen recognition and signaling in adapter CAR technologies alone facilitates to control the unleashed CARs in the patient (20, 65). In summary, the development of effective CAR immunotherapies requires decoding the immunological processes in the tumor and converting them into effective and safe additional effector functions.
On the adapter molecule side, pharmacokinetic and pharmacodynamic variables need to be implemented to ensure high functionality of the adapter CAR system in all body compartments. Besides the critical antigen density (12), other factors like antigen shedding (231), antigen turnover and internalization kinetics (232) require the mindful selection of adapter molecule formats. They shall reach all various immune compartments. To overcome the blood-brain-barrier, adapter molecules either must be applied into the central nervous system (CNS) which is a viable administration route (233, 234) or be able to penetrate into the CNS (235). Certainly, they need to reach sufficient concentrations in the primary tumor niche and in metastases (236).
By nature, adapter CAR T will always have a lower avidity to the targeted antigen than conventional CARs, illustrated and outlined in Figure 4. However, multitargeted strategies and performance tuning can compensate for the reduced initial formation of the cytolytic synapse and turn the disadvantage into dominance. Additionally, adapter CAR T cell technologies facilitate a more physiological CAR recruitment and engagement compared to conventional CAR design and allow cells to recover from the massive CAR stimulation according to the administration regimen of the adapter molecules (237).
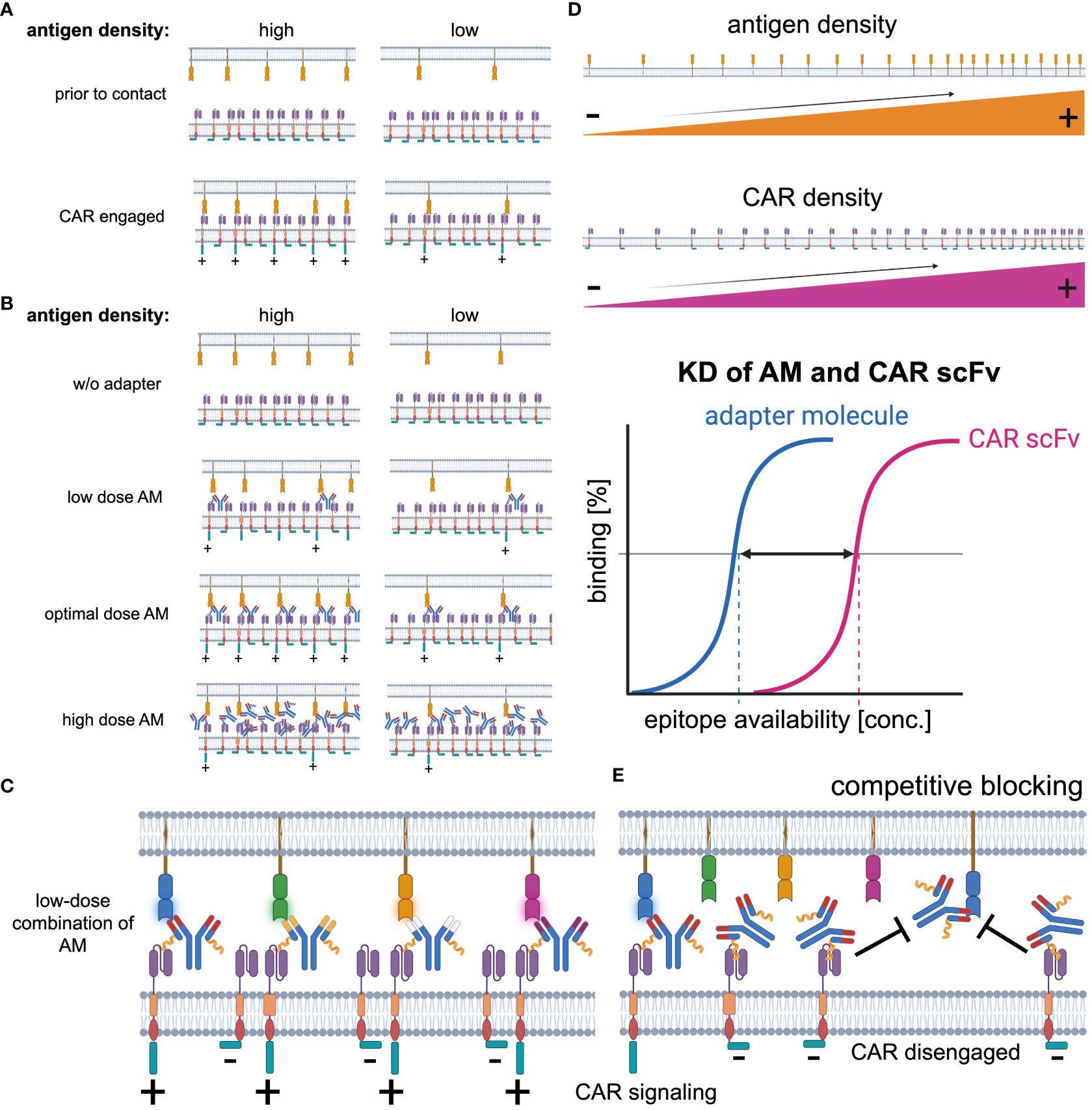
Figure 4 Key determinants of CAR signaling. (A) The CAR receptor-target antigen interaction in conventional CAR technologies is primarily determined by the antigen expression. Most target antigens are expressed at lower levels than the CAR receptor on the effector cell population. Thus, in antigen high expressing target cells, the avidity is higher than in antigen low expressing targets and as the CAR signaling is proportional to the CAR receptor-target antigen interaction, it is enhanced in antigen high expressing targets. (B) Besides the target antigen density, in adapter CAR technologies the adapter molecule concentration significantly impacts on the CAR engagement with the target cells. Without any adapter molecule available, there is no CAR engagement possible. With increasing concentrations, the CAR engagement becomes more efficient and reaches an optimum before inhibitory effects start to reduce the CAR-target interaction at supra-optimal adapter molecule concentrations. (C) Synchronic multitargeting, utilizing a combination of adapter molecules at low concentrations, can increase the CAR-target interaction while the blocking effects are reduced. (D) Key determinants of CAR-target interaction are i) the antigen expression density on the target cells, ii) the CAR receptor expression density on the CAR+ effector cell population, and the iii) interplay of the avidity and the affinity (KD) of the adapter molecule to the targeted antigen as well as the CAR recognition domain, e.g., CAR-scFv to the CAR-target moiety on the adapter molecules. (E) At supra-optimal concentrations, competitive blocking effects reduce the probability of CAR-target interaction and therefore pharmacokinetic as well as pharmacodynamic aspects must be considered optimizing the adapter molecule dosing.
While there are numerous advantages of iPSC-derived adapter CAR products, summarized in Table 2, there are limitations and challenges in the creation of universal (iPSC-derived) adapter CAR effector cells, referred to as universal CAR 2.0.
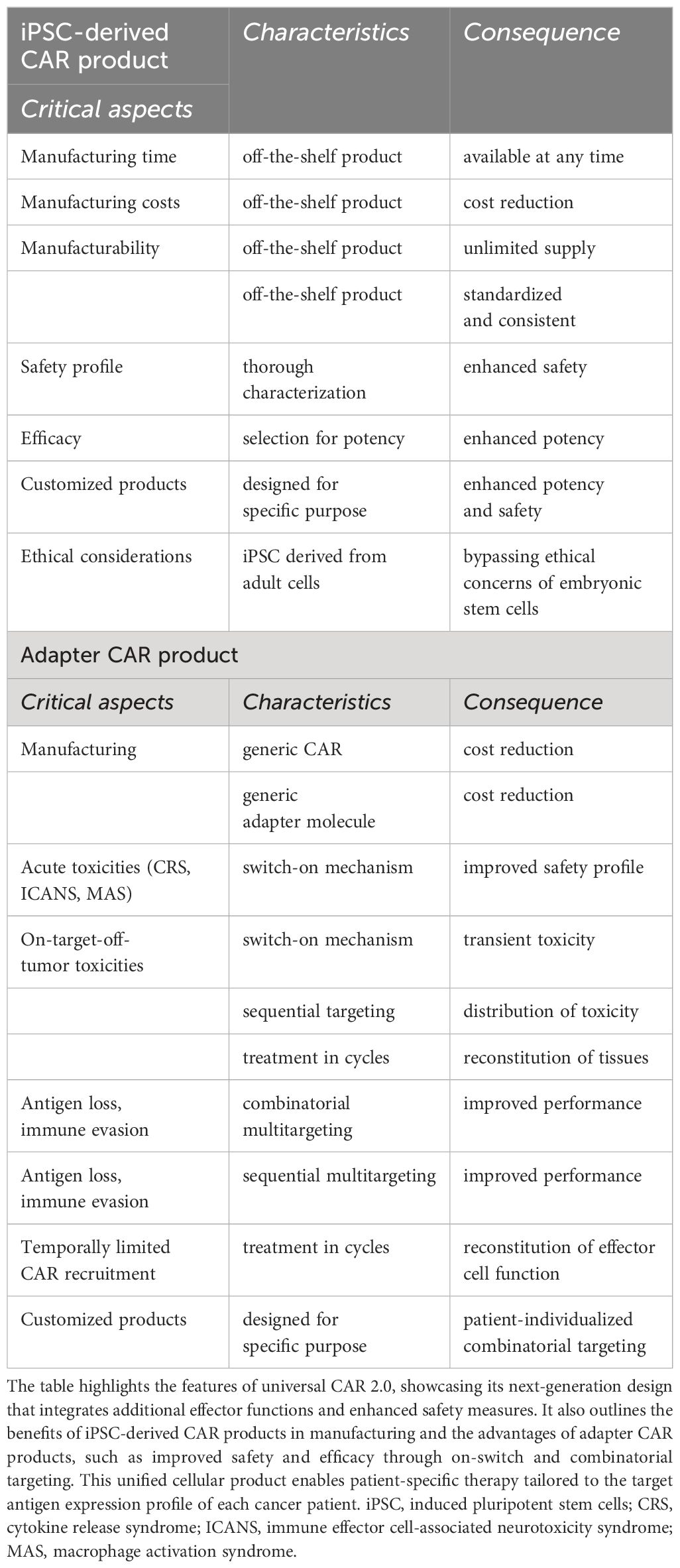
Table 2 Features of universal CAR products.
A significant current challenge with the use of artificially engineered third-party effector cells is their notably shorter lifespan compared to autologous CAR therapies (238). Despite sophisticated design efforts, there remains a substantial gap in our understanding of the potential long-term risks associated with these therapies. Concerns particularly focus on the possibility of malignant transformations due to unintended genetic modifications and contamination with undifferentiated stem cells (239), as well as autoimmune or hyperinflammatory reactions. These adverse effects may arise from specific combinations of genetic alterations that promote inflammatory responses via activating receptors and cytokines while simultaneously deactivating inhibitory receptors, e.g. PD-1, disrupting the immune system’s natural regulatory balance (240).
The primary challenge of the adapter CAR function is largely influenced by the biodistribution of adapter molecules and their ability to penetrate less vascularized and perfused body compartments, such as the testicles, the central nervous system (CNS), and particularly fibrotic and encapsulated solid tumors (241). Additionally, the pharmacokinetics and compatibility of specific adapter molecules can complicate the design and outcomes of clinical trials. It is improbable that a uniform adapter molecule format will be effective across different types of cancers. Consequently, in clinical trials, the format of the adapter molecule will dictate the administration route and regimen. Furthermore, combinatorial targeting will necessitate intricate protocols and meticulous monitoring during the optimization of treatment regimens.
CAR products are designed to equip immune cells with novel effector functions and combine these with their natural capacity for adaptation to everchanging circumstances. These multifaceted adaptive functions allow the CARs to migrate efficiently to any part of the body via chemotaxis (242), then to mediate a potent but considerate immune response that does not lead to detrimental effects and unrepairable tissue damage (243). The immune response is complemented by the recruitment of accessory immune cells via the secretion of proinflammatory and immunosuppressive cytokines, chemokines as well as the presentation of immunomodulatory ligands to shape and orchestrate the immune response (244). During the acute phase of the response the vertical differentiation and exponential multiplication of the effector cells is imminently important (4). However, to maintain a strong immune response the horizontal proliferation (maintenance), differentiation and maturation of immune cells provide the ability for self-renewal and long-lived immune memory function (245) which are the prerequisite for the comprehensive elimination of cancer and subsequent protection from recurrence (4).
In consequence, the most promising effector cells of the future are a reflection of the complex natural cellular immunity (246) in artificially designed effector cells that are equipped with versatile synthetic receptors (57, 110, 175, 247). Therefore, the appreciation and incorporation of natural, multilayered immune functions will lead to the most advanced cellular products equipped with the versatile biological plasticity, capable of autonomously regulated adaptability, self-renewal, and the persistence of immune memory (245). Long-lived immune memory allows resting memory T cells to awaken and protect humans from harm (virus, bacteria, fungi and cancer) after re-encounter with invaders even with the latency of many years and displays one of the most impressive evolutionary immune functions (114).
All of the named immune functions have only been partially understood but have guaranteed immune protection and thus certainly shall be considered in the design of novel cell products. The practical challenge in creating cellular medicines is to retain these features while adding beneficial effector functions which is supported by shorter manufacturing protocols (248).
Presently, the scope for clinical applications is confined to meticulously defined cellular products prevailing from the legislative landscape and the regulatory authorities’ inclination towards stringent product specifications (249). The encounter with cellular therapies has underscored the intricacies within the immune compartment. Yet, the remarkable diversity exhibited by T cells stands as a valuable asset in tackling immunological challenges. This resilience has been notably exemplified in the realm of CAR T cells, laying the groundwork for future explorations in the design of next-generation CARs.
In conclusion, universal allogeneic CAR T cell products hold the promise to overcome the economic hurdles of personalized cellular medicines with regards to infrastructure, logistics as well as manufacturing and distribution costs (250). Additionally, they support to broaden the applicability of CAR T cell therapy in patients that do not meet the basic CAR manufacturing requirements, e.g., reduced general condition, low lymphocyte count, poor immune status or simply lack the time required for the manufacturing of autologous CAR T cell products before initiating the treatment (28). Whilst allogeneic universal iPSC-derived cellular therapies are promising, they reside at a very early stage of development and are mainly compromised by allorejection that limits persistence and their overall potency of the anticancer response (251). In the coming years, preclinical and clinical research will need to provide more insight into both safety and efficacy of iPSC-derived CAR products to improve this novel, potentially cost-effective and life-saving technology (28, 251) that may become a complementary treatment to “conventional CD19- and BCMA-specific CAR T cell therapies” in the future (4, 23, 101).
Numerous adapter CAR technologies have been developed with the aim to introduce a safety on-switch to control early onset toxicities and on-target-off-tumor toxicities as well as to establish versatile patient-individualized combinatorial immunotargeting (20, 65). Incorporating next-generation CAR design (209) may catapult adapter CAR technologies to outcompete conventional CAR designs while retaining safety (237). The fusion of these distinct two universal CAR technologies unites key features of two concepts that might resolve our current skepticism for clinical potency and safety as well as the significant costs required for the development. In summary, groundbreaking advancements remain pivotal to usher in this new era of two-component universal CAR cellular therapies, based on bespoke patient-individualized treatments, derived from readily available off-the-shelf products.
LS: Conceptualization, Validation, Visualization, Writing – original draft, Writing – review & editing. CW: Writing – original draft, Writing – review & editing. MB: Writing – original draft, Writing – review & editing, Resources. PS: Conceptualization, Formal analysis, Funding acquisition, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The figures were created with BioRender.
PS and CW are inventors of patents in the field of indirect CAR technologies.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. (2013) 342:1432–3. doi: 10.1126/science.342.6165.1432
2. Jayaraman J, Mellody MP, Hou AJ, Desai RP, Fung AW, Pham AHT, et al. CAR-T design: Elements and their synergistic function. EBioMedicine. (2020) 58:102931. doi: 10.1016/j.ebiom.2020.102931
3. Boettcher M, Joechner A, Li Z, Yang SF, Schlegel P. Development of CAR T cell therapy in children-A comprehensive overview. J Clin Med. (2022) 11(8). doi: 10.3390/jcm11082158
4. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. New Engl J Med. (2018) 378:439–48.
5. Nasiri F, Safarzadeh Kozani P, Rahbarizadeh F. T-cells engineered with a novel VHH-based chimeric antigen receptor against CD19 exhibit comparable tumoricidal efficacy to their FMC63-based counterparts. Front Immunol. (2023) 14. doi: 10.3389/fimmu.2023.1063838
6. Wang G, Zhou X, Fucà G, Dukhovlinova E, Shou P, Li H, et al. Fully human antibody VH domains to generate mono and bispecific CAR to target solid tumors. J ImmunoTher Cancer. (2021) 9:e002173. doi: 10.1136/jitc-2020-002173
7. Lee L, Lim WC, Galas-Filipowicz D, Fung K, Taylor J, Patel D, et al. Limited efficacy of APRIL CAR in patients with multiple myeloma indicate challenges in the use of natural ligands for CAR T-cell therapy. J ImmunoTher Cancer. (2023) 11:e006699. doi: 10.1136/jitc-2023-006699
8. Lee L, Draper B, Chaplin N, Philip B, Chin M, Galas-Filipowicz D, et al. An APRIL-based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood. (2018) 131:746–58. doi: 10.1182/blood-2017-05-781351
9. Qin H, Edwards JP, Zaritskaya L, Gupta A, Mu CJ, Fry TJ, et al. Chimeric antigen receptors incorporating D domains targeting CD123 direct potent mono- and bi-specific antitumor activity of T cells. Mol Ther. (2019) 27:1262–74. doi: 10.1016/j.ymthe.2019.04.010
10. Kozani PS, Kozani PS, O'Connor RS. Humanized chimeric antigen receptor (CAR) T cells. J Cancer Immunol (Wilmington). (2021) 3:183–7. doi: 10.33696/cancerimmunol
11. Vincke C, Loris R, Saerens D, Martinez-Rodriguez S, Muyldermans S, Conrath K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem. (2009) 284:3273–84. doi: 10.1074/jbc.M806889200
12. Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discovery. (2020) 10:702–23. doi: 10.1158/2159-8290.CD-19-0945
13. Liu CSC, Ganguly D. Mechanical cues for T cell activation: role of piezo1 mechanosensors. Crit Rev Immunol. (2019) 39:15–38. doi: 10.1615/CritRevImmunol.v39.i1
14. Liu CSC, Raychaudhuri D, Paul B, Chakrabarty Y, Ghosh AR, Rahaman O, et al. Cutting edge: piezo1 mechanosensors optimize human T cell activation. J Immunol. (2018) 200:1255–60. doi: 10.4049/jimmunol.1701118
15. Lewis RS. Store-operated calcium channels: from function to structure and back again. Cold Spring Harb Perspect Biol. (2020) 12(5). doi: 10.1101/cshperspect.a035055
16. Dutta D, Barr VA, Akpan I, Mittelstadt PR, Singha LI, Samelson LE, et al. Recruitment of calcineurin to the TCR positively regulates T cell activation. Nat Immunol. (2017) 18:196–204. doi: 10.1038/ni.3640
17. Zhou Y, Ramachandran S, Oh-hora M, Rao A, Hogan PG. Pore architecture of the ORAI1 store-operated calcium channel. Proc Natl Acad Sci. (2010) 107:4896–901. doi: 10.1073/pnas.1001169107
18. Watanabe K, Terakura S, Martens AC, van Meerten T, Uchiyama S, Imai M, et al. Target antigen density governs the efficacy of anti–CD20-CD28-CD3 ζ Chimeric antigen receptor–modified effector CD8+ T cells. J Immunol. (2015) 194:911–20. doi: 10.4049/jimmunol.1402346
19. Furukawa Y, Hamano Y, Shirane S, Kinoshita S, Azusawa Y, Ando J, et al. Advances in allogeneic cancer cell therapy and future perspectives on "Off-the-shelf" T cell therapy using iPSC technology and gene editing. Cells. (2022) 11(2). doi: 10.3390/cells11020269
20. Seitz CM, Mittelstaet J, Atar D, Hau J, Reiter S, Illi C, et al. Novel adapter CAR-T cell technology for precisely controllable multiplex cancer targeting. Oncoimmunology. (2021) 10:2003532. doi: 10.1080/2162402X.2021.2003532
21. Benjamin R, Jain N, Maus MV, Boissel N, Graham C, Jozwik A, et al. UCART19, a first-in-class allogeneic anti-CD19 chimeric antigen receptor T-cell therapy for adults with relapsed or refractory B-cell acute lymphoblastic leukaemia (CALM): a phase 1, dose-escalation trial. Lancet Haematol. (2022) 9:e833–43.
22. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
23. Cappell KM, Sherry RM, Yang JC, Goff SL, Vanasse DA, McIntyre L, et al. Long-term follow-up of anti-CD19 chimeric antigen receptor T-cell therapy. J Clin Oncol. (2020) 38:3805–15. doi: 10.1200/JCO.20.01467
24. Khan AN, Chowdhury A, Karulkar A, Jaiswal AK, Banik A, Asija S, et al. Immunogenicity of CAR-T cell therapeutics: evidence, mechanism and mitigation. Front Immunol. (2022) 13:886546. doi: 10.3389/fimmu.2022.886546
25. Smith M, Zakrzewski J, James S, Sadelain M. Posttransplant chimeric antigen receptor therapy. Blood. (2018). doi: 10.1182/blood-2017-08-752121
26. Ukrainskaya V, Molostova O, Shelikhova L, Pershin D, Kulakovskaya E, Volkov D, et al. Haploidentical donor-derived memory CAR T cells: first in human experience and in vitro correlative study. Blood Adv. (2022) 6:5582–8. doi: 10.1182/bloodadvances.2021006262
27. Tang TCY, Xu N, Nordon R, Haber M, Micklethwaite K, Dolnikov A. Donor T cells for CAR T cell therapy. biomark Res. (2022) 10:14. doi: 10.1186/s40364-022-00359-3
28. Ueda T, Shiina S, Iriguchi S, Terakura S, Kawai Y, Kabai R. Optimization of the proliferation and persistency of CAR T cells derived from human induced pluripotent stem cells. Nat Biomed Eng. (2023) 7:24–37. doi: 10.1038/s41551-022-00969-0
29. Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. (2017) 4:92–101. doi: 10.1016/j.omtm.2016.12.006
30. Mehta PH, Fiorenza S, Koldej RM, Jaworowski A, Ritchie DS, Quinn KM. T cell fitness and autologous CAR T cell therapy in haematologic Malignancy. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.780442
31. Jiang W, Clancy LE, Avdic S, Sutrave G, Street J, Simms R, et al. Third-party CMV- and EBV-specific T-cells for first viral reactivation after allogeneic stem cell transplant. Blood Adv. (2022) 6:4949–66. doi: 10.1182/bloodadvances.2022007103
32. Withers B, Clancy L, Burgess J, Simms R, Brown R, Micklethwaite K, et al. Establishment and operation of a third-party virus-specific T cell bank within an allogeneic stem cell transplant program. Biol Blood Marrow Transplant. (2018) 24:2433–42. doi: 10.1016/j.bbmt.2018.08.024
33. Blache U, Popp G, Dünkel A, Koehl U, Fricke S. Potential solutions for manufacture of CAR T cells in cancer immunotherapy. Nat Commun. (2022) 13:5225. doi: 10.1038/s41467-022-32866-0
34. Jo T, Yoshihara S, Okuyama Y, Fujii K, Henzan T, Kahata K, et al. Risk factors for CAR-T cell manufacturing failure among DLBCL patients: A nationwide survey in Japan. Br J Haematol. (2023) 202:256–66. doi: 10.1111/bjh.18831
35. Jo T, Arai Y, Yoshihara S, Kojima M, Ikemoto J, Uemura T, et al. Thrombocytopenia and low CD4/CD8 ratio at apheresis along with prior bendamustine use increase risk of CAR-T cell manufacturing failure. Blood. (2022) 140:1608–9. doi: 10.1182/blood-2022-165613
36. Bethge WA, Martus P, Schmitt M, Holtick U, Subklewe M, von Tresckow B, et al. GLA/DRST real-world outcome analysis of CAR T-cell therapies for large B-cell lymphoma in Germany. Blood. (2022) 140:349–58. doi: 10.1182/blood.2021015209
37. Reyes KR, Huang C-Y, Lo M, Arora S, Chung A, Wong SW, et al. Safety and efficacy of BCMA CAR-T cell therapy in older patients with multiple myeloma. Transplant Cell Therapy Off Publ Am Soc Transplant Cell Ther. (2023) 29:350–5. doi: 10.1016/j.jtct.2023.03.012
38. Kenderian SS, Porter DL, Gill S. Chimeric antigen receptor T cells and hematopoietic cell transplantation: how not to put the CART before the horse. Biol Blood Marrow Transplant. (2017) 23:235–46. doi: 10.1016/j.bbmt.2016.09.002
39. Leen AM, Bollard CM, Mendizabal AM, Shpall EJ, Szabolcs P, Antin JH, et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood. (2013) 121:5113–23. doi: 10.1182/blood-2013-02-486324
40. van der Zwan M, Baan CC, van Gelder T, Hesselink DA. Review of the clinical pharmacokinetics and pharmacodynamics of alemtuzumab and its use in kidney transplantation. Clin Pharmacokinet. (2018) 57:191–207. doi: 10.1007/s40262-017-0573-x
41. Krawczyk M, Peyraud N, Rybtsova N, Masternak K, Bucher P, Barras E, et al. Long distance control of MHC class II expression by multiple distal enhancers regulated by regulatory factor X complex and CIITA. J Immunol. (2004) 173:6200–10. doi: 10.4049/jimmunol.173.10.6200
42. Liu X, Zhang Y, Cheng C, Cheng AW, Zhang X, Li N, et al. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. (2017) 27:154–7. doi: 10.1038/cr.2016.142
43. Caldwell KJ, Gottschalk S, Talleur AC. Allogeneic CAR cell therapy-more than a pipe dream. Front Immunol. (2020) 11:618427.
44. Poiré X, van Besien K. Alemtuzumab in allogeneic hematopoetic stem cell transplantation. Expert Opin Biol Ther. (2011) 11:1099–111. doi: 10.1517/14712598.2011.592824
45. Mo F, Watanabe N, McKenna MK, Hicks MJ, Srinivasan M, Gomes-Silva D, et al. Engineered off-the-shelf therapeutic T cells resist host immune rejection. Nat Biotechnol. (2021) 39:56–63. doi: 10.1038/s41587-020-0601-5
46. Benjamin R, et al. Preliminary data on safety, cellular kinetics and anti-leukemic activity of UCART19, an allogeneic anti-CD19 CAR T-cell product, in a pool of adult and pediatric patients with high-risk CD19+ Relapsed/refractory B-cell acute lymphoblastic leukemia. Blood. (2018) 132:896–6. doi: 10.1182/blood-2018-99-111356
47. Look A, Burns D, Tews I, Roghanian A, Mansour S. Towards a better understanding of human iNKT cell subpopulations for improved clinical outcomes. Front Immunol. (2023) 14. doi: 10.3389/fimmu.2023.1176724
48. Dwivedi A, et al. Engineering off-the-shelf gamma delta CAR T cells for the treatment of acute myeloid leukemia. Blood. (2023) 142:4827–7. doi: 10.1182/blood-2023-190357
49. Ureña-Bailén G, et al. Preclinical evaluation of CRISPR-edited CAR-NK-92 cells for off-the-shelf treatment of AML and B-ALL. Int J Mol Sci. (2022) 23:12828. doi: 10.3390/ijms232112828
50. Montagner IM, Penna A, Fracasso G, Carpanese D, Dalla Pietà A, Barbieri V, et al. Anti-PSMA CAR-engineered NK-92 cells: an off-the-shelf cell therapy for prostate cancer. Cells. (2020) 9(6). doi: 10.20944/preprints202005.0259.v1
51. Cutmore LC, Marshall JF. Current perspectives on the use of off the shelf CAR-T/NK cells for the treatment of cancer. Cancers (Basel). (2021) 13(8). doi: 10.3390/cancers13081926
52. Sadeqi Nezhad M, Abdollahpour-Alitappeh M, Rezaei B, Yazdanifar M, Seifalian AM. Induced pluripotent stem cells (iPSCs) provide a potentially unlimited T cell source for CAR-T cell development and off-the-shelf products. Pharm Res. (2021) 38:931–45. doi: 10.1007/s11095-021-03067-z
53. Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. (2018) 9:1911. doi: 10.1038/s41467-018-04252-2
54. Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. (2018) 559:405–9.
55. Blaeschke F, Chen YY, Apathy R, Daniel B, Chen AY, Chen PA, et al. Modular pooled discovery of synthetic knockin sequences to program durable cell therapies. Cell. (2023) 186:4216–4234.e4233.
56. Yu X, Liang X, Xie H, Kumar S, Ravinder N, Potter J, et al. Improved delivery of Cas9 protein/gRNA complexes using lipofectamine CRISPRMAX. Biotechnol Lett. (2016) 38:919–29. doi: 10.1007/s10529-016-2064-9
57. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. (2017) 543:113–7. doi: 10.1038/nature21405
58. Dong W, Kantor B. Lentiviral vectors for delivery of gene-editing systems based on CRISPR/Cas: current state and perspectives. Viruses. (2021) 13. doi: 10.3390/v13071288
59. Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. (2020) 38:824–44. doi: 10.1038/s41587-020-0561-9
60. Cichocki F, van der Stegen SJC, Miller JS. Engineered and banked iPSCs for advanced NK- and T-cell immunotherapies. Blood. (2023) 141:846–55. doi: 10.1182/blood.2022016205
61. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. (2013) 31:822–6. doi: 10.1038/nbt.2623
62. Bishop DC, Clancy LE, Simms R, Burgess J, Mathew G, Moezzi L, et al. Development of CAR T-cell lymphoma in 2 of 10 patients effectively treated with piggyBac-modified CD19 CAR T cells. Blood. (2021) 138:1504–9. doi: 10.1182/blood.2021010813
63. Li YR, Dunn ZS, Yu Y, Li M, Wang P, Yang L. Advancing cell-based cancer immunotherapy through stem cell engineering. Cell Stem Cell. (2023) 30:592–610. doi: 10.1016/j.stem.2023.02.009
64. Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol. (2018) 11:132. doi: 10.1186/s13045-018-0677-2
65. Arndt C, Fasslrinner F, Loureiro LR, Koristka S, Feldmann A, Bachmann M. Adaptor CAR platforms-next generation of T cell-based cancer immunotherapy. Cancers (Basel). (2020) 12(5). doi: 10.3390/cancers12051302
66. Liu D, Zhao J, Song Y. Engineering switchable and programmable universal CARs for CAR T therapy. J Hematol Oncol. (2019) 12:69. doi: 10.1186/s13045-019-0763-0
67. Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell Malignancies. Proc Natl Acad Sci. (2016) 113:E459–68. doi: 10.1073/pnas.1524155113
68. Cartellieri M, Loff S, von Bonin M, Bejestani EP, Ehninger A, Feldmann A, et al. Unicar: A novel modular retargeting platform technology for CAR T cells. Blood. (2015) 126:5549–9. doi: 10.1182/blood.V126.23.5549.5549
69. Borrok MJ, Li Y, Harvilla PB, Vellalore Maruthachalam B, Tamot N, Prokopowitz C, et al. Conduit CAR: redirecting CAR T-cell specificity with A universal and adaptable bispecific antibody platform. Cancer Res Commun. (2022) 2:146–57. doi: 10.1158/2767-9764.CRC-21-0150
70. Rennert P, Su L, Dufort F, Birt A, Sanford T, Wu L, et al. A novel CD19-anti-CD20 bridging protein prevents and reverses CD19-negative relapse from CAR19 T cell treatment in vivo. Blood. (2019) 134:252–2. doi: 10.1182/blood-2019-130654
71. Atar D, Mast A-S, Scheuermann S, Ruoff L, Seitz CM, Schlegel P. Adapter CAR T cell therapy for the treatment of B-lineage lymphomas. Biomedicines. (2022) 10:2420. doi: 10.3390/biomedicines10102420
72. Seidel UJE, Schlegel P, Grosse-Hovest L, Hofmann M, Aulwurm S, Pyz E, et al. Reduction of minimal residual disease in pediatric B-lineage acute lymphoblastic leukemia by an Fc-optimized CD19 antibody. Mol Ther. (2016). doi: 10.1038/mt.2016.141
73. Kudo K, Imai C, Lorenzini P, Kamiya T, Kono K, Davidoff AM, et al. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. (2014) 74:93–103. doi: 10.1158/0008-5472.CAN-13-1365
74. Stock S, Benmebarek M-R, Kluever A-K, Darowski D, Jost C, Stubenrauch K-G, et al. Chimeric antigen receptor T cells engineered to recognize the P329G-mutated Fc part of effector-silenced tumor antigen-targeting human IgG1 antibodies enable modular targeting of solid tumors. J ImmunoTher Cancer. (2022) 10:e005054. doi: 10.1136/jitc-2022-005054
75. Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, et al. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res. (2012) 18:6436–45. doi: 10.1158/1078-0432.CCR-12-1449
76. Lohmueller JJ, Ham JD, Kvorjak M, Finn OJ. mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. Oncoimmunology. (2017) 7:e1368604. doi: 10.1080/2162402X.2017.1368604
77. Urbanska K, Powell DJ. Development of a novel universal immune receptor for antigen targeting: To Infinity and beyond. Oncoimmunology. (2012) 1:777–9. doi: 10.4161/onci.19730
78. Wermke M, Metzelder S, Kraus S, Sala E, Vucinic V, Fiedler W, et al. Updated results from a phase I dose escalation study of the rapidly-switchable universal CAR-T therapy UniCAR-T-CD123 in relapsed/refractory AML. Blood. (2023) 142:3465–5. doi: 10.1182/blood-2023-177867
79. Nikolaenko L, Mulroney C, Riedell PA, Flinn IW, Cruz JC, Vaidya R, et al. First in human study of an on/off switchable CAR-T cell platform targeting CD19 for B cell Malignancies (CLBR001 + SWI019). Blood. (2021) 138:2822. doi: 10.1182/blood-2021-151727
80. Edwards JP, Swers JS, Buonato JM, Zaritskaya L, Mu CJ, Gupta A, et al. Controlling CAR-T cell activity and specificity with synthetic SparX adapters. Mol Ther.
81. Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. (2018) 173:1426–1438.e1411.
82. Minutolo NG, Sharma P, Poussin M, Shaw LC, Brown DP, Hollander EE, et al. Quantitative control of gene-engineered T-cell activity through the covalent attachment of targeting ligands to a universal immune receptor. J Am Chem Soc. (2020) 142:6554–68. doi: 10.1021/jacs.9b11622
83. Ruffo E, Butchy AA, Tivon Y, So V, Kvorjak M, Parikh A, et al. Post-translational covalent assembly of CAR and synNotch receptors for programmable antigen targeting. Nat Commun. (2023) 14:2463. doi: 10.1038/s41467-023-37863-5
84. Urbanska K, Lynn RC, Stashwick C, Thakur A, Lum LG, Powell DJ Jr, et al. Targeted cancer immunotherapy via combination of designer bispecific antibody and novel gene-engineered T cells. J Transl Med. (2014) 12:347. doi: 10.1186/s12967-014-0347-2
85. Karches CH, Benmebarek M-R, Schmidbauer ML, Kurzay M, Klaus R, Geiger M, et al. Bispecific antibodies enable synthetic agonistic receptor-transduced T cells for tumor immunotherapy. Clin Cancer Res. (2019) 25:5890–900. doi: 10.1158/1078-0432.CCR-18-3927
86. Svoboda J, Gerson JN, Landsburg DJ, Chong EA, Barta SK, Dwivedy Nasta S, et al. Interleukin-18 secreting autologous anti-CD19 CAR T-cells (huCART19-IL18) in patients with non-Hodgkin lymphomas relapsed or refractory to prior CAR T-cell therapy. Blood. (2022) 140:4612–4. doi: 10.1182/blood-2022-162393
87. Fischer J, Paret C, El Malki K, Alt F, Wingerter A, Neu MA, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. (2017) 40:187–95. doi: 10.1097/CJI.0000000000000169
88. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. New Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
89. Shalabi H, Kraft IL, Wang HW, Yuan CM, Yates B, Delbrook C, et al. Sequential loss of tumor surface antigens following chimeric antigen receptor T-cell therapies in diffuse large B-cell lymphoma. Haematologica. (2018) 103:e215–8. doi: 10.3324/haematol.2017.183459
90. Frey NV, Gill S, Hexner EO, Schuster S, Nasta S, Loren A, et al. Long-term outcomes from a randomized dose optimization study of chimeric antigen receptor modified T cells in relapsed chronic lymphocytic leukemia. J Clin Oncol. (2020) 38:2862–71. doi: 10.1200/JCO.19.03237
91. Mikkilineni L, Kochenderfer JN. CAR T cell therapies for patients with multiple myeloma. Nat Rev Clin Oncol. (2021) 18:71–84. doi: 10.1038/s41571-020-0427-6
92. Munshi NC, Anderson LD Jr., Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. New Engl J Med. (2021) 384:705–16. doi: 10.1056/NEJMoa2024850
93. Wang Z, Chen C, Wang L, Jia Y, Qin Y. Chimeric antigen receptor T-cell therapy for multiple myeloma. Front Immunol. (2022) 13:1050522. doi: 10.3389/fimmu.2022.1050522
94. Giri S, Bal S, Godby KN, Ravi G, Clark D, Ubersax C, et al. Real-world applicability of commercial chimeric antigen receptor T-cell therapy among older adults with relapsed and/or refractory multiple myeloma. Am J Hematol. (2022) 97:E153–e155. doi: 10.1002/ajh.26472
95. Bufalo FD, Angelis BD, Caruana I, Baldo GD, Ioris MAD, Serra A, et al. GD2-CART01 for relapsed or refractory high-risk neuroblastoma. New Engl J Med. (2023) 388:1284–95. doi: 10.1056/NEJMoa2210859
96. Majzner RG, Ramakrishna S, Yeom KW, Patel S, Chinnasamy H, Schultz LM, et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. (2022) 603:934–41. doi: 10.1038/s41586-022-04489-4
97. Adusumilli PS, Zauderer MG, Rivière I, Solomon SB, Rusch VW, O'Cearbhaill RE, et al. A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with Malignant pleural disease, in combination with the anti-PD-1 agent pembrolizumab. Cancer Discovery. (2021) 11:2748–63. doi: 10.1158/2159-8290.CD-21-0407
98. Zhong G, Zhang X, Guo Z, Gao Y, Zhao B, Liu X, et al. Complete remission of advanced pancreatic cancer induced by claudin18.2-targeted CAR-T cell therapy: a case report. Front Immunol. (2024) 15. doi: 10.3389/fimmu.2024.1325860
99. Bagley SJ, Logun M, Fraietta JA, Wang X, Desai AS, Bagley LJ, et al. Intrathecal bivalent CAR T cells targeting EGFR and IL13Rα2 in recurrent glioblastoma: phase 1 trial interim results. Nat Med. (2024) 30:1320–9. doi: 10.1038/s41591-024-02893-z
100. Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. (2019) 36:471–82. doi: 10.1016/j.ccell.2019.09.006
101. Rodriguez-Otero P, Ailawadhi S, Arnulf B, Patel K, Cavo M, Nooka AK, et al. Ide-cel or standard regimens in relapsed and refractory multiple myeloma. New Engl J Med. (2023) 388:1002–14. doi: 10.1056/NEJMoa2213614
102. Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med. (2019) 25:1341–55. doi: 10.1038/s41591-019-0564-6
103. Qi T, McGrath K, Ranganathan R, Dotti G, Cao Y. Cellular kinetics: A clinical and computational review of CAR-T cell pharmacology. Adv Drug Delivery Rev. (2022) 188:114421. doi: 10.1016/j.addr.2022.114421
104. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. (2015) 125:4017–23. doi: 10.1182/blood-2014-12-580068
105. Ruella M, Maus MV. Catch me if you can: Leukemia Escape after CD19-Directed T Cell Immunotherapies. Comput Struct Biotechnol J. (2016) 14:357–62. doi: 10.1016/j.csbj.2016.09.003
106. Shabaneh TB, Stevens AR, Stull SM, Shimp KR, Seaton BW, Gad EA, et al. Systemically administered low-affinity HER2 CAR T cells mediate antitumor efficacy without toxicity. J ImmunoTher Cancer. (2024) 12:e008566. doi: 10.1136/jitc-2023-008566
107. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
108. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. (2019) 25:1408–14. doi: 10.1038/s41591-019-0549-5
109. Zhang T, He X, Tsang TC, Harris DT. Transgenic TCR expression: comparison of single chain with full-length receptor constructs for T-cell function. Cancer Gene Ther. (2004) 11:487–96. doi: 10.1038/sj.cgt.7700703
110. Mansilla-Soto J, Eyquem J, Haubner S, Hamieh M, Feucht J, Paillon N, et al. HLA-independent T cell receptors for targeting tumors with low antigen density. Nat Med. (2022) 28:345–52. doi: 10.1038/s41591-021-01621-1
111. Golubovskaya V, Wu L. Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers. (2016) 8:36. doi: 10.3390/cancers8030036
112. Sallusto F, Lanzavecchia A, Mackay CR. Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol Today. (1998) 19:568–74. doi: 10.1016/S0167-5699(98)01346-2
113. Kortekaas Krohn I, Aerts JL, Breckpot K, Goyvaerts C, Knol E, Van Wijk F, et al. T-cell subsets in the skin and their role in inflammatory skin disorders. Allergy. (2022) 77:827–42. doi: 10.1111/all.15104
114. Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. (2014) 14:24–35. doi: 10.1038/nri3567
115. St. Paul M, Ohashi PS. The roles of CD8+ T cell subsets in antitumor immunity. Trends Cell Biol. (2020) 30:695–704. doi: 10.1016/j.tcb.2020.06.003
116. Corthay A. How do regulatory T cells work? Scand J Immunol. (2009) 70:326–36. doi: 10.1111/j.1365-3083.2009.02308.x
117. Ryan D, Frezza C. IL-10-mediated refueling of exhausted T cell mitochondria boosts anti-tumour immunity. Immunometabolism. (2021) 3:e210030. doi: 10.20900/immunometab20210030
118. de Jong VMT, Wang Y, Ter Hoeve ND, Opdam M, Stathonikos N, Jóźwiak K, et al. Prognostic value of stromal tumor-infiltrating lymphocytes in young, node-negative, triple-negative breast cancer patients who did not receive (neo)Adjuvant systemic therapy. J Clin Oncol. (2022) 40:2361–74. doi: 10.1200/JCO.21.01536
119. Fu Q, Chen N, Ge C, Li R, Li Z, Zeng B, et al. Prognostic value of tumor-infiltrating lymphocytes in melanoma: a systematic review and meta-analysis. Oncoimmunology. (2019) 8:1593806. doi: 10.1080/2162402X.2019.1593806
120. Watanabe N, Mo F, McKenna MK. Impact of manufacturing procedures on CAR T cell functionality. Front Immunol. (2022) 13:876339. doi: 10.3389/fimmu.2022.876339
121. Dickinson MJ, Barba P, Jäger U, Shah NN, Blaise D, Briones J, et al. A novel autologous CAR-T therapy, YTB323, with preserved T-cell stemness shows enhanced CAR T-cell efficacy in preclinical and early clinical development. Cancer Discovery. (2023) 13:1982–97. doi: 10.1158/2159-8290.CD-22-1276
122. Klysz DD, Fowler C, Malipatlolla M, Stuani L, Freitas KA, Chen Y, et al. Inosine induces stemness features in CAR-T cells and enhances potency. Cancer Cell. (2024) 42:266–282.e268.
123. Chan JD, Scheffler CM, Munoz I, Sek K, Lee JN, Huang Y-K, et al. FOXO1 enhances CAR T cell stemness, metabolic fitness and efficacy. Nature. (2024) 629:201–10. doi: 10.1038/s41586-024-07242-1
124. Yunis J, Short KR, Yu D. Severe respiratory viral infections: T-cell functions diverging from immunity to inflammation. Trends Microbiol. (2023) 31:644–56. doi: 10.1016/j.tim.2022.12.008
125. Fajgenbaum DC, June CH. Cytokine storm. New Engl J Med. (2020) 383:2255–73. doi: 10.1056/NEJMra2026131
126. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
127. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
128. Asaadi Y, Jouneghani FF, Janani S, Rahbarizadeh F. A comprehensive comparison between camelid nanobodies and single chain variable fragments. biomark Res. (2021) 9:87. doi: 10.1186/s40364-021-00332-6
129. Wagner DL, Fritsche E, Pulsipher MA, Ahmed N, Hamieh M, Hegde M, et al. Immunogenicity of CAR T cells in cancer therapy. Nat Rev Clin Oncol. (2021) 18:379–93. doi: 10.1038/s41571-021-00476-2
130. Korell F, Laier S, Sauer S, Veelken K, Hennemann H, Schubert ML, et al. Current challenges in providing good leukapheresis products for manufacturing of CAR-T cells for patients with relapsed/refractory NHL or ALL. Cells. (2020) 9. doi: 10.3390/cells9051225
131. Lythe G, Callard RE, Hoare RL, Molina-París C. How many TCR clonotypes does a body maintain? J Theor Biol. (2016) 389:214–24. doi: 10.1016/j.jtbi.2015.10.016
132. Jackson Z, Roe A, Sharma AA, Lopes FBTP, Talla A, Kleinsorge-Block S, et al. Automated manufacture of autologous CD19 CAR-T cells for treatment of non-Hodgkin lymphoma. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.01941
133. Roon J, Glaudemans KAFM, Bijlsma JWJ. & Lafeber, F.P.J.G. Interleukin 7 stimulates tumour necrosis factor α and Th1 cytokine production in joints of patients with rheumatoid arthritis. Ann Rheum Dis. (2003) 62:113–9.
134. Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo CF, et al. IL15 enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol Res. (2019) 7:759–72. doi: 10.1158/2326-6066.CIR-18-0466
135. Jin J, Gkitsas N, Fellowes VS, Ren J, Feldman SA, Hinrichs CS, et al. Enhanced clinical-scale manufacturing of TCR transduced T-cells using closed culture system modules. J Trans Med. (2018) 16:13. doi: 10.1186/s12967-018-1384-z
136. Calderon H, Mamonkin M, Guedan S. Analysis of CAR-mediated tonic signaling. Methods Mol Biol. (2020) 2086:223–36.
137. Dabiri H, Safarzadeh Kozani P, Habibi Anbouhi M, Mirzaee Godarzee M, Haddadi MH, Basiri M, et al. Site-specific transgene integration in chimeric antigen receptor (CAR) T cell therapies. biomark Res. (2023) 11:67. doi: 10.1186/s40364-023-00509-1
138. Wu L, Wei Q, Brzostek J, Gascoigne NRJ. Signaling from T cell receptors (TCRs) and chimeric antigen receptors (CARs) on T cells. Cell Mol Immunol. (2020) 17:600–12. doi: 10.1038/s41423-020-0470-3
139. Improving non-viral knock-in with modified single-stranded DNAs and small molecules. Nat Biotechnol. (2023) 41:478–9. doi: 10.1038/s41587-022-01420-0
140. Eshhar Z, Waks T, Gross G. The emergence of T-bodies/CAR T cells. Cancer J. (2014) 20. doi: 10.1097/PPO.0000000000000027
141. Wei J, Han X, Bo J, Han W. Target selection for CAR-T therapy. J Hematol Oncol. (2019) 12:62. doi: 10.1186/s13045-019-0758-x
142. Anurathapan U, Chan RC, Hindi HF, Mucharla R, Bajgain P, Hayes BC, et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol Ther. (2014) 22:623–33. doi: 10.1038/mt.2013.262
143. Benmebarek MR, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20061283
144. Spiegel JY, Patel S, Muffly L, Hossain NM, Oak J, Baird JH, et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell Malignancies: a phase 1 trial. Nat Med. (2021) 27:1419–31.
145. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discovery. (2015) 5:1282–95. doi: 10.1158/2159-8290.CD-15-1020
146. Upadhyay R, Boiarsky JA, Pantsulaia G, Svensson-Arvelund J, Lin MJ, Wroblewska A, et al. A critical role for Fas-mediated off-target tumor killing in T-cell immunotherapy. Cancer Discovery. (2021) 11:599–613. doi: 10.1158/2159-8290.CD-20-0756
147. Klampatsa A, Leibowitz MS, Sun J, Liousia M, Arguiri E, Albelda SM. Analysis and augmentation of the immunologic bystander effects of CAR T cell therapy in a syngeneic mouse cancer model. Mol Ther Oncolytics. (2020) 18:360–71. doi: 10.1016/j.omto.2020.07.005
148. Silveira CRF, Corveloni AC, Caruso SR, Macêdo NA, Brussolo NM, Haddad F, et al. Cytokines as an important player in the context of CAR-T cell therapy for cancer: Their role in tumor immunomodulation, manufacture, and clinical implications. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.947648
149. Wong HH, Halford S. Dose-limiting toxicity and maximum tolerated dose: still fit for purpose? Lancet Oncol. (2015) 16:1287–8. doi: 10.1016/S1470-2045(15)00248-X
150. Dempke WCM, Zielinski R, Winkler C, Silberman S, Reuther S, Priebe W. Anthracycline-induced cardiotoxicity — are we about to clear this hurdle? Eur J Cancer. (2023) 185:94–104. doi: 10.1016/j.ejca.2023.02.019
151. Atilla E, Benabdellah K. The black hole: CAR T cell therapy in AML. Cancers (Basel). (2023) 15. doi: 10.20944/preprints202304.0482.v1
152. Mount CW, Majzner RG, Sundaresh S, Arnold EP, Kadapakkam M, Haile S, et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat Med. (2018) 24:572–9. doi: 10.1038/s41591-018-0006-x
153. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor–modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol. (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
154. Flugel CL, Majzner RG, Krenciute G, Dotti G, Riddell SR, Wagner DL, et al. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat Rev Clin Oncol. (2023) 20:49–62. doi: 10.1038/s41571-022-00704-3
155. Wang Y, Li C, Xia J, Li P, Cao J, Pan B, et al. Humoral immune reconstitution after anti-BCMA CAR T-cell therapy in relapsed/refractory multiple myeloma. Blood Adv. (2021) 5:5290–9. doi: 10.1182/bloodadvances.2021004603
156. Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. (2019) 34:45–55. doi: 10.1016/j.blre.2018.11.002
157. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
158. Mougiakakos D, Krönke G, Völkl S, Kretschmann S, Aigner M, Kharboutli S, et al. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. New Engl J Med. (2021) 385:567–9. doi: 10.1056/NEJMc2107725
159. Perna F, Berman SH, Soni RK, Mansilla-Soto J, Eyquem J, Hamieh M, et al. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell. (2017) 32:506–519.e505.
160. Tambaro FP, Singh H, Jones E, Rytting M, Mahadeo KM, Thompson P, et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia. (2021) 35:3282–6. doi: 10.1038/s41375-021-01232-2
161. Qin H, Yang L, Chukinas JA, Shah N, Tarun S, Pouzolles M, et al. Systematic preclinical evaluation of CD33-directed chimeric antigen receptor T cell immunotherapy for acute myeloid leukemia defines optimized construct design. J Immunother Cancer. (2021) 9(9). doi: 10.1136/jitc-2021-003149
162. Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. (2018) 173:1439–1453.e1419.
163. Wellhausen N, O’Connell RP, Lesch S, Engel NW, Rennels AK, Gonzales D, et al. Epitope base editing CD45 in hematopoietic cells enables universal blood cancer immune therapy. Sci Trans Med. (2023) 15:eadi1145. doi: 10.1126/scitranslmed.adi1145
164. Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, et al. Phase I escalating-dose trial of CAR-T therapy targeting CEA(+) metastatic colorectal cancers. Mol Ther. (2017) 25:1248–58. doi: 10.1016/j.ymthe.2017.03.010
165. Vitanza NA, Wilson AL, Huang W, Seidel K, Brown C, Gustafson JA, et al. Intraventricular B7-H3 CAR T cells for diffuse intrinsic pontine glioma: preliminary first-in-human bioactivity and safety. Cancer Discovery. (2023) 13:114–31. doi: 10.1093/neuonc/noad073.315
166. Ye H, Huang RR, Shuch BM, Chen Z, Said JW, Pantuck AJ, et al. CD70 is a promising CAR-T cell target in patients with advanced renal cell carcinoma. J Clin Oncol. (2022) 40:384–4. doi: 10.1200/JCO.2022.40.6_suppl.384
167. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. New Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
168. Arroyo Hornero R, Georgiadis C, Hua P, Trzupek D, He L-Z, Qasim W, et al. CD70 expression determines the therapeutic efficacy of expanded human regulatory T cells. Commun Biol. (2020) 3:375. doi: 10.1038/s42003-020-1097-8
169. Flieswasser T, Van den Eynde A, Van Audenaerde J, De Waele J, Lardon F, Riether C, et al. The CD70-CD27 axis in oncology: the new kids on the block. J Exp Clin Cancer Res. (2022) 41:12. doi: 10.1186/s13046-021-02215-y
170. Yi KH, Chen L. Fine tuning the immune response through B7-H3 and B7-H4. Immunol Rev. (2009) 229:145–51. doi: 10.1111/j.1600-065X.2009.00768.x
171. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
172. Duan Y, Chen R, Huang Y, Meng X, Chen J, Liao C, et al. Tuning the ignition of CAR: optimizing the affinity of scFv to improve CAR-T therapy. Cell Mol Life Sci. (2021) 79:14. doi: 10.1007/s00018-021-04089-x
173. Park S, Shevlin E, Vedvyas Y, Zaman M, Park S, Hsu Y-MS, et al. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep. (2017) 7:14366. doi: 10.1038/s41598-017-14749-3
174. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. (2015) 75:3596–607. doi: 10.1158/0008-5472.CAN-15-0159
175. Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. (2019) 25:82–8. doi: 10.1038/s41591-018-0290-5
176. Zhang E, Yang P, Gu J, Wu H, Chi X, Liu C, et al. Recombination of a dual-CAR-modified T lymphocyte to accurately eliminate pancreatic Malignancy. J Hematol Oncol. (2018) 11:102. doi: 10.1186/s13045-018-0646-9
177. Curio S, Jonsson G, Marinović S. A summary of current NKG2D-based CAR clinical trials. Immunother Adv. (2021) 1:ltab018. doi: 10.1093/immadv/ltab018
178. Murad JM, Graber DJ, Sentman CL. Advances in the use of natural receptor- or ligand-based chimeric antigen receptors (CARs) in haematologic Malignancies. Best Pract Res Clin Haematol. (2018) 31:176–83. doi: 10.1016/j.beha.2018.03.003
179. Jain MD, Smith M, Shah NN. How I treat refractory CRS and ICANS after CAR T-cell therapy. Blood. (2023) 141:2430–42. doi: 10.1182/blood.2022017414
180. Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer. (2018) 6:56. doi: 10.1186/s40425-018-0343-9
181. Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. (2022) 22:85–96. doi: 10.1038/s41577-021-00547-6
182. Sievers S, Watson G, Johncy S, Adkins S. Recognizing and grading CAR T-cell toxicities: an advanced practitioner perspective. Front Oncol. (2020) 10:885. doi: 10.3389/fonc.2020.00885
183. Sterner RC, Sterner RM. Immune effector cell associated neurotoxicity syndrome in chimeric antigen receptor-T cell therapy. Front Immunol. (2022) 13:879608. doi: 10.3389/fimmu.2022.879608
184. Gust J, Ponce R, Liles WC, Garden GA, Turtle CJ. Cytokines in CAR T cell-associated neurotoxicity. Front Immunol. (2020) 11:577027. doi: 10.3389/fimmu.2020.577027
185. Martín-Rojas RM, Gómez-Centurión I, Bailén R, Bastos M, Diaz-Crespo F, Carbonell D, et al. Hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS) following treatment with tisagenlecleucel. Clin Case Rep. (2022) 10:e05209. doi: 10.1002/ccr3.5209
186. Henderson LA, Cron RQ. Macrophage activation syndrome and secondary hemophagocytic lymphohistiocytosis in childhood inflammatory disorders: diagnosis and management. Paediatr Drugs. (2020) 22:29–44. doi: 10.1007/s40272-019-00367-1
187. Grom AA, Horne A, De Benedetti F. Macrophage activation syndrome in the era of biologic therapy. Nat Rev Rheumatol. (2016) 12:259–68. doi: 10.1038/nrrheum.2015.179
188. Kankeu Fonkoua LA, Sirpilla O, Sakemura R, Siegler EL, Kenderian SS. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol Ther Oncolytics. (2022) 25:69–77. doi: 10.1016/j.omto.2022.03.009
189. Anderson NM, Simon MC. The tumor microenvironment. Curr Biol. (2020) 30:R921–5. doi: 10.1016/j.cub.2020.06.081
190. Gumber D, Wang LD. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. eBioMedicine. (2022) 77:103941. doi: 10.1016/j.ebiom.2022.103941
191. Yang ZZ, Grote DM, Xiu B, Ziesmer SC, Price-Troska TL, Hodge LS, et al. TGF-beta upregulates CD70 expression and induces exhaustion of effector memory T cells in B-cell non-Hodgkin's lymphoma. Leukemia. (2014) 28:1872–84. doi: 10.1038/leu.2014.84
192. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. (2006) 439:682–7. doi: 10.1038/nature04444
193. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. (2009) 114:1537–44. doi: 10.1182/blood-2008-12-195792
194. Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.133977
195. Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. (2019) 68:365–77. doi: 10.1007/s00262-018-2281-2
196. Narayan V, Barber-Rotenberg JS, Jung IY, Lacey SF, Rech AJ, Davis MM, et al. PSMA-targeting TGFbeta-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat Med. (2022) 28:724–34.
197. Liu X, Zhang Y, Li K, Liu Y, Xu J, Ma J, et al. A novel dominant-negative PD-1 armored anti-CD19 CAR T cell is safe and effective against refractory/relapsed B cell lymphoma. Transl Oncol. (2021) 14:101085. doi: 10.1016/j.tranon.2021.101085
198. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, Van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. (2018) 36:847–56. doi: 10.1038/nbt.4195
199. Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. (2016) 76:1578–90. doi: 10.1158/0008-5472.CAN-15-2524
200. Noh K-E, Lee J-H, Choi S-Y, Jung N-C, Nam J-H, Oh J-S, et al. TGF-β/IL-7 chimeric switch receptor-expressing CAR-T cells inhibit recurrence of CD19-positive B cell lymphoma. Int J Mol Sci. (2021) 22:8706. doi: 10.3390/ijms22168706
201. Hawkins ER, D'Souza RR, Klampatsa A. Armored CAR T-cells: the next chapter in T-cell cancer immunotherapy. Biologics: Targets Ther Volume. (2021) 15:95–105. doi: 10.2147/BTT.S291768
202. Li L, Li Q, Yan Z-X, Sheng L-S, Fu D, Xu P, et al. Transgenic expression of IL-7 regulates CAR-T cell metabolism and enhances in vivo persistence against tumor cells. Sci Rep. (2022) 12:12506. doi: 10.1038/s41598-022-16616-2
203. Du L, Nai Y, Shen M, Li T, Huang J, Han X, et al. IL-21 optimizes the CAR-T cell preparation through improving lentivirus mediated transfection efficiency of T cells and enhancing CAR-T cell cytotoxic activities. Front Mol Biosci. (2021) 8:675179. doi: 10.3389/fmolb.2021.675179
204. Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T cells expressing IL-12 eradicate lymphoma in fully lymphoreplete mice through induction of host immunity. Mol Ther Oncolytics. (2018) 8:41–51. doi: 10.1016/j.omto.2017.12.003
205. Kubin M, Kamoun M, Trinchieri G. Interleukin 12 synergizes with B7/CD28 interaction in inducing efficient proliferation and cytokine production of human T cells. J Exp Med. (1994) 180:211–22. doi: 10.1084/jem.180.1.211
206. Ferlazzo G, Pack M, Thomas D, Paludan C, Schmid D, Strowig T, et al. Distinct roles of IL-12 and IL-15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs. Proc Natl Acad Sci U.S.A. (2004) 101:16606–11. doi: 10.1073/pnas.0407522101
207. Zhao J, Zhao J, Perlman S. Differential effects of IL-12 on Tregs and non-Treg T cells: roles of IFN-γ, IL-2 and IL-2R. PloS One. (2012) 7:e46241. doi: 10.1371/journal.pone.0046241
208. Lu X. Impact of IL-12 in cancer. Curr Cancer Drug Targets. (2017) 17:682–97. doi: 10.2174/1568009617666170427102729
209. Chmielewski M, Abken H. CAR T cells transform to trucks: chimeric antigen receptor-redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol Immunother. (2012) 61:1269–77. doi: 10.1007/s00262-012-1202-z
210. Glienke W, Dragon AC, Zimmermann K, Martyniszyn-Eiben A, Mertens M, Abken H, et al. GMP-compliant manufacturing of TRUCKs: CAR T cells targeting GD(2) and releasing inducible IL-18. Front Immunol. (2022) 13:839783. doi: 10.3389/fimmu.2022.839783
211. Rudek LS, Zimmermann K, Galla M, Meyer J, Kuehle J, Stamopoulou A, et al. Generation of an NFκB-driven alpharetroviral “All-in-one” Vector construct as a potent tool for CAR NK cell therapy. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.751138
212. Gu X, He D, Li C, Wang H, Yang G. Development of inducible CD19-CAR T cells with a tet-on system for controlled activity and enhanced clinical safety. Int J Mol Sci. (2018) 19. doi: 10.3390/ijms19113455
213. Fraessle SP, Tschulik C, Effenberger M, Cletiu V, Gerget M, Schober K, et al. Activation-inducible CAR expression enables precise control over engineered CAR T cell function. Commun Biol. (2023) 6:604. doi: 10.1038/s42003-023-04978-w
214. Jan M, Scarfò I, Larson RC, Walker A, Schmidts A, Guirguis AA, et al. Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci Transl Med. (2021), 13. doi: 10.1126/scitranslmed.abb6295
215. Wermke M, Kraus S, Ehninger A, Bargou RC, Goebeler M-E, Middeke JM, et al. Proof of concept for a rapidly switchable universal CAR-T platform with UniCAR-T-CD123 in relapsed/refractory AML. Blood. (2021) 137:3145–8. doi: 10.1182/blood.2020009759
216. Qin S, Tian W, Li M, Wei H, Sun L, Xie Q, et al. 1054P A phase Ia study to evaluate the safety, tolerability, pharmacokinetics and preliminary efficacy of a modular CLDN18.2-targeting PG CAR-T therapy (IBI345) in patients with CLDN18.2+ solid tumors. Ann Oncol. (2023) 34:S638.
217. Cunningham D, Hawkes EA, Jack A, Qian W, Smith P, Mouncey P, et al. Rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone in patients with newly diagnosed diffuse large B-cell non-Hodgkin lymphoma: a phase 3 comparison of dose intensification with 14-day versus 21-day cycles. Lancet. (2013) 381:1817–26. doi: 10.1016/S0140-6736(13)60313-X
218. Tabernero J, Pfeiffer P, Cervantes A. Administration of cetuximab every 2 weeks in the treatment of metastatic colorectal cancer: an effective, more convenient alternative to weekly administration? Oncologist. (2008) 13:113–9. doi: 10.1634/theoncologist.2007-0201
219. Flinn IW, Cohen JB, Akard LP, Jaglowski S, Vasconcelles M, Ranger A, et al. Preliminary clinical results of a phase 1 study evaluating the safety and anti-tumor activity of ACTR707 in combination with rituximab in subjects with relapsed or refractory CD20+ B-cell lymphoma. Blood. (2018) 132:2966. doi: 10.1182/blood-2018-99-111622
220. Munoz J, Flinn IW, Cohen JB, Sachs J, Exter B, Ranger A, et al. Results from a phase 1 study of ACTR707 in combination with rituximab in patients with relapsed or refractory CD20+ B cell lymphoma. Transplant Cell Therapy Off Publ Am Soc Transplant Cell Ther. (2024) 30:241.e241–241.e248.
221. Mehta A, Farooq U, Chen A, McGuirk JP, Ly T, Wong L, et al. Interim phase I clinical data of FT819-101, a study of the first-ever, off-the-shelf, iPSC-derived TCR-less CD19 CAR T-cell therapy for patients with relapsed/refractory B-cell Malignancies. Blood. (2022) 140:4577–8. doi: 10.1182/blood-2022-167194
222. Schneider D, Xiong Y, Wu D, Hu P, Alabanza L, Steimle B, et al. Trispecific CD19-CD20-CD22-targeting duoCAR-T cells eliminate antigen-heterogeneous B cell tumors in preclinical models. Sci Trans Med. (2021) 13. doi: 10.1126/scitranslmed.abc6401
223. Chong EA, Alanio C, Svoboda J, Nasta SD, Landsburg DJ, Lacey SF, et al. Pembrolizumab for B-cell lymphomas relapsing after or refractory to CD19-directed CAR T-cell therapy. Blood. (2022) 139:1026–38. doi: 10.1182/blood.2021012634
224. Cao X, Jin X, Zhang X, Utsav P, Zhang Y, Guo R, et al. Small-molecule compounds boost CAR-T cell therapy in hematological Malignancies. Curr Treat Options Oncol. (2023) 24:184–211. doi: 10.1007/s11864-023-01049-4
225. Wang AX, Ong XJ, D'Souza C, Neeson PJ, Zhu JJ. Combining chemotherapy with CAR-T cell therapy in treating solid tumors. Front Immunol. (2023) 14:1140541. doi: 10.3389/fimmu.2023.1140541
226. Cercek A, Lumish M, Sinopoli J, Weiss J, Shia J, Lamendola-Essel M, et al. PD-1 blockade in mismatch repair–deficient, locally advanced rectal cancer. New Engl J Med. (2022) 386:2363–76. doi: 10.1056/NEJMoa2201445
227. D'Huyvetter M, Vos J, Caveliers V, Vaneycken I, Heemskerk J, Duhoux FP, et al. Phase I trial of (131)I-GMIB-anti-HER2-VHH1, a new promising candidate for HER2-targeted radionuclide therapy in breast cancer patients. J Nucl Med. (2021) 62:1097–105.
228. Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature. (2023) 618:144–50.
229. Foeng J, Comerford I, McColl SR. Harnessing the chemokine system to home CAR-T cells into solid tumors. Cell Rep Med. (2022) 3:100543. doi: 10.1016/j.xcrm.2022.100543
230. Yoon DH, Osborn MJ, Tolar J, Kim CJ. Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR-Ts): combination or built-in CAR-T. Int J Mol Sci. (2018) 19. doi: 10.3390/ijms19020340
231. Sun L, Gao F, Gao Z, Ao L, Li N, Ma S. Shed antigen-induced blocking effect on CAR-T cells targeting Glypican-3 in Hepatocellular Carcinoma. J Immunother Cancer. (2021) 9. doi: 10.1136/jitc-2020-001875
232. Schmidt MM, Thurber GM, Wittrup KD. Kinetics of anti-carcinoembryonic antigen antibody internalization: effects of affinity, bivalency, and stability. Cancer Immunol Immunother. (2008) 57:1879–90. doi: 10.1007/s00262-008-0518-1
233. Wahl AS, Correa D, Imobersteg S, Maurer MA, Kaiser J, Augath MA, et al. Targeting therapeutic antibodies to the CNS: a comparative study of intrathecal, intravenous, and subcutaneous anti-Nogo A antibody treatment after stroke in rats. Neurotherapeutics. (2020) 17:1153–9. doi: 10.1007/s13311-020-00864-z
234. Perissinotti AJ, Reeves DJ. Role of intrathecal rituximab and trastuzumab in the management of leptomeningeal carcinomatosis. Ann Pharmacother. (2010) 44:1633–40. doi: 10.1345/aph.1P197
235. Freskgård PO, Urich E. Antibody therapies in CNS diseases. Neuropharmacology. (2017) 120:38–55. doi: 10.1016/j.neuropharm.2016.03.014
236. Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv Drug Delivery Rev. (2008) 60:1421–34. doi: 10.1016/j.addr.2008.04.012
237. Pennell CA, Campbell H, Storlie MD, Bolivar-Wagers S, Osborn MJ, Refaeli Y, et al. Human CD19-specific switchable CAR T-cells are efficacious as constitutively active CAR T-cells but cause less morbidity in a mouse model of human CD19(+) Malignancy. J Immunother Cancer. (2022) 10. doi: 10.1136/jitc-2022-005934
238. van der Stegen SJC, Rivière I. Unraveling barriers to iPSC-derived CAR-T cell differentiation. Cell Stem Cell. (2023) 30:248–9. doi: 10.1016/j.stem.2023.02.004
239. Ando M, Nishimura T, Yamazaki S, Yamaguchi T, Kawana-Tachikawa A, Hayama T, et al. A safeguard system for induced pluripotent stem cell-derived rejuvenated T cell therapy. Stem Cell Rep. (2015) 5:597–608. doi: 10.1016/j.stemcr.2015.07.011
240. Dötsch S, Svec M, Schober K, Hammel M, Wanisch A, Gökmen F, et al. Long-term persistence and functionality of adoptively transferred antigen-specific T cells with genetically ablated PD-1 expression. Proc Natl Acad Sci. (2023) 120:e2200626120. doi: 10.1073/pnas.2200626120
241. Tabrizi M, Bornstein GG, Suria H. Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J. (2010) 12:33–43. doi: 10.1208/s12248-009-9157-5
242. DeRenzo C, Krenciute G, Gottschalk S. The landscape of CAR T cells beyond acute lymphoblastic leukemia for pediatric solid tumors. Am Soc Clin Oncol Educ Book. (2018), 830–7. doi: 10.1200/EDBK_200773
243. Good Z, Spiegel JY, Sahaf B, Malipatlolla MB, Ehlinger ZJ, Kurra S, et al. Post-infusion CAR TReg cells identify patients resistant to CD19-CAR therapy. Nat Med. (2022) 28:1860–71. doi: 10.1038/s41591-022-01960-7
244. Guerra E, Di Pietro R, Basile M, Trerotola M, Alberti S. Cancer-homing CAR-T cells and endogenous immune population dynamics. Int J Mol Sci. (2021) 23. doi: 10.3390/ijms23010405
245. Omilusik KD, Goldrath AW. Remembering to remember: T cell memory maintenance and plasticity. Curr Opin Immunol. (2019) 58:89–97. doi: 10.1016/j.coi.2019.04.009
246. Waldman AD, Fritz JM. & Lenardo, M.J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
247. Goodman DB, Azimi CS, Kearns K, Talbot A, Garakani K, Garcia J, et al. Pooled screening of CAR T cells identifies diverse immune signaling domains for next-generation immunotherapies. Sci Transl Med. (2022) 14:eabm1463. doi: 10.1126/scitranslmed.abm1463
248. Ghassemi S, Durgin JS, Nunez-Cruz S, Patel J, Leferovich J, Pinzone M, et al. Rapid manufacturing of non-activated potent CAR T cells. Nat Biomed Eng. (2022) 6:118–28. doi: 10.1038/s41551-021-00842-6
249. Wang V, Gauthier M, Decot V, Reppel L, Bensoussan D. Systematic review on CAR-T cell clinical trials up to 2022: academic center input. Cancers (Basel). (2023) 15. doi: 10.3390/cancers15041003
250. Heine R, Thielen FW, Koopmanschap M, Kersten MJ, Einsele H, Jaeger U, et al. Health economic aspects of chimeric antigen receptor T-cell therapies for hematological cancers: present and future. Hemasphere. (2021) 5:e524. doi: 10.1097/HS9.0000000000000524
Keywords: CAR (chimeric antigen receptor) T-cell therapy, antibody therapies, iPSC (induced pluripotent stem cell), cancer immune cell therapy, individualized cancer therapy
Citation: Schlegel LS, Werbrouck C, Boettcher M and Schlegel P (2024) Universal CAR 2.0 to overcome current limitations in CAR therapy. Front. Immunol. 15:1383894. doi: 10.3389/fimmu.2024.1383894
Received: 08 February 2024; Accepted: 03 June 2024;
Published: 19 June 2024.
Edited by:
Claudia Arndt, Helmholtz Center Dresden-Rossendorf, GermanyReviewed by:
Yaya Chu, New York Medical College, United StatesCopyright © 2024 Schlegel, Werbrouck, Boettcher and Schlegel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick Schlegel, cGF0cmljay5zY2hsZWdlbEBnbWFpbC5jb20=
†ORCID: Lara Sophie Schlegel, orcid.org/0009-0001-6726-8357
Coralie Werbrouck, orcid.org/0009-0000-4797-0619
Michael Boettcher, orcid.org/0000-0003-4972-9866
Patrick Schlegel, orcid.org/0000-0002-5425-2049
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.