 Hiam Naiditch
Hiam Naiditch Michael R. Betts2
Michael R. Betts2 H. Benjamin Larman
H. Benjamin Larman Avi Z. Rosenberg
Avi Z. Rosenberg
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 February 2025
Sec. Viral Immunology
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1376654
The emergence of the COVID-19 pandemic made it critical to understand the immune and inflammatory responses to the SARS-CoV-2 virus. It became increasingly recognized that the immune response was a key mediator of illness severity and that its mechanisms needed to be better understood. Early infection of both tissue and immune cells, such as macrophages, leading to pyroptosis-mediated inflammasome production in an organ system critical for systemic oxygenation likely plays a central role in the morbidity wrought by SARS-CoV-2. Delayed transcription of Type I and Type III interferons by SARS-CoV-2 may lead to early disinhibition of viral replication. Cytokines such as interleukin-1 (IL-1), IL-6, IL-12, and tumor necrosis factor α (TNFα), some of which may be produced through mechanisms involving nuclear factor kappa B (NF-κB), likely contribute to the hyperinflammatory state in patients with severe COVID-19. Lymphopenia, more apparent among natural killer (NK) cells, CD8+ T-cells, and B-cells, can contribute to disease severity and may reflect direct cytopathic effects of SARS-CoV-2 or end-organ sequestration. Direct infection and immune activation of endothelial cells by SARS-CoV-2 may be a critical mechanism through which end-organ systems are impacted. In this context, endovascular neutrophil extracellular trap (NET) formation and microthrombi development can be seen in the lungs and other critical organs throughout the body, such as the heart, gut, and brain. The kidney may be among the most impacted extrapulmonary organ by SARS-CoV-2 infection owing to a high concentration of ACE2 and exposure to systemic SARS-CoV-2. In the kidney, acute tubular injury, early myofibroblast activation, and collapsing glomerulopathy in select populations likely account for COVID-19-related AKI and CKD development. The development of COVID-19-associated nephropathy (COVAN), in particular, may be mediated through IL-6 and signal transducer and activator of transcription 3 (STAT3) signaling, suggesting a direct connection between the COVID-19-related immune response and the development of chronic disease. Chronic manifestations of COVID-19 also include systemic conditions like Multisystem Inflammatory Syndrome in Children (MIS-C) and Adults (MIS-A) and post-acute sequelae of COVID-19 (PASC), which may reflect a spectrum of clinical presentations of persistent immune dysregulation. The lessons learned and those undergoing continued study likely have broad implications for understanding viral infections’ immunologic and inflammatory consequences beyond coronaviruses.
On March 11, 2020, as healthcare systems worldwide saw rising SARS-CoV-2 cases, COVID-19 was declared a pandemic (1). The infectivity of the virus and the severity of COVID-19 in many patients led to the accelerated study of the disease (2). It became increasingly apparent that the immune milieu generated by SARS-CoV-2 was unique, even among similar coronaviruses (3). Peripheral lymphopenia, elevated inflammatory markers (4), endothelial damage, and microthrombosis characterized some early findings in COVID-19 (5). In the years that followed, the immune response to COVID-19 has been further elucidated, allowing for the identification of effective vaccines (6, 7), monoclonal antibodies (8), and other therapies (9, 10). The emergence of syndromes associated with post-infectious immune dysregulation such as Multisystem Inflammatory Syndrome in children (MIS-C) (11, 12) or adults (MIS-A) (13, 14) further propelled the study of the disease. A compelling association with autoimmune disease, perhaps related to the development of autoantibodies in the presence of SARS-CoV-2-mediated pyroptosis, further exemplified the complex interplay between the immunologic response in COVID-19 and chronic disease (15).
Among the organ systems studied, a high concentration of ACE2 in a highly vascular structure readily exposed to systemic pathogens highlights the human kidney as a unique model for the systemic effects of SARS-CoV-2 (16). Prior to infection, there is a paucity of immune cells in the human kidney; most of these are CD4+ and CD8+ T cells, with a smaller percentage of NK cells, B cells (17), and CD14+, CD16+ and CD68+ myeloid cells (18). Following infection with SARS-CoV-2, an upregulation in proinflammatory genes such as HSPA1A in podocytes and JUN1 in mesenchymal clusters (19) can accompany selective immune suppression of lymphocytes mediated through T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) and Programmed cell death protein 1 (PD-1) (20). Resident immune cells may mediate inflammation by TNFα release, IL-34-mediated necrosis, and NLRP3 inflammasome production (266). Myeloid cell activation is a hallmark of COVID-19 and is associated with immune dysregulation in COVID-19, particularly in severe disease (21). Moreover, early post-mortem studies revealed compelling evidence for direct infection of predominantly ACE2-positive renal tubular cells by SARS-CoV-2 (16). Intriguingly, the upregulation of TGF-β, PI3K/Akt, MAPK, and WNT signaling can be associated with tubule interstitial fibrosis and may also point to a mechanism of COVID-19-related CKD (19).
The following represents a comprehensive review of the immunologic and inflammatory consequences of SARS-CoV-2 infection, synthesizing the molecular immune response to the acute and chronic end-organ dysfunction in both acute and chronic forms of COVID-19. Among the organ systems studied, particular attention is paid to the kidney and recently described pathophysiology in COVID-19.
SARS-CoV-2 infection occurs predominantly in the upper respiratory tract via ACE2 and TMPRSS2.
Coronaviruses likely originated in bats and rodents and eventually evolved to affect other animals and humans . Their pathogenicity was not fully appreciated until 2002, when the sudden acute respiratory syndrome (SARS) broke out in Guangdong Province, China (22). Since that time, coronaviruses have been implicated in Middle East respiratory syndrome (MERS) and other less overtly severe clinical entities (22). Approximately seven years after MERS, SARS-CoV-2 emerged as a public health threat, spurring research into its structure, infectivity, and the immune response it provokes.
SARS-CoV-2 is a betacoronavirus with an envelope containing an envelope (E) protein, a membrane (M) protein, and a spike (S) protein interspersed within a lipid membrane (5). The nucleus contains a positive-sense single-stranded RNA molecule bound to the nucleocapsid (N) protein (23). The bulk of the ssRNA has two open reading frames at the 5’ end, ORF1a and ORF1b, which are the transcriptional precursors of the viral replication and transcription complex (RTC) (23). SARS-CoV-2 is transmitted by respiratory and aerosolized droplets from infected individuals actively shedding the virus (24–26). Virions can contact areas of high angiotensin-converting enzyme 2 (ACE2) and transmembrane protease serine 2 (TMPRSS2) co-expression, namely in goblet secretory cells in the nasopharynx, epithelial cells of the oral mucosa, airway, and alveoli including Type II pneumocytes, and lung macrophages (27, 28). Viral entry into host cells is facilitated by binding the S protein to ACE2, followed by proteolytic cleavage at the S1/S2 and S’ sites by TMPRSS2, a process that allows for virus-host membrane fusion (29). Expression of ACE2 and TMPRSS2 has also been noted in endothelial cells, enterocytes (27), and podocytes in the kidney glomerulus (5).
The importance of ACE2 has been confirmed through GWAS identification of variants (such as X-linked rs190509934) associated with reduced ACE2 expression and reduced likelihood of infection with SARS-CoV-2 (30, 31). Likewise, increased susceptibility to infection is associated with SLC6A20 on chromosome 3p21.1, which encodes a protein [sodium-imino acid transporter 1 (SIT1)] that is associated with ACE2 (30, 31). Dipeptidyl peptidase-4 (DPP-4) has been identified as another possible candidate for SARS-CoV-2 binding in silico, albeit of unclear clinical significance (32–36). In addition to TMPRSS2, proteolysis can also be performed by endosomal cathepsin B (catB) and cathepsin L (catL) (23). Following fusion, the release and subsequent translation of ORF1a and ORF1b lead to the production of polypeptides pp1a and pp1ab, which are eventually processed into 16 non-structural proteins that comprise the viral RTC (23) (Figures 1A, B). Structural proteins are encoded from interspersed ORFs at the 3’ end of the ssRNA and are eventually processed in the endoplasmic reticulum and Golgi apparatus with modifications including N- and O-glycosylation, which are thought to be critical to virion infectivity (23, 37). Among the proteins produced during acute infection are replicase proteins, which help form replication complexes within the endoplasmic reticulum (38).
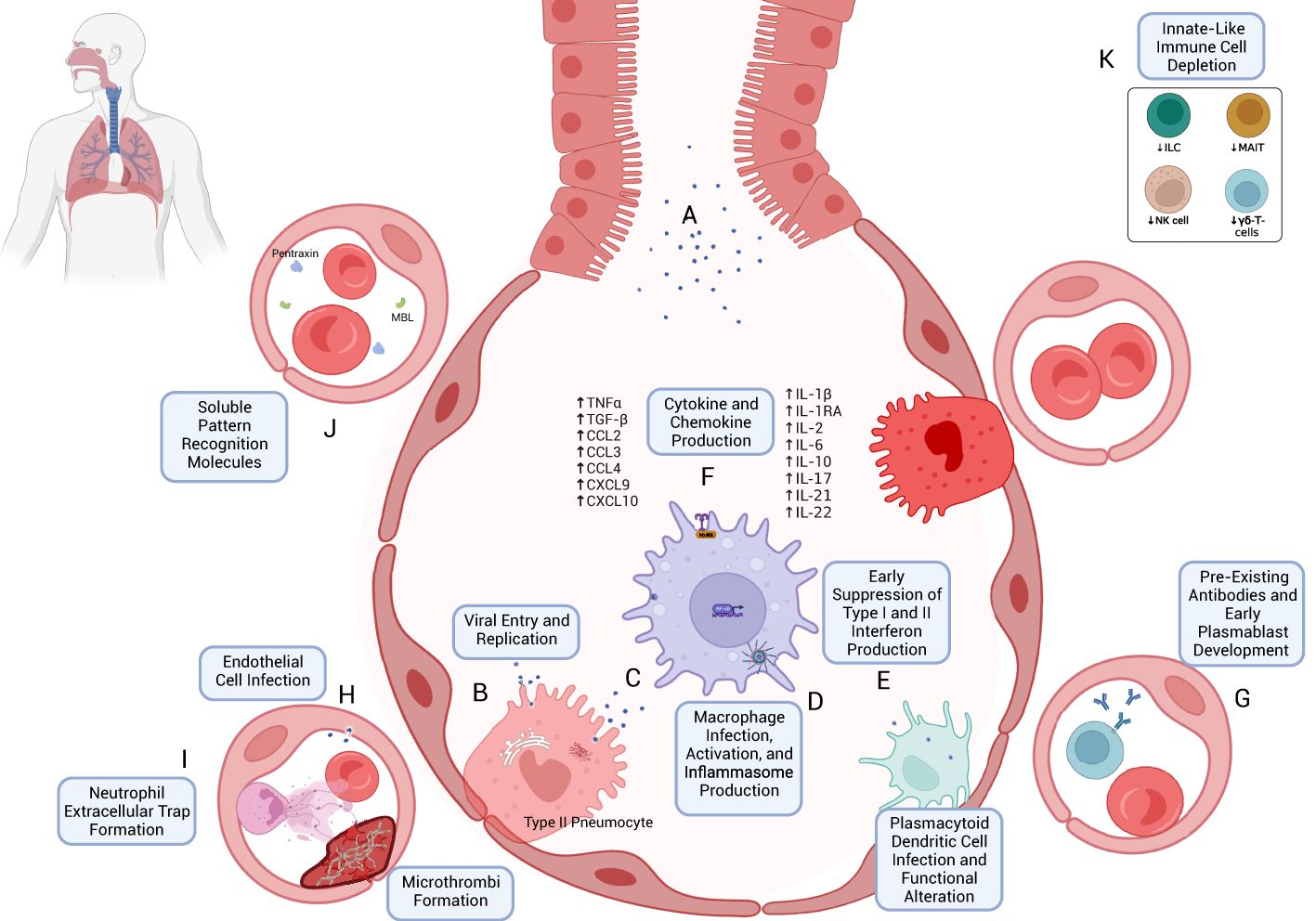
Figure 1. Early immune response to SARS-CoV-2 infection. (A) SARS-CoV-2 virions are inhaled through respiratory and aerosolized droplets (24–26). Infection of susceptible cell types, particularly those bearing ACE2 and TMPRSS, such as Type II pneumocytes, can occur (27, 28). (B) Viral entry can occur through mechanisms including host-membrane fusion (29). Entry is followed by the release and translation of ORF1a and ORF1b of SARS-CoV-2 ssRNA, formation of the viral replication and transcription complex (23), and production of new SARS-CoV-2 virions, which are eventually exocytosed. (C) Resident macrophages are likely among the first immune cells to encounter SARS-CoV-2 through both direct infection and indirect immune activation. As with other cells, TLR- and RLR-mediated recognition are accompanied by downstream effects through Myd88 activation, TRIF binding (43, 45, 50), MAVS activation (45), and NF-κB activation (45, 52). (D) Production of Type I and Type III interferons appears to be blunted in the early immune response (42, 89) despite their antiviral potential. (E) Infection and activation with SARS-CoV-2 may induce inflammasome production and macrophage pyroptosis, which may be a key early driver of a heightened immune response following SARS-CoV-2 infection (57). (F) One such mechanism is through cytokine and chemokine release (70, 103, 104), including TNFα, TGF-β, CCL2/3, CXCL9/10, IL-1β, IL-2, IL-6, IL-10, IL-17, IL-21, and IL-22 (see text for references). (G) The early immune response may also be mediated by pre-existing cross-reactive antibodies (174–177) and plasmablast development (173). (H) Endothelial cell infection may also occur (207); endothelial cell activation may be associated with a hypercoagulable state (210, 211). (I) Early neutrophil responses include the production of immature neutrophils through emergency myelopoiesis (115, 400) and neutrophil extracellular trap (NET) formation, which may also contribute to microvascular thrombosis (111, 117). (J) Soluble pattern recognition molecules such as mannose-binding lectin (MBL) and pentraxin three can bind to SARS-CoV-2 spike and nucleocapsid proteins, respectively (96). (K) Lymphopenia, which often accompanies COVID-19 (408), can include the peripheral depletion of cell types such as Innate Lymphoid Cells (ILC) (126, 127), MAIT (127), NK-cells (124, 398), and γδ-T-cells (136).
One mechanism of protection from pattern recognition receptors (PRRs), such as melanoma differentiation-associated protein 5 (MDA5), includes the formation of double-membrane vesicles (38). Relatively unchecked viral replication and an immune response that can be in disarray in severe disease can lead to the eventual dissemination through deeper inhalation of upper respiratory virus-containing particles or contiguous spread from the site of infection (38). Further dissemination of SARS-CoV-2 to other organ systems can be mediated through viremia, which is associated with adverse clinical outcomes (39). SARS-CoV-2 can evolve within a host to develop enhanced infectivity, immune evasion, and transmissibility, all of which can translate to pathogenicity and disease severity on the individual and population levels (40). A mutation rate of approximately 1 × 10–6 to 2 × 10–6 mutations per nucleotide per replication cycle is one characteristic of SARS-CoV-2 evolution (40). Selection pressures such as innate immune defense mechanisms like those mediated by the apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) family provoke the formation of C->U mutations, which can alter phenotypic properties of the virus , which can, in turn, lead to the development of variants of concern (VOC). Transmissibility resulting from molecular evolution represents a bottleneck for mutations to pass from one host to another and in turn the likelihood of a virus like SARS-CoV-2 to cause an epidemic or pandemic (40). As a virus enters and uses the host cell for its own propagation, a response is elicited in the human host to fight for its own preservation. In the subsequent sections, that response—characterized by distinct but often overlapping cellular and extracellular mechanisms that accompany each stage of infection with SARS-CoV-2—will be elucidated.
The innate immune response to SARS-CoV-2 is the first line of defense against the virus (41) and also represents a crossroad at which immune cell recruitment and coordination can be pathologically skewed. Inflammatory cytokine disarray with concomitantly delayed IFN-I and IFN-III responses may be primary mediators of immune dysregulation in severe disease (42). Dysregulated innate immune responses comprise one of the critical features of COVID-19 (42).
Early pattern recognition receptor and Toll-like receptor activation in SARS-CoV-2.
In normal physiologic conditions, the entry of viral RNA into a host cell does not go unnoticed. The newly released viral ssRNA contains pathogen-associated molecular patterns (PAMPs) recognized as foreign entities by pattern recognition receptors (PRRs) in the cytoplasm and along the cell membrane and endosomal surfaces (43–45). PRRs include cytoplasmic retinoic acid-inducible gene I-like receptors (RLRs) and membrane-bound Toll-like receptors (TLRs), with subclasses that recognize a wide array of viral nucleic acid assortments (44, 45). As a positive-sense ssRNA, the viral RNA from SARS-CoV-2 is recognized by the RLRs retinoic acid-inducible gene (RIG-I) and MDA5 by its uncapped 5’ triphosphate terminus (43). The activation of RIG-I and MDA5 and their association with mitochondrial antiviral signaling protein (MAVS) eventually leads to the transcription of type I interferons, which are likely dependent on nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) (45).
Positive-sense ssRNA also activates endosomal TLR7 (as well as TLR8), which, in conjunction with myeloid differentiation primary-response gene 88 (MyD88), typically leads to the production of both type I and type III interferons (IFNs) (46, 47). The TLR7/MyD88 pathway mediates immunity in plasmacytoid dendritic cells (pDCs) in multiple viral infections and is also likely relevant in airway epithelial cells infected with SARS-CoV-2 (48, 49). Furthermore, the severity of illness in young male patients with loss-of-function variants in the X-linked TLR7, as well as the associated defects in type I and type II interferon production, suggest a critical role of this pathway in the innate response to SARS-CoV-2 infection (46). Endosomal TLR3 may also be activated in the presence of SARS-CoV-2, perhaps due to transient dsRNA produced in the viral replication process (43, 49). TLR3 binds to TIR-domain-containing adapter-inducing interferon-β (TRIF), which also can lead to type I and type III interferon production via IFN regulatory factor 3 (IRF3) as well as robust NF-κB activation (43, 45, 50). MyD88 activation can polarize macrophages towards a pro-inflammatory M1 phenotype in mouse models (51). In addition to the above mechanisms, human airway epithelial cells infected with SARS-CoV-2 have also been shown to have decreased transcription of dual-specificity phosphatase 1 and 5 (DUSP1 and DUSP5), which may lead to unbalanced activation of NF-κB (52).
SARS-CoV-2 provokes the release of pro-inflammatory cytokines via NF-κB, inflammasome production, and pyroptosis.
Cytopathic viral infection of airway epithelial cells can trigger inflammasome activation, pyroptosis, necroptosis, and apoptosis (28, 53–55). Early studies during the pandemic revealed intense NLRP3 inflammasome expression in the lungs of patients with fatal COVID-19 (56), suggesting a pivotal role for pyroptosis in severe COVID-19 (57). SARS-CoV-2 may promote pyroptosis through non-structural protein 6 (nsp6) (58). The subsequent release of damage-associated molecular patterns (DAMPs) and activation of IL-1β may be one of the first events leading to the inflammatory cascade that characterizes COVID-19 (28). The release of DAMPs and IL-1β promotes the release of additional inflammatory cytokines such as macrophage inflammatory protein 1α (MIP1α), MIP1β, IL-6, and interferon gamma-induced protein 10 (IP-10) from adjacent alveolar macrophages, epithelial cells, and endothelial cells (28, 59). TGF-β production may also be enhanced in severe disease (60).
An essential function of ACE2 is to degrade angiotensin II (Ang II) into Ang- (1, 7, 61). As SARS-CoV-2 binds to ACE2 and the complex is endocytosed or incorporated through other mechanisms (62), less ACE2 is available to perform its typical functions, and levels of Ang II rise. The increasing levels of Ang II provide excess stimulation to the Ang II Type 1 receptor (AT1R), which—in addition to vasoconstriction—promotes the activation of NF-ĸβ through ADAM17 cleavage of TNFα, membrane-bound EGF (63), and IL-6Rα into their soluble forms (61). Subsequent activation of TNFαR and EGFR also leads to downstream NF-κB activation (61, 63). The activation of gp130 by the IL6R-α-IL-6 complex further activates a JAK/STAT pathway, amplifying the NF-κB response via STAT3 (63).
NF-κB plays a pivotal role in the upregulation of the immune response (64). It has been hypothesized that the benefits of corticosteroids such as dexamethasone may be partially due to the downregulation of NF-κB pathways (65). In airway epithelial cells, NF-κB promotes the production of IL-6, G-CSF, GM-CSF, and MIP2 kβ (66). In M1 alveolar macrophages, NF-κB promotes the production of an array of inflammatory cytokines, including IL-1β, IL-6, IL-12, and TNFα (64). NF-κB in SARS-CoV-2-infected hosts has also been shown to trigger NLRP3 (57) inflammasome formation and pyroptosis in monocytes through a caspase-1 mechanism with resultant production of DAMPs and cytokines such as IL-1β (67, 68). DAMP-mediated signaling itself leads to a conformational change in NLRP3 and caspase-1-mediated cleavage of the inhibitory C-terminus of gasdermin D, which is the main effector of pyroptosis (57). Gasdermin D can then traffic to the cell membrane and polymerize into β-barrel pores, promoting ion (namely, Ca2+) and protein flux and the eventual loss of the mitochondrial membrane potential and IL-1 and IL-18 activation and IL-1β release (57).
Other cytokines include IL-2, IL-17, IL-8, macrophage inflammatory protein 1 (MIP-1), monocyte chemoattractant protein 1 (MCP-1), and chemokine ligands (e.g. CCL2, CCL3, and CCL7) including C-X-C motif chemokine ligands (e.g. CXCL2 and CXCL10) (65). The clinical significance of cytokine release was demonstrated in a 2021 single-cell and bulk RNA-seq analysis of specimens from patients with COVID-19, which showed that upregulation of IL-2, IL-6, IL-8, IL-17A, and NF-κB was correlated with the severity of COVID-19 (69). The function of several such cytokines and chemokines in COVID-19 are summarized in Table 1. The release of cytokines in COVID-19 is associated with a hyperinflammatory state, leading to what some have called a “cytokine storm” that characterizes severe COVID-19 and its associated end-organ dysfunction syndromes (61, 63), similar to that seen in other hyperimmune responses characterized by cytokine release syndromes (CRS) related to chimeric antigen receptor-modified T (CAR-T) cell therapy, macrophage activation syndrome (MAS), and hemophagocytic lymphohistiocytosis (HLH) (70, 71).

Table 1. Cytokine and chemokine response in COVID-19.
Hyperinflammatory immune responses are not necessarily unique to SARS-CoV-2 (72, 73): High levels of IL-6 have been noted in patients with acute respiratory distress (ARDS) before the COVID-19 pandemic (74). Others have suggested that lymphocyte depletion and exhaustion, delayed interferon response, and decreased TNFα production in certain phases of the disease are indicators of immune suppression (75). Although a more precise depiction of the cytokine-mediated inflammatory response in COVID-19 may be suggested by the term “cytokine disharmony” or other terms denoting dysregulation, by and large, the immune response in severe forms of COVID-19 appears to skew towards a hyperinflammatory cytokine dysregulation (76–81), with early outcomes in severe disease perhaps attributable to cytokines preferentially elevated in SARS-CoV-2 (73, 82), interferon I dysregulation (83), and local microthrombi formation (74, 84), as will be discussed in further detail.
Interferon release is suppressed early in the course of SARS-CoV-2 infection.
Interferons are integral to the antiviral immune armamentarium and have been shown to have potent anti-SARS-CoV-2 activity (85). Critical illness in COVID-19 is associated with inborn errors of genes involved in Type I interferon production (86). While IFN-α and IFN-β can contribute to inflammation in SARS-CoV-2 through IFNR-mediated downstream activation of NF-κB, the simultaneous stimulation of interferon-stimulated genes (ISGs) can promote the development of a potent antiviral state (87, 88). First, IFN acts to increase PRRs, which can improve viral detection (88). In tandem, many proteins are produced via ISGs, which can inhibit nearly every step in viral infection and replication (88).
Despite their seemingly potent anti-SARS-CoV-2 activity, the transcription of crucial interferon genes for type I and type III interferons (IFNB, IFNK, IFNA5, and IFNL1-5) is suppressed early in the course of SARS-CoV-2 infection, particularly in severe disease (42, 89). Mechanisms may include suppression of sequential steps along the pathway of ISG production, similar to that mediated by other coronaviruses such as SARS-CoV-1 [mediated in part by Nsp1 (90)] and MERS-CoV in humans and murine hepatitis virus (MHV) in mice (91–93). This phenomenon, among others, may help to explain the pathogenicity and associated immune dysregulation of SARS-CoV-2 (85, 89, 94, 95).
Soluble pattern recognition molecules (PRMs) have antibody-like functions representing a key component of humoral immunity (96). These include collectins (e.g., mannose-binding lectin (MBL), which activates the complement system via the lectin pathway), pentraxins [e.g., C-reactive protein (CRP)], C1q, and ficolins (96). Systemic complement activation was upregulated in patients with COVID-19, with sC5b-9 and C4d found to be higher in those patients with respiratory failure (97). A 2022 study revealed that MBL and pentraxin 3 (PTX3) play critical roles in humoral immunity against SARS-CoV-2, with MBL binding to spike (S) protein in a glycan-dependent manner prompting complement activation via the lectin pathway and with PTX3 binding to nucleocapsid (96) (Figure 1J). Furthermore, single nucleotide polymorphisms (SNPs) of the MBL2 gene, particularly rs10824845, suggest an association with the severity of COVID-19 (96). Interestingly, despite its apparent association with clinical outcomes, CRP was not shown to bind to any of the SARS-CoV-2 proteins tested, which included S, N, and E (96, 98).
Monocytes and macrophages play a prominent role in the dysregulated immune response to SARS-CoV-2 (71). Macrophages express ACE2 receptors and represent targets for SARS-CoV-2 viral entry [which may also occur via the Fc receptor CD16 (99)], viral replication (99) and spread (100, 101), and associated immune responses (102). Responsiveness to TLR associated with suppressing IL-1R-associated kinase (IRAK)-M expression points to hyperinflammatory phenotype (102). Macrophages mediate cytokine release in COVID-19 through STAT1 signaling and pDC activation and may be associated with a dampened IFN-I response (103, 104). Once activated during SARS-CoV-2 infection, macrophages showed signs of inflammasome activation as evidenced by an associated speck-like protein containing a CARD (ASC) co-localization with NLR family pyrin domain containing 3 (NLRP3) and activated caspase-1, as shown in a recently published in vivo murine model with a humanized immune system (99). IL-18 and IL-1RA (considered downstream evidence of inflammasome activation) were upregulated (with IL-1β also evident in vitro), as was chemokine CXCL10 expression (99). Finally, pyroptosis was also increased in this model as measured by LDH and gasdermin D levels (99). Similar results were seen in in vitro studies of monocytes (67). IL-6 further promotes the differentiation of monocytes to macrophages (70). Perhaps owing to the ubiquity of macrophages in tissues (105), macrophage-induced inflammation—mediated in part by direct viral infection, inflammasome activation, pyroptosis (57), and cytokine release (70, 103, 104)—appears to be a prominent driver of the SARS-CoV-2-mediated immune response (101, 106) (Figures 1C–F).
Plasmacytoid dendritic cells (pDCs) help drive the immune response to viral infections such as coronaviruses (107). pDCs are more likely to be directly infected with SARS-CoV-2 than macrophages (104), and emerging evidence has shown that pDCs are the predominant cell type involved in the production of Type I and Type III interferons (108). COVID-19 severity is inversely correlated with a pDC-driven IFNα response (108). The presence of pDCs was seen in the lungs of patients with SARS-CoV-2 and was associated with IFN-I signaling, itself correlated with macrophage inflammatory response (104). pDCs have been shown to enhance TLR signaling in macrophages via IFN-1; RNAseq studies have additionally shown mediation of transcriptional changes in macrophages that correlate with a more robust inflammatory response to SARS-CoV-2 than to LPS (104). A 2021 study showed that both myeloid and plasmacytoid dendritic cells show an alteration in DC homing and activation markers such as PDL-1, CD86, and CCR7 that correlated with hospitalization status (109). Given the central importance of macrophages in COVID-19, the mechanistic pathways involving pDCs are likely a critical first step in developing the hyperinflammatory cytokine response accompanying macrophage activation in severe disease. Functional alterations of DCs and deficiency of pDCs (as well as CD1c+ mDCs) may persist seven months after acute SARS-CoV-2 infection (109).
In addition to their overt role in bacterial and fungal infections, neutrophils can contribute to the immune response to viruses (110). They may additionally exhibit enhanced trafficking to highly vascularized organs such as the lungs and kidneys (111). Neutrophilia (112) and a high neutrophil-to-lymphocyte ratio are associated with severe COVID-19 (113, 114). Early studies using single-cell transcriptomics and proteomics from peripheral blood mononuclear cells (PBMC) of patients with COVID-19 showed altered myelopoiesis in patients with severe disease (115). Immature neutrophils expressing CD24, DEFA3, DEFA4, and PGLYRP1 were a prominent cell population in that study (115). Another study showed a reduction in the neutrophil maturation marker CD-10, associated with emergency myelopoiesis (400) and poor clinical outcomes (116). Among the mechanisms studied, the formation of neutrophil extracellular traps (NETs) (aggregates of extracellular DNA, histones, neutrophil elastase, myeloperoxidase, and other molecules such as tissue factor) (111) are thought to play a prominent role in COVID-19 pathogenesis, mediated in part through target-organ microvascular occlusion (111) and associated immunothrombosis (111, 117) (Figure 1I).
Acknowledging the innate-adaptive overlap exhibited by natural killer (NK) cells (118), NK cells play an essential role in innate antiviral responses (119, 120). While an increased frequency of activated NKG2C+CD57+ CD56dim NK cell phenotypes may be seen in severe COVID-19 (121), severe infection is associated with peripheral NK cell depletion and an exhausted phenotype based on LAG3, PDCD1, and HAVCR2 expression, which may be mediated by aberrant TGF-β production and homing to lung tissue (121, 122). NK cell quiescence manifesting as decreased perforin and granzyme production has also been demonstrated in COVID-19. This may be mediated by increased IL-6 (123) and TNFα, both part of the cytokine milieu characterizing the syndrome (124).
Innate lymphoid cells (ILC) are more recently described subsets of lymphocytes that can participate in innate immune responses that mirror type 1, type 2, and type 3 immune responses (125). Studies have shown that illness severity in COVID-19 is inversely associated with ILC count (126, 127). ILC-mediated amphiregulin production has been proposed as one mechanism of immune tolerance (126). Of note, a proportionally higher number of amphiregulin-producing ILC was seen in healthy controls vs patients hospitalized with COVID-19 and in females versus males (126).
Mucosal-associated invariant T-cells (MAIT) are innate-like T cells that recognize non-specific riboflavin metabolites of bacteria at mucosal surfaces, including those lining the bronchial tree, which are also activated during viral infections (128, 129)and can inhibit viral replication (130). While the specificity and significance of this cell type in COVID-19 is still under study, heightened activation of circulating CD8+ MAIT cells (127, 128) may correlate with illness severity as measured by Simplified Acute Physiology (SAPS) II score (128).
γδ-T-cells are another subset of T-lymphocytes with innate-like functionality with the potential to induce strong antiviral responses independent of MHC-mediated antigen presentation (122, 131, 132). Their use in cancer immunotherapy (122, 133) and presence at mucosal barrier sites (134) stimulated interest in their role in COVID-19 (133, 134). In SARS-CoV-1, they appear to be active members of the immune milieu (133, 135). However, their role in SARS-CoV-2 appears to be muted (131) compared to SARS-CoV-1120. In addition to their peripheral depletion, as with other T-cell subsets (136), γδ-T cells could not mount a potent immune response to spike or nucleocapsid antigen in vitro studies (131). The depletion of various innate-like lymphocyte subtypes is portrayed as a component of the early immune response to SARS-CoV-2 in Figure 1K.
Components of the innate immune response to SARS-CoV-2 infection can help in the immediacy of their response while simultaneously perpetuating detrimental non-specific inflammatory signaling. The adaptive immune response generally follows the innate immune response and provides an additional layer of nuanced and potentially potent protection in patients with and recovering from COVID-19. In addition, the memory adaptive response induced by both natural infection and vaccination, as discussed below, plays a crucial role in protection from reinfection, limiting disease severity and hastening resolution in the event of reinfection.
The adaptive immune response plays an important role in viral infections, primarily consisting of CD4+ T-cells, CD8+ T-cells, and B-cells (137). Despite this role, lymphopenia is a hallmark of COVID-19 and may correlate with disease severity and severe lung injury (127, 138, 139) (Figure 2A). In addition to the direct cytopathic effects of SARS-CoV-2 (140), early studies showed significant damage to lymphoid tissue and lymphocyte apoptosis in the lymph nodes and spleen of deceased patients with COVID-19 (141). Additional mechanisms may involve sequestration or recruitment of lymphocytes to actively inflamed tissues such as the lung and other target organs (127, 142). Lymphopenia also appears to be inversely related to levels of IL-6 and therapeutically targeted IL-6 inhibition can lead to the correction of circulating lymphocyte counts (143, 144).
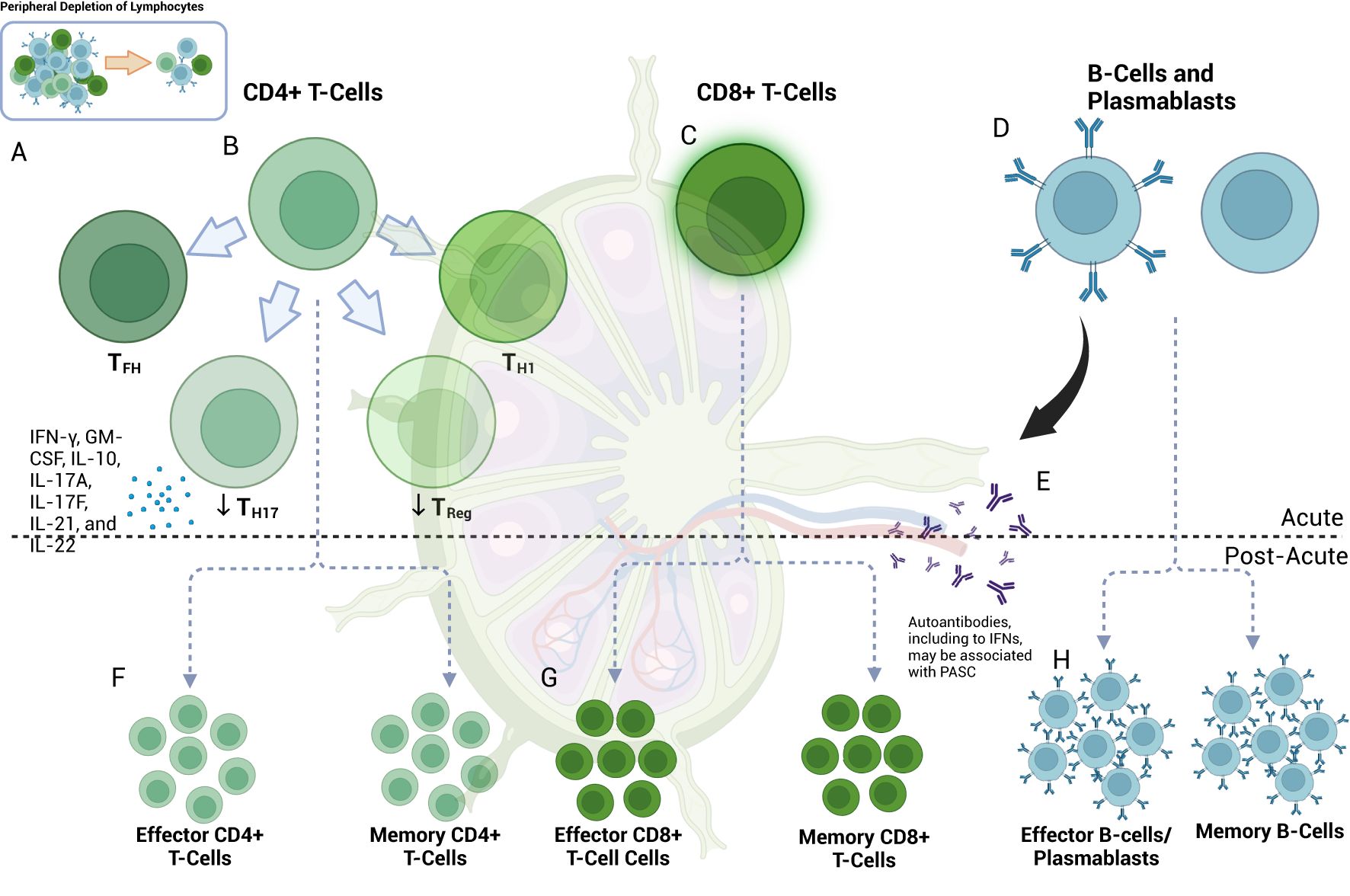
Figure 2. Lymphocyte response in acute and post-acute SARS-COV-2. (A) Lymphopenia is a hallmark of COVID-19 (408). (B) Among the cell types present, CD4+ T-cells skew towards TH1 and TFH phenotypes with associated effector functions (145, 151); germinal center TFH may be decreased (151). Upregulation of TH17-cells can occur [in contrast to Treg (156)] and may be accompanied by the pro-inflammatory release of IFN-γ, GM-CSF, IL-10, IL-17A, IL-17F, IL-21, and IL-22 (155). (C) CD8+ cells exhibit activated phenotypes with preserved effector function and a potential role in macrophage-mediated cytokine release in acute COVID-19 (137, 161, 163) despite showing peripheral depletion (139, 144, 159, 160). (D) B-cells are also peripherally depleted in COVID-19 (173, 181). Plasmablast development occurs at first in extrafollicular zones (173), initially with less sophisticated targeting (173, 181) via DN2 and DN3 B-cells (181), but later [at approximately seven days post-infection (181)] with more robust class-switching, somatic hypermutation and affinity maturation (173) from germinal center (GC) cells. (E) Autoantibody development, including those directed towards interferons, can develop at this time (309) and may later be associated with PASC (285). (F) In the post-acute phase, effector CD4+ T-cell subtypes may remain active for at least 2-3 months (285). Decreased levels of naïve CD4+ T-cells suggest ongoing activation (301). Memory T-cells (which can develop early in the course of infection) can include those of the CD4+ and CD8+ T-cells and subtypes (165, 406), and may be detected at least one year post-infection (165). S-specific CXCR3-/CXCR5-/CCR6+ TH17-cells can have a half-life of 4.9 years (405). Memory T-cells may be important drivers of vaccine-mediated immunity (194, 195). (G) CD8+ T-cells may continue to exhibit markers of activation and exhaustion (PD-1 and TIM-3) at three months, particularly in patients with PASC, and have been observed to persist for up to 8 months (301). Memory CD8+ T-cells may have a half-life of one year (405); this can vary by disease severity (405). (H) Depletion of naïve B-cells suggests ongoing activation at eight months (301). Memory B-cell (MBC) frequency may correlate with decreased symptom duration (403) and may also have a role in vaccine-mediate immunity, even as antibody levels subside (192). MBC associated with autoantibody or SARS-CoV-2-directed antibody production may also be associated with varying PASC symptomatology (285). Of note, autoantibody production may occur early and may precede COVID-19 diagnosis (285).
CD4+ T-cells are integral to developing a targeted adaptive immune response by recognizing foreign antigens on MHC Class II molecules and activating other immune cells, including CD8+ T-cells, B-cells, and NK cells (145). Specific viral targets of CD4+ T-cells include M, N, and S, nsp3 and ORF3, and others, albeit to a smaller degree (137, 146). Tan et al. and others showed that a robust CD4+ T-cell response is important for viral clearance in SARS-CoV-2 (147) and that the relative absence of such a response could be fatal (148). Early studies showed a more prominent CD4+ T-cell response than CD8+ T-cells (137), with increased expression of CD38 in moderate-to-severe disease indicative of an activated phenotype (149). An increased proportion of cytotoxic CD4+ T helper (CTL-TH) cells and follicular TFH-cells (which have a role in B-cell affinity maturation and antibody production) were seen in hospitalized patients with COVID-19 relative to regulatory T-cell (Treg) populations (145). Curiously, another study showed a positive correlation of specifically CD8+ Treg cells with increased CD4+ cells in patients with acute SARS-CoV-2 infection (150), a finding potentially indicative of a compensatory anti-inflammatory program (89).
As noted, depletion of T-cells, particularly in the peripheral blood, is a hallmark of severe COVID-19 (144, 149). T-cells exhibit enhanced expression of apoptotic pathways and T-cell immunoglobulin and mucin-domain containing-3 (Tim-3) and programmed cell death protein 1 (PD-1), which—while expressed in activated states—can also be markers of T-cell exhaustion (139). In addition, germinal centers—which are important sites of T-cell and B-cell interaction—are lost in thoracic lymph nodes of patients with acute COVID-19, with an associated loss of germinal center CD4+ Bcl-6+ TFH-cells and an increased proportion of TH1-cells (151), which are responsible for IFNγ production. Bcl-6 is a transcriptional repressor that can allow for the development of high-affinity immunoglobulins in both T and B-cells (152). Taken together, T-cell lymphopenia, exhaustion, and associated immune sequelae likely contribute to the immune dysregulation in COVID-19 (Figure 2B).
TH17 cells are a distinct lineage of CD4+ T-cells that are typically involved in the clearance of extracellular pathogens (153). Their differentiation is mediated through TGF-β and IL-6 (153), both of which are upregulated in severe COVID-19 (60, 154). TH17 cells may contribute to acute inflammation in COVID-19 through the release of IFN-γ, GM-CSF, IL-10, IL-17A, IL-17F, IL-21, and IL-22 (155) (Figure 2B). An increased TH17/Treg ratio and their associated factors (e.g., RAR-related orphan receptor gamma (RORγt)/Forkhead box protein P3 (FoxP3) appear to correlate with SARS-CoV-2 infection as well as clinical outcome (156). As will be discussed, TH17 cells have been implicated in chronic autoimmune and inflammatory conditions and thus may represent an important link between acute and chronic immunity in patients with COVID-19 (153).
CD8+ T-cells target cells infected with intracellular pathogens, including viruses such as SARS-CoV-2, by detecting foreign antigens on MHC-1 (137, 157). CD8+ T-cells can target numerous proteins derived from SARS-CoV-2, depending on expressed HLA class I types (146). While widely viewed as critical for disease resolution and subsequent protection from re-infection (137), SARS-CoV-2-specific CD8+ T cell responses in the blood tend to be relatively low in frequency (compared to influenza or Epstein-Barr virus) in most infected or resolved individuals (158). Several studies have confirmed a decrease in circulating CD8+ T-cells in COVID-19 (139, 144, 159, 160). Those CD8+ T-cells that were present were found to be highly activated in a subset of COVID-19 patients as measured by KI67 and HLA-DR+CD38 expression (161) and appear to retain their cytotoxic capacity as measured by IFNγ, granzyme B, perforin, and CD107a production or expression (137) (Figure 2C). In addition, pre-existing cross-reactive nucleocapsid-specific memory CD8+ T-cells that recognize seasonal coronaviruses (162) may provide some level of protection from primary SARS-CoV-2 infection or disease severity, depending on the specificity and HLA haplotype. CD8+ T-cells have also been implicated in non-COVID cytokine release syndromes such as macrophage activation syndrome (MAS) (163) and may play a similar role in the exacerbated immune response in COVID-19.
Memory T-cells can recognize and aid in clearing viruses to which a host may be re-exposed (164). Given the novelty of SARS-CoV-2, the duration of detectable memory T-cells responsive to SARS-CoV-2 remains the subject of ongoing study. Highly reactive, polyclonal CD8+ T-cells have been detected in the serum of patients with a history of mild COVID-19 up to one year after infection (165), with recent studies supporting at least this duration for SARS-CoV-2-specific CD8+ T-cells expressing CD45RA, IL-7R-α, T cell factor 1, and low CCR7 (166). This phenotype suggests tissue recirculating effector memory CD8+ T cells that retain high proliferative potential yet traffic through lymphoid and non-lymphoid tissues (167). Of note, memory T-cells have been detected in patients with SARS-CoV-1 beyond 17 years (164). Importantly, SARS-CoV-2-specific stem-cell-like memory T-cell populations appear to be maintained regardless of disease severity (164) (Figures 2F, G). While the clinical significance of memory cell phenotypes remains to be determined, particularly in the context of viral variants (89), evidence suggests that CD8+ T-cells generated through natural immunity (168) or vaccination (169) can cross-recognize viral antigens despite a significant number of mutations in the viral genome (169).
B-lymphocytes are a critical component of adaptive immunity. Antibodies secreted by B-cell-derived plasma cells can serve multiple roles through antigen binding: Neutralization by preventing pathogen entry, immune cell recruitment and/or activation, and enhancement of antigen uptake and processing by antigen-presenting cells to other immune cells (170). Antibodies have also historically provided a measurable immune response that can serve as an imperfect but established benchmark for immunity (170–172).
Pre-existing antibodies to SARS-CoV-2 in humans were predominantly non-neutralizing, directed towards the N-protein and, to a lesser extent, the largely conserved S2 component of the S protein, which does not contain the receptor binding domain (RBD) (173, 174), with the latter suggesting some degree of cross-reactivity from endemic coronaviruses (174, 175) (Figure 1G). Anti-glycan antibodies—by and large of the IgM class (176)—may also be found in humans at baseline and can be targeted towards glycans found on enveloped viruses (176). Lower risk of COVID-19 in patients with blood type O (177) may have contributed to the hypothesis that certain blood types may be more protective against SARS-CoV-2 infection, perhaps by anti-glycan ABO antibodies (177). Early GWAS of COVID-19 patients provided a biologically plausible mechanism for this proposed phenomenon by identifying a reproducible association of susceptibility to COVID-19 with locus 9q34.2, which coincides with the ABO locus (178, 179). However, observational data since the discovery of the virus have been mixed (176, 180), perhaps owing to the difference between susceptibility and clinical severity of the disease (179).
The acute phase of SARS-CoV-2 infection is accompanied by B-cell lymphopenia and early plasmablast development from extrafollicular sites, a response typically occurring within 3-5 days (173, 181). In contrast to other infections, early antibody responses to SARS-CoV-2 include IgG, IgA, and IgM production (173) directed predominantly toward the N and S proteins (173). Somatic hypermutation and affinity maturation are less likely to occur at this stage (173, 181), and plasma cells produced during this time tend to be short-lived (173). The degree of extrafollicular B cell activation is characterized by an increase in CD11c+ activated naive (aN) B-cells which are precursors to IgD-CD27-, C-X-C chemokine receptor type 5 (CXCR5)-CD21- [double-negative 2 (DN2)] B-cells in addition to more recently discovered Cd11-CD21- (DN3) B-cell populations in severe COVID-19, which correlates with an increase in antibody-secreting cell populations (181) (Figure 2D). The differentiation of these cells is TLR7-independent and is mediated through IFNγ-IL-21 interactions, with the increase in DN2 B-cells additionally found to correlate with levels of IL-6 and IP-10 (181). Notably, an increase in endogenous neutralizing antibodies correlates with severe disease (181) and thus may suggest a raging battle rather than a victorious immune response. Curiously, a similar B-cell activation phenotype has been found in active SLE (181).
Approximately seven days after infection (181), a more nuanced and durable B-cell follicular response begins to develop from germinal center (GC) cells in lymphoid tissue via class-switching, affinity maturation, and somatic hypermutation (173). IgM and IgG anti-N, anti-S (182), and specifically anti-RBD (183)-defined seroconversion typically occur at a median of 11-13 days after symptom onset (173, 182, 183). In severe COVID-19, however, the GC response may be markedly blunted: Post-mortem specimens revealed an absence of germinal centers in the thoracic lymph nodes of patients with acute COVID-19 (151). A reduction of Bcl-6+ GC B-cells was seen in the same patient population (151). As previous research points to a quantitatively robust and seemingly adequate neutralizing capacity of the early extrafollicular antibody response (181), it is conceivable that the sustained production of meticulously targeted antibodies may be adversely impacted. However, the mechanism underlying this relationship likely requires further study (184).
Memory B-cells (MBCs) provide an immune reservoir for clonal expansion after infection (185, 186). In acute SARS-CoV-2 infection, the extrafollicular B-cell response includes the development of transient non-class-switched MBC with low rates of somatic hypermutation and class-switching (173). In tandem, pre-existing MBC populations with cross-reactivity to conserved S2 regions can proliferate, as evidenced by a robust initial IgG response to S2 (174). B-cells that enter germinal centers of secondary lymphoid organs can develop a more nuanced immune memory through class switching and somatic hypermutation later in the course of infection (173). Patients who recover from COVID-19 have an increased proportion of tissue-like memory (TLM) IgG+ S1-specific MBC (187) and circulating resting IgDloCD20+ MBC sustained for at least seven months after acute infection (188) (Figure 2H). The process of MBC generation likely requires CD4+ T-cells and can be affected by the severity of illness (187). Among the various immunoglobulin subtypes, IgG MBC eventually predominates in recovered individuals, along with a minor population of IgA MBC (171, 173).
Long-lived CD19−CD38hiCD138+ bone marrow plasma cells (BMPC) are another component of the humoral memory (173, 189) and may be the primary source of circulating antibodies years after a viral infection (188). Their presence has been detected at least seven months following infection with SARS-CoV-2 (188).
A summary of several important cell types and their function in acute and post-acute COVID-19 is shown in Table 2. Of note, while neutralizing antibody titers may suggest protective immunity early in the post-infectious period (172), the relative contributions of various immune memory cells to sterilizing or protective immunity in SARS-CoV-2 remain the subject of further study, making such immunity challenging to define at this time.
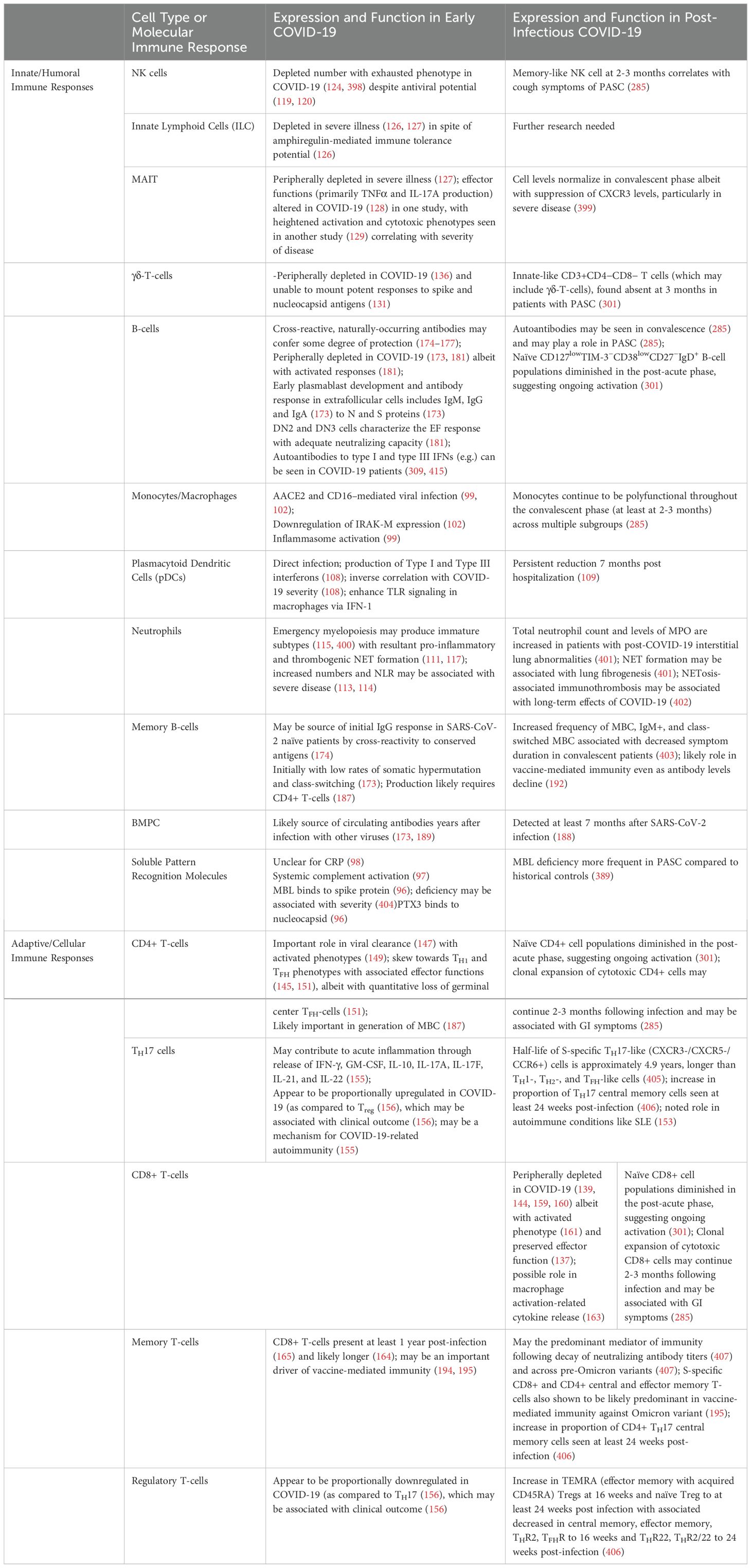
Table 2. Innate and adaptive immune responses to COVID-19.
The severity of the COVID-19 pandemic spurred the development of highly effective vaccines with the benefit of low-risk immunity across a population. Currently, available SARS-CoV-2 vaccines contain mRNA (or DNA in adenovirus vaccines) encoding the S protein, which can be further cleaved into its subunits S1 and S2 (190). Initial studies with mRNA vaccines established immunogenicity based on clinical efficacy (6), IgG response to RBD or S1, and at least 50% neutralization titers (191). Later studies further showed the durability of vaccine-induced immune memory, as evidenced by an increase in CD71+ MBC cells 3-6 months post-vaccination, even as antibody levels to S and the RBD declined (192). Notably, MBCs were cross-reactive to alpha, beta, and delta variants per data available then (192). More recent findings highlight the effect of sequential vaccine boosters as promoting refined MBC antibody-mediated immunity that targets both the ACE2 binding site of SARS-CoV-2 spike protein as well as more conserved components of the RBD (i.e., Type 1/4 antibodies), which correlate with increasing potency of SARS-CoV-2 neutralization (185).
While not suited for neutralizing and preventing viral entry into host cells, T-cell responses can help prevent the spread of infection within a host and consequently can impact illness severity and virus transmissibility (193). Omicron variant vaccine efficacy has been shown to be adversely affected by Omicron-specific CD8+ T-cell responses despite moderate levels of neutralizing antibodies in macaques (194). More recent studies also suggest conserved vaccine-mediated cellular immunity across variants, including Omicron, despite a diminished neutralizing antibody response (195). While the authors would be remiss if we did not mention vaccine-mediated immunity, the topic itself warrants further exploration beyond this paper’s scope.
The immune response evoked by infection with SARS-CoV-2 at the molecular level is often accompanied by the clinical syndrome known as COVID-19. COVID-19 is widely recognized as a predominantly respiratory disease manifesting as fever and cough (196). It is also recognized that the virus and its inflammatory response can affect multiple organ systems outside of the respiratory tract, as described in the following section.
Acute SARS-CoV-2 infection can lead to end organ damage through direct viral infection associated with localized immune activation and systemic immune responses. In addition to the lung, ACE2 receptors can be found in the heart (197), brain (198), kidney (199), testis (199), liver (200), colon (199, 201), and other organs (199, 202) (Figure 3A). While the clinical significance of such expression has not yet been fully elucidated, SARS-CoV-2 viremia can lead to end-organ infection and associated dysfunction by developing local pathologic inflammatory responses. Furthermore, the presence of ACE2 on endothelial cells (203, 204) and the known association with endotheliitis (197, 205) and microthrombi (203, 206) may allow for SARS-CoV-2 to affect multiple organs through disruption of the normal functioning of the microvasculature (197).
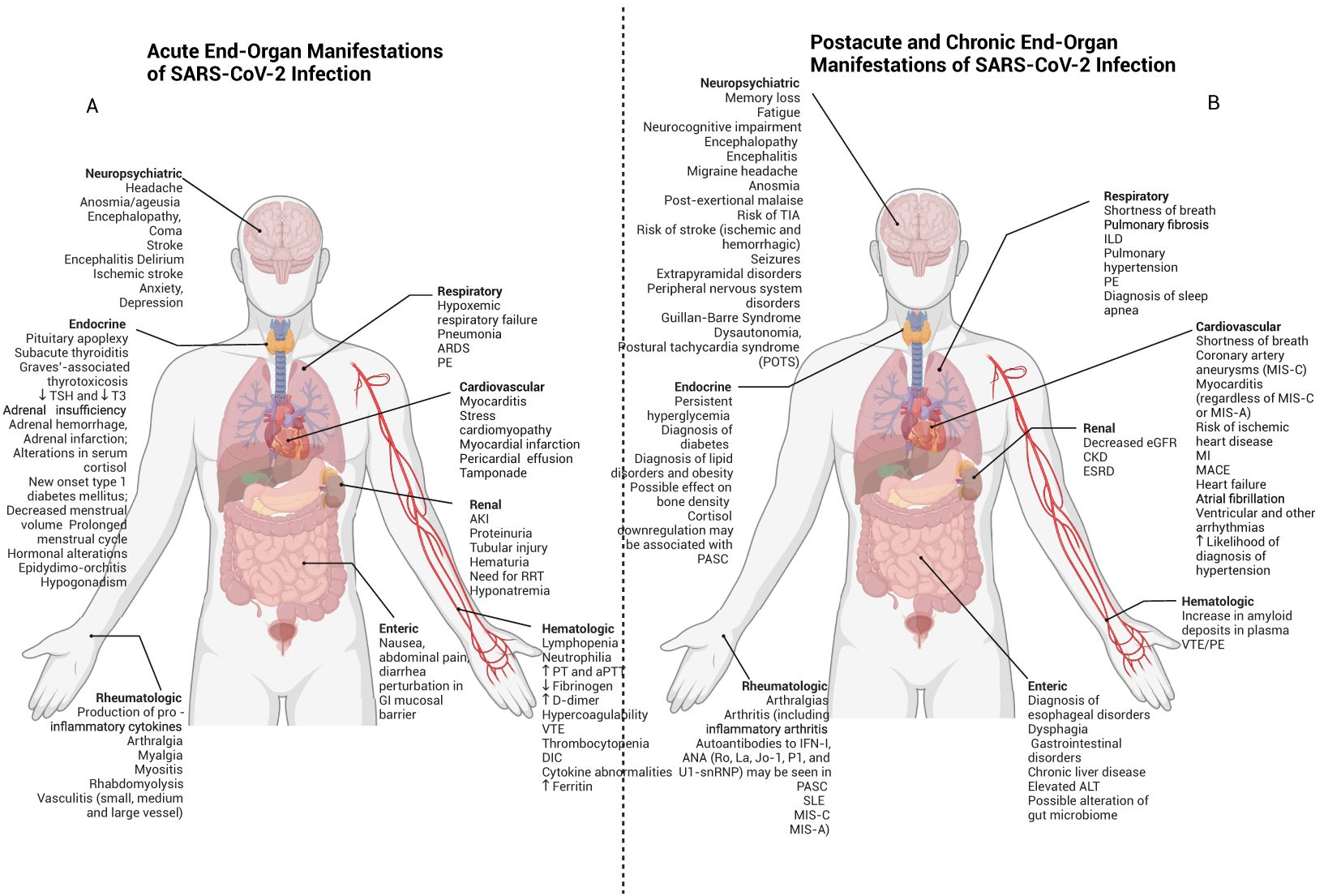
Figure 3. Acute, post-acute and chronic end organ manifestations of SARS-CoV-2 infection. SARS-CoV-2 may have adverse clinicopathophysiological impacts on multiple organ systems throughout the body. (A) Early effects are shown here and may include delirium, stroke, cardiomyopathy, myocardial infarction, pneumonia, ARDS, and AKI. (B) Postacute and chronic end-organ manifestations of SARS-CoV-2 infection can include neuropsychiatric symptoms such as fatigue and POTS (which may be features of PASC), in addition to myocarditis, interstitial lung disease and CKD.
ACE2 expression on endothelial cells may allow for viral infection to impact multiple organs, including the lung and kidney (207). Early pathologic specimens from COVID-19 patients revealed direct viral infection of endothelial cells in a transplanted kidney, with inflammatory lymphocytic infiltration noted in microvasculature of the heart, lung, kidney, and liver (207). While endotheliitis and hypercoagulation can be found with other viral infections, additional studies revealed the presence of intracapillary microthrombi in the pulmonary vasculature, as well as intussusceptive angiogenesis likely resulting from loss of normal microvascular architecture, which was seen more often in SARS-CoV-2 when compared to H1N1 influenza A (208, 209). Endothelial cell activation is associated with a hypercoagulable state in COVID-19 (210, 211), with proposed mechanisms including exocytosis of granules containing von Willebrand factor (vWF) and P-selectin (211) in addition to alteration in plasminogen activator inhibitor 1 (PAI-1) and tissue factor pathway inhibitor (TFPI) levels and upregulating cell adhesion molecules like VCAM and ICAM, which facilitates extravasation of circulating white blood cells (212) (Figure 1H).
The hypercoagulable state of COVID-19 may be a function of the complex interplay between immune cells and endothelial cells. Early polymorphonuclear (PMN) cell activation may lead to neutrophil extracellular traps (NETs) forming via NETosis, as suggested by increased plasma NET formation in intubated patients with COVID-19 (213). Colocalization of citrullinated histone H3+ neutrophils with platelets in blood vessels suggests that NET formation is implicated in the development of microthrombi in COVID-19 patients (213) (Figure 1I). Macrophages and monocytes in which inflammasomes and pyroptosis are activated may release microparticles containing tissue factor (TF) (214, 215), a critical first step in the coagulation cascade (216). IL-6 may further enhance the release of TF from monocytes (217). Of note, while not unique to COVID-19, severe infections can also promote the development of disseminated intravascular coagulation (DIC), the mechanisms of which have been well-studied (218).
The lungs are among the earliest and most overtly affected organs in COVID-19 (219), with symptoms ranging from mild [and sometimes relatively asymptomatic (220, 221)] to severe hypoxemic respiratory failure requiring mechanical ventilation (222). Despite comparatively low ACE2 expression in the lung overall, type II alveolar epithelial cells (223) are preferentially targeted by SARS-CoV-2 (69). Early infection is typically characterized by viral pneumonia of varying severity (219, 224), which may progress to COVID-19-related acute respiratory distress syndrome (ARDS) (224) with a predominance of CD-3+ and CD-4+ T lymphocytes in precapillary and postcapillary blood vessels (208). As with other organ systems, the anatomic and clinical sequelae of viral infections within the lungs are related both to the infection and to the inflammatory response with which it is accompanied (225). Findings in early COVID-19 typically manifest as bilateral peripheral ground-glass opacities and consolidations, which may reflect diffuse alveolar damage when examined histologically (225). As with other viruses, organizing pneumonia can also be seen, as well as bronchiolitis, manifesting as bronchial wall thickening, centrilobular nodules, and tree-in-bud opacities (225). While early studies hypothesized a unique trajectory of lung compliance changes in COVID-19 ARDS (226), later studies suggest that COVID-19 ARDS is a heterogeneous entity composed of pathophysiologic phenotypes that may be managed similarly to previously described phenotypes of ARDS (227–231). The overlap with typical ARDS in severe COVID-19 may explain the benefit of dexamethasone in hospitalized patients with an increased supplemental oxygen requirement (232), similar to a recent trial studying dexamethasone in ARDS (233). Of note, the development of endotheliitis and microthrombi (208, 234) in COVID-19 does appear to be unique to SARS-CoV-2 as compared to other viral illnesses such as influenza H1N1 (208, 235), a finding which may reflect SARS-CoV-2 tropism for ACE2 on endothelial cells (197, 203, 205, 206) exacerbated by an inflammation-related hypercoagulable state (236). In line with such pathophysiology, the initiation of therapeutic-dose anticoagulation in non-critically ill patients has been shown to be effective in preventing the need for mechanical ventilation and improving survival to hospital discharge (237). Initiation of anticoagulation during critical illness, in contrast, has not been shown to improve survival, which may be due to the timing of microthrombi development and the momentum of the inflammation-related hypercoagulable state (238). Of note, overt pulmonary emboli (PE) have a pooled incidence of 21% in hospitalized patients with COVID-19 (239); treatment for PE in such scenarios involves standard anticoagulation.
Although the phenomenon itself is probably rare (240), numerous studies have revealed associations of COVID-19 to myocarditis (197, 240–243). One US hospital administrative database study among patients hospitalized from March 2020 to January 2021 showed that the risk for myocarditis was 0.146% for patients with COVID-19 compared to 0.009%, with an estimated 15.7 times increased risk for the syndrome (244). In several such cases, SARS-CoV-2 mRNA was shown to be present in the myocardium and was associated with an intense inflammatory response comprised of macrophages and CD8+ cytotoxic T-cells (245, 246). At least one patient exhibited no overt evidence of pulmonary SARS-CoV-2 infection (245). Other mechanisms of acute cardiac involvement may include stress-related (Takutsobu) cardiomyopathy (242) and myocardial infarction (240, 247) related to supply-and-demand mismatch (248) or overt coronary occlusion (249). COVID-19-associated IL-6 production can also affect cardiac function: IL-6 has been shown to exacerbate viral myocarditis (240), and a recently submitted study using cardiac organoids stimulated with IL-1β suggests cardiac dysfunction may be due to downregulation of sarcomere components, reduced sarcomere width, contraction amplitude, increased cardiac fibrosis, and prothrombotic vasculature, irrespective of direct infection with SARS-CoV-2 (250).
Neurological manifestations of COVID-19 appear to portend a worse outcome in terms of in-hospital mortality, hospital length of stay, and persistent functional disability (251). Neurological manifestations were estimated to occur in as many as 80% of patients hospitalized with COVID-19 (252), with the most common self-reported symptoms including headache (37%) and anosmia/ageusia (26%) and the most common syndromes including encephalopathy (49%), coma (17%) and stroke (6%) (252). Ischemic stroke appears to be more prevalent in SARS-CoV-2 infection (1.6%; 95% CI 1.1-2.3%) (253) than in influenza (0.2%; 95% CI, 0.0%-0.6%) (253, 254). COVID-19 patients with co-morbid pre-existing dementia are at exceptionally high risk for in-hospital delirium, itself associated with prolonged hospitalization, intensive care unit (ICU) admission, or in-hospital mortality (251). Other disorders include seizures, hypoxic/ischemic brain injury (251), encephalitis (255), critical illness neuropathy/myopathy, myalgias, and dizziness (251). Psychiatric disorders including psychosis (255), anxiety, depression and post-traumatic stress disorder (PTSD) were also reported in COVID-19 patients (256). Single-cell CSF analysis in patients with neurological manifestations demonstrated a dampened interferon response, an increase in de-differentiated monocyte populations, and exhausted CD4+ T-cell phenotypes (257). Other proposed mechanisms of neurological dysfunction include endothelial damage, local inflammation (258), direct effect on choroid plexus epithelium, glial cell tropism, retrograde transportation into the CNS from the olfactory bulb (256), and hypoxic-ischemic injury to the CNS (256).
The kidney is unique in its high concentration of ACE2, its increased vascularity, and its exposure to systemic pathogens (16). ACE2 is co-expressed with TMPRSS2 in podocytes and tubule epithelial cells as it is in the lungs (259). Furin and CD147, which bind SARS-CoV-2, are likewise expressed in the kidney (260). Immune cells in the healthy human kidney are sparse and are primarily CD4+ and CD8+ T cells, with a smaller percentage of NK cells, B cells (17), and CD14+, CD16+ and CD68+ myeloid cells (18). Infection with SARS-CoV-2, a shift occurs from an upregulation in proinflammatory genes such as HSPA1A in podocytes and JUN1 in mesenchymal clusters (19) can accompany selective immune suppression of lymphocytes mediated through T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) and Programmed cell death protein 1 (PD-1) (20). A 2022 review suggests that resident immune cells may mediate inflammation by TNFα release, IL-34-mediated necrosis, and NLRP3 inflammasome production (266). ICAM-1, E- and P-selectin upregulation may be associated with neutrophil and circulating immune cell recruitment, platelet activation, NETosis (266), and immunothrombosis. While tropism for the kidney had previously been debated (255), the detection of SARS-CoV-2 RNA in a case series of kidney autopsies (264) suggests a role for direct infection. One elegant study built on the finding of increased extracellular matrix (ECM) in pathological specimens to explore an association with tubule-interstitial nephrosis (260). In this study, single-cell RNA sequencing of SARS-CoV-2-infected human-induced pluripotent stem-cell-derived kidney organoids confirmed the presence of SARS-CoV-2 in podocytes, proximal tubular cells, and myofibroblasts. Increased collagen one protein expression was seen in SARS-CoV-2 infected organoids compared to controls, a finding abrogated with a TGF-β blocker (SB431542). Moreover, the upregulation of HSPA1A, NRF21, S100A9, and TMSB10 proinflammatory genes was seen in podocytes, as was CCN1, JUN, and NFKBIA in mesenchymal clusters. Pathways upregulated among SARS-CoV-2-infected cells included TGF-β, PI3K/Akt, MAPK, and WNT signaling in proximal tubular cells and mesenchymal clusters (260). Upregulated IL-6 and STAT3 signaling may be important in COVAN-mediated disease (265). These results support a hypothesis that SARS-CoV-2 promotes tubule interstitial fibrosis through profibrotic pathways, at least in severe disease (Figure 4).

Figure 4. Inflammatory and immunologic consequences of SARS-CoV-2 in the kidney. (A) Histological and pathological mechanisms of acute kidney injury (AKI) in SARS-CoV-2 infection include collapsing glomerulonephritis, myoglobin cast nephropathy, proliferative glomerulonephritis with monoclonal IgG deposits, sequelae of nephrotoxic agents, acute tubular injury and necrosis (267), and perfusion pressure changes. More recent studies suggest that early tubule-interstitial fibrosis can occur in the setting of upregulated pro-fibrotic and pro-inflammatory mechanisms accompanied by myofibroblast activation, collagen, and ECM deposition. (B) Mechanisms of the development of chronic kidney disease (CKD) can include natural progression from AKI (414), which may occur as a result of maladaptive repair manifesting as early myofibroblast activation through proinflammatory pathways such as TGF-β, decreased Ang 1-7 production, and concomitant ongoing ECM and collagen deposition through mechanisms set off during the COVID-19 AKI phase. Direct damage to podocytes and proximal convoluted tubules via SARS-CoV-2 colocalization with Lex and sialyl-Lex (CD15s) in addition to ACE2 in the setting of chronic replication of SARS-CoV-2 may occur in immunocompromised hosts (365). Progression of pre-existing CKD may occur following COVID-19. In renal PASC, mildly decreased eGFR is likely to occur as a less clinically significant manifestation of SARS-CoV-2 infection. Additional mechanisms include CKD as a sequela of the array of etiologies of AKI in acute infection (414). Long-term sequelae of COVID-19 may also include subclinical decreased creatinine clearance (366), presumably through the mechanisms described for more overt CKD phenotypes. Persistent viral RNA may be present in immunocompetent patients and has been suggested as one mechanism for PASC (291).
Of note, patients with ESRD may already exhibit exhausted T-cell phenotypes (259). As discussed above, infection with SARS-CoV-2 is likewise associated with lymphopenia and exhausted T-cell phenotypes (260), which may indicate exacerbated immune dysregulation in such patients. Patients with ESRD have been shown to produce durable antibody anti-nucleocapsid and anti-RBD antibodies at least 6 months post-infection (261, 262), although this was not found to correlate with a decreased likelihood of reinfection in patients undergoing hemodialysis (263). While not the subject of this review, a more recent study noted that patients with both mRNA-1273 vaccination as well as prior infection with SARS-CoV-2 exhibited more robust neutralizing antibody production and S-specific B-cells were higher among patients with chronic kidney disease, and that the percentage of S-specific memory CD4+ and CD8+ T-cells were higher among patients who were dialysis-dependent than healthy hybrid controls (264).
Early studies in China suggested an incidence of AKI in 4.7% of all patients presenting with SARS-CoV-2 infection by Kidney Disease: Improving Global Outcomes (KDIGO) Criteria (265, 266). In another study, AKI was reported in approximately 25% of hospitalized patients with COVID-19 (267). The International Severe Acute Respiratory and Emerging Infections Consortium (ISARIC) World Health Organization (WHO) Clinical Characterisation Protocol UK (CCP-UK) for Severe Emerging Infections was among the largest epidemiological studies performed during the COVID-19 pandemic (268). Among the 85687 patients in this multicenter cohort, 2198 (2.6%) ultimately underwent acute renal replacement therapy (RRT) (long-term dialysis patients were excluded.) Among 41294 patients with available serum creatinine, 13000 (31.5%) had biochemical AKI: 8562 had stage 1 (65.9%), 2609 stage 2 (20.1%) and 1829 stage 3 (14.1%), with a concurrent increased risk in 28-day mortality by AKI severity (stage 1: aOR 1.58 (1.49–1.67); stage 2: aOR 2.41 (2.20–2.64); stage 3 aOR 3.50 (3.14–3.91); and RRT aOR 3.06 (2.75–3.39) (268). As summarized in the 2022 study, predominant risk factors for RRT were chronic kidney disease (CKD) (aOR 3.41; 95% CI=3.06–3.81), male sex (aOR 2.43; CI=2.18–2.71) and Black race (aOR 2.17; CI=1.79–2.63). Primary risk factors for biochemical AKI were admission respiratory rate >30 breaths per minute (aOR 1.68; CI=1.56–1.81), CKD (aOR 1.66; CI=1.57–1.76), and Black race (aOR 1.44; CI=1.28–1.61) (268).
In addition to the acute effect of COVID-19 and its consequences on the kidney, an early phenome-wide association study (PheWAS) of patients showed a significant increase in the likelihood of hospitalization among those patients with stage 4 CKD and above (Stage 4 CKD: OR 2.90, 95% CI: 1.47, 5.74), stage 5 CKD or dialysis (OR 8.83, 95% CI: 2.76, 28.27) (269). The odds of hospitalization were especially high among kidney transplant recipients (OR 14.98, 95% CI: 2.77, 80.8) (269), with plausible mechanisms including transplant rejection as seen in a 2021 biopsy series (270).
In the largest biopsy series among patients with SARS-CoV-2 infection (45.4% with AKI and 42.6% for proteinuria with or without AKI), among whom 44.6% were African American, the most common diagnosis was collapsing glomerulopathy (25.8%) (270). Of those patients, 91.7% had high-risk APOL1 genotypes (270) [G1/G1, G1/G2, or G2/G2 (271)]. Increased rates of myoglobin cast nephropathy (3.3%), proliferative glomerulonephritis with monoclonal IgG deposits (PMIG) (1.7%), and rejection among transplant patients (61.4% of transplant patients) were also seen when compared to historical controls (Figure 4A) (270). Lower rates of arterionephrosclerosis, diabetic nephropathy, and IgA nephropathy were also noted within this cohort (270). Only 3.7% of biopsy specimens within this series exhibited SARS-CoV-2 N protein by immunohistochemistry, none of which were positive by in situ hybridization (272).
The unique association of COVID-19 and collapsing glomerulopathy has led to the identification of an entity known as COVID-19 associated nephropathy (COVAN), which is thought to be similar to HIV-associated nephropathy (HIVAN) (273). Mechanisms of COVID-19-induced collapsing glomerulopathy—a variant of focal segmental glomerulosclerosis (FSGS) characterized by collapse of the glomerular capillaries with hypertrophy of the overlying podocytes (271)—may include direct infection as well as thrombotic microangiopathy or other ischemic insults (271). In addition to the above-mentioned immunologic processes, mechanisms of COVID-19-related AKI include acute tubular injury (ATI) and acute tubular necrosis (ATN) from various etiologies, including systemic hemodynamic instability (267). Proteinuria can occur in COVID-19-related AKI and is typically low molecular weight, indicative of tubular injury (267). Other proposed mechanisms include intravascular hypovolemia, increased right heart filling pressures secondary to pulmonary pathology with associated renal venous vascular congestion, rhabdomyolysis, glomerulonephritis, and collapsing glomerulopathy (267). Additional patho-etiologies include endothelial inflammation, microthrombi formation, and thrombotic microangiopathy (267). NET-related immunothrombosis may be implicated, particularly in patients with proteinuria (111). Nephrotoxic agents administered during the course of hospitalization may also cause AKI through varied mechanisms (267) (Figure 4).
Up to 50% of patients with COVID-19 develop GI symptoms such as nausea, abdominal pain, and diarrhea (274). SARS-CoV-2 can be detected in the stool, suggesting a role for fecal-oral transmission (275). Enterocytes can be directly infected with SARS-CoV-2 (276). Inflammatory infiltration of the intestine comprised of neutrophils, macrophages, and lymphocytes may be seen in severe COVID-19 (276). Studies in human small intestinal organoids reveal a response similar to that seen in the lungs (277), with low levels of interferons I and III and high levels of chemokines such as IP-10 and CXCL10 (278) skewing the immune response towards inflammation with less potent antiviral activity (274, 277). Systemic inflammation, including cytokine release, can induce leakiness in the gut mucosal barrier, leading to exposure to LPS and β-D-glucan and the development of neutrophil extracellular traps (NETs) (279). Alterations in the gut microbiome in SARS-CoV-2 infection are thought to contribute to COVID-19-mediate inflammation, including through increased gut permeability and associated endotoxemia (280). In one study of metagenomic and metaproteomic profiles of COVID-19 patients, an increase in opportunistic pathogenic species such as Burkholderia contaminans was seen along with a decrease in commensal bacteria and was associated with illness severity, highlighting the role of the microbiome (281). Other studies showed increases in Escherichia coli, Klebsiella pneumoniae and Enterococcus faecalis (282). Antibiotic use during the pandemic is also likely to have contributed to microbial dysbiosis (282). The reduction in short-chain fatty acids, metabolites of a normal microbiome that can serve as fuel for enterocytes and regulators of innate and adaptive immunity, may help to explain COVID-19-related microbial dysbiosis (282).
Early waves of the pandemic were followed by the growth of a population of patients who continued to suffer from physical and neuropsychiatric symptoms (283) as well as overt organ damage (284). In the UK’s National Health System (NHS), for instance, approximately one-third of patients who were hospitalized with COVID-19 were readmitted to the hospital within 5 months (284), implying a substantial individual and public health burden. A collection of symptoms, including shortness of breath, fatigue, memory loss, GI distress, and anosmia, has come to be called “long COVID” or post-acute sequelae of COVID-19 (PASC) (285). In a recent meta-analysis that alluded to substantial study heterogeneity (I2 = 100%, P < 0.001), global pooled prevalence of any post-COVID-19 condition is estimated to be approximately 43% (95% CI, 39-46%) (283), with fatigue being the most common symptom reported (23%; 95% CI, 17-30%) and with symptoms more likely to be present in patients who were hospitalized with COVID-19 (283). Other studies reported at least one PASC symptom in half of all survivors of COVID-19, extending to at least 6 months following hospitalization (median 54.0% (IQR 31.0%-67.0%) (286). Pre-existing comorbidities likely predispose patients to PASC: A recent study showed a prevalence of 2.8% to 5.5% in people with pre-existing health conditions as compared with 1.8% in healthy controls (287). Mechanisms of post-COVID organ dysfunction and PASC are an area of current and active interest (285, 288) with definitions of PASC being in a state of flux likely accounting for the heterogeneous findings (283).
Pathological studies have shown that SARS-CoV-2 viral RNA can persist and replicate throughout the body in multiple organs, including the heart, lung, brain, small intestine, and adrenal gland, for at least seven months post-infection (289). Circulating spike protein has been detected in approximately 60% of patients diagnosed with PASC for up to 12 months post-infection compared to those not diagnosed with PASC (290). While the clinical implications are unclear (289), the above findings and several other studies have fueled the hypothesis that PASC may be related to viral persistence (291). The presence of antibodies to spike antigen in 60% of COVID-19 survivors with PASC, in contrast to 0% of such antibodies in patients without PASC, indirectly points to a viral reservoir promoting persistent inflammation (290, 292). Of note, although less common than with DNA viruses, the phenomenon of persistent RNA virus is not unique to SARS-CoV-2 (293, 294) and has been seen with adenovirus, enterovirus, parvovirus B19 (293, 294), rhinovirus, respiratory syncytial virus (RSV) and others, and correlated with cardiomyopathy, asthma, and chronic pulmonary disease (293). The attention to virus-associated clinicopathology that the SARS-CoV-2 pandemic has spurred may thus prove impactful for many chronic illnesses.
Among the mechanisms studied for persistent symptoms in patients who have recovered from acute COVID-19 is altered immune activation to co-persistent Epstein-Barr Virus (EBV) (291). A 2021 study found that those patients who suffered from PASC symptoms were more likely to have evidence of EBV reactivation manifesting as early antigen diffuse (EA-D) IgG or viral capsid IgM (295). A later study showed that detection of EBV DNA was more than twice as common (27.1% vs. 12.5%) in patients diagnosed with COVID-19 (296). In another study, EBV viremia was seen in 14% of patients diagnosed with acute COVID-19 (285). In that same study, EBV viremia was associated with specific symptoms of PASC such as fatigue (285). Given the association of EBV with a number of clinical conditions ranging from chronic fatigue syndrome and multiple sclerosis (297), it’s role in acute and chronic forms of COVID-19 should not be ignored. Of note, re-activation of dormant infections is likely not unique to EBV in COVID-19. Similar phenomena have been suggested with tuberculosis (298) and toxoplasmosis (299), and may yet be the subject of further study.
Cytokines such as IL-2, IL-6, IL-17, TNFα, and IFN-γ may be persistently elevated for weeks during active acute COVID-19 (139) and may continue to be elevated in the early post-acute phase of COVID-19 (300, 301). In particular, IL-6, IFN-β, IFN-γ, and IFN-λ2/3 are associated with symptoms of PASC at least eight months after acute infection (301). Macrophages and monocytes may be responsible for the ongoing release of IL-1β, IL-6, and TNF (302). Hypotheses about the relevance of persistently elevated cytokines in PASC include low-grade chronic peripheral inflammation leading to microglia dysfunction and neuro-inflammation, which may explain “brain fog” and similar symptoms. This mechanism has yet to be proven (303). Among the mechanisms of cytokine release, the inflammasome-mediated pyroptosis pathway has been implicated in COVID-19 (67) and may involve NLRP3 (304). NLRP3 is associated with the development of such autoimmune conditions as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), systemic sclerosis (SSc), and inflammatory bowel disease (IBD) (305). Therefore, persistent NLRP3-mediated inflammation via IL-1β may be at play in patients with PASC (306). In a study among PASC patients with lung fibrosis, activation of the Absent in Melanoma 2 (AIM2) inflammasome was associated with the release of IL-1α, IFN-α and TGF-β, a finding not seen in non-PASC patients (306). Of note, in that same study, the provocation of NLRP3 with LPS or ATP did not induce IL-1α release (306). The significance of inflammasomes in acute and chronic forms of COVID-19 remains the subject of ongoing study (307).
Autoantibody development may be associated with the emergence of PASC (285). Several patients can develop autoantibodies early in the course of illness (308), as well as convalescent patients (285) (Figure 2E). Among a cohort of patients with life-threatening COVID-19, 10.2% were found to have IgG autoantibodies to type I IFNs IFN-α2 and IFN-ω (309). A unique sequencing technique called Molecular Indexing of Proteins by Self-Assembly (MIPSA) further identified type III-anti-IFN-λ3 autoantibodies in patients with severe COVID-19 (415). Another study revealed that one out of every four hospitalized patients with COVID-19 in their cohort had anti-nuclear antibodies (ANA) of various titers and patterns (308). Following acute infection, autoantibodies can linger or develop anew. In a recent study, 44% of patients with symptoms of PASC were found to have autoantibodies, including the ANAs Jo-1, Ro/SS-A, La/SS-B, U1-snRNP, P1, and anti-IFN-α2, approximately 2-3 months after initial symptom onset (285). Intriguingly, immune profiles of patients with PASC may be similar to those with SLE (285, 310). While it is difficult to ignore the possibility of pre-existing autoantibodies in certain patients before the development of COVID-19, a 2022 study points out that few of the patients found to have autoantibodies early in the course of illness had clinical evidence of autoimmune disease before COVID-19, suggesting a pre-existing subclinical autoimmune process that COVID-19 exacerbates (285).
In the early post-recovery phase, lymphopenia may persist (300). Following acute infection, CD4+ and CD8+ T-cell populations that upregulate genes associated with inflammatory regulation are preferentially expanded, and those associated with effector functions are contracted (285). In some patients, however—particularly those experiencing GI symptoms of PASC—populations of cytotoxic CD4+ and CD8+ T-cells continue to evolve into new clones, suggesting a role for T-cell-mediated inflammation in PASC (285). Additional studies have shown that naive CD127lowGzmB−CCR7+CD45RA+CD27+CD8+ T-cells, naive CD127lowTIM-3−CCR7+CD45 RA+CD27+CD4+ T-cells, and naive CD127lowTIM-3−CD38lowCD27−IgD+ B cells are absent eight months post-infection in patients with PASC, suggesting ongoing activation of normally naïve T-cell and B-cell subsets (301). As noted above, TH17 cells may be preferentially expressed in the hyperinflammatory environment in COVID-19, which includes TGF-β and IL-6 (153, 311). TH17 cells have been associated with several autoimmune diseases, including asthma, multiple sclerosis, SLE, and RA (153). Specific cell signatures in T-cells, B-cells, and NK cells have defined different immune endophenotypes among patients with PASC (285). The interplay between T-cells and other viruses in a patient’s virome (312), such as EBV and CMV, may also affect the development of PASC symptoms (285). Additional research can help further describe clinical and endophenotypes following infection with SARS-CoV-2 and elucidate the significance of these findings.
Multisystem inflammatory syndrome in children (MIS-C) and adults (MIS-A) are distinct acute hyperinflammatory illnesses involving end-organ dysfunction (313) that can occur 2-12 weeks after acute infection with SARS-CoV-2 (314). Diagnostic criteria for MIS-C vary but typically include age <21 years, persistent fever, involvement of ≥2 organ systems [e.g., cardiac dysfunction (seen in 86.5% in one study), dermatologic or mucocutaneous involvement (70.9%), and GI involvement (90.9%) (315)] and inflammatory markers in the context of a preceding SARS-CoV-2 infection (313). Diagnostic criteria for MIS-A presently include severe cardiac illness, dermatologic and conjunctival manifestations, and elevated inflammatory markers in the context of recently positive SARS-CoV-2 PCR, antigen, or antibody testing in patients >21 (316). It is important to note that these two syndromes, while similar in name, likely reflect distinct pathological processes. MIS-C is more common than MIS-A and exhibits strong similarities to Kawasaki Disease (313). Both MIS-C and MIS-A exhibit similarities to macrophage activation syndrome (MAS), secondary hemophagocytic lymphohistiocytosis (HLH), and toxic shock syndrome (317) and may be equivalent to an immunologic aftershock following a sometimes-benign initial infection (317). MIS-C may be distinguishable from severe COVID-19 by cytokine profile differences such as increased IL-10 in MIS-C (317). Coronary artery aneurysms have been reported in MIS-C (318); both MIS-C and MIS-A can be associated with myocarditis (319–321), which can progress to cardiogenic shock (321) and may respond to immunosuppressive agents (13, 320, 321).
The early effects of SARS-CoV-2 infection and the associated immune response can have a long-term impact on multiple organ systems. Post-acute sequelae of COVID-19 (PASC), also called Long COVID, long-haul COVID, and other names, is now widely recognized as a clinical entity carrying its own International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM) diagnosis (U09.9 Post COVID-19 condition, unspecified) (322). The convenience of a unifying diagnostic code belies the sundry manifestations of this condition. Patients who have recovered from COVID-19 may display clinical and pathological evidence of end-organ dysfunction, which may overlap those underlying PASC (Figure 3B). Pre-existing factors such as older age, female gender, higher BMI, and previous hospitalization are associated with the development of symptoms of PASC (323).
A large US Veterans’ study found that the risk of cardiovascular disease, including inflammatory heart disease, was increased beyond 30 days after acute SARS-CoV-2 infection, even when mild (324). COVID-19 in the acute phase (defined as ≤ 21 days) and in the post-acute phase (> 21 days) were shown to be associated with an increased risk of cardiovascular disease than contemporary controls [hazard ratio (HR) for acute phase: 4.3 (CI=2.6-6.9); HR for post-acute phase: 1.4 (CI=1.2-1.8)], findings which also held for historical controls whose population health was not overtly impacted by COVID-19 as a whole (325, 326). The risk of ischemic heart disease, heart failure, myocardial infarction, MACE, myocarditis, and pericarditis were all increased in the 12 months following acute SARS-CoV-2 infection (324). The risks of atrial fibrillation, ventricular arrhythmias, and other dysrhythmias were also higher (324). Myocarditis carried the highest risk (HR 5.38; CI: 3.80-7.59) of all such outcomes, which remained increased for COVID-19 patients even when vaccination was accounted for (324). A retrospective cohort study from a US health plan reported similarly increased risks in several of the outcomes mentioned above, as well as an increase in the diagnosis of hypertension (327). The risk of cerebrovascular disorders like stroke and TIA was also increased (324). While the impact of pre-existing comorbidities should not be ignored (287), subgroup analyses of the 2022 Veteran study suggested that COVID-19 was itself a risk factor for future cardiovascular disease regardless of pre-COVID-19 risk (324, 328). Moreover, COVID-19 disrupted the care of patients with cardiovascular disease and was associated with an overall increase in mortality due to these conditions (326).
Pulmonary hypertension (and associated right-sided cardiac dysfunction) (329) can be observed as a result of pulmonary fibrosis (330) and prior pulmonary emboli (i.e., Group 3 and Group 4 pulmonary hypertension, respectively) (331). Patients with pulmonary emboli may exhibit functional limitations and dyspnea (332) even in the absence of overt pulmonary hypertension. Notably, the risk of new-onset PE [as defined by hazard ratio (HR) (324)] and a new diagnosis of pulmonary hypertension (327) have also been found to be higher in patients who have had COVID-19. On imaging, early infection with SARS-CoV-2 can give way to air trapping in patients with PASC seen in a post-COVID-19 clinic 42-204 days after diagnosis (333). This CT finding suggests persistent small airway disease (333), presumably reflective of post-viral constrictive bronchiolitis, and is typically not associated with large airway obstruction on pulmonary function tests (PFTs) (333, 334). Patients with more severe infections (i.e., those requiring an ICU stay) were additionally found to have GGOs, architectural distortion, honeycombing, scar, or traction bronchiectasis accompanied by restrictive lung physiology and a reduction in DLCO (333, 335). Pulmonary fibrosis and interstitial lung disease (ILD) following COVID-19 (336–338) may be due to an initial hyperproliferation of pathologic fibroblasts acting via TGF-β signaling (339). Post-COVID pulmonary complications are likely to be related to disease severity, as suggested by an association with the inflammatory markers ESR and CRP (340), as well as to pre-existing comorbidities (287). Observational studies are now underway to elucidate the mechanisms and clinical trajectory of post-COVID-19 ILD (336). The risk of a new diagnosis of sleep apnea appears to be higher following hospitalization for COVID-19 (327).
Neuropsychiatric symptoms can be a prominent component of PASC and are typically characterized by fatigue, cognitive dysfunction, and post-exertional malaise (341). A recent survey study confirmed similarities between fibromyalgia and chronic fatigue syndrome with respect to self-reported fatigue, cognitive function, anxiety, depression, kinesiophobia, pain, and physical dysfunction, although patients with PASC had lower levels of fatigue and pain (342). Neurocognitive testing confirmed impairments in 46% of this preselected group of patients (343) and has been reported in other populations (344). Persistent neuroinflammation and endothelial injury are among the proposed mechanisms for this phenomenon (345).
Laboratory abnormalities among patients with PASC include reduced cortisol levels (346) and a reduced circulating serotonin level (347). A compensatory increase in ACTH was not seen among patients with low cortisol, which may reflect a blunted stress response in patients with PASC (346). The correlation between viral-RNA-induced type I interferons and reduced serotonin is thought to be mediated by reduced tryptophan uptake and storage and hypercoagulability, as shown in a recent elegant study from 2023 (347). In that same study—which opened the door to a compelling mechanism underlying PASC—cognitive dysfunction correlated with peripheral serotonin depletion (347).
In post-hospital discharge patients with symptoms of PASC, the most common MRI finding was scattered white matter lesions (343). An increased risk of incident ischemic and hemorrhagic stroke, encephalitis, encephalopathy, migraine, seizures, sensory disorders, peripheral nervous system disorders, musculoskeletal disorders, Guillain-Barre Syndrome, and extrapyramidal disorders was seen among patients followed for 12 months following acute SARS-CoV-2 infection, with an estimated HR for any neurologic sequela of 1.42 (1.38-1.47) (344). In one comprehensive NIH study of 12 patients with neurologic sequelae of SARS-CoV-2 infection, mild cognitive impairment (Montreal Cognitive Assessment Score (MoCA) < 26) was seen in half of the patients, with deficits predominantly seen in short-term memory (348). No MRI abnormalities were seen in those patients. Immunophenotyping of CSF revealed higher frequencies of antibody-secreting B cells and PD-L1-expressing monocytes and lower frequencies of CD4+ and CD8+ effector memory cells (348). It should be noted that one-third of patients included in the study had a prior history of resolved long-term disability due to a preceding infection (amoebiasis due to Entamoeba histolytica, infectious mononucleosis, Lyme disease, and severe sepsis due to group A Streptococcus) (348).
In a US health plan cohort, renal dysfunction, as defined by a composite of AKI and CKD, was found to be more likely after a diagnosis of COVID-19 in hospitalized patients with a pre-existing condition (327). Other studies reported an increased likelihood of a new diagnosis of CKD within the first 4-6 months following COVID-19 in addition to AKI (284, 349). A large VA health system study also revealed an increase in the diagnosis of CKD following COVID-19 infection in hospitalized and intensive care unit patients (HR, hospitalized patients=1.36 (CI=1.24-1.49), and HR for ICU patients=1.88 (1.66-2.13) (350).
CKD is known to be associated with altered immunity. ESRD has likewise been defined in part as a state of “acquired immunodeficiency” by Vanholder and Ringoir in 1993 (351), although simultaneously heightened levels of inflammation and immune activation have also been described noted (352). In ESRD patients, CD14+CD16+ monocyte populations are expanded, cytokine production and chemokine expression is increased, as is basal ROS production (352, 353). LDL likewise elicits a stronger pro-inflammatory response (354). In contrast, a decreased number of DCs with impaired function (355) are seen, circulating PMNs have decreased phagocytic capacity, naïve and central memory T cells are depleted (with a reduced CD4+/CD8+ T cell ratio), CD4+CD25bright+FoxP3+ Treg populations are both diminished and impaired, and B cell populations are likewise diminished (352).
An early PheWAS study cited above pointed to a strong association between pre-existing Stage 4 CKD, Stage 5 CKD, and dialysis with the likelihood of hospitalization with even higher odds among kidney transplant recipients , suggesting a disproportionate impact among those with pre-existing renal disease as well as the bidirectional interplay between COVID-19 and CKD (356). Among 758 patients in the Health Outcome Predictive Evaluation of COVID-19 (HOPE COVID-19) Study, an eGFR of < 60 ml/min/1.73m2 was associated with a higher risk of in-hospital mortality (eGFR > 60: 18.4%; eGFR 30-60: 56.5%; and eGFR < 30: 65.5%; p < 0.001), multi-organ failure, and sepsis (356, 357). Notably, while only 8.5% of the HOPE-COVID-19 cohort had documented CKD on admission, 30.6% (N=322) presented with eGFR < 60 ml/min/1.73m2, suggesting a role in the development of AKI as described above (356, 357).
Mechanisms of the development of CKD in the general population include the progression of changes set in motion through COVID-19 AKI, particularly as the development of AKI itself can be associated with the eventual development of CKD (358). Acknowledging the unique etiological mechanisms of the initial AKI insult (359, 360), injury of proximal tubule cells in severe AKI may result in cell cycle arrest in the G2/M phase, which leads to the secretion of TGF-β and connective tissue growth factor, both of which mediate fibrosis through c-jun NH2-terminal kinase (JNK) signaling (359, 361). Following AKI, animal models have shown that aberrant activation of developmental pathways such as Hedgehog and Wnt/β-catenin can also promote fibrosis (359). Persistent mitochondrial dysfunction following the initial insult may also play a role in developing fibrosis and persistent inflammation (359). Resident P0-Cre fibroblasts in the renal cortex and medulla (359) may transdifferentiate into αSMA-positive myofibroblasts and promote fibrosis (359); Gli1-Cre fibroblasts are likewise related to end-organ fibrosis and may be therapeutically targeted (362).
A 2022 study reviewed a biopsy series and confirmed the presence of tubule interstitial fibrosis in patients with COVID-19 as compared to age, sex, and comorbidity-matched controls ; in an elegant study described above for AKI, infection of human iPSC-derived organoids elucidated SARS-CoV-2-specific pathways of tubule-interstitial fibrosis mediated through direct infection (19). Early pro-inflammatory, fibroblast activation, and myofibroblast differentiation, including those mediated by TGF-β, NFκB, and JAK-STAT, were upregulated in podocytes, proximal tubule cells (PTC), and fibroblasts, with a concomitant increase in ECM and collagen deposition (19). Additional mechanisms are described above. Further, the initial binding of SARS-CoV-2 with ACE2 in the kidney in acute infection limits its bioavailability for Ang1-7 production, leading to fibrosis (363) and maladaptive repair (359, 364). Mechanisms in immunocompromised patients may involve direct damage to podocytes and proximal convoluted tubules via SARS-CoV-2 colocalization with Lex and sialyl-Lex (CD15s) in addition to ACE2 in the setting of chronic replication of SARS-CoV-2 as seen on biopsy from a patient with splenic marginal cell lymphoma (365).
Much of the research on COVID-19 AKI and CKD has been performed among patients with more severe illness. A 2022 study of a primarily ambulatory cohort of patients in Hamburg, Germany, showed a decreased eGFR (regression estimate -2.35mL/min/1.73m2 (CI= -4.28, -0.42); Bonferroni adjusted p=0.019) as compared to matched controls (366). Mechanisms or histological specimens were not described in this study. However, it is conceivable that SARS-CoV-2-mediated AKI and subsequent CKD exist on a spectrum, and likely that if so, the above mechanisms are at play in what may be considered “renal PASC.” As noted above, persistent viral RNA may be among the mechanisms in renal PASC (291).
In a large cohort of post-discharge patients in the UK’s NHS, the rate of new diagnoses of chronic liver disease increased following hospitalization for COVID-19 (284). Diagnosis of diabetes was also increased (284). A large VA cohort identified an excess burden of diabetes beyond thirty days after COVID-19 diagnosis in non-hospitalized patients, in addition to lipid disorders and obesity (350). An increased burden of esophageal disorders, gastrointestinal disorders, and dysphagia was also seen, as was incident elevated alanine aminotransferase (ALT) levels (350). As described above in the context of acute infection, mechanisms of excess inflammation in post-infectious symptoms include alterations in the gut microbiome, with acute and post-infectious inflammation possibly reflecting perturbations in the gastrointestinal mucosal barrier (367). An extra burden of skin disorders, arthralgias, and arthritis was also seen in the VA cohort (350). Thyrotoxicosis related to both subacute thyroiditis and Graves’ Disease has been reported (368). Reports of hormonal changes in male and female patients vary; an increase in menstrual cycle has been reported (368, 369). Orchitis and epidydimo-orchitis may occur in acute COVID-19 (368). The immunologic changes that accompany SARS-CoV-2 may also prove to be associated with long-term pathophysiology in bone (370). Finally, vasculitis (371), myositis (371, 372), and rhabdomyolysis (371) have been reported in acute COVID-19; arthritis (350), including inflammatory arthritis (371), has been reported in the post-infectious period. A comprehensive summary of the end-organ effects on various organ systems affected in both acute and post-infectious COVID-19 is shown in Table 3 and Figure 3.
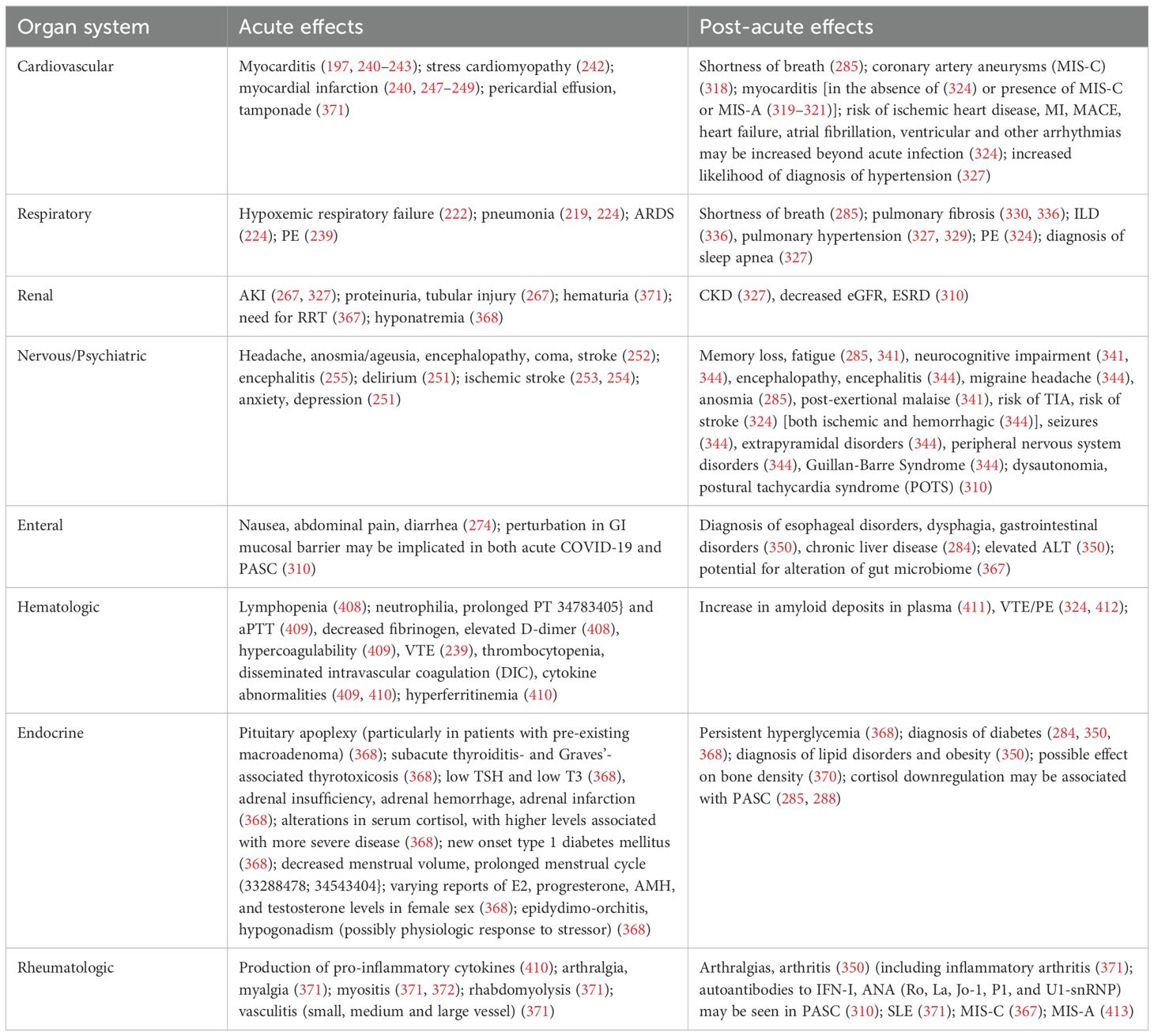
Table 3. Acute and post-acute end-organ dysfunction in COVID-19.
The impact of variants throughout the pandemic is a subject of ongoing interest, particularly concerning ongoing vaccine development (373). Mutations in the S protein are likely to be the most important in determining viral tropism, infectivity, and mortality (374). However, mutations in other proteins, such as R203K and G204R in N, may also impact viral fitness (375). A 2022 study compared the impact of Delta and Omicron SARS-CoV-2 variants in a cohort of 65 patients admitted to an intensive care unit. A higher mortality rate was seen with Omicron (52.9% vs. 41.9% with Delta), although the study acknowledges a higher rate of comorbidities among Omicron patients (376). Similar PaO2/FiO2 ratios (partial pressure of oxygen in arteries to the fraction of inspired oxygen) were seen [Omicron: 156.57, SD 65.98 vs. Delta: 157.31, SD 84.56 (p=0.971)], suggesting a role for extrapulmonary pathophysiology. Organ-specific sequelae may also relate to SARS-CoV-2 variants: Cardiovascular mortality was higher during the pandemic period, ranging from March to June 2020 (likely the D614G variant) and during the Delta wave (June to December 2021) (326). Rates of AKI were higher among Omicron patients than Delta (13 (38.24%) vs. 3 (9.7%), OR 5.78; CI 1.46–22.9, p=0.0172) (376), which may be due to a higher rate of comorbidities among Omicron patients admitted to the ICU rather than being reflective of virus-specific effects.
One meta-analysis examined the impact of variants on post-COVID symptomatology by pooled estimates of several studies, reporting CT abnormalities (60.5%; 95% CI: 40.4-80.6%) and sleep difficulty (24.5%; 95%: 17.5-31.5%) to be the most common PASC symptom following infection with wild-type SARS-CoV-2, fatigue most common among survivors of the Alpha variant, and myalgia among survivors of the Omicron variant (11.7% (95% CI: 8.3-15.1%) in Omicron compared to 9.4% (95% CI: 6.3-12.5%) in wild-type) (377). In that same study, the Alpha and Gamma variants had higher rates of dyspnea (34.2% (8.3-60.1%) and 43.0% (35.3, 50.8%), respectively). The Alpha variant was found to have a higher rate of patients with greater than one general symptom of PASC and fatigue (377). As noted by the authors and as suggested by recent meta-analyses, vaccination is likely to have attenuated the risk of PASC among survivors of COVID-19 (323, 378, 379). As pointed out by the authors of a recent study on pooled survivors of COVID-19 across SARS-CoV-2 variants (323), however, it is difficult to parse out the interaction of vaccines with individual variants, which may be the subject of future studies.
Mechanisms underlying responses to variants of concern may vary. Studies in zebrafish models showed that the wild-type (WT)/Wuhan SARS-CoV-2 variant activated emergency myelopoiesis and recruited neutrophils and macrophages through inflammasome production (380). Deficiency of ACE2 led to exacerbation of inflammation, which was reversed with Ang 1-7 injection, suggesting a role for this pathway in WT-SARS-CoV-2 (380). S1 from the Gamma (P.1) variant (S1γ) and from the Beta variant (S1β from B.1.351) produced higher levels of inflammation, and S1δ (Delta variant; B.1.617.2) produced lower levels of inflammation as measured by macrophage and neutrophil recruitment as well as NF-κB activity in this study (380). A study published in November 2021 showed evidence of microvascular dysfunction among patients infected with SARS-CoV-2 (exact variants unknown) (381), which was not seen in a follow-up study among patients infected with Omicron (382). A complete discussion regarding variant-specific mechanisms of virulence is outside of the scope of this review and will likely be the subject of ongoing study [see, e.g. (375)].
In conclusion, the immune response to novel SARS-CoV-2 involves a complex milieu of cytokines, macrophages, lymphocytes, and other immune cells skewed towards a pathologically hyperactivated response. Organized but immunostimulatory pyroptosis and inflammasome production may be at play in an environment that lacks crucial early antiviral interferon production, which, along with the development of immune-mediated microthrombi, may be associated with the severity of disease in COVID-19. In the kidney, these mechanisms, as well as acute tubular injury, early myofibroblast activation, and collapsing glomerulopathy in select populations, are likely to account for COVID-19-related AKI and CKD development.
Among the limitations of this review is a thorough discussion regarding variant-specific mechanisms of immune responses, particularly as subsequent waves of the pandemic met longitudinal populations of varying degrees of immunity mediated through vaccination and natural infection. Typical immune responses to SARS-CoV-2 continue to change as the novelty of the virus changes.
It is still unclear whether the acute and long-term sequelae and associated pathophysiology following SARS-CoV-2 infection are unique to the virus or whether this reflects the unprecedented focus of research brought about by the pandemic. In any case, the pace of research in the COVID-19 era has led to a greater understanding of the components of a regulated immune response to SARS-CoV-2 and perhaps to viral infections more generally.
HN: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. MB: Writing – review & editing. HL: Writing – review & editing. ML: Writing – review & editing. AR: Conceptualization, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. HN is the recipient of T32 grant 5T32HL007563-35 and F32 grant 1F32MD019534-01.
Images were created using BioRender.com.
The authors declare that the research was conducted without any commercial or financial relationships that could potentially create a conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Cucinotta D, Vanelli M. WHO declares COVID-19 a pandemic. Acta Biomed. (2020) 91:157–60. doi: 10.23750/abm.v91i1.9397
2. Mather N. How we accelerated clinical trials in the age of coronavirus. Nature. (2020) 584:326. doi: 10.1038/d41586-020-02416-z
3. Wu Z, Harrich D, Li Z, Hu D, Li D. The unique features of SARS-CoV-2 transmission: Comparison with SARS-CoV, MERS-CoV and 2009 H1N1 pandemic influenza virus. Rev Med Virol. (2021) 31:e2171. doi: 10.1002/rmv.2171
4. Ji P, Zhu J, Zhong Z, Li H, Pang J, Li B, et al. Association of elevated inflammatory markers and severe COVID-19: A meta-analysis. Med (Baltimore). (2020) 99:e23315. doi: 10.1097/MD.0000000000023315
5. Perico L, Benigni A, Casiraghi F, Ng LFP, Renia L, Remuzzi G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat Rev Nephrol. (2021) 17:46–64. doi: 10.1038/s41581-020-00357-4
6. Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and efficacy of the BNT162b2 mRNA covid-19 vaccine. N Engl J Med. (2020) 383:2603–15. doi: 10.1056/NEJMoa2034577
7. Anderson EJ, Rouphael NG, Widge AT, Jackson LA, Roberts PC, Makhene M, et al. Safety and immunogenicity of SARS-coV-2 mRNA-1273 vaccine in older adults. N Engl J Med. (2020) 383:2427–38. doi: 10.1056/NEJMoa2028436
8. Taylor PC, Adams AC, Hufford MM, de la Torre I, Winthrop K, Gottlieb RL. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat Rev Immunol. (2021) 21:382–93. doi: 10.1038/s41577-021-00542-x
9. Owen DR, Allerton CMN, Anderson AS, Aschenbrenner L, Avery M, Berritt S, et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science. (2021) 374:1586–93. doi: 10.1126/science.abl4784
10. Jayk Bernal A, Gomes da Silva MM, Musungaie DB, Kovalchuk E, Gonzalez A, Delos Reyes V, et al. Molnupiravir for oral treatment of covid-19 in nonhospitalized patients. N Engl J Med. (2022) 386:509–20. doi: 10.1056/NEJMoa2116044
11. Aldawas A, Ishfaq M. COVID-19: multisystem inflammatory syndrome in children (MIS-C). Cureus. (2022) 14:e21064. doi: 10.7759/cureus.21064
12. Kashyap H, Kumar RNS, Gautam S, Gupta A, Gupta S, Tiwari PK. Multisystem inflammatory syndrome in children (MIS-C) associated with COVID-19 infection. Indian J Pediatr. (2021) 88:1053. doi: 10.1007/s12098-021-03832-3
13. Cattaneo P, Volpe A, Cardellino CS, Riccardi N, Bertoli G, Ursini T, et al. Multisystem inflammatory syndrome in an adult (MIS-A) successfully treated with anakinra and glucocorticoids. Microorganisms. (2021) 9:1393. doi: 10.3390/microorganisms9071393
14. Patel P, DeCuir J, Abrams J, Campbell AP, Godfred-Cato S, Belay ED. Clinical characteristics of multisystem inflammatory syndrome in adults: A systematic review. JAMA Netw Open. (2021) 4:e2126456. doi: 10.1001/jamanetworkopen.2021.26456
15. Gracia-Ramos AE, Martin-Nares E, Hernandez-Molina G. New onset of autoimmune diseases following COVID-19 diagnosis. Cells. (2021) 10:3592. doi: 10.3390/cells10123592
16. Diao B, Wang C, Wang R, Feng Z, Zhang J, Yang H, et al. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. Nat Commun. (2021) 12:2506. doi: 10.1038/s41467-021-22781-1
17. McEvoy CM, Murphy JM, Zhang L, Clotet-Freixas S, Mathews JA, An J, et al. Single-cell profiling of healthy human kidney reveals features of sex-based transcriptional programs and tissue-specific immunity. Nat Commun. (2022) 13:7634. doi: 10.1038/s41467-022-35297-z
18. Park JG, Na M, Kim MG, Park SH, Lee HJ, Kim DK, et al. Immune cell composition in normal human kidneys. Sci Rep. (2020) 10:15678. doi: 10.1038/s41598-020-72821-x
19. Jansen J, Reimer KC, Nagai JS, Varghese FS, Overheul GJ, de Beer M, et al. SARS-CoV-2 infects the human kidney and drives fibrosis in kidney organoids. Cell Stem Cell. (2022) 29:217–31.e8. doi: 10.1016/j.stem.2021.12.010
20. Wu H, He P, Ren Y, Xiao S, Wang W, Liu Z, et al. Postmortem high-dimensional immune profiling of severe COVID-19 patients reveals distinct patterns of immunosuppression and immunoactivation. Nat Commun. (2022) 13:269. doi: 10.1038/s41467-021-27723-5
21. Qin G, Liu S, Yang L, Yu W, Zhang Y. Myeloid cells in COVID-19 microenvironment. Signal Transduct Target Ther. (2021) 6:372. doi: 10.1038/s41392-021-00792-0
22. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. (2019) 17:181–92. doi: 10.1038/s41579-018-0118-9
23. V’Kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. (2021) 19:155–70. doi: 10.1038/s41579-020-00468-6
24. Hawks SA, Prussin AJ 2nd, Kuchinsky SC, Pan J, Marr LC, Duggal NK. Infectious SARS-coV-2 is emitted in aerosol particles. mBio. (2021) 12:e0252721. doi: 10.1128/mBio.02527-21
25. Richard M, Kok A, de Meulder D, Bestebroer TM, Lamers MM, Okba NMA, et al. SARS-CoV-2 is transmitted via contact and via the air between ferrets. Nat Commun. (2020) 11:3496. doi: 10.1038/s41467-020-17367-2
26. Azimi P, Keshavarz Z, Cedeno Laurent JG, Stephens B, Allen JG. Mechanistic transmission modeling of COVID-19 on the Diamond Princess cruise ship demonstrates the importance of aerosol transmission. Proc Natl Acad Sci U S A. (2021) 118:e2015482118. doi: 10.1073/pnas.2015482118
27. Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-coV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. (2020) 181:1016–35 e19. doi: 10.1016/j.cell.2020.04.035
28. Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. (2020) 20:363–74. doi: 10.1038/s41577-020-0311-8
29. Senapati S, Banerjee P, Bhagavatula S, Kushwaha PP, Kumar S. Contributions of human ACE2 and TMPRSS2 in determining host-pathogen interaction of COVID-19. J Genet. (2021) 100:12. doi: 10.1007/s12041-021-01262-w
30. Horowitz JE, Kosmicki JA, Damask A, Sharma D, Roberts GHL, Justice AE, et al. Genome-wide analysis provides genetic evidence that ACE2 influences COVID-19 risk and yields risk scores associated with severe disease. Nat Genet. (2022) 54:382–92. doi: 10.1038/s41588-021-01006-7
31. Karlsen TH. Understanding COVID-19 through genome-wide association studies. Nat Genet. (2022) 54:368–9. doi: 10.1038/s41588-021-00985-x
32. Scheen AJ. DPP-4 inhibition and COVID-19: From initial concerns to recent expectations. Diabetes Metab. (2021) 47:101213. doi: 10.1016/j.diabet.2020.11.005
33. Eleftheriou P, Amanatidou D, Petrou A, Geronikaki A. In silico evaluation of the effectivity of approved protease inhibitors against the main protease of the novel SARS-coV-2 virus. Molecules. (2020) 25:2529. doi: 10.3390/molecules25112529
34. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. (2020) 5:562–9. doi: 10.1038/s41564-020-0688-y
35. Xiong Y, Delic D, Zeng S, Chen X, Chu C, Hasan AA, et al. Regulation of SARS CoV-2 host factors in the kidney and heart in rats with 5/6 nephrectomy-effects of salt, ARB, DPP4 inhibitor and SGLT2 blocker. BMC Nephrol. (2022) 23:117. doi: 10.1186/s12882-022-02747-1
36. Xi CR, Di Fazio A, Nadvi NA, Patel K, Xiang MSW, Zhang HE, et al. A novel purification procedure for active recombinant human DPP4 and the inability of DPP4 to bind SARS-coV-2. Molecules. (2020) 25:5392. doi: 10.3390/molecules25225392
37. Reis CA, Tauber R, Blanchard V. Glycosylation is a key in SARS-CoV-2 infection. J Mol Med (Berl). (2021) 99:1023–31. doi: 10.1007/s00109-021-02092-0
38. Lamers MM, Haagmans BL. SARS-coV-2 pathogenesis. Nat Rev Microbiol. (2022) 20:270–84. doi: 10.1038/s41579-022-00713-0
39. Jacobs JL, Bain W, Naqvi A, Staines B, Castanha PMS, Yang H, et al. Severe acute respiratory syndrome coronavirus 2 viremia is associated with coronavirus disease 2019 severity and predicts clinical outcomes. Clin Infect Dis. (2022) 74:1525–33. doi: 10.1093/cid/ciab686
40. Markov PV, Ghafari M, Beer M, Lythgoe K, Simmonds P, Stilianakis NI, et al. The evolution of SARS-coV-2. Nat Rev Microbiol. (2023) 21:361–79. doi: 10.1038/s41579-023-00878-2
41. Schiuma G, Beltrami S, Bortolotti D, Rizzo S, Rizzo R. Innate immune response in SARS-coV-2 infection. Microorganisms. (2022) 10:501. doi: 10.3390/microorganisms10030501
42. Lowery SA, Sariol A, Perlman S. Innate immune and inflammatory responses to SARS-CoV-2: Implications for COVID-19. Cell Host Microbe. (2021) 29:1052–62. doi: 10.1016/j.chom.2021.05.004
43. Choi H, Shin EC. Roles of type I and III interferons in COVID-19. Yonsei Med J. (2021) 62:381–90. doi: 10.3349/ymj.2021.62.5.381
44. Iwasaki A. A virological view of innate immune recognition. Annu Rev Microbiol. (2012) 66:177–96. doi: 10.1146/annurev-micro-092611-150203
45. Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. Pattern recognition receptors and the innate immune response to viral infection. Viruses. (2011) 3:920–40. doi: 10.3390/v3060920
46. van der Made CI, Simons A, Schuurs-Hoeijmakers J, van den Heuvel G, Mantere T, Kersten S, et al. Presence of genetic variants among young men with severe COVID-19. JAMA. (2020) 324:663–73. doi: 10.1001/jama.2020.13719
47. Akamatsu MA, de Castro JT, Takano CY, Ho PL. Off balance: Interferons in COVID-19 lung infections. EBioMedicine. (2021) 73:103642. doi: 10.1016/j.ebiom.2021.103642
48. Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. (2008) 8:594–606. doi: 10.1038/nri2358
49. Bortolotti D, Gentili V, Rizzo S, Schiuma G, Beltrami S, Strazzabosco G, et al. TLR3 and TLR7 RNA sensor activation during SARS-coV-2 infection. Microorganisms. (2021) 9:1820. doi: 10.3390/microorganisms9091820
50. Vabret N, Britton GJ, Gruber C, Hegde S, Kim J, Kuksin M, et al. Immunology of COVID-19: current state of the science. Immunity. (2020) 52:910–41. doi: 10.1016/j.immuni.2020.05.002
51. Yu M, Zhou H, Zhao J, Xiao N, Roychowdhury S, Schmitt D, et al. MyD88-dependent interplay between myeloid and endothelial cells in the initiation and progression of obesity-associated inflammatory diseases. J Exp Med. (2014) 211:887–907. doi: 10.1084/jem.20131314
52. Goel S, Saheb Sharif-Askari F, Saheb Sharif Askari N, Madkhana B, Alwaa AM, Mahboub B, et al. SARS-coV-2 switches ‘on’ MAPK and NFkappaB signaling via the reduction of nuclear DUSP1 and DUSP5 expression. Front Pharmacol. (2021) 12:631879. doi: 10.3389/fphar.2021.631879
53. Li S, Zhang Y, Guan Z, Li H, Ye M, Chen X, et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct Target Ther. (2020) 5:235. doi: 10.1038/s41392-020-00334-0
54. Nainu F, Shiratsuchi A, Nakanishi Y. Induction of apoptosis and subsequent phagocytosis of virus-infected cells as an antiviral mechanism. Front Immunol. (2017) 8:1220. doi: 10.3389/fimmu.2017.01220
55. Ren Y, Wang A, Fang Y, Shu T, Wu D, Wang C, et al. SARS-coV-2 membrane glycoprotein M triggers apoptosis with the assistance of nucleocapsid protein N in cells. Front Cell Infect Microbiol. (2021) 11:706252. doi: 10.3389/fcimb.2021.706252
56. Toldo S, Bussani R, Nuzzi V, Bonaventura A, Mauro AG, Cannatà A, et al. Inflammasome formation in the lungs of patients with fatal COVID-19. Inflamm Res. (2021) 70:7–10. doi: 10.1007/s00011-020-01413-2
57. Bittner ZA, Schrader M, George SE, Amann R. Pyroptosis and its role in SARS-coV-2 infection. Cells. (2022) 11:1717. doi: 10.3390/cells11101717
58. Sun X, Liu Y, Huang Z, Xu W, Hu W, Yi L, et al. SARS-CoV-2 non-structural protein 6 triggers NLRP3-dependent pyroptosis by targeting ATP6AP1. Cell Death Differ. (2022) 29:1240–54. doi: 10.1038/s41418-021-00916-7
59. Hu G, Christman JW. Editorial: alveolar macrophages in lung inflammation and resolution. Front Immunol. (2019) 10:2275. doi: 10.3389/fimmu.2019.02275
60. Barros-Martins J, Forster R, Bosnjak B. NK cell dysfunction in severe COVID-19: TGF-beta-induced downregulation of integrin beta-2 restricts NK cell cytotoxicity. Signal Transduct Target Ther. (2022) 7:32. doi: 10.1038/s41392-022-00892-5
61. El-Arif G, Khazaal S, Farhat A, Harb J, Annweiler C, Wu Y, et al. Angiotensin II type I receptor (AT1R): the gate towards COVID-19-associated diseases. Molecules. (2022) 27:2048. doi: 10.3390/molecules27072048
62. Knyazev E, Nersisyan S, Tonevitsky A. Endocytosis and transcytosis of SARS-coV-2 across the intestinal epithelium and other tissue barriers. Front Immunol. (2021) 12:636966. doi: 10.3389/fimmu.2021.636966
63. Hojyo S, Uchida M, Tanaka K, Hasebe R, Tanaka Y, Murakami M, et al. How COVID-19 induces cytokine storm with high mortality. Inflammation Regen. (2020) 40:37. doi: 10.1186/s41232-020-00146-3
64. Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
65. Attiq A, Yao LJ, Afzal S, Khan MA. The triumvirate of NF-kappaB, inflammation and cytokine storm in COVID-19. Int Immunopharmacol. (2021) 101:108255.
66. Cheng DS, Han W, Chen SM, Sherrill TP, Chont M, Park GY, et al. Airway epithelium controls lung inflammation and injury through the NF-kappa B pathway. J Immunol. (2007) 178:6504–13. doi: 10.4049/jimmunol.178.10.6504
67. Ferreira AC, Soares VC, de Azevedo-Quintanilha IG, Dias S, Fintelman-Rodrigues N, Sacramento CQ, et al. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discovery. (2021) 7:43. doi: 10.1038/s41420-021-00428-w
68. Rodrigues TS, de Sa KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L, et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J Exp Med. (2021) 218:e20201707. doi: 10.1084/jem.20201707
69. Bass A, Liu Y, Dakshanamurthy S. Single-cell and bulk RNASeq profiling of COVID-19 patients reveal immune and inflammatory mechanisms of infection-induced organ damage. Viruses. (2021) 13:2418. doi: 10.3390/v13122418
70. Gomez-Rial J, Rivero-Calle I, Salas A, Martinon-Torres F. Role of monocytes/macrophages in covid-19 pathogenesis: implications for therapy. Infect Drug Resist. (2020) 13:2485–93. doi: 10.2147/IDR.S258639
71. Knoll R, Schultze JL, Schulte-Schrepping J. Monocytes and macrophages in COVID-19. Front Immunol. (2021) 12:720109. doi: 10.3389/fimmu.2021.720109
72. Ryabkova VA, Churilov LP, Shoenfeld Y. Influenza infection, SARS, MERS and COVID-19: Cytokine storm - The common denominator and the lessons to be learned. Clin Immunol. (2021) 223:108652. doi: 10.1016/j.clim.2020.108652
73. Pacheco-Hernandez LM, Ramirez-Noyola JA, Gomez-Garcia IA, Ignacio-Cortes S, Zuniga J, Choreno-Parra JA. Comparing the cytokine storms of COVID-19 and pandemic influenza. J Interferon Cytokine Res. (2022) 42:369–92. doi: 10.1089/jir.2022.0029
74. Sinha P, Matthay MA, Calfee CS. Is a “Cytokine storm” Relevant to COVID-19? JAMA Intern Med. (2020) 180:1152–4. doi: 10.1001/jamainternmed.2020.3313
75. Remy KE, Mazer M, Striker DA, Ellebedy AH, Walton AH, Unsinger J, et al. Severe immunosuppression and not a cytokine storm characterizes COVID-19 infections. JCI Insight. (2020) 5:e140329. doi: 10.1172/jci.insight.140329
76. Montazersaheb S, Hosseiniyan Khatibi SM, Hejazi MS, Tarhriz V, Farjami A, Ghasemian Sorbeni F, et al. COVID-19 infection: an overview on cytokine storm and related interventions. Virol J. (2022) 19:92. doi: 10.1186/s12985-022-01814-1
77. Gajjela BK, Zhou MM. Calming the cytokine storm of COVID-19 through inhibition of JAK2/STAT3 signaling. Drug Discovery Today. (2022) 27:390–400. doi: 10.1016/j.drudis.2021.10.016
78. Cron RQ, Caricchio R, Chatham WW. Calming the cytokine storm in COVID-19. Nat Med. (2021) 27:1674–5. doi: 10.1038/s41591-021-01500-9
79. Yang Y, Shen C, Li J, Yuan J, Wei J, Huang F, et al. Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. J Allergy Clin Immunol. (2020) 146:119–27 e4. doi: 10.1016/j.jaci.2020.04.027
80. Han H, Ma Q, Li C, Liu R, Zhao L, Wang W, et al. Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerg Microbes Infect. (2020) 9:1123–30. doi: 10.1080/22221751.2020.1770129
81. Del Valle-Mendoza J, Tarazona-Castro Y, Merino-Luna A, Carrillo-Ng H, Kym S, Aguilar-Luis MA, et al. Comparison of cytokines levels among COVID-19 patients living at sea level and high altitude. BMC Infect Dis. (2022) 22:96. doi: 10.1186/s12879-022-07079-x
82. Buszko M, Nita-Lazar A, Park JH, Schwartzberg PL, Verthelyi D, Young HA, et al. Lessons learned: new insights on the role of cytokines in COVID-19. Nat Immunol. (2021) 22:404–11. doi: 10.1038/s41590-021-00901-9
83. Olbei M, Hautefort I, Modos D, Treveil A, Poletti M, Gul L, et al. SARS-coV-2 causes a different cytokine response compared to other cytokine storm-causing respiratory viruses in severely ill patients. Front Immunol. (2021) 12:629193. doi: 10.3389/fimmu.2021.629193
84. Fahmy OH, Daas FM, Salunkhe V, Petrey JL, Cosar EF, Ramirez J, et al. Is microthrombosis the main pathology in coronavirus disease 2019 severity?-A systematic review of the postmortem pathologic findings. Crit Care Explor. (2021) 3:e0427. doi: 10.1097/CCE.0000000000000427
85. Hatton CF, Botting RA, Duenas ME, Haq IJ, Verdon B, Thompson BJ, et al. Delayed induction of type I and III interferons mediates nasal epithelial cell permissiveness to SARS-CoV-2. Nat Commun. (2021) 12:7092. doi: 10.1038/s41467-021-27318-0
86. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. (2020) 370:eabd4570. doi: 10.1126/science.abd4570
87. Kehrer T, Garcia-Sastre A, Miorin L. Control of innate immune activation by severe acute respiratory syndrome coronavirus 2 and other coronaviruses. J Interferon Cytokine Res. (2021) 41:205–19. doi: 10.1089/jir.2021.0060
88. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
89. Moss P. The T cell immune response against SARS-CoV-2. Nat Immunol. (2022) 23:186–93. doi: 10.1038/s41590-021-01122-w
90. Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T, Hirschenberger M, et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science. (2020) 369:1249–55. doi: 10.1126/science.abc8665
91. Park A, Iwasaki A. Type I and type III interferons - induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe. (2020) 27:870–8. doi: 10.1016/j.chom.2020.05.008
92. Rose KM, Elliott R, Martinez-Sobrido L, Garcia-Sastre A, Weiss SR. Murine coronavirus delays expression of a subset of interferon-stimulated genes. J Virol. (2010) 84:5656–69. doi: 10.1128/JVI.00211-10
93. Zhang R, Li Y, Cowley TJ, Steinbrenner AD, Phillips JM, Yount BL, et al. The nsp1, nsp13, and M proteins contribute to the hepatotropism of murine coronavirus JHM. WU. J Virol. (2015) 89:3598–609. doi: 10.1128/JVI.03535-14
94. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. (2020) 369:718–24. doi: 10.1126/science.abc6027
95. Hoagland DA, Moller R, Uhl SA, Oishi K, Frere J, Golynker I, et al. Leveraging the antiviral type I interferon system as a first line of defense against SARS-CoV-2 pathogenicity. Immunity. (2021) 54:557–70 e5. doi: 10.1016/j.immuni.2021.01.017
96. Stravalaci M, Pagani I, Paraboschi EM, Pedotti M, Doni A, Scavello F, et al. Recognition and inhibition of SARS-CoV-2 by humoral innate immunity pattern recognition molecules. Nat Immunol. (2022) 23:275–86. doi: 10.1038/s41590-021-01114-w
97. Holter JC, Pischke SE, de Boer E, Lind A, Jenum S, Holten AR, et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc Natl Acad Sci U S A. (2020) 117:25018–25. doi: 10.1073/pnas.2010540117
98. Smilowitz NR, Kunichoff D, Garshick M, Shah B, Pillinger M, Hochman JS, et al. C-reactive protein and clinical outcomes in patients with COVID-19. Eur Heart J. (2021) 42:2270–9. doi: 10.1093/eurheartj/ehaa1103
99. Sefik E, Qu R, Junqueira C, Kaffe E, Mirza H, Zhao J, et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature. (2022) 606:585–93. doi: 10.1038/s41586-022-04802-1
100. Park MD. Macrophages: a trojan horse in COVID-19? Nat Rev Immunol. (2020) 20:351. doi: 10.1038/s41577-020-0317-2
101. Abassi Z, Knaney Y, Karram T, Heyman SN. The lung macrophage in SARS-coV-2 infection: A friend or a foe? Front Immunol. (2020) 11:1312. doi: 10.3389/fimmu.2020.01312
102. Pantazi I, Al-Qahtani AA, Alhamlan FS, Alothaid H, Matou-Nasri S, Sourvinos G, et al. SARS-coV-2/ACE2 interaction suppresses IRAK-M expression and promotes pro-inflammatory cytokine production in macrophages. Front Immunol. (2021) 12:683800. doi: 10.3389/fimmu.2021.683800
103. Yang D, Chu H, Hou Y, Chai Y, Shuai H, Lee AC, et al. Attenuated interferon and proinflammatory response in SARS-coV-2-infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. J Infect Dis. (2020) 222:734–45. doi: 10.1093/infdis/jiaa356
104. Laurent P, Yang C, Rendeiro AF, Nilsson-Payant BE, Carrau L, Chandar V, et al. Sensing of SARS-CoV-2 by pDCs and their subsequent production of IFN-I contribute to macrophage-induced cytokine storm during COVID-19. Sci Immunol. (2022) 7:eadd4906. doi: 10.1126/sciimmunol.add4906
105. Gordon S, Pluddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol. (2017) 15:53. doi: 10.1186/s12915-017-0392-4
106. Xu Q, Yang Y, Zhang X, Cai JJ. Association of pyroptosis and severeness of COVID-19 as revealed by integrated single-cell transcriptome data analysis. Immunoinformatics (Amst). (2022) 6:100013. doi: 10.1016/j.immuno.2022.100013
107. Cervantes-Barragan L, Zust R, Weber F, Spiegel M, Lang KS, Akira S, et al. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood. (2007) 109:1131–7. doi: 10.1182/blood-2006-05-023770
108. Venet M, Ribeiro MS, Decembre E, Bellomo A, Joshi G, Nuovo C, et al. Severe COVID-19 patients have impaired plasmacytoid dendritic cell-mediated control of SARS-CoV-2. Nat Commun. (2023) 14:694. doi: 10.1038/s41467-023-36140-9
109. Perez-Gomez A, Vitalle J, Gasca-Capote C, Gutierrez-Valencia A, Trujillo-Rodriguez M, Serna-Gallego A, et al. Dendritic cell deficiencies persist seven months after SARS-CoV-2 infection. Cell Mol Immunol. (2021) 18:2128–39. doi: 10.1038/s41423-021-00728-2
110. Galani IE, Andreakos E. Neutrophils in viral infections: Current concepts and caveats. J Leukoc Biol. (2015) 98:557–64. doi: 10.1189/jlb.4VMR1114-555R
111. Ackermann M, Anders HJ, Bilyy R, Bowlin GL, Daniel C, De Lorenzo R, et al. Patients with COVID-19: in the dark-NETs of neutrophils. Cell Death Differ. (2021) 28:3125–39. doi: 10.1038/s41418-021-00805-z
112. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in wuhan, China. JAMA Intern Med. (2020) 180:934–43. doi: 10.1001/jamainternmed.2020.0994
113. Prozan L, Shusterman E, Ablin J, Mitelpunkt A, Weiss-Meilik A, Adler A, et al. Prognostic value of neutrophil-to-lymphocyte ratio in COVID-19 compared with Influenza and respiratory syncytial virus infection. Sci Rep. (2021) 11:21519. doi: 10.1038/s41598-021-00927-x
114. Liu Y, Du X, Chen J, Jin Y, Peng L, Wang HHX, et al. Neutrophil-to-lymphocyte ratio as an independent risk factor for mortality in hospitalized patients with COVID-19. J Infect. (2020) 81:e6–e12. doi: 10.1016/j.jinf.2020.04.002
115. Schulte-Schrepping J, Reusch N, Paclik D, Bassler K, Schlickeiser S, Zhang B, et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell. (2020) 182:1419–40 e23. doi: 10.1016/j.cell.2020.08.001
116. O’Driscoll DN. Emergency myelopoiesis in critical illness: lessons from the COVID-19 pandemic. Ir J Med Sci. (2022) 192:831–2. doi: 10.1007/s11845-022-03068-w
117. Arcanjo A, Logullo J, Menezes CCB, de Souza Carvalho Giangiarulo TC, Dos Reis MC, de Castro GMM, et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci Rep. (2020) 10:19630. doi: 10.1038/s41598-020-76781-0
118. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science. (2011) 331:44–9. doi: 10.1126/science.1198687
119. Pituch-Noworolska AM. NK cells in SARS-CoV-2 infection. Cent Eur J Immunol. (2022) 47:95–101. doi: 10.5114/ceji.2022.113078
120. Diaz-Salazar C, Sun JC. Coordinated viral control by cytotoxic lymphocytes ensures optimal adaptive NK cell responses. Cell Rep. (2020) 32:108186. doi: 10.1016/j.celrep.2020.108186
121. Maucourant C, Filipovic I, Ponzetta A, Aleman S, Cornillet M, Hertwig L, et al. Natural killer cell immunotypes related to COVID-19 disease severity. Sci Immunol. (2020) 5:eabd6832. doi: 10.1126/sciimmunol.abd6832
122. Bjorkstrom NK, Ponzetta A. Natural killer cells and unconventional T cells in COVID-19. Curr Opin Virol. (2021) 49:176–82. doi: 10.1016/j.coviro.2021.06.005
123. Cifaldi L, Prencipe G, Caiello I, Bracaglia C, Locatelli F, De Benedetti F, et al. Inhibition of natural killer cell cytotoxicity by interleukin-6: implications for the pathogenesis of macrophage activation syndrome. Arthritis Rheumatol. (2015) 67:3037–46. doi: 10.1002/art.39295
124. Gustine JN, Jones D. Immunopathology of hyperinflammation in COVID-19. Am J Pathol. (2021) 191:4–17. doi: 10.1016/j.ajpath.2020.08.009
125. Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science. (2015) 348:aaa6566. doi: 10.1126/science.aaa6566
126. Silverstein NJ, Wang Y, Manickas-Hill Z, Carbone C, Dauphin A, Boribong BP, et al. Innate lymphoid cells and COVID-19 severity in SARS-CoV-2 infection. Elife. (2022) 11:e74681. doi: 10.7554/eLife.74681
127. Kuri-Cervantes L, Pampena MB, Meng W, Rosenfeld AM, Ittner CAG, Weisman AR, et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci Immunol. (2020) 5:eabd7114. doi: 10.1126/sciimmunol.abd7114
128. Deschler S, Kager J, Erber J, Fricke L, Koyumdzhieva P, Georgieva A, et al. Mucosal-associated invariant T (MAIT) cells are highly activated and functionally impaired in COVID-19 patients. Viruses. (2021) 13:241. doi: 10.3390/v13020241
129. Flament H, Rouland M, Beaudoin L, Toubal A, Bertrand L, Lebourgeois S, et al. Outcome of SARS-CoV-2 infection is linked to MAIT cell activation and cytotoxicity. Nat Immunol. (2021) 22:322–35. doi: 10.1038/s41590-021-00870-z
130. Toubal A, Nel I, Lotersztajn S, Lehuen A. Mucosal-associated invariant T cells and disease. Nat Rev Immunol. (2019) 19:643–57. doi: 10.1038/s41577-019-0191-y
131. Singh K, Cogan S, Elekes S, Murphy DM, Cummins S, Curran R, et al. SARS-CoV-2 spike and nucleocapsid proteins fail to activate human dendritic cells or gammadelta T cells. PloS One. (2022) 17:e0271463. doi: 10.1371/journal.pone.0271463
132. Lo Presti E, Dieli F, Meraviglia S. Lymphopenia in COVID-19: gammadelta T cells-based therapeutic opportunities. Vaccines (Basel). (2021) 9:562. doi: 10.3390/vaccines9060562
133. von Massow G, Oh S, Lam A, Gustafsson K. Gamma delta T cells and their involvement in COVID-19 virus infections. Front Immunol. (2021) 12:741218. doi: 10.3389/fimmu.2021.741218
134. Yazdanifar M, Mashkour N, Bertaina A. Making a case for using gammadelta T cells against SARS-CoV-2. Crit Rev Microbiol. (2020) 46:689–702. doi: 10.1080/1040841X.2020.1822279
135. Poccia F, Agrati C, Castilletti C, Bordi L, Gioia C, Horejsh D, et al. Anti-severe acute respiratory syndrome coronavirus immune responses: the role played by V gamma 9V delta 2 T cells. J Infect Dis. (2006) 193:1244–9. doi: 10.1086/502975
136. Lei L, Qian H, Yang X, Zhang X, Zhang D, Dai T, et al. The phenotypic changes of gammadelta T cells in COVID-19 patients. J Cell Mol Med. (2020) 24:11603–6. doi: 10.1111/jcmm.15620
137. Sette A, Crotty S. Adaptive immunity to SARS-coV-2 and COVID-19. Cell. (2021) 184:861–80. doi: 10.1016/j.cell.2021.01.007
138. Tan L, Wang Q, Zhang D, Ding J, Huang Q, Tang YQ, et al. Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Signal Transduct Target Ther. (2020) 5:33. doi: 10.1038/s41392-020-0148-4
139. Yao C, Bora SA, Parimon T, Zaman T, Friedman OA, Palatinus JA, et al. Cell-type-specific immune dysregulation in severely ill COVID-19 patients. Cell Rep. (2021) 34:108590. doi: 10.1016/j.celrep.2020.108590
140. Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, et al. Extrapulmonary manifestations of COVID-19. Nat Med. (2020) 26:1017–32. doi: 10.1038/s41591-020-0968-3
141. Xiang Q, Feng Z, Diao B, Tu C, Qiao Q, Yang H, et al. SARS-coV-2 induces lymphocytopenia by promoting inflammation and decimates secondary lymphoid organs. Front Immunol. (2021) 12:661052. doi: 10.3389/fimmu.2021.661052
142. Huang I, Pranata R. Lymphopenia in severe coronavirus disease-2019 (COVID-19): systematic review and meta-analysis. J Intensive Care. (2020) 8:36. doi: 10.1186/s40560-020-00453-4
143. Xu X, Han M, Li T, Sun W, Wang D, Fu B, et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci U S A. (2020) 117:10970–5. doi: 10.1073/pnas.2005615117
144. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol. (2020) 11:827. doi: 10.3389/fimmu.2020.00827
145. Meckiff BJ, Ramirez-Suastegui C, Fajardo V, Chee SJ, Kusnadi A, Simon H, et al. Imbalance of regulatory and cytotoxic SARS-coV-2-reactive CD4(+) T cells in COVID-19. Cell. (2020) 183:1340–53 e16. doi: 10.1016/j.cell.2020.10.001
146. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T cell responses to SARS-coV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell. (2020) 181:1489–501 e15. doi: 10.1016/j.cell.2020.05.015
147. Tan AT, Linster M, Tan CW, Le Bert N, Chia WN, Kunasegaran K, et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. (2021) 34:108728. doi: 10.1016/j.celrep.2021.108728
148. Rydyznski Moderbacher C, Ramirez SI, Dan JM, Grifoni A, Hastie KM, Weiskopf D, et al. Antigen-specific adaptive immunity to SARS-coV-2 in acute COVID-19 and associations with age and disease severity. Cell. (2020) 183:996–1012 e19. doi: 10.1016/j.cell.2020.09.038
149. Sekine T, Perez-Potti A, Rivera-Ballesteros O, Stralin K, Gorin JB, Olsson A, et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell. (2020) 183:158–68 e14. doi: 10.1016/j.cell.2020.08.017
150. Gao M, Liu Y, Guo M, Wang Q, Wang Y, Fan J, et al. Regulatory CD4(+) and CD8(+) T cells are negatively correlated with CD4(+)/CD8(+) T cell ratios in patients acutely infected with SARS-CoV-2. J Leukoc Biol. (2021) 109:91–7. doi: 10.1002/JLB.5COVA0720-421RR
151. Kaneko N, Kuo HH, Boucau J, Farmer JR, Allard-Chamard H, Mahajan VS, et al. Loss of bcl-6-expressing T follicular helper cells and germinal centers in COVID-19. Cell. (2020) 183:143–57 e13. doi: 10.1016/j.cell.2020.08.025
152. Basso K, Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunol Rev. (2012) 247:172–83. doi: 10.1111/j.1600-065X.2012.01112.x
153. Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol. (2012) 181:8–18. doi: 10.1016/j.ajpath.2012.03.044
154. Jones SA, Hunter CA. Is IL-6 a key cytokine target for therapy in COVID-19? Nat Rev Immunol. (2021) 21:337–9. doi: 10.1038/s41577-021-00553-8
155. Martonik D, Parfieniuk-Kowerda A, Rogalska M, Flisiak R. The role of th17 response in COVID-19. Cells. (2021) 10:1550. doi: 10.3390/cells10061550
156. Sadeghi A, Tahmasebi S, Mahmood A, Kuznetsova M, Valizadeh H, Taghizadieh A, et al. Th17 and Treg cells function in SARS-CoV2 patients compared with healthy controls. J Cell Physiol. (2021) 236:2829–39. doi: 10.1002/jcp.30047
157. Rha MS, Shin EC. Activation or exhaustion of CD8(+) T cells in patients with COVID-19. Cell Mol Immunol. (2021) 18:2325–33. doi: 10.1038/s41423-021-00750-4
158. Nguyen THO, Rowntree LC, Petersen J, Chua BY, Hensen L, Kedzierski L, et al. CD8(+) T cells specific for an immunodominant SARS-CoV-2 nucleocapsid epitope display high naive precursor frequency and TCR promiscuity. Immunity. (2021) 54:1066–82 e5. doi: 10.1016/j.immuni.2021.04.009
159. Varchetta S, Mele D, Oliviero B, Mantovani S, Ludovisi S, Cerino A, et al. Unique immunological profile in patients with COVID-19. Cell Mol Immunol. (2021) 18:604–12. doi: 10.1038/s41423-020-00557-9
160. Song JW, Zhang C, Fan X, Meng FP, Xu Z, Xia P, et al. Immunological and inflammatory profiles in mild and severe cases of COVID-19. Nat Commun. (2020) 11:3410. doi: 10.1038/s41467-020-17240-2
161. Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, Wu JE, et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. (2020) 369:eabc8511. doi: 10.1126/science.abc8511
162. Swadling L, Diniz MO, Schmidt NM, Amin OE, Chandran A, Shaw E, et al. Pre-existing polymerase-specific T cells expand in abortive seronegative SARS-CoV-2. Nature. (2022) 601:110–7. doi: 10.1038/s41586-021-04186-8
163. Grom AA, Horne A, De Benedetti F. Macrophage activation syndrome in the era of biologic therapy. Nat Rev Rheumatol. (2016) 12:259–68. doi: 10.1038/nrrheum.2015.179
164. Jung JH, Rha MS, Sa M, Choi HK, Jeon JH, Seok H, et al. SARS-CoV-2-specific T cell memory is sustained in COVID-19 convalescent patients for 10 months with successful development of stem cell-like memory T cells. Nat Commun. (2021) 12:4043. doi: 10.1038/s41467-021-24377-1
165. Wagner KI, Mateyka LM, Jarosch S, Grass V, Weber S, Schober K, et al. Recruitment of highly cytotoxic CD8(+) T cell receptors in mild SARS-CoV-2 infection. Cell Rep. (2022) 38:110214. doi: 10.1016/j.celrep.2021.110214
166. Adamo S, Michler J, Zurbuchen Y, Cervia C, Taeschler P, Raeber ME, et al. Signature of long-lived memory CD8(+) T cells in acute SARS-CoV-2 infection. Nature. (2022) 602:148–55. doi: 10.1038/s41586-021-04280-x
167. Buggert M, Vella LA, Nguyen S, Wu VH, Chen Z, Sekine T, et al. The identity of human tissue-emigrant CD8(+) T cells. Cell. (2020) 183:1946–61 e15. doi: 10.1016/j.cell.2020.11.019
168. Redd AD, Nardin A, Kared H, Bloch EM, Pekosz A, Laeyendecker O, et al. CD8+ T-cell responses in COVID-19 convalescent individuals target conserved epitopes from multiple prominent SARS-coV-2 circulating variants. Open Forum Infect Dis. (2021) 8:ofab143. doi: 10.1093/ofid/ofab143
169. Keeton R, Tincho MB, Ngomti A, Baguma R, Benede N, Suzuki A, et al. T cell responses to SARS-CoV-2 spike cross-recognize Omicron. Nature. (2022) 603:488–92. doi: 10.1038/s41586-022-04460-3
170. Lu LL, Suscovich TJ, Fortune SM, Alter G. Beyond binding: antibody effector functions in infectious diseases. Nat Rev Immunol. (2018) 18:46–61. doi: 10.1038/nri.2017.106
171. Dan JM, Mateus J, Kato Y, Hastie KM, Yu ED, Faliti CE, et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science. (2021) 371:eabf4063. doi: 10.1126/science.abf4063
172. Lau EHY, Tsang OTY, Hui DSC, Kwan MYW, Chan WH, Chiu SS, et al. Neutralizing antibody titres in SARS-CoV-2 infections. Nat Commun. (2021) 12:63. doi: 10.1038/s41467-020-20247-4
173. Roltgen K, Boyd SD. Antibody and B cell responses to SARS-CoV-2 infection and vaccination. Cell Host Microbe. (2021) 29:1063–75. doi: 10.1016/j.chom.2021.06.009
174. Embong AK, Nguyen-Contant P, Wang J, Kanagaiah P, Chaves FA, Fitzgerald TF, et al. Formation and expansion of memory B cells against coronavirus in acutely infected COVID-19 individuals. Pathogens. (2022) 11:186. doi: 10.3390/pathogens11020186
175. Morgenlander WR, Henson SN, Monaco DR, Chen A, Littlefield K, Bloch EM, et al. Antibody responses to endemic coronaviruses modulate COVID-19 convalescent plasma functionality. J Clin Invest. (2021) 131:e146927. doi: 10.1172/JCI146927
176. Breiman A, Ruven-Clouet N, Le Pendu J. Harnessing the natural anti-glycan immune response to limit the transmission of enveloped viruses such as SARS-CoV-2. PloS Pathog. (2020) 16:e1008556. doi: 10.1371/journal.ppat.1008556
177. Pendu JL, Breiman A, Rocher J, Dion M, Ruvoen-Clouet N. ABO blood types and COVID-19: spurious, anecdotal, or truly important relationships? A reasoned review of available data. Viruses. (2021) 13:160. doi: 10.3390/v13020160
178. Severe Covid GG, Ellinghaus D, Degenhardt F, Bujanda L, Buti M, Albillos A, et al. Genomewide association study of severe covid-19 with respiratory failure. N Engl J Med. (2020) 383:1522–34. doi: 10.1056/NEJMoa2020283
179. Roberts GHL, Partha R, Rhead B, Knight SC, Park DS, Coignet MV, et al. Expanded COVID-19 phenotype definitions reveal distinct patterns of genetic association and protective effects. Nat Genet. (2022) 54:374–81. doi: 10.1038/s41588-022-01042-x
180. Boechat JL, Chora I, Morais A, Delgado L. The immune response to SARS-CoV-2 and COVID-19 immunopathology - Current perspectives. Pulmonology. (2021) 27:423–37. doi: 10.1016/j.pulmoe.2021.03.008
181. Woodruff MC, Ramonell RP, Nguyen DC, Cashman KS, Saini AS, Haddad NS, et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat Immunol. (2020) 21:1506–16. doi: 10.1038/s41590-020-00814-z
182. Long QX, Liu BZ, Deng HJ, Wu GC, Deng K, Chen YK, et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat Med. (2020) 26:845–8. doi: 10.1038/s41591-020-0897-1
183. Iyer AS, Jones FK, Nodoushani A, Kelly M, Becker M, Slater D, et al. Persistence and decay of human antibody responses to the receptor binding domain of SARS-CoV-2 spike protein in COVID-19 patients. Sci Immunol. (2020) 5:eabe0367. doi: 10.1126/sciimmunol.abe0367
184. Laidlaw BJ, Ellebedy AH. The germinal centre B cell response to SARS-CoV-2. Nat Rev Immunol. (2022) 22:7–18. doi: 10.1038/s41577-021-00657-1
185. Muecksch F, Wang Z, Cho A, Gaebler C, Ben Tanfous T, DaSilva J, et al. Increased memory B cell potency and breadth after a SARS-CoV-2 mRNA boost. Nature. (2022) 607:128–34. doi: 10.1038/s41586-022-04778-y
186. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. (2022) 40:413–42. doi: 10.1146/annurev-immunol-120419-022408
187. Pusnik J, Richter E, Schulte B, Dolscheid-Pommerich R, Bode C, Putensen C, et al. Memory B cells targeting SARS-CoV-2 spike protein and their dependence on CD4(+) T cell help. Cell Rep. (2021) 35:109320. doi: 10.1016/j.celrep.2021.109320
188. Turner JS, Kim W, Kalaidina E, Goss CW, Rauseo AM, Schmitz AJ, et al. SARS-CoV-2 infection induces long-lived bone marrow plasma cells in humans. Nature. (2021) 595:421–5. doi: 10.1038/s41586-021-03647-4
189. Halliley JL, Tipton CM, Liesveld J, Rosenberg AF, Darce J, Gregoretti IV, et al. Long-lived plasma cells are contained within the CD19(-)CD38(hi)CD138(+) subset in human bone marrow. Immunity. (2015) 43:132–45. doi: 10.1016/j.immuni.2015.06.016
190. Heinz FX, Stiasny K. Distinguishing features of current COVID-19 vaccines: knowns and unknowns of antigen presentation and modes of action. NPJ Vaccines. (2021) 6:104. doi: 10.1038/s41541-021-00369-6
191. Walsh EE, Frenck RW Jr., Falsey AR, Kitchin N, Absalon J, Gurtman A, et al. Safety and immunogenicity of two RNA-based covid-19 vaccine candidates. N Engl J Med. (2020) 383:2439–50. doi: 10.1056/NEJMoa2027906
192. Goel RR, Painter MM, Apostolidis SA, Mathew D, Meng W, Rosenfeld AM, et al. mRNA vaccines induce durable immune memory to SARS-CoV-2 and variants of concern. Science. (2021) 374:abm0829. doi: 10.1126/science.abm0829
193. Wherry EJ, Barouch DH. T cell immunity to COVID-19 vaccines. Science. (2022) 377:821–2. doi: 10.1126/science.add2897
194. Chandrashekar A, Yu J, McMahan K, Jacob-Dolan C, Liu J, He X, et al. Vaccine protection against the SARS-CoV-2 Omicron variant in macaques. Cell. (2022) 185:1549–55 e11. doi: 10.1016/j.cell.2022.03.024
195. Liu J, Chandrashekar A, Sellers D, Barrett J, Jacob-Dolan C, Lifton M, et al. Vaccines elicit highly conserved cellular immunity to SARS-CoV-2 Omicron. Nature. (2022) 603:493–6. doi: 10.1038/s41586-022-04465-y
196. Wong CKH, Wong JYH, Tang EHM, Au CH, Wai AKC. Clinical presentations, laboratory and radiological findings, and treatments for 11,028 COVID-19 patients: a systematic review and meta-analysis. Sci Rep. (2020) 10:19765. doi: 10.1038/s41598-020-74988-9
197. Ho HT, Peischard S, Strutz-Seebohm N, Klingel K, Seebohm G. Myocardial damage by SARS-coV-2: emerging mechanisms and therapies. Viruses. (2021) 13:1880. doi: 10.3390/v13091880
198. Cui H, Su S, Cao Y, Ma C, Qiu W. The altered anatomical distribution of ACE2 in the brain with alzheimer’s disease pathology. Front Cell Dev Biol. (2021) 9:684874. doi: 10.3389/fcell.2021.684874
199. Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol. (2022) 23:3–20. doi: 10.1038/s41580-021-00418-x
200. Thakur V, Ratho RK, Kumar P, Bhatia SK, Bora I, Mohi GK, et al. Multi-organ involvement in COVID-19: beyond pulmonary manifestations. J Clin Med. (2021) 10:446. doi: 10.3390/jcm10030446
201. Hikmet F, Mear L, Edvinsson A, Micke P, Uhlen M, Lindskog C. The protein expression profile of ACE2 in human tissues. Mol Syst Biol. (2020) 16:e9610. doi: 10.15252/msb.20209610
202. Xu J, Chu M, Zhong F, Tan X, Tang G, Mai J, et al. Digestive symptoms of COVID-19 and expression of ACE2 in digestive tract organs. Cell Death Discovery. (2020) 6:76. doi: 10.1038/s41420-020-00307-w
203. Albini A, Calabrone L, Carlini V, Benedetto N, Lombardo M, Bruno A, et al. Preliminary evidence for IL-10-induced ACE2 mRNA expression in lung-derived and endothelial cells: implications for SARS-cov-2 ARDS pathogenesis. Front Immunol. (2021) 12:718136. doi: 10.3389/fimmu.2021.718136
204. Guney C, Akar F. Epithelial and endothelial expressions of ACE2: SARS-coV-2 entry routes. J Pharm Pharm Sci. (2021) 24:84–93. doi: 10.18433/jpps31455
205. Maccio U, Zinkernagel AS, Shambat SM, Zeng X, Cathomas G, Ruschitzka F, et al. SARS-CoV-2 leads to a small vessel endotheliitis in the heart. EBioMedicine. (2021) 63:103182. doi: 10.1016/j.ebiom.2020.103182
206. Bois MC, Boire NA, Layman AJ, Aubry MC, Alexander MP, Roden AC, et al. COVID-19-associated nonocclusive fibrin microthrombi in the heart. Circulation. (2021) 143:230–43. doi: 10.1161/CIRCULATIONAHA.120.050754
207. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
208. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in covid-19. N Engl J Med. (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
209. Ackermann M, Mentzer SJ, Kolb M, Jonigk D. Inflammation and intussusceptive angiogenesis in COVID-19: everything in and out of flow. Eur Respir J. (2020) 56:2003147. doi: 10.1183/13993003.03147-2020
210. Kichloo A, Dettloff K, Aljadah M, Albosta M, Jamal S, Singh J, et al. COVID-19 and hypercoagulability: A review. Clin Appl Thromb Hemost. (2020) 26:1076029620962853. doi: 10.1177/1076029620962853
211. Lowenstein CJ, Solomon SD. Severe COVID-19 is a microvascular disease. Circulation. (2020) 142:1609–11. doi: 10.1161/CIRCULATIONAHA.120.050354
212. Norooznezhad AH, Mansouri K. Endothelial cell dysfunction, coagulation, and angiogenesis in coronavirus disease 2019 (COVID-19). Microvasc Res. (2021) 137:104188. doi: 10.1016/j.mvr.2021.104188
213. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. (2020) 136:1169–79. doi: 10.1182/blood.2020007008
214. Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity. (2019) 50:1401–11 e4. doi: 10.1016/j.immuni.2019.04.003
215. Gedefaw L, Ullah S, Leung PHM, Cai Y, Yip SP, Huang CL. Inflammasome activation-induced hypercoagulopathy: impact on cardiovascular dysfunction triggered in COVID-19 patients. Cells. (2021) 10:916. doi: 10.3390/cells10040916
216. Grover SP, Mackman N. Tissue factor: an essential mediator of hemostasis and trigger of thrombosis. Arterioscler Thromb Vasc Biol. (2018) 38:709–25. doi: 10.1161/ATVBAHA.117.309846
217. Patra T, Ray R. IL-6 induction and signaling: horizons of COVID-19-related pathogenesis. DNA Cell Biol. (2021) 40:639–42. doi: 10.1089/dna.2021.0152
218. Popescu NI, Lupu C, Lupu F. Disseminated intravascular coagulation and its immune mechanisms. Blood. (2022) 139:1973–86. doi: 10.1182/blood.2020007208
219. Torres Acosta MA, Singer BD. Pathogenesis of COVID-19-induced ARDS: implications for an ageing population. Eur Respir J. (2020) 56:2002049. doi: 10.1183/13993003.02049-2020
220. Fuehner T, Renger I, Welte T, Freundt T, Gottlieb J. Silent hypoxia in COVID-19: A case series. Respiration. (2022) 101:376–80. doi: 10.1159/000520083
221. Simonson TS, Baker TL, Banzett RB, Bishop T, Dempsey JA, Feldman JL, et al. Silent hypoxaemia in COVID-19 patients. J Physiol. (2021) 599:1057–65. doi: 10.1113/JP280769
222. Karagiannidis C, Hentschker C, Westhoff M, Weber-Carstens S, Janssens U, Kluge S, et al. Observational study of changes in utilization and outcomes in mechanical ventilation in COVID-19. PloS One. (2022) 17:e0262315. doi: 10.1371/journal.pone.0262315
223. Monteil V, Kwon H, Prado P, Hagelkruys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-coV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. (2020) 181:905–13 e7. doi: 10.1016/j.cell.2020.04.004
224. Hsu CY, Lai CC, Yeh YP, Chang-Chuan C, Chen HH. Progression from pneumonia to ARDS as a predictor for fatal COVID-19. J Infect Public Health. (2021) 14:504–7. doi: 10.1016/j.jiph.2020.12.026
225. Elicker BM. What are the long-term pulmonary sequelae of COVID-19 infection? Radiology. (2022) 304:193–4. doi: 10.1148/radiol.220449
226. Gattinoni L, Chiumello D, Caironi P, Busana M, Romitti F, Brazzi L, et al. COVID-19 pneumonia: different respiratory treatments for different phenotypes? Intensive Care Med. (2020) 46:1099–102. doi: 10.1007/s00134-020-06033-2
227. Bos LDJ, Sjoding M, Sinha P, Bhavani SV, Lyons PG, Bewley AF, et al. Longitudinal respiratory subphenotypes in patients with COVID-19-related acute respiratory distress syndrome: results from three observational cohorts. Lancet Respir Med. (2021) 9:1377–86. doi: 10.1016/S2213-2600(21)00365-9
228. Haudebourg AF, Perier F, Tuffet S, de Prost N, Razazi K, Mekontso Dessap A, et al. Respiratory mechanics of COVID-19- versus non-COVID-19-associated acute respiratory distress syndrome. Am J Respir Crit Care Med. (2020) 202:287–90. doi: 10.1164/rccm.202004-1226LE
229. Bain W, Yang H, Shah FA, Suber T, Drohan C, Al-Yousif N, et al. COVID-19 versus non-COVID-19 acute respiratory distress syndrome: comparison of demographics, physiologic parameters, inflammatory biomarkers, and clinical outcomes. Ann Am Thorac Soc. (2021) 18:1202–10. doi: 10.1513/AnnalsATS.202008-1026OC
230. Bos LDJ. COVID-19-related acute respiratory distress syndrome: not so atypical. Am J Respir Crit Care Med. (2020) 202:622–4. doi: 10.1164/rccm.202004-1423LE
231. Konopka KE, Nguyen T, Jentzen JM, Rayes O, Schmidt CJ, Wilson AM, et al. Diffuse alveolar damage (DAD) resulting from coronavirus disease 2019 Infection is Morphologically Indistinguishable from Other Causes of DAD. Histopathology. (2020) 77:570–8. doi: 10.1111/his.14180
232. Group RC, Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, et al. Dexamethasone in hospitalized patients with covid-19. N Engl J Med. (2021) 384:693–704. doi: 10.1056/NEJMoa2021436
233. Villar J, Ferrando C, Martinez D, Ambros A, Munoz T, Soler JA, et al. Dexamethasone treatment for the acute respiratory distress syndrome: a multicentre, randomised controlled trial. Lancet Respir Med. (2020) 8:267–76. doi: 10.1016/S2213-2600(19)30417-5
234. Hattori Y, Hattori K, Machida T, Matsuda N. Vascular endotheliitis associated with infections: Its pathogenetic role and therapeutic implication. Biochem Pharmacol. (2022) 197:114909. doi: 10.1016/j.bcp.2022.114909
235. Haberecker M, Schwarz EI, Steiger P, Frontzek K, Scholkmann F, Zeng X, et al. Autopsy-based pulmonary and vascular pathology: pulmonary endotheliitis and multi-organ involvement in COVID-19 associated deaths. Respiration. (2022) 101:155–65. doi: 10.1159/000518914
236. Abou-Ismail MY, Diamond A, Kapoor S, Arafah Y, Nayak L. The hypercoagulable state in COVID-19: Incidence, pathophysiology, and management. Thromb Res. (2020) 194:101–15. doi: 10.1016/j.thromres.2020.06.029
237. Investigators A, Investigators AC-a, Investigators R-C, Lawler PR, Goligher EC, Berger JS, et al. Therapeutic anticoagulation with heparin in noncritically ill patients with covid-19. N Engl J Med. (2021) 385:790–802. doi: 10.1056/NEJMoa2105911
238. Langner RO, Fuller GC. Collagen synthesis in thoracic aortas of rabbits with epinephrine-thyroxine induced arteriosclerosis. Atherosclerosis. (1973) 17:463–9. doi: 10.1016/0021-9150(73)90036-1
239. Gong X, Yuan B, Yuan Y. Incidence and prognostic value of pulmonary embolism in COVID-19: A systematic review and meta-analysis. PloS One. (2022) 17:e0263580. doi: 10.1371/journal.pone.0263580
240. Fraser M, Agdamag ACC, Maharaj VR, Mutschler M, Charpentier V, Chowdhury M, et al. COVID-19-associated myocarditis: an evolving concern in cardiology and beyond. Biol (Basel). (2022) 11:520. doi: 10.3390/biology11040520
241. Peretto G, Villatore A, Rizzo S, Esposito A, De Luca G, Palmisano A, et al. The spectrum of COVID-19-associated myocarditis: A patient-tailored multidisciplinary approach. J Clin Med. (2021) 10:1974. doi: 10.3390/jcm10091974
242. Haussner W, DeRosa AP, Haussner D, Tran J, Torres-Lavoro J, Kamler J, et al. COVID-19 associated myocarditis: A systematic review. Am J Emerg Med. (2022) 51:150–5. doi: 10.1016/j.ajem.2021.10.001
243. Oleszak F, Maryniak A, Botti E, Abrahim C, Salifu MO, Youssef M, et al. Myocarditis associated with COVID-19. Am J Med Case Rep. (2020) 8:498–502. doi: 10.12691/ajmcr-8-12-19
244. Boehmer TK, Kompaniyets L, Lavery AM, Hsu J, Ko JY, Yusuf H, et al. Association between COVID-19 and myocarditis using hospital-based administrative data - United States, march 2020-january 2021. MMWR Morb Mortal Wkly Rep. (2021) 70:1228–32. doi: 10.15585/mmwr.mm7035e5
245. Gauchotte G, Venard V, Segondy M, Cadoz C, Esposito-Fava A, Barraud D, et al. SARS-Cov-2 fulminant myocarditis: an autopsy and histopathological case study. Int J Legal Med. (2021) 135:577–81. doi: 10.1007/s00414-020-02500-z
246. Wenzel P, Kopp S, Gobel S, Jansen T, Geyer M, Hahn F, et al. Evidence of SARS-CoV-2 mRNA in endomyocardial biopsies of patients with clinically suspected myocarditis tested negative for COVID-19 in nasopharyngeal swab. Cardiovasc Res. (2020) 116:1661–3. doi: 10.1093/cvr/cvaa160
247. Katsoularis I, Fonseca-Rodriguez O, Farrington P, Lindmark K, Fors Connolly AM. Risk of acute myocardial infarction and ischaemic stroke following COVID-19 in Sweden: a self-controlled case series and matched cohort study. Lancet. (2021) 398:599–607. doi: 10.1016/S0140-6736(21)00896-5
248. Talanas G, Dossi F, Parodi G. Type 2 myocardial infarction in patients with coronavirus disease 2019. J Cardiovasc Med (Hagerstown). (2021) 22:603–5. doi: 10.2459/JCM.0000000000001136
249. Stefanini GG, Montorfano M, Trabattoni D, Andreini D, Ferrante G, Ancona M, et al. ST-elevation myocardial infarction in patients with COVID-19: clinical and angiographic outcomes. Circulation. (2020) 141:2113–6. doi: 10.1161/CIRCULATIONAHA.120.047525
250. Arhontoulis DC, Kerr C, Richards D, Tjen K, Hyams N, Jones JA, et al. Human cardiac organoids to model COVID-19 cytokine storm induced cardiac injuries. bioRxiv. (2022) 16:799–811. doi: 10.1101/2022.01.31.478497
251. Beghi E, Giussani G, Westenberg E, Allegri R, Garcia-Azorin D, Guekht A, et al. Acute and post-acute neurological manifestations of COVID-19: present findings, critical appraisal, and future directions. J Neurol. (2022) 269:2265–74. doi: 10.1007/s00415-021-10848-4
252. Chou SH, Beghi E, Helbok R, Moro E, Sampson J, Altamirano V, et al. Global incidence of neurological manifestations among patients hospitalized with COVID-19-A report for the GCS-neuroCOVID consortium and the ENERGY consortium. JAMA Netw Open. (2021) 4:e2112131. doi: 10.1001/jamanetworkopen.2021.12131
253. Merkler AE, Parikh NS, Mir S, Gupta A, Kamel H, Lin E, et al. Risk of ischemic stroke in patients with coronavirus disease 2019 (COVID-19) vs patients with influenza. JAMA Neurol. (2020) 77:1–7. doi: 10.1001/jamaneurol.2020.2730
254. Nersesjan V, Amiri M, Christensen HK, Benros ME, Kondziella D. Thirty-Day Mortality and Morbidity in COVID-19 Positive vs. COVID-19 Negative Individuals and vs. Individuals Tested for Influenza A/B: A Population-Based Study. Front Med (Lausanne). (2020) 7:598272. doi: 10.3389/fmed.2020.598272
255. Varatharaj A, Thomas N, Ellul MA, Davies NWS, Pollak TA, Tenorio EL, et al. Neurological and neuropsychiatric complications of COVID-19 in 153 patients: a UK-wide surveillance study. Lancet Psychiatry. (2020) 7:875–82. doi: 10.1016/S2215-0366(20)30287-X
256. Han Y, Yuan K, Wang Z, Liu WJ, Lu ZA, Liu L, et al. Neuropsychiatric manifestations of COVID-19, potential neurotropic mechanisms, and therapeutic interventions. Transl Psychiatry. (2021) 11:499. doi: 10.1038/s41398-021-01629-8
257. Heming M, Li X, Rauber S, Mausberg AK, Borsch AL, Hartlehnert M, et al. Neurological manifestations of COVID-19 feature T cell exhaustion and dedifferentiated monocytes in cerebrospinal fluid. Immunity. (2021) 54:164–75 e6. doi: 10.1016/j.immuni.2020.12.011
258. Franceschi AM, Ahmed O, Giliberto L, Castillo M. Hemorrhagic posterior reversible encephalopathy syndrome as a manifestation of COVID-19 infection. AJNR Am J Neuroradiol. (2020) 41:1173–6. doi: 10.3174/ajnr.A6595
259. Hartzell S, Bin S, Cantarelli C, Haverly M, Manrique J, Angeletti A, et al. Kidney failure associates with T cell exhaustion and imbalanced follicular helper T cells. Front Immunol. (2020) 11:583702. doi: 10.3389/fimmu.2020.583702
260. Cantarelli C, Angeletti A, Perin L, Russo LS, Sabiu G, Podesta MA, et al. Immune responses to SARS-CoV-2 in dialysis and kidney transplantation. Clin Kidney J. (2022) 15:1816–28. doi: 10.1093/ckj/sfac174
261. Banham GD, Godlee A, Faustini SE, Cunningham AF, Richter A, Harper L, et al. Hemodialysis patients make long-lived antibodies against SARS-coV-2 that may be associated with reduced reinfection. J Am Soc Nephrol. (2021) 32:2140–2. doi: 10.1681/ASN.2021020188
262. Clarke CL, Prendecki M, Dhutia A, Gan J, Edwards C, Prout V, et al. Longevity of SARS-CoV-2 immune responses in hemodialysis patients and protection against reinfection. Kidney Int. (2021) 99:1470–7. doi: 10.1016/j.kint.2021.03.009
263. Shankar S, Beckett J, Tipton T, Ogbe A, Kasanyinga M, Dold C, et al. SARS-coV-2-specific T cell responses are not associated with protection against reinfection in hemodialysis patients. J Am Soc Nephrol. (2022) 33:883–7. doi: 10.1681/ASN.2021121587
264. Aguilar-Bretones M, den Hartog Y, van Dijk LLA, Malahe SRK, Dieterich M, Mora HT, et al. SARS-CoV-2-specific immune responses converge in kidney disease patients and controls with hybrid immunity. NPJ Vaccines. (2024) 9:93. doi: 10.1038/s41541-024-00886-0
265. Pei G, Zhang Z, Peng J, Liu L, Zhang C, Yu C, et al. Renal involvement and early prognosis in patients with COVID-19 pneumonia. J Am Soc Nephrol. (2020) 31:1157–65. doi: 10.1681/ASN.2020030276
266. Faour WH, Choaib A, Issa E, Choueiry FE, Shbaklo K, Alhajj M, et al. Mechanisms of COVID-19-induced kidney injury and current pharmacotherapies. Inflammation Res. (2022) 71:39–56. doi: 10.1007/s00011-021-01520-8
267. Legrand M, Bell S, Forni L, Joannidis M, Koyner JL, Liu K, et al. Pathophysiology of COVID-19-associated acute kidney injury. Nat Rev Nephrol. (2021) 17:751–64. doi: 10.1038/s41581-021-00452-0
268. Sullivan MK, Lees JS, Drake TM, Docherty AB, Oates G, Hardwick HE, et al. Acute kidney injury in patients hospitalized with COVID-19 from the ISARIC WHO CCP-UK Study: a prospective, multicentre cohort study. Nephrol Dial Transplant. (2022) 37:271–84. doi: 10.1093/ndt/gfab303
269. Oetjens MT, Luo JZ, Chang A, Leader JB, Hartzel DN, Moore BS, et al. Electronic health record analysis identifies kidney disease as the leading risk factor for hospitalization in confirmed COVID-19 patients. PloS One. (2020) 15:e0242182. doi: 10.1371/journal.pone.0242182
270. May RM, Cassol C, Hannoudi A, Larsen CP, Lerma EV, Haun RS, et al. A multi-center retrospective cohort study defines the spectrum of kidney pathology in Coronavirus 2019 Disease (COVID-19). Kidney Int. (2021) 100:1303–15. doi: 10.1016/j.kint.2021.07.015
271. Nasr SH, Kopp JB. COVID-19-associated collapsing glomerulopathy: an emerging entity. Kidney Int Rep. (2020) 5:759–61. doi: 10.1016/j.ekir.2020.04.030
272. Hassler L, Batlle D. Potential SARS-CoV-2 kidney infection and paths to injury. Nat Rev Nephrol. (2022) 18:275–6. doi: 10.1038/s41581-022-00551-6
273. Velez JCQ, Caza T, Larsen CP. COVAN is the new HIVAN: the re-emergence of collapsing glomerulopathy with COVID-19. Nat Rev Nephrol. (2020) 16:565–7. doi: 10.1038/s41581-020-0332-3
274. Guimaraes Sousa S, Kleiton de Sousa A, Maria Carvalho Pereira C, Sofia Miranda Loiola Araujo A, de Aguiar Magalhaes D, Vieira de Brito T, et al. SARS-CoV-2 infection causes intestinal cell damage: Role of interferon’s imbalance. Cytokine. (2022) 152:155826. doi: 10.1016/j.cyto.2022.155826
275. Xu Y, Li X, Zhu B, Liang H, Fang C, Gong Y, et al. Characteristics of pediatric SARS-CoV-2 infection and potential evidence for persistent fecal viral shedding. Nat Med. (2020) 26:502–5. doi: 10.1038/s41591-020-0817-4
276. Feng Y, Zeng D, Hu L, Yang Y, Song S, Shi Y, et al. Case report: histopathology and molecular pathology analysis on enteric tissue of a COVID-19 patient. Diagn Pathol. (2021) 16:40. doi: 10.1186/s13000-021-01082-7
277. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced host response to SARS-coV-2 drives development of COVID-19. Cell. (2020) 181:1036–45 e9. doi: 10.1016/j.cell.2020.04.026
278. Lamers MM, Beumer J, van der Vaart J, Knoops K, Puschhof J, Breugem TI, et al. SARS-CoV-2 productively infects human gut enterocytes. Science. (2020) 369:50–4. doi: 10.1126/science.abc1669
279. Saithong S, Worasilchai N, Saisorn W, Udompornpitak K, Bhunyakarnjanarat T, Chindamporn A, et al. Neutrophil extracellular traps in severe SARS-coV-2 infection: A possible impact of LPS and (1–>3)-beta-D-glucan in blood from gut translocation. Cells. (2022) 11:1103. doi: 10.3390/cells11071103
280. Kruglikov IL, Scherer PE. Preexisting and inducible endotoxemia as crucial contributors to the severity of COVID-19 outcomes. PloS Pathog. (2021) 17:e1009306. doi: 10.1371/journal.ppat.1009306
281. Sun Z, Song ZG, Liu C, Tan S, Lin S, Zhu J, et al. Gut microbiome alterations and gut barrier dysfunction are associated with host immune homeostasis in COVID-19 patients. BMC Med. (2022) 20:24. doi: 10.1186/s12916-021-02212-0
282. Zhang F, Lau RI, Liu Q, Su Q, Chan FKL, Ng SC. Gut microbiota in COVID-19: key microbial changes, potential mechanisms and clinical applications. Nat Rev Gastroenterol Hepatol. (2023) 20:323–37. doi: 10.1038/s41575-022-00698-4
283. Chen C, Haupert SR, Zimmermann L, Shi X, Fritsche LG, Mukherjee B. Global prevalence of post-coronavirus disease 2019 (COVID-19) condition or long COVID: A meta-analysis and systematic review. J Infect Dis. (2022) 226:1593–607. doi: 10.1093/infdis/jiac136
284. Ayoubkhani D, Khunti K, Nafilyan V, Maddox T, Humberstone B, Diamond I, et al. Post-covid syndrome in individuals admitted to hospital with covid-19: retrospective cohort study. BMJ. (2021) 372:n693. doi: 10.1136/bmj.n693
285. Su Y, Yuan D, Chen DG, Ng RH, Wang K, Choi J, et al. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell. (2022) 185:881–95 e20. doi: 10.1016/j.cell.2022.01.014
286. Groff D, Sun A, Ssentongo AE, Ba DM, Parsons N, Poudel GR, et al. Short-term and long-term rates of postacute sequelae of SARS-coV-2 infection: A systematic review. JAMA Netw Open. (2021) 4:e2128568. doi: 10.1001/jamanetworkopen.2021.28568
287. Russell CD, Lone NI, Baillie JK. Comorbidities, multimorbidity and COVID-19. Nat Med. (2023) 29:334–43. doi: 10.1038/s41591-022-02156-9
288. Klein J, Wood J, Jaycox JR, Dhodapkar RM, Lu P, Gehlhausen JR, et al. Distinguishing features of Long COVID identified through immune profiling. Nature. (2023) 623:139–48. doi: 10.1038/s41586-023-06651-y
289. Stein SR, Ramelli SC, Grazioli A, Chung JY, Singh M, Yinda CK, et al. SARS-CoV-2 infection and persistence in the human body and brain at autopsy. Nature. (2022) 612:758–63. doi: 10.1038/s41586-022-05542-y
290. Swank Z, Senussi Y, Manickas-Hill Z, Yu XG, Li JZ, Alter G, et al. Persistent circulating SARS-CoV-2 spike is associated with post-acute COVID-19 sequelae. Clin Infect Dis. (2022) 76:e487-90. doi: 10.1101/2022.06.14.22276401
291. Mantovani A, Morrone MC, Patrono C, Santoro MG, Schiaffino S, Remuzzi G, et al. Long Covid: where we stand and challenges ahead. Cell Death Differ. (2022) 29:1891–900. doi: 10.1038/s41418-022-01052-6
292. Davis HE, McCorkell L, Vogel JM, Topol EJ. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol. (2023) 21:133–46. doi: 10.1038/s41579-023-00896-0
293. Griffin DE. Why does viral RNA sometimes persist after recovery from acute infections? PloS Biol. (2022) 20:e3001687. doi: 10.1371/journal.pbio.3001687
294. Kuhl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M, Poller W, et al. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation. (2005) 112:1965–70. doi: 10.1161/CIRCULATIONAHA.105.548156
295. Gold JE, Okyay RA, Licht WE, Hurley DJ. Investigation of long COVID prevalence and its relationship to epstein-barr virus reactivation. Pathogens. (2021) 10:763. doi: 10.3390/pathogens10060763
296. Bernal KDE, Whitehurst CB. Incidence of Epstein-Barr virus reactivation is elevated in COVID-19 patients. Virus Res. (2023) 334:199157. doi: 10.1016/j.virusres.2023.199157
297. Sepulveda N, Malato J, Sotzny F, Grabowska AD, Fonseca A, Cordeiro C, et al. Revisiting igG antibody reactivity to epstein-barr virus in myalgic encephalomyelitis/chronic fatigue syndrome and its potential application to disease diagnosis. Front Med (Lausanne). (2022) 9:921101. doi: 10.3389/fmed.2022.921101
298. Falzon D, Zignol M, Bastard M, Floyd K, Kasaeva T. The impact of the COVID-19 pandemic on the global tuberculosis epidemic. Front Immunol. (2023) 14:1234785. doi: 10.3389/fimmu.2023.1234785
299. Habib S, Hamza E, El-Gamal R, Nosser NA, Aboukamar WA, Abdelsalam S, et al. Clinical and immunological impacts of latent toxoplasmosis on COVID-19 patients. Cureus. (2023) 15:e45989. doi: 10.7759/cureus.45989
300. Hasichaolu, Zhang X, Li X, Li X, Li D. Circulating cytokines and lymphocyte subsets in patients who have recovered from COVID-19. BioMed Res Int. (2020) 2020:7570981. doi: 10.1155/2020/7570981
301. Phetsouphanh C, Darley DR, Wilson DB, Howe A, Munier CML, Patel SK, et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat Immunol. (2022) 23:210–6. doi: 10.1038/s41590-021-01113-x
302. Schultheiss C, Willscher E, Paschold L, Gottschick C, Klee B, Henkes SS, et al. The IL-1beta, IL-6, and TNF cytokine triad is associated with post-acute sequelae of COVID-19. Cell Rep Med. (2022) 3:100663. doi: 10.1016/j.xcrm.2022.100663
303. Low RN, Low RJ, Akrami A. A review of cytokine-based pathophysiology of Long COVID symptoms. Front Med (Lausanne). (2023) 10:1011936. doi: 10.3389/fmed.2023.1011936
304. Zhao N, Di B, Xu LL. The NLRP3 inflammasome and COVID-19: Activation, pathogenesis and therapeutic strategies. Cytokine Growth Factor Rev. (2021) 61:2–15. doi: 10.1016/j.cytogfr.2021.06.002
305. Shen HH, Yang YX, Meng X, Luo XY, Li XM, Shuai ZW, et al. NLRP3: A promising therapeutic target for autoimmune diseases. Autoimmun Rev. (2018) 17:694–702. doi: 10.1016/j.autrev.2018.01.020
306. Potere N, Del Buono MG, Caricchio R, Cremer PC, Vecchie A, Porreca E, et al. Interleukin-1 and the NLRP3 inflammasome in COVID-19: Pathogenetic and therapeutic implications. EBioMedicine. (2022) 85:104299. doi: 10.1016/j.ebiom.2022.104299
307. Wang M, Yu F, Chang W, Zhang Y, Zhang L, Li P. Inflammasomes: a rising star on the horizon of COVID-19 pathophysiology. Front Immunol. (2023) 14:1185233. doi: 10.3389/fimmu.2023.1185233
308. Chang SE, Feng A, Meng W, Apostolidis SA, Mack E, Artandi M, et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat Commun. (2021) 12:5417. doi: 10.1038/s41467-021-25509-3
309. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. (2020) 370:eabd4585. doi: 10.1126/science.abd4585
310. Ahamed J, Laurence J. Long COVID endotheliopathy: hypothesized mechanisms and potential therapeutic approaches. J Clin Invest. (2022) 132:e161167. doi: 10.1172/JCI161167
311. De Biasi S, Meschiari M, Gibellini L, Bellinazzi C, Borella R, Fidanza L, et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat Commun. (2020) 11:3434. doi: 10.1038/s41467-020-17292-4
312. Cao J, Wang C, Zhang Y, Lei G, Xu K, Zhao N, et al. Integrated gut virome and bacteriome dynamics in COVID-19 patients. Gut Microbes. (2021) 13:1–21. doi: 10.1080/19490976.2021.1887722
313. Molloy EJ, Nakra N, Gale C, Dimitriades VR, Lakshminrusimha S. Multisystem inflammatory syndrome in children (MIS-C) and neonates (MIS-N) associated with COVID-19: optimizing definition and management. Pediatr Res. (2023) 93:1499–508. doi: 10.1038/s41390-022-02263-w
314. Ahmad F, Ahmed A, Rajendraprasad SS, Loranger A, Gupta S, Velagapudi M, et al. Multisystem inflammatory syndrome in adults: A rare sequela of SARS-CoV-2 infection. Int J Infect Dis. (2021) 108:209–11. doi: 10.1016/j.ijid.2021.05.050
315. Godfred-Cato S, Bryant B, Leung J, Oster ME, Conklin L, Abrams J, et al. COVID-19-associated multisystem inflammatory syndrome in children - United States, march-july 2020. MMWR Morb Mortal Wkly Rep. (2020) 69:1074–80. doi: 10.15585/mmwr.mm6932e2
316. Available online at: https://www.cdc.gov/mis/mis-a/hcp.html (Accessed December 02, 2022).
317. Vogel TP, Top KA, Karatzios C, Hilmers DC, Tapia LI, Moceri P, et al. Multisystem inflammatory syndrome in children and adults (MIS-C/A): Case definition & guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine. (2021) 39:3037–49. doi: 10.1016/j.vaccine.2021.01.054
318. Sperotto F, Friedman KG, Son MBF, VanderPluym CJ, Newburger JW, Dionne A. Cardiac manifestations in SARS-CoV-2-associated multisystem inflammatory syndrome in children: a comprehensive review and proposed clinical approach. Eur J Pediatr. (2021) 180:307–22. doi: 10.1007/s00431-020-03766-6
319. Shen M, Milner A, Foppiano Palacios C, Ahmad T. Multisystem inflammatory syndrome in adults (MIS-A) associated with SARS-CoV-2 infection with delayed-onset myocarditis: case report. Eur Heart J Case Rep. (2021) 5:ytab470. doi: 10.1093/ehjcr/ytab470
320. Kaufmann CC, Simon A, Reinhart-Mikocki D, Publig S, Huber K, Freynhofer MK. Multisystem inflammatory syndrome in adults in a young male following severe acute respiratory syndrome coronavirus-2 infection: a case report. Eur Heart J Case Rep. (2022) 6:ytab521. doi: 10.1093/ehjcr/ytab521
321. Aldeghaither S, Qutob R, Assanangkornchai N, Issa-Chergui B, Tam M, Larotondo R, et al. Clinical and histopathologic features of myocarditis in multisystem inflammatory syndrome (Adult)-associated COVID-19. Crit Care Explor. (2022) 10:e0630. doi: 10.1097/CCE.0000000000000630
322. McGrath LJ, Scott AM, Surinach A, Chambers R, Benigno M, Malhotra D. Use of the postacute sequelae of COVID-19 diagnosis code in routine clinical practice in the US. JAMA Netw Open. (2022) 5:e2235089. doi: 10.1001/jamanetworkopen.2022.35089
323. Tsampasian V, Elghazaly H, Chattopadhyay R, Debski M, Naing TKP, Garg P, et al. Risk factors associated with post-COVID-19 condition: A systematic review and meta-analysis. JAMA Intern Med. (2023) 183:566–80. doi: 10.1001/jamainternmed.2023.0750
324. Xie Y, Xu E, Bowe B, Al-Aly Z. Long-term cardiovascular outcomes of COVID-19. Nat Med. (2022) 28:583–90. doi: 10.1038/s41591-022-01689-3
325. Wan EYF, Mathur S, Zhang R, Yan VKC, Lai FTT, Chui CSL, et al. Association of COVID-19 with short- and long-term risk of cardiovascular disease and mortality: a prospective cohort in UK Biobank. Cardiovasc Res. (2023) 119:1718–27. doi: 10.1093/cvr/cvac195
326. Han L, Zhao S, Li S, Gu S, Deng X, Yang L, et al. Excess cardiovascular mortality across multiple COVID-19 waves in the United States from March 2020 to March 2022. Nat Cardiovasc Res. (2023) 2:322–33. doi: 10.1038/s44161-023-00220-2
327. Daugherty SE, Guo Y, Heath K, Dasmarinas MC, Jubilo KG, Samranvedhya J, et al. Risk of clinical sequelae after the acute phase of SARS-CoV-2 infection: retrospective cohort study. BMJ. (2021) 373:n1098. doi: 10.1136/bmj.n1098
328. Abbasi J. The COVID heart-one year after SARS-coV-2 infection, patients have an array of increased cardiovascular risks. JAMA. (2022) 327:1113–4. doi: 10.1001/jama.2022.2411
329. Scudiero F, Silverio A, Muraca I, Russo V, Di Maio M, Silvestro A, et al. Long-term prognostic impact of right ventricular dysfunction in patients with COVID-19. J Pers Med. (2022) 12:162. doi: 10.3390/jpm12020162
330. Ruffenach G, Hong J, Vaillancourt M, Medzikovic L, Eghbali M. Pulmonary hypertension secondary to pulmonary fibrosis: clinical data, histopathology and molecular insights. Respir Res. (2020) 21:303. doi: 10.1186/s12931-020-01570-2
331. Simonneau G, Galie N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. (2004) 43:5S–12S. doi: 10.1016/j.jacc.2004.02.037
332. Klok FA, van der Hulle T, den Exter PL, Lankeit M, Huisman MV, Konstantinides S. The post-PE syndrome: a new concept for chronic complications of pulmonary embolism. Blood Rev. (2014) 28:221–6. doi: 10.1016/j.blre.2014.07.003
333. Cho JL, Villacreses R, Nagpal P, Guo J, Pezzulo AA, Thurman AL, et al. Quantitative chest CT assessment of small airways disease in post-acute SARS-coV-2 infection. Radiology. (2022) 304:185–92. doi: 10.1148/radiol.212170
334. Lewis KL, Helgeson SA, Tatari MM, Mallea JM, Baig HZ, Patel NM. COVID-19 and the effects on pulmonary function following infection: A retrospective analysis. EClinicalMedicine. (2021) 39:101079. doi: 10.1016/j.eclinm.2021.101079
335. Faverio P, Luppi F, Rebora P, D’Andrea G, Stainer A, Busnelli S, et al. One-year pulmonary impairment after severe COVID-19: a prospective, multicenter follow-up study. Respir Res. (2022) 23:65. doi: 10.1186/s12931-022-01994-y
336. Wild JM, Porter JC, Molyneaux PL, George PM, Stewart I, Allen RJ, et al. Understanding the burden of interstitial lung disease post-COVID-19: the UK Interstitial Lung Disease-Long COVID Study (UKILD-Long COVID). BMJ Open Respir Res. (2021) 8:e001049. doi: 10.1136/bmjresp-2021-001049
337. Ambardar SR, Hightower SL, Huprikar NA, Chung KK, Singhal A, Collen JF. Post-COVID-19 pulmonary fibrosis: novel sequelae of the current pandemic. J Clin Med. (2021) 10:2452. doi: 10.3390/jcm10112452
338. Tale S, Ghosh S, Meitei SP, Kolli M, Garbhapu AK, Pudi S. Post-COVID-19 pneumonia pulmonary fibrosis. QJM. (2020) 113:837–8. doi: 10.1093/qjmed/hcaa255
339. Melms JC, Biermann J, Huang H, Wang Y, Nair A, Tagore S, et al. A molecular single-cell lung atlas of lethal COVID-19. Nature. (2021) 595:114–9. doi: 10.1038/s41586-021-03569-1
340. Li F, Deng J, Song Y, Wu C, Yu B, Wang G, et al. Pulmonary fibrosis in patients with COVID-19: A retrospective study. Front Cell Infect Microbiol. (2022) 12:1013526. doi: 10.3389/fcimb.2022.1013526
341. Davis HE, Assaf GS, McCorkell L, Wei H, Low RJ, Re’em Y, et al. Characterizing long COVID in an international cohort: 7 months of symptoms and their impact. EClinicalMedicine. (2021) 38:101019. doi: 10.1016/j.eclinm.2021.101019
342. Haider S, Janowski AJ, Lesnak JB, Hayashi K, Dailey DL, Chimenti R, et al. A comparison of pain, fatigue, and function between post-COVID-19 condition, fibromyalgia, and chronic fatigue syndrome: a survey study. Pain. (2023) 164:385–401. doi: 10.1097/j.pain.0000000000002711
343. Hellgren L, Birberg Thornberg U, Samuelsson K, Levi R, Divanoglou A, Blystad I. Brain MRI and neuropsychological findings at long-term follow-up after COVID-19 hospitalisation: an observational cohort study. BMJ Open. (2021) 11:e055164. doi: 10.1136/bmjopen-2021-055164
344. Xu E, Xie Y, Al-Aly Z. Long-term neurologic outcomes of COVID-19. Nat Med. (2022) 28:2406–15. doi: 10.1038/s41591-022-02001-z
345. Spudich S, Nath A. Nervous system consequences of COVID-19. Science. (2022) 375:267–9. doi: 10.1126/science.abm2052
346. Klein J, Wood J, Jaycox JR, Dhodapkar RM, Lu P, Gehlhausen JR, et al. Distinguishing features of long COVID identified through immune profiling. Nature. (2023) 623:139–48. doi: 10.1038/s41586-023-06651-y
347. Wong AC, Devason AS, Umana IC, Cox TO, Dohnalova L, Litichevskiy L, et al. Serotonin reduction in post-acute sequelae of viral infection. Cell. (2023) 186:4851–67 e20. doi: 10.1016/j.cell.2023.09.013
348. Mina Y, Enose-Akahata Y, Hammoud DA, Videckis AJ, Narpala SR, O’Connell SE, et al. Deep phenotyping of neurologic postacute sequelae of SARS-coV-2 infection. Neurol Neuroimmunol Neuroinflamm. (2023) 10:e200097. doi: 10.1212/NXI.0000000000200097
349. Greer N, Bart B, Billington CJ, Diem SJ, Ensrud KE, Kaka A, et al. COVID-19 postacute care major organ damage: a systematic review. BMJ Open. (2022) 12:e061245. doi: 10.1136/bmjopen-2022-061245
350. Al-Aly Z, Xie Y, Bowe B. High-dimensional characterization of post-acute sequelae of COVID-19. Nature. (2021) 594:259–64. doi: 10.1038/s41586-021-03553-9
351. Vanholder R, Ringoir S. Infectious morbidity and defects of phagocytic function in end-stage renal disease: a review. J Am Soc Nephrol. (1993) 3:1541–54. doi: 10.1681/ASN.V391541
352. Vaziri ND, Pahl MV, Crum A, Norris K. Effect of uremia on structure and function of immune system. J Ren Nutr. (2012) 22:149–56. doi: 10.1053/j.jrn.2011.10.020
353. Yoon JW, Pahl MV, Vaziri ND. Spontaneous leukocyte activation and oxygen-free radical generation in end-stage renal disease. Kidney Int. (2007) 71:167–72. doi: 10.1038/sj.ki.5002019
354. Yuan Q, Tang B, Zhang C. Signaling pathways of chronic kidney diseases, implications for therapeutics. Signal Transduct Target Ther. (2022) 7:182. doi: 10.1038/s41392-022-01036-5
355. Verkade MA, van Druningen CJ, Op de Hoek CT, Weimar W, Betjes MG. Decreased antigen-specific T-cell proliferation by moDC among hepatitis B vaccine non-responders on haemodialysis. Clin Exp Med. (2007) 7:65–71. doi: 10.1007/s10238-007-0127-x
356. Schiffl H, Lang SM. Long-term interplay between COVID-19 and chronic kidney disease. Int Urol Nephrol. (2023) 55:1977–84. doi: 10.1007/s11255-023-03528-x
357. Uribarri A, Nunez-Gil IJ, Aparisi A, Becerra-Munoz VM, Feltes G, Trabattoni D, et al. Impact of renal function on admission in COVID-19 patients: an analysis of the international HOPE COVID-19 (Health Outcome Predictive Evaluation for COVID 19) Registry. J Nephrol. (2020) 33:737–45. doi: 10.1007/s40620-020-00790-5
358. Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. (2012) 81:442–8. doi: 10.1038/ki.2011.379
359. Sato Y, Takahashi M, Yanagita M. Pathophysiology of AKI to CKD progression. Semin Nephrol. (2020) 40:206–15. doi: 10.1016/j.semnephrol.2020.01.011
360. Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest. (2014) 124:2299–306. doi: 10.1172/JCI72267
361. Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. (2010) 16:535–43. doi: 10.1038/nm.2144
362. Kramann R, Schneider RK, DiRocco DP, MaChado F, Fleig S, Bondzie PA, et al. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell. (2015) 16:51–66. doi: 10.1016/j.stem.2014.11.004
363. Maksimowski N, Williams VR, Scholey JW. Kidney ACE2 expression: Implications for chronic kidney disease. PloS One. (2020) 15:e0241534. doi: 10.1371/journal.pone.0241534
364. Azinheira Nobrega Cruz N, Goncalves de Oliveira LC, Tedesco Silva Junior H, Osmar Medina Pestana J, Casarini DE. Angiotensin-converting enzyme 2 in the pathogenesis of renal abnormalities observed in COVID-19 patients. Front Physiol. (2021) 12:700220. doi: 10.3389/fphys.2021.700220
365. Tarris G, de Rougemont A, Estienney MA, Journet J, Lariotte AC, Aubignat D, et al. Chronic kidney disease linked to SARS-CoV-2 infection: a case report. BMC Nephrol. (2021) 22:278. doi: 10.1186/s12882-021-02490-z
366. Petersen EL, Gossling A, Adam G, Aepfelbacher M, Behrendt CA, Cavus E, et al. Multi-organ assessment in mainly non-hospitalized individuals after SARS-CoV-2 infection: The Hamburg City Health Study COVID programme. Eur Heart J. (2022) 43:1124–37. doi: 10.1093/eurheartj/ehab914
367. Nalbandian A, Sehgal K, Gupta A, Madhavan MV, McGroder C, Stevens JS, et al. Post-acute COVID-19 syndrome. Nat Med. (2021) 27:601–15. doi: 10.1038/s41591-021-01283-z
368. Clarke SA, Abbara A, Dhillo WS. Impact of COVID-19 on the endocrine system: A mini-review. Endocrinology. (2022) 163:bqab203. doi: 10.1210/endocr/bqab203
369. Li K, Chen G, Hou H, Liao Q, Chen J, Bai H, et al. Analysis of sex hormones and menstruation in COVID-19 women of child-bearing age. Reprod BioMed Online. (2021) 42:260–7. doi: 10.1016/j.rbmo.2020.09.020
370. Sapra L, Saini C, Garg B, Gupta R, Verma B, Mishra PK, et al. Long-term implications of COVID-19 on bone health: pathophysiology and therapeutics. Inflammation Res. (2022) 71:1025–40. doi: 10.1007/s00011-022-01616-9
371. Zacharias H, Dubey S, Koduri G, D’Cruz D. Rheumatological complications of covid 19. Autoimmun Rev. (2021) 20:102883. doi: 10.1016/j.autrev.2021.102883
372. Beydon M, Chevalier K, Al Tabaa O, Hamroun S, Delettre AS, Thomas M, et al. Myositis as a manifestation of SARS-coV-2. Ann Rheum Dis. (2021) 80:e42. doi: 10.1136/annrheumdis-2020-217573
373. Fiolet T, Kherabi Y, MacDonald CJ, Ghosn J, Peiffer-Smadja N. Comparing COVID-19 vaccines for their characteristics, efficacy and effectiveness against SARS-CoV-2 and variants of concern: a narrative review. Clin Microbiol Infect. (2022) 28:202–21. doi: 10.1016/j.cmi.2021.10.005
374. Forchette L, Sebastian W, Liu T. A comprehensive review of COVID-19 virology, vaccines, variants, and therapeutics. Curr Med Sci. (2021) 41:1037–51. doi: 10.1007/s11596-021-2395-1
375. Mendiola-Pastrana IR, Lopez-Ortiz E, Rio de la Loza-Zamora JG, Gonzalez J, Gomez-Garcia A, Lopez-Ortiz G. SARS-coV-2 variants and clinical outcomes: A systematic review. Life (Basel). (2022) 12:170. doi: 10.3390/life12020170
376. Corriero A, Ribezzi M, Mele F, Angrisani C, Romaniello F, Daleno A, et al. COVID-19 variants in critically ill patients: A comparison of the delta and omicron variant profiles. Infect Dis Rep. (2022) 14:492–500. doi: 10.3390/idr14030052
377. Du M, Ma Y, Deng J, Liu M, Liu J. Comparison of long COVID-19 caused by different SARS-coV-2 strains: A systematic review and meta-analysis. Int J Environ Res Public Health. (2022) 19:16010. doi: 10.3390/ijerph192316010
378. Notarte KI, Catahay JA, Velasco JV, Pastrana A, Ver AT, Pangilinan FC, et al. Impact of COVID-19 vaccination on the risk of developing long-COVID and on existing long-COVID symptoms: A systematic review. EClinicalMedicine. (2022) 53:101624. doi: 10.1016/j.eclinm.2022.101624
379. Byambasuren O, Stehlik P, Clark J, Alcorn K, Glasziou P. Effect of covid-19 vaccination on long covid: systematic review. BMJ Med. (2023) 2:e000385. doi: 10.1136/bmjmed-2022-000385
380. Tyrkalska SD, Martinez-Lopez A, Arroyo AB, Martinez-Morcillo FJ, Candel S, Garcia-Moreno D, et al. Differential proinflammatory activities of Spike proteins of SARS-CoV-2 variants of concern. Sci Adv. (2022) 8:eabo0732. doi: 10.1126/sciadv.abo0732
381. Nishijima Y, Hader SN, Hanson AJ, Zhang DX, Sparapani R, Gutterman DD, et al. Prolonged endothelial-dysfunction in human arterioles following infection with SARS-CoV-2. Cardiovasc Res. (2022) 118:18–9. doi: doi.org/10.1093/cvr/cvab339
382. Nishijima Y, Hader SN, Beyer AM. Differential impacts of COVID-19 variants on human microvascular function. Cardiovasc Res. (2023) 119:e115–e7. doi: 10.1093/cvr/cvad006
383. Jiang Y, Rubin L, Peng T, Liu L, Xing X, Lazarovici P, et al. Cytokine storm in COVID-19: from viral infection to immune responses, diagnosis and therapy. Int J Biol Sci. (2022) 18:459–72. doi: 10.7150/ijbs.59272
384. Henderson LA, Canna SW, Schulert GS, Volpi S, Lee PY, Kernan KF, et al. On the alert for cytokine storm: immunopathology in COVID-19. Arthritis Rheumatol. (2020) 72:1059–63. doi: 10.1002/art.41285
385. Chen X, Zhao B, Qu Y, Chen Y, Xiong J, Feng Y, et al. Detectable serum severe acute respiratory syndrome coronavirus 2 viral load (RNAemia) is closely correlated with drastically elevated interleukin 6 level in critically ill patients with coronavirus disease 2019. Clin Infect Dis. (2020) 71:1937–42. doi: 10.1093/cid/ciaa449
386. Luporini RL, Rodolpho JMA, Kubota LT, Martin A, Cominetti MR, Anibal FF, et al. IL-6 and IL-10 are associated with disease severity and higher comorbidity in adults with COVID-19. Cytokine. (2021) 143:155507. doi: 10.1016/j.cyto.2021.155507
387. Bekele Y, Sui Y, Berzofsky JA. IL-7 in SARS-coV-2 infection and as a potential vaccine adjuvant. Front Immunol. (2021) 12:737406. doi: 10.3389/fimmu.2021.737406
388. Ma A, Zhang L, Ye X, Chen J, Yu J, Zhuang L, et al. High levels of circulating IL-8 and soluble IL-2R are associated with prolonged illness in patients with severe COVID-19. Front Immunol. (2021) 12:626235. doi: 10.3389/fimmu.2021.626235
389. Kedor C, Freitag H, Meyer-Arndt L, Wittke K, Hanitsch LG, Zoller T, et al. A prospective observational study of post-COVID-19 chronic fatigue syndrome following the first pandemic wave in Germany and biomarkers associated with symptom severity. Nat Commun. (2022) 13:5104. doi: 10.1038/s41467-022-32507-6
390. Shabgah AG, Navashenaq JG, Shabgah OG, Mohammadi H, Sahebkar A. Interleukin-22 in human inflammatory diseases and viral infections. Autoimmun Rev. (2017) 16:1209–18. doi: 10.1016/j.autrev.2017.10.004
391. Fang S, Ju D, Lin Y, Chen W. The role of interleukin-22 in lung health and its therapeutic potential for COVID-19. Front Immunol. (2022) 13:951107. doi: 10.3389/fimmu.2022.951107
392. Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, et al. Functional role of type I and type II interferons in antiviral defense. Science. (1994) 264:1918–21. doi: 10.1126/science.8009221
393. Lee JS, Shin EC. The type I interferon response in COVID-19: implications for treatment. Nat Rev Immunol. (2020) 20:585–6. doi: 10.1038/s41577-020-00429-3
394. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-alpha and IFN-gamma triggers inflammatory cell death, tissue damage, and mortality in SARS-coV-2 infection and cytokine shock syndromes. Cell. (2021) 184:149–68 e17. doi: 10.1016/j.cell.2020.11.025
395. Rubtsova K, Rubtsov AV, Cancro MP, Marrack P. Age-associated B cells: A T-bet-dependent effector with roles in protective and pathogenic immunity. J Immunol. (2015) 195:1933–7. doi: 10.4049/jimmunol.1501209
396. Ferreira-Gomes M, Kruglov A, Durek P, Heinrich F, Tizian C, Heinz GA, et al. SARS-CoV-2 in severe COVID-19 induces a TGF-beta-dominated chronic immune response that does not target itself. Nat Commun. (2021) 12:1961. doi: 10.1038/s41467-021-22210-3
397. Witkowski M, Tizian C, Ferreira-Gomes M, Niemeyer D, Jones TC, Heinrich F, et al. Untimely TGFbeta responses in COVID-19 limit antiviral functions of NK cells. Nature. (2021) 600:295–301. doi: 10.1038/s41586-021-04142-6
398. Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martinez-Colon GJ, McKechnie JL, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med. (2020) 26:1070–6. doi: 10.1038/s41591-020-0944-y
399. Parrot T, Gorin JB, Ponzetta A, Maleki KT, Kammann T, Emgard J, et al. MAIT cell activation and dynamics associated with COVID-19 disease severity. Sci Immunol. (2020) 5:eabe1670. doi: 10.1101/2020.08.27.20182550
400. Townsend L, Dyer AH, Naughton A, Imangaliyev S, Dunne J, Kiersey R, et al. Severe COVID-19 is characterised by inflammation and immature myeloid cells early in disease progression. Heliyon. (2022) 8:e09230. doi: 10.1016/j.heliyon.2022.e09230
401. George PM, Reed A, Desai SR, Devaraj A, Faiez TS, Laverty S, et al. A persistent neutrophil-associated immune signature characterizes post-COVID-19 pulmonary sequelae. Sci Transl Med. (2022) 14:eabo5795. doi: 10.1126/scitranslmed.abo5795
402. Zhu Y, Chen X, Liu X. NETosis and neutrophil extracellular traps in COVID-19: immunothrombosis and beyond. Front Immunol. (2022) 13:838011. doi: 10.3389/fimmu.2022.838011
403. Newell KL, Clemmer DC, Cox JB, Kayode YI, Zoccoli-Rodriguez V, Taylor HE, et al. Switched and unswitched memory B cells detected during SARS-CoV-2 convalescence correlate with limited symptom duration. PloS One. (2021) 16:e0244855. doi: 10.1371/journal.pone.0244855
404. Speletas M, Dadouli K, Syrakouli A, Gatselis N, Germanidis G, Mouchtouri VA, et al. MBL deficiency-causing B allele (rs1800450) as a risk factor for severe COVID-19. Immunobiology. (2021) 226:152136. doi: 10.1016/j.imbio.2021.152136
405. Terahara K, Sato T, Adachi Y, Tonouchi K, Onodera T, Moriyama S, et al. SARS-CoV-2-specific CD4(+) T cell longevity correlates with Th17-like phenotype. iScience. (2022) 25:104959. doi: 10.1016/j.isci.2022.104959
406. Ryan FJ, Hope CM, Masavuli MG, Lynn MA, Mekonnen ZA, Yeow AEL, et al. Long-term perturbation of the peripheral immune system months after SARS-CoV-2 infection. BMC Med. (2022) 20:26. doi: 10.1186/s12916-021-02228-6
407. Guo L, Wang G, Wang Y, Zhang Q, Ren L, Gu X, et al. SARS-CoV-2-specific antibody and T-cell responses 1 year after infection in people recovered from COVID-19: a longitudinal cohort study. Lancet Microbe. (2022) 3:e348–e56. doi: 10.1016/S2666-5247(22)00036-2
408. Al-Saadi E, Abdulnabi MA. Hematological changes associated with COVID-19 infection. J Clin Lab Anal. (2022) 36:e24064. doi: 10.1002/jcla.24064
409. Terpos E, Ntanasis-Stathopoulos I, Elalamy I, Kastritis E, Sergentanis TN, Politou M, et al. Hematological findings and complications of COVID-19. Am J Hematol. (2020) 95:834–47. doi: 10.1002/ajh.25829
410. Halpert G, Shoenfeld Y. SARS-CoV-2, the autoimmune virus. Autoimmun Rev. (2020) 19:102695. doi: 10.1016/j.autrev.2020.102695
411. Pretorius E, Vlok M, Venter C, Bezuidenhout JA, Laubscher GJ, Steenkamp J, et al. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc Diabetol. (2021) 20:172. doi: 10.1186/s12933-021-01359-7
412. Salisbury R, Iotchkova V, Jaafar S, Morton J, Sangha G, Shah A, et al. Incidence of symptomatic, image-confirmed venous thromboembolism following hospitalization for COVID-19 with 90-day follow-up. Blood Adv. (2020) 4:6230–9. doi: 10.1182/bloodadvances.2020003349
413. Munipalli B, Seim L, Dawson NL, Knight D, Dabrh AMA. Post-acute sequelae of COVID-19 (PASC): a meta-narrative review of pathophysiology, prevalence, and management. SN Compr Clin Med. (2022) 4:90. doi: 10.1007/s42399-022-01167-4
414. Jewell PD, Bramham K, Galloway J, Post F, Norton S, Teo J, et al. COVID-19-related acute kidney injury; incidence, risk factors and outcomes in a large UK cohort. BMC Nephrol. (2021) 22:359. doi: 10.1186/s12882-021-02557-x
Keywords: SARS-CoV-2, COVID-19, PASC, long COVID, inflammasome, inflammation, AKI
Citation: Naiditch H, Betts MR, Larman HB, Levi M and Rosenberg AZ (2025) Immunologic and inflammatory consequences of SARS-CoV-2 infection and its implications in renal disease. Front. Immunol. 15:1376654. doi: 10.3389/fimmu.2024.1376654
Received: 30 January 2024; Accepted: 23 December 2024;
Published: 12 February 2025.
Edited by:
Stelvio Tonello, University of Eastern Piedmont, ItalyReviewed by:
Xavier Contreras, Université Toulouse III Paul Sabatier, FranceCopyright © 2025 Naiditch, Betts, Larman, Levi and Rosenberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Avi Z. Rosenberg, YXJvc2VuMzRAamhtaS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.