 Hanna Chowaniec1*
Hanna Chowaniec1* Antonina Ślubowska2
Antonina Ślubowska2 Magdalena Mroczek3
Magdalena Mroczek3 Martyna Borowczyk4
Martyna Borowczyk4 Małgorzata Braszka5Grzegorz Dworacki1,6
Małgorzata Braszka5Grzegorz Dworacki1,6 Paula Dobosz7,8Mateusz Wichtowski9
Paula Dobosz7,8Mateusz Wichtowski9- 1Department of Immunology, Poznan University of Medical Sciences, Poznan, Poland
- 2Department of Biostatistics and Research Methodology, Faculty of Medicine, Collegium Medicum, Cardinal Stefan Wyszynski University of Warsaw, Warsaw, Poland
- 3Department of Neurology, University Hospital Basel, Univeristy of Basel, Basel, Switzerland
- 4Department of Endocrinology, Metabolism and Internal Medicine, Poznan University of Medical Sciences, Poznan, Poland
- 5Faculty of Medical Sciences, University College London Medical School, London, United Kingdom
- 6Chair of Patomorphology and Clinical Immunology, Poznań University of Medical Sciences, Poznan, Poland
- 7University Centre of Cancer Diagnostics, Poznan University of Medical Sciences, Poznan, Poland
- 8Institute of Genetics and Biotechnology, Faculty of Biology, University of Warsaw, Warsaw, Poland
- 9Surgical Oncology Clinic, Institute of Oncology, Poznan University of Medical Sciences, Poznan, Poland
Oncolytic virus (OV) therapy has emerged as a promising frontier in cancer treatment, especially for solid tumours. While immunotherapies like immune checkpoint inhibitors and CAR-T cells have demonstrated impressive results, their limitations in inducing complete tumour regression have spurred researchers to explore new approaches targeting tumours resistant to current immunotherapies. OVs, both natural and genetically engineered, selectively replicate within cancer cells, inducing their lysis while sparing normal tissues. Recent advancements in clinical research and genetic engineering have enabled the development of targeted viruses that modify the tumour microenvironment, triggering anti-tumour immune responses and exhibiting synergistic effects with other cancer therapies. Several OVs have been studied for breast cancer treatment, including adenovirus, protoparvovirus, vaccinia virus, reovirus, and herpes simplex virus type I (HSV-1). These viruses have been modified or engineered to enhance their tumour-selective replication, reduce toxicity, and improve oncolytic properties.Newer generations of OVs, such as Oncoviron and Delta-24-RGD adenovirus, exhibit heightened replication selectivity and enhanced anticancer effects, particularly in breast cancer models. Clinical trials have explored the efficacy and safety of various OVs in treating different cancers, including melanoma, nasopharyngeal carcinoma, head and neck cancer, and gynecologic malignancies. Notably, Talimogene laherparepvec (T-VEC) and Oncorine have. been approved for advanced melanoma and nasopharyngeal carcinoma, respectively. However, adverse effects have been reported in some cases, including flu-like symptoms and rare instances of severe complications such as fistula formation. Although no OV has been approved specifically for breast cancer treatment, ongoing preclinical clinical trials focus on four groups of viruses. While mild adverse effects like low-grade fever and nausea have been observed, the effectiveness of OV monotherapy in breast cancer remains insufficient. Combination strategies integrating OVs with chemotherapy, radiotherapy, or immunotherapy, show promise in improving therapeutic outcomes. Oncolytic virus therapy holds substantial potential in breast cancer treatment, demonstrating safety in trials. Multi-approach strategies combining OVs with conventional therapies exhibit more promising therapeutic effects than monotherapy, signalling a hopeful future for OV-based breast cancer treatments.
1 Introduction
Oncolytic viruses (OVs) are the main subject of interest in multiple ongoing clinical trials in many cancer types. Effects in the field of immunotherapy for solid tumours are promising and mainly focussed on the field of immune checkpoint inhibitors and CAR-T cells (1). Despite these therapies having the potential to achieve full tumour regression, there is a number of patients whose response to the treatment is limited (2), fostering the development of new solutions targeting tumours resistant to the currently available forms of immunotherapy (3). Although the first OV gained the approval of the US Food and Drug Administration (FDA) only in 20151, researchers have been working on developing this form of therapy for decades (4). OV therapy is currently known as ,,a major breakthrough in cancer treatment” (4). What makes OVs so clinically useful is their ability to affect the cancer cells through several different mechanisms (3) as well as their selective, destructible impact only on cancer cells, without destroying physiological tissues in the human body (5, 6). For the past twenty years, many clinical trials have been carried out, with the most used OVs being adenovirus, HSV-1, reovirus, vaccinia virus, and Newcastle disease virus, and only a few of them having been approved for commercial use (7).
2 Mechanism of action
OVs may be natural or artificially engineered viruses. They can replicate in cancer cells, leading to the lysis (8). This phenomenon has been observed for years as a spontaneous tumour regression after viral infection in some patients. Recent advances in clinical research and genetic engineering enabled the development of specifically targeted viruses, performing various types of anticancer activity (8). Unlike chemotherapy and radiotherapy, OVs kill cancer cells without harm to normal tissues, which makes them a promising alternative to traditional methods of treatment. OVs have also played a significant role in cancer immunotherapy. They can modify the tumour microenvironment (TME) by triggering an antitumour immune response and having synergistic effects with other anticancer therapies (8).
Oncolytic viruses are divided into two groups, natural weak (wild-type OVs) and genetically modified virus strains. As some mutations typical for cancers, such as in P53, RB1, PTEN, DCC, RAS, P16, and VHL genes, impair the antiviral abilities of cells; they are often suitable targets of OV attacks. Some natural virus strains prefer tumour cells. However, their anticancer effectiveness is limited, and the pathogenicity might be challenging to control. Both the safety and performance of viruses can be increased by genetic manipulations, including gene element regulation, and inserting exogenous genes in engineered recombinant OVs. The examples of possible modifications augmenting the anti-tumour efficacy of OVs are presented in Table 1. All of them have been already considered as a potential therapy for breast cancer or its metastases.
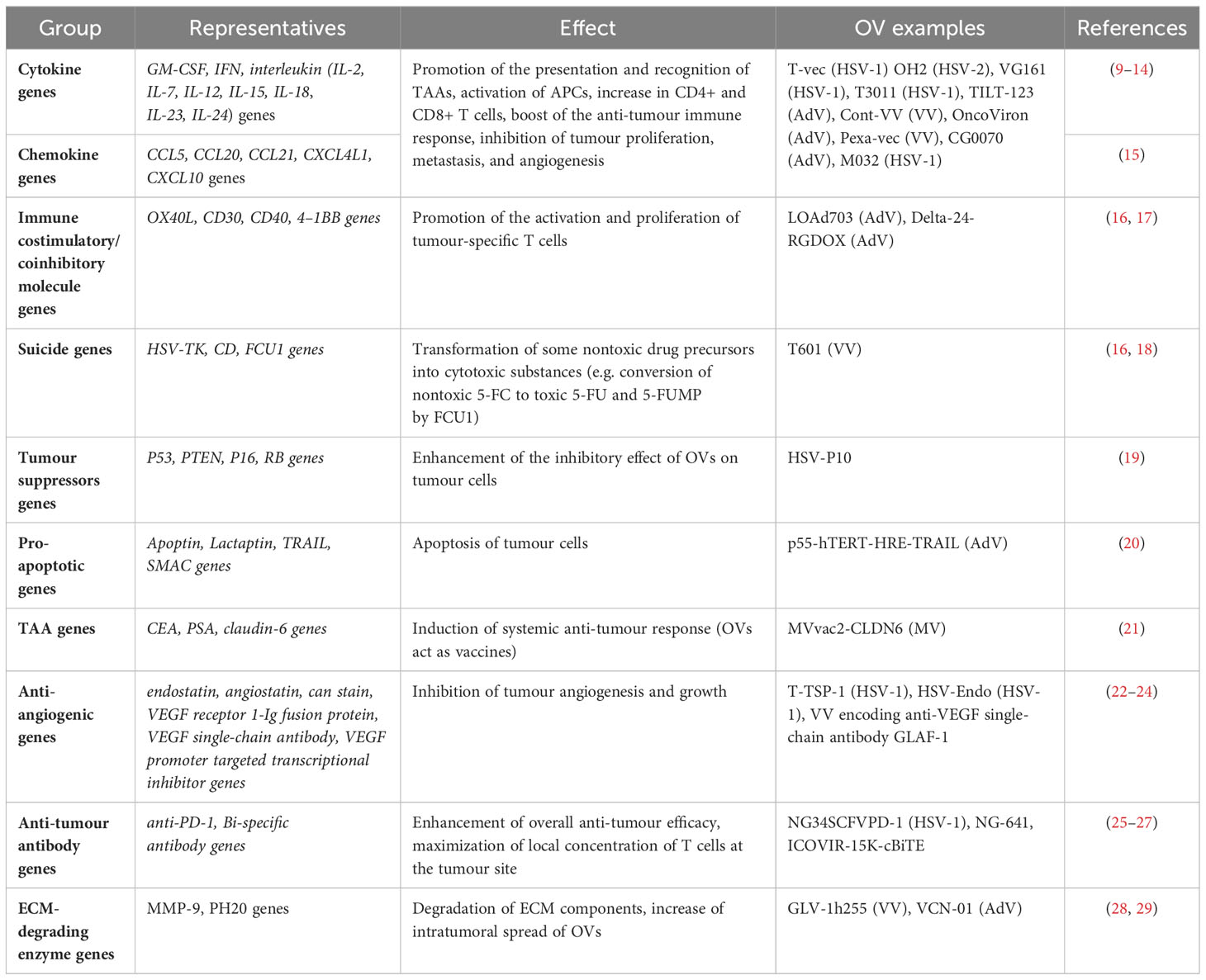
Table 1 Genes expressed by the genetically engineered OVs that improve their anti-cancer efficacy.
3 Types of viruses used in oncolytic trials
Each oncolytic virus has its characteristics that imply the safety profile, influence the possible therapeutic use, and suggest which areas require modifications in a particular case (8). In this review, we decided to focus on selected oncolytic virus strains that are used in breast cancer trials in particular. Their mechanisms of tumour targeting and antitumour activity are presented in Table 2.
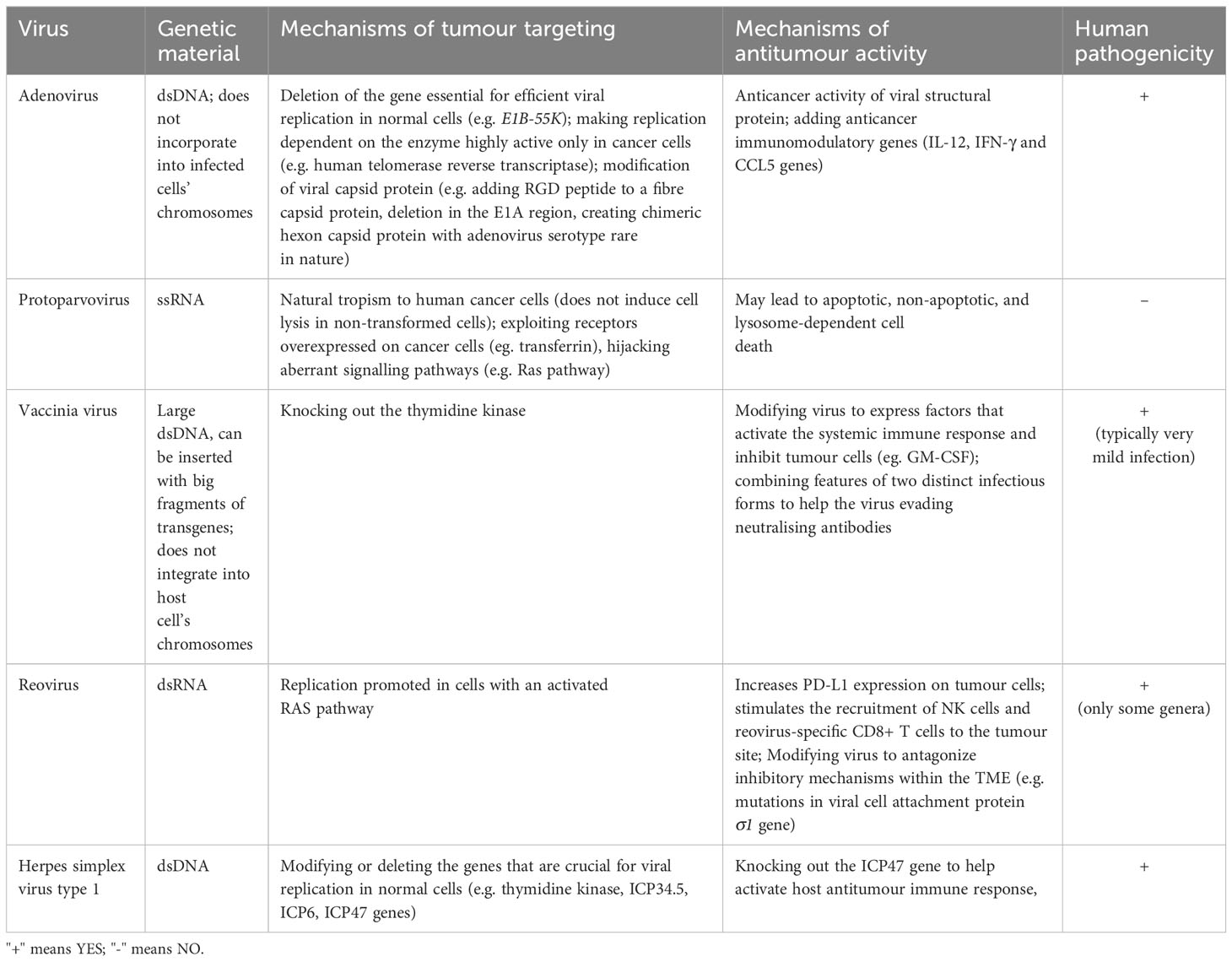
Table 2 Mechanism of action of selected OVs.
3.1 Adenovirus
Adenovirus is a DNA double-stranded virus that can enter the cells in a receptor-mediated way or through endolysis. Its genomic DNA is released and transferred to the nucleus, where it is replicated but not incorporated into chromosomes. The main efforts in the optimisation design of oncolytic adenoviruses concern restricting their replication selectively to cancer cells together with reducing viral toxicity and enhancing their oncolytic properties.
Selective replication was achieved in ONYX-050 and H101, the prototypes for oncolytic adenoviral therapy, by deletion of the viral E1B-55K gene, which is essential for efficient viral replication in normal cells but not in tumour cells, where this process is regulated differently (30). Another approach is to make the adenovirus replication dependent on the enzymes highly active only in cancer cells (31). A good example is tumour-specific replication-competent adenovirus OBP-301, in which the human telomerase reverse transcriptase (the catalytic subunit of telomerase quiescent in healthy tissues) promoter element drives the gene expressions of E1A and E1B, proteins linked to the internal ribosome entry site (31). It has been shown that OBP-301 replicates effectively exclusively in human cancer cells (31).
The possible changes also include fibre viral capsid protein (32). The initial step of the adenoviral infection is the attachment of the virus to the Coxackie-adenovirus receptor (CAR). OBP- 405, a telomerase-specific replication-selective adenoviral agent, is a version of OBP-301 with a fibre modified to contain an RGD peptide that binds with high affinity to integrins on the cell surface, facilitating the CAR-independent virus entry (32). Delta-24-RGD adenovirus undergoes the 24 base pairs deletion in the E1A region that is responsible for binding the Rb protein. This deletion renders viral replication dependent on the inactivation of Rb and generates a tumour-selective, replication-competent virus that has been shown to induce an anticancer effect in some types of gliomas (33). Modification of viral hexon capsid protein into chimeric hexon with adenovirus serotype rare in nature has the potential to reduce hepatotoxicity, uptake in the liver and spleen, and innate immune response (34).
In recent years, several Oncolytic Adenoviruses (OAVs) armed with multiple regulatory elements combined, were developed. This further increased their specificity, efficacy, and ability to escape from patients’ immune systems, and made them the most remarkable among all OVs (15).
Currently, attempts are made to use OAVs as vectors carrying anticancer genes, the local expression that may result in tumour suppression, without affecting other cells, ultimately resulting in OAVs’ increased antitumour activity. One of the representatives of the newest generation of OAVs is OncoViron. Due to numerous modifications, its replication selectivity is regulated at both transcriptional and translational levels. The anticancer activity of viral structural proteins, the ability to infect cancer cells and avoid the neutralising antibodies, and the adsorption by hepatocytes are enhanced, and the killing effect on cancer cells is boosted by adding three types of anticancer immunomodulatory genes (15). OncoViron showed significant anticancer effects on its own and in combination with programmed death 1 (PD-1) antibody and chimeric antigen receptor (CAR) T cells on a variety of implanted solid tumour models, including breast cancer, in immunodeficient, immunocompetent, and humanized mice (15).
3.2 Protoparvovirus
H-1PV is a small single-stranded rat RNA virus that presents natural tropism to human cancer cells but does not replicate or induce cell lysis in non-transformed cells. No pre-existing immunity to H-1PV has been found in humans that acts as its advantage over OVs based on human pathogens (15). Various factors that are overexpressed in cancer cells are known to control H-1PV nuclear transfer. The research focussed on the identification of new cellular modulators has the potential to further favour the outcome of H-1PV treatment (15). Cancer cell lines derived from multiple tumours, including brain, pancreas, lung, cervical, colorectal, and breast cancers, as well as melanoma and osteosarcoma, are indeed susceptible to H-1PV infection and oncolysis (35). H-1 PV efficiency has also been shown in haematological diseases. H-1PV may cause apoptotic, non-apoptotic, and lysosome-dependent cell death. The latter is essential in glioma cells, resistant to conventional cytotoxic agents. Besides tumour lysis, the oncosuppressive effect of H-1PV results in the stimulation of both innate and adaptive immune responses (36). H-1PV has been tested in combination with conventional treatment, epigenetic modulators, apoptosis inducers, and angiogenic and immune-modulating drugs. The potential of some other rodent protoparvoviruses as anticancer therapeutics is also currently investigated in preclinical studies (37). Overall, the combination of enhanced viral entry, exploitation of dysregulated cellular pathways, defective antiviral responses, altered cell cycle regulation, and the tumour microenvironment contributes to the selective targeting and efficient oncolysis of cancer cells by Protoparvovirus H-1PV (38).
Protoparvovirus H-1PV targets cancer cells by exploiting overexpressed receptors like the transferrin and Heparan sulphate proteoglycan receptors, making them more susceptible to infection than non-cancerous cells (37).
Dysfunctional innate immune pathways and defective interferon responses in cancer cells create a favourable environment for viral replication and oncolysis (39).
By hijacking aberrant signalling pathways such as the Ras pathway, Protoparvovirus H-1PV facilitates its replication and spread within the tumour microenvironment, selectively killing cancer cells while sparing normal cells (40).
Factors like hypoxia, acidic pH, and immunosuppression in the tumour microenvironment enhance H-1PV replication in cancer cells while minimizing its impact on non-tumour cells (37).
Dysregulated cell cycle progression in cancer cells increases their susceptibility to viral infection and replication, with Protoparvovirus H-1PV preferring actively dividing cancer cells for infection and oncolysis (41).
3.3 Vaccinia virus
Vaccinia virus (VV) is a double-stranded DNA virus with a large genome that can be inserted with big fragments of transgenes and does not integrate into host cell chromosomes. Oncolytic VVs (OVVs) are engineered by knocking out the thymidine kinase (TK) gene and they can replicate exclusively in cancer cells (42). The focus of the research is to augment its oncolytic efficacy, which is naturally relatively low. Pexa-vec is an OVV that can activate the systemic immune response and inhibit tumour cells by expressing granulocyte-macrophage colony-stimulating factor (GM- CSF). It possesses features of two distinct infectious forms - intracellular mature virus (IMV) and extracellular enveloped virus (EEV). Such a characteristic allows its simultaneous intravenous and intratumoral injection, as well as evading neutralising antibodies (43). OVV has been recently used as a vector for personalised neoantigen immunotherapy against triple-negative breast cancer in a study assessing such a therapeutic approach (44).
3.4 Reovirus
Reovirus is a double-stranded RNA virus. Its replication is promoted in cells with an activated RAS pathway. The gain-of-function mutations, activating RAS signalling, are prevalent in cancers. Therefore, reovirus is a natural candidate for a therapeutic agent (45). Reovirus has oncolytic activity in vitro against multiple solid tumour types, including breast cancer (46). Reolysin is an unmodified wild- type oncolytic reovirus. In 2017, it received FDA approval for the treatment of metastatic breast cancer (46). Data from the Phase II trial for the treatment of advanced metastatic breast cancer showed that the combination of Reolysin and Paclitaxel significantly increased overall survival for about seven months (47). Clinical studies have demonstrated its effectiveness in combination with systemic anti-programmed cell death protein 1 (PD-1). In a murine breast cancer model, intratumoral reovirus increased PD-L1 expression on tumour cells, and combination reovirus/anti-PD-1 treatment improved survival by reducing Treg numbers and ameliorating tumour-specific cytotoxic T lymphocyte responses (48). Reovirus has also been used in combination with CD3-bispecific antibodies. Reovirus-induced IFN stimulated the recruitment of NK cells and reovirus-specific CD8+ T cells to the tumour site, while reovirus-specific effector T cells acted synergistically with CD3- bispecific antibodies, reducing the in vivo growth of several tumour types, including breast (46). Interestingly, this combination treatment was also effective against distant lesions that were not previously injected with reovirus. It suggests the possibility of using this therapy in case of metastatic disease (45). The advances in reovirus engineering have enabled the creation of oncolytic reoviruses that can antagonize inhibitory mechanisms within the TME. In particular, mutations in viral cell attachment protein σ1 gene, have been incorporated to prevent proteolytic cleavage and inactivation of σ1 by breast cancer-associated proteases (49).
3.5 Herpes simplex virus type I (HSV-1)
HSV is a DNA double-stranded neurotropic virus with a highly effective ability to infect. It is divided into two types - HSV-1 and HSV-2. The first one is commonly used for OV therapy. It has been widely used in cancer treatment. It is recognised as a potent activator of innate and adaptive immunity. Therapeutic forms of HSV-1 are obtained by modifying or deleting the genes that are crucial for viral replication in normal cells but not in tumour ones, such as thymidine kinase (TK), ICP34.5 (required for viral replication in nerve cells), ICP6 (coding the large subunit of HSV-1 ribonucleotide reductase), and ICP47 (50–52). In addition, the knockout of ICP47 prevents from inhibiting antigen presentation by MHC-1 and helps activate the host antitumour immune response (53). The examples of HSV-1 OVs are T-vec, with a knockout of ICP34.5 and ICP47, and HSV1716, with deletion of double copies of ICP34.5. HSV1716 was approved for clinical trials in Europe in 1996 and has been used with satisfying results in the treatment of glioblastoma multiforme, melanoma, and head and neck squamous cell carcinoma (54). T-vec, also known as IMLYGIC or OncoVEX GM-CSF, proved to be effective and safe in a few Phase 1 clinical trials in patients with refractory breast cancer (55). It has been suggested that the retention of ICP34.5 may be beneficial in some situations of IFN-dependent antiviral tumour status, as it can enhance the oncolytic effect and end the overall efficacy of OV. The alternative for direct deletion of the ICP34.5 gene, proposed by researchers, is to control its expression by inserting into HSV a microRNA-responsive target element (56).
4 Discussion
4.1 Clinical use and observed adverse effects
The first OV was registered in 2004 in Latvia as a melanoma treatment. The OV is composed of the genetically unmodified Picarnoviridae family Enterovirus genus – Rigivir (57). Despite the standard procedure in treating melanoma with surgical resection, in metastatic melanoma, oncolytic viruses are showing promising effects. Melanoma has a heterogeneous presentation and can cause distant, dermal as well as visceral metastasis. The mechanism of OVs is appropriate for this type of cancer spread, as it causes the lysis of cancer cells and the lysis of infected malignant cells (58, 59). The retrospective studies from 2015 were performed in Latvia on 79 patients with stage IB, IIA, IIB, and IIC of melanoma after surgery (54). There was no statistically significant difference in the period of time for the patients to remain disease-free, however, the overall survival was prolonged among patients treated with Rigvir (54). In this study there were no significant side effects reported (54). Additionally, previous clinical trials reported side effects that were mild, completely reversible, and not causing an interruption in treatment, such as subfebrile temperature, pain in the location of the tumour, fatigue, sleepiness, and dyspepsia (55). Despite its registration in Latvia, Georgia, and Armenia, Rigvir was discontinued in mid-2019 due to manufacturing issues and then suspended for marketing authorization (60). Currently, there is a new product on the market - Imlygic registered by FDA in 2015 for advanced melanoma (7). Talimogene laherparepvec (T-VEC; Imlygic™), is a genetically modified herpes simplex virus, type 1. This is also the first OV to be approved by the FDA2 and EMA3. In the Phase III trial, 436 patients were enrolled, with unresected stage IIIB to IV melanoma but no metastases to the brain or bones. The durable response rate, overall survival, and objective response rate in patients treated with Imlygic were higher compared to the GM-CSF group (61). Adverse effects of using T-VEC were not severe, mostly presenting as flu-like mild symptoms: fatigue, chills, pyrexia, nausea, and local injection site reactions. Based on these significantly beneficial clinical results T-VEC was registered in monotherapy for curing advanced melanoma with unresectable cutaneous, subcutaneous, or nodal lesions after initial surgery by the FDA2 and the ATGA and for the treatment of stage III and IV M1a melanoma by the EMA3 (61–63).
In China in 2005 another OV, Oncorine, was approved by the National Medical Products Administration (NMPA) of China for treating nasopharyngeal carcinoma and advanced head and neck cancer (64). Oncorine is a genetically modified (deletion of E1b55K) human adenovirus type 5 called H101, which was developed by the Chinese company Sunway Biotech (65). In 2021 results of a new retrospective cohort study were published, with very promising clinical outcomes, using Oncorine as a treatment for advanced gastric carcinoma. 95 patients were divided into 3 groups (A=30, B=33, C= 32) and treated with Oncorine only (A), chemotherapy, and a combination of H101 and chemotherapy only (C). The results showed that control of lesions, progression-free survival, and overall survival were significantly higher in patients treated both with chemotherapy and Oncorine. What also was noted, is that side effects typical for chemotherapy such as nausea, vomiting, granulocytopenia, anemia, and hair loss occurred in patients from groups B and C (with no statistically significant difference between them) and more commonly than in group A. Pyrexia was observed mostly in patients from groups A and B. The study showed that Oncorine may be a therapeutic option for patients with gastric carcinoma in combination with chemotherapy (66). In 2022 the next retrospective study was carried out on the efficacy and safety of Oncorine in 29 patients with persistent, recurrent, or metastatic gynecologic malignancies: cervical cancer (22 cases), vaginal cancer (2 cases), vulvar cancer (3 cases) and ovarian cancer (2 cases). 22 patients responded to the treatment - significant tumour regression and reduction in necrotic tissue were observed as well as longer progression-free survival. Additionally, the objective response rate for radiotherapy was 72.4%, suggesting that Oncorine may increase the radiotherapy sensitivity (64). Side effects were similar to those that were previously reported, such as pyrexia, nausea, vomiting, fatigue, and pain with sometimes bleeding in the place of injection (60). There was one case reported of severe side effects - a rectovaginal fistula after Oncorine combined brachytherapy (60). Oncorine’s potential in the treatment of liver cancer, MPE, and pancreatic cancer is currently being investigated.
The newest registered OV is Delytac. This is a genetically engineered replication-competent herpes simplex virus type 1, called G47Δ or teserpaturev, approved in 2021 by the Japanese Ministry of Health, Labor and Welfare and developed by Daiichi Sankyo Co (67). Of 19 patients enrolled in this trial, 13 met the primary criterion of 1-year survival and the study was terminated earlier because of high efficacy achieved (68). 3 patients developed pyrexia but also severe side effects were reported such as death, cerebral infarction, hemiplegia, syncope, urinary tract infection, postprocedural infection, and subcutaneous abscess each in 1 patient (68).
Breast cancer is the most common cancer among women in Poland and the second cause of cancer-related deaths4. It has a relatively good prognosis if it is diagnosed and treated in early stages. On the other hand, when it is advanced and with metastases the treatment methods are limited and mortality rises due to complications of cancer itself as well as due to the treatment-related toxicity (69)5. The research in OV for breast cancer is now in the clinical trials phase and it showcases very promising effects as a future cancer treatment due to its ability to target only tumour cells (70) (71). In the preclinical and clinical trials, four virus groups (from seven groups of Baltimore classification) are mostly researched: group I (double-stranded DNA viruses), group III (double-stranded RNA viruses), group IV (single-stranded RNA viruses – positive-sense), and group V (single-stranded RNA viruses – negative-sense). Among these viruses there are naturally anti-neoplastic, those that are designed for tumour-selective replication, and those genetically modified to activate the immune system (71).
Currently, there is no OV registered for breast cancer treatment. Table 3 represents OVs used in clinical trials (in monotherapy and combined therapy) tested on patients with breast cancer, however, there are a number of ongoing preclinical trials focusing on a variety of viruses from those four groups.
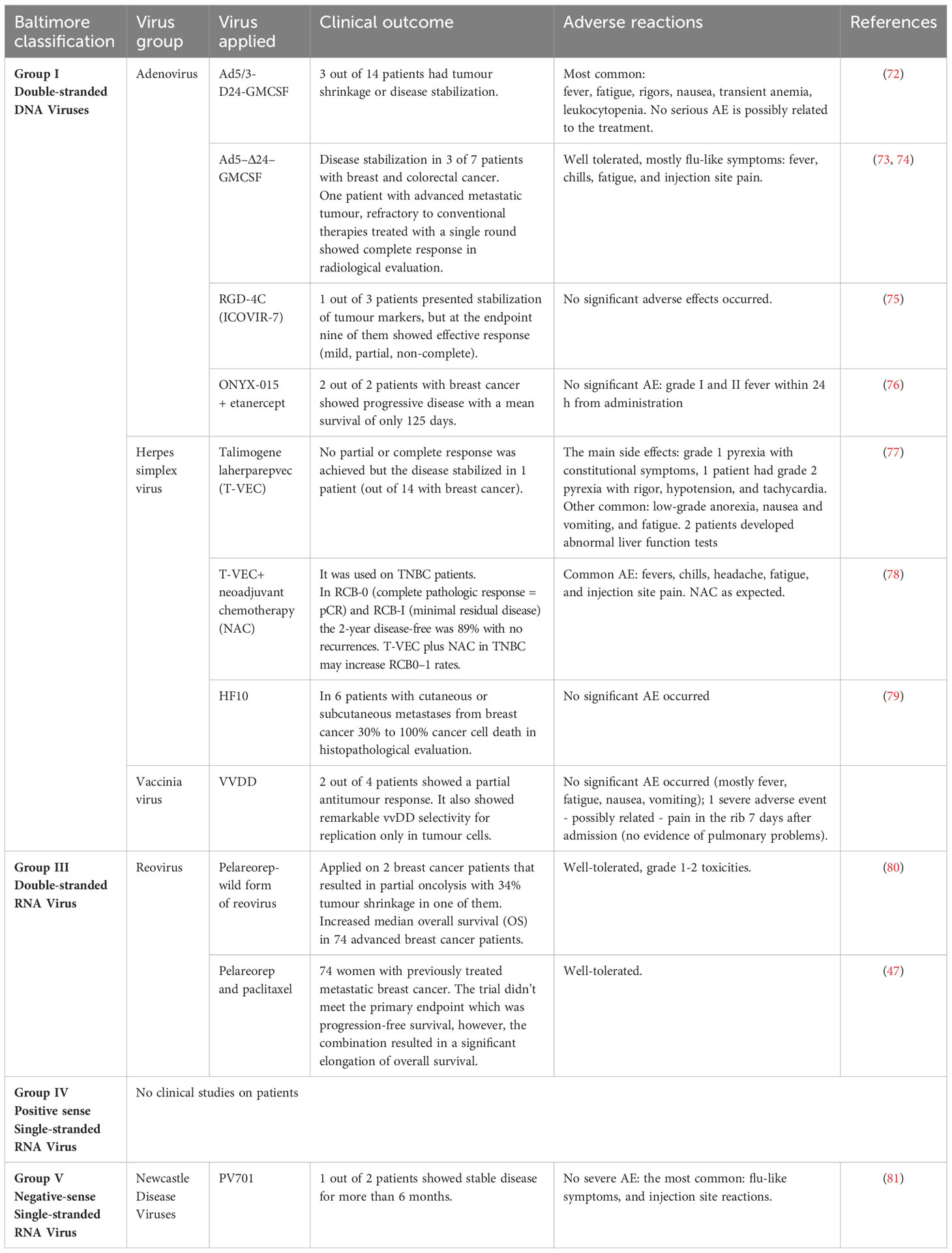
Table 3 Oncolytic viruses used in clinical trials (up to date, Dec 2023).
Many preclinical trials focus on finding the OV against TNBC. TNBC is a highly aggressive cancer with a very poor response to treatment due to the lack of receptors for estrogen, progesterone, and human epidermal growth factor 2. Although the patients are currently treated with chemotherapy - the side effects are devastating, drug resistance occurs often, and the prognosis is poor. The difficulties in treatment and high heterogeneity in TNBC led to the beginning of many studies in order to find a more efficient and safer treatment (82).
Adenoviruses are the most studied OVs in breast cancer, so preclinical studies -with additional modification (antitumour and immune regulatory genes were inserted to enhance effects) - were performed also against TNBC (83). One of them is a recombinant type five adenovirus containing IL-24 gene (CNHK600-IL24). It significantly suppressed tumour growth in the nude mice model and improved survival in the metastatic model (84). Another OV which showed high efficiency against MDA-MB-435 cancer cells was G47Δ - oncolytic HSV (registered for Malignant Glioma). It presented high cytotoxicity against human breast cancer cells in vitro and in tumour xenografts in vivo (85). As for MDA-MB-231 TNBC cells VG9-IL-24 recombinant Vaccina virus presented promising effects. In the xenograft mouse model it showed efficiency in infecting and selectively killing breast cancer cells with no strong cytotoxicity to physiologic cells (86).
Recently (November 2023) a case study was published of a previously treated patient with mTNBC. The purpose was to evaluate the safety and efficiency of CHECKvacc - an oncolytic virus composed of CF33, a chimeric vaccinia poxvirus. The first intratumoral administration showed no immediate response, but later the patient underwent T-Dxd treatment and the tumour regressed significantly also disease-free survival was 10 months (87). This is just one of many examples where combined therapy shows the best effects. There is also an ongoing phase 1 clinical trial on Codalytic, this is the first codon-modified virus. In the preclinical trial, it was tested on a mouse model with implanted TNBC cells in monotherapy. After 3 weeks the tumour was reduced by 76% and the cure rate was 66% (88).
In clinical trials (listed in Table 3) the adverse effects in treating breast cancer and other types of cancer are mostly mild: flu-like symptoms such as low-grade fever, chills, nausea, and vomiting; with single severe adverse effects occurring.
Overall, OV therapy has been proven safe in many trials. Unfortunately, the inadequately effective outcomes of these trials and incomplete responses are not sufficient enough to use OVs as a monotherapy treatment for breast cancer. On the other hand, multi-approach strategies - combining OVs and chemotherapy, radiotherapy, or immunotherapy - provide better therapeutic effects and show great potential in future approaches to breast cancer treatment (69, 89, 90).
4.2 Immunological response to the oncolytic virus therapy
Cancer is not just a collection of rebellious cells. They need the support of a specific compartment of the tumour stroma called the TME. It provides many vital signals to support tumour growth and progression. We have long thought of the tumour stroma as a rather passive element of the bulky cancerous tumour, but it seems that its effects go far beyond blood supply and mechanical stabilization. It is now well established that the TME can influence almost every step of cancer growth. It plays a role in the initiation of cancer transformation, its growth, invasion, and ultimately metastasis. In rare cases, it can also induce spontaneous tumour regression.
The TME consists of cellular and non-cellular compartments. The tumour-associated stromal cell compartment contains immune cells as antigen-processing cells as macrophages (M1, M2), and antigen- presenting cells as dendritic cells (DC), and finally antigen-specific cells as T cells, both CD4 and CD8 subsets and B cells. In addition, other cells such as blood and lymphatic vessels with all their variety of cellular elements such as endothelial cells, cancer-associated fibroblasts, pericytes but also adjacent neuronal cells and adipocytes (91) play their role in the growth rate of cancer (92).
The non-cellular components of the TME are mainly matrix proteins, but also the often-neglected microbiome (93), which can be exploited by the tumour. Now that we know that the TME is involved in almost every step of cancer development and progression, it is not surprising that the development of therapies targeting the TME machinery is an emerging target for future cancer research. The cellular composition and dynamic function of the TME are not permanent but can vary greatly over time, depending on many local tissue factors reflecting the actual status of cancer growth. It depends on the tissue in which the cancer arises as well as the characteristics of the cancer cells, the stage of the tumour, and the clinical status of the patient.
TME usually reflects a normal immune response when the immune system is unable to eliminate an antigen. Under physiological conditions, the immune response initially attempts to eliminate the antigen. This situation occurs when a high burden of inflammatory cells can eliminate antigens at the site of immune action. It is associated with the presence of immunocompetent cells involved in antigen elimination, rich in CD4 Th1, T cytotoxic T, CD8, and NK cells. This situation is usually not present in TME without additional manipulations since the tumour grows when immune surveillance is impaired. If, over time, an antigen remains and the immune effort to eliminate it is useless, the immune response is redirected. It first changes to tolerate the antigen. The situation is equivalent to chronic inflammation, with an impaired ability to eliminate antigen but still control its spread. The cost is reduced immune surveillance. Finally, in the worst-case scenario, to protect the host, the immune system tries to physically isolate the source of the dangerous antigen, forming a physical barrier with extensive fibrosis. These last two situations are also indicative of an immunosuppressive microenvironment that could in the long term be the soil for cancer initiation and progression. The typical example is the so-called scar cancer in the lung, but many others can arise on the soil of chronic inflammation, e.g. liver cancer in hepatitis B infection, lung cancer following chronic irritations such as asbestosis and silicosis, and so on. These last two situations occur in cancers, therefore TME can finally be divided into distinct separate groups currently playing a significant role in clinical outcomes and response to therapy. The pro- and anti-tumourigenic effects of tumour-infiltrating immune competent cells can profoundly determine tumour progression and the success or failure of anti-cancer therapies.
Currently, based on the immunomorphological response to immunotherapy, TME can be classified into three main groups, referred to as: immune inflamed, immune excluded, and immune desert (94, 95). However, the response to immune checkpoint blockade immunotherapy has been linked to the degree of T cell infiltration (96, 97) in tumours with high tumour mutation burden (98) and neoantigen load (99), as well as tumour antigenicity (100, 101), which led to the elucidation of four distinct TME subgroups: immune-enriched fibrotic, immune-enriched non-fibrotic, fibrotic and immune-depleted (86–88).
Oncolytic virus treatment is an emerging and promising therapy that not only directly targets tumour cells but may also modify the TME towards its more immune-eliminating rather than immunosuppressive properties. Data confirms that these types of manipulations in an experimental model increased tumour vascular permeability, host leukocyte infiltration into tumours, and ultimately, tumour inflammation (102). A combination of OV and CAR-T cell therapy may stimulate naive T cells and enhance CAR-T efficacy in mice (103). A better understanding of the complex interactions between tumour cells and their stroma determines disease progression and is critical for the rational development of effective cancer therapy.
4.3 Side effects on non-tumour cells
The application of oncolytic viruses is not without potential side effects on non-tumour cells (104). While oncolytic viruses are designed to selectively target and destroy cancer cells, they may inadvertently impact neighbouring healthy cells (105). The mechanism of cytotoxic effects on non-tumour cells may be direct but can be mediated also by immunological responses (39). The activation of the immune system by viral infection can lead to the release of pro-inflammatory cytokines and chemokines, resulting in a local inflammatory response. While inflammation is a critical component of the antitumour immune response, excessive or prolonged inflammation can cause tissue damage and exacerbate pre-existing pathological conditions. Moreover, the activation of innate immune pathways, such as toll-like receptor signalling, may trigger immune-mediated cytotoxicity against non-infected cells, contributing to bystander effects (39). The risk is put also in off-target viral replication (106).
Moreover, oncolytic viruses may alter the tumour microenvironment, impacting the function and phenotype of stromal cells, endothelial cells, and immune cells, which can influence tumour progression and treatment outcomes (107).
Integrating strategies to mitigate off-target effects, such as engineering viruses with improved tumour selectivity or combining virotherapy with immunomodulatory agents, represents a promising approach to enhance the therapeutic index of oncolytic virus-based treatments (108, 109) The successes of no histological signs of viral induced toxicity for non-tumour bearing organs have been announced for the urokinase receptor (uPAR) retargeted oncolytic measles virus in syngeneic cancer models (110), Vstat120-expressing (RAMBO) oncolytic herpes simplex virus (oHSV) (111), novel combination oncolytic adenoviral gene therapy armed with Dm-dNK and CD40L for Breast Cancer (112), or cancer-specific targeting of a conditionally replicative adenovirus using mRNA translational control (105).
4.4 Potential biomarkers of response
The emergence of OV as a promising therapeutic approach in breast cancer has generated interest in identifying predictive biomarkers for treatment response (113). Biomarkers to predict response to OV in breast cancer patients are currently lacking but are essential for selecting patients who will most likely benefit from the treatment. Moreover, the rapidly expanding combination strategies force the finding of the biomarkers to match individual patients to their most promising treatment option (114).
One of the key determinants of response to OV in breast cancer treatment appears to be the immune composition of TME (115–117). Tumours exhibiting high infiltration of tumour- infiltrating lymphocytes (TILs) demonstrate a propensity for better responses to OV. TILs are indicative of pre-existing immunity which is essential for OV efficacy. Specifically, a robust presence of CD8+ T cells within the TME indicates a potentially favourable response (118, 119). Important to predict a response are not only higher levels of TILs but also the spatial distribution of TILs and their functional state (120). Moreover, the expression of immune checkpoint molecules like PD-L1 on tumour cells can indicate the likelihood of a favourable response when OVs are combined with immune checkpoint inhibitors (119).
Tumour Mutational Burden (TMB) serves as another potential biomarker influencing treatment outcomes. Tumours with increased mutational burdens harbour more neoantigens, rendering them more susceptible to immune recognition, thus potentially enhancing the response to oncolytic viruses (98, 121).
The infectivity and replication capacity of the oncolytic virus within tumour cells represent critical aspects for predicting treatment response. Techniques monitoring viral presence or replication within the tumour tissue might serve as valuable biomarkers (83, 122).
Genetic signatures linked to viral replication, immune response pathways, or susceptibility to viral infection might also contribute to predicting response to OVs in breast cancer. An example of this approach is the identification of realistic interferon (IFN)-mediated biomarkers to identify patients who most likely respond to virotherapy (123) as replication of OV is usually limited to cancer cells that have interferon (IFN) signalling defects. Upregulation of protein biomarkers such as IFN gamma may reflect immune induction and become an OV efficacy biomarker to improve the ability to select patients who do not exhibit resistance to virotherapy (124).
Analysing the cytokine profile within the TME provides insights into the immune response elicited by OVs. Elevated levels of specific cytokines may indicate a more favourable response to treatment. They may play a role in T-cell helper polarization in viral tolerability. The previous research described that the Th1 cytokine profile was expressed in pleural effusions of patients that responded to HSV1716 treatment for malignant pleural mesothelioma with low side effects, to be investigated as a biomarker for predictive response (125).
Serum markers such as carcinoembryonic antigen (CEA) have been also used to investigate the antitumour potential of a novel viral agent, an attenuated strain of measles virus deriving from the Edmonston vaccine lineage, genetically engineered to produce CEA against breast cancer. CEA production as the virus replicates can serve as a marker of viral gene expression (126). Therefore, CEA may serve as a low-risk method of detecting viral gene expression during treatment and could allow dose optimisation and individualization of treatment (126).
Advanced imaging techniques offer a non-invasive approach to monitor changes in the tumour microenvironment post-oncolytic virus treatment, potentially serving as a valuable tool for assessing treatment response. Examples of those may be organ-on-chip and tumour-on-chip microfluidic cell cultures (127).
Further research is imperative to establish the reliability and efficacy of these biomarkers in guiding the selection of patients likely to benefit from oncolytic virus therapy. The studies should be aimed at finding out how the ability of specific OVs to replicate in individual tumour cells is affected, and if and how it influences antitumour and antiviral action (89, 128).
4.5 Physical barriers
Several physical barriers can limit the delivery and effectiveness of the OVs and in our opinion, this topic requires a separate chapter in this review. Most of the therapies are delivered directly to the tumour. Such examples are adenovirus, poxvirus, HSV-1, measles, and reovirus which are delivered intramurally (129, 130). However, not all the tumours are available for direct delivery, because of their location. For example, the Seneca Valley Virus can be delivered to the bloodstream directly as it does not cause hemagglutination (131). Parvovirus H- 1PV can go through the blood-brain barrier and is applied in the treatment of glioblastoma multiforme (131). Another type of physical barrier is the extracellular matrix (ECM) in tumours is dense and contains impermeable for the viruses’ components, such as collagen and elastic fibres. For example, in pancreatic ductal adenocarcinomas (PDACs) ECM is so dense that can lead to interstitial hypertension (130) and can form a physical barrier for the delivery of the OVs (132). Other types of physical barriers associated with the TME are necrosis, calcification, hypoxia, acidosis, and increased proteolytic activity (130).
4.6 General risks of oncolytic virus therapy
4.6.1 Immunogenicity
Immunogenicity is a critical consideration in the context of OV therapy, representing the capacity of the introduced viruses to stimulate an immune response within the host. Oncolytic viruses are designed to selectively replicate within cancer cells, triggering cell lysis and the release of tumour- associated antigens. This process is intended to provoke an immune response, engaging components such as T cells and natural killer (NK) cells. However, the effectiveness of this response depends on the ability of the immune system to recognise and mount a robust reaction against cancer cells. Successful oncolytic virus therapy relies on the activation of adaptive immune responses, particularly cytotoxic T lymphocytes (CTLs). These immune effectors play a central role in recognising and eliminating cancer cells. However, factors such as pre-existing immunity to the viral vector may influence the magnitude and efficacy of these responses (133).
4.6.2 Off-target effects
Off-target effects refer to the unintended impact of OVs on non-cancerous or healthy cells within the body. Despite the selectivity engineered into these viruses, factors such as imperfect targeting mechanisms, interactions with the host immune system, or unexpected viral behaviour may lead to unintended consequences in off-target tissues. Off-target effects may manifest as localised toxicity in tissues surrounding the site of viral administration. This can include inflammation, tissue damage, or discomfort near the treatment site. In some cases, OVs may enter the bloodstream and disseminate throughout the body, potentially affecting distant organs and tissues. Systemic off-target effects can lead to more widespread adverse events. The activation of the immune system in response to viral infection may extend beyond the tumour site, leading to immune-mediated effects in healthy tissues. This could result in autoimmune-like reactions or inflammation in non-cancerous areas. This may be especially dangerous in immunocompetent patients (134).
4.6.3 Inflammatory response
The introduction of OVs elicits an immune response aimed at recognising and eliminating foreign entities. While this response is essential for combating cancer, excessive or uncontrolled inflammatory reactions may lead to adverse effects. The inflammatory response can manifest both locally at the site of viral administration and systemically throughout the body. Locally, inflammation may cause redness, swelling, and pain at the injection site. Systemically, the release of inflammatory mediators into the bloodstream can lead to flu-like symptoms, fever, and malaise. The delicate balance between inducing an immune response against cancer cells and minimizing collateral damage to healthy tissues is pivotal for the therapy’s success. Excessive inflammation may not only compromise patient comfort but also impact the therapeutic effectiveness of oncolytic viruses by diverting the immune system’s attention away from cancer cells (135).
4.6.4 Virus resistance
Virus resistance poses a significant challenge in oncolytic virus therapy, where cancer cells may develop mechanisms to evade viral infection and subsequent destruction. Cancer cells can develop resistance to oncolytic viruses through various mechanisms. These may include alterations in viral entry receptors, inhibition of viral replication, interference with the apoptotic pathways triggered by viral infection, and the evolution of antiviral immune responses within the TME (136).
4.7 Breast cancer-specific risks
Breast cancer exhibits substantial molecular and genetic heterogeneity, encompassing diverse subtypes such as luminal A/B, HER2-positive, and triple-negative breast cancer (TNBC). This heterogeneity influences disease progression, treatment response, and the overall clinical outcome. The diverse molecular landscape of breast cancer subtypes poses a challenge in designing oncolytic viruses with universal efficacy. Different subtypes may have distinct vulnerabilities and response patterns to viral infection, necessitating tailored approaches for each breast cancer subtype (90).
4.7.1 Impact on healthy breast tissue
The proximity of healthy breast tissue to cancerous lesions raises concerns about potential off-target effects. Ensuring the selectivity of OVs for cancer cells while sparing normal tissue is critical in minimizing adverse effects and enhancing the safety profile of the therapy.
4.7.2 Hormone receptor status
Hormone receptor status, including estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) expression, further complicates the landscape of oncolytic virus therapy. Subtypes with specific hormone receptor profiles may exhibit differential responses to viral infection, necessitating a nuanced approach to treatment planning (137).
4.7.3 Combination therapies
OV therapy is often combined with other modalities such as chemotherapy, immunotherapy, or targeted therapies. Evaluating potential interactions and cumulative toxicities of these combined approaches is essential to mitigate risks and enhance therapeutic outcomes (55).
4.8 Limitations of the oncoviral therapy
OV therapy holds great potential in modern cancer therapy, however, the therapy with genetically modified viruses apart from its potential also shows limitations to overcome. One of the first obstacles is the delivery of the OVs. OVs can be administered intravenously, but it brings other barriers. OVs circulating in the bloodstream can be neutralised.
Major problems with systematic delivery are preexisting antibodies due to immunisation or previous oncolytic treatment. As Reovirus is commonly found in the environment, many people have antibodies against it which causes immunity to Reovirus and recombined OVs (138). In order to overcome the host’s immunity for different types of viruses, a lot of work still needs to be performed (139). Apart from antibodies circulating in the blood, there also are factors of the complement system, which after contact with the pathogen - activate and start the protease cascade, which leads to the deposition of the membrane attack complex (140).
There is also a risk of them not being guided directly into the tumour and nonspecific uptake by the lungs, liver or spleen, so only a small payload may be delivered to the tumour (139).
The next problem of intravenous administration is collapsed vasculature in the tumour, so the penetration can be insufficient, thus, the therapeutic dose will not be met (90). Currently, most of the OVs are injected directly into the tumour. The intratumoral administration can be difficult for the operator and painful for the patient, especially if the tumour is hardly accessible, as well as if the cancer cells are in several nodes dispersed in large areas - hence, not every metastasis might be equally reached (141). As for breast cancer treatment, the intratumoral administration of the OV is difficult only if the tumour is in a hardly accessed location or has already metastasized. The most optimal way of administration has not been defined yet, and also if OV should be used in monotherapy or in combination with other available forms of therapy.
Another difficulty in administration in solid tumours apart from delivering the OV to the patient itself, is the need to reach the tumour and spread in it, which can also cause limited efficacy against cancer cells (90). First, the OVs have to overcome the physical barriers (tissues) to get to the tumour. Secondly, intratumoral hypertension (caused by abnormal lymphatic networks, vascular hyperpermeability, and dense extracellular matrix) may obstruct viral infiltration, thus, the effectiveness of the OVs. (132, 136, 142).
The tumour is constructed with a great amount of extracellular matrix. Viruses, which are passively diffusing, may not fit through the strands. Limited penetration enables further tumour regions to grow regardless of administered OV (139). To improve tumour penetration various strategies are being developed such as pretreatment with enzymes or protein effectors (for example protease and relaxin) (143).
The next obstacle in OV therapy, which has been already recognised in the ongoing clinical trials, is the inadequate effectiveness in some types of tumours, when OVs are administered in monotherapy. In this case, combined therapy seems to be the best option, as confirmed by a growing number of studies with positive results (142). That has been reflected in a clinical trial on Oncorine, where the combination of Oncorine with chemotherapy resulted in better control of lesions, and elongated progression-free survival and overall survival in comparison to patients treated only with chemotherapy or only with Oncorine (121). Similarly, ONYX-015 with 5-fluorouracil used in clinical trials as a treatment for patients with recurrent head and neck cancer showed that after 6 months there was no progression in the tumours that responded to the OV treatment, while all of the tumours, treated only with chemotherapy, further progressing (144). ONYX-015 was also used in combination with etanercept for the treatment of patients with solid tumours. Patients with colon cancer achieved stable disease, while patients with breast cancer presented progression of the disease (76). The mechanism of OVs and drug combinations must be further understood and executed to find the best treatment solution. Currently, more research is required to present the most effective and safest treatment since most of the OV therapies are still in the early stages of development (136).
5 Future directions
Oncolytic virus therapy has emerged as a promising avenue in breast cancer treatment, showcasing remarkable potential in preclinical and early clinical trials. However, there are still challenges to overcome (Table 4).
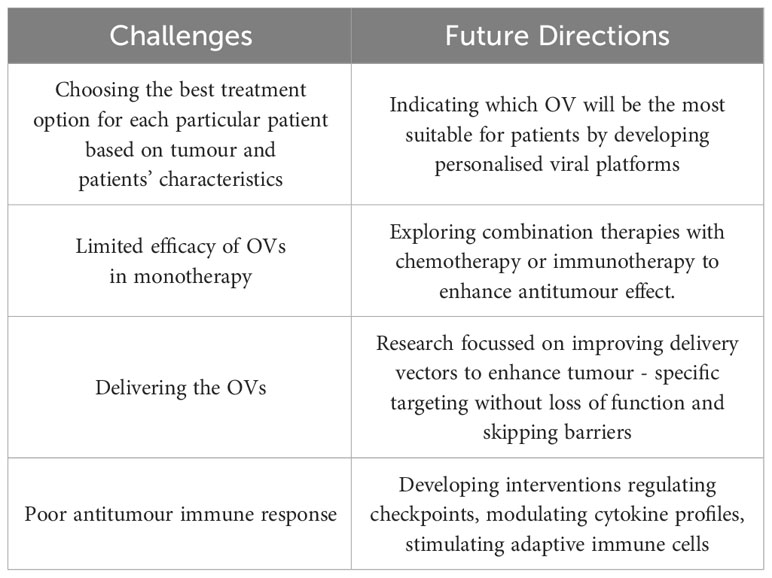
Table 4 Challenges and future directions of developing OVs.
The advent of precision medicine calls for tailoring OVs to individual patient profiles. Developing personalised viral platforms could involve genetic modifications or viral engineering to enhance tumour specificity, replication efficiency, and immune activation while mitigating adverse effects. Advancements in identifying predictive biomarkers for treatment response remain also urgent. Identifying biomarkers associated with the efficacy of OVs can aid in patient stratification, ensuring targeted therapies for individuals most likely to benefit.
An exciting frontier remains the synergy between OVs and conventional treatments like chemotherapy, radiotherapy, and immunotherapy. Exploring combination therapies can capitalize on their complementary mechanisms, potentially amplifying therapeutic efficacy and overcoming resistance.
It is also crucial to find strategies that restore the tumour microenvironment and bolster anti- tumour immune responses. This includes interventions that regulate immune checkpoints, modulate cytokine profiles, or stimulate adaptive immune cells within the tumour milieu.
Innovations in delivery systems to ensure efficient viral dissemination and penetration into tumour sites are also imperative. Engineering improved delivery vectors that enhance tumour-specific targeting while reducing off-target effects remains an active area of research.
A glycoprotein from others that has an affinity to the receptor can be inserted directly for enveloped viruses (145). An alternative approach is to use adapters that can bind both to the OV and the receptor (146). A promising approach are genetically engineered OV, with modification including deletions in the E1B region (147), E3B gene or for the PV deleting P1 coding region (replicons), A133G mutation in cis-acting replication element (CRE) (145). Off target effects are a concern especially for adenoviruses that have a high affinity to the liver (135). It has been reported that coagulation factor X (FX) binds to Ad5-hexon and enables transduction to the liver (148). Several modifications have been developed to circumferent the issue, such as constructing FX-binding ablated adenoviruses serotype 5 vectors (149). In the recent years. Recently the 3rd generation of OV has been introduced, e.g. OV with truncated CD19 (CD19t) protein for tumour-selective delivery (150).
As for many other research areas, a fast and efficient transition from preclinical success to clinical applicability remains the clue. Streamlining regulatory pathways and conducting robust clinical trials are essential steps towards obtaining approvals for OV therapies in breast cancer treatment.
The progress requires caring about the treatment’s long-term safety profiles. Establishing comprehensive monitoring mechanisms for potential adverse effects post-treatment is essential for ensuring patient safety and treatment optimisation.
The collaboration between researchers, clinicians, and pharmaceutical entities can facilitate resource-sharing, accelerate discoveries, and promote a collective effort towards advancing OV therapy. The future of oncolytic virus therapy in breast cancer treatment holds immense promise. Realizing these potential demands concerted interdisciplinary efforts, innovative strategies, and a commitment to translational research to revolutionize breast cancer management.
Author contributions
HCh: Supervision, Writing – original draft, Writing – review & editing. AŚ: Writing – original draft, Writing – review & editing, Supervision. MM: Writing – original draft, Writing – review & editing. MBo: Writing – original draft, Writing – review & editing. MBr: Writing – original draft, Writing – review & editing. GD: Writing – original draft, Writing – review & editing. PD: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. MW: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
- ^ Food and Drug Administration. (2024). Alphabetical List of Licensed Establishments Including Product Approval Dates as of 01-JAN-2024. https://www.fda.gov/media/76356/download [Accessed January 16, 2024].
- ^ Food and Drug Administration. (2024). Alphabetical List of Licensed Establishments Including Product Approval Dates as of 01-JAN-2024. https://www.fda.gov/media/76356/download [Accessed January 16, 2024].
- ^ European Medicines Agency. (2022). Imlygic. https://www.ema.europa.eu/en/medicines/human/EPAR/imlygic [Accessed January 16, 2024]
- ^ Krajowy Rejest Nowotworów. Raporty. https://onkologia.org.pl/pl/raporty [Accessed January 16, 2024]
- ^ American Cancer Society. (2017). Breast Cancer. Facts & Figures 2017-2018. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/breast-cancer-facts-and-figures/breast-cancer-facts-and-figures-2017-2018.pdf [Access January 16, 2024]
References
1. Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. (2015) 125:3335–7. doi: 10.1172/JCI83871
2. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. New Engl J Med. (2017) 377:1345–56. doi: 10.1056/NEJMoa1709684
3. Yun CO, Hong JW, Yoon AR. Current clinical landscape of oncolytic viruses as novel cancer immunotherapeutic and recent preclinical advancements. Front Immunol. (2022) 13:953410. doi: 10.3389/fimmu.2022.953410
4. Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. (2016) 107:1373–9. doi: 10.1111/cas.13027
5. Russell SJ, Peng KW, Bell JC. ONCOLYTIC VIROTHERAPY. Nat Biotechnol. (2012) 30:658. doi: 10.1038/nbt.2287
6. Russell SJ, Peng KW. Viruses as anticancer drugs. Trends Pharmacol Sci. (2007) 28:326–33. doi: 10.1016/j.tips.2007.05.005
7. Li K, Zhao Y, Hu X, Jiao J, Wang W, Yao H. Advances in the clinical development of oncolytic viruses. Am J Transl Res. (2022) 14:4192.
8. Su Y, Su C, Qin L. Current landscape and perspective of oncolytic viruses and their combination therapies. Transl Oncol. (2022) 25:101530. doi: 10.1016/j.tranon.2022.101530
9. Andtbacka RHI, Collichio F, Harrington KJ, Middleton MR, Downey G, Öhrling K, et al. Final analyses of OPTiM: a randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III–IV melanoma. J Immunother Cancer. (2019) 7:145. doi: 10.1186/s40425-019-0623-z
10. Wang Y, Zhou X, Wu Z, Hu H, Jin J, Hu Y, et al. Preclinical safety evaluation of oncolytic herpes simplex virus type 2. Hum Gene Ther. (2019) 30(5):651–60. doi: 10.1089/hum.2018.170
11. Chouljenko DV, Ding J, Lee IF, Murad YM, Bu X, Liu G, et al. Induction of durable antitumour response by a novel oncolytic herpesvirus expressing multiple immunomodulatory transgenes. Biomedicines. (2020) 8:484. doi: 10.3390/biomedicines8110484
12. Havunen R, Kalliokoski R, Siurala M, Sorsa S, Santos JM, Cervera-carrascon V, et al. Cytokine-coding oncolytic adenovirus TILT-123 is safe, selective, and effective as a single agent and in combination with immune checkpoint inhibitor anti-PD-1. Cells. (2021) 10:246. doi: 10.3390/cells10020246
13. Nakao S, Arai Y, Tasaki M, Yamashita M, Murakami R, Kawase T, et al. Intratumoral expression of IL-7 and IL-12 using an oncolytic virus increases systemic sensitivity to immune checkpoint blockade. Sci Transl Med. (2020) 12:7992. doi: 10.1126/scitranslmed.aax7992
14. Patel DM, Foreman PM, Nabors LB, Riley KO, Gillespie GY, Markert JM. Design of a phase I clinical trial to evaluate M032, a genetically engineered HSV-1 expressing IL-12, in patients with recurrent/progressive glioblastoma multiforme, anaplastic astrocytoma, or gliosarcoma. Hum Gene Ther Clin Dev. (2016) 27(2):69–78. doi: 10.1089/humc.2016.031
15. Su Y, Li J, Ji W, Wang G, Fang L, Zhang Q, et al. Triple-serotype chimeric oncolytic adenovirus exerts multiple synergistic mechanisms against solid tumours. J Immunother Cancer. (2022) 10:e004691. doi: 10.1136/jitc-2022-004691
16. Eriksson E, Milenova I, Wenthe J, Hle MS, Leja-Jarblad J, Ullenhag G, et al. Shaping the tumour stroma and sparking immune activation by CD40 and 4-1BB signaling induced by an armed oncolytic virus. Clin Cancer Res. (2017) 23:5846–57. doi: 10.1158/1078-0432.CCR-17-0285
17. Jiang H, Shin DH, Nguyen TT, Fueyo J, Fan X, Henry V, et al. Localized treatment with oncolytic adenovirus delta-24-RGDOX induces systemic immunity against disseminated subcutaneous and intracranial melanomas. Clin Cancer Res. (2019) 25:6801–14. doi: 10.1158/1078-0432.CCR-19-0405
18. Carter ME, Hartkopf AD, Wagner A, Volmer LL, Brucker SY, Berchtold S, et al. A three-dimensional organoid model of primary breast cancer to investigate the effects of oncolytic virotherapy. Front Mol Biosci. (2022) 9:826302. doi: 10.3389/fmolb.2022.826302
19. Russell L, Swanner J, Jaime-Ramirez AC, Wang Y, Sprague A, Banasavadi-Siddegowda Y, et al. PTEN expression by an oncolytic herpesvirus directs T-cell mediated tumour clearance. Nat Commun. (2018) 9:1–16. doi: 10.1038/s41467-018-07344-1
20. Zhu W, Zhang H, Shi Y, Song M, Zhu B, Wei L. Oncolytic adenovirus encoding tumour necrosis factor-related apoptosis inducing ligand (TRAIL) inhibits the growth and metastasis of triple-negative breast cancer. Cancer Biol Ther. (2013) 14:1016–23. doi: 10.4161/cbt.26043
21. Hutzler S, Erbar S, Jabulowsky RA, Hanauer JRH, Schnotz JH, Beissert T, et al. Antigen-specific oncolytic MV-based tumour vaccines through presentation of selected tumour-associated antigens on infected cells or virus-like particles. Sci Rep. (2017) 7:1–15. doi: 10.1038/s41598-017-16928-8
22. Tsuji T, Nakamori M, Iwahashi M, Nakamura M, Ojima T, Iida T, et al. An armed oncolytic herpes simplex virus expressing thrombospondin-1 has an enhanced in vivo antitumour effect against human gastric cancer. Int J Cancer. (2013) 132:485–94. doi: 10.1002/ijc.27681
23. Goodwin JM, Schmitt AD, McGinn CM, Fuchs BC, Kuruppu D, Tanabe KK, et al. Angiogenesis inhibition using an oncolytic herpes simplex virus expressing endostatin in a murine lung cancer model. Cancer Invest. (2012) 30:243–50. doi: 10.3109/07357907.2012.654870
24. Frentzen A, Yu YA, Chen N, Zhang Q, Weibel S, Raab V, et al. Anti-VEGF single-chain antibody GLAF-1 encoded by oncolytic vaccinia virus significantly enhances antitumour therapy. Proc Natl Acad Sci U.S.A. (2009) 106:12915–20. doi: 10.1073/pnas.0900660106
25. Passaro C, Alayo Q, De Laura I, McNulty J, Grauwet K, Ito H, et al. Arming an oncolytic herpes simplex virus type 1 with a single-chain fragment variable antibody against PD-1 for experimental glioblastoma therapy. Clin Cancer Res. (2019) 25:290–9. doi: 10.1158/1078-0432.CCR-18-2311
26. Heidbuechel JPW, Engeland CE. Oncolytic viruses encoding bispecific T cell engagers: a blueprint for emerging immunovirotherapies. J Hematol Oncol. (2021) 14:1–24. doi: 10.1186/s13045-021-01075-5
27. Guo ZS, Lotze MT, Zhu Z, Storkus WJ, Song XT. Bi- and tri-specific T cell engager-armed oncolytic viruses: next-generation cancer immunotherapy. Biomedicines. (2020) 8:204. doi: 10.3390/biomedicines8070204
28. Rodríguez-García A, Giménez-Alejandre M, Rojas JJ, Moreno R, Bazan-Peregrino M, Cascalló M, et al. Safety and efficacy of VCN-01, an oncolytic adenovirus combining fiber HSG-binding domain replacement with RGD and hyaluronidase expression. Clin Cancer Res. (2015) 21:1406–18. doi: 10.1158/1078-0432.CCR-14-2213
29. Schäfer S, Weibel S, Donat U, Zhang Q, Aguilar RJ, Chen NG, et al. Vaccinia virus-mediated intra-tumoural expression of matrix metalloproteinase 9 enhances oncolysis of PC-3 xenograft tumours. BMC Cancer. (2012) 12:1–9. doi: 10.1186/1471-2407-12-366
30. Ries SJ. Elucidation of the molecular mechanism underlying tumour-selective replication of the oncolytic adenovirus mutant ONYX-015. Future Oncol. (2005) 1(6):763–6. doi: 10.2217/14796694.1.6.763
31. Umeoka T, Kawashima T, Kagawa S, Teraishi F, Taki M, Nishizaki M, et al. Visualization of intrathoracically disseminated solid tumours in mice with optical imaging by telomerase-specific amplification of a transferred green fluorescent protein gene. Cancer Res. (2004) 64:6259–65. doi: 10.1158/0008-5472.CAN-04-1335
32. Taki M, Kagawa S, Nishizaki M, Mizuguchi H, Hayakawa T, Kyo S, et al. Enhanced oncolysis by a tropism-modified telomerase-specific replication-selective adenoviral agent OBP-405 (‘Telomelysin-RGD’). Oncogene. (2005) 24:3130–40. doi: 10.1038/sj.onc.1208460
33. Fueyo J, Alemany R, Gomez-Manzano C, Fuller GN, Khan A, Conrad CA, et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. JNCI: J Natl Cancer Institute. (2003) 95:652–60. doi: 10.1093/jnci/95.9.652
34. Xu W, Zhang Z, Yang Y, Hu Z, Wang CH, Morgan M, et al. Ad5/48 hexon oncolytic virus expressing sTGFβRIIFc produces reduced hepatic and systemic toxicities and inhibits prostate cancer bone metastases. Mol Ther. (2014) 22:1504–17. doi: 10.1038/mt.2014.80
35. Marchini A, Bonifati S, Scott EM, Angelova AL, Rommelaere J. Oncolytic parvoviruses: From basic virology to clinical applications. Virol J. (2015) 12:1–16. doi: 10.1186/s12985-014-0223-y
36. Angelova AL, Witzens-Harig M, Galabov AS, Rommelaere J. The oncolytic virotherapy era in cancer management: Prospects of applying H-1 parvovirus to treat blood and solid cancers. Front Oncol. (2017) 7:268562. doi: 10.3389/fonc.2017.00093
37. Bretscher C, Marchini A. H-1 parvovirus as a cancer-killing agent: past, present, and future. Viruses. (2019) 11:562. doi: 10.3390/v11060562
38. Hartley A, Kavishwar G, Salvato I, Marchini A. A roadmap for the success of oncolytic parvovirus-based anticancer therapies. Annu Rev Virol. (2020) 7:537–57. doi: 10.1146/annurev-virology-012220-023606
39. Lemos de Matos A, Franco LS, McFadden G. Oncolytic viruses and the immune system: the dynamic duo. Mol Ther Methods Clin Dev. (2020) 17:349–58. doi: 10.1016/j.omtm.2020.01.001
40. Shi J, Pei Y, Yu Q, dong H. Progress in the study of parvovirus entry pathway. Virol J. (2023) 20:1–9. doi: 10.1186/s12985-023-02016-z
41. Cristi F, Gutiérrez T, Hitt MM, Shmulevitz M. Genetic modifications that expand oncolytic virus potency. Front Mol Biosci. (2022) 9:831091. doi: 10.3389/fmolb.2022.831091
42. Ahmed J, Chard LS, Yuan M, Wang J, Howells A, Li Y, et al. A new oncolytic V accinia virus augments antitumour immune responses to prevent tumour recurrence and metastasis after surgery. J Immunother Cancer. (2020) 8:e000415. doi: 10.1136/jitc-2019-000415
43. Kim MK, Breitbach CJ, Moon A, Heo J, Lee YK, Cho M, et al. Oncolytic and immunotherapeutic vaccinia induces antibody-mediated complement-dependent cancer cell lysis in humans. Sci Transl Med. (2013) 5. doi: 10.1126/scitranslmed.3005361
44. Brito Baleeiro R, Liu P, Chard Dunmall LS, Di Gioia C, Nagano A, Cutmore L, et al. Personalized neoantigen viro-immunotherapy platform for triple-negative breast cancer. J Immunother Cancer. (2023) 11:e007336. doi: 10.1136/jitc-2023-007336
45. Müller L, Berkeley R, Barr T, Ilett E, Errington-Mais F. Past, present and future of oncolytic reovirus. Cancers. (2020) 12:3219. doi: 10.3390/cancers12113219
46. Norman KL, Coffey MC, Hirasawa K, Demetrick DJ, Nishikawa SG, DiFrancesco LM, et al. Reovirus oncolysis of human breast cancer. Hum Gene Ther. (2004) 13(5):641–52. doi: 10.1089/10430340252837233
47. Bernstein V, Ellard SL, Dent SF, Tu D, Mates M, Dhesy-Thind SK, et al. A randomized phase II study of weekly paclitaxel with or without pelareorep in patients with metastatic breast cancer: final analysis of Canadian Cancer Trials Group IND.213. Breast Cancer Res Treat. (2018) 167:485–93. doi: 10.1007/s10549-017-4538-4
48. Mostafa AA, Meyers DE, Thirukkumaran CM, Liu PJ, Gratton K, Spurrell J, et al. Oncolytic reovirus and immune checkpoint inhibition as a novel immunotherapeutic strategy for breast cancer. Cancers. (2018) 10(6):205. doi: 10.3390/cancers10060205
49. Fernandes JP, Cristi F, Eaton HE, Chen P, Haeflinger S, Bernard I, et al. Breast tumour-associated metalloproteases restrict reovirus oncolysis by cleaving the σ1 cell attachment protein and can be overcome by mutation of σ1. J Virol. (2019) 93. doi: 10.1128/jvi.01380-19
50. Koch MS, Lawler SE, Antonio Chiocca E. HSV-1 oncolytic viruses from bench to bedside: an overview of current clinical trials. Cancers. (2020) 12:3514. doi: 10.3390/cancers12123514
51. Streby KA, Geller JI, Currier MA, Warren PS, Racadio JM, Towbin AJ, et al. Intratumoral injection of HSV1716, an oncolytic herpes virus, is safe and shows evidence of immune response and viral replication in young cancer patients. Clin Cancer Res. (2017) 23:3566–74. doi: 10.1158/1078-0432.CCR-16-2900
52. Todo T. Oncolytic virus therapy using genetically engineered herpes simplex viruses. Front Bioscience. (2008) 13:2060–4. doi: 10.2741/2823
53. Thomas S, Kuncheria L, Roulstone V, Kyula JN, Mansfield D, Bommareddy PK, et al. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J Immunother Cancer. (2019) 7:214. doi: 10.1186/s40425-019-0682-1
54. Streby KA, Currier MA, Triplet M, Ott K, Dishman DJ, Vaughan MR, et al. First-in-human intravenous seprehvir in young cancer patients: A phase 1 clinical trial. Mol Ther. (2019) 27:1930–8. doi: 10.1016/j.ymthe.2019.08.020
55. Javanbakht M, Tahmasebzadeh S, Cegolon L, Gholami N, Kashaki M, Nikoueinejad H, et al. Oncolytic viruses: A novel treatment strategy for breast cancer. Genes Dis. (2023) 10:430–46. doi: 10.1016/j.gendis.2021.11.011
56. Kennedy EM, Farkaly T, Grzesik P, Lee J, Denslow A, Hewett J, et al. Design of an interferon-resistant oncolytic HSV-1 incorporating redundant safety modalities for improved tolerability. Mol Ther Oncolytics. (2020) 18:476–90. doi: 10.1016/j.omto.2020.08.004
57. Alberts P, Tilgase A, Rasa A, Bandere K, Venskus D. The advent of oncolytic virotherapy in oncology: The Rigvir® story. Eur J Pharmacol. (2018) 837:117–26. doi: 10.1016/j.ejphar.2018.08.042
58. Babiker HM, Riaz IB, Husnain M, Borad MJ. Oncolytic virotherapy including Rigvir and standard therapies in Malignant melanoma. Oncolytic Virother. (2017) 6:11–8. doi: 10.2147/OV
59. Robinson C, Xu MM, Nair SK, Beasley GM, Rhodin KE. Oncolytic viruses in melanoma. Front Bioscience - Landmark. (2022) 27:63. doi: 10.31083/j.fbl2702063/htm
60. Onnockx S, Baldo A, Pauwels K. Oncolytic viruses: an inventory of shedding data from clinical trials and elements for the environmental risk assessment. Vaccines (Basel). (2023) 11:1448. doi: 10.3390/vaccines11091448
61. Bommareddy PK, Patel A, Hossain S, Kaufman HL. Talimogene laherparepvec (T-VEC) and other oncolytic viruses for the treatment of melanoma. Am J Clin Dermatol. (2017) 18:1–15. doi: 10.1007/s40257-016-0238-9
62. Ferrucci PF, Pala L, Conforti F, Cocorocchio E. Talimogene laherparepvec (T-VEC): an intralesional cancer immunotherapy for advanced melanoma. Cancers (Basel). (2021) 13:1–14. doi: 10.3390/cancers13061383
63. Rutkowski P, Zdzienicki M. Talimogene laherparepvec (T-VEC), review of a new therapy of cutaneous melanoma with genetically modified oncolytic virus. Nowotwory J Oncol. (2016) 66:234–7. doi: 10.5603/NJO.2016.0039
64. Zhang J, Zhang Q, Liu Z, Wang J, Shi F, Su J, et al. Efficacy and safety of recombinant human adenovirus type 5 (H101) in persistent, recurrent, or metastatic gynecologic Malignancies: A retrospective study. Front Oncol. (2022) 12:877155. doi: 10.3389/fonc.2022.877155
65. Cheng PH, Wechman SL, McMasters KM, Zhou HS. Oncolytic replication of E1b-deleted adenoviruses. Viruses. (2015) 7:5767–79. doi: 10.3390/v7112905
66. Zhang R, Cui Y, Guan X, Jiang X. A recombinant human adenovirus type 5 (H101) combined with chemotherapy for advanced gastric carcinoma: A retrospective cohort study. Front Oncol. (2021) 11:752504. doi: 10.3389/fonc.2021.752504
67. Frampton JE. Teserpaturev/G47Δ: first approval. BioDrugs. (2022) 36:667–72. doi: 10.1007/s40259-022-00553-7
68. Maruyama Y, Sakurai A, Noda S, Fujiwara Y, Okura N, Takagi T, et al. Regulatory issues: PMDA – review of sakigake designation products: oncolytic virus therapy with delytact injection (Teserpaturev) for Malignant glioma. Oncologist. (2023) 28:664–70. doi: 10.1093/oncolo/oyad041
69. O’Bryan SM, Mathis JM. Oncolytic virotherapy for breast cancer treatment. Curr Gene Ther. (2018) 18:192. doi: 10.2174/1566523218666180910163805
70. Singh PK, Doley J, Ravi Kumar G, Sahoo AP, Tiwari AK. Oncolytic viruses & their specific targeting to tumour cells. Indian J Med Res. (2012) 136:571.
71. Carter ME, Koch A, Lauer UM, Hartkopf AD. Clinical trials of oncolytic viruses in breast cancer. Front Oncol. (2021) 11:803050. doi: 10.3389/fonc.2021.803050
72. Bramante S, Koski A, Liikanen I, Vassilev L, Oksanen M, Siurala M, et al. Oncolytic virotherapy for treatment of breast cancer, including triple-negative breast cancer. Oncoimmunology. (2016) 5. doi: 10.1080/2162402X.2015.1078057
73. Cerullo V, Pesonen S, Diaconu I, Escutenaire S, Arstila PT, Ugolini M, et al. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoural immunity in cancer patients. Cancer Res. (2010) 70:4297–309. doi: 10.1158/0008-5472.CAN-09-3567
74. Cawood R, Hills T, Wong SL, Alamoudi AA, Beadle S, Fisher KD, et al. Recombinant viral vaccines for cancer. Trends Mol Med. (2012) 18:564–74. doi: 10.1016/j.molmed.2012.07.007
75. Nokisalmi P, Pesonen S, Escutenaire S, Särkioja M, Raki M, Cerullo V, et al. Oncolytic adenovirus ICOVIR-7 in patients with advanced and refractory solid tumours. Clin Cancer Res. (2010) 16:3035–43. doi: 10.1158/1078-0432.CCR-09-3167
76. Nemunaitis J, Senzer N, Sarmiento S, Zhang YA, Arzaga R, Sands B, et al. A phase I trial of intravenous infusion of ONYX-015 and enbrel in solid tumour patients. Cancer Gene Ther. (2007) 14:885–93. doi: 10.1038/sj.cgt.7701080
77. Hu JCC, Coffin RS, Davis CJ, Graham NJ, Groves N, Guest PJ, et al. A phase I study of oncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. (2006) 12:6737–47. doi: 10.1158/1078-0432.CCR-06-0759
78. Soliman H, Hogue D, Han H, Mooney B, Costa R, Lee MC, et al. Oncolytic T-VEC virotherapy plus neoadjuvant chemotherapy in nonmetastatic triple-negative breast cancer: a phase 2 trial. Nat Med. (2023) 29:450–7. doi: 10.1038/s41591-023-02210-0
79. Kimata H, Imai T, Kikumori T, Teshigahara O, Nagasaka T, Goshima F, et al. Pilot study of oncolytic viral therapy using mutant herpes simplex virus (HF10) against recurrent metastatic breast cancer. Ann Surg Oncol. (2006) 13:1078–84. doi: 10.1245/ASO.2006.08.035
80. McCrudden CM, McCarthy HO. Current status of gene therapy for breast cancer: progress and challenges. Appl Clin Genet. (2014) 7:209–20. doi: 10.2147/TACG
81. Laurie SA, Bell JC, Atkins HL, Roach J, Bamat MK, O’Neil JD, et al. A phase 1 clinical study of intravenous administration of PV701, an oncolytic virus, using two-step desensitization. Clin Cancer Res. (2006) 12:2555–62. doi: 10.1158/1078-0432.CCR-05-2038
82. Jin S, Wang Q, Wu H, Pang D, Xu S. Oncolytic viruses for triple negative breast cancer and beyond. biomark Res. (2021) 9:1–16. doi: 10.1186/s40364-021-00318-4
83. Wei W, Thórsson V, Tang Y, Monaco ML, Idris OA, Essani K. Triple-negative breast cancer: basic biology and immuno-oncolytic viruses. Cancers. (2023) 15:2393. doi: 10.3390/cancers15082393
84. Zhu W, Wei L, Zhang H, Chen J, Qin X. Oncolytic adenovirus armed with IL-24 Inhibits the growth of breast cancer in vitro and in vivo. J Exp Clin Cancer Res. (2012) 31:1–10. doi: 10.1186/1756-9966-31-51
85. Gao YG, Ge ZC, Zhang ZT, Bai ZG, Ma XM, Wang Y. Vascular endothelial growth inhibitor affects the invasion, apoptosis and vascularisation in breast cancer cell line MDA-MB-231. Chin Med J (Engl). (2014) 127:1947–53. doi: 10.3760/cma.j.issn.0366-6999.20130794
86. Deng L, Fan J, Ding Y, Yang X, Huang B, Hu Z. Target therapy with vaccinia virus harboring IL-24 for human breast cancer. J Cancer. (2020) 11:1017–26. doi: 10.7150/jca.37590
87. Yuan Y, Egelston C, Colunga Flores O, Chaurasiya S, Lin D, Chang H, et al. CF33-hNIS-anti-PD-L1 oncolytic virus followed by trastuzumab-deruxtecan in a patient with metastatic triple negative breast cancer: a case study. Ther Adv Med Oncol. (2023) 15. doi: 10.1177/17588359231210675
88. Stawowczyk M, Zhao Y, Pfeffer K, Tafrova J, Rodriguez J, Yang C, et al. Abstract PD2-04: Preclinical development of CodaLytic™, a codon-modified influenza virus, as a novel virotherapeutic agent for breast cancer immunotherapy. Cancer Res. (2023) 83:PD2–04. doi: 10.1158/1538-7445.SABCS22-PD2-04
89. Eissa IR, Bustos-Villalobos I, Ichinose T, Matsumura S, Naoe Y, Miyajima N, et al. The current status and future prospects of oncolytic viruses in clinical trials against melanoma, glioma, pancreatic, and breast cancers. Cancers (Basel). (2018) 10:356. doi: 10.3390/cancers10100356
90. Kwan A, Winder N, Muthana M. Oncolytic virotherapy treatment of breast cancer: barriers and recent advances. Viruses. (2021) 13:1128. doi: 10.3390/v13061128
91. Iyengar NM, Gucalp A, Dannenberg AJ, Hudis CA. Obesity and cancer mechanisms: tumour microenvironment and inflammation. J Clin Oncol. (2016) 34:4270. doi: 10.1200/JCO.2016.67.4283
92. Leong SP, Witz IP, Sagi-Assif O, Izraely S, Sleeman J, Piening B, et al. Cancer microenvironment and genomics: evolution in process. Clin Exp Metastasis. (2022) 39:85–99. doi: 10.1007/s10585-021-10097-9
93. Wong-Rolle A, Wei HK, Zhao C, Jin C. Unexpected guests in the tumour microenvironment: microbiome in cancer. Protein Cell. (2021) 12:426–35. doi: 10.1007/s13238-020-00813-8
94. Fridman WH, Pagès F, Saut̀s-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. (2012) 12:298–306. doi: 10.1038/nrc3245
95. Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res. (2016) 22:1865–74. doi: 10.1158/1078-0432.CCR-15-1507
96. Rodriguez AB, Peske JD, Engelhard VH. Identification and characterization of tertiary lymphoid structures in murine melanoma. Methods Mol Biol. (2018) 1845:241–57. doi: 10.1007/978-1-4939-8709-2_14
97. Tang H, Qiao J, Fu YX. Immunotherapy and tumour microenvironment. Cancer Lett. (2016) 370:85–90. doi: 10.1016/j.canlet.2015.10.009
98. Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumour mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. (2019) 51:202–6. doi: 10.1038/s41588-018-0312-8
99. Luksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, Solovyov A, et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature. (2017) 551:517–20. doi: 10.1038/nature24473
100. Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. (2018) 359:582. doi: 10.1126/science.aao4572
101. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. New Engl J Med. (2016) 375:819–29. doi: 10.1056/nejmoa1604958
102. Kurozumi K, Hardcastle J, Thakur R, Yang M, Christoforidis G, Fulci G, et al. Effect of tumour microenvironment modulation on the efficacy of oncolytic virus therapy. JNCI: J Natl Cancer Institute. (2007) 99:1768–81. doi: 10.1093/jnci/djm229
103. Evgin L, Kottke T, Tonne J, Thompson J, Huff AL, van Vloten J, et al. Oncolytic virus–mediated expansion of dual-specific CAR T cells improves efficacy against solid tumours in mice. Sci Transl Med. (2022) 14:2231. doi: 10.1126/scitranslmed.abn2231
104. Omole RK, Oluwatola O, Akere MT, Eniafe J, Agboluaje EO, Daramola OB, et al. Comprehensive assessment on the applications of oncolytic viruses for cancer immunotherapy. Front Pharmacol. (2022) 13:1082797. doi: 10.3389/fphar.2022.1082797
105. Stoff-Khalili MA, Rivera AA, Nedeljkovic-Kurepa A, DeBenedetti A, Li XL, Odaka Y, et al. Cancer-specific targeting of a conditionally replicative adenovirus using mRNA translational control. Breast Cancer Res Treat. (2008) 108:43–55. doi: 10.1007/s10549-007-9587-7
106. Kesari S, Lasner TM, Balsara KR, Randazzo BP, Lee VMY, Trojanowski JQ, et al. A neuroattenuated ICP34.5-deficient herpes simplex virus type 1 replicates in ependymal cells of the murine central nervous system. J Gen Virol. (1998) 79(Pt 3)(3):525–36. doi: 10.1099/0022-1317-79-3-525
107. Marchini A, Daeffler L, Pozdeev VI, Angelova A, Rommelaere J. Immune conversion of tumour microenvironment by oncolytic viruses: The protoparvovirus H-1PV case study. Front Immunol. (2019) 10:460251. doi: 10.3389/fimmu.2019.01848
108. Shen Z, Liu X, Fan G, Na J, Liu Q, Lin F, et al. Improving the therapeutic efficacy of oncolytic viruses for cancer: targeting macrophages. J Trans Med. (2023) 21:1–14. doi: 10.1186/s12967-023-04709-z
109. Wan PKT, Ryan AJ, Seymour LW. Beyond cancer cells: Targeting the tumour microenvironment with gene therapy and armed oncolytic virus. Mol Ther. (2021) 29:1668–82. doi: 10.1016/j.ymthe.2021.04.015
110. Jing Y, Zaias J, Duncan R, Russell SJ, Merchan JR. In vivo safety, biodistribution and antitumour effects of uPAR retargeted oncolytic measles virus in syngeneic cancer models. Gene Ther. (2014) 21:289–97. doi: 10.1038/gt.2013.84
111. Bolyard C, Meisen WH, Banasavadi-Siddegowda Y, Hardcastle J, Yoo JY, Wohleb ES, et al. BAI1 orchestrates macrophage inflammatory response to HSV infection-implications for oncolytic viral therapy. Clin Cancer Res. (2017) 23:1809–19. doi: 10.1158/1078-0432.CCR-16-1818
112. Wang Q, Yang M, Zhang Y, Zhong L, Zheng X. Novel combination oncolytic adenoviral gene therapy armed with dm-dNK and CD40L for breast cancer. Curr Gene Ther. (2019) 19:54–65. doi: 10.2174/1566523219666190307094713
113. Subhan MA, Parveen F, Shah H, Yalamarty SSK, Ataide JA, Torchilin VP. Recent advances with precision medicine treatment for breast cancer including triple-negative sub-type. Cancers (Basel). (2023) 15. doi: 10.3390/cancers15082204
114. Geurts V, Kok M. Immunotherapy for metastatic triple negative breast cancer: current paradigm and future approaches. Curr Treat Options Oncol. (2023) 24:628–43. doi: 10.1007/s11864-023-01069-0
115. Chon HJ, Lee WS, Yang H, Kong SJ, Lee NK, Moon ES, et al. Tumour microenvironment remodeling by intratumoral oncolytic vaccinia virus enhances the efficacy of immune-checkpoint blockade. Clin Cancer Res. (2019) 25:1612–23. doi: 10.1158/1078-0432.CCR-18-1932
116. Sahin TT, Kasuya H, Nomura N, Shikano T, Yamamura K, Gewen T, et al. Impact of novel oncolytic virus HF10 on cellular components of the tumour microenviroment in patients with recurrent breast cancer. Cancer Gene Ther. (2012) 19:229–37. doi: 10.1038/cgt.2011.80
117. Groeneveldt C, Kinderman P, Van Den Wollenberg DJM, Van Den Oever RL, Middelburg J, Mustafa DAM, et al. Original research: Preconditioning of the tumour microenvironment with oncolytic reovirus converts CD3-bispecific antibody treatment into effective immunotherapy. J Immunother Cancer. (2020) 8:1191. doi: 10.1136/jitc-2020-001191
118. Brito Baleeiro R, Liu P, Chard Dunmall LS, Di Gioia C, Nagano A, Cutmore L, et al. Original research: Personalized neoantigen viro-immunotherapy platform for triple-negative breast cancer. J Immunother Cancer. (2023) 11:7336. doi: 10.1136/jitc-2023-007336
119. Nabi R, Musarrat F, Jose JC, Langohr IM, Chouljenko VN, Kousoulas KG. The Oncolytic herpes simplex virus type-1 (HSV-1) vaccine strain VC2 causes intratumour infiltration of functionally active T cells and inhibition of tumour metastasis and pro-tumour genes VEGF and PDL1 expression in the 4T1/Balb/c mouse model of stage four breast cancer. Front Mol Biosci. (2023) 10:1199068. doi: 10.3389/fmolb.2023.1199068
120. Lisi L, Lacal PM, Martire M, Navarra P, Graziani G. Clinical experience with CTLA-4 blockade for cancer immunotherapy: From the monospecific monoclonal antibody ipilimumab to probodies and bispecific molecules targeting the tumour microenvironment. Pharmacol Res. (2022) 175:105997. doi: 10.1016/j.phrs.2021.105997
121. O’Meara TA, Tolaney SM, Meara TA, Tolaney SM. Tumour mutational burden as a predictor of immunotherapy response in breast cancer. Oncotarget. (2021) 12:394–400. doi: 10.18632/oncotarget.27877
122. Luo C, Wang P, He S, Zhu J, Shi Y, Wang J. Progress and prospect of immunotherapy for triple-negative breast cancer. Front Oncol. (2022) 12:919072. doi: 10.3389/fonc.2022.919072
123. Nejatipour Z, Teimoori-Toolabi L, Forooshani RS, Barough MS, Farahmand M, Biglari A, et al. Looking for biomarkers in interferon response pathway to predict response to oncolytic HSV-1 in breast cancer: An ex vivo study. Cancer Biomarkers. (2023) 38:37–47. doi: 10.3233/CBM-230033
124. Cousin S, Toulmonde M, Kind M, Guegan JP, Bessede A, Cantarel C, et al. Phase 2 trial of intravenous oncolytic virus JX-594 combined with low-dose cyclophosphamide in patients with advanced breast cancer. Exp Hematol Oncol. (2022) 11:1–4. doi: 10.1186/s40164-022-00338-2
125. Howard F, Conner J, Danson S, Muthana M. Inconsistencies in modeling the efficacy of the oncolytic virus HSV1716 reveal potential predictive biomarkers for tolerability. Front Mol Biosci. (2022) 9:889395. doi: 10.3389/fmolb.2022.889395
126. McDonald CJ, Erlichman C, Ingle JN, Rosales GA, Allen C, Greiner SM, et al. A measles virus vaccine strain derivative as a novel oncolytic agent against breast cancer. Breast Cancer Res Treat. (2006) 99:177–84. doi: 10.1007/s10549-006-9200-5
127. Mencattini A, Lansche C, Veith I, Erbs P, Balloul JM, Quemeneur E, et al. Direct imaging and automatic analysis in tumour-on-chip reveal cooperative antitumoural activity of immune cells and oncolytic vaccinia virus. Biosens Bioelectron. (2022) 215:114571. doi: 10.1016/j.bios.2022.114571
128. Kaufman HL. Can biomarkers guide oncolytic virus immunotherapy? Clin Cancer Res. (2021) 27:3278–9. doi: 10.1158/1078-0432.CCR-21-0660
129. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discovery. (2015) 14:642–62. doi: 10.1038/nrd4663
130. Nguyen A, Ho L, Wan Y. Chemotherapy and oncolytic virotherapy: Advanced tactics in the war against cancer. Front Oncol. (2014) 4:91394. doi: 10.3389/fonc.2014.00145
131. Rudin CM, Poirier JT, Senzer NN, Stephenson J, Loesch D, Burroughs KD, et al. Phase I clinical study of seneca valley virus (SVV-001), a replication-competent picornavirus, in advanced solid tumours with neuroendocrine features. Clin Cancer Res. (2011) 17:888. doi: 10.1158/1078-0432.CCR-10-1706
132. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumours. Nat Rev Clin Oncol. (2010) 7:653–64. doi: 10.1038/nrclinonc.2010.139
133. He B, Gao R, Lv S, Chen A, Huang J, Wang L, et al. Cancer cell employs a microenvironmental neural signal trans-activating nucleus-mitochondria coordination to acquire stemness. Signal Transduction Targeted Ther. (2023) 8:1–19. doi: 10.1038/s41392-023-01487-4
134. Lawler SE, Speranza MC, Cho CF, Chiocca EA. Oncolytic viruses in cancer treatment: A review. JAMA Oncol. (2017) 3:841–9. doi: 10.1001/jamaoncol.2016.2064
135. Lin D, Shen Y, Liang T. Oncolytic virotherapy: basic principles, recent advances and future directions. Signal Transduction Targeted Ther. (2023) 8:1–29. doi: 10.1038/s41392-023-01407-6
136. Zheng M, Huang J, Tong A, Yang H. Oncolytic viruses for cancer therapy: barriers and recent advances. Mol Ther Oncolytics. (2019) 15:234. doi: 10.1016/j.omto.2019.10.007
137. Suryawanshi YR, Zhang T, Essani K. Oncolytic viruses: emerging options for the treatment of breast cancer. Med Oncol. (2017) 34:1–11. doi: 10.1007/s12032-017-0899-0
138. Tai JH, Williams JV, Edwards KM, Wright PF, Crowe JE, Dermody TS. Prevalence of reovirus-specific antibodies in young children in Nashville, Tennessee. J Infect Dis. (2005) 191:1221–4. doi: 10.1086/428911
139. Ferguson MS, Lemoine NR, Wang Y. Systemic delivery of oncolytic viruses: hopes and hurdles. Adv Virol. (2012). doi: 10.1155/2012/805629
140. Stoermer KA, Morrison TE. Complement and viral pathogenesis. Virology. (2011) 411:362–73. doi: 10.1016/j.virol.2010.12.045
141. Roy DG, Bell JC. Cell carriers for oncolytic viruses: current challenges and future directions. Oncolytic Virother. (2013) 2:47–56. doi: 10.2147/OV
142. Kuczynski EA, Vermeulen PB, Pezzella F, Kerbel RS, Reynolds AR. Vessel co-option in cancer. Nat Rev Clin Oncol. (2019) 16:469–93. doi: 10.1038/s41571-019-0181-9
143. Huang D, Jin YH, Weng H, Huang Q, Zeng XT, Wang XH. Combination of intravesical bacille calmette-guérin and chemotherapy vs. Bacille calmette-guérin alone in non-muscle invasive bladder cancer: A meta-analysis. Front Oncol. (2019) 9. doi: 10.3389/fonc.2019.00121
144. Khuri FR, Nemunaitis J, Ganly I, Arseneau J, Tannock IF, Romel L, et al. a controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat Med. (2000) 6:879–85. doi: 10.1038/78638
145. Jayawardena N, Poirier JT, Burga LN, Bostina M, Virotherapy O. Virus–receptor interactions and virus neutralization: insights for oncolytic virus development. Oncolytic Virother. (2020) 9:1–15. doi: 10.2147/OV
146. Gail MH, Brinton LA, Byar DP, Corle DK, Green SB, Schairer C, et al. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst. (1989) 81:1879–86. doi: 10.1093/jnci/81.24.1879
147. Dix B, O’Carroll S, Myers C, Edwards S, Braithwaite A. Efficient induction of cell death by adenoviruses requires binding of E1B55k and p53. Cancer Res. (2000) 60(10):2666–72.
148. Waddington SN, McVey JH, Bhella D, Parker AL, Barker K, Atoda H, et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell. (2008) 132:397–409. doi: 10.1016/j.cell.2008.01.016
149. Alba R, Bradshaw AC, Coughlan L, Denby L, McDonald RA, Waddington SN, et al. Biodistribution and retargeting of FX-binding ablated adenovirus serotype 5 vectors. Blood. (2010) 116:2656–64. doi: 10.1182/blood-2009-12-260026
150. Park AK, Fong Y, Kim SI, Yang J, Murad JP, Lu J, et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumours. Sci Transl Med. (2020) 12. doi: 10.1126/scitranslmed.aaz1863
Glossary
Keywords: breast cancer, immunotherapy, oncolytic virus therapy, tumour microenvironment, TME
Citation: Chowaniec H, Ślubowska A, Mroczek M, Borowczyk M, Braszka M, Dworacki G, Dobosz P and Wichtowski M (2024) New hopes for the breast cancer treatment: perspectives on the oncolytic virus therapy. Front. Immunol. 15:1375433. doi: 10.3389/fimmu.2024.1375433
Received: 23 January 2024; Accepted: 11 March 2024;
Published: 21 March 2024.
Edited by:
Don J. Diamond, City of Hope National Medical Center, United StatesReviewed by:
Peng Qu, National Institutes of Health (NIH), United StatesGuohao Wang, National Institutes of Health (NIH), United States
Copyright © 2024 Chowaniec, Ślubowska, Mroczek, Borowczyk, Braszka, Dworacki, Dobosz and Wichtowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanna Chowaniec, aGFubmEuY2hvd2FuaWVjQGdtYWlsLmNvbQ==