
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 03 April 2024
Sec. Vaccines and Molecular Therapeutics
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1373367
This article is part of the Research TopicCommunity Series in Fighting an Elusive Enemy: Staphylococcus aureus and its Antibiotic Resistance, Immune-Evasion and Toxic Mechanisms: Volume 2View all 5 articles
 Yu Wang1†
Yu Wang1† Ipsita Mukherjee2†
Ipsita Mukherjee2† Arundhathi Venkatasubramaniam2
Arundhathi Venkatasubramaniam2 Dustin Dikeman1
Dustin Dikeman1 Nicholas Orlando1Jing Zhang1
Nicholas Orlando1Jing Zhang1 Roger Ortines2
Roger Ortines2 Mark Mednikov2Shardulendra P. Sherchand2
Mark Mednikov2Shardulendra P. Sherchand2 Tulasikumari Kanipakala2Thao Le3
Tulasikumari Kanipakala2Thao Le3 Sanjay Shukla3Mark Ketner4Rajan P. Adhikari2
Sanjay Shukla3Mark Ketner4Rajan P. Adhikari2 Hatice Karauzum2M. Javad Aman2
Hatice Karauzum2M. Javad Aman2 Nathan K. Archer1*
Nathan K. Archer1*Staphylococcus aureus is the leading cause of skin and soft tissue infections (SSTIs) in the U.S. as well as more serious invasive diseases, including bacteremia, sepsis, endocarditis, surgical site infections, osteomyelitis, and pneumonia. These infections are exacerbated by the emergence of antibiotic-resistant clinical isolates such as methicillin-resistant S. aureus (MRSA), highlighting the need for alternatives to antibiotics to treat bacterial infections. We have previously developed a multi-component toxoid vaccine (IBT-V02) in a liquid formulation with efficacy against multiple strains of Staphylococcus aureus prevalent in the industrialized world. However, liquid vaccine formulations are not compatible with the paucity of cold chain storage infrastructure in many low-to-middle income countries (LMICs). Furthermore, whether our IBT-V02 vaccine formulations are protective against S. aureus isolates from LMICs is unknown. To overcome these limitations, we developed lyophilized and spray freeze-dried formulations of IBT-V02 vaccine and demonstrated that both formulations had comparable biophysical attributes as the liquid formulation, including similar levels of toxin neutralizing antibodies and protective efficacy against MRSA infections in murine and rabbit models. To enhance the relevancy of our findings, we then performed a multi-dimensional screen of 83 S. aureus clinical isolates from LMICs (e.g., Democratic Republic of Congo, Palestine, and Cambodia) to rationally down-select strains to test in our in vivo models based on broad expression of IBT-V02 targets (i.e., pore-forming toxins and superantigens). IBT-V02 polyclonal antisera effectively neutralized toxins produced by the S. aureus clinical isolates from LMICs. Notably, the lyophilized IBT-V02 formulation exhibited significant in vivo efficacy in various preclinical infection models against the S. aureus clinical isolates from LMICs, which was comparable to our liquid formulation. Collectively, our findings suggested that lyophilization is an effective alternative to liquid vaccine formulations of our IBT-V02 vaccine against S. aureus infections, which has important implications for protection from S. aureus isolates from LMICs.
Staphylococcus aureus is a major health burden worldwide, being the predominant cause of skin and soft tissue infections (SSTIs), bacteremia, and pyomyositis (1–3). Furthermore, a global antibiotic resistance study found that methicillin-resistant S. aureus (MRSA) is now the major clinical isolate recovered in infected patients from both industrialized and low-to-middle income countries (LMICs) (4). This is particularly concerning in LMICs, which are disproportionately affected by antibiotic-resistant bacterial infections due to limited access to healthcare resources, decreased surveillance of antibiotic-resistance, and challenges to widespread vaccination distribution (5–9). For instance, in-hospital mortality due to intensive care unit-acquired infections and S. aureus bacteremia is significantly higher in LMICs than industrialized countries (10, 11). Thus, there is an unmet need for alternative therapeutic strategies to circumvent the emergence of antibiotic-resistant S. aureus infections, especially in LMICs.
We have developed IBT-V02 (12), a novel multi-component toxoid vaccine against S. aureus infections that neutralizes extracellular pore-forming toxins α-hemolysin (Hla), Panton-Valentine leukocidin (PVL), leukocidin AB (LukAB), and the superantigens toxic shock syndrome toxin-1 (TSST-1) and staphylococcal enterotoxins A and B (SEA and SEB). These toxins are important in S. aureus pathogenesis as they selectively kill or manipulate immune cells to evade host immunity (13, 14). This is especially pertinent as S. aureus cell-associated antigens have failed as vaccine targets in clinical trials (15–17). A liquid formulation of IBT-V02 was previously shown to have marked efficacy against S. aureus clinical isolates from the industrialized world (e.g., USA100, USA300, and USA1000) in a murine intradermal infection model (12). However, it is unclear whether dry formulations of IBT-V02 (e.g., lyophilized and spray freeze-dried), which may overcome the limitations of maintaining the cold chain and storage in LMICs (18), can recapitulate efficacy of the liquid IBT-V02 formulation. Furthermore, whether dry and liquid formulations have comparable efficacy against S. aureus clinical isolates from LMICs is unknown.
To address these gaps in knowledge, we first developed and biochemically tested the protein integrity, long-term stability, and immunogenicity of dry formulations of IBT-V02 in comparison to the liquid formulation. Moreover, we performed comparative efficacy studies of the dry and liquid formulations using a murine intradermal infection model and a novel rabbit pyomyositis infection model. Furthermore, we collected S. aureus clinical isolates from LMICs and used a rational down-selection strategy to identify isolates with broad toxin and superantigen production. Finally, we used the selected LMIC isolates to test the comparative efficacy of dry and liquid formulations of IBT-V02 in bacteremia and intradermal infection models in mice.
Bioluminescent S. aureus strain USA300 (SF8300) was kindly provided by Dr. Binh Diep at UCSF (19). S. aureus strains referred to as ‘Cambodia isolates’ were kindly provided by Dr. Catherine Moore at University of Oxford, UK. S. aureus strains referred to as ‘Kinshasa isolates’ were kindly provided by Dr. Frieder Schaumburg at University Hospital of Munster, Germany. S. aureus strains referred to as ‘Gaza isolates’ were kindly provided by Dr. Barry Kreiswirth at Center for Discovery & Innovation, HMH, NJ, USA.
The HL-60 cells, from ATCC (atcc.org), Manassas, VA were cultured in RPMI media containing 82 U/ml of penicillin and streptomycin with 16% FBS. Cells were differentiated in media with 1.5% dimethyl sulfoxide (DMSO), as previously described (20).
For mouse infections with USA300 (SF8300) and LMIC isolates (KCO075, Pyo603, or Gaza209) and for rabbit infections with USA300 (SF8300), S. aureus strains were cultured in tryptic soy broth (TSB) as previously described (21, 22). Briefly, S. aureus was streaked onto a tryptic soy agar (TSA) plate (TSB plus 1.5% bacto agar (BD Biosciences)) and grown overnight at 37°C in a bacterial incubator. Two-to-three colonies were picked and cultured in TSB at 37°C in a shaking incubator (MaxQ HP 420, ThermoFisher) (240 rpm) overnight (16 h), followed by a 1:50 subculture at 37°C for 2 h to obtain mid-logarithmic phase bacteria. The bacteria were pelleted and washed in sterile phosphate-buffered saline (PBS). The absorbance at 600 nm (A600) was measured to estimate the number of colony-forming units (CFU) for inoculation, which was verified in each experiment after overnight culture on TSA plates.
For mouse experiments, 6-week-old female BALB/c mice obtained from Charles River were used. For rabbit experiments, 16-week-old female Dutch Belted Rabbits obtained from Robinson Services were used. All animals were maintained under the same specific pathogen-free conditions, with air-isolated cages at an American Association for the Accreditation of Laboratory Animal Care (AAALAC)-accredited animal facility at Johns Hopkins University and Integrated BioTherapeutics (IBT). They were handled according to procedures described in the Guide for the Care and Use of Laboratory Animals as well as Johns Hopkins University’s policies and procedures as outlined in the Johns Hopkins University Animal Care and Use Training Manual. Studies at JHU were approved by the Johns Hopkins Animal Care and Use Committee (Protocol #: MO21M378 for mice, Protocol#: RB22M400 for rabbit). Mouse studies at IBT were approved by institutional animal care and use committees (IACUC) (Immunogenicity Protocol# AP160805 and Efficacy against bacterial challenge Protocol#: AP-161007).
Mice and rabbits were immunized intramuscularly (IM) or subcutaneously (SC) on each side of the tail base or thigh muscle (50 µl each side) three times, two weeks apart, with a total of 50 µg of IBT-V02 (10 µg each antigen) or 50 µg of BSA in 200 µg Alhydrogel in 100 µl histidine-borate buffer (buffer reconstituted in sterile water in the case of lyophilized vaccine) for each immunization. For mouse serological analyses, mice were bled via the retro-orbital (RO) route prior to and 10 days after the final immunization. For rabbit serological analyses, rabbits were bled via the central ear artery prior to, 10 days after the final immunization, and 7 days after infection.
Two weeks after the last immunization, the ID infection was performed as previously described (12, 23, 24). Briefly, the dorsal backs of anesthetized mice (2% isoflurane) were shaved and intradermally injected with S. aureus (8×106 CFU of SF8300, 1×109 CFU of KCO075, 3×108 CFU of Pyo603, 2×108 CFU of Gaza209) in 100 µl of PBS using a 29-gauge insulin syringe. Inocula were chosen to achieve an ~2 cm2 lesion size for each S. aureus isolate. Total lesion size (cm2) was measured by analyzing digital photographs using Image J ((https://imagej.nih.gov/ij/) and a millimeter ruler as a reference. For the mice infected with bioluminescent SF8300, in vivo bioluminescence (BLI) was performed using a Lumina III IVIS (PerkinElmer) and total flux (photons/s) was measured within a 1.78×1.78 cm circular region of interest using Living Image software (PerkinElmer). Ex vivo CFU were enumerated from overnight cultures of serially diluted 10 mm lesioned skin punch biopsy specimen homogenized at 4°C (Pro200 Series homogenizer; Pro Scientific).
Two weeks after the last immunization, the IM infection was performed on the right thigh. Rabbits were anesthetized with IM ketamine (20-30 mg/kg) and xylazine (1-2 mg/kg) and general anesthesia was maintained with inhalation isoflurane (∼1.5%). Sterile ophthalmic ointment (Rugby, Livonia, MI, USA) was applied to the eyes. Metoclopramide (Teva) (0.3 mg/kg) was injected subcutaneously as a gastrointestinal promotility agent and sustained-release buprenorphine (ZooPharm) (0.2 mg/kg) was injected subcutaneously for analgesia. For infection, 6-8 ×109 CFU of bioluminescent SF8300 in 100 µl of PBS was injected into the muscle located in the middle of femur with depth of 0.5 cm using a 29-gauge insulin syringe. In vivo BLI was performed using a Lumina III IVIS (PerkinElmer) and maximum flux (photons/s/cm2/steradian) was measured within an 8.1×6.4 cm circular region of interest using Living Image software (PerkinElmer). Thigh width was measured by Pittsburgh digital caliper (Harbor Freight Tools). Ex vivo CFU was enumerated from overnight cultures of serially diluted muscle abscess specimen homogenized at 4°C (Pro200 Series homogenizer; Pro Scientific).
Lethal and sublethal doses of the three LMIC strains in BALB/c mice were first established prior to testing the efficacy of IBT-V02 in a systemic infection model. Mice were challenged with different doses of either KCO075, Pyo603 or Gaza209 via the intraperitoneal (IP) route and infective dose resulting in 60-80% mortality was selected for testing efficacy of IBT-V02 in the IP infection model. For bacteremia, six weeks after the last immunization, mice were infected intraperitoneally (IP) with a 200 µl suspension of S. aureus (8x107 CFU/mouse of KCO075, 4x107 CFU/mouse of Pyo603 or 2x107 CFU/mouse of Gaza209), as modified from previously described protocols (20). The animals were monitored daily for 14 days for survival.
Differential Scanning Calorimetry was performed to determine the glass transition temperature (Tg’), collapse temperature (Tc), and impact of freezing rate for the liquid formulation (HlaH35LH48L, LukSmut9, LukFmut1, TBA225 and LukABmut50 formulated at total protein 0.2mg/ml in 20mM histidine-borate, 1.2mM phosphate buffer with 8% sucrose and 0.02% Tween 80). The average values for Tg’ and Tc obtained from several experiments were used to design the lyophilization process, with primary drying set at -40°C (5°C below Tc) and annealing at -25°C (at least 5°C above the Tg’). Traditional lyophilization was accomplished by freezing the liquid vaccine and then reducing the surrounding pressure and applying enough heat to allow the frozen water in the material to sublime. During primary drying, the pressure in the surrounding environment was lowered to facilitate sublimation. In secondary drying the remaining water molecules were removed, bringing the lyophilized vaccine down to a final moisture content of between 3 and 5%. The result of the process was a “cake” of powder that could be reconstituted in water at time of use. In addition to traditional lyophilization, a spray freeze-drying process was also incorporated where a carefully formulated liquid solution was atomized into specifically sized spherical droplets (determined by spray nozzle orifice) and immediately frozen, locking in the size and shape of each individual particle. The particles were collected and dried in bulk in a standard lyophilization chamber. Both traditional lyophilization and spray freeze-drying processes were run at Engineered BioPharmaceuticals to produce small-scale batches to determine feasibility followed by production of large-scale batches. For long-term storage studies, lyophilized IBT-V02 formulations were kept at -20°C and 30°C for 1, 3, 6, 9, or 12 months.
The lyophilized and spray freeze-dried (SFD) IBT-V02 were characterized by several biophysical/biochemical assays. Briefly, Bicinchoninic acid protein assay (BCA) was used to confirm the protein concentrations after reconstitution of lyophilized/SFD IBT-V02 in sterile water. SDS-PAGE was run at protein loads as indicated in Figure 1A using a 12% Bis-Tris gel. For WB, primary antibodies which were antigen-specific and goat anti-rabbit IgG alkaline phosphatase conjugate (Bio-Rad) secondary antibody (1:3000, v/v) were used. For SEC-HPLC, 50µg of Lyo, SFD, or liquid IBT-V02 were injected in an Agilent Technologies 1260 Infinity Series instrument using an AdvanceBio SEC 300Å 7.8x300 mm LC column with a mobile phase of 50 mM sodium phosphate buffer + 150 mM NaCl, pH 7.0 running at a flow rate of 0.5 mL/min. The chromatogram generated by the Agilent OpenLabs software plots absorbance at 280 nm as a function of retention time. All analysis of the peaks was performed by the auto-integrate function in the OpenLabs software.
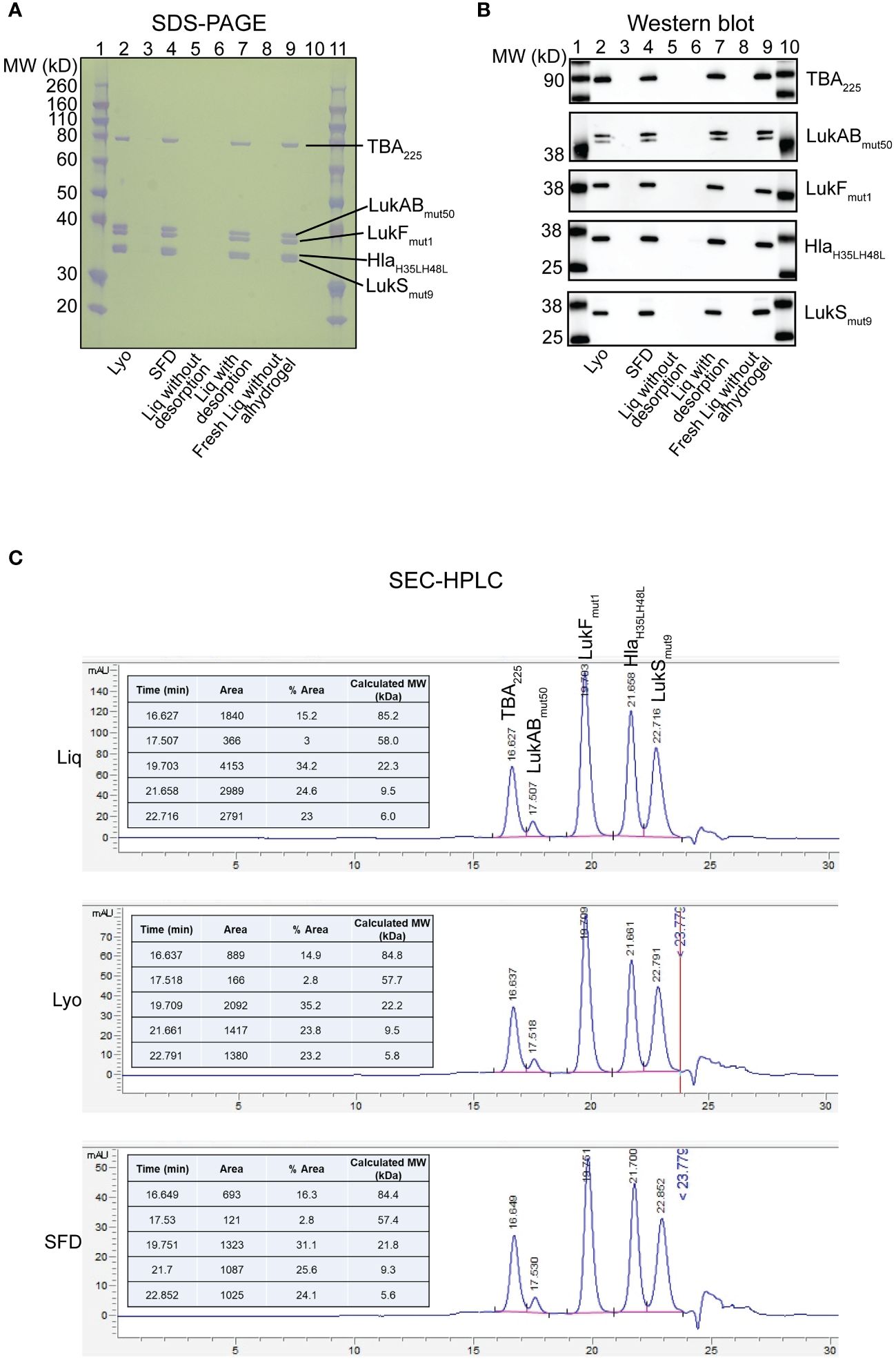
Figure 1 Biochemical characterization of IBT-V02 components in Lyo, SFD and Liq vaccine formulations. (A) SDS-PAGE and (B) Western Blot analysis of Lyo (Lane 2), SFD (Lane 4), Liq without desorption (Lane 6), Liq with desorption (Lane 7) and freshly prepared Liq without alhydrogel (Lane 9). Samples were loaded at 1µg total protein for SDS-PAGE and 100ng for all individual proteins in Western Blot, except LukABmut50 which was loaded at 250ng. (C) SEC-HPLC of Liq (Top), Lyo (Middle) and SFD (Bottom).
Bacterial cells were streaked on sheep blood agar plates and incubated at 37°C overnight for 16 hours. Next, 7ml of BHI broth were inoculated with 4-6 colonies from each plate, incubated at 37°C at 230rpm for 16 hours, and OD600 was measured. Cells were centrifuged at 3500rpm, and supernatant was collected. The volume of supernatants among different cultures was normalized to a bacterial OD600 = 6.0 with BHI broth. The normalized supernatants were then sterile-filtered and stored at -80°C.
Qualitative α-, β- and δ-hemolysin production was evaluated on sheep blood agar plates, as previously described (25). Briefly, bacterial strains were streaked perpendicular to a β-hemolysin producing tester strain, RN4220. The inoculated plate was incubated at 37°C for 16-18 hours. Results were determined as follows: δ-hemolysin and β-hemolysin act synergistically, which produces a clearing zone at their intersection. α-hemolysin, and β-hemolysin inhibit each other, which results in a V-shaped zone where they intersect. When a strain is β-hemolytic, the clearing blends in with the tester strain. When a strain is both α- and δ-hemolytic, there is a stronger clearing on the characteristic V-shaped zone.
Induced HL-60 cells were harvested by centrifugation at 1200 rpm for 10 minutes at 20°C (12, 20). Cells were washed and resuspended with phenol red-free RPMI 1640 (Gibco) supplemented with 2% FBS to a final concentration of 5 x 106 cells/ml. Supernatants were serially diluted 2-fold across 96-well plates (50 µl/well) and 100µl of cells were added to each well. Plates were incubated at 37°C, 5% CO2, 95% humidity for 3 hours. After 3 hours, 50 μl of reconstituted CellTiter Glo reagent (Promega) was added to each well. The plate was shaken on an orbital shaker for 10-15 min at room temperature followed by measurement of luminescence (emission at 560 nm) using Cytation 5 imaging reader (Biotek) and Gen5 2.09 software to determine cell viability.
Polyclonal rabbit antibody against IBT-V02 was diluted 2-fold in phenol-red RPMI across 96-well plate starting at 500µg/ml (25 µl/well) (12, 20). Equal volumes of supernatants were added at determined concentrations. 100µl of prepared induced HL-60 cells were added to each well and incubated at 37°C, 5% CO2, 95% humidity. Cell viability was determined with CellTiter Glo reagent, as described above.
10ml PBS was added to 5ml of rabbit blood (Colorado Springs) and centrifuged at 1500rpm for 10 minutes at 20°C (12, 20). Supernatant was discarded and pellet was washed with 13ml PBS. The final pellet was weighed and diluted in PBS to 8% w/v for rabbit blood.
Bacterial supernatants were serially diluted 2-fold in PBS across 96-well plates (100 µl/well). 100 µl of prepared red blood cells were added to each well. The plate was incubated at 37°C for 30 minutes and then centrifuged at 3500rpm for 3 minutes at 20°C. Supernatants were transferred to 96-well plates compatible with plate reader. Toxicity was determined by measuring absorbance at 416nm using Spectramax 190 plate reader (Molecular Devices) and Softmax 5.4.5 software.
Polyclonal rabbit antibody against IBT-V02 was diluted 2-fold in PBS across 96-well plate starting at 500µg/ml (50 µl/well) (12, 20). Equal volumes of supernatants were added at determined concentrations. 100µl of prepared rabbit red blood cells were added to each well and incubated at 37°C, 5% CO2, 95% humidity. Neutralization was determined by measuring absorbance at 416nm using Spectramax 190 plate reader (Molecular Devices) and Softmax 5.4.5 software.
Supernatant dilutions consisted of 75% supernatant and 25% 4X Laemli’s reducing buffer (Boston BioProducts). Dilutions were denatured at 70°C for 10 minutes or at 100°C for 5 minutes. Gels were run for 35 minutes at 165V. Blots were transferred using a standard 7-minute procedure using iBlot 2 (Thermo Fisher Scientific).
Blots were blocked with StartingBlock (TBS) blocking buffer (Thermo Fisher Scientific) for 10 minutes and incubated in primary antibodies specific to different Staphylococcal toxins: Hla (Cat# Hla-ID-P), LukS-PV(Cat# LukS-ID-P), LukAB (cat# LukAB-ID-P), LukF-PV (Cat# LukF-IP-T01), TSST-1 (cat# TSST-1-ID-P-02), SEB (Cat-SEB-ID-P), and SEA (cat# SEA_ID-02) (all generated by IBT Bioservices) for 18 +/- 2 hours (26). This was followed by 2 rapid washes and a third 5-minute wash in 1X TBS-T (Thermo Fisher Scientific). Secondary antibody was diluted at 1:3000, and blots were incubated in it for 50-60 minutes. The blots were finally washed three times as described earlier. Blots incubated in AP- or HRP-conjugated secondary antibodies were washed in 1X TBS for 5 minutes. When using AP-conjugated secondary antibody (Bio-Rad), blots were developed using AP detection buffer (Bio-Rad) and imaged with a camera (Azure system). The blots incubated in HRP-conjugated secondary antibodies (KPL) were developed with ECL reagent (Azure) and imaged with Azure 600 imager.
QiAmp DNA Mini Kit (Qiagen, Cat# 51306) was used to extract and purify DNA from the bacterial isolates. A small number of pure colonies (5, 6) bacteria were mixed with 200 µL of purified water as a starting material. The DNA was extracted by following the manufacture’s protocol. The following are the PCR reagents and conditions used for PCRs. PCR Reagents: 15µl of HotStarTaq ™ Master Mix, 1µl of Forward primer (20 pmol/µl), 1µl of Reverse primer (20 pmol/µl), 3µl of extracted DNA for the template, 0.6 µl of 50 mM Mg2+, 9.4 µl of dH2O PCR Conditions: i.) Initial denaturation 95°C for 15 minutes; ii.) 35 cycles of Denaturation 94°C for 30 seconds, Annealing 55°C for 30seconds, and Elongation 72°C for 45 seconds; iii.) Final Elongation 72°C for 10 minutes; and iv.) Hold at 4°C. Genotyping primers are listed in Supplementary Table 1. Genotyping data were generated at Marshfield clinic under the supervision of Dr. Shukla.
A multiplex assay to detect serum IgG titers to S. aureus antigens has been previously developed at IBT using the Luminexⓒ xMAP® technology (20). Briefly, IBT-V02 target antigens Hla, LukS-PV, LukF-PV, LukAB, SEA, SEB, and TSST-1 were coupled to carboxylated MagPlex microsphere beads with distinct spectral regions via a carbodiimide reaction. Antigen-coupled beads were incubated with serum samples (mouse or rabbit) at a starting dilution of 1:40 in a two-fold 8-point dilution series at room temperature (RT) for 2 hours. Samples were washed and incubated with a PE-conjugated goat anti-mouse or donkey anti-rabbit IgG Antibody (Biolegend, San Diego, CA.) for one hour at RT. The samples were washed and acquired using a Luminex200. Data were analyzed using a 4-parameter (4PL) curve fit in XLFit (Microsoft). IgG titers were expressed as the effective dilution at the point of the 4PL curve where 50% (ED50) of antigen was detected by toxin-specific antibodies present in the serum sample.
Hla TNAs were performed as previously described (27). In brief, 4% rabbit red blood cells (RRBCs) were co-cultured with wild-type Hla ± serially diluted serum samples. Cells were centrifuged after 30 min at 37°C, and absorbance determined at OD416 nm. PVL and LukAB TNAs were performed with human promyelocytic leukemia (HL-60) cells as previously described (28). In brief, differentiated HL-60 cells were incubated with either PVL or LukAB ± serially diluted serum samples for 3 hours at 37°C, and CellTiter Glo reagent (Promega) was added to the culture to measure cell viability. Superantigen (SAg) TNAs were performed with human PBMCs isolated from Leukopaks (StemExpress). Cells were co-cultured with SEA, SEB, or TSST-1 in the presence or absence of serially diluted serum samples for 48 hours; supernatants were collected, and IFNγ was measured in the supernatants as a readout of superantigenicity as we previously described (29). Data was analyzed using a 4-parameter (4PL) curve fit with XLFit (Microsoft). Toxin neutralizing activity was defined as the effective dilution of sera at the point of the 4PL curve at which 50% of toxin activity was neutralized (ND50).
Data between more than two groups for longitudinal comparisons (in vivo BLI, lesion size) were compared using a two-way analysis of variance (ANOVA) multiple comparisons test with Dunnett correction. For single time point comparisons (abscess weight and ex vivo CFU), groups were compared using a one-way ANOVA-Kruskal-Wallis multiple comparisons test with Dunn’s correction, and survival rates were compared by log rank (Mantel-Cox) test, as indicated in the figure legends. All statistical analyses were calculated with Prism software (GraphPad 10.3 Software, La Jolla, California). Data are presented as mean or geometric mean (for ex vivo CFU data) ± standard error of the mean (SEM) and values of P <0.05 were considered statistically significant.
To determine the biochemical characterization of the IBT-V02 toxoid vaccine components in our solid lyophilized (Lyo) and spray freeze-dried (SFD) formulations compared to our liquid (Liq) formulation, we performed SDS-PAGE, Western Blot, and SEC-HPLC analyses (Figure 1). First, we examined the formulations by SDS-PAGE and found that overall protein integrity was intact in the Lyo (lane 2), SFD (lane 4), Liq without desorption (lane 6), Liq with desorption (Lane 7), and freshly prepared Liq without alhydrogel (Lane 9) formulations (Figure 1A). Next, we performed a Western blot on vaccine toxoid components from the same conditions described above and verified that neutralizing antibody binding specificity was maintained in all the formulations tested (Figure 1B). Finally, we performed SEC-HPLC, which confirmed our SDS-PAGE and Western blot results that protein constitution of the IBT-V02 vaccine toxoid components is comparable between the Liq, Lyo, and SFD formulations tested (Figure 1C).
Given the difficulty in maintaining cold chain storage in LMICs (30), we next examined the stability of the Lyo and SFD IBT-V02 formulations at varying storage times and temperatures, and compared the stability to the freshly prepared Liq formulation. First, we tested the storage stability of the IBT-V02 formulations at -20°C and 30°C for 1- and 3-month durations. Excitingly, we found that vaccine components of both Lyo and SFD formulations had comparable stability to the freshly prepared Liq formulation by SDS-PAGE (Supplementary Figures 1A, C) and SEC-HPLC (Supplementary Figures 1B, D) analyses. We next tested the storage stability of the Lyo formulation at -20°C and 30°C for 6-, 9-, and 12-month durations. Similar to our findings at 1 and 3 months, the Lyo formulation had comparable biochemical stability as freshly prepared Liq IBT-V02 formulation at 6, 9, and 12 months of storage (Supplementary Figure 2). Collectively, our findings suggested that the Lyo and SFD IBT-V02 formulations effectively maintain long-term stability at room temperature storage.
After testing the long-term biochemical stability of the solid formulations, we next set out to compare the immunogenicity of the solid and liquid IBT-V02 formulations in vaccinated mice. To this end, mice were immunized with 50 µg of either Liq, Lyo, or SFD formulations in 200µg of Alhydrogel (Al(OH)3) on days 0, 14 and 28 (Figure 2A). On day 38, 10 days after the final immunization, sera were collected to assess toxin binding IgG titers by Luminex, and toxin neutralizing antibody titers in cell-based assays, as previously described (12). We found that all three formulations of IBT-V02 induced similar levels of toxin neutralizing capacity when tested against target antigens, which were increased over BSA-controls (Figure 2B). Importantly, IBT-V02 immunogenicity was maintained in the Lyo formulation after long-term storage (6 months) at -20C and 30C compared to fresh preparations (Supplementary Figure 3), indicating the viability of the solid formulations as an alternative to IBT-V02 vaccination with the liquid formulation in mice.
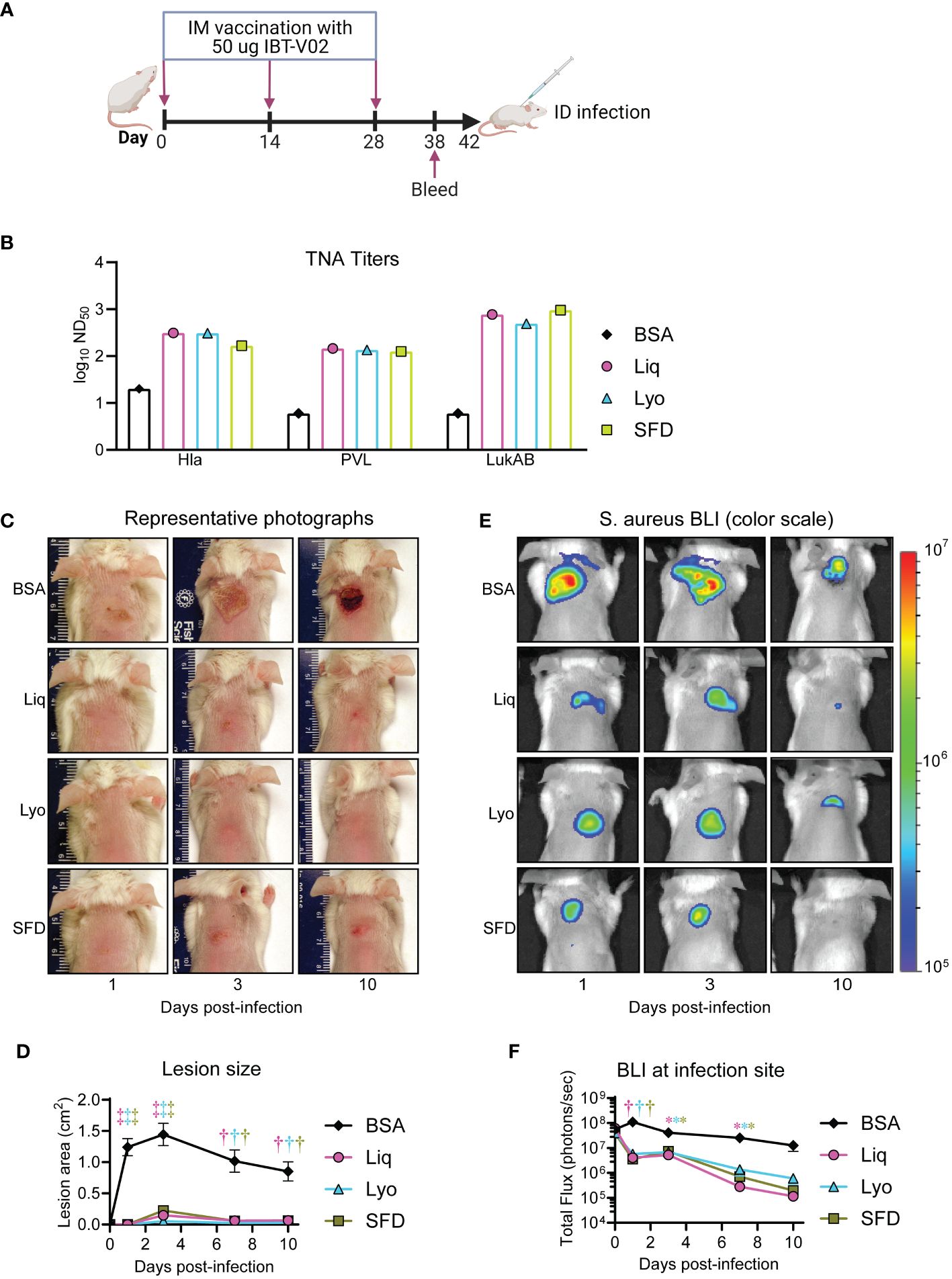
Figure 2 Comparative immunogenicity and efficacy in a murine intradermal infection model. Mice (n=10 mice per group) were immunized with 50 µg of either Liq, Lyo, or SFD formulations of IBT-V02 or 50 µg of BSA in 200µg of Alhydrogel (Al(OH)3) on days 0, 14 and 28. On day 38, sera were collected to assess toxin neutralizing antibody titers in cell-based assays. On day 42, MRSA (8×106 CFU of the bioluminescent MRSA strain SF8300) skin infections were performed on immunized mice and bacterial burden and lesion size monitored until the experiment was arbitrarily ended after 10 days. (A) Model timeline. (B) Toxin neutralizing antibody titers. (C) Representative photographs of skin lesion. (D) Mean total lesion size (cm2) ± SEM. (E) Representative in vivo BLI (photons/s) (log10 scale). (F) Mean total flux (photons/s) ± SEM. *P < 0.05, †P < 0.01, ‡P < 0.001, as calculated by a two-way analysis of variance (ANOVA) Dunnett’s multiple comparisons test (D, F). Results are combined from two independent experiments.
We next compared the protective efficacy of Liq, Lyo, and SFD IBT-V02 formulations compared to BSA controls in a mouse model of MRSA skin infection, whereby mice were injected i.d. with 8×106 CFU of the bioluminescent MRSA strain, SF8300, and bacterial burden and lesion size monitored until the experiment was arbitrarily ended after 10 days. We used in vivo bioluminescence imaging (BLI) to non-invasively monitor bacterial burden, which we have previously shown to correlate with ex vivo CFUs (R2 = 0.9996) (31). We discovered that Liq, Lyo, and SFD vaccinated mice had significantly reduced lesion sizes (Figures 2C, D) and bacterial burdens (Figures 2E, F) compared to BSA-treated controls. Taken together, our findings indicated that Liq, Lyo, and SFD IBT-V02 formulations had significant efficacy against MRSA skin infections in mice.
Since S. aureus pore-forming toxins that are neutralized by the IBT-V02 vaccine have higher affinity to rabbit immune cells than mice (32), we next wanted to compare the immunogenicity of the dry and liquid IBT-V02 formulations in vaccinated rabbits. Given the similar outcomes between SFD and Lyo formulations in the murine intradermal infection model and the successful use of lyophilization in commercialized vaccines in LMICs (e.g., MenAfriVac) (18, 33), we herein focused our comparative studies on the Lyo IBT-V02 formulation. To this end, rabbits were immunized with 50 µg of either Liq or Lyo formulations in 200µg of Alhydrogel (Al(OH)3) on days 0, 14 and 28 (Figure 3A). On day 38, 10 days after the final immunization, sera were collected to assess toxin binding IgG titers by Luminex, and toxin neutralizing antibody titers in cell-based assays, as previously described (12). We found that the Liq and Lyo IBT-V02 formulations had similar trends in the increased levels of toxin-binding IgG titers (Figure 3B) and toxin-neutralizing antibodies (Figure 3C) compared to BSA-treated rabbits.
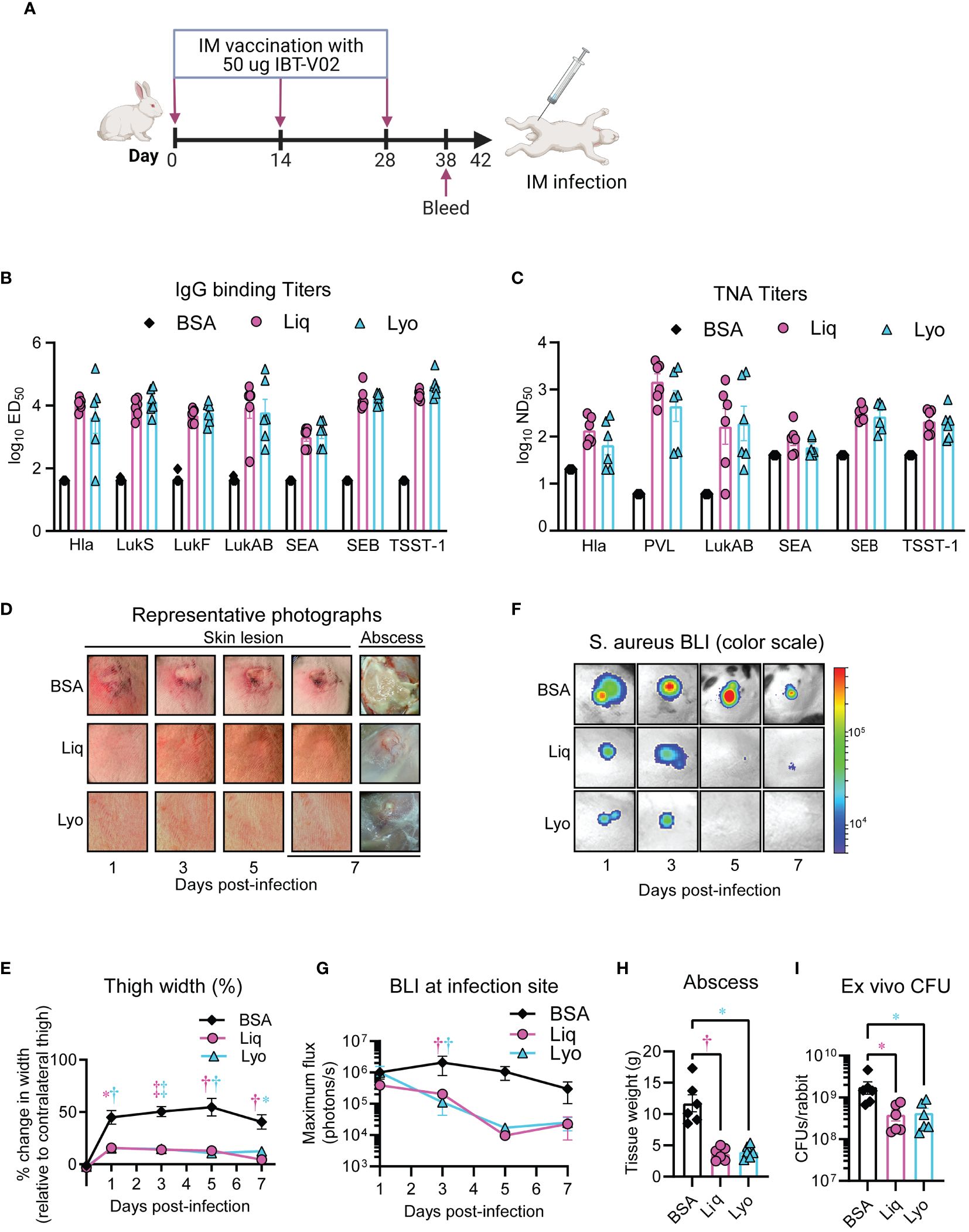
Figure 3 Comparative immunogenicity and efficacy in rabbit pyomyositis. Rabbits (n=6 rabbits per group) were immunized with 50 µg of either Liq or Lyo formulations of IBT-V02 or 50 µg of BSA in 200µg of Alhydrogel (Al(OH)3) on days 0, 14 and 28. On day 38, sera were collected to assess toxin binding IgG titers by Luminex, and toxin neutralizing antibody titers in cell-based assays. On day 42, MRSA (6-8 ×109 CFU of the bioluminescent MRSA strain SF8300) pyomyositis infections were performed on immunized rabbits and skin lesions and thigh width monitored until the experiment was arbitrarily ended after 7 days, when abscesses were imaged, weighted, and harvested for ex vivo CFUs. (A) Model timeline. (B) Toxin-binding IgG titers. (C) Toxin neutralizing antibody titers. (D) Representative photographs of skin lesion and abscess. (E) Mean thigh width change (% of contralateral thigh) ± SEM. (F) Representative in vivo BLI images. (G) Mean in vivo BLI signals quantified as maximum flux (photons/s/cm2/steradian) ± SEM. (H) Mean tissue weight of abscess ± SEM. (I) Geometric mean of ex vivo CFUs ± SD. *P < 0.05, †P < 0.01, ‡P < 0.001, as calculated by a two-way analysis of variance (ANOVA) Dunnett’s multiple comparisons test (E, G) or Kruskal-Wallis Dunn’s multiple comparisons test (H, I). Results are combined from two independent experiments.
Pyomyositis is a major health burden in LMICs and increasing in prevalence in high-income countries (3), with S. aureus as the predominant etiological agent (2). Therefore, we developed a rabbit pyomyositis infection model to compare the protective efficacy of Lyo and liquid IBT-V02 formulations whereby rabbit thighs were injected intra-muscularly (IM) with 6-8 ×109 CFU of S. aureus strain SF8300 and skin lesions and thigh width monitored until the experiment was arbitrarily ended after 7 days, when abscesses were imaged, weighed, and harvested for ex vivo CFUs. Lyo and Liq treated rabbits had marked reductions in gross skin lesion and abscess sizes (Figure 3D) and thigh widths (Figure 3E) compared to BSA-treated rabbits. Furthermore, there was a decrease in BLI in vaccinated rabbits compared to BSA-treated controls (Figures 3F, G), which correlated with significant reductions in abscess weights (Figure 3H) and ex vivo CFUs (Figure 3I). Thus, our data suggested that Lyo and Liq IBT-V02 formulations had comparable and significant efficacy in a novel rabbit pyomyositis infection model.
Since we only tested efficacy of IBT-V02 against S. aureus isolates from the U.S. in our preclinical infection models (12), we wanted to confirm the efficacy of the Lyo and Liq IBT-V02 formulations against S. aureus isolates from LMICs. Thus, we acquired a total of 83 clinical isolates of S. aureus from pyomyositis patients in the Democratic Republic of Congo, Palestine, and Cambodia. Since testing 83 clinical isolates in vivo is cost-prohibitive, we created a rational multi-dimensional down-selection strategy to screen isolates that exhibited broad toxin production for testing the efficacy of our vaccine formulations (Figure 4A). First, we measured toxin release via Western blot to down-select to 29 clinical isolates.
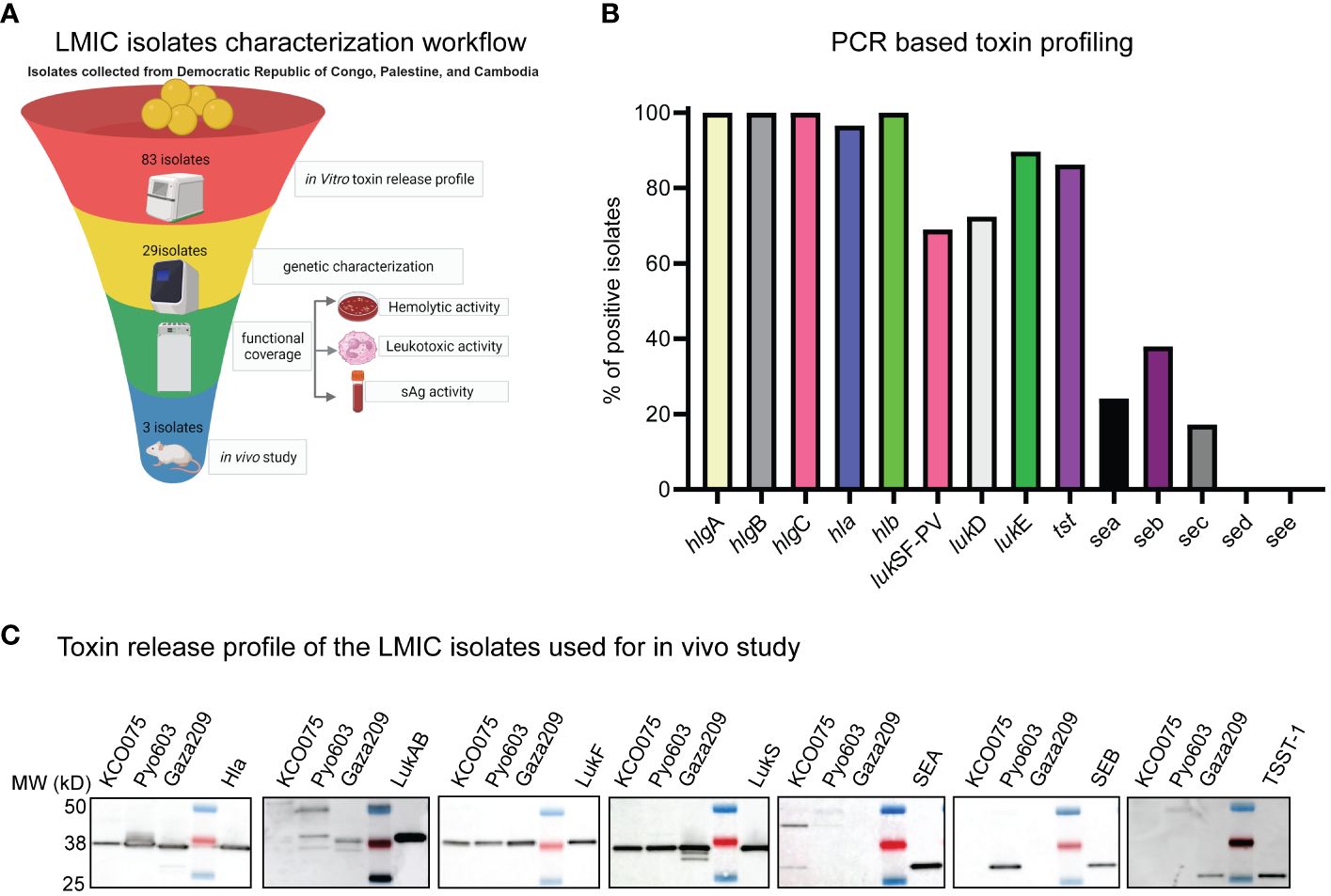
Figure 4 Characterization of LMIC isolates in vitro. (A) LMIC isolates characterization workflow. (B) PCR based toxin profiling. (C) Toxin release profile of the LMIC isolates used for in vivo study.
Next, we performed a toxin profile PCR assay to identify isolates that were positive for toxins neutralized by IBT-V02 (Figure 4B; Supplementary Table 2). We discovered that 100% of the isolates had the hla, hlgA, B, C, and hlb genes, 90% of the isolates had the LukE and tst, 60-70% of isolates had the PVL and LukD genes, and less than 40% of isolates had the Sea, Seb, and Sec genes (Figure 4B). However, we did not detect Sed and See in any of the isolates (Figure 4B). Out of the 29 isolates analyzed, 25 of them had more than 4 leukotoxins genes other than PVL. Furthermore, 22 of the isolates had 1-2 superantigen (SAg) genes (Supplementary Table 2). Only one isolate had 4 SAg genes, 4 isolates had 3, and 3 isolates did not have any SAg genes. Furthermore, we typed these isolates using 18 different genes as typing markers. Among them, 14 were toxin genes, and 4 (SPA type, ST, CC, and SCCmec) were the established epidemiological typing genes. Out of 29 isolates, 15 were MSSA isolates, and 14 were MRSA isolates. Among MRSA isolates, SCCmec type IVA was the most predominant (12 out of 14), and 2 were SCCmec type III. The most common CC types were CC22 and CC121 whereas the SPA types were highly diverse.
To confirm toxin protein production by the down-selected isolates, we performed in vitro hemolytic and leukocytic activity assays using rabbit red blood cells and neutrophils, respectively. From our multi-dimensional screen, we identified 3 clinical isolates to use in in vivo studies, one from each of the Democratic Republic of Congo, Palestine, and Cambodia that expressed the lytic toxins and at least one superantigen. These strains were named KCO075, Pyo603, and Gaza209, which in addition to the lytic toxins produced SEA, SEB, and TSST-1, respectively (Figure 4C).
To assess the potential efficacy of IBT-V02 against the down-selected isolates from LMICs, we explored the neutralizing potential via in vitro hemolytic and leukocytic activity assays. The toxicity of each bacterial supernatant (SUP) was first determined in rabbit red blood cells and HL-60 cells (Supplementary Figure 4). SUPs were prepared as described previously (20). Starting at neat, cells were incubated with 11-point dilutions of bacterial SUPs and toxicity was measured. Data were analyzed using a 4-parameter (4PL) curve fit in XLFit (Microsoft) and the effective dilution at the point of the 4PL curve where ED85 toxicity occurred was calculated. Cells were then incubated with the ED85 dilution of SUPs in the presence or absence of decreasing concentrations of IBT-V02 rabbit pAbs. The neutralizing concentration (NC) at the point of the 4PL curve where 50% neutralization of SUP toxicity occurred (NC50) was then calculated. As shown in Supplementary Figure 4, pAbs raised against IBT-V02 showed neutralizing capacity towards the SUPs of all three LMIC isolates at different concentrations. For RRBCs the NC50 ranged from 19.3-26.4µg/ml and for HL60 cells from 20.5-60.9µg/ml.
S. aureus bacteremia has a 20-30% mortality rate in high-income countries (34, 35), which is elevated to over 50% in LMICs (36, 37). Thus, to evaluate the in vivo efficacy of our IBT-V02 formulations against LMIC isolates we immunized mice with 50 µg of either Liq or Lyo formulations in 200µg of Alhydrogel (Al(OH)3) on days 0, 14 and 28 (Figure 5A). On day 60, we performed a S. aureus bacteremia infection model in mice whereby KCO075, Pyo603, and Gaza209 isolates were injected intraperitoneally into BSA, Liq, or Lyo treated mice and survival monitored over time until day 14 when the experiments were arbitrarily ended. Mice infected with the KCO075 isolate had comparably low reductions in survival rates between all groups tested (Figure 5B). However, mice treated with the Liq and Lyo IBT-V02 formulations had significant increases in survival compared to BSA-treated mice when infected with the Pyo603 isolate (Figure 5C). Lastly, we found a similar trend in mice infected with the Gaza209 isolate, whereby Liq and Lyo vaccinated mice had markedly increased survival rates compared to BSA control mice (Figure 5D). Collectively, our data suggested that Liq and Lyo IBT-V02 formulations have protective efficacy against LMIC isolates in a S. aureus bacteremia infection model.
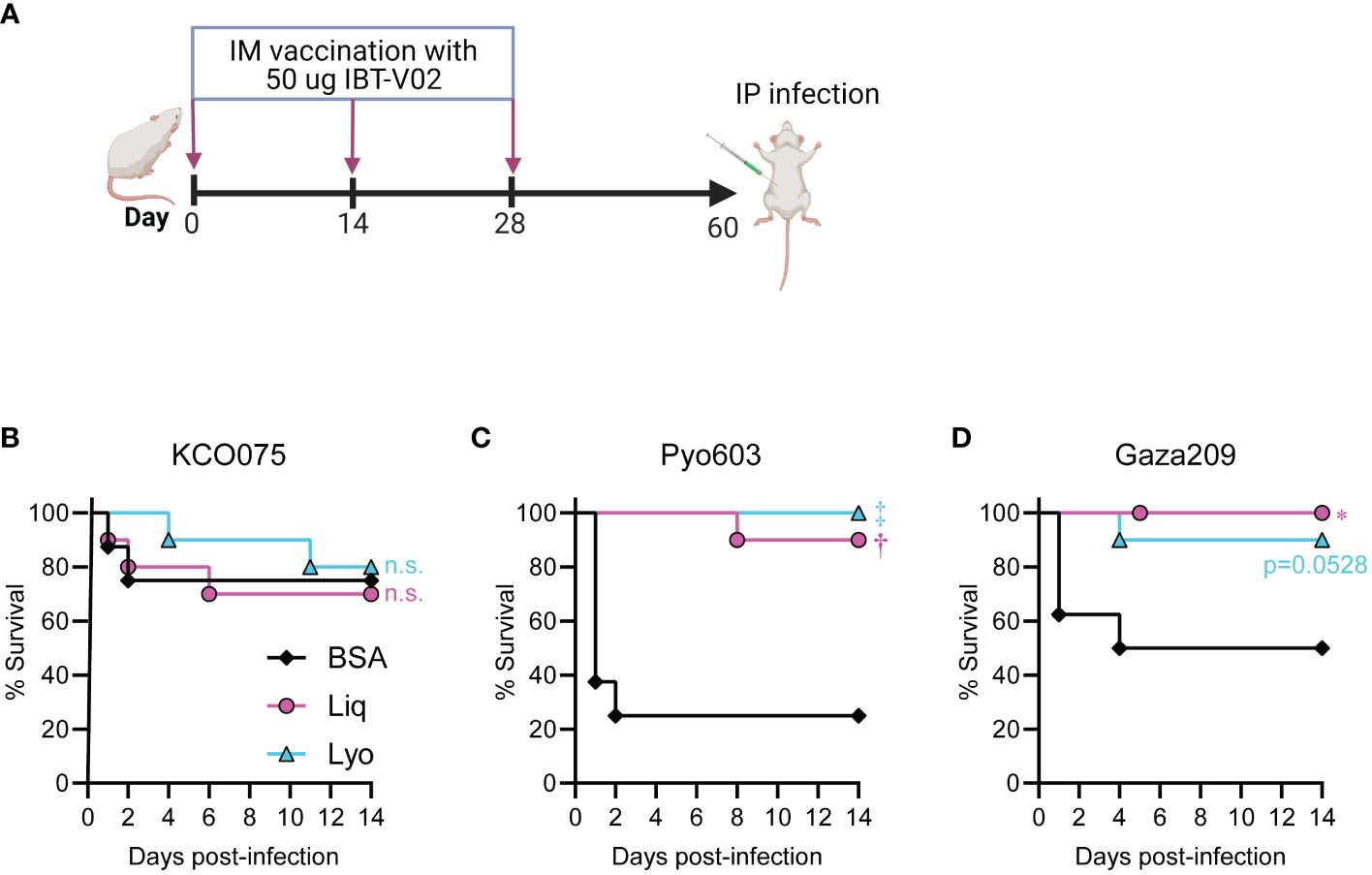
Figure 5 Protective efficacy of IBT-V02 against LMIC isolates in a mouse bacteremia model. Mice (n=10 mice per group) were immunized with 50 µg of either Liq or Lyo formulations of IBT-V02 or 50 µg of BSA in 200µg of Alhydrogel (Al(OH)3) on days 0, 14 and 28. Six weeks post-immunization, the LMIC bacteremia infection was performed on immunized mice. (A) Model timeline. (B) Survival (%) of mice infected with 8x107 CFU of KCO075. (C) Survival (%) of mice infected with 4x107 CFU of Pyo603. (D) Survival (%) of mice infected with 2x107 CFU of Gaza209. *P < 0.05, †P < 0.01, ‡P < 0.001, BSA versus Liq or Lyo as calculated by log rank (Mantel-Cox) test. n.s., not significant.
We next tested the efficacy of the Liq and Lyo IBT-V02 formulations against LMIC isolates KCO075, Pyo603, and Gaza 209 in the mouse intradermal infection model as described above. Mice were immunized with 50 µg of either Liq or Lyo formulations in 200µg of Alhydrogel (Al(OH)3) on days 0, 14 and 28 (Figure 6A). Mice infected with S. aureus isolate KCO075 and vaccinated with Liq or Lyo formulations had significant reductions in lesions sizes compared to BSA-treated mice (Figures 6B, C). These results corresponded with markedly reduced ex vivo CFUs from the skin and kidneys of Liq and Lyo treated mice compared with BSA-treated mice (Figures 6D, E). Similar to mice infected with KCO075, Liq and Lyo treated mice infected with Pyo603 had markedly reduced lesion sizes and ex vivo CFUs from skin compared to BSA controls (Figures 6F–H). However, Pyo603-infected mice did not have differences in ex vivo CFUs from kidneys between all the groups tested (Figure 6I). Lastly, Liq and Lyo treated mice infected with Gaza209 had significantly dampened lesion sizes and ex vivo skin CFUs compared to BSA-treated mice (Figures 6J–L), with ex vivo kidney CFUs significantly lessened only in the Lyo treated mice (Figure 6M). Taken together, Liq and Lyo IBT-V02 formulations were effective against S. aureus isolates from LMICs in a mouse intradermal infection model.
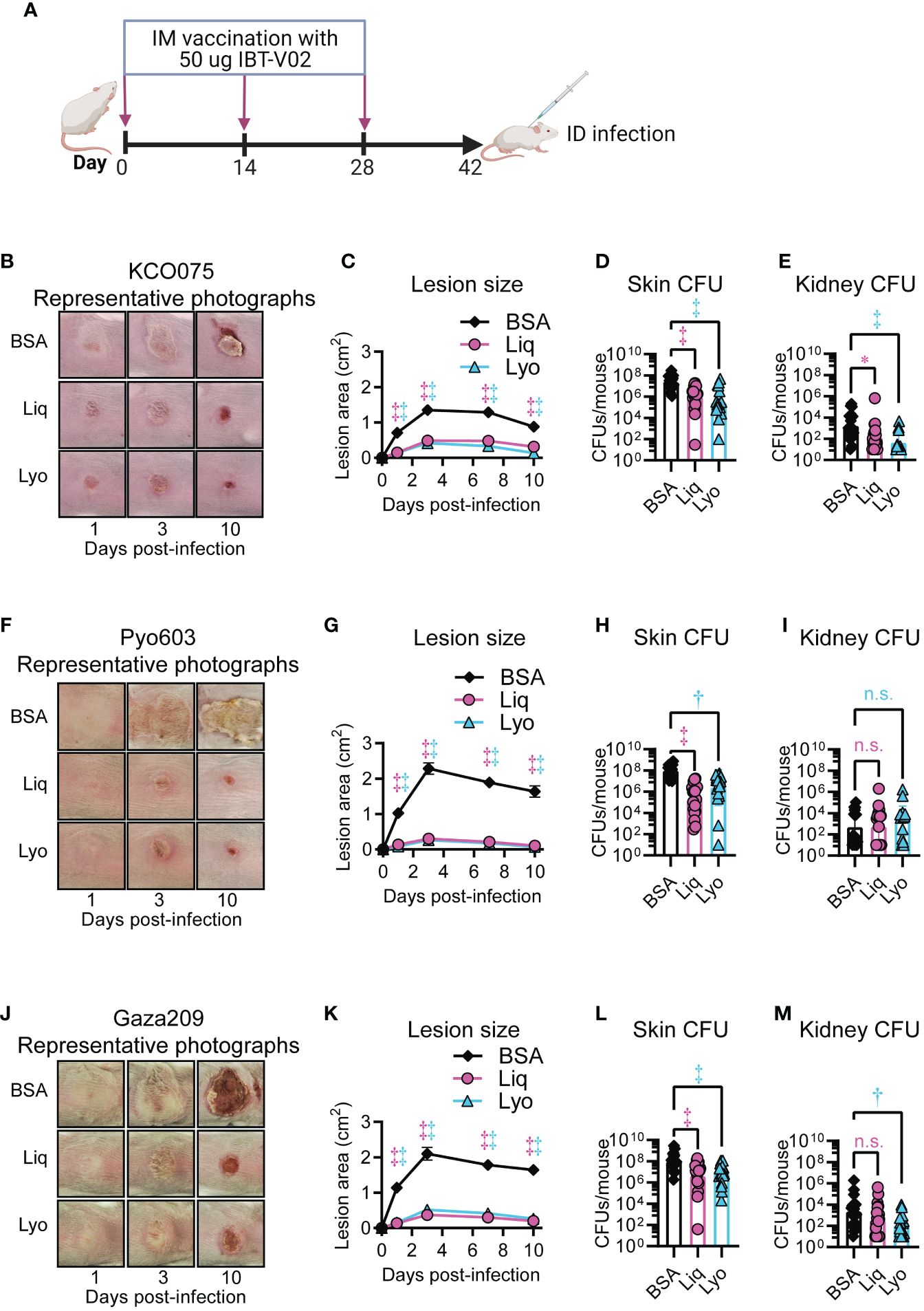
Figure 6 Protective efficacy of IBT-V02 against LMIC isolates in a mouse intradermal infection model. Mice (n=10 mice per group) were immunized with 50 µg of either Liq or Lyo formulations of IBT-V02 or 50 µg of BSA in 200µg of Alhydrogel (Al(OH)3) on days 0, 14 and 28. On day 42, intradermal infections with 1x109 CFU of KCO075 (B–E), 3x108 CFU of Pyo603 (F–I), and 2x108 CFU of Gaza209 (J–M) were performed on immunized mice and skin lesions monitored until the experiment was arbitrarily ended after 10 days, when skin and kidney were harvested for ex vivo CFUs. (A) Model timeline. (B, F, J) Representative photographs of skin lesion. (C, G, K) Mean total lesion size (cm2) ± SEM. (D, H, L) Geometric mean of skin lesion ex vivo CFUs ± SD. (E, I, M) Geometric mean of kidney ex vivo CFUs ± SD. *P < 0.05, †P < 0.01, ‡P < 0.001, as calculated by a two-way analysis of variance (ANOVA) Dunnett’s multiple comparisons test (C, G, K) or Kruskal-Wallis Dunn’s multiple comparisons test (D, E, H, I, L, M). Results are combined from two independent experiments. n.s., not significant.
The emergence of antibiotic-resistant S. aureus is a global health burden (4), which disproportionately affects LMICs. Although we have previously shown efficacy of IBT-V02 in liquid formulations against S. aureus isolates from the industrialized world (12), whether protection is maintained in dry formulations against S. aureus isolates from LMICs has not been established. Therefore, we developed and tested lyophilized and spray freeze-dried formulations of IBT-V02 compared to the liquid formulation for protein stability, immunogenicity, and in vivo efficacy in multiple preclinical models against S. aureus isolates from the U.S. and LMICs. Our investigation revealed that the dry formulations of IBT-V02 are comparable to the liquid formulation in protein stability during long-term storage at room temperature, immunogenicity against S. aureus toxins, and protective efficacy against S. aureus isolates from LMICs in murine and rabbit infection models. These results provide several important insights into the protective efficacy of IBT-V02 during S. aureus infections.
First, we found that the lyophilized and freeze-dried IBT-V02 formulations had preserved protein stability after reconstitution, even after long-term storage at room temperature. These findings recapitulated the prevailing literature that dry vaccine formulations have decreased sensitivity to temperature-induced degradation (38). Given the difficulty in maintaining cold chain storage in LMICs, our results suggested that dry formulations of IBT-V02 have potential to be distributed in warm and tropical climates without reductions in vaccine component integrity. This was further corroborated in our immunogenicity studies, which found that mice and rabbits vaccinated with the liquid and dry formulations had comparable induction of anti-toxin neutralizing titers compared to BSA-treated controls. Our findings are similar to the modified vaccinia Ankara vaccine for smallpox, whereby a lyophilized vaccine formulation had comparable immunogenicity as a liquid formulation (39). Collectively, our results indicated that lyophilized and spray freeze-dried IBT-V02 are viable formulations for maintaining vaccine immunogenicity during long-term storage at room temperature.
We also discovered that the dry and liquid IBT-V02 formulations had comparable protective efficacy against a S. aureus USA300 strain in an intradermal infection model in mice, which replicates our previous findings using the liquid IBT-V02 formulation against S. aureus isolates from the U.S (12). Moreover, the lyophilized and liquid IBT-V02 formulations had comparable protective efficacy in a pyomyositis infection model in rabbits. This is important, as S. aureus pore-forming toxin leukocytic activity in rabbits is more comparable to humans than mice (32), enhancing the relevancy of our results. However, studies to determine whether IBT-V02 sustains protective efficacy during S. aureus infections in humans are warranted and will be the focus of future clinical trials.
Our findings revealed that dry and liquid IBT-V02 formulations promoted survival during bacteremia infections with multiple isolates of S. aureus from LMICs. Although we did not find a benefit for IBT-V02 in mice infected with LMIC isolate KCO075, this may be due to reduced toxin expression of the KCO075 isolate, which had lower RRBC and HL-60 toxicity compared to Pyo603 and Gaza209 strains (Supplementary Figure 4). Similar to our current and published results with U.S. isolates of S. aureus (12), dry and liquid IBT-V02 formulations had protective efficacy against multiple isolates from LMICs in an intradermal infection model in mice. However, IBT-V02 vaccination did not reduce the bacterial dissemination to the kidneys of Pyo603-infected mice, despite markedly decreased lesion sizes and bacterial burdens in the skin. A possible explanation is that the Pyo603 isolate is a ST121 subset strain, which have been shown to require prolonged hospitalizations and antimicrobial therapy in humans (40), and have heightened virulence in rabbits (41). Understanding the strain level variation between LMIC isolates and vaccination outcomes will be interrogated in our future studies.
There were several limitations to our study. First, we only tested the immunogenicity and protective efficacy of IBT-V02 using the IM/subcutaneous delivery method. To improve the potential vaccination rate of IBT-V02 in LMICs, we will examine vaccination delivery methods that have reduced training requirements (e.g., oral, nasal, dermal) compared to IM delivery in our future work (42, 43). Furthermore, we did not determine whether prior S. aureus exposure caused decreased protective efficacy of lyophilized IBT-V02, which has been reported to influence vaccine responses in other S. aureus infection models (44). This will be investigated in our subsequent studies.
In conclusion, dry and liquid formulations of IBT-V02 had comparable stability, immunogenicity, and protective efficacy in preclinical infection models with S. aureus isolates from the U.S. Importantly, dry and liquid formulations of IBT-V02 mediated protection against LMIC strains of S. aureus. Collectively, these findings indicated that vaccination with IBT-V02 dry formulations is a viable strategy to overcome cold chain storage limitations to protect against S. aureus isolates from LMICs.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
All animals were maintained under the same specific pathogen-free conditions, with air-isolated cages at an American Association for the Accreditation of Laboratory Animal Care (AAALAC)-accredited animal facility at Johns Hopkins University and Integrated BioTherapeutics. They were handled according to procedures described in the Guide for the Care and Use of Laboratory Animals as well as Johns Hopkins University’s policies and procedures as outlined in the Johns Hopkins University Animal Care and Use Training Manual. Studies at JHU were approved by the Johns Hopkins Animal Care and Use Committee (Protocol #: MO21M378 for mice, Protocol#: RB22M400 for rabbit). Mouse studies at IBT were approved by institutional animal care and use committees (IACUC) (Immunogenicity Protocol# AP160805 and Efficacy against bacterial challenge Protocol#: AP-161007). The study was conducted in accordance with the local legislation and institutional requirements.
YW: Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. IM: Data curation, Formal analysis, Investigation, Visualization, Writing – original draft, Writing – review & editing. AV: Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. DD: Data curation, Investigation, Writing – original draft. NO: Data curation, Investigation, Writing – original draft. JZ: Data curation, Investigation, Writing – original draft. RO: Data curation, Formal analysis, Investigation, Writing – original draft. MM: Data curation, Formal analysis, Investigation, Writing – original draft. SPS: Data curation, Formal analysis, Investigation, Writing – original draft. TK: Data curation, Formal analysis, Investigation, Writing – original draft. TL: Data curation, Resources, Writing – original draft. SS: Data curation, Resources, Writing – original draft. MK: Data curation, Resources, Writing – original draft. RA: Conceptualization, Investigation, Methodology, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing. HK: Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing. MJ: Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing. NA: Conceptualization, Funding acquisition, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded in part by grants from R01AR073665 (NA), R01AI111205 (MJ, NA), and R01AI146177 (MJ, NA) from the United States National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) and National Institute of Allergy and Infectious Diseases (NIAID). Research reported in this publication was also supported by CARB-X. CARB-X’s funding for this project was provided by the UK Government Department of Health and Social Care as part of the Global Antimicrobial Resistance Innovation Fund (GAMRIF). The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of CARB-X or any of its funders.
We would like to thank Dr. Catherine Moore at the University of Oxford, for ‘Cambodia isolates; Dr. Frieder Schaumburg at the University Hospital of Munster, Germany for ‘Kinshasa isolates’ and to Dr. Barry Kreiswirth at the Center for Discovery & Innovation, HMH, NJ, USA for Gaza isolates of S. aureus. Figure 2A, 3A, 4A, 5A, and 6A were created with BioRender.com.
Authors IM, AV, RO, MM, SS, TK, RA, HK and MJ were employed by company Integrated Biotherapeutics Inc. Author MK was employed by company Engineered Biopharmaceuticals. Author NA has received previous grant support from Pfizer and Boehringer Ingelheim and has been a paid consultant for Janssen Pharmaceuticals. MJ and RA have stocks in Razi Holdings, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1373367/full#supplementary-material
Supplementary Figure 1 | Biochemical characterization of IBT-V02 formulations after storage for 1 and 3 months. (A, C) SDS-PAGE analysis of 1µg Lyo IBT-V02 stored at -20°C (Lane 2) and 30°C (Lane 3), 1µg SFD IBT-V02 stored at -20°C (Lane 5) and 30°C (Lane 6), 1µg Liq IBT-V02 stored at 30°C without desorption (Lane 8) and with desorption (Lane 9) and 1µg freshly prepared Liq IBT-V02 without alhydrogel without desorption (Lane 11) and with desorption (Lane 12) after storage for 1 month (A) and 3 months (C). (B and D) SEC-HPLC of Lyo (-20°C, 30°C), SFD (-20°C, 30°C), Liq (30°C, ± desorption) and freshly prepared liq (± desorption) after storage for 1 month (B) and 3 months (D).
Supplementary Figure 2 | Biochemical characterization of the Lyo IBT-V02 formulation after storage for 6, 9, and 12 months. (A, C, E) SDS-PAGE analysis of 1µg Lyo IBT-V02 stored at -20°C (Lane 2) and 30°C (Lane 3) and 1µg freshly prepared Liq IBT-V02 without alhydrogel (6mo: Lanes 5, 9; 12 mo: Lane 4) after storage for 6 months (A), 9 months (C), and 12 months (E). (B, D, F) SEC-HPLC of Lyo IBT-V02 stored at -20°C and 30°C and freshly prepared Liq without alhydrogel after storage for 6 months (B), 9 months (D), and 12 months (F).
Supplementary Figure 3 | Immunogenicity of the Lyo IBT-V02 formulation after long-term storage. Serum toxin neutralizing titers to toxins Hla, PVL, LukAB, SEA, SEB, TSST-1 for individual BALB/c mice (n = 5/group) immunized with 50µg of Lyo IBT-V02 (as described in Methods) on Day 0 of stability study or after 6 months storage of vaccine at either -20°C or 30°C. Bars indicate mean log-transformed ND50, the serum dilution that elicits 50% of neutralizing titer. Dotted lines indicate the limit of detection for each toxin (LOD). Statistical analysis performed with the Mann-Whitney test.
Supplementary Figure 4 | Toxin neutralization of LMIC supernatants in vitro. Toxicity of bacterial supernatants and neutralizing efficacy of IBT-V02 pAbs tested with rabbit red blood cells (RRBCs) (A) and induced HL-60 cells (B). Effective dilutions of bacterial supernatants causing 85% toxicity (ED85) against RRBCs, and HL-60 cells were calculated for each LMIC isolate. Bacterial SUP dilutions [1:17.3 (KCO075); 1:171 (Pyo603); 1:32.3 (Gaza209)] resulting in ED85 were used to determine concentration of IBT-V02 Rb pAb required to neutralize 50% of bacterial SUP toxicity (NC50).
Supplementary Table 1 | List of PCR targets and primers. List of PCR gene targets and primers for enterotoxins, protein A (Spa), SCCmec, and multilocus sequence typing (MLST).
Supplementary Table 2 | Characterization of LMIC isolates in vitro. Genotyping was conducted on the strains at Marshfield clinic and results reported in the form of a table. (P, Positive; N, Negative; NA, CC not available; RED, New Sequence Type; NT, Non-Typeable).
1. Turner NA, Sharma-Kuinkel BK, Maskarinec SA, Eichenberger EM, Shah PP, Carugati M, et al. Methicillin-resistant Staphylococcus aureus: an overview of basic and clinical research. Nat Rev Microbiol. (2019) 17:203–18. doi: 10.1038/s41579-018-0147-4
2. Habeych ME, Trinh T, Crum-Cianflone NF. Purulent infectious myositis (formerly tropical pyomyositis). J Neurol Sci. (2020) 413:116767. doi: 10.1016/j.jns.2020.116767
3. Ngor C, Hall L, Dean JA, Gilks CF. Factors associated with pyomyositis: A systematic review and meta-analysis. Trop Med Int Health. (2021) 26:1210–9. doi: 10.1111/tmi.13669
4. Collaborators AR. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet. (2022) 399(10325):629–55. doi: 10.1016/S0140-6736(21)02724-0
5. Iskandar K, Molinier L, Hallit S, Sartelli M, Hardcastle TC, Haque M, et al. Surveillance of antimicrobial resistance in low- and middle-income countries: a scattered picture. Antimicrob Resist Infect Control. (2021) 10:63. doi: 10.1186/s13756-021-00931-w
6. Pokharel S, Raut S, Adhikari B. Tackling antimicrobial resistance in low-income and middle-income countries. BMJ Glob Health. (2019) 4:e002104. doi: 10.1136/bmjgh-2019-002104
7. Sulis G, Sayood S, Gandra S. Antimicrobial resistance in low- and middle-income countries: current status and future directions. Expert Rev Anti Infect Ther. (2022) 20:147–60. doi: 10.1080/14787210.2021.1951705
8. Allel K, Stone J, Undurraga EA, Day L, Moore CE, Lin L, et al. The impact of inpatient bloodstream infections caused by antibiotic-resistant bacteria in low- and middle-income countries: A systematic review and meta-analysis. PloS Med. (2023) 20:e1004199. doi: 10.1371/journal.pmed.1004199
9. Sharma A, Singh A, Dar MA, Kaur RJ, Charan J, Iskandar K, et al. Menace of antimicrobial resistance in LMICs: Current surveillance practices and control measures to tackle hostility. J Infect Public Health. (2022) 15:172–81. doi: 10.1016/j.jiph.2021.12.008
10. Bai AD, Lo CK, Komorowski AS, Suresh M, Guo K, Garg A, et al. Staphylococcus aureus bacteremia mortality across country income groups: A secondary analysis of a systematic review. Int J Infect Dis. (2022) 122:405–11. doi: 10.1016/j.ijid.2022.06.026
11. Saharman YR, Karuniawati A, Severin JA, Verbrugh HA. Infections and antimicrobial resistance in intensive care units in lower-middle income countries: a scoping review. Antimicrob Resist Infect Control. (2021) 10:22. doi: 10.1186/s13756-020-00871-x
12. Karauzum H, Venkatasubramaniam A, Adhikari RP, Kort T, Holtsberg FW, Mukherjee I, et al. IBT-V02: A multicomponent toxoid vaccine protects against primary and secondary skin infections caused by. Front Immunol. (2021) 12:624310. doi: 10.3389/fimmu.2021.624310
13. Thammavongsa V, Kim HK, Missiakas D, Schneewind O. Staphylococcal manipulation of host immune responses. Nat Rev Microbiol. (2015) 13:529–43. doi: 10.1038/nrmicro3521
14. Howden BP, Giulieri SG, Wong Fok Lung T, Baines SL, Sharkey LK, Lee JYH, et al. Staphylococcus aureus host interactions and adaptation. Nat Rev Microbiol. (2023) 21:380–95. doi: 10.1038/s41579-023-00852-y
15. McNeely TB, Shah NA, Fridman A, Joshi A, Hartzel JS, Keshari RS, et al. Mortality among recipients of the Merck V710 Staphylococcus aureus vaccine after postoperative S. aureus infections: an analysis of possible contributing host factors. Hum Vaccin Immunother. (2014) 10:3513–6. doi: 10.4161/hv.34407
16. Hassanzadeh H, Baber J, Begier E, Noriega DC, Konishi H, Yato Y, et al. Efficacy of a 4-Antigen Staphylococcus aureus Vaccine in Spinal Surgery: The STaphylococcus aureus suRgical Inpatient Vaccine Efficacy (STRIVE) Randomized Clinical Trial. Clin Infect Dis. (2023) 77:312–20. doi: 10.1093/cid/ciad218
17. Fattom A, Matalon A, Buerkert J, Taylor K, Damaso S, Boutriau D. Efficacy profile of a bivalent Staphylococcus aureus glycoconjugated vaccine in adults on hemodialysis: Phase III randomized study. Hum Vaccin Immunother. (2015) 11:632–41. doi: 10.4161/hv.34414
18. Preston KB, Randolph TW. Stability of lyophilized and spray dried vaccine formulations. Adv Drug Delivery Rev. (2021) 171:50–61. doi: 10.1016/j.addr.2021.01.016
19. Wang Y, Ahmadi MZ, Dikeman DA, Youn C, Archer NK. γδ T cell-intrinsic IL-1R promotes survival during. Front Immunol. (2023) 14:1171934. doi: 10.3389/fimmu.2023.1171934
20. Han X, Ortines R, Mukherjee I, Kanipakala T, Kort T, Sherchand SP, et al. Hyperimmune targeting staphylococcal toxins effectively protect against USA 300 MRSA infection in mouse bacteremia and pneumonia models. Front Immunol. (2022) 13:893921. doi: 10.3389/fimmu.2022.893921
21. Wang Y, Dikeman D, Zhang J, Ackerman N, Kim S, Alphonse MP, et al. CCR2 contributes to host defense against Staphylococcus aureus orthopedic implant-associated infections in mice. J Orthop Res. (2021) 40(2):409–19. doi: 10.1002/jor.25027
22. Wang Y, Cheng LI, Helfer DR, Ashbaugh AG, Miller RJ, Tzomides AJ, et al. Mouse model of hematogenous implant-related. Proc Natl Acad Sci U.S.A. (2017) 114:E5094–E102. doi: 10.1073/pnas.1703427114
23. Youn C, Pontaza C, Wang Y, Dikeman DA, Joyce DP, Alphonse MP, et al. Neutrophil-intrinsic TNF receptor signaling orchestrates host defense against. Sci Adv. (2023) 9:eadf8748. doi: 10.1126/sciadv.adf8748
24. Marchitto MC, Dillen CA, Liu H, Miller RJ, Archer NK, Ortines RV, et al. Clonal Vgamma6+Vdelta4+ T cells promote IL-17-mediated immunity against Staphylococcus aureus skin infection. Proc Natl Acad Sci U.S.A. (2019) 116:10917–26. doi: 10.1073/pnas.1818256116
25. Adhikari RP, Arvidson S, Novick RP. A nonsense mutation in agrA accounts for the defect in agr expression and the avirulence of Staphylococcus aureus 8325-4 traP::kan. Infect Immun. (2007) 75:4534–40. doi: 10.1128/IAI.00679-07
26. Brignoli T, Douglas E, Duggan S, Fagunloye OG, Adhikari R, Aman MJ, et al. Wall teichoic acids facilitate the release of toxins from the surface of staphylococcus aureus. Microbiol Spectr. (2022) 10:e0101122. doi: 10.1128/spectrum.01011-22
27. Adhikari RP, Karauzum H, Sarwar J, Abaandou L, Mahmoudieh M, Boroun AR, et al. Novel structurally designed vaccine for S. aureus alpha-hemolysin: protection against bacteremia and pneumonia. PloS One. (2012) 7:e38567. doi: 10.1371/journal.pone.0038567
28. Kailasan S, Kort T, Mukherjee I, Liao GC, Kanipakala T, Williston N, et al. Rational design of toxoid vaccine candidates for staphylococcus aureus leukocidin AB (LukAB). Toxins (Basel). (2019) 11:399. doi: 10.3390/toxins11060339
29. Venkatasubramaniam A, Adhikari RP, Kort T, Liao GC, Conley S, Abaandou L, et al. TBA225, a fusion toxoid vaccine for protection and broad neutralization of staphylococcal superantigens. Sci Rep. (2019) 9:3279. doi: 10.1038/s41598-019-39890-z
30. Reza HM, Sultana F, Bari R, Cole J, Baqui AH. Local distribution infrastructure and robust vaccine manufacturing facilities in LMICs should be prioritised to tackle ongoing and future pandemic risk. Lancet Reg Health Southeast Asia. (2023) 11:100158. doi: 10.1016/j.lansea.2023.100158
31. Guo Y, Ramos RI, Cho JS, Donegan NP, Cheung AL, Miller LS. In vivo bioluminescence imaging to evaluate systemic and topical antibiotics against community-acquired methicillin-resistant Staphylococcus aureus-infected skin wounds in mice. Antimicrob Agents Chemother. (2013) 57:855–63. doi: 10.1128/AAC.01003-12
32. Diep BA, Le VT, Visram ZC, Rouha H, Stulik L, Dip EC, et al. Improved Protection in a Rabbit Model of Community-Associated Methicillin-Resistant Staphylococcus aureus Necrotizing Pneumonia upon Neutralization of Leukocidins in Addition to Alpha-Hemolysin. Antimicrob Agents Chemother. (2016) 60:6333–40. doi: 10.1128/AAC.01213-16
33. Zipursky S, Djingarey MH, Lodjo JC, Olodo L, Tiendrebeogo S, Ronveaux O. Benefits of using vaccines out of the cold chain: delivering meningitis A vaccine in a controlled temperature chain during the mass immunization campaign in Benin. Vaccine. (2014) 32:1431–5. doi: 10.1016/j.vaccine.2014.01.038
34. Kaasch AJ, Barlow G, Edgeworth JD, Fowler VG, Hellmich M, Hopkins S, et al. Staphylococcus aureus bloodstream infection: a pooled analysis of five prospective, observational studies. J Infect. (2014) 68:242–51. doi: 10.1016/j.jinf.2013.10.015
35. Anantha RV, Jegatheswaran J, Pepe DL, Priestap F, Delport J, Haeryfar SM, et al. Risk factors for mortality among patients with Staphylococcus aureus bacteremia: a single-centre retrospective cohort study. CMAJ Open. (2014) 2:E352–9. doi: 10.9778/cmajo.20140018
36. Nickerson EK, Hongsuwan M, Limmathurotsakul D, Wuthiekanun V, Shah KR, Srisomang P, et al. Staphylococcus aureus bacteraemia in a tropical setting: patient outcome and impact of antibiotic resistance. PloS One. (2009) 4:e4308. doi: 10.1371/journal.pone.0004308
37. Eshwara VK, Munim F, Tellapragada C, Kamath A, Varma M, Lewis LE, et al. Staphylococcus aureus bacteremia in an Indian tertiary care hospital: observational study on clinical epidemiology, resistance characteristics, and carriage of the Panton-Valentine leukocidin gene. Int J Infect Dis. (2013) 17:e1051–5. doi: 10.1016/j.ijid.2013.06.002
38. Ghaemmaghamian Z, Zarghami R, Walker G, O'Reilly E, Ziaee A. Stabilizing vaccines via drying: Quality by design considerations. Adv Drug Delivery Rev. (2022) 187:114313. doi: 10.1016/j.addr.2022.114313
39. Frey SE, Wald A, Edupuganti S, Jackson LA, Stapleton JT, El Sahly H, et al. Comparison of lyophilized versus liquid modified vaccinia Ankara (MVA) formulations and subcutaneous versus intradermal routes of administration in healthy vaccinia-naïve subjects. Vaccine. (2015) 33:5225–34. doi: 10.1016/j.vaccine.2015.06.075
40. Rao Q, Shang W, Hu X, Rao X. Staphylococcus aureus ST121: a globally disseminated hypervirulent clone. J Med Microbiol. (2015) 64:1462–73. doi: 10.1099/jmm.0.000185
41. Viana D, Comos M, McAdam PR, Ward MJ, Selva L, Guinane CM, et al. A single natural nucleotide mutation alters bacterial pathogen host tropism. Nat Genet. (2015) 47:361–6. doi: 10.1038/ng.3219
42. Wallis J, Shenton DP, Carlisle RC. Novel approaches for the design, delivery and administration of vaccine technologies. Clin Exp Immunol. (2019) 196:189–204. doi: 10.1111/cei.13287
43. Peyraud N, Zehrung D, Jarrahian C, Frivold C, Orubu T, Giersing B. Potential use of microarray patches for vaccine delivery in low- and middle- income countries. Vaccine. (2019) 37:4427–34. doi: 10.1016/j.vaccine.2019.03.035
Keywords: vaccine development, Staphylococcus aureus, preclinical infection models, toxins, neutralizing antibodies, LMIC, IBT-V02
Citation: Wang Y, Mukherjee I, Venkatasubramaniam A, Dikeman D, Orlando N, Zhang J, Ortines R, Mednikov M, Sherchand SP, Kanipakala T, Le T, Shukla S, Ketner M, Adhikari RP, Karauzum H, Aman MJ and Archer NK (2024) Dry and liquid formulations of IBT-V02, a novel multi-component toxoid vaccine, are effective against Staphylococcus aureus isolates from low-to-middle income countries. Front. Immunol. 15:1373367. doi: 10.3389/fimmu.2024.1373367
Received: 19 January 2024; Accepted: 18 March 2024;
Published: 03 April 2024.
Edited by:
Isabelle Bekeredjian-Ding, University of Marburg, GermanyReviewed by:
Isidora Del Carmen Suazo, College of Pontifical Catholic University of Chile, ChileCopyright © 2024 Wang, Mukherjee, Venkatasubramaniam, Dikeman, Orlando, Zhang, Ortines, Mednikov, Sherchand, Kanipakala, Le, Shukla, Ketner, Adhikari, Karauzum, Aman and Archer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathan K. Archer, bmFyY2hlcjJAamhtaS5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.