
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 08 April 2024
Sec. Alloimmunity and Transplantation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1372862
 Leonard Knoedler1,2*
Leonard Knoedler1,2* Jillian Dean3Fortunay Diatta2Noelle Thompson4
Jillian Dean3Fortunay Diatta2Noelle Thompson4 Samuel Knoedler1
Samuel Knoedler1 Richmond Rhys1Khalil Sherwani5
Richmond Rhys1Khalil Sherwani5 Tobias Ettl6Simon Mayer4Florian Falkner5Katja Kilian5
Tobias Ettl6Simon Mayer4Florian Falkner5Katja Kilian5 Adriana C. Panayi5
Adriana C. Panayi5 Jasper Iske7,8
Jasper Iske7,8 Ali-Farid Safi9,10Stefan G. Tullius11
Ali-Farid Safi9,10Stefan G. Tullius11 Siba Haykal1Bohdan Pomahac1†
Siba Haykal1Bohdan Pomahac1† Martin Kauke-Navarro1*†
Martin Kauke-Navarro1*†Balancing the immune response after solid organ transplantation (SOT) and vascularized composite allotransplantation (VCA) remains an ongoing clinical challenge. While immunosuppressants can effectively reduce acute rejection rates following transplant surgery, some patients still experience recurrent acute rejection episodes, which in turn may progress to chronic rejection. Furthermore, these immunosuppressive regimens are associated with an increased risk of malignancies and metabolic disorders. Despite significant advancements in the field, these IS related side effects persist as clinical hurdles, emphasizing the need for innovative therapeutic strategies to improve transplant survival and longevity. Cellular therapy, a novel therapeutic approach, has emerged as a potential pathway to promote immune tolerance while minimizing systemic side-effects of standard IS regiments. Various cell types, including chimeric antigen receptor T cells (CAR-T), mesenchymal stromal cells (MSCs), regulatory myeloid cells (RMCs) and regulatory T cells (Tregs), offer unique immunomodulatory properties that may help achieve improved outcomes in transplant patients. This review aims to elucidate the role of cellular therapies, particularly MSCs, T cells, Tregs, RMCs, macrophages, and dendritic cells in SOT and VCA. We explore the immunological features of each cell type, their capacity for immune regulation, and the prospective advantages and obstacles linked to their application in transplant patients. An in-depth outline of the current state of the technology may help SOT and VCA providers refine their perioperative treatment strategies while laying the foundation for further trials that investigate cellular therapeutics in transplantation surgery.
Over the last thirty years, the field of plastic surgery has adopted principles from solid organ transplantation (SOTs), particularly as vascularized composite allotransplantation (VCA) has become more prominent (1–3). This biotechnology offers unique clinical benefits, especially in restoring form and functionality in patients with complex and devastating deformities that cannot be adequately reconstructed using conventional strategies such as local tissue rearrangement or free flap reconstruction (4). One of the major challenges of transplantation is rejection of the allograft and the need for life-long multidrug immunosuppression that often has significant systemic side effects.
Current standard immunosuppressive regimens include corticosteroids, tacrolimus, and mycophenolate mofetil (MMF), among other immunosuppressants (4–6). While modern immunosuppressive drugs have effectively reduced rejection rates, prolonged use has been implicated with increased risk of metabolic disorders, infections, and cancer (5). Tacrolimus has been linked with pancreatic beta cell toxicity (7), increasing the risk of post-transplant diabetes by 25% compared to cyclosporine-based regimens (8). Antithymocyte globulin (ATG) has also been shown to predispose post-transplant patients to opportunistic infections, such as cytomegalovirus (CMV) or fungal infections (9). In this context, a 2019 study enrolled 2,495 liver transplant patients receiving tacrolimus-based immunosuppression and revealed that 19.7% of study participants developed malignancies over a mean follow-up period of 5.6 years (10).
Recent advances suggest cellular therapy as a promising solution to address these ongoing challenges in transplantation surgery (11). This strategy involves administering viable cell subsets stemming from autologous, allogeneic, and xenogeneic sources (11–13). In contrast to conventional immunosuppressants that are non-specific modulators of the immune response (14), cellular therapies are intended to be more specific and targeted, thereby reducing systemic side effects (1). The main goal of these therapies is to establish a stable state of immune homeostasis, inhibiting both graft-versus-host and host-versus-graft reactions, while reducing the need for long-term conventional immunosuppressive treatments (15). This review is intended to provide a comprehensive overview of cellular therapies for the treatment of SOT and VCA patients, reviewing their key functions, clinical use cases, and potential applications. Ultimately, this line of research may unlock untapped therapeutic potential and help SOT and VCA providers optimize their perioperative treatment protocols.
Options for induction therapy include ATGs, anti-IL2 receptor antibodies, and anti-CD3 antibodies (16), all of which have been shown to significantly reduce acute rejection rates in the short-term perioperative setting (17, 18). The 2021 Organ Procurement and Transplantation Network (OPTN) Report indicated that 67.5% of adult transplant patients were discharged on a triple therapy maintenance regimen consisting of tacrolimus, MMF, and corticosteroids, while the remaining 25.6% of patients were discharged on dual therapy (tacrolimus-MMF) (19). For VCAs, recent research has shed light on the primary targets of rejection reactions, identifying the mucosa (20) and skin (21) as key areas of concern. Induction therapy, akin to SOTs, is frequently based on ATG (22). Despite the adoption of SOT triple therapy as the standard maintenance regimen for the majority of VCA patients, the occurrence of acute rejection within the first-year post-transplant remains high, exceeding 80% (23). Furthermore, while chronic rejection rates in VCA patients have not been definitively established, approximately 85% of VCA recipients experience at least one episode of acute rejection during their first year of transplantation (24, 25).
Over the past three decades, cellular therapies have emerged as a novel approach to address the persisting challenges linked to multidrug and lifelong immunosuppression (26, 27). The concept of cellular therapies is centered around the infusion of immunomodulating cells. Such cell populations target specific immunologic pathways linked with allograft rejection and off-target systemic side effects (28). In preclinical VCA models, Tregs (29), CAR-T cells (30, 31), MSCs (32, 33), and RMCs (34) have demonstrated to improve allograft acceptance and minimize systemic toxicity.
Tregs are a specialized subset of T cells that play a crucial role in modulating the immune response to promote anti-inflammatory outcomes, serving as one of the primary mediators of physiological immune suppression (35). Tregs are broadly classified into thymus-derived Tregs (nTregs), which develop in the thymus, and induced Tregs (iTregs), which differentiate from conventional T cells in the periphery under specific conditions, such as exposure to antigens in a tolerogenic context (36). Peripheral Tregs (pTregs) are a type of iTreg that arises in the periphery following antigenic stimulation, and can be distinguished by specific markers like neuropilin-1 and Helios in conjunction with Foxp3 expression, the defining transcription factor for regulatory T cell lineage (36). Contrary to the previous classification based on CD45RA and Foxp3 expression, recent findings suggest that the expression of neuropilin-1 (Nrp-1) and Helios can more accurately distinguish nTregs from pTregs in vivo, with the former typically expressing higher levels of Nrp-1 (37).
Tregs are characterized by the transcription factor Foxp3, which is crucial for their suppressive function and the maintenance of immune homeostasis (38). pTregs are increasingly recognized for their role in peripheral tolerance (39), especially at mucosal sites and during certain inflammatory conditions (40). It is important to note that not all regulatory T cells express Foxp3 (41). Type 1 regulatory T (Tr1) cells, for instance, do not express Foxp3 but instead secrete high levels of IL-10 and contribute to immune tolerance primarily through IL-10 and TGF-β dependent pathways, contributing to their regulatory capabilities (42). Thus, the functional mechanisms of Tregs are more diverse than solely suppressive interactions, encompassing a range of anti-inflammatory pathways. These characteristics make Tr1 cells pivotal for the maintenance of long-term tolerance, particularly in SOT (43), however the production of antigen-specific Tr1 cells alone is inadequate, and it is also essential to reduce the activity of effector T cells (Teffs) (44). The clinical significance of Tr1 cells is highlighted by emerging evidence and ongoing clinical trials that are exploring their potential in inducing operational tolerance. Tr1 cells’ ability to be induced and expanded ex vivo (45), along with their stable phenotype in inflammatory post-transplant environments (46), positions them as promising candidates for achieving and sustaining this state of tolerance.
Tr1 cells have been associated with positive outcomes in transplantation, including stable graft function and tolerance (47). The induction of Tr1 cells can be achieved through several methods, often involving cytokines like IL-10 and IL-27 (48), signaling through specific surface molecules (47), and interactions with dendritic cells (DCs) (49). This versatility makes Tr1 cells a valuable target for clinical applications, particularly in transplantation where they can potentially prevent organ rejection and reduce the need for long-term immunosuppression.
In a study conducted by Jofra et al., Tr1 cell immunotherapy was employed to enhance transplant tolerance in a mouse model of pancreatic islet transplantation (50). They discovered that the LAG3+ cells from tolerant mice, which were enriched in Tr1 cells, expressed high levels of IL-10, with approximately 17.5 ± 2.2% FoxP3+ cells. This indicated a significant presence of Tr1 cells. The role of these Tr1 cells in maintaining transplant tolerance was further elucidated as blocking these cells with an anti-LAG3 mAb led to the loss of transplant tolerance, highlighting their crucial role. Moreover, when Tr1 cell therapy was administered to IL-10-deficient mice, the mice failed to develop transplant tolerance, suggesting that host IL-10 is essential for the induction of tolerance (50).
Another key finding was the de novo generation of Tr1 cells in the recipients. The study showed that transferred Tr1 cells did not home to the graft or its draining lymph nodes due to the lack of CD62L expression, which is necessary for adhesion to high endothelial venules. An increase in host LAG3+ CD4 T cells in Tr1-treated recipient mice indicated that the therapy led to an induction of Tr1 cells in the host. This was further supported by the increased number of IL-10+ cells expressing LAG-3 in the spleens of IL-10-reporter mice that received Tr1 cell therapy (50). The study also addressed concerns about the safety and stability of Tr1 cell therapy during acute viral infections. To test this, researchers infected Tr1-treated tolerant mice with lymphocytic choriomeningitis virus (LCMV) and observed that the tolerance induced by Tr1 cell therapy was not detrimental to the host’s ability to control the viral infection. The Tr1 treated mice successfully survived the infection and maintained the graft. Importantly, the tolerogenic state did not compromise the development of effective antiviral immune responses, as evidenced by the maintenance of both CD8 and CD4 anti-LCMV responses (50). This suggests that Tr1 cell therapy can establish a stable and safe transplant tolerance that does not inhibit the host’s ability to respond to acute viral infections.
A study of Tr1 cell therapy for type 1 diabetes in the context of islet transplantation presents compelling data across different experimental models (51). In the less stringent transplant scenario involving BALB/c mice receiving C57BL/6 islets, polyclonal Tr1 cells demonstrated notable efficacy. Specifically, after the transfer of these cells into diabetic BALB/c mice, 62.5% (5 out of 8) did not reject the graft within 25-days post-transplant. Furthermore, a long-term graft survival rate of 50% was observed at the 100-day mark. This contrasted sharply with the outcomes in a more stringent model, where C57BL/6 mice received BALB/c islets. In this setting, all recipient mice, regardless of receiving polyclonal Tr1 cells or Th0 cells, uniformly rejected the graft promptly, underscoring the limited effectiveness of polyclonal Tr1 cells in more challenging transplant environments (51).
A significant enhancement in outcomes was noted with the use of antigen (Ag)-specific Tr1 cells in the so-called stringent model (51). All mice receiving these Ag-specific Tr1 cells maintained functional grafts 25-days post-transplant, and 40% sustained long-term graft survival up to 100 days. The study also highlighted the critical role of IL-10 in sustaining transplant tolerance. This was particularly evident when all mice treated with an αIL-10 receptor monoclonal antibody experienced rapid graft rejection, confirming the IL-10-dependent nature of the tolerance induced by Tr1-cell therapy. An important aspect of the study was the emphasis on the donor antigen specificity of the Tr1 cells. When BALB-specific Tr1 cells were transferred into B6 mice receiving C3H islets, the grafts were rejected, illustrating that the therapeutic advantage of Tr1 cells is closely tied to their donor antigen specificity. Taken together, these findings suggest that Tr1-cell therapy, especially when tailored to the donor antigen, can effectively induce IL-10–dependent tolerance in challenging models of islet transplantation (51).
The findings from the study on Tr1 cell therapy in the context of type 1 diabetes have broader implications for SOTs. By producing high levels of IL-10 and suppressing immune responses against specific antigens, Tr1 cells could reduce the need for generalized immunosuppression (52). This approach is particularly valuable in SOTs where the risk of chronic rejection and drug-induced complications remains a significant challenge (53). The potential to tailor Tr1 cells to the specific antigens of the transplanted organ opens a pathway to more personalized and effective transplant therapies, enhancing graft survival while maintaining overall immune system integrity (54). Clinically, protocols have been developed for the in vitro expansion of Tr1 cells (55, 56), enabling their use as therapeutic products in immune-mediated diseases and transplantation (42). However, challenges remain, such as the need for specific biomarkers to isolate pure Tr1 cells and the limited expansion capacity of current protocols (47).
As of now, clinical trials have established the safety and effectiveness of Tr1 cell therapy in certain contexts. A single-center, non-randomized, and prospective phase I–II trial (ALT-TEN protocol, IS/11/6172/8309/8391) has been conducted, where patients with high-risk or advanced-stage hematologic malignancies received both haploidentical hematopoietic stem cell transplantation (haplo-HSCT) and Tr1-cell therapy (57). This combined treatment has shown potential in enhancing immune system reconstitution and reducing both the risk of disease relapse and the occurrence of GVHD following HSCT. Additionally, a new Phase I trial (NCT03198234) is underway, testing the use of T-allo10 cells to prevent GVHD after mismatched related or unrelated hematopoietic stem cell transplantation (HSCT) without any pre-treatment in cases of hematologic malignancies (58). Preliminary results from this trial have been encouraging and deemed safe, yet further analysis is necessary to fully assess the clinical outcomes.
The clinical trials investigating Tr1-cell therapy, particularly those in the context of HSCT (59), provide a foundational understanding that can be applied to SOT and VCA. In SOT, the goal of Tr1 cell therapy would be to induce specific tolerance to the transplanted organ, thereby minimizing the risk of organ rejection while potentially reducing the need for long-term systemic immunosuppressive drugs. The ability of Tr1 cells to regulate the immune response by producing anti-inflammatory cytokines, and their stability in the presence of immunosuppressive drugs, as observed in kidney transplant patients, are particularly promising for SOT (47). These properties may help to maintain the delicate balance necessary to prevent organ rejection while avoiding the complications of over-suppression, such as susceptibility to infections and malignancies. In VCA, the immunological challenges are even greater due to the diversity of tissue types involved, each with different antigenic profiles (10, 60). The immunomodulatory capabilities of Tr1 cells could be harnessed to promote graft tolerance and reduce the incidence of both acute and chronic rejection, which is vital for the long-term success of these grafts. The preliminary results of Tr1-cell therapy in preventing GvHD in HSCT patients indicates its potential for application in SOT and VCA, where similar mechanisms of immune-mediated rejection occur (61). By extending the application of Tr1-cell therapy to these areas, there is a potential to significantly improve patient outcomes through targeted immune regulation.
Looking ahead, the potential of genetically engineered Tr1 cells, such as those modified with CAR constructs (CAR-Tr1 cells), represents a promising avenue of research (62). CAR-Tr1 cells could offer a more targeted approach to immunomodulation in transplantation, possibly enhancing the specificity and efficacy of cellular therapies (47). Despite the progress made, further research is essential to fully understand Tr1 cells’ regulatory mechanisms and to harness their therapeutic potential reliably. Further, the induction of Tr1 cells could be a therapeutic lever that offers advantages over traditional FoxP3+ Treg therapies (40). While FoxP3+ Tregs are essential for establishing tolerance (63), Tr1 cells have been identified as key players in sustaining this tolerance long-term (59). This understanding may reshape therapeutic strategies in SOT, with an emphasis on harnessing the unique properties of Tr1 cells to improve SOT outcomes.
Tregs have become crucial in the field of SOT, presenting new therapeutic possibilities for enhancing graft acceptance and decreasing the need for immunosuppressive medications (64, 65). Investigations into Foxp3+CD4+CD25+ splenic nTregs have shown the critical importance of ex vivo expansion modifications in preserving Foxp3+ expression in SOT (66). Optimal Foxp3+ preservation was achieved through preconditioning CD4+CD25+ cells with TGF-β/IL-12, not only bolstering donor suppression but also effectively preventing acute heart allograft rejection (66) (Figure 1A). These findings laid the foundation for subsequent trials, such as the Autologous Polyclonal Expanded Tregs Adoptive Cell Therapy (TRACT) study (69). Patients who received autologous polyclonal expanded Tregs at 60-days post-transplant, following standard induction and daily maintenance immunosuppression, demonstrated an absence of infection or rejection for up to 2 years in a phase I clinical trial (69). Roemhild et al. further validated these outcomes by revealing that a combination of Tregs and conventional immunosuppressants enabled 90% of patients to maintain a low tacrolimus dose within the target range of 6-8 ng/mL over 48 weeks, with 72% displaying stable graft function when treated with nTregs monotherapy (70).
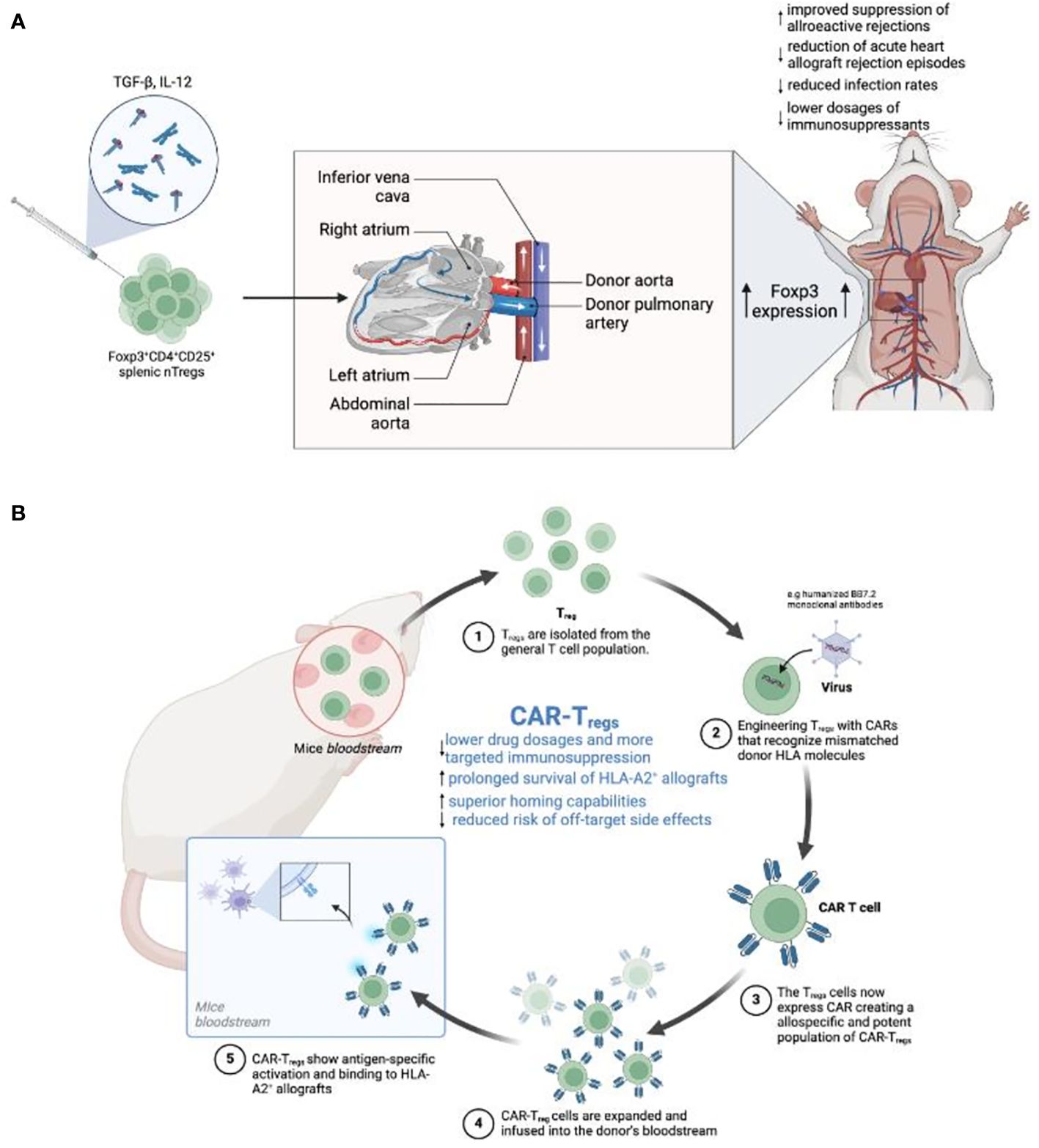
Figure 1 (A) Enhancing Tregs to promote tolerance in heart transplantation. This figure illustrates the sequential steps from the in vitro induction of Tregs using TGF-β and IL-12 cytokines to the maturation of Foxp3+CD4+CD25+ Tregs. Additionally, the diagram provides a schematic representation of a human heart transplant, indicating the flow of blood through major vessels and heart chambers. This enhancement process is expected to improve transplant tolerance, reduce rejection rates, and potentially lower the need for immunosuppressive drugs (66). (B) Development of CAR-Tregs for targeted immunosuppression in a mouse model. The process begins with the extraction of Tregs from the mouse bloodstream, followed by their engineering to express CARs recognizing specific HLA molecules on transplanted tissues. The diagram depicts the binding of modified Tregs (CAR-Tregs) to HLA-A2+ allografts, showcasing antigen-specific activation. The expanded CAR-Tregs are then reintroduced into the mouse’s circulatory system. This targeted approach aims to minimize drug dosages, extend allograft survival, and reduce the risk of off-target side effects (67, 68).
Clinical trials have highlighted the impact of Tregs cell therapy on liver transplant recipients. Fueyo et al. conducted a phase 1 study assessing the safety and tolerability of polyclonal Tregs immunotherapy in patients undergoing liver transplantation from deceased donors (71). Results indicated that infusion of Tregs significantly reduced alloreactive T cell activity by over 20%, suggesting the potential for Tregs based therapy in liver transplantation (71). Further, Todo et al. demonstrated the potential of Tregs-enriched cell products in liver transplantation, where patients received Tregs infusions after living donor liver transplantation, resulting in prolonged transplant survival of more than 15% (72).
“The ONE Study,” an EU-funded consortium, conducted a series of non-randomized, single-arm trials across several international sites to assess the safety and potential benefits of various regulatory cell-based medicinal products (CBMPs) in living-donor kidney transplant recipients (73). The study aimed to reduce reliance on general immunosuppression by employing CBMPs that included regulatory T cells (both polyclonal and donor-antigen reactive), tolerogenic dendritic cells, and regulatory macrophages. The trials demonstrated that this approach is feasible and safe, with a primary focus on the safety of regulatory CBMPs when combined with a reduced immunosuppressive regimen. Results showed similar rates of biopsy-confirmed acute rejection (BCAR) between the cell therapy groups and the reference group, which received standard immunosuppressive treatment. Furthermore, the study found that regulatory cell therapy was associated with fewer infectious complications, suggesting a potential benefit in reducing the burden of general immunosuppression in kidney transplant recipients (73). The ONE Study’s findings support the continued exploration of regulatory cell therapy as a therapeutic strategy in organ transplantation, with a focus on minimizing the adverse effects of long-term immunosuppression.
In another groundbreaking study, Tang et al. explored the use of donor-alloantigen-reactive regulatory T cells (darTregs) to induce graft-specific immunosuppression in liver transplant recipients, potentially reducing the need for broad immunosuppressive drugs (74). This phase I/II trial, named ARTEMIS (NCT02474199), aimed to test the safety and efficacy of darTregs in maintaining stable liver function while allowing significant reduction in immunosuppression. Although the infusion of darTregs in five out of ten participants who initiated immunosuppression withdrawal did not lead to adverse events, they faced challenges in manufacturing sufficient darTreg doses and in achieving the primary efficacy endpoint—a 75% reduction in calcineurin inhibitor dose with stable liver function for at least 12 weeks (74). Only two participants managed to reach this endpoint, and mechanistic studies suggested darTregs became dysfunctional after liver transplantation, affecting their efficacy. Mechanistic insights revealed a reduction in Treg donor reactivity after liver transplantation, suggesting a need for further research to understand and potentially counteract Treg dysfunction in this context (74). This study highlights several critical areas for future research, including overcoming manufacturing challenges, understanding the mechanisms behind Treg dysfunction post-transplant, and exploring alternative strategies for inducing transplant tolerance.
A current phase I/IIa trial is investigating the safety and efficacy of inducing transient chimerism through a novel combination cell therapy in kidney transplant recipients without requiring myelosuppressive conditioning (75). Conducted in HLA-mismatched living donor kidney transplants, the approach involves administering in vitro expanded recipient Tregs, donor BM cells, tocilizumab, and an anti-IL6R monoclonal antibody shortly after transplantation, with a regimen of thymoglobulin, belatacept, sirolimus, and steroids. Starting 6-months post-transplant, sirolimus and steroids are gradually withdrawn in stable patients, aiming for belatacept monotherapy. The control group receives a standard immunosuppressive regimen without Tregs, anti-IL6R, or BM infusion (75).
Primary outcomes include safety measures and the assessment of donor chimerism within the first month post-transplant (75). Secondary outcomes focus on the frequency of acute and subclinical rejection episodes, graft function, and pro-tolerogenic immunomodulatory effects. This trial is driven by the hypothesis that recipient Tregs and donor BM infusion, combined with tocilizumab and a belatacept-based immunosuppressive regimen, can safely induce transient chimerism and a pro-tolerogenic immune environment in kidney transplant recipients (75). If successful, this innovative treatment could represent a significant advance in transplant immunomodulation, offering a safer alternative to current myelosuppressive chimerism-based tolerance protocols and potentially improving long-term transplant outcomes.
Studies on the immunological dynamics during VCA graft rejection revealed a novel link between Foxp3 and IDO, an immune-regulatory enzyme that promotes an immunosuppressive environment (76). Over a period of 6 years, a total of 104 skin biopsies were obtained from bilateral hands of 3 human hand allograft recipients and assessed by hematoxylin-eosin histology and immunohistochemistry. The analysis revealed a temporally synchronized elevation of Foxp3 and IDO during graft rejection episodes (76). The simultaneous rise within the allograft suggested a coordinated function of Foxp3 and IDO to balance immune responses. This synchronization may provide a robust dual marker for detecting graft rejection and a potential lever for orchestrating targeted therapies to the rejection site.
Tregs, with their intrinsic ability to modulate immune responses, hold promise in transplant immunology for their role in fostering graft tolerance and preventing rejection (77). However, despite their potential, there are numerous immunomodulatory limitations of Tregs, such as the stability of Tregs in the inflammatory transplant environment (78). The inflammatory cytokines and post-transplantation milieu can lead to a phenomenon known as Treg plasticity (79), where Tregs can lose their regulatory phenotype and potentially convert into pro-inflammatory effector T cells, thus contributing to graft rejection rather than prevention (79, 80). Furthermore, the scale-up of Tregs for therapeutic purposes has its challenges, including maintaining their suppressive function after expansion and ensuring that they retain their stability and functionality once infused back into patients (81). The in vivo survival, homing, and persistence of Tregs are critical factors that need optimization to ensure long-term graft tolerance (82).
Lastly, the regulatory pathways and mechanisms through which human Tregs exert their function are complex and not fully understood (83). This makes it challenging to predict and control their behavior within the immune system of a transplant recipient. The interactions between Tregs and other immune cells, such as dendritic cells, B cells, and Teffs, are crucial for graft tolerance (84), but can also lead to unpredictable outcomes if not properly regulated (85). While Tregs present a significant therapeutic opportunity for inducing tolerance in SOT, their immunomodulatory limitations, such as lack of antigen specificity, stability, and complex regulatory mechanisms present hurdles that need to be addressed to fully harness their potential (86–88). Advances in genetic engineering, like the development of CAR-Tregs, are promising strategies to overcome some of these limitations by enhancing specificity and control over Treg activity.
CAR-T cells represent a class of adoptive cellular therapies exhibiting promising efficacy in the management of hematological malignancies like multiple myelomas (89). These specialized T cells are engineered with synthetic CAR constructs, comprising an extracellular antigen recognition domain derived from the single-chain variable fragment (scFv) of a monoclonal antibody, linked to intracellular signaling domains that induce T cell activation. This configuration allows CAR-T cells to directly bind to target antigens (TAAs) on the cell surface (67) (Figure 1B).
The structure of a CAR is segmented into several distinct components. The extracellular domain confers antigen specificity and is connected via a hinge region to a transmembrane domain that anchors the receptor in the T cell membrane (90). Intracellularly, the CAR possesses at least one signaling domains derived from the CD3ζ chain of the TCR complex and co-stimulatory molecules such as CD28, 4-1BB (CD137), or OX40 (CD134) (91, 92). The first generation of CARs only contained the CD3ζ signaling domain and, therefore, showed limited clinical efficacy due to inadequate T cell persistence and expansion (93). Second-generation CARs, which include an additional co-stimulatory domain, demonstrated improved T cell proliferation, cytokine production, and CAR-T cell survival (94). Third-generation CARs further augment the signal by incorporating two co-stimulatory domains, while fourth-generation CARs, also known as T cells redirected for universal cytokine-mediated killing (TRUCKs), are engineered to induce the secretion of transgenic cytokines upon antigen engagement, amplifying and sustaining the immune response (94). Different types of cells such as Tregs, Natural Killer (NK) cells, and macrophages can be equipped with CAR constructs (95).
The effector functions of CAR-T cells are multifaceted, encompassing the secretion of cytotoxic granules such as perforin and granzyme (96), the production of inflammatory cytokines like IL-6, IFN-γ, TNF-α (97), and the induction of apoptosis in target cells (98). Upon antigen recognition, CAR-T cells undergo expansion and induce a broader immune response through the release of pro-inflammatory cytokines, which can recruit and activate additional immune effector cells (99, 100).
Turning to CAR-Tregs, recent research has demonstrated the therapeutic potential of CAR-Tregs in dampening the pro-inflammatory responses of Teffs and demonstrating a stable phenotype even in a pro-inflammatory microenvironment (101, 102). Further, CAR-Tregs have been shown to secrete anti-inflammatory cytokines that promote immunotolerance (35). Research has shown promising potential in engineering CAR-Tregs to further exploit key pathways, such as TGF-β and IL-2 to improve the cellular stamina and therapeutic effectiveness of CAR-Tregs (64, 82, 90).
To date, SOT and VCA research has mainly focused on CAR-Tregs by directing them against HLA (103, 104). By engineering Tregs with CARs that recognize mismatched donor HLA molecules, it is possible to create a population of Tregs that are both allospecific and potent, capable of homing to and protecting allografts from the host immune system with higher efficacy than polyclonal Tregs (103). Studies have demonstrated the viability of HLA-A2 specific CAR Tregs, which show antigen-specific activation and demonstrate superior localization to HLA-A2 positive allografts, with the potential to substantially prolong allograft survival in pre-clinical humanized mouse models (104, 105).
The specificity of these CAR-Tregs enables them to outperform polyclonal Tregs in terms of suppression of the immune response against the transplant (103). This specificity is a crucial advantage because it may pave the way for lower dosages, yet more potent and targeted immunosuppression. In preclinical models, HLA-A2 specific CAR-Tregs have been able to prolong the survival of HLA-A2+ allografts (106–108). In these models, CAR-Tregs have demonstrated superior homing capabilities (103), preferentially migrating to HLA-A2+ graft sites when compared to polyclonal Tregs (106–108). Ultimately, this targeted homing ensures local and targeted immunosuppression, thereby reducing the risk of systemic immunosuppression and off-target side effects.
Specifically, Sun et al. described the development of CAR-Tregs tailored to identify the HLA-A2 antigen, which has been implicated in SOT transplant rejection (68). These cells, when engineered with the humanized BB7.2 monoclonal antibody, effectively migrated to HLA-A2 positive grafts while demonstrating reduced binding to other HLA alleles. Additionally, donor-specific (ds) CAR-Tregs administered to non-sensitized mice with HLA-A2+ skin grafts prolonged the graft lifespan by more than 30% (68). However, these findings were not reproducible in HLA-2 sensitized recipients, suggesting that previous immune exposure to the HLA-A2 antigen can influence the therapeutic efficacy of dsCAR-Tregs.
MacDonald et al. took this approach further and designed HLA-A02 antigen-specific antibody (A2-CAR) CD8+ Tregs, which effectively mitigated GvHD and skin rejection in NSG mice without inducing harmful cytotoxicity or altering the immunotolerant state of the endothelial cells (106). Of note, CAR-Tregs cells seem to be more efficient in inducing transplant tolerance compared to conventional Tregs (62, 109), with lower CAR-Tregs dosages (1.5×108 CAR-Tregs versus 1×109 Tregs) showing similar immunosuppressive potency, when combined with T cell depleting agents (110). However, the application of CAR-Tregs is not without risks as complex genetic modifications have been implicated with neurologic and hematologic side effects (111, 112). Furthermore, the long-term stability of these cells, precise dosage requirements, and the potential for off-target effects remain areas of ongoing research (90).
Further preclinical research has shown the stability and persistence of CAR-Tregs, which are critical determinants for long-term therapy success. However, the exact half-life of CAR-T cell therapeutics is still under investigation, with recent studies reporting CAR-Tregs infiltration in murine grafts for up to 40 days compared to conventional Tregs (113). An ongoing phase I/IIa clinical trial is set to provide more concrete data on the safety and efficacy of CAR-Tregs in living donor renal transplant recipients (NCT04817774)120. While this trial may help determine the optimal dosing, frequency of infusion, and long-term stability of these cells in renal transplant patients, future research is needed to standardize CAR-Treg for additional types of SOT and VCA transplants. Moreover, safety concerns such as tonic signaling, leading to T cell exhaustion and the phenotypic stability of Tregs, remain to be further investigated (114).
Kauke-Navarro & Knoedler proposed an innovative strategy to mitigate transplant rejection, by harnessing and enhancing the immunoregulatory capabilities of Tregs (115). Recognizing the adverse effects associated with lifelong immunosuppressive therapy, the strategy emphasizes the bioengineering of Tregs to promote immune tolerance. These engineered Tregs, potentially modified to express CAR constructs or specific T cell receptors, aim to target specific antigens, thereby minimizing off-target effects (115). The approach builds on the natural role of Tregs in maintaining immune homeostasis, including self-tolerance and antimicrobial resistance, and leverages their capacity for direct immunosuppression and modulation of the immune response. By focusing on antigen-specific Tregs, the strategy seeks to provide a more targeted, effective means of preventing graft rejection while reducing the reliance on broad-spectrum immunosuppressive drugs. Further, they discuss the potential for employing advanced genomic editing techniques, such as CRISPR/Cas9, to enhance the specificity and safety of engineered Tregs (115).
Despite their potential, the utilization of CAR-T cell therapies in SOTs and VCA has a spectrum of limitations and challenges warranting consideration. Among these limitations is the protracted timeline inherent in the engineering and production of CAR-T cell products (116). The process encompasses several steps, beginning with the retrieval of patient-derived T cells, genetic manipulation to incorporate CAR constructs, ex vivo expansion of modified T cells, quality assessment procedures, patient conditioning, followed by infusion (117). This can take several weeks to months and is both costly and resource intensive (118), which may not align with the immediate needs of transplant patients who require rapid immune modulation to prevent rejection or manage complications like PTLD.
Unlike hematological malignancies where tumor cells express well-defined TAAs, the identification of suitable target antigens for CAR-T cell therapy in SOTs and VCA is more challenging (90). To our knowledge, there is limited research focused on this area, highlighting a significant gap in understanding and development of effective CAR-T cell therapies. Further, while CAR-T cell therapy offers the advantage of precise targeting of donor-specific HLA molecules, the potential for inadvertently targeting analogous antigens expressed on healthy tissues or vital cells presents a substantial challenge (119). Such off-target effects hold the potential to precipitate inadvertent tissue injury, autoimmune responses, or adverse events, thereby necessitating exhaustive preclinical evaluation of CAR-T cell specificity and safety profiles (120).
The protracted persistence and activity of CAR-T cells in vivo pose inherent risks of on-target side effects, such as sustained immune activation, CRS, and immune-related neuro-toxicities (121). Additionally, the prospect of CAR-T cell anergy or dysfunction over time remains a pertinent concern, as prolonged exposure to antigen stimulation may impair CAR-T cell functionality and constrain their efficacy in sustaining transplant tolerance or regulating alloimmune responses (122).
Moreover, the optimization of dosing regimens, frequency of administration, and long-term management of CAR-T cell therapies in transplant recipients necessitates further elucidation and standardization. The delineation of precise dosing and administration schedules for CAR-T cells may exhibit variability contingent on individual patient parameters, transplant modalities, and immunological status, thereby mandating personalized therapeutic approaches and monitoring. Given the potential variability, creating a standardized protocol and manufacturing process presents a challenge. Another concern is the potential interplay between CAR-T cells and standard immunosuppressive medications commonly used in transplant recipients introduces queries regarding pharmacological compatibility, drug-drug interactions, and the risk of additive or synergistic toxicities. Thus, we need a more comprehensive and in-depth understanding of the safety profile and long-term effects of CAR-T in transplant patients.
MSCs can differentiate into different tissue types (cartilage, bone, fat, muscle) and have been shown to possess potent self-renewal capacities in vitro (123). They can be derived from various tissues including adipose tissue, umbilical cord, dental pulp, and bone marrow (124). Interestingly, MSCs do not express B7-1, B7-2, CD40, or CD40L, which are key co-stimulatory molecules for effective immune responses (125). The secretome of MSCs, a crucial paracrine regulator, has been demonstrated to impact fibrosis formation and wound healing through the release of vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), nerve growth factor (NGF), transforming growth factor-beta (TGF-β), and keratinocyte growth factor (KGF) (126), while orchestrating chemotaxis via CCL5, CCL8, and CXCL12 (127). The MSC secretome also includes factors with anti-viral [indoleamine 2,3-dioxygenase (IDO), prostaglandin E2 (PGE2), interferon-γ (IFN-γ)] and anti-bacterial effects [hepcidin AMP (HAMP), lipocalin 2 (LCN2), and beta-defensin-2 (BD2)] (128). Studies have shown that MSCs can release factors such as hepatocyte growth factor (HGF) (129), tissue inhibitor of metalloproteinase 1 and 2 (TIMP-1 and TIMP-2) (130), and brain-derived neurotrophic factor (BDNF) (131), which all contribute to the MSC-induced anti-apoptotic effects on tissue cells.
The multifaceted role of MSCs in tissue regeneration and immune modulation is further underscored by their capability to expand Tregs, linking their general cellular characteristics to potential therapeutic applications in immunotherapy (132). The absence of co-stimulatory molecules complements their role in immune regulation, particularly in the expansion of Tregs (133). This property is instrumental in preventing autoimmunity and in promoting immune tolerance, making MSCs a promising candidate for treating various autoimmune diseases and in transplant medicine (134). The secretome of MSCs plays a pivotal role in this context by mediating the immunosuppressive environment that fosters Treg expansion (135). These interactions between MSCs and Tregs highlight a therapeutic synergy that leverages the inherent regenerative and immunomodulatory capacities of MSCs. This could pave the way for innovative treatments that combine tissue repair and immunological balance, addressing the complex needs of patients with autoimmune disorders or those undergoing organ transplantation.
The unique properties of MSCs position them as allies in modulating immune responses in SOT patients. Pre-clinical studies have highlighted the ability of MSCs to induce graft tolerance in both kidney and heart transplantation. For example, the presence of MSCs promoted the expression of IL-17A+Foxp3+ cells and IL-17-producing IL-17A+Foxp3+ T cells (136). These cells are potent counterparts to proinflammatory Th17 cells, which have been implicated with transplant rejection (137). Further, IL-17A+Foxp3+ subsets have been shown to drive Tregs expansion, ultimately suppressing rejection reactions and promoting graft tolerance (136). Overall, this MSC-induced transformation led to reduced inflammation, which correlated with extended graft survival (Figure 2) (136).
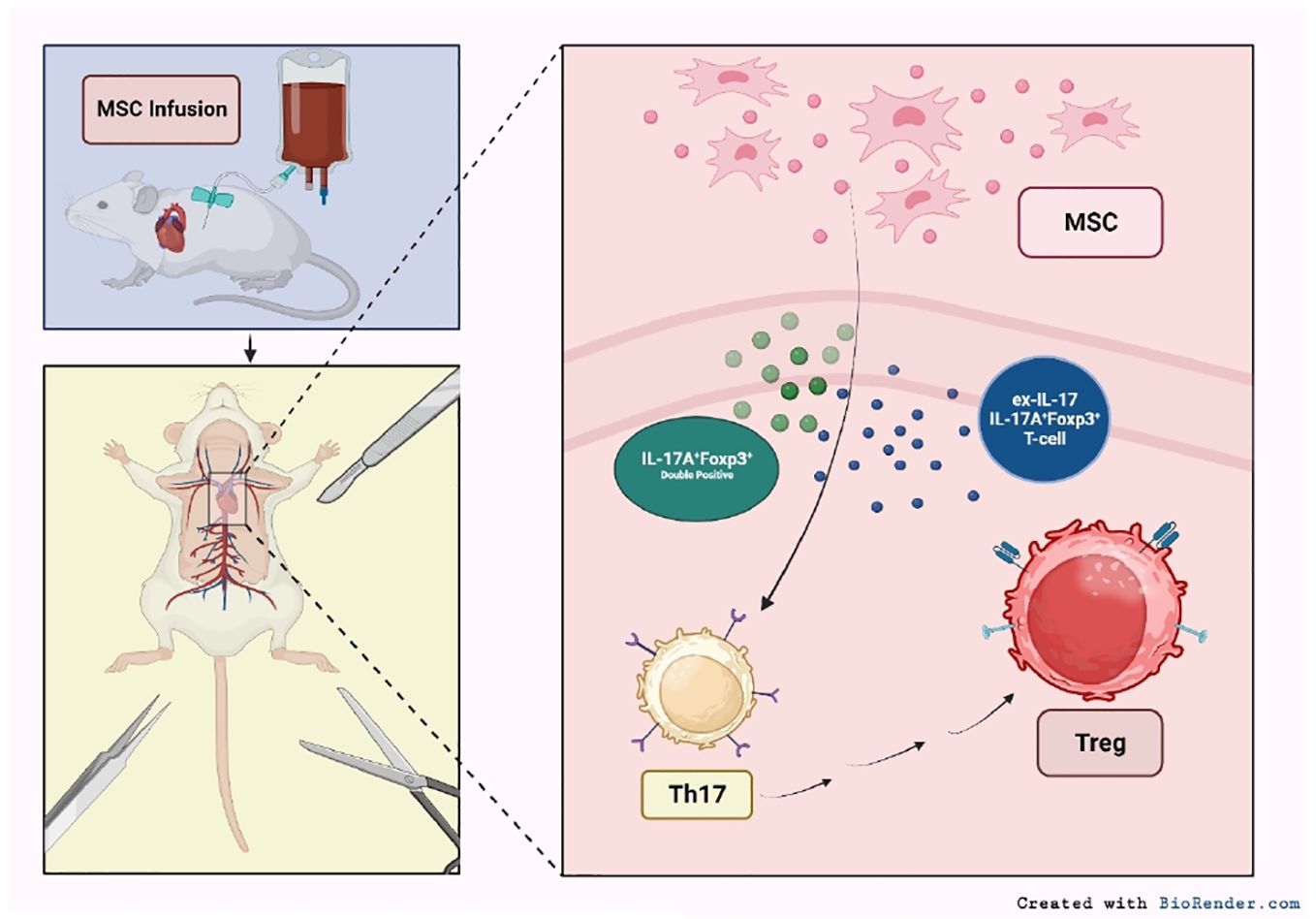
Figure 2 Mechanisms of MSC-mediated immune modulation in SOT. Infused MSCs interact with various immune cell populations, impacting Th17 and Tregs cell dynamics. The presence of MSCs further fosters the emergence of IL-17A+Foxp3+ cells and transitions IL-17-producing T cells to an IL-17A-Foxp3+ phenotype. This MSC-induced alteration facilitates the direct conversion of pro-inflammatory Th17 cells to anti-inflammatory Tregs (92).
Building upon these pre-clinical observations, research has expanded into the nuanced roles of MSCs in immune modulation. A review by English et al. explored the effects of BM-MSCs on the innate immune system, particularly that BM-MSCs inhibited dendritic cell migration by promoting the transformation of mature dendritic cells to a pro-tolerogenic form (138). When dendritic cells were co-cultured with BM-MSCs and tumor necrosis factor alpha (TNF-α), there was a significant suppression of CD86 and major histocompatibility complex class II (MHCII), both pivotal drivers of immune activation (139). Meanwhile, expression levels of CCR7 and E-cadherin mRNA diminished, which have been shown to impact dendritic cell motility and adhesion (140). However, the effects of BM-MSCs on dendritic cells were transient: once dendritic cells were separated from the co-culture and reintroduced to TNF-α, they elicited an immune response while showing weak immunosuppressive effects (140). This work underscores the temporary nature of MSC-mediated effects, highlighting the intricacy of their therapeutic application. Such studies mark a crucial step forward, bridging fundamental research with the complex realities of immune interactions in a clinical setting.
A living-donor kidney transplant recipient was successfully induced into immune tolerance through the remote administration of autologous BM-MSCs (141). This groundbreaking case, believed to be the first of its kind, demonstrated that modulating the host’s immune system with MSCs enabled the complete withdrawal of maintenance immunosuppressive drugs while preserving optimal long-term kidney allograft function. The patient, a 37-year-old male with end-stage renal disease, received a kidney transplant from his father. Before transplantation, he was administered a dose of autologous BM-MSCs and underwent a low-dose rabbit antithymocyte globulin (RATG) induction therapy post-transplant. Maintenance immunosuppression was initially managed with CsA, MMF, and steroids, which were gradually tapered off. Eighteen-months post-withdrawal, the patient remained free from antirejection therapy with stable kidney function (141). This report is significant as it not only showcases the potential of MSCs to induce a sustained pro-tolerogenic immune environment in kidney transplant recipients but also illustrates the feasibility of safely discontinuing antirejection drugs. This achievement marks a significant advancement in transplant medicine, offering insights into the mechanisms of immune regulation and tolerance induction. The patient’s immune profile showed a high ratio of Tregs to memory CD8+ T cells, expansion of naïve and transitional B cells, and donor-specific T cell unresponsiveness, indicating a strong and lasting pro-tolerogenic milieu (141).
Additionally, Azevedo et al. explored the capability of MSCs to expand Tregs in vitro, aiming to support future clinical trials for immune tolerance therapies (132). Utilizing allogeneic BM-MSCs co-cultured with human peripheral blood mononuclear cells from healthy donors, the research found a significant increase in CD4+CD25high Foxp3+ CD127low Treg cells after 14 days. This increase is attributed not to the proliferation of natural Tregs but to the conversion of conventional CD4 T cells into Treg-like cells in the presence of MSCs (132). These Treg-like cells demonstrated suppressive capabilities akin to natural Tregs and exhibited a DNA methylation profile closer to natural Tregs, suggesting a stable conversion. The study points to mechanisms involving TGF-β and/or PD-1/PDL-1 expression in the induction process. This MSC-induced Treg population closely resembles natural Tregs in phenotype, suppressive ability, and methylation profile (132).
Interestingly, another study investigating the immunosuppressive properties of MSCs derived from fetal liver (FL) and adult BM, reveals a compelling differentiation in the capabilities of FL-MSCs (142). While both FL and BM-MSCs exhibit similar phenotypic profiles and differentiation capacities, FL-MSCs display significantly higher proliferative abilities and more potent suppression of CD4+ and CD8+ T cell proliferation. This suppression is accompanied by a notable increase in functional CD4+CD25+Foxp3+ Tregs when compared to BM-MSCs (142). The findings underscore the unique immunosuppressive activity of FL-MSCs, which not only directly suppress T cell proliferation but also indirectly enhance Treg induction. This dual mechanism suggests FL-MSCs as a promising cell therapy source for treating immune-mediated diseases and preventing allograft rejection (142). The study highlights the advanced proliferative and immunosuppressive capabilities of FL-MSCs over adult BM-MSCs, emphasizing the potential of FL-MSCs in creating a favorable immunomodulatory environment for Treg expansion, which could be pivotal for future clinical applications in immune tolerance.
Transitioning to clinical investigations, the safety and therapeutic potential of umbilical cord-derived MSCs (UC-MSCs) were studied in a cohort of 27 liver transplant patients (143). Upon receiving a single infusion of UC-MSCs (1x106/kg body weight), the patients demonstrated a pronounced expansion of peripheral Tregs over 4 weeks. Notably, there was an elevation in the levels of PGE2 and TGFβ-1, key mediators for attenuating immune responses and fostering tissue repair. Histological analysis post-infusion showed a significant reduction in the total rejection activity index (RAI) scores in 43% of patients (143). Further findings from this study revealed that UC-MSC treatment led to marked declines in liver damage indicators such as alanine transaminase (ALT), aspartate aminotransferase (AST), and total bilirubin (TBIL). Additionally, there was an upregulation in Tregs, a downregulation in Th17 cells, and a suppression of CD4+ T cell activation, as evidenced by decreased HLA-DR expression. Plasma levels of TGF-β1 and PGE2, both integral to immunoregulation, substantially increased 4-weeks post-infusion (143). These findings underscore the potential therapeutic benefits of UC-MSCs, indicating not only their safety but also their effectiveness in alleviating liver damage and modulating immune responses in liver transplant recipients with acute rejection.
Reinders et al. conducted additional clinical investigations to assess the effectiveness and safety of MSC therapy in renal transplantation (144). They carried out a randomized, prospective, single-center, open-label trial to compare the outcomes of post-transplant MSC infusion with those of a control group receiving standard tacrolimus doses.
This investigation, designed to address the need for immunosuppressive regimens that effectively prevent allograft rejection without compromising renal function or leading to significant side effects, involved 70 patients who were initially randomized, with 57 ultimately included in the final analysis (29 in the MSC group and 28 in the control group) (144). The study’s primary endpoint was the quantitative assessment of interstitial fibrosis from biopsies taken at 4 and 24 weeks after transplantation, aiming to discern the potential benefits of MSC therapy in mitigating fibrosis compared to conventional immunosuppression strategies. Secondary endpoints encompassed a range of critical outcomes, including rates of acute rejection, graft loss, patient survival, renal function metrics, adverse events, and immunological responses, particularly focusing on the regulatory T cell populations as indicators of the immunomodulatory effects of MSC therapy (144). The results revealed that MSC therapy, combined with early tacrolimus withdrawal, did not demonstrate a statistically significant benefit in reducing interstitial fibrosis compared to the control group (p=.014). Renal function remained stable across both cohorts, with only one instance of acute rejection documented in the MSC group. Interestingly, regulatory T cell numbers were significantly higher in the MSC group at the 24-week mark, suggesting a potential immunomodulatory benefit of MSC therapy that warrants further investigation (144).
The Neptune study, a phase I single-center trial, explored the safety and feasibility of using allogeneic MSCs in renal transplantation, focusing on a novel approach of HLA-selected allogeneic MSC therapy combined with adjusted immunosuppression (145). This nonrandomized, prospective study involved ten patients who underwent renal transplantation from living donors. 6-months post-transplantation, these patients received two doses of 1.5×106/kg allogeneic MSCs. The immunosuppressive regimen included a reduction in tacrolimus to a trough level of 3 ng/mL, supplemented with everolimus and prednisone (145). The primary goal was to assess safety, particularly looking at biopsy-proven acute rejection (BPAR) and graft loss over a 12-month period following MSC infusion. Remarkably, the study reported no instances of BPAR or graft loss, and renal function remained stable throughout the follow-up period. Immune monitoring showed no significant alterations in T and B cell populations or plasma cytokine levels post-MSC infusion (145). Interestingly, one patient had pre-existing donor-specific antibodies (DSAs) against the MSCs, indicating sensitization prior to infusion. However, this did not impact the overall safety profile or renal function. The MSCs were carefully selected to prevent repeated HLA mismatches, minimizing the risk of an immune response against the allograft (145). This pioneering trial demonstrates that administering HLA-selected allogeneic MSCs, in conjunction with a tailored immunosuppressive regimen, is safe within the first year after renal transplantation. It lays the groundwork for further research into the efficacy and long-term outcomes of third-party MSC therapy in transplant medicine, offering a potential new avenue for enhancing graft survival and patient outcomes.
Subsequent research efforts have aimed to further elucidate the safety and efficacy of MSC therapy in kidney transplantation (146). An open-label phase I-II clinical trial involving 20 kidney transplant recipients who received kidneys from deceased donors were divided into two groups: one group received a single infusion of third-party bone marrow-derived MSCs (approximately 2-3 million MSCs per kilogram) on day 3 post-transplant, while a concurrent control group did not receive MSC therapy. The study monitored patients for adverse effects related to MSC infusion, opportunistic infections, acute rejection incidents, renal function, and the development of antibodies against MSCs or shared kidney-MSC HLA antigens (146). One of the key findings was the absence of significant adverse effects at MSC injection, although one participant with a history of cardiac disease experienced a non-ST-elevation myocardial infarction shortly after the MSC infusion. The incidence of opportunistic infections and acute rejections did not differ significantly between the MSC-treated group and the control group, suggesting that MSC therapy did not increase the risk of such complications (146). A notable observation was the early improvement in estimated glomerular filtration rate (eGFR) among MSC-treated recipients, reaching 48.6 ml/min/1.73m2 by day 7 post-transplant, compared to 32.5 ml/min/1.73m2 in controls and 29.3 ml/min/1.73m2 in the overall cohort of kidney transplant recipients at the same institution (146). This early improvement suggests a potential nephroprotective effect of MSC therapy, although no significant difference in eGFR was found at the one-year mark. Immunologically, the MSC-treated group exhibited increased frequencies of Tregs at day 30 post-transplant, without significant changes in B cell frequencies (146).
This observation points towards an immunomodulatory benefit of MSC therapy, possibly contributing to a more tolerant and less inflammatory post-transplant environment. Moreover, four participants in the MSC-treated group developed antibodies against MSC or shared kidney-MSC HLA antigens, with only one showing a mean fluorescence intensity (MFI) greater than 1500, indicating a relatively low level of immunization against the infused MSCs (147). The study concludes that a single infusion of third-party MSC following kidney transplantation is safe and feasible, with an observed cardiac event of unclear relation to the intervention. The MSC therapy was associated with improved early allograft function and increased regulatory T cell proportions, suggesting potential immunomodulatory benefits (148).
Expanding upon this, in another prospective, randomized control trial, 159 end-stage renal disease patients undergoing ABO-compatible, cross-match–negative kidney transplants received kidneys from living-related donors (148). The intervention involved inoculating patients with marrow-derived autologous MSCs (1-2 × 106/kg) at the time of kidney reperfusion and again two weeks later. The study was designed to assess whether autologous MSCs could replace antibody-based induction therapy, traditionally combined with CNIs, to reduce acute rejection rates while mitigating the risks of opportunistic infections and toxic effects associated with CNIs (15). The participants were divided into three groups: one received autologous MSCs plus standard-dose CNIs, another received MSCs plus low-dose CNIs (80% of the standard dose), and the control group was treated with anti–IL-2 receptor antibodies plus standard-dose CNIs (148). The primary outcome measured was the incidence of acute rejection and renal function, as indicated by the eGFR, within the first-year post-transplant. Secondary measures included patient and graft survival rates and the incidence of adverse events. The findings revealed compelling advantages of using autologous MSCs over traditional induction therapies (148). Acute rejection within six-months post-transplant was significantly lower in both MSC-treated groups compared to the control group, with 7.5% and 7.7% in the MSC groups versus 21.6% in the control group. Notably, none of the patients in the MSC groups experienced glucocorticoid-resistant rejection, which was observed in 7.8% of the control group (148). Furthermore, renal function recovery was faster among the MSC-treated patients, demonstrated by significantly increased eGFR levels in the first month post-surgery compared to the control group. The study also noted a reduced risk of opportunistic infections in the MSC-treated groups, highlighting the immunomodulatory benefits of MSC therapy (148). Patient and graft survival rates at 13 to 30 months were comparable across all groups, emphasizing the safety and feasibility of autologous MSC therapy in kidney transplantation. The incidence of adverse events was lower in the MSC groups, particularly regarding opportunistic infections, further underscoring the potential of MSC therapy to enhance transplant outcomes while minimizing the risks associated with standard immunosuppressive protocols (148).
Of note, the therapeutic utility of MSCs in SOTs depends on multiple factors including timing, the expansion behavior of MSCs, cell-cell interactions, and their transient effects on other immune cells (149, 150). Studies have shown that the timing of MSC administration could significantly influence their immunomodulatory abilities and transplant outcomes (147, 151). In fact, participants who were administered MSCs prior to kidney transplantation exhibited lower serum creatinine levels, ranging between 1.35-0.97 mg/dL and 1.37-1.49 mg/dL, in contrast to those who received post-transplant infusions, where levels ranged between 1.83-2.27 mg/dL and 2.38-2.21 mg/dL (147). Serum creatinine is a marker often used to evaluate kidney function—elevated levels can indicate impaired kidney function or kidney disease (147). Paralleling this finding, Casiraghi et al. have shown that introducing MSCs prior to transplantation could elevate their immunosuppressive effects, which may directly suppress acute graft complications (152).
Besides transplantation timing, the cellular profile of MSCs has been demonstrated to influence transplant outcomes. In a study using a murine kidney transplant model, mice underwent a single MSC infusion (0.5×106 cells) either before or after the transplant procedure (153). The post-transplant group (n=7) exhibited higher blood urea nitrogen (BUN) levels than the pre-transplant mice, pointing to possible renal dysfunction. Further, the authors observed significantly higher levels of neutrophils, complement C3 deposition, interleukin 6 (IL-6), and TNF-α in the post-transplant group on days 2 and 7 following MSC-infusion, indicating a prolonged inflammatory immune response (153). Conversely, pre-transplantation MSC administration resulted in an upregulation of Tregs and extended graft survival by more than 60 days. Furthermore, the study shed light on the MSC homing behavior. After infusion, MSCs first positioned themselves in the spleen and later migrated to the allograft (153). The migration process may influence immune modulation, potentially promoting graft tolerance and impacting cell-cell interactions crucial for graft acceptance or rejection via the Tregs axis.
Allogeneic BM-MSC transplantation, either co-transplanted with vascularized bone marrow or alone in a hemi-facial allotransplant model, significantly extended the survival of pig limb allografts when combined with irradiation and cyclosporine A (CsA) treatment (154). Moreover, repetitive high-dose BM-MSC treatment (>1x106 cells) extended the survival of pig hemi-facial transplant recipients by over 20 days, even in the absence of conditioning therapy; these grafts experienced only mild rejection (Grade I to II), which improved under CsA treatment (155). The beneficial effects of BM-MSCs on rejection grades were also associated with upregulated IL-10 expression and the induction of Tregs, underscoring the dampening effects BM-MSCs on the recipient immune response (156).
In another approach, Kuo et al. highlighted the effects of repeated AD-MSCs doses in promoting tolerance in hind limb rat VCA model and driving the proliferation of tolerance-inducing CD4+CD25+Foxp3+ Tregs (157). Conversely, a single dose of syngeneic AD-MSCs (2x106 cells) was shown to induce tolerance in a rat VCA model, even in the presence of donor-recipient MHC mismatch when administered preoperatively in combination with CsA and anti-lymphocyte globulin (157). Currently, there is no consensus on the dosing and timing of MSC administration in VCA protocols. Experimental studies have shown beneficial results with MSC dosages ranging from 5×105 to 5×107 cells per kg body weight, administered at various time points relative to transplantation (151). Overall, future research work is needed to standardize MSC-based therapies for VCA and SOT patients and balance the various cellular interactions of MSC populations. The complex cellular and cytokine network involving MSCs is shown in Figure 3.
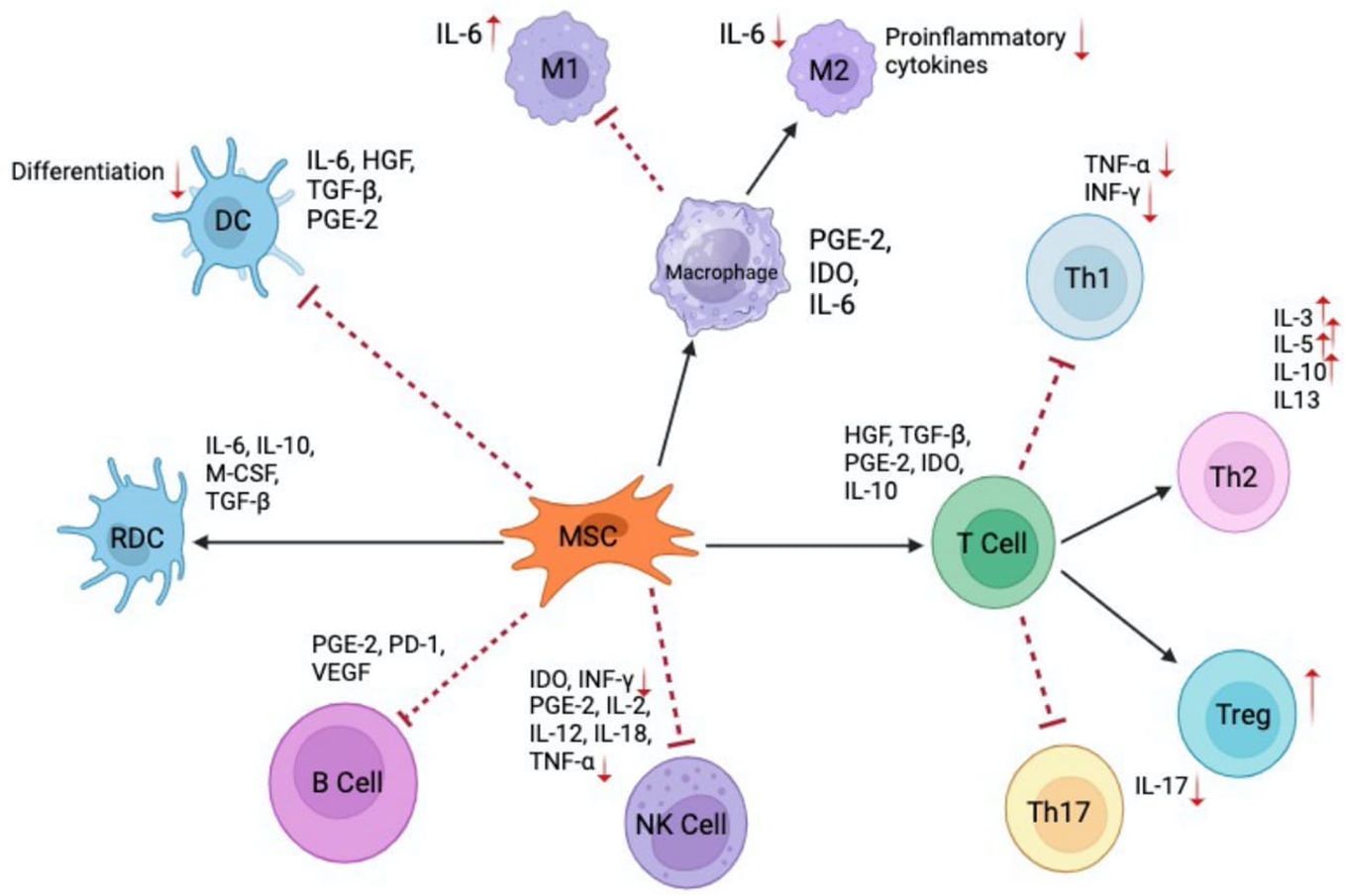
Figure 3 Interactions of MSCs with immune cells and their mediated cytokines. MSC illustrates the role in modulating various immune responses through cytokine secretion and direct interactions with multiple immune cell types. Dendritic cells (DC) and regulatory dendritic cells (RDC) and their differentiation modulated by cytokines such as 1L-6, HGF, TGF-β, and PGE2 (131). Macrophages (MI and M2) showcase the polarization of macrophages into Ml (pro-inflammatory) and M2 (anti-inflammatory) phenotypes in the presence of 1L-6 and other modulating factors. MSCs play a role in this differentiation through the release of cytokines like PGE-2, IDO, and 1L-6 (132). T Cells illustrates the various T cell subsets - Th1, Th2, Treg, and Th17, and their interactions with MSCs. Cytokines like HGF, TGF-β1, PGE-2, 1DO, and IL-10 play a role in modulating these T cell responses (133). MSCs impact B cells and natural killer (NK) cells through the secretion of mediators such as PGE-2, PD-1, VEGF for B cells and IDO, INF-γ, PGE-2, IL-2, IL-12, IL-18, and TNF-α for NK cells (134).
Another cellular strategy to mitigate alloreaction and improve transplant acceptance is based on the hypothesis that the co-transplantation of vascularized bone marrow (VBM) compartments (mandible, ulna, radius) generates a continuous source of donor-derived hematopoietic stem cells (158). In fact, this approach has shown promising potential to modulate the recipient’s immune system in different animal models. In one notable study, researchers investigated the effects of co-transplantation in rats, dividing them into groups where Group I received syngeneic transplants as a control, and Groups II-V underwent allogeneic transplants with receiving antilymphocyte globulin (5 mg) and tacrolimus (1 mg/kg), along with total body irradiation of 0, 600, 400, and 200 cGy respectively, prior to transplantation (159). On day 0, each Lewis rat in Groups II-V received a Brown Norway hind-limb osteomyocutaneous flap. The presence of different donor hematopoietic cell lineages in recipients’ blood was assessed on days 15, 30, 60, 90, 120, and 150 post-transplantation using flow cytometry. The researchers showed that allotransplanted hind-limb flaps in Groups III, IV, and V led to chimerism and donor multilineage hematopoietic cells, with acceptance rates of 37.5%, 16.7%, and 0% respectively (159).
Building on these findings, Barth et al. developed a nonhuman primate model of facial segment allotransplantation in cynomolgus monkeys (155). Heterotopically transplanted facial segment VCA with VBM (n=4) treated with tacrolimus and MMF demonstrated prolonged rejection-free survival between 205 and 430 days, compared to VCA without VBM that demonstrated early rejection episodes and graft loss by postoperative days 7–15168. Further investigation in a rat model that combined hemiabdominal wall and hindlimb osteomyocutaneous flaps with VBM transplantation revealed that a subset of recipients showed long-term allograft survival, developed donor-specific tolerance, and exhibited significantly higher levels of peripheral chimerism (160). This study also pointed out that a balanced ratio of allograft to VBM mass is crucial for optimizing chimerism and enhancing graft survival. The authors found that 3 of 8 VBM recipients showed long-term allograft survival of more than 100 days and developed donor-specific tolerance. Further, VBM groups demonstrated significantly higher peripheral chimerism, analyzed through flow cytometry. Interestingly, the authors reported that higher ratios of allograft to VBM mass correlated with lower levels of chimerism and reduced graft survival, underscoring the need for balancing VBM against VCA transplant weight (160).
However, translating these animal model successes to human clinical practice has been challenging. To date, evidence of long-term donor chimerism in patients undergoing hand, forearm, arm, or face transplantation, even with the inclusion of facial bone, is limited (161). There has been one reported case of a face transplant patient who, after receiving a mandible co-transplantation along with an infusion of hematopoietic stem cells, exhibited transient chimerism of CD34+ T-cells (162).
Beyond the cotransplantation of VBM, infusion of donor BM cells have shown positive outcomes in animal models (163, 164). Based on these experimental findings, Schneeberger et al. established the Pittsburgh Protocol (161). The authors enrolled 5 patients who either received a bilateral hand (n=2), a bilateral hand/forearm (n=1), or a unilateral (n=2) hand transplant. All patients were treated with alemtuzumab and methylprednisolone for induction therapy, followed by tacrolimus monotherapy as maintenance treatment. On day 14, patients received an infusion of donor BM cells isolated from the patients’ vertebral bodies. Immunomonitoring through flow cytometry revealed transient moderate levels of donor-specific antibodies, adequate immunocompetence, but no peripheral blood chimerism (161). While the authors concluded that the Pittsburgh Protocol represented a safe and well tolerated approach for low-dose tacrolimus monotherapy following upper extremity transplantation, their findings remain to be confirmed in long-term follow-up studies (161). Overall, the clinical effects of donor-derived bone marrow cells, either infused or as VBM cotransplant, still stand in sharp contrast to the promising in vitro and in vivo data, therefore warranting future research work.
RMCs, including macrophages, dendritic cells, and MDSCs, play pivotal roles in both immune regulation and tissue repair (165). They originate from diverse sources, ranging from the embryonic yolk sac to hematopoietic stem cells and bone marrow-derived monocytes (166). Beyond their immune-modulating functions, RMCs are instrumental in tissue regeneration, particularly within the TNFR1/TNF-α and IL-8 signaling pathways (165). They exhibit clinical promise in mitigating excessive inflammation through the release of IL-10 and TGF-β, orchestrating angiogenesis, and facilitating tissue remodeling via the modulation of pro-inflammatory and anti-inflammatory cytokines at different stages (34). This dual capacity of immune suppression and tissue repair positions RMCs as a promising avenue for advancements in transplantation research. A deeper understanding of RMC biology and function could unveil strategies to optimize VCA outcomes, bridging the gap between rejection and tolerance.
RMCs encompass subsets like Mregs, DCregs, and MDSCs. Mregs, akin to M1 macrophages, differentiate from monocytes in response to M-CSF and IFN-γ (167). They are characterized by their secretion of critical cytokines, including IL-10 and TGF-β (168). Conversely, DCregs play a central role in immune regulation by promoting Tregs development, balancing T cell responses and inducing T cell apoptosis, particularly in lactate-enriched microenvironments (49). MDSCs, known for their potent immunosuppressive capabilities, comprise multiple subtypes, each with distinct functional attributes (169). These subtypes, ranging from polymorphonuclear (pmnMDSC) to monocytic (mMDSC) and early-stage (eMDSC) cells, employ various mechanisms for immune suppression (170). Such mechanisms include cytokine secretion, depletion of essential amino acids like arginine and tryptophan, and the production of reactive oxygen and nitrogen species. Additionally, they demonstrate a dual role by inhibiting the actions of T cells and NK cells, while enhancing the functions of Tregs (171).
Researchers investigating the optimization of SOT have explored cellular mechanisms influencing rejection and tolerance. Central to this inquiry is the role of Mregs, which have been associated with significant advancements in graft acceptance and reduction of immunosuppression (172). Conde et al. identified critical determinants and signals altering RMC response following SOTs (173). They observed that the regulation of dendritic cell C-type lectin receptor (DC-SIGN) in macrophages through IFN-γ and colony stimulating factors (CSFs) led to an upregulation in CD4+ T cell expansion and a decline in CD8+ T cell populations. This effect can be attributed to either the manipulation of Fcγ-receptor (FcγR) ligation on activated macrophages or the modulation of DC-SIGN+ macrophages through IFN-γ and CSF1 (173). Further exploring this pathway, IFN-γ and macrophage colony stimulating factor (M-CSF) were crucial in generating Mregs that suppress polyclonal T cell proliferation via an inducible nitric oxide synthase (iNOS)-dependent mechanism (174).
To manipulate RMCs, researchers used double-stranded oligodeoxynucleotides (ODNs) to decrease nuclear factor kappa B (NF-κB)-driven gene activity and IL-4 production (175). Combining these modified cells with recombinant adenovirus (rAd) vectors created a hybrid termed rAd CTLA4-Ig/NF-κB ODN DCs. Compared to untreated allografts, these hybrid cells led to a significant 71-day extension in graft survival and decreased inflammation levels (measured by Th1 and Th2 cytokine levels) at a concentration of 2x106 cells per mouse (175). In parallel with these findings, administering a combination of 2x106 MDSCs and 3 mg/kg of rapamycin on postoperative days 0, 2, 4, and 6, resulted in markedly extended graft survival in mice with heart transplants (172). Interestingly, Mregs monotherapy led to significantly shorter graft survivals compared to the combination treatment (170).
Beyond their direct impact, Mregs can also modulate the activity of other immune cells. For instance, they have been shown to induce the conversion CD4+ T cells into Mregs-induced Tregs (miTregs), which release IL-10, thereby promoting the proliferation of Tregs populations (176). This distinct cell subset is characterized by butyrophilin like 8 (BTNL8) gene expression (177). Moreover, Mregs can stimulate the expression of T cell immunoreceptor with Ig and ITIM domains known as TIGIT+iTregs populations (177). Additionally, Mregs can secrete PGE2 serving as a crucial mediator for macrophages by promoting anti-inflammatory cytokine synthesis while suppressing pro-inflammatory molecules like TNF-α and IL-12 (178).
On the dendritic cell front, Cai et al. created immature DCregs derived from induced pluripotent stem cells (iPSCs) (179). They showed that a dosage of 1x106 iPS-DCregs sustained permanent allograft tolerance in a murine cardiac transplant model (179). In a phase I/II trial involving 12 liver transplant recipients, researchers discovered that injections of donor-derived DCregs decreased T cell proliferation, increased Tregs populations, and balanced the IL-10-to-IL-12 ratio post CD40Ld activation, ultimately driving transplant tolerance (84). Histological samples from study participants (n=10) showed a programmed death ligand-1 (PD-L1) to CD86 ratio exceeding 2.5, which has been proposed as another marker of transplant tolerance (84).
The role of MDSCs in regulating immune responses is well documented. They suppress the proliferation of Teffs without affecting their activation, mediated by the iNOS enzyme (180). Furthermore, there is an association between MDSCs and increased Treg levels, alongside decreased IL-17 production (136). Additional research has confirmed the correlation between MDSCs and Treg accumulation, with a specific MDSC subset, CD11b+CD33+HLA-DR-/lo, promoting the expansion of naive CD4+Foxp3+ Tregs in vitro (181). This expansion also correlated with a rise in cytokines such as tumor growth factor beta-1 (TGF-β1) and IL-10 (41). Interestingly, the immunosuppressive ability of this MDSC subset was only observed in renal transplant recipients but not in non-transplant controls (182). They further reported that, among MDSCs, CD14+ mononuclear (M)-MDSCs exhibited more potent immunosuppressive effects than CD14+Granulocyte-like MDSCs (G-MDSCs), even though both subsets increased in transplant recipients (182).
Immediate post-transplant introduction of MDSCs has been associated with prolonged graft survival of more than 19 days when combined with anti-CD154 monoclonal antibodies (183). However, MDSC therapy has also been implicated with increased risk of malignancies. Utero-Rico et al. examined 229 renal transplant recipients, focusing on the relationship between certain myeloid cells and cancer risk (184). The results indicated that high M-MDSC levels, especially surpassing >179.2 per microliter on day 14 post-transplant, correlated with a sevenfold increase in all-types cancer risk theoretically by predisposing to an autoimmunoreactive environment (184). Interestingly, prolonged elevated M-MDSC levels were also correlated with stronger immunosuppressive action and reduced antioxidant activity (184). Thus, while MDSCs seem promising for early post-transplant care, their extended application warrants further investigation and closer perioperative monitoring.
Cellular therapies have emerged as promising alternatives to current immunosuppressive drugs in transplantation. Their versatility and specificity offer an opportunity to precisely regulate the immune system, potentially extend graft longevity, and reduce the dosages of conventional immunosuppressive regimens. These therapies, representing a fusion of immunological insights and bioengineering innovations, mark a paradigm shift from broad-spectrum immunosuppression to a tailored immunomodulatory strategy, potentially minimizing the adverse effects associated with conventional regimens.
Within the expanding framework of SOTs and VCAs, cellular therapies are illuminating pathways towards optimizing perioperative care in transplantation. From augmenting the immunoregulatory capabilities of Tregs, to leveraging the potent immunosuppressive effects of MSCs, and capitalizing on the distinctive capabilities of CAR-T cells, these methodologies offer a sophisticated approach to inducing immune tolerance, averting graft rejection, and facilitating robust graft integration. Furthermore, certain microenvironmental factors implicated with poor transplant function, such as elevated lactate levels, could be leveraged to potentiate the immune-modulating properties of these cell therapeutics (49). The current body of knowledge suggests that the multifaceted immunomodulatory roles of cellular therapies may advance perioperative patient management following SOTs and VCAs.
As discussed, the combination of various cell types may strengthen the therapeutic effects by compensating for cell-specific limitations and leveraging cell-specific tolerance-inducing capacities. The transplant microenvironment and timing of cell therapeutics represent understudied but promising angles for advancing peri-transplant care (147, 149–151). Broadening the pool of cell subsets with immunosuppressive effects could further pave the way towards more individualized cell therapeutics. While these strategies carry promising potential for both SOT and VCA, future VCA-specific trials are needed to bridge the research gap between SOT and the emerging field of VCA surgery.
Despite these promising advances, the clinical translation of cellular therapies into routine transplantation care faces significant hurdles. The challenges span the need for standardized protocols for cell isolation, expansion, and infusion, to a deeper understanding of the molecular and cellular mechanisms underpinning graft rejection and tolerance to further advance cellular therapeutics. Moreover, the identification of reliable biomarkers for monitoring therapeutic efficacy, understanding the optimal timing for therapy administration, and unraveling the synergistic potential between cellular and conventional therapies remain critical areas for future research to determine the most effective ratio of cellular to conventional immunosuppressive regimens.
Tr1 cell therapy has shown promising potential in transplantation to improve graft acceptance and patient outcomes, representing a significant step toward more targeted and less toxic immunomodulation strategies (37, 42). However, the need for specific Tr1-cell biomarkers, enhanced in vitro expansion techniques, and a deeper understanding of their regulatory mechanisms pose challenges that future research must address to fully harness the therapeutic potential of Tr1 cells in clinical practice. The innovative concept of incorporating Tr1 cells into CAR constructs could further refine their specificity and functionality (38, 47), representing an exciting frontier for future therapeutic development in this field.
As CAR-T cell therapies emerge as promising and innovative modalities for enhancing transplant acceptance and reducing reliance on conventional immunosuppression, their integration into transplant care paradigms requires careful consideration and mitigation of multifaceted challenges and limitations (62, 68, 103–115, 185). Rigorous preclinical investigations, well-designed clinical trials, and ongoing research is imperative to comprehensively elucidate the therapeutic potential, safety profiles, and outcomes of CAR-T cell therapies in SOTs and VCA. Additionally, the careful selection of target antigens is critical to minimizing the risk of adverse events and ensuring the safety of CAR-T cell-based treatments.
Further, the complex pharmacological interplay between the patient’s standard immunosuppressants and CAR-T cells remains to be fully elucidated. Personalized therapies, such as CAR constructs, offers both benefits and limitations. While personalized therapy holds the potential to tailor treatments to individual patients, providing more targeted and effective interventions, it also requires time for customization that off-the-shelf therapies do not. As medicine increasingly adopts personalized therapeutic approaches, balancing the benefits of tailored treatments with the time required for their development becomes essential.
In this comprehensive review, we analyzed the dual impact of cellular therapies on SOTs and VCA. Overall, this advanced understanding of cellular therapies could lead to more targeted perioperative treatment strategies in SOTs and VCAs. While these therapies have demonstrated positive clinical outcomes in transplant patients, their standardized integration into transplant care requires further investigation in future trials. This review underscores the potential of cellular therapies to revolutionize transplant surgery, emphasizing the need for rigorous scientific inquiry and systematic evaluation to validate their efficacy, safety, and feasibility on a broader scale. Moving forward, continued research efforts are essential to elucidate the precise role of cellular therapies in transplant medicine and to address the logistical and ethical challenges associated with their widespread adoption.
LK: Conceptualization, Writing – original draft, Writing – review & editing. JD: Writing – original draft, Writing – review & editing. FD: Writing – original draft. NT: Writing – original draft. SK: Writing – original draft, Writing – review & editing. RR: Data curation, Writing – review & editing. KS: Writing – original draft. TE: Writing – original draft. SM: Writing – original draft. FF: Writing – original draft. KK: Writing – original draft. AP: Writing – original draft. JI: Writing – original draft. A-FS: Writing – original draft. ST: Writing – review & editing, Supervision, Validation. SH: Writing – review & editing. BP: Conceptualization, Project administration, Supervision, Writing – review & editing. MK-N: Conceptualization, Project administration, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer GM declared a shared affiliation, though no other collaboration, with one of the authors JI at the time of the review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kauke-Navarro M, Noel OF, Knoedler L, Knoedler S, Panayi AC, et al. Novel strategies in transplantation: genetic engineering and vascularized composite allotransplantation. J Surg Res. (2023) 291:176–86. doi: 10.1016/j.jss.2023.04.028
2. Starzl R, Brandacher G, Lee WPA, Carbonell J, Zhang W, et al. Review of the early diagnoses and assessment of rejection in vascularized composite allotransplantation. Clin Dev Immunol. (2013) 2013:1–9. doi: 10.1155/2013/402980
3. Chim H, Amer H, Mardini S, Moran SL. Vascularized composite allotransplant in the realm of regenerative plastic surgery. Mayo Clinic Proc. (2014) 89:1009–20. doi: 10.1016/j.mayocp.2014.05.009
4. Kauke M, Safi A-F, Panayi AC, Palmer WJ, Haug V, et al. A systematic review of immunomodulatory strategies used in skin-containing preclinical vascularized composite allotransplant models. J Plastic Reconstructive Aesthetic Surg. (2022) 75:586–604. doi: 10.1016/j.bjps.2021.11.003
5. Ruiz R, Kirk AD. Long-term toxicity of immunosuppressive therapy. Transplant Liver. (2015), 1354–63. doi: 10.1016/B978-1-4557-0268-8.00097-X
6. Kauke M, Panayi AC, Safi AF, Haug V, Perry B, et al. Full facial retransplantation in a female patient—Technical, immunologic, and clinical considerations. Am J Transplant. (2021) 21:3472–80. doi: 10.1111/ajt.16696
7. Kauke-Navarro M, Knoedler S, Panayi AC, Knoedler L, Haller B, et al. Correlation between facial vascularized composite allotransplantation rejection and laboratory markers: Insights from a retrospective study of eight patients. J Plastic Reconstructive Aesthetic Surg. (2023) 83:155–64. doi: 10.1016/j.bjps.2023.04.050
8. Rodriguez-Rodriguez AE, Porrini E, Torres A. Beta-cell dysfunction induced by tacrolimus: A way to explain type 2 diabetes? IJMS. (2021) 22:10311. doi: 10.3390/ijms221910311
9. Luan FL, Steffick DE, Ojo AO. New-onset diabetes mellitus in kidney transplant recipients discharged on steroid-free immunosuppression. Transplantation. (2011) 91:334–41. doi: 10.1097/TP.0b013e318203c25f
10. Issa NC, Fishman JA. Infectious complications of antilymphocyte therapies in solid organ transplantation. Clin Infect Dis. (2009) 48:772–86. doi: 10.1086/597089
11. Rodríguez-Perálvarez M, Colmenero J, González A, Gastaca M, Curell A, et al. Cumulative exposure to tacrolimus and incidence of cancer after liver transplantation. Am J Transplant. (2022) 22:1671–82. doi: 10.1111/ajt.17021
12. Knoedler L, Knoedler S, Panayi AC, Lee CAA, Sadigh S, et al. Cellular activation pathways and interaction networks in vascularized composite allotransplantation. Front Immunol. (2023) 14:1179355. doi: 10.3389/fimmu.2023.1179355
13. Hussain Y, Khan H. Immunosuppressive drugs. Encyclopedia Infection Immun. (2022), 726–40. doi: 10.1016/B978-0-12-818731-9.00068-9
14. Geissler EK, Hutchinson JA. Cell therapy as a strategy to minimize maintenance immunosuppression in solid organ transplant recipients. Curr Opin Organ Transplant. (2013) 18:408–15. doi: 10.1097/MOT.0b013e328363319d
15. Chouhan KK, Zhang R. Antibody induction therapy in adult kidney transplantation: A controversy continues. World J Transplant. (2012) 2:19–26. doi: 10.5500/wjt.v2.i2.19
16. Delgado JF, Vaqueriza D, Sánchez V, Escribano P, Ruiz-Cano M, et al. Induction treatment with monoclonal antibodies for heart transplantation. Transplant Rev (Orlando Fla.). (2011) 25:21–6. doi: 10.1016/j.trre.2010.10.002
17. Bamoulid J, Staeck O, Crépin T, Halleck F, Saas P, et al. Anti-thymocyte globulins in kidney transplantation: focus on current indications and long-term immunological side effects. Nephrology dialysis transplantation: Off Publ Eur Dialysis Transplant Assoc - Eur Renal Assoc. (2017) 32:1601–8. doi: 10.1093/ndt/gfw368
18. Penaranda C, Tang Q, Bluestone JA. Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. J Immunol. (2011) 187:2015–22. doi: 10.4049/jimmunol.1100713
19. Organ Procurement and Transplantation Network (OPTN), Scientific Registry of Transplant Recipients (SRTR). OPTN/SRTR 2021 Annual Data Report. U.S. Department of Health and Human Services, Durham, NC, USA: Health Resources and Services Administration (2023).
20. Kauke M, Safi AF, Zhegibe A, Haug V, Kollar B, et al. Mucosa and rejection in facial vascularized composite allotransplantation: A systematic review. Transplantation. (2020) 104:2616–24. doi: 10.1097/TP.0000000000003171
21. Leonard DA, Amin KR, Giele H, Fildes JE, Wong JKF. Skin immunology and rejection in VCA and organ transplantation. Curr Transpl Rep. (2020) 7:251–9. doi: 10.1007/s40472-020-00310-1
22. Marghoob B, Rahimian N, Ataiepour Y, Mahdifarani M, Nejatifar M, Kabir A. Comparing the effect of induction therapy with or without antithymocyte globulin on renal allograft outcomes in live-donor kidney transplant recipients. Med J Islamic Republic Iran. (2019) 33:141. doi: 10.34171/mjiri.33.141
23. Kueckelhaus M, Fischer S, Seyda M, Bueno EM, Aycart MA, Alhefzi M, et al. Vascularized composite allotransplantation: current standards and novel approaches to prevent acute rejection and chronic allograft deterioration. Transplant international: Off J Eur Soc Organ Transplant. (2016) 29:655–62. doi: 10.1111/tri.12652
24. Yang JH, Johnson AC, Colakoglu S, Huang CA, Mathes DW. Clinical and preclinical tolerance protocols for vascularized composite allograft transplantation. Arch Plast Surg. (2021) 48:703–13. doi: 10.5999/aps.2021.00927
25. Sarhane KA, Tuffaha SH, Broyles JM, Ibrahim AE, Khalifian S, Baltodano P, et al. A critical analysis of rejection in vascularized composite allotransplantation: clinical, cellular and molecular aspects, current challenges, and novel concepts. Front Immunol. (2013) 4:406. doi: 10.3389/fimmu.2013.00406
26. Hoogduijn MJ, Issa F, Casiraghi F, Reinders MEJ. Cellular therapies in organ transplantation. Transpl Int. (2021) 34:233–44. doi: 10.1111/tri.13789
27. Siemionow M, Cwykiel J, Brodowska S, Chambily L. Human multi-chimeric cell (HMCC) therapy as a novel approach for tolerance induction in transplantation. Stem Cell Rev Rep. (2023) 19:2741–55. doi: 10.1007/s12015-023-10608-z
28. Chua CYX, Jiang AY, Eufrásio-da-Silva T, Dolatshahi-Pirouz A, Langer R, et al. Emerging immunomodulatory strategies for cell therapeutics. Trends Biotechnol. (2023) 41:358–73. doi: 10.1016/j.tibtech.2022.11.008
29. Matar AJ, Crepeau RL, Duran-Struuck R. Cellular immunotherapies in preclinical large animal models of transplantation. Transplant Cell Ther. (2021) 27:36–44. doi: 10.1016/j.bbmt.2020.09.032
30. Kline K, Chen W, Kallen ME, Koka R, Omili D, Fan X, et al. Chimeric antigen receptor (CAR) T cells for the treatment of a kidney transplant patient with post-transplant lymphoproliferative disorder (PTLD). Hum Vaccines immunotherapeutics. (2023) 19:2216116. doi: 10.1080/21645515.2023.2216116
31. Oren D, DeFilippis EM, Lotan D, Clerkin KJ, Fried J, Reshef R, et al. Successful CAR T cell therapy in a heart and kidney transplant recipient with refractory PTLD. JACC CardioOncology. (2022) 4:713–6. doi: 10.1016/j.jaccao.2022.09.002
32. Podestà MA, Remuzzi G, Casiraghi F. Mesenchymal stromal cells for transplant tolerance. Front Immunol. (2019) 10:1287. doi: 10.3389/fimmu.2019.01287
33. Podestà MA, Remuzzi G, Casiraghi F. Mesenchymal stromal cell therapy in solid organ transplantation. Front Immunol. (2021) 11:618243. doi: 10.3389/fimmu.2020.618243
34. Rosborough BR, Raïch-Regué D, Turnquist HR, Thomson AW. Regulatory myeloid cells in transplantation. Transplantation. (2014) 97:367–79. doi: 10.1097/TP.0b013e3182a860de
35. Lei H, Schmidt-Bleek K, Dienelt A, Reinke P, Volk HD. Regulatory T cell-mediated anti-inflammatory effects promote successful tissue repair in both indirect and direct manners. Front Pharmacol. (2015) 6:184. doi: 10.3389/fphar.2015.00184
36. Workman CJ, Szymczak-Workman AL, Collison LW, Pillai MR, Vignali DA. The development and function of regulatory T cells. Cell Mol Life sciences: CMLS. (2009) 66:2603–22. doi: 10.1007/s00018-009-0026-2
37. Szurek E, Cebula A, Wojciech L, Pietrzak M, Rempala G, et al. Differences in expression level of helios and neuropilin-1 do not distinguish thymus-derived from extrathymically-induced CD4+Foxp3+ Regulatory T cells. PloS One. (2015) 10:e0141161. doi: 10.1371/journal.pone.0141161
38. Wing JB, Tanaka A, Sakaguchi S. Human FOXP3+ Regulatory T cell heterogeneity and function in autoimmunity and cancer. Immunity. (2019) 50:302–16. doi: 10.1016/j.immuni.2019.01.020
39. Jørgensen N, Persson G, Hviid TVF. The tolerogenic function of regulatory T cells in pregnancy and cancer. Front Immunol. (2019) 10:911. doi: 10.3389/fimmu.2019.00911
40. Yadav M, Stephan S, Bluestone JA. Peripherally induced tregs - role in immune homeostasis and autoimmunity. Front Immunol. (2013) 4:232. doi: 10.3389/fimmu.2013.00232
41. Rocamora-Reverte L, Melzer FL, Würzner R, Weinberger B. The complex role of regulatory T cells in immunity and aging. Front Immunol. (2021) 11:616949. doi: 10.3389/fimmu.2020.616949
42. Freeborn RA, Strubbe S, Roncarolo MG. Type 1 regulatory T cell-mediated tolerance in health and disease. Front Immunol. (2022) 13:1032575. doi: 10.3389/fimmu.2022.1032575
43. Stern JNH, Keskin DB, Zhang H, Lv H, Kato Z, et al. Amino acid copolymer-specific IL-10-secreting regulatory T cells that ameliorate autoimmune diseases in mice. Proc Natl Acad Sci United States America. (2008) 105:5172–6. doi: 10.1073/pnas.0712131105
44. Bluestone J, Thomson A, Shevach E, Weiner HL. What does the future hold for cell-based tolerogenic therapy? Nat Rev Immunol. (2007) 7:650–4. doi: 10.1038/nri2137
45. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M, Gagliani N. The biology of T regulatory type 1 cells and their therapeutic application in immune-mediated diseases. Immunity. (2018) 49:1004–19. doi: 10.1016/j.immuni.2018.12.001
46. Arteaga-Cruz S, Cortés-Hernández A, Alvarez-Salazar EK, Rosas-Cortina K, Aguilera-Sandoval C, Morales-Buenrostro LE, et al. Highly purified and functionally stable in vitro expanded allospecific Tr1 cells expressing immunosuppressive graft-homing receptors as new candidates for cell therapy in solid organ transplantation. Front Immunol. (2023) 14:1062456. doi: 10.3389/fimmu.2023.1062456
47. Song Y, Wang N, Chen L, Fang L. Tr1 cells as a key regulator for maintaining immune homeostasis in transplantation. Front Immunol. (2021) 12:671579. doi: 10.3389/fimmu.2021.671579
48. Chihara N, Madi A, Karwacz K, Awasthi A, Kuchroo VK. Differentiation and characterization of tr1 cells. Curr Protoc Immunol. (2016) 113:113. doi: 10.1002/0471142735.im0327s113
49. Kushwah R, Hu J. Role of dendritic cells in the induction of regulatory T cells. Cell bioscience. (2011) 1:20. doi: 10.1186/2045-3701-1-20
50. Jofra T, Fonte R, Galvani G, Kuka M, Iannacone M, et al. Tr1 cell immunotherapy promotes transplant tolerance via de novo Tr1 cell induction in mice and is safe and effective during acute viral infection. Eur J Immunol. (2018) 48:1389–99. doi: 10.1002/eji.201747316
51. Gagliani N, Jofra T, Stabilini A, Valle A, Atkinson M, et al. Antigen-specific dependence of Tr1-cell therapy in preclinical models of islet transplant. Diabetes. (2010) 59:433–9. doi: 10.2337/db09-1168
52. Martínez-Pasamar S, Martínez-Forero I, Villoslada P. Tr1 cells in the neat sighting-pole. Inmunología. (2008) 27:78–84. doi: 10.1016/S0213-9626(08)70054-4
53. Etxebarria A, Díez-Martín E, Astigarraga E, Barreda-Gómez G. Role of the immune system in renal transplantation, types of response, technical approaches and current challenges. Immuno. (2022) 2:548–70. doi: 10.3390/immuno2040035
54. Tang Q, Bluestone JA. Regulatory T-cell therapy in transplantation: moving to the clinic. Cold Spring Harbor Perspect Med. (2013) 3:a015552. doi: 10.1101/cshperspect.a015552
55. Gregori S, Roncarolo MG, Bacchetta R. Methods for in vitro generation of human type 1 regulatory T cells. Methods Mol Biol. (2011) 677:31–46. doi: 10.1007/978-1-60761-869-0_3
56. Gregori S, Roncarolo MG. Engineered T regulatory type 1 cells for clinical application. Front Immunol. (2018) 9:233. doi: 10.3389/fimmu.2018.00233
57. Bacchetta R, Lucarelli B, Sartirana C, Gregori S, Lupo Stanghellini MT, Miqueu P, et al. Immunological outcome in haploidentical-HSC transplanted patients treated with IL-10-anergized donor T cells. Front Immunol. (2014) 5:16. doi: 10.3389/fimmu.2014.00016
58. Clinicaltrials.gov (2017). Available online at: https://clinicaltrials.gov/study/NCT03198234.
59. Liu JM-H, Chen P, Uyeda MJ, Cieniewicz B, Sayitoglu EC, et al. Pre-clinical development and molecular characterization of an engineered type 1 regulatory T-cell product suitable for immunotherapy. Cytotherapy. (2021) 23:1017–28. doi: 10.1016/j.jcyt.2021.05.010
60. Iske J, Nian Y, Maenosono R, Maurer M, Sauer IM, Tullius SG. Composite tissue allotransplantation: opportunities and challenges. Cell Mol Immunol. (2019) 16:343–9. doi: 10.1038/s41423-019-0215-3
61. Sayitoglu EC, Freeborn RA, Roncarolo MG. The yin and yang of type 1 regulatory T cells: from discovery to clinical application. Front Immunol. (2021) 12:693105. doi: 10.3389/fimmu.2021.693105
62. Arjomandnejad M, Kopec AL, Keeler AM. CAR-T regulatory (CAR-treg) cells: engineering and applications. Biomedicines. (2022) 10:287. doi: 10.3390/biomedicines10020287
63. Sanders JM, Jeyamogan S, Mathew JM, Leventhal JR. Foxp3+ regulatory T cell therapy for tolerance in autoimmunity and solid organ transplantation. Front Immunol. (2022) 13:1055466. doi: 10.3389/fimmu.2022.1055466
64. Steiner R, Pilat N. The potential for Treg-enhancing therapies in transplantation. Clin Exp Immunol. (2023) 211:122–37. doi: 10.1093/cei/uxac118
65. Juneja T, Kazmi M, Mellace M, Saidi RF. Utilization of treg cells in solid organ transplantation. Front Immunol. (2022) 13:746889. doi: 10.3389/fimmu.2022.746889
66. Karlsson F, Robinson-Jackson SA, Gray L, Zhang S, Grisham MB. Ex vivo generation of regulatory T cells: characterization and therapeutic evaluation in a model of chronic colitis. Methods Mol Biol. (2011) 677:7–61. doi: 10.1007/978-1-60761-869-0_4
67. Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. biomark Res. (2017) 5:22. doi: 10.1186/s40364-017-0102-y
68. Sun M-Y, Li W, Chen W. Chimeric antigen receptor T cell and regulatory T cell therapy in non-oncology diseases: A narrative review of studies from 2017 to 2023. Hum Vaccines Immunotherapeutics. (2023) 19:2251839. doi: 10.1080/21645515.2023.2251839
69. Mathew JM, H.-Voss J, LeFever A, Konieczna I, Stratton C, et al. A phase I clinical trial with ex vivo expanded recipient regulatory T cells in living donor kidney transplants. Sci Rep. (2018) 8:7428. doi: 10.1038/s41598-018-25574-7
70. Roemhild A, Otto NM, Moll G, Abou-El-Enein M, Kaiser D, et al. Regulatory T cells for minimising immune suppression in kidney transplantation: phase I/IIa clinical trial. BMJ. (2020), m3734. doi: 10.1136/bmj.m3734
71. Sánchez-Fueyo A, Whitehouse G, Grageda N, Cramp ME, Lim TY, Romano M, et al. Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. Am J transplantation: Off J Am Soc Transplant Am Soc Transplant Surgeons. (2020) 20:1125–36. doi: 10.1111/ajt.15700
72. Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology. (2016) 64:632–43. doi: 10.1002/hep.28459
73. Sawitzki B, Harden PN, Reinke P, Moreau A, Hutchinson JA, Game DS, et al. Regulatory cell therapy in kidney transplantation (The ONE Study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet. (2020) 395:1627–39. doi: 10.1016/S0140-6736(20)30167-7
74. Tang Q, Leung J, Peng Y, Sanchez-Fueyo A, Lozano J-J, et al. Selective decrease of donor-reactive Tregs after liver transplantation limits Treg therapy for promoting allograft tolerance in humans. Sci Trans Med. (2022) 14:eabo2628. doi: 10.1126/scitranslmed.abo2628
75. Oberbauer R, Edinger M, Berlakovich G, Kalhs P, Worel N, et al. A prospective controlled trial to evaluate safety and efficacy of in vitro expanded recipient regulatory T cell therapy and tocilizumab together with donor bone marrow infusion in HLA-mismatched living donor kidney transplant recipients (Trex001). Front Med. (2021) 7:634260. doi: 10.3389/fmed.2020.634260
76. Hautz T, Brandacher G, Zelger B, Müller HG, Lee AW, et al. Indoleamine 2,3-dioxygenase and foxp3 expression in skin rejection of human hand allografts. Transplant Proc. (2009) 41:509–12. doi: 10.1016/j.transproceed.2009.01.008
77. Walsh PT, Taylor DK, Turka LA. Tregs and transplantation tolerance. J Clin Invest. (2004) 114:1398–403. doi: 10.1172/JCI23238
78. Guo H, Xun L, Zhang R, Hu F, Luan J, Lao K, et al. Stability and inhibitory function of Treg cells under inflammatory conditions in vitro. Exp Ther Med. (2019) 18:2443–50. doi: 10.3892/etm.2019.7873
79. Goswami TK, Singh M, Dhawan M, Mitra S, Emran TB, Rabaan AA, et al. Regulatory T cells (Tregs) and their therapeutic potential against autoimmune disorders - Advances and challenges. Hum Vaccines immunotherapeutics. (2022) 18:2035117. doi: 10.1080/21645515.2022.2035117
80. Hua J, Inomata T, Chen Y, Foulsham W, Stevenson W, Shiang T, et al. Pathological conversion of regulatory T cells is associated with loss of allotolerance. Sci Rep. (2018) 8:7059. doi: 10.1038/s41598-018-25384-x
81. Baeten P, Van Zeebroeck L, Kleinewietfeld M, Hellings N, Broux B. Improving the efficacy of regulatory T cell therapy. Clinic Rev Allerg Immunol. (2022) 62:363–81. doi: 10.1007/s12016-021-08866-1
82. Stucchi A, Maspes F, Montee-Rodrigues E, Fousteri G. Engineered Treg cells: The heir to the throne of immunotherapy. J Autoimmun. (2023), 102986. doi: 10.1016/j.jaut.2022.102986
83. Wieckiewicz J, Goto R, Wood KJ. T regulatory cells and the control of alloimmunity: from characterisation to clinical application. Curr Opin Immunol. (2010) 22:662–8. doi: 10.1016/j.coi.2010.08.011
84. Ni X, Wang Q, Gu J, Lu L. Clinical and basic research progress on Treg-induced immune tolerance in liver transplantation. Front Immunol. (2021) 12:535012. doi: 10.3389/fimmu.2021.535012
85. Li R, Li H, Yang X, Hu H, Liu P, Liu H. Crosstalk between dendritic cells and regulatory T cells: Protective effect and therapeutic potential in multiple sclerosis. Front Immunol. (2022) 13:970508. doi: 10.3389/fimmu.2022.970508
86. McGovern JL, Wright GP, Stauss HJ. Engineering specificity and function of therapeutic regulatory T cells. Front Immunol. (2017) 8:1517. doi: 10.3389/fimmu.2017.01517
87. Shi H, Hongbo C. Metabolic control of Treg cell stability, plasticity, and tissue-specific heterogeneity. Front Immunol. (2019) 10:2716. doi: 10.3389/fimmu.2019.02716
88. Sojka DK, Huang YH, Fowell DJ. Mechanisms of regulatory T-cell suppression - a diverse arsenal for a moving target. Immunology. (2008) 124:13–22. doi: 10.1111/j.1365-2567.2008.02813.x
89. Zhang X, Zhu L, Zhang H, Chen S, Xiao Y. CAR-T cell therapy in hematological Malignancies: current opportunities and challenges. Front Immunol. (2022) 13:927153. doi: 10.3389/fimmu.2022.927153
90. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. (2020) 17:147–67. doi: 10.1038/s41571-019-0297-y
91. Meng X, Jing R, Qian L, Zhou C, Sun J. Engineering cytoplasmic signaling of CD28ζ CARs for improved therapeutic functions. Front Immunol. (2020) 11:1046. doi: 10.3389/fimmu.2020.01046
92. Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Trans Immunol. (2019) 8:e1049. doi: 10.1002/cti2.1049
93. Stoiber S, Cadilha BL, Benmebarek MR, Lesch S, Endres S, Kobold S. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells. (2019) 8:472. doi: 10.3390/cells8050472
94. Dagar G, Gupta A, Masoodi T, Nisar S, Merhi M, et al. Harnessing the potential of CAR-T cell therapy: progress, challenges, and future directions in hematological and solid tumor treatments. J Transl Med. (2023) 21:449. doi: 10.1186/s12967-023-04292-3
95. Cho JH, Okuma A, Sofjan K, Lee S, Collins JJ, et al. Engineering advanced logic and distributed computing in human CAR immune cells. Nat Commun. (2021) 12:792. doi: 10.1038/s41467-021-21078-7
96. Kimman T, Slomp A, Martens A, Grabherr S, Li S, van Diest E, et al. Serpin B9 controls tumor cell killing by CAR T cells. J immunotherapy Cancer. (2023) 11:e006364. doi: 10.1136/jitc-2022-006364
97. Xiao X, Huang S, Chen S, Wang Y, Sun Q, et al. Mechanisms of cytokine release syndrome and neurotoxicity of CAR T-cell therapy and associated prevention and management strategies. J Exp Clin Cancer Res. (2021) 40:367. doi: 10.1186/s13046-021-02148-6
98. Benmebarek MR, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci. (2019) 20:1283. doi: 10.3390/ijms20061283
99. Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med. (2019) 25:1341–55. doi: 10.1038/s41591-019-0564-6
100. Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. (2016) 4:92–101. doi: 10.1016/j.omtm.2016.12.006
101. Beheshti SA, Shamsasenjan K, Ahmadi M, Abbasi B. CAR Treg: A new approach in the treatment of autoimmune diseases. Int Immunopharmacol. (2022) 102:108409. doi: 10.1016/j.intimp.2021.108409
102. Kaljanac M, Abken H. Do treg speed up with CARs? Chimeric antigen receptor treg engineered to induce transplant tolerance. Transplantation. (2023) 107:74–85. doi: 10.1097/TP.0000000000004316
103. Gille I, Claas FHJ, Haasnoot GW, Heemskerk MHM, Heidt S. Chimeric antigen receptor (CAR) regulatory T-cells in solid organ transplantation. Front Immunol. (2022) 13:874157. doi: 10.3389/fimmu.2022.874157
104. Wagner JC, Tang Q. CAR-tregs as a strategy for inducing graft tolerance. Curr Transplant Rep. (2020) 7:205–14. doi: 10.1007/s40472-020-00285-z
105. Wagner JC, Ronin E, Ho P, Peng Y, Tang Q. Anti-HLA-A2-CAR Tregs prolong vascularized mouse heterotopic heart allograft survival. Am J transplantation: Off J Am Soc Transplant Am Soc Transplant Surgeons. (2022) 22:2237–45. doi: 10.1111/ajt.17063
106. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MAA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant. (2017) 17:931–43. doi: 10.1111/ajt.14185
107. Dawson NAJ, Lamarche C, Hoeppli RE, Bergqvist P, Fung VCW, McIver E, et al. Systematic testing and specificity mapping of alloantigen-specific chimeric antigen receptors in regulatory T cells. JCI Insight. (2019) 4:e123672. doi: 10.1172/jci.insight.123672
108. Sicard A, Lamarche C, Speck M, Wong M, Rosado-Sanchez I, Blois M, et al. Donor-specific chimeric antigen receptor Tregs limit rejection in naive but not sensitized allograft recipients. Am J Transplant. (2020) 20:1562–73. doi: 10.1111/ajt.15787
109. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. (2016) 126:1413–24. doi: 10.1172/JCI82771
110. Lee K, Nguyen V, Lee K-M, Kang S-M, Tang Q. Attenuation of donor-reactive T cells allows effective control of allograft rejection using regulatory T cell therapy. Am J Transplant. (2014) 14:27–38. doi: 10.1111/ajt.12509
111. Tang Q, Lee K. Regulatory T-cell therapy for transplantation: How many cells do we need? Curr Opin Organ Transplant. (2012) 17:349–54. doi: 10.1097/MOT.0b013e328355a992
112. Abraham AR, Maghsoudlou P, Copland DA, Nicholson LB, Dick AD. CAR-Treg cell therapies and their future potential in treating ocular autoimmune conditions. Front Ophthalmol. (2023) 3:1184937. doi: 10.3389/fopht.2023.1184937
113. Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of allograft rejection by use of regulatory T cells with an Mhc-specific chimeric antigen receptor. Am J Transplant. (2017) 17:917–30. doi: 10.1111/ajt.14175
114. Lamarche C, Ward-Hartstonge K, Mi T, Lin DTS, Huang Q, et al. Tonic-signaling chimeric antigen receptors drive human regulatory T cell exhaustion. Proc Natl Acad Sci United States America. (2023) 120:e2219086120. doi: 10.1073/pnas.2219086120
115. Kauke-Navarro M, Knoedler S, Panayi AC, Knoedler L, Noel OF, et al. Regulatory T cells: liquid and living precision medicine for the future of VCA. Transplantation. (2023) 107:86–97. doi: 10.1097/TP.0000000000004342
116. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. (2019) 16:372–85. doi: 10.1038/s41571-019-0184-6
117. Aparicio C, Acebal C, González-Vallinas M. Current approaches to develop “off-the-shelf” chimeric antigen receptor (CAR)-T cells for cancer treatment: a systematic review. Exp Hematol Oncol. (2023) 12:73. doi: 10.1186/s40164-023-00435-w
118. Gajra A, Zalenski A, Sannareddy A, Jeune-Smith Y, Kapinos K, Kansagra A. Barriers to chimeric antigen receptor T-cell (CAR-T) therapies in clinical practice. Pharm Med. (2022) 36:163–71. doi: 10.1007/s40290-022-00428-w
119. Holzinger A, Abken H. CAR T cells: A snapshot on the growing options to design a CAR. HemaSphere. (2019) 3:e172. doi: 10.1097/HS9.0000000000000172
120. Miao L, Zhang Z, Ren Z, Li Y. Reactions related to CAR-T cell therapy. Front Immunol. (2021) 12:663201. doi: 10.3389/fimmu.2021.663201
121. Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. (2019) 34:45–55. doi: 10.1016/j.blre.2018.11.002
122. Gumber D, Wang LD. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. EBioMedicine. (2022) 77:103941. doi: 10.1016/j.ebiom.2022.103941
123. Rosenbaum AJ, Grande DA, Dines JS. The use of mesenchymal stem cells in tissue engineering: A global assessment. Organogenesis. (2008) 4:23–7. doi: 10.4161/org.6048
124. Costela-Ruiz VJ, Melguizo-Rodríguez L, Bellotti C, Illescas-Montes R, Stanco D, Arciola CR, et al. Different sources of mesenchymal stem cells for tissue regeneration: A guide to identifying the most favorable one in orthopedics and dentistry applications. Int J Mol Sci. (2022) 23:6356. doi: 10.3390/ijms23116356
125. Han Z, Jing Y, Zhang S, Liu Y, Shi Y, Wei L. The role of immunosuppression of mesenchymal stem cells in tissue repair and tumor growth. Cell bioscience. (2012) 2:8. doi: 10.1186/2045-3701-2-8
126. Chang C, Yan J, Yao Z, Zhang C, Li X, Mao HQ. Effects of mesenchymal stem cell-derived paracrine signals and their delivery strategies. Advanced healthcare materials. (2021) 10:e2001689. doi: 10.1002/adhm.202001689
127. Zeng Z, Lan T, Wei Y, Wei X. CCL5/CCR5 axis in human diseases and related treatments. Genes Dis. (2022) 9:12–27. doi: 10.1016/j.gendis.2021.08.004
128. Su Y, Xu C, Cheng W, Zhao Y, Sui L, Zhao Y. Pretreated mesenchymal stem cells and their secretome: enhanced immunotherapeutic strategies. Int J Mol Sci. (2023) 24:1277. doi: 10.3390/ijms24021277
129. Bai L, Lennon DP, Caplan AI, DeChant A, Hecker J, Kranso J, et al. Hepatocyte growth factor mediates mesenchymal stem cell–induced recovery in multiple sclerosis models. Nat Neurosci. (2012) 15:862–70. doi: 10.1038/nn.3109
130. Lozito TP, Rocky ST. Mesenchymal stem cells inhibit both endogenous and exogenous MMPs via secreted TIMPs. J Cell Physiol. (2011) 226:385–96. doi: 10.1002/jcp.22344
131. Kim HJ, Bayarsaikhan D, Lee J, Bayarsaikhan G, Lee B. Brain-derived neurotrophic factor secreting human mesenchymal stem cells improve outcomes in rett syndrome mouse models. Front Neurosci. (2021) 15:725398. doi: 10.3389/fnins.2021.725398
132. Azevedo RI, Minskaia E, Fernandes-Platzgummer A, Vieira AIS, da Silva CL, et al. Mesenchymal stromal cells induce regulatory T cells via epigenetic conversion of human conventional CD4 T cells in vitro. Stem Cells. (2020) 38:1007–19. doi: 10.1002/stem.3185
133. Ryan JM, Barry FP, Murphy JM, Mahon BP. Mesenchymal stem cells avoid allogeneic rejection. J Inflammation. (2005) 2:8. doi: 10.1186/1476-9255-2-8
134. Li SW, Cai Y, Mao XL, He SQ, Chen YH, Yan LL, et al. The immunomodulatory properties of mesenchymal stem cells play a critical role in inducing immune tolerance after liver transplantation. Stem Cells Int. (2021), 6930263. doi: 10.1155/2021/6930263
135. Han Y, Yang J, Fang J, Zhou Y, Candi E, Wang J, et al. The secretion profile of mesenchymal stem cells and potential applications in treating human diseases. Signal transduction targeted Ther. (2022) 7:92. doi: 10.1038/s41392-022-00932-0
136. Downs-Canner S, Berkey S, Delgoffe GM, Edwards RP, Curiel T, et al. Suppressive IL-17A+Foxp3+ and ex-Th17 IL-17AnegFoxp3+ Treg cells are a source of tumour-associated Treg cells. Nat Commun. (2017) 8:14649. doi: 10.1038/ncomms14649
137. Heidt S, Segundo DS, Chadha R, Wood KJ. The impact of Th17 cells on transplant rejection and the induction of tolerance. Curr Opin Organ Transplant. (2010) 15:456–61. doi: 10.1097/MOT.0b013e32833b9bfb
138. English K, French A, Wood KJ. Mesenchymal stromal cells: facilitators of successful transplantation? Cell Stem Cell. (2010) 7:431–42. doi: 10.1016/j.stem.2010.09.009
139. Wang Y, Qi Z, Yan Z, Ji N, Yang X, Gao D, et al. Mesenchymal stem cell immunomodulation: A novel intervention mechanism in cardiovascular disease. Front Cell Dev Biol. (2022) 9:742088. doi: 10.3389/fcell.2021.742088
140. English K, Barry FP, Mahon BP. Murine mesenchymal stem cells suppress dendritic cell migration, maturation and antigen presentation. Immunol Lett. (2008) 115:50–8. doi: 10.1016/j.imlet.2007.10.002
141. Casiraghi F, Perico N, Gotti E, Todeschini M, Mister M, et al. Kidney transplant tolerance associated with remote autologous mesenchymal stromal cell administration. Stem Cells Trans Med. (2020) 9:427–32. doi: 10.1002/sctm.19-0185
142. Yu Yi, Valderrama AV, Han Z, Uzan G, Naserian S, et al. Human fetal liver MSCs are more effective than adult bone marrow MSCs for their immunosuppressive, immunomodulatory and Foxp3+ T regs induction capacity. (2020). doi: 10.21203/rs.3.rs-40561/v2
143. Shi M, Liu Z, Wang Y, Xu R, Sun Y, et al. A pilot study of mesenchymal stem cell therapy for acute liver allograft rejection. Stem Cells Trans Med. (2017) 6:12. doi: 10.1002/sctm.17-0134
144. Reinders MEJ, Groeneweg KE, Hendriks SH, Bank JR, Dreyer GJ, et al. Autologous bone marrow-derived mesenchymal stromal cell therapy with early tacrolimus withdrawal: The randomized prospective, single-center, open-label TRITON study. Am J transplantation: Off J Am Soc Transplant Am Soc Transplant Surgeons. (2021) 21:3055–65. doi: 10.1111/ajt.16528
145. Dreyer GJ, Groeneweg KE, Heidt S, Roelen DL, van Pel M, et al. Human leukocyte antigen selected allogeneic mesenchymal stromal cell therapy in renal transplantation: The Neptune study, a phase I single-center study. Am J transplantation: Off J Am Soc Transplant Am Soc Transplant Surgeons. (2020) 20:2905–15. doi: 10.1111/ajt.15910
146. Erpicum P, Weekers L, Detry O, Bonvoisin C, Delbouille MH, et al. Infusion of third-party mesenchymal stromal cells after kidney transplantation: a phase I-II, open-label, clinical study. Kidney Int. (2019) 95:693–707. doi: 10.1016/j.kint.2018.08.046
147. Ban TH, Lee S, Kim HD, Ko EJ, Kim BM, Kim KW, et al. Clinical trial of allogeneic mesenchymal stem cell therapy for chronic active antibody-mediated rejection in kidney transplant recipients unresponsive to rituximab and intravenous immunoglobulin. Stem Cells Int. (2021), 6672644. doi: 10.1155/2021/6672644
148. Tan J, Wu W, Xu X, Liao L, Zheng F, et al. Induction therapy with autologous mesenchymal stem cells in living-related kidney transplants: a randomized controlled trial. JAMA vol. (2012) 307:1169–77. doi: 10.1001/jama.2012.316
149. Musiał-Wysocka A, Kot M, Majka M. The pros and cons of mesenchymal stem cell-based therapies. Cell Transplant. (2019) 28:801–12. doi: 10.1177/0963689719837897
150. Miclau K, Hambright WS, Huard J, Stoddart MJ, Bahney CS. Cellular expansion of MSCs: Shifting the regenerative potential. Aging Cell. (2023) 22:e13759. doi: 10.1111/acel.13759
151. Plock JA, Schnider JT, Solari MG, Zheng XX, Gorantla VS. Perspectives on the use of mesenchymal stem cells in vascularized composite allotransplantation. Front Immunol. (2013) 4:175. doi: 10.3389/fimmu.2013.00175
152. Casiraghi F, Azzollini N, Cassis P, Imberti B, Morigi M, et al. Pretransplant infusion of mesenchymal stem cells prolongs the survival of a semiallogeneic heart transplant through the generation of regulatory T cells. J Immunol. (2008) 181:3933–46. doi: 10.4049/jimmunol.181.6.3933
153. Tseng WC, Lee PY, Tsai MT, Chang FP, Chen NJ, Chien CT, et al. Hypoxic mesenchymal stem cells ameliorate acute kidney ischemia-reperfusion injury via enhancing renal tubular autophagy. Stem Cell Res Ther. (2021) 12:367. doi: 10.1186/s13287-021-02374-x
154. Kuo YR, Goto S, Shih HS, Wang FS, Lin CC, et al. Mesenchymal stem cells prolong composite tissue allotransplant survival in a swine model Transplantation. (2009) 87:1769–77. doi: 10.1097/TP.0b013e3181a664f1
155. Barth RN, Rodriguez ED, Mundinger GS, Nam AJ, Ha JS, Hui–Chou H, et al. Vascularized bone marrow-based immunosuppression inhibits rejection of vascularized composite allografts in nonhuman primates. Am J Transpl. (2011) 11:1407–16. doi: 10.1111/j.1600-6143.2011.03551.x
156. Kuo YR, Chen CC, Goto S, Huang YT, Wang CT, Tsai CC, et al. Immunomodulatory effects of bone marrow-derived mesenchymal stem cells in a swine hemi-facial allotransplantation model. PloS One. (2012) 7:e35459. doi: 10.1371/journal.pone.0035459
157. Kuo YR, Chen CC, Goto S, Lee IT, Huang CW, Tsai CC, et al. Modulation of immune response and T-cell regulation by donor adipose-derived stem cells in a rodent hind-limb allotransplant model. Plast Reconstr Surg. (2011) 128:661e–72. doi: 10.1097/PRS.0b013e318230c60b
158. Sarhane KA, Ibrahim Z, Barone AAL, Cooney DS, Lee WPA, et al. Minimization of immunosuppression and tolerance induction in reconstructive transplantation. Curr Surg Rep. (2013) 1:40–6. doi: 10.1007/s40137-012-0008-4
159. Huang W-C, Lin JY, Wallace CG, Chuang WY, Wei FC, et al. Vascularized Bone Grafts within Composite Tissue Allotransplants Can Autocreate Tolerance through Mixed Chimerism with Partial Myeloablative Conditioning: An Experimental Study in Rats. Plast Reconstructive Surg. (2010) 125:1095–103. doi: 10.1097/PRS.0b013e3181d0ab80
160. Ramirez AE, Cheng HY, Lao WW, Wang YL, Wen CJ, et al. A novel rat full-thickness hemi-abdominal wall/hindlimb osteomyocutaneous combined flap: influence of allograft mass and vascularized bone marrow content on vascularized composite allograft survival. Transpl Int. (2014) 27:977–86. doi: 10.1111/tri.2014.27.issue-9
161. Schneeberger S, Gorantla VS, Brandacher G, Zeevi A, Demetris AJ, et al. Upper-extremity transplantation using a cell-based protocol to minimize immunosuppression. Ann Surg. (2013) 257:345–51. doi: 10.1097/SLA.0b013e31826d90bb
162. Petruzzo P, Kanitakis J, Testelin S, Pialat JB, Buron F, et al. Clinicopathological findings of chronic rejection in a face grafted patient. Transplantation. (2015) 99:2644–50. doi: 10.1097/TP.0000000000000765
163. Prabhune KA, Gorantla VS, Perez-Abadia G, Francois CG, Vossen M, et al. Composite tissue allotransplantation in chimeric hosts part II. A clinically relevant protocol to induce tolerance in a rat model. Transplantation. (2003) 76:1548–55. doi: 10.1097/01.TP.0000085288.12571.65
164. Leto Barone AA, Angelo A, Kurtz JM, Albritton A, Mallard CA, Shanmugarajah K, et al. Effects of transient donor chimerism on rejection of MHC-mismatched vascularized composite allografts in swine. Vascularized Composite Allotransplantation. (2015) 2:1–8. doi: 10.1080/23723505.2015.1039692
165. Amodio G, Cichy J, Conde P, Matteoli G, Moreau A, Ochando J, et al. Role of myeloid regulatory cells (MRCs) in maintaining tissue homeostasis and promoting tolerance in autoimmunity, inflammatory disease and transplantation. Cancer immunology immunotherapy: CII. (2019) 68:661–72. doi: 10.1007/s00262-018-2264-3
166. Zhuang J, Hou J. The role of regulatory myeloid cell therapy in renal allograft rejection. Front Immunol. (2021) 12:625998. doi: 10.3389/fimmu.2021.625998
167. Boyette LB, Macedo C, Hadi K, Elinoff BD, Walters JT, et al. Phenotype, function, and differentiation potential of human monocyte subsets. PloS One. (2017) 12:e0176460. doi: 10.1371/journal.pone.0176460
168. MacKenzie KF, Clark K, Naqvi S, McGuire VA, Nöehren G, et al. PGE2 induces macrophage IL-10 production and a regulatory-like phenotype via a protein kinase A–SIK–CRTC3 pathway. J Immunol. (2013) 190:565–77. doi: 10.4049/jimmunol.1202462
169. Zhang W, Li J, Qi G, Tu G, Yang C, et al. Myeloid-derived suppressor cells in transplantation: the dawn of cell therapy. J Transl Med. (2018) 16:19. doi: 10.1186/s12967-018-1395-9
170. Nakamura T, Nakao T, Yoshimura N, Ashihara E. Rapamycin prolongs cardiac allograft survival in a mouse model by inducing myeloid-derived suppressor cells. Am J transplantation: Off J Am Soc Transplant Am Soc Transplant Surgeons. (2015) 15:2364–77. doi: 10.1111/ajt.13276
171. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. (2018) 19:108–19. doi: 10.1038/s41590-017-0022-x
172. Zhang F, Zhang J, Cao P, Sun Z, Wang W. The characteristics of regulatory macrophages and their roles in transplantation. Int Immunopharmacol. (2021) 91:107322. doi: 10.1016/j.intimp.2020.107322
173. Conde P, Rodriguez M, van der Touw W, Jimenez A, Burns M. DC-SIGN+ Macrophages control the induction of transplantation tolerance. Immunity. (2015) 42:1143–58. doi: 10.1016/j.immuni.2015.05.009
174. Navasardyan I, Bonavida B. Regulation of T cells in cancer by nitric oxide. Cells. (2021) 10:2655. doi: 10.3390/cells10102655
175. Bonham CA, Peng L, Liang X, Chen Z, Wang L. Marked prolongation of cardiac allograft survival by dendritic cells genetically engineered with NF-κB oligodeoxyribonucleotide decoys and adenoviral vectors encoding CTLA4-Ig. J Immunol. (2002) 169:3382–91. doi: 10.4049/jimmunol.169.6.3382
176. O’Garra A, Vieira PL, Vieira P, Goldfeld AE. IL-10–producing and naturally occurring CD4+ Tregs: limiting collateral damage. J Clin Invest. (2004) 114:1372–8. doi: 10.1172/JCI23215
177. Riquelme P, Hutchinson JA. Novel molecules mediate specialized functions of human regulatory macrophages. Curr Opin Organ Transplant. (2018) 23:533–7. doi: 10.1097/MOT.0000000000000560
178. Chen S, Saeed AFUH, Liu Q, Jiang Q, Xu H, Xiao GG, et al. Macrophages in immunoregulation and therapeutics. Signal transduction targeted Ther. (2023) 8:207. doi: 10.1038/s41392-023-01452-1
179. Cai S, Hou J, Fujino M, Zhang Q, Ichimaru N, et al. iPSC-derived regulatory dendritic cells inhibit allograft rejection by generating alloantigen-specific regulatory T cells. Stem Cell Rep. (2017) 8:1174–89. doi: 10.1016/j.stemcr.2017.03.020
180. Calderon JJ, Prieto K, Lasso P, Fiorentino S, Barreto A. Modulation of myeloid-derived suppressor cells in the tumor microenvironment by natural products. Arch Immunol Ther Exp. (2023) 71:17. doi: 10.1007/s00005-023-00681-0
181. Gabriely G, Ma D, Siddiqui S, Sun L, Skillin NP, et al. Myeloid cell subsets that express latency-associated peptide promote cancer growth by modulating T cells. iScience. (2021) 24:103347. doi: 10.1016/j.isci.2021.103347
182. Dugast A-S, Haudebourg T, Coulon F, Heslan M, Haspot F, et al. Myeloid-derived suppressor cells accumulate in kidney allograft tolerance and specifically suppress effector T cell expansion. J Immunol. (2008) 180:7898–906. doi: 10.4049/jimmunol.180.12.7898
183. Kucirka LM, Durand CM, Bae S, Avery RK, Locke JE, et al. Induction immunosuppression and clinical outcomes in kidney transplant recipients infected with human immunodeficiency virus. Am J Transplant. (2016) 16:2368–76. doi: 10.1111/ajt.13840
184. Utrero-Rico A, Laguna-Goya R, Cano-Romero F, Chivite-Lacaba M, Gonzalez-Cuadrado C, et al. Early posttransplant mobilization of monocytic myeloid-derived suppressor cell correlates with increase in soluble immunosuppressive factors and predicts cancer in kidney recipients. Transplantation. (2020) 104:2599–608. doi: 10.1097/TP.0000000000003179
Keywords: solid organ transplantation, SOT, vascularized composite allotransplantation, VCA, cellular therapies
Citation: Knoedler L, Dean J, Diatta F, Thompson N, Knoedler S, Rhys R, Sherwani K, Ettl T, Mayer S, Falkner F, Kilian K, Panayi AC, Iske J, Safi A-F, Tullius SG, Haykal S, Pomahac B and Kauke-Navarro M (2024) Immune modulation in transplant medicine: a comprehensive review of cell therapy applications and future directions. Front. Immunol. 15:1372862. doi: 10.3389/fimmu.2024.1372862
Received: 18 January 2024; Accepted: 22 March 2024;
Published: 08 April 2024.
Edited by:
Alessandro Alessandrini, Harvard Medical School, United StatesReviewed by:
Martin Johannes Hoogduijn, Erasmus University Medical Center Rotterdam, NetherlandsCopyright © 2024 Knoedler, Dean, Diatta, Thompson, Knoedler, Rhys, Sherwani, Ettl, Mayer, Falkner, Kilian, Panayi, Iske, Safi, Tullius, Haykal, Pomahac and Kauke-Navarro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leonard Knoedler, bGVvbmFyZGtub2VkbGVyQGdtYWlsLmNvbQ==; Martin Kauke-Navarro, a2F1a2UtbmF2YXJyby5tYXJ0aW5AeWFsZS5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.