
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 March 2024
Sec. Immunological Tolerance and Regulation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1371089
This article is part of the Research Topic Novel Insights into Liver Injury: Mechanisms, Pathophysiology, and Therapeutic Strategies View all 7 articles
 Ananya Ajith1
Ananya Ajith1 Makram Merimi2
Makram Merimi2 Mandana Kazem Arki3Nikoo Hossein-khannazer4
Mandana Kazem Arki3Nikoo Hossein-khannazer4 Mehdi Najar5,6
Mehdi Najar5,6 Massoud Vosough7,8Etienne Marc Sokal1
Massoud Vosough7,8Etienne Marc Sokal1 Mustapha Najimi1*
Mustapha Najimi1*CD4+ CD25+ FOXP3+ T regulatory cells (Tregs) are a subset of the immunomodulatory cell population that can inhibit both innate and adaptive immunity by various regulatory mechanisms. In hepatic microenvironment, proliferation, plasticity, migration, and function of Tregs are interrelated to the remaining immune cells and their secreted cytokines and chemokines. In normal conditions, Tregs protect the liver from inflammatory and auto-immune responses, while disruption of this crosstalk between Tregs and other immune cells may result in the progression of chronic liver diseases and the development of hepatic malignancy. In this review, we analyze the deviance of this protective nature of Tregs in response to chronic inflammation and its involvement in inducing liver fibrosis, cirrhosis, and hepatocellular carcinoma. We will also provide a detailed emphasis on the relevance of Tregs as an effective immunotherapeutic option for autoimmune diseases, liver transplantation, and chronic liver diseases including liver cancer.
Tregs are a minor population of T subset cells, representing about 5-10% of the whole cluster of differentiation (CD) 4+ T lymphocyte population (1, 2). Tregs maintain immune homeostasis by controlling autoimmune reactions and imparting self-tolerance in tissues (3). In both Humans and other mammals, Tregs prevent self-antigen reactions by suppressing immune cells of innate lineages like monocytes, macrophages, dendritic cells (DCs), natural killer cells (NKs), and natural killer T cells (NKTs) as well as immune cells of adaptive lineage such as T and B lymphocytes (4, 5). Treg family comprises of natural Tregs (nTregs) that express the nuclear forkhead or winged-helix family of transcription factor P3 (FoxP3), along with cell surface proteins cytotoxic T lymphocyte antigen-4 (CTLA-4) and CD25, as well as peripherally derived Tregs (pTregs) or those generated in vitro, known as induced Tregs (iTregs) (6). nTregs are characterized as CD4+ T cells expressing high levels of CD25 and FOXP3, and low levels of CD127 surface marker (7–9). CD25 is the α-chain of Interleukin-2 (IL-2) receptor expressed on the cell surface of Tregs and activated T effector cells. Transcription factor FOXP3 is crucial for the development, function, and lineage commitment of Tregs (7, 10). It has been reported as a specific marker required for the development of thymic CD4+ CD25+ Tregs and is directly correlated with the cell surface expression of CD25 receptor (11). Mutations or deficiency of the FOXP3 gene can lead to IPEX syndrome (immune dysregulation, polyendocrinopathy, enteropathy, X-linked genetic trait) in humans, and in scurfy mutant mice, it causes lymphoproliferation and multiorgan autoimmunity (11, 12).
Regulatory T cells are classified into Tregs expressing Foxp3 (Foxp3+ Tregs), Type 1 regulatory T (Tr1) cells, T helper (Th3) cells, CD8+CD28− T cells, human leukocyte antigen (HLA)‐G+CD4+ T cells and HLA‐E‐specific CD8+ T cells (13). In this review we will focus on the CD4+ Tregs. FoxP3 expressing Tregs are a highly heterogenous population of immune cells. Based on their site of development, Tregs are subdivided into naturally occurring Tregs (nTregs) and adaptive or induced Tregs (pTregs). While nTregs develop in the thymus, pTregs develop in the peripheral lymphoid organs (14) (Figure 1). Strong T-cell Receptor (TCR) signaling and CD28 co-stimulation result in the nTreg lineage, whereas pTregs are generated from naïve, mature T cells under weak TCR stimulation along with other factors like IL-2, Transforming growth factor-β (TGF-β) or retinoic acid (RA) (2, 15). High demethylation of Treg-specific demethylated region (TSDR), a highly conserved CpG-rich region in the FOXP3 enhancer, is specific for nTregs while in pTregs, it is only partially demethylated (2). Th-3 and Th1-like or Tr1 are Treg subsets arising from pTregs in the periphery. The presence of cytokines like TGF-β and IL-4 promotes the proliferation of Th3 Tregs while IL-10 and Interferon (IFN)-γ promote the proliferation of the Th1-like or Tr1 lineages of Treg cell types. Th-3 Tregs are FoxP3+ and they largely maintain the immunosuppressive microenvironment by secreting TGF-β. Th1 Tregs lack FoxP3 expression, but express Lymphocyte-activation gene 3 (LAG-3) and secrete IL-10 into their microenvironment (16).
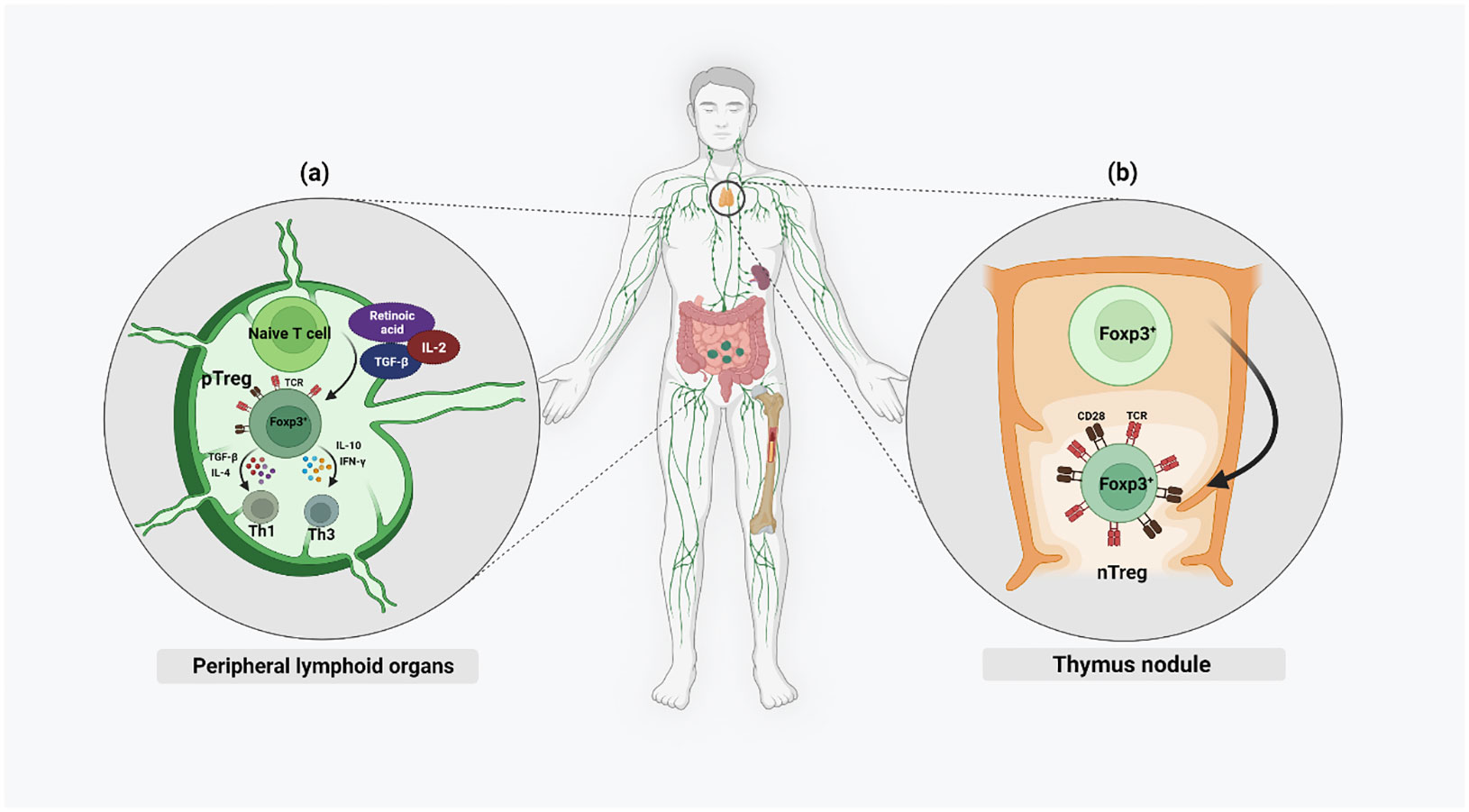
Figure 1 Different chemokines and cytokines activate both nTregs and pTregs in the microenvironment of both thymus and the periphery. Naïve nTregs are already FoxP3+ and their activation is mainly by strong TCR stimulation and CD28 co-stimulation. Only activated pTregs express FoxP3 and this is mainly by weak TCR stimulation along with higher levels of IL-2, TGF-β, and RA.
Furthermore, Tregs can be subdivided based on their differentiation potential into naïve Tregs (nTregs), effector Tregs (eTregs), effector memory cells (emTregs), and central memory cells (cmTregs) (Table 1) (18). Peripheral self-antigens activate nTregs to increase their cell number and suppressive function, thereby differentiating them into eTregs, which are recruited toward the inflammation site. A proportion of these cells remain in the non-lymphoid tissue after antigen expression cessation and survive as effector memory Tregs (emTregs) with an enhanced capacity to suppress autoimmunity in these tissues (21). Apart from this, memory Tregs may be located in the secondary lymphoid organs as the central memory Tregs (cmTregs). A re-exposure to the antigen can result in an expansion of these memory Tregs into a more effective immune response than during the primary exposure (17).
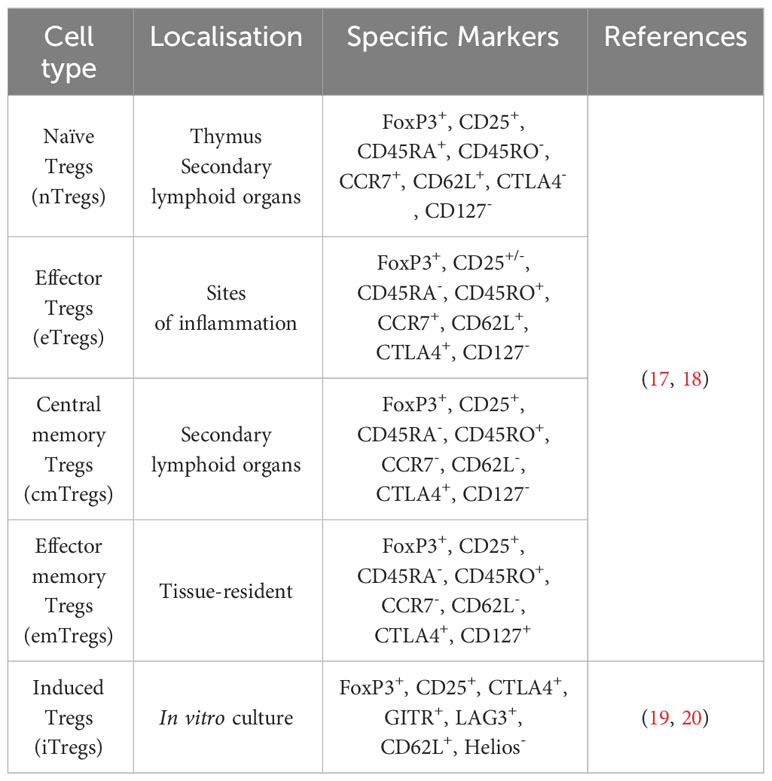
Table 1 Site of localization and marker heterogeneity in different Treg subsets.
In homeostatic conditions, Tregs show reduced recirculation or tissue infiltration in comparison to conventional T cells. During pathological conditions, Treg recirculation through the lymphoid tissues for entering the inflamed, tumors, and infectious sites, increases (22). Tregs induce immunosuppression by inhibiting the proliferation, differentiation, cytokine or antibody production, of their target cells. Tregs can suppress a large number of target immune cells by direct cell-cell contact inhibition/killing, secretion of anti-inflammatory cytokines, and disruption of the metabolic pathways competing for growth factors (23). Direct cell-cell contact elimination is a strategy used by nTregs to directly kill cytotoxic cells by releasing perforin or serine protease granzyme (24). Activated Tregs may express cell surface molecules like Galectin1 which bind the receptors of effector T cells to induce cell cycle arrest, apoptosis, or inhibition of pro-inflammatory cytokine secretions. Cell-cell contact of Tregs with target cells can also induce several inhibitory pathways by the modulation of molecules such as cyclic adenosine monophosphate (cAMP), resulting in the inhibition of cellular proliferation, differentiation, inhibition of cytokines IL-2, IFNγ or the activation of transcriptional repressor inducible cAMP early repressor (ICER). In trans-well system studies, Tregs failed to suppress effector T target cells, which suggests that it is necessary for Tregs to be in close proximity of the target cells to impart their inhibitory action (25). However, recent studies showed that the secretion of extracellular vesicles (EVs) by Tregs has emerged as a novel suppressive mechanism with the ability to modulate immunity in a cell-contact independent and targeted manner. This phenomenon has been identified in various autoimmune and cancer pathologies. Yu et al., demonstrated that Tregs-derived EVs effectively suppressed T cell proliferation in a dose-dependent manner (26). In another study by Okoye et al., Treg-derived EVs (Treg-EVs) were shown to suppress T cell-mediated responses through the transfer of packaged Let-7d miRNA to Th1 cells, leading to an inhibition of their proliferation and IFN-γ secretion (27). Aiello et al. further demonstrated that EVs derived from Tregs possessed the ability to convert T cells into regulatory cells, upon delivery of miR-503, miR-330, miR-9 (28). Naïve T cells exposed to these Treg-EVs exhibited an increased IL-10 secretion and expressed T cell immunoglobulin and mucin-domain containing-3 (Tim3), indicating the regulatory effects of these EVs on T cell behavior. Such studies collectively shed light on the immunomodulatory potential of Treg-EVs, offering insights into their therapeutic implications in immune-related disorders (29). Notably, Treg-EVs appear to exhibit a dual role, either protecting or inducing cell death, contingent upon the target cells involved. These EVs demonstrate the capability to inhibit apoptosis in myocardial cells during acute myocardial infarction, while concurrently inducing apoptosis in conventional T cells.
DCs represent a major antigen-presenting cell (APC) type, so targeting this cell population can indirectly inhibit T effector cell activation and immune response. LAG-3 (CD223) expressed on Tregs, binds to the Major histocompatibility complex class II (MHC class II) in immature DCs and induces an inhibitory signal which leads to a suppression of DC maturation and their capability for immune stimulation. Prolonged interaction of Neuropilin 1 (Nrp1), present in Tregs, with immature DCs, restricts the antigen-presenting capabilities of DCs (Figure 2) (30, 31). Thereby, only mature DCs promote T effector cell proliferation, while immature/tolerogenic DCs induce the activity of Tregs. Thus, tolerogenic DCs-induced Tregs and the Treg-maintained immunosuppressive nature of DCs are linked (32, 33). As FOXP3 regulates the high expression of cytotoxic T lymphocyte antigen 4 (CTLA-4) required for immunosuppressive activity of Tregs (34), another strategy for DC suppression is the binding of CTLA-4 to DC surface proteins CD80/86 (Figure 2). Formed complex is trans-endocytosed by Tregs thereby inhibiting the ability of DCs to interact with effector T cells. Thereafter, CD80/86 is fused with the lysosome and CTLA-4 is recycled back to the cell surface for further APC suppression (35). Another mechanism of Treg-induced maintenance of tolerogenic DCs is by inducing the secretion of indoleamine 2,3-dioxygenase (IDO). Studies have shown that both in mice and Humans, Tregs induce DCs to produce IDO, in order to suppress the T cell response through CTLA-4 dependent mechanisms (36). Cheng et al. reported that the DCs recruited by the carcinoma-associated fibroblasts (CAFs) in hepatocellular carcinoma (HCC) get converted to IDO producing regulatory DCs which exhibits an increased ability to suppress T cell proliferation, and upregulation of Tregs through IL-6 mediated Signal transducer and activator of transcription 3 (STAT3) activation (37).
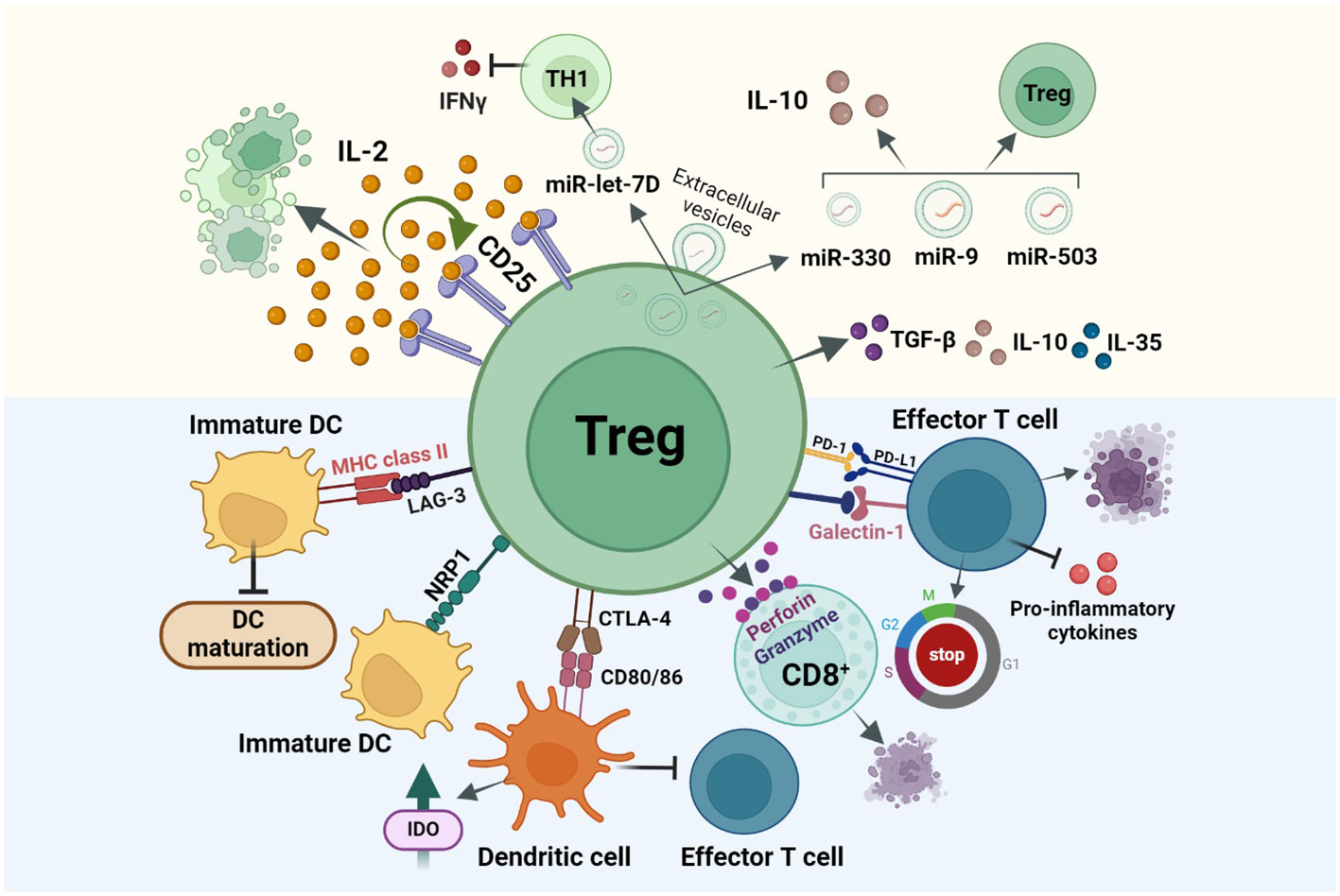
Figure 2 Treg immunosuppression is achieved via three main mechanisms: direct cell-cell contact inhibition or cell killing (Blue background), secretion of immunosuppressive cytokines, and disruption of metabolic pathways (Yellow background). (i) Treg cell surface molecules like, CTLA-4, LAG-3, NRP1 bind to the cell surface receptors of tolerogenic DCs resulting the DCs from maturing for antigen presentation and thereby inhibiting T effector cell activation and functioning. (ii) Secretion of immunosuppressive cytokines like IL-35, IL-10, TGF-β results in cell cycle arrest in other immune cells (iii) Tregs over-express CD25 receptor which has a higher affinity for IL-2, this in turn will suppress other cell types which require IL-2 for activation like T effector cells from effectively functioning. Other mechanisms include Ca2+ suppression in T effector cells, IDO based and PD-1/PDL-1 interaction-based cell suppressions.
Programmed death ligand 1/programmed death receptor 1 (PDL-1/PD-1) based interaction between tumor cells and T lymphocytes, plays an important role in suppressing the cytotoxic effects of T cell immunity in the tumor microenvironment. Likewise, Tregs also express PD-1 and PDL-1 on their cell surface and have the potential to interact with the PDL-1-expressing T and B lymphocytes, DCs, macrophages, and bone marrow-derived mast cells. During cancer, PD-1 is upregulated in Tregs resulting in an increased interaction of PDL-1 in T effector cells thereby resulting in their inhibition and suppression of immune responses. While PDL-1/PD-1 suppresses the immunogenic activity of T lymphocytes, this interaction in Tregs may activate and induce their differentiation (38, 39). Tregs can also secrete suppressive cytokines like IL-10, TGF-β, and IL-35, which could induce cell cycle arrest in other immune and myeloid cells (Figure 2) (31). Studies show that deficiency of membrane-bound and soluble TGF-β produced by Tregs, results in T-cell mediated autoimmunity in mice and that IL-35 directly influences T cells by inhibiting their proliferation (40).
Key features of FoxP3+ Tregs are high expression of CTLA-4, CD25 cell surface marker and absence of IL-2 production (41). IL-2 is an important cytokine signal required for the proliferation, differentiation, survival and activity of T-helper cells, memory T cells and Tregs. Naive Tregs do not produce IL-2, but the cell surface receptor CD25 displays high affinity for IL-2. By competitive consumption, Tregs use the IL-2 produced by activated T cells and suppress them via a negative feed-back loop (34, 35). In IL-2 deprived T cells, Tregs induce apoptosis through BCL2 Associated Agonist of cell Death (Bad)/BCL-2 interacting mediator of cell death (Bim) driven caspase activation (42). Inducible co-stimulator (ICOS) is a member of CD-28/CTLA-4 family, widely expressed on activated CD4+ T cells and Tregs. The pathway activation occurs during the binding of ICOS with its ligand ICOSL, expressed on APCs (43). According to the expression of ICOS, Tregs can be divided into ICOS+ and ICOS- Tregs. In vitro studies have proven that cytokine secretion is different in these 2 Treg subsets, i.e. ICOS+ Tregs tend to secrete more IL-10 and moderate amounts of TGF-β1, while ICOS- Tregs play a suppressive function by secreting TGF-β1 alone. In mice, studies have shown that ICOS+ Tregs have better survival and superior suppressive properties as compared to ICOS- subset. In a study by Tu et al., HCC patients have been shown to display a higher prevalence of tumor infiltrating ICOS+ Tregs. Due to higher proportion of FoxP3+ Tregs/CD4+ T cells and ICOS+ FoxP3+ Tregs/total FoxP3+ Tregs, their study concludes that ICOS+ FoxP3+ Tregs may be the main immunosuppressive cell type in HCC microenvironment (44–46). Another mechanism of Treg suppression is the deprivation of calcium levels in T effector cells. Calcium is essential for the proliferation of lymphocytes, their expression of activation-associated genes, production of chemokines and cytokines, and activation and differentiation of naïve T cells into effector or memory T cells. Tregs have been reported to suppress the calcium signaling in T conventional cells (47).
Tregs promote immune tolerance by modulating both innate and adaptive immunity (48). CD4+ CD25+ Tregs are the natural suppressor cells of the immune system. They function during early innate and adaptive immune responses even prior to antigen activation, indicating their capability to differentiate self from non-self-cells during immune responses (49). CD4+ CD25+ Tregs maintain immune tolerance by suppressing the activity of both CD8+ and CD4+ T cell populations during inflammatory responses (50, 51). To understand their interactions with CD8+ and CD4+ cells, Treg population was depleted in mouse. The study demonstrated that CD4+ T cell population remained unchanged suggesting other regulatory factors, while a significant increase in the CD8+ T population was shown (5). The liver is renowned for its exceptional regenerative potential and is recognized for its ability to sequester auto-reactivated T cells, facilitating their apoptotic clearance. In neonatal liver, the transcriptomic profile of thymus-derived Tregs was thoroughly examined, revealing that their intra-hepatic accumulation plays a crucial role in preserving self-tolerance and promoting liver maturation. However, adult mice livers harbor a reduced number of tissue-resident Tregs, exhibiting distinct phenotypic characteristics while retaining increased proliferation, which suggests a lineage instability (52). Indeed, the liver’s unique immunological characteristics, resulting from its constant exposure to foreign antigens, are essential for its proper function. However, this constant exposure also renders the liver susceptible to immune-mediated injuries, as any disruption in the delicate balance of immune responses can lead to inflammation and damage of the liver tissue. The innate immune system in the liver consists of NKs, NKTs, Kupffer cells (KCs), DCs, and mast cells. In both human and mouse, Tregs can inhibit the NK effector function. TGF-β produced by Tregs inhibits the cytolytic activity of NKs. Tregs kill NKs by releasing granzyme-B and cell-cell contact mechanism (53). In an autoimmune condition, NKTs can secrete both anti-inflammatory and pro-inflammatory cytokines and regulate the recruitment of Tregs to the inflammation site (54). Tregs exert a negative effect on the proliferation and cytokine production of NKTs while NKTs promote the proliferation and homing of Tregs in the liver (55). KCs are involved in the pathogenesis of viral hepatitis, steatohepatitis, alcoholic liver disease, intrahepatic cholestasis, organ rejection in liver transplantation and liver fibrosis. During fibrosis, KCs induce the activation of hepatic stellate cells (HSCs) into myofibroblasts which leads to an increase in collagen and proteoglycans production (56). Depletion of KCs has been shown to induce a complete inhibition of IL-10 production by Tregs suggesting an interaction of KCs with Tregs for inducing tolerance (57). Tregs have also a role in blocking the antigen presenting capabilities of DCs and these immature DCs can promote the immune tolerance by Tregs or by inactivating T cells whereas, mature DCs trigger the development of naïve CD4+ and CD8+ T cell populations (58).
The human liver receives 70-80% of blood from the portal vein rich in nutrients and the other 20-30% from the hepatic artery rich in oxygen. Tregs can either directly use the metabolites present in the liver or from the environmental metabolism of DCs. The intrahepatic microenvironment which is, deprived of oxygen and enriched with microbes, inflammatory cytokines and hormones, plays a major role in the differentiation and function of Tregs (35). Activated Tregs generate energy using glucose, amino acids, fatty acids (FAs) and vitamins. In vitro, lipid biosynthesis, aerobic glycolysis and FAO were also used for the generation of ATP (59). FoxP3 expression in Tregs is correlated with higher mitochondrial mass and increased reactive oxygen species (ROS) production as demonstrated in both nTregs and iTregs. Oxidative phosphorylation (OXPHOS) and ROS play an important role in Treg signaling and homeostasis (60). Unlike other T cells, Tregs rely on FAs oxidation and OXPHOS for cell differentiation and function (61). Tregs proliferation is dependent on glycolysis and FAs oxidation. Upregulated insulin receptor INSR increases the glucose uptake in Tregs, required for their migration. Mammalian target of rapamycin (mTOR) complex 2 (mTORC2) regulates the Treg motility through Phosphoinositide 3 kinase- protein kinase B (PI3K-Akt) pathway and modulation of cytoskeleton reorganization by glycogen synthase kinase (GSK) activity. PI3K modulation is likely to alter cellular metabolism and FOXP3 expression (62). Metabolites like purine, tryptophan, RA, and glutamine maintain the induction of FOXP3 gene (60). Liver is enriched with metabolites like vitamin A, vitamin D, RA, etc. required for the generation and trafficking of Tregs (59). Vitamin A, C and D3 increase and stabilize the expression and activity of FOXP3 in Tregs (63).
Epigenetic modifications like DNA methylation, histone modifications, and microRNA regulations may significantly control the proliferation of Tregs, as well as development and facilitation of their suppressive properties. Epigenetic modification of its promoter, enhancer and CpG rich region, primarily through methylation and demethylation mechanisms, modulate the expression of FOXP3 in Tregs. In response to TCR signals, FOXP3 promoter gets activated consequently to the binding of transcription factors Nuclear factor of activate T cells (NFAT) and activator protein 1 (AP1). The enhancer region of FOXP3 is a TGF-β sensor with binding sites for NFAT and SMADs (64). TSDR is demethylated in Tregs while methylated in other T cells. Such TSDR demethylation determines the stability of FOXP3 expression in Tregs (65). Inhibition or mutations occurring in DNA methyl transferase 1 gene, results in hypomethylation of FOXP3 gene, thereby increasing its expression levels in Tregs (66). In HCC, lower methylation of FOXP3 promoter via DNA methyl transferase (DNMT1) is positively linked to a higher percentage of intra-tumoral Treg levels and tumor growth (64). Like DNA demethylation, histone modifications namely N terminal lysine methylation and acetylation are other important epigenetic alterations that can result in gene activation. Histone acetyl transferase (HAT) 300 is an important factor for the immune suppressive function of Tregs and pTregs induction (67). According to this study, p300 is required for Treg activity and maintenance of homeostasis both in vivo and in vitro. Acetylation of P300 has been shown to increase FOXP3 protein levels and FOXP3 mediated transcriptional repression of IL-2 production. Apart from p300, other HATs like Tat-interactive protein 60 kD (TIP60) and CREB-binding protein (CBP) are also involved in the development, activity and lineage specificity of Tregs (68).
MicroRNAs are another epigenetic regulator of several genes implicated in Tregs polarization and play an important role in their thymic development and function (Table 2) (65). miR-155 is an important epigenetic factor required for an enhanced responsiveness of Tregs to IL-2. miR-155 and miR-21 are positive enhancers of the FOXP3 activity in Tregs (76). Other miRNAs like miR-17a, miR-18a, miR-19a, miR-20a, miR19b and miR-92-1, miR-31 are negatively regulating Tregs (66). In HCC patients, the modulated expression of several miRNAs hsa-miR-182-5p, hsa-miR-214-3p, hsa-miR-129-5p and hsa-miR-30b-5p in Tregs, is targeting several signaling pathways including cytokine, chemotaxis and adhesion, thereby supporting the critical role of epigenetic regulation of this immune cell population.
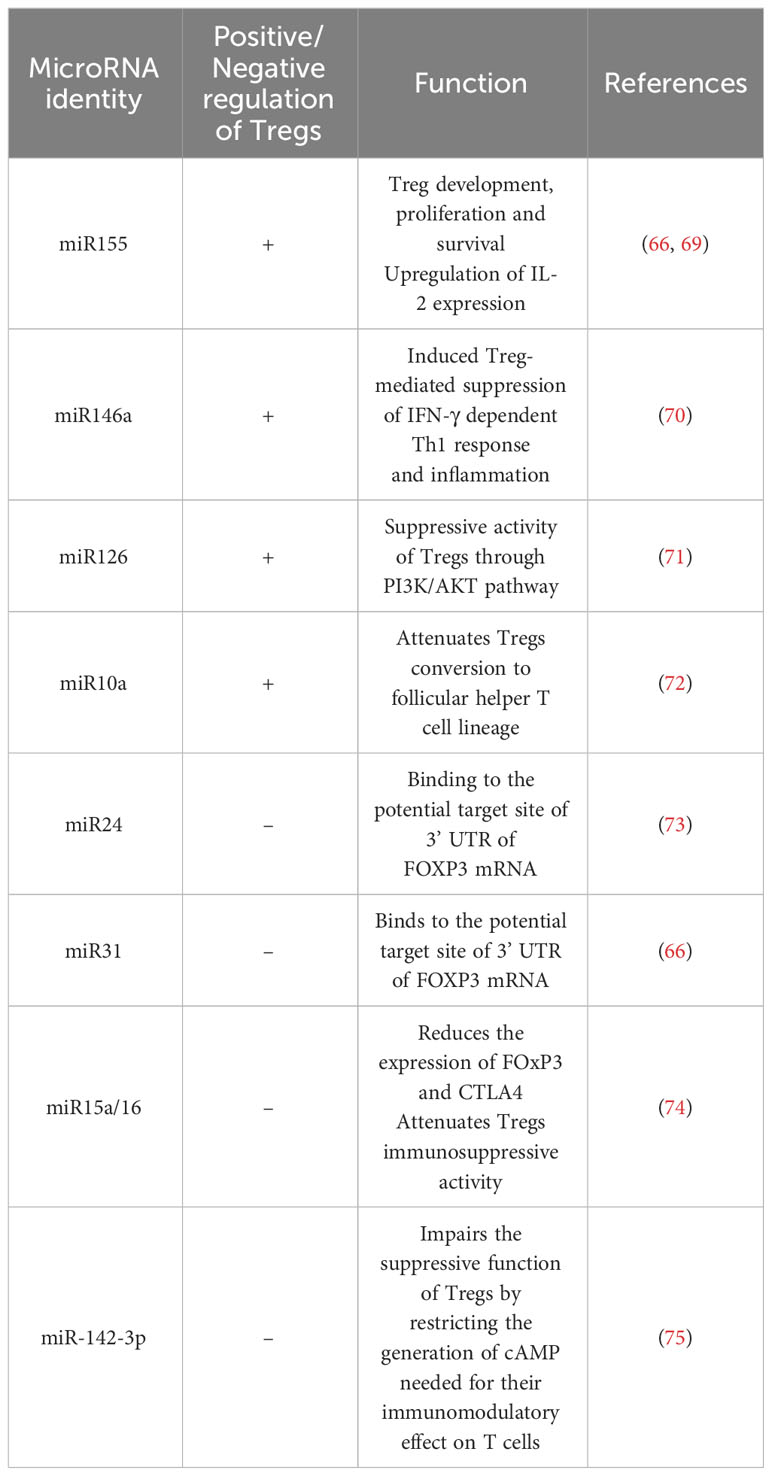
Table 2 miRNAs critically involved in Treg cell regulation.
Chronic wound healing process in response to sustained inflammation is a trigger for liver fibrosis and development of cirrhosis (77). Chronic alcohol consumption, non-alcoholic steatohepatitis (NASH), viral infections like hepatitis B (HBV) and C (HCV), autoimmune hepatitis (AIH), non-alcoholic fatty liver disease (NAFLD), cholestatic liver diseases, result in the inflammatory response causing fibrosis (78, 79). There is an increased risk for NASH to progress into liver fibrosis, cirrhosis and then to HCC. In NAFLD mouse model, the expression of FoxP3 mRNA is low in liver, indicating a decreased activity of Tregs. This has a deleterious effect resulting in NAFLD progression, resulting in the imbalance of Treg/Th17 cell levels due to increased inflammatory response by Th17 cells (80). A clinical study in children revealed an association between pronounced hepatic inflammation due to NAFLD and elevated number of Foxp3+ lymphocytes within the lobules. Conversely, adults showcased a decline in Foxp3+ lymphocytes alongside increased IL-17A+ lymphocytes in the portal/periportal tracts (81). Steatohepatitis was induced in mice fed with high fat diet and treated with endotoxin, resulted in Treg deficiency, while adaptive transfer of Tregs to these mice showed reduced disease progression and liver injury (82). The impact of Tregs on NASH remains unclear while an increased count of intrahepatic Tregs was reported in a related mouse model. Surprisingly, attempts at adoptive Treg transfer or anti-CD3 therapy yielded an unforeseen outcome escalated steatosis and elevated alanine aminotransferase levels—without any other discernible impact on NASH (83). In alcoholic steatohepatitis, alcoholic hepatitis and autoimmune liver diseases like primary biliary cirrhosis and autoimmune hepatitis, the population of Tregs infiltrating the liver is higher than the circulating one (84). In this setting, sustained inflammation induces necrosis and apoptosis of hepatocytes, therefore the anti-inflammatory function of Tregs has a protective effect against the inflammatory liver responses (85). Tregs have been reported to support the progression of liver fibrosis by targeting KCs through the TGF-β pathway. Furthermore, HSCs can selectively induce Tregs by the production of RA or in an IL-2 dependent manner (86). However, IL-10 released by Tregs has been reported to inhibit the ECM production by HSCs (87, 88).
Tregs are essential for wound healing and cessation of inflammation. Yet, studies report that reduction in Treg numbers can promote fibrosis regression. Activated HSCs secrete IL-2, resulting in the abundance of Tregs in fibrotic tissue. In healthy liver, matrix metalloproteinases (MMPs) released by KCs degrade and regulate the liver ECM. TGFβ secreted by Tregs can repress the MMP secretion by KCs which may disrupt the fibrosis regression (89). Study by (79) demonstrated that depletion of Tregs was associated with liver fibrosis progression in DEREG transgenic (DEpletion of REGulatory T cells) mice. This was mainly due to an increase of CD8+ and IL-17A+ T cell populations and enhanced secretion of pro-inflammatory cytokines and chemokines (Roh et al, 2015).
TGFβ is an important cytokine required for the determination of the differentiation of Tregs and Th17 cells. Indeed, low concentrations of TGFβ synergized with IL-6, induce Th-17 cell differentiation, while high concentrations of TGFβ induce Treg differentiation from the naïve CD4+ T cell precursors. During liver fibrosis, secretion of higher levels of IL-6 and TGFβ occurs which in turn activates the HSCs to produce ECM proteins, leading to increased Th-17 cells and an imbalance in the Th17/Treg ratio (78).
During viral infection, immune cells release cytokines needed for the termination of infection and the eradication of infected cells. Tregs regulate a delicate balance by preventing the immune cells from destroying non-infected self-cells. Studies have shown that during HBV infection, the number of CD4+ CD25+ Tregs increases in the liver and peripheral blood as compared to normal conditions (90). The analysis of Tregs in acute, chronic and chronic severe HBV infections, showed that their numbers were higher in chronic severe HBV infection than in acute or chronic HBV infections (91). HBV induced liver fibrosis also showed a similar trend where the number of Tregs was increased in advanced HBV related liver fibrosis than in early HBV-fibrosis (87). Persistent HBV infection has an increased risk for the progression of chronic hepatitis B (CHB) to hepatic cirrhosis and HCC. The role of Tregs played during HBV-induced fibrosis is dependent on disease stage at which they are activated. If the Tregs get activated by the HBV antigen in earlier stage, they might protect the HBV from host immune response, while if the host elicited Treg response occurs in the later stage of infection, Tregs will protect against excessive inflammation and prevent further liver injury. However, for progression into fibrosis from CHB infection, immune tolerance should decrease, and inflammatory response ratio should increase (92). Frequencies of Th-17 cells, IL-22 and IL-17A were found to increase with the severity of infection while the frequency of Tregs decreased in HBV associated cirrhosis than in CHB or HCC cases. IL-17 and IL-22 activate the proliferation of HSCs and the progression of fibrosis through PI3K/AKT pathway (93). When the immune suppressive properties of Tregs in the presence of HCV inoculum were studied, a significant increase of the of CD4, CD25, FOXP3 expression and inhibitory markers like CTLA-4, LAG-3, was noticed in parallel to a decreased expression of CD127. This confirms that Treg population increases with the number of HBV antigens (94). Treg immunosuppressive activity in HBV and HCV infection has dual consequences on the disease prognosis by i) restraining the CD8+ and CD4+ T cell activity which is beneficial for the host since it prevents the inflammatory liver damage and ii) persisting the infection since it provides protection for the virus from the immunogenic activities of CD8+ and CD4+ T cells (95, 96).
The ability of Tregs to migrate to the site of inflammation is critical for their capacity to regulate inflammation, and this is mainly achieved by the expression of adhesion molecules and chemokine receptors. Like conventional T cells, Tregs also undergo trans-endothelial migration by interacting with pro-adhesive signals. In general, during their migration when ligands bind to the receptors, leukocytes alter their cell surface integrin conformation (like β2 and α4 families). This allows the adhesion molecules to bind to the endothelial cell expressed ligands like intracellular adhesion molecule-1,2,3 (ICAM) and Vascular cell adhesion molecule-1 (VCAM)/mucosal addressin cell adhesion mol-1 (MAdCAM-1). This interaction with the adhesion molecules, enables the arrest of rolling leukocytes, resulting in the trans-endothelial migration of leukocytes (97). Similarly, interaction between liver sinusoidal endothelial cells (LSECs) and Tregs, leads to the activation of integrins lymphocyte function associated antigen 1 (LFA-1) and very late antigen 1 (VLA-4) on Treg surface thereby interacting with the cell adhesion molecules like intercellular adhesion molecule 1 (ICAM), vascular cell adhesion molecule (VACM) and Vascular adhesion protein-1 (VAP-1) expressed by inflamed LSECs and resulting in the trans-endothelial migration of Tregs into liver (98). In inflamed liver, Shetty and colleagues demonstrated that the homing of Tregs is also mediated by their interaction with the common lymphatic endothelial and vascular endothelial receptor (CLEVER-1) expressed by LSECs. ICAM-1 and VAP-1 along with common lymphatic endothelial and vascular endothelial receptor-1 (CLEVER-1) support the transcellular migration of Tregs in the liver (99).
Migration of immune cells to the site of inflammation is an important process, mediated by secreted cytokines and chemokines, for an effective immune response. The homing receptors expressed by Tregs are specific to the location and microenvironment at which they are exposed to, during their activation. Homing of Tregs in non-lymphoid tissue is mediated via higher expression levels of cell surface receptors like chemokine receptor 3 (CXCR3), CD103, C-C chemokine receptor type 5 (CCR5), CCR4, and CCR6. Recruitment of Tregs to the site of inflammation is facilitated by CXCR3, CCR5, and CCR6 receptors. Chemokine receptors CXCR3, CCR6 and CCR4 overlap with those of other effector T cells like Th1, CD8 T cells, Th17 cells and enable Tregs to co-localize with them (100). During Tregs infiltration, CXCR3 helps to undergo trans-endothelial migration across the hepatic sinusoidal endothelium. Naïve CXCR3 deficient mice displayed lower number of CD4+ CD25+ Tregs than in wild type mice. Accordingly, CXCR3 deficient mice tend to develop severe liver injury due to a lower infiltration of Tregs (101, 102). CCR4 helps Tregs to co-localize with liver infiltrating DCs by responding to chemokines CCL17 and CCL22 secreted by those cells (Figure 3). In chronic inflammatory liver diseases, liver infiltrating Tregs express both CXCR3 and CCR4. CCL17 and CCL22 secreted by activated intrahepatic DCs guide the CCR4 receptor in Tregs for the subsequent migration within the inflamed liver (103). Infected primary human hepatocytes secrete chemokines CCL20 and CCL22, which activate CCR6 and CCR2 present on Tregs thereby recruiting them to the site of inflammation (94). Crosstalk between the other liver immune cells and Tregs also regulates their infiltration into the inflammation site. The recruitment and the ability to maintain Tregs in the inflamed liver is influenced by several parameters, including intricate interactions with other extra-hepatic immune cell populations. Indeed, the role of GATA-Binding protein-6+ peritoneal macrophages in this complex orchestration has been recently unveiled, in the dual modulation of hepatic immunopathogenic genesis and the concurrent augmentation of Treg assemblages (104).
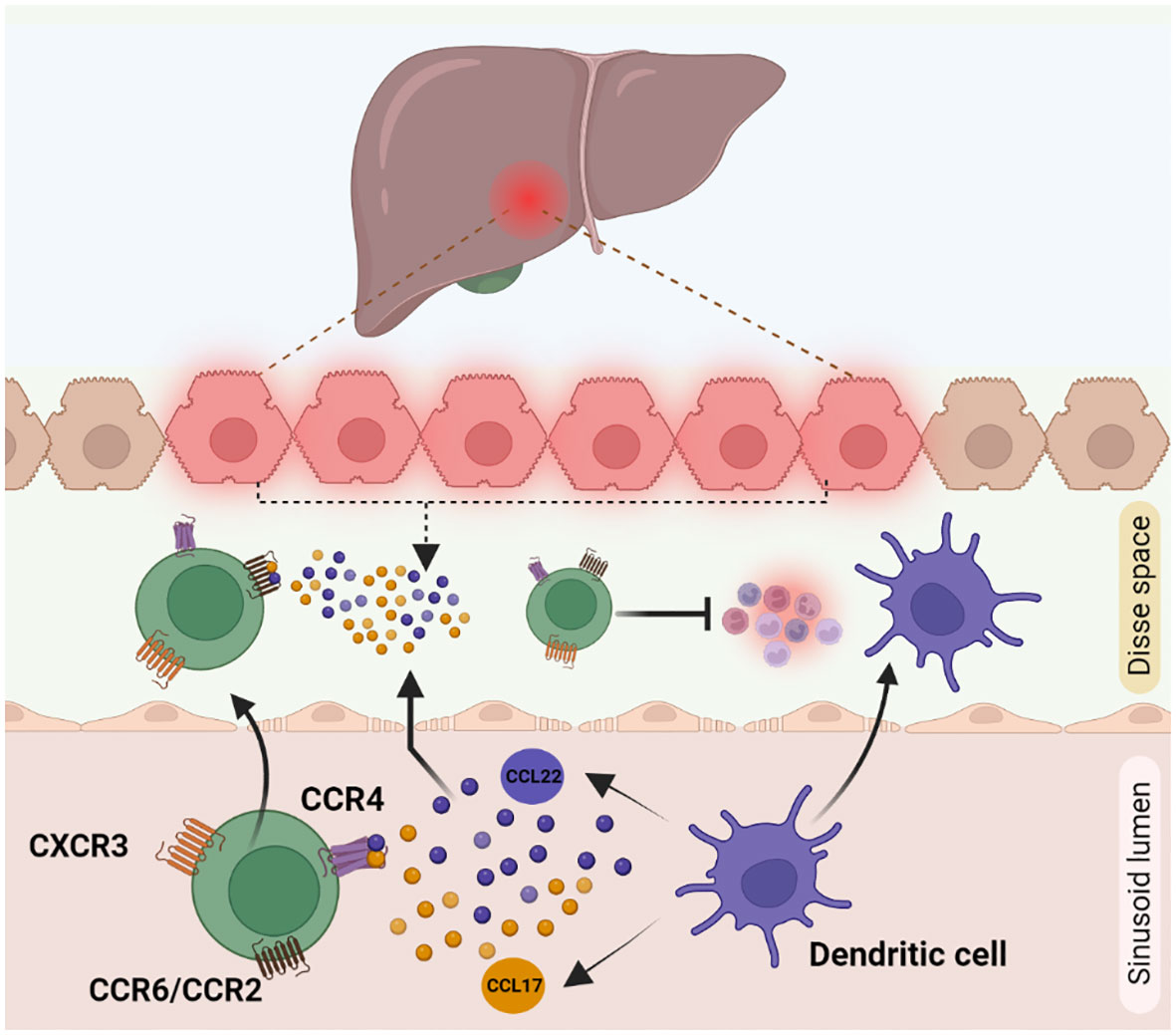
Figure 3 Cytokine mediated Treg recruitment during inflammation in liver: Treg recruitment towards the site of inflammation is required for other immune cell suppression during inflammation. This phenomenon is mainly mediated by receptors like CXCR3, CCR4 and CCR6 present on Treg cell surface. (1) CXCR3 helps Tregs in trans-endothelial migration across hepatic sinusoidal endothelium (2) CCR4 receptor, helps to respond to the chemokines CCL17 and CCL22 secreted by dendritic cells to co-localise with them to the inflamed liver. (3) CCL22 and CCL17 secretions by damaged hepatocytes are identified by CCR6/CCR2 receptors on Tregs, which helps in the active migration of Tregs.
Cancer immune response is dependent on the balance between tumor antigenicity and the microenvironment of the tumor tissue. Immune cells like Th1 cells, Th2 cells, Tregs, myeloid derived suppressor cells, tumor associated macrophages, DCs, NKs, CD4+ and CD8+ T cells are all involved in the regulation of immune response in the tumor microenvironment (105). Tregs in the HCC microenvironment, have a suppressive activity. An expansion of CD4+ CD25+ T cell population is observed in both peripheral blood and tumor microenvironment of HCC patients (106, 107). An increased number of Tregs in tumor microenvironment can have an unfavorable outcome on immunoregulatory activities. Treg population has been shown to increase in HBV infected HCC patients (108). The two possible pathways for the recruitment of Tregs to the tumor microenvironment are either influenced by the priming of naive CD4+ T cells to differentiate into CD4+ CD25+ Tregs or by inducing the selective migration of Tregs via specific chemokine secretion (46) (Figure 4). In tumor microenvironment, CD4+ CD25low T cells and CD4+ CD25- T cells may get induced for FOXP3 expression and converted to CD4+ CD25+ Tregs. Tumor derived TGFβ plays an important role in this promotion of FoxP3 expressing Tregs from naïve CD4+ T cells. In mouse, the principal mechanism for increasing CD4+ CD25+ presenting FOXP3+ population in the tumor site is via the conversion of CD4+ CD25- T cells (109, 110). Upregulation of chemokine receptors like CXCR3, CCR5, CCR4 and CCR8 is involved in both the activation and differentiation of Tregs in HCC. CCL22 produced by tumor derived macrophages and CCL17 regulate the infiltration of Tregs into the tumor sites. The CCR6-CCL20 axis plays an important role in recruiting Tregs to the tumor site. High levels of CCL20 are secreted by tumor cells and KCs to which CCR6 expressed in Tregs gets attracted inducing the migration towards the tumor site (46, 111). Secretion of the chemokine SDF-1 through the activation of CXCR4/CXCL12 signaling also has a positive correlation with increased Treg number in the tumor microenvironment (44). Thus, lack of proper tumor immunity is not due to the absence of immunogenicity but due to the increased infiltration and conversion of CD4+ CD25- T cells to CD4+ CD25+ T cells in HCC.
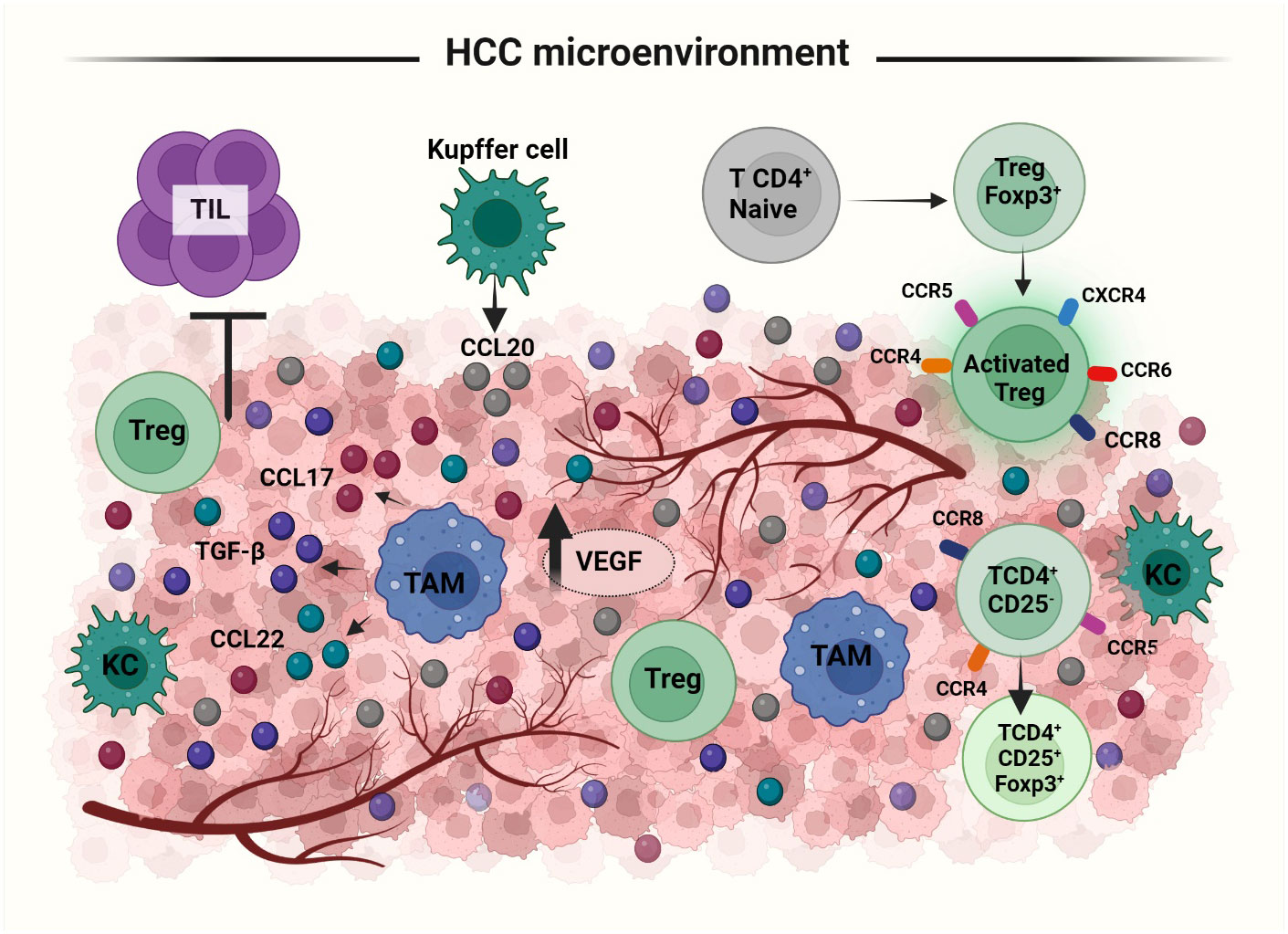
Figure 4 Recruitment and activation of Tregs to HCC tumor microenvironment (TME): Treg recruitment into TME happens by two mechanisms: priming of naïve T cells to differentiate into FoxP3 expressing Tregs or selective migration of Tregs by cytokines. In response to the cytokine secretion from TME, Tregs upregulate the expression of several receptors like, CXCR3, CCR5, CCR4, CCR8 etc. Treg migration is facilitated via the secretion of i) CCL20 by Kupffer and tumor cells, ii) CCL22 and CCL17 by tumor associated macrophages and iii) SDF1, TGFβ from tumor cells. TAMs; Tumor Associated Macrophages, KC; Kupffer Cells, TILs; Tumor Infiltrating Lymphocytes.
The intra-tumoral balance between Tregs and cytotoxic T cells, determines the recurrence and survival rate of HCC. The number of Tregs in HCC microenvironment is proportional to the tumor size (112). Therefore, an increased incidence of CD4+ Tregs is associated with HCC invasiveness and poor prognosis (113). Higher grade of the tumor is correlated with both higher Treg and lower CD8+ T cell infiltrations (114). In the tumor microenvironment, CD4+ CD25+ Tregs have a negative impact on cytokine secretion and proliferation of CD4+ CD25- anti-tumor T cells (44). CD8+ cells have been reported to accumulate in the peritumoral region, thereby proving that Tregs not only suppress CD8+ activity but also block their migration into the tumor. Besides inhibiting CD8+ T cell proliferation and activation, Tregs also suppress the production and release of the cytolytic enzymes perforin, granzyme A and granzyme B that mediate CD8+ T cell effector functions (109). Apart from CD4+ T cells not expressing CD25, Tregs were also documented for their ability to eliminate DCs, one of the most important surveillance mechanisms for the removal of cancer cells in the tumor microenvironment (115). Furthermore, the proportion of Tregs in the tumor microenvironment predicts HCC recurrence and survival rate. Indeed, the prevalence and migration of Tregs have been reported to enhance with the progression of HCC from an early to a later stage (44). A steady increase in the number of CD4+ has been noticed from hepatitis to liver cirrhosis to HCC (107, 111). HCC invasiveness is also associated with the number of Tregs. Indeed, HCC patients with lower intra-tumoral Tregs population had a higher overall survival and disease-free survival rates of 70 and 69 months when compared to patients with higher intra-tumoral Tregs population. Lower intra-tumoral Tregs with high CTL population in the HCC tumor are associated with an improved survival as well as a reduced recurrence in HCC (114). Analysis of specific surface markers of Tregs [GITR, HLA-DR, CD45RO, CD152, and CD45RA and FOXP3] also showed that there is a steady increase in the number of Tregs in both peripheral blood and tumor microenvironment (116).
Angiogenesis is another important feature of tumor progression and Tregs are known to play a positive role in HCC angiogenesis. increased levels of intra-tumoral FOXP3 cells was correlated to higher expression of vascular endothelial growth factor (VEGF) and microvascular density (117). HCC cells produce TGF-β1 which could generate Tregs from CD4+ CD25- T cells. This tumor derived TGF-β1 negatively affects the tumor infiltrating leukocytes (TILs) thereby suppressing the immunosurveillance activity in tumor microenvironment. Another disadvantage correlated with TGF-β in HCC is that it induces epithelial-mesenchymal transition in HCC. Indeed, silencing TGF-β1 prolongs the survival rate in mouse models of HCC (1, 118). The levels of growth differentiation factor 15 (GDF15), which belongs to the family of TGF-β, are upregulated during inflammatory conditions or cancer as compared to normal physiological conditions. GDF15 promotes and modulates the proliferation of pTregs and the suppressive function of nTregs through CD48 T cell receptor and by post-transcriptional regulation of FoxP3 in humans (119). Constructing and validating an angiogenesis-related scoring (ARGs) model holds great promise for prognostication, tumor immune microenvironment assessment, and therapeutic stratification in HCC. A recent study harnessed the TCGA dataset to identify 97 differentially expressed ARGs linked to the prognosis of HCC patients. Nine-gene signature, accurately predicted unfavorable clinical outcomes, standing out as an independent prognostic indicator for HCC. Furthermore, associations with diverse immune cell enrichment, encompassing CD4+ T cells, Tregs, macrophages, neutrophils, and DCs have been raised, showcasing an inherently immunosuppressive phenotype. Accordingly, a higher ARGs Score reflects an elevated expression of immune checkpoint genes and an ineffective response to immunotherapy (120). In short, there are numerous regulatory mechanisms that initiate the activation and modulation of the immunosuppressive nature of Tregs resulting in the progression of HCC condition. Increasing evidences supported the involvement of new immune cell subtypes in orchestrating the recruitment of Tregs within the tumor microenvironment, particularly in HCC, including IFNγ- Tc17 cells, a distinct subset of CD8 cells producing IL-17. These cells display characteristics that foster tumor progression, marked by elevated CCL20 expression. Consequently, this triggered an escalation in the Tregs infiltration in the tumor microenvironment, correlating with an unfavorable prognosis (121). by using single-cell RNA sequencing (scRNA-seq) and flow cytometry, a specific subset of the innate-like mucosa-associated invariant T cells termed regulatory Mucosal associated invariant T cells (MAITregs) were detected within HCC patients. These MAITregs possess robust immunosuppressive abilities and are derived from a precursor reservoir exhibiting mild Treg-associated characteristics. Their development is triggered under Treg-promoting conditions like β1 adrenergic receptor signaling. Intriguingly, intra-tumoral MAITregs exert immune suppressive effects and are linked to unfavorable clinical outcomes in HCC patients (122).
Alteration of Tregs expression may have deleterious responses in tissues like i) suppression of effector immune response resulting in cancer progression ii) impaired autoimmune and inflammatory responses, and iii) protection against immune cells in viral infection. This suppressive population of immune cells has to be finely tuned to maintain the delicate balance between inflammatory insults, autoimmunity induction and facilitating protection against infection and tumor. Thus, selective purification and expansion of FOXP3+ Tregs is a potent cellular immunotherapy mechanism that could potentially treat T cell mediated inflammatory liver injuries and liver transplantation rejection. For successful Tregs-mediated cell therapy, strategies have been developed to maintain their phenotypic stability and suppressive capabilities post-infusion into the inflamed liver microenvironment. Neutralization of pro-inflammatory cytokines, IL-12 and IL-6 present in the inflamed intrahepatic microenvironment, can stabilize the Tregs (123). In vitro, the mTOR inhibitor rapamycin, in presence of TCR and CD28 or IL-2, can influence rapid expansion and proliferation of Tregs and increase their activity against effector T cells than Tregs grown in the absence of rapamycin (124). TX527, a vitamin D analogue, triggered the induction and migration of Tregs to the site of inflammation and induced the Tregs-based suppression of effector T cell functions. This Tregs’ immunosuppressive activity can be exploited for their therapeutic applicability in inflammatory and autoimmune disorders (125).
In the liver, crosstalk between leukocytes, hepatocytes, HSCs, LSECs, and cholangiocytes is important for maintaining the balance between immune reactivity and immune tolerance. For patient survival after liver transplantation, lifelong intake of immunosuppressants is a necessity. As an alternative approach, a small group of patients underwent Tregs-based therapy. Cells were generated ex vivo by co-culturing recipient lymphocytes with irradiated donor cells in the presence of anti-CD80/CD86 antibodies and were administrated back into the patients. The study was found to be a safer and an effective approach in maintaining immune tolerance without the intake of immunosuppressants (126), thereby proving the important role of Tregs in the induction and maintenance of immune tolerance after liver transplantation (127). During organ transplantation and due to graft-versus-host disease (GvHD), donor T cells get continuously activated and thus react with the host, resulting in tissue damage and extreme complications. Preclinical trials have demonstrated that adoptive transfer of Tregs can regulate GvHD (16). The first clinical trial in the treatment of acute and chronic GvHD using Tregs was performed in 2009. In chronic GvHD, reduction in symptoms and increase in the number of Tregs were observed while in acute GvHD, no significant conclusion was drawn (128).
A breakdown in immune homeostasis mediated by a balance between effector T cells and Tregs results in autoimmune liver diseases (AILDs) like AIH, primary biliary cholangitis and primary sclerosing cholangitis in liver. The current strategies of AILDs are not fully effective and require long term immunosuppressive medications leading to a poor control over hepatic and biliary inflammations. Tregs are highly sensitive to IL-2 which is required for Tregs survival (129). Trials on administration of low doses of IL-2 as an immunomodulatory agent into AIH patients showed positive therapeutic results (130). In animal models, gene transfer for the ectopic expression of autoantigens to liver cells has successfully used Tregs as a therapeutic option for inflammatory and autoimmune diseases. Selective delivery of autoantigens using nanoparticles to hepatic cells like LSECs helps in the generation of auto-antigen specific Tregs in the liver. Such approach has been proved to be effective at least in mice (131). In humans, Tregs therapy in autoimmune liver diseases (AILD) is not yet tested, but similar tests in Type 1 diabetes mellitus was found to be successful suggesting a possibility for AILD based Tregs therapy in the future (35).
HCC is the most prevalent form of primary liver cancer with various causes for tumorigenesis. Immunocompromised tumor environment due to natural suppressor cells like CD4+ CD25+ T cells is one of the major reasons for tumor progression and malignancy. TILs have a negative impact on the progression of solid tumors and a positive impact in anti-cancer therapies and prognosis of cancer (132). The two strategies employed for cancer immunotherapy aim at i) enhancing the number and function of immune effector cells and ii) blocking the immune suppressor cells like Tregs in the tumor microenvironment. Depletion and prevention of Tregs proliferation in the tumor microenvironment, represent accordingly a potential therapeutic measurement for HCC (133). In a mouse model of HCC, treatment with anti-CD25 antibody [PC-61] resulted in the suppression of Tregs thereby reducing tumor growth. Treatment with antibody for chemokine receptor CCR4 reduced the intra-tumoral population of Tregs as well as the tumor growth (Table 3) (134–136). Furthermore, targeting OX40, an immune checkpoint extensively expressed in Tregs, could result in the depletion of HCC tumor cells. A combination therapy using anti PD-1 antibody Nivolumab and anti CTLA-4 antibody ipilimumab resulted in tumor remission in 29% of patients with HCC within 6 weeks of treatment (Table 3) (133, 137). Sirolimus and sorafenib induce apoptosis of Tregs when activated through T cell receptors. Therefore, these molecules can be used to reduce the tumor recurrence rate by keeping the Tregs population regulated (Table 3) (112). A potential mechanism underlying the increased intra-tumoral Treg accumulation induced by anti-PD-1 treatment in HCC, has been proposed. By using single-cell transcriptomic approach, the study reveals that Nrp-1 facilitates the migration behavior of Tregs, and the genes Crem and Tnfrsf9 governing the activity of the terminal suppressive Tregs. As Tregs transition from lymphoid tissues to the tumor, a progression from Nrp-1 + 4-1BB- Tregs to Nrp-1- 4-1BB+ Tregs was reported. Depleting Nrp-1 specifically in Tregs curtailed the heightened intra-tumoral Tregs resulting from anti-PD-1 therapy, and its combination with a 4-1BB agonist enhances the antitumor response. Importantly, employing a Nrp-1 inhibitor alongside a 4-1BB agonist in humanized HCC models demonstrates favorable outcomes and safety, augmenting the anti-tumor effects of PD-1 blockade (6). Administration of daclizumab, a blocking antibody that reduces the Treg numbers, is a potential immunotherapy option for HCC. Depleting the local Treg population while preserving the host immune response, is therefore an important strategic approach for successful immunotherapy in HCC (138).
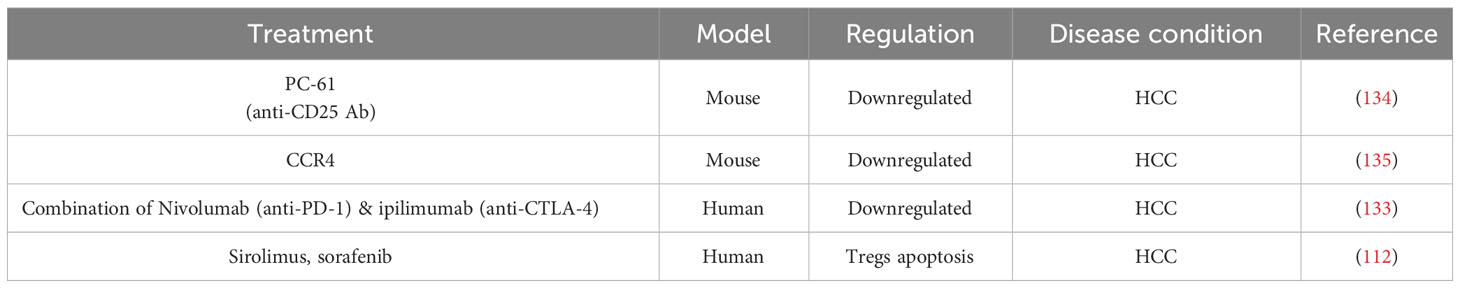
Table 3 Treg based immunotherapy trials in HCC.
Notably, Tregs have a detrimental effect in HCC malignancy due to their anti-tumor immune cell suppression. Thus, immunotherapy targeting the Tregs is an effective way for preventing the progression of non-cancerous lesions into HCC. In this context, focusing on miRNAs to decrease Tregs is an alternative strategy. Recent research illustrates the efficacy of miR-22 delivery via adeno-associated virus in treating HCC, showing superior survival outcomes and lower toxicity when compared to FDA-approved lenvatinib. In liver, miR-22 reduces IL17-producing T cells, resulting in the attenuation of IL17 signaling via HIF1α. Moreover, miR-22 fosters the expansion of cytotoxic T cells while concurrently reducing the presence of Tregs (139).
Adoptive Tregs therapy is an alternative approach for harnessing a Tregs-induced immune suppressive activity. For this approach, Tregs are isolated from the patient, expanded in vitro and reinfused back to the patient. The advantage of this approach is that, the phenotype and functionality of the Tregs can be analyzed prior to their administration (140). Cell surface markers CD4, CD25high CD127low help in the identification of Tregs from non-Tregs (141). However, the functional instability of Tregs pose a challenge for adoptive Tregs therapy, as there is a chance for these infused Tregs to lose FOXP3 expression due to the influence of pro-inflammatory factors and to shift towards effector T phenotypes secreting IL-17 and interferon-γ (48). Firstly, Peripheral blood mononuclear cells (PBMCs) isolated from peripheral blood are subjected to density gradient centrifugation. Via CliniMACS selection, PBMCs were isolated and subjected to negative selection of CD4+ using antibodies against CD8, CD14, CD16, CD19, CD33, CD238a and a positive selection using anti-CD25 antibody for obtaining a high purity CD4+ CD25+ T cell fraction. For expansion and selective enrichment for CD25+, T cells were activated with anti-CD3/CD28 beads and occasionally re-stimulated with rapamycin, IL-2 and RA. During harvest of the expanded Tregs, IL-17 and IFNγ expression is analyzed by intracellular staining and indirect sandwich ELISA methods to deplete CD127+ cells and selectively culture CD4+ CD25+ T cells (142). The suppressive capacity of cultured Tregs is studied in co-culture assays (143). In phase I clinical trial conducted at King’s College Hospital London and University Hospitals Plymouth (UK), Tregs expanded under Good Manufacturing Practices (GMP), were administrated back into the patient at two dosages of 0.5-1 million Tregs/kg and 3-4.5 million Tregs/kg. The study showed that in patients administered with 4.5 million Tregs/kg, the transfer increased the circulating Tregs pool for 1 month as compared to the Treg pool before infusion (144). Cell tracking and dose escalating studies to monitor the safety and efficacy of Tregs are necessary to confirm the accumulation of Tregs in the site of inflammation. However, Tregs choice and antigen specificity, adequate cell number, timing and frequency of administration are still to be considered for achieving adoptive cell therapy (123).
The major disadvantage of adoptive Treg cell therapy is that, Tregs fail to maintain their phenotypic characters for immunosuppression at longer time periods. To overcome this shortcoming, a vector encoding TCR or chimeric antigen receptor (CAR) with defined antigen specificity Tregs, was developed. TCR expressed on Tregs surface determines their specificity. Tregs encoding the genes with TCR or CAR can be genetically engineered to increase the Tregs specificity to antigens present during autoimmunity and not in normal cells. CARs have MHC independent recognition, so they are applicable to patients irrespective of their genotypes (145). CARs have an extracellular antigen recognition site, transmembrane domain and hinge site connected to the intracellular signaling domain. CAR based mechanism of suppression is similar to Tregs mediated immune cell suppression. Suppression of DCs is accomplished by binding to its CD80/CD86 cell surface protein. It can interfere with the metabolism of lymphocytes, via the secretion of anti-inflammatory cytokines like IL-10, IL-35 and TGFβ or direct cell-to-cell interaction induced inhibition and apoptosis (146). Delivery of CAR is by viral vectors like lentiviruses, adeno-associated viruses or adenoviruses or it can be through non-viral vectors like transposons or plasmid vectors (147). Using nanocarriers as vectors can be a safer option than conventional viral and non-viral vectors. The next step in Tregs engineering can be using newer techniques as clustered regularly interspaced short palindromic repeats (CRISPR) by removing genes for cytokine signals like IL-6, TNFγ, and IL-17. Preclinical trials in immunosuppressive diseases have been successful using CAR expressing Tregs (148). These encouraging findings have paved the way for the first clinical trial involving CAR Tregs sponsored by Sangamo Therapeutics, authorized by the UK MHRA and the US (NCT04817774), and targeting kidney transplanted patients. Several other biopharmaceutical companies are closely pursuing similar initiatives, with Quell Therapeutics focusing on CAR Tregs for liver transplanted recipients, indicative of the growing interest and potential in this field (149). An additional study involving HLA-A2-specific CAR Tregs, termed LIBERATE (Phase I/II), is started in 2022 and its primary completion estimated in 2025. Notably, this study will uniquely investigate both clinical outcomes and immunosuppression in the context of liver transplantation (QEL-001). This forward-looking initiative aims to provide valuable insights into the potential efficacy and broader implications of CAR Treg therapy in the realm of liver transplantation (150).
During liver transplantation and autoimmune liver diseases, Tregs are important to prevent GvHD and self-cell destruction. Thus, the future prospects would be the development and administration of Tregs in-vitro cultured or genetically engineered for expressing specific antigens required for self-cell destruction by other immune cells. While, during HCC progression, it is necessary that the Treg numbers are under control for the cytotoxic T cell population to eliminate tumor cells. To summarize, the dysregulation of Tregs is deleterious on the liver disease.
Tregs play an important role in the monitoring of immune balance and have a protective role against excessive inflammation. Tregs are heterogenous cell populations differentiated based on their site of localization, cell markers, homing receptors and chemokine secretions. Tregs play different roles in chronic liver diseases and their progression towards HCC. During liver fibrosis, Tregs have a protective role in preventing chronic inflammation, but in liver tumor microenvironment, the increase in Tregs’ numbers can be detrimental because Tregs target the T effector and NK cells which are required for killing tumor cells and preventing tumor progression and metastasis. Thus, it could be concluded that Tregs play an important role in the progression of chronic liver diseases toward carcinogenesis. Also due to the recent advances in the field of immunotherapy, studies are focusing on using these cells in preventing autoimmune diseases, while using specific blockers to inhibit their activity or altering their phenotype to reduce the immunosuppressive properties in tumor microenvironment. Thus, deeply understanding the mechanism of Tregs behavior in chronic liver diseases can be useful for elucidating the therapeutic options to adopt for preventing the progression of the chronic liver disease to HCC.
AA: Writing – review & editing, Writing – original draft. MM: Writing – review & editing. MA: Writing – review & editing. NH: Writing – review & editing. MeN: Writing – review & editing. MV: Writing – review & editing. ES: Writing – review & editing. MN: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study has received support from “Fonds National de la Recherche Scientifique (FNRS)”, Télévie, and the Research Project (PDR) (Ref: PDR-TLV 7.8513.19).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Yang XH, Yamagiwa S, Ichida T, Matsuda Y, Sugahara S, Watanabe H, et al. Increase of CD4+ CD25+ regulatory T-cells in the liver of patients with hepatocellular carcinoma. J Hepatol. (2006) 45:254–62. doi: 10.1016/j.jhep.2006.01.036
2. Wawman RE, Bartlett H, Oo YH. Regulatory T cell metabolism in the hepatic microenvironment. Front Immunol. (2018) 8:1889. doi: 10.3389/fimmu.2017.01889
3. Lutsiak MC, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H. Inhibition of CD4 + 25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood. (2005) 105:2862–8. doi: 10.1182/blood-2004-06-2410
4. Kobayashi N, Hiraoka N, Yamagami W, Ojima H, Kanai Y, Kosuge T, et al. FOXP3+ regulatory T cells affect the development and progression of hepatocarcinogenesis. Clin Cancer Res. (2007) 13:902–11. doi: 10.1158/1078-0432.CCR-06-2363
5. Zhang X, Lou J, Bai L, Chen Y, Zheng S, Duan Z. Immune regulation of intrahepatic regulatory T cells in fibrotic livers of mice. Med Sci Monitor: Int Med J Exp Clin Res. (2017) 23:1009. doi: 10.12659/MSM.899725
6. Wu Q, Pan C, Zhou Y, Wang S, Xie L, Zhou W, et al. Targeting neuropilin-1 abolishes anti-PD-1-upregulated regulatory T cells and synergizes with 4-1BB agonist for liver cancer treatment. Hepatology. (2023) 78:1402–17. doi: 10.1097/HEP.0000000000000320
7. Sun L, Xu G, Liao W, Yang H, Xu H, Du S, et al. Clinicopathologic and prognostic significance of regulatory T cells in patients with hepatocellular carcinoma: a meta-analysis. Oncotarget. (2017) 8:39658. doi: 10.18632/oncotarget.v8i24
8. Dunham RM, Cervasi B, Brenchley JM, Albrecht H, Weintrob A, Sumpter B, et al. CD127 and CD25 expression defines CD4+ T cell subsets that are differentially depleted during HIV infection. J Immunol (Baltimore Md.: 1950). (2008) 180:5582–92. doi: 10.4049/jimmunol.180.8.5582
9. Li Z, Li D, Tsun A, Li B. FOXP3+ regulatory T cells and their functional regulation. Cell Mol Immunol. (2015) 12:558–65. doi: 10.1038/cmi.2015.10
10. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. (2015) 74:5–17. doi: 10.1016/j.cyto.2014.09.011
11. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+ CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
12. Allan SE, Passerini L, Bacchetta R, Crellin N, Dai M, Orban PC, et al. The role of 2 FOXP3 isoforms in the generation of human CD4+ Tregs. J Clin Invest. (2005) 115:3276–84. doi: 10.1172/JCI24685
13. Song Y, Wang N, Chen L, Fang L. Tr1 cells as a key regulator for maintaining immune homeostasis in transplantation. Front Immunol. (2021) 12:671579. doi: 10.3389/fimmu.2021.671579
14. Revilla-Nuin B, de Bejar Á., Martínez-Alarcón L, Herrero JI, Martínez-Cáceres CM, Ramírez P, et al. Differential profile of activated regulatory T cell subsets and microRNAs in tolerant liver transplant recipients. Liver Transplant. (2017) 23:933–45. doi: 10.1002/lt.24691
15. Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, et al. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity. (2011) 35:109–22. doi: 10.1016/j.immuni.2011.03.029
16. Romano M, Tung SL, Smyth LA, Lombardi G. Treg therapy in transplantation: a general overview. Transplant Int. (2017) 30:745–53. doi: 10.1111/tri.2017.30.issue-8
17. Golubovskaya V, Wu L. Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers. (2016) 8:36. doi: 10.3390/cancers8030036
18. Shevyrev D, Tereshchenko V. Treg heterogeneity, function, and homeostasis. Front Immunol. (2020) 10:3100. doi: 10.3389/fimmu.2019.03100
19. Workman CJ, Szymczak-Workman AL, Collison LW, Pillai MR, Vignali DA. The development and function of regulatory T cells. Cell Mol Life Sci. (2009) 66:2603–22. doi: 10.1007/s00018-009-0026-2
20. Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev. (2014) 259:88–102. doi: 10.1111/imr.12160
21. Gratz IK, Truong HA, Yang SHY, Maurano MM, Lee K, Abbas AK, et al. Cutting Edge: memory regulatory t cells require IL-7 and not IL-2 for their maintenance in peripheral tissues. J Immunol. (2013) 190:4483–7. doi: 10.4049/jimmunol.1300212
22. Huehn J, Hamann A. Homing to suppress: address codes for Treg migration. Trends Immunol. (2005) 26:632–6. doi: 10.1016/j.it.2005.10.001
23. Liberal R, Grant CR, Longhi MS, Mieli-Vergani G, Vergani D. Regulatory T cells: mechanisms of suppression and impairment in autoimmune liver disease. IUBMB Life. (2015) 67:88–97. doi: 10.1002/iub.1349
24. Askenasy N, Kaminitz A, Yarkoni S. Mechanisms of T regulatory cell function. Autoimmun Rev. (2008) 7:370–5. doi: 10.1016/j.autrev.2008.03.001
25. Sojka DK, Huang YH, Fowell DJ. Mechanisms of regulatory T-cell suppression–a diverse arsenal for a moving target. Immunology. (2008) 124:13–22. doi: 10.1111/j.1365-2567.2008.02813.x
26. Yu X, Huang C, Song B, Xiao Y, Fang M, Feng J, et al. CD4+CD25+ regulatory T cells-derived exosomes prolonged kidney allograft survival in a rat model. Cell Immunol. (2013) 285:62–8. doi: 10.1016/j.cellimm.2013.06.010
27. Okoye IS, Coomes SM, Pelly VS, Czieso S, Papayannopoulos V, Tolmachova T, et al. MicroRNA-containing T-regulatory-cell-derived exosomes suppress pathogenic T helper 1 cells. Immunity. (2014) 41:503. doi: 10.1016/j.immuni.2014.08.008
28. Aiello S, Rocchetta F, Longaretti L, Faravelli S, Todeschini M, Cassis L, et al. Extracellular vesicles derived from T regulatory cells suppress T cell proliferation and prolong allograft survival. Sci Rep. (2017) 7:11518. doi: 10.1038/s41598-017-08617-3
29. Lin C, Guo J, Jia R. Roles of regulatory T cell-derived extracellular vesicles in human diseases. Int J Mol Sci. (2022) 23:11206. doi: 10.3390/ijms231911206
30. Sarris M, Andersen KG, Randow F, Mayr L, Betz AG. Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition. Immunity. (2008) 28:402–13. doi: 10.1016/j.immuni.2008.01.012
31. Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. (2009) 30:636–45. doi: 10.1016/j.immuni.2009.04.010
32. Mahnke K, Johnson TS, Ring S, Enk AH. Tolerogenic dendritic cells and regulatory T cells: a two-way relationship. J Dermatol Sci. (2007) 46:159–67. doi: 10.1016/j.jdermsci.2007.03.002
33. Matta BM, Castellaneta A, Thomson AW. Tolerogenic plasmacytoid DC. Eur J Immunol. (2010) 40:2667–76. doi: 10.1002/eji.201040839
34. Wing JB, Tanaka A, Sakaguchi S. Human FOXP3+ regulatory T cell heterogeneity and function in autoimmunity and cancer. Immunity. (2019) 50:302–16. doi: 10.1016/j.immuni.2019.01.020
35. Jeffery HC, Braitch MK, Brown S, Oo YH. Clinical potential of regulatory T cell therapy in liver diseases: an overview and current perspectives. Front Immunol. (2016) 7:334. doi: 10.3389/fimmu.2016.00334
36. Terness P, Chuang JJ, Opelz G. The immunoregulatory role of IDO-producing human dendritic cells revisited. Trends Immunol. (2006) 27:68–73. doi: 10.1016/j.it.2005.12.006
37. Cheng JT, Deng YN, Yi HM, Wang GY, Fu BS, Chen WJ, et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis. (2016) 5:e198–8. doi: 10.1038/oncsis.2016.7
38. Chen L, Han X. Anti–PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. (2015) 125:3384–91. doi: 10.1172/JCI80011
39. Gianchecchi E, Fierabracci A. Inhibitory receptors and pathways of lymphocytes: the role of PD-1 in Treg development and their involvement in autoimmunity onset and cancer progression. Front Immunol. (2018) 9:2374. doi: 10.3389/fimmu.2018.02374
40. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol. (2012) 3:51. doi: 10.3389/fimmu.2012.00051
41. Wing JB, Sakaguchi S. Multiple treg suppressive modules and their adaptability. Front Immunol. (2012) 3:178. doi: 10.3389/fimmu.2012.00178
42. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+ CD25+ Foxp3+ regulatory T cells induce cytokine deprivation–mediated apoptosis of effector CD4+ T cells. Nat Immunol. (2007) 8:1353–62. doi: 10.1038/ni1536
43. Zheng J, Chan PL, Liu Y, Qin G, Xiang Z, Lam KT, et al. ICOS regulates the generation and function of human CD4+ Treg in a CTLA-4 dependent manner. PloS One. (2013) 8:e82203. doi: 10.1371/journal.pone.0082203
44. Shen X, Li N, Li H, Zhang T, Wang F, Li Q. Increased prevalence of regulatory T cells in the tumor microenvironment and its correlation with TNM stage of hepatocellular carcinoma. J Cancer Res Clin Oncol. (2010) 136:1745–54. doi: 10.1007/s00432-010-0833-8
45. Simpson TR, Quezada SA, Allison JP. Regulation of CD4 T cell activation and effector function by inducible costimulator (ICOS). Curr Opin Immunol. (2010) 22:326–32. doi: 10.1016/j.coi.2010.01.001
46. Tu JF, Ding YH, Ying XH, Wu FZ, Zhou XM, Zhang DK, et al. Regulatory T cells, especially ICOS+ FOXP3+ regulatory T cells, are increased in the hepatocellular carcinoma microenvironment and predict reduced survival. Sci Rep. (2016) 6:35056. doi: 10.1038/srep35056
47. Oh-hora M, Rao A. Calcium signaling in lymphocytes. Curr Opin Immunol. (2008) 20:250–8. doi: 10.1016/j.coi.2008.04.004
48. Singer BD, King LS, D’Alessio FR. Regulatory T cells as immunotherapy. Front Immunol. (2014) 5:46. doi: 10.3389/fimmu.2014.00046
49. Jiang H, Chess L. An integrated view of suppressor T cell subsets in immunoregulation. J Clin Invest. (2004) 114:1198–208. doi: 10.1172/JCI200423411
50. Wiegard C, Frenzel C, Herkel J, Kallen KJ, Schmitt E, Lohse AW. Murine liver antigen presenting cells control suppressor activity of CD4+ CD25+ regulatory T cells. Hepatology. (2005) 42:193–9. doi: 10.1002/(ISSN)1527-3350
51. Sawant DV, Vignali DAA. Once a treg, always a treg? Immunol Rev. (2014) 259:173–91. doi: 10.1111/imr.12173
52. Kurt AS, Strobl K, Ruiz P, Osborn G, Chester T, Dawson L, et al. IL-2 availability regulates the tissue specific phenotype of murine intra-hepatic Tregs. Front Immunol. (2022) 13:1040031. doi: 10.3389/fimmu.2022.1040031
53. Bergmann C, Wild CA, Narwan M, Lotfi R, Lang S, Brandau S. Human tumor-induced and naturally occurring Treg cells differentially affect NK cells activated by either IL-2 or target cells. Eur J Immunol. (2011) 41:3564–73. doi: 10.1002/eji.201141532
54. Santodomingo-Garzon T, Swain MG. Role of NKT cells in autoimmune liver disease. Autoimmun Rev. (2011) 10:793–800. doi: 10.1016/j.autrev.2011.06.003
55. Hua J, Liang S, Ma X, Webb TJ, Potter JP, Li Z. The interaction between regulatory T cells and NKT cells in the liver: a CD1d bridge links innate and adaptive immunity. PloS One. (2011) 6:e27038. doi: 10.1371/journal.pone.0027038
56. Kolios G, Valatas V, Kouroumalis E. Role of Kupffer cells in the pathogenesis of liver disease. World J gastroenterol: WJG. (2006) 12:7413. doi: 10.3748/wjg.v12.i46.7413
57. Breous E, Somanathan S, Vandenberghe LH, Wilson JM. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology. (2009) 50:612–21. doi: 10.1002/hep.23043
58. Schon HT, Weiskirchen R. Immunomodulatory effects of transforming growth factor-β in the liver. Hepatobiliary Surg Nutr. (2014) 3:386. doi: 10.3978/j.issn.2304-3881.2014.11.06
59. Wang H, Lu CH, Ho PC. Metabolic adaptation orchestrates tissue context-dependent behavior in regulatory T cells. Immunol Rev. (2020) 295:126–39. doi: 10.1111/imr.12844
60. Kempkes RW, Joosten I, Koenen HJ, He X. Metabolic pathways involved in regulatory T cell functionality. Front Immunol. (2019) 10:2839. doi: 10.3389/fimmu.2019.02839
61. Wang H, Franco F, Ho PC. Metabolic regulation of Tregs in cancer: opportunities for immunotherapy. Trends Cancer. (2017) 3:583–92. doi: 10.1016/j.trecan.2017.06.005
62. Galgani M, De Rosa V, La Cava A, Matarese G. Role of metabolism in the immunobiology of regulatory T cells. J Immunol. (2016) 197:2567–75. doi: 10.4049/jimmunol.1600242
63. Shi H, Chi H. Metabolic control of Treg cell stability, plasticity, and tissue-specific heterogeneity. Front Immunol. (2019) 10. doi: 10.3389/fimmu.2019.02716
64. Liu Q, Du F, Huang W, Ding X, Wang Z, Yan F, et al. Epigenetic control of Foxp3 in intratumoral T-cells regulates growth of hepatocellular carcinoma. Aging (Albany NY). (2019) 11:2343. doi: 10.18632/aging.v11i8
65. Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol. (2009) 9:83–9. doi: 10.1038/nri2474
66. Barbi J, Pardoll D, Pan F. Treg functional stability and its responsiveness to the microenvironment. Immunol Rev. (2014) 259:115–39. doi: 10.1111/imr.12172
67. Liu Y, Wang L, Predina J, Han R, Beier UH, Wang LCS, et al. Inhibition of p300 impairs Foxp3+ T regulatory cell function and promotes antitumor immunity. Nat Med. (2013) 19:1173–7. doi: 10.1038/nm.3286
68. Deng G, Song X, Fujimoto S, Piccirillo CA, Nagai Y, Greene MI. Foxp3 post-translational modifications and Treg suppressive activity. Front Immunol. (2019) 10:2486. doi: 10.3389/fimmu.2019.02486
69. Kohlhaas S, Garden OA, Scudamore C, Turner M, Okkenhaug K, Vigorito E. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J Immunol. (2009) 182:2578–82. doi: 10.4049/jimmunol.0803162
70. Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, et al. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. (2010) 142:914–29. doi: 10.1016/j.cell.2010.08.012
71. Qin A, Wen Z, Zhou Y, Li Y, Li Y, Luo J, et al. Micro RNA-126 regulates the induction and function of CD 4+ Foxp3+ regulatory T cells through PI 3K/AKT pathway. J Cell Mol Med. (2013) 17:252–64. doi: 10.1111/jcmm.12003
72. Takahashi H, Kanno T, Nakayamada S, Hirahara K, Sciumè G, Muljo SA, et al. TGF-β and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat Immunol. (2012) 13:587–95. doi: 10.1038/ni.2286
73. Fayyad-Kazan H, Rouas R, Fayyad-Kazan M, Badran R, El Zein N, Lewalle P, et al. MicroRNA profile of circulating CD4-positive regulatory T cells in human adults and impact of differentially expressed microRNAs on expression of two genes essential to their function. J Biol Chem. (2012) 287:9910–22. doi: 10.1074/jbc.M111.337154
74. Singh Y, Garden OA, Lang F, Cobb BS. MicroRNA-15b/16 enhances the induction of regulatory T cells by regulating the expression of Rictor and mTOR. J Immunol. (2015) 195:5667–77. doi: 10.4049/jimmunol.1401875
75. Huang B, Zhao J, Lei Z, Shen S, Li D, Shen GX, et al. miR-142-3p restricts cAMP production in CD4+CD25- T cells and CD4+CD25+ TREG cells by targeting AC9 mRNA. EMBO Rep. (2009) 10:180–5. doi: 10.1038/embor.2008.224
76. Fayyad-Kazan M, Rouas R, Merimi M, Najar M, Badran B, Lewalle P, et al. Human CD4+CD25+CD127lowFOXP3+ regulatory T lymphocytes and CD4+CD25-FOXP3- conventional T lymphocytes: a differential transcriptome profile. Nucleosides Nucleotides Nucleic Acids. (2023) 29:1–11. doi: 10.1080/15257770.2023.2216226
77. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest. (2017) 127:55–64. doi: 10.1172/JCI88881
78. Sun XF, Gu L, Deng WS, Xu Q. Impaired balance of T helper 17/T regulatory cells in carbon tetrachloride-induced liver fibrosis in mice. World J Gastroenterol: WJG. (2014) 20:2062. doi: 10.3748/wjg.v20.i8.2062
79. Roh YS, Park S, Lim CW, Kim B. Depletion of Foxp3+ regulatory T cells promotes profibrogenic milieu of cholestasis-induced liver injury. Digestive Dis Sci. (2015) 60:2009–18. doi: 10.1007/s10620-014-3438-2
80. He B, Wu L, Xie W, Shao Y, Jiang J, Zhao Z, et al. The imbalance of Th17/Treg cells is involved in the progression of nonalcoholic fatty liver disease in mice. BMC Immunol. (2017) 18:1–9. doi: 10.1186/s12865-017-0215-y
81. Cairoli V, De Matteo E, Rios D, Lezama C, Galoppo M, Casciato P, et al. Hepatic lymphocytes involved in the pathogenesis of pediatric and adult non-alcoholic fatty liver disease. Sci Rep. (2021) 11:5129. doi: 10.1038/s41598-021-84674-z
82. Rau M, Schilling AK, Meertens J, Hering I, Weiss J, Jurowich C, et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of Th17 cells in the liver and an increased Th17/resting regulatory T cell ratio in peripheral blood and in the liver. J Immunol. (2016) 196:97–105. doi: 10.4049/jimmunol.1501175
83. Dywicki J, Buitrago-Molina LE, Noyan F, Davalos-Misslitz AC, Hupa-Breier KL, Lieber M, et al. The detrimental role of regulatory T cells in nonalcoholic steatohepatitis. Hepatol Commun. (2022) 6:320–33. doi: 10.1002/hep4.1807
84. Almeida J, Polvorosa MA, Gonzalez-Quintela A, Marcos M, Pastor I, Hernandez Cerceno ML, et al. Decreased peripheral blood CD 4+/CD 25+ Regulatory T cells in patients with alcoholic hepatitis. Alcoholism: Clin Exp Res. (2013) 37:1361–9. doi: 10.1111/acer.12095
85. Czaja AJ. Hepatic inflammation and progressive liver fibrosis in chronic liver disease. World J gastroenterol: WJG. (2014) 20:2515. doi: 10.3748/wjg.v20.i10.2515
86. Zhang X, Feng M, Liu X, Bai L, Kong M, Chen Y, et al. Persistence of cirrhosis is maintained by intrahepatic regulatory T cells that inhibit fibrosis resolution by regulating the balance of tissue inhibitors of metalloproteinases and matrix metalloproteinases. Trans Res. (2016) 169:67–79. doi: 10.1016/j.trsl.2015.10.008
87. Li X, Su Y, Hua X, Xie C, Liu J, Huang Y, et al. Levels of hepatic Th17 cells and regulatory T cells upregulated by hepatic stellate cells in advanced HBV-related liver fibrosis. J Trans Med. (2017) 15:1–11. doi: 10.1186/s12967-017-1167-y
88. Claassen MA, de Knegt RJ, Tilanus HW, Janssen HL, Boonstra A. Abundant numbers of regulatory T cells localize to the liver of chronic hepatitis C infected patients and limit the extent of fibrosis. J Hepatol. (2010) 52:315–21. doi: 10.1016/j.jhep.2009.12.013
89. Wu KJ, Qian QF, Zhou JR, Sun DL, Duan YF, Zhu X, et al. Regulatory T cells (Tregs) in liver fibrosis. Cell Death Discovery. (2023) 9:53. doi: 10.1038/s41420-023-01347-8
90. Sugimoto K, Ikeda F, Stadanlick J, Nunes FA, Alter HJ, Chang KM. Suppression of HCV-specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology. (2003) 38:1437–48. doi: 10.1016/j.hep.2003.09.026
91. Alatrakchi N, Koziel M. Regulatory T cells and viral liver disease. J Viral hepatitis. (2009) 16:223–9. doi: 10.1111/j.1365-2893.2009.01081.x
92. Li J, Qiu SJ, She WM, Wang FP, Gao H, Li L, et al. Significance of the balance between regulatory T (Treg) and T helper 17 (Th17) cells during hepatitis B virus related liver fibrosis. PloS One. (2012) 7:e39307. doi: 10.1371/journal.pone.0039307
93. Zhang H, Yan X, Yang C, Zhan Q, Fu Y, Luo H, et al. Intrahepatic T helper 17 cells recruited by hepatitis B virus X antigen-activated hepatic stellate cells exacerbate the progression of chronic hepatitis B virus infection. J Viral hepatitis. (2020) 27:1138–49. doi: 10.1111/jvh.13352
94. Ouaguia L, Moralès O, Aoudjehane L, Wychowski C, Kumar A, Dubuisson J, et al. Hepatitis C virus improves human tregs suppressive function and promotes their recruitment to the liver. Cells. (2019) 8:1296. doi: 10.3390/cells8101296
95. Chang KM. Regulatory T cells and the liver: a new piece of the puzzle. Hepatology. (2005) 41:700–2. doi: 10.1002/(ISSN)1527-3350
96. Cheng LS, Liu Y, Jiang W. Restoring homeostasis of CD4+ T cells in hepatitis-B-virus-related liver fibrosis. World J Gastroenterol: WJG. (2015) 21:10721. doi: 10.3748/wjg.v21.i38.10721
97. Chow Z, Banerjee A, Hickey MJ. Controlling the fire—tissue-specific mechanisms of effector regulatory T-cell homing. Immunol Cell Biol. (2015) 93:355–63. doi: 10.1038/icb.2014.117
98. Richardson N, Wootton GE, Bozward AG, Oo YH. Challenges and opportunities in achieving effective regulatory T cell therapy in autoimmune liver disease. Semin immunopathol. (2022) 44:461–74. doi: 10.1007/s00281-022-00940-w
99. Fu H, Ward EJ, Marelli-Berg FM. Mechanisms of T cell organotropism. Cell Mol Life Sci. (2016) 73:3009–33. doi: 10.1007/s00018-016-2211-4
100. Osei-Bordom D, Bozward AG, Oo YH. The hepatic microenvironment and regulatory T cells. Cell Immunol. (2020) 357:104195. doi: 10.1016/j.cellimm.2020.104195
101. Erhardt A, Wegscheid C, Claass B, Carambia A, Herkel J, Mittrücker HW, et al. CXCR3 deficiency exacerbates liver disease and abrogates tolerance in a mouse model of immune-mediated hepatitis. J Immunol. (2011) 186:5284–93. doi: 10.4049/jimmunol.1003750
102. Mempel TR, Marangoni F. Guidance factors orchestrating regulatory T cell positioning in tissues during development, homeostasis, and response. Immunol Rev. (2019) 289:129–41. doi: 10.1111/imr.12761
103. Oo YH, Shetty S, Adams DH. The role of chemokines in the recruitment of lymphocytes to the liver. Digestive Dis. (2010) 28:31–44. doi: 10.1159/000282062
104. Yang ZQ, Yang ZY, Zhang LD, Ping-Bie, Wang SG, Ma KS, et al. Increased liver-infiltrating CD8+FoxP3+ regulatory T cells are associated with tumor stage in hepatocellular carcinoma patients. Hum Immunol. (2010) 71(12):1180–6. doi: 10.1016/j.humimm.2010.09.011
105. Nishida N, Kudo M. Immunological microenvironment of hepatocellular carcinoma and its clinical implication. Oncology. (2017) 92:40–9. doi: 10.1159/000451015
106. Thakur S, Singla A, Chawla Y, Rajwanshi A, Kalra N, Arora SK. Expansion of peripheral and intratumoral regulatory T-cells in hepatocellular carcinoma: a case-control study. Indian J Pathol Microbiol. (2011) 54:448. doi: 10.4103/0377-4929.85073
107. Chen KJ, Zhou L, Xie HY, Ahmed TE, Feng XW, Zheng SS. Intratumoral regulatory T cells alone or in combination with cytotoxic T cells predict prognosis of hepatocellular carcinoma after resection. Med Oncol. (2012) 29:1817–26. doi: 10.1007/s12032-011-0006-x
108. Lim CJ, Lee YH, Pan L, Lai L, Chua C, Wasser M, et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut. (2019) 68:916–27. doi: 10.1136/gutjnl-2018-316510
109. Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. (2007) 132:2328–39. doi: 10.1053/j.gastro.2007.03.102
110. Feng H, Zhuo Y, Zhang X, Li Y, Li Y, Duan X, et al. Tumor microenvironment in hepatocellular carcinoma: key players for immunotherapy. J Hepatocell Carcinoma. (2022) 9:1109–25. doi: 10.2147/JHC.S381764
111. Li W, Han J, Wu H. Regulatory T-cells promote hepatitis B virus infection and hepatocellular carcinoma progression. Chronic Dis Trans Med. (2016) 2:67–80. doi: 10.1016/j.cdtm.2016.09.001
112. Lee WC, Wu TJ, Chou HS, Yu MC, Hsu PY, Hsu HY, et al. The impact of CD4+ CD25+ T cells in the tumor microenvironment of hepatocellular carcinoma. Surgery. (2012) 151:213–22. doi: 10.1016/j.surg.2011.07.029
113. Chang H, Ni Y, Shen C, Li C, He K, Zhu X, et al. Peritoneal GATA6+ macrophage drives hepatic immunopathogenesis and maintains the Treg cell niche in the liver. Immunology. (2022) 167(1):77–93. doi: 10.1111/imm.13519
114. Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol. (2007) 25:2586–93. doi: 10.1200/JCO.2006.09.4565
115. Huang Q, Lam AJ, Boardman DA, Dawson NAJ, Levings MK. Suppression of human dendritic cells by regulatory T cells. Bio Protoc. (2021) 11:e4217. doi: 10.21769/BioProtoc.4217
116. Ormandy LA, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. (2005) 65:2457–64. doi: 10.1158/0008-5472.CAN-04-3232
117. Huang Y, Wang FM, Wang T, Wang YJ, Zhu ZY, Gao YT, et al. Tumor-infiltrating FoxP3+ Tregs and CD8+ T cells affect the prognosis of hepatocellular carcinoma patients. Digestion. (2012) 86:329–37. doi: 10.1159/000342801
118. Wang Y, Liu T, Tang W, Deng B, Chen Y, Zhu J, et al. Hepatocellular carcinoma cells induce regulatory T cells and lead to poor prognosis via production of transforming growth factor-β1. Cell Physiol Biochem. (2016) 38:306–18. doi: 10.1159/000438631
119. Wang Z, He L, Li W, Xu C, Zhang J, Wang D, et al. GDF15 induces immunosuppression via CD48 on regulatory T cells in hepatocellular carcinoma. J ImmunoTherapy Cancer. (2021) 9(9):e002787. doi: 10.1136/jitc-2021-002787
120. Chen BJ, Zhao JW, Zhang DH, Zheng AH, Wu GQ. Immunotherapy of cancer by targeting regulatory T cells. Int Immunopharmacol. (2022) 104:108469. doi: 10.1016/j.intimp.2021.108469
121. Lee YH, Chuah S, Nguyen PHD, Lim CJ, Lai HLH, Wasser M, et al. IFNγ-IL-17+ CD8 T cells contribute to immunosuppression and tumor progression in human hepatocellular carcinoma. Cancer Lett. (2023) 552:215977. doi: 10.1016/j.canlet.2022.215977
122. Fu S, Liu M, Zhu C, Zhang H, Zhao C, Xie Y, et al. Regulatory mucosa-associated invariant T cells controlled by β1 adrenergic receptor signaling contribute to hepatocellular carcinoma progression. Hepatol. (2023) 78:72–87. doi: 10.1097/HEP.0000000000000014
123. Oo YH, Sakaguchi S. Regulatory T-cell directed therapies in liver diseases. J Hepatol. (2013) 59:1127–34. doi: 10.1016/j.jhep.2013.05.034
124. Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+ CD25+ Foxp3+ regulatory T cells cultured with rapamycin. J Immunol. (2007) 178:320–9. doi: 10.4049/jimmunol.178.1.320
125. Baeke F, Korf H, Overbergh L, Verstuyf A, Thorrez L, Van Lommel L, et al. The vitamin D analog, TX527, promotes a human CD4+ CD25highCD127low regulatory T cell profile and induces a migratory signature specific for homing to sites of inflammation. J Immunol. (2011) 186:132–42. doi: 10.4049/jimmunol.1000695
126. Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology. (2016) 64:632–43. doi: 10.1002/hep.28459
127. Lei H, Reinke P, Volk HD, Lv Y, Wu R. Mechanisms of immune tolerance in liver transplantation-crosstalk between alloreactive T cells and liver cells with therapeutic prospects. Front Immunol. (2019) , 10:2667. doi: 10.3389/fimmu.2019.02667
128. Trzonkowski P, Bieniaszewska M, Juścińska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+ CD25+ CD127– T regulatory cells. Clin Immunol. (2009) 133:22–6. doi: 10.1016/j.clim.2009.06.001
129. Terry LV, Oo YH. The next frontier of regulatory T cells: Promising immunotherapy for autoimmune diseases and organ transplantations. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.565518
130. Lim TY, Martinez-Llordella M, Kodela E, Gray E, Heneghan MA, Sanchez-Fueyo A. Low-dose interleukin-2 for refractory autoimmune hepatitis. Hepatology. (2018) 68:1649–52. doi: 10.1002/hep.30059
131. Herkel J. Regulatory T cells in hepatic immune tolerance and autoimmune liver diseases. Digestive Dis. (2015) 33.Suppl. 2:70–4. doi: 10.1159/000440750
132. Atanasov G, Dino K, Schierle K, Dietel C, Aust G, Pratschke J, et al. Immunologic cellular characteristics of the tumour microenvironment of hepatocellular carcinoma drive patient outcomes. World J Surg Oncol. (2019) 17:97. doi: 10.1186/s12957-019-1635-3
133. Zongyi Y, Xiaowu L. Immunotherapy for hepatocellular carcinoma. Cancer Lett. (2020) 470:8–17. doi: 10.1016/j.canlet.2019.12.002
134. Cany J, Tran L, Gauttier V, Judor JP, Vassaux G, Ferry N, et al. Immunotherapy of hepatocellular carcinoma: is there a place for regulatory T-lymphocyte depletion? Immunotherapy. (2011) 3:32–4. doi: 10.2217/imt.11.29
135. Mukaida N, Nakamoto Y. Emergence of immunotherapy as a novel way to treat hepatocellular carcinoma. World J Gastroenterol. (2018) 24:1839. doi: 10.3748/wjg.v24.i17.1839
136. Chaudhary B, Elkord E. Regulatory T cells in the tumour microenvironment and cancer progression: role and therapeutic targeting. Vaccines. (2016) 4:28. doi: 10.3390/vaccines4030028
137. Hong YP, Li ZD, Prasoon P, Zhang Q. Immunotherapy for hepatocellular carcinoma: From basic research to clinical use. World J Hepatol. (2015) 7:980. doi: 10.4254/wjh.v7.i7.980
138. Hofmann J, Hackl V, Esser H, Meszaros AT, Fodor M, Öfner D, et al. Cell-based regeneration and treatment of liver diseases. Int J Mol Sci. (2021) 22:10276. doi: 10.3390/ijms221910276
139. Hu Y, Setayesh T, Vaziri F, Wu X, Hwang ST, Chen X, et al. miR-22 gene therapy treats HCC by promoting anti-tumor immunity and enhancing metabolism. Mol Ther. (2023) 31:1829–45. doi: 10.1016/j.ymthe.2023.04.019
140. Riley JL, June CH, Blazar BR. Human T regulatory cell therapy: take a billion or so and call me in the morning. Immunity. (2009) 30:656–65. doi: 10.1016/j.immuni.2009.04.006
141. Whitehouse GP, Hope A, Sanchez-Fueyo A. Regulatory T-cell therapy in liver transplantation. Transplant Int. (2017) 30:776–84. doi: 10.1111/tri.2017.30.issue-8
142. Safinia N, Vaikunthanathan T, Fraser H, Thirkell S, Lowe K, Blackmore L, et al. Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget. (2016) 7:7563. doi: 10.18632/oncotarget.v7i7
143. Peters JH, Preijers FW, Woestenenk R, Hilbrands LB, Koenen HJ, Joosten I. Clinical grade Treg: GMP isolation, improvement of purity by CD127 pos depletion, Treg expansion, and Treg cryopreservation. PloS One. (2008) 3:e3161. doi: 10.1371/journal.pone.0003161
144. Sánchez-Fueyo A, Whitehouse G, Grageda N, Cramp ME, Lim TY, Romano M, et al. Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. Am J Transplant. (2020) 20:1125–36. doi: 10.1111/ajt.15700
145. McGovern JL, Wright GP, Stauss HJ. Engineering specificity and function of therapeutic regulatory T cells. Front Immunol. (2017) 8:1517. doi: 10.3389/fimmu.2017.01517
146. Yi LI, Weifan Y, Huan Y. Chimeric antigen receptor–engineered regulatory T lymphocytes: promise for immunotherapy of autoimmune disease. Cytotherapy. (2019) 21:925–34. doi: 10.1016/j.jcyt.2019.04.060
147. Mohseni YR, Tung SL, Dudreuilh C, Lechler RI, Fruhwirth GO, Lombardi G. The future of regulatory T cell therapy: promises and challenges of implementing CAR technology. Front Immunol. (2020) 11:1608. doi: 10.3389/fimmu.2020.01608
148. Henschel P, Landwehr-Kenzel S, Engels N, Schienke A, Kremer J, Riet T, et al. Supraphysiological FOXP3 expression in human CAR-Tregs results in improved stability, efficacy, and safety of CAR-Treg products for clinical application. J Autoimmun. (2023) 138:103057. doi: 10.1016/j.jaut.2023.103057
149. Arjomandnejad M, Kopec AL, Keeler AM. CAR-T regulatory (CAR-treg) cells: engineering and applications. Biomedicines. (2022) 10:287. doi: 10.3390/biomedicines10020287
Keywords: T regulatory cells, foxp3, hepatic microenvironment, liver fibrosis, liver cirrhosis, HCC, Treg immunotherapy
Citation: Ajith A, Merimi M, Arki MK, Hossein-khannazer N, Najar M, Vosough M, Sokal EM and Najimi M (2024) Immune regulation and therapeutic application of T regulatory cells in liver diseases. Front. Immunol. 15:1371089. doi: 10.3389/fimmu.2024.1371089
Received: 16 January 2024; Accepted: 28 February 2024;
Published: 20 March 2024.
Edited by:
Yang Xie, Harvard Medical School, United StatesReviewed by:
Jingyuan Wang, University of Pittsburgh, United StatesCopyright © 2024 Ajith, Merimi, Arki, Hossein-khannazer, Najar, Vosough, Sokal and Najimi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mustapha Najimi, bXVzdGFwaGEubmFqaW1pQHVjbG91dmFpbi5iZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.