 Brian M. Lozinski
Brian M. Lozinski Samira Ghorbani
Samira Ghorbani V. Wee Yong
V. Wee Yong- Hotchkiss Brain Institute and the Department of Clinical Neuroscience, University of Calgary, Calgary, AB, Canada
Tissue damage elicits a wound healing response of inflammation and remodeling aimed at restoring homeostasis. Dysregulation of wound healing leads to accumulation of effector cells and extracellular matrix (ECM) components, collectively termed fibrosis, which impairs organ functions. Fibrosis of the central nervous system, neurofibrosis, is a major contributor to the lack of neural regeneration and it involves fibroblasts, microglia/macrophages and astrocytes, and their deposited ECM. Neurofibrosis occurs commonly across neurological conditions. This review describes processes of wound healing and fibrosis in tissues in general, and in multiple sclerosis in particular, and considers approaches to ameliorate neurofibrosis to enhance neural recovery.
1 Introduction: impaired wound healing contributes to tissue fibrosis
A proper wound healing response is vital for tissue regeneration and involves discrete stages of inflammation, tissue remodeling, and their resolution (1). The inflammatory response is critical for the removal of pathogens and debris from the injury site. It also recruits and activates tissue specific effector cells required for remodeling. Immune and effector cells deposit a variety of extracellular matrix (ECM) molecules to reconstitute matrix lost to injury (1). Successful wound healing recapitulates the tissue environment and restores function (2). Dysregulation of wound healing following repeated, chronic, or pronounced single injury leads to fibrotic scarring due to excessive build-up of cells and ECM (1, 3).
Fibrosis is characterized by increased tissue stiffness and disrupted tissue architecture (Figure 1) (3). It is identified by the accumulation of ECM components, altered protease expression, increased levels of pro-fibrotic signaling molecules, and the presence of pro-fibrotic cells such as fibroblasts and their differentiated forms, myofibroblasts (1). Even in conditions not classically associated with fibrosis, disease outcomes such as hypoxia and epigenetic reprogramming of cells are described with fibrosis-linked responses (4) such as progressive scarification and exacerbated tissue injury (5).
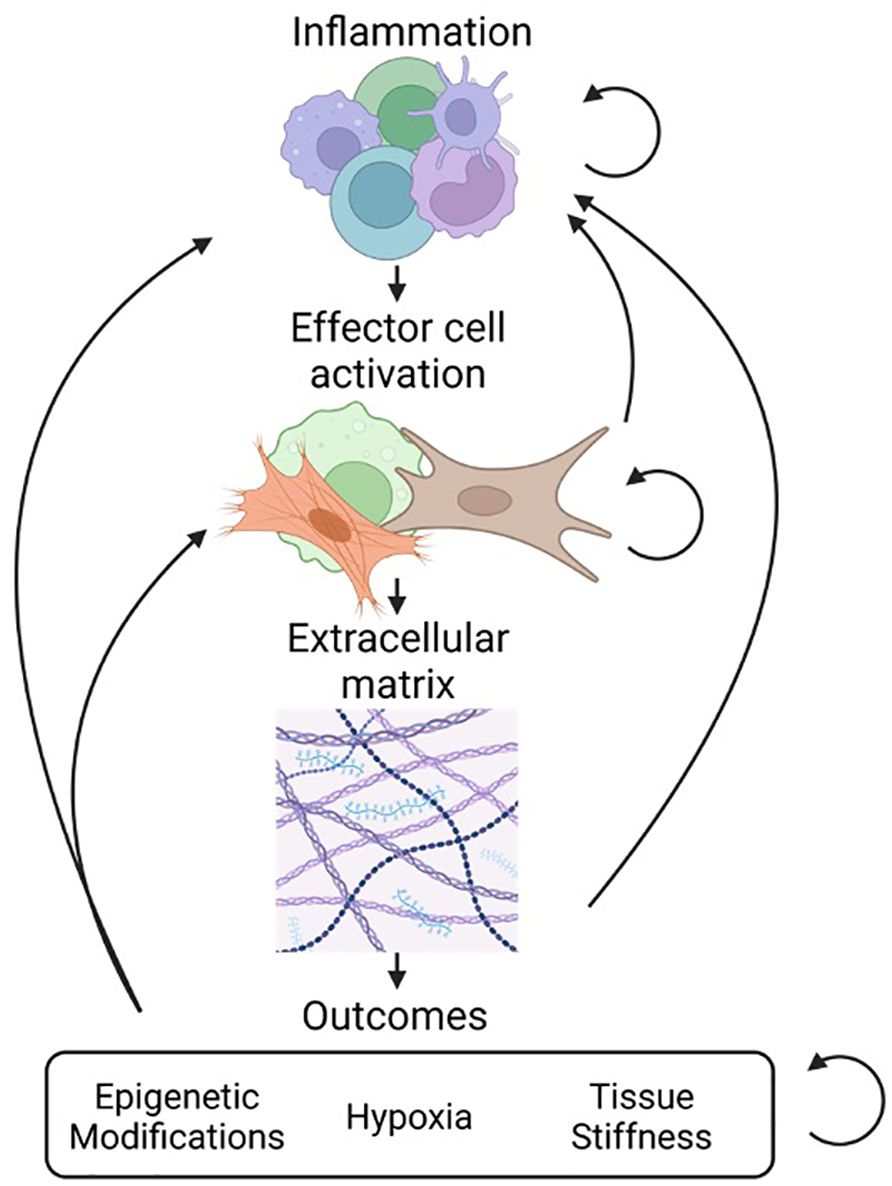
Figure 1 Overview of fibrosis and interconnectivity of stages. Fibrosis progresses through several stages. The first stage of inflammation involves accumulation of a heterogeneous population of cells such as macrophages, dendritic cells, and lymphocytes (top). This is followed by the activation of effector cells such as fibroblasts and myofibroblasts that drive the process of tissue remodeling (middle). The result of effector cell activation is the production and deposition of ECM components including collagen, fibronectin, laminins, and proteoglycans (bottom). As a result of fibrosis there can be secondary injury that results from epigenetic modification of cells, tissue hypoxia, and increased tissue stiffness.
In the central nervous system (CNS) fibroblasts occupy border regions such as the meninges and perivascular space (6). Following injury they become elevated in the parenchyma where they, along with astrocytes, microglia, infiltrating immune cells and potentially pericytes contribute to tissue reorganization and ECM accumulation (7, 8). This neurofibrosis response occurs commonly in CNS pathologies such as spinal cord injury (SCI), stroke and multiple sclerosis (MS) (9). Here we discuss general wound healing and fibrosis related responses so as to instruct concepts of neurofibrosis, and we then consider fibrosis in the CNS with a focus on MS. Finally, we evaluate the therapeutic potential of targeting neurofibrosis to improve outcomes from CNS injuries.
2 Wound healing and fibrosis related responses
Fibrosis occurs in many organs and has shared and unique tissue specific characteristics (4). The initiating injury varies based on anatomical location and trigger (5). For instance, liver and kidney fibrosis can occur due to viral hepatitis and diabetes, respectively. Resident and infiltrating immune cells contribute to fibrosis during initial innate and later adaptive responses (3) (Figure 2). Two primary signaling axes, interleukin (IL)-4/IL-13 and IL-1/IL-17A/transforming growth factor-β (TGFβ), are core pro-fibrosis pathways (5), although IL-17 may also be anti-fibrotic depending on the triggering insult and the organ affected (10). Macrophages, the most abundant immune cell type in fibrosis, are recruited by damage associated molecular patterns (DAMPs), pathogen associated molecular patterns (PAMPs), and chemokines (11). Early arriving macrophages possess an inflammatory phenotype and secrete pro-inflammatory tumor necrosis factor-α (TNFα) and IL-1β which fuel inflammation (11). Later stage macrophages assume a tissue remodeling regulatory phenotype characterized by the expression of TGFβ, IL-10, and mannose receptor 1(MRC1/CD206) (11). Inflammatory macrophages contribute to fibrosis by exacerbating the injury, while regulatory macrophages stimulate effector cells to deposit ECM and express other tissue remodeling genes (5). It is not clear what dictates IL-4/IL-13 or IL-1 mediated fibrosis although the chronicity of the injury may be a determinant. Single injections of bleomycin produce fibrosis in an IL-1/IL-17/TGFβ dependent manner; conversely, repeated injections of bleomycin over a longer period elicits IL-4R signaling (5). This has repercussions for signaling to effector cells and the transition to the tissue remodeling stage of repair.
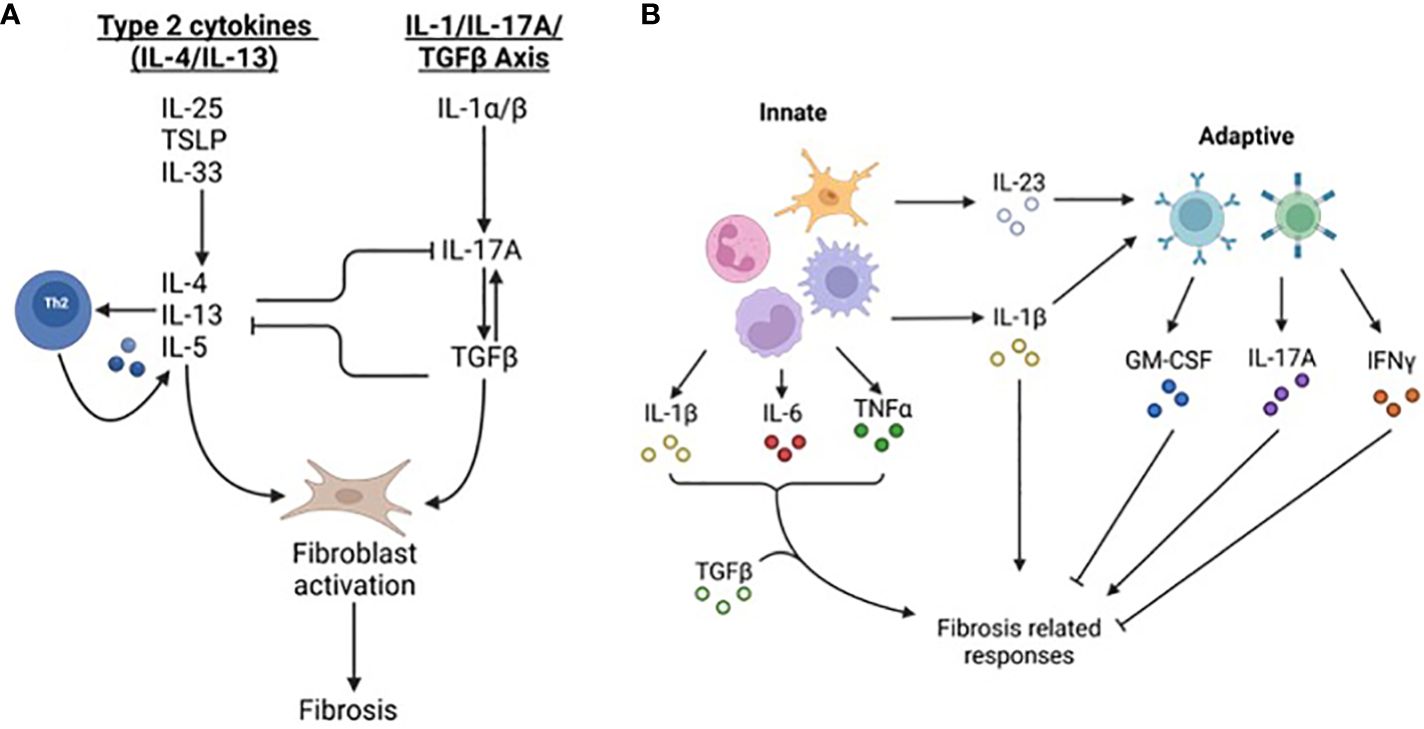
Figure 2 Fibrosis-related signaling pathways. (A) Fibrosis canonically occurs through ‘type 2 cytokines’ including IL-4 and IL-13 and the IL-1/IL-17/TGF-β signaling axis. Danger-associated molecular patterns including thymic stromal lymphopoietin (TSLP), IL-25 and IL-33 stimulate the production of IL-4, IL-5, and IL-13. These cytokines cause Th2 cells to produce more cytokines including IL-4/IL-13, macrophages to produce TGF-β, and stromal cells, including fibroblasts, to elevate and deposit ECM components. The second pathway stimulating fibrosis is the IL-1/IL-17/TGF-β axis. IL-1, along with other inflammatory cytokines such as IL-6 and TNF-α, stimulate IL-17A expression in neutrophils and T cells. IL-17A increases expression of TGF-β and fibroblast expression of TGFβRIII leading to greater fibrosis. (B) Fibrosis is regulated by innate and adaptive immunity. Early phases of fibrosis are influenced more by innate immune cells including macrophages, neutrophils, monocytes, and microglia (in the CNS). Cytokines such as IL-1β, IL-6, and IL23 affect both fibrosis and immune responses. During later stages of fibrosis lymphocyte derived IL-17A, IFN-γ, and GM-CSF promote and impede fibrosis-related responses. IFN-γ may impede fibrosis by limiting Th2 differentiation and type 2 cytokine signaling but can contribute to fibrosis by stimulating immune infiltration leading to greater tissue injury. GM-CSF is anti-fibrotic and dampens pro-fibrotic cytokine production and alternatively activated macrophage polarization.
Effector cells responsible for tissue remodeling originate from resident and/or recruited fibroblasts, epithelial cells, endothelial cells, and other tissue resident cells and immune cells (4). Effector cells are recruited by chemokines and growth factors elaborated at sites of injury by the initial-arriving immune cells where they then proliferate and upregulate ECM, proteases, and growth factors (12). Positive feedback loops between effector and immune cells and the fibrotic environment promote further fibrosis by elevation of pro-fibrotic genes (13, 14). For example, fibroblasts upregulate ECM genes when cultured on ECM derived from idiopathic pulmonary fibrosis tissue (14). This can lead to greater scarring of the tissue and disruption of function.
Organ failure and increased morbidity are outcomes of fibrosis-related disorders such as cardiac fibrosis following myocardial infarction (5). Prominently, fibrosis-related disorders are responsible for 45% of fatalities in the United States of America (5). This highlights the severity of fibrosis for tissue function and recovery which is made more significant in a tissue environment that is not prone to regeneration, such as the CNS.
3 Neurofibrosis
Functional regeneration of the CNS does not occur effectively in adult humans (15) due to factors including lack of available stem cells, age-related changes in neural cells and the tissue environment, and the formation of inhibitory scar tissue after injury (16, 17). Scarring of the CNS results from the accumulation of astrocytes, microglia and macrophages, and fibroblasts and potentially pericytes. The formation of neurofibrosis contributes to impairment of axonal regrowth due to elevated deposition of collagens and chondroitin sulfate proteoglycans (CSPGs), amongst others, into the lesion (7, 8).
In SCI astrocytes become reactive and form a protective boundary to prevent spread of the injury while microglia/macrophages and fibroblast-like cells occupy the center (Figure 3). In MS lesions microglia/macrophages and astrocytes are more intermingled while fibroblast-like cells are expanded in the perivascular compartments (18). Conflicting descriptions of the beneficial and harmful roles of reactive astrocytes exist and have been elaborated on elsewhere (17). Though reactive astrocytes restrict the spread of injury and attempt to maintain homeostatic functions (17), they have also been described to contribute to chronic neuroinflammation and they upregulate many ECM components detrimental for regeneration (19). Conversely, depletion of reactive astrocytes impairs the ability of axons to regrow after SCI (20). Instructively, studies such as these suggest that certain levels of regeneration of the CNS are possible under permissive conditions.
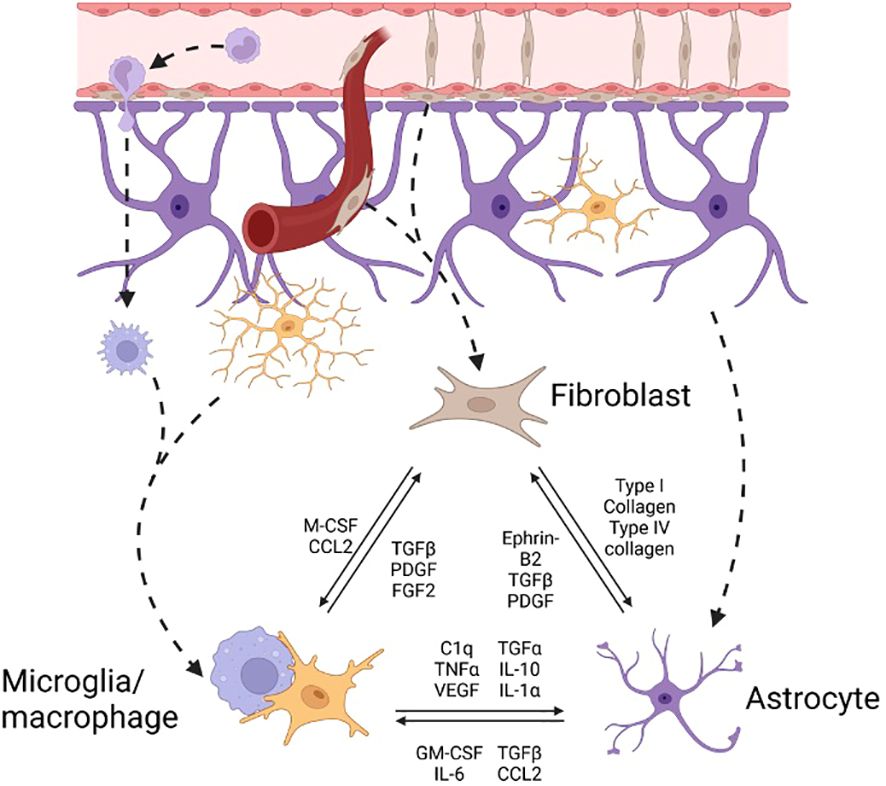
Figure 3 Effector cell interactions in neurofibrosis The pool of effector cells in CNS fibrosis is drawn from circulating monocyte derived macrophages, border derived meningeal and perivascular fibroblasts, and resident microglia and astrocytes. There is significant interconnectedness between cell types. cell-cell, cell-ECM-cell, and soluble factor signals between effector cells leading to reduced or greater stimulation of fibrosis-related pathways such as TGF-β signaling in fibroblasts or collagen-1 activation of astrocytes.
CNS-associated fibroblasts identified by PDGFRβ and Col1a1 driven reporter in mice and antibody labeling are increasingly implicated in neurofibrosis (8, 21). Their complete depletion in SCI results in an open wound defect (8) while their partial depletion leads to greater axon regeneration (22, 23). The identification and ontogeny of fibroblasts in neurofibrosis remain to be better resolved, and the use of different reporter mouse lines is partly responsible for the controversy. Due to the use of PDGFRβ based reporter mice and PDGFRβ as a common marker across several cell types, it is likely that fibroblasts, pericytes, and smooth muscle cells have been conflated with each other (24). Transcriptional profiling of murine mural cells has shown that all these three cell types express common markers including PDGFRβ and NG2 (5, 24). While these studies identified homeostatic gene expression patterns that differentiate pericytes (Kcnj8), fibroblasts (Col1a1), and smooth muscle cells (Acta2), it is unclear how expression of these are affected by injury or inflammation. Further adding to the controversy, type A pericytes identified by a GLAST driven reporter line have been described in SCI (8, 25) but GLAST also labels populations of astrocytes. As well, Dorrier et al. showed using NG2 and SMA inducible reporters that cells basally positive for these genes do not significantly contribute to fibrotic cells in neuroinflammatory lesions (21, 22). However, this does not preclude subpopulations of pericytes or other cell types from contributing to neurofibrosis. Additionally, other lines of evidence suggest that pericytes and smooth muscle cells do not contribute to the fibroblast populations in neurofibrosis although the data is inconclusive (8, 21). Based on the uncertainty of the ontogeny or type of fibroblasts in CNS injuries, we shall refer to them as fibroblast-like cells where indicated.
The molecular impediments to CNS regeneration within fibrotic lesions include inhibitory molecules, reduced levels of growth signals and products of particular inflammatory cells (8, 17). Microglia are CNS resident macrophages and are critical for maintaining tissue homeostasis. Monocyte derived macrophages become elevated following injury and, together with microglia, participate in cellular and lipid debris removal. As well, they upregulate inflammatory and tissue remodeling genes during early and late stages of injury. The ability of microglia/macrophages to metabolize debris becomes impaired with age leading to formation of foamy cells that contribute to chronic inflammation and impaired tissue remodeling (16). In neonatal mice infiltration by peripheral macrophages is resolved rapidly compared to adult mice. Depleting microglia using PLX3397 or CX3CR1-CSF1Rfl/fl reversed spontaneous neonatal axonal regeneration implying that it is microglia that act to promote recovery following optic nerve crush injury (26). Additionally, there is a clear interconnectedness of the cells within CNS lesions as highlighted by altered levels of astrocytes, macrophages, and fibroblasts following depletion of each individually (22, 25, 27). Indeed, both astrocytes and fibroblasts interact reciprocally with immune cells informing each cell’s phenotype (Figure 3) (21, 28). This emphasizes the importance of the cellular components of CNS injury to the success of regeneration.
A theme that has been topical in peripheral fibrosis is that of fibroblast senescence and resistance to apoptosis (29, 30). Thus far, a systematic analysis of age-related neurofibrosis has not been thoroughly conducted. Studies focusing on the effects of age on traumatic brain injury described increased collagen levels in the injured aged meninges (31). Whether this is consistent in humans is not clear though proteomic analysis of CSF in people found increased levels of collagen with aging (32). Further determinations of changes with aging would include fibroblast density and susceptibility to apoptosis in lesions, accumulation of neural ECM in neurological disorders, and tissue rigidity in healthy aging or disease CNS.
While tissue stiffness generally increases in non-CNS fibrosis-related disorders, many forms of CNS injury are reported to result in reduced stiffness (33). One explanation for this phenomenon may be that most animal models of MS are studied during acute stages of injury and tissue stiffness increases during chronic stages of injury. Indeed, CNS lesions become increasingly stiff with chronicity (34). Thus, neurofibrosis is akin to fibrosis present in other organs and presents further questions about the role of fibrosis-related responses in CNS pathologies including MS.
4 MS and its regenerative processes
MS is an inflammatory disease characterized by demyelination and neuroaxonal degeneration in the brain, spinal cord, and optic nerve (35). Demyelination occurs in the context of inflammation involving CD8+ T lymphocytes, CD4+ T lymphocytes, B cells, plasma cells, macrophages, microglia and reactive astrocytes (35–37). Lesions are categorized as active, chronic active, inactive, and remyelinating based on immune and remyelinating phenotypes (16). Found in the brain and spinal cord white and gray matter, the location of lesions contributes to symptom presentation and onset (35).
Axonal regeneration can occur under ideal conditions, but it is not common and has only been minimally examined in MS. Conversely, remyelination occurs spontaneously to varying degrees in MS, in all types of lesions except inactive ones, and has been extensively examined (16, 38). During remyelination, oligodendrocyte progenitor cells (OPCs) migrate to the site of injury, proliferate, and differentiate into mature oligodendrocytes capable of producing new compact myelin (16). Remyelination promotes axonal neuroprotection and functional recovery making it an important and regularly occurring regenerative process (16, 38).
Fibrosis affects remyelination in MS (7). OPC migration, proliferation, and maturation are positively and negatively regulated by particular ECM components deposited in lesions (7). Specific effects of different ECM components have been reviewed in detail previously (39). Examples include beneficial effects of some isoforms of laminins on OPC proliferation while ECM molecules such as CSPGs and fibronectin impair OPC activity (7, 40). Inhibition of CSPG synthesis following injury increased oligodendrocyte numbers and remyelination (41). Furthermore, depletion of astrocytes and fibroblasts in models of MS leads to greater density of oligodendrocytes (21, 22). Thus, several elements of fibrosis impair regenerative processes in MS, to which we now turn our attention.
5 Biology of neurofibrosis in MS
Many processes dysregulated in fibrosis are present in MS lesions including chronic inflammation, effector cell recruitment, and excessive ECM deposition (5, 16). As well, outcomes of fibrosis such as tissue stiffness, hypoxia, cellular senescence, and impaired repair processes impact MS pathology (42, 43). Here we describe neurofibrosis in MS focusing on the inflammation, and remodeling and progression stages.
5.1 Inflammation
Lesion-associated cells in MS include microglia and astrocytes, infiltrated leukocytes, and CNS barrier-associated cells such as meningeal and perivascular macrophages and fibroblast-like cells (6, 35–37). CD8+ and, to a lesser extent, CD4+ T cells are present in MS lesions while B cells are within the perivascular and meningeal borders (37). Lymphocytes are contributors to MS pathology and have been detailed elsewhere (36, 37, 44).
The inflammatory milieu within MS lesions is diverse and shares many inflammatory characteristics seen in fibrosis. Pro-inflammatory cytokines including GM-CSF, IL-17, and IFNγ are elevated in MS and contribute to pathological processes (44). As well, TGFβ, the master regulator of fibrosis and important regulatory cytokine, is highly expressed in MS lesions (45). Both IL-4/IL-13 and IL-1 mediated fibrosis pathways converge through TGFβ. As mentioned previously, IL-4/IL-13 seem to be associated with persistent injury such as that seen in MS although the prominent pro-inflammatory nature of MS leads IL-4 and IL-13 to be often minimized in its pathophysiology. IL-1 on the other hand is highly expressed in MS lesions, as are Th17 cells and IL-17. Thus, the IL-1/IL-17/TGFβ pathway constituents are more prevalent in MS (46, 47). The increased IL-17 levels are important in sustaining inflammation by recruitment of myeloid cells (48), potentially producing a positive feedback loop for neurofibrosis. As well, IL-17 upregulates and stabilizes TGFβR (R: receptor) expression on fibroblasts (8, 17). This allows for TGFβ signaling through SMAD2/3 to promote activation of astrocytes and differentiation of fibroblasts into myofibroblasts positive for αSMA, collagen, and fibronectin (4, 20).
Microglia/macrophages are the most prevalent immune cells in MS lesions. Fibrosis-associated microglia/macrophages expressing arginase-1, MRC1/CD206, and MerTK (49, 50) have been described in MS lesions (35, 51). Important for the onset of fibrosis, microglia and macrophages become polarized to arginase-producing cells by their local environment after initially expressing nitric oxide synthase (51, 52). Arginase 1 converts arginine into precursors of proline essential for collagen production (4, 50). As well, macrophages upregulate proteases in response to Th1 derived cytokines such as IFNγ (53). IFNγ may also impair collagen synthesis by fibroblasts and reduce pro-fibrotic IL-17 expression (1, 44). However, in the presence of IL-6, IFNγ can help promote fibrosis in a STAT1 dependent manner (54). Thus, it is not clear whether IFNγ will behave as a pro- or anti-fibrotic cytokine in MS though IFNγ and IL-6 are commonly described in MS and may favor an avenue to progression of fibrosis (55).
Astrocytes express pro- and anti-inflammatory cytokines in a context dependent manner and contribute to neurofibrosis. In chronic experimental autoimmune encephalomyelitis (EAE), a model of MS, astrocytes promote inflammation, immune cell recruitment, and further astrocyte reactivity by production of GM-CSF, CCL2 and lactosylceramide (19). The promotion of chronic inflammation during late stage EAE by reactive astrocytes contributes to worse disease scores and is reversed following depletion of reactive astrocytes (19). As well, IL-17-NFκB signaling in astrocytes results in expression of pro-inflammatory cytokines, chemokines, and proteases (Figure 4A). Astrocytes are also a source of IL-6 and vascular endothelial growth factor (VEGF) which stimulate macrophages and fibroblasts towards pro-fibrotic phenotypes (57). It is reasonable to surmise that while astrocytes may limit fibrosis early in EAE (8), they contribute to injurious neuroinflammation that promotes fibrosis during chronic stages of EAE and likely MS.
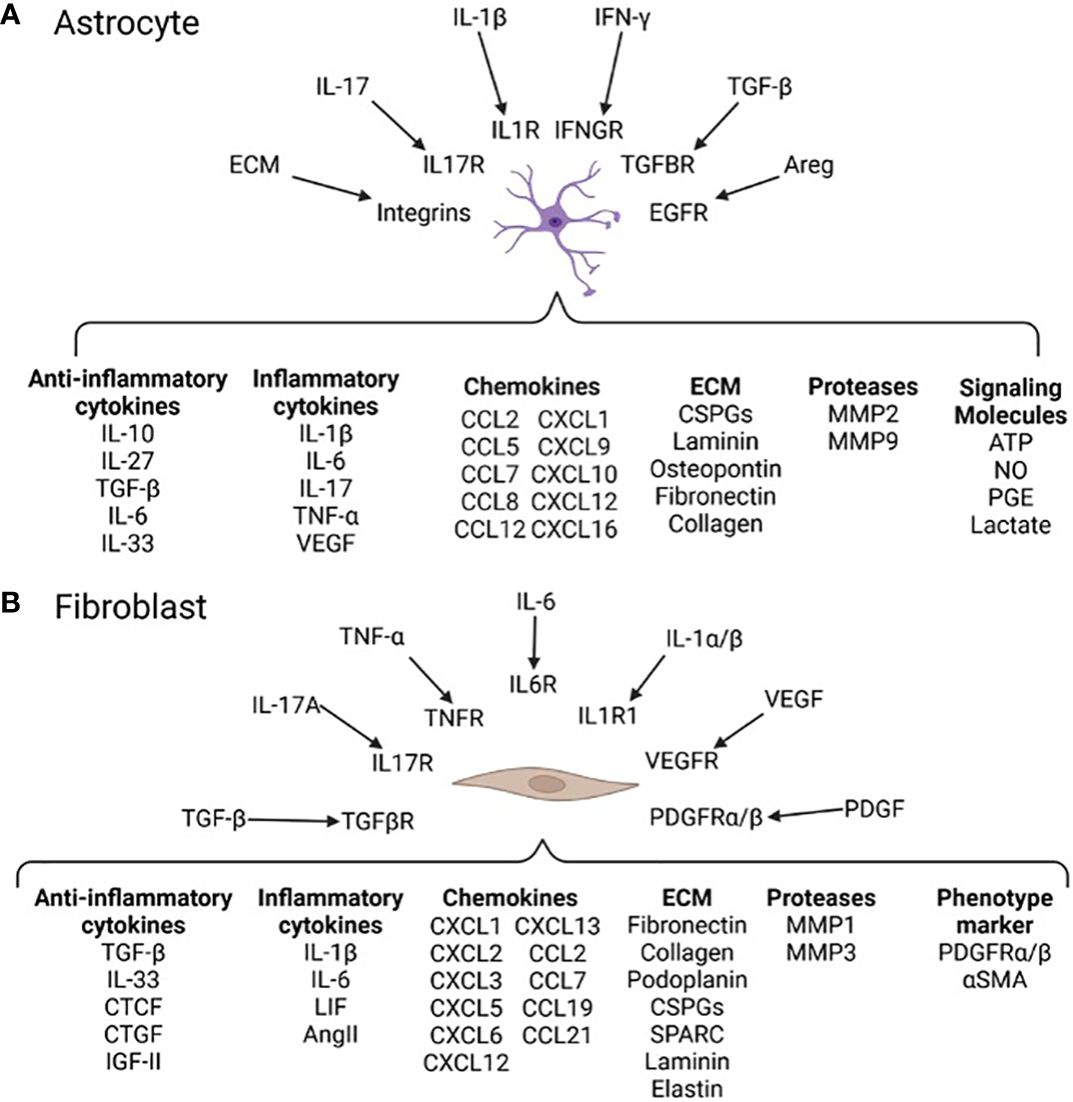
Figure 4 Neurofibrosis related signals and responses in astrocytes and fibroblasts (A) Soluble factors and ECM components such as IL-1β and collagen can be recognized by astrocytes through cell surface receptors (17). Activation of secondary signaling cascades polarize astrocyte phenotypes affecting production of pro- and anti-fibrotic proteins. For instance, autocrine LacCer signaling induces astrocyte expression of NFkB and downstream genes such as CCL2 and GM-CSF leading to greater inflammation (19). (B) Fibroblasts reside in regions of the CNS in close proximity to immune infiltrates allowing them to sample inflammatory factors such as IL-17A and TNF-α (8). Fibroblasts express a range of genes related to immune modulation, contractility, and tissue remodeling (8, 56). Activated fibroblasts transition to a tissue remodeling myofibroblast phenotype expressing contractility proteins (e.g. αSMA) and producing ECM such as collagen and fibronectin (8). The specific outcomes of this in MS and other neurological diseases is not well understood.
5.2 Tissue remodeling and progression of fibrosis in MS
Tissue remodeling is driven by effector cell recruitment and activation (4). Effector cells in MS lesions include microglia/macrophages, astrocytes, perhaps pericytes, and perivascular and meningeal fibroblasts (7, 21). Indeed, these cells are associated with ECM deposition in MS and animal models of MS (7, 8). Proteomic and histological analysis of active, chronic active, and inactive MS lesions highlight the involvement of fibrosis and ECM components (42, 55). The latter include fibronectin, proteoglycans and thrombospondin particularly in chronic active lesions (55). Importantly, molecular network analyses implies integrin signaling is involved within both chronic active and inactive lesions indicating a role for integrin-interacting ECM in resolving or following the resolution of inflammation (55). As noted above, key regulators of ECM expression such as TNFα, IL-17, TGFβ, IFNγ, IL-1, and platelet derived growth factor (PDGF) are elevated in MS lesions (55, 58).
Microglia/macrophages are found in MS lesions in close proximity of accumulated ECM (7). As well, microglia/macrophages undergo transition from inflammatory to remodeling phenotypes in MS and models of MS that coincides with a transition from inflammation to tissue remodeling (16). Depleting microglia/macrophages during tissue remodeling impairs remyelination in part due to the loss of Activin A mediated OPC maturation (51). During early inflammation, depleting microglia/macrophages causes myelin debris accumulation and reduced OPC proliferation (28, 51). Interestingly, type 1 interferons associated with viral infection and aging cause microglia/macrophages to express fibrosis related genes (37). A direct connection between tissue stiffness and disease progression in MS in not known, but increasing tissue stiffness is associated with age and lesion chronicity and may suggest a role of altered ECM (43, 59).
Astrocytes tile the CNS making them ideal effector cells (17). Following injury they upregulate a plethora of tissue remodeling and ECM components including hyaluronan, fibulin-2, CSPGs, and laminins (18, 60). Expression of these molecules relies in part on TGFβ and EGF signaling (7, 61). During chronic EAE, astrocytes upregulate tissue remodeling associated genes Arg1, Spp1 (which encodes the matrix associated protein osteopontin), and Vegf (19) that may lead to enhanced fibrosis.
In MS, fibroblasts are associated with the perivascular space and increased expression of basement membrane and mesenchymal cell markers (21, 62). During EAE they are found in regions high in microglia/macrophages and express tissue remodeling related genes for ECM, proteases, and cytoskeleton proteins (21). Lesion-associated fibroblasts express many molecules (Figure 4B) including high levels of collagens and fibronectin (21), which impair the function of OPCs (7). As well, these, and other, ECM components can feedback into fibrosis-related pathways by acting as agonists for TLR signaling of microglia/macrophages, exacerbating inflammation and subsequent fibrosis (7).
Despite the prominence of fibroblasts in non-CNS fibrosis disorders, their description in MS lesions has thus far been limited. Reports identify fibroblasts in brains of people with MS in the meninges and perivascular space, and sparsely in the parenchyma (9, 21). This may be due to poor availability of fibroblast markers, altered marker expression, or they may be transient populations within the lesion environment. Further work is clearly needed to characterize and understand the contribution of fibroblasts to MS lesions.
6 Emerging/potential therapeutics
No therapies currently are used to directly affect fibrosis in MS. Thus, this section (Figure 5) begins with an overview of medications that treat fibrosis in non-CNS disorders, and addresses whether these could be applied for MS. We then discuss whether disease modifying therapies (DMTs) used in MS have unintended effects on ameliorating fibrosis in MS, and end with a forward-looking view of potential therapeutics that could be applied to counter fibrosis in MS.
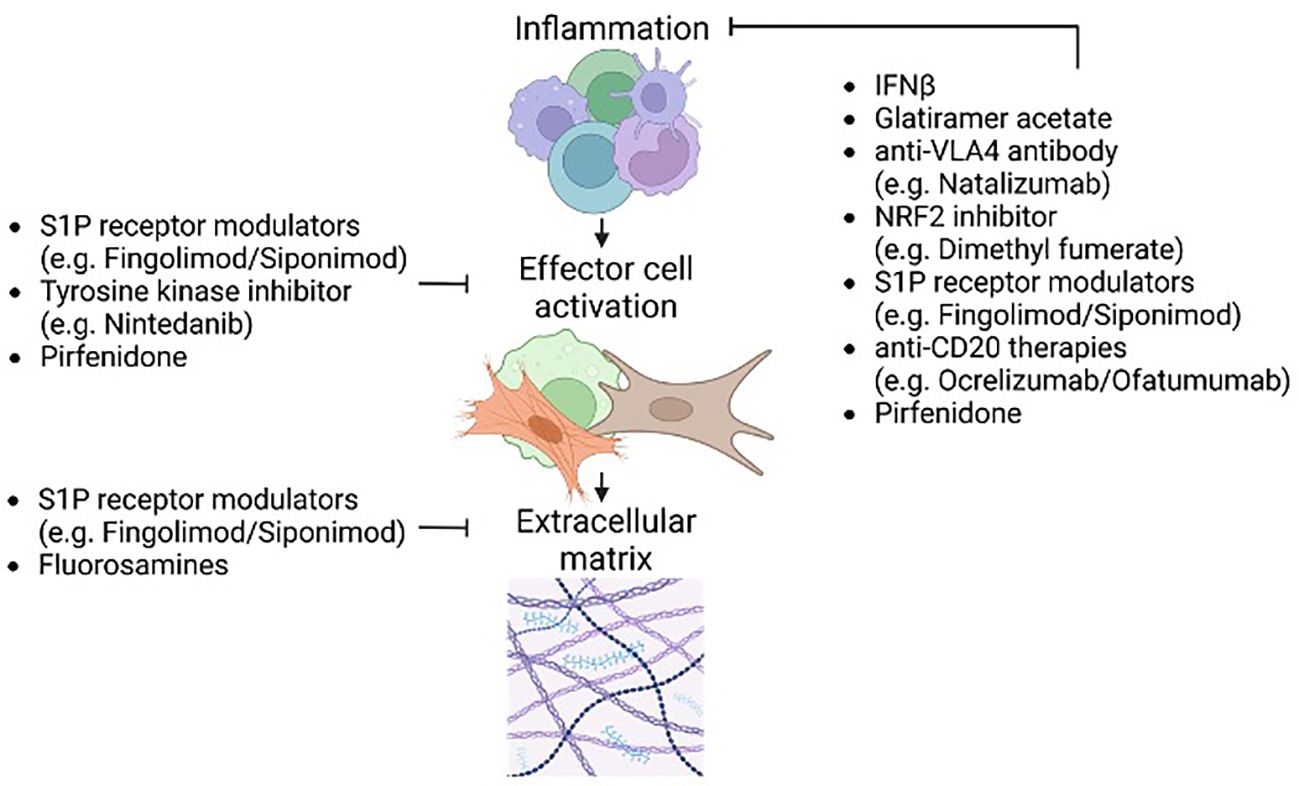
Figure 5 Effects of existing MS disease-modifying therapies (DMTs) on fibrosis pathways. Existing MS related DMTs may affect all stages of fibrosis. Due to the neuroinflammatory nature of MS the inflammatory component of fibrosis is most affected by DMTs. Impeding inflammation serves to limit inflammatory tissue injury and the potential for unchecked remodeling during later lesion stages. Furthermore, impeding the initial inflammatory stages of fibrosis has downstream effects on the later fibrosis-related processes such as ECM deposition and remodeling. Some current and potential DMTs including S1P receptor modulators and pirfenidone target activation of effector cell populations. This includes but is not limited to affecting TGFβ signaling important for effector cell activation and the development of fibrosis. Affecting effector cell activation directly affects ECM deposition and secondary injury caused by elevated stiffness and hypoxia. Preliminary studies on therapeutics that target the final stages of fibrosis such as ECM synthesis (e.g. fluorosamines) highlight the benefits of altering later stages of neurofibrosis.
Only two medications are approved to treat fibrosis-related disorders, specifically for lung fibrosis: pirfenidone and nintendanib (5). Pirfenidone acts as an immunomodulator and antagonist of the TGFβ pathway that promotes fibrosis (4, 63). In people with idiopathic pulmonary fibrosis pirfenidone reduced disease progression, improved lung function, and increased exercise tolerance (64). Pirfenidone has undergone phase I and II clinical trials in secondary progressive MS reporting reduced incidence of relapse and improved bladder function (65, 66). However, these trials were small, and a larger study is needed to determine the potential of pirfenidone for people with progressive MS.
When inhibiting TGFβ signaling, it is important to consider its role as a regulatory cytokine and its function for the maintenance of microglia homeostasis in the CNS (44). Another consideration for drugs to overcome CNS fibrosis is the additional challenge that these compounds will have to cross the blood-brain barrier.
The other approved drug for lung fibrosis, nintendanib, is a tyrosine kinase inhibitor (4, 67). Pharmacological inhibition of tyrosine kinases by other drugs in animal models of MS reduced clinical scores, demyelination, inflammation, astrocyte reactivity, and vascular injury (68–71). However, tyrosine kinases are broadly expressed making off-target effects of systemic administration difficult to isolate from potential anti-fibrotic effects within the CNS. Nonetheless, two clinical trials using the tyrosine kinase inhibitor masitinib in progressive MS have been conducted (72, 73). The first, a Phase I trial, showed masitinib to be safe (72). The second, a Phase IIb/III trial, reported reduced elevation of EDSS disability scores and delayed the time to EDSS of 7.0 (73). The extent to which the promising clinical result could be attributed to amelioration of CNS fibrosis is unknown.
Next, we consider whether the approximately (country-dependent) 20 DMTs used in MS have potential impact on evolution of fibrosis (Figure 5). While such data is lacking, the immunomodulatory nature (74, 75) of MS DMTs conceivably can alter the immune stimulation of fibrogenesis. Either by reducing the content of pro-inflammatory cytokines or elevating levels of Th2 cells (e.g. IFNβ, glatiramer acetate), sequestering leukocytes in secondary lymphoid tissues (e.g. sphingosine-1-phosphate receptor modulators such as siponimod), inhibiting leukocyte trafficking across the blood-brain barrier (natalizumab), or by depleting B cells (anti-CD20 monoclonal antibodies) and the larger leukocyte populations (alemtuzumab, cladribine), the effect would be reduced recruitment and activation of effector cells of CNS fibrosis. Thus, MS DMTs may have unintended and indirect effects on reducing neurofibrosis, although the possibility of this occurring will have to be established. Future work may consider whether the extent of neurofibrosis is ameliorated in people with MS treated with DMTs, and such studies will be highly reliant on evolution of biomarkers with the capacity to detect neurofibrosis.
Recently, inhibitors targeting the intracellular signaling enzyme, Bruton’s tyrosine kinase (BTK), have grown in interest in the treatment of MS (76). Inhibition of BTK in models of graft versus host disease resulted in reduced dermal fibrosis. As well, the first generation BTK inhibitor, ibrutinib, has been approved for treatment of graft versus host disease (77). BTK inhibitors affect B cells but also microglia proliferation, activation, and survival (76). BTK inhibitors have not been trialed for fibrotic disorders, and the preclinical literature is heterogeneous. Some evidence suggests that loss of BTK can reduce fibrosis following cardiac injury while other studies show that BTK inhibitors either have no effect or detrimental effects in kidney and lung fibrosis (78, 79). However, BTK inhibitors have been approved for treatment of several cancers such as chronic lymphocyte leukemia and mantle-cell lymphoma (77). As well, they have been tested in a number of autoimmune diseases leading to many clinical trials including in MS. Five BTK inhibitors have entered late-stage clinical trials including evobrutinib, fenebrutinib, tolebrutinib, remibrutinib and orelabrutanib (76). Unfortunately, evobrutinib failed to meet its primary endpoint in the phase 3 EVOLUTION trials (80). At this time it is unknown whether BTK inhibitors have a role for treatment of fibrosis in MS.
Finally, another means to reduce CNS fibrosis is to prevent or limit the deposition of ECM molecules that occurs after injury. Pharmacological manipulation of ECM and its production have improved recovery in both SCI and MS models (41, 81). Both impairing CSPG synthesis with fluorosamines (4-F- or 4,4-difluoro-N-acetylglucosamine) and digestion of existing CSPGs with chondroitinase-ABC led to significant recovery (41, 81). In EAE, 4,4,-difluoro-N-acetylglucosamine reduced the frequency of cytotoxic Th17 and improved remyelination (41). Altering expression of other common ECM components such as hyaluronan improved EAE outcomes (82).
In summary, targeting fibrosis-related responses and outcomes in preclinical studies shows promise as therapeutic strategies for MS. Translation from preclinical into clinically relevant therapies requires more work to understand the implications of these mechanisms on MS and to determine safety and efficacy.
7 Conclusions
Tissue regeneration is necessary to restore function (2). Altered immune and tissue remodeling results in fibrosis blocking functional recovery, and increases morbidity (5). The adult CNS does not regenerate well leading to accumulation of injury and disability (16, 83). Inability to repair successfully is partly due to remnant cells and ECM components of fibrosis within the injured CNS (9, 18, 41, 84). Similarities exist between CNS pathologies such as MS and peripheral fibrosis and the benefits of targeting these fibrosis-related processes in MS have been highlighted by a growing body of preclinical research (9, 21, 41, 68) as well as some early clinical trials (65, 66). Fibroblasts are beginning to be recognized as components of neurofibrosis and deserve more studies. Questions remain regarding safety and efficacy of targeting neurofibrosis, but its successful treatment would represent an important step forward in the promotion of CNS regeneration and recovery.
Author contributions
BL: Conceptualization, Writing – original draft, Writing – review & editing. SG: Writing – original draft, Writing – review & editing. VY: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors’ research on neurofibrosis is supported by operating grants from the Canadian Institutes of Health Research (number 1049959), Multiple Sclerosis Canada (number 3527) and the USA Department of Defense MS Research program (contract number W81XWH2210468).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Talbott HE, Mascharak S, Griffin M, Wan DC, Longaker MT. Wound healing, fibroblast heterogeneity, and fibrosis. Cell Stem Cell. (2022) 29:1161–80. doi: 10.1016/j.stem.2022.07.006
2. Rodrigues M, Kosaric N, Bonham CA, Gurtner GC. Wound healing: A cellular perspective. Physiol Rev. (2019) 99:665–706. doi: 10.1152/physrev.00067.2017
3. Soliman H, Theret M, Scott W, Hill L, Underhill TM, Hinz B, et al. Multipotent stromal cells: One name, multiple identities. Cell Stem Cell. (2021) 28:1690–707. doi: 10.1016/j.stem.2021.09.001
4. Distler JHW, Györfi AH, Ramanujam M, Whitfield ML, Königshoff M, Lafyatis R. Shared and distinct mechanisms of fibrosis. Nat Rev Rheumatol. (2019) 15:705–30. doi: 10.1038/s41584-019-0322-7
5. Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature. (2020) 587:555–66. doi: 10.1038/s41586-020-2938-9
6. Ampie L, McGavern DB. Immunological defense of CNS barriers against infections. Immunity. (2022) 55:781–99. doi: 10.1016/j.immuni.2022.04.012
7. Ghorbani S, Yong VW. The extracellular matrix as modifier of neuroinflammation and remyelination in multiple sclerosis. Brain. (2021) 144:1958–73. doi: 10.1093/brain/awab059
8. Dorrier CE, Jones HE, Pintarić L, Siegenthaler JA, Daneman R. Emerging roles for CNS fibroblasts in health, injury and disease. Nat Rev Neurosci. (2022) 23:23–34. doi: 10.1038/s41583-021-00525-w
9. Dias DO, Kalkitsas J, Kelahmetoglu Y, Estrada CP, Tatarishvili J, Holl D, et al. Pericyte-derived fibrotic scarring is conserved across diverse central nervous system lesions. Nat Commun. (2021) 12:5501. doi: 10.1038/s41467-021-25585-5
10. Ramani K, Biswas PS. Interleukin-17: Friend or foe in organ fibrosis. Cytokine. (2019) 120:282–8. doi: 10.1016/j.cyto.2018.11.003
11. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
12. Pakshir P, Alizadehgiashi M, Wong B, Coelho NM, Chen X, Gong Z, et al. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nat Commun. (2019) 10:1–17. doi: 10.1038/s41467-019-09709-6
13. Long Y, Niu Y, Liang K, Du Y. Mechanical communication in fibrosis progression. Trends Cell Biol. (2022) 32:70–90. doi: 10.1016/j.tcb.2021.10.002
14. Parker MW, Rossi D, Peterson M, Smith K, Sikstromü K, White ES, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest. (2014) 124:1622–35. doi: 10.1172/JCI71386
15. Rentsch NH, Rust R. “Scary” pericytes: the fibrotic scar in brain and spinal cord lesions. Trends Neurosci. (2022) 45:6–7. doi: 10.1016/j.tins.2021.10.013
16. Franklin RJM, Simons M. CNS remyelination and inflammation: From basic mechanisms to therapeutic opportunities. Neuron. (2022) 110:3549–65. doi: 10.1016/j.neuron.2022.09.023
17. Sofroniew MV. Astrocyte reactivity: subtypes , states , and functions in CNS innate immunity. Trends Immunol. (2020) 41:758–70. doi: 10.1016/j.it.2020.07.004
18. Anderson MA, O’Shea TM, Burda JE, Ao Y, Barlatey SL, Bernstein AM, et al. Required growth facilitators propel axon regeneration across complete spinal cord injury. Nature. (2018) 561:396–400. doi: 10.1038/s41586-018-0467-6
19. Mayo L, Trauger SA, Blain M, Nadeau M, Patel B, Alvarez JI, et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat Med. (2014) 20:1147–56. doi: 10.1038/nm.3681
20. Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature. (2016) 532:195–200. doi: 10.1038/nature17623
21. Dorrier CE, Aran D, Haenelt EA, Sheehy RN, Hoi KK, Pintarić L, et al. CNS fibroblasts form a fibrotic scar in response to immune cell infiltration. Nat Neurosci. (2021) 24:234–44. doi: 10.1038/s41593-020-00770-9
22. Dias DO, Kim H, Holl D, Solnestam BW, Lundeberg J, Carlen M, et al. Reducing pericyte-derived scarring promotes recovery after spinal cord injury. Cell. (2018) 173:153–65. doi: 10.1016/j.cell.2018.02.004
23. Goritz C, Dias D, Tomilin N, Barbacid M, Shupliakov O, Frisen J. A pericyte origin of spinal cord scar tissue. Science. (2011) 333:238–43. doi: 10.1126/science.1203165
24. Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature. (2018) 554:475–80. doi: 10.1038/nature25739
25. Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, et al. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci. (2013) 33:12870–86. doi: 10.1523/JNEUROSCI.2121-13.2013
26. Li Y, He X, Kawaguchi R, Zhang Y, Wang Q, Monavarfeshani A, et al. Microglia-organized scar-free spinal cord repair in neonatal mice. Nature. (2020) 587:613–8. doi: 10.1038/s41586-020-2795-6
27. Zhu Y, Soderblom C, Krishnan V, Ashbaugh J, Bethea JR, Lee JK. Hematogenous macrophage depletion reduces the fibrotic scar and increases axonal growth after spinal cord injury. Neurobiol Dis. (2015) 74:114–25. doi: 10.1016/j.nbd.2014.10.024
28. Rawji KS, Young AMH, Ghosh T, Michaels NJ, Mirzaei R, Kappen J, et al. Niacin-mediated rejuvenation of macrophage/microglia enhances remyelination of the aging central nervous system. Acta Neuropathol (Berl). (2020) 139:893–909. doi: 10.1007/s00401-020-02129-7
29. Mebratu YA, Soni S, Rosas L, Rojas M, Horowitz JC, Nho R. The aged extracellular matrix and the profibrotic role of senescence-associated secretory phenotype. Am J Physiol-Cell Physiol. (2023) 325:C565–79. doi: 10.1152/ajpcell.00124.2023
30. Hinz B, Lagares D. Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases. Nat Rev Rheumatol. (2020) 16:11–31. doi: 10.1038/s41584-019-0324-5
31. Bolte AC, Shapiro DA, Dutta AB, Ma WF, Bruch KR, Kovacs MA, et al. The meningeal transcriptional response to traumatic brain injury and aging. eLife. (2023) 12:1–38. doi: 10.7554/eLife.81154
32. Shuken SR, Rutledge J, Iram T, Losada PM, Wilson EN, Andreasson KI, et al. Limited proteolysis – mass spectrometry reveals aging-associated changes in cerebrospinal fluid protein abundances and structures. Nat Aging. (2022) 2:379–88. doi: 10.1038/s43587-022-00196-x
33. Wuerfel J, Paul F, Beierbach B, Hamhaber U, Klatt D, Papazoglou S, et al. MR-elastography reveals degradation of tissue integrity in multiple sclerosis. NeuroImage. (2010) 49:2520–5. doi: 10.1016/j.neuroimage.2009.06.018
34. Cooper JG, Sicard D, Sharma S, Van Gulden S, McGuire TL, Cajiao MP, et al. Spinal cord injury results in chronic mechanical stiffening. J Neurotrauma. (2020) 37:494–506. doi: 10.1089/neu.2019.6540
35. Charabati M, Wheeler MA, Weiner HL, Quintana FJ. Multiple sclerosis: Neuroimmune crosstalk and therapeutic targeting. Cell. (2023) 186:1309–27. doi: 10.1016/j.cell.2023.03.008
36. Dong Y, Yong VW. When encephalitogenic T cells collaborate with microglia in multiple sclerosis. Nat Rev Neurol. (2019) 15:704–17. doi: 10.1038/s41582-019-0253-6
37. Jain RW, Yong VW. B cells in central nervous system disease: diversity, locations and pathophysiology. Nat Rev Immunol. (2022) 22:513–24. doi: 10.1038/s41577-021-00652-6
38. Mei F, Lehmann-Horn K, Shen YAA, Rankin KA, Stebbins KJ, Lorrain DS, et al. Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. eLife. (2016) 5:1–21. doi: 10.7554/eLife.18246
39. Pu A, Stephenson EL, Yong VW. The extracellular matrix: Focus on oligodendrocyte biology and targeting CSPGs for remyelination therapies. Glia. (2018) 66:1–17. doi: 10.1002/glia.23333
40. van Schaik PEM, Zuhorn IS, Baron W. Targeting fibronectin to overcome remyelination failure in multiple sclerosis: the need for brain- and lesion-targeted drug delivery. Int J Mol Sci. (2022) 23:8418. doi: 10.3390/ijms23158418
41. Ghorbani S, Jelinek E, Jain R, Buehner B, Li C, Lozinski BM, et al. Versican promotes T helper 17 cytotoxic inflammation and impedes oligodendrocyte precursor cell remyelination. Nat Commun. (2022) 13:1–18. doi: 10.1038/s41467-022-30032-0
42. Mohan H, Krumbholz M, Sharma R, Eisele S, Junker A, Sixt M, et al. Extracellular matrix in multiple sclerosis lesions: fibrillar collagens , biglycan and decorin are upregulated and associated with infiltrating immune cells. Brain Pathol. (2010) 20:966–75. doi: 10.1111/j.1750-3639.2010.00399.x
43. Urbanski MM, Brendel MB, Melendez-Vasquez CV. Acute and chronic demyelinated CNS lesions exhibit opposite elastic properties. Sci Rep. (2019) 9:1–13. doi: 10.1038/s41598-018-37745-7
44. Becher B, Spath S, Goverman J. Cytokine networks in neuroinflammation. Nat Rev Immunol. (2017) 17:49–59. doi: 10.1038/nri.2016.123
45. Cekanaviciute E, Fathali N, Doyle KP, Williams AM, Han J, Buckwalter MS. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia. (2014) 62:1227–40. doi: 10.1002/glia.22675
46. Burm SM, Peferoen LAN, Zuiderwijk-Sick EA, Haanstra KG, ‘t Hart BA, van der Valk P, et al. Expression of IL-1β in rhesus EAE and MS lesions is mainly induced in the CNS itself. J Neuroinflamm. (2016) 13:138. doi: 10.1186/s12974-016-0605-8
47. van Horssen J, Bö L, Dijkstra CD, de Vries HE. Extensive extracellular matrix depositions in active multiple sclerosis lesions. Neurobiol Dis. (2006) 24:484–91. doi: 10.1016/j.nbd.2006.08.005
48. McGinley AM, Sutton CE, Edwards SC, Leane CM, DeCourcey J, Teijeiro A, et al. Interleukin-17A serves a priming role in autoimmunity by recruiting IL-1β-producing myeloid cells that promote pathogenic T cells. Immunity. (2020) 52:342–356.e6. doi: 10.1016/j.immuni.2020.01.002
49. Cai B, Dongiovanni P, Corey KE, Wang X, Shmarakov IO, Zheng Z, et al. Macrophage MerTK promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metab. (2020) 31:406–421.e7. doi: 10.1016/j.cmet.2019.11.013
50. Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, Kasmi KCE, Smith AM, et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PloS Pathog. (2009) 5:e1000371. doi: 10.1371/journal.ppat.1000371
51. Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. (2013) 16:1211–8. doi: 10.1038/nn.3469
52. Locatelli G, Theodorou D, Kendirli A, Jordão MJC, Staszewski O, Phulphagar K, et al. Mononuclear phagocytes locally specify and adapt their phenotype in a multiple sclerosis model. Nat Neurosci. (2018) 21:1196–208. doi: 10.1038/s41593-018-0212-3
53. Zhang M, Zhang S. T cells in fibrosis and fibrotic diseases. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.01142
54. Fielding CA, Jones GW, McLoughlin RM, McLeod L, Hammond VJ, Uceda J, et al. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity. (2014) 40:40–50. doi: 10.1016/j.immuni.2013.10.022
55. Satoh JI, Tabunoki H, Yamamura T. Molecular network of the comprehensive multiple sclerosis brain-lesion proteome. Mult Scler. (2009) 15:531–41. doi: 10.1177/1352458508101943
56. Plikus MV, Wang X, Sinha S, Forte E, Thompson SM, Herzog EL, et al. Fibroblasts: Origins, definitions, and functions in health and disease. Cell. (2021) 184:3852–72. doi: 10.1016/j.cell.2021.06.024
57. Colombo E, Farina C. Astrocytes: key regulators of neuroinflammation. Trends Immunol. (2016) 37:608–20. doi: 10.1016/j.it.2016.06.006
58. Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol. (2010) 10:712–23. doi: 10.1038/nri2852
59. Kaufmann M, Schaupp AL, Sun R, Coscia F, Dendrou CA, Cortes A, et al. Identification of early neurodegenerative pathways in progressive multiple sclerosis. Nat Neurosci. (2022) 25:944–55. doi: 10.1038/s41593-022-01097-3
60. Wheeler MA, Clark IC, Tjon EC, Li Z, Zandee SEJ, Couturier CP, et al. MAFG-driven astrocytes promote CNS inflammation. Nature. (2020) 578:593–9. doi: 10.1038/s41586-020-1999-0
61. McKeon RJ, Schreiber RC, Rudge JS, Silver J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci. (1991) 11:3398–411. doi: 10.1523/JNEUROSCI.11-11-03398.1991
62. Iacobaeus E, Sugars RV, Törnqvist Andrén A, Alm JJ, Qian H, Frantzen J, et al. Dynamic changes in brain mesenchymal perivascular cells associate with multiple sclerosis disease duration, active inflammation, and demyelination. Stem Cells Transl Med. (2017) 6:1840–51. doi: 10.1002/sctm.17-0028
63. Richeldi L, Azuma A, Cottin V, Hesslinger C, Stowasser S, Valenzuela C, et al. Trial of a preferential phosphodiesterase 4B inhibitor for idiopathic pulmonary fibrosis. N Engl J Med. (2022) 386:2178–87. doi: 10.1056/NEJMoa2201737
64. King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. (2014) 370:2083–92. doi: 10.1056/NEJMoa1402582
65. Walker JE, Giri SN, Margolin SB. A double-blind, randomized, controlled study of oral pirfenidone for treatment of secondary progressive multiple sclerosis. Mult Scler J. (2005) 11:149–58. doi: 10.1191/1352458505ms1134oa
66. Walker JE, Margolin SB. Pirfenidone for chronic progressive multiple sclerosis. Mult Scler. (2001) 7:305–12. doi: 10.1177/135245850100700506
67. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. (2019) 381:1718–27. doi: 10.1056/NEJMoa1908681
68. Crespo O, Kang SC, Daneman R, Lindstrom TM, Ho PP, Sobel RA, et al. Tyrosine kinase inhibitors ameliorate autoimmune encephalomyelitis in a mouse model of multiple sclerosis. J Clin Immunol. (2011) 31:1010–20. doi: 10.1007/s10875-011-9579-6
69. Azizi G, Haidari MR, Khorramizadeh M, Naddafi F, Sadria R, Javanbakht MH, et al. Effects of imatinib mesylate in mouse models of multiple sclerosis and in vitro determinants. Iran J Allergy Asthma Immunol. (2014) 13:198–206.
70. Adzemovic M Z, Zeitelhofer M, Eriksson U, Olsson T, Nilsson I. Imatinib ameliorates neuroinflammation in a rat model of multiple sclerosis by enhancing blood-brain barrier integrity and by modulating the peripheral immune response. PloS One. (2013) 8:e56586. doi: 10.1371/journal.pone.0056586
71. Zeitelhofer M, Adzemovic MZ, Moessinger C, Stefanitsch C, Strell C, Muhl L, et al. Blocking PDGF-CC signaling ameliorates multiple sclerosis-like neuroinflammation by inhibiting disruption of the blood–brain barrier. Sci Rep. (2020) 10:22383. doi: 10.1038/s41598-020-79598-z
72. Vermersch P, Benrabah R, Schmidt N, Zéphir H, Clavelou P, Vongsouthi C, et al. Masitinib treatment in patients with progressive multiple sclerosis: a randomized pilot study. BMC Neurol. (2012) 12:36. doi: 10.1186/1471-2377-12-36
73. Vermersch P, Brieva-Ruiz L, Fox RJ, Paul F, Ramio-Torrenta L, Schwab M, et al. Efficacy and safety of masitinib in progressive forms of multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2022) 9:e1148. doi: 10.1212/NXI.0000000000001148
74. Galetta SL, Markowitz C, Lee AG. Immunomodulatory agents for the treatment of relapsing multiple sclerosis: a systematic review. Arch Intern Med. (2002) 162:2161–9. doi: 10.1001/archinte.162.19.2161
75. Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. (2017) 376:209–20. doi: 10.1056/NEJMoa1606468
76. Krämer J, Bar-Or A, Turner TJ, Wiendl H. Bruton tyrosine kinase inhibitors for multiple sclerosis. Nat Rev Neurol. (2023) 19:289–304. doi: 10.1038/s41582-023-00800-7
77. Zhu S, Jung J, Victor E, Arceo J, Gokhale S, Xie P. Clinical trials of the BTK inhibitors ibrutinib and acalabrutinib in human diseases beyond B cell Malignancies. Front Oncol. (2021) 11:737943. doi: 10.3389/fonc.2021.737943
78. Dubovsky JA, Flynn R, Du J, Harrington BK, Zhong Y, Kaffenberger B, et al. Ibrutinib treatment ameliorates murine chronic graft-versus-host disease. J Clin Invest. (2014) 124:4867–76. doi: 10.1172/JCI75328
79. Gu Y, Huang B, Yang Y, Qi M, Lu G, Xia D, et al. Ibrutinib exacerbates bleomycin-induced pulmonary fibrosis via promoting inflammation. Inflammation. (2018) 41:904–13. doi: 10.1007/s10753-018-0745-3
80. Evobrutinib Phase III. Darmstadt, Germany: Merck KGaA (2024). Available at: https://www.emdgroup.com/en/news/evobrutinib-phase-lll.html.
81. Bradbury EJ, Moon LDF, Popat RJ, King VR, Bennett GS, Patel PN, et al. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. (2002) 416:636–40. doi: 10.1038/416636a
82. Kuipers HF, Rieck M, Gurevich I, Nagy N, Butte MJ, Negrin RS, et al. Hyaluronan synthesis is necessary for autoreactive T-cell trafficking, activation, and Th1 polarization. Proc Natl Acad Sci. (2016) 113:1339–44. doi: 10.1073/pnas.1525086113
83. O’Shea TM, Burda JE, Sofroniew MV. Cell biology of spinal cord injury and repair. J Clin Invest. (2017) 127:3259–70. doi: 10.1172/JCI90608
Keywords: neurofibrosis, fibrosis, CNS, scarring, ECM, MS
Citation: Lozinski BM, Ghorbani S and Yong VW (2024) Biology of neurofibrosis with focus on multiple sclerosis. Front. Immunol. 15:1370107. doi: 10.3389/fimmu.2024.1370107
Received: 13 January 2024; Accepted: 12 March 2024;
Published: 26 March 2024.
Edited by:
Benjamin Knier, Technical University of Munich, GermanyReviewed by:
Claudia Cantoni, Barrow Neurological Institute (BNI), United StatesCarlo Chizzolini, University of Geneva, Switzerland
Copyright © 2024 Lozinski, Ghorbani and Yong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: V. Wee Yong, dnlvbmdAdWNhbGdhcnkuY2E=