 Derek A. Wiggins
Derek A. Wiggins Jack N. Maxwell
Jack N. Maxwell David E. Nelson
David E. Nelson- Department of Biology, Middle Tennessee State University, Murfreesboro, TN, United States
Macrophages are tissue resident innate phagocytic cells that take on contrasting phenotypes, or polarization states, in response to the changing combination of microbial and cytokine signals at sites of infection. During the opening stages of an infection, macrophages adopt the proinflammatory, highly antimicrobial M1 state, later shifting to an anti-inflammatory, pro-tissue repair M2 state as the infection resolves. The changes in gene expression underlying these transitions are primarily governed by nuclear factor kappaB (NF-κB), Janus kinase (JAK)/signal transducer and activation of transcription (STAT), and hypoxia-inducible factor 1 (HIF1) transcription factors, the activity of which must be carefully controlled to ensure an effective yet spatially and temporally restricted inflammatory response. While much of this control is provided by pathway-specific feedback loops, recent work has shown that the transcriptional co-regulators of the CBP/p300-interacting transactivator with glutamic acid/aspartic acid-rich carboxy-terminal domain (CITED) family serve as common controllers for these pathways. In this review, we describe how CITED proteins regulate polarization-associated gene expression changes by controlling the ability of transcription factors to form chromatin complexes with the histone acetyltransferase, CBP/p300. We will also cover how differences in the interactions between CITED1 and 2 with CBP/p300 drive their contrasting effects on pro-inflammatory gene expression.
Introduction
Macrophages are innate immune cells that exhibit remarkable phenotypic plasticity, adopting contrasting polarization states over the course of an infection to best contribute to tissue defense and repair (1). During an infection’s opening stages, exposure to interferon-γ (IFNγ) and/or pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS), promotes M1 polarization of naïve (M0) macrophages (2). In this state, macrophages secrete pro-inflammatory cytokines and exhibit heightened antimicrobial activity, partially driven by inducible nitric oxide synthase (iNOS) expression, which produces nitric oxide (NO) as a precursor for microbicidal reactive nitrogen species (RNS) (3–5). As an infection resolves, rising levels of interleukin (IL)-4 and IL-13 repolarize macrophages to the M2 state, which is associated with tissue repair and anti-inflammatory cytokine secretion. Additionally, the production of cytotoxic RNS is attenuated through arginase-1 (Arg1) expression, which competes with iNOS for arginine, the common substrate of both enzymes (6).
The transition between polarization states involves broad transcriptional reprogramming affecting >1,000 genes (7, 8). The molecular mechanisms governing these changes are of particular importance to human health as the failure to downregulate pro-inflammatory gene expression programs or activate them results in adverse outcomes. Specifically, inappropriate M1 activation contributes to inflammatory disorders, such as atherosclerosis, asthma, diet-induced obesity, and insulin resistance (9–13). Conversely, intracellular pathogens that colonize macrophages, including Toxoplasma gondii and Salmonella enterica serovar Typhimurium, utilize effectors that target these same mechanisms to block M1 or promote M2 polarization of host cells, creating a more permissive environment for growth and persistence (14–19).
While multiple transcription factors, including Krüppel-like factors (KLFs), hypoxia-inducible factor 1 (HIF1), and p53 (20–27) contribute to gene expression changes during polarization, the nuclear factor kappaB (NF-κB) and Janus kinase (JAK)/signal transducer and activation of transcription (STAT):interferon regulated factor (IRF) pathways are considered the primary controllers (28–30). Each is regulated by their own pathway-specific feedbacks that amplify or attenuate the response to polarizing stimuli. These include reciprocal positive feedback loops between Stat1 and Irf1 (31), and the various negative feedbacks (i.e., inhibitor-kappaB (IκB)α, ϵ, and A20) that prevent premature or prolonged NF-κB activity (32–39). However, these are supplemented by feedbacks that impact the activity of multiple polarization regulatory pathways, providing a higher level of control and coordination (40). For example, suppressor of cytokine signaling 1 (SOCS1) attenuates STAT1 activity by inhibiting JAKs and blocking NF-κB activity through recruitment to specific NF-κB-regulated gene promoters, stimulating nuclear p65 (RelA) NF-κB protein degradation (41–43). In this review, we will bring together recent findings showing how the CBP/p300-interacting transactivator with glutamic acid/aspartic acid-rich carboxy-terminal domain (CITED) family of transcriptional co-regulators contribute to this broader level of control. Specifically, we will describe how CITED1 and 2 proteins operate as common regulators of NF-κB, HIF1, IRF1, and the STAT family of transcription factors and explore how the antagonistic activities of these proteins shape the transcriptional response of macrophages to polarizing signals.
The CITED family of transcriptional co-regulators
Originally named melanocyte-specific gene 1 (msg1) following its discovery in highly pigmented B16-F1 murine melanoma cells, Cited1 was the first member of the Cited gene family to be described (44), with the same study also identifying Cited2, initially dubbed msg-related gene 1 (mrg1) based on sequence homology. This was quickly followed by Cited3, present in avian, amphibian, and fish species (45, 46) but absent in mice and humans, and Cited4, the only other family member expressed in mammalian systems (47, 48). These genes participate in different and diverse biological processes, such as melanocyte pigmentation (44), placentation (49), embryonic development (50), and differentiation (48). They are even implicated in different sets of pathological conditions, including congenital heart disease and cancer (51–64). In all of these biological processes, they function as transcriptional co-regulators (48, 65, 66).
As transcriptional co-regulators, CITED proteins lack DNA binding domains and are not known to form dimers or multimers. Instead, these proteins contain sets of conserved region (CR) motifs including the centrally located CR1 motif (~14 aa) present in CITED1, 2, and 3, a shorter CR3 motif (~6 aa) at the N-terminus of CITED2, 3, and 4, and the longer C-terminal CR2 domain (48-55 aa) common to all CITED proteins (Figure 1A) (47, 65). Outside these regions, CITED proteins exhibit minimal sequence similarity and are largely unstructured (Figure 1B), but can contain protein interaction motifs, such as the Smad-interacting domain (SID) identified in CITED1 (65).
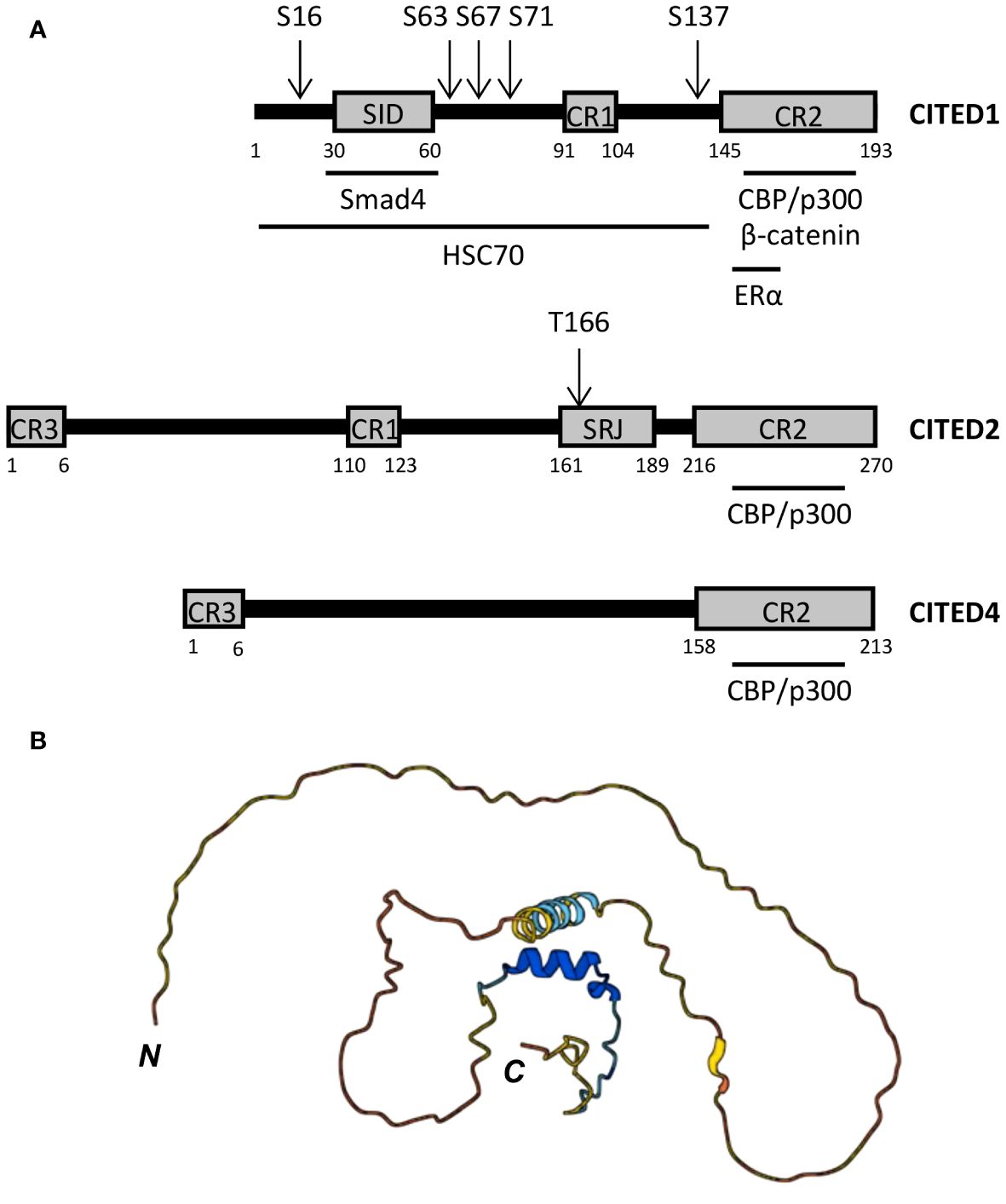
Figure 1 CITED protein structure. (A) Domain structure of human CITED1, 2, and 4. CR, conserved region; SID, Smad-interacting domain; SRJ, serine-rich junction. Black horizontal bars below each peptide map mark the regions required for the indicated protein:protein interactions, including the minimal CBP/p300-binding region, which is entirely contained within the CR2 domain of each CITED protein. Known phosphorylation sites are marked by arrows above each peptide map. (B) Predicted structure of human CITED1 generated in AlphaFold (67, 68). N- and C-termini are marked. Per-residue model confidence scores (pLDDT) on a 1-100 scale are encoded as follows: Dark blue, very high (pLDDT > 90); light blue, high (90 > pLDDT > 70); yellow, low (70 > pLDDT > 50); orange, very low (pLDDT < 50). The prediction indicates that CITED1 is largely unstructured.
While no clear function has been ascribed to CR1 and 3, the CR2 domain is essential for transcriptional co-regulatory activity (65, 69). Analogous to the transactivation domains (TADs) of STAT, NF-κB, and HIF1 proteins, CR2 enables CITED proteins to associate with one or more of the three cysteine-histidine-rich (CH) domains within the histone acetyltransferase (HAT), CREB binding protein (CBP) and its paralogue, p300 (65, 70–72) (Figure 2A). In this way, CITED proteins can inhibit or facilitate the formation of transcription factor:CBP/p300 complexes at gene cis-regulatory sites by blocking access to specific CH domains or functioning as a co-factor for transcription factors that weakly associate with CBP/p300 (66, 73–76) (Figures 2B, C).
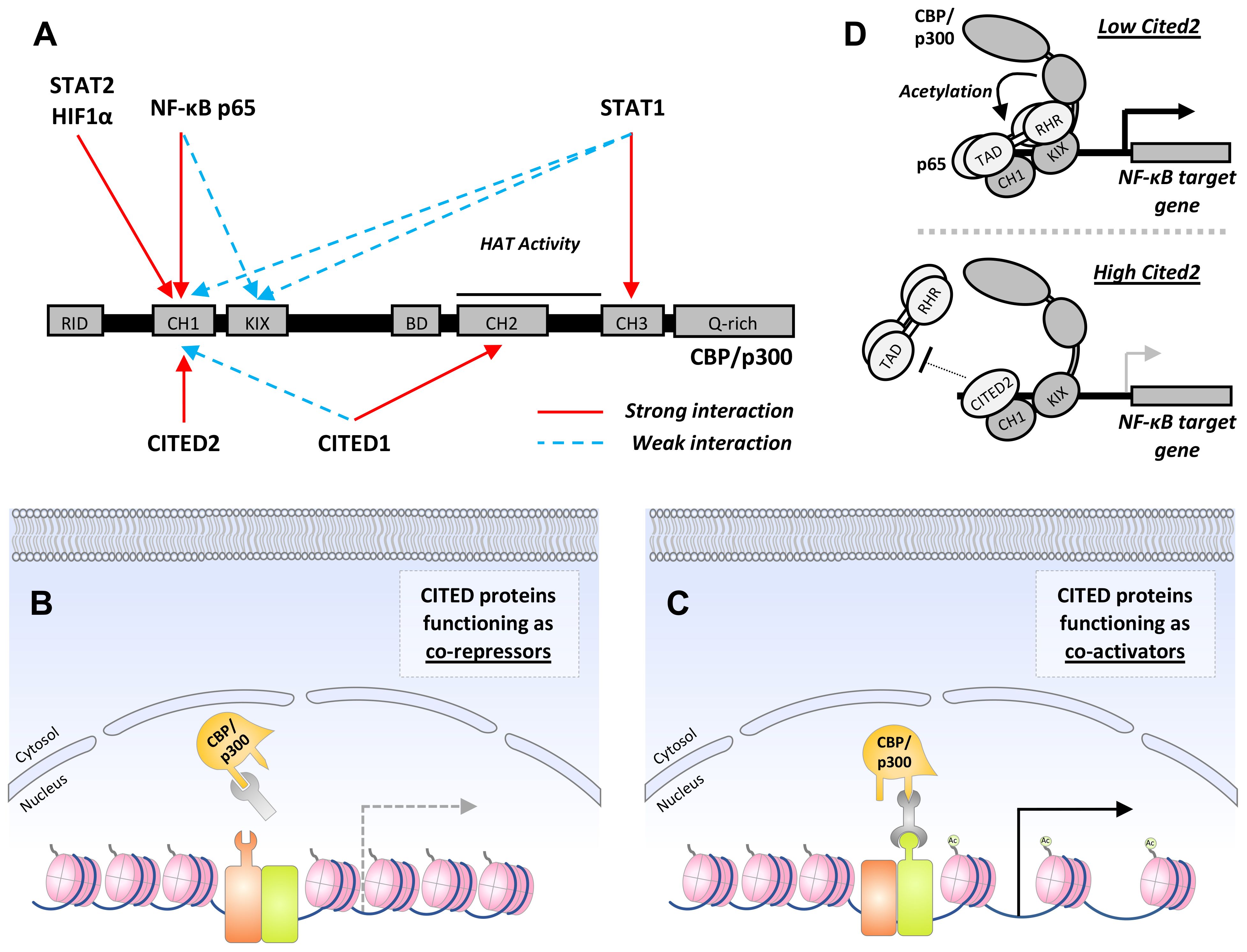
Figure 2 CITED proteins function as co-activators or repressors by regulating CBP/p300:transcription factor complex formation. (A) Schematic showing domains common to CBP and p300 with the interaction sites utilized by transcription factors that regulate macrophage M1 polarization, and the CITED transcriptional co-regulators are marked above and below, respectively. Strong and weak interactions are encoded in red and blue, respectively. RID, receptor-interacting domain; CH1-3, cysteine and histidine-rich regions; KIX, CREB-binding domain; BD, bromodomain; Q-rich, glutamine-rich domain. In some cases, interactions between a transcription factor and CITED protein with a common CH domain are known to be mutually exclusive (i.e., CITED2 and HIF1α with CH1). (B+C) General mechanisms for CITED protein co-repressor and co-activator activity. (B) By associating with the same docking site used by a transcription factor, CITED proteins can operate as co-repressors through a simple competitive inhibition mechanism. This is the mechanism by which CITED2 suppresses HIF1α-dependent gene expression. (C) Alternatively, CITED proteins can facilitate CBP/p300:transcription factor complex formation by operating as co-activators or adaptors. This results in the recruitment of CBP/p300 to cis-regulatory sites, histone acetylation, and chromatin remodeling that favors gene expression. Examples of this include CITED1:Smad4 and CITED1:STAT3:ERα complex formation Ac represents histone acetylation. (D) A specific example of CITED2 co-repressor activity: The p65 subunit of canonical NF-κB transcription factors makes bipartite interactions with CBP/p300, contacting both the CH1 and KIX domain, recruiting CBP/p300 to κB sites in target gene promoters. By disrupting the stronger of the two interactions, which occurs between the p65-TA2 subdomain and CBP/p300 CH1 domain, CITED2 inhibits CBP/p300-mediated acetylation of p65 itself, reducing NF-κB DNA-binding activity, and destabilizing the CBP/p300: NF-κB chromatin complex, thereby reduced expression of pro-inflammatory NF-κB target genes.
CITED2 interactions with CBP/p300
The first indication that CITED proteins function as transcriptional co-repressors was provided by Bhattacharya et al. (77). In this study, p35srj, a CITED2 isoform containing a 39 aa serine-glycine-rich junction (SRJ) between CR1 and 2 (Figure 1A), was identified in a screen for proteins interacting with p300-CH1 (TAZ1). Endogenous p35srj was shown to be constitutively nuclear in U2OS human osteosarcoma cells with almost the entire cellular pool found in CBP/p300 complexes, and the minimal binding region necessary for p300-CH1 interaction completely contained within the CR2 domain (224-255 aa). This study also showed that p35srj could inhibit HIF1-dependent gene transactivation by competing with the C-terminal TAD (CAD) of HIF1α for CH1 binding (Figures 2A, B). This is consistent with subsequent work showing synthetic membrane-permeable peptides, based on the CITED2 CR2 domain, inhibit HIF1α:p300 complex formation and HIF1-dependent gene expression (73).
One of the more important outcomes of the Bhattacharya study was the demonstration that CITED2 preferentially binds the CH1 domain of CBP/p300, distinguishing it from CITED1, which primarily associates with CH2 (65) (Figure 2A). Specifically, Bhattacharya et al. showed that p35srj did not reduce the activity of transcriptional regulators binding p300 outside the CH1 domain, including SREBP2 and src-1, which associate with the CREB-binding domain (KIX) and glutamine-rich domains of CBP/p300, respectively (78, 79). This result was supported by subsequent studies showing that CITED2 blocks the binding of the HIF1α N-terminal TAD (NAD) to p300-CH1 but not CH3 (74). More recently, NMR spectroscopy was used to identify the specific CH1 binding surface utilized by CITED2 and it was found that while the CITED2 CR2 domain and the HIF1α CAD bind to CH1 with similar affinity, they occupy distinct but overlapping binding positions (70). Collectively, these data indicate that CITED2 proteins may function as general inhibitors of transcription factors that use the CH1 domain for docking with CBP/p300.
CITED2 as a modulator of macrophage pro-inflammatory gene expression
Regulation of HIF1α activity
While HIF1α signaling is most closely associated with the hypoxia response, it also plays a significant role in macrophage polarization under normoxic conditions. Induced by M1 polarizing stimuli, including LPS (23, 26), HIF1α modulates the expression of almost half of all IFNγ-responsive genes (80). It also provides a protective role during Leishmaina donovani and Mycobacterium tuberculosis infection (81, 82) by regulating the shift to aerobic glycolysis and increasing macrophage microbicidal activity (80, 83, 84).
In 2018, the Mahabeleshwar lab demonstrated that CITED2 is present in human and murine macrophages and functions as a general inhibitor of pro-inflammatory gene expression and macrophage activation (85). This was found to be dependent, in part, on the ability of CITED2 to suppress HIF1α-regulated gene expression, demonstrated using CITED2-deficient bone marrow-derived macrophages (BMDMs). While this study did not discount the competitive inhibition model (Figure 2B), whereby CITED2 blocks HIF1α:CBP/p300 complex formation (74, 77), it showed that CITED2 enhances the expression of Egln3, a HIF1α negative regulator, in turn destabilizing the transcription factor (86). Furthermore, this study also showed that CITED2 expression is dynamic and is temporarily down-regulated in LPS or IFNγ stimulated BMDMs, presumably to disinhibit or permit expression of M1-associated genes after appropriate signals have been encountered.
Regulation of NF-κB activity
Canonical p65-containing NF-κB transcription factors play a central role in regulating M1-associated gene expression. Activated by cytokines (e.g., TNFα) and PAMPs, NF-κB regulates genes associated with cell survival, proliferation, and inflammation (87). Many of these genes are co-regulated by HIF1α and STAT1, including a principal M1 marker, Nos2, which exhibits synergistic expression following NF-κB and STAT1 activation (88). While the signaling cascades that trigger NF-κB activity vary between stimuli, most converge at the level of the inhibitor-kappaB kinase (IKK) signalosome (89). Once activated, the IKK signalosome phosphorylates the IκB proteins that keep dimeric NF-κB transcription factors locked in an inactive state in the cytoplasm (90), marking them for destruction by the proteasome (91, 92). This allows NF-κB proteins to translocate to the nucleus, and transactivate NF-κB-responsive gene promoters (93).
Like HIF1α, NF-κB also recruits CBP/p300 to target gene cis-regulatory sites to initiate gene transcription and does so in a bipartite fashion (94, 95). The TA2 subdomain of the p65 C-terminal TAD interacts strongly with the CBP/p300 CH1 domain, whereas the N-terminal Rel homology region, when phosphorylated at Ser276, forms a weaker interaction with the KIX domain (Figure 2A). The p65 TA2 subdomain is intrinsically disordered, and it associates with interlinked hydrophobic grooves on the surface of CH1 (95). Experiments using p65 TA2 and Ser276 mutants, ablating KIX or CH1 binding activity, respectively, show that these interactions are functionally separable and regulate different NF-κB target gene sets. Mutation of the TA2 domain affects a broader set of genes, including Ptgs2 and Cxcl1, than the loss of Ser276 phosphorylation, suggestive of the important role played by the p65-TA2:CBP-CH1 interaction. Additionally, expression of the main NF-κB negative regulators, IκBα and A20, encoded by Nfkbia and Tnfaip3, required both interactions (95).
Given the importance of the p65-TA2:CBP-CH1 interaction for target gene transactivation, it is unsurprising that CITED2 operates as a negative regulator of macrophage NF-κB signaling (96, 97). This was first shown by Lou et al, demonstrating that CITED2 attenuates NF-κB target gene expression in J774 macrophage-like cells and BMDMs (97). Their data was consistent with CITED2 operating as a competitive inhibitor of p65:CBP/p300 interactions, as i) CITED2 expression had no effect on up-stream processes, including IκBα phosphorylation and degradation, and ii) mutations within the CITED2 CR2 domain ablating p300 binding resulted in the complete loss of CITED2 co-repressor activity. Lou et al. also showed that CITED2 expression reduced p300-dependent acetylation of p65. This likely explains the reduction in p65 and RNA polymerase II detected at Tnfaip3 and Cxcl8 promoters by ChIP as acetylation of p65 at Lys310 is required for DNA binding (98). These findings are largely corroborated by Pong et al, which used complementary gain- and loss-of-function manipulations coupled with RNA sequencing and gene set enrichment analysis (GSEA) to investigate the role of CITED2 in a mouse model of lung inflammation (96). BMDMs derived from Cited2fl/fl:Lyz2cre mice exhibited enhanced expression of multiple pro-inflammatory NF-κB targets following LPS stimulation, including Il1β, Cxcl2, Tnf, and Klf6. In agreement with Lou et al, the investigators found that CITED2 inhibited LPS-induced p65-promoter binding without affecting the release of NF-κB from IκB complexes. Additionally, Pong et al. showed that loss of myeloid CITED2 expression significantly increased macrophage recruitment to lung tissue following intratracheal delivery of zymosan in a murine model of acute lung inflammation. Collectively, these studies support the notion that CITED2 functions as an NF-κB co-repressor by preventing productive p65:CBP/p300 interactions, reducing p65 acetylation and DNA binding activity. This hampers NF-κB:CBP/p300 complex recruitment to κB sites and attenuates pro-inflammatory gene expression (Figure 2D).
Regulation of JAK-STAT activity
As an additional finding of the Pong et al. study, data from their GSEA analysis indicated that CITED2 loss enhanced expression of IFNγ-responsive genes in LPS-stimulated BMDMs, raising the possibility that CITED2 also operates as a suppressor of STAT1-IRF1 signaling. This was explored by the Mahabeleshwar lab in Zahar et al. using the same Cited2fl/fl:Lyz2cre mouse model (99). Here, they showed that CITED2 loss enhanced expression of both STAT1 and IRF1 gene targets in IFNγ-stimulated BMDMs, including members of the Isg, Ifit, and Oasl gene families. As observed for NF-κB, the effect of CITED2 expression on STAT1 signaling was confined to the nucleus as CITED2 loss had no effect on STAT1 protein levels or phosphorylation at Tyr701 following IFNγ stimulation. Rather, CITED2 deficient BMDMs showed increased STAT1-chromatin binding. Most significantly, these cells showed enrichment of STAT1 at the Irf1 promoter, correlating with increased transcription of the Irf1 gene and heightened IRF1 protein levels. This supports a model where CITED2 is interrupting the positive feedback between STAT1 and IRF1, thereby attenuating the STAT1-IRF1 transcriptional program that is crucial for macrophage M1 polarization.
Quite how CITED2 inhibits STAT1 activity has not been fully resolved and is likely to be distinct from the HIF1 and NF-κB competitive inhibition mechanisms due to differences in how STAT1 interacts with CBP/p300. While there are some variations in how this interaction is reported (100–102), there appears to be two contact points between these proteins involving the STAT1 N- and C-termini. The N-terminus, which plays a role in cooperative binding interactions with DNA (103), interacts with the CBP/p300 KIX domain (104), whereas the C-terminal TAD binds both CH1 and CH3 (72). However, it exhibits a strong preference for CH3, with the CH1 binding interaction being 100-fold weaker. Additionally, Zafar et al. show that STAT1 and CITED2 are both recruited to the IRF1 promoter in IFNγ-stimulated BMDMs (99). Taken together, these data bring a CITED2:STAT1 competitive inhibition model into question.
Interestingly, STAT2 proteins, which have a slightly longer C-terminal TAD than STAT1, preferentially bind CH1, interacting with the same surfaces as HIF1α and CITED2 (72, 105). This indicates that CITED2 may also function as a STAT2 co-repressor following type I interferon stimulation, which is consistent with the Pong et al. transcriptome data showing enhancement of the IFNα-response gene sets in LPS-stimulated BMDMs from Cited2fl/fl:Lyz2cre mice (96). As a small caveat, STAT2 is known to recruit other HATs, such as GCN5 rather than CBP/p300, to some ISG enhancers (106), which may limit the impact of CITED2 on STAT2 targets.
The interplay between CITED2 and the JAK/STAT signaling pathway is not restricted to STAT1 and STAT2. Clear genetic and protein interactions with other STAT family members that contribute to the shaping of the M1 and M2 transcriptome have also been demonstrated. Within the past year new data generated using the Cited2fl/fl:Lyz2cre mouse model has identified a relationship between CITED2, STAT3, and STAT5 that modulates the macrophage gene expression programs and contributes to the inflammation and insulin resistance observed during diet-induced obesity (107). In this system, CITED2 was found to suppress STAT3 and STAT5 target gene expression in BMDMs. For STAT5, this was shown to involve the suppression of LPS-induced Tyr694 phosphorylation, a modification necessary for STAT5a nuclear accumulation, dimerization, and activity. An important downstream effect of this was increased expression of B cell lymphoma 6 (BCL6), a transcriptional repressor that restrains the macrophage pro-inflammatory response and is itself inhibited at the transcriptional level by STAT5 (108, 109). In effect, this CITED2-STAT5-BCL6 axis enables CITED2 to control broad swaths of the M1 transcriptional program in a manner that is at least partially separable from its own role as a co-repressor of transcription factor:CBP/p300 interactions.
In addition to acting as a general inhibitor of M1-associated pro-inflammatory gene expression, the relationship between STAT6 and CITED2 enables it to make meaningful contributions to the M2 transcriptome. Just as CITED2 expression is inhibited by M1-polarizaing signals, an M2-polarizing environment (IL-4 or IL-13 stimulation) promotes a modest increase in CITED2 expression, which was found to be regulated at a transcriptional level by STAT6 (85). Under these conditions, CITED2 proteins form complexes with peroxisome proliferator-activated receptor gamma (PPARγ), a nuclear receptor that collaborates with STAT6 to regulate IL-4-stimulated anti-inflammatory gene expression (110, 111). Not only was CITED2 shown to enhance PPARγ-dependent gene expression in macrophages, but it was also found to stabilize the interaction between PPARγ and the Arg1 enhancer in IL-4-stimulated BMDMs and was associated with increased expression on this M2 marker. Interestingly, IL-4 has also been shown to promote the STAT6-dependent transcription of Cited1 in human adipocytes with CITED1 potentially functioning as a thermogenic protein in this context (112). Whether it is similarly regulated in myeloid cells has yet to be investigated.
Collectively, these studies show that the contrasting regulation of CITED2 expression under M1 or M2-stimulating conditions creates an elegant switching mechanism that ensures clear transitions between polarization states. While this may primarily utilize the canonical transcriptional co-repressor activities of CITED2, its ability to influence HIF1α and BCL6 levels, as well as facilitating PPARγ-promoter interactions, extends the influence of CITED2 over a wider set of pro- and anti-inflammatory genes.
Cited1, a newly discovered regulator of macrophage pro-inflammatory gene expression
While Cited2 expression is ubiquitous, Cited1 is largely restricted to a small number of tissues (113), and was thought to be absent in macrophages (85). This perception was changed in 2020 when Cited1 transcripts were detected in M1-polarized RAW264.7 murine macrophage-like cells infected with the fungal pathogen, Cryptococcus neoformans (114). It was subsequently determined that exposure of these cells to IFNγ for periods greater than 24 hours stimulated expression of CITED1 proteins, regulated by both STAT1 and IRF1 at the transcriptional level. Cited1 expression was unaffected by other M1 polarizing stimuli (i.e., LPS), indicating that it operates as an interferon-stimulated gene (ISG) (113). This pattern of regulation is in stark contrast to Cited2, which is transiently downregulated following short periods of exposure (6 hours) to M1 polarizing stimuli (LPS or IFNγ) and upregulated by M2 stimuli (IL-4 or IL-13) (85), indicating that the circumstances under which CITED1 and 2 operate may be minimal or non-overlapping.
A further regulatory difference between CITED1 and 2 is their subcellular localization under varying polarizing conditions. While CITED2 has been shown to be constitutively nuclear (97), CITED1 is predominantly cytoplasmic in unstimulated cells but becomes enriched in the nucleus following IFNγ stimulation (113). Quite how this shift in subcellular distribution is controlled is presently unclear, but it may be connected to the increased phosphorylation of the protein observed in IFNγ-stimulated macrophages as CITED1 phosphorylation at Ser79 following parathyroid-hormone-stimulation in murine osteoblastic cells is known to be associated with its nuclear translocation (115, 116).
The differences between CITED1 and 2 go beyond the regulation of their expression and localization and extends to their function with the two transcriptional regulators exerting opposing effects on macrophage pro-inflammatory gene expression. This was shown by Subramani et al. using a complementary loss- and gain-of-function approach, where it was found that CITED1 enhances STAT1- and IRF1-regulated gene expression in IFNγ-stimulated macrophages, increasing transcript levels of Ccl, Isg, and Ifit family members amongst many others (113). These data suggest that CITED1 is functioning as a transcriptional co-enhancer (Figure 2C), versus the largely co-inhibitory effects of CITED2 in the same context. This is perhaps unsurprising as (i) neither STAT1 or IRF1 bind CBP/p300 via CH2, which is thought to be the preferred CITED1 interaction site (72, 117), and (ii) CITED1 has been shown to function as a co-enhancer in other contexts (65, 69, 118). For example, CITED1 enhances Smad4-dependent gene expression by stabilizing CBP/p300:Smad4 complexes, interacting with both proteins (65). Similarly, CITED1 acts as a co-factor for the assembly of STAT3:estrogen receptor-α (ERα) transcriptional complexes in neurons as part of leptin signaling, contributing to whole-body energy homeostasis (118). This is of relevance as STAT3 contributes to IFNγ-stimulated gene expression in macrophages (119, 120). It also illustrates the close relationship that both CITED1 and 2 share with the STAT family of transcriptional regulators.
Concluding remarks and future directions
CBP/p300 serves as a convergence point for the principal signaling pathways that control macrophage polarization, with their associated transcription factors either competing for shared CBP/p300 binding surfaces or acting cooperatively to recruit it to enhancers. The CITED family of transcriptional regulators largely operate at this level and their differing binding preference for the three CH domains within CBP/p300 enable them to hinder or facilitate this process, shaping macrophage pro- and anti-inflammatory gene expression and phenotype. This process is assisted by the contrasting regulation of Cited1 and 2, which results in the highest expression of CITED2 in the absence of M1 or presence of M2-polarizing stimuli, dampening pro-inflammatory gene expression. Whereas CITED1 is only expressed at detectable levels in cells exposed to IFNγ for extended periods, enhancing ISG expression and maintaining the pro-inflammatory response.
The contributions of CITED2 to macrophage polarization and its role in a variety of inflammatory disorders, including atherosclerosis and diet-induced obesity, has been well explored using mouse models and primary cells (85, 96, 97, 99, 107). Additionally, the molecular interactions of this protein with CBP/p300 are well understood to the extent where the CITED2 CR2 domain has been used as the basis for cell-permeable peptide inhibitors of HIF1:CBP/p300 interaction with clinical potential (73). Far less is known about CITED1 in this context and our current knowledge is based exclusively on studies in murine macrophage cell lines (113, 114). Those in vivo studies that have been performed have instead focused on the impact of Cited1 gene deletion or the effect of CITED1 mutants on development and cancer rather than its role in myeloid cells (60, 121). Perhaps most importantly, the CITED1:CBP/p300 interaction has not been fully resolved with the proposed CH2-binding preference of CITED1 based only on qualitative in vitro pull-down mapping experiments using large GST-fused portions of p300 containing more than just the CH domains (65). While the CR2 domains of CITED1 and 2 are highly similar, (82% amino acid identity based on aa 161-209 of human CITED2 (44);), there are notable differences, including a 7 aa insert at the end of the domain that is absent from CITED1, indicating that a difference in CH binding preference is at least plausible. Future in-depth structural studies will provide clarity in this area. Should these differences in binding preference prove to be valid then their contributions to the seemingly opposite effects of CITED1 and 2 on IFNγ-stimulated gene expression could be explored using CITED mutants where the CR2 domains have been exchanged. Hypothetically, this would convert CITED1 from a transcriptional co-enhancer to a co-repressor.
A further unresolved question concerns the potentially differing relationships between the CITED proteins and the HATs, CBP and p300. It is currently unclear whether they exhibit a binding preference for one or the other, which is potentially significant as while the activities of CBP and p300 are at least partially overlapping, they are known to have separate functions and regulate distinct collections of genes (102). This is relevant within the context of macrophage polarization as CBP and p300 interact differently with STAT1. As an example of this, CBP, but not p300, has been shown to directly acetylate the STAT1 DNA binding domain at Lys410 and 413 (122). This has two important consequences; i) acetylated STAT1 binds nuclear NF-κB dimers, reducing their DNA-binding and transcriptional regulatory activity, and ii) signals for the nuclear export and dephosphorylation of STAT1 as part of a phosphorylation-acetylation switch that deactivates the protein (123). This would suggest that by interfering with the STAT1:CBP interaction, CITED proteins could, perhaps counterintuitively, extend STAT1 nuclear occupancy and enhance NF-κB activity.
As a similar concern, our understanding of how post-translational modifications (PTMs) of the CITED proteins themselves impact their function and activity remains incomplete. Most of those reported to date are not known to occur within myeloid cells and include the MAPK1-dependent phosphorylation of CITED2 at T166 at the N-terminal end of the SRJ domain (124), and the cell cycle-regulated phosphorylation of CITED1 mapped to five serine residues throughout the protein (125), with these modifications altering the coactivator functions of the proteins by influencing CBP/p300 binding. So far only CITED1 has been shown to be phosphorylated in macrophages in response to polarizing signals (in this case, IFNγ), but the modification sites, kinases responsible, and the functional consequences have yet to be elucidated. Additionally, just as many of the transcription factors that interact with CBP/p300 are acetylated by these HATs as an important part of their regulation [e.g., STAT1 and p65 NF-κB (98, 122, 123)], it seems plausible that the CITED proteins will also be acetylation targets or subject to other PTMs.
In closing, much remains to be discovered about how CITED proteins contribute to the control of macrophage polarization. Continued efforts in this area will help us better understand the various CBP/p300-dependent and independent mechanisms used by these proteins to tune pro- and anti-inflammatory gene expression and how the CITED proteins themselves are regulated.
Author contributions
DW: Writing – original draft, Writing – review & editing. JM: Writing – original draft, Writing – review & editing. DN: Writing – original draft, Writing – review & editing, Conceptualization, Funding acquisition, Supervision.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by funds from the National Institutes of Health (NIAID 1R15AI178461-01) to DN, the Molecular Biosciences (MOBI) doctoral program at Middle Tennessee State University (MTSU) to DW, and the MTSU Foundation to JM.
Acknowledgments
The authors would like to acknowledge members of the DEN lab and Drs. Rebecca Seipelt-Thiemann (Middle Tennessee State University) and Erin E. McClelland (Marian University) for useful discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Murray PJ. Macrophage polarization. Annu Rev Physiol. (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
2. Dileepan KN, Page JC, Li Y, Stechschulte DJ. Direct activation of murine peritoneal macrophages for nitric oxide production and tumor cell killing by interferon-gamma. J Interferon Cytokine Res. (1995) 15:387–94. doi: 10.1089/jir.1995.15.387
3. de Vera ME, Shapiro RA, Nussler AK, Mudgett JS, Simmons RL, Morris SM Jr., et al. Transcriptional regulation of human inducible nitric oxide synthase (NOS2) gene by cytokines: initial analysis of the human NOS2 promoter. Proc Natl Acad Sci USA. (1996) 93:1054–9. doi: 10.1073/pnas.93.3.1054
4. Granger DL, Hibbs JB Jr., Perfect JR, Durack DT. Specific amino acid (L-arginine) requirement for the microbiostatic activity of murine macrophages. J Clin Invest. (1988) 81:1129–36. doi: 10.1172/JCI113427
5. MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. (1995) 81:641–50. doi: 10.1016/0092-8674(95)90085-3
6. Rath M, Muller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. (2014) 5:532. doi: 10.3389/fimmu.2014.00532
7. Beyer M, Mallmann MR, Xue J, Staratschek-Jox A, Vorholt D, Krebs W, et al. High-resolution transcriptome of human macrophages. PloS One. (2012) 7:e45466. doi: 10.1371/journal.pone.0045466
8. Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S, et al. Novel markers to delineate murine M1 and M2 macrophages. PloS One. (2015) 10:e0145342. doi: 10.1371/journal.pone.0145342
9. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. (2017) 542:177–85. doi: 10.1038/nature21363
10. Heilbronn LK, Campbell LV. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr Pharm Des. (2008) 14:1225–30. doi: 10.2174/138161208784246153
11. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. (2013) 13:709–21. doi: 10.1038/nri3520
12. van der Veen TA, de Groot LES, Melgert BN. The different faces of the macrophage in asthma. Curr Opin Pulm Med. (2020) 26:62–8. doi: 10.1097/MCP.0000000000000647
13. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. (2013) 496:445–55. doi: 10.1038/nature12034
14. Nelson RH, Nelson DE. Signal distortion: how intracellular pathogens alter host cell fate by modulating NF-kappaB dynamics. Front Immunol. (2018) 9:2962. doi: 10.3389/fimmu.2018.02962
15. Luder CG, Walter W, Beuerle B, Maeurer MJ, Gross U. Toxoplasma gondii down-regulates MHC class II gene expression and antigen presentation by murine macrophages via interference with nuclear translocation of STAT1alpha. Eur J Immunol. (2001) 31:1475–84. doi: 10.1002/1521-4141(200105)31:5<1475::AID-IMMU1475>3.0.CO;2-C
16. Seabra SH, de Souza W, DaMatta RA. Toxoplasma gondii partially inhibits nitric oxide production of activated murine macrophages. Exp Parasitol. (2002) 100:62–70. doi: 10.1006/expr.2001.4675
17. Zimmermann S, Murray PJ, Heeg K, Dalpke AH. Induction of suppressor of cytokine signaling-1 by Toxoplasma gondii contributes to immune evasion in macrophages by blocking IFN-gamma signaling. J Immunol. (2006) 176:1840–7. doi: 10.4049/jimmunol.176.3.1840
18. Panagi I, Jennings E, Zeng J, Gunster RA, Stones CD, Mak H, et al. Salmonella effector steE converts the mammalian serine/Threonine kinase GSK3 into a tyrosine kinase to direct macrophage polarization. Cell Host Microbe. (2020) 27:41–53.e46. doi: 10.1016/j.chom.2019.11.002
19. Stapels DAC, Hill PWS, Westermann AJ, Fisher RA, Thurston TL, Saliba AE, et al. Salmonella persisters undermine host immune defenses during antibiotic treatment. Science. (2018) 362:1156–60. doi: 10.1126/science.aat7148
20. Sanceau J, Kaisho T, Hirano T, Wietzerbin J. Triggering of the human interleukin-6 gene by interferon-gamma and tumor necrosis factor-alpha in monocytic cells involves cooperation between interferon regulatory factor-1, NF kappa B, and Sp1 transcription factors. J Biol Chem. (1995) 270:27920–31. doi: 10.1074/jbc.270.46.27920
21. Blanco JC, Contursi C, Salkowski CA, DeWitt DL, Ozato K, Vogel SN. Interferon regulatory factor (IRF)-1 and IRF-2 regulate interferon gamma-dependent cyclooxygenase 2 expression. J Exp Med. (2000) 191:2131–44. doi: 10.1084/jem.191.12.2131
22. Kamijo R, Harada H, Matsuyama T, Bosland M, Gerecitano J, Shapiro D, et al. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science. (1994) 263:1612–5. doi: 10.1126/science.7510419
23. Mahabeleshwar GH, Kawanami D, Sharma N, Takami Y, Zhou G, Shi H, et al. The myeloid transcription factor KLF2 regulates the host response to polymicrobial infection and endotoxic shock. Immunity. (2011) 34:715–28. doi: 10.1016/j.immuni.2011.04.014
24. Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, et al. Kruppel-like factor 4 regulates macrophage polarization. J Clin Invest. (2011) 121:2736–49. doi: 10.1172/JCI45444
25. Takeda N, O’Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, et al. Differential activation and antagonistic function of HIF-alpha isoforms in macrophages are essential for NO homeostasis. Genes Dev. (2010) 24:491–501. doi: 10.1101/gad.1881410
26. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, et al. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. (2005) 115:1806–15. doi: 10.1172/JCI23865
27. Li L, Ng DS, Mah WC, Almeida FF, Rahmat SA, Rao VK, et al. A unique role for p53 in the regulation of M2 macrophage polarization. Cell Death Differ. (2015) 22:1081–93. doi: 10.1038/cdd.2014.212
28. Mussbacher M, Derler M, Basilio J, Schmid JA. NF-kappaB in monocytes and macrophages - an inflammatory master regulator in multitalented immune cells. Front Immunol. (2023) 14:1134661. doi: 10.3389/fimmu.2023.1134661
29. Darnell JE Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. (1994) 264:1415–21. doi: 10.1126/science.8197455
30. Varinou L, Ramsauer K, Karaghiosoff M, Kolbe T, Pfeffer K, Muller M, et al. Phosphorylation of the Stat1 transactivation domain is required for full-fledged IFN-gamma-dependent innate immunity. Immunity. (2003) 19:793–802. doi: 10.1016/S1074-7613(03)00322-4
31. Wong LH, Sim H, Chatterjee-Kishore M, Hatzinisiriou I, Devenish RJ, Stark G, et al. Isolation and characterization of a human STAT1 gene regulatory element. Inducibility by interferon (IFN) types I and II and role of IFN regulatory factor-1. J Biol Chem. (2002) 277:19408–17. doi: 10.1074/jbc.M111302200
32. Dorrington MG, Fraser IDC. NF-kappaB signaling in macrophages: dynamics, crosstalk, and signal integration. Front Immunol. (2019) 10:705. doi: 10.3389/fimmu.2019.00705
33. Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, et al. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. (2004) 306:704–8. doi: 10.1126/science.1099962
34. Kearns JD, Basak S, Werner SL, Huang CS, Hoffmann A. IkappaBepsilon provides negative feedback to control NF-kappaB oscillations, signaling dynamics, and inflammatory gene expression. J Cell Biol. (2006) 173:659–64. doi: 10.1083/jcb.200510155
35. Brown K, Park S, Kanno T, Franzoso G, Siebenlist U. Mutual regulation of the transcriptional activator NF-kappa B and its inhibitor, I kappa B-alpha. Proc Natl Acad Sci USA. (1993) 90:2532–6. doi: 10.1073/pnas.90.6.2532
36. Scott ML, Fujita T, Liou HC, Nolan GP, Baltimore D. The p65 subunit of NF-kappa B regulates I kappa B by two distinct mechanisms. Genes Dev. (1993) 7:1266–76. doi: 10.1101/gad.7.7a.1266
37. Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science. (1993) 259:1912–5. doi: 10.1126/science.8096091
38. Song HY, Rothe M, Goeddel DV. The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-kappaB activation. Proc Natl Acad Sci USA. (1996) 93:6721–5. doi: 10.1073/pnas.93.13.6721
39. Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. (2010) 327:1135–9. doi: 10.1126/science.1182364
40. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. (2014) 5:614. doi: 10.3389/fimmu.2014.00614
41. Strebovsky J, Walker P, Lang R, Dalpke AH. Suppressor of cytokine signaling 1 (SOCS1) limits NFκB signaling by decreasing p65 stability within the cell nucleus. FASEB J. (2011) 25:863–74. doi: 10.1096/fj.10-170597
42. Liau NPD, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E, et al. The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun. (2018) 9:1558. doi: 10.1038/s41467-018-04013-1
43. Coelho DR, Palma FR, Paviani V, LaFond KM, Huang Y, Wang D, et al. SOCS1 regulates a subset of NFkappaB-target genes through direct chromatin binding and defines macrophage functional phenotypes. iScience. (2023) 26:106442. doi: 10.1016/j.isci.2023.106442
44. Shioda T, Fenner MH, Isselbacher KJ. msg1, a novel melanocyte-specific gene, encodes a nuclear protein and is associated with pigmentation. Proc Natl Acad Sci USA. (1996) 93:12298–303. doi: 10.1073/pnas.93.22.12298
45. Andrews JE, O’Neill MJ, Binder M, Shioda T, Sinclair AH. Isolation and expression of a novel member of the CITED family. Mech Dev. (2000) 95:305–8. doi: 10.1016/S0925-4773(00)00362-2
46. Ng PK, Chiu SK, Kwong TF, Yu RM, Wong MM, Kong RY. Functional characterization of two CITED3 homologs (gcCITED3a and gcCITED3b) in the hypoxia-tolerant grass carp, Ctenopharyngodon idellus. BMC Mol Biol. (2009) 10:101. doi: 10.1186/1471-2199-10-101
47. Braganca J, Swingler T, Marques FI, Jones T, Eloranta JJ, Hurst HC, et al. Human CREB-binding protein/p300-interacting transactivator with ED-rich tail (CITED) 4, a new member of the CITED family, functions as a co-activator for transcription factor AP-2. J Biol Chem. (2002) 277:8559–65. doi: 10.1074/jbc.M110850200
48. Yahata T, Takedatsu H, Dunwoodie SL, Braganca J, Swingler T, Withington SL, et al. Cloning of mouse Cited4, a member of the CITED family p300/CBP-binding transcriptional coactivators: induced expression in mammary epithelial cells. Genomics. (2002) 80:601–13. doi: 10.1006/geno.2002.7005
49. Kuna M, Dhakal P, Iqbal K, Dominguez EM, Kent LN, Muto M, et al. CITED2 is a conserved regulator of the uterine-placental interface. Proc Natl Acad Sci USA. (2023) 120:e2213622120. doi: 10.1073/pnas.2213622120
50. Buaas FW, Val P, Swain A. The transcription co-factor CITED2 functions during sex determination and early gonad development. Hum Mol Genet. (2009) 18:2989–3001. doi: 10.1093/hmg/ddp237
51. Sperling S, Grimm CH, Dunkel I, Mebus S, Sperling HP, Ebner A, et al. Identification and functional analysis of CITED2 mutations in patients with congenital heart defects. Hum Mutat. (2005) 26:575–82. doi: 10.1002/(ISSN)1098-1004
52. Yang XF, Wu XY, Li M, Li YG, Dai JT, Bai YH, et al. [Mutation analysis of Cited2 in patients with congenital heart disease]. Zhonghua Er Ke Za Zhi. (2010) 48:293–6.
53. Mattes K, Berger G, Geugien M, Vellenga E, Schepers H. CITED2 affects leukemic cell survival by interfering with p53 activation. Cell Death Dis. (2017) 8:e3132. doi: 10.1038/cddis.2017.548
54. Jayaraman S, Doucet M, Kominsky SL. CITED2 attenuates macrophage recruitment concordant with the downregulation of CCL20 in breast cancer cells. Oncol Lett. (2018) 15:871–8. doi: 10.3892/ol
55. Jayaraman S, Doucet M, Kominsky SL. Down-regulation of CITED2 attenuates breast tumor growth, vessel formation and TGF-beta-induced expression of VEGFA. Oncotarget. (2017) 8:6169–78. doi: 10.18632/oncotarget.v8i4
56. Bai L, Merchant JL. A role for CITED2, a CBP/p300 interacting protein, in colon cancer cell invasion. FEBS Lett. (2007) 581:5904–10. doi: 10.1016/j.febslet.2007.11.072
57. Sun HB, Zhu YX, Yin T, Sledge G, Yang YC. MRG1, the product of a melanocyte-specific gene related gene, is a cytokine-inducible transcription factor with transformation activity. Proc Natl Acad Sci USA. (1998) 95:13555–60. doi: 10.1073/pnas.95.23.13555
58. Chou YT, Hsieh CH, Chiou SH, Hsu CF, Kao YR, Lee CC, et al. CITED2 functions as a molecular switch of cytokine-induced proliferation and quiescence. Cell Death Differ. (2012) 19:2015–28. doi: 10.1038/cdd.2012.91
59. Jayaraman S, Doucet M, Lau WM, Kominsky SL. CITED2 modulates breast cancer metastatic ability through effects on IKKalpha. Mol Cancer Res. (2016) 14:730–9. doi: 10.1158/1541-7786.MCR-16-0081
60. Murphy AJ, Pierce J, de Caestecker C, Ayers GD, Zhao A, Krebs JR, et al. CITED1 confers stemness to Wilms tumor and enhances tumorigenic responses when enriched in the nucleus. Oncotarget. (2014) 5:386–402. doi: 10.18632/oncotarget.v5i2
61. Murphy AJ, Pierce J, de Caestecker C, Taylor C, Anderson JR, Perantoni AO, et al. SIX2 and CITED1, markers of nephronic progenitor self-renewal, remain active in primitive elements of Wilms’ tumor. J Pediatr Surg. (2012) 47:1239–49. doi: 10.1016/j.jpedsurg.2012.03.034
62. Murphy AJ, de Caestecker C, Pierce J, Boyle SC, Ayers GD, Zhao Z, et al. CITED1 expression in liver development and hepatoblastoma. Neoplasia. (2012) 14:1153–63. doi: 10.1593/neo.12958
63. Nasu T, Oku Y, Takifuji K, Hotta T, Yokoyama S, Matsuda K, et al. Predicting lymph node metastasis in early colorectal cancer using the CITED1 expression. J Surg Res. (2013) 185:136–42. doi: 10.1016/j.jss.2013.05.041
64. Yadav ML, Jain D, Neelabh, Agrawal D, Kumar A, Mohapatra B, et al. A gain-of-function mutation in CITED2 is associated with congenital heart disease. Mutat Res. (2021) 822:111741. doi: 10.1016/j.mrfmmm.2021.111741
65. Yahata T, de Caestecker MP, Lechleider RJ, Andriole S, Roberts AB, Isselbacher KJ, et al. The MSG1 non-DNA-binding transactivator binds to the p300/CBP coactivators, enhancing their functional link to the Smad transcription factors. J Biol Chem. (2000) 275:8825–34. doi: 10.1074/jbc.275.12.8825
66. Leung MK, Jones T, Michels CL, Livingston DM, Bhattacharya S. Molecular cloning and chromosomal localization of the human CITED2 gene encoding p35srj/Mrg1. Genomics. (1999) 61:307–13. doi: 10.1006/geno.1999.5970
67. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. (2021) 596:583–9. doi: 10.1038/s41586-021-03819-2
68. Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. (2022) 50:D439–44. doi: 10.1093/nar/gkab1061
69. Yahata T, Shao W, Endoh H, Hur J, Coser KR, Sun H, et al. Selective coactivation of estrogen-dependent transcription by CITED1 CBP/p300-binding protein. Genes Dev. (2001) 15:2598–612. doi: 10.1101/gad.906301
70. Chu WT, Chu X, Wang J. Investigations of the underlying mechanisms of HIF-1alpha and CITED2 binding to TAZ1. Proc Natl Acad Sci USA. (2020) 117:5595–603. doi: 10.1073/pnas.1915333117
71. Hobor F, Hegedus Z, Ibarra AA, Petrovicz VL, Bartlett GJ, Sessions RB, et al. Understanding p300-transcription factor interactions using sequence variation and hybridization. RSC Chem Biol. (2022) 3:592–603. doi: 10.1039/D2CB00026A
72. Wojciak JM, Martinez-Yamout MA, Dyson HJ, Wright PE. Structural basis for recruitment of CBP/p300 coactivators by STAT1 and STAT2 transactivation domains. EMBO J. (2009) 28:948–58. doi: 10.1038/emboj.2009.30
73. Qin X, Chen H, Tu L, Ma Y, Liu N, Zhang H, et al. Potent inhibition of HIF1alpha and p300 interaction by a constrained peptide derived from CITED2. J Med Chem. (2021) 64:13693–703. doi: 10.1021/acs.jmedchem.1c01043
74. Yoon H, Lim JH, Cho CH, Huang LE, Park JW. CITED2 controls the hypoxic signaling by snatching p300 from the two distinct activation domains of HIF-1alpha. Biochim Biophys Acta. (2011) 1813:2008–16. doi: 10.1016/j.bbamcr.2011.08.018
75. Freedman SJ, Sun ZY, Kung AL, France DS, Wagner G, Eck MJ. Structural basis for negative regulation of hypoxia-inducible factor-1alpha by CITED2. Nat Struct Biol. (2003) 10:504–12. doi: 10.1038/nsb936
76. De Guzman RN, Martinez-Yamout MA, Dyson HJ, Wright PE. Interaction of the TAZ1 domain of the CREB-binding protein with the activation domain of CITED2: regulation by competition between intrinsically unstructured ligands for non-identical binding sites. J Biol Chem. (2004) 279:3042–9. doi: 10.1074/jbc.M310348200
77. Bhattacharya S, Michels CL, Leung MK, Arany ZP, Kung AL, Livingston DM. Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev. (1999) 13:64–75. doi: 10.1101/gad.13.1.64
78. Oliner JD, Andresen JM, Hansen SK, Zhou S, Tjian R. SREBP transcriptional activity is mediated through an interaction with the CREB-binding protein. Genes Dev. (1996) 10:2903–11. doi: 10.1101/gad.10.22.2903
79. Yao TP, Ku G, Zhou N, Scully R, Livingston DM. The nuclear hormone receptor coactivator SRC-1 is a specific target of p300. Proc Natl Acad Sci USA. (1996) 93:10626–31. doi: 10.1073/pnas.93.20.10626
80. Braverman J, Sogi KM, Benjamin D, Nomura DK, Stanley SA. HIF-1alpha is an essential mediator of IFN-gamma-dependent immunity to mycobacterium tuberculosis. J Immunol. (2016) 197:1287–97. doi: 10.4049/jimmunol.1600266
81. McGettrick AF, O’Neill LAJ. The role of HIF in immunity and inflammation. Cell Metab. (2020) 32:524–36. doi: 10.1016/j.cmet.2020.08.002
82. Schatz V, Strussmann Y, Mahnke A, Schley G, Waldner M, Ritter U, et al. Myeloid cell-Derived HIF-1alpha promotes control of leishmania major. J Immunol. (2016) 197:4034–41. doi: 10.4049/jimmunol.1601080
83. Gleeson LE, Sheedy FJ, Palsson-McDermott EM, Triglia D, O’Leary SM, O’Sullivan MP, et al. Cutting edge: mycobacterium tuberculosis induces aerobic glycolysis in human alveolar macrophages that is required for control of intracellular bacillary replication. J Immunol. (2016) 196:2444–9. doi: 10.4049/jimmunol.1501612
84. Knight M, Braverman J, Asfaha K, Gronert K, Stanley S. Lipid droplet formation in Mycobacterium tuberculosis infected macrophages requires IFN-gamma/HIF-1alpha signaling and supports host defense. PloS Pathog. (2018) 14:e1006874. doi: 10.1371/journal.ppat.1006874
85. Kim GD, Das R, Rao X, Zhong J, Deiuliis JA, Ramirez-Bergeron DL, et al. CITED2 restrains proinflammatory macrophage activation and response. Mol Cell Biol. (2018) 38:1–18. doi: 10.1128/MCB.00452-17
86. Kiss J, Mollenhauer M, Walmsley SR, Kirchberg J, Radhakrishnan P, Niemietz T, et al. Loss of the oxygen sensor PHD3 enhances the innate immune response to abdominal sepsis. J Immunol. (2012) 189:1955–65. doi: 10.4049/jimmunol.1103471
87. Taniguchi K, Karin M. NF-kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. (2018) 18:309–24. doi: 10.1038/nri.2017.142
88. Farlik M, Reutterer B, Schindler C, Greten F, Vogl C, Muller M, et al. Nonconventional initiation complex assembly by STAT and NF-kappaB transcription factors regulates nitric oxide synthase expression. Immunity. (2010) 33:25–34. doi: 10.1016/j.immuni.2010.07.001
89. Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. (2010) 2:a000158. doi: 10.1101/cshperspect.a000158
90. Gilmore TD, Morin PJ. The I kappa B proteins: members of a multifunctional family. Trends Genet. (1993) 9:427–33. doi: 10.1016/0168-9525(93)90106-R
91. Suzuki H, Chiba T, Suzuki T, Fujita T, Ikenoue T, Omata M, et al. Homodimer of two F-box proteins betaTrCP1 or betaTrCP2 binds to IkappaBalpha for signal-dependent ubiquitination. J Biol Chem. (2000) 275:2877–84. doi: 10.1074/jbc.275.4.2877
92. Traenckner EB, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. (1995) 14:2876–83. doi: 10.1002/embj.1995.14.issue-12
93. Natoli G, Saccani S, Bosisio D, Marazzi I. Interactions of NF-kappaB with chromatin: the art of being at the right place at the right time. Nat Immunol. (2005) 6:439–45. doi: 10.1038/ni1196
94. Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. (1998) 1:661–71. doi: 10.1016/S1097-2765(00)80066-0
95. Mukherjee SP, Behar M, Birnbaum HA, Hoffmann A, Wright PE, Ghosh G. Analysis of the RelA : CBP/p300 interaction reveals its involvement in NF-kappaB-driven transcription. PloS Biol. (2013) 11:e1001647. doi: 10.1371/journal.pbio.1001647
96. Pong Ng H, Kim GD, Ricky Chan E, Dunwoodie SL, Mahabeleshwar GH. CITED2 limits pathogenic inflammatory gene programs in myeloid cells. FASEB J. (2020) 34:12100–13. doi: 10.1096/fj.202000864R
97. Lou X, Sun S, Chen W, Zhou Y, Huang Y, Liu X, et al. Negative feedback regulation of NF-kappaB action by CITED2 in the nucleus. J Immunol. (2011) 186:539–48. doi: 10.4049/jimmunol.1001650
98. Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. (2002) 21:6539–48. doi: 10.1093/emboj/cdf660
99. Zafar A, Pong Ng H, Diamond-Zaluski R, Kim GD, Ricky Chan E, Dunwoodie SL, et al. CITED2 inhibits STAT1-IRF1 signaling and atherogenesis. FASEB J. (2021) 35:e21833. doi: 10.1096/fj.202100792R
100. Karamouzis MV, Konstantinopoulos PA, Papavassiliou AG. Roles of CREB-binding protein (CBP)/p300 in respiratory epithelium tumorigenesis. Cell Res. (2007) 17:324–32. doi: 10.1038/cr.2007.10
101. Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. (2000) 14:1553–77. doi: 10.1101/gad.14.13.1553
102. Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem. (2001) 276:13505–8. doi: 10.1074/jbc.R000025200
103. Xu X, Sun YL, Hoey T. Cooperative DNA binding and sequence-selective recognition conferred by the STAT amino-terminal domain. Science. (1996) 273:794–7. doi: 10.1126/science.273.5276.794
104. Zhang JJ, Vinkemeier U, Gu W, Chakravarti D, Horvath CM, Darnell JE Jr. Two contact regions between Stat1 and CBP/p300 in interferon gamma signaling. Proc Natl Acad Sci USA. (1996) 93:15092–6. doi: 10.1073/pnas.93.26.15092
105. Bhattacharya S, Eckner R, Grossman S, Oldread E, Arany Z, D’Andrea A, et al. Cooperation of Stat2 and p300/CBP in signalling induced by interferon-alpha. Nature. (1996) 383:344–7. doi: 10.1038/383344a0
106. Paulson M, Press C, Smith E, Tanese N, Levy DE. IFN-Stimulated transcription through a TBP-free acetyltransferase complex escapes viral shutoff. Nat Cell Biol. (2002) 4:140–7. doi: 10.1038/ncb747
107. Zafar A, Ng HP, Chan ER, Dunwoodie SL, Mahabeleshwar GH. Myeloid-CITED2 deficiency exacerbates diet-induced obesity and pro-inflammatory macrophage response. Cells. (2023) 12:1–19. doi: 10.3390/cells12172136
108. Barish GD, Yu RT, Karunasiri M, Ocampo CB, Dixon J, Benner C, et al. Bcl-6 and NF-kappaB cistromes mediate opposing regulation of the innate immune response. Genes Dev. (2010) 24:2760–5. doi: 10.1101/gad.1998010
109. Walker SR, Nelson EA, Yeh JE, Pinello L, Yuan GC, Frank DA. STAT5 outcompetes STAT3 to regulate the expression of the oncogenic transcriptional modulator BCL6. Mol Cell Biol. (2013) 33:2879–90. doi: 10.1128/MCB.01620-12
110. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. (2007) 447:1116–20. doi: 10.1038/nature05894
111. Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity. (2010) 33:699–712. doi: 10.1016/j.immuni.2010.11.009
112. Lizcano F, Vargas D, Gomez A, Torrado A. Human ADMC-Derived adipocyte thermogenic capacity is regulated by IL-4 receptor. Stem Cells Int. (2017) 2017:2767916. doi: 10.1155/2017/2767916
113. Subramani A, Hite MEL, Garcia S, Maxwell J, Kondee H, Millican GE, et al. Regulation of macrophage IFNgamma-stimulated gene expression by the transcriptional coregulator CITED1. J Cell Sci. (2023) 136:1–15. doi: 10.1242/jcs.260529
114. Subramani A, Griggs P, Frantzen N, Mendez J, Tucker J, Murriel J, et al. Intracellular Cryptococcus neoformans disrupts the transcriptome profile of M1- and M2-polarized host macrophages. PloS One. (2020) 15:e0233818. doi: 10.1371/journal.pone.0233818
115. Lin Z, Feng R, Li J, Meng Y, Yuan L, Fu Z, et al. Nuclear translocation of CBP/p300-interacting protein CITED1 induced by parathyroid hormone requires serine phosphorylation at position 79 in its 63-84 domain. Cell Signal. (2014) 26:2436–45. doi: 10.1016/j.cellsig.2014.06.015
116. Yang D, Guo J, Divieti P, Shioda T, Bringhurst FR. CBP/p300-interacting protein CITED1 modulates parathyroid hormone regulation of osteoblastic differentiation. Endocrinology. (2008) 149:1728–35. doi: 10.1210/en.2007-0826
117. Dornan D, Eckert M, Wallace M, Shimizu H, Ramsay E, Hupp TR, et al. Interferon regulatory factor 1 binding to p300 stimulates DNA-dependent acetylation of p53. Mol Cell Biol. (2004) 24:10083–98. doi: 10.1128/MCB.24.22.10083-10098.2004
118. Gonzalez-Garcia I, Garcia-Clave E, Cebrian-Serrano A, Le Thuc O, Contreras RE, Xu Y, et al. Estradiol regulates leptin sensitivity to control feeding via hypothalamic Cited1. Cell Metab. (2023) 35:438–455.e437. doi: 10.1016/j.cmet.2023.02.004
119. Qing Y, Stark GR. Alternative activation of STAT1 and STAT3 in response to interferon-gamma. J Biol Chem. (2004) 279:41679–85. doi: 10.1074/jbc.M406413200
120. Yu T, Zuo Y, Cai R, Huang X, Wu S, Zhang C, et al. SENP1 regulates IFN-gamma-STAT1 signaling through STAT3-SOCS3 negative feedback loop. J Mol Cell Biol. (2017) 9:144–53. doi: 10.1093/jmcb/mjw042
121. Howlin J, McBryan J, Napoletano S, Lambe T, McArdle E, Shioda T, et al. CITED1 homozygous null mice display aberrant pubertal mammary ductal morphogenesis. Oncogene. (2006) 25:1532–42. doi: 10.1038/sj.onc.1209183
122. Kramer OH, Baus D, Knauer SK, Stein S, Jager E, Stauber RH, et al. Acetylation of Stat1 modulates NF-kappaB activity. Genes Dev. (2006) 20:473–85. doi: 10.1101/gad.364306
123. Kramer OH, Knauer SK, Greiner G, Jandt E, Reichardt S, Guhrs KH, et al. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev. (2009) 23:223–35. doi: 10.1101/gad.479209
124. Chen CM, Bentham J, Cosgrove C, Braganca J, Cuenda A, Bamforth SD, et al. Functional significance of SRJ domain mutations in CITED2. PloS One. (2012) 7:e46256. doi: 10.1371/journal.pone.0046256
Keywords: CITED2, CITED1, STAT1, macrophage, innate immunity, inflammation, NF-κB, CBP/p300
Citation: Wiggins DA, Maxwell JN and Nelson DE (2024) Exploring the role of CITED transcriptional regulators in the control of macrophage polarization. Front. Immunol. 15:1365718. doi: 10.3389/fimmu.2024.1365718
Received: 04 January 2024; Accepted: 25 March 2024;
Published: 05 April 2024.
Edited by:
Chunling Liang, Guangzhou University of Chinese Medicine, ChinaReviewed by:
Andrew B. Caldwell, University of California, San Diego, United StatesZhiqiang Huang, Nanjing University, China
Copyright © 2024 Wiggins, Maxwell and Nelson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David E. Nelson, ZGF2aWQuZS5uZWxzb25AbXRzdS5lZHU=