
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 06 March 2024
Sec. T Cell Biology
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1365074
 Carola Amormino1
Carola Amormino1 Emanuela Russo1
Emanuela Russo1 Valentina Tedeschi1
Valentina Tedeschi1 Maria Teresa Fiorillo1
Maria Teresa Fiorillo1 Alessandro Paiardini2
Alessandro Paiardini2 Francesco Spallotta1,3Laura Rosanò4
Francesco Spallotta1,3Laura Rosanò4 Loretta Tuosto1*
Loretta Tuosto1* Martina Kunkl1,5
Martina Kunkl1,5Staphylococcus aureus is a gram-positive bacterium that may cause intestinal inflammation by secreting enterotoxins, which commonly cause food-poisoning and gastrointestinal injuries. Staphylococcal enterotoxin B (SEB) acts as a superantigen (SAg) by binding in a bivalent manner the T-cell receptor (TCR) and the costimulatory receptor CD28, thus stimulating T cells to produce large amounts of inflammatory cytokines, which may affect intestinal epithelial barrier integrity and functions. However, the role of T cell-mediated SEB inflammatory activity remains unknown. Here we show that inflammatory cytokines produced by T cells following SEB stimulation induce dysfunctions in Caco-2 intestinal epithelial cells by promoting actin cytoskeleton remodelling and epithelial cell-cell junction down-regulation. We also found that SEB-activated inflammatory T cells promote the up-regulation of epithelial-mesenchymal transition transcription factors (EMT-TFs) in a nuclear factor-κB (NF-κB)- and STAT3-dependent manner. Finally, by using a structure-based design approach, we identified a SEB mimetic peptide (pSEB116-132) that, by blocking the binding of SEB to CD28, dampens inflammatory-mediated dysregulation of intestinal epithelial barrier.
Staphylococcus aureus (S. aureus) is a commensal opportunistic gram-positive bacterium that colonizes around 20-30% of people, but it can become pathogenic for the presence of virulence and immune evasion factors, causing different diseases ranging from mild skin infections to toxic shock syndrome (TSS) and multi-organ failure (1–4). The bacterium is a component of the nasopharynx microbiota that may colonize secondary sites including skin, throat, armpit, groin and intestine (2), where it can survive thanks to factors and enzymes that allow its adherence to the epithelial surfaces and proliferation (5). The persistent proliferation and survival of S. aureus at the site of infection often lead to dysbiosis with serious life-threatening conditions, including chronic inflammation (6). For example, S. aureus infections may cause chronic intestinal inflammation, such as staphylococcal enteritis, an inflammatory disorder common in infants and older people (7), which can also occur in healthy adults exposed to methicillin-resistant S. aureus (MRSA) (8, 9). The enteropathic effects and intestinal epithelial dysfunctions observed in staphylococcal enteritis have been also related to the activity of staphylococcal enterotoxins (SEs) (10), the most common cause of food-poisoning and gastrointestinal injuries (11, 12).
SEs may act as superantigens (SAgs) and activate a large population of T lymphocytes to produce inflammatory cytokines (13). In particular, SEB directly activate T cells bearing TCRVβ3, 12, 13.2, 14, 17 and 20 (14), widely expressed in the human population (15), and CD28 costimulatory molecules either in the presence or absence of the major histocompatibility complex class II (MHC-II) or B7.1/CD80 and B7.2/CD86 molecules on antigen presenting cells (APCs) (16–23). Indeed, we recently highlighted that both SEB and SEA can directly bind the TCR and CD28, activating inflammatory signalling in the absence of APCs expressing B7 and/or MHC-II (23). Staphylococcal SAg-induced T cell activation leads to a massive uncontrolled production of inflammatory cytokines such as TNF-α, IL-6, IFN-γ, IL-17A and IL-22 (23) that may affect the integrity and functions of the intestinal mucosa.
The intestinal mucosa is composed of three main interconnected layers that provide a physical barrier. These include the mucus layer, which is the body’s first-line defence, the epithelial and the inner layers that form the mucosal immune system (24–26). The epithelial layer consists of epithelial cells interconnected and linked each other by cell-cell adhesion molecules, which provide the epithelium with the integrity and cellular activity required for its specific functions. The apical tight junctions (TJs) comprise integral transmembrane proteins, including occludins and claudins, linked to the cytosolic zona occludens (ZO) proteins (27). TJs create a permeable barrier that generally limits paracellular transport and maintains the polarity of epithelial cells by interacting with filamentous actin (F-actin) and myosin (28). In addition, cell-cell adhesion is also maintained by adherent junctions such as E-cadherin that form a complex with β- and p120-catenin determining its connection to the actin cytoskeleton (29, 30). Alterations in intestinal permeability as well as in TJs and E-cadherin organization and functions have been observed following the exposure of the intestinal epithelium to staphylococcal SEs including SEB (31–33). However, it remains unknown whether the enterotoxic activities are mediated by T-cell dependent SAg inflammatory activity of SEs (34).
Here, we show that inflammatory cytokines elicited by SEB-mediated stimulation of T cells are able to dysregulate the intestinal epithelial barrier function of Caco-2 through the downregulation of cell-cell adherent junctions and the up-regulation of transcription factors associated to epithelial mesenchymal transition (EMT-TFs) in a nuclear factor-κB (NF-κB)- and STAT3-dependent manner. By using computational design, we also identified a novel SEB mimetic peptide that, by blocking SEB/CD28 interaction, dampens inflammatory-mediated dysregulation of the intestinal epithelial barrier.
Human primary T cells or CD4+ T cells were isolated from peripheral blood mononuclear cells (PBMC) of buffy coats of anonymous healthy donors (HD) (Policlinico Umberto I, Sapienza University of Rome, Italy) by using a EasySep™ isolation kits (#17951 and #17952, STEMCELL Technology, CAN). HD signed the informed consent and the Ethic Committee of Policlinico Umberto I approved the procedure (ethical code N., 1061bis/2019, 13/09/2019). T cells were cultured in RPMI-1640 containing 5% human serum (Euroclone, UK), L-glutamine, penicillin and streptomycin. The sorted population was > 95% pure, as evidenced by flow cytometry.
Caco-2 epithelial cell line (ATCC, Maryland, USA) was cultured in DMEM supplemented with 10% FBS (Euroclone, UK), L-glutamine, penicillin and streptomycin.
The following antibodies were used: rabbit anti-human vimentin (#5741), rabbit anti-human NF-κB/p65 (#8242), rabbit anti-human phosphoTyr705-STAT3 (#9145) were from Cell Signalling Technologies (MA, USA); mouse anti-human E-cadherin (#610182), mouse anti-human N-cadherin (#610254) were from BD Biosciences (Milan, Italy); rabbit anti-human FAS (#sc-715), mouse anti-human GAPDH (#sc-47724) were from Santa Cruz Biotechnology (Texas, USA); rabbit anti-human ZO-1 (#40-2200), Alexa Fluor 594 phalloidin (#A12381), goat anti-mouse Alexa-flour 488 (#A11070), goat anti-rabbit Alexa Fluor 647 (#A21245) were from Thermo Fisher Scientific (MA, USA). Inflammatory cytokines neutralising Abs (NAbs) used were mouse anti-TNF-α (#MAB610), anti-IFN-γ (#MAB2852), anti-IL6 (#MAB206), anti-IL-17A (#MAB317) and anti-IL-22 (#MAB7822) (R&D Systems, USA). All NAbs were used at 2.5 μg ml-1 final concentration.
Staphylococcal Enterotoxin B (SEB, #54881) was purchased by Merck (Milan, Italy). 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, #H-1200) was from Vector Laboratories, Inc. (Burlingame, CA, USA). pSEB116-132 mimetic peptide (GGVTEHNGNQLDKYRSI) containing D-Ala at both termini to enhance protease resistance was purchased by BIO-FAB research (Rome, Italy). Peptide was > 95% pure and used in culture at 10 μM final concentration. PS1145 (#P6624, Merck) and S31-201 (#sc-204304, Santa Cruz Biotechnology) inhibitory drugs were used at 10 μM and 100 μm, respectively.
T cells (0.5 x 106 ml-1) were cultured in 24-well plate or 24-well plate inserts when co-cultured with Caco-2 cells (2.5 x 105 ml-1) and stimulated for different times with 0.1 μg ml-1 SEB. Secretion of inflammatory cytokines (TNF-α, IL-6, IL-17A, IL-22 and IFN-γ) in culture supernatants was measured by ELISA using human TNF-α (#DY-210), IL-6 (#DY-206), IFN-γ (#DY285), IL-17A (#DY-317) and IL-22 (#DY-782) kits (R&D Systems). The assays were performed in duplicate and data analyses was performed on a Bio-Plex (Bio-Rad, Hercules, CA, USA). The sensitivity of the assays was as follow: IL-6 and IFN-γ = 9.4 pg ml-1, TNF-α and IL-17A = 15.6 pg ml-1 and IL-22 = 31.2 pg ml-1.
Caco-2 cells (2.5 × 105 ml−1) were co-cultured with T cells (0.5 x 106 ml−1) in 24 trans-well plate inserts for different times at 37°C. At the end of incubation, cells were lysed for 30 min on ice in 1% Nonidet P-40 lysis buffer (0.15 M NaCl, 0.02 M Tris-HCl pH 7.5, 0.001 mM EGTA and MgCl2, 0.05 M NaF, 0.01 M Na4P2O7) containing proteases and phosphatases inhibitors (10 μg ml-1 leupeptin, 10 μg ml-1 aprotinin, 1 mM NaVO4, 1 mM Pefablock-SC). Proteins were resolved by SDS-PAGE and blotted onto nitrocellulose membranes. Blots were incubated with anti-E-cadherin, anti-ZO-1, anti-N-cadherin or anti-vimentin (1:1000 dilution) or anti-GAPDH (1:400 dilution) Abs, extensively washed and after incubation with horseradish peroxidase (HRP)-labelled goat anti-rabbit (#NA934V, 1:5000 dilution) or HRP-labelled goat anti-mouse (#NA931V, 1:5000 dilution) developed with the enhanced chemiluminescence’s detection system (GE Healthcare Life Sciences, Italy) and analysed by ChemiDoc XRS+ (Bio-Rad Laboratories, Italy). Protein levels were quantified by densitometric analysis using the ImageJ 1.50i program (NIH, USA)
Caco-2 (2.5 x 105 ml-1) were co-cultured with T cells (0.5 x 106 ml−1) unstimulated or stimulated with 0.1 μg ml-1 SEB in 24-well inserts plates. After 72 hours, Caco-2 cells were stained with 10 μg ml-1 propidium iodide (PI) and the percentage of PI-positive cells was quantified by flow cytometry (FACScalibur, BD Biosciences, Mountain View, CA).
Caco-2 cells (2.5 × 105 ml−1) were co-cultured with T cells (0.5 x 106 ml−1) in 24 trans-well plate inserts for different times. After fixing (2% paraformaldehyde) and permeabilization (0.1% saponin), E-cadherin and ZO-1 staining was performed by using mouse anti-human E-cadherin (1:100 dilution) followed by goat anti-mouse Alexa flour 488 (1:100 dilution), and rabbit anti-human ZO-1 (followed by goat anti-rabbit Alexa flour 647 (1:100 dilution). Alexa fluor 594 phalloidin (1:1000 dilution) was used to stain filamentous actin (F-actin) and DAPI was used to stain nuclei. Observations were performed with a Nikon Eclipse Ti2 confocal microscope (63X oil objective). The same acquisition settings were used to process the Z stack images by NIS Elements AR 5.30 software (Nikon Europe B.V.).
Caco-2 cells were treated as indicated and the nuclear translocation of RelA/NF-κB and pSTAT3 was analysed by using anti- RelA (1:300 dilution) and anti- pSTAT3 (1:300 dilution) Abs followed by goat anti-rabbit Alexa-flour 488 (1:100 dilution). Nuclei were stained with DAPI. Images of RelA and pSTAT3 localization were obtained with a computer-controlled Nikon Eclipse 50i epifluorescence microscope and a plan achromat microscope objective 100XA/1.25Oil OFN22 WD 0.2 and QImaging QICAM Fast, 1394 Digital Camera, 12-bit-Mono (Minato, Tokyo, Japan). The fluorescence intensities of nuclear RelA and pSTAT3, overlapping with DAPI, were quantified by using Fiji ImageJ software and intensities ratio were calculated for each cell. At least one hundred cells were examined quantitatively for each condition in three independent experiments.
Total RNA was extracted using Trizol (Thermo Fisher Scientific) from 2.5 × 105 Caco-2 cells, reverse-transcribed into cDNA, and real-time PCR was performed by Luna® Universal qPCR Master Mix (NEB, M3003E). The relative quantification was performed using the comparative cycle threshold (CT) method. Specific primer/probe sets (Table 1) to quantify the expression level of ZO-1, CDH1, SNAIL-1, TWIST-1, ZEB-1 and GAPDH mRNA levels were purchased by Metabion International AG (Planegg, Germany).

Table 1 Primer sequences of investigated genes and promoters.
Caco-2 cells were cultured for 8 h with the supernatant of primary T cells unstimulated or stimulated for 72 h with 0.1 μg ml-1 SEB. ChIP assays were performed as previously described (35). Briefly, after fixing in 1% formaldehyde, Caco-2 cells were lysed and chromatin sheared by sonication. Lysates were incubated at 4°C with anti-RelA (1:100 dilution), anti pSTAT3 (1:100 dilution), or anti-FAS (1:100 dilution) Abs as control, and immune complexes were collected with sperm–saturated Protein-A Sepharose beads. After protein-DNA cross-links reversion, DNA was extracted by phenol-chloroform and analysed by real-time PCR with Luna® Universal qPCR Master Mix. Specific primer/probe sets for the human SNAIL-1, TWIST- and ZEB-1 promoters were used (Table 1). Specific enrichment was calculated by using the CT: 2(controlChIP−controlInput)/2(specificChIP−specificInput) as previously described (36).
Structures and complexes modelling were performed as previously described (23). The following experimentally determined structures was used to this purpose: SEB (PDB: 1SEB) (37); CD28 extracellular domain (PDB: 1YJD) (38); TCR (TRAV22/TRBV19) (PDB:4C56) (39). Briefly, full-length proteins were modelled with AlphaFold2 (40) and docked with ClusPro 2.0 (41), HADDOCK (42), and MultiLZerD (43). Protein sequence manipulations, superpositions and modelling were carried out using PyMod 3.0 (44).
The sample size was chosen based on previous studies to ensure adequate power. Statistical analysis (mean and standard error of mean, SEM) was performed through Prism 8.0 (GraphPad Software, San Diego, CA), by using Student’s t test or one-way ANOVA with the Fisher’s LSD test for multiple comparisons. For all tests, P values < 0.05 were considered significant.
We have recently demonstrated that staphylococcal SAgs may trigger inflammatory cytokine production by binding the TCR and CD28 in the absence of APCs expressing MHC-II- and/or B7 (23). Consistently with these data and with the dose-response production of inflammatory cytokines (Supplementary Figure S1A), stimulation of T cells with 0.1 μg ml-1 SEB induced a significant secretion of IFN-γ (Figure 1A), IL-6 (Figure 1B), TNF-α (Figure 1C), IL-17A (Figure 1D) and IL-22 (Figure 1E) after 48-72 hours (h). Since inflammatory cytokines produce by SEB-stimulated T cells have been described to alter the homeostasis and integrity of intestinal epithelial cells (45), we co-cultured SEB-activated inflammatory T cells with Caco-2 cells in trans-well plates in time-course experiments. Caco-2 is a cell line derived from a human colorectal adenocarcinoma that acquires in vitro the morphological and functional features of mature enterocytes of the small intestine, representing a good model of intestinal barrier (46, 47). Thereafter, both Caco-2 cell viability (Figure 1F) and intestinal epithelial barrier integrity (Figure 1G) were assessed. While no change in Caco-2 cell viability was observed (Figure 1F), confocal microscopy analysis revealed changes in F-actin organization in Caco-2 cells co-cultured with SEB-activated T cells (Figure 1G). For instance, unstimulated Caco-2 cells (CTR) displayed an extensive actin network with F-actin mainly organised into cortical bundles closely associated with cell-cell contacts. Neither SEB alone nor unstimulated T cells (T) significantly affected the steady-state of actin cytoskeleton organization as well as Caco-2 cell morphology. On the contrary, within 24 h of co-culture, Caco-2 cells exhibited para-cellular gaps, which further increased after 48-72 h (Figure 1G). Moreover, after 48 h of co-culture, SEB-activated T cells induced changes in F-actin dynamics in Caco-2 cells with the formation of tight parallel bundles and/or stress-like fibers (white arrows), which further increased after 72 h (Figure 1G).
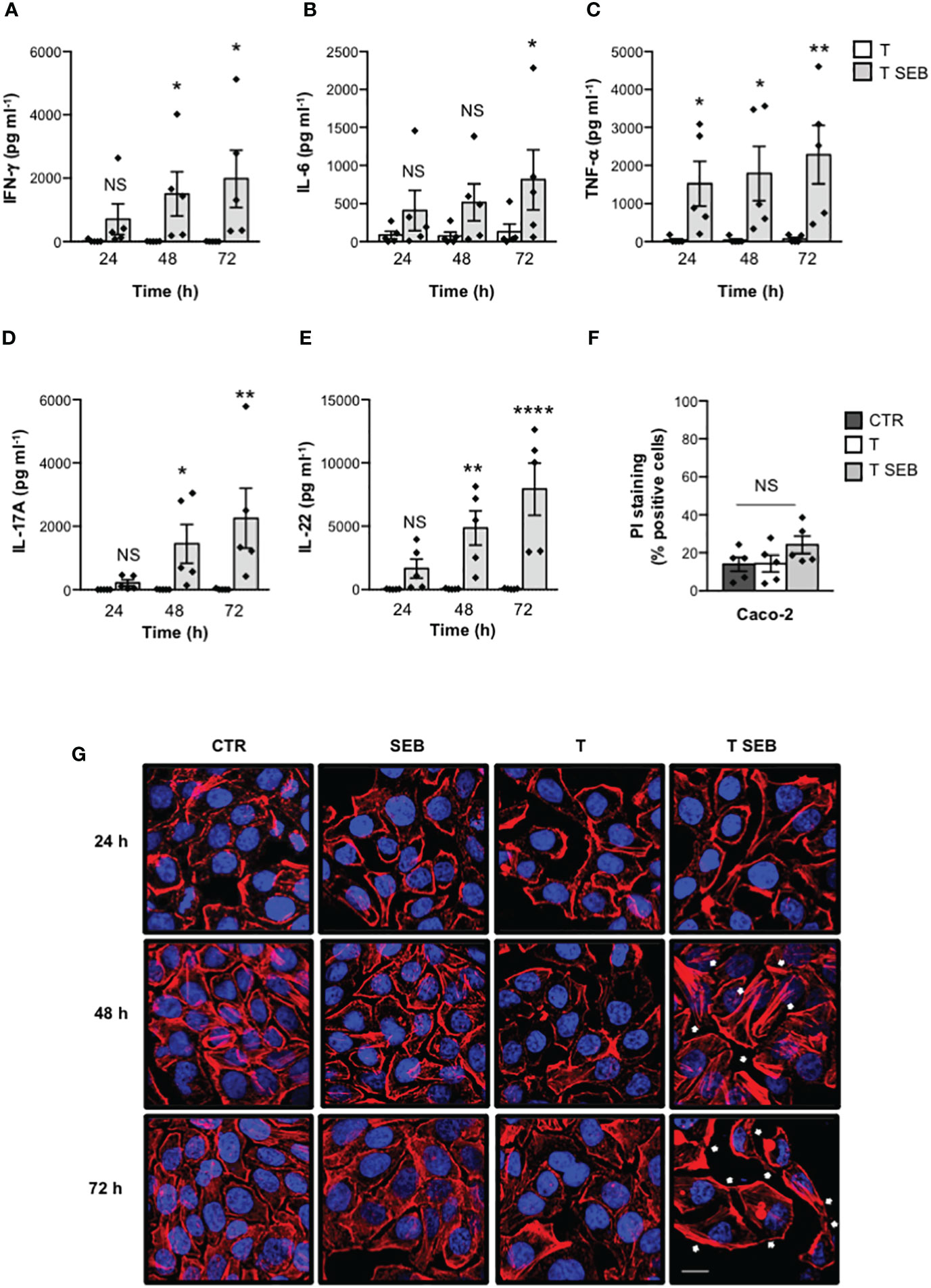
Figure 1 SEB-stimulated T cells secrete inflammatory cytokines and induce alterations of both permeability and morphology in Caco-2 cells. (A–E) Human T cells isolated from the peripheral blood of healthy donors (HD) were unstimulated (T) or stimulated for different times with 0.1 μg ml-1 SEB (T SEB). IFN-γ (A), IL-6 (B), TNF-α (C), IL-17A (D) and IL-22 (E) levels in culture supernatant were measured by ELISA. Data show the mean ± SEM (standard error of the mean) of different HD (n = 5). Statistical significance was calculated by one-way ANOVA. Mean (pg ml-1) ± SEM values (n = 5): IFN-γ (A) 24 h, T = 20.74 ± 17.93, T SEB = 708.3 ± 486.1; 48 h, T = 5.29 ± 3.29, T SEB = 1505 ± 698; 72 h, T = 4.34 ± 2.52, T SEB = 1983 ± 903.5; IL-6 (B) 24 h, T = 86.96 ± 49.57, T SEB = 408.7 ± 267.0; 48 h, T = 74.8 ± 51.27, T SEB = 516.2 ± 243.7; 72 h, T = 129.4 ± 100.3, T SEB = 812.7 ± 394.7; TNF-α (C) 24 h, T = 38.55 ± 35.91, T SEB = 1522 ± 587.7; 48 h, T = 35.81 ± 33.88, T SEB = 1792 ± 712.7; 72 h, T = 69.56 ± 41.9, T SEB = 2285 ± 767.3; IL-17A (D) 24 h, T = 0 ± 0, T SEB = 218.8 ± 96.34; 48 h, T = 3.51 ± 3.51, T SEB = 1448 ± 609.3; 72 h, T = 9.07 ± 9.07, T SEB = 2252 ± 944.3; IL-22 (E) 24h, T = 28.82 ± 10.46, T SEB = 1665 ± 758; 48 h, T = 48.88 ± 14.99, T SEB = 4852 ± 1363; 72 h, T = 55.07 ± 14.38, T SEB = 7929 ± 2055. (F) Caco-2 cells were cultured in 24 trans-well plates for 72 h with medium alone (CTR) or T cells (T), or T cells stimulated with 0.1 μg ml-1 SEB (T SEB). Cell death was analysed by flow cytometry by quantifying the ability of cells to incorporate propidium iodide (PI). Data show the mean % PI positive cells ± SEM of different HD (n = 5). Statistical significance was calculated by one-way ANOVA. Mean (%) ± SEM values (n = 5); CTR = 14.06 ± 3.65, T = 14.38 ± 4.38, T SEB = 24.22 ± 4.55. (*) p < 0.05, (**) p < 0.01, (****) p < 0.0001, NS = not significant. (G) Caco-2 cells were cultured in 24 trans-well plates for the indicated times with medium alone (CTR) or T cells (T), or T cells stimulated with 0.1 μg ml-1 SEB (T SEB) for 24, 48 or 72 h. After fixing and permeabilization, F-actin was stained with 594-conjugated phalloidin (red) and analysed by confocal microscopy. Nucleus was stained with DAPI (blue). The white arrows indicate the tight parallel bundles or stress-like fibers. Scale bar = 20 μm.
These data suggest a potential role of inflammatory cytokines produced by SEB-stimulated T cells in altering F-actin dynamic and intestinal barrier integrity.
Since F-actin remodelling observed in Caco-2 cells exposed to SEB-activated inflammatory T cells resembled those occurring during epithelial-mesenchymal transition (EMT) (48, 49), we analysed the expression of E-cadherin (E-CAD) and ZO-1 (epithelial markers) as well as of N-cadherin (N-CAD) and vimentin (mesenchymal markers). The kinetic analysis pointed out that both ZO-1 and E-CAD were strongly down-regulated after 72 h of co-culture of Caco-2 cells with T cells activated with SEB (Figures 2A, B). On the contrary, no detectable expression of N-CAD and vimentin was observed (Figure 2A). The reduction of both E-CAD and ZO-1 protein content in Caco-2 cells cultured with SEB-activated T cells was also accompanied by the inhibition of ZO-1 (Figure 2C) and E-CAD mRNA levels (CDH1, Figure 2D), which started to decrease within 24 h and reached a peak of inhibition after 72 h (Figure 2E).
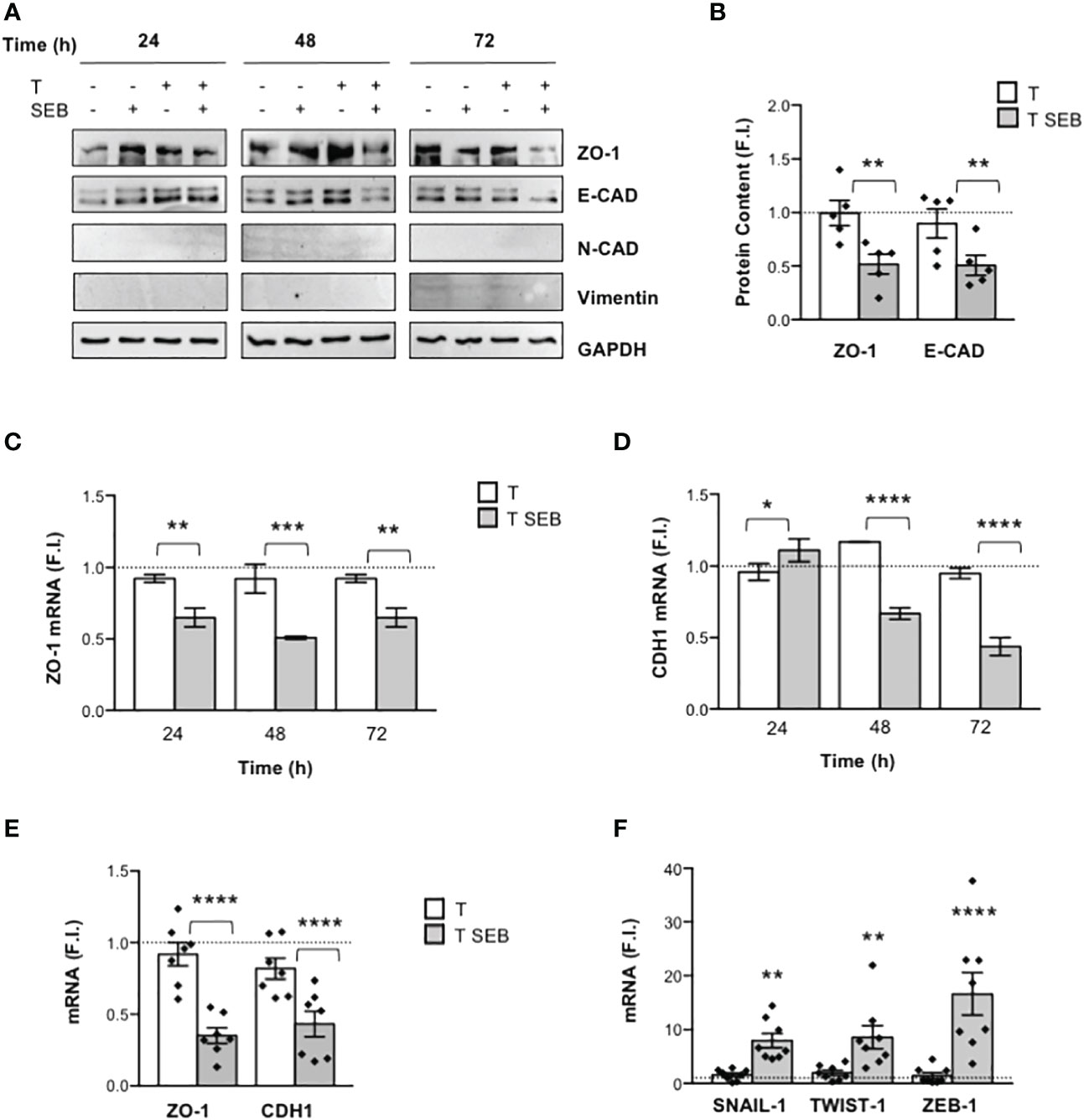
Figure 2 SEB-stimulated T cells induce the down-regulation of E-CAD and ZO-1, and the up-regulation of EMT-TFs in Caco-2 cells. (A) Representative anti-ZO-1, anti-E-cadherin (E-CAD), anti-N-cadherin (N-CAD), anti-vimentin and anti-GAPDH western blotting of Caco-2 cells cultured in 24 trans-well plates for the indicated times with medium alone or with T cells (T), or T cells stimulated with 0.1 μg ml-1 SEB (T SEB). (B) E-CAD and ZO-1 proteins levels of Caco-2 cells cultured with medium alone or with T cells from HD (n = 5) or with SEB-stimulated T cells for 72 h were quantified by densitometric analysis. Fold inductions (F.I.) over the basal level of Caco-2 cells cultured with medium alone were calculated after normalization to GAPDH levels. Data show the mean F.I. ± SEM and statistical significance was calculated by one-way ANOVA. Mean (F.I.) ± SEM values (n = 5): ZO-1, T = 0.99 ± 0.11, T SEB = 0.51 ± 0.09; E-CAD, T = 0.89 ± 0.13, T SEB = 0.50 ± 0.09. (C, D) Caco-2 cells were cultured in triplicates in 24 trans-well plates with medium alone or T cells (T), or SEB-stimulated T cells (T SEB) for the indicated times. ZO-1 (C) and E-CAD (CDH1) mRNA levels (D) in Caco-2 cells were measured by real-time PCR and values, normalized to GAPDH, expressed as fold inductions (F.I.) over the basal level of Caco-2 cultured with medium alone. Data show the mean F.I. ± SEM of one out of three HD. Statistical significance was calculated by one-way ANOVA. Mean (F.I.) ± SEM values (n = 2): ZO-1 (C) 24 h, T = 0.92 ± 0.02, T SEB = 0.64 ± 0.06; 48 h, T = 0.92 ± 0.10, T SEB = 0.50 ± 0.01; 72 h, T = 0.92 ± 0.02, T SEB = 0.64 ± 0.06; CDH1 (D) 24 h, T = 0.95 ± 0.05, T SEB = 1.10 ± 0.07; 48 h, T = 1.16 ± 0.002, T SEB = 0.66 ± 0.04; 72 h, T = 0.94 ± 0.03, T SEB = 0.43 ± 0.06. (E, F) ZO-1 and CDH1 (E) and the EMT-TFs SNAIL-1, TWIST-1 and ZEB-1 (F) mRNA levels in Caco-2 cells cultured for 72 h with medium alone or with T cells from HD unstimulated (T) or SEB-stimulated (T SEB). Values, normalized to GAPDH, were expressed as fold inductions (F.I.) over the basal level of Caco-2 cultured with medium alone. Data show the mean F.I.± SEM and statistical significance was calculated by one-way ANOVA. Mean (F.I.) ± SEM values; [(E), n = 7] ZO-1, T = 0.92 ± 0.08, T SEB = 0.35 ± 0.05; CDH1, T = 0.81 ± 0.07, T SEB = 0.43 ± 0.08; [(F), n = 8] SNAIL-1, T = 1.56 ± 0.36, T SEB = 7.97 ± 1.33; TWIST-1, T = 1.95 ± 0.47, T SEB = 8.59 ± 2.14; ZEB-1, T = 1.47 ± 0.54, T SEB = 16.66 ± 3.93. (*) p < 0.05, (**) p < 0.01, (***) p < 0.001, (****) p < 0.0001.
The transcriptional downregulation of ZO-1 and E-CAD observed in Caco-2 cells co-cultured with SEB-stimulated T cells prompted us to analyse the expression of the EMT-transcription factors (TFs) SNAIL, TWIST and ZEB, known epithelial marker repressors (50). Interestingly, we found a strong up-regulation of SNAIL-1, TWIST-1 and ZEB-1 expression in Caco-2 cells co-cultured for 72 h with SEB-stimulated T cells (Figure 2F), suggesting that these effects might be linked to E-CAD and ZO-1 downregulation. Of note, the comparison of Caco-2 cells co-cultured with total T cells or CD4+ T cells evidenced no significant differences in cell-cell junction downregulation and EMT-TF upregulation (Supplementary Figure S1B), supporting our previous data showing that most of the inflammatory cytokines produced by T cells following SEB stimulation derive from CD4+ T cells (23).
Finally, to assess whether Caco-2 cell dysfunctions were mediated by the inflammatory cytokines produced by SEB-stimulated T cells, Caco-2 cells were cultured with anti-TNF-α, anti-IFN-γ, anti-IL-6, anti-IL-17A and anti-IL-22 neutralising antibodies. Indeed, Caco-2 cells express all the receptors for these inflammatory cytokines and are susceptible to their activity (51–54). The neutralization of each cytokine did not significantly affect neither ZO-1 and CDH1 downregulation (Supplementary Figure S2A) nor SNAIL-1, TWIST-1 and ZEB-1 up-regulation (Supplementary Figure S2B) induced in Caco-2 cells by SEB-activated T cells. However, when a cocktail of all neutralising Abs (NAbs) was added in culture, we observed a significant impairment of F-actin remodelling as well as E-CAD and ZO-1 delocalization in Caco-2 cells co-cultured with SEB-stimulated T cells (Figure 3A). Moreover, neutralization of inflammatory cytokines also restored ZO-1 and CDH1 gene expression (Figure 3B) and inhibited SNAIL-1, TWIST-1 and ZEB-1 expression (Figure 3C) induced in Caco-2 cells by SEB-activated T cells.
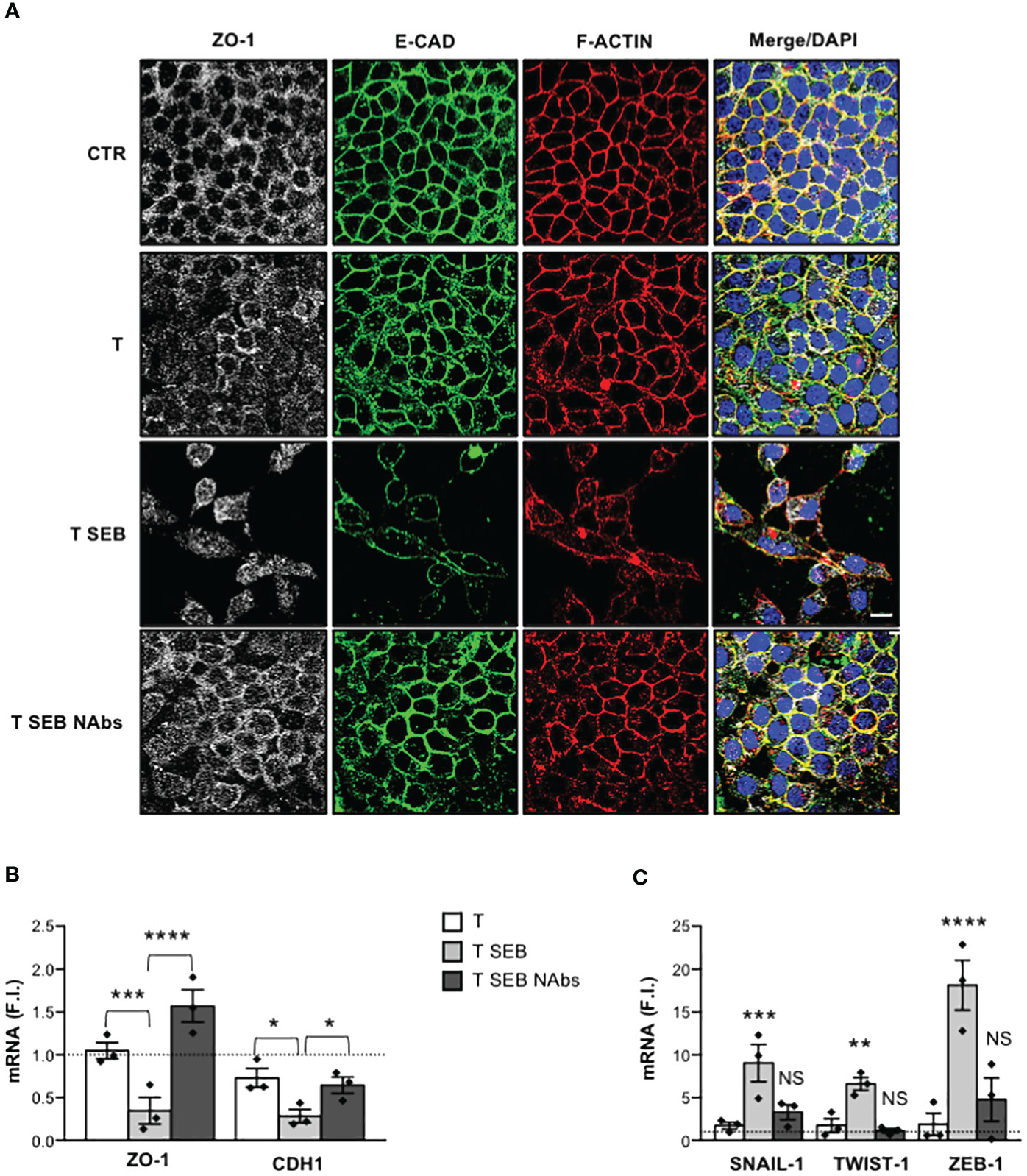
Figure 3 Inflammatory cytokines produced by SEB-stimulated T cells mediate Caco-2 cell dysfunctions. (A) Caco-2 cells were cultured in 24 trans-well plates for 72 h with medium alone (CTR) or T cells (T), or T cells stimulated with 0.1 μg ml-1 SEB (T SEB) in the presence of 2.5 μg ml-1 isotype control or 2.5 μg ml-1 anti-IL-6, anti-TNF-α, anti-IFN-γ, anti-IL-17A Abs neutralising Abs (NAbs). After fixing and permeabilization Caco-2 cells were stained with anti-ZO-1 followed Alexa Fluor 647 Abs (white), anti-E-CAD followed by Alexa Fluor 488 Abs (green), and F-actin was stained with Alexa Fluor 594 phalloidin (red). Nucleus was stained with DPI (blue). Scale bar = 20 μm. (B, C) ZO-1 and CDH1 (B), and SNAIL-1, TWIST-1 and ZEB-1 (C) mRNA levels in Caco-2 cells cultured for 72 h with medium alone or with T cells from HD (n =3) or SEB-stimulated T cells (T SEB) in the presence of isotype control or cytokine NAbs. Values, normalized to GAPDH, were expressed as fold inductions (F.I.) over the basal level of Caco-2 cultured with medium alone. Data show the mean F.I. ± SEM and statistical significance was calculated by one-way ANOVA. Mean (F.I.) ± SEM values (n = 3); (B) ZO-1, T = 1.05 ± 0.09, T SEB = 0.34 ± 0.15, T SEB NAbs = 1.57 ± 0.18; CDH1, T = 0.73 ± 0.10, T SEB = 0.28 ± 0.07, T SEB NAbs = 0.65 ± 0.09; (C) SNAIL-1, T = 1.73 ± 0.40, T SEB = 9.04 ± 2.18, T SEB NAbs = 3.30 ± 0.87; TWIST-1, T = 1.75 ± 0.80, T SEB = 6.60 ± 0.76, T SEB NAbs = 1.13 ± 0.25; ZEB-1, T = 1.88 ± 1.30, T SEB = 18.13 ± 2.91, T SEB NAbs = 4.78 ± 2.53. (*) p < 0.05, (**) p < 0.01, (***) p < 0.001, (****) p < 0.0001. NS, not significant.
Thus, inflammatory cytokines produced by SEB-stimulated T cells disrupt Caco-2 intestinal epithelial barrier and cell-cell adhesion.
Most of the inflammatory cytokines produced by T cells following SEB bivalent binding to the TCR and CD28 exert their functions by activating two main TFs, RelA/NF-κB and STAT3 (23, 35, 55). Consistently, a significant nuclear translocation of RelA/NF-κB (Figures 4B, C) and Tyr705 phosphorylated STAT3 (pSTAT3) (56) (Figures 4D, E) was observed in Caco-2 cells cultured for 8 h with the inflammatory milieu of SEB-stimulated T cells (Figure 4A). Treatment of Caco-2 cells with the NF-κB inhibitor PS1145 (57) or the selective STAT3 inhibitor S31-201 (58) restored both ZO-1 and CDH1 gene expression (Figure 4F) and impaired the up-regulation of SNAIL-1, TWIST-1 and ZEB-1 (Figure 4G) induced by the inflammatory milieu of SEB-activated T cells.
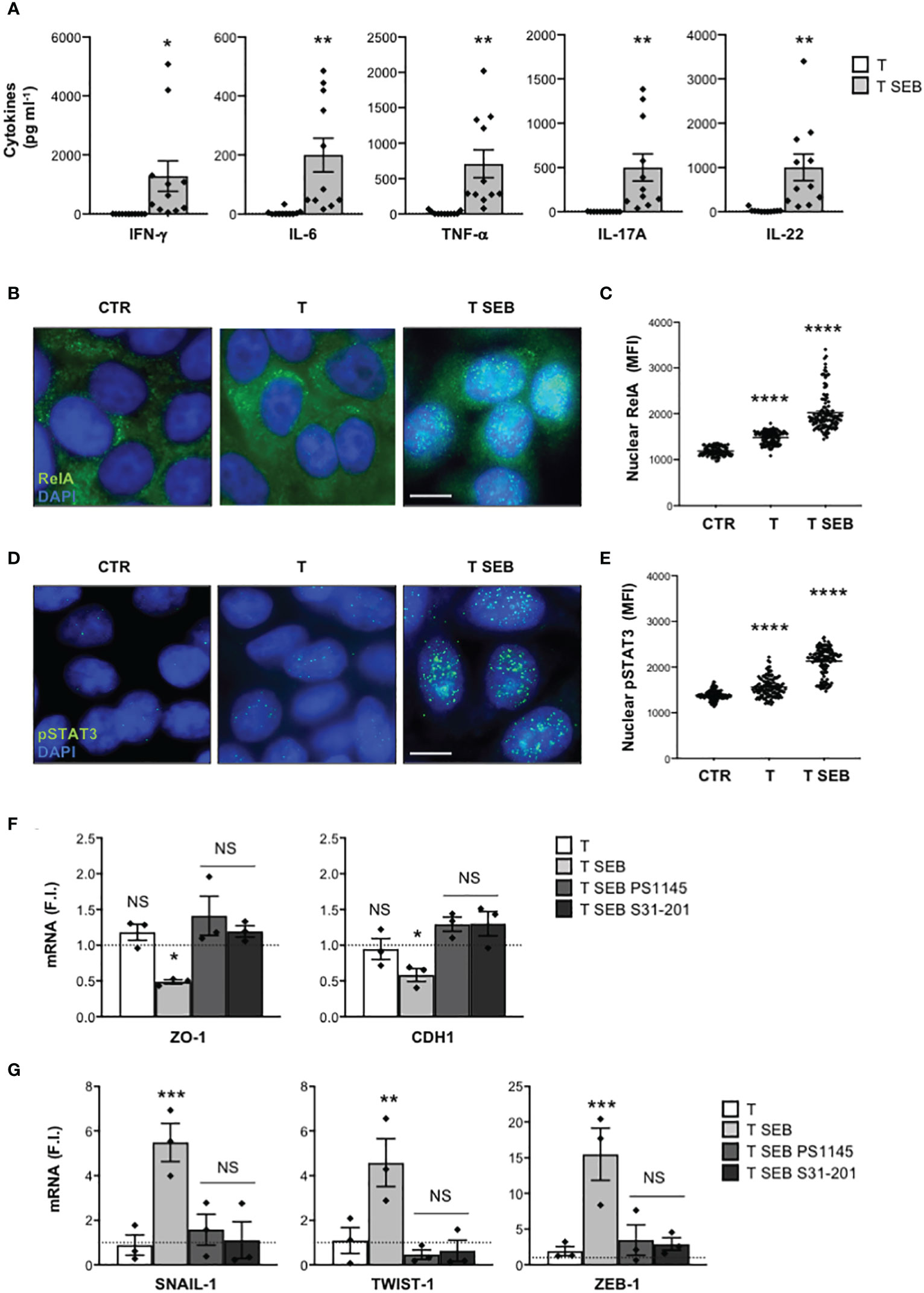
Figure 4 The down-regulation of adhesion molecules and the up-regulation of EMT-TFs in Caco-2 cells exposed to the inflammatory milieu of SEB-stimulated T cells depend on pSTAT3 and RelA/NF-κB TFs. (A) Inflammatory cytokine levels in the supernatants of peripheral T cells from HD (n = 11) stimulated with SEB for 72 h. Data show the mean ± SEM and statistical significance was calculated by Student’s t test. Mean (pg ml-1) ± SEM values (n = 11): IFN-γ, T = 1.71 ± 0.73, T SEB = 1289 ± 519.4; IL-6, T = 4.618 ± 3.04, T SEB = 200.2 ± 57.1; TNF-α, T = 15.54 ± 7.31, T SEB = 709.5 ± 196.6; IL-17A, T = 0.49 ± 0.49, T SEB = 500.4 ± 152.7; IL-22, T = 24.28 ± 12.12, T SEB = 1004 ± 297.8. (B–E) Fluorescence microscopy imaging of RelA (B) or pSTAT3 (D) nuclear translocation in Caco-2 cells cultured for 8 h with medium alone (CTR) or the culture supernatants (1:4 dilution) of T cells (T) or T cells stimulated with SEB for 72 h (T SEB). Mean fluorescence intensity (MFI) fold increase of nuclear RelA (C) or pSTAT3 (E) over unstimulated cells was calculated. Data show the mean MFI ± SEM and statistical significance was calculated by one-way ANOVA. Mean MFI ± SEM values: RelA (C) CTR (n = 96) = 1183 ± 9.8, T (n = 115) = 1419 ± 12.8, T SEB (n = 121) = 2024 ± 37.56; pSTAT3 (E), CTR (n = 114) = 1384 ± 9.8, T (n = 120) = 1562 ± 19.39, T SEB (n = 130) = 2129 ± 26.63. (F, G) ZO-1 and CDH1 (F), and SNAIL-1, TWIST-1 AND ZEB-1 (G) mRNA levels in Caco-2 cells untreated or treated with DMSO, as vehicle control, or 10 μM PS1145, or 100 μM S31-201 and cultured for 24 h with medium alone (CTR) or the culture supernatants (1:4 dilution) of T cells (T) or T cells stimulated with SEB for 72 h (T SEB). Values, normalized to GAPDH, were expressed as fold inductions (F.I.) over the basal level of Caco-2 cultured with medium alone. Data show the mean F.I. ± SEM of three independent experiments. Statistical significance was calculated by one-way ANOVA. Mean (F.I.) ± SEM values (n = 3); (F) ZO-1, T = 1.18 ± 0.11, T SEB = 0.48 ± 0.02, T SEB PS1145 = 1.41 ± 0.27, T SEB S31-201 = 1.19 ± 0.07; CDH1, T = 0.94 ± 0.14, T SEB = 0.58 ± 0.09, T SEB PS1145 = 1.29 ± 0.10, T SEB S31-201 = 1.30 ± 0.17; (G) SNAIL-1, T = 0.89 ± 0.45, T SEB = 5.48 ± 0.84, T SEB PS1145 = 1.58 ± 0.69, T SEB S31-201 = 1.13 ± 0.82; TWIST-1, T = 1.09 ± 0.58, T SEB = 4.57 ± 1.06, T SEB PS1145 = 0.46 ± 0.21, T SEB S31-201 = 0.63 ± 0.47; ZEB-1, T = 1.92 ± 0.64, T SEB = 15.49 ± 3.64, T SEB PS1145 = 3.48 ± 2.13, T SEB S31-201 = 2.91 ± 0.86. (*) p < 0.05, (**) p < 0.01, (***) p < 0.001, (****) p < 0.0001. NS, not significant.
Altogether, these data highlight a critical role of RelA/NF-κB and STAT3 in inducing epithelial barrier dysfunctions and associated EMT-TFs up-regulation following exposure of Caco-2 cells to SEB-activated inflammatory T cells.
Several putative binding sites for RelA/NF-κB and STAT3 have been identified within the promoters of SNAIL-1, TWIST-1 and ZEB1 (59–63). In particular, SNAIL-1 promoter contains three putative RelA/NF-κB binding sites, with NF-κB2 and NF-κB3 sites mainly involved in promoting SNAIL-1 expression (63), and one STAT3 binding site (62) near the NF-κB3 site (Figure 5A). Two STAT3 (59) and one NF-κB consensus sequence (63) were identified in the promoter of TWIST-1 (Figure 5C). Similarly, ZEB-1 promoter contains one NF-κB (61) and two STAT3 binding sites (60) (Figure 5E). To assess whether the up-regulation of EMT-TFs induced by the inflammatory milieu of SEB-activated T cells could depend on RelA/NF-κB and/or pSTAT3, we performed chromatin immunoprecipitations (ChIPs) in Caco-2 cells cultured with the supernatants of SEB-activated T cells using specific oligonucleotide probes (Table 1). The results evidenced a specific recruitment of both pSTAT3 and RelA on the promoters of SNAIL-1, with RelA that preferentially bound NF-κB2 but not NF-κB3 site (Figure 5B). Similarly, selective and specific recruitment of both RelA and pSTAT3 was also observed on TWIST-1 (Figure 5D) and ZEB-1 promoters (Figure 5F).
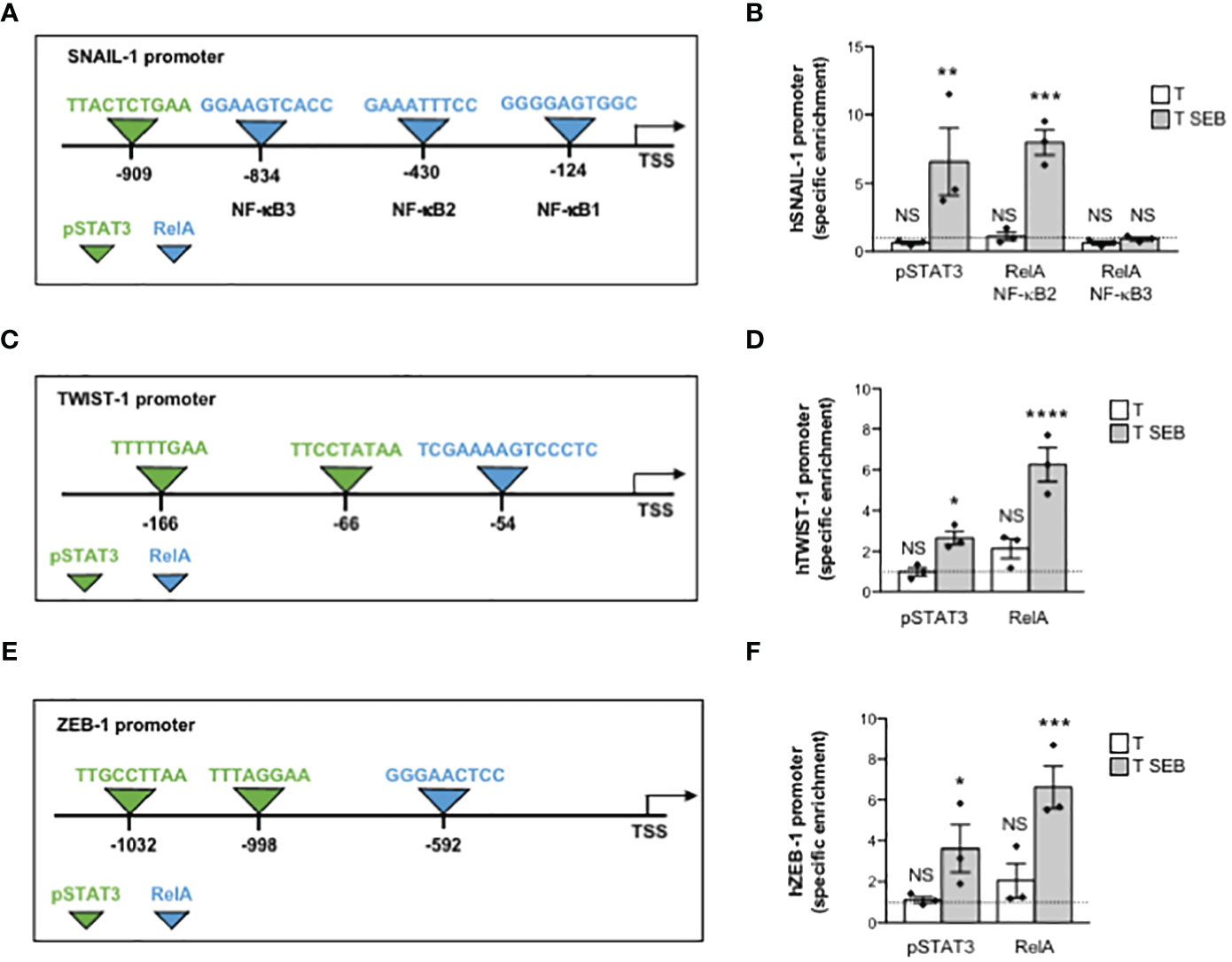
Figure 5 The inflammatory milieu of SEB-stimulated T cells induces the recruitment of RelA/NF-κB and pSTAT3 to the promoters of EMT-TFs. (A, C, E) Schematic sequence of the region upstream of the transcriptional starting site (TSS) of human SNAIL-1 (A), TWIST-1 (C) and ZEB-1 (E) promoter genes. The binding sites of NF-κB (light blue) and STAT3 (yellow) are indicated. (B, D, F) Real time PCR for SNAIL-1 (B), TWIST-1 (D) and ZEB-1 (F) promoters from anti-pSTAT3 and anti-RelA ChIPs performed on Caco-2 cells cultured for 24 h with the culture supernatant (1:4 dilution) of T cells from HD (n = 3) unstimulated (T) or stimulated for 72 h with SEB (T SEB). Specific enrichment over isotype control Abs was calculated by the Cτ method. Data express the mean ± SEM and statistical significance was calculated by one-way ANOVA. Mean ± SEM values (n = 3): (B) SNAIL, pSTAT3, T = 0.63 ± 0.10, T SEB = 6.58 ± 2.46; RelA (NF-κB2), T = 1.12 ± 0.30, T SEB = 7.97 ± 0.91; RelA (NF-κB3), T = 0.62 ± 0.14, T SEB = 0.92 ± 0.12; (D) TWIST-1, pSTAT3, T = 0.98 ± 0.20, T SEB = 2.66 ± 0.32; RelA, T = 2.14 ± 0.48, T SEB = 6.26 ± 0.83; (F) ZEB-1, pSTAT3, T = 1.13 ± 0.15, T SEB = 3.62 ± 1.16 (*) p < 0.05; RelA, T = 2.05 ± 0.83, T SEB = 6.62 ± 1.03. (*) p < 0.05, (**) p < 0.01, (***) p < 0.001, (****) p < 0.0001. NS, not significant.
Altogether these data evidence that SEB activates T cells to secrete inflammatory cytokines, which in turn promote the activation of RelA/NF-κB and STAT3 and their cooperation in up-regulating EMT-TFs expression.
We have recently demonstrated that SEB may bind the TCR and CD28 in a bivalent manner, thus stimulating the production of inflammatory cytokines independently of APCs (23). Based on the proposed complex between SEB the TCR and CD28, we identified a solvent exposed loop of SEB (residues 116-132) interacting with CD28, and evolutionarily well conserved in other SAgs (Figure 6A). We then tested the capability of a mimetic peptide of this region (N-GGVTEHNGNQLDKYRSI-C; hereinafter pSEB116-132) to dampen SEB inflammatory activity. Treatment of T cells with pSEB116-132 mimetic peptide significantly impaired inflammatory cytokine production induced by SEB (Supplementary Figure S2). Moreover, when Caco-2 cells were co-cultured with SEB-stimulated T cells in the presence of pSEB116-132 mimetic peptide, F-actin remodelling as well as E-CAD and ZO-1 down-regulation were impaired (Figure 6B). Finally, pSEB116-132 mimetic peptide also restored ZO-1 and CDH1 gene expression (Figure 6C) and inhibited the up-regulation of SNAIL-1, TWIST-1 and ZEB-1 mRNA levels (Figure 6D).
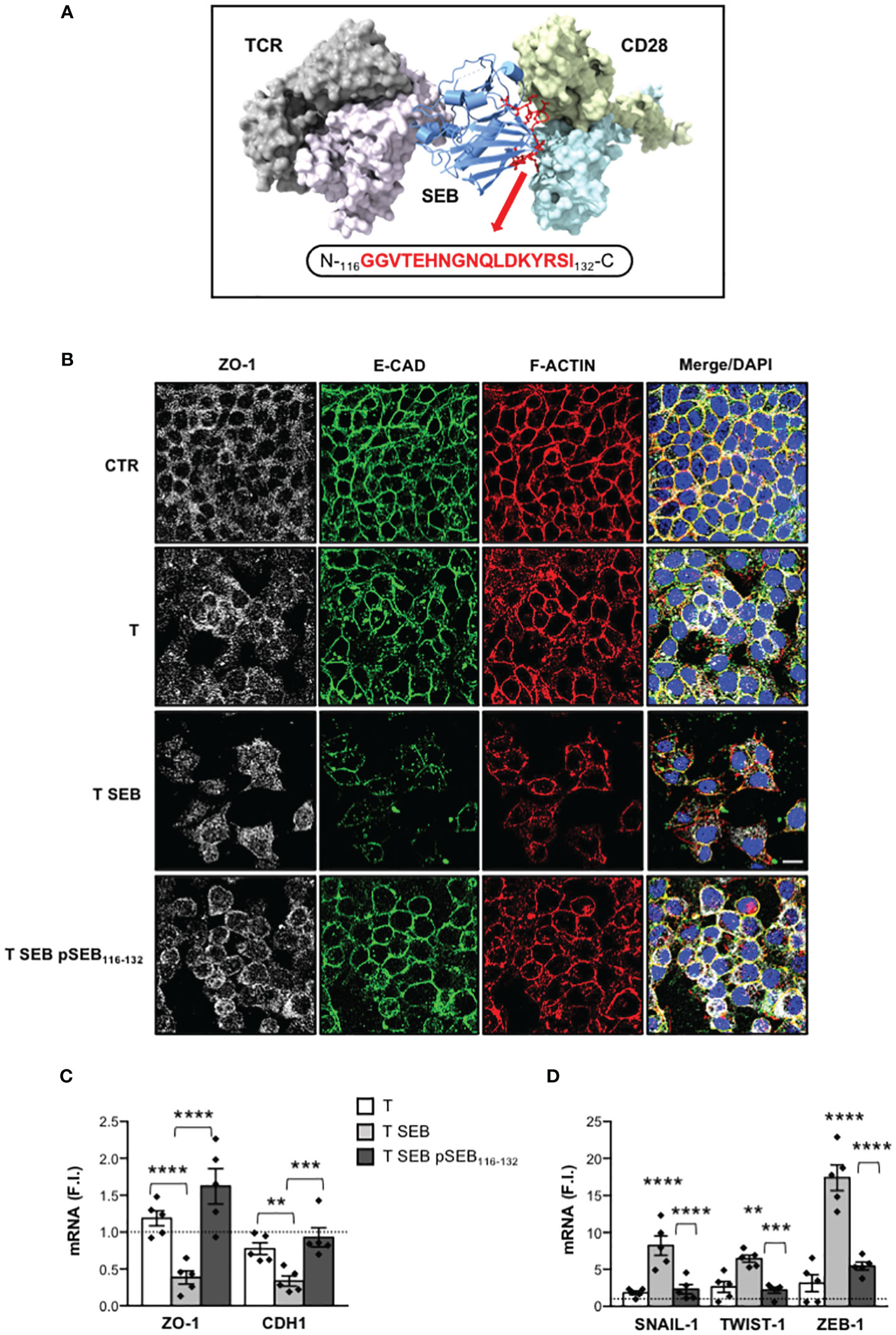
Figure 6 pSEB116-132 mimetic peptide targeting the homodimer interface of CD28 restores epithelial barrier integrity and adhesion molecule expression and inhibits EMT-TFs up-regulation. (A) Structural model of the TCR–SEB–CD28 complex. The TCR (TCRVβ in light purple and TCRVα in grey) and CD28 dimers (light blue and yellow) are represented as solid surface, SEB in cartoon ribbons (blue). The sequence of pSEB116-132 mimetic peptide targeting the region of SEB interacting with the homodimer interface of CD28 is shown in red ball-and-sticks models. (B) Caco-2 cells were cultured in 24 trans-well plates for 72 h with medium alone (CTR) or T cells (T), or T cells stimulated with 0.1 μg ml-1 SEB (T SEB) in the presence or absence of 10 μM pSEB116-132 mimetic peptide. After fixing and permeabilization Caco-2 cells were stained with anti-ZO-1 followed Alexa Fluor 647 Abs (white), anti-E-CAD followed by Alexa Fluor 488 Abs (green), and F-actin was stained with Alexa Fluor 594 phalloidin (red). Nucleus was stained with DPI (blue). Scale bar = 20 μm. (C, D) ZO-1 and CDH1 (C), and SNAIL-1, TWIST-1 and ZEB-1 (D) mRNA levels in Caco-2 cells cultured for 72 h with medium alone or with T cells from HD (n = 5) or SEB-stimulated T cells (T SEB) in the presence or absence of pSEB116-132 mimetic peptide. Values, normalized to GAPDH, were expressed as F.I. over the basal level of Caco-2 cultured with medium alone. Data show the mean F.I. ± SEM and statistical significance was calculated by one-way ANOVA. Mean F.I. ± SEM values (n = 5); (C) ZO-1, T = 1.18 ± 0.10, T SEB = 0.38 ± 0.08, T SEB pSEB116-132 = 1.62 ± 0.23; CDH1, T = 0.77 ± 0.08, T SEB = 0.33 ± 0.06, T SEB pSEB116-132 = 0.92 ± 0.12; (D) SNAIL.1, T = 1.83 ± 0.25, T SEB = 8.23 ± 1.32, T SEB pSEB116-132 = 2.31 ± 0.63; TWIST-1, T = 2.63 ± 0.75, T SEB = 6.45 ± 0.48, T SEB pSEB116-132 = 2.18 ± 0.40; ZEB-1, T = 3.13 ± 1.15, T SEB = 17.39 ± 1.72, T SEB pSEB116-132 = 5.46 ± 0.53. (**) p < 0.01, (***) p < 0.001, (****) p < 0.0001. NS, not significant.
Altogether these data disclose a novel pSEB116-132 mimetic peptide that by competing with SEB for its binding to the homodimer interface of CD28 is able to dampen inflammatory cytokine production and associated intestinal epithelial cell dysfunctions.
Intestinal epithelial cells are usually exposed to several pro-inflammatory and anti-inflammatory cytokines during chronic inflammation and in inflammatory bowel diseases (IBD) (45). These cytokines are mainly produced by immune cells of the local microenvironment, healing the intestinal mucosa and favouring chronic inflammation, which in some cases may lead to tumour development and progression (52, 64–66). S. aureus infections may contribute to intestinal inflammation by secreting SAgs such as SEB that penetrate the epithelial barrier through a transcytosis pathway (33), thus reaching the lamina propria where they stimulate T cells to produce pathogenic inflammatory cytokines (20, 23). Consistently, we found that, by directly binding the TCR and CD28, SEB stimulates T cells to produce inflammatory cytokines such as IFN-γ, IL-6, TNFα, IL-17A and IL-22 (Figure 1) (23), which potentially alter the homeostasis and integrity of intestinal epithelial cells (45, 64).
Several inflammatory cytokines may negatively regulate intestinal barrier integrity by modifying the expression and/or localization of cadherins and/or TJs (45). In intestinal epithelial cells, including Caco-2, TNF-α was shown to increase para-cellular permeability by remodelling F-actin cytoskeleton and downregulating both ZO-1 and E-cadherin expressions at both mRNA and protein levels (52, 67–69). The decrease of ZO-1 protein levels induced by TNF-α in Caco-2 cells has been associated to NF-κB-dependent up-regulation of myosin light chain kinase (MLCK) (67, 70) that, by inducing F-actin rearrangement, promotes the dissociation of ZO-1 from TJs ultimately leading to its degradation (52). IFN-γ was found to synergize with TNF-α in inducing TJs disruption and ZO-1 deregulation, favouring both MLCK expression and activation in Caco-2 cells (53, 71, 72). IL-6, a critical pathogenic cytokine in several inflammatory diseases, including IBD (73), enhances intestinal epithelial permeability by impairing TJs integrity and downregulating E-cadherin expression (51, 74, 75). IL-22, a cytokine of the IL-10 family, whose levels increase in colonic tissues and in the peripheral blood of IBD patients (76), was found to increase TJs permeability (54, 77) by downregulating the expression of both ZO-1 and E-cadherin (78). IL-17A was capable to inhibit the expression of E-cadherin in intestinal epithelial cells in vitro (79). Interestingly, T lymphocytes stimulated with SEB produced high amounts of TNF-α, IL-6, IFN-γ, IL-22 and IL-17A (23) (Figures 1A–F, Figure 3A). In Caco-2 cells exposed to the inflammatory milieu of SEB-stimulated T cells, actin cytoskeleton reorganized into actin stress-like fibers (Figure 1G) and both ZO-1 and E-cadherin were down-regulated at both protein and mRNA level (Figure 2). The neutralization of inflammatory cytokines by NAbs restored Caco-2 cell integrity (Figure 3), suggesting that the enterotoxic effects of SEB strongly depend on its T-cell mediated SAg inflammatory activity.
The downregulation of E-CAD and ZO-1 is often associated to the expression of EMT-TFs such as SNAIL, TWIST and ZEB (49, 80, 81). Indeed, the promoter of E-CAD gene (CDH1) contains specific E-box sequences for binding SNAIL-1 (82), TWIST-1 (83) and ZEB-1 (84), which act by repressing CDH1 transcription. Similarly, ZEB-1 acts as a transcriptional repressor of ZO-1 (50, 85, 86). The expression of EMT-TFs is regulated by several signals, including those activated by inflammatory cytokines (80). In this context, Cohen et al. found that T cell-induced inflammatory cytokines mediated the up-regulation of both ZEB-1 and SNAIL-1 in inflammatory breast cancer (87). Goebel et al. showed that activated CD4+ T cells induced ZEB-1 expression in a TNF-α- and IL-6-dependent manner, in both pre-malignant and malignant pancreatic ductal epithelial cells (88). IL-17A was found at high levels in the colonic mucosal tissues of patients with Chron’s disease, where it mediated the upregulation of SNAIL-1 while inhibiting E-cadherin expression (79). Similarly, IL-22 has been proven to act on intestinal epithelial cells by inducing the expression of SNAIL-1 and impairing TJs expression and distribution (78). Interestingly, we found a strong up-regulation of SNAIL-1, TWIST-1 and ZEB-1 (Figure 2F) in Caco-2 cells co-cultured with SEB-activated T cells, which were inhibited by inflammatory cytokine NAbs (Figure 3C). These data suggest an involvement of SEB-induced inflammatory cytokines in the induction of EMT-TFs and the repression of E-Cadherin and ZO-1 expressions. Indeed, many of the inflammatory cytokines secreted by SEB-stimulated T cells signal through NF-κB and STAT3, which are also involved in EMT-TFs expression (89). For example, through the activation and nuclear translocation of pSTAT3 (90), IL-6 up-regulates the expression of SNAIL, TWIST and ZEB in several cancer cell types (91). STAT3 is also activated by IL-22 (92) and IFN-γ (93), suggesting a potential role in amplifying EMT-TFs expression (89). TNF-α (94) and IL-17A (95) mainly activate NF-κB that cooperates with pSTAT3 to induce EMT-TFs expression (50, 80). Consistently with this evidence, the inflammatory milieu of SEB-stimulated T cells (Figure 4A) induced a significant nuclear translocation of both RelA/NF-κB (Figures 4B, C) and pSTAT3 in Caco-2 cells (Figures 4D, E). Treatment of Caco-2 cells with the NF-κB inhibitor PS1145 (57) or the STAT3 inhibitor S31-201 (58, 96) restored ZO-1 and E-cadherin expression (Figure 4F) impairing SNAIL-1, TWIST-1 and ZEB-1 up-regulation induced by SEB-stimulated T cells (Figure 4G). These data suggest a cooperation of both NF-κB and STAT3 in the transcriptional activation of EMT-TFs promoters.
The promoters of SNAIL-1, TWIST-1 and ZEB-1 contain several putative binding sites for both NF-κB and pSTAT3 (59–63). Notably, by using in silico tools, Pires et al. identified four (-124, -430, -834 and -1119 bp) and six (-54, -249, -870, -956, -983 and -997 bp) putative NF-κB binding sites in the promoter of SNAIL-1 and TWIST-1, respectively (63). A functional consensus sequence for NF-κB binding on ZEB-1 promoter has been also identified by Rajabi et al. at position -592 bp (61). We extended these data by showing that the inflammatory milieu of SEB-activated T cells induced in Caco-2 cells a strong recruitment of RelA/NF-κB on the SNAIL-1 (-430 bp, GGAAATTTCC), TWIST-1 (-54 bp, TCGAAAAGTCCCTC) and ZEB-1 (-592 bp, GGGAACTCC) promoters (Figure 5). According to Liu et al. (62), Cheng et al. (59) and Xiong et al. (60), who identified functional pSTAT3 consensus sequences on the promoters of EMT-TFs, we also found that pSTAT3 was recruited to the promoters of SNAIL-1 (-909 bp, TTACTCTGAA), TWIST-1 (-66 bp, TTCCTATAA), and ZEB-1 (-1032 bp, TTGCCTTAA and -998 bp, TTTAGGAA) in Caco-2 cells exposed to SEB-activated T cell secretome (Figure 5). Thus, by binding in a bivalent manner the TCR and CD28 on the surface of T cells (23), SEB stimulates the secretion of inflammatory cytokines that in turn mediate the expression of EMT-TFs in a NF-κB- and STAT3-dependent manner.
The inflammatory activity of SEB depends on its capability to stimulate T cells by engaging specific TCRVβ chain elements and the CD28/B7 costimulatory axis either in the presence or absence of MHC-II molecules on APCs (16–22). In particular, extensive studies from Kaempfer and co-workers pointed out a crucial role of CD28 and its co-ligand B7 in binding SEB and in stimulating T cells to produce inflammatory cytokines. Indeed, they identified a 12 amino-acid β-strand (8)/hinge/α-helix (4) conserved region in staphylococcal SAgs that, by engaging the dimer interface of CD28 and B7, enhances their interaction triggering the cytokine storm (16, 18, 19). Furthermore, they also showed that short mimetic peptides targeting the homodimer interface of CD28 were able to attenuate inflammatory cytokine production by interfering with CD28/B7 engagement and signalling induced by SEB (16, 17, 19, 22, 97, 98). More recently, we demonstrated that SEB is able to elicit massive cytokine production by engaging the TCR and CD28 in a bivalent manner and in the absence of APCs expressing B7 molecules (22). By using the cryo-EM structure of SEB-MHCII-TCR-CD3 complex (99) and CD28 (38, 100) together with the previously published spatial-restrained protein-protein docking approach (39, 101, 102), we were able to define that SEB may bind the TCR and CD28 simultaneously by adopting a wedge-like conformation (23). Notably, in our modelling, we identified residues 116-132 of SEB forming a loop that acts as a crucial linker between the two subdomains of SEB (Figure 6A). Moreover, this region was mainly responsible for SEB binding to the CD28 homodimer interface. A comparison with the experimentally available structures of other SAgs evidenced that SEB116-132 loop is highly exposed and very well conserved from an evolutionary and structural standpoint. Intrigued by these observations, we tested the capability of a short SEB mimetic peptide (pSEB116-132) to dampen both inflammatory cytokine production and intestinal epithelial cell dysregulation. Our data demonstrated that pSEB116-132 mimetic peptide strongly impaired SEB-mediated inflammatory cytokine production in T cells (Supplementary Figure S2) and restored Caco-2 cell integrity (Figure 6B) as well as ZO-1 and E-CAD expression (Figure 6C) by inhibiting the up-regulation of EMT-TFs (Figure 6D).
Overall, our results provide new insights into the enterotoxic activity of SEB during S. aureus infections, which may lead to the exacerbation of chronic inflammatory diseases such as IBD (103), especially following infections by MRSA (8, 9, 104), and the identification of a novel mimetic peptide able to attenuate inflammatory-dependent epithelial barrier dysfunctions. In vivo experiments to test the efficacy of pSEB116-132 mimetic peptide in attenuating or protecting rats or mice from S. aureus-induced foodborne intoxication or intestinal inflammation (31, 105, 106) will be seminal to assess its therapeutic potential in S. aureus-associated inflammatory diseases.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The studies involving humans were approved by Ethical Committee of the Policlinico Umberto I (Sapienza University, Rome, Italy). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
CA: Writing – review & editing, Data curation, Investigation, Methodology. ER: Investigation, Methodology, Writing – review & editing. VT: Methodology, Writing – review & editing. MF: Writing – review & editing, Funding acquisition. AP: Funding acquisition, Writing – review & editing, Conceptualization, Data curation, Methodology, Software. FS: Funding acquisition, Methodology, Writing – review & editing, Investigation. LR: Funding acquisition, Writing – review & editing, Conceptualization. LT: Conceptualization, Funding acquisition, Writing – review & editing, Supervision, Writing – original draft. MK: Conceptualization, Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The study was supported by: Sapienza University of Rome, “Progetto Ateneo” to LT, MTF, FS, MK and AP; AIRC (Italian Association for Cancer Research) to AP (grant number MFAG, 20447), FS (grant number MFAG, 23099) and LR (grant number AIRC, 21372); POR FESR Lazio, 2014-2020 “Gruppi di Ricerca, 2020” to LR (grant ID A0375-2020-36596 “ORGANOVA”); “Ceschina Foundation” to MTF; PRIN, 2022 PNRR (European Union -Next Generation EU) to LT and LR (project number P2022Z7TEC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1365074/full#supplementary-material
1. Ramachandran G. Gram-positive and gram-negative bacterial toxins in sepsis: a brief review. Virulence. (2014) 5:213–8. doi: 10.4161/viru.27024
2. Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev. (2015) 28:603–61. doi: 10.1128/CMR.00134-14
3. Wozniak JM, Mills RH, Olson J, Caldera JR, Sepich-Poore GD, Carrillo-Terrazas M, et al. Mortality risk profiling of staphylococcus aureus bacteremia by multi-omic serum analysis reveals early predictive and pathogenic signatures. Cell. (2020) 182:1311–1327.e1314. doi: 10.1016/j.cell.2020.07.040
4. Howden BP, Giulieri SG, Wong Fok Lung T, Baines SL, Sharkey LK, Lee JYH, et al. Staphylococcus aureus host interactions and adaptation. Nat Rev Microbiol. (2023) 21:380–95. doi: 10.1038/s41579-023-00852-y
5. Fitzgerald JR. Evolution of Staphylococcus aureus during human colonization and infection. Infect Genet Evol. (2014) 21:542–7. doi: 10.1016/j.meegid.2013.04.020
6. Chen H, Zhang J, He Y, Lv Z, Liang Z, Chen J, et al. Exploring the role of staphylococcus aureus in inflammatory diseases. Toxins (Basel). (2022) 14:464. doi: 10.3390/toxins14070464
7. Lin Z, Kotler DP, Schlievert PM. and Sordillo, E.M. Staphylococcal enterocolitis: forgotten but not gone? Dig Dis Sci. (2010) 55:1200–7. doi: 10.1007/s10620-009-0886-1
8. Ogawa Y, Saraya T, Koide T, Kikuchi K, Ohkuma K, Araki K, et al. Methicillin-resistant Staphylococcus aureus enterocolitis sequentially complicated with septic arthritis: a case report and review of the literature. BMC Res Notes. (2014) 7:21. doi: 10.1186/1756-0500-7-21
9. Takeuchi K, Tsuzuki Y, Ando T, Sekihara M, Hara T, Yoshikawa M, et al. Clinical studies of enteritis caused by methicillin-resistant Staphylococcus aureus. Eur J Surg. (2001) 167:293–6. doi: 10.1080/110241501300091507
10. Edwards LA, O'Neill C, Furman MA, Hicks S, Torrente F, Perez-MaChado M, et al. Enterotoxin-producing staphylococci cause intestinal inflammation by a combination of direct epithelial cytopathy and superantigen-mediated T-cell activation. Inflammation Bowel Dis. (2012) 18:624–40. doi: 10.1002/ibd.21852
11. Pinchuk IV, Beswick EJ, Reyes VE. Staphylococcal enterotoxins. Toxins (Basel). (2010) 2:2177–97. doi: 10.3390/toxins2082177
12. Principato M. and Qian, B.F. Staphylococcal enterotoxins in the etiopathogenesis of mucosal autoimmunity within the gastrointestinal tract. Toxins (Basel). (2014) 6:1471–89. doi: 10.3390/toxins6051471
13. Kaempfer R. Bacterial superantigen toxins, CD28, and drug development. Toxins (Basel). (2018) 10:459–65. doi: 10.3390/toxins10110459
14. Thomas D, Dauwalder O, Brun V, Badiou C, Ferry T, Etienne J, et al. Staphylococcus aureus superantigens elicit redundant and extensive human Vbeta patterns. Infect Immun. (2009) 77:2043–50. doi: 10.1128/IAI.01388-08
15. Ochsenreither S, Fusi A, Busse A, Nagorsen D, Schrama D, Becker J, et al. Relative quantification of TCR Vbeta-chain families by real time PCR for identification of clonal T-cell populations. J Transl Med. (2008) 6:34–42. doi: 10.1186/1479-5876-6-34
16. Arad G, Levy R, Nasie I, Hillman D, Rotfogel Z, Barash U, et al. Binding of superantigen toxins into the CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock. PLoS Biol. (2011) 9:e1001149–1001162. doi: 10.1371/journal.pbio.1001149
17. Ramachandran G, Kaempfer R, Chung CS, Shirvan A, Chahin AB, Palardy JE, et al. CD28 homodimer interface mimetic peptide acts as a preventive and therapeutic agent in models of severe bacterial sepsis and gram-negative bacterial peritonitis. J Infect Dis. (2015) 211:995–1003. doi: 10.1093/infdis/jiu556
18. Popugailo A, Rotfogel Z, Supper E, Hillman D, Kaempfer R. Staphylococcal and streptococcal superantigens trigger B7/CD28 costimulatory receptor engagement to hyperinduce inflammatory cytokines. Front Immunol. (2019) 10:942. doi: 10.3389/fimmu.2019.00942
19. Levy R, Rotfogel Z, Hillman D, Popugailo A, Arad G, Supper E, et al. Superantigens hyperinduce inflammatory cytokines by enhancing the B7-2/CD28 costimulatory receptor interaction. Proc Natl Acad Sci U.S.A. (2016) 113:E6437–46. doi: 10.1073/pnas.1603321113
20. Kunkl M, Amormino C, Caristi S, Tedeschi V, Fiorillo MT, Levy R, et al. Binding of staphylococcal enterotoxin B (SEB) to B7 receptors triggers TCR- and CD28-mediated inflammatory signals in the absence of MHC class II molecules. Front Immunol. (2021) 12:723689. doi: 10.3389/fimmu.2021.723689
21. Kaempfer R, Popugailo A, Levy R, Arad G, Hillman D, Rotfogel Z. Bacterial superantigen toxins induce a lethal cytokine storm by enhancing B7-2/CD28 costimulatory receptor engagement, a critical immune checkpoint. Receptors Clin Investig. (2017) 4:e1500.
22. Popugailo A, Rotfogel Z, Levy M, Turgeman O, Hillman D, Levy R, et al. The homodimer interfaces of costimulatory receptors B7 and CD28 control their engagement and pro-inflammatory signaling. J BioMed Sci. (2023) 30:49. doi: 10.1186/s12929-023-00941-3
23. Kunkl M, Amormino C, Spallotta F, Caristi S, Fiorillo MT, Paiardini A, et al. Bivalent binding of staphylococcal superantigens to the TCR and CD28 triggers inflammatory signals independently of antigen presenting cells. Front Immunol. (2023) 14:1170821. doi: 10.3389/fimmu.2023.1170821
24. Vancamelbeke M, Vermeire S. The intestinal barrier: a fundamental role in health and disease. Expert Rev Gastroenterol Hepatol. (2017) 11:821–34. doi: 10.1080/17474124.2017.1343143
25. Chelakkot C, Ghim J. and Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med. (2018) 50:1–9. doi: 10.1038/s12276-018-0126-x
26. Schoultz I, Keita AV. The intestinal barrier and current techniques for the assessment of gut permeability. Cells. (2020) 9:1909. doi: 10.3390/cells9081909
27. Kuo WT, Odenwald MA, Turner JR, Zuo L. Tight junction proteins occludin and ZO-1 as regulators of epithelial proliferation and survival. Ann N Y Acad Sci. (2022) 1514:21–33. doi: 10.1111/nyas.14798
28. Otani T, Furuse M. Tight junction structure and function revisited. Trends Cell Biol. (2020) 30:805–17. doi: 10.1016/j.tcb.2020.08.004
29. Garcia MA, Nelson WJ, Chavez N. Cell-cell junctions organize structural and signaling networks. Cold Spring Harb Perspect Biol. (2018) 10. doi: 10.1101/cshperspect.a029181
30. Brunner J, Ragupathy S, Borchard G. Target specific tight junction modulators. Adv Drug Delivery Rev. (2021) 171:266–88. doi: 10.1016/j.addr.2021.02.008
31. Perez-Bosque A, Moreto M. A rat model of mild intestinal inflammation induced by Staphylococcus aureus enterotoxin B. Proc Nutr Soc. (2010) 69:447–53. doi: 10.1017/S0029665110001849
32. Moreto M, Perez-Bosque A. Dietary plasma proteins, the intestinal immune system, and the barrier functions of the intestinal mucosa. J Anim Sci. (2009) 87:E92–100. doi: 10.2527/jas.2008-1381
33. Danielsen EM, Hansen GH, Karlsdottir E. Staphylococcus aureus enterotoxins A- and B: binding to the enterocyte brush border and uptake by perturbation of the apical endocytic membrane traffic. Histochem Cell Biol. (2013) 139:513–24. doi: 10.1007/s00418-012-1055-8
34. Fisher EL, Otto M, Cheung GYC. Basis of virulence in enterotoxin-Mediated staphylococcal food poisoning. Front Microbiol. (2018) 9:436. doi: 10.3389/fmicb.2018.00436
35. Kunkl M, Mastrogiovanni M, Porciello N, Caristi S, Monteleone E, Arcieri S, et al. CD28 individual signaling up-regulates human IL-17A expression by promoting the recruitment of relA/NF-kappaB and STAT3 transcription factors on the proximal promoter. Front Immunol. (2019) 10:864. doi: 10.3389/fimmu.2019.00864
36. Northrop JK, Thomas RM, Wells AD, Shen H. Epigenetic remodeling of the IL-2 and IFN-gamma loci in memory CD8 T cells is influenced by CD4 T cells. J Immunol. (2006) 177:1062–9. doi: 10.4049/jimmunol.177.2.1062
37. Papageorgiou AC, Tranter HS, Acharya KR. Crystal structure of microbial superantigen staphylococcal enterotoxin B at 1.5 A resolution: implications for superantigen recognition by MHC class II molecules and T-cell receptors. J Mol Biol. (1998) 277:61–79. doi: 10.1006/jmbi.1997.1577
38. Evans EJ, Esnouf RM, Manso-Sancho R, Gilbert RJ, James JR, Yu C, et al. Crystal structure of a soluble CD28-Fab complex. Nat Immunol. (2005) 6:271–9. doi: 10.1038/ni1170
39. Rodstrom KE, Elbing K, Lindkvist-Petersson K. Structure of the superantigen staphylococcal enterotoxin B in complex with TCR and peptide-MHC demonstrates absence of TCR-peptide contacts. J Immunol. (2014) 193:1998–2004. doi: 10.4049/jimmunol.1401268
40. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. (2021) 596:583–9. doi: 10.1038/s41586-021-03819-2
41. Brenke R, Hall DR, Chuang GY, Comeau SR, Bohnuud T, Beglov D, et al. Application of asymmetric statistical potentials to antibody-protein docking. Bioinformatics. (2012) 28:2608–14. doi: 10.1093/bioinformatics/bts493
42. Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. (2003) 125:1731–7. doi: 10.1021/ja026939x
43. Christoffer C, Chen S, Bharadwaj V, Aderinwale T, Kumar V, Hormati M, et al. LZerD webserver for pairwise and multiple protein-protein docking. Nucleic Acids Res. (2021) 49:W359–65. doi: 10.1093/nar/gkab336
44. Janson G, Paiardini A. PyMod 3: a complete suite for structural bioinformatics in PyMOL. Bioinformatics. (2021) 37:1471–2. doi: 10.1093/bioinformatics/btaa849
45. Andrews C, McLean MH, Durum SK. Cytokine tuning of intestinal epithelial function. Front Immunol. (2018) 9:1270. doi: 10.3389/fimmu.2018.01270
46. Sambuy Y, De Angelis I, Ranaldi G, Scarino ML, Stammati A, Zucco F. The Caco-2 cell line as a model of the intestinal barrier: influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol Toxicol. (2005) 21:1–26. doi: 10.1007/s10565-005-0085-6
47. Lopez-Escalera S, Wellejus A. Evaluation of Caco-2 and human intestinal epithelial cells as in vitro models of colonic and small intestinal integrity. Biochem Biophys Rep. (2022) 31:101314. doi: 10.1016/j.bbrep.2022.101314
48. Leggett SE, Hruska AM, Guo M, Wong IY. The epithelial-mesenchymal transition and the cytoskeleton in bioengineered systems. Cell Commun Signal. (2021) 19:32. doi: 10.1186/s12964-021-00713-2
49. Suarez-Carmona M, Lesage J, Cataldo D, Gilles C. EMT and inflammation: inseparable actors of cancer progression. Mol Oncol. (2017) 11:805–23. doi: 10.1002/1878-0261.12095
50. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. (2014) 15:178–96. doi: 10.1038/nrm3758
51. Al-Sadi R, Ye D, Boivin M, Guo S, Hashimi M, Ereifej L, et al. Interleukin-6 modulation of intestinal epithelial tight junction permeability is mediated by JNK pathway activation of claudin-2 gene. PLoS One. (2014) 9:e85345. doi: 10.1371/journal.pone.0085345
52. Al-Sadi R, Guo S, Ye D, Ma TY. TNF-alpha modulation of intestinal epithelial tight junction barrier is regulated by ERK1/2 activation of Elk-1. Am J Pathol. (2013) 183:1871–84. doi: 10.1016/j.ajpath.2013.09.001
53. Cao M, Wang P, Sun C, He W, Wang F. Amelioration of IFN-gamma and TNF-alpha-induced intestinal epithelial barrier dysfunction by berberine via suppression of MLCK-MLC phosphorylation signaling pathway. PLoS One. (2013) 8:e61944. doi: 10.1371/journal.pone.0061944
54. Wang Y, Mumm JB, Herbst R, Kolbeck R, Wang Y. IL-22 increases permeability of intestinal epithelial tight junctions by enhancing claudin-2 expression. J Immunol. (2017) 199:3316–25. doi: 10.4049/jimmunol.1700152
55. Kunkl M, Amormino C, Frascolla S, Sambucci M, De Bardi M, Caristi S, et al. CD28 autonomous signaling orchestrates IL-22 expression and IL-22-regulated epithelial barrier functions in human T lymphocytes. Front Immunol. (2020) 11:590964. doi: 10.3389/fimmu.2020.590964
56. Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. (1998) 394:145–51. doi: 10.1038/28101
57. Yemelyanov A, Gasparian A, Lindholm P, Dang L, Pierce JW, Kisseljov F, et al. Effects of IKK inhibitor PS1145 on NF-kappaB function, proliferation, apoptosis and invasion activity in prostate carcinoma cells. Oncogene. (2006) 25:387–98. doi: 10.1038/sj.onc.1209066
58. Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U.S.A. (2007) 104:7391–6. doi: 10.1073/pnas.0609757104
59. Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola D, Mansour M, et al. Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J Biol Chem. (2008) 283:14665–73. doi: 10.1074/jbc.M707429200
60. Xiong H, Hong J, Du W, Lin YW, Ren LL, Wang YC, et al. Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial-mesenchymal transition. J Biol Chem. (2012) 287:5819–32. doi: 10.1074/jbc.M111.295964
61. Rajabi H, Alam M, Takahashi H, Kharbanda A, Guha M, Ahmad R, et al. MUC1-C oncoprotein activates the ZEB1/miR-200c regulatory loop and epithelial-mesenchymal transition. Oncogene. (2014) 33:1680–9. doi: 10.1038/onc.2013.114
62. Liu WH, Chen MT, Wang ML, Lee YY, Chiou GY, Chien CS, et al. Cisplatin-selected resistance is associated with increased motility and stem-like properties via activation of STAT3/Snail axis in atypical teratoid/rhabdoid tumor cells. Oncotarget. (2015) 6:1750–68. doi: 10.18632/oncotarget.2737
63. Pires BR, Mencalha AL, Ferreira GM, de Souza WF, Morgado-Diaz JA, Maia AM, et al. NF-kappaB is involved in the regulation of EMT genes in breast cancer cells. PLoS One. (2017) 12:e0169622. doi: 10.1371/journal.pone.0169622
64. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. (2014) 14:329–42. doi: 10.1038/nri3661
65. McLean MH, Neurath MF. and Durum, S.K. Targeting interleukins for the treatment of inflammatory bowel disease-what lies beyond anti-TNF therapy? Inflammation Bowel Dis. (2014) 20:389–97. doi: 10.1097/01.MIB.0000437616.37000.41
66. Borowczak J, Szczerbowski K, Maniewski M, Kowalewski A, Janiczek-Polewska M, Szylberg A, et al. The role of inflammatory cytokines in the pathogenesis of colorectal carcinoma-recent findings and review. Biomedicines. (2022) 10:1670. doi: 10.3390/biomedicines10071670
67. He F, Peng J, Deng XL, Yang LF, Camara AD, Omran A, et al. Mechanisms of tumor necrosis factor-alpha-induced leaks in intestine epithelial barrier. Cytokine. (2012) 59:264–72. doi: 10.1016/j.cyto.2012.04.008
68. Xu P, Elamin E, Elizalde M, Bours P, Pierik MJ, Masclee AAM, et al. Modulation of intestinal epithelial permeability by plasma from patients with crohn's disease in a three-dimensional cell culture model. Sci Rep. (2019) 9:2030. doi: 10.1038/s41598-018-38322-8
69. Wang H, Wang HS, Zhou BH, Li CL, Zhang F, Wang XF, et al. Epithelial-mesenchymal transition (EMT) induced by TNF-alpha requires AKT/GSK-3beta-mediated stabilization of snail in colorectal cancer. PLoS One. (2013) 8:e56664. doi: 10.1371/journal.pone.0056664
70. Watari A, Sakamoto Y, Hisaie K, Iwamoto K, Fueta M, Yagi K, et al. Rebeccamycin attenuates TNF-alpha-induced intestinal epithelial barrier dysfunction by inhibiting myosin light chain kinase production. Cell Physiol Biochem. (2017) 41:1924–34. doi: 10.1159/000472367
71. Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT. and Turner, J.R. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. (2005) 166:409–19. doi: 10.1016/S0002-9440(10)62264-X
72. Matsuhisa K, Watari A, Iwamoto K, Kondoh M, Yagi K. Lignosulfonic acid attenuates NF-kappaB activation and intestinal epithelial barrier dysfunction induced by TNF-alpha/IFN-gamma in Caco-2 cells. J Nat Med. (2018) 72:448–55. doi: 10.1007/s11418-017-1167-5
73. Rose-John S, Jenkins BJ, Garbers C, Moll JM, Scheller J. Targeting IL-6 trans-signalling: past, present and future prospects. Nat Rev Immunol. (2023) 23:666–81. doi: 10.1038/s41577-023-00856-y
74. Suzuki T, Yoshinaga N, Tanabe S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J Biol Chem. (2011) 286:31263–71. doi: 10.1074/jbc.M111.238147
75. Huang YH, Chen HK, Hsu YF, Chen HC, Chuang CH, Huang SW, et al. Src-FAK signaling mediates interleukin 6-induced HCT116 colorectal cancer epithelial-mesenchymal transition. Int J Mol Sci. (2023) 24:6650. doi: 10.3390/ijms24076650
76. Keir M, Yi Y, Lu T, Ghilardi N. The role of IL-22 in intestinal health and disease. J Exp Med. (2020) 217:e20192195. doi: 10.1084/jem.20192195
77. Tsai PY, Zhang B, He WQ, Zha JM, Odenwald MA, Singh G, et al. IL-22 upregulates epithelial claudin-2 to drive diarrhea and enteric pathogen clearance. Cell Host Microbe. (2017) 21:671–681.e674. doi: 10.1016/j.chom.2017.05.009
78. Delbue D, Lebenheim L, Cardoso-Silva D, Dony V, Krug SM, Richter JF, et al. Reprogramming intestinal epithelial cell polarity by interleukin-22. Front Med (Lausanne). (2021) 8:656047. doi: 10.3389/fmed.2021.656047
79. Zhang HJ, Zhang YN, Zhou H, Guan L, Li Y. and sun, M.J. IL-17A promotes initiation and development of intestinal fibrosis through EMT. Dig Dis Sci. (2018) 63:2898–909. doi: 10.1007/s10620-018-5234-x
80. Dongre A. and Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. (2019) 20:69–84. doi: 10.1038/s41580-018-0080-4
81. Bracken CP. and Goodall, G.J. The many regulators of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. (2022) 23:89–90. doi: 10.1038/s41580-021-00442-x
82. Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. (2000) 2:76–83. doi: 10.1038/35000025
83. Vesuna F, van Diest P, Chen JH, Raman V. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem Biophys Res Commun. (2008) 367:235–41. doi: 10.1016/j.bbrc.2007.11.151
84. Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. (2001) 7:1267–78. doi: 10.1016/S1097-2765(01)00260-X
85. Sanchez-Tillo E, Lazaro A, Torrent R, Cuatrecasas M, Vaquero EC, Castells A, et al. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. (2010) 29:3490–500. doi: 10.1038/onc.2010.102
86. Liu M, Yang J, Zhang Y, Zhou Z, Cui X, Zhang L, et al. ZIP4 promotes pancreatic cancer progression by repressing ZO-1 and claudin-1 through a ZEB1-dependent transcriptional mechanism. Clin Cancer Res. (2018) 24:3186–96. doi: 10.1158/1078-0432.CCR-18-0263
87. Cohen EN, Gao H, Anfossi S, Mego M, Reddy NG, Debeb B, et al. Inflammation mediated metastasis: immune induced epithelial-to-mesenchymal transition in inflammatory breast cancer cells. PLoS One. (2015) 10:e0132710. doi: 10.1371/journal.pone.0132710
88. Goebel L, Grage-Griebenow E, Gorys A, Helm O, Genrich G, Lenk L, et al. CD4(+) T cells potently induce epithelial-mesenchymal-transition in premalignant and Malignant pancreatic ductal epithelial cells-novel implications of CD4(+) T cells in pancreatic cancer development. Oncoimmunology. (2015) 4:e1000083. doi: 10.1080/2162402X.2014.1000083
89. Jain SM, Deka D, Das A, Paul S, Pathak S, Banerjee A. Role of interleukins in inflammation-mediated tumor immune microenvironment modulation in colorectal cancer pathogenesis. Dig Dis Sci. (2023) 68:3220–36. doi: 10.1007/s10620-023-07972-8
90. Jones SA, Jenkins BJ. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat Rev Immunol. (2018) 18:773–89. doi: 10.1038/s41577-018-0066-7
91. Abaurrea A, Araujo AM, Caffarel MM. The role of the IL-6 cytokine family in epithelial-mesenchymal plasticity in cancer progression. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22158334
92. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. (2015) 33:747–85. doi: 10.1146/annurev-immunol-032414-112123
93. Qing Y, Stark GR. Alternative activation of STAT1 and STAT3 in response to interferon-gamma. J Biol Chem. (2004) 279:41679–85. doi: 10.1074/jbc.M406413200
94. Hayden MS, Ghosh S. Regulation of NF-kappaB by TNF family cytokines. Semin Immunol. (2014) 26:253–66. doi: 10.1016/j.smim.2014.05.004
95. Monin L, Gaffen SL. Interleukin 17 family cytokines: Signaling mechanisms, biological activities, and therapeutic implications. Cold Spring Harb Perspect Biol. (2018) 10. doi: 10.1101/cshperspect.a028522
96. Gunning PT, Katt WP, Glenn M, Siddiquee K, Kim JS, Jove R, et al. Isoform selective inhibition of STAT1 or STAT3 homo-dimerization via peptidomimetic probes: structural recognition of STAT SH2 domains. Bioorg Med Chem Lett. (2007) 17:1875–8. doi: 10.1016/j.bmcl.2007.01.077
97. Bulger EM, Maier RV, Sperry J, Joshi M, Henry S, Moore FA, et al. A novel drug for treatment of necrotizing soft-tissue infections: A randomized clinical trial. JAMA Surg. (2014) 149:528–36. doi: 10.1001/jamasurg.2013.4841
98. Ramachandran G, Tulapurkar ME, Harris KM, Arad G, Shirvan A, Shemesh R, et al. A peptide antagonist of CD28 signaling attenuates toxic shock and necrotizing soft-tissue infection induced by Streptococcus pyogenes. J Infect Dis. (2013) 207:1869–77. doi: 10.1093/infdis/jit104
99. Dong D, Zheng L, Lin J, Zhang B, Zhu Y, Li N, et al. Structural basis of assembly of the human T cell receptor-CD3 complex. Nature. (2019) 573:546–52. doi: 10.1038/s41586-019-1537-0
100. Wu H, Cao R, Wen M, Xue H, OuYang B. Structural characterization of a dimerization interface in the CD28 transmembrane domain. Structure. (2022) 30:803–812.e805. doi: 10.1016/j.str.2022.03.004
101. Rodstrom KEJ, Regenthal P, Bahl C, Ford A, Baker D, Lindkvist-Petersson K. Two common structural motifs for TCR recognition by staphylococcal enterotoxins. Sci Rep. (2016) 6:25796. doi: 10.1038/srep25796
102. Kaempfer R, Arad G, Levy R, Hillman D, Nasie I, Rotfogel Z. CD28: direct and critical receptor for superantigen toxins. Toxins (Basel). (2013) 5:1531. doi: 10.3390/toxins5091531
103. Seyyed Mousavi MN, Mehramuz B, Sadeghi J, Alizadeh N, Oskouee MA. and Kafil, H.S. The pathogenesis of Staphylococcus aureus in autoimmune diseases. Microb Pathog. (2017) 111:503–7. doi: 10.1016/j.micpath.2017.09.028
104. Bettenworth D, Nowacki TM, Friedrich A, Becker K, Wessling J, Heidemann J. Crohn's disease complicated by intestinal infection with methicillin-resistant Staphylococcus aureus. World J Gastroenterol. (2013) 19:4418–21. doi: 10.3748/wjg.v19.i27.4418
105. Lu J, Wang A, Ansari S, Hershberg RM, McKay DM. Colonic bacterial superantigens evoke an inflammatory response and exaggerate disease in mice recovering from colitis. Gastroenterology. (2003) 125:1785–95. doi: 10.1053/j.gastro.2003.09.020
Keywords: staphylococcal enterotoxin B (SEB), superantigen (s), T cells, CD28, inflammation, intestinal epithelial barrier dysfunctions
Citation: Amormino C, Russo E, Tedeschi V, Fiorillo MT, Paiardini A, Spallotta F, Rosanò L, Tuosto L and Kunkl M (2024) Targeting staphylococcal enterotoxin B binding to CD28 as a new strategy for dampening superantigen-mediated intestinal epithelial barrier dysfunctions. Front. Immunol. 15:1365074. doi: 10.3389/fimmu.2024.1365074
Received: 03 January 2024; Accepted: 26 February 2024;
Published: 06 March 2024.
Edited by:
Takashi Saito, RIKEN, JapanCopyright © 2024 Amormino, Russo, Tedeschi, Fiorillo, Paiardini, Spallotta, Rosanò, Tuosto and Kunkl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Loretta Tuosto, bG9yZXR0YS50dW9zdG9AdW5pcm9tYTEuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.