 Emily A. Henderson1
Emily A. Henderson1 Abby Ivey2Soo Jeon Choi1Stell Santiago3Dudley McNitt1
Abby Ivey2Soo Jeon Choi1Stell Santiago3Dudley McNitt1 Tracy W. Liu1,2
Tracy W. Liu1,2 Slawomir Lukomski1,2*†
Slawomir Lukomski1,2*† Brian A. Boone1,2,4*†
Brian A. Boone1,2,4*†- 1Department of Microbiology, Immunology and Cell Biology, School of Medicine, West Virginia University, Morgantown, WV, United States
- 2West Virginia University Cancer Institute, School of Medicine, West Virginia University, Morgantown, WV, United States
- 3Department of Pathology, School of Medicine, West Virginia University, Morgantown, WV, United States
- 4Department of Surgery, West Virginia University, Morgantown, WV, United States
Introduction: Pancreatic ductal adenocarcinoma (PDAC) is a lethal cancer associated with an immunosuppressive environment. Neutrophil extracellular traps (NETs) were initially described in the context of infection but have more recently been implicated in contributing to the tolerogenic immune response in PDAC. Thus, NETs are an attractive target for new therapeutic strategies. Group A Streptococcus (GAS) has developed defensive strategies to inhibit NETs.
Methods: In the present work, we propose utilizing intra-tumoral GAS injection to stimulate anti-tumor activity by inhibiting cancer-promoting NETs. Mice harboring Panc02 or KPC subcutaneous tumors injected with three different M-type GAS strains. Tumors and spleens were harvested at the endpoint of the experiments to assess bacterial colonization and systemic spread, while sera were analyzed for humoral responses toward the streptococcal antigens, especially the M1 and Scl1 proteins. Role of the streptococcal collagen-like protein 1 (Scl1) in anti-PDAC activity was assessed in vivo after intratumoral injection with M1 GAS wild-type, an isogenic mutant strain devoid of Scl1, or a complemented mutant strain with restored scl1 expression. In addition, recombinant Scl1 proteins were tested for NET inhibition using in vitro and ex vivo assays assessing NET production and myeloperoxidase activity.
Results: Injection of three different M-type GAS strains reduced subcutaneous pancreatic tumor volume compared to control in two different murine PDAC models. Limitation of tumor growth was dependent on Scl1, as isogenic mutant strain devoid of Scl1 did not reduce tumor size. We further show that Scl1 plays a role in localizing GAS to the tumor site, thereby limiting the systemic spread of bacteria and off-target effects. While mice did elicit a humoral immune response to GAS antigens, tested sera were weakly immunogenic toward Scl1 antigen following intra-tumoral treatment with Scl1-expressing GAS. M1 GAS inhibited NET formation when co-cultured with neutrophils while Scl1-devoid mutant strain did not. Recombinant Scl1 protein inhibited NETs ex vivo in a dose-dependent manner by suppressing myeloperoxidase activity.
Discussion: Altogether, we demonstrate that intra-tumoral GAS injections reduce PDAC growth, which is facilitated by Scl1, in part through inhibition of cancer promoting NETs. This work offers a novel strategy by which NETs can be targeted through Scl1 protein and potentiates its use as a cancer therapeutic.
1 Introduction
Pancreatic adenocarcinoma (PDAC) is one of the most lethal cancers with a five-year survival rate of only 12% (1, 2). Late diagnosis and resistance to current treatment modalities contribute to this poor survival (3, 4). PDAC is an immunologically “cold” tumor, with minimal response to checkpoint immunotherapy (5, 6). Therefore, there is a growing need for the development of strategies to induce an anti-tumor immune response and generate novel cancer therapies in this deadly disease.
William Coley, a bone osteosarcoma surgeon influenced by the work of German physician Wilhelm Busch, performed the first systematic study using streptococcal organisms as a cancer treatment strategy in 1891 leading to the development of Coley’s toxin, a concoction of heat-killed Streptococcus pyogenes (group A Streptococcus) and Serratia marcescens. Though his work fell out of favor with implementation of radiation and chemotherapy, evidence suggests Coley’s intuition of stimulating the immune system to elicit an anti-cancer response is effective (7). Bacteria harbor a variety of structures, toxins and mechanisms that make them attractive candidates as therapeutic agents with some evidence of preclinical efficacy in PDAC (8). Evidence suggests that group A Streptococcus (GAS) peptides (9) and superantigens (10) can elicit tumor regression. GAS therapy showed promise in instances of bladder cancer (11), though was less successful in preclinical trial of other cancers (12, 13). Still, GAS utilizes several compounds to evade innate immunity that offer mechanisms by which GAS can stimulate an anti-tumor response. One mechanism is the inhibition of neutrophil extracellular trap (NET) release (14). NETs were initially described as a method for neutrophils to trap and kill bacteria and other infectious agents (15), however NETs are also upregulated in sterile inflammatory diseases including PDAC (16, 17). Recent evidence suggests the significance of NETs in contributing to PDAC tumor growth, progression, metastasis, and promotion of an immunosuppressive tumor environment (18–21). Moreover, NET levels are a prognostic indicator of PDAC patient survival and treatment outcomes (22, 23). GAS has the capacity to evade NET degradation through the activity of streptococcal collagen-like 1 protein which prevents NET release (14, 24), though the use of GAS to limit NETs has yet to be explored in the context of cancer.
Herein, we demonstrate GAS as a potential therapeutic agent in two murine models of PDAC. We also propose the therapeutic significance of streptococcal collagen-like-1 (Scl1) protein as an essential factor in GAS anti-tumor response that supports tumor colonization, and acts as a potent inhibitor of NET activity through reduction of myeloperoxidase activity. These findings have significant implications in the exploration of recombinant Scl1 as a novel therapy to limit PDAC progression.
2 Materials and methods
2.1 Animals
Age and gender matched C57BL/6 wild-type (WT) mice (8-weeks old) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). All animal experiments were approved by the Institutional Animal Care and Use Committee of West Virginia University, protocol number 1602000144_R1.5 and 180918204 and complied with the use of experimental animals published by the National Institutes of Health.
2.2 Cell lines and cell-culture
Panc02 cells derived from a pancreatic adenocarcinoma model in male C57BL/6 mice were obtained from the National Cancer Institute. KPCY6422c1 cells derived from mouse pancreatic neoplasms were purchased from Kerafast (Kerafast, MA, USA). Panc02 cells were cultured in RPMI 1640 media supplemented with 10% fetal bovine serum and 1% Penicillin-Streptomycin (Hyclone, Logan, UT, USA). KPCY6422c1 cells were cultured with Dulbecco’s Modified Eagle Medium with 10% fetal bovine serum and 1% glutamine. Cells were grown at 37°C in a humidified 5% CO2 atmosphere.
2.3 Murine subcutaneous tumor model
Age and gender-matched 8-week-old C57BL/6 WT mice were injected with 5x105 Panc02 or KPCY6422c1 cells into the right flank. Once tumors reached 7 mm (10-12 days post tumor implantation), tumors were injected with group A Streptococcus as outlined below or with PBS. Caliper measurements of tumors were taken every other day until experimental endpoint. Length (L) and width (W) of tumor were measured and tumor volume in mm3 was calculated using equation for spherical ellipsoid (VT=0.5*L*W2) (25). Tumors and spleens were harvested, weighed, homogenized, and cultured on blood agar (Supplementary Figure S1). Tissues were processed for H&E and Gram stains in the West Virginia University Pathology Research Histology Laboratory. Neutrophil index scores from tumor tissues were determined and scored by a blinded pathologist.
2.4 Generation of recombinant proteins
Recombinant rScl1 proteins were generated as described previously (26–28). Briefly, rScl1-encoding clones were generated in the Strep-tag II cloning, expression, and purification system using the E. coli vector pASK-IBA2 designed for periplasmic expression (IBA-GmbH, Geottingen, Germany); rScl1.1 was derived from the Scl1 protein in M1-type strain, rScl1.28 in M28-type strain, and rScl1.41 in M41-type strain. Clones were verified by DNA Sanger sequencing and rScl1 polypeptides by N-terminal sequencing. Bacterial cultures were grown in Luria-Bertani medium (LB broth, Miller) and protein expression was induced with anhydrotetracycline (0.2 μg/mL) for three hours. The periplasmic fraction was collected following treatment with CellLytic B Cell Lysis reagent (Sigma, B7435) and purified on Strep-Tactin Sepharose.
rEDA was produced intracellularly using the pQE-30 His tag purification system (Qiagen) in E. coli JM-109 (29). E. coli was cultured in LB broth supplemented with ampicillin (100 μg/mL) and protein expression was induced with 1 mM isopropyl β-D-thiogalactopyranoside for three hours. Cells were harvested and lysed in lysis buffer (50 mM Tris/HCL, pH 8.0, 50 mM NaCl, 2 mM MgCl2, 2% Triton X-100, 10 mM beta-mercaptoethanol, 2 mg lysozyme, 1 mM EDTA-free protease inhibitor cocktail tablet, 1 mM phenylmethylsulfonyl fluoride (PMSF), 100 U DNase I). Supernatant was collected following centrifugation, and rEDA was purified using nickel-nitrilotriacetic acid-agarose resin (Qiagen).
Following purification, rScl1 proteins and rEDA were analyzed by SDS-PAGE (Supplementary Figure S2). Proteins were dialyzed in 25 mM HEPES pH 8.0 and stored at -20°C.
2.5 Group A Streptococcus strain preparation and injection
The M1-type GAS strain MGAS5005 Δscl1 mutant was generated by allelic replacement mutagenesis as described previously (30). Briefly, scl1.1 allele was cloned into an E. coli vector and coding sequence was replaced with a nonpolar spectinomycin resistance cassette spc2, resulting in pSL134 plasmid suicide in GAS. MGAS5005 cells were electroporated with pSL134 and spectinomycin-resistant colonies were screened by PCR for a single amplification product of mutated size indicating double cross-over event; mutant candidates were then verified by sequencing and loss of Scl1 protein was confirmed by western immunoblotting with specific anti-Scl1.1 Abs.
Complementation of M1 Δscl1 mutant was performed as previously described (26) using GAS-E. coli shuttle plasmid. The DNA fragment encoding scl1.1 allele was PCR-amplified from genomic DNA of MGAS5005 and cloned into pSB027 generating plasmid pSL620. pSL620 was introduced into MGAS5005 Δscl1.1 mutant by electroporation and selected on Brain Heart Infusion (BHI) agar with 10 μg/mL chloramphenicol; Scl1.1 expression was confirmed by western immunoblotting as above.
M1 wild type (WT), Δscl1.1 isogenic mutant, Δscl1.1::620 complemented mutant, M41 strain MGAS6183, and M28 strain MGAS6143 group A Streptococcus were grown overnight at 37°C with 5% CO2 on BHI agar solid media supplemented with antibiotics, as needed. Plated bacteria were inoculated into Todd-Hewitt Yeast (THY) broth and incubated until OD600 reached ~0.5. Following centrifugation, bacterial cells were washed once with PBS, centrifuged again, and diluted in PBS to designated concentrations (M1; 1x105, 1x106, 1x107; M41 and M28; 1x104, 1x105, 1x106). Indicated inocula of bacteria in 0.1-mL volume were injected into subcutaneous tumors established in C57BL/6 mice. Control mice received intra-tumoral PBS injections at equal volume.
To assess colonization by GAS, tumors collected at the endpoint of experiments were homogenized and plated serially diluted on blood agar; bacteria were enumerated after overnight incubation. In addition, bacteria recovered from tumors injected with Δscl1.1 isogenic mutant were assessed for mutated genotype after 14 days in vivo colonization without antibiotic selection. Serially diluted tumor homogenates were plated on blood agar as above to produce single colonies. Next, 100 streptococcal colonies were re-streaked on blood agar and on BHI agar supplemented with 100 μg/mL of spectinomycin, and incubated overnight (Supplementary Figure S3A). In addition, GAS colonies were screened by PCR with primers flanking scl1 gene and analyzed on 1% agarose gel. The amplicon sizes expected differ between wild-type allele and mutated allele (Supplementary Figure S3B).
2.6 ELISA and western blot analysis
To assess mouse seroconversion to streptococcal antigens, blood was collected from tumor burdened mice 14 days following GAS injection or PBS controls. For whole-cell ELISA, M1 wild-type bacteria were harvested at OD600 ~0.5 and washed as above in PBS. Cells were finally suspended in bicarbonate buffer, pH 9.6; for coating, 1x107 CFU were added into wells of 96-well high binding microplates and incubated at 4°C overnight (31). Wells were washed with tris-buffered saline (TBS)-1% BSA and blocked in this reagent at 37°C for 2 hours. Primary mouse sera were added at a 1:100 dilution in TBS-1% BSA. Anti-M1 and anti-Scl1.1 rabbit antibodies were used as positive controls. Plates were incubated at 37°C for 2 hours. Anti-mouse or anti-rabbit HRP-conjugated secondary antibodies at 1:1000 dilution were added and incubated at room temperature for 1 hour; then, wells were washed and ABST substrate was added, and immunoreactivity was read at 415 nm. To detect anti-Scl1.1 specific seroconversion, ELISA was performed using Strep-Tactin coated wells immobilized with rScl1.1 protein, as previously described (32); rScl1.1 construct contains a strep-tag II at the C-terminus, which binds to Strep-Tactin and thus reflecting the natural orientation of the Scl1 protein as on the GAS bacterial surface.
Western immunoblotting was carried out against GAS cell wall-associated protein fraction prepared from MGAS5005 and separated on 15% SDS–PAGE, as described (30). Log-phase GAS cells were harvested and resuspended in 20% sucrose–10 mM Tris (pH 8.0), 1mM ethylene diamine tetra-acetic acid buffer containing 5 U of mutanolysin,100 μg of lysozyme, and 100 mM PMSF. After digestion at 37°C for 1 h and centrifugation, the supernatants were used for subsequent analyses. The primary antibodies were the same sera (1:500 dil.) used above in whole cell ELISA prepared from mice bled at the endpoint of experiments. The control anti-GAS-antigen antibodies include: (i) anti-M1 protein rabbit Ab (1:2500 dil.) (33) and anti-Scl1.1 affinity purified rabbit Ab (1:1000 dil.) (30). Secondary Abs used were, accordingly, goat anti-mouse IgG(H+L)- or goat anti-rabbit IgG(H+L)-HRP conjugated (Bio-Rad, 1721019), followed by chemiluminescent detection with Clarity Western ECL Substrates (Bio-Rad, 1705060). Western immunoblotting to detect specific anti-rScl1.1 antibodies was performed as described (30). Briefly, rScl1.1 protein was separated by SDS-PAGE, transferred onto a membrane, and probed with mouse sera (1:100 dil.); anti-Scl1.1 rabbit antibodies were used in control well.
2.7 Neutrophil extracellular trap assay and quantification
Bone marrow neutrophils were isolated from the femur of euthanized C57BL/6 mice as described previously (16, 34). Bone marrow was collected, and cells were washed with RPMI-10 with 10% FBS and 1% PenStrep. Cells were separated using a density gradient centrifugation with histopaque 1077 and 1099. The layer between 1077 and 1099 was collected and plated onto a 96-well plate at 1.5x104 cells per well in Hank’s Balanced Salt Solution. Neutrophils were incubated for thirty minutes at 37°C to allow for cell attachment. Recombinant Scl1 protein, HEPES, sterile PBS, or whole-cell bacteria were added at the designated concentration and incubated for 5-10 minutes. 50 μM platelet activating factor (PAF) (511075, Sigma Aldrich) was added for activation for 3 hours. Cells were fixed with 4% paraformaldehyde and stained for DNA with Hoechst reagent. NETs were visualized using an EVOS microscope at 10-40x objective. NETs were quantified by collecting the supernatant and measuring cellular free DNA using Quant-iT Picogreen (Invitrogen, Grand Island, NY) according to manufacturer’s protocol. 500,000 U/mL polymyxin B sulfate was added to the recombinant protein for all in vitro experiments to account for LPS contamination.
2.8 Myeloperoxidase assays and quantification
Recombinant Scl1 proteins were examined for MPO activity using the Myeloperoxidase Inhibitor Screening Assay Kit (700170, Cayman Chemical, Ann Arbor, MI). The proteins were reconstituted in PBS and used in the assay according to the manufacturer’s protocol.
MPO activity was also assessed by luminol bioluminescence imaging of isolated bone marrow neutrophils as described previously (35, 36). Briefly, 1x105 isolated neutrophils were added to a 96-well black walled plate and stained with 100mM luminol (Sigma-Aldrich, MO, USA) followed by stimulation with 500nM phorbol 12-myristate 13-acetate, 99+% (Thermo Scientific Chemicals). Immediately following stimulation, cells were imaged for 60 minutes using the Kino imaging system (Spectral Instruments Imaging, AZ, USA) at 37°C under 5% CO2 flow for 12 total acquisitions. Data was analyzed by ROI measurements using Aura software (Spectral Instruments Imaging, AZ, USA) imported into Excel (Microsoft Corp., WA, USA). Data was represented as total flux (photons/second) by averaging triplicate wells and calculating the area under the curve from images taken at 5-60 min.
2.9 Statistical analysis
All statistical analysis was performed on GraphPad Prism 10.1.0 (316). Experiments were analyzed for significance using one-way ANOVA with or without repeated measures with multiple comparison post hoc tests as indicated in figure legends. Significance is designated as *=p<0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001.
3 Results
3.1 GAS limits subcutaneous Panc02 and KPC murine PDAC tumor growth
S. pyogenes serotype M49 has been shown to elicit anti-tumor growth activity in vivo against Panc02 tumors (37, 38). We first sought to substantiate these findings and investigate whether this effect was observable in other M-types or was restricted to M49. We utilized three different strains of S. pyogenes (M1, M28, and M41) at three different inocula injected into subcutaneous murine PDAC tumors. One week following injection of 5x105 Panc02 cells into the right flank, male and female mice were randomized to intra-tumoral injection with low, medium, and high inocula of each GAS M type or PBS control at equal volume. Tumor burden was measured on day eight post-infection.
Intra-tumoral injection with all three strains exhibited minimal skin pathology on day 8 (Figure 1A), while eliciting significant and comparable reductions in tumor volume and weight using all three strains compared to the PBS-treated tumors (Figures 1B, C). Tumor reduction was most pronounced at medium and high inoculum, although there were no significant differences between strains and inoculum sizes. Only two mice inoculated with the high inoculum died from systemic spread of bacteria indicated by the growth of β-hemolytic GAS colonies on blood agar inoculated with blood and spleen homogenates from these mice (Figure S1). Overall, we established that GAS of various M-types are capable to reduce Panc02-tumor growth in vivo.
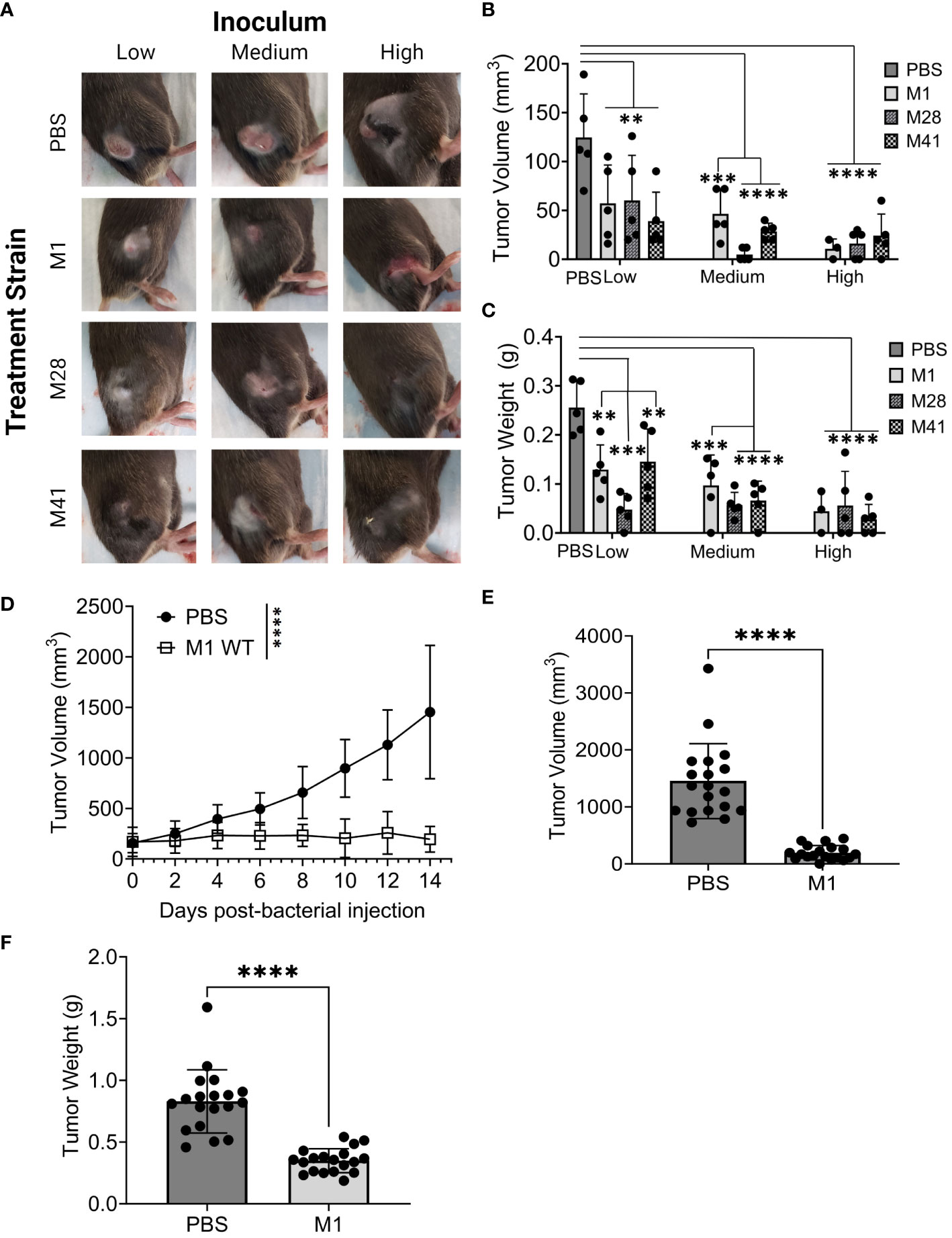
Figure 1 GAS injection limits the growth of subcutaneous murine Panc02 and KPC tumors. (A) Representative images of tumors and skin pathology associated with GAS injection. Pre-formed Panc02 tumors were injected intratumorally with GAS strains at low, medium, and high inocula (M1: 105, 106, 107 CFU or M41/M28: 106, 107, 108 CFU) or PBS at equal volume. Data shown were recorded on day 8 post-injection. (B) Comparison of tumor volume between experimental groups. Tumor dimensions were measured with a caliper on day 8 post-injection of GAS compared to PBS control. (C) Comparison of tumor weight between the groups. Tumors were collected and weighed on day 8 post-injection of GAS or PBS. n=5/group. (D) Changes in tumor volume following GAS injection. Measurements were taken by caliper during fourteen days following single-dose treatment of GAS or PBS into murine KPC tumors. Results are representative of two independent experiments. Significance is determined by one-way repeated measures ANOVA. n=19 per group total. (E) Tumor volume 14 days post-injection of M1 GAS or PBS. (F) Tumor weight at experimental endpoint of M1 GAS treated tumors and PBS control as done in (B) Significance determined by student’s t-test. **p<0.01, ***p<0.001, ****p<0.0001.
The KRAS oncogene encodes the protein KRAS involved in regulating cell proliferation and survival. Mutations in KRAS can disrupt the switch between active and inactive states thus promoting cancer development (39). Virtually all PDAC tumors harbor KRAS mutations and a majority also have mutations in P53, a tumor suppressor protein (3, 40). Therefore, we investigated whether GAS M1 strain had similar anti-tumor effects using KPCY6422c1 cells (abbreviated as KPC), a PDAC tumor lineage that mirrors the genetics of human PDAC (41). We injected 106 CFU, the medium-size inoculum in Figure 1, in further experiments to avoid systemic infection with GAS. Mice received a subcutaneous injection of 5x105 KPC cells and were monitored until tumor volumes were within a range of 150-175 mm3, around 10 to 12 days, followed by a single intra-tumoral dose of M1 GAS or PBS injection, represented as day 0 post-injection. Tumors were measured every other day for 14 days, e.g., a time, which required to scarify the control PBS-injected mice.
Tumor volume and weight were significantly reduced following treatment with M1 GAS in comparison to PBS control (Figures 1D–F). These results indicate that M1 GAS has a persistent therapeutic effect in more aggressive and clinically relevant subcutaneous KPC-PDAC tumors at medium-size inoculum without systemic spread. Based on this data, we established this dosing to be utilized for further studies.
3.2 GAS streptococcal collagen-like protein 1 is critical for reduction in PDAC tumor burden and localized tumor colonization
Streptococcal collagen-like protein 1 (Scl1) is expressed by majority of GAS strains tested (42–46) and is an important adhesion molecule binding to selected type III repeats of cellular fibronectin (47, 48), a component of the tumoral extracellular matrix also known as oncofetal fibronectin (49). Given that all three-experimental GAS-M-types used express Scl1, which resulted in tumor regression, we sought to explore if Scl1.1 (Scl1 variant in M1-type GAS) was mediating these effects. Following a similar model as above, mice carried subcutaneous KPC-PDAC tumors were administered with a single-dose intra-tumoral injection of either GAS M1 wild-type (WT), an isogenic Δscl1.1 mutant strain lacking Scl1.1 protein, or a Δscl1.1::620 mutant strain complemented for Scl1.1 expression. Tumor burden was monitored for fourteen days, then tumors were collected for analyses (Figure 2).
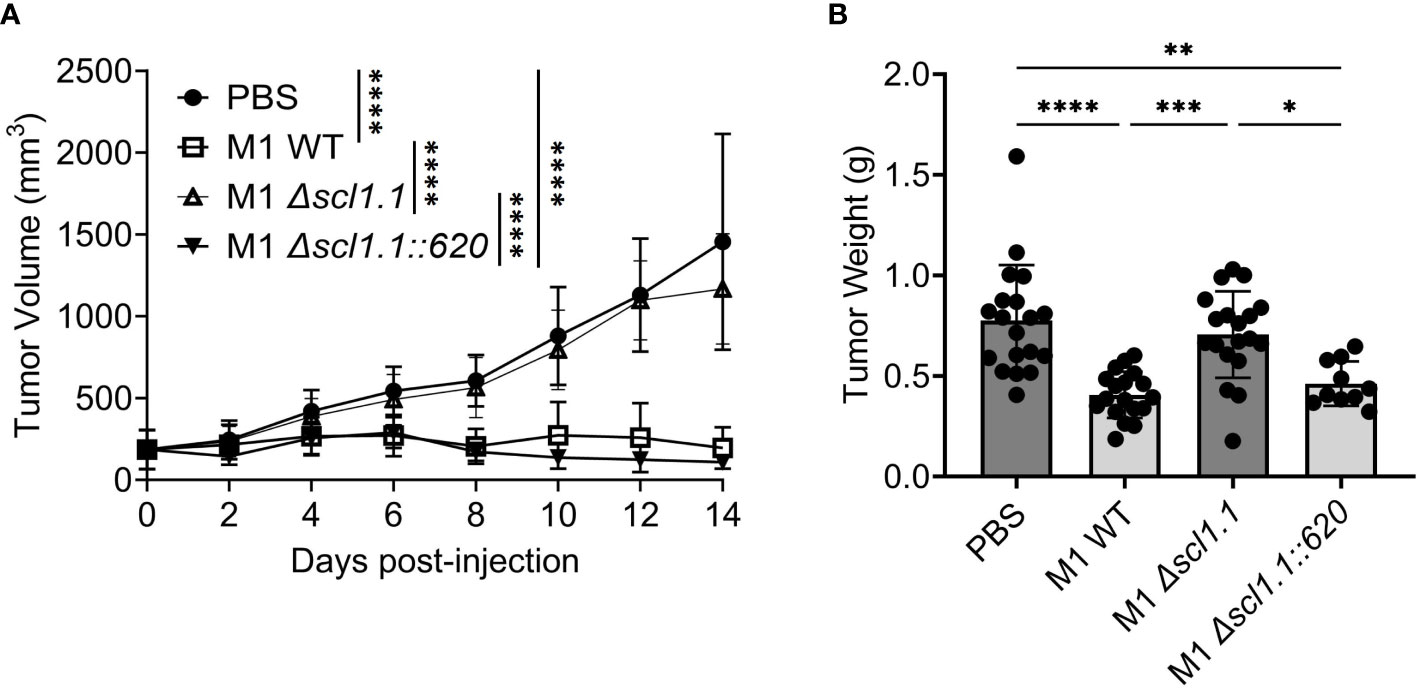
Figure 2 Injection of GAS expressing Scl1 limits the growth of subcutaneous PDAC tumor in mice. (A) Changes in tumor volume following GAS injection. Measurements were taken by caliper during fourteen days following single-dose treatment of indicated GAS strains or PBS control in murine KPC tumors. Results are representative of two independent experiments. Significance is determined by one-way repeated measures ANOVA. ****p<0.0001. n=10-20 per group total. (B) Tumor weight at experimental endpoint. Mouse tumors treated with indicated strains of GAS or PBS control were extracted and weighed (grams) at day 14. Results are representative of two independent experiments. Each dot represents a single murine tumor. Significance determined by one-way ANOVA with Tukey’s post-hoc test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Mice treated with the Scl1-expressing WT bacteria exhibited significant decreases in tumor growth over time as well as decreased tumor weights (Figures 2A, B). In contrast, tumor volumes in mice injected with the Δscl1.1 mutant strain showed no such effect and followed the volume pattern of the PBS control group, suggesting that anti-PDAC activity is contingent on Scl1 expression. When Scl1 expression was complemented back into the mutant strain in mice treated with Δscl1.1::620, tumor growth was again diminished and similar to injection with the WT strain, validating a critical role for Scl1 in GAS anti-tumor response.
To confirm that the GAS recovered from the tumors treated with the Δscl1.1 mutant strain did not revert to the wild-type genotype during replication within the tumor, homogenates prepared from two tumors treated with the isogenic Δscl1.1 mutant strain were serially diluted and plated onto blood agar. 100 colonies from each tumor were streaked in duplicate onto blood agar and BHI supplemented with spectinomycin (Δscl1.1 mutant was generated using a spectinomycin-resistant cassette) (Supplementary Figure S3A) - all 200 colonies grew on the selective medium, thereby, verifying Δscl1.1-mutant phenotype. 10 colonies from tumor 1 and 7 colonies from tumor 2 were arbitrarily selected and analyzed by colony PCR for the size of the amplicon corresponding to either to the scl1.1 wild-type or the Δscl1.1 mutant allele. All selected colonies produced the same single amplicons corresponding in size to the Δscl1.1 mutant but not the wild-type allele (Supplementary Figure S3B). Taken together, the Δscl1.1 mutant strain recovered from tumor homogenates retains the mutant genotype and validates the anti-tumor effects seen with GAS-expressing Scl1 during tumor colonization.
Based on known specific Scl1 interactions with oncofetal fibronectin in the tumor extracellular matrix (47), we hypothesized that Scl1 facilitates a focused nidus of GAS infection, localizing bacteria to the tumor site. Tumor-bearing mice were injected with either GAS M1 WT, an isogenic Δscl1.1 mutant, or a Δscl1.1::620 complemented mutant. Fourteen days post-injection, tumors and spleens were harvested for histopathological analysis and bacterial culture on blood agar (Figure 3). Tumors were locally colonized by GAS strains, as evidenced on Gram-stained histograms and on significant recovery of GAS in cultures from tumor homogenates plated on blood agar at the 14-day endpoint post-injection (Figures 3A, B). There were no statistically significant differences in tumor colonization levels (CFU per mL) between GAS isogenic strains. However, 5 out of 19 (26%) spleen homogenates recovered from mice treated with the isogenic Δscl1.1 mutant strain yielded bacterial growth, while no growth occurred from the spleens of mice treated with the WT and Δscl1.1::620 complemented strains (Figure 3C). These data suggest that Scl1 plays a role in localizing GAS to the tumor microenvironment (TME), thereby limiting systemic infection with bacteria.
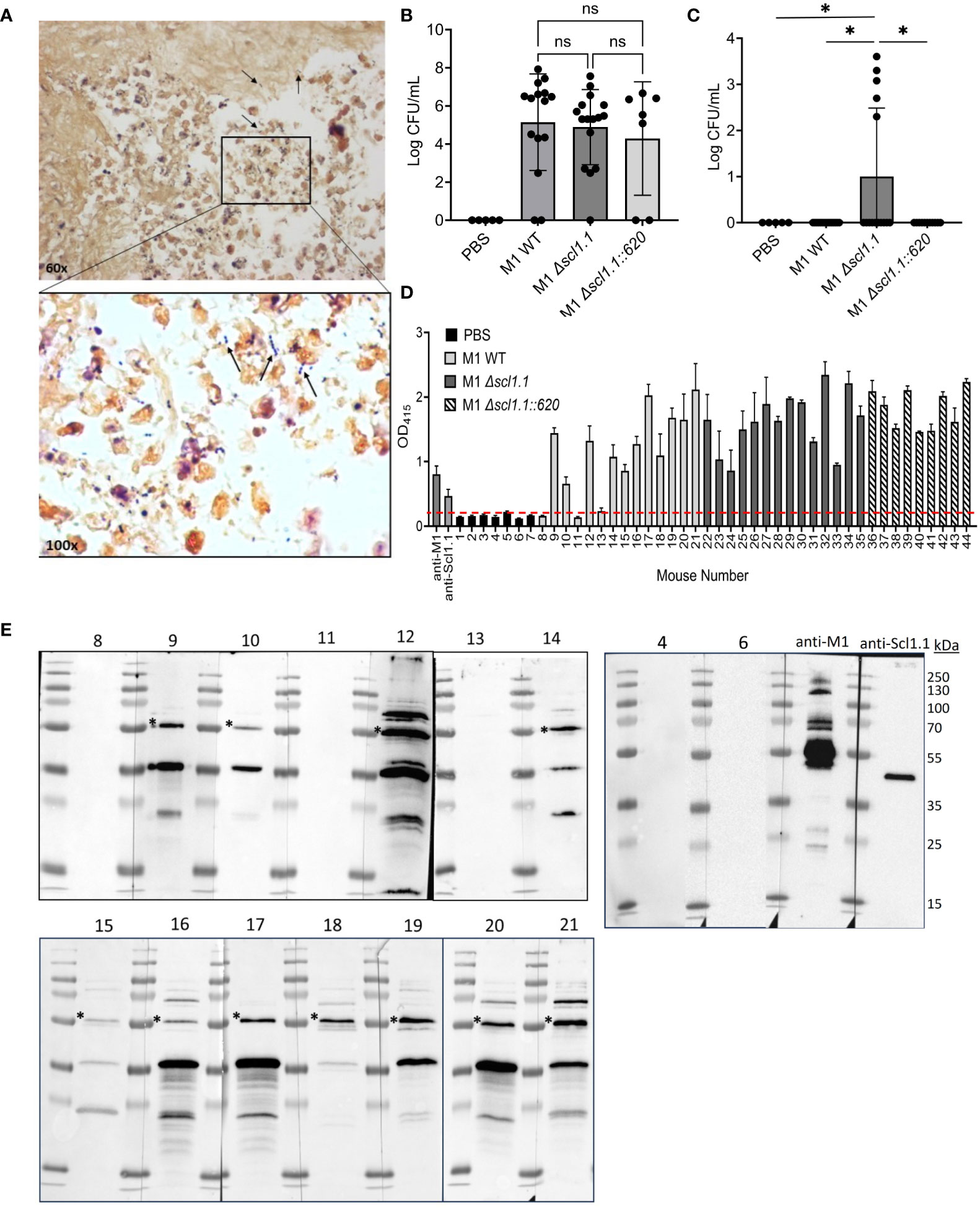
Figure 3 Scl1 promotes localized tumor colonization by GAS without eliciting strong humoral response. (A) Histopathology of GAS-treated KPC tumors. Representative Gram-stained image from the KPC tumor treated with GAS (600x magnification). Arrows indicate GAS (1000x magnification) viewed under immersion oil. (B) Detection of GAS in KPC tumors. Homogenates were prepared from tumors collected from mice treated with GAS or PBS and serial dilutions were cultured on blood agar, then enumerated. (C) Detection of GAS in spleen homogenates. Spleens were processed and analyzed for GAS presence as above (panel B) for tumor samples. Results are from 2-3 independent experiments. Significance is determined by one-way ANOVA. *p<0.05. (D) Mouse seropositivity for streptococcal cells developed during tumor colonization by whole-cell ELISA. Mouse sera (1:100 dil.) were from blood collected 14 days after single-dose treatment with either GAS M1 WT (Nos. 8-21; light gray bars), M1 Δscl1.1 (Nos. 22-35; dark gray bars), M1 Δscl1.1::620 (Nos. 36-44; striped bars) or PBS (Nos. 1-7; black bars) and tested for immunodetection of GAS surface antigens; specific anti-M1 (1:1000 dil.) and anti-Scl1.1 (1:1000 dil.) Abs were used as positive controls. Each bar represents the average of three technical replicates plus SD. Threshold is indicated by dashed line and was determined by averaging reading of PBS-treated mice at OD 415 nm. (E) Mouse seroconversion to GAS antigens developed during tumor colonization by western blot analysis. The same mouse sera (1:500 dil.) tested in D were reacted against the blots of the M1-GAS cell-wall protein fraction resolved by 15% SDS-PAGE. Asterisk indicates presumed M1-protein immunoreactive band. Specific anti-M1 (1:5000 dil.) and anti-Scl1.1 (1:1000 dil.) Abs were used as positive controls. ns, not statistically significant.
Consequently, we next asked whether 14-day-long tumor colonization was sufficient to induce host seroconversion toward M1 bacteria, which would demonstrate immune response to the bacterial injection. Mouse sera (1:100 dil.) collected at the endpoint of the experiment (day 14) following a single-dose injection of GAS or PBS control were screened for anti-M1 GAS immunoreactivity by whole-cell ELISA (Figure 3D; Supplementary Figure S4A). 11 out of 14 (79%) mouse sera collected from mice treated with wild-type bacteria (mice Nos. 8-21) – and none of 7 PBS controls (Nos. 1-7) - exhibited seroconversion to whole bacteria. Similarly, all sera collected from 14 mice treated with the Δscl1.1 mutant strain (Nos. 22-35) and 9 with Δscl1.1::620 complemented strain (Nos. 36-44) exhibited seroconversion to whole bacteria. However, there were no significant differences in responses to whole-cell bacteria between GAS-treated groups (Supplementary Figure S4A). All 14 sera Nos. 8-21 (1:500 dil.) from mice treated with the wild-type GAS plus 2 sera from PBS controls (Nos. 4 and 6) (Figure 3E), as well as sera from mice treated with Δscl1.1 mutant and with Δscl1.1::620 complemented strains (8 each) (Supplementary Figure S4B), were subsequently assessed for antibody reactivity against GAS WT cell-wall protein fraction by western immunoblotting. The same 11 mouse sera from the wild-type group, previously indicating anti-GAS seropositivity in whole-cell ELISA, consistently produced three major distinct immunoreactive bands of about 55 kDa, 35 kDa, and 20 kDa, with the 55-kDa band co-migrating with M1-protein control. As expected, PBS control sera and the remaining 3 sera from GAS-injected mice that were negative in whole-cell ELISA did not show immunoreactive bands against GAS cell wall antigen preparations. Sera from mice treated with the Δscl1.1 mutant strain and Δscl1.1::620 complemented strain also produced immunoreactive bands; however, band patterns were more variable, although nearly all showed a band co-migrating with M1-protein control. The lack of clear seroconversion to Scl1 antigen in sera from the wild-type group was surprising given that Scl1.1 expression, which is controlled by the positive GAS transcriptional regulator Mga (30, 50), is highly transcribed in M1-GAS (44) and has previously demonstrated to elicit humoral responses in both humans and mice (32). Therefore, we hypothesized that mouse sera may not react strongly to the preparation of the cell-wall protein fraction.
To test this hypothesis, representative mouse sera obtained from GAS-WT- and PBS-treated groups were analyzed (1:100 dil.) against purified rScl1.1 protein, instead of the cell-wall protein fraction (Supplementary Figure S5). In an ELISA assay, sera from several mice demonstrated immunogenic responses to Scl1.1 antigen, while the PBS mice did not (Supplementary Figure S5A). Sera (1:100 dil.) showing positivity in rScl1.1-coated wells, as well as combined control PBS sera, were subsequently analyzed at this concentration (higher than 1:500 dil. in Figure 3E) by western immunoblotting following rScl1.1 separation by SDS-PAGE. Under these experimental conditions we could detect bands immunoreactive for rScl1.1 protein (Supplementary Figure S5B), in-line with responses seen in the ELISA. Altogether, our data indicate that mice elicit humoral responses against M1-GAS antigens in vivo, which includes a relatively weak response against Scl1.1 antigen expressed during tumor colonization.
3.3 Intratumoral GAS infection is associated with neutrophil infiltration and Scl1-mediated inhibition of neutrophil extracellular trap formation
Given the profound Scl1-dependent anti-tumor effects of GAS bacteria in murine PDAC, we began to consider potential immunostimulatory mechanisms that could be driving this treatment response. We observed that tumors treated with M1 WT and Δscl1.1::620 complemented strains had higher neutrophil inflammatory indices of 2-3, scored by a blinded pathologist, compared to indices 1-2 recorded for the Δscl1.1 mutant strain and PBS control (Figures 4A, B). These group differences, e.g., Scl1.1-positive (WT and complemented strains) and Scl1.1-negative (PBS and Scl1.1-devoid mutant), may indicate that higher neutrophil indices observed in Scl1-positive groups could result from either higher PMN infiltration or the possibility of fewer NETs in these experimental groups (since NETs are a form of neutrophil cell death). Previous studies demonstrated that NETs can enhance pancreatic tumor growth (21) and promote metastasis (18). We hypothesized that Scl1.1 may be working, in part, through a neutrophil mediated mechanism to exert its anti-PDAC effects. Previously, Scl1.1 was found to reduce NET formation and killing in vitro (24). Therefore, we sought to further explore the role of Scl1.1 in cancer-promoting NETs. Bone marrow neutrophils were isolated and incubated with whole cell bacteria (M1 wild-type, an isogenic Scl1.1-devoid mutant Δscl1.1, and a complemented mutant strain Δscl1.1::620) followed by stimulation with the NET inducer platelet activating factor (PAF). NET production was quantified as a measurement of cell-free DNA (cfDNA) released into the supernatant. Neutrophils co-incubated with the Scl1.1-expressing bacteria produced significantly less cell-free DNA as compared to the Scl1.1-lacking mutant and PAF-only control, consistent with reduced NET formation (Figure 4C).
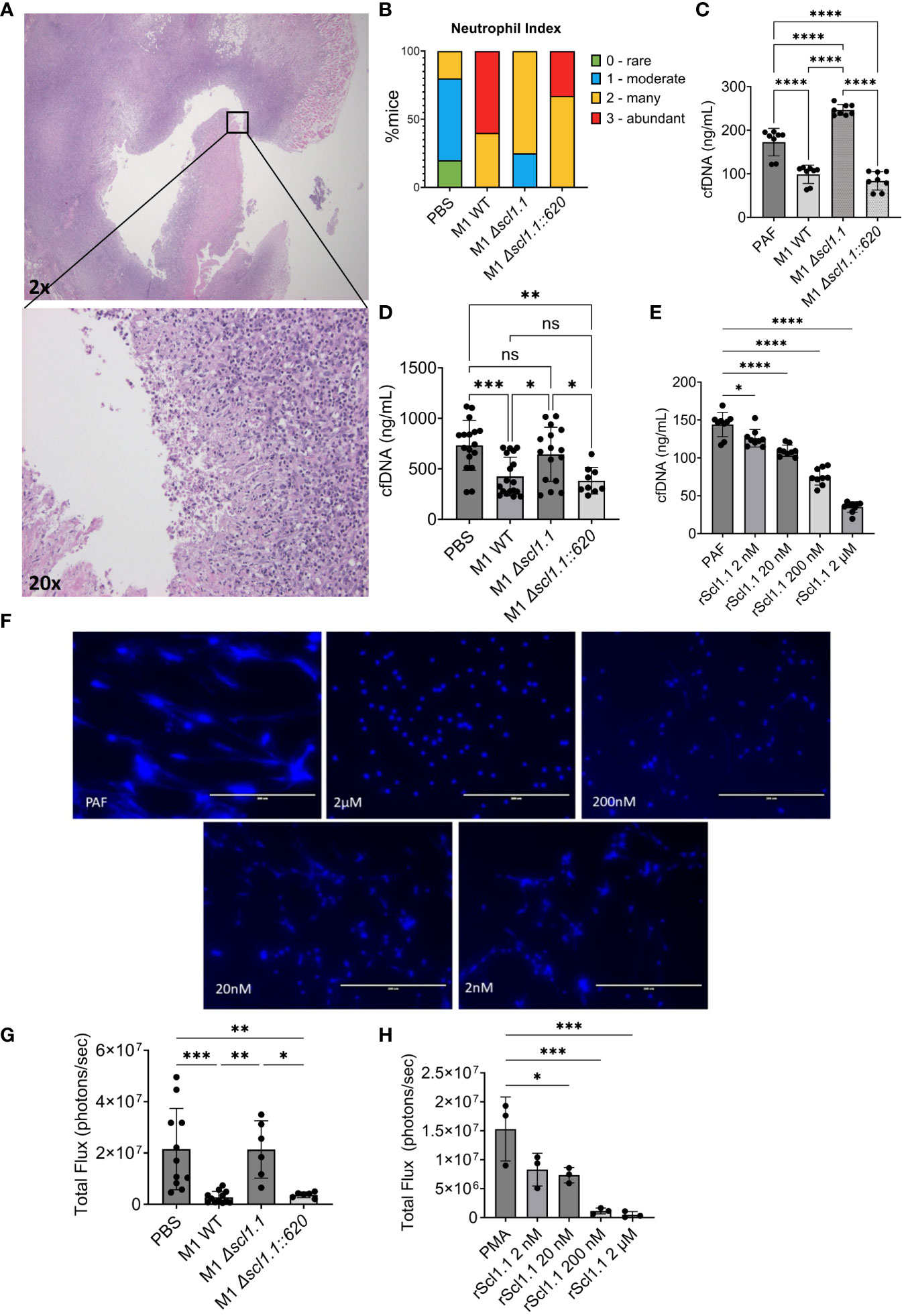
Figure 4 GAS-Scl1.1 impacts intratumoral neutrophil density in vivo while rScl1.1 inhibits neutrophil extracellular trap formation in vitro. (A) Neutrophil infiltration into M1 GAS treated KPC tumors. Representative H&E stains from tumor specimen at low magnification (top image, 20x total magnification) shows neutrophil infiltration at higher magnification (bottom image, 200x total magnification). (B) Neutrophil index in KPC tumors. PMN infiltration was scored in tumors from experimental groups harvested 14 days post-injection of a single dose of Scl1 GAS compared to PBS controls (C) NET inhibition induced by Scl1.1-expressing GAS. Ex vivo NET assay measuring cell-free DNA (cfDNA) concentration following co-incubation of neutrophils and designated bacterial strains quantified by Picogreen assay. Results are representative of three-independent experiments (eight technical replicates). Significance determined by one-way ANOVA with multiple comparison test. (D) NET inhibition induced by Scl1.1-expressing GAS in vivo. NET assay measuring cfDNA concentration in serum collected from mice treated with indicated strains of GAS or PBS control. Each point represents an individual mouse from two-independent experiments (n=9-18 total/group). Significance determined by one-way ANOVA with multiple comparisons test. (E) Quantification of NET inhibition by recombinant proteins. cfDNA concentration measured via Picogreen assay of neutrophils co-incubated with indicated concentrations of rScl1.1 followed by stimulation with PAF. Results are representative of two to three independent experiments (eight to nine technical replicates total. Significance determined by one-way ANOVA with multiple comparison test. (F) NET inhibition induced by rScl1.1 in vitro. Representative images of neutrophils co-incubated with or without rScl1.1 and stimulated with PAF. NETs are indicated by Hoechst staining. (G) MPO activity of neutrophils isolated from treated, tumor-bearing mice. Total flux as measurement of MPO activity of BMNs isolated from mice 14 days after injection of a single dose GAS treatment or PBS control, demonstrating that Scl1 mediates MPO activity from isolated neutrophils. Results are representative of two independent experiments (n=6 total). Significance is determined by one-way ANOVA and multiple comparison test. (H) MPO activity of neutrophils treated with rScl1.1. Total flux as measurement of MPO activity of murine BMNs co-incubated with rScl1.1 at indicated concentrations, showing a dose dependent inhibition of MPO. Results are representative of three independent experiments (9 technical replicates total). Each point represents the average area under curve of three technical replicates. Significance is determined by one-way ANOVA and multiple comparison test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ns, not statistically significant.
We sought to determine the efficacy of treatment with Scl1.1-expressing bacteria on circulating NETs in vivo. Sera were prepared from blood collected from mice 14 days following treatment with a single-dose intra-tumoral injection of M1 wild-type, an isogenic Δscl1.1 mutant, the complemented Δscl1.1::620 mutant strain, or PBS control. Sera (1:10) were assessed for cfDNA quantification, a validated marker of in vivo NET formation in murine PDAC (17). Mice treated with the wild-type and complemented strains had significantly reduced cfDNA levels in comparison to PBS and Scl1.1-lacking mutant, suggesting reduced circulating NETs (Figure 4D).
Because GAS has multiple antigenic influences and mechanisms with the potential to inhibit NET formation (14, 51–53), we deemed it necessary to confirm Scl1 as the major driver of NET evasion by employing recombinant Scl1 (rScl1) proteins (Supplementary Figure S2) derived from GAS M1 (rScl1.1), M28 (rScl1.28), and M41 (rScl1.41) strains that inhibited the growth of Panc02 tumors (Figure 1). Bone marrow neutrophils were isolated and incubated with indicated rScl1 proteins or rEDA control recombinant protein. NET release was assessed by measuring the cfDNA and microscopic imaging following fluorescent staining of DNA with Hoechst reagent. Co-incubation of neutrophils with all three distinct rScl1 protein variants significantly inhibited NET formation (Supplementary Figure S6). rScl1.1 had more potent NET reduction in comparison to other rScl1 proteins tested. Moreover, rScl1.1 inhibited NETs in a concentration-dependent manner reaching ~70% inhibition at 2 μM (Figures 4E, F). Overall, we demonstrate for the first time that recombinant Scl1 can inhibit NET formation by the same capability as whole cell bacteria, suggesting strong potential as a useful therapeutic approach for NET inhibition.
We next sought to determine the mechanism of Scl1.1 NET inhibition. Myeloperoxidase (MPO) activity is a critical step in NETosis whereby MPO stored in the azurophilic granules of neutrophils interacts with neutrophil elastase to induce chromatin decondensation and subsequent NET release (54). Previous studies demonstrated that Scl1.1 expressed on M1-type GAS suppresses NET formation through inhibition of MPO activity in vitro (24). However, at the current time of publication, these results have not been established in cancer models. Here, we hypothesized that treatment with Scl1-expressing GAS limits MPO activity and subsequent NET formation from neutrophils harvested from our murine model using a novel ex vivo bioluminescence assay. Bone marrow neutrophils isolated from mice treated with GAS M1-wild type, the isogenic Scl1.1-negative mutant and a complemented Scl1.1-positive strain, or PBS control were stained with the bioluminescence compound luminol, which measures MPO activity (55) and stimulated with phorbol myristate acetate (PMA), a compound known to induce activation of neutrophil NADPH-oxidase and trigger degranulation (55). Neutrophils isolated from mice treated with the Scl1.1 expressing wild-type and complemented strains exhibited significantly less MPO activity compared to neutrophils from PBS controls or treated with the Scl1.1-negative mutant strain (Figure 4G), validating that Scl1.1 reduces MPO activity.
We next assessed if rScl1.1 protein could elicit similar reductions in MPO activity as seen in our murine model using live bacteria. As described, bone marrow neutrophils were isolated and co-incubated with rScl1.1 at different concentrations followed by luminol staining and PMA stimulation. MPO activity was significantly reduced in neutrophils treated with rScl1.1 in a concentration-dependent manner (Figure 4H). To validate these results, we assessed rScl1.1 in a commercially available MPO inhibitor screening assay, demonstrating that rScl1.1 inhibited MPO production even at low concentrations of protein (Supplementary Figure S7). Taken together, these results indicate a potential mechanism by which Scl1 inhibits NET formation by hindering MPO activity and offers therapeutic value to rScl1.1.
4 Discussion
PDAC is an aggressive cancer characterized by low survival due to failure of early detection and marked treatment resistance. Despite recent advances in cytotoxic regimens, current therapeutic interventions offer minimal treatment response or survival benefit (56), which necessitates exploration into alternative strategies. William Coley established the significance of GAS in treating bone and soft-tissue sarcomas. However, discrepancies in Coley’s work and the development of radiation and chemotherapy brought disfavor towards the use of Coley’s Toxin (7). In the present work, we elucidate the significance of GAS as a potent anti-cancer agent in PDAC through inhibition of cancer promoting NETosis and re-examine the therapeutic value of bacteriotherapy using GAS.
GAS and other bacteria are attractive candidates for cancer therapy in their capabilities to be recognized by immune cells even in immunosuppressive tumor environments (8). This recognition has the capacity to switch this “immunologically cold” environment to a more immunostimulatory one (57). As demonstrated by Maletzki et al. (2008) using Panc02 subcutaneous model, a single intra-tumoral injection of M49-type GAS can mediate anti-tumor effects through direct tumor lysis and stimulation of anti-tumor immunity by active infection. In the present study, injection with M1, M28, and M41 GAS each significantly reduced Panc02 tumor growth. Moreover, a subsequent use of the M1 GAS led to tumor reduction of a more aggressive and clinically relevant cell lineage such as the KPCY6422c1 line, which solidifies this concept and offers greater potential for clinical translation.
A hallmark of PDAC is an immunosuppressive tumor micro-environment that renders tumors resistant to current immunotherapies (58, 59). Existing literature demonstrates the contribution of NETs to this immunologically cold state (60, 61) and their enhancement to cancer progression (17, 62, 63) and reduced patient survival (23, 64). Thus, NETs are encouraging targets for further cancer therapies. The current work strengthens existing data generated in a non-cancer inflammatory model to suggest that Scl1 on GAS cells is a potent NET inhibitor through reduction in MPO activity (14, 24). Based on these findings, we tested three recombinant Scl1 proteins - derived from GAS M types above inhibiting PDAC growth - rScl1.1, rScl.28, and rScl1.41 for potential NET inhibition. Neutrophils co-incubated with those rScl1 constructs had a decreased propensity to form NETs, as indicated by reduced cell-free DNA concentrations similar to the effects obtained with Scl1-expressing live bacteria. Further, rScl1.1 inhibited MPO activity indicated by an in vitro luminol assay. Taken together these results suggest that rScl1 reagents function in a comparable manner as their native proteins expressed in live bacteria. This original finding potentiates rScl1 as a conceivable PDAC therapeutic agent to combat concerns accompanying live and whole bacterial therapy and represents an innovative advance in the field, as recombinant Scl1 has not been previously tested to inhibit cancer-associated NET formation.
Our previous work reported that Scl1 selectively binds to cancer-associated fibroblast (CAF)-deposited oncofetal fibronectin (47, 48, 65), which can modulate the tumor extracellular matrix microenvironment (49). Here, we observed that injection of Scl1.1-expressing GAS was associated with tumor-localized nidus of infection persisting for an extended time, whereas Scl1.1-lacking isogenic mutant had greater systemic bacterial dissemination. Despite these encouraging results in preclinical models in the current study, injection of live GAS may not be a translatable long-term strategy in humans given the potential for pathogenic infection or would require extensive investigations to generate and test attenuated strains still retaining anti-PDAC activity. Our findings implicated Scl1 in the therapeutic efficacy of GAS, with Scl1-devoid mutant strain demonstrating no significant treatment response comparable to PBS control. Taken together with the evidence demonstrating robust neutrophil infiltration at sites of tumor colonization by GAS, these results potentiate the capacity of Scl1 to counteract the immunotolerant environment and function as an anti-cancer therapeutic. Given the observed activity of Scl1 from our previous work (47, 65), the anti-tumor effects observed by Scl1 are likely to be multifactorial. Therefore, further analysis isolating the role of NET inhibition and elucidating specific immune infiltrates and inflammatory queues will offer a more mechanistic approach to characterizing immunomodulatory responses within the tumor microenvironment in response to GAS bacteriotherapy.
Of interest when considering future therapeutic strategies, we observed host immunity to GAS antigens, thus, increasing the likelihood of resolution of inflammation and the need for additional treatment administration to induce a durable response. Repeat treatment within 3 weeks is well in line with currently utilized cytotoxic treatment regimens and is clinically realistic. Experimental treatments utilizing other bacteria, such as Listeria (66), show promise and require multiple rounds of injection as well. However, this observation expresses a need to understand specific host-immune responses to bacterial antigens to identify proper dosing. Previous work demonstrating antigenicity and humoral responses against Scl1.1 (32) in addition to the capacity of M1-type GAS to strongly transcribe the scl1.1 gene in vitro (44–46) suggests that injection with M1-type strain could induce robust immune responses to both M1 and Scl1.1 proteins, although this concept was not explored in a tumoral setting. Herein, we report that mice seroconvert to live bacteria, as evidenced by the positive immunoreactivity detected in sera prepared from mouse blood collected 14 days post-GAS injection via whole-cell ELISA and western immunoblotting. While this finding is not surprising given the prominent antigenicity of M proteins (33, 67) and Scl1.1 (32), it poses potential challenges to future studies examining the efficacy of repeated GAS therapy. However, highly immunogenic antigens are beneficial in the context of vaccines (68). Therefore, we cannot exclude the possibility of utilizing these proteins in cancer vaccine development to elicit potent antitumor immunity. We expected Scl1.1 being expressed in vivo, as a Mga-regulated protein (30, 50). Surprisingly, western blot analysis did not detect immunoreactive band corresponding to Scl1.1 control in serum (1:500 dil.) sample set during initial testing against M1-GAS cell-wall preparation. However, subsequent analysis utilizing a purified rScl1.1 protein and higher concentration of mouse immune sera (1:100 dil.) demonstrated that mice do seroconvert to Scl1.1 antigen, though this response seemed weakly immunogenic under these experimental conditions. The tumor microenvironment is highly complex and immunosuppressive, and the anti-tumor effects demonstrated herein are suggested to be Scl1-mediated. Therefore, identifying the complex immunological responses within the tumor microenvironment is necessary to determine immunotolerance at later intervals of treatment. Given the lack of seroconversion demonstrated in these early studies, future studies investigating the therapeutic efficacy of Scl1.1 can be conducted utilizing rScl1.1 or engineered non-pathogenic or attenuated bacteria expressing Scl1.1, as opposed to live M1-WT GAS.
In conclusion, we demonstrate a profound anti-tumor effect of several GAS strains, validated in two PDAC model cell lines. We also identify a novel role for Scl1.1 in PDAC therapy and provide insight on future applications utilizing rScl1.1 to target cancer-promoting NETs, which is the subject of ongoing research. We also address limitations of current and future bacteriotherapies through analysis of seropositivity. Taken together, Scl1.1 could be a beneficial therapeutic approach in PDAC that warrants additional studies.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The animal study was approved by Institutional Animal Care and Use Committee of West Virginia University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
EAH: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. AI: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. SJC: Investigation, Writing – review & editing. SS: Visualization, Writing – review & editing, Conceptualization. DHM: Methodology, Writing – review & editing. TL: Methodology, Writing – original draft, Writing – review & editing, Conceptualization. SL: Conceptualization, Funding acquisition, Investigation, Supervision, Writing – original draft, Writing – review & editing. BB: Conceptualization, Funding acquisition, Investigation, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work is supported by US DHHS-NIH-National Cancer Institute R21CA267302 (SL, BB). Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number 5U54GM104942-04 (BB) and 5P20GM121322 (BB). This work was supported in part by the West Virginia University Health Sciences Center, Office of Research and Graduate Education and the Office of the Chancellor and Executive Dean, HSC Multi-Investigator Synergy Grant (BB and SL). Tissue processing is supported by the WVU Department of Pathology, Anatomy, & Laboratory Medicine and NIH grant U54GM1049420.
Acknowledgments
We acknowledge James (Jim) Dale, Division of Infectious Diseases at the University of Tennessee Health Science Center for the anti-M protein 30-valent antibodies used as controls in the ELISA and western blots. We also thank all additional members of the Liu, Lukomski, and Boone labs for their assistance in experiments. Some figures were configured using Biorender.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1363962/full#supplementary-material
References
1. Cancer of the Pancreas - Cancer Stat Facts . SEER. Available online at: https://seer.cancer.gov/statfacts/html/pancreas.html (Accessed November 9, 2023).
2. Cronin KA, Scott S, Firth AU, Sung H, Henley SJ, Sherman RL, et al. Annual report to the nation on the status of cancer, part 1: national cancer statistics. Cancer. (2022) 128:4251–84. doi: 10.1002/cncr.34479
3. Fesinmeyer MD, Austin MA, Li CI, De Roos AJ, Bowen DJ. Differences in survival by histologic type of pancreatic cancer. Cancer Epidemiol Biomarkers Prev. (2005) 14:1766–735. doi: 10.1158/1055-9965.EPI-05-0120
4. Sarantis P, Koustas E, Papadimitropoulou A, Papavassiliou AG, Karamouzis MV. Pancreatic ductal adenocarcinoma: treatment hurdles, tumor microenvironment and immunotherapy. World J Gastrointest Oncol. (2020) 12:173–815. doi: 10.4251/wjgo.v12.i2.173
5. Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med. (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
6. Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 trial of single agent ipilimumab (Anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother (Hagerstown Md. : 1997). (2010) 33:828–33. doi: 10.1097/CJI.0b013e3181eec14c
7. McCarthy EF. The toxins of william B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthopaedic J. (2006) 26:154–58.
8. Henderson EA, Lukomski S, Boone BA. Emerging applications of cancer bacteriotherapy towards treatment of pancreatic cancer. Front Oncol. (2023) 13:1217095. doi: 10.3389/fonc.2023.1217095
9. Chernov AN, Tsapieva A, Alaverdian DA, Filatenkova TA, Galimova ES, Suvorova M, et al. In vitro evaluation of the cytotoxic effect of Streptococcus pyogenes strains, protegrin PG-1, cathelicidin LL-37, nerve growth factor and chemotherapy on the C6 glioma cell line. Molecules. (2022) 27:5695. doi: 10.3390/molecules27020569
10. Patterson KG, Pittaro JLD, Bastedo PS, Hess DA, Haeryfar SMM, McCormick JK. Control of established colon cancer xenografts using a novel humanized single chain antibody-streptococcal superantigen fusion protein targeting the 5T4 oncofetal antigen. PloS One. (2014) 9:e952005. doi: 10.1371/journal.pone.0095200
11. Tian Y-F, Tang K, Guan W, Yang T, Xu H, Zhuang Q-Y, et al. OK-432 suppresses proliferation and metastasis by tumor associated macrophages in bladder cancer. Asian Pacific J Cancer Prev. (2015) 16:4537–425. doi: 10.7314/APJCP.2015.16.11.4537
12. Ludwig Institute for Cancer Research. Safety and Immunogenicity of Cholesterol-bearing Hydrophobized Pullulan HER2 Protein 146 (CHP-HER2) and NY-ESO-1 Protein (CHP-NY-ESO-1) in Combination With OK-432 in HER2- and/or NY-ESO-1-Expressing Cancers. Clinical trial registration NCT00291473. Mie University Hospital, Tsu, Mie, Japan (2009). Available at: https://clinicaltrials.gov/study/NCT00291473.
13. Kenjo A. Phase I/II Study on Intratumor Dendritic Cell Injection Immunotherapy Using Immature Dendritic Cells with S. pyogenes Preparation (OK-432) for Patients With Resectable Pancreatic Cancer. Clinical trial registration NCT00795977. Fukushima Medical University Hospital, Fukushima, Japan. (2011). Available at: https://clinicaltrials.gov/study/NCT00795977.
14. Ríos-López AL, González GM, Hernández-Bello R, Sánchez-González A. Avoiding the trap: mechanisms developed by pathogens to escape neutrophil extracellular traps. Microbiol. Res. (2021) 243:126644. doi: 10.1016/j.micres.2020.126644
15. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–35. doi: 10.1126/science.1092385
16. Boone BA, Murthy P, Miller-Ocuin J, Doerfler WR, Ellis JT, Liang X, et al. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer. (2018) 18:678. doi: 10.1186/s12885-018-4584-2
17. Boone BA, Orlichenko L, Schapiro NE, Loughran P, Gianfrate GC, Ellis JT, et al. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther. (2015) 22:326–34. doi: 10.1038/cgt.2015.21
18. Chen Y, Hu H, Tan S, Dong Q, Fan X, Wang Yi, et al. The role of neutrophil extracellular traps in cancer progression, metastasis and therapy. Exp Hematol Oncol. (2022) 11:995. doi: 10.1186/s40164-022-00345-3
19. Jung HS, Gu J, Kim J-E, Nam Y, Song JW, Kim HK. Cancer cell–induced neutrophil extracellular traps promote both hypercoagulability and cancer progression. PloS One. (2019) 14:e02160555. doi: 10.1371/journal.pone.0216055
20. Mitsis M, Drosou P, Tatsis V, Markopoulos GS. Neutrophil extracellular traps and pancreatic cancer development: A vicious cycle. Cancers. (2022) 14:33395. doi: 10.3390/cancers14143339
21. Miller-Ocuin JL, Liang X, Boone BA, Doerfler WR, Singhi AD, Tang D, et al. DNA released from neutrophil extracellular traps (NETs) activates pancreatic stellate cells and enhances pancreatic tumor growth. Oncoimmunology. (2019) 8:e16058225. doi: 10.1080/2162402X.2019.1605822
22. Chen X, Ma H, Mo S, Yu S, Lu Z, Chen J. Intratumoral neutrophil extracellular traps are associated with unfavorable clinical outcomes and immunogenic context in pancreatic ductal adenocarcinoma. Front Immunol. (2022) 13:1027459. doi: 10.3389/fimmu.2022.1027459
23. Jin W, Xu H-X, Zhang S-R, Li H, Wang W-Q, Gao H-L, et al. Tumor-infiltrating NETs predict postsurgical survival in patients with pancreatic ductal adenocarcinoma. Ann Surg Oncol. (2019) 26:635–43. doi: 10.1245/s10434-018-6941-4
24. Döhrmann S, Anik S, Olson J, Anderson EL, Etesami N, No H, et al. Role for streptococcal collagen-like protein 1 in M1T1 group A Streptococcus resistance to neutrophil extracellular traps. Infect Immun. (2014) 82:4011–205. doi: 10.1128/IAI.01921-14
25. Tumor Volume Measurements by Calipers . Biopticon. Available online at: https://biopticon.com/resources/tumor-volume-measurements-by-calipers/ (Accessed December 6, 2023).
26. McNitt DH, Choi SJ, Keene DR, De Water LV, Squeglia F, Berisio R, et al. Surface-exposed loops and an acidic patch in the scl1 protein of group A Streptococcus enable scl1 binding to wound-associated fibronectin. J Biol Chem. (2018) 293:7796–78105. doi: 10.1074/jbc.RA118.002250
27. Xu Y, Keene DR, Bujnicki JM, Höök M, Lukomski S. Streptococcal scl1 and scl2 proteins form collagen-like triple helices. J Biol Chem. (2002) 277:27312–185. doi: 10.1074/jbc.M201163200
28. Lukomski S, McNitt DH. Expression and purification of collagen-like proteins of group A Streptococcus. Methods Mol Biol (Clifton N.J.). (2020) 2136:163–79. doi: 10.1007/978-1-0716-0467-0_12
29. Shinde AV, Bystroff C, Wang C, Vogelezang MG, Vincent PA, Hynes RO, et al. Identification of the peptide sequences within the EIIIA (EDA) segment of fibronectin that mediate integrin α9β1-dependent cellular activities. J Biol Chem. (2008) 283:2858–705. doi: 10.1074/jbc.M708306200
30. Lukomski S, Nakashima K, Abdi I, Cipriano VJ, Ireland RM, Reid SD, et al. Identification and characterization of the scl gene encoding a group A Streptococcus extracellular protein virulence factor with similarity to human collagen. Infect Immun. (2000) 68:6542–535. doi: 10.1128/IAI.68.12.6542-6553.2000
31. Elder BL, Boraker DK, Fives-Taylor PM. Whole-bacterial cell enzyme-linked immunosorbent assay for Streptococcus sanguis fimbrial antigens. J Clin Microbiol. (1982) 16:141–44. doi: 10.1128/jcm.16.1.141-144.1982
32. Hoe NP, Lukomska E, Musser JM, Lukomski S. Characterization of the immune response to collagen-like proteins scl1 and scl2 of serotype M1 and M28 group A Streptococcus. FEMS Microbiol Lett. (2007) 277:142–495. doi: 10.1111/j.1574-6968.2007.00955.x
33. Dale JB, Penfound TA, Chiang EY, Walton WJ. New 30-valent M protein-based vaccine evokes cross-opsonic antibodies against non-vaccine serotypes of group A streptococci. Vaccine. (2011) 29:8175–785. doi: 10.1016/j.vaccine.2011.09.005
34. Swamydas M, Luo Y, Dorf ME, Lionakis MS. Isolation of mouse neutrophils. Curr Protoc Immunol. (2015) 110:3.20.1–3.20.15. doi: 10.1002/0471142735.im0320s110
35. Liu TW, Gammon ST, Yang P, Ma W, Wang J, Piwnica-Worms D. Inhibition of myeloperoxidase enhances immune checkpoint therapy for melanoma. J Immunother Cancer. (2023) 11:e0058375. doi: 10.1136/jitc-2022-005837
36. Gross S, Gammon ST, Moss BL, Rauch D, Harding J, Heinecke JW, et al. Bioluminescence imaging of myeloperoxidase activity in vivo. Nat Med. (2009) 15:455–615. doi: 10.1038/nm.1886
37. Linnebacher M, Maletzki C, Emmrich J, Kreikemeyer B. Lysates of S. pyogenes serotype M49 induce pancreatic tumor growth delay by specific and unspecific antitumor immune responses. J Immunother. (2008) 31:704–7135. doi: 10.1097/CJI.0b013e3181829f62
38. Maletzki C, Linnebacher M, Kreikemeyer B, Emmrich J. Pancreatic cancer regression by intratumoural injection of live streptococcus pyogenes in a syngeneic mouse model. Gut. (2008) 57:4835. doi: 10.1136/gut.2007.125419
39. Jančík S, Drábek J, Radzioch D, Hajdúch M. Clinical relevance of KRAS in human cancers. J Biomed Biotechnol. (2010) 2010:150960. doi: 10.1155/2010/150960
40. McCubrey JA, Yang L, Abrams SL, Steelman LS, Follo MY, Cocco L, et al. Effects of TP53 mutations and miRs on immune responses in the tumor microenvironment important in pancreatic cancer progression. Cells. (2022) 11:21555. doi: 10.3390/cells11142155
41. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and krasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. (2005) 7:469–835. doi: 10.1016/j.ccr.2005.04.023
42. Lukomski S, Bachert BA, Squeglia F, Berisio R. Collagen-like proteins of pathogenic streptococci. Mol Microbiol. (2017) 103:919–305. doi: 10.1111/mmi.13604
43. Rasmussen M, Edén A, Björck L. SclA, a novel collagen-like surface protein of streptococcus pyogenes. Infect Immun. (2000) 68:6370–775. doi: 10.1128/IAI.68.11.6370-6377.2000
44. Bachert BA, Choi SJ, LaSala PR, Harper TI, McNitt DH, Boehm DT, et al. Unique footprint in the scl1.3 locus affects adhesion and biofilm formation of the invasive M3-type group A streptococcus. Front Cell Infect Microbiol. (2016) 6:90. doi: 10.3389/fcimb.2016.00090
45. Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. Genome-wide analysis of group a streptococci reveals a mutation that modulates global phenotype and disease specificity. PloS Pathog. (2006) 2:e55. doi: 10.1371/journal.ppat.0020005
46. Tsatsaronis JA, Hollands A, Cole JN, Maamary PG, Gillen CM, Zakour NLB, et al. Streptococcal collagen-like protein A and general stress protein 24 are immunomodulating virulence factors of group A streptococcus. FASEB J. (2013) 27:2633–43. doi: 10.1096/fj.12-226662
47. McNitt DH, Choi SJ, Allen JL, Hames RA, Weed SA, De Water LV, et al. Adaptation of the group A Streptococcus adhesin scl1 to bind fibronectin type III repeats within wound-associated extracellular matrix: implications for cancer therapy. Mol Microbiol. (2019) 112:800–8195. doi: 10.1111/mmi.14317
48. Oliver-Kozup H, Martin KH, Schwegler-Berry D, Green BJ, Betts C, Shinde AV, et al. The group A streptococcal collagen-like protein-1, scl1, mediates biofilm formation by targeting the extra domain A-containing variant of cellular fibronectin expressed in wounded tissue. Mol Microbiol. (2013) 87:672–895. doi: 10.1111/mmi.12125
49. Gopal S, Veracini L, Grall D, Butori C, Schaub S, Audebert S, et al. Fibronectin-guided migration of carcinoma collectives. Nat Commun. (2017) 8:14105. doi: 10.1038/ncomms14105
50. Almengor AC, McIver KS. Transcriptional activation of sclA by mga requires a distal binding site in streptococcus pyogenes. J Bacteriol. (2004) 186:7847–575. doi: 10.1128/JB.186.23.7847-7857.2004
51. Kristian SA, Datta V, Weidenmaier C, Kansal R, Fedtke I, Peschel A, et al. D-alanylation of teichoic acids promotes group A streptococcus antimicrobial peptide resistance, neutrophil survival, and epithelial cell invasion. J Bacteriol. (2005) 187:6719–255. doi: 10.1128/jb.187.19.6719-6725.2005
52. Cole JN, Pence MA, Köckritz-Blickwede Mv, Hollands A, Gallo RL, Walker MJ, et al. M protein and hyaluronic acid capsule are essential for in vivo selection of covRS mutations characteristic of invasive serotype M1T1 group A Streptococcus. mBio. (2010) 1:105. doi: 10.1128/mbio.00191-10
53. Uchiyama S, Andreoni F, Schuepbach RA, Nizet V, Zinkernagel AS. DNase sda1 allows invasive M1T1 group A Streptococcus to prevent TLR9-dependent recognition. PloS Pathog. (2012) 8:e10027365. doi: 10.1371/journal.ppat.1002736
54. Rizo-Téllez SA, Sekheri M, Filep JG. Myeloperoxidase: regulation of neutrophil function and target for therapy. Antioxidants. (2022) 11:23025. doi: 10.3390/antiox11112302
55. Damascena HL, Silveira WAA, Castro MS, Fontes W. Neutrophil activated by the famous and potent PMA (Phorbol myristate acetate). Cells. (2022) 11:28895. doi: 10.3390/cells11182889
56. Principe DR, Underwood PW, Korc M, Trevino JG, Munshi HG, Rana A. The current treatment paradigm for pancreatic ductal adenocarcinoma and barriers to therapeutic efficacy. Front Oncol. (2021) 11:688377. doi: 10.3389/fonc.2021.688377
57. Huang X, Pan J, Xu F, Shao B, Wang Y, Guo X, et al. Bacteria-based cancer immunotherapy. Adv Sci. (2021) 8:20035725. doi: 10.1002/advs.202003572
58. Avşar G, Pi̇r P. An integrated study to decipher immunosuppressive cellular communication in the PDAC environment. NPJ Syst Biol Appl. (2023) 9:1–13. doi: 10.1038/s41540-023-00320-6
59. Steele NG, Carpenter ES, Kemp SB, Sirihorachai V, The S, Delrosario L, et al. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nat Cancer. (2020) 1:1097–112. doi: 10.1038/s43018-020-00121-4
60. Fang Q, Stehr AM, Naschberger E, Knopf J, Herrmann M, Stürzl M. No NETs no TIME: crosstalk between neutrophil extracellular traps and the tumor immune microenvironment. Front Immunol. (2022) 13:1075260. doi: 10.3389/fimmu.2022.1075260
61. Kaltenmeier C, Yazdani HO, Morder K, Geller DA, Simmons RL, Tohme S. Neutrophil extracellular traps promote T cell exhaustion in the tumor microenvironment. Front Immunol. (2021) 12:785222. doi: 10.3389/fimmu.2021.785222
62. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. (2013) 123:3446–585. doi: 10.1172/JCI67484
63. Spicer JD, McDonald B, Cools-Lartigue JJ, Chow SC, Giannias B, Kubes P, et al. Neutrophils promote liver metastasis via mac-1–mediated interactions with circulating tumor cells. Cancer Res. (2012) 72:3919–275. doi: 10.1158/0008-5472.CAN-11-2393
64. Schoeps B, Eckfeld C, Prokopchuk O, Bottcher J, Haubler D, Steiger K, et al. TIMP1 triggers neutrophil extracellular trap formation in pancreatic cancer. Cancer Res. (2021) 81:3568–79. doi: 10.1158/0008-5472.CAN-20-4125
65. Caswell CC, Oliver-Kozup H, Han R, Lukomska E, Lukomski S. Scl1, the multifunctional adhesin of group A Streptococcus, selectively binds cellular fibronectin and laminin, and mediates pathogen internalization by human cells. FEMS Microbiol Lett. (2010) 303:61–685. doi: 10.1111/j.1574-6968.2009.01864.x
66. Quispe-Tintaya W, Chandra D, Jahangir A, Harris M, Casadevall A, Dadachova E, et al. Nontoxic radioactive listeria(at) is a highly effective therapy against metastatic pancreatic cancer. Proc Natl Acad Sci USA. (2013) 110:8668–735. doi: 10.1073/pnas.1211287110
67. Metzgar D, Zampolli A. The M protein of group A Streptococcus is a key virulence factor and a clinically relevant strain identification marker. Virulence. (2011) 2:402–4125. doi: 10.4161/viru.2.5.16342
Keywords: pancreatic cancer, neutrophil extracellular traps, bacteriotherapy, group A Streptococcus, myeloperoxidase
Citation: Henderson EA, Ivey A, Choi SJ, Santiago S, McNitt D, Liu TW, Lukomski S and Boone BA (2024) Group A streptococcal collagen-like protein 1 restricts tumor growth in murine pancreatic adenocarcinoma and inhibits cancer-promoting neutrophil extracellular traps. Front. Immunol. 15:1363962. doi: 10.3389/fimmu.2024.1363962
Received: 31 December 2023; Accepted: 14 February 2024;
Published: 07 March 2024.
Edited by:
Ping Wang, Michigan State University, United StatesReviewed by:
Hamza Yazdani, University of Pittsburgh, United StatesJerry Malayer, Oklahoma State University, United States
Copyright © 2024 Henderson, Ivey, Choi, Santiago, McNitt, Liu, Lukomski and Boone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian A. Boone, YnJpYW4uYm9vbmVAaHNjLnd2dS5lZHU=; Slawomir Lukomski, c2x1a29tc2tpQGhzYy53dnUuZWR1
†These authors have contributed equally to this work