 Jonatan J. Carvajal1†
Jonatan J. Carvajal1† Valeria García-Castillo1†
Valeria García-Castillo1† Shelsy V. Cuellar1Claudia P. Campillay-Véliz2
Shelsy V. Cuellar1Claudia P. Campillay-Véliz2 Camila Salazar-Ardiles3
Camila Salazar-Ardiles3 Andrea M. Avellaneda1,4
Andrea M. Avellaneda1,4 Christian A. Muñoz5,6,7
Christian A. Muñoz5,6,7 Angello Retamal-Díaz1,5,7
Angello Retamal-Díaz1,5,7 Susan M. Bueno8
Susan M. Bueno8 Pablo A. González8
Pablo A. González8 Alexis M. Kalergis8,9
Alexis M. Kalergis8,9 Margarita K. Lay1,5,7*
Margarita K. Lay1,5,7*- 1Department of Biotechnology, Faculty of Marine Sciences and Biological Resources, University of Antofagasta, Antofagasta, Chile
- 2Department of Pharmacology, Faculty of Biological Sciences, University of Concepción, Concepción, Chile
- 3Center for Research in Physiology and Altitude Medicine (FIMEDALT), Biomedical Department, Faculty of Health Sciences, University of Antofagasta, Antofagasta, Chile
- 4Department of Basic Sciences, Faculty of Sciences, Universidad Santo Tomás, Antofagasta, Chile
- 5Research Center in Immunology and Biomedical Biotechnology of Antofagasta (CIIBBA), University of Antofagasta, Antofagasta, Chile
- 6Department of Medical Technology, Faculty of Health Sciences, University of Antofagasta, Antofagasta, Chile
- 7Millennium Institute on Immunology and Immunotherapy, Department of Biotechnology, Faculty of Marine Sciences and Biological Resources, Department of Medical Technology, Faculty of Health Sciences, University of Antofagasta, Antofagasta, Chile
- 8Millennium Institute on Immunology and Immunotherapy, Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile, Santiago, Chile
- 9Departamento de Endocrinología, Facultad de Medicina, Pontificia Universidad Católica de Chile, Santiago, Chile
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is responsible for the respiratory distress condition known as COVID-19. This disease broadly affects several physiological systems, including the gastrointestinal, renal, and central nervous (CNS) systems, significantly influencing the patient’s overall quality of life. Additionally, numerous risk factors have been suggested, including gender, body weight, age, metabolic status, renal health, preexisting cardiomyopathies, and inflammatory conditions. Despite advances in understanding the genome and pathophysiological ramifications of COVID-19, its precise origins remain elusive. SARS-CoV-2 interacts with a receptor-binding domain within angiotensin-converting enzyme 2 (ACE2). This receptor is expressed in various organs of different species, including humans, with different abundance. Although COVID-19 has multiorgan manifestations, the main pathologies occur in the lung, including pulmonary fibrosis, respiratory failure, pulmonary embolism, and secondary bacterial pneumonia. In the post-COVID-19 period, different sequelae may occur, which may have various causes, including the direct action of the virus, alteration of the immune response, and metabolic alterations during infection, among others. Recognizing the serious adverse health effects associated with COVID-19, it becomes imperative to comprehensively elucidate and discuss the existing evidence surrounding this viral infection, including those related to the pathophysiological effects of the disease and the subsequent consequences. This review aims to contribute to a comprehensive understanding of the impact of COVID-19 and its long-term effects on human health.
1 Introduction
In December 2019, a novel coronavirus was identified by sequencing nasopharyngeal samples from patients experiencing severe pneumonia in Wuhan City, China. Initially designated as “2019-nCoV,” subsequent sequence analysis classified it as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). This virus is the cause of Coronavirus Acute Respiratory Disease 2019 (COVID-19). Due to its heightened transmissibility and asymptomatic cases, SARS-CoV-2 rapidly disseminated globally, prompting its declaration as a pandemic on March 11, 2020 (1–3). This virus enters the body through the upper respiratory tract, mainly affecting the lungs. However, this pathogen can affect different systems, such as the gastrointestinal, renal, and Central Nervous Systems (CNS). Four years after the first reported case, the World Health Organization (WHO) has notified more than 771,820,937 confirmed cases; 6,978,175 deaths due to SARS-CoV-2 infection; and 13,534,474,309 doses of COVID-19 vaccines distributed globally. The fatality rate of SARS-CoV-2 is estimated to be between 1 to 5%, which is significantly lower when compared to the rate of severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1) and the Middle East Respiratory Syndrome coronavirus (MERS-CoV), which are 9.7% and 34%, respectively (4).
Most recently, in May 2023, the World Health Organization (WHO) declared “The end of COVID-19 as a public health emergency.” This declaration signifies a substantial global improvement in the situation. However, it’s crucial to note that SARS-CoV-2 persists actively within the population, leading to ongoing transmission and the unfortunate loss of human lives (5, 6). Nonetheless, this WHO statement does not eliminate the possibility of experiencing future COVID-19 pandemic events, which underlines the need for continued surveillance and preparation for possible epidemic outbreaks or pandemics caused by SARS-CoV-2 in the future.
Coronaviruses (CoV) are classified within the order Nidovirales, suborder Cornidovirineae, family Coronaviridae, and subfamily Orthocoronavirinae. This subfamily is further divided into four genera: Alphacoronavirus (αCoV), Betacoronavirus (βCoV), Gammacoronavirus (γCoV), and Deltacoronavirus (δCoV). SARS-CoV-2, the causative agent of COVID-19, falls into the βCoV genus, sharing this classification with other clinically significant viruses, including SARS-CoV and MERS-CoV (6–9).
Over the last century, the emergence of several clinically relevant CoVs has had a dramatic impact on public health. Furthermore, their ability to “cross host barriers” and infect various organisms poses a future risk that requires careful consideration. Cross-species transmission can occur due to proximity between organisms, shared ecological niches, predation, or interspecies contact. Another mechanism facilitating the transmission across host barriers is the occurrence of random mutations in binding receptors. Transmission from more than one host also occurs in CoVs, such as MERS-CoV (10). Understanding the dynamics and mechanisms behind these cross-species transmissions is crucial to proactively assessing and mitigating the risk of new diseases or pandemics derived from CoVs (9, 11, 12).
The origin of SARS-CoV-2 remains unknown. There are multiple hypotheses about it, some of which are as follows: 1) SARS-CoV-2 comes from a mutation of the bat coronavirus RaTG13, which shares 96.2% similarity of its genome. It is thought that this virus mutated due to constant interaction with humans (predation of bats by humans), which forced it to adapt and thus be able to infect this new host; 2) SARS-CoV-2 comes from the mutation of human SARS-CoV (which would explain its ability to infect humans naturally) and through a series of random mutations resulting from selective and immunological pressures. In this scenario, it is thought that SARS-CoV mutated as a defense mechanism against these pressures, giving rise to a new type of virus. Finally, SARS-CoV-2 comes from the Malayan pangolin, with a similarity of 85.5 to 92.4%. Similar to the case of the bat hypothesis, the virus adapted to this new host due to the constant predation by humans against these animals. In summary, the exact origin of SARS-CoV-2 remains to be elucidated, making it difficult to determine whether the origin of this viral infection is zoonotic or abiotic. The ongoing investigation into these theories is essential for a comprehensive understanding of the virus’s origins and potential implications for public health (10–16).
This new infectious agent uses its Receptor Binding Domain (RBD) to bind, with high affinity, to Angiotensin Converting Enzyme 2 (ACE2) and Type II Transmembrane Serine Protease Coreceptor (TMPRSS2) to enter the host cells. The importance of ACE2 arises from the fact that it is an enzyme expressed in various organs and at different quantities (11). This enzyme plays a fundamental role in regulating blood pressure, kidney function, and electrolyte balance. In short, it is essential to maintain physiological homeostasis (17). In the context of infection, SARS-CoV-2 is associated with high levels of ACE2 (the higher the concentration of this enzyme, the greater the susceptibility to infection) (18). ACE2 can be elevated due to stress factors, low blood pressure, diet, or metabolic disorders (18, 19). Understanding the physiological effects of ACE2 concentrations in the body is crucial to elucidate the pathophysiological consequences of SARS-CoV-2 infection and its systemic effects.
The symptoms of COVID-19 are many and vary significantly among individuals. However, initial manifestations typically include fever, anosmia, and ageusia (20, 21). As SARS-CoV-2 infection progresses, additional signs and symptoms such as myalgia, fatigue, nasal congestion, sore throat, rash, and diarrhea may manifest. However, the latter set of signs and symptoms are comparatively less common (22). Despite COVID-19 being a disease with a mortality rate lower than SARS-CoV-1, the severity of the illness differed significantly from person to person. For some individuals, it presented as a mild cold, while for others, it posed a severe threat to their lives. Initial reports indicated that children and young adults with COVID-19 were often asymptomatic, while older adults experienced more severe forms of the disease. Notably, the presence of comorbidities increased the risk of death among individuals affected by COVID-19 (5, 23). Understanding the diverse clinical presentations of COVID-19 is essential for effective management and public health interventions.
As SARS-CoV-2 continues to circulate, the emergence of variants poses an ongoing health challenge. Each variant exhibits unique characteristics, contributing to distinct clinical courses, increased immune resistance, or heightened efficiency in dissemination (Table 1). These variations among strains underscore the dynamic nature of the virus and the importance of continuous monitoring and research to understand and address the evolving landscape of the pandemic (68, 69).
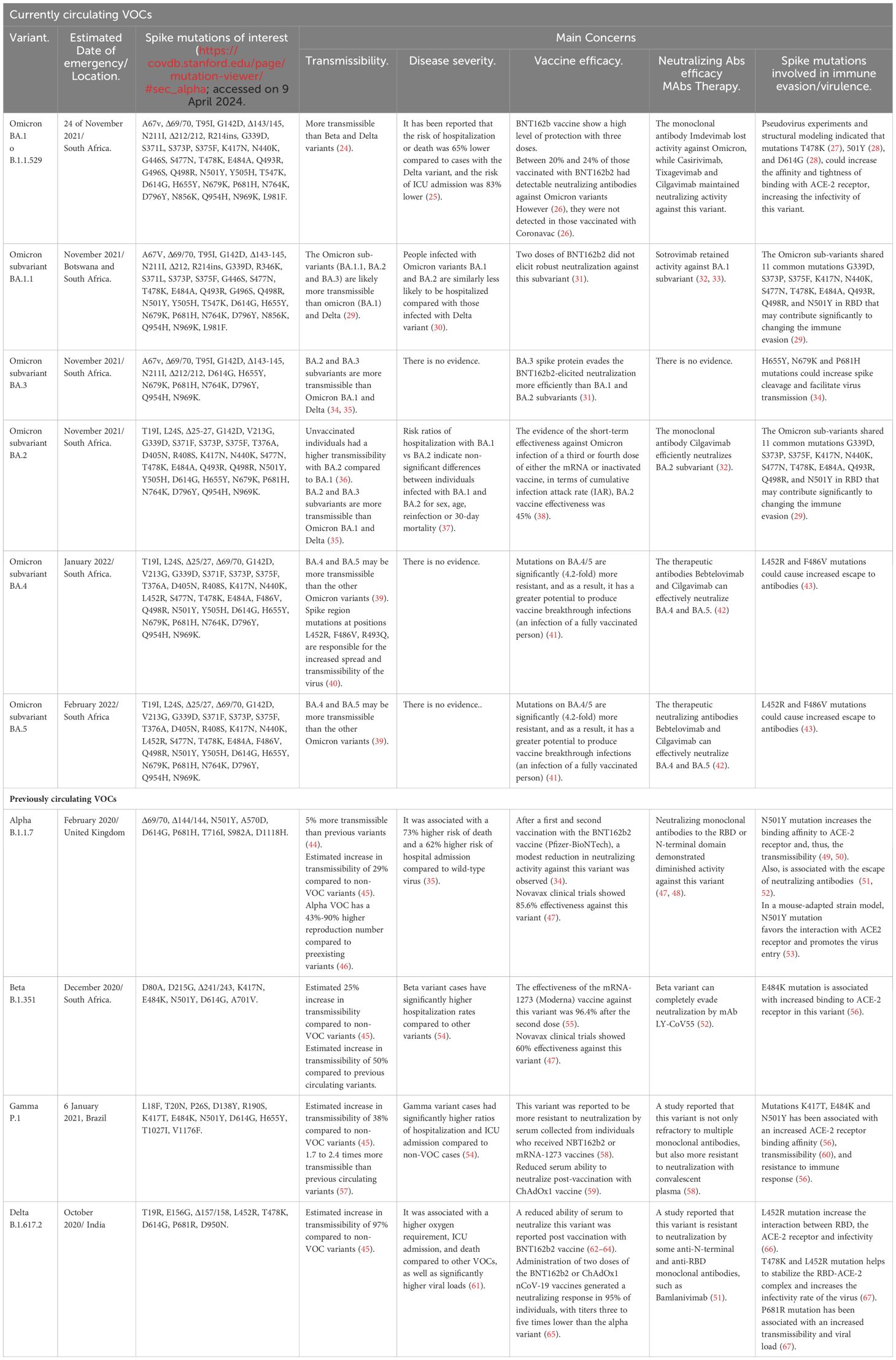
Table 1 List of SARS-CoV-2 variants of concern worldwide.
Although the clinical course of the infection by SARS-CoV-2 varies from person to person, it has been reported that after COVID-19, sequelae of varying durations range from one week to prolonged periods, possibly lifetime (70, 71). Although the mechanism by which these sequelae originate is not entirely clarified, it has been postulated that they may occur due to: 1) direct damage of the virus to the tissue or cells; 2) cellular damage caused by the immune system when fighting the infection; 3) direct damage to tissues resulting from a deregulation of the immune response; or 4) metabolic alterations produced by the SARS-CoV-2 infection. This phenomenon has been described in the literature with various terms, “Long-COVID-19”, “long-term COVID-19”, “post-acute COVID-19 syndrome (PACS)” or “chronic COVID-19”. For consistency, this review adopts the terminology proposed by the World Health Organization (WHO), referring to these conditions as “Long-COVID-19” (72, 73).
This review aims to delineate the viral and host components that facilitated SARS-CoV-2 infection and influenced the severity of COVID-19 during the pandemic. Additionally, the focus will be on elucidating the significance of “Long-COVID-19,” exploring distinctions between variants, examining various immunization strategies to combat this pathogen, and evaluating host conditions contributing to infection severity in the post-pandemic period (74–77). Given the challenges posed by the high transmission rate, immune evasion, and mutation rate of SARS-CoV-2, it remains uncertain whether the virus will evolve into an endemic state. Consequently, sustained vigilance and continued research efforts are imperative to mitigate the severity of infections, reduce mortality rates, and address potential long-term effects (78).
2 SARS-CoV-2 infection onset, progression, and pathophysiology of COVID-19
The initiation of infection and the progression of the pathophysiology of COVID-19 have been the focus of numerous investigations. Some studied characteristics of SARS-CoV-2 focus on its resistance to environmental pressures, the affinity of its receptor to ACE2, and the general clinical course. Nevertheless, significant gaps in our understanding of this agent persist, emphasizing the need for further research to uncover additional information.
2.1 SARS-CoV-2 and its infection cycle
SARS-CoV-2 has a single-stranded positive-sense RNA genome (ssRNA+) with an approximate size of 29,891 nucleotides (~30 kb), containing 15 ORFs (Figure 1). During the infection process, replication, and virulence of SARS-CoV-2, different viral proteins intervene, such as structural proteins (SP) and non-structural proteins (NSP). Both kinds of proteins play multiple roles and functions, which are shown in the Table 2 and Figures 1–3 (163, 164).
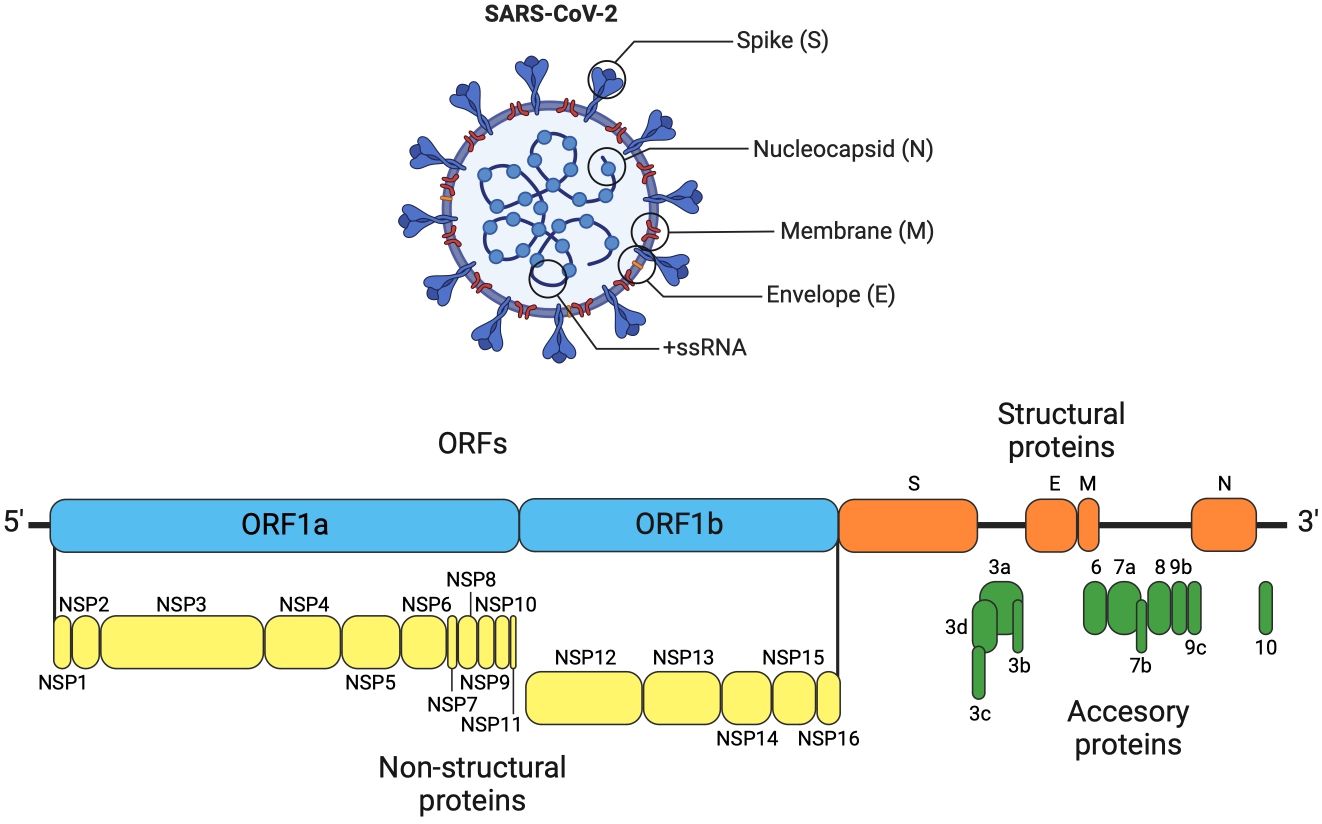
Figure 1 SARS-CoV-2 genome organization. The genome of SARS-CoV-2 is organized similarly to other coronaviruses, consisting of the following elements: a 5´ untranslated region, ORF1a, which encodes two overlapping polyproteins (pp1a and pp1ab). The pp1a non-structural protein corresponds to nsp1 to nsp10, while pp1ab non-structural protein comprises nsp12 to nsp16, which are further cleaved into 16 non-structural proteins (nsp1–16). The 3´ untranslated region contains the structural genes that encode four structural proteins: Spike Surface glycoprotein (S), Envelope glycoprotein (E), Nucleocapsid (N), and main protein or Matrix (M). The accessory genes are interspersed among the structural genes (79, 80).

Table 2 Functions of SARS-CoV-2 proteins related to viral pathogenicity.
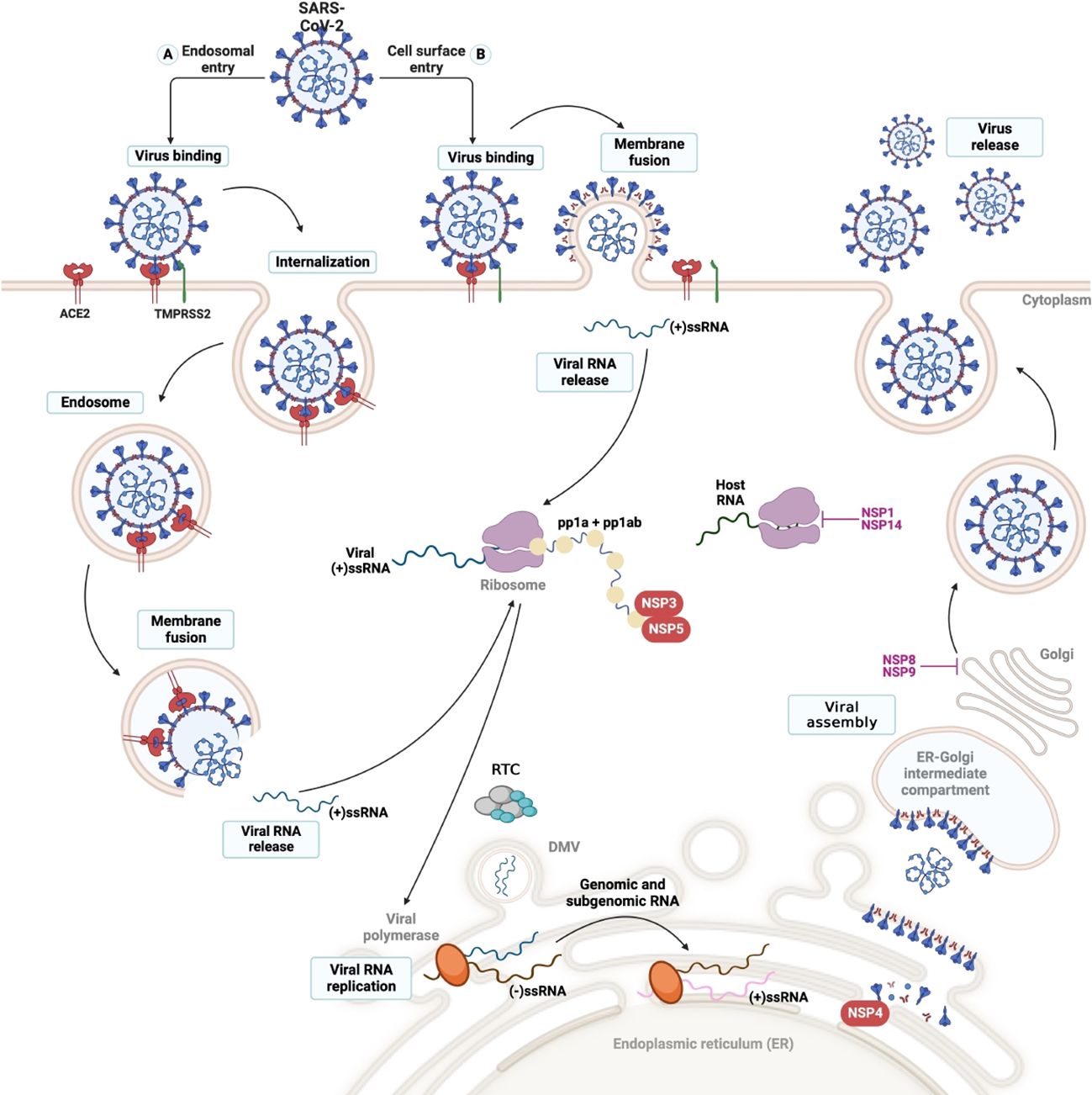
Figure 2 SARS-CoV-2 cell entry and viral cycle. The viral cycle commences with the binding of the Spike (S) protein to the ACE-2 receptor, facilitated by either cell surface or endosomal entry. (2) The TMPRRS2 protease (transmembrane protease serine) mediates the fusion of the virus-cell membranes through the cleavage of protein S, allowing the virus to enter the cytosol of the host cell. (3) Once internalized, the virus undergoes uncoating, releasing the viral genome into the cytosol, where it undergoes replication and translation. (4) This process generates two polyproteins: pp1a and pp1b, which are subsequently cleaved by the viral protease present in the nonstructural proteins (nsps) encoded by the virus. (5) In endoplasmic reticulum (ER)-derived double-membrane vesicles (DMVs), the negative-stranded genome serves as a template to generate the entire positive-stranded genome and subgenomic RNA (sgRNA). Translation of sgRNA in the ER results in the synthesis of the structural glycoproteins N, S, M, and E, which are utilized for viral assembly in the ER-Golgi intermediate compartment (ERGIC). (6) Finally, the entire positive-stranded genome is encapsulated in newly synthesized virions, which are released from the cell via exocytosis.
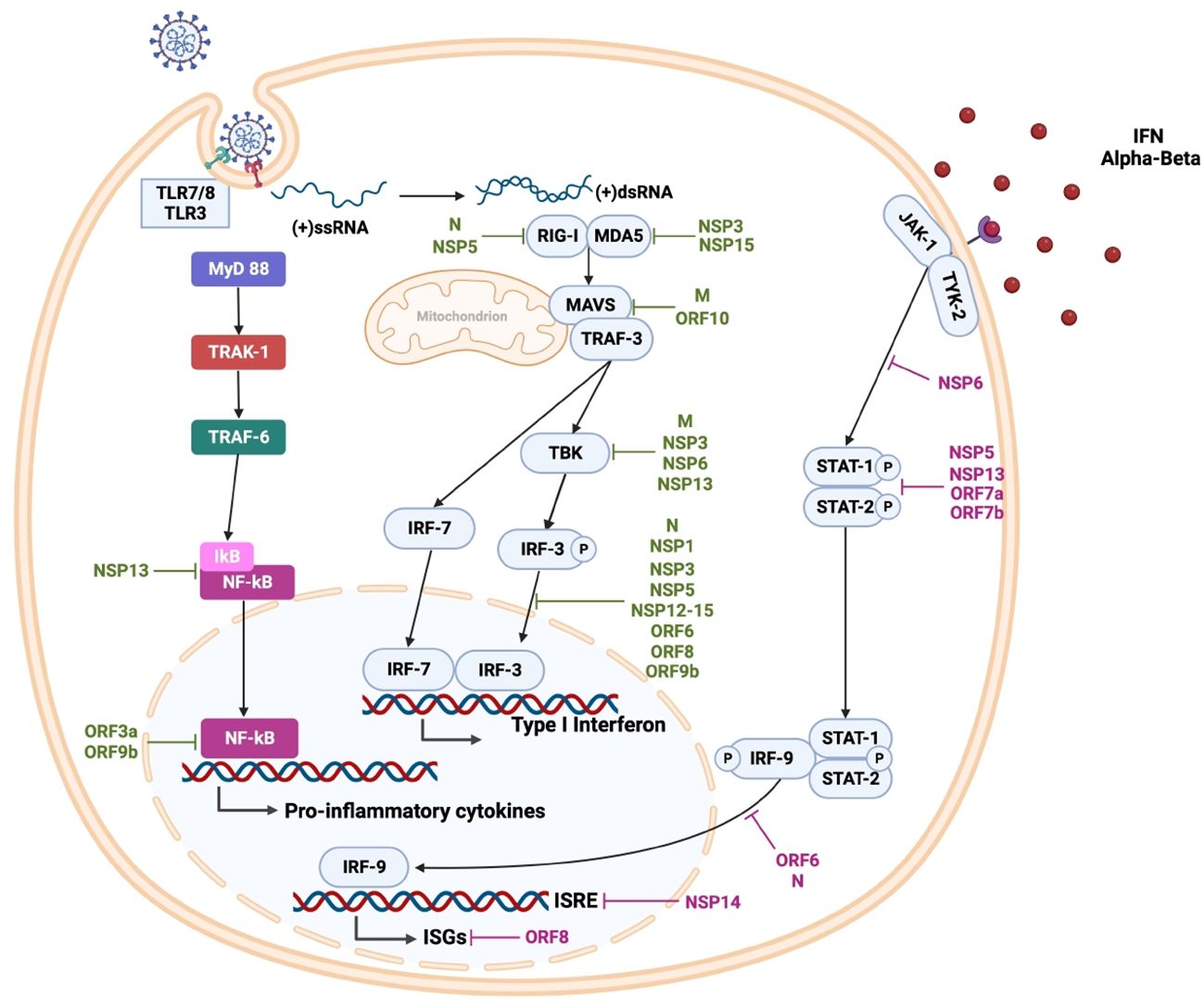
Figure 3 SARS-CoV-2 innate immune response and viral evasion. During viral replication, cytosolic double-stranded RNA is detected by retinoic acid-inducible gene I (RIG-1) and melanoma differentiation-associated protein 5 (MDA5). Viral genomic single-stranded RNA is detected by toll-like receptor (TLR) 7 and TLR8 in the endosomes of plasmacytoid dendritic cells and B cells, as well as myeloid cells, respectively. Additionally, endosomal TLR3 recognizes double-stranded RNA in various cells, and TLR4 participates in the detection of oxidized phospholipids (OxPLs) induced by SARS-CoV-2 infection. These interactions lead to the activation of downstream transcription factors, most notably IRF3 and NF-κB, promoting the expression of type I and III interferons (IFNs) and proinflammatory cytokines such as TNF-α, transforming Growth Factor β (TGF-β), interleukins (IL-1β, IL-6, IL-8, IL-12, IL-18), and chemokines like chemokine (C-C motif) ligand 2 (CCL2), CCL3, CCL5, C-X-C motif chemokine ligand 8 (CXCL8), CXCL9, CXCL10. Furthermore, endosomal TLR3 can recognize double-stranded RNA in various cells, while TLR4 detects oxidized phospholipids (OxPL) induced by SARS-CoV-2 infection. SARS-CoV-2 antagonizes the innate immune response at different steps; specific viral proteins interfere with type I IFN production (shown in green) and interferon signaling (shown in purple), impairing the early antiviral state.
SARS-CoV-2 SPs participate in the pathogenicity of the virus, which includes receptor recognition, replication process, viral resistance, and others (165). On the other hand, NSPs of this virus are involved in genome replication and regulation of the early stages of transcription (166). Additionally, the different SARS-CoV-2 proteins exhibit participation as virulence factors that are resistant to the immune response, among others (167–169). These biological traits of SARS-CoV-2 allow it to infect different types of cells. The virus first enters the upper respiratory tract through respiratory droplets or viral particles and then progresses to the lungs. Within the lung, the virus prefers to infect the surface of alveolar epithelial cells and cells expressing ACE2 and possessing coreceptors such as TMPRSS2 and neuropilin-1 (NRP1), among others (170). These proteins are not exclusive to the lung but are expressed on the surface of various cells in different tissues, including the ciliated epithelium, nasopharyngeal tract, upper respiratory tract, bronchial epithelium, type II pneumocytes, alveolar monocytes, macrophages, liver, kidneys, among others (171–174). This widespread expression pattern has been postulated as a potential explanation for the diverse multisystem damages observed in COVID-19 (175–178).
In the lung, the virus enters the cell through the Spike protein (Protein S), which is composed of 2 subunits (S1 and S2). The initial step of infection involves the binding of S1 to the host RBD. Subsequently, TMPRSS2 cleaves the S1/S2 region, inducing fusion of the virus with the membrane through the S2 domain (although other proteases such as endosomal cysteine proteases, cathepsin B and L, furin, basin, and neuropilin-1 are relevant, TMPRSS2 has been proved to be essential for SARS-CoV-2 pathogenesis) (179). The virus’ entry mechanism into the cell depends on multiple factors, such as cell-type physiological conditions. SARS-CoV-2 entry into the cell can be through membrane fusion or endocytic pathways (Figure 2) (165, 175, 180). Upon completion of viral entry, the ACE2 receptor detaches from the cell membrane, increasing serum concentrations of angiotensin II and causing vasoconstriction, inflammation, and thrombosis, as explained below in the section “Complications of organs and body systems after SARS-CoV-2 infection” (178, 180).
2.2 Viral progression: recognition of SARS-CoV-2 by the immune system and pathophysiology of COVID-19
After membrane fusion, the viral nucleocapsid of SARS-CoV-2 is removed, and the viral genome is released into the host cell’s cytoplasm. Viral infection generates Pathogen-Associated Molecular Patterns (PAMPS) and Damage-Associated Molecular Patterns (DAMPS), which are detected by different Pattern Recognition Receptors (PRRs) (Figure 3). Activation of these PRR receptors leads to activation of downstream transcription factors, such as Interferon Regulatory Factor (IRF) 3 and Kappa Light Chains of Activated B-cell Nuclear Enhancer Factor (NF-κB), which express type I and III Interferons (IFNs), along with pro-inflammatory agents, cytokines, Tumor Necrosis Factor α (TNF-α), Transforming Growth Factor β (TGF-β), Interleukin (IL)-1β, IL-6, IL-8, IL-12, IL-18 and chemokines such as Chemokine Ligand (CCL) CCL2, CCL3, CCL5, and Chemokine Ligand Motif C-X-C (CXCL) 8, CXCL9 and CXCL10 (181–183). The secreted IFNs bind to the cell surface receptor on neighboring cells to increase the expression of IFN-Stimulated Genes (ISGs) via the Signal Transducer and Activator of Transcription (STAT) 1/2/IRF9 complex, driving an antiviral state (184).
The release of these pro-inflammatory substances induces the recruitment of monocytes, neutrophils, and macrophages into the infected tissue, which in turn secrete IL-6, IL-1β, IL-8, TNF-α (181, 185), generating a concentrated medium of Reactive Oxygen Substances (ROS), which contribute to viral pathogenesis and by producing damage in the tissue surrounding the infection (185). This leads to an inflammatory feedback loop so that the more cytokines and chemokines are released, the greater the recruitment of immune cells, and with it, the greater the number of cytokines and chemokines, favoring tissue damage and increasing the severity of SARS-CoV-2 infection (186–189). As the infection progresses and innate and adaptive immune responses are intensified, highly glycosylated viral S protein with L-fucose and D-mannose-rich residues activates the complement system via the lectin pathway along with the major complement opsonin H regulatory factor (C3b) (190–192). Moreover, although complement activation is essential in the initial immune response, its sustained activity contributes to viral pathogenesis, leading to plasma clotting and tissue damage.
Thus, SARS-CoV-2 drives an exacerbated immune response, influencing the activity of natural killer (NK) cells, macrophages, and neutrophils, among other cells. So, all this immune response dysfunction continuously increases TGF-β concentrations, resulting in sustained damage to surrounding tissue and nerves in the affected area. As the damage proceeds, the cells affected by the harm release substances that further alter the affected area, compromising the permeability of the tissues and allowing the invasion of more immune cells into the tissues and organs. This sustained infiltration of immune cells, such as neutrophils, leads to the formation of Neutrophil Extracellular Traps (NETs), which increases tissue damage by forming a feedback loop (193–196).
CoVs employ diverse strategies to evade the immune response, primarily by interfering with IFN signaling, production, and resistance. The virus subsequently conceals itself within various types of cells to evade immune recognition, thus subverting the initial stages of immune defense. Furthermore, congenital defects in the host’s immune response have been linked to increased severity of COVID-19.
An early response by the host to the cytokine storm is relevant to reducing the replication and severity of SARS-CoV-2 infection, thus enabling it to promptly prepare an adaptive response. The damage generated by SARS-CoV-2 in tissues may be due to the destruction of infected cells by CD8+ T cells and the production of pro-inflammatory cytokines by other immune cells, which can produce collateral damage to tissues when fighting the viral infection (197). Some reports of this self-tolerance and cross-reactivity effect have shown that they are due to antiphospholipid antibodies, antitype I interferon, and antinuclear polymorphs, among others (198–200). This, in turn, increases the risk of contracting autoimmune diseases such as Guillain-Barré syndrome and systemic lupus erythematosus. Furthermore, according to some reports, the fact that the immune system attacks the organism simultaneously while it defends itself from the invading agent is due to the imitation or similarity of epitopes between the pathogen and parts of the organism. This could be one of the possible reasons for the severity of COVID-19 upon infections by SARS-CoV-2 (201).
Thus, considering the systemic effects of viral recognition by the immune system, long-term consequences of SARS-CoV-2 infection have been postulated to occur by viral persistence, which produces damage by viral replication products and tissue damage by chronic inflammation, where the dysregulated activation of proinflammatory pathways, lead to multiple organ system conditions (202). Some of these physiological alterations will be discussed further below.
It has been proposed that more than one of these mechanisms leads to diverse clinical manifestations. However, the greatest damage and consequences reported in Long-COVID-19 are due to indirect damage caused by the cytokine storm. In that regard, key cytokines implicated in the onset of disease severity include TNF-α, IFN-γ, IL-6, IL-1β, granulocyte-macrophage colony-stimulating factor (GM-CSF), G-CSF, and TSLP (1, 136, 203–207). To ameliorate this cytokine storm, the use of specific monoclonal antibodies for cytokines antagonistic to the JAK-STAT pathway inhibitory receptors, plasmapheresis, and the use of mesenchymal stem cells has been proposed (206, 208, 209).
2.3 Viral evasion and immune response to SARS-CoV-2 infection
The virus, at the beginning of the infection, hides in various types of cells to avoid immune recognition, counteracting the initial stages of immune defense, to subsequently interfere with the signaling pathways for the production and secretion of antiviral proteins, providing to SARS-CoV-2 a resistance to the IFN systems. In addition, congenital defects in the host’s immune response are associated with greater severity of COVID-19 (134, 198). The non-structural proteins Nsp1, Nsp5, Nsp13, Nsp14, and structural proteins ORF6, and ORF7 of SARS CoV-2 counteract antiviral immune response both prior to and following assembly. Moreover, as described above, SARS-CoV-2 affects the antiviral immune response by interfering with the type I IFN induction pathway and the production of other proinflammatory cytokines (Figure 3 and Table 2) (133, 134, 185).
SARS-CoV-2 promotes a T helper (Th) 1 cell response and an early CD8+ T cell response, which initially helps eliminate the virus. However, in the long run, it can produce a cytokine storm (210–212). The CD4+ T cell type 1 (Th1) phenotype has been associated with promoting a state of viral control, favoring an antiviral response. In contrast, the type 2 phenotype (Th2) is associated with a greater severity of infection (213). On the other hand, circulating T follicular helper cells (cTfh) contribute to the development of neutralizing antibodies, and they have been associated with a reduction in COVID-19 severity. In addition, there are notable differences between asymptomatic and symptomatic COVID-19 patients regarding cytokine profile. Indeed, asymptomatic individuals have been shown to have a “coordinated” secretion of pro-inflammatory and regulatory cytokines, while symptomatic patients present with a polarized inflammatory reaction and a pro-inflammatory cytokine storm (214, 215).
SARS-CoV-2 has been described as using cell-to-cell transmission to spread, so the cellular response plays a role in viral control when antibody neutralization is impaired (216). On the other hand, host factors can affect the adaptive immune response. For example, it is recognized that naïve T cells are reduced in older people, which is associated with worse outcomes during infection (217). The humoral adaptive response is also relevant for controlling viral load, disease severity, and reinfection (213, 218).
When the responses of surviving and deceased patients were compared, a delay in the production of neutralizing antibodies was observed in patients with worse post-disease outcomes, which is associated with a higher viral load. The lifespan of the protective immune response produced by natural infection and vaccination is still under study and depends on multiple factors, such as the type of vaccine for the SARS-CoV-2 strain. Likewise, reports demonstrate that the immune response persists for at least 10 months up to one year upon SARS-CoV-2 infection (213, 219). Moreover, immunological memory after SARS-CoV-2 infection is observed at multiple sites, preferably in the lungs and associated lymphoid nodes (220).
In addition, studies have suggested the relationship between autoantibody levels and COVID-19 severity, so it has been proposed that self-tolerance may be affected in SARS-CoV-2 infection, leading to reactivity against host cells. Several autoantibodies have been found in patients with COVID-19, including antiphospholipid, anti- type I interferons, anti-nuclear polymorph anti-antibodies, and anti-neutrophil cytoplasmic antibodies (200, 221). In turn, some autoantibodies have been linked to a potential risk of autoimmune diseases, such as Guillain-Barré syndrome, systemic lupus erythematosus, Kawasaki disease, and autoimmune hemolytic anemia, among others (201). Although the exact mechanism underlying the development of these complications remains a topic of debate, it is hypothesized that they may result from hyperstimulation of the immune system and viral molecular mimicry. In a controlled immune response, specific T cells and neutralizing antibodies limit infection, and apoptotic cells die by phagocytosis, preventing lung damage. Conversely, a dysregulated immune response leads to an overproduction of pro-inflammatory cytokines known as cytokine storm, which directly, indirectly, or synergistically can lead to lung injury, acute respiratory distress syndrome (ARDS), and multi-organ damage in COVID-19 patients (222, 223).
In the context of SARS-CoV-2 infection, it has been postulated that T cells might have a pivotal role in the severity of clinical outcomes. Less severe disease has been observed when a prompt T cell response occurs, but this correlation is still debated (224). During natural infection by SARS-CoV-2, the whole viral particle is recognized, inducing T cell responses to several epitopes that might persist for up to 6–8 months (225). In that regard, epitope studies have identified the structural proteins ORF3, ORF8, and non-structural proteins nsp3, 4, 6, 7, 12, and 13 as dominant targets of CD4+ and CD8+ SARS-CoV-2 T cell responses (226). Likewise, T cell responses induced by vaccines have been demonstrated to have different ranges of protection. In that regard, mRNA vaccines (mRNA-1273 and BNT162b2), protein recombinant vaccine Novavax (NVX-CoV2373), or Janssen vector vaccine (Ad26.COV2.S) have been shown to induce CD4+ and CD8+ T cells that recognize a median of 11 CD4+ and 10 CD8+ SARS-CoV-2 Spike protein epitopes. Similar results have been described for inactivated vaccines, which induce T cells responses against epitopes of the Spike proteins, as well as other SARS-CoV-2 proteins included in the inactivated viral particle, both in adults and children (227–231). Particularly, the consequent T cell response against the Omicron variant persisted after 6 months: 84% for CD4+T cells and 85% for CD8+ T cells, determined by activation-induced markers (232–234). Consistently, activation of specific CD4+ T cells was detected in individuals vaccinated with Coronavac® for 4 weeks after the second booster, and these cells demonstrated to be responsive against the SARS-CoV-2 Wild type and Omicron variants. Furthermore, cell-mediated immunity was shown to be more perdurable than the humoral immunity (228). Thus, considering the emergence of viral variants that evade neutralizing immunity, T-cell responses have gained importance in understanding protective immunity in SARS-CoV-2 infection (143). Observations of recovered COVID-19 patients, vaccinated and exposed individuals, support the role of memory T cells in reducing viral loads, in the prevention of SARS-CoV-2 infections and reinfections, and early presentation of multiple viral epitopes, avoiding viral escape by variants (233).
In that regard, it has been studied that the population has developed immunity through infection or vaccination after four years of spreading the SARS-CoV-2 virus worldwide. It has previously been discussed that heterogeneous immune imprinting repertoires would depend on several factors, including whether the population was vaccinated before infection, the type of vaccine, the infectious strain, and the particular conditions of the host (235, 236). Despite the above, epidemiological studies have shown that antibody levels gradually decrease over time, so they do not offer long-lasting protection against SARS-CoV-2 infection (233, 237). Moreover, considering the above, the emergence of viral variants with a repertoire of mutations in the Spike protein and the receptor binding domain (RBD) allows the virus to evade the binding and neutralization of antibodies, leading to outbreaks of reinfection around the world (237). In this context, considering the emergence of the Omicron lineage (Table 1), bivalent mRNA booster vaccines were developed and implemented in 2022. Booster with bivalent vaccines has demonstrated greater vaccine efficacy than monovalent vaccines against the Omicron subvariants in all age groups, especially against severe clinical outcomes (238, 239). A systematic evidence synthesis and meta-analysis, conducted up to December 2022, summarize the evidence regarding the long-term effectiveness of COVID-19 vaccines in preventing infections, hospitalizations, and mortality (240). The WHO defined adequate levels of protection of vaccines against COVID-19 for infection. In the first series of primary pandemic vaccines, protection against infection was 83% and 62% after 42 days and 139 days post-immunization, respectively. This study revealed that the baseline levels of effectiveness of the vaccine against the Omicron variant were 70% against infection, and these decreased to 43%. Of note, protection levels against hospitalization also decreased from 89% at baseline to 71%, and there was insufficient evidence on effectiveness against Omicron wave mortality (240). Therefore, it is necessary to address new COVID-19 vaccination strategies to improve protection levels among the population.
3 Organ and body system complications upon SARS-CoV-2 infection
As mentioned above, SARS-CoV-2 mainly infects the lungs but can also affect other organ systems. SARS-CoV-2 infection to various host organs has been proposed due to the heterogeneous distribution of ACE2 and TMPRSS2. In addition, the damage caused by SARS-CoV-2 infection and the post-infection sequelae produced by COVID-19 can be highlighted, which includes lung, kidney, and liver damage, among others (241–243). The most common sequelae and complications of severe COVID-19 are fatigue, dyspnea, cognitive impairment, anxiety, depression, anosmia, and ageusia (243). In contrast, in mild or asymptomatic COVID-19, fatigue, myalgia, headache, cough, and mucus are present (Table 3) (243).
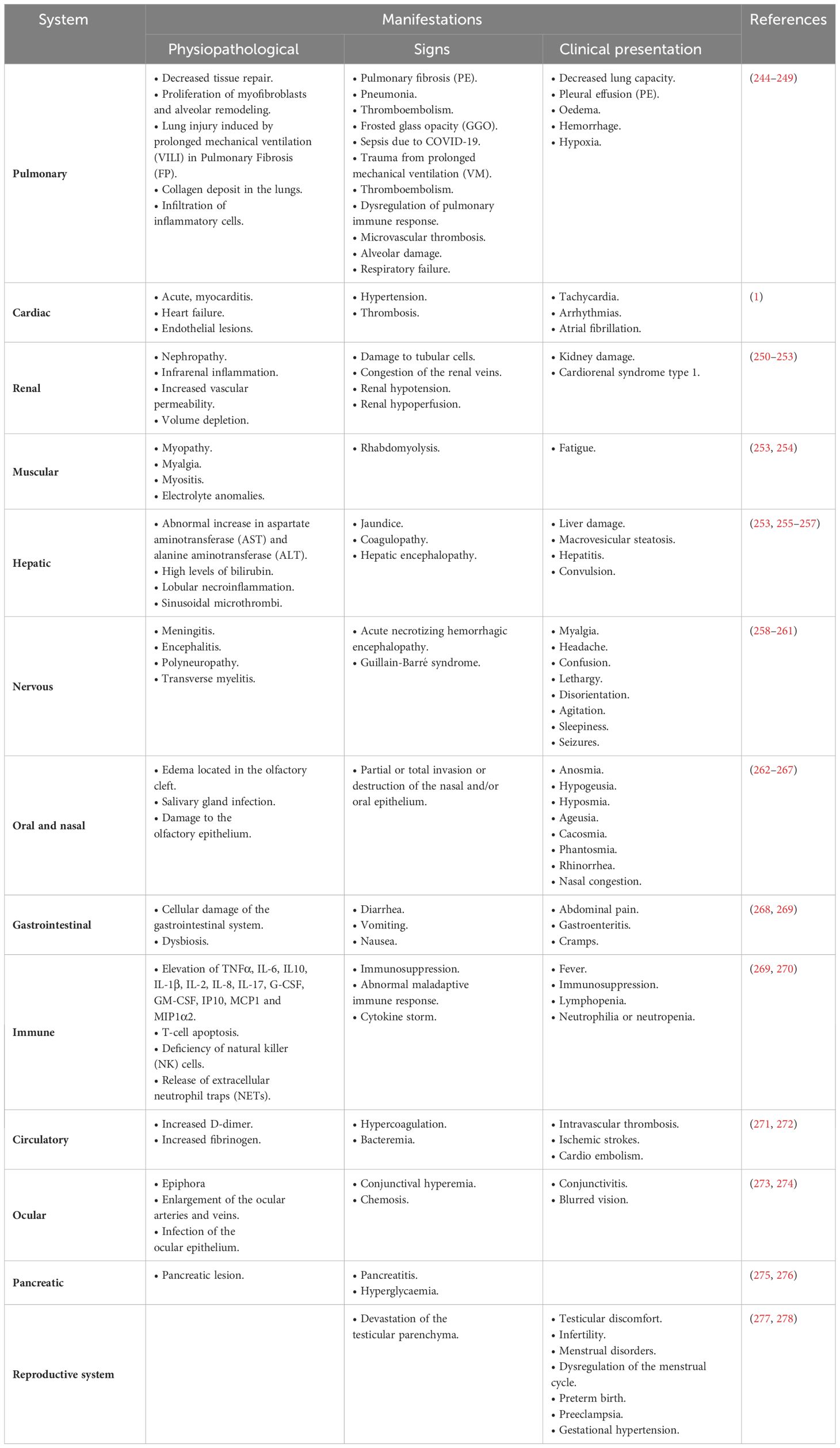
Table 3 Pathophysiological manifestations, clinical signs, and clinical presentation and/or sequelae of SARS-CoV-2 infection in different systems.
3.1 Renal complications
The kidneys express a high concentration of ACE2, TMPRSS2, and furins. At the same time, the kidneys play an important role in controlling blood pressure through different mechanisms, such as the Renin Angiotensin Aldosterone System (RAAS). SARS-CoV-2 can be distributed throughout the body through the blood, infecting organs and tissues susceptible to the presence or absence of the receptors necessary for viral entry (279–281). Kidney damage is not only produced by the direct action of the virus by destroying tissues, but it can also result from an overactivation of the immune system (281).
SARS-CoV-2 damages renal tubules by inducing the infiltration of macrophages and complement proteins such as C3a and C5b-9, leading to tissue fibrosis (282). This damage causes waste to accumulate in the kidney, producing different multisystem effects. Additionally, damage to the kidney generates an imbalance of the ACE 2/Ang 1–7/MasR axis, which leads to an imbalance of the RAAS (281, 282). If kidney damage occurs together with lung damage, hypercapnic acidosis occurs, which contributes to aggravating multisystem damage. By lowering the price and not being able to activate the RAAS adequately, it causes kidney and heart damage, generating a hypovolemic state, which in turn produces vasoconstriction, leading to a procoagulant state, injuring small-caliber arteries and ducts (282–284).
The cumulative damage caused by SARS-CoV-2 results in toxins and organic waste accumulation. As it destroys tissues, the waste products enter the bloodstream, accumulating in the kidneys already compromised by the viral infection and the hyperactivated immune system. This exacerbates a detrimental loop, reaching a critical juncture where the abundance of toxins and waste in the organism compromises additional tissues, ultimately culminating in renal and/or hepatic failure (285–287).
3.2 Cardiovascular complications
When SARS-CoV-2 reaches the heart, it causes heart damage, myocardial injury, arrhythmias, and heart failure. This damage to the heart can be generated directly by the virus or by the exacerbated immune response. As SARS-CoV-2 enters and destroys cells, their functions are altered, leading to cardiac arrhythmias, deep vein thrombosis, pulmonary embolisms, and decreased lung volume (288). People with heart disease before infection tend to have more severe symptoms or develop complications during and after COVID-19, including the development of hypoxemia, coagulopathies, cardiac arrest, and damage (289). During infection, cardiac cells release troponin, and troponin concentrations have been reported to correlate with disease severity and can be considered a biomarker (284, 290–293). Synergistically, due to cardiac damage, a decrease in blood pressure occurs, leading to the activation of the RAAS system, which in turn triggers vasoconstriction, vascular inflammation, and an increase in Angiotensin 2 levels, contributing to the pathogenesis and cardiovascular complications of the infected individual. In addition, it has been shown that SARS-CoV-2 causes coagulation disorders, producing hyper coagulopathies and forming thrombi, which can trigger embolisms in different organs, causing the death of tissues or cells that release toxins and subsequently travel through the blood. These cellular wastes accumulate in the kidneys, which will hinder their functions. Likewise, the released toxins accumulate in the blood, potentially causing peritonitis (294–297).
3.3 Metabolic complications
SARS-CoV-2 can induce widespread metabolic effects, including insulin resistance, dyslipidemia, diabetes, and systemic inflammation. In particular, alterations in the pancreas and liver of individuals infected by SARS-CoV-2 can lead to a state of hyperglycemia due to metabolic stress, leading to the development of “New Onset Diabetes (NOD). This condition can significantly impact the course of the infection. However, the precise mechanisms underlying the emergence of these complications and new pathologies have not been fully elucidated (298–300).
One explanation for this is that SARS-CoV-2 infection in adipose cells can cause a decrease in insulin sensitivity, which can lead to hyperglycemia, leading to an increase in lipids in the body, which can lead to dyslipidemia (301), this affects the pancreas and liver, causing a deterioration in the function of these organs. Furthermore, since SARS-CoV-2 can infect the CNS and the Peripheral Nervous System (PNS), it has been reported that damage to either of these two systems, either at the nervous level or at the functional level, produces a cascade effect altering normal metabolism (302, 303).
3.4 Pulmonary complications
Pulmonary complications are prevalent conditions during and after SARS-CoV-2 infection. Common complications include pulmonary fibrosis, respiratory failure, pulmonary embolism, and secondary bacterial pneumonia. The damage to the pulmonary epithelium is due to either a direct action of the virus or the effects of the immune system in a small space. This damage and effects lead to pulmonary fibrosis, making breathing difficult. Moreover, immune cells are recruited, which release TNF-α, IL-1β, IFN-γ, and GM-CSF (304, 305). Additionally, as immune cell recruitment occurs at the site of injury, these cells also secrete Proteolytic Enzymes such as Matrix Metalloproteinase (MMP) that degrade the extracellular matrix, producing the release of cytokines, TGF-β and Chemoattractant Protein (MCP) 1, which in turn induces the detachment of fibroblasts, collagen, and cellular matrix components, aggravating pulmonary fibrosis (268, 306–308).
3.5 Gastrointestinal complications
COVID-19 has been reported to produce gastrointestinal complications due to damage to gut epithelial cells, which activates the innate immune response. Indeed, activated dendritic cells, macrophages, and NK cells target the site of infection and secrete IL-1β, IL-6, and TNF-α, while activated CD4+ T and CD8+ T cells produce IFN-γ among other inflammatory cytokines that aggravate tissue damage (309, 310). As damage increases in the intestine, cell death, cellular alterations, and pathological complications increase, hindering the absorption of nutrients, increasing intestinal permeability that induces a greater filtration of immune cells, repeating the cycle and damaging the gastrointestinal mucosal barriers. This, in turn, leads to less water reabsorption in the kidneys and intestines, which could explain the persistent diarrhea and increased nutritional disorders that occur in COVID-19 patients. Moreover, intestinal damage impairs gastrointestinal motility, fluid, and electrolyte secretion, contributing to nausea, vomiting, and abdominal discomfort (311, 312). Gastrointestinal damage drives macrophages and T lymphocytes to the injury site, secreting IL-1β, IL-6, IL-8, IFN-γ and CCL2, CCL5, and CXCL8, which can act synergistically to recruit more inflammatory cells, thus aggravating intestinal inflammation (313–315).
3.6 Hematological complications
The most common hematologic complications caused by SARS-CoV-2 are thrombocytopenia, thrombosis, disseminated intravascular coagulation (DIC), and platelet dysfunction. While these disorders have been seen in repeated cases of COVID-19, the underlying causes of these abnormalities have not been fully elucidated. Some hypotheses suggest that these effects result from the destruction of platelets in the circulation due to an antibody cross-reaction resulting from mimicry of viral epitopes with those of platelets. This hypothesis is based on the fact that the antibodies generated to fight the virus attack the platelets by mistake, or the latter is in the “crossfire” of the immune system against the virus (316–318).
On the other hand, other hypotheses suggest that this effect is because the virus could infect platelet precursor cells in the bone marrow, which would alter their function (317). Furthermore, it is known that proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 can alter the coagulation factor system by increasing the production of fibrinogen, Von Willebrand Factor (vWF), Factor VIII, and Factor XII, which increase the risk of coagulopathies (319–321).
3.7 Neurological complications
It has been postulated that SARS-CoV-2 could infect CNS cells, which explains the cognitive and sensory symptoms patients experience. It has also been proposed that the virus could cross the blood-brain barrier, affecting the recognition of signals traveling from the PNS to the CNS (302). Alternatively, based on cerebral Positron Emission Tomography (PET), it has been proposed that patients with prolonged COVID-19 who change their brain metabolism have a functional alteration of cells, which do not process or transmit information properly. This effect is greater in the bilateral brainstem, which causes hyposmia, anosmia, memory impairment, cognitive impairment, and pain (303). Other hypotheses have proposed that the virus, by damaging nerves and receptors for olfactory and gustatory signals, causes ageusia and anosmia. This damage to the receptors could be due to the direct action of the virus or collateral damage from the overreactive immune system in the nervous tissue environment (322–325). Other complications observed in the brain are encephalopathy, confusion, dizziness and alterations in memory or perceptions, Cerebrovascular Accidents (CVA), transverse myelitis, memory loss, decreased reflexes, neuropathies, myopathies or Guillain-Barré syndrome (322–325).
3.8 Post COVID-19 condition
It has been described that the risk of long-term sequelae after the resolution of COVID-19 is proportional to the age and health status of the patient. Similarly, patients with a more severe infection during COVID-19 tended to have a higher probability of developing sequelae of varying severity and durability (326–328). The post-COVID-19 sequelae can be generated by various factors, such as the fact that SARS-CoV-2 uses some cells as a reservoir, entering a latency period and affecting cell function while waiting for regrowth or activation (183, 328). Other theories suggest that these sequelae are generated by damage to nerves and cells due to a direct action of the virus on nerve cells and receptors or by indirect damage produced by an exacerbated immune system that, in its function of counteracting the infection, can cause collateral diseases, affecting nerve cells and receptors (1, 183). The most common complications are chronic fatigue syndrome, sleep disorders, breathing problems or asthma, heart problems, neurological or psychological problems, anosmia, and ageusia (329, 330). Finally, other theories point out that due to SARS-CoV2 infection, the cellular and organic metabolism is altered. Specifically, the body and systems try to readapt to these new conditions, and it is during this readjustment that some functions are partially or permanently changed, thus generating a loss of tastes, sensations, and confusion of tastes or smells (330–332). In the latter hypothesis, the receptors and nerve cells remain intact. However, they can no longer process information as before SARS-CoV-2 infection (333, 334).
It is known that, in order of frequency, the symptoms of COVID-19 are fever (98%), cough (82%), and, in a lower percentage of cases, difficulty breathing (to be determined). On the contrary, the severity and symptoms of Long-COVID-19 are diverse, and their frequency is highly variable. Particularly, it is challenging to elucidate the frequency of long-term COVID-19 symptoms. However, it has been proposed, based on the number of cases, that the frequency of these, from highest to lowest, would-be cardiovascular conditions, thrombotic conditions, cerebrovascular conditions, diabetes, encephalomyelitis, dysautonomia and metabolic alterations, myalgic encephalomyelitis (ME) and chronic fatigue syndrome (CFS), which is the least common (335–341). It is important to note that ME and CFS, along with postural orthostatic tachycardia syndrome (POTS), can be lifelong (342). It has been reported that between 2 to 14% of patients who had COVID-19 present with conditions similar to or compatible with POTS during Long-COVID-19. These conditions include tachycardia, orthostatic intolerance, fatigue, cognitive and muscular impairment from 6 to 10 months (343, 344). Moreover, it has been postulated that these symptoms develop due to autoimmune disorders, autonomic dysfunction, direct damage from SARS-CoV-2 infection to nerve cells, and invasion of the virus into the CNS (343–346). At the same time, due to the different mechanisms that SARS-CoV-2 has to induce damage in the body, as well as affect the bioavailability of ACE2 and its presence in the various organ systems, other conditions can develop, such as metabolic diseases and those that affect the thyroid, kidney, and liver (347).
In addition, it has been reported that Graves’ disease and Hashimoto’s thyroiditis are among the thyroid conditions of long-term COVID-19. These conditions are thought to be induced by the cytokine storm during COVID-19 and by the molecular mimicry of components resulting from the viral infection with those of the host (347–351). Other conditions that have been reported are lymphopenia, neutropenia, and oxidative damage. However, these sequelae do not have a high frequency, so they have not been directly attributed to a Long-COVID-19 pathology but rather to late COVID-19 symptomatology due to SARS-CoV-2 viremia disseminated (352, 353).
Despite the consequences of Long-COVID-19, there is not enough information to estimate the frequency and severity of each pathology present in this disease. To overcome these limitations, studies with a larger number of patients are needed to assess the frequency, and exhaustive studies of each clinical manifestation are required to estimate severity.
3.9 Viral factors
The susceptibility to SARS-CoV-2 infection and the severity of COVID-19 are contingent upon various variables and diverse factors pertaining to both the host and the virus. Essential factors encompass viral load, replication rate, immune evasion capacity, and the pathogen’s virulence. Concurrently, host-related factors, including age, sex, immune status, comorbidities, smoking habits, and genetics, among others, play crucial roles in determining the infection’s outcome and the disease’s progression.
At the beginning of the pandemic, SARS-CoV-2 had high fidelity in the replication of its genome, but as it spread, it interacted with many other hosts, which caused this virus to mutate and generate new high-impact variants (354–358). In this sense, the reproduction number (R0) value for the first variants was ~2.9, but as it spread and generated new variants, the R0 increased, becoming more significant than 7.0 in the later variants.
SARS-CoV-2 variants tend to have different distinctive features, such as better affinity with ACE2, more resistance to the environment, and greater ability to evade the immune response, among other effects. There is still the possibility of mutations that alter the course of infection, leading to lower efficacy of current vaccines and prophylactic measures, which may ultimately lead to a new variant not affected by current vaccines (Table 1) (359–361). It has been described that the Alpha variant contains mutations in sub-genomic RNA, improving efficiency and immune evasion. Other mutations, such as those associated with ORF 3a, ORF 6, and ORF 7a, provide a better ability to evade the immune response, highlighting the need to keep this viral agent under continuous investigation (362, 363).
Although SARS-CoV-2 encodes an RdRp complex and additional non-structural protein complexes that provide corrective activities, increasing the fidelity of viral replication, this fidelity does not always fulfill its corrective function by producing mutations in the genome and generating new variants with different capabilities (354–358). In the Delta and Omicron variants, the R0 was ~2.79 and ~7.0, respectively, suggesting that each variant’s mutation rate is increasing (357, 364). Other factors that modulate circulation and the appearance of variants include immunocompromised people and the implementation of COVID-19 vaccination.
4 Host-related risk factors
According to the literature, commonly reported risk factors in severe cases of COVID-19 include comorbidities such as kidney injury, chronic obstructive pulmonary disease (COPD), hypertension, diabetes, cardiovascular diseases, cancer, and obesity. Also, the gender and age of the patients are considered risk factors (365, 366).
4.1 Gender
It is known that there are immunological differences between men and women in susceptibility to certain infections due to, among other factors, anatomic characteristics of each gender (such as the width of the respiratory tract), metabolism, etc. (366). Although SARS-CoV-2 has been reported to affect men and women equally, more severe cases of COVID-19 occur in men more frequently compared to women, possibly due to the anatomy and physiology of each gender (367). Genetics may also be involved in the pathological differences and their respective severities. It is known that the X chromosome encodes genes related to innate and adaptive immune responses, since the genes for chemokine receptors (CXCR) 3, Toll-like receptors (TLR) 7 and CD40 ligand (CD40L) are expressed in it (368, 369). On the other hand, unlike women, a generalized inflammatory response can occur in men, probably due to immune plasticity associated with patterns of expression of genes encoded on the X chromosome (370, 371). These differences are also reflected at the level of hormones, where androgens, mostly produced in men, can increase the expression of ACE2 and TMPRSS2 in lung epithelial cells. Moreover, testosterone has been shown to decrease the immune response (370–372). On the contrary, estrogen, considered one of the female sex hormones, has anti-inflammatory properties that lead to a higher degree of immune modulation and induces a downregulation of ACE2 expression on type 2 pneumocytes, endothelial cells and kidney cells, among others (372–375). Immunological differences between men and women have been shown to affect susceptibility to infectious diseases, the immune response produced against pathogens and the effectiveness that certain vaccines can provide (366–369). In conclusion, the role of gender in the severity of COVID-19 is still unclear, so it is essential to continue studying it.
4.2 Obesity
It is important to consider that the underlying characteristics of individuals will dictate the physiological response to infection. Clinical evidence has shown that obesity is a factor to consider when defining the susceptibility and severity of COVID-19. Specifically, there is a Body Mass Index (BMI) scale, in which individuals with: a BMI of 18.5 to 24.9 kg/m2 are not obese; a BMI of 25 to 29.9 kg/m2 are overweight; a BMI of 30 to 34.9 kg/m2 have a type I obesity; a BMI of 35 to 34.9 kg/m2 have a type II obesity; and a BMI ≥ 40 kg/m2 have a grade III obesity. Likewise, degrees of obesity have been related to a less favorable prognosis during COVID-19 compared to those individuals with a BMI less than 29.9 kg/m2. Moreover, patients with a BMI ≥ 30 kg/m2 have shown greater admission rates to the Intensive Care Unit (ICU) compared to those with a lower BMI (376, 377).
Indeed, a high BMI has been correlated with complications such as increased respiratory symptoms, kidney damage, coagulopathies, thromboembolisms, and increased mortality in patients with COVID-19. Furthermore, obesity is commonly associated with metabolic alterations such as insulin resistance, alteration of serum glucose levels, dyslipidemia, etc. These health alterations, combined with the stress caused by the SARS-CoV-2 infection itself, can lead to a worsening of comorbidities or the appearance of new pathologies, such as NOD (376–378).
Furthermore, obesity is also characterized by a dysfunction of the immune system. In fact, it has been reported that patients with a high BMI have been correlated with a higher frequency of the anti-inflammatory CD4+ T cell subsets Th2 and regulatory T cells that can make it difficult to resolve a viral infection, such as that caused by SARS-CoV-2 (376, 378). Moreover, SARS-CoV-2-specific IgG antibodies have been reported to be negatively associated with BMI in COVID-19 patients. Likewise, there has also been shown a positive correlation between serum C-reactive protein (CRP), serum amyloid protein, and ferritin levels with BMI, which could be used as predictive biomarkers for dysfunctional immunity and severity of COVID-19 (378, 379). Besides that, the excess of adipose tissue has been defined as a chronic state of inflammation or “low-grade inflammation”, where proinflammatory cells (macrophages, dendritic cells, cytotoxic T cells, and Th1 cells) accumulate in it, particularly in visceral fat. Therefore, this pro-inflammatory environment in patients with higher BMI favors the amplification of the COVID-19 cytokine storm and, in turn, the exacerbation of the severity of the disease (379–381). Specifically, secretion of IL-6 and high levels of leptin, TNF-α, CXCL-10 have been observed in obese patients with a severe COVID-19 disease.
In addition, ACE-2 is highly expressed in visceral fat, suggesting an important role of adipose tissue in the severity of COVID-19. However, studies on the effect of overexpression of ACE2 in this type of tissue are still inconclusive (381). Nevertheless, it has been suggested that due to ACE-2 expression, adipocytes act as a reservoir of SARS-CoV-2, making more difficult viral clearance.
Furthermore, adipocytes can secrete TNF-α, CXCL10 and leptin. The latter is an immunoregulatory protein since several immune cells express the receptor for this protein. Leptin, when is activated, produces the induction of different downstream signaling pathways such as: the Janus Kinase (JAK) signal transducer and the STAT pathway; the phosphatidylinositol-3-kinase (PI3K) pathway; and the Mitogen-Activated Kinase (MAPK) pathway. As a result, an inflammatory phenotype is induced, including the secretion of IL-6, IL-12, and TNF-α by monocytes and ROS production by neutrophils, which stimulates activators of CD4+ T and CD8+ T cells. Additionally, adiponectin decreases in obesity, favoring a dysregulation of endothelial homeostasis and an exaggerated inflammatory response. On the other hand, weight gain could lead to leptin resistance, which, in turn, increases the frequency of Treg cells in tissues, leading to an immunosuppressive phenotype. Consequently, the effective elimination of SARS-CoV-2 infection is affected, increasing the severity of the disease (382, 383). Therefore, adipokine dynamics in obese individuals contribute to an immunological imbalance, which is relevant to the outcome of COVID-19 patients with this condition. Taken together, ACE2 overexpression in visceral fat, metabolic alterations, and immune imbalance may contribute to a poor prognosis during and after SARS-CoV-2 infection in obese COVID-19 patients (377, 383, 384).
4.3 Age
Some reports suggest that the severity of COVID-19 varies with age, with older adults facing an increased risk and severity of the disease, as well as a higher likelihood of experiencing sequelae and death compared to children and young adults. However, it is worth noting that the impact of COVID-19 on children remains a topic of debate and ongoing research (377, 384, 385). It has been postulated that the variation in the severity of COVID-19, between different age groups, could be attributed to immune immaturity in children and immune senescence in older adults (386, 387). Interestingly, although both age groups receive similar viral loads after SARS-CoV-2 infection, children tend to be asymptomatic or present mild symptoms (unless they develop a pathology known as Chronic Multisystem Inflammatory Syndrome, which is associated with unfavorable outcomes in children under 5 years of age) (388). In patients under 20 years of age, increased levels of CXCL10 and GM-CSF, together with T and B cells in the blood, produce a hyperreactive immune response, causing damage to different tissues (386).
4.4 Diabetes
It has been reported that high blood glucose levels impair the immune response. Therefore, diabetes favors infectious diseases, which are directly related to an increase in the severity and mortality of COVID-19 patients (389). In fact, patients with abnormal blood glucose levels tend to have coagulation imbalance, endothelial dysfunction, and alveolar and extrapulmonary macroangiopathy (390–392). In patients with type 2 diabetes, neutrophils, macrophages, monocytes and NK cells have altered function, decreasing the release of cytokines and IFN-α necessary to eliminate viral infections (393–396). Furthermore, metabolic alteration of glucose and lipids leads to a pro-inflammatory state by altering the synthesis of pro-inflammatory cytokines of immune cells (396, 397). Particularly, it has been postulated that diabetes affects antibodies at the glycosylation level of immunoglobulins, which increases the action of the Major Histocompatibility Complex (MHC) Class I on myeloid cells, contributing to the development of a hyperreactive immune response (24, 398, 399). Although the evidence on whether glucose levels increase susceptibility to SARS-CoV-2 infection is inconclusive, greater severity of COVID-19 has been reported in hyperglycemic patients (390). A possibility for this increase in disease severity may be that these patients have hypercoagulopathies, leukocytosis, and neutrophilia, among others (25, 26). In addition, patients with hyperglycemia increase the expression of ACE2 in the pulmonary, renal, and cardiac systems (391, 392). Of particular interest is NOD, which, as mentioned above, has been associated with SARS-CoV-2. Although the exact mechanisms by which it develops are not entirely clear, the main theories point to undiagnosed (latent) diabetes that worsens after COVID-19. Other theories point to hyperglycemia due to metabolic stress and damage to the β cells of the pancreas due to the direct action of the virus or the indirect action of the immune system (298, 392). While scientific evidence is inconclusive on susceptibility to SARS-CoV-2 infection in diabetic patients, there is a correlation between the severity of COVID-19 disease and blood glucose and insulin levels (27, 400). In addition, insulin affects blood pressure, which in turn causes activation of the RAAS, further increasing this multisystem effect and ACE2 concentrations in the body.
4.5 Nephropathy
Patients with kidney problems have been observed to present significant clinical manifestations during COVID-19, often resulting in complications such as acute kidney injury (AKI). This nephrological complication is characterized by altered values of the estimated Glomerular Filtration Rate (eGFR), serum creatinine, proteinuria, hematuria, and abnormal blood urea (BUN) levels. In particular, people with AKI are more likely to require ICU admission and tend to experience less favorable outcomes after SARS-CoV-2 infection (28, 392). One hypothesis explaining this phenomenon suggests that as the virus infects various cells and tissues, subsequent cell death releases waste materials, further contributing to the overactive immune response. These waste products and substances reach the kidney, causing this organ to function at an accelerated rate. Likewise, since the virus can still infect kidney cells and compromise their normal function, the organ becomes stressed and eventually develops AKI (28, 30, 401). Clinical management is crucial to mitigate the severity of kidney injury, which requires careful monitoring of kidney function in patients with COVID-19. Reports indicate patients may continue to shed viral loads for prolonged periods after COVID-19. This persistence could result from a dysfunctional immune response during SARS-CoV-2 infection. Alternatively, the virus may remain dormant in the system or adopt a latent form in airway epithelial cells, which could explain the epithelial damage and rearrangement observed in the post-COVID-19 period. Another possibility is that the virus persists in the cells of the nervous system, which could explain the consequences and damage observed after SARS-CoV-2 infection in sensory functions of the body, such as taste or smell (31, 32). Patients with acute renal failure (ARF) have also been reported to have a higher rate of SARS-CoV-2 infection than those without ARF. In addition, patients undergoing ARF, or chronic kidney disease (CKD) are reported to have a higher frequency of ICU admission and death (33, 36).
4.6 Cardiovascular disease
SARS-CoV-2 has the potential to directly and indirectly affect the cardiovascular system. On the one hand, direct effects include the destruction of cells of the cardiovascular system that express ACE2 and TMPRRS2. On the other hand, the virus indirectly affects the circulatory system. As the infection of SARS-CoV-2 progresses, there is an increased risk of cardiovascular complications, such as elevated coagulopathies due to alterations in the coagulation cascade and an elevated likelihood of inflammation in various tissues, particularly in the endothelial and vascular systems. This heightened inflammation contributes to cardiovascular events, including myocardial infarction, venous thrombosis, and coronary damage. Individuals with pre-existing cardiovascular conditions during COVID-19 have been reported to exhibit altered levels of biomarkers such as C-reactive protein (CRP), D-dimer (DD), tissue factor (TF), and Von Willebrand factor (VWF). These changes in biomarker levels indicate the potential impact of SARS-CoV-2 on the cardiovascular system, emphasizing the need for comprehensive monitoring and treatment in individuals with cardiovascular conditions (34, 402, 403).
It has been shown that people with pre-existing conditions such as coronary heart disease, high blood pressure, history of cardiovascular disease, or coronary heart disease are more likely to develop post-COVID-19 sequelae (43–45). As previously discussed, in the renal section, cell death results in the release of potassium into the bloodstream. Under normal circumstances, the kidneys filter potassium. However, when the kidneys are overloaded, their functioning is impaired. The increased potassium levels in the blood activate voltage-gated potassium channels (KV), inducing an altered electrical response in muscle fibers. This alteration affects blood pressure by causing a decrease in adenosine triphosphate (ATP), subsequently activating ATP-dependent potassium channels (KATP). Activation of KATP leads to a reduction in heart muscle contractions, which contributes to a decrease in blood pressure. This decrease in blood pressure activates the RAAS system, causing the synthesis of ACE2. When SARS-CoV-2 induces the activation of the RAAS system, it triggers the depletion of ACE2 on the cell surface, increasing angiotensin I in the blood, which could contribute to the development of atherosclerosis (43–45).
Atherosclerosis can manifest itself through several mechanisms: a) First, ACE2, by converting angiotensin I to angiotensin II, helps raise blood pressure. However, if there are low levels of ACE2, angiotensin II is not produced, consequently angiotensin I levels will increase; b) Secondly, ACE2 is an enzyme that facilitates the removal of cholesterol from the arteries. In the absence or reduction of ACE2, cholesterol can accumulate in the arteries, leading to plaque formation; and c) Third, ACE2 functions as an enzyme that plays a protective role for endothelial cells, which line the inside of arteries. Therefore, when ACE2 is deficient or decreased, endothelial cells may become more vulnerable to injury, contributing to plaque formation. The reduction in ACE2 expression induced by the virus may thus contribute to the onset of atherosclerosis through multiple pathways, further complicating the symptomatology of COVID-19 (34, 35, 46).
4.7 Immunocompromised conditions
Immunocompromised individuals who may have this condition due to a genetic disorder or having an underlying pathology, such as one caused by a pathogen or having received a transplant, constitute a group of particular interest. This is attributed to the alteration observed in immune responses within this population, which subsequently influences the clinical course of various pathologies.
a) Immune disorders due to congenital problems: More than 95 inborn errors of immunity, also called primary immunodeficiency syndromes, have been reported (47). These genetic disorders are a group of conditions that impact the immune system from birth. These disorders compromise the immune system’s ability to effectively fight pathogens, making people more susceptible to recurrent infections and, in some cases, ending up with autoimmune conditions. In the context of COVID-19, people with these genetic disorders have been reported to experience prolonged durations of SARS-CoV-2 infection. Prolonged virus shedding times after infection cause a higher likelihood of severe illness, less favorable prognoses for resolution of infection, and a higher risk of developing serious complications after COVID-19, including pneumonia and sepsis, among others (49, 50, 404).
b) Immune disorders due to infectious agents: Certain infectious agents, such as the human immunodeficiency virus (HIV) or bacteria such as Streptococcus pneumoniae, can weaken the immune system, either by suppressing or altering the body’s immune response. Theoretically, it has been postulated that individuals in this group may experience greater severity of COVID-19, due to the combined impact of the damage caused by their existing pathogens, the effects of the virus itself, and the compromised immune system. Additionally, other opportunistic infectious agents may take advantage of this depressed immune state. This complex condition poses challenges for healthcare, as treatments must target not only the SARS-CoV-2 infection, but also the coexisting pathogens that contribute to comorbidities in these patients (49–52, 404).
c) Immunological disorders due to immunosuppressants: Patients who undergo organ or cell transplants, such as hematopoietic cells (HTC), or solid transplants (SOT), have a higher rate of admission to the ICU and a greater severity of COVID-19 (50). It is thought that in these patients, SARS-CoV-2 has a longer viral replication time and, therefore, a greater viral load is produced, leading to more significant and sustained pathology (49). However, the results of this type of research that have been carried out so far remain ambiguous. For example, other studies have reported low rates of SARS-CoV-2 infection among transplant patients. Although, this can be attributed to the strict health measures implemented for these patients (53).
Therefore, more extensive investigations are required to elucidate the mechanisms behind the pathogenesis of SARS-CoV-2 infection in immunocompromised patients.
4.8 Microbiota
It is well known that several microorganisms live in a symbiotic relationship in the human body, called Microbiota. These microorganisms produce molecules and vitamins, such as vitamin K, which are used by the human body and, in turn, provide nutrients and protection to these microorganisms (54). The Microbiota is characteristic and specific to everyone. This microbiota aids digestion and protects the host from possible pathogenic microorganisms (55). It has been reported that the microbiota has immunoregulatory functions. In fact, changes in this microbiota induce a dysbiosis that affects the host by exposing it to opportunistic pathogens, or triggering altered immune responses such as allergies or inflammatory bowel disease (IBD), which increases the probability of secondary diseases, such as obesity and diabetes (54–56).
During SARS-CoV-2 infection, bacterial co-infections or dysbiosis may occur, contributing to increased mortality and morbidity in individuals. This effect has been previously reported in CoVs such as SARS-CoV and MERS-CoV, with co-infections of each of the respective viruses together with respiratory tract pathogenic bacteria such as Streptococcus pneumoniae and Staphylococcus aureus (57). Concentrations and types of microbial populations vary upon a viral infection. Specifically, it has been shown that 3 days after SARS-CoV-2 infection, Fusobacterium periodonticum in the oral cavity decreased significantly (58). It has also been reported that Faecalibacterium prausnitzii, a vital bacterium involved in anti-inflammatory processes, is negatively altered as the severity of COVID-19 increases (58, 59). Additionally, a reduction in host-typical commensal bacteria, along with increased presence of pathogens, has been correlated with increased severity of COVID-19. This association may be related to the role of commensal bacteria in shaping the immune system response, influencing the outcome of viral infections. However, the specific association between the upper respiratory tract Microbiota and the severity of COVID-19 remains to be elucidated. Therefore, further studies in this field are warranted to provide a comprehensive understanding of these relationships (58–60, 62, 405).
5 Conclusions
Humanity has undergone a protracted journey of nearly four years since the start of the COVID-19 pandemic, and fortunately, the landscape has undergone significant positive changes. Despite the WHO declaring the “end of COVID-19 as a public health emergency,” the virus persists, circulating and causing loss of lives while undergoing mutations, albeit to a lesser extent due to widespread vaccination and prophylactic measures. This underscores that SARS-CoV-2 remains a threat, albeit reduced from its earlier impact. Despite the wealth of current evidence and information, it remains uncertain whether the virus could stage a resurgence and evolve into an endemic or seasonal presence. Clinical and laboratory biomarkers have played a crucial role in assessing infection progression, enabling improved clinical management, and facilitating tailored therapies based on individual patient conditions (63–65). Although the fatality and infection rate of SARS-CoV-2 has decreased compared to the beginning of the pandemic and previous years thanks to the efficiency of vaccines, strains, and variants that are capable of avoiding this therapeutic measure still continue to emerge, which raises doubts about whether future booster doses are necessary despite the appearance of large epidemiological outbreaks of cases, and highlights the need to establish continuous surveillance systems against this pathogen and others with pandemic potential (66, 67). Currently, the most pressing concern lies in the aftermath of COVID-19, with common sequelae including pulmonary fibrosis, respiratory distress, pulmonary thromboembolism, damage to the epithelium, and embolisms. This underscores the need for a comprehensive understanding of the consequences of SARS-CoV-2 infection and the management of post-COVID complications (Table 3) (10).
In conclusion, this review presents a comprehensive overview of the factors leading to severe or chronic COVID-19. We have explored the disease from its pathophysiology, considering both viral and host factors, to the complications caused by SARS-CoV-2 infection, particularly in the context of the post-pandemic period of COVID-19. Additionally, this work emphasizes the importance of remaining vigilant to the circulation of variants and the overall impact of SARS-CoV-2 infection on the global population, especially in people with comorbidities or at high risk.
Author contributions
JC: Writing – original draft, Writing – review & editing. VG-C: Writing – original draft, Writing – review & editing. SC: Writing – original draft. CC-V: Writing – original draft. CS-A: Writing – review & editing. AA: Writing – original draft. CM: Writing – review & editing. AR-D: Writing – review & editing. SB: Writing – review & editing. PG: Writing – review & editing. AK: Writing – review & editing. ML: Writing – original draft, Writing – review & editing.
Funding
The author(s) declared financial support was received for the research, authorship, and/or publication of this article. We would like to thank the Regional Government of Antofagasta through the Innovation Fund for Competitiveness FIC-R 2017 (BIP Code: 30488811–0); Copec-UC 2020.R.001; National Research and Development Agency (ANID) - Millennium Science Initiative Program, Millennium Institute on Immunology and Immunotherapy (ICN09_016/ICN 2021_045; former P09/016-F); “Fondecyt Regular #1240971; and the VRIIP Strengthening Plan code ANT20992”.
Acknowledgments
Dr. Darian K. González, Faculty of Sciences, Natural Resources and Veterinary Medicine, Universidad Santo Tomás, Viña del Mar, Chile. For his contribution to this manuscript with her expertise. Sw. Elías R. Navea, scholarships, and credits department, AIEP professional institute, Calama. For your support and help at different stages of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
2. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. (2020) 382:727–33. doi: 10.1056/NEJMoa2001017
3. Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Drosten C, Gulyaeva AA, et al. Severe acute respiratory syndrome-related coronavirus: The species and its viruses–a statement of the Coronavirus Study Group. BioRxiv. (2020). doi: 10.1101/2020.02.07.937862
4. Petersen E, Koopmans M, Go U, Hamer DH, Petrosillo N, Castelli F, et al. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect Dis. (2020) 20:e238–44. doi: 10.1016/S1473-3099(20)30484-9
5. Hu B, Guo H, Zhou P, Shi Z-L. Characteristics of SARS-coV-2 and COVID-19. Nat Rev Microbiol. (2021) 19:141–54. doi: 10.1038/s41579-020-00459-7
6. Zhou Z, Qiu Y, Ge X. The taxonomy, host range and pathogenicity of coronaviruses and other viruses in the Nidovirales order. Anim Dis. (2021) 1:5. doi: 10.1186/s44149-021-00005-9
7. Wu Y, Ho W, Huang Y, Jin D-Y, Li S, Liu S-L, et al. SARS-CoV-2 is an appropriate name for the new coronavirus. Lancet. (2020) 395:949–50. doi: 10.1016/S0140-6736(20)30557-2
9. Flerlage T, Boyd DF, Meliopoulos V, Thomas PG, Schultz-Cherry S. Influenza virus and SARS-CoV-2: pathogenesis and host responses in the respiratory tract. Nat Rev Microbiol. (2021) 19:425–41. doi: 10.1038/s41579-021-00542-7
10. Abdelrahman Z, Li M, Wang X. Comparative review of SARS-CoV-2, SARS-CoV, MERS-CoV, and influenza A respiratory viruses. Front Immunol. (2020) 11:552909. doi: 10.3389/fimmu.2020.552909
11. Clayton E, Ackerley J, Aelmans M, Ali N, Ashcroft Z, Ashton C, et al. Structural bases of zoonotic and zooanthroponotic transmission of SARS-CoV-2. Viruses. (2022) 14. doi: 10.3390/v14020418
12. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. (2020) 395:565–74. doi: 10.1016/S0140-6736(20)30251-8
13. Benvenuto D, Angeletti S, Giovanetti M, Bianchi M, Pascarella S, Cauda R, et al. Evolutionary analysis of SARS-CoV-2: how mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J Infect. (2020) 81:e24–7. doi: 10.1016/j.jinf.2020.03.058
14. Frutos R, Pliez O, Gavotte L, Devaux CA. There is no “origin” to SARS-CoV-2. Environ Res. (2022) 207:112173. doi: 10.1016/j.envres.2021.112173
15. Lam TT, Jia N, Zhang YW, Shum MH, Jiang JF, Zhu HC, et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature. (2020) 583:282–5. doi: 10.1038/s41586-020-2169-0
16. Jo WK, de Oliveira‐Filho EF, Rasche A, Greenwood AD, Osterrieder K, Drexler JF. Potential zoonotic sources of SARS-CoV-2 infections. Transboundary emerging Dis. (2021) 68:1824–34. doi: 10.1111/tbed.13872
17. Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. (2002) 417:822–8. doi: 10.1038/nature00786
18. Peron JPS, Nakaya H. Susceptibility of the elderly to SARS-CoV-2 infection: ACE-2 overexpression, shedding, and antibody-dependent enhancement (ADE). Clinics. (2020) 75:e1912. doi: 10.6061/clinics/2020/e1912
19. Liu LP, Zhang XL, Li J. New perspectives on angiotensin-converting enzyme 2 and its related diseases. World J Diabetes. (2021) 12:839–54. doi: 10.4239/wjd.v12.i6.839
20. Vargas-Gandica J, Winter D, Schnippe R, Rodriguez-Morales AG, Mondragon J, Escalera-Antezana JP, et al. Ageusia and anosmia, a common sign of COVID-19? A case series from four countries. J Neurovirol. (2020) 26:785–9. doi: 10.1007/s13365-020-00875-8
21. Acosta RAH, Garrigos ZE, Marcelin JR, Vijayvargiya P. COVID-19 pathogenesis and clinical manifestations. Infect Dis Clinics. (2022) 36:231–49. doi: 10.1016/j.idc.2022.01.003
22. Kadirvelu B, Burcea G, Quint JK, Costelloe CE, Faisal AA. Covid-19 does not look like what you are looking for: clustering symptoms by nation and multi-morbidities reveal substantial differences to the classical symptom triad. medRxiv. (2021). doi: 10.1101/2021.04.02.21254818
23. Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. New Engl J Med. (2020) 382:1199–207. doi: 10.1056/NEJMoa2001316
24. Hao W, Gladstone P, Engardt S, Greenbaum C, Palmer JP. Major histocompatibility complex class I molecule expression is normal on peripheral blood lymphocytes from patients with insulin-dependent diabetes mellitus. J Clin Invest. (1996) 98:1613–8. doi: 10.1172/JCI118955
25. Lui DTW, Li TK, Lee CH, Chow ES, Lee ACH, Tam AR, et al. A prospective study of the impact of glycaemic status on clinical outcomes and anti-SARS-CoV-2 antibody responses among patients with predominantly non-severe COVID-19. Diabetes Res Clin Pract. (2022) 185:109232. doi: 10.1016/j.diabres.2022.109232
26. Lampasona V, Secchi M, Scavini M, Bazzigaluppi E, Brigatti C, Marzinotto I, et al. Antibody response to multiple antigens of SARS-CoV-2 in patients with diabetes: an observational cohort study. Diabetologia. (2020) 63:2548–58. doi: 10.1007/s00125-020-05284-4
27. Huang Y, Yang C, Xu XF, Xu W, Liu SW. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin. (2020) 41:1141–9. doi: 10.1038/s41401-020-0485-4
28. Cai R, Zhang J, Zhu Y, Liu L, Liu Y, He Q. Mortality in chronic kidney disease patients with COVID-19: a systematic review and meta-analysis. Int Urol Nephrol. (2021) 53:1623–9. doi: 10.1007/s11255-020-02740-3
29. Kumar S, Karuppanan K, Subramaniam G. Omicron (BA. 1) and sub-variants (BA. 1.1, BA. 2, and BA. 3) of SARS-CoV-2 spike infectivity and pathogenicity: A comparative sequence and structural-based computational assessment. J Med Virol. (2022) 94(10):4780–91.
30. Carlson N, Nelveg-Kristensen KE, Ballegaard Freese E, Feldt-Rasmussen B, Hornum M, Kamper AL, et al. Increased vulnerability to COVID-19 in chronic kidney disease. J Intern Med. (2021) 290:166–78. doi: 10.1111/joim.13239
31. Rincon-Arevalo H, Choi M, Stefanski AL, Halleck F, Weber U, Szelinski F, et al. Impaired humoral immunity to SARS-CoV-2 BNT162b2 vaccine in kidney transplant recipients and dialysis patients. Sci Immunol. (2021) 6:eabj1031. doi: 10.1126/sciimmunol.abj1031
32. Sanders JF, Bemelman FJ, Messchendorp AL, Baan CC, van Baarle D, van Binnendijk R, et al. The RECOVAC immune-response study: the immunogenicity, tolerability, and safety of COVID-19 vaccination in patients with chronic kidney disease, on dialysis, or living with a kidney transplant. Transplantation. (2022) 106:821–34. doi: 10.1097/TP.0000000000003983
33. Dessie ZG, Zewotir T. Mortality-related risk factors of COVID-19: a systematic review and meta-analysis of 42 studies and 423,117 patients. BMC Infect Dis. (2021) 21:855. doi: 10.1186/s12879-021-06536-3
34. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. (2020) 8:475–81. doi: 10.1016/S2213-2600(20)30079-5
35. Inciardi RM, Adamo M, Lupi L, Cani DS, Di Pasquale M, Tomasoni D, et al. Characteristics and outcomes of patients hospitalized for COVID-19 and cardiac disease in Northern Italy. Eur Heart J. (2020) 41:1821–9. doi: 10.1093/eurheartj/ehaa600
36. Meng B, Abdullahi A, Ferreira I, Goonawardane N, Saito A, Kimura I, et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature. (2022) 603:706–14. doi: 10.1038/s41586-022-04474-x
37. Bansal M. Cardiovascular disease and COVID-19. Diabetes Metab Syndrome: Clin Res Rev. (2020) 14:247–50. doi: 10.1016/j.dsx.2020.03.013
38. Lau JJ, et al. Real-world COVID-19 vaccine effectiveness against the Omicron BA. 2 variant in a SARS-CoV-2 infection-naive population. Nat Med. (2023) 29(2):348–57.
39. Tegally H, et al. Emergence of SARS-CoV-2 omicron lineages BA. 4 and BA. 5 in South Africa. Nat Med. (2022) 28(9):1785–90.
40. Sabbatucci M, et al. Omicron variant evolution on vaccines and monoclonal antibodies. Inflammopharmacology. (2023) 31(4):1779–88.
41. Wang Q, et al. Antibody evasion by SARS-CoV-2 Omicron subvariants BA. 2.12. 1, BA. 4 and BA. 5. Nature. (2022) 608(7923):603–8.
42. Cao Y, et al. BA. 2.12. 1, BA. 4 and BA. 5 escape antibodies elicited by Omicron infection. Nature. (2022) 608(7923):593–602.
43. Sardu C, Gambardella J, Morelli MB, Wang X, Marfella R, Santulli G, et al. Hypertension, thrombosis, kidney failure, and diabetes: is COVID-19 an endothelial disease? A comprehensive evaluation of clinical and basic evidence. J Clin Med. (2020) 9:1417. doi: 10.3390/jcm9051417
44. Nishiga M, Wang DW, Han Y, Lewis DB, Wu JC. COVID-19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat Rev Cardiol. (2020) 17:543–58. doi: 10.1038/s41569-020-0413-9
45. Li Y, Li M, Wang M, Zhou Y, Chang J, Xian Y, et al. Acute cerebrovascular disease following COVID-19: a single center, retrospective, observational study. Stroke Vasc Neurol. (2020) 5:279–84. doi: 10.1136/svn-2020-000431
46. Khera A, Baum SJ, Gluckman TJ, Gulati M, Martin SS, Michos ED, et al. Continuity of care and outpatient management for patients with and at high risk for cardiovascular disease during the COVID-19 pandemic: A scientific statement from the American Society for Preventive Cardiology. Am J Prev Cardiol. (2020) 1:100009. doi: 10.1016/j.ajpc.2020.100009
47. Barreto ICDP, Barreto BAP, Cavalcante EGN, Neto AC. Immunological deficiencies: more frequent than they seem to be. Jornal Pediatria. (2021) 97:49–58. doi: 10.1016/j.jped.2020.10.009
48. Suryadevara N, Shrihari S, Gilchuk P, VanBlargan LA, Binshtein E, Zost SJ, et al. Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell. (2021) 184:2316–2331.e15. doi: 10.1016/j.cell.2021.03.029
49. Milota T, Sobotkova M, Smetanova J, Bloomfield M, Vydlakova J, Chovancova Z, et al. Risk factors for severe COVID-19 and hospital admission in patients with inborn errors of immunity-results from a multicenter nationwide study. Front Immunol. (2022) 13:835770. doi: 10.3389/fimmu.2022.835770
50. Belsky JA, Tullius BP, Lamb MG, Sayegh R, Stanek JR, Auletta JJ, et al. COVID-19 in immunocompromised patients: A systematic review of cancer, hematopoietic cell and solid organ transplant patients. J Infect. (2021) 82:329–38. doi: 10.1016/j.jinf.2021.01.022
51. Mitiku H, Weldegebreal F, Teklemariam Z. Magnitude of opportunistic infections and associated factors in HIV-infected adults on antiretroviral therapy in eastern Ethiopia. In: HIV/AIDS-Research and Palliative Care (2015). p. 137–44.
52. Chepkondol GK, Jolly PE, Yatich N, Mbowe O, Jaoko WG. Types and prevalence of HIV-related opportunistic infections/conditions among HIV-positive patients attending Kenyatta National Hospital in Nairobi, Kenya. Afr Health Sci. (2020) 20:615–24. doi: 10.4314/ahs.v20i2.9
53. Groban L, Wang H, Sun X, Ahmad S, Ferrario CM. Is sex a determinant of COVID-19 infection? Truth myth? Curr Hypertension Rep. (2020) 22:1–12. doi: 10.1007/s11906-020-01073-x
54. Berg G, Rybakova D, Fischer D, Cernava T, Verges MC, Charles T, et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome. (2020) 8:1–22. doi: 10.1186/s40168-020-00875-0
55. Mousa WK, Chehadeh F, Husband S. Recent advances in understanding the structure and function of the human microbiome. Front Microbiol. (2022) 13:825338. doi: 10.3389/fmicb.2022.825338
56. Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB, et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature. (2017) 550:61–6. doi: 10.1038/nature23889
57. Guarner J. Three emerging coronaviruses in two decades: the story of SARS, MERS, and now COVID-19. Oxford University Press US (2020) p. 420–1.
58. Moore SC, Penrice-Randal R, Alruwaili M, Dong X, Pullan ST, Carter DP, et al. Amplicon based MinION sequencing of SARS-CoV-2 and metagenomic characterisation of nasopharyngeal swabs from patients with COVID-19. MedRxiv. (2020). doi: 10.1101/2020.03.05.20032011
59. Zuo T, Zhang F, Lui GCY, Yeoh YK, Li AYL, Zhan H, et al. Alterations in gut microbiota of patients with COVID-19 during time of hospitalization. Gastroenterology. (2020) 159:944–955 e8. doi: 10.1053/j.gastro.2020.05.048
60. Ventero MP, Cuadrat RRC, Vidal I, Andrade BGN, Molina-Pardines C, Haro-Moreno JM, et al. Nasopharyngeal microbial communities of patients infected with SARS-CoV-2 that developed COVID-19. Front Microbiol. (2021) 12:637430. doi: 10.3389/fmicb.2021.637430
61. Shulman RJ. Distinct systemic and mucosal immune responses during acute SARS-coV-2 infection. Israel Med Assoc J. (2021), 751–1.
62. Shilts MH, Rosas-Salazar C, Strickland BA, Kimura KS, Asad M, Sehanobish E, et al. Severe COVID-19 is associated with an altered upper respiratory tract microbiome. Front Cell infection Microbiol. (2022) 11:781968. doi: 10.3389/fcimb.2021.781968
63. Danwang C, Endomba FT, Nkeck JR, Wouna DLA, Robert A, Noubiap JJ, et al. A meta-analysis of potential biomarkers associated with severity of coronavirus disease 2019 (COVID-19). biomark Res. (2020) 8:1–13. doi: 10.1186/s40364-020-00217-0
64. Kumar D, Pandit R, Sharma S, Raval J, Patel Z, Joshi M, et al. Nasopharyngeal microbiome of COVID-19 patients revealed a distinct bacterial profile in deceased and recovered individuals. Microb Pathog. (2022) 173:105829. doi: 10.1016/j.micpath.2022.105829
65. Mølhave M, Agergaard J, Wejse C. Clinical management of COVID-19 patients–an update. In: Seminars in nuclear medicine. Elsevier (2022) 2(1):4–10.
66. Daria S, Islam MR. The SARS-CoV-2 omicron wave is indicating the end of the pandemic phase but the COVID-19 will continue. J Med Virol. (2022) 94:2343. doi: 10.1002/jmv.27635
67. Darmarajan T, Paudel KJ, Candasamy M, Chellian J, Madheswaran T, Sakthivel LP, et al. Autoantibodies and autoimmune disorders in SARS-CoV-2 infection: pathogenicity and immune regulation. Environ Sci pollut Res. (2022) 29:54072–87. doi: 10.1007/s11356-022-20984-7
68. Antonelli M, Pujol JC, Spector TD, Ourselin S, Steves CJ. Risk of long COVID associated with delta versus omicron variants of SARS-CoV-2. Lancet. (2022) 399:2263–4. doi: 10.1016/S0140-6736(22)00941-2
69. Vihta KD, Pouwels KB, Peto TE, Pritchard E, House T, Studley R, et al. Omicron-associated changes in SARS-CoV-2 symptoms in the United Kingdom. Clin Infect Dis. (2022) 76:e133–41. doi: 10.1101/2022.01.18.22269082
70. Han Q, Zheng B, Daines L, Sheikh A. Long-term sequelae of COVID-19: a systematic review and meta-analysis of one-year follow-up studies on post-COVID symptoms. Pathogens. (2022) 11:269. doi: 10.3390/pathogens11020269
71. Nalbandian A, Sehgal K, Gupta A, Madhavan MV, McGroder C, Stevens JS, et al. Post-acute COVID-19 syndrome. Nat Med. (2021) 27:601–15. doi: 10.1038/s41591-021-01283-z
72. Mehandru S, Merad M. Pathological sequelae of long-haul COVID. Nat Immunol. (2022) 23:194–202. doi: 10.1038/s41590-021-01104-y
73. Proal AD, VanElzakker MB. Long COVID or post-acute sequelae of COVID-19 (PASC): an overview of biological factors that may contribute to persistent symptoms. Front Microbiol. (2021) 12:698169. doi: 10.3389/fmicb.2021.698169
74. Gomez-Zorita S, Milton-Laskibar I, García-Arellano L, González M, Portillo MP, et al. An overview of adipose tissue ACE2 modulation by diet and obesity. Potential implications in COVID-19 infection and severity. Int J Mol Sci. (2021) 22:7975. doi: 10.3390/ijms22157975
75. Marik PE, Iglesias J, Varon J, Kory P. A scoping review of the pathophysiology of COVID-19. Int J immunopathology Pharmacol. (2021) 35:20587384211048026. doi: 10.1177/20587384211048026
76. Acharya KP, Ghimire TR, Subramanya SH. Access to and equitable distribution of COVID-19 vaccine in low-income countries. NPJ Vaccines. (2021) 6:54. doi: 10.1038/s41541-021-00323-6
77. Nguyen KV. Problems associated with antiviral drugs and vaccines development for COVID-19: approach to intervention using expression vectors via GPI anchor. Nucleosides Nucleotides Nucleic Acids. (2021) 40:665–706. doi: 10.1080/15257770.2021.1914851
78. Phillips N. The coronavirus is here to stay–here’s what that means. Nature. (2021) 590:382–5. doi: 10.1038/d41586-021-00396-2
79. Michel CJ, Mayer C, Poch, Thompson JD. Characterization of accessory genes in coronavirus genomes. Virology J. (2020) 17:1–13.
80. Wu A, Peng Y, Huang B, Ding X, Wang X, Niu P, et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe. (2020) 27(3):325–8. doi: 10.1016/j.chom.2020.02.001
81. Fu Y-Z, Wang S-Y, Zheng Z-Q, Huang Y, Li W-W, Xu Z-S, et al. SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cell Mol Immunol. (2021) 18:613–20. doi: 10.1038/s41423-020-00571-x
82. Mu J, Xu J, Zhang L, Shu T, Wu D, Huang M, et al. SARS-CoV-2-encoded nucleocapsid protein acts as a viral suppressor of RNA interference in cells. Sci China Life Sci. (2020) 63:1413–6. doi: 10.1007/s11427-020-1692-1
83. Oh SJ, Shin OS. SARS-CoV-2 nucleocapsid protein targets RIG-I-like receptor pathways to inhibit the induction of interferon response. Cells. (2021) 10:530. doi: 10.3390/cells10030530
84. Zheng Y, Deng J, Han L, Zhuang M-W, Xu Y, Zhang J, et al. SARS-CoV-2 NSP5 and N protein counteract the RIG-I signaling pathway by suppressing the formation of stress granules. Signal transduction targeted Ther. (2022) 7:22. doi: 10.1038/s41392-022-00878-3
85. Mitchell TJ, John S. Signal transducer and activator of transcription (STAT) signalling and T-cell lymphomas. Immunology. (2005) 114:301–12. doi: 10.1111/j.1365-2567.2005.02091.x
86. Lu S, Ye Q, Singh D, Cao Y, Diedrich JK, Yates JR III, et al. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat Commun. (2021) 12:502. doi: 10.1038/s41467-020-20768-y
87. Bhat EA, Sajjad N, Ali A, Aldakheel FM, Mateen A, Alqahtani MS, et al. SARS-CoV-2: insight in genome structure, pathogenesis and viral receptor binding analysis–an updated review. Int Immunopharmacol. (2021) 95:107493. doi: 10.1016/j.intimp.2021.107493
88. Zhou S, Lv P, Li M, Chen Z, Xin H, Reilly S, et al. SARS-CoV-2 E protein: Pathogenesis and potential therapeutic development. Biomedicine Pharmacotherapy. (2023) 159:114242. doi: 10.1016/j.biopha.2023.114242
89. Shepley-McTaggart A, Sagum CA, Oliva I, Rybakovsky E, DiGuilio K, Liang J, et al. SARS-CoV-2 Envelope (E) protein interacts with PDZ-domain-2 of host tight junction protein ZO1. PloS One. (2021) 16:e0251955. doi: 10.1371/journal.pone.0251955
90. Schubert K, Karousis ED, Jomaa A, Scaiola A, Echeverria B, Gurzeler L-A, et al. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat Struct Mol Biol. (2020) 27:959–66. doi: 10.1038/s41594-020-0511-8
91. Kumar A, Ishida R, Strilets T, Cole J, Lopez-Orozco J, Fayad N, et al. SARS-CoV-2 nonstructural protein 1 inhibits the interferon response by causing depletion of key host signaling factors. J Virol. (2021) 95. doi: 10.1128/jvi.00266–21
92. Kim N-E, Kim D-K, Song Y-J. SARS-CoV-2 nonstructural proteins 1 and 13 suppress caspase-1 and the NLRP3 inflammasome activation. Microorganisms. (2021) 9:494. doi: 10.3390/microorganisms9030494
93. Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun. (2020) 11:3810. doi: 10.1038/s41467-020-17665-9
94. Angeletti S, Benvenuto D, Bianchi M, Giovanetti M, Pascarella S, Ciccozzi M. COVID-2019: the role of the nsp2 and nsp3 in its pathogenesis. J Med Virol. (2020) 92:584–8. doi: 10.1002/jmv.25719
95. Ma J, Chen Y, Wu W, Chen Z. Structure and function of N-terminal zinc finger domain of SARS-CoV-2 NSP2. Virologica Sin. (2021) 36:1104–12. doi: 10.1007/s12250-021-00431-6
96. Gupta M, Azumaya CM, Moritz M, Pourmal S, Diallo A, Merz GE, et al. CryoEM and AI reveal a structure of SARS-CoV-2 Nsp2, a multifunctional protein involved in key host processes. Res square. (2021). doi: 10.21203/rs.3.rs-515215/v1
97. Kasuga Y, Zhu B, Jang K-J, Yoo J-S. Innate immune sensing of coronavirus and viral evasion strategies. Exp Mol Med. (2021) 53:723–36. doi: 10.1038/s12276-021-00602-1
98. Lei J, Ma‐Lauer Y, Han Y, Thoms M, Buschauer R, Jores J, et al. The SARS-unique domain (SUD) of SARS-CoV and SARS-CoV-2 interacts with human Paip1 to enhance viral RNA translation. EMBO J. (2021) 40:e102277. doi: 10.15252/embj.2019102277
99. Lavigne M, Helynck O, Rigolet P, Boudria-Souilah R, Nowakowski M, Baron B, et al. SARS-CoV-2 Nsp3 unique domain SUD interacts with guanine quadruplexes and G4-ligands inhibit this interaction. Nucleic Acids Res. (2021) 49:7695–712. doi: 10.1093/nar/gkab571
100. Liu G, Du W, Sang X, Tong Q, Wang Y, Chen G, et al. RNA G-quadruplex in TMPRSS2 reduces SARS-CoV-2 infection. Nat Commun. (2022) 13:1444. doi: 10.1038/s41467-022-29135-5
101. Wolff G, Limpens RW, Zevenhoven-Dobbe JC, Laugks U, Zheng S, de Jong AW, et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science. (2020) 369:1395–8. doi: 10.1126/science.abd3629
102. Liu G, Lee J-H, Parker ZM, Acharya D, Chiang JJ, van Gent M, et al. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nat Microbiol. (2021) 6:467–78. doi: 10.1038/s41564-021-00884-1
103. Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. (2020) 587:657–62. doi: 10.1038/s41586-020-2601-5
104. Russo LC, Tomasin R, Matos IA, Manucci AC, Sowa ST, Dale K, et al. The SARS-CoV-2 Nsp3 macrodomain reverses PARP9/DTX3L-dependent ADP-ribosylation induced by interferon signaling. J Biol Chem. (2021) 297. doi: 10.1101/2021.04.06.438552
105. Hackstadt T, Chiramel AI, Hoyt FH, Williamson BN, Dooley CA, Beare PA, et al. Disruption of the Golgi apparatus and contribution of the endoplasmic reticulum to the SARS-CoV-2 replication complex. Viruses. (2021) 13:1798. doi: 10.3390/v13091798
106. Sakai Y, Kawachi K, Terada Y, Omori H, Matsuura Y, Kamitani W. Two-amino acids change in the nsp4 of SARS coronavirus abolishes viral replication. Virology. (2017) 510:165–74. doi: 10.1016/j.virol.2017.07.019
107. Chen J, Li Z, Guo J, Xu S, Zhou J, Chen Q, et al. SARS-CoV-2 nsp5 exhibits stronger catalytic activity and interferon antagonism than its SARS-CoV ortholog. J Virol. (2022) 96:e00037–22. doi: 10.1128/jvi.00037-22
108. Lu J-L, Zhou X-L. SARS-CoV-2 main protease Nsp5 cleaves and inactivates human tRNA methyltransferase TRMT1. J Mol Cell Biol. (2023) 15:mjad024. doi: 10.1093/jmcb/mjad024
109. Fung S-Y, Siu K-L, Lin H, Yeung ML, Jin D-Y. SARS-CoV-2 main protease suppresses type I interferon production by preventing nuclear translocation of phosphorylated IRF3. Int J Biol Sci. (2021) 17:1547. doi: 10.7150/ijbs.59943
110. Rui Y, Su J, Shen S, Hu Y, Huang D, Zheng W, et al. Unique and complementary suppression of cGAS-STING and RNA sensing-triggered innate immune responses by SARS-CoV-2 proteins. Signal transduction targeted Ther. (2021) 6:123. doi: 10.1038/s41392-021-00515-5
111. Wu Y, Ma L, Zhuang Z, Cai S, Zhao Z, Zhou L, et al. Main protease of SARS-CoV-2 serves as a bifunctional molecule in restricting type I interferon antiviral signaling. Signal transduction targeted Ther. (2020) 5:221. doi: 10.1038/s41392-020-00332-2
112. Moustaqil M, Ollivier E, Chiu H.-P, Tol Van S, Rudolffi-Soto P, Stevens C, et al. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): implications for disease presentation across species. Emerging Microbes infections. (2021) 10:178–95. doi: 10.1080/22221751.2020.1870414
113. Vela JM. Repurposing sigma-1 receptor ligands for COVID-19 therapy? Front Pharmacol. (2020) 11:582310. doi: 10.3389/fphar.2020.582310
114. Xia H, Cao Z, Xie X, Zhang X, Chen JY-C, Wang H, et al. Evasion of type I interferon by SARS-CoV-2. Cell Rep. (2020) 33. doi: 10.1016/j.celrep.2020.108234
115. Sun X, Liu Y, Huang Z, Xu W, Hu W, Yi L, et al. SARS-CoV-2 non-structural protein 6 triggers NLRP3-dependent pyroptosis by targeting ATP6AP1. Cell Death Differentiation. (2022) 29:1240–54. doi: 10.1038/s41418-021-00916-7
116. Zhao J, Guo S, Yi D, Li Q, Ma L, Zhang Y, et al. A cell-based assay to discover inhibitors of SARS-CoV-2 RNA dependent RNA polymerase. Antiviral Res. (2021) 190:105078. doi: 10.1016/j.antiviral.2021.105078
117. Xia H, Shi P-Y. Antagonism of type I interferon by severe acute respiratory syndrome coronavirus 2. J Interferon Cytokine Res. (2020) 40:543–8. doi: 10.1089/jir.2020.0214
118. Suryawanshi RK, Koganti R, Agelidis A, Patil CD, Shukla D. Dysregulation of cell signaling by SARS-CoV-2. Trends Microbiol. (2021) 29:224–37. doi: 10.1016/j.tim.2020.12.007
119. Yang Z, Zhang X, Wang F, Wang P, Kuang E, Li X. Suppression of MDA5-mediated antiviral immune responses by NSP8 of SARS-CoV-2. BioRxiv. (2020). doi: 10.1101/2020.08.12.247767
120. Banerjee AK, Blanco MR, Bruce EA, Honson DD, Chen LM, Chow A, et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell. (2020) 183:1325–1339.e21. doi: 10.1016/j.cell.2020.10.004
121. Astuti I. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab Syndrome: Clin Res Rev. (2020) 14:407–12. doi: 10.1016/j.dsx.2020.04.020
122. Yan L, Huang Y, Ge J, Liu Z, Lu P, Huang B, et al. A mechanism for SARS-CoV-2 RNA capping and its inhibition by nucleotide analog inhibitors. Cell. (2022) 185:4347–4360.e17. doi: 10.1016/j.cell.2022.09.037
123. Rogstam A, Nyblom M, Christensen S, Sele C, Talibov VO, Lindvall T, et al. Crystal structure of non-structural protein 10 from severe acute respiratory syndrome coronavirus-2. Int J Mol Sci. (2020) 21:7375. doi: 10.3390/ijms21197375
124. Hardenbrook NJ, Zhang P. A structural view of the SARS-CoV-2 virus and its assembly. Curr Opin Virol. (2022) 52:123–34. doi: 10.1016/j.coviro.2021.11.011
125. Hsu JC-C, Laurent-Rolle M, Pawlak JB, Wilen CB, Cresswell P. Translational shutdown and evasion of the innate immune response by SARS-CoV-2 NSP14 protein. Proc Natl Acad Sci. (2021) 118:e2101161118. doi: 10.1073/pnas.2101161118
126. Gadhave K, Kumar P, Kumar A, Bhardwaj T, Garg N, Giri R. Conformational dynamics of 13 amino acids long NSP11 of SARS-CoV-2 under membrane mimetics and different solvent conditions. Microbial Pathogenesis. (2021) 158:105041. doi: 10.1016/j.micpath.2021.105041
127. Yan L, Ge J, Zheng L, Zhang Y, Gao Y, Wang T, et al. Cryo-EM structure of an extended SARS-CoV-2 replication and transcription complex reveals an intermediate state in cap synthesis. Cell. (2021) 184:184–193.e10. doi: 10.1016/j.cell.2020.11.016
128. Wang W, Zhou Z, Xiao X, Tian Z, Dong X, Wang C, et al. SARS-CoV-2 nsp12 attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell Mol Immunol. (2021) 18:945–53. doi: 10.1038/s41423-020-00619-y
129. Chen J, Malone B, Llewellyn E, Grasso M, Shelton PM, Olinares PDB, et al. Structural basis for helicase-polymerase coupling in the SARS-CoV-2 replication-transcription complex. Cell. (2020) 182:1560–1573.e13. doi: 10.1016/j.cell.2020.07.033
130. Shu T, Huang M, Wu D, Ren Y, Zhang X, Han Y, et al. SARS-coronavirus-2 Nsp13 possesses NTPase and RNA helicase activities that can be inhibited by bismuth salts. Virologica Sin. (2020) 35:321–9. doi: 10.1007/s12250-020-00242-1
131. Vazquez C, Swanson SE, Negatu SG, Dittmar M, Miller J, Ramage HR, et al. SARS-CoV-2 viral proteins NSP1 and NSP13 inhibit interferon activation through distinct mechanisms. PloS One. (2021) 16:e0253089. doi: 10.1371/journal.pone.0253089
132. Ogando NS, Zevenhoven-Dobbe JC, van Der Meer Y, Bredenbeek PJ, Posthuma CC, Snijder EJ. The enzymatic activity of the nsp14 exoribonuclease is critical for replication of MERS-CoV and SARS-CoV-2. J Virol. (2020) 94. doi: 10.1128/jvi.01246–20
133. Yuen C-K, Lam J-Y, Wong W-M, Mak L-F, Wang X, Chu H, et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerging Microbes infections. (2020) 9:1418–28. doi: 10.1080/22221751.2020.1780953
134. Hayn M, Hirschenberger M, Koepke L, Nchioua R, Straub JH, Klute S, et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. (2021) 35:109126. doi: 10.1016/j.celrep.2021.109126
135. Hackbart M, Deng X, Baker SC. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc Natl Acad Sci. (2020) 117:8094–103. doi: 10.1073/pnas.1921485117
136. Fajgenbaum DC, June CH. Cytokine storm. New Engl J Med. (2020) 383:2255–73. doi: 10.1056/NEJMra2026131
137. Krafcikova P, Silhan J, Nencka R, Boura E. Structural analysis of the SARS-CoV-2 methyltransferase complex involved in RNA cap creation bound to sinefungin. Nat Commun. (2020) 11:3717. doi: 10.1038/s41467-020-17495-9
138. Kern DM, Sorum B, Mali SS, Hoel CM, Sridharan S, Remis JP, et al, et al. Cryo-EM structure of SARS-CoV-2 ORF3a in lipid nanodiscs. Nat Struct Mol Biol. (2021) 28:573–82. doi: 10.1038/s41594-021-00619-0
139. Chen D, Zheng Q, Sun L, Ji M, Li Y, Deng H, Zhang H, et al, et al. ORF3a of SARS-CoV-2 promotes lysosomal exocytosis-mediated viral egress. Dev Cell. (2021) 56:3250–3263.e5. doi: 10.1016/j.devcel.2021.10.006
140. Miao G, Zhao H, Li Y, Ji M, Chen Y, Shi Y, Bi Y, et al. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev Cell. (2021) 56:427–442.e5. doi: 10.1016/j.devcel.2020.12.010
141. Ren Y, Shu T, Wu D, Mu J, Wang C, Huang M, et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol Immunol. (2020) 17:881–3. doi: 10.1038/s41423-020-0485-9
142. Zhang S, Wang L, Cheng G. The battle between host and SARS-CoV-2: Innate immunity and viral evasion strategies. Mol Ther. (2022) 30:1869–84. doi: 10.1016/j.ymthe.2022.02.014
143. Hurme A, Jalkanen P, Heroum J, Liedes O, Vara S, Melin M, et al. Long-lasting T cell responses in BNT162b2 COVID-19 mRNA vaccinees and COVID-19 convalescent patients. Front Immunol. (2022) 13:869990. doi: 10.3389/fimmu.2022.869990
144. Azad GK, Khan PK. Variations in Orf3a protein of SARS-CoV-2 alter its structure and function. Biochem biophysics Rep. (2021) 26:100933. doi: 10.1016/j.bbrep.2021.100933
145. Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. (2020) 32. doi: 10.1101/2020.05.11.088179
146. Xia J, Tong J, Liu M, Shen Y, Guo D. Evaluation of coronavirus in tears and conjunctival secretions of patients with SARS-CoV-2 infection. J Med Virol. (2020) 92:589–94. doi: 10.1002/jmv.25725
147. Li J-Y, Liao C.-H, Wang Q, Tan Y.-J, Luo R, Qiu Y, et al. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. (2020) 286:198074. doi: 10.1016/j.virusres.2020.198074
148. Miorin L, Kehrer T, Sanchez-Aparicio MT, Zhang K, Cohen P, Patel RS, et al, et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci. (2020) 117:28344–54. doi: 10.1073/pnas.2016650117
149. Addetia A, Lieberman NA, Phung Q, Hsiang T-Y, Xie H, Roychoudhury P, et al. SARS-CoV-2 ORF6 disrupts bidirectional nucleocytoplasmic transport through interactions with Rae1 and Nup98. MBio. (2021) 12. doi: 10.1128/mbio.00065–21
150. Cao Z, Xia H, Rajsbaum R, Xia X, Wang H, Shi P-Y, et al. Ubiquitination of SARS-CoV-2 ORF7a promotes antagonism of interferon response. Cell Mol Immunol. (2021) 18:746–8. doi: 10.1038/s41423-020-00603-6
151. Zhou Z, Huang C, Zhou Z, Huang Z, Su L, Kang S, et al. Structural insight reveals SARS-CoV-2 ORF7a as an immunomodulating factor for human CD14+ monocytes. IScience. (2021) 24. doi: 10.1016/j.isci.2021.102187
152. Wang X, Lam J-Y, Wong W-M, Yuen C-K, Cai J-P, Au S, et al. Accurate diagnosis of COVID-19 by a novel immunogenic secreted SARS-CoV-2 orf8 protein. MBio. (2020) 11. doi: 10.1128/mbio.02431–20
153. Rashid F, Dzakah EE, Wang H, Tang S. The ORF8 protein of SARS-CoV-2 induced endoplasmic reticulum stress and mediated immune evasion by antagonizing production of interferon beta. Virus Res. (2021) 296:198350. doi: 10.1016/j.virusres.2021.198350
154. Geng H, Subramanian S, Wu L, Bu H.-F, Wang X, Du C, et al. SARS-CoV-2 ORF8 forms intracellular aggregates and inhibits IFNγ-induced antiviral gene expression in human lung epithelial cells. Front Immunol. (2021) 12:679482. doi: 10.3389/fimmu.2021.679482
155. Zhang Y, Chen Y, Li Y, Huang F, Luo B, Yuan Y, et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-I. Proc Natl Acad Sci. (2021) 118:e2024202118. doi: 10.1073/pnas.2024202118
156. Sirera Pérez R. Las nuevas variantes del SARS-CoV-2 que nadie busca y que están haciendo al virus más resistente a la respuesta inmunitaria. Inmunología. (2021) 40:14–21.
157. Lin X, Fu B, Yin S, Li Z, Liu H, Zhang H, et al. ORF8 contributes to cytokine storm during SARS-CoV-2 infection by activating IL-17 pathway. Iscience. (2021) 24. doi: 10.1016/j.isci.2021.102293
158. Wu J, Shi Y, Pan X, Wu S, Hou R, Zhang Y, et al. SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep. (2021) 34. doi: 10.1016/j.celrep.2021.108761
159. Gao X, Zhu K, Qin B, Olieric V, Wang M, Cui S Crystal structure of SARS-CoV-2 Orf9b in complex with human TOM70 suggests unusual virus-host interactions. Nat Commun. (2021) 12:2843. doi: 10.1038/s41467-021-23118-8
160. Han L, Zhuang MW, Deng J, Zheng Y, Zhang J, Nan ML, et al. SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways. J Med Virol. (2021) 93:5376–89. doi: 10.1002/jmv.27050
161. Dominguez Andres A, Feng Y, Campos A.R, Yin J, Yang C-C, James B, et al. SARS-CoV-2 ORF9c is a membrane-associated protein that suppresses antiviral responses in cells. BioRxiv. (2020). doi: 10.1101/2020.08.18.256776
162. Li X, Hou P, Ma W, Wang X, Wang H, Yu Z, et al. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell Mol Immunol. (2022) 19:67–78. doi: 10.1038/s41423-021-00807-4
163. Chan JFW, Kok K-H, Zhu Z, Chu H, To KK-W, Yuan S, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. (2020) 9:221–36. doi: 10.1080/22221751.2020.1719902
164. Kannan SR, Spratt AN, Sharma K, Chand HS, Byrareddy SN, Singh K. Omicron SARS-CoV-2 variant: Unique features and their impact on pre-existing antibodies. J Autoimmun. (2022) 126:102779. doi: 10.1016/j.jaut.2021.102779
165. Gusev E, Sarapultsev A, Solomatina L, Chereshnev V. SARS-CoV-2-specific immune response and the pathogenesis of COVID-19. Int J Mol Sci. (2022) 23(3):1716. doi: 10.3390/ijms23031716
166. Baggen J, Vanstreels E, Jansen S, Daelemans D. Cellular host factors for SARS-CoV-2 infection. Nat Microbiol. (2021) 6:1219–32. doi: 10.1038/s41564-021-00958-0
167. Li J, Guo M, Tian X, Wang X, Yang X, Wu P, et al. Virus-host interactome and proteomic survey reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. Med. (2021) 2:99–112 e7. doi: 10.1016/j.medj.2020.07.002
168. Papanikolaou V, Chrysovergis A, Ragos V, Tsiambas E, Katsinis S, Manoli A, et al. From delta to Omicron: S1-RBD/S2 mutation/deletion equilibrium in SARS-CoV-2 defined variants. Gene. (2022) 814:146134. doi: 10.1016/j.gene.2021.146134
169. Redondo N, Zaldívar-López S, Garrido JJ, Montoya M. SARS-CoV-2 accessory proteins in viral pathogenesis: knowns and unknowns. Front Immunol. (2021) 12:708264. doi: 10.3389/fimmu.2021.708264
170. Anderson MR, Easthausen I, Gallagher G, Udupa J, Tong Y, Torigian D, et al. Skeletal muscle adiposity and outcomes in candidates for lung transplantation: a lung transplant body composition cohort study. Thorax. (2020) 75:801–4. doi: 10.1136/thoraxjnl-2019-214461
171. Malik JR, Acharya A, Avedissian SN, Byrareddy SN, Fletcher CV, Podany AT, et al. ACE-2, TMPRSS2, and neuropilin-1 receptor expression on human brain astrocytes and pericytes and SARS-CoV-2 infection kinetics. Int J Mol Sci. (2023) 24(10):8622. doi: 10.3390/ijms24108622
172. Huang Y, Wang Y, Xu D, Xiao L, Qin W, Liu B, et al. Characterization of the SARS-CoV-2 co-receptor NRP1 expression profiles in healthy people and cancer patients: Implication for susceptibility to COVID-19 disease and potential therapeutic strategy. Front Genet. (2022) 13:995736. doi: 10.3389/fgene.2022.995736
173. Hopkins C, Lechien JR, Saussez S. More that ACE2? NRP1 may play a central role in the underlying pathophysiological mechanism of olfactory dysfunction in COVID-19 and its association with enhanced survival. Med Hypotheses. (2021) 146:110406. doi: 10.1016/j.mehy.2020.110406
174. Zhang F, Li W, Feng J, Ramos da Silva S, Ju E, Zhang H, et al. SARS-CoV-2 pseudovirus infectivity and expression of viral entry-related factors ACE2, TMPRSS2, Kim-1, and NRP-1 in human cells from the respiratory, urinary, digestive, reproductive, and immune systems. J Med Virol. (2021) 93:6671–85. doi: 10.1002/jmv.27244
175. Eslami N, Aghbash PS, Shamekh A, Entezari-Maleki T, Nahand JS, Sales AJ, et al. SARS-CoV-2: receptor and co-receptor tropism probability. Curr Microbiol. (2022) 79:133. doi: 10.1007/s00284-022-02807-7
176. Lukassen S, Chua R.L, Trefzer T, Kahn NC, Schneider MA, Muley T, et al. SARS-CoV-2 receptor ACE 2 and TMPRSS 2 are primarily expressed in bronchial transient secretory cells. EMBO J. (2020) 39:e105114. doi: 10.15252/embj.2020105114
177. Salamanna F, Maglio M, Landini MP, Fini M. Body localization of ACE-2: on the trail of the keyhole of SARS-CoV-2. Front Med (Lausanne). (2020) 7:594495. doi: 10.3389/fmed.2020.594495
178. Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Internal Med. (2020) 76:14–20. doi: 10.1016/j.ejim.2020.04.037
179. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. cell. (2020) 181:271–280.e8. doi: 10.1016/j.cell.2020.02.052
180. Matsuyama S, Nao N, Shirato K, Kawase M, Saito S, Takayama I, et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc Natl Acad Sci. (2020) 117:7001–3. doi: 10.1073/pnas.2002589117
181. Birra D, Benucci M, Landolfi L, Merchionda A, Loi G, Amato P, et al. COVID 19: a clue from innate immunity. Immunologic Res. (2020) 68:161–8. doi: 10.1007/s12026-020-09137-5
182. Madden EA, Diamond MS. Host cell-intrinsic innate immune recognition of SARS-CoV-2. Curr Opin Virol. (2022) 52:30–8. doi: 10.1016/j.coviro.2021.11.002
183. Merad M, Diamond MS. The immunology and immunopathology of COVID-19. Science. (2022) 375:1122–7. doi: 10.1126/science.abm8108
184. Cheemarla NR, Watkins TA, Mihaylova VT, Wang B, Zhao D, Wang G, et al. Dynamic innate immune response determines susceptibility to SARS-CoV-2 infection and early replication kinetics. J Exp Med. (2021) 218(8):e20210583. doi: 10.1084/jem.20210583
185. Bhardwaj A, Sapra L, Saini C, Azam Z, Mishra PK, Verma B, et al. COVID-19: immunology, immunopathogenesis and potential therapies. Int Rev Immunol. (2022) 41:171–206. doi: 10.1080/08830185.2021.1883600
186. Land WG. Role of DAMPs in respiratory virus-induced acute respiratory distress syndrome-with a preliminary reference to SARS-CoV-2 pneumonia. Genes Immun. (2021) 22:141–60. doi: 10.1038/s41435-021-00140-w
187. Singh A, Strobbe D, Campanella M. Pyroptosis targeting via mitochondria: An educated guess to innovate COVID-19 therapies. Br J Pharmacol. (2022) 179:2081–5. doi: 10.1111/bph.15670
188. Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LF. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. (2020) 20:363–74. doi: 10.1038/s41577-020-0311-8
189. McGill AR, Khalil R, Dutta R, Green R, Howell M, Mohapatra S, et al. SARS-CoV-2 immuno-pathogenesis and potential for diverse vaccines and therapies: opportunities and challenges. Infect Dis Rep. (2021) 13:102–25. doi: 10.3390/idr13010013
190. Polycarpou A, Howard M, Farrar CA, Greenlaw R, Fanelli G, Wallis R, et al. Rationale for targeting complement in COVID-19. EMBO Mol Med. (2020) 12:e12642. doi: 10.15252/emmm.202012642
191. Wong NA, Saier MH Jr. The SARS-coronavirus infection cycle: a survey of viral membrane proteins, their functional interactions and pathogenesis. Int J Mol Sci. (2021) 22:1308. doi: 10.3390/ijms22031308
192. Afzali B, Noris M, Lambrecht BN, Kemper C. The state of complement in COVID-19. Nat Rev Immunol. (2022) 22:77–84. doi: 10.1038/s41577-021-00665-1
193. Khosroshahi LM, Rokni M, Mokhtari T, Noorbakhsh F. Immunology, immunopathogenesis and immunotherapeutics of COVID-19; an overview. Int Immunopharmacol. (2021) 93:107364. doi: 10.1016/j.intimp.2020.107364
194. Wool GD, Miller JL. The impact of COVID-19 disease on platelets and coagulation. Pathobiology. (2021) 88:15–27. doi: 10.1159/000512007
195. Witkowski M, Tizian C, Ferreira-Gomes M, Niemeyer D, Jones TC, Heinrich F, et al. Untimely TGFbeta responses in COVID-19 limit antiviral functions of NK cells. Nature. (2021) 600:295–301. doi: 10.1038/s41586-021-04142-6
196. Boechat JL, Chora I, Morais A, Delgado L. The immune response to SARS-CoV-2 and COVID-19 immunopathology - Current perspectives. Pulmonology. (2021) 27:423–37. doi: 10.1016/j.pulmoe.2021.03.008
197. Zlei M, Sidorov IA, Joosten SA, Heemskerk MH, Myeni SK, Pothast CR, et al. Immune determinants of viral clearance in hospitalised COVID-19 patients: reduced circulating naive CD4+ T cell counts correspond with delayed viral clearance. Cells. (2022) 11. doi: 10.3390/cells11172743
198. Sette A, Crotty S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell. (2021) 184:861–80. doi: 10.1016/j.cell.2021.01.007
199. Liu Y, Sawalha AH, Lu Q. COVID-19 and autoimmune diseases. Curr Opin Rheumatol. (2021) 33:155–62. doi: 10.1097/BOR.0000000000000776
200. Sacchi MC, Tamiazzo S, Stobbione P, Agatea L, De Gaspari P, Stecca A, et al. SARS-CoV-2 infection as a trigger of autoimmune response. Clin Trans Sci. (2021) 14:898–907. doi: 10.1111/cts.12953
201. Dotan A, Muller S, Kanduc D, David P, Halpert G, Shoenfeld Y, et al. The SARS-CoV-2 as an instrumental trigger of autoimmunity. Autoimmun Rev. (2021) 20:102792. doi: 10.1016/j.autrev.2021.102792
202. Klein J, Wood J, Jaycox JR, Dhodapkar RM, Lu P, Gehlhausen JR, et al. Distinguishing features of long COVID identified through immune profiling. Nature. (2023) 623:139–48. doi: 10.1038/s41586-023-06651-y
203. Pertiwi D, Hafiz I, Illian DN. Dendritic cell-based COVID-19 vaccines: A mini review. Clin trials. (2021) 6:9. doi: 10.29228/jrp.78
204. Hu B, Huang S, Yin L. The cytokine storm and COVID-19. J Med Virol. (2021) 93:250–6. doi: 10.1002/jmv.26232
205. Tang Y, Liu J, Zhang D, Xu Z, Ji J, Wen C. Cytokine storm in COVID-19: the current evidence and treatment strategies. Front Immunol. (2020) 11:1708. doi: 10.3389/fimmu.2020.01708
206. Dharra R, Sharma AK, Datta S. Emerging aspects of cytokine storm in COVID-19: The role of proinflammatory cytokines and therapeutic prospects. Cytokine. (2023) 156287. doi: 10.1016/j.cyto.2023.156287
207. Gerla L, Moitra S, Pink D, Govindasamy N, Duchesne M, Reklow E, et al. SARS-CoV-2-induced TSLP is associated with duration of hospital stay in COVID-19 patients. Viruses. (2023) 15:556. doi: 10.3390/v15020556
208. Lai CC, Shih T-P, Ko W-C, Tang H-J, Hsueh P-R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int J Antimicrob Agents. (2020) 55:105924. doi: 10.1016/j.ijantimicag.2020.105924
209. Shimizu M. Clinical features of cytokine storm syndrome. In: Cytokine storm syndrome (2019). p. 31–41.
210. Niessl J, Sekine T, Buggert M. T cell immunity to SARS-CoV-2. In: Seminars in immunology. Elsevier (2021) 55:101505. doi: 10.1016/j.smim.2021.101505
211. Israelow B, Mao T, Klein J, Song E, Menasche B, Omer SB, et al. Adaptive immune determinants of viral clearance and protection in mouse models of SARS-CoV-2. bioRxiv. (2021). doi: 10.1101/2021.05.19.444825
212. McMahan K, Yu J, Mercado NB, Loos C, Tostanoski LH, Chandrashekar A, et al. Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature. (2021) 590:630–4. doi: 10.1038/s41586-020-03041-6
213. Notarbartolo S, Ranzani V, Bandera A, Gruarin P, Bevilacqua V, Putignano AR, et al. Integrated longitudinal immunophenotypic, transcriptional and repertoire analyses delineate immune responses in COVID-19 patients. Sci Immunol. (2021) 6. doi: 10.1126/sciimmunol.abg5021
214. Le Bert N, Clapham HE, Tan AT, Chia WN, Tham CY, Lim JM, et al. Highly functional virus-specific cellular immune response in asymptomatic SARS-CoV-2 infection. J Exp Med. (2021) 218. doi: 10.1084/jem.20202617
215. Moss P. The T cell immune response against SARS-CoV-2. Nat Immunol. (2022) 23:186–93. doi: 10.1038/s41590-021-01122-w
216. Zeng C, Evans JP, King T, Zheng Y-M, Oltz EM, Whelan SP, et al. SARS-CoV-2 spreads through cell-to-cell transmission. Proc Natl Acad Sci. (2022) 119:e2111400119. doi: 10.1073/pnas.2111400119
217. Moderbacher CR, Ramirez SI, Dan JM, Grifoni A, Hastie KM, Weiskopf D, et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell. (2020) 183:996–1012 e19. doi: 10.1016/j.cell.2020.09.038
218. Shin E-C, Jung JH, Rha M-S, Sa M, Choi HK, Jeon JH, et al. SARS-CoV-2-specific T cell memory is sustained in COVID-19 convalescents for 8 months with successful development of stem cell-like memory T cells. (preprint). (2021).
219. Jung JH, Rha M.-S, Sa M, Choi HK, Jeon JH, Seok H, et al. SARS-CoV-2-specific T cell memory is sustained in COVID-19 convalescent patients for 10 months with successful development of stem cell-like memory T cells. Nat Commun. (2021) 12:4043. doi: 10.1038/s41467-021-24377-1
220. Wang M, Pan W, Xu Y, Zhang J, Wan J, Jiang H. Microglia-mediated neuroinflammation: A potential target for the treatment of cardiovascular diseases. J Inflammation Res. (2022) 15:3083–94. doi: 10.2147/JIR.S350109
221. Gao Z-w, Zhang H.-z, Liu C, Dong K. Autoantibodies in COVID-19: frequency and function. Autoimmun Rev. (2021) 20:102754. doi: 10.1016/j.autrev.2021.102754
222. Migliorini F, Torsiello E, Spiezia F, Oliva F, Tingart M, Maffulli N. Association between HLA genotypes and COVID-19 susceptibility, severity and progression: a comprehensive review of the literature. Eur J Med Res. (2021) 26:1–9. doi: 10.1186/s40001-021-00563-1
223. Rastogi M, Pandey N, Shukla A, Singh SK. SARS coronavirus 2: from genome to infectome. Respir Res. (2020) 21:318. doi: 10.1186/s12931-020-01581-z
224. Sette A, Sidney J, Crotty S. T cell responses to SARS-CoV-2. Annu Rev Immunol. (2023) 41:343–73. doi: 10.1146/annurev-immunol-101721-061120
225. Dan JM, Mateus J, Kato Y, Hastie KM, Yu ED, Faliti CE, et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science. (2021) 371. doi: 10.1126/science.abf4063
226. Grifoni A, Sidney J, Vita R, Peters B, Crotty S, Weiskopf D, et al. SARS-CoV-2 human T cell epitopes: Adaptive immune response against COVID-19. Cell Host Microbe. (2021) 29:1076–92. doi: 10.1016/j.chom.2021.05.010
227. Soto JA, Melo-González F, Gutierrez-Vera C, Schultz BM, Berríos-Rojas RV, Rivera-Pérez D, et al. Inactivated vaccine-induced SARS-CoV-2 variant-specific immunity in children. mBio. (2022) 13:e0131122. doi: 10.1128/mbio.01311-22
228. Mendez C, Peñaloza HF, Schultz BM, Piña-Iturbe A, Ríos M, Moreno-Tapia D, et al. Humoral and cellular response induced by a second booster of an inactivated SARS-CoV-2 vaccine in adults. EBioMedicine. (2023) 91:104563. doi: 10.1016/j.ebiom.2023.104563
229. Schultz BM, Melo-González F, Duarte LF, Gálvez NM, Pacheco GA, Soto JA, et al. A booster dose of CoronaVac increases neutralizing antibodies and T cells that recognize Delta and Omicron variants of concern. MBio. (2022) 13:e01423–22. doi: 10.1128/mbio.01423-22
230. Bueno SM, Abarca K, González PA, Gálvez NM, Soto JA, Duarte LF, et al. Safety and immunogenicity of an inactivated severe acute respiratory syndrome coronavirus 2 vaccine in a subgroup of healthy adults in Chile. Clin Infect Dis. (2022) 75:e792–804. doi: 10.1093/cid/ciab823
231. Galvez NM, Pacheco GA, Schultz BM, Melo-González F, Soto JA, Duarte LF, et al. Differences in the immune response elicited by two immunization schedules with an inactivated SARS-CoV-2 vaccine in a randomized phase 3 clinical trial. Elife. (2022) 11. doi: 10.7554/eLife.81477
232. Stieber F, Allen N, Carpenter K, Hu P, Alagna R, Rao S, et al. Durability of COVID-19 vaccine induced T-cell mediated immune responses measured using the QuantiFERON SARS-CoV-2 assay. Pulmonology. (2023) 29:151–3. doi: 10.1016/j.pulmoe.2022.09.006
233. Wang L, Nicols A, Turtle L, Richter A, Duncan CJ, Dunachie SJ, et al. T cell immune memory after covid-19 and vaccination. BMJ Med. (2023) 2:e000468. doi: 10.1136/bmjmed-2022-000468
234. Wang L, Nicols A, Turtle L, Richter A, Duncan CJ, et al. T cell immune memory after covid-19 and vaccination. BMJ Medicine. (2023) 2(1).
235. Reynolds CJ, Gibbons JM, Pade C, Lin K.-M, Sandoval DM, Pieper F, et al. Heterologous infection and vaccination shapes immunity against SARS-CoV-2 variants. Science. (2022) 375:183–92. doi: 10.1126/science.abm0811
236. Wang J, Li K, Mei X, Cao J, Zhong J, Huang P, et al. SARS-CoV-2 vaccination-infection pattern imprints and diversifies T cell differentiation and neutralizing response against Omicron subvariants. Cell Discovery. (2022) 8:136. doi: 10.1038/s41421-022-00501-3
237. Röltgen K, Boyd SD. Antibody and B cell responses to SARS-CoV-2 infection and vaccination: the end of the beginning. Annu Rev Pathology: Mech Dis. (2024) 19:69–97. doi: 10.1146/annurev-pathmechdis-031521-042754
238. Rzymski P, Pokorska-Śpiewak M, Jackowska T, Kuchar E, Nitsch-Osuch A, Pawłowska M, et al. Key considerations during the transition from the acute phase of the COVID-19 pandemic: A narrative review. Vaccines (Basel). (2023) 11. doi: 10.3390/vaccines11091502
239. Song S, Madewell ZJ, Liu M, Miao Y, Xiang S, Huo Y, et al. A systematic review and meta-analysis on the effectiveness of bivalent mRNA booster vaccines against Omicron variants. Vaccine. (2024). doi: 10.1016/j.vaccine.2024.04.049
240. Wu N, Joyal-Desmarais K, Ribeiro PA, Vieira AM, Stojanovic J, Sanuade C, et al. Long-term effectiveness of COVID-19 vaccines against infections, hospitalisations, and mortality in adults: findings from a rapid living systematic evidence synthesis and meta-analysis up to December, 2022. Lancet Respir Med. (2023) 11:439–52. doi: 10.1016/S2213-2600(23)00015-2
241. Morin L, Savale L, Pham T, Colle R, Figueiredo S, Harrois A, et al. Four-month clinical status of a cohort of patients after hospitalization for COVID-19. JAMA. (2021) 325:1525–34. doi: 10.1001/jama.2021.3331
242. Choutka J, Jansari V, Hornig M, Iwasaki A. Unexplained post-acute infection syndromes. Nat Med. (2022) 28:911–23. doi: 10.1038/s41591-022-01810-6
243. Huang C, Huang L, Wang Y, Li X, Ren L, Gu X, et al. 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. Lancet. (2023) 401:e21–33. doi: 10.1016/S0140-6736(23)00810-3
244. Kalchiem-Dekel O, Galvin JR, Burke AP, Atamas SP, Todd NW. Interstitial lung disease and pulmonary fibrosis: a practical approach for general medicine physicians with focus on the medical history. J Clin Med. (2018) 7:476. doi: 10.3390/jcm7120476
245. Lechowicz K, Drożdżal S, Machaj F, Rosik J, Szostak B, Zegan-Barańska M, et al. COVID-19: the potential treatment of pulmonary fibrosis associated with SARS-CoV-2 infection. J Clin Med. (2020) 9:1917. doi: 10.3390/jcm9061917
246. Todd NW, Luzina IG, Atamas SP. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair. (2012) 5:1–24. doi: 10.1186/1755-1536-5-11
247. Gao J-W, Rizzo S, Ma L-H, Qiu X-Y, Warth A, Seki N, et al. Pulmonary ground-glass opacity: computed tomography features, histopathology and molecular pathology. Trans Lung Cancer Res. (2017) 6:68. doi: 10.21037/tlcr
248. Venkataraman T, Frieman MB. The role of epidermal growth factor receptor (EGFR) signaling in SARS coronavirus-induced pulmonary fibrosis. Antiviral Res. (2017) 143:142–50. doi: 10.1016/j.antiviral.2017.03.022
249. Ambardar SR, Hightower SL, Huprikar NA, Chung KK, Singhal A, Collen JF. Post-COVID-19 pulmonary fibrosis: novel sequelae of the current pandemic. J Clin Med. (2021) 10:2452. doi: 10.3390/jcm10112452
250. Ronco C, Reis T. Kidney involvement in COVID-19 and rationale for extracorporeal therapies. Nat Rev Nephrol. (2020) 16:308–10. doi: 10.1038/s41581-020-0284-7
251. Kunutsor SK, Laukkanen JA. Renal complications in COVID-19: a systematic review and meta-analysis. Ann Med. (2020) 52:345–53. doi: 10.1080/07853890.2020.1790643
252. Armaly Z, Kinaneh S, Skorecki K. Renal manifestations of Covid-19: physiology and pathophysiology. J Clin Med. (2021) 10:1216. doi: 10.3390/jcm10061216
253. Ashraf O, Young M, Malik KJ, Cheema T. Systemic complications of COVID-19. Crit Care Nurs Q. (2020) 43:390–9. doi: 10.1097/CNQ.0000000000000324
254. Paghdar S, Khan TM, Patel NP, Chandrasekaran S, De Sousa JFM, Tsouklidis N, et al. Doxycycline therapy for abdominal aortic aneurysm: inhibitory effect on matrix metalloproteinases. Cureus. (2021) 13. doi: 10.7759/cureus.14966
255. Xu L, Liu J, Lu M, Yang D, Zheng X. Liver injury during highly pathogenic human coronavirus infections. Liver Int. (2020) 40:998–1004. doi: 10.1111/liv.14435
256. Bangash MN, Patel J, Parekh D. COVID-19 and the liver: little cause for concern. Lancet Gastroenterol Hepatol. (2020) 5:529–30. doi: 10.1016/S2468-1253(20)30084-4
257. Lagana SM, Kudose S, Iuga AC, Lee MJ, Fazlollahi L, Remotti HE, et al. Hepatic pathology in patients dying of COVID-19: a series of 40 cases including clinical, histologic, and virologic data. Modern Pathol. (2020) 33:2147–55. doi: 10.1038/s41379-020-00649-x
258. Arabi Y, Harthi A, Hussein J, Bouchama A, Johani S, Hajeer A, et al. Severe neurologic syndrome associated with Middle East respiratory syndrome corona virus (MERS-CoV). Infection. (2015) 43:495–501. doi: 10.1007/s15010-015-0720-y
259. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. jama. (2020) 323:1061–9. doi: 10.1001/jama.2020.1585
260. Zhao K, Huang J, Dai D, Feng Y, Liu L, Nie S. Acute myelitis after SARS-CoV-2 infection: a case report. MedRxiv. (2020). doi: 10.1101/2020.03.16.20035105
261. Toscano G, Palmerini F, Ravaglia S, Ruiz L, Invernizzi P, Cuzzoni MG, et al. Guillain–Barré syndrome associated with SARS-CoV-2. New Engl J Med. (2020) 382:2574–6. doi: 10.1056/NEJMc2009191
262. Rocke J, Hopkins C, Philpott C, Kumar N. Is loss of sense of smell a diagnostic marker in COVID-19: a systematic review and meta-analysis. Clin Otolaryngol. (2020) 45:914–22. doi: 10.1111/coa.13620
263. Mao L, Wang M, Chen S, He Q, Chang J, Hong C, et al. Neurological manifestations of hospitalized patients with COVID-19 in Wuhan, China: a retrospective case series study. MedRxiv. (2020). doi: 10.1101/2020.02.22.20026500
264. Giacomelli A, Pezzati L, Conti F, Bernacchia D, Siano M, Oreni L, et al. Self-reported olfactory and taste disorders in patients with severe acute respiratory coronavirus 2 infection: a cross-sectional study. Clin Infect Dis. (2020) 71:889–90. doi: 10.1093/cid/ciaa330
265. Moein ST, Hashemian SM, Mansourafshar B, Khorram‐Tousi A, Tabarsi P, Doty RL. Smell dysfunction: a biomarker for COVID-19. In: International forum of allergy & rhinology. Wiley Online Library (2020) 10(8):944–50. doi: 10.1002/alr.22587
266. Lechien JR, Cabaraux P, Chiesa-Estomba CM, Khalife M, Plzak J, Hans S, et al. Objective olfactory testing in patients presenting with sudden onset olfactory dysfunction as the first manifestation of confirmed COVID-19 infection. Medrxiv. (2020). doi: 10.1101/2020.04.15.20066472
267. Kaye R, Chang CD, Kazahaya K, Brereton J, Denneny JC III. COVID-19 anosmia reporting tool: initial findings. Otolaryngology–Head Neck Surg. (2020) 163:132–4. doi: 10.1177/0194599820922992
268. Guan W-j, Ni Z-y, Hu Y, Liang W-h, Ou C-q, He J-x, et al. Clinical characteristics of coronavirus disease 2019 in China. New Engl J Med. (2020) 382:1708–20. doi: 10.1056/NEJMoa2002032
269. Carvalho A, Alqusairi R, Adams A, Paul M, Kothari N, Peters S, et al. SARS-CoV-2 gastrointestinal infection causing hemorrhagic colitis: implications for detection and transmission of COVID-19 disease. Off J Am Coll Gastroenterology| ACG. (2020) 115:942–6. doi: 10.14309/ajg.0000000000000667
270. Tian S, Hu N, Lou J, Chen K, Kang X, Xiang Z, et al. Characteristics of COVID-19 infection in Beijing. J infection. (2020) 80:401–6. doi: 10.1016/j.jinf.2020.02.018
271. Goyal P, Choi JJ, Pinheiro LC, Schenck EJ, Chen R, Jabri A, et al. Clinical characteristics of Covid-19 in New York city. New Engl J Med. (2020) 382:2372–4. doi: 10.1056/NEJMc2010419
272. Coolen T, Lolli V, Sadeghi N, Rovai A, Trotta N, Taccone FS, et al. Early postmortem brain MRI findings in COVID-19 non-survivors. Neurology. (2020) 95:e2016–27. doi: 10.1212/WNL.0000000000010116
273. Shemer A, Einan-Lifshitz A, Itah A, Dubinsky-Pertzov B, Pras E, Hecht I, et al. Ocular involvement in coronavirus disease 2019 (COVID-19): a clinical and molecular analysis. Int Ophthalmol. (2021) 41:433–40. doi: 10.1007/s10792-020-01592-1
274. Pérez-Bartolomé F, Sánchez-Quirós J. Ocular manifestations of SARS-CoV-2: literature review. Archivos la Sociedad Española Oftalmología (English Edition). (2021) 96:32–40. doi: 10.1016/j.oftale.2020.07.003
275. Wang F, Wang H, Fan J, Zhang Y, Wang H, Zhao Q. Pancreatic injury patterns in patients with coronavirus disease 19 pneumonia. Gastroenterology. (2020) 159:367–70. doi: 10.1053/j.gastro.2020.03.055
276. Hadi A, Werge M, Kristiansen KT, Pedersen UG, Karstensen JG, Novovic S, et al. Coronavirus disease-19 (COVID-19) associated with severe acute pancreatitis: case report on three family members. Pancreatology. (2020) 20:665–7. doi: 10.1016/j.pan.2020.04.021
277. Jing Y, Run-Qian L, Hao-Ran W, Hao-Ran C, Ya-Bin L, Yang G, et al. Potential influence of COVID-19/ACE2 on the female reproductive system. Mol Hum Reprod. (2020) 26:367–73. doi: 10.1093/molehr/gaaa030
278. Di Mascio D, Khalil A, Saccone G, Rizzo G, Buca D, Liberati M, et al. Outcome of coronavirus spectrum infections (SARS, MERS, COVID-19) during pregnancy: a systematic review and meta-analysis. Am J obstetrics gynecology MFM. (2020) 2:100107. doi: 10.1016/j.ajogmf.2020.100107
279. Zhou L, Niu Z, Jiang X, Zhang Z, Zheng Y, Wang Z, et al. SARS-CoV-2 targets by the pscRNA profiling of ACE2, TMPRSS2 and furin proteases. iScience. (2020) 23:101744. doi: 10.1016/j.isci.2020.101744
280. Diao B, Wang C, Wang R, Feng Z, Zhang J, Yang H, et al. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. Nat Commun. (2021) 12:2506. doi: 10.1038/s41467-021-22781-1
281. Wichmann D. Autopsy findings and venous thromboembolism in patients with COVID-19: A prospective cohort study. Ann Intern Med. (2020) 173:268–77. doi: 10.7326/L20-1206
282. He W, Liu X, Hu B, Li D, Chen L, Li Y, et al. Mechanisms of SARS-coV-2 infection-induced kidney injury: A literature review. Front Cell Infect Microbiol. (2022) 12:838213. doi: 10.3389/fcimb.2022.838213
283. Ronco C, Reis T, Husain-Syed F. Management of acute kidney injury in patients with COVID-19. Lancet Respir Med. (2020) 8:738–42. doi: 10.1016/S2213-2600(20)30229-0
284. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel A.S, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
285. Qian JY, Wang B, Lv L-L, Liu B-C. Pathogenesis of acute kidney injury in coronavirus disease 2019. Front Physiol. (2021) 12:586589. doi: 10.3389/fphys.2021.586589
286. Shimojo M, Condon J, Whorwood C, Stewart P. Adrenal 11β-hydroxysteroid dehydrogenase. Endocrine Res. (1996) 22:771–80. doi: 10.1080/07435809609043775
287. Akwii RG, Sajib MS, Zahra FT, Mikelis CM. Role of angiopoietin-2 in vascular physiology and pathophysiology. Cells. (2019) 8. doi: 10.3390/cells8050471
288. Sato K, Sinclair JE, Sadeghirad H, Fraser JF, Short KR, Kulasing A. Cardiovascular disease in SARS-CoV-2 infection. Clin Transl Immunol. (2021) 10:e1343. doi: 10.1002/cti2.1343
289. Fernandez F, Vazquez-Muñoz M, Canals A, Arce-Álvarez A, Salazar-Ardiles C, Alvarez C, et al. Intrahospital supervised exercise training improves survival rate among hypertensive patients with COVID-19. J Appl Physiol. (2023) 134:678–84. doi: 10.1152/japplphysiol.00544.2022
290. VasanthiDharmalingam P, Karuppagounder V, Watanabe K, Karmouty‐Quintana H, Palaniyandi SS, Guha A, et al. SARS–CoV-2 mediated hyperferritinemia and cardiac arrest: preliminary insights. Drug Discovery Today. (2021) 26:1265–74. doi: 10.1016/j.drudis.2021.01.014
291. Zheng Y-Y, Ma Y-T, Zhang J-Y, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol. (2020) 17:259–60. doi: 10.1038/s41569-020-0360-5
292. Wu Q, Zhou L, Sun X, Yan Z, Hu C, Wu J, et al. Altered lipid metabolism in recovered SARS patients twelve years after infection. Sci Rep. (2017) 7:9110. doi: 10.1038/s41598-017-09536-z
293. Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. (2020) 5:802–10. doi: 10.1001/jamacardio.2020.0950
294. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
295. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. (2020) 5:811–8. doi: 10.1001/jamacardio.2020.1017
296. Libby P, Lüscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. (2020) 41:3038–44. doi: 10.1093/eurheartj/ehaa623
297. Liu PP, Blet A, Smyth D, Li H. The science underlying COVID-19: implications for the cardiovascular system. Circulation. (2020) 142:68–78. doi: 10.1161/CIRCULATIONAHA.120.047549
298. Bornstein SR, Dalan R, Hopkins D, Mingrone G, Boehm BO. Endocrine and metabolic link to coronavirus infection. Nat Rev Endocrinol. (2020) 16:297–8. doi: 10.1038/s41574-020-0353-9
299. Khunti K, Del Prato S, Mathieu C, Kahn SE, Gabbay RA, Buse JB. COVID-19, hyperglycemia, and new-onset diabetes. Diabetes Care. (2021) 44:2645–55. doi: 10.2337/dc21-1318
300. Unnikrishnan R, Misra A. Diabetes and COVID19: a bidirectional relationship. Nutr Diabetes. (2021) 11:21. doi: 10.1038/s41387-021-00163-2
301. Hussain A, Bhowmik B, do Vale Moreira NC. COVID-19 and diabetes: Knowledge in progress. Diabetes Res Clin Pract. (2020) 162:108142. doi: 10.1016/j.diabres.2020.108142
302. Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci. (2021) 24:168–75. doi: 10.1038/s41593-020-00758-5
303. Guedj E, Campion J, Dudouet P, Kaphan E, Bregeon F, Dupont Tissot- H, et al. 18F-FDG brain PET hypometabolism in patients with long COVID. Eur J Nucl Med Mol Imaging. (2021) 48:2823–33. doi: 10.1007/s00259-021-05215-4
304. Wuyts WA, Agostini C, Antoniou KM, Bouros D, Chambers RC, Cottin V, et al. The pathogenesis of pulmonary fibrosis: a moving target. Eur Respir J. (2013) 41:1207–18. doi: 10.1183/09031936.00073012
305. She YX, Yu QY, Tang XX. Role of interleukins in the pathogenesis of pulmonary fibrosis. Cell Death Discovery. (2021) 7:52. doi: 10.1038/s41420-021-00437-9
306. Wynn T. Cellular and molecular mechanisms of fibrosis. J Pathology: A J Pathological Soc Great Britain Ireland. (2008) 214:199–210. doi: 10.1002/path.2277
307. Carsana L, Sonzogni A, Nasr A, Rossi RS, Pellegrinelli A, Zerbi P, et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect Dis. (2020) 20:1135–40. doi: 10.1016/S1473-3099(20)30434-5
308. Chen T, Wu D, Chen H, Yan W, Yang D, Chen G, et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ. (2020) 368:m1091. doi: 10.1136/bmj.m1091
309. Chai X, Hu L, Zhang Y, Han W, Lu Z, Ke A, et al. Specific ACE2 expression in cholangiocytes may cause liver damage after 2019-nCoV infection. biorxiv. (2020). doi: 10.1101/2020.02.03.931766
310. Dhar D, Mohanty A. Gut microbiota and Covid-19- possible link and implications. Virus Res. (2020) 285:198018. doi: 10.1016/j.virusres.2020.198018
311. Gou W, Fu Y, Yue L, Chen G.-d, Cai X, Shuai M, et al. Gut microbiota may underlie the predisposition of healthy individuals to COVID-19. MedRxiv. (2020). doi: 10.1101/2020.04.22.20076091
312. He L-H, Ren L.-F, Li J.-F, Wu Y.-N, Li X, Zhang L, et al. Intestinal flora as a potential strategy to fight SARS-CoV-2 infection. Front Microbiol. (2020) 11:1388. doi: 10.3389/fmicb.2020.01388
313. Parasa S, Desai M, Chandrasekar VT, Patel HK, Kennedy KF, Roesch T, et al. Prevalence of gastrointestinal symptoms and fecal viral shedding in patients with coronavirus disease 2019: A systematic review and meta-analysis. JAMA Netw Open. (2020) 3:e2011335. doi: 10.1001/jamanetworkopen.2020.11335
314. Lim JK, Chang AY, Zaman A, Martin P, Rodriguez Fernandez- CM, Korkmaz M, et al. Clinical outcome event adjudication in a 10-year prospective study of nucleos(t)ide analogue therapy for chronic hepatitis B. J Clin Transl Hepatol. (2020) 8:377–84. doi: 10.14218/JCTH.2020.00039
315. Zuo T, Liu Q, Zhang F, Lui GC-Y, Tso EY, Yeoh YK, et al. Depicting SARS-CoV-2 faecal viral activity in association with gut microbiota composition in patients with COVID-19. Gut. (2021) 70:276–84. doi: 10.1136/gutjnl-2020-322294
316. Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clinica chimica Acta. (2020) 506:145–8. doi: 10.1016/j.cca.2020.03.022
317. Levi M, Thachil J, Iba T, Levy JH. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematology. (2020) 7:e438–40. doi: 10.1016/S2352-3026(20)30145-9
318. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet gene expression and function in patients with COVID-19. Blood. (2020) 136:1317–29. doi: 10.1182/blood.2020007214
319. Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb haemostasis. (2020) 18:1094–9. doi: 10.1111/jth.14817
320. Levi M, van der Poll T. Coagulation and sepsis. Thromb Res. (2017) 149:38–44. doi: 10.1016/j.thromres.2016.11.007
321. Al-Samkari H, Karp Leaf RS, Dzik WH, Carlson JC, Fogerty AE, Waheed A, et al. COVID-19 and coagulation: bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood J Am Soc Hematol. (2020) 136:489–500. doi: 10.1182/blood.2020006520
322. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. (2020) 77:683–90. doi: 10.1001/jamaneurol.2020.1127
323. Ellul MA, Benjamin L, Singh B, Lant S, Michael BD, Easton A, et al. Neurological associations of COVID-19. Lancet Neurol. (2020) 19:767–83. doi: 10.1016/S1474-4422(20)30221-0
324. Liotta EM, Batra A, Clark JR, Shlobin NA, Hoffman SC, Orban ZS, et al. Frequent neurologic manifestations and encephalopathy-associated morbidity in Covid-19 patients. Ann Clin Transl Neurol. (2020) 7:2221–30. doi: 10.1002/acn3.51210
325. Paterson RW, Brown RL, Benjamin L, Nortley R, Wiethoff S, Bharucha T, et al. The emerging spectrum of COVID-19 neurology: clinical, radiological and laboratory findings. Brain. (2020) 143:3104–20. doi: 10.1093/brain/awaa240
326. Daugherty SE, Guo Y, Heath K, Dasmariñas MC, Jubilo KG, Samranvedhya J, et al. Risk of clinical sequelae after the acute phase of SARS-CoV-2 infection: retrospective cohort study. BMJ. (2021) 373:n1098. doi: 10.1136/bmj.n1098
327. Nalbandian A, Desai AD, Wan EY. Post-COVID-19 condition. Annu Rev Med. (2023) 74:55–64. doi: 10.1146/annurev-med-043021-030635
328. Herman JD, Atyeo C, Zur Y, Cook CE, Patel NJ, Vanni KM, et al. Impact of cross-coronavirus immunity in post-acute sequelae of COVID-19. medRxiv. (2022). doi: 10.1101/2022.09.25.22280335
329. Huang C, Huang L, Wang Y, Li X, Ren L, Gu X, et al. 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. Lancet. (2021) 397:220–32. doi: 10.1016/S0140-6736(20)32656-8
330. Greenhalgh T, Knight M, Buxton M, Husain L. Management of post-acute covid-19 in primary care. BMJ. (2020) 370:m3026. doi: 10.1136/bmj.m3026
331. Lancet T. Retraction and republication: 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. Lancet. (2023) 401:2025. doi: 10.1016/S0140-6736(23)01175-3
332. Zhao YM, Shang Y.-m, Song W.-b, Li Q.-q, Xie H, Xu Q.-f, et al. Follow-up study of the pulmonary function and related physiological characteristics of COVID-19 survivors three months after recovery. EClinicalMedicine. (2020) 25:100463. doi: 10.1016/j.eclinm.2020.100463
333. Wu Y, Xu X, Chen Z, Duan J, Hashimoto K, Yang L, et al. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav Immun. (2020) 87:18–22. doi: 10.1016/j.bbi.2020.03.031
334. Puelles VG, Lütgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, et al. Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med. (2020) 383:590–2. doi: 10.1056/NEJMc2011400
335. Davis HE, Assaf GS, McCorkell L, Wei H, Low RJ, Re'em Y, et al. Characterizing long COVID in an international cohort: 7 months of symptoms and their impact. EClinicalMedicine. (2021) 38. doi: 10.1016/j.eclinm.2021.101019
336. Xie Y, Xu E, Bowe B, Al-Aly Z. Long-term cardiovascular outcomes of COVID-19. Nat Med. (2022) 28:583–90. doi: 10.1038/s41591-022-01689-3
337. Xie Y, Al-Aly Z. Risks and burdens of incident diabetes in long COVID: a cohort study. Lancet Diabetes Endocrinol. (2022) 10:311–21. doi: 10.1016/S2213-8587(22)00044-4
338. Mancini DM, Brunjes DL, Lala A, Trivieri MG, Contreras JP, Natelson BH. Use of cardiopulmonary stress testing for patients with unexplained dyspnea post–coronavirus disease. Heart Failure. (2021) 9:927–37. doi: 10.1016/j.jchf.2021.10.002
339. Kedor C, Freitag, Meyer-Arndt L, Wittke K, Hanitsch LG, Zoller T, et al. A prospective observational study of post-COVID-19 chronic fatigue syndrome following the first pandemic wave in Germany and biomarkers associated with symptom severity. Nat Commun. (2022) 13:5104. doi: 10.1038/s41467-022-32507-6
340. Larsen NW, Stiles LE, Shaik R, Schneider L, Muppidi S, Tsui CT, et al. Characterization of autonomic symptom burden in long COVID: A global survey of 2,314 adults. Front Neurol. (2022) 13:1012668. doi: 10.3389/fneur.2022.1012668
341. Demko ZO, Yu T, Mullapudi SK, Varela Heslin MG, Dorsey CA, Payton CB, et al. Post-acute sequelae of SARS-CoV-2 (PASC) impact quality of life at 6, 12 and 18 months post-infection. MedRxiv. (2022). doi: 10.1101/2022.08.08.22278543
342. Cairns R, Hotopf M. A systematic review describing the prognosis of chronic fatigue syndrome. Occup Med. (2005) 55:20–31. doi: 10.1093/occmed/kqi013
343. Grubb BP. Postural tachycardia syndrome. Circulation. (2008) 117:2814–7. doi: 10.1161/CIRCULATIONAHA.107.761643
344. Karas B, Grubb BP, Boehm K, Kip K. The postural orthostatic tachycardia syndrome: a potentially treatable cause of chronic fatigue, exercise intolerance, and cognitive impairment in adolescents. Pacing Clin Electrophysiology. (2000) 23:344–51. doi: 10.1111/j.1540-8159.2000.tb06760.x
345. Raj SR. Postural tachycardia syndrome (POTS). Circulation. (2013) 127:2336–42. doi: 10.1161/CIRCULATIONAHA.112.144501
346. Bryarly M, Phillips LT, Fu Q, Vernino S, Levine BD. Postural orthostatic tachycardia syndrome: JACC focus seminar. J Am Coll Cardiol. (2019) 73:1207–28. doi: 10.1016/j.jacc.2018.11.059
347. Scappaticcio L, Pitoia F, Esposito K, Piccardo A, Trimboli P. Impact of COVID-19 on the thyroid gland: an update. Rev Endocr Metab Disord. (2021) 22:803–15. doi: 10.1007/s11154-020-09615-z
348. Lania A, Sandri MT, Cellini M, Mirani M, Lavezzi E, Mazziotti G. Thyrotoxicosis in patients with COVID-19: the THYRCOV study. Eur J Endocrinol. (2020) 183:381–7. doi: 10.1530/EJE-20-0335
349. Desailloud R, Hober D. Viruses and thyroiditis: an update. Virol J. (2009) 6:5. doi: 10.1186/1743-422X-6-5
350. Aemaz Ur Rehman M, Farooq H, Ali MM, Ebaad Ur Rehman M, Dar QA, Hussain A. The association of subacute thyroiditis with COVID-19: a systematic review. SN Compr Clin Med. (2021) 3:1515–27. doi: 10.1007/s42399-021-00912-5
351. Khatri A, Charlap E, Kim A. Subacute thyroiditis from COVID-19 infection: A case report and review of literature. Eur Thyroid J. (2021) 9:324–8. doi: 10.1159/000511872
352. Pierce JD, Shen Q, Cintron SA, Hiebert JB. Post-COVID-19 syndrome. Nurs Res. (2022) 71:164–74. doi: 10.1097/NNR.0000000000000565
353. Bouslama B, Pierret C, Khelfaoui F, Bellanné-Chantelot C, Donadieu J, Héritier S. Post-COVID-19 severe neutropenia. Pediatr Blood Cancer. (2021) 68. doi: 10.1002/pbc.28866
354. Denison MR, Graham RL, Donaldson EF, Eckerle LD, Baric RS. Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. (2011) 8:270–9. doi: 10.4161/rna.8.2.15013
355. Gupta RK. Will SARS-CoV-2 variants of concern affect the promise of vaccines? Nat Rev Immunol. (2021) 21:340–1. doi: 10.1038/s41577-021-00556-5
356. Hillen HS, Kokic G, Farnung L, Dienemann C, Tegunov D, Cramer P. Structure of replicating SARS-CoV-2 polymerase. Nature. (2020) 584:154–6. doi: 10.1038/s41586-020-2368-8
357. Tregoning JS, Flight KE, Higham SL, Wang Z, Pierce BF. Progress of the COVID-19 vaccine effort: viruses, vaccines and variants versus efficacy, effectiveness and escape. Nat Rev Immunol. (2021) 21:626–36. doi: 10.1038/s41577-021-00592-1
358. Gomez CE, Perdiguero B, Esteban M. Emerging SARS-CoV-2 variants and impact in global vaccination programs against SARS-CoV-2/COVID-19. Vaccines (Basel). (2021) 9. doi: 10.3390/vaccines9030243
359. Kumar A, Parashar R, Kumar S, Faiq MA, Kumari C, Kulandhasamy M, et al. Emerging SARS-CoV-2 variants can potentially break set epidemiological barriers in COVID-19. J Med Virol. (2022) 94:1300–14. doi: 10.1002/jmv.27467
360. Tao K, Tzou PL, Nouhin J, Gupta RK, Oliveira T, Kosakovsky Pond SL, et al. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat Rev Genet. (2021) 22:757–73. doi: 10.1038/s41576-021-00408-x
361. Thorne LG, Bouhaddou M, Reuschl A-K, Zuliani-Alvarez L, Polacco B, Pelin A, et al. Evolution of enhanced innate immune evasion by SARS-CoV-2. Nature. (2022) 602:487–95. doi: 10.1038/s41586-021-04352-y
362. Leary S, Gaudieri S, Parker MD, Chopra A, James I, Pakala S, et al. Generation of a novel SARS-CoV-2 sub-genomic RNA due to the R203K/G204R variant in nucleocapsid: homologous recombination has potential to change SARS-CoV-2 at both protein and RNA level. Pathog Immun. (2021) 6:27. doi: 10.20411/pai.v6i2.460
363. Mourier T, Shuaib M, Hala S, Mfarrej S, Alofi F, Naeem R, et al. Saudi Arabian SARS-CoV-2 genomes implicate a mutant Nucleocapsid protein in modulating host interactions and increased viral load in COVID-19 patients. medRxiv. (2021). doi: 10.1101/2021.05.06.21256706
365. Boppana S, Qin K, Files JK, Russell RM, Stoltz R, Bibollet- Ruche F, et al. SARS-CoV-2-specific circulating T follicular helper cells correlate with neutralizing antibodies and increase during early convalescence. PloS Pathog. (2021) 17:e1009761. doi: 10.1371/journal.ppat.1009761
366. Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. (2016) 16:626–38. doi: 10.1038/nri.2016.90
367. Osuchowski MF, Winkler MS, Skirecki T, Cajander S, Shankar-Hari M, Lachmann G, et al. The COVID-19 puzzle: deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir Med. (2021) 9:622–42. doi: 10.1016/S2213-2600(21)00218-6
368. Tramontana F, Battisti S, Napoli N, Strollo R. Immuno-endocrinology of COVID-19: the key role of sex hormones. Front Endocrinol (Lausanne). (2021) 12:726696. doi: 10.3389/fendo.2021.726696
369. Gadi N, Wu SC, Spihlman AP, Moulton VR. What’s sex got to do with COVID-19? Gender-based differences in the host immune response to coronaviruses. Front Immunol. (2020) 11:562631. doi: 10.3389/fimmu.2020.02147
370. Arnold CG, Libby A, Vest A, Hopkinson A, Monte AA. Immune mechanisms associated with sex-based differences in severe COVID-19 clinical outcomes. Biol Sex Differ. (2022) 13:7. doi: 10.1186/s13293-022-00417-3
371. Spiering AE, de Vries TJ. Why females do better: the X chromosomal TLR7 gene-dose effect in COVID-19. Front Immunol. (2021) 12:756262. doi: 10.3389/fimmu.2021.756262
372. Mohamed MS, Moulin TC, Schiöth HB. Sex differences in COVID-19: the role of androgens in disease severity and progression. Endocrine. (2021) 71:3–8. doi: 10.1007/s12020-020-02536-6
373. Beacon TH, Delcuve GP, Davie JR. Epigenetic regulation of ACE2, the receptor of the SARS-CoV-2 virus(1). Genome. (2021) 64:386–99. doi: 10.1139/gen-2020-0124
374. Saengsiwaritt W, Jittikoon J, Chaikledkaew U, Udomsinprasert W. Genetic polymorphisms of ACE1, ACE2, and TMPRSS2 associated with COVID-19 severity: A systematic review with meta-analysis. Rev Med Virol. (2022) 32:e2323. doi: 10.1002/rmv.2323
375. da Silva JS, Montagnoli TL, Rocha BS, Tacco ML, Marinho SC, Zapata-Sudo G. Estrogen receptors: therapeutic perspectives for the treatment of cardiac dysfunction after myocardial infarction. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22020525
376. Popkin BM, Du S, Green WD, Beck MA, Algaith T, Herbst CH, et al. Individuals with obesity and COVID-19: a global perspective on the epidemiology and biological relationships. Obes Rev. (2020) 21:e13128. doi: 10.1111/obr.13128
377. Frasca D, Reidy L, Romero M, Diaz A, Cray C, Kahl K, et al. The majority of SARS-CoV-2-specific antibodies in COVID-19 patients with obesity are autoimmune and not neutralizing. Int J Obes (Lond). (2022) 46:427–32. doi: 10.1038/s41366-021-01016-9
378. Sanchis-Gomar F, Lavie CJ, Mehra MR, Henry BM, Lippi G. Obesity and outcomes in COVID-19: when an epidemic and pandemic collide. In: Mayo Clinic Proceedings. Elsevier (2020) 95(7):1445–53. doi: 10.1016/j.mayocp.2020.05.006
379. Frasca D, Reidy L, Cray C, Diaz A, Romero M, Kahl K, et al. Influence of obesity on serum levels of SARS-CoV-2-specific antibodies in COVID-19 patients. PloS One. (2021) 16:e0245424. doi: 10.1371/journal.pone.0245424
380. Drucker DJ. Diabetes, obesity, metabolism, and SARS-CoV-2 infection: the end of the beginning. Cell Metab. (2021) 33:479–98. doi: 10.1016/j.cmet.2021.01.016
381. Krams IA, Luoto S, Rantala MJ, Jõers P, Krama T. Covid-19: fat, obesity, inflammation, ethnicity, and sex differences. Pathogens. (2020) 9. doi: 10.3390/pathogens9110887
382. Pugliese G, Liccardi A, Graziadio C, Barrea L, Muscogiuri G, Colao A. Obesity and infectious diseases: pathophysiology and epidemiology of a double pandemic condition. Int J Obes (Lond). (2022) 46:449–65. doi: 10.1038/s41366-021-01035-6
383. Sudhakar M, Winfred SB, Meiyazhagan G, Venkatachalam DP. Mechanisms contributing to adverse outcomes of COVID-19 in obesity. Mol Cell Biochem. (2022) 477:1155–93. doi: 10.1007/s11010-022-04356-w
384. Zimmermann P, Curtis N. Why is COVID-19 less severe in children? A review of the proposed mechanisms underlying the age-related difference in severity of SARS-CoV-2 infections. Arch Dis childhood. (2021) 106:429–39. doi: 10.1136/archdischild-2020-320338
385. Yang HS, Costa V, Racine-Brzostek SE, Acker KP, Yee J, Chen Z, et al. Association of age with SARS-CoV-2 antibody response. JAMA network Open. (2021) 4:e214302–e214302. doi: 10.1001/jamanetworkopen.2021.4302
386. Zimmermann P, Curtis N. Why does the severity of COVID-19 differ with age?: understanding the mechanisms underlying the age gradient in outcome following SARS-CoV-2 infection. Pediatr Infect Dis J. (2022) 41:e36–45. doi: 10.1097/INF.0000000000003413
387. Bartleson JM, Radenkovic D, Covarrubias AJ, Furman D, Winer DA, Verdin E. SARS-CoV-2, COVID-19 and the ageing immune system. Nat Aging. (2021) 1:769–82. doi: 10.1038/s43587-021-00114-7
388. Vono M, Huttner A, Lemeille S, Martinez-Murillo P, Meyer B, Baggio S, et al. Robust innate responses to SARS-CoV-2 in children resolve faster than in adults without compromising adaptive immunity. Cell Rep. (2021) 37. doi: 10.1016/j.celrep.2021.109773
389. Casqueiro J, Casqueiro J, Alves C. Infections in patients with diabetes mellitus: A review of pathogenesis. Indian J Endocrinol Metab. (2012) 16:S27–36. doi: 10.4103/2230-8210.94253
390. Mazucanti CH, Egan JM. SARS-CoV-2 disease severity and diabetes: why the connection and what is to be done? Immun Ageing. (2020) 17:21. doi: 10.1186/s12979-020-00192-y
391. Rahimi L, Malek M, Ismail-Beigi F, Khamseh ME. Challenging issues in the management of cardiovascular risk factors in diabetes during the COVID-19 pandemic: A review of current literature. Adv Ther. (2020) 37:3450–62. doi: 10.1007/s12325-020-01417-8
392. Herman-Edelstein M, Guetta T, Barnea A, Waldman M, Ben- Dor N, Barac YD, et al. Expression of the SARS-CoV-2 receptorACE2 in human heart is associated with uncontrolled diabetes, obesity, and activation of the renin angiotensin system. Cardiovasc Diabetol. (2021) 20:90. doi: 10.1186/s12933-021-01275-w
393. Delamaire M, Maugendre D, Moreno M, Le Goff MC, Allannic H, Genetet B, et al. Impaired leucocyte functions in diabetic patients. Diabetes Med. (1997) 14:29–34. doi: 10.1002/(ISSN)1096-9136
394. Summers KL, Marleau AM, Mahon JL, McManus R, Hramiak I, Singh B. Reduced IFN-alpha secretion by blood dendritic cells in human diabetes. Clin Immunol. (2006) 121:81–9. doi: 10.1016/j.clim.2006.05.015
395. Berbudi A, Rahmadika N, Tjahjadi AI, Ruslami R. Type 2 diabetes and its impact on the immune system. Curr Diabetes Rev. (2020) 16:442–9. doi: 10.2174/18756417MTAxgODQqy
396. Hodgson K, Morris J, Bridson T, Govan B, Rush C, Ketheesan N. Immunological mechanisms contributing to the double burden of diabetes and intracellular bacterial infections. Immunology. (2015) 144:171–85. doi: 10.1111/imm.12394
397. Meshkani R, Vakili S. Tissue resident macrophages: Key players in the pathogenesis of type 2 diabetes and its complications. Clin Chim Acta. (2016) 462:77–89. doi: 10.1016/j.cca.2016.08.015
398. Matuschik L, Riabov V, Schmuttermaier C, Sevastyanova T, Weiss C, Klüter H, et al. Hyperglycemia induces inflammatory response of human macrophages to CD163-mediated scavenging of hemoglobin-haptoglobin complexes. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23031385
399. Restrepo BI, Twahirwa M, Jagannath C. Hyperglycemia and dyslipidemia: Reduced HLA-DR expression in monocyte subpopulations from diabetes patients. Hum Immunol. (2021) 82:124–9. doi: 10.1016/j.humimm.2020.11.005
400. Ilias I, Jahaj E, Kokkoris S, Zervakis D, Temperikidis P, Magira E, et al. Clinical study of hyperglycemia and SARS-CoV-2 infection in intensive care unit patients. In Vivo. (2020) 34:3029–32. doi: 10.21873/invivo.12136
401. Betjes MG. Immune cell dysfunction and inflammation in end-stage renal disease. Nat Rev Nephrol. (2013) 9:255–65. doi: 10.1038/nrneph.2013.44
402. Figueroa Triana JF, Márquez Salas DA, Cabrera Silva JS, Alvarado Castro CC, Buitrago Sandoval AF. COVID-19 y enfermedad cardiovascular. Rev Colombiana Cardiología. (2020) 27:166–74. doi: 10.1016/j.rccar.2020.04.004
403. Gómez-Tejeda JJ, Hernández-Pérez C, Aguilera-Velázquez Y. Afectación del sistema cardiovascular en la infección por SARS-CoV-2. Universidad Médica Pinareña. (2020) 16:1–11.
404. Goudouris ES, Pinto-Mariz F, Mendonca LO, Aranda CS, Guimaraes RR, Kokron C, et al. Outcome of SARS-CoV-2 infection in 121 patients with inborn errors of immunity: a cross-sectional study. J Clin Immunol. (2021) 41:1479–89. doi: 10.1007/s10875-021-01066-8
Keywords: SARS-CoV-2, COVID-19, pathogenesis, immune response, pathophysiology, variants, host factors, long-COVID-19
Citation: Carvajal JJ, García-Castillo V, Cuellar SV, Campillay-Véliz CP, Salazar-Ardiles C, Avellaneda AM, Muñoz CA, Retamal-Díaz A, Bueno SM, González PA, Kalergis AM and Lay MK (2024) New insights into the pathogenesis of SARS-CoV-2 during and after the COVID-19 pandemic. Front. Immunol. 15:1363572. doi: 10.3389/fimmu.2024.1363572
Received: 31 December 2023; Accepted: 24 April 2024;
Published: 07 June 2024.
Edited by:
Tanushree Dangi, Northwestern University, United StatesReviewed by:
Mohan Tulapurkar, University of Maryland, United StatesEvangelia Koukaki, Sotiria General Hospital, Greece
Copyright © 2024 Carvajal, García-Castillo, Cuellar, Campillay-Véliz, Salazar-Ardiles, Avellaneda, Muñoz, Retamal-Díaz, Bueno, González, Kalergis and Lay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margarita K. Lay, bWFyZ2FyaXRhLmxheUB1YW50b2YuY2w=
†These authors have contributed equally to this work