 Jordan Mattke1
Jordan Mattke1 Carly M. Darden2
Carly M. Darden2 Michael C. Lawrence3
Michael C. Lawrence3 Jayachandra Kuncha3
Jayachandra Kuncha3 Yumna Ali Shah3
Yumna Ali Shah3 Robert R. Kane1
Robert R. Kane1 Bashoo Naziruddin2*
Bashoo Naziruddin2*- 1Baylor University, Institute of Biomedical Studies, Waco, TX, United States
- 2Baylor University Medical Center, Annette C. and Harold C. Simmons Transplant Institute, Dallas, TX, United States
- 3Islet Cell Laboratory, Baylor Scott and White Research Institute, Dallas, TX, United States
Acute pancreatitis is a complex inflammatory disease resulting in extreme pain and can result in significant morbidity and mortality. It can be caused by several factors ranging from genetics, alcohol use, gall stones, and ductal obstruction caused by calcification or neutrophil extracellular traps. Acute pancreatitis is also characterized by immune cell infiltration of neutrophils and M1 macrophages. Toll-like receptor 4 (TLR4) is a pattern recognition receptor that has been noted to respond to endogenous ligands such as high mobility group box 1 (HMGB1) protein and or exogenous ligands such as lipopolysaccharide both of which can be present during the progression of acute pancreatitis. This receptor can be found on a variety of cell types from endothelial cells to resident and infiltrating immune cells leading to production of pro-inflammatory cytokines as well as immune cell activation and maturation resulting in the furthering of pancreatic damage during acute pancreatitis. In this review we will address the various mechanisms mediated by TLR4 in the advancement of acute pancreatitis and how targeting this receptor could lead to improved outcomes for patients suffering from this condition.
1 Introduction
The pancreas is an organ comprised of exocrine acinar tissue and endocrine islet tissue. Inflammation targeting the endocrine islets of the pancreas results in type 1 diabetes mellitus, which can be managed through the use of exogenous insulin to replace insulin production lost following beta cell destruction. Acute pancreatitis is an inflammatory condition within the pancreas characterized by damage to the organ in the form of exocrine acinar cell death and local and systemic inflammation (1). Acute pancreatitis may present in mild, moderate, severe forms. Mild pancreatitis presents with no local or systemic complications and no organ failure while moderate and severe pancreatitis both present with local and systemic complications and organ failure. Patients with acute pancreatitis present with epigastric pain radiating to the back, elevated serum amylase and lipase activity, and characteristics of pancreatitis based on ultrasonography, computed tomography, or magnetic resonance imaging (2, 3). The most common causes of acute pancreatitis are gallstones and alcohol, but other causes include hypertriglyceridemia, medication toxicity, trauma, hypercalcemia, various infections, autoimmune, ischemia, and hereditary causes (2). One of the other hallmarks of acute pancreatitis is the infiltration of immune cells such as neutrophils and macrophages following initial insult of pancreatitis, which generates further pancreatic damage (4, 5).
Although mortality due to pancreatitis has decreased over time, hospitalizations and cost of care have significantly increased for patients suffering from acute pancreatitis (6). It is also estimated that ~18% of patients presenting with acute pancreatitis will have recurrence or develop chronic pancreatitis (1). The current therapeutic strategies for managing acute pancreatitis include intravenous fluid resuscitation, nutritional support, and administration of analgesics (1). However, there is currently no targeted therapy for the treatment of acute pancreatitis.
Many cell types are implicated in the progression of acute pancreatitis. It is believed that acute pancreatitis is initiated by one of many factors that results in dysregulation of acinar cells resulting in the production of proinflammatory cytokines and chemokines (7). Following the induction of pancreatitis, neutrophils have been found to infiltrate the pancreas as soon as 1 hour after induction of pancreatitis (4). Proinflammatory M1 macrophages can be found infiltrating the pancreas during the early inflammatory stages of pancreatitis while M2 macrophages have been noted to be more prevalent during the resolution of phases of pancreatitis several days after pancreatitis induction (8). Infiltration of CD4+ T cells can be noted as soon as 6 hours following cerulein challenge in mice further contributing to pancreatic damage (9).
Toll-like receptors (TLRs) are the best characterized class of pattern recognition receptor. There are currently 11 recognized TLRs in humans while there are 13 TLRs in mice. These receptors can be found on the cell surface (TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10) as well as intracellularly localized to endosomes (TLR3, TLR7, TLR8, TLR9, TLR11, TLR12, and TLR13) (10).
TLR1 associates with TLR2 to recognize mycobacterial lipoprotein as well as triacylated lipopeptides (11). TLR6 also forms a heterodimer with TLR2 recognizing macrophage-activating lipopeptide-2 and other diacylated lipopeptides (12). Although TLR1, TLR2, and TLR6 have been shown to be expressed on the surface, Motoi et al. have demonstrated that signaling of TLR1/TLR2 and TLR2/TLR6 actually happens in the endolysosomes (13). TLR5 is known to recognize bacterial flagellin and is also able to form a physical complex with TLR4 (14, 15). TLR10 is a unique toll-like receptor for several reasons. First, the ligands for TLR10 are currently unknown as it is a disrupted pseudogene in mice. TLR10 has been shown to be most homologous to TLR1 and TLR6 and is able to bind to the TLR1/2 ligand, PAM3CSK4 (16). TLR10 is also the only toll-like receptor to demonstrate anti-inflammatory properties (17). TLR4 is one of the most extensively studied TLRs, which responds to a variety of ligands and will be covered in more detail in the next section.
While the previously discussed toll-like receptors have been shown to the be localized to the plasma membrane of cells, several toll-like receptors have been shown to be localized to the endosomal compartments of cells. TLR3 recognizes and binds double-stranded RNA, which is usually the byproduct of viral infection (18). TLR7 and TLR8 recognize guanosine- and uridine-rich single-stranded RNA as well as imidazoquinoline-like molecules (19, 20). Imidazoquinoline is a tricyclic organic molecule known for enhancing immune responses to viruses and cancers (21, 22) TLR9 recognizes and is activated in response to microbial DNA sequences containing unmethylated CpG dinucleotides (23). Mouse TLR11 has been noted to recognize unknown components of uropathogenic bacteria and a profilin-like molecule of a protozoan parasite (10). TLR12 also plays a major role in the recognition of profilin of Toxoplasma gondii (24). TLR13 has been shown to be a key receptor for detecting bacterial RNA (25).
Bacterial translocation is a phenomenon which live bacteria that colonize the intestines or their products are able to cross the intestinal barrier into neighboring organs or the circulatory system resulting in remote organ inflammation and complications. This has been noted in both animal models and human trials during the progression of severe acute pancreatitis (26). Pancreatitis can also occur following viral infection with viral hepatitis, coxsackie and echoviruses, hemorrhagic fever viruses, CMV, and VZV being the most common (27). Because TLRs play a major role in bacterial and viral recognition, the activation of these receptors could play a protective role in the progression of pancreatitis following bacterial and viral infection.
Up to this point all of the discussed ligands for toll-like receptors are related to bacterial and viral infection and serve a role in inflammation in order to protect the body from infection. However, toll-like receptors are also able to respond to markers of sterile inflammation and cell apoptosis. For example, it has been demonstrated that mRNA released from necrotic cells stimulates the activation of TLR3 (28). High mobility group box 1 (HMGB1) released by innate immune cells in response to TNF or IL-1β is able to activate both TLR2 and TLR4 (29).
Heat shock proteins (HSP) are a class of protein originally found to be produced in response to a sudden increase in temperature. However, over time many different types of stresses have been associated with the upregulation of these proteins. These proteins fall into 3 main categories based on their size. HSP60 and HSP70 are both implicated in protein folding and unfolding as well as protein assembly while HSP90 has been shown to prevent steroid binding to DNA (30). Ethridge et al. demonstrated both HSP70 and HSP27 are significantly upregulated during the induction of pancreatitis in mice (31). Cao et al. also showed that HSP60 and HSP70 were significantly upregulated following the induction of pancreatitis in mice and the inhibition of p38 caused a decrease in expression of these heat shock proteins (32). In humans HSP60 serves to activate both TLR2 and TLR4 to promote proliferation of venous smooth muscle cells (33). HSP27 released after global ischemia is able to stimulate both TLR2 and TLR4 resulting in increased NF-ΚB signaling, which upregulates monocyte chemoattractant protein (MCP)-1 and intercellular adhesion molecule (ICAM)-1 as well as cytokine IL-6 (34).
2 Toll-like receptor 4 signaling and pathological relevance in acute pancreatitis
Toll-like receptor 4 (TLR4) is a receptor expressed on a multitude of cell types recognizing bacterial lipopolysaccharides (LPS), viral RNA, saturated fatty acids, and damage associated molecular patterns (DAMPs) such as high mobility group box 1 (HMGB1) protein and heat shock proteins 60 and 70 (Figure 1) (33, 35–39). HMGB1 has been found to be a key marker of many inflammatory conditions such as Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis in the nervous system (40). HMGB1 is also implicated in inflammatory heart disease as well as well as several vascular inflammatory diseases (41, 42). HMGB1 has even been implicated in musculoskeletal diseases such as osteoarthritis (43). The production of HMGB1 and its signaling through TLR4 has been shown to be especially important in the progression of severe acute pancreatitis as administration of HMGB1 to mice resulted in increased pancreatic injury and NF-ΚB signaling that could be attenuated in TLR4-deficient mice (44). Pancreatitis patients with higher HMGB1 levels have also corresponded to increased disease severity (45). TLR4 can also be activated by pancreatic elastase, which has been shown to be elevated during acute pancreatitis (46). A comprehensive list of TLR4 ligands and their effects on different cell types within the pancreas is listed in Table 1.
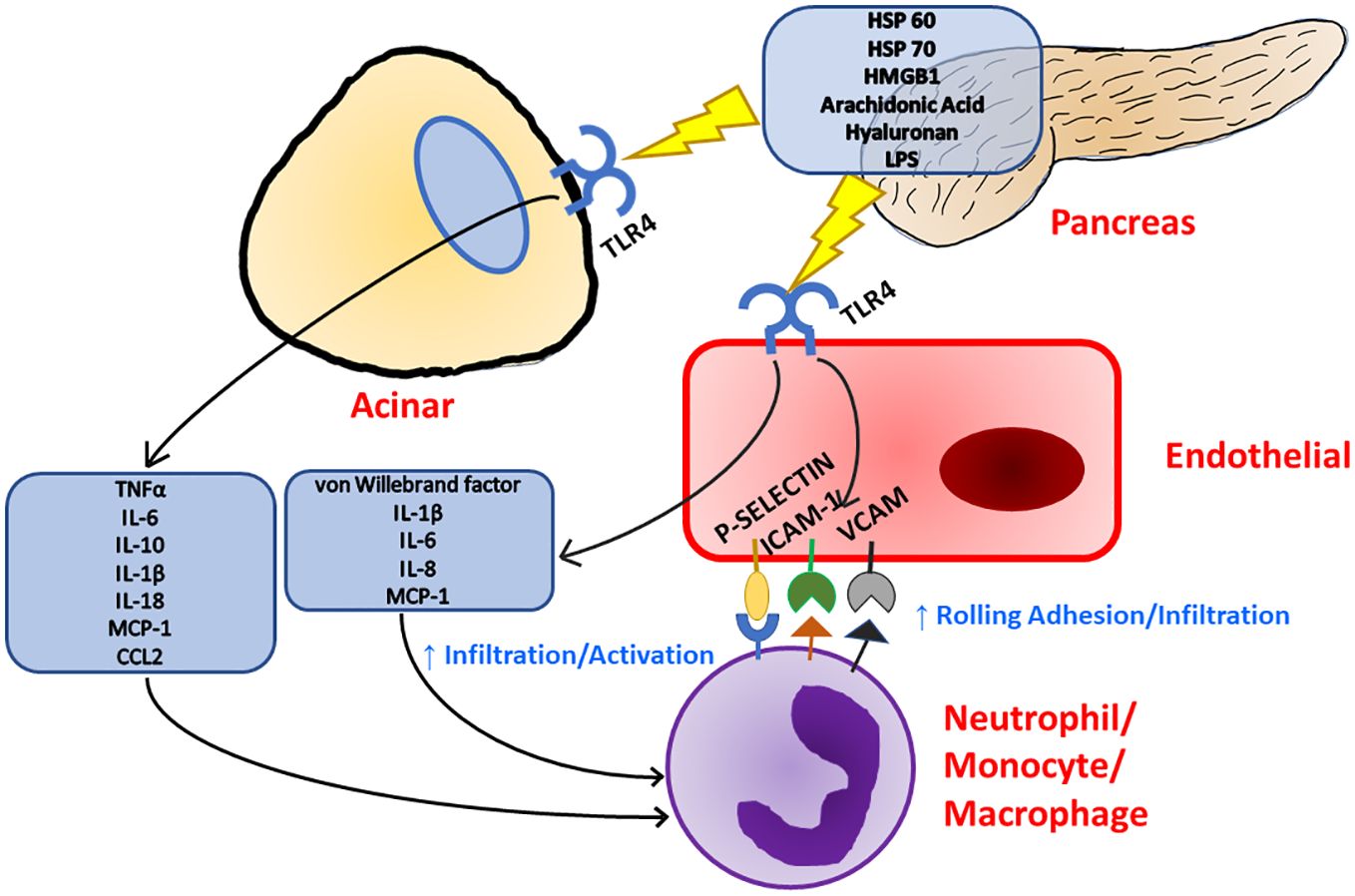
Figure 1 Inflammation in the pancreas can be initiated with TLR4 signaling as TLR4 has been known to respond to a multitude of ligands. Upon TLR4 stimulation, both acinar and endothelial cells release pro-inflammatory cytokines and chemokines which leads to immune cell recruitment and activation. Immune cell recruitment is further supported by TLR4 stimulation as endothelial cells upregulate the expression of surface adhesion molecules.
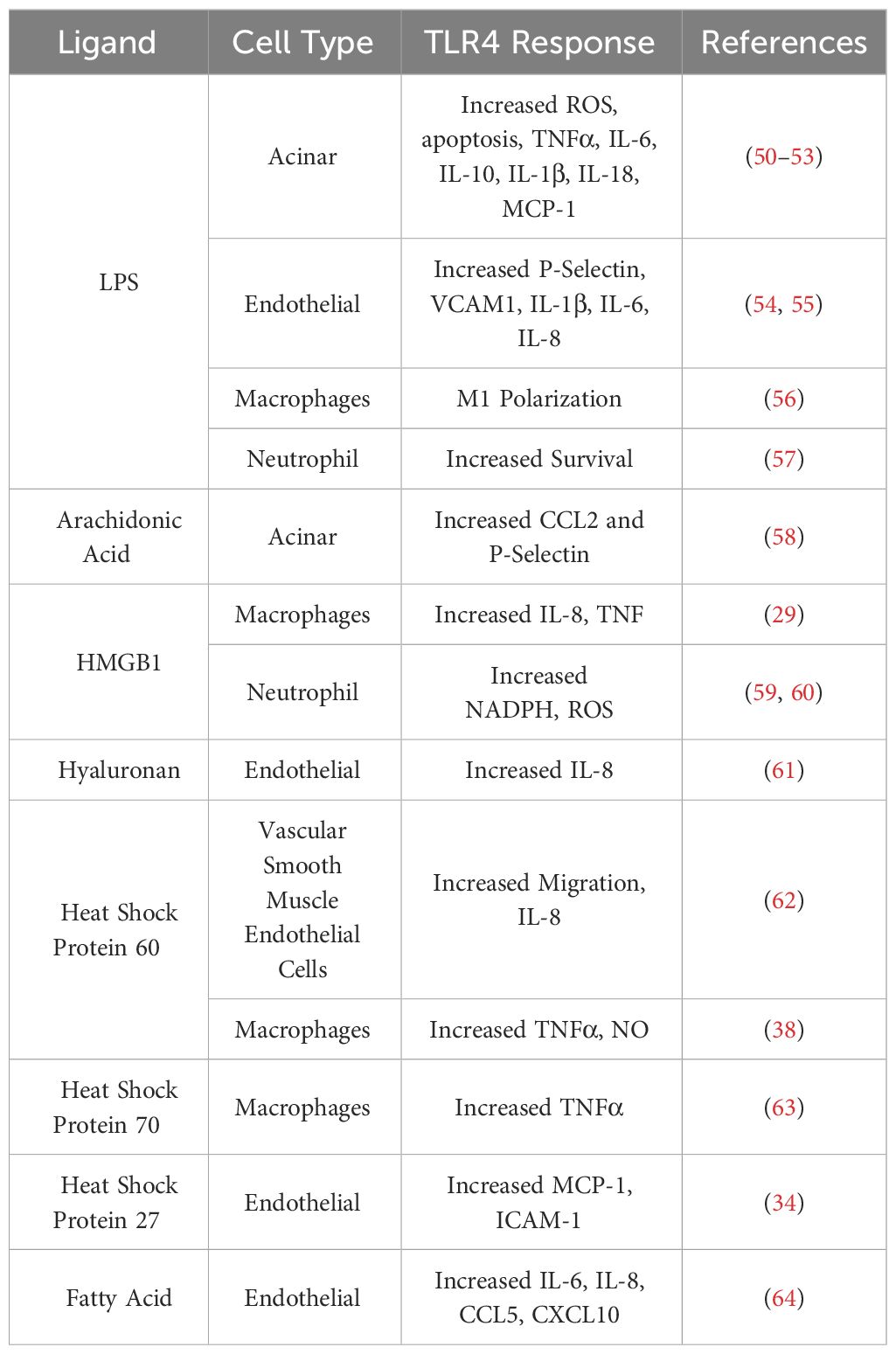
Table 1 TLR4 responses to various ligands within pancreatic tissue.
Within the pancreas Li et al. demonstrated TLR4 is localized to pancreatic ductal epithelium, vascular endothelium, and islets while being absent in exocrine acinar cells of rats (Figure 2) (47). However, TLR4 signaling has proven to play a significant role in acinar cell inflammation during acute pancreatitis, which will be discussed in a later section. TLR4 expression is increased on monocytes 24 hours after the onset of acute pancreatitis and reduces to normal levels after 7 days (48). During severe acute pancreatitis, TLR4 is found to be elevated in liver, kidney, and intestinal tissue following induction of pancreatitis (49).
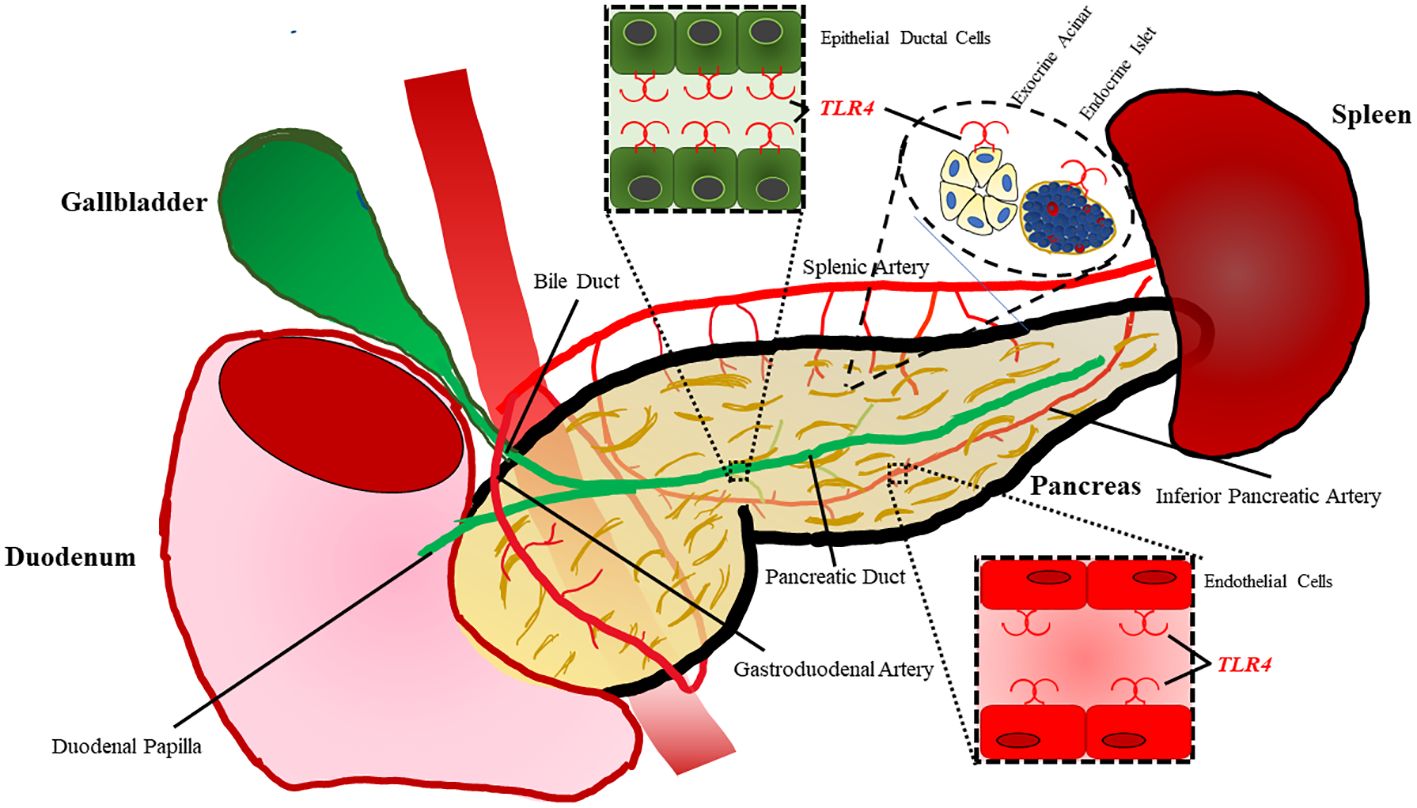
Figure 2 TLR4 is expressed on a variety of cell types within the pancreas. Epithelial cells lining the ductal tissue of the bile and main pancreatic duct are known to express TLR4. TLR4 is also expressed by exocrine acinar tissue as well as endocrine islets. The pancreas also has vasculature composed of endothelial cells with also have been shown to express TLR4.
The process of TLR4 signaling by LPS begins with LPS binding protein (LBP) binding and escorting LPS to CD14. CD14 then transfers LPS to the TLR4-MD2 complex (65). Concentrations of LBP have been found to be significantly elevated in patients suffering from severe acute pancreatitis indicating the process of TLR4 signaling may have a role in systemic complications associated with severe acute pancreatitis (66, 67). Upon stimulation with LPS, TLR4-MD2 oligomerizes and is able to signal through the MyD88 dependent pathway to activate NF-κB and produce proinflammatory cytokines such as TNFα, IL-1β, IL-6, and IL12 (35). As markers such as IL-6 has proven to be significantly altered when looking at the severity of pancreatitis, TLR4 signaling could have a role in the progression of this disease (Table 2) (79). TLR4 is also able to signal through the MyD88 independent pathway through TRIF activating NF-κB and IRF3 to produce type I interferons (35). Inhibition of NF-κB signaling using Withaferin A has led to more beneficial outcomes in cerulein induced pancreatitis as leukocyte infiltration was decreased as well as fibrosis (80). Therefore, indirectly decreasing NF-κB signaling through inhibition of TLR4 could result in beneficial outcomes in the study of acute pancreatitis. An important downstream target of TLR4 signaling in pancreatitis is TRAF6, which is involved in both the TLR4 dependent and TLR4 independent pathways. This plays a significant role in the progression of pancreatitis as TLR4 deficient mice demonstrated much slower progression of pancreatic inflammation when compared to WT mice. TRAF6 was significantly higher in TLR4 deficient mice when compared to WT and TRAF6 was localized to pancreatic acinar cells (81). TRAF6 has also demonstrated a protective role in the progression of acute pancreatitis in acinar cells as stimulation of TLR4 lead to increased SOCS1 and SOCS3 expression, which are responsible for the degradation of TRAF6 through polyubiquitination. Knockout of TLR4 prevented the progression of acute edematous pancreatitis to acute necrotizing pancreatitis with the administration of cerulein and LPS (82).
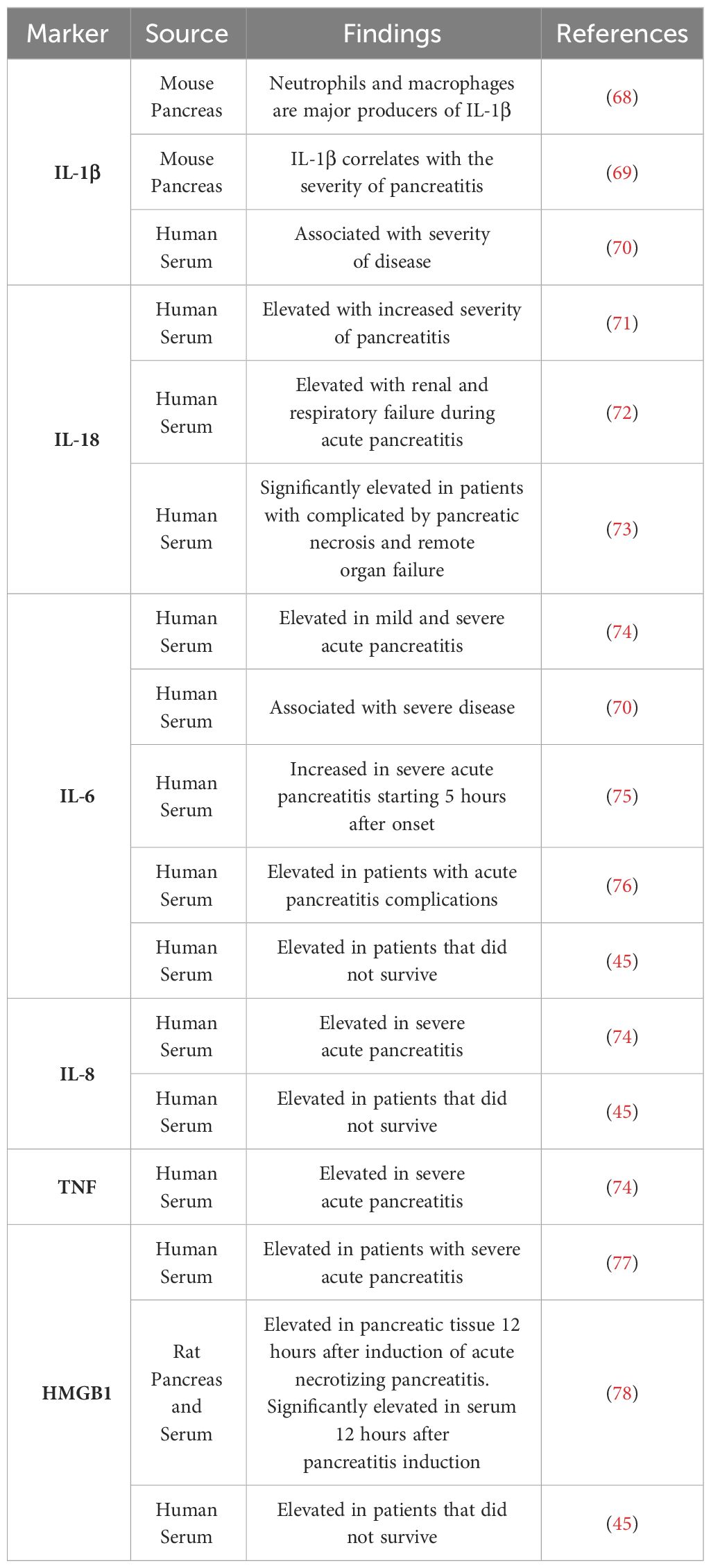
Table 2 TLR4 related molecules used as diagnostic biomarkers.
Another key outcome of TLR4 signaling via NF-κB is the activation of the NOD-leucine rich repeat family pyrin domain containing protein 3 (NLRP3) inflammasome. Upon activation, NLRP3 forms a complex with ASC and pro-caspase-1. This complex formation and activation results in the conversion of pro-caspase-1 to active caspase-1, which will then cleave pro-IL-1β and IL-18 to active IL-1β and IL-18 that will be released from the cell promoting further inflammation. This process also results in the cleavage of gasdermin D. The N-terminal fragment of this cleavage will then form a pore in the plasma membrane allowing for further release of IL-1β and IL-18. This inflammatory process mediated by the NLRP3 inflammasome has been termed pyroptosis (Figure 3) (83). NLRP3, ASC, and caspase-1 are involved in inflammation associated with acute pancreatitis as loss of any of these signaling components has been shown to reduce the severity of acute pancreatitis (84). In a study focusing on patients with acute pancreatitis, it was demonstrated that higher serum IL-18 levels corresponded to pancreatitis complicated by pancreatic necrosis and remote organ failure showing that pyroptosis was increased in the organs of these patients (73). Targeting of the TLR4/NLRP3 axis with emodin, rhein, baicalin, and chrysin significantly reduced acinar cell necrosis and decreased nitric oxide production in macrophages demonstrating more beneficial outcomes in mice by targeting this pathway (85). NLRP3 is also a promising target in severe acute pancreatitis in that Sendler et al. propose that activation of this pathway in macrophages initiates innate inflammatory responses such as neutrophil recruitment and maturation as well as acts as a Th2-cell mediator for the adaptive immune system (86). It has also been shown that inhibition of caspase-1 leads to reduced acinar cell death by necrosis in severe acute pancreatitis (87). Therefore, targeting of TLR4 upstream of caspase-1 could at least partially inhibit cleaving and activation of caspase-1, which would result in more beneficial outcomes to those suffering from pancreatitis. Taken further, TLR4 on platelets can also signal through the NLRP3-ASC inflammasome leading to activation of caspase-1 promoting increased neutrophil extracellular trap (NET) formation leading to increased production of TLR4 agonist, S100A8/A9, which creates a positive feedback loop of inflammation during pyroptosis (88).
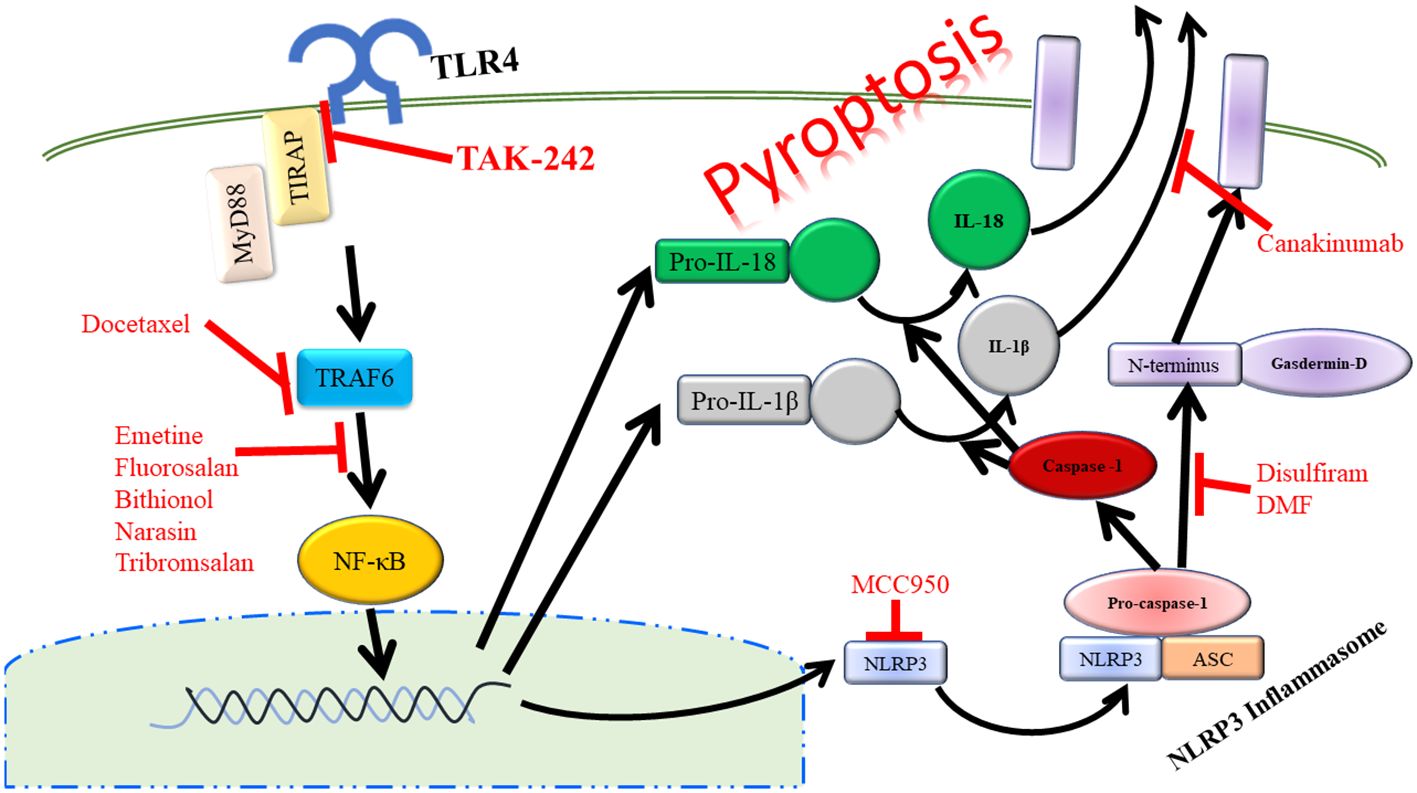
Figure 3 Activation of TLR4 can lead to the inflammatory process of pyropotosis. Many therapeutic drugs have been developed to target the different components of pyroptosis pathway.
Another outcome of TLR4 signaling is the production of macrophage migration inhibitory factor (MIF), which has been shown to be a significant indicator of the severity of pancreatitis in both humans and mice (89–91). Inhibition of MIF has led to alleviated damage in pancreatic and renal tissues in a severe acute pancreatitis model through the attenuation of the NLRP3 pathway (92). MIF plays a key role in the NLRP3 inflammasome assembly leading the production and release of IL-1β in macrophages (93).
Activation of TLR4 can also lead to the activation of RIP3 and the induction of necroptosis (94). While apoptosis is usually associated with an orderly process of cell disassembly with little or no DAMP release, necroptosis is associated with massive release of DAMP molecules resulting in enhanced inflammation as a result of cell death (95). Inhibition of TLR4 signaling using small molecule inhibitor of, TAK-242, has led to attenuated necroptosis in an acute pancreatitis model as evidenced by a reduced expression of RIP3 within the pancreas of treated mice (96).
3 TLR4 in acinar cells
TLR4 plays a significant role in inflammation within acinar cells of the pancreas. Treatment with LPS has been shown to significantly increase the amount of apoptotic acinar cells in a cerulein induced pancreatitis (50). Upon stimulation with LPS, primary pancreatic acinar cells were found to have a significant increase in intracellular reactive oxygen species (ROS) as viability was decreased (51). LPS also induces apoptosis and expression of TNF-α, IL-1β, and IL-18 mRNA in AR4-2J cells (derived from azaserine-induced malignant nodules in rat pancreas) (52). Another study found that TLR4-positive acinar cells respond to LPS by activating the inflammasome and producing TNF-α, IL-6, IL-10, IL-1β, IL-18, and MCP-1 during acute pancreatitis, and these effects could be exacerbated by alcohol (Figure 1) (53). Vona-Davis et al. investigated the effects of LPS and TNFα treatment on AR4-2J cells and noted that there was an activation of both STAT3 and SOCS3 in response to this treatment with significant increases in SOCS3 expression with treatment of LPS and IL-6 while TNFα enhanced expression of STAT3, which also stimulated SOCS3 expression (97). This highlights the importance of SOCS3 in the resolution of inflammation and a possible role of TLR4 in not only the initiation of inflammation, but the resolution of inflammation within acinar cells.
Isolated pancreatic acini are able to respond to arachidonic acid via TLR4 resulting in the upregulation of monocyte chemoattractant protein 1 (CCL2) and P-selectin, which would cause increased immune recruitment and adhesion in the pancreas (58). In a study carried out by Sztefko and Panek it was noted that elevated levels of arachidonic acid could be involved in the development of complications in acute pancreatitis (98). In vivo TLR4-/- and CD14-/- mice showed reduced acinar atrophy in a severe acute pancreatitis model (99). Small molecule inhibitor of TLR4, TAK-242, increased the viability of pancreatic acinar cells, decreased lactate dehydrogenase, and reduced apoptotic cell death following exposure to taurocholate. This was accompanied by decreased release of cytochrome c into the cytoplasm, reduced mitochondrial swelling, and decreased mitochondrial Ca2+ buffering capacity following exposure to taurocholate (100). Administration of natural product, biochanin A, resulted in reduced pancreas damage through decreased TLR4 and NLRP3 signaling (101). This is further evidenced in a study that showed knockout of NLRP3 or gasdermin D in acinar tissue of cerulein treated mice resulted in reduced pyroptosis (102). In TLR4 deficient mice there is significantly less acinar cell necrosis (103).
4 TLR4 in endothelial cells
TLR4 plays a key role in the inflammation of endothelial cells, which could contribute to the infiltration of immune cells during the progression of acute pancreatitis. First, stimulation of TLR4 in endothelial cells leads to significant increases in surface adhesion proteins as well as increases in P-selectin and von Willebrand factor expression (Figure 1) (104). P-selectin plays a significant role in the adhesion and infiltration of neutrophils as inhibition of P-selectin has resulted in reduced damage and neutrophil infiltration in experimental pancreatitis (105, 106). Elevated von Willebrand factor has also proven to be a useful biomarker for severe acute necrotizing pancreatitis (107). TLR4 signaling in endothelial cells can lead to Weibel-Palade body degranulation, NF-κB activation, and vaso-occlusion (108). It has been further demonstrated that TLR4 participates in HLA class I signaling to upregulate P-selectin and von Willebrand factor expression leading to increased rolling adhesion and infiltration by monocytes (109). Vein endothelial cells have been shown to respond to LPS via TLR4 signaling to produce cytokines such as IL-1β, IL-6, and IL-8 further contributing to immune infiltration and activation (54). Increased IL-8 concentrations in serum are correlated to more complicated pancreatitis as well as neutrophil elastase (110). These results implicate serum IL-8 as a biomarker for neutrophil activation leading to pancreatitis complications. This also shows that reduction in production of IL-8 through the inhibition of TLR4 signaling could result in improved pancreatitis outcomes.
Within the pancreas, intraperitoneal administration of LPS lead to significant increases in P-selectin and vascular cell adhesion molecule-1 (VCAM-1) expression within the pancreas (55). As VCAM-1 and P-selectin are both associated with increased immune cell adhesion and infiltration, a reduction of these markers through inhibition of TLR4 signaling could serve as a therapeutic strategy to attenuate damage due to immune cell infiltration and activation. TLR4 of endothelial cells is also able to sense components of the extracellular matrix such as hyaluronan to trigger inflammation and the initial stages of would defense and repair (61). As hyaluronan has been shown to be accumulated in the edematous interstium during acute pancreatitis, this could lead to the activation of TLR4 in endothelial cells further contributing to inflammation and immune cell recruitment to the pancreas (111).
5 TLR4 in macrophages
TLR4 and the associated pathways are essential to the maturation and polarization of macrophages (Figure 4). Macrophages can be divided into different categories based on stimulus. M0 macrophages are non-activated macrophages, which can differentiate into proinflammatory M1 macrophages or anti-inflammatory M2 macrophages. M1 macrophages develop following exposure to inflammatory signals such as IFNγ, LPS, or TNFα, which activates STAT1, NF-κB, p65/p50, and IRF5 resulting in the production of IL-6, TNFα, IL-23, and iNOS. M2 macrophages can be further subdivided into M2a, M2b, M2c, and M2d subtypes all of which play a role in resolution of inflammation and wound healing and form in response to different stimuli (56). As mentioned previously, the activation of TLR4 not only signals through TRAF6 to promote inflammation and survival signals, but it also signals through SOCS1 and SOCS3 to provide negative feedback for this process. SOCS1 has proven to be especially important in dampening immune infiltration and inflammation responses as mice deficient in SOCS1 showed increases in inducible nitric oxide synthase expression in the pancreases of these mice, which appeared to preferentially damage exocrine over endocrine tissue (112). Qin et al. also show that deficiency of SOCS3 in macrophages resulted in higher levels of M1 macrophage genes (113). However, this contrasts to a study carried out by Gordon et al. who found that silencing of SOCS3 promoted a shift of activated M1 macrophage markers to increased expression of M2 macrophage markers. This same study also noted that silencing of SOCS3 resulted in increased phagocytic capacity of these macrophages (114). In a study conducted by Arnold et al. focusing on renal and peritoneal inflammation, SOCS3 expression correlated strongly with disease severity and renal injury. SOCS3 was also shown to colocalize with markers of M1 macrophage polarization (115). The role of SOCS3 in the activation of macrophages in pancreatitis is further supported by a study showing that macrophage-specific deletion of SOCS3 developed less severe pancreatitis and produced less TNFα in response to cerulein injections. This same study also highlighted the importance of CCL2 in the migration of macrophages from the bone marrow to the pancreas during the induction of pancreatitis (116). This further emphasizes the importance of TLR4 response in acinar cells as CCL2 has been found to be upregulated by TLR4 in response to arachidonic acid. NLRP3 signaling also plays a large role in the activation of macrophages and associated pro-inflammatory as well as the compensatory anti-inflammatory responses. In a study where bone marrow-derived macrophages were incubated with pancreatic acini, macrophages were found to secrete increased IL-1β and IL-18 showing an upregulation of the NLRP3 pathway. In a study conducted by Sendler et al. NLRP3 activation was shown to have a significant role in both the hyperinflammation responses as macrophages are activated promoting enhanced recruitment and activation of other immune cells. Inhibition of the NLRP3 inflammasome using MCC950 resulted in significantly reduced neutrophil infiltration, T cell activation, and disease severity in mice. They also showed that NLRP3 signaling has a role in the anti-inflammatory response syndrome in which Th2 and Tregs are activated by IL-18 in the absence of IL-12, which results in enhanced pancreatic fibrosis and permits bacterial translocation into pancreatic necrosis or severe sepsis (86). Since TLR4 is known to activate the NLRP3 inflammasome, inhibition of TLR4 could also lead to some of these beneficial effects.
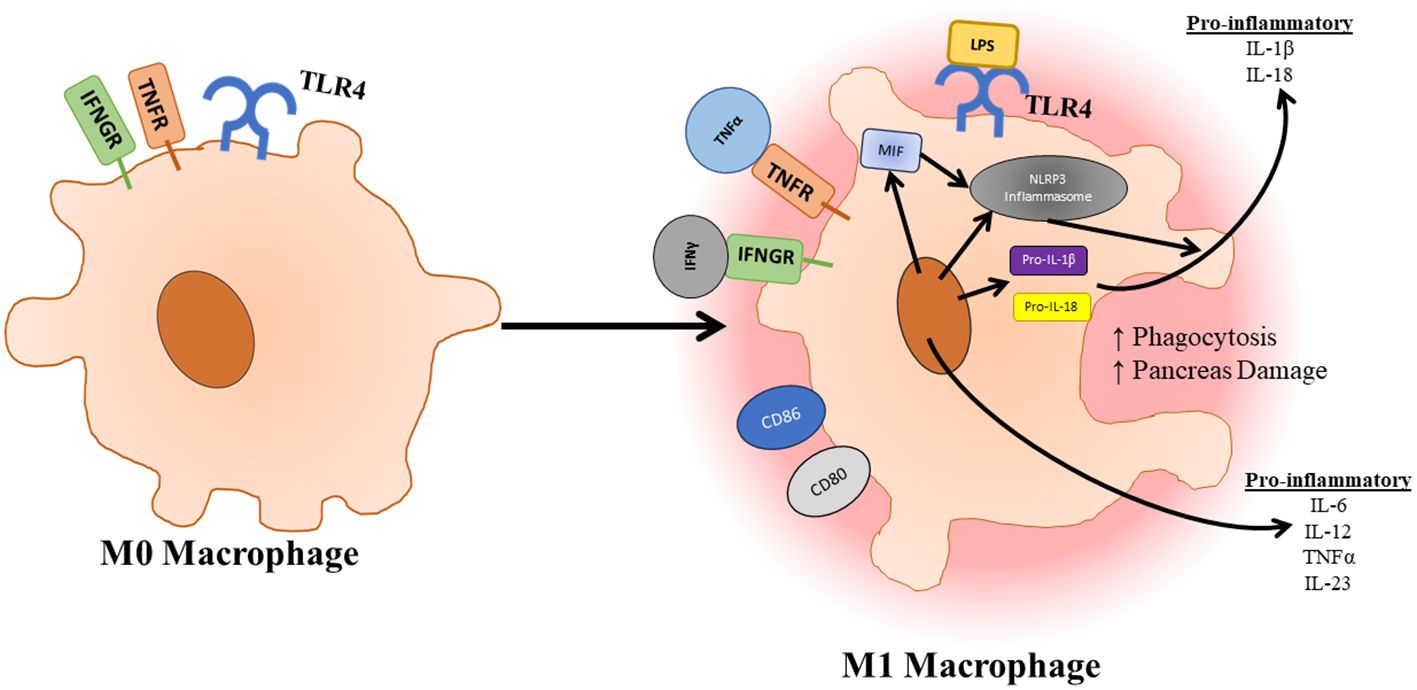
Figure 4 Once macrophages have infiltrated the pancreas, they can be activated and polarized to an M1 state through TLR4 signaling. Activation of macrophages is further supported by factors released by pancreatic tissue. Upon activation, polarized macrophages increase their phagocytic capacity as well as release pro-inflammatory cytokines which further immune infiltration and destruction.
Macrophages play an important role in the progression of pancreatitis as a delicate balance between M1 and M2 macrophages is needed in order for pathogen clearing and wound healing to occur. During acute pancreatitis, infiltrating macrophages are mainly activated and differentiated into an M1 phenotype (5). This is further confirmed by Sendler et al. who showed M2 macrophages were found in non-necrotic areas of pancreatic tissue while M1 macrophages were found in necrotic fields during induction of pancreatitis. Co-culture of bone marrow derived macrophages with acinar cells showed significant increases in IL-6 and TNFα as well as anti-inflammatory IL-10 (86). TLR4 signaling plays a large role in the differentiation of M1 macrophages. In macrophages advanced glycation end products have been found to stimulate TLR4 signaling resulting in an M1 phenotype, which could be inhibited by treatment with TAK-242 (117). The fungal protein paracoccin has also been found to stimulate TLR4 resulting in a pro-inflammatory M1 phenotype (118). Treatment of macrophages with berberine results in a reduction in the production of inflammatory factors and reduced polarization of macrophages to an M1 phenotype by inhibiting the binding of MyD88 to TLR4 (119). Blocking of TLR4 and TNFR1 promotes of a shift of macrophages to an M2 phenotype (120). Therefore, inhibition of TLR4 could serve as a therapeutic strategy to dampen inflammation induced by macrophages by promoting a shift from inflammatory to anti-inflammatory macrophages.
Administration of pancreatic elastase results in increased inflammatory responses in THP-1 cells mediated by TLR4 (46, 121). High fat diet in acute pancreatitis rats aggravated infiltration of activated, inflammatory macrophages by signaling of TLR4 which could be inhibited by the administration of TAK-242 (96). Carbon monoxide releasing molecule has been found to inhibit TLR4 signaling in macrophages leading to reduced production of TNFα and proinflammatory cytokines in cerulein-induced pancreatitis (122).
6 TLR4 in neutrophils
TLR4 signaling plays a significant role in the activation and lifespan of neutrophils. Stimulation of TLR4 using purified LPS almost completely prevents neutrophil apoptosis at early timepoints by signaling through NF-κB (57). TLR4 also signals through MEK kinase leading to the inhibition of G-protein-coupled receptor kinases 2 and 5, which are responsible for the internalization or surface chemokine receptor CXCR2, resulting in increased neutrophil activation and migration (123). TLR4 signaling is also unique in neutrophils in that neutrophils only utilize the MyD88-dependent signaling pathway when stimulated with ligands such as LPS (124). HMGB1 during hemorrhagic shock/resuscitation results in increased TLR4 signaling resulting in increased NADPH oxidase activity in neutrophils leading to an increased production of reactive oxygen species (59). The activity of NADPH oxidase and production of reactive oxygen species by neutrophils results in increased TLR2 and ICAM-1 expression on endothelial cells, which creates a positive feedback loop of inflammation and cellular adhesion (125). High fat diet in acute pancreatitis induced rats results in increased activation and infiltration of neutrophils into the pancreas by signaling through TLR4 (96). TLR4-/- mice have demonstrated significantly less neutrophil infiltration in an acute pancreatitis model (103, 126).
One unique feature of neutrophils is their ability to produce web-like structures composed of decondensed chromatin fragments wrapped in histones, proteases, granules, and cytoplasmic proteins referred to as neutrophil extracellular traps (NETs) (Figure 4) (127). NETs have been found to play a significant role in the progression of a taurocholate induced acute pancreatitis model as neutrophil depletion and administration of DNase I lead to attenuated pancreas damage (128). In patients with acute pancreatitis increases in platelet microparticles were observed. When these platelet microparticles were mixed with healthy neutrophils, there was an increase in myeloperoxidase, neutrophil elastase, and histone H3 production as well as the release of NETs. This indicates that the production of NETs also plays a role in the progression of pancreatitis in humans (129). TLR4 on platelets induces platelet binding to neutrophils resulting in neutrophil activation and NET formation (130). The presence of free radicals has been shown to play a role in the progression of pancreatitis and the production of NETs (131). These free radicals such as superoxide can lead to an increased production of NETs at the site of sterile inflammation through signaling of TLR4 (132). Treatment of neutrophils with HMGB1 (elevated during severe acute pancreatitis) results in increased production of NETs through increased signaling of TLR4 (60). The production of NETs is also supported by the activation of the NLRP3 inflammasome under sterile conditions as NLRP3 was found to support both nuclear envelope and plasma membrane rupture during the release of NETs (133). Not only are NETs produced directly through TLR4 signaling, but NET release is also supported by cytokines produced in other tissues during the progression of acute pancreatitis. IL-8 (elevated following TLR4 signaling in endothelial cells) promotes the production of NETs through its interaction with CXCR2, which is upregulated on neutrophils in response to TLR4 signaling (134). IL-1β (upregulated in response to TLR4 signaling in acinar and endothelial cells in response to TLR4 signaling)has also shown the ability to stimulate the production of NETs in neutrophils as well (135).
7 TLR4 in remote organ complications during severe acute pancreatitis
TLR4 has a demonstrated role in pancreatic inflammation and in the progression of acute pancreatitis. In addition, previous studies have also demonstrated a role for TLR4 in remote organ complications as well. It has long been recognized that severe acute pancreatitis can lead to lung damage, which in turn leads to increased mortality of patients with pancreatitis (136). During the induction of acute pancreatitis mice utilizing LPS, an upregulation of both TLR4 and MIF were observed in the lungs of treated mice. The inhibition or loss of MIF resulted in lower TLR4 expression in the lungs of mice following the induction of pancreatitis as well as increased survival outcomes further implicating TLR4 in respiratory distress following the onset of pancreatitis (137). TLR4-/- mice also showed significantly less lung myeloperoxidase activity following the cerulein induction of pancreatitis (99). Induction of pancreatitis in rats using L-arginine also resulted in increased serum creatinine and BUN indicating increased renal injury during this induction of pancreatitis. This same study also showed increased liver biomarkers aspartate transaminase (AST) and alanine transaminase (ALT) during the induction of pancreatitis. Down regulation of HMGB1/TLR4/NF-ΚB signaling using protocatechuic acid resulted in significant reduction in levels of pancreatic amylase and lipase as well as reduced AST, ALT, creatinine, and BUN (138). The trends of reduced renal and hepatic damage were also noted in TLR4 deficient mice as well as significantly lower serum levels of interleukin-1 and tumor necrosis factor (49). Taken together, not only could inhibition of TLR4 affect immune infiltration and damage within the pancreas during the onset of acute pancreatitis, but inhibition of TLR4 could also alleviate remote organ complications during severe acute pancreatitis such as adult respiratory distress syndrome.
8 Conclusions and future directions
TLR4 is a pattern recognition receptor expressed on a diverse population of cells and plays a multitude of roles in the progression in acute pancreatitis. The initial insult of pancreatitis results in the production of key ligands for TLR4 such as HMGB1 and heat shock proteins associated with sterile inflammation. These ligands can then stimulate localized inflammation in acinar and endothelial tissue through TLR4 to promote the production of many inflammatory factors which increase infiltration and activation of innate immune cells (Figure 1). Once these innate immune cells enter the pancreas, TLR4 signaling stimulates the polarization of macrophages to an inflammatory M1 phenotype, which will lead to further destruction of exocrine tissue as well as continued infiltration of immune cells (Figure 4). However, further study is warranted in the area of TLR4 signaling and the polarization of macrophages as overproduction of M2 macrophages can result in chronic pancreatitis (139). Clarification of the role of SOCS3 and TLR4 in macrophage polarization is also of interest in the progression of pancreatitis as deletion of SOCS3 resulted in more beneficial outcomes following induction of pancreatitis. SOCS3 also colocalized with M1 macrophage markers. However, conflicting information states that SOCS3 deficiency results in increased M1 gene expression. TLR4 also has a significant role in the infiltration and activation of neutrophils as TLR4 deficient mice show reduced neutrophil infiltration and pancreas damage. TLR4 has a role in the production of NETs, which are shown to be increased in the progression of pancreatitis and play a role in destruction of exocrine tissue (Figure 5). We would also like to acknowledge that very little research has been carried out investigated TLR4 signaling in ductal epithelial cells. TLR4 has been identified in cells of the ductal epithelium of the pancreas (47). As TLR4 has been shown to be upregulated in epithelial cells of the bowel during inflammatory conditions such as Crohn’s disease and ulcerative colitis, it would also be worth investigating TLR4 expression and signaling of the pancreatic ductal epithelial cells during the progression of acute pancreatitis as this could also contribute to immune cell infiltration and activation (140). Investigation of TLR4 inhibition has been shown to be clinically feasible as small molecule inhibitor of TLR4, TAK-242, has been used in a clinical trial for use in the treatment of sepsis and has proven to be safe for use in humans (141). Because of the diversity of roles TLR4 plays in the progression of pancreatitis and pancreas damage, the inhibition of TLR4 could be worth investigating for the treatment of acute pancreatitis in humans.
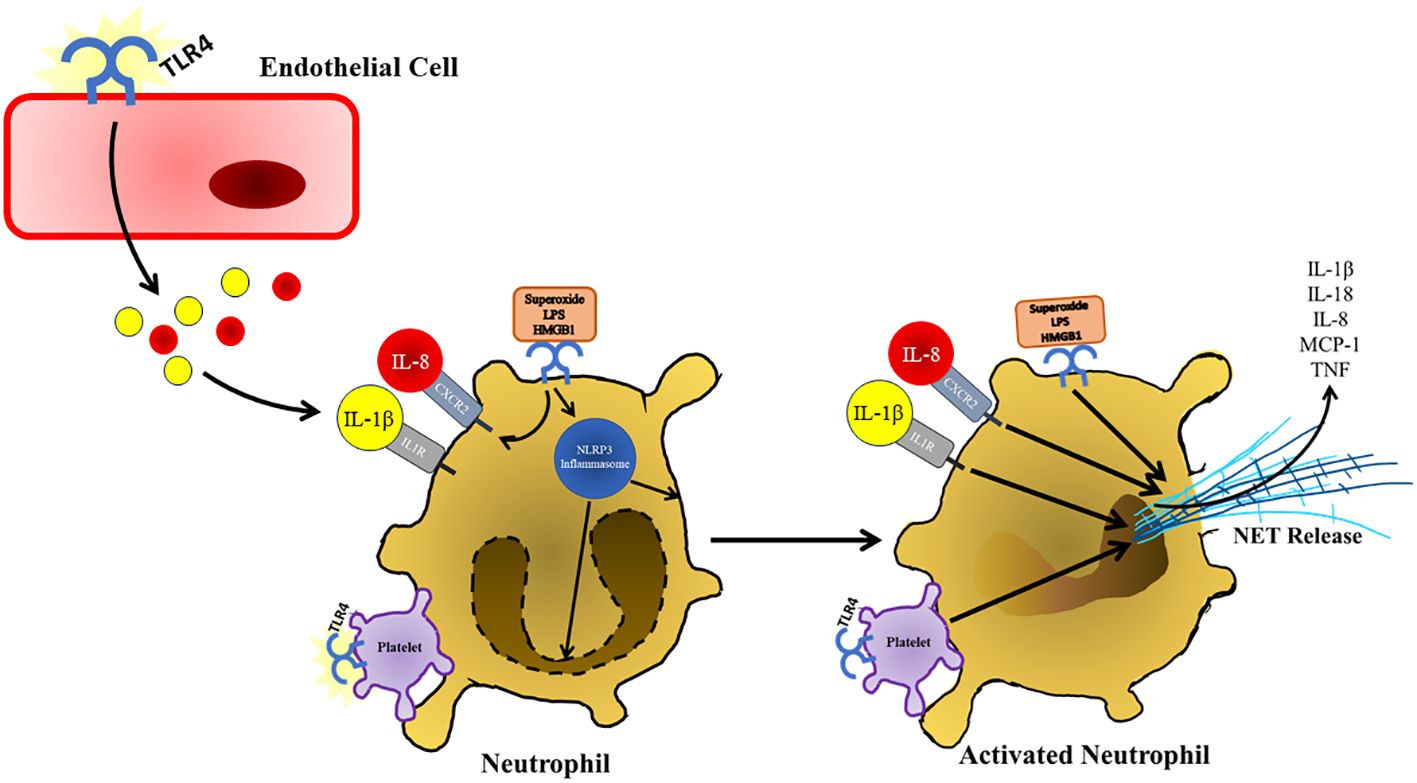
Figure 5 Following TLR4 stimulation and other damage associated signals present in the pancreas, neutrophils respond by secreting NETs, which further contribute to pancreatic damage and immune infiltration during the progression of acute pancreatitis. NET formation is also supported by TLR4 activation and binding of platelets. TLR4 stimulation also supports the production of NETs by upregulating of CXCL2, which is stimulated by IL-8 that presents at increased concentrations during the progression of acute pancreatitis. NETs are also released in response to increased IL-1β, which is also increased in response to TLR4 stimulation in various cell types in response to TLR4 signaling.
Author contributions
JM: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. CD: Writing – review & editing. ML: Writing – review & editing. JK: Writing – review & editing. YA: Writing – review & editing. BK: Writing – review & editing. BN: Writing – review & editing, Conceptualization.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors would like to express their gratitude to Baylor University Medical Center for continued funding support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lee PJ, Papachristou GI. New insights into acute pancreatitis. Nat Rev Gastroenterol Hepatol. (2019) 16:479–96. doi: 10.1038/s41575-019-0158-2
2. Cappell MS. Acute pancreatitis: etiology, clinical presentation, diagnosis, and therapy. Common Hepatic Emergencies. (2008) 92:889–923. doi: 10.1016/j.mcna.2008.04.013
3. Walkowska J, Zielinska N, Tubbs RS, Podgórski M, Dłubek-Ruxer J, Olewnik Ł. Diagnosis and treatment of acute pancreatitis. Diagnostics. (2022) 12(8):1974. doi: 10.3390/diagnostics12081974
4. Folch E, Closa D, Prats N, Gelpí E, Roselló-Catafau J. Leukotriene generation and neutrophil infiltration after experimental acute pancreatitis. Inflammation. (1998) 22:83–93. doi: 10.1023/A:1022399824880
5. Hu F, Lou N, Jiao J, Guo F, Xiang H, Shang D. Macrophages in pancreatitis: Mechanisms and therapeutic potential. BioMed Pharmacother. (2020) 131:110693. doi: 10.1016/j.biopha.2020.110693
6. Brindise E, Elkhatib I, Kuruvilla A, Silva R. Temporal trends in incidence and outcomes of acute pancreatitis in hospitalized patients in the United States from 2002 to 2013. Pancreas. (2019) 48(2):169–75. doi: 10.1097/MPA.0000000000001228
7. Halangk W, Lerch MM. Early events in acute pancreatitis. Gastroenterol Clin. (2004) 33:717–31. doi: 10.1016/j.gtc.2004.07.009
8. Wu J, Zhang L, Shi J, He R, Yang W, Habtezion A, et al. Macrophage phenotypic switch orchestrates the inflammation and repair/regeneration following acute pancreatitis injury. eBioMedicine. (2020) 58:102920. doi: 10.1016/j.ebiom.2020.102920
9. Demols A, Le Moine O, Desalle F, Quertinmont E, van Laethem JL, Devière J. CD4+ T cells play an important role in acute experimental pancreatitis in mice. Gastroenterology. (2000) 118:582–90. doi: 10.1016/S0016-5085(00)70265-4
10. Kawai T, Akira S. TLR signaling. Cell Death Differ. (2006) 13:816–25. doi: 10.1038/sj.cdd.4401850
11. Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, et al. Cutting edge: role of toll-like receptor 1 in mediating immune response to microbial lipoproteins1. J Immunol. (2002) 169:10–4. doi: 10.4049/jimmunol.169.1.10
12. Takeuchi O, Kawai T, Mühlradt PF, Morr M, Radolf JD, Zychlinsky A, et al. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol. (2001) 13:933–40. doi: 10.1093/intimm/13.7.933
13. Motoi Y, Shibata T, Takahashi K, Kanno A, Murakami Y, Li X, et al. Lipopeptides are signaled by Toll-like receptor 1, 2 and 6 in endolysosomes. Int Immunol. (2014) 26:563–73. doi: 10.1093/intimm/dxu054
14. Yoon S, Kurnasov O, Natarajan V, Hong M, Gudkov AV, Osterman AL, et al. Structural basis of TLR5-flagellin recognition and signaling. Science. (2012) 335:859–64. doi: 10.1126/science.1215584
15. Hussain S, Johnson CG, Sciurba J, Meng X, Stober VP, Liu C, et al. TLR5 participates in the TLR4 receptor complex and promotes MyD88-dependent signaling in environmental lung injury. eLife. (2020) 9:e50458. doi: 10.7554/eLife.50458
16. Guan Y, Ranoa DRE, Jiang S, Mutha SK, Li X, Baudry J, et al. Human TLRs 10 and 1 share common mechanisms of innate immune sensing but not signaling. J Immunol. (2010) 184:5094–103. doi: 10.4049/jimmunol.0901888
17. Jiang S, Li X, Hess NJ, Guan Y, Tapping RI. TLR10 is a negative regulator of both myD88-dependent and -independent TLR signaling. J Immunol. (2016) 196:3834–41. doi: 10.4049/jimmunol.1502599
18. Leonard JN, Ghirlando R, Askins J, Bell JK, Margulies DH, Davies DR, et al. The TLR3 signaling complex forms by cooperative receptor dimerization. Proc Natl Acad Sci. (2008) 105:258–63. doi: 10.1073/pnas.0710779105
19. Gibson SJ, Lindh JM, Riter TR, Gleason RM, Rogers LM, Fuller AE, et al. Plasmacytoid dendritic cells produce cytokines and mature in response to the TLR7 agonists, imiquimod and resiquimod. Cell Immunol. (2002) 218:74–86. doi: 10.1016/S0008-8749(02)00517-8
20. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. (2004) 303:1526–9. doi: 10.1126/science.1093620
21. Hengge UR, Cusini M. Topical immunomodulators for the treatment of external genital warts, cutaneous warts and molluscum contagiosum. Br J Dermatol. (2003) 149:15–9. doi: 10.1046/j.0366-077X.2003.05623.x
22. Smith KJ, Hamza S, Skelton H. Topical imidazoquinoline therapy of cutaneous squamous cell carcinoma polarizes lymphoid and monocyte/macrophage populations to a Th1 and M1 cytokine pattern. Clin Exp Dermatol. (2004) 29:505–12. doi: 10.1111/j.1365-2230.2004.01593.x
23. Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. (2004) 5:190–8. doi: 10.1038/ni1028
24. Koblansky AA, Jankovic D, Oh H, Hieny S, Sungnak W, Mathur R, et al. Recognition of profilin by toll-like receptor 12 is critical for host resistance to toxoplasma gondii. Immunity. (2013) 38:119–30. doi: 10.1016/j.immuni.2012.09.016
25. Hidmark A, von Saint Paul A, Dalpke AH. Cutting edge: TLR13 is a receptor for bacterial RNA. J Immunol. (2012) 189:2717–21. doi: 10.4049/jimmunol.1200898
26. Liu J, Huang L, Luo M, Xia X. Bacterial translocation in acute pancreatitis. Crit Rev Microbiol. (2019) 45:539–47. doi: 10.1080/1040841X.2019.1621795
27. Simons-Linares CR, Imam Z, Chahal P. Viral-attributed acute pancreatitis: A systematic review. Dig Dis Sci. (2021) 66:2162–72. doi: 10.1007/s10620-020-06531-9
28. Karikó K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for toll-like receptor 3 *. J Biol Chem. (2004) 279:12542–50. doi: 10.1074/jbc.M310175200
29. Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, et al. HMGB1 SIGNALS THROUGH TOLL-LIKE RECEPTOR (TLR) 4 AND TLR2. Shock. (2006) 26(2):174–9. doi: 10.1097/01.shk.0000225404.51320.82
30. Kaufmann SH. Heat shock proteins and the immune response. Immunol Today. (1990) 11:129–36. doi: 10.1016/0167-5699(90)90050-J
31. Ethridge RT, Ehlers RA, Hellmich MR, Rajaraman S, Evers BM. Acute pancreatitis results in induction of heat shock proteins 70 and 27 and heat shock factor-1. Pancreas. (2000) 21(3):248–56. doi: 10.1097/00006676-200010000-00005
32. Cao MH, Xu J, Cai HD, Lv ZW, Feng YJ, Li K, et al. p38 MAPK inhibition alleviates experimental acute pancreatitis in mice. Hepatobiliary Pancreat Dis Int. (2015) 14:101–6. doi: 10.1016/S1499-3872(15)60327-7
33. de Graaf R, Kloppenburg G, Kitslaar PJHM, Bruggeman CA, Stassen F. Human heat shock protein 60 stimulates vascular smooth muscle cell proliferation through Toll-like receptors 2 and 4. Microbes Infect. (2006) 8:1859–65. doi: 10.1016/j.micinf.2006.02.024
34. Jin C, Cleveland JC, Ao L, Li J, Zeng Q, Fullerton DA, et al. Human Myocardium Releases Heat Shock Protein 27 (HSP27) after Global Ischemia: The Proinflammatory Effect of Extracellular HSP27 through Toll-like Receptor (TLR)-2 and TLR4. Mol Med. (2014) 20:280–9. doi: 10.2119/molmed.2014.00058
35. Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. (2008) 42:145–51. doi: 10.1016/j.cyto.2008.01.006
36. Yang H, Wang H, Andersson U. Targeting inflammation driven by HMGB1. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.00484
37. Laird MHW, Rhee SH, Perkins DJ, Medvedev AE, Piao W, Fenton MJ, et al. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J Leukoc Biol. (2009) 85:966–77. doi: 10.1189/jlb.1208763
38. Ohashi K, Burkart V, Flohé S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex1. J Immunol. (2000) 164:558–61. doi: 10.4049/jimmunol.164.2.558
39. Rocha DM, Caldas AP, Oliveira LL, Bressan J, Hermsdorff HH. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis. (2016) 244:211–5. doi: 10.1016/j.atherosclerosis.2015.11.015
40. Fang P, Schachner M, Shen YQ. HMGB1 in development and diseases of the central nervous system. Mol Neurobiol. (2012) 45:499–506. doi: 10.1007/s12035-012-8264-y
41. Bangert A, Andrassy M, Müller AM, Bockstahler M, Fischer A, Volz CH, et al. Critical role of RAGE and HMGB1 in inflammatory heart disease. Proc Natl Acad Sci. (2016) 113:E155–64. doi: 10.1073/pnas.1522288113
42. de Souza AWS, Westra J, Limburg PC, Bijl M, Kallenberg CGM. HMGB1 in vascular diseases: Its role in vascular inflammation and atherosclerosis. Autoimmun Rev. (2012) 11:909–17. doi: 10.1016/j.autrev.2012.03.007
43. Shao B, Xu Y, Jia M, Li C, Gong Zc. Association of HMGB1 levels in synovial fluid with the severity of temporomandibular joint osteoarthritis. BMC Musculoskelet Disord. (2023) 24:183. doi: 10.1186/s12891-023-06208-0
44. Li G, Wu X, Yang L, He Y, Liu Y, Jin X, et al. TLR4-mediated NF-κB signaling pathway mediates HMGB1-induced pancreatic injury in mice with severe acute pancreatitis. Int J Mol Med. (2016) 37:99–107. doi: 10.3892/ijmm.2015.2410
45. Arriaga-Pizano L, Boscó-Gárate I, Martínez-Ordaz JL, Wong-Baeza I, Gutiérrez-Mendoza M, Sánchez-Fernandez P, et al. High serum levels of high-mobility group box 1 (HMGB1) and low levels of heat shock protein 70 (Hsp70) are associated with poor prognosis in patients with acute pancreatitis. Arch Med Res. (2018) 49:504–11. doi: 10.1016/j.arcmed.2019.02.003
46. Hietaranta A, Mustonen H, Puolakkainen P, Haapiainen R, Kemppainen E. Proinflammatory effects of pancreatic elastase are mediated through TLR4 and NF-κB. Biochem Biophys Res Commun. (2004) 323:192–6. doi: 10.1016/j.bbrc.2004.08.077
47. Li Y, Zhou ZG, Xia QJ, Zhang J, Li HG, Cao GQ, et al. Toll-like receptor 4 detected in exocrine pancreas and the change of expression in cerulein-induced pancreatitis. Pancreas. (2005) 30(4):375–81. doi: 10.1097/01.mpa.0000160959.21580.41
48. Li HG, Zhou ZG, Li Y, Zheng XL, Lei S, Zhu L, et al. Alterations of toll-like receptor 4 expression on peripheral blood monocytes during the early stage of human acute pancreatitis. Dig Dis Sci. (2007) 52:1973–8. doi: 10.1007/s10620-006-9211-4
49. Sawa H, Ueda T, Takeyama Y, Yasuda T, Shinzeki M, Nakajima T, et al. Role of toll-like receptor 4 in the pathophysiology of severe acute pancreatitis in mice. Surg Today. (2007) 37:867–73. doi: 10.1007/s00595-007-3520-x
50. Kimura K, Shimosegawa T, Abe R, Masamune A, Satoh A, Takasu A, et al. Low doses of lipopolysaccharide upregulate acinar cell apoptosis in cerulein pancreatitis. Pancreas. (1998) 17(1):38–50. doi: 10.1097/00006676-199808000-00002
51. Pan L, Yu L, Wang L, He J, Sun J, Wang X, et al. Inflammatory stimuli promote oxidative stress in pancreatic acinar cells via Toll-like receptor 4/nuclear factor-κB pathway. Int J Mol Med. (2018) 42:3582–90. doi: 10.3892/ijmm
52. Vaccaro MI, Calvo EL, Suburo AM, Sordelli DO, Lanosa G, Iovanna JL. Lipopolysaccharide directly affects pancreatic acinar cells. Dig Dis Sci. (2000) 45:915–26. doi: 10.1023/A:1005521007609
53. Gu H, Werner J, Bergmann F, Whitcomb DC, Büchler MW, Fortunato F. Necro-inflammatory response of pancreatic acinar cells in the pathogenesis of acute alcoholic pancreatitis. Cell Death Dis. (2013) 4:e816–6. doi: 10.1038/cddis.2013.354
54. Wang W, Deng M, Liu X, Ai W, Tang Q, Hu J. TLR4 activation induces nontolerant inflammatory response in endothelial cells. Inflammation. (2011) 34:509–18. doi: 10.1007/s10753-010-9258-4
55. Andonegui G, Goyert SM, Kubes P. Lipopolysaccharide-induced leukocyte-endothelial cell interactions: A role for CD14 versus toll-like receptor 4 within microvessels1. J Immunol. (2002) 169:2111–9. doi: 10.4049/jimmunol.169.4.2111
56. Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev. (2014) 262:153–66. doi: 10.1111/imr.12218
57. Sabroe I, Prince LR, Jones EC, Horsburgh MJ, Foster SJ, Vogel SN, et al. Selective roles for toll-like receptor (TLR)2 and TLR4 in the regulation of neutrophil activation and life span1. J Immunol. (2003) 170:5268–75. doi: 10.4049/jimmunol.170.10.5268
58. Mateu A, Ramudo L, Manso MA, De Dios I. Cross-talk between TLR4 and PPARγ pathways in the arachidonic acid-induced inflammatory response in pancreatic acini. Int J Biochem Cell Biol. (2015) 69:132–41. doi: 10.1016/j.biocel.2015.10.022
59. Fan J, Li Y, Levy RM, Fan JJ, Hackam DJ, Vodovotz Y, et al. Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: role of HMGB1-TLR4 signaling1. J Immunol. (2007) 178:6573–80. doi: 10.4049/jimmunol.178.10.6573
60. Tadie JM, Bae HB, Jiang S, Park DW, Bell CP, Yang H, et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am J Physiol-Lung Cell Mol Physiol. (2013) 304:L342–9. doi: 10.1152/ajplung.00151.2012
61. Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4 *. J Biol Chem. (2004) 279:17079–84. doi: 10.1074/jbc.M310859200
62. Zhao Y, Zhang C, Wei X, Li P, Cui Y, Qin Y, et al. Heat shock protein 60 stimulates the migration of vascular smooth muscle cells via Toll-like receptor 4 and ERK MAPK activation. Sci Rep. (2015) 5:15352. doi: 10.1038/srep15352
63. Luong M, Zhang Y, Chamberlain T, Zhou T, Wright JF, Dower K, et al. Stimulation of TLR4 by recombinant HSP70 requires structural integrity of the HSP70 protein itself. J Inflammation. (2012) 9:11. doi: 10.1186/1476-9255-9-11
64. Chen L, Yu CX, Song B, Cai W, Liu C, Guan QB. Free fatty acids mediates human umbilical vein endothelial cells inflammation through toll-like receptor-4. Eur Rev Med Pharmacol Sci. (2018) 22(8):2421–2431. doi: 10.26355/eurrev_201804_14835
65. Ryu JK, Kim SJ, Rah SH, Kang JI, Jung HE, Lee D, et al. Reconstruction of LPS transfer cascade reveals structural determinants within LBP, CD14, and TLR4-MD2 for efficient LPS recognition and transfer. Immunity. (2017) 46:38–50. doi: 10.1016/j.immuni.2016.11.007
66. Erwin PJ, Lewis H, Dolan S, Tobias PS, Schumann RR, Lamping N, et al. Lipopolysaccharide binding protein in acute pancreatitis. Crit Care Med. (2000) 28(1):104–9. doi: 10.1097/00003246-200001000-00017
67. Rau B, Steinbach G, Krüger CM, Baumgart K, Schilling M, Beger HG. Clinical value of lipopolysaccharide-binding protein (LBP) determinations in acute pancreatitis. Langenbecks Arch Surg. (2003) 388:181–8. doi: 10.1007/s00423-003-0390-6
68. Fink GW, Norman JG. Intrapancreatic interleukin-1β Gene expression by specific leukocyte populations during acute pancreatitis. J Surg Res. (1996) 63:369–73. doi: 10.1006/jsre.1996.0278
69. Fink GW, Norman JG. Specific changes in the pancreactic expression of the interleukin 1 family of genes during experimental acute pancreatitis. Cytokine. (1997) 9:1023–7. doi: 10.1006/cyto.1997.0260
70. Sternby H, Hartman H, Thorlacius H, Regnér S. The initial course of IL1β, IL-6, IL-8, IL-10, IL-12, IFN-γ and TNF-α with regard to severity grade in acute pancreatitis. Biomolecules. (2021) 11(4). doi: 10.3390/biom11040591
71. Wereszczynska-Siemiatkowska U, Mroczko B, Siemiatkowski A. Serum profiles of interleukin-18 in different severity forms of human acute pancreatitis. Scand J Gastroenterol. (2002) 37:1097–102. doi: 10.1080/003655202320378310
72. Malmstrøm ML, Hansen MB, Andersen AM, Ersbøll AK, Nielsen OH, Jørgensen LN, et al. Cytokines and organ failure in acute pancreatitis: inflammatory response in acute pancreatitis. Pancreas. (2012) 41(2):271–7. doi: 10.1097/MPA.0b013e3182240552
73. Rau B, Baumgart K, Paszkowski AS, Mayer JM, Beger HG. Clinical relevance of caspase-1 activated cytokines in acute pancreatitis: High correlation of serum interleukin-18 with pancreatic necrosis and systemic complications. Crit Care Med. (2001) 29(8):1556–62. doi: 10.1097/00003246-200108000-00010
74. Pooran N, Indaram A, Singh P, Bank S. Cytokines (IL-6, IL-8, TNF): early and reliable predictors of severe acute pancreatitis. J Clin Gastroenterol. (2003) 37(3):263–6. doi: 10.1097/00004836-200309000-00013
75. Inagaki T, Hoshino M, Hayakawa T, Ohara H, Yamada T, Yamada H, et al. Interleukin-6 is a useful marker for early prediction of the severity of acute pancreatitis. Pancreas. (1997) 14(1):1–8. doi: 10.1097/00006676-199701000-00001
76. Fisic E, Poropat G, Bilic-Zulle L, Licul V, Milic S, Stimac D. The role of IL-6, 8, and 10, sTNFr, CRP, and pancreatic elastase in the prediction of systemic complications in patients with acute pancreatitis. Gastroenterol Res Pract. (2013) 2013:282645. doi: 10.1155/2013/282645
77. Yang R, Tenhunen J, Tonnessen TI. HMGB1 and histones play a significant role in inducing systemic inflammation and multiple organ dysfunctions in severe acute pancreatitis. Int J Inflammation. (2017) 2017:1817564. doi: 10.1155/2017/1817564
78. Yu C, Yu X, Zhu H, Li X, Huang L, Li Z, et al. Expression pattern of HMGB1 and its association with autophagy in acute necrotizing pancreatitis. Mol Med Rep. (2016) 14:5507–13. doi: 10.3892/mmr.2016.5945
79. van den Berg FF, de Bruijn AC, van Santvoort HC, Issa Y, Boermeester MA. Early laboratory biomarkers for severity in acute pancreatitis; A systematic review and meta-analysis. Pancreatology. (2020) 20:1302–11. doi: 10.1016/j.pan.2020.09.007
80. Kanak MA, Shahbazov R, Yoshimatsu G, Levy MF, Lawrence MC, Naziruddin B. A small molecule inhibitor of NFκB blocks ER stress and the NLRP3 inflammasome and prevents progression of pancreatitis. J Gastroenterol. (2017) 52:352–65. doi: 10.1007/s00535-016-1238-5
81. Zhou XY, Zhou ZG, Ding JL, Wang L, Wang R, Zhou B, et al. TRAF6 as the key adaptor of TLR4 signaling pathway is involved in acute pancreatitis. Pancreas. (2010) 39(3):359–66. doi: 10.1097/MPA.0b013e3181bb9073
82. Zhou X, Liu Z, Cheng X, Zheng Y, Zeng F, He Y. Socs1 and Socs3 degrades Traf6 via polyubiquitination in LPS-induced acute necrotizing pancreatitis. Cell Death Dis. (2015) 6:e2012–2. doi: 10.1038/cddis.2015.342
83. Danielski LG, Giustina AD, Bonfante S, Barichello T, Petronilho F. The NLRP3 inflammasome and its role in sepsis development. Inflammation. (2020) 43:24–31. doi: 10.1007/s10753-019-01124-9
84. Hoque R, Sohail M, Malik A, Sarwar S, Luo Y, Shah A, et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology. (2011) 141:358–69. doi: 10.1053/j.gastro.2011.03.041
85. Wen Y, Han C, Liu T, Wang R, Cai W, Yang J, et al. Chaiqin chengqi decoction alleviates severity of acute pancreatitis via inhibition of TLR4 and NLRP3 inflammasome: Identification of bioactive ingredients via pharmacological sub-network analysis and experimental validation. Phytomedicine. (2020) 79:153328. doi: 10.1016/j.phymed.2020.153328
86. Sendler M, van den Brandt C, Glaubitz J, Wilden A, Golchert J, Weiss FU, et al. NLRP3 inflammasome regulates development of systemic inflammatory response and compensatory anti-inflammatory response syndromes in mice with acute pancreatitis. Gastroenterology. (2020) 158:253–269.e14.
87. Rau B, Paszkowski A, Lillich S, Baumgart K, Möller P, Beger HG. Differential effects of caspase-1/interleukin-1β–converting enzyme on acinar cell necrosis and apoptosis in severe acute experimental pancreatitis. Lab Invest. (2001) 81:1001–13. doi: 10.1038/labinvest.3780312
88. Su M, Chen C, Li S, Li M, Zeng Z, Zhang Y, et al. Gasdermin D-dependent platelet pyroptosis exacerbates NET formation and inflammation in severe sepsis. Nat Cardiovasc Res. (2022) 1:732–47. doi: 10.1038/s44161-022-00108-7
89. Roger T, David J, Glauser MP, Calandra T. MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature. (2001) 414:920–4. doi: 10.1038/414920a
90. Sakai Y, Masamune A, Satoh A, Nishihira J, Yamagiwa T, Shimosegawa T. Macrophage migration inhibitory factor is a critical mediator of severe acute pancreatitis. Gastroenterology. (2003) 124:725–36. doi: 10.1053/gast.2003.50099
91. Zhu C, Liu Y, Song Y, Wang Q, Liu Y, Yang S, et al. Deletion of macrophage migration inhibitory factor ameliorates inflammation in mice model severe acute pancreatitis. BioMed Pharmacother. (2020) 125:109919. doi: 10.1016/j.biopha.2020.109919
92. Liu Y, Liu Y, Wang Q, Song Y, Chen S, Cheng B, et al. MIF inhibitor ISO-1 alleviates severe acute pancreatitis-associated acute kidney injury by suppressing the NLRP3 inflammasome signaling pathway. Int Immunopharmacol. (2021) 96:107555. doi: 10.1016/j.intimp.2021.107555
93. Lang T, Lee JPW, Elgass K, Pinar AA, Tate MD, Aitken EH, et al. Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat Commun. (2018) 9:2223. doi: 10.1038/s41467-018-04581-2
94. Su Y, Hong Y, Mei Fc, Wang Cy, Li M, Zhou Y, et al. High-fat diet aggravates the intestinal barrier injury via TLR4-RIP3 pathway in a rat model of severe acute pancreatitis. Mediators Inflamm. (2019) 2019:2512687. doi: 10.1155/2019/2512687
95. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. (2015) 517:311–20. doi: 10.1038/nature14191
96. Hong Y, Yu J, Su Y, Mei Fc, Li M, Zhao Kl, et al. High-fat diet aggravates acute pancreatitis via TLR4-mediated necroptosis and inflammation in rats. Oxid Med Cell Longev. (2020) 2020:8172714. doi: 10.1155/2020/8172714
97. Vona-Davis LC, Frankenberry KA, Waheed U, Peterson E, McFadden DW. Expression of STAT3 and SOCS3 in pancreatic acinar cells1,2. J Surg Res. (2005) 127:14–20. doi: 10.1016/j.jss.2005.03.019
98. Sztefko K, Panek J. Serum free fatty acid concentration in patients with acute pancreatitis. Pancreatology. (2001) 1:230–6. doi: 10.1159/000055816
99. Sharif R, Dawra R, Wasiluk K, Phillips P, Dudeja V, Kurt-Jones E, et al. Impact of toll-like receptor 4 on the severity of acute pancreatitis and pancreatitis-associated lung injury in mice. Gut. (2009) 58:813. doi: 10.1136/gut.2008.170423
100. Pan LF, Yu L, Wang LM, He JT, Sun JL, Wang XB, et al. The toll-like receptor 4 antagonist transforming growth factor-β-activated kinase(TAK)-242 attenuates taurocholate-induced oxidative stress through regulating mitochondrial function in mice pancreatic acinar cells. J Surg Res. (2016) 206:298–306. doi: 10.1016/j.jss.2016.08.011
101. Pan X, Ye L, Ren Z, Li J, Li B, Pan LL, et al. Biochanin A ameliorates caerulein-induced acute pancreatitis and associated intestinal injury in mice by inhibiting TLR4 signaling. J Nutr Biochem. (2023) 113:109229. doi: 10.1016/j.jnutbio.2022.109229
102. Gao L, Dong X, Gong W, Huang W, Xue J, Zhu Q, et al. Acinar cell NLRP3 inflammasome and gasdermin D (GSDMD) activation mediates pyroptosis and systemic inflammation in acute pancreatitis. Br J Pharmacol. (2021) 178:3533–52. doi: 10.1111/bph.15499
103. Awla D, Abdulla A, Regnér S, Thorlacius H. TLR4 but not TLR2 regulates inflammation and tissue damage in acute pancreatitis induced by retrograde infusion of taurocholate. Inflammation Res. (2011) 60:1093–8. doi: 10.1007/s00011-011-0370-1
104. Beckman JD, Abdullah F, Chen C, Kirchner R, Rivera-Rodriguez D, Kiser ZM, et al. Endothelial TLR4 expression mediates vaso-occlusive crisis in sickle cell disease. Front Immunol. (2021) 11. doi: 10.3389/fimmu.2020.613278
105. Hackert T, Sperber R, Hartwig W, Fritz S, Schneider L, Gebhard MM, et al. P-selectin inhibition reduces severity of acute experimental pancreatitis. Pancreatology. (2009) 9:369–74. doi: 10.1159/000212098
106. Hartman H, Abdulla A, Awla D, Lindkvist B, Jeppsson B, Thorlacius H, et al. P-selectin mediates neutrophil rolling and recruitment in acute pancreatitis. Br J Surg. (2012) 99:246–55. doi: 10.1002/bjs.7775
107. Reuken PA, Brozat JF, Quickert S, Ibidapo-obe O, Reißing J, Franz A, et al. Soluble mannose receptor CD206 and von Willebrand factor are early biomarkers to identify patients at risk for severe or necrotizing acute pancreatitis. J Intensive Care. (2022) 10:28. doi: 10.1186/s40560-022-00619-2
108. Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. (2014) 123:377–90. doi: 10.1182/blood-2013-04-495887
109. Jin YP, Nevarez-Mejia J, Terry AQ, Sosa RA, Heidt S, Valenzuela NM, et al. Cross-talk between HLA class I and TLR4 mediates P-selectin surface expression and monocyte capture to human endothelial cells. J Immunol. (2022) 209:1359–69. doi: 10.4049/jimmunol.2200284
110. Gross V, Andreesen R, Leser HG, Ceska M, Liehl E, Lausen M, et al. lnterleukin-8 and neutrophil activation in acute pancreatitis. Eur J Clin Invest. (1992) 22:200–3. doi: 10.1111/j.1365-2362.1992.tb01826.x
111. Johnsson C, Hällgren R, Tufveson G. Role of hyaluronan in acute pancreatitis. Surgery. (2000) 127:650–8. doi: 10.1067/msy.2000.106587
112. Chen Y, Chong MMW, Darwiche R, Thomas HE, Kay TWH. Severe pancreatitis with exocrine destruction and increased islet neogenesis in mice with suppressor of cytokine signaling-1 deficiency. Am J Pathol. (2004) 165:913–21. doi: 10.1016/S0002-9440(10)63353-6
113. Qin H, Holdbrooks AT, Liu Y, Reynolds SL, Yanagisawa LL, Benveniste EN. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J Immunol. (2012) 189:3439–48. doi: 10.4049/jimmunol.1201168
114. Gordon P, Okai B, Hoare JI, Erwig LP, Wilson HM. SOCS3 is a modulator of human macrophage phagocytosis. J Leukoc Biol. (2016) 100:771–80. doi: 10.1189/jlb.3A1215-554RR
115. Arnold CE, Whyte CS, Gordon P, Barker RN, Rees AJ, Wilson HM. A critical role for suppressor of cytokine signalling 3 in promoting M 1 macrophage activation and function in vitro and in vivo. Immunology. (2014) 141:96–110. doi: 10.1111/imm.12173
116. Saeki K, Kanai T, Nakano M, Nakamura Y, Miyata N, Sujino T, et al. CCL2-induced migration and SOCS3-mediated activation of macrophages are involved in cerulein-induced pancreatitis in mice. Gastroenterology. (2012) 142:1010–1020.e9. doi: 10.1053/j.gastro.2011.12.054
117. Liu Z, Ma Y, Cui Q, Xu J, Tang Z, Wang Y, et al. Toll-like receptor 4 plays a key role in advanced glycation end products-induced M1 macrophage polarization. Biochem Biophys Res Commun. (2020) 531:602–8. doi: 10.1016/j.bbrc.2020.08.014
118. Freitas MS, Oliveira AF, da Silva TA, Fernandes FF, Gonçales RA, Almeida F, et al. Paracoccin induces M1 polarization of macrophages via interaction with TLR4. Front Microbiol. (2016) 7:1003. doi: 10.3389/fmicb.2016.01003
119. Gong J, Li J, Dong H, Chen G, Qin X, Hu M, et al. Inhibitory effects of berberine on proinflammatory M1 macrophage polarization through interfering with the interaction between TLR4 and MyD88. BMC Complement Altern Med. (2019) 19:314. doi: 10.1186/s12906-019-2710-6
120. Sawoo R, Dey R, Ghosh R, Bishayi B. TLR4 and TNFR1 blockade dampen M1 macrophage activation and shifts them towards an M2 phenotype. Immunol Res. (2021) 69:334–51. doi: 10.1007/s12026-021-09209-0
121. Hietaranta A, Mustonen H, Puolakkainen P, Haapiainen R, Kemppainen E. Pancreatic elastase induced tnf-α secretion and cd11b expression are mediated by tlr4 receptor on cultured human myeloid cells. Pancreas. (2004) 29(4):328–9. doi: 10.1097/00006676-200411000-00024
122. Xue J, Habtezion A. Carbon monoxide–based therapy ameliorates acute pancreatitis via TLR4 inhibition. J Clin Invest. (2014) 124:437–47. doi: 10.1172/JCI71362
123. Fan J, Malik AB. Toll-like receptor-4 (TLR4) signaling augments chemokine-induced neutrophil migration by modulating cell surface expression of chemokine receptors. Nat Med. (2003) 9:315–21. doi: 10.1038/nm832
124. Tamassia N, Le Moigne V, Calzetti F, Donini M, Gasperini S, Ear T, et al. The MYD88-independent pathway is not mobilized in human neutrophils stimulated via TLR41. J Immunol. (2007) 178:7344–56. doi: 10.4049/jimmunol.178.11.7344
125. Fan J, Frey RS, Malik AB. TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. J Clin Invest. (2003) 112:1234–43. doi: 10.1172/JCI200318696
126. Akbarshahi H, Axelsson JB, Said K, Malmström A, Fischer H, Andersson R. TLR4 dependent heparan sulphate-induced pancreatic inflammatory response is IRF3-mediated. J Transl Med. (2011) 9:219. doi: 10.1186/1479-5876-9-219
127. Li H, Zhao L, Wang Y, Zhang MC, Qiao C. Roles, detection, and visualization of neutrophil extracellular traps in acute pancreatitis. Front Immunol. (2022) 13:974821. doi: 10.3389/fimmu.2022.974821
128. Merza M, Hartman H, Rahman M, Hwaiz R, Zhang E, Renström E, et al. Neutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis. Gastroenterology. (2015) 149:1920–1931.e8. doi: 10.1053/j.gastro.2015.08.026
129. Qi Q, Yang B, Li H, Bao J, Li H, Wang B, et al. Platelet microparticles regulate neutrophil extracellular traps in acute pancreatitis. Pancreas. (2020) 49(8):1099–1103. doi: 10.1097/MPA.0000000000001631
130. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
131. Guice KS, Miller DE, Oldham KT, Townsend CM, Thompson JC. Superoxide dismutase and catalase: A possible role in established pancreatitis. Am J Surg. (1986) 151:163–9. doi: 10.1016/0002-9610(86)90027-9
132. Al-Khafaji AB, Tohme S, Yazdani HO, Miller D, Huang H, Tsung A. Superoxide induces neutrophil extracellular trap formation in a TLR-4 and NOX-dependent mechanism. Mol Med. (2016) 22:621–31. doi: 10.2119/molmed.2016.00054
133. Münzer P, Negro R, Fukui S, di Meglio L, Aymonnier K, Chu L, et al. NLRP3 inflammasome assembly in neutrophils is supported by PAD4 and promotes NETosis under sterile conditions. Front Immunol. (2021) 12:683803. doi: 10.3389/fimmu.2021.683803
134. An Z, Li J, Yu J, Wang X, Gao H, Zhang W, et al. Neutrophil extracellular traps induced by IL-8 aggravate atherosclerosis via activation NF-κB signaling in macrophages. Cell Cycle. (2019) 18:2928–38. doi: 10.1080/15384101.2019.1662678
135. Meher AK, Spinosa M, Davis JP, Pope N, Laubach VE, Su G, et al. Novel role of IL (Interleukin)-1β in neutrophil extracellular trap formation and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. (2018) 38:843–53. doi: 10.1161/ATVBAHA.117.309897
136. Interiano B, Stuard ID, Hyde RW. Acute respiratory distress syndrome in pancreatitis. Ann Intern Med. (1972) 77:923–6. doi: 10.7326/0003-4819-77-6-923
137. Matsuda N, Nishihira J, Takahashi Y, Kemmotsu O, Hattori Y. Role of macrophage migration inhibitory factor in acute lung injury in mice with acute pancreatitis complicated by endotoxemia. Am J Respir Cell Mol Biol. (2006) 35:198–205. doi: 10.1165/rcmb.2005-0272OC
138. Abdelmageed ME, Nader MA, Zaghloul MS. Targeting HMGB1/TLR4/NF-κB signaling pathway by protocatechuic acid protects against l-arginine induced acute pancreatitis and multiple organs injury in rats. Eur J Pharmacol. (2021) 906:174279. doi: 10.1016/j.ejphar.2021.174279
139. Xue J, Sharma V, Hsieh MH, Chawla A, Murali R, Pandol SJ, et al. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat Commun. (2015) 6:7158. doi: 10.1038/ncomms8158
140. Cario E, Podolsky Daniel K. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. (2000) 68:7010–7. doi: 10.1128/IAI.68.12.7010-7017.2000
Keywords: TLR4, pancreatitis, inflammation, macrophage, neutrophil, NET
Citation: Mattke J, Darden CM, Lawrence MC, Kuncha J, Shah YA, Kane RR and Naziruddin B (2024) Toll-like receptor 4 in pancreatic damage and immune infiltration in acute pancreatitis. Front. Immunol. 15:1362727. doi: 10.3389/fimmu.2024.1362727
Received: 28 December 2023; Accepted: 11 March 2024;
Published: 22 March 2024.
Edited by:
Wandong Hong, First Affiliated Hospital of Wenzhou Medical University, ChinaReviewed by:
Ivana Kawikova, University of Hartford, United StatesVenkatraman M, VIT University, India
Copyright © 2024 Mattke, Darden, Lawrence, Kuncha, Shah, Kane and Naziruddin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bashoo Naziruddin, QmFzaG9vLk5hemlydWRkaW5AQlNXSGVhbHRoLm9yZw==