
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 05 March 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1360291
This article is part of the Research Topic Beyond PD-1: novel checkpoint receptors and ligands as targets for immunotherapy View all 18 articles
 Laura Tiberio1
Laura Tiberio1 Mattia Laffranchi2
Mattia Laffranchi2 Giovanni Zucchi2
Giovanni Zucchi2 Valentina Salvi1
Valentina Salvi1 Tiziana Schioppa1,3
Tiziana Schioppa1,3 Silvano Sozzani2,4
Silvano Sozzani2,4 Annalisa Del Prete1,3
Annalisa Del Prete1,3 Daniela Bosisio1*
Daniela Bosisio1*Plasmacytoid dendritic cells (pDCs) are the major producers of type I interferons (IFNs), which are essential to mount antiviral and antitumoral immune responses. To avoid exaggerated levels of type I IFNs, which pave the way to immune dysregulation and autoimmunity, pDC activation is strictly regulated by a variety of inhibitory receptors (IRs). In tumors, pDCs display an exhausted phenotype and correlate with an unfavorable prognosis, which largely depends on the accumulation of immunosuppressive cytokines and oncometabolites. This review explores the hypothesis that tumor microenvironment may reduce the release of type I IFNs also by a more pDC-specific mechanism, namely the engagement of IRs. Literature shows that many cancer types express de novo, or overexpress, IR ligands (such as BST2, PCNA, CAECAM-1 and modified surface carbohydrates) which often represent a strong predictor of poor outcome and metastasis. In line with this, tumor cells expressing ligands engaging IRs such as BDCA-2, ILT7, TIM3 and CD44 block pDC activation, while this blocking is prevented when IR engagement or signaling is inhibited. Based on this evidence, we propose that the regulation of IFN secretion by IRs may be regarded as an “innate checkpoint”, reminiscent of the function of “classical” adaptive immune checkpoints, like PD1 expressed in CD8+ T cells, which restrain autoimmunity and immunopathology but favor chronic infections and tumors. However, we also point out that further work is needed to fully unravel the biology of tumor-associated pDCs, the neat contribution of pDC exhaustion in tumor growth following the engagement of IRs, especially those expressed also by other leukocytes, and their therapeutic potential as targets of combined immune checkpoint blockade in cancer immunotherapy.
The concept of “immune checkpoint” is currently extending to proteins others than CTLA-4 and PD1, provided their capability to limit immune responses to a physiologic range while minimizing tissue damage. As a consequence, the number of novel potential targets for checkpoint inhibition to awake the immune system against tumors is rapidly growing, together with the envisioned combinations to develop more effective and patient-tailored cancer therapies (1–3). A particular emphasis is being placed on checkpoints expressed by innate immune cells. Indeed, the combined targeting of innate and adaptive checkpoints would unleash T-cell–mediated tumor killing also by rescuing the activation of the innate arm of immunity (4, 5). Also, innate checkpoint targeting could turn strategical when genetic instability prevents the success of T-cell-targeted checkpoint blockade, given that innate activation is independent of neoantigen recognition (6). In this scenario, a thorough understanding of the biology of novel immune checkpoints is a fundamental need for the definition of innovative therapeutic strategies.
Plasmacytoid dendritic cells (pDCs) represent a rare subset of dendritic cells characterized by the ability to secrete massive amounts of type-I interferons (IFNs), thus eliciting antiviral and antitumor responses (7). This review explores the hypothesis that tumor microenvironment, similar to chronic viral infections, may reduce the release of type-I IFNs by engaging inhibitory receptors (IRs) expressed by pDCs (Figure 1, left panel): following a brief overview of pDC biology and of general mechanisms of pDC impairment in tumors, we will review the panel of IRs, collect evidence concerning their contribution in the generation of exhausted tumor-associated pDCs (TA-pDCs) and describe existing blocking strategies to rescue the anticancer potential of this cell type. In this light, we hypothesize that IRs should be regarded as “innate immune checkpoints” and further studied as potential targets for checkpoint blockade in cancer immunotherapy (Figure 1, right panel).
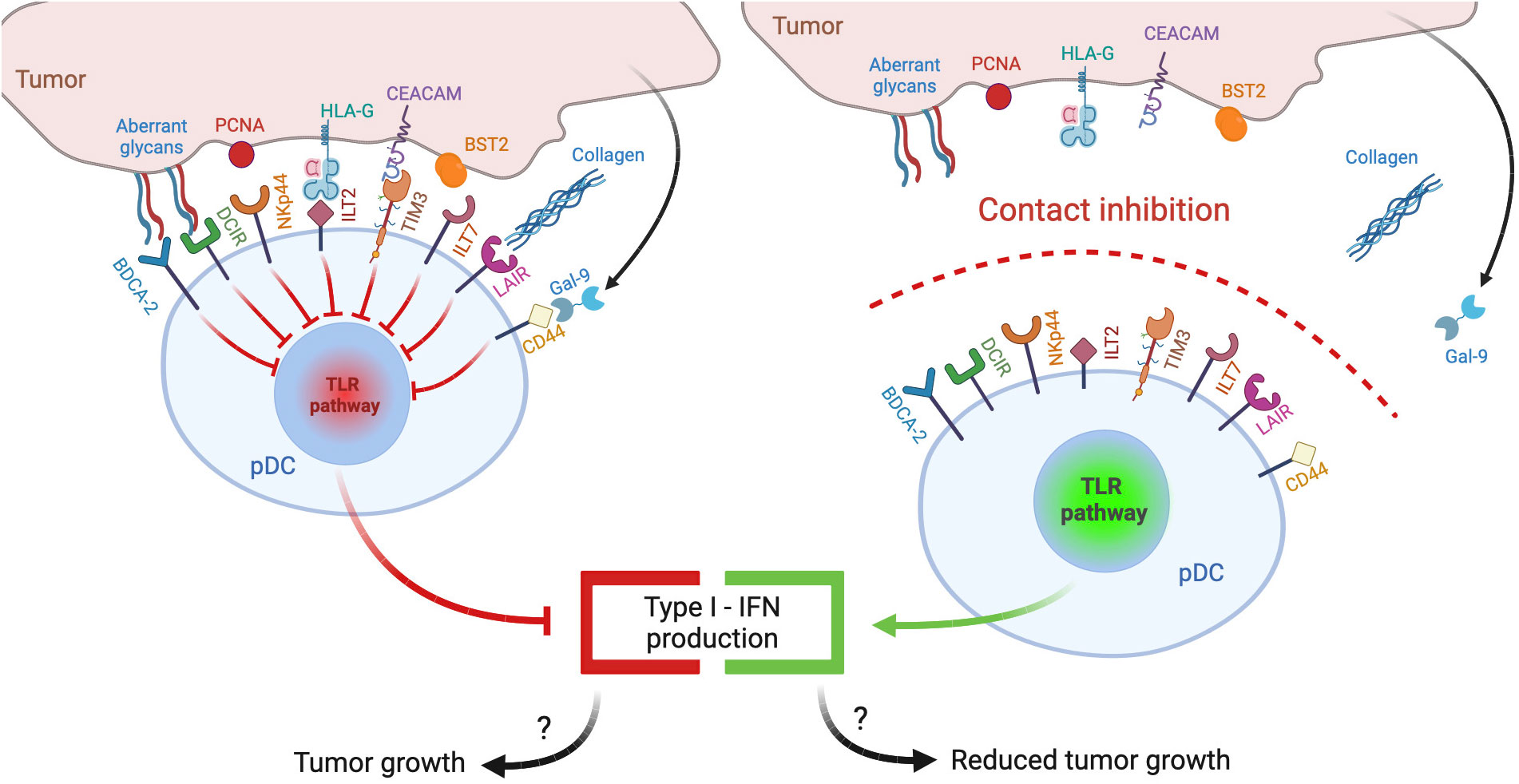
Figure 1 pDC inhibitory receptors as immune checkpoints and potential targets for anticancer immunotherapy. Different ligands from tumor cells may engage inhibitory receptors (IRs) expressed on pDCs and reduce pDC activation and type I IFN production, potentially leading to poor antitumor responses (left panel). Therapeutic strategies aimed at preventing IR engagement by ligand-bearing tumors may restore antitumoral pDC activity (right panel).
Articles referenced in the text specifically dealing with pDC IRs were searched in PubMed database from inception to December 2023 using as search terms: (“name of the receptor”[MeSH Terms] OR “name of the receptor”[All Fields] OR “alternative name/s of the receptor”[All Fields]) AND (“human plasmacytoid dendritic cells”[MeSH Terms] OR “human plasmacytoid dendritic cells”[All Fields] OR “human pDCs”[All Fields]). Retrieved papers where manually screened and were selected if related to IR characterization/biology or cancer or autoimmunity. Only inhibitory receptors of human pDCs were analyzed. Additional literature was added to draw up the more general parts of the review, concerning pDC pathophysiology and IR categorization.
Human pDCs, a rare population of innate cells accounting for 0.1-0.5% of mononuclear cells (8), are continuously produced in the bone marrow by both myeloid and lymphoid precursors (9). Very recently, the ontogeny of pDC has been debated and a proposal of a reclassification of their name was formulated (10, 11). Mouse data and studies on patients with combined immunodeficiencies highlighted the role of the transcription factors TCF4 (also known as E2-2), IRF8 and Ikaros family zinc finger 1 (IKZF1) for pDC differentiation (12–14). Phenotypic markers of human pDCs are blood DC antigen 2 (BDCA-2/CD303; also known as C-type lectin 4C -CLEC4C-), blood DC antigen 4 (BDCA-4/CD304; also known as C-type lectin 4A -CLEC4A-), Immunoglobulin -like transcript 7 (ILT7, also known as leukocyte immunoglobulin-like receptor subfamily A member 4 -LILRA4) and the receptor for IL7; in addition, human pDCs express other non-specific markers such as CD4, CD45RA, CD68, ILT3 and CD123 (IL-3 receptor) (15). In mice, pDCs are characterized by the expression of surface markers CD45R (B220), CD45RA, Ly-6C, Siglec-H, and BST2 (CD317/PDCA-1) (16).
Under physiologic conditions, human pDCs recirculate through lymphoid organs via peripheral blood (8). Lymph node entry occurs across high endothelial venules that express the ligands of L-selectin, CXCR4 and CMKLR1 (recently named Chemerin1, (17)) that are constitutively expressed by resting, immature pDC (18). Upon inflammatory conditions, human pDCs can enter the lymph nodes draining the target tissues guided by the acquired responsiveness to CCR7 ligands (16, 19–21). The functional role of CMKLR1- or CCR6/CCR10-mediated recruitment of human pDCs to non-lymphoid tissues has been documented during pathological conditions such as autoimmune, allergic and infectious diseases as well as in tumors (18, 22, 23). Human pDC can also migrate in response to chemotactic molecules released after tissue damage such as adenosine, formyl peptides and C3a and C5a anaphylotoxins (24–26).
pDCs were initially characterized as “natural interferon producing cells” due to their unique capability to secrete massive levels of type I IFNs (especially IFN-α) but also type III IFN (27, 28). Indeed, type I and type III IFNs account for about 60% of novel transcripts of activated pDCs (29). Moreover, pDCs also secrete proinflammatory cytokines and chemokines and were reported to present antigens to T lymphocytes (30, 31). Recent studies suggest that pDCs are a heterogeneous population, although several questions regarding pDC subsets and functional plasticity remain unanswered (15). In humans, the expression of CD2 was proposed to discriminate two different IFN-α producing pDCs subsets according to CD2 expression, with the CD2high subset being more effective in IL-12 secretion, in triggering naïve T lymphocyte proliferation and with a significant survival advantage over CD2low expressing pDC during stress conditions (32, 33). A CD5+CD81+CD2high human pDC subset, defined as Axl+ DC, was also identified. This “non-canonical” pDC subset was found unable to produce type I IFNs but endowed with the ability to stimulate B cells and promote the development of T regulatory (Treg) cells (34). However, these observations were recently challenged by a different view, supporting the idea that pDC diversification and functional specialization could occur upon activation and independently of pre-existing heterogeneity (35).
The ability to secrete huge amounts of type I IFN makes pDCs crucial in antiviral immune responses (31) against both RNA and DNA viruses (36–38). Of note, impaired secretion of type I and III IFNs caused by heterozygous null mutations in IRF7, a nonredundant transcription factor for IFN production, was associated to life-threatening H1N1 influenza A virus or SARS-CoV2 infections (39, 40). To accomplish this role, pDCs are equipped with innate immune receptors, primarily represented by elevated levels of TLR7 and TLR9. In particular, TLR7 detects ssRNA viruses, but also endogenous RNA and synthetic oligoribonucleotides or imidazoquinoline compounds. TLR9 recognizes DNA containing unmethylated CpG-rich DNA sequences, endogenous DNA and synthetic CpG DNA. The engagement of TLR7 and TLR9 activates the recruitment of the adapter protein MyD88 leading to the IRF7-mediated secretion of type I IFN and to the NF-kB-mediated secretion of proinflammatory cytokines (41). Studies using synthetic oligonucleotides demonstrated that the two pathways are spatially and temporally distinct, depending on the subcellular compartments in which these TLRs encounter their ligands (42, 43). In addition to TLRs, functional activation of cytosolic DNA-sensors including cyclic GMP-AMP (cGAMP) synthase (cGAS), stimulator of IFN gene (STING) and the dsRNA-sensor RIG-I in human pDCs has been recently described (44, 45). Virus-activated human pDCs can sustain NK cell functions by inducing NK cell migration and promoting IFN-γ secretion and NK cell cytotoxicity (46–48).
Besides their role in innate immunity, pDCs also regulate the activation of adaptive immune responses. Upon activation, pDCs increase the expression of major histocompatibility complex (MHC) and costimulatory molecules and were described to present antigens, both particulate and cell-associated, to CD4+ T cells and cross-present antigens to CD8+ T cells (49, 50). These functional pDC properties were demonstrated by specifically targeting receptors involved in antigen delivery, such as members of the C-type lectin family (CLR, such as BDCA-2, DEC-205 and DCIR, see further) or the immunoglobulin receptor FcγRII (CD32), with specific antibodies coupled to antigens, which were properly endocytosed, processed and presented (51–55). Activated pDCs secrete T-cell recruiting chemokines (18) and promote Th-polarization and differentiation (19, 56–58). Finally, type I IFNs and IL-6 released by pDCs contribute to drive memory B cell differentiation into effector plasma cell (59).
pDCs also potentially play a relevant role also in eliciting antitumor responses, which share many functional similarities with antiviral immunity (7). Indeed, type I IFNs enhance NK cell cytotoxicity against tumor cells (60, 61), modulate the activity and/or survival of lymphocytes (62, 63), suppress the generation of tumor associated macrophages (64) and also display direct antitumoral activities by inducing apoptosis and inhibiting the release of proangiogenic factors (65–68). However, the timing and duration of type-I IFN release critically condition the efficacy of antitumor responses, as recently reviewed elsewhere (69, 70), suggesting that pDC activation needs to be tightly regulated.
Besides being directly associated with two major types of primary liquid neoplasia, namely Blastic pDC Neoplasm (BPDCN) and Mature pDC Proliferation (MPDCP) (71), tumor infiltration by pDCs is reported in several human solid malignancies including melanoma, head and neck cancer, ovarian carcinoma and breast cancer (72, 73). Yet, tumor-associated pDCs (TA-pDCs) generally present a dysfunctional immature phenotype, with decreased secretion of IFN-α and inability to induce appropriate T cell responses and were described as negative prognostic markers in oral, ovarian, melanoma breast cancers and others human malignancies (74–78). The following paragraphs will briefly overview the general mechanisms of pDC induction of immunosuppression and exhaustion (Figure 2).
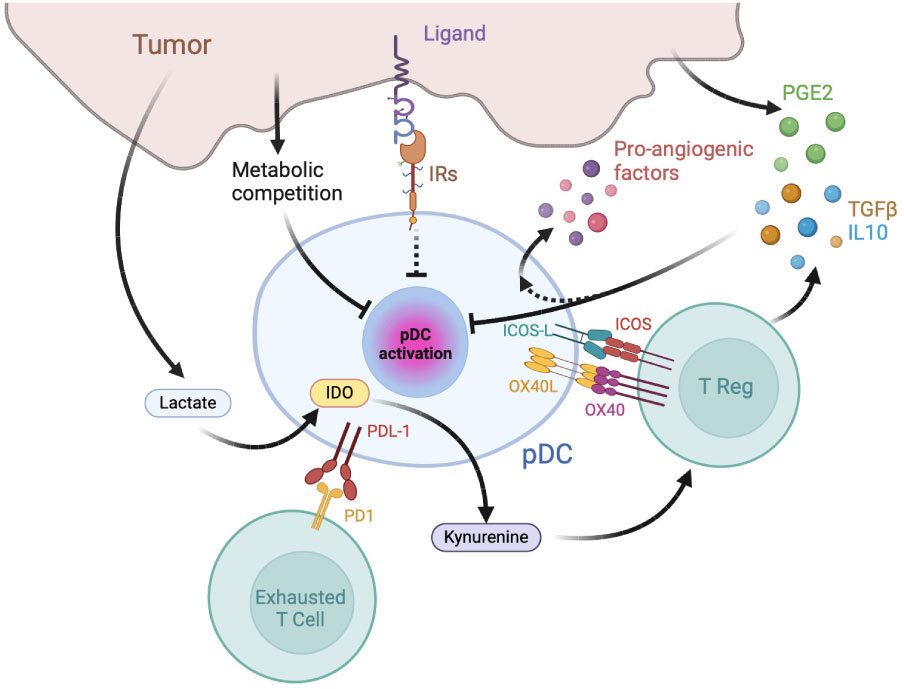
Figure 2 Mechanisms of pDC exhaustion and pDC-dependent immune-suppression in tumors. The tumor microenvironment is enriched in immunosuppressive cytokines, catabolites and hormones capable of inhibiting pDC maturation and type I IFN production, such as prostaglandin E2 (PGE2), TGFβ, and IL-10. pDCs favor tumor growth by inducing effector T cell exhaustion, T reg activation and by secreting pro-angiogenic factors. IR: inhibitory receptors; IDO: Indoleamine 2,3-dioxygenase.
TA-pDCs exploit several immunosuppressive molecular mechanisms contributing to the establishment of a tolerogenic, protumor microenvironment (73). Among the best characterized, the expression of OX40L and ICOSL, two surface molecules involved in Th2/Treg activation (79–81), were described to promote an immunosuppressive milieu by secreting TGF-β and IL-10 (82–84). Indeed, Treg activation by ICOSL+ pDCs was reported in several human cancers, including melanoma (85), gastric (86), ovarian (80), glioma (87), breast (81), liver (88, 89) and thyroid gland cancers (90). Increased frequencies of OX40L+ pDC and Th2 T cells were detected in the circulation of melanoma patients (85), consistent with the Th2-skewing role OX40L expressed by TA-pDCs, as demonstrated in a melanoma mouse model (85). Moreover, circulating pDCs from multiple myeloma patients were found to express high levels of the immune checkpoint ligand PDL1 (91). On the contrary, in the presence of PDL1-blocking antibodies, pDCs promoted T cell proliferation and NK cytotoxicity in patients (91). In accordance, in non-small cell lung cancer patients undergoing anti-PDL1 therapy, a high intra-tumoral pDC signature was associated to improved survival (92). TA-pDCs were also shown to secrete immunosuppressive and tumor-promoting mediators. Indoleamine 2,3-dioxygenase expressing (IDO+) pDCs from melanoma-draining lymph nodes mediated active immunosuppression in vitro and caused profound local T cell anergy in vivo through the direct activation of Foxp3+ Tregs which, in turn, upregulated the expression of PDL1 on mouse DCs (93). TA-pDCs recovered in ascites from ovarian tumor patients secreted the proangiogenetic factors CXCL8 and TNF-α (94), while in non-small cell lung cancer patients, tumor-infiltrating pDCs were reported to cause tumor proliferation via the pro-angiogenic effects of IL-1α (95). A direct demonstration of the detrimental role of TA-pDCs comes from a glioma mouse model, where pDC depletion increased survival by reducing the number of infiltrating Tregs and their ability to secrete IL-10 (87). Similarly, in mouse models of breast cancer bone metastasis, pDC depletion resulted in an overall decreased tumor burden and bone loss via the activation of CD8+ T cells and a Th1-oriented immune response (96).
The above described tolerogenic/hypo-functional state of TA-pDCs is induced by complex and often tumor-type specific molecular mechanisms (97, 98). Generally, however, the tumor microenvironment is enriched in immunosuppressive cytokines and hormones capable of inhibiting pDC maturation and type I IFN production, such as prostaglandin E2 (PGE2), TGFβ, and IL-10 (79, 82, 84, 99). These mediators are produced both by tumor cells and infiltrating immune cells, including Tregs that pDCs contribute to foster at the tumor site (83), thus establishing a feedback loop favoring tumor progression. Tumor-derived PGE2 and TGF-β were shown to act in synergy to inhibit the production of IFN-α and TNF-α induced in TLR7- and TLR9-triggered pDCs, by decreasing TLR membrane expression or by blocking TLR downstream signaling (99). This finding is consistent with the reduced capability of TA-pDCs in head and neck cancer patients to secrete type I IFNs as compared to circulating pDCs (100). The reduced expression of TLR7 and TLR9 induced by the immunosuppressive tumor microenvironment in pDCs was also demonstrated in ovarian and breast cancers (79, 81, 83, 84, 101). Conversely, PGE2-exposed pDCs release CXCL8, a chemokine that promotes tumor cell proliferation, migration/invasion and stimulates angiogenesis (73, 102). Indeed, pDCs recruited in malignant ascites from ovarian cancer patients can induce angiogenesis through the production of TNF-α and CXCL8 (94). In addition, increased serum levels of IL-10 in hepatocellular carcinoma patients were reported to induce a substantial reduction in circulating pDCs, which also displayed an immature phenotype with decreased HLA-DR, CD80, and CD86 expression (73, 103). Aberrant release of DAMPs and proinflammatory cytokines, especially TNF-α, contributes to human pDC hypo-functionality as well (94). For example, in virus-associated human cervical cancer, the production of type-I IFNs was impaired by HMGB1 secreted by transformed keratinocytes (104). Persistent stimulation of TLRs by nucleic acids released by tumor necrotic cells may also contribute to TA-pDC exhaustion like in chronic viral infections (105, 106).
Oncometabolites, such as lactate, create a microenvironment that is metabolically disadvantageous for several immune cells including pDCs (107). In mouse breast cancer, elevated lactate levels impaired the production of type I IFNs by pDCs and increased tryptophan metabolism and kynurenine, which participate in the activation of Tregs (108). In addition, in some tumor microenvironments, pDCs have to compete with tumor cells for nutrients, which are crucial for the highly metabolically demanding production of IFNs (109, 110).
All the above cited mechanisms affect different immune cells within tumors. Paragraph 4 of this review will explore the hypothesis that an additional, pDC-specific mechanism, may exist, namely the engagement of inhibitory receptors by ligand-expressing tumors.
pDCs express a large variety of membrane receptors, either specifically expressed or shared with other immune and non-immune cells, conveying inhibitory signals that decrease the production of type I IFNs (Figure 3). The physiological significance of these receptors is preventing aberrant immune activation. Indeed, a deregulated and prolonged exposure to IFNs not only can increase the risk of autoimmunity, but can also interfere with haematopoiesis leading to lymphopenia (111, 112). Therefore, in homeostatic conditions, the engagement of IRs ensures a specific and brief IFN secretion and is crucial to maintain efficient immune responses while preventing immune-mediated tissue damage. However, IRs can be hijacked by pathogens or tumour cells, thus hindering pDC activation. Here, we will describe pDC-expressed IRs, emphasizing available evidence for their hijacking in cancer as well as novel blocking strategies aimed at rescuing the anticancer potential of pDCs.
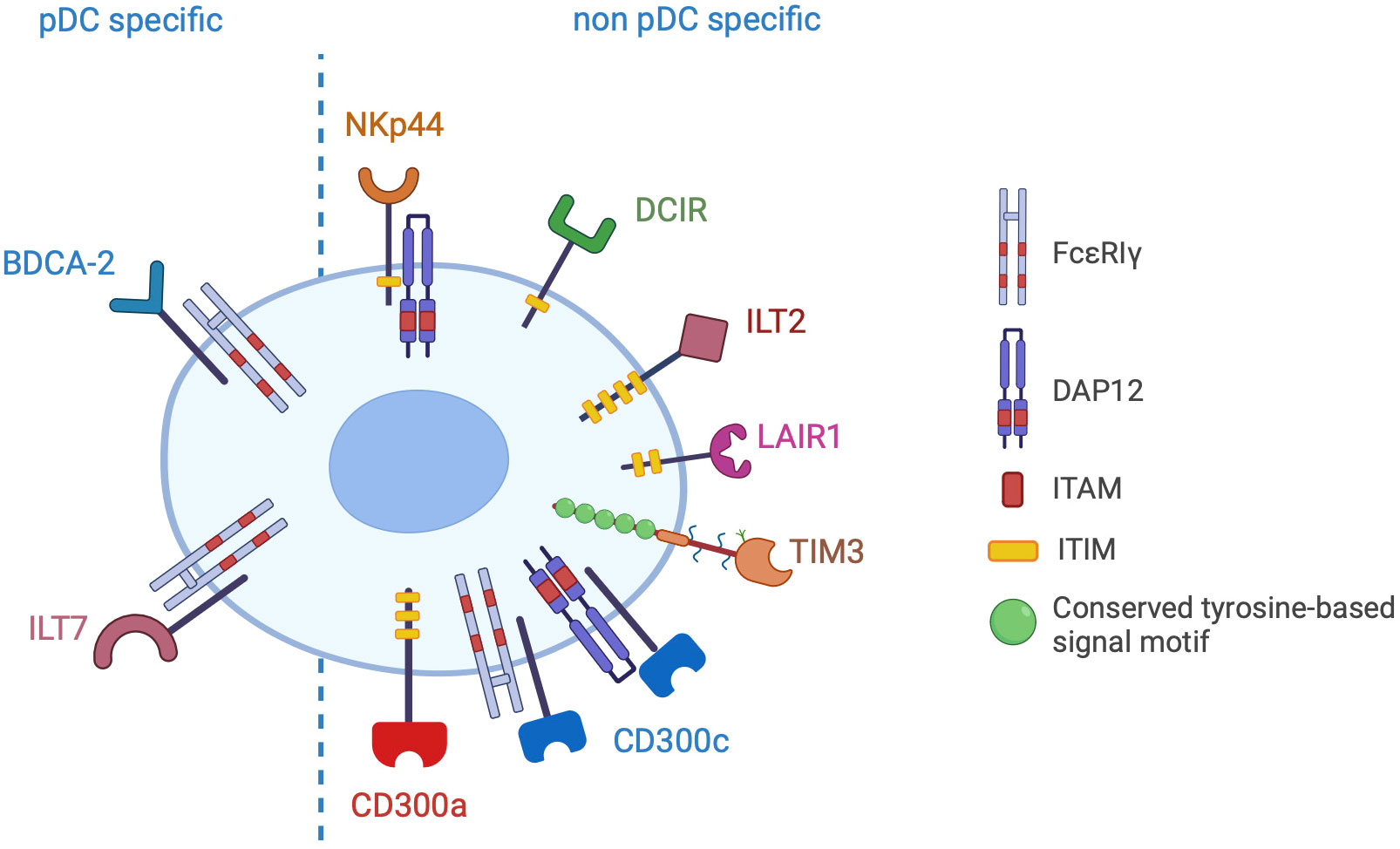
Figure 3 Inhibitory receptors of pDCs. The picture shows the inhibitory receptors (IRs) specifically expressed by pDCs (pDC specific) or shared with other cell types (non pDC specific). IRs signal through ITAM/ITIM motives present in the cytoplasmic domain of the receptor or in the associated adapter proteins (FcεRIγ or DAP12).
BDCA-2/CD303/CLEC4C is a human pDC-specific phenotypic marker (Figure 3), downregulated upon pDC maturation and TLR7/TLR9 triggering (113) and upregulated by IFN-α (37), and was the first receptor identified to negatively regulate the IFN response of pDCs (114). Lately, it was also shown to inhibit the TLR-mediated induction of TNF-related apoptosis-inducing ligand (TRAIL), thus impairing the capability of activated pDCs to kill TRAIL receptor-expressing neoplastic or infected cells (115).
BDCA-2 belongs to C-type lectin receptor (CLR) superfamily, as named after the Calcium-dependent binding of the first identified member. To date, CLRs are subdivided in 17 subgroups according to their structure, ligands and phylogeny (116). BDCA-2 belongs to the group II of CLRs that also includes, in humans, the closely related Dectin-2, dendritic cell immunoreceptor (DCIR, described further), DC immune-activating receptor (DCAR), and other members (117). These receptors are type II transmembrane proteins, with an extracellular C-terminal domain containing the carbohydrate recognition domain (CRD), and a short intracellular tail. In general, CLRs bind glycosylated molecules, a capability exploited by immune cells to recognize glyco-conjugated structures in non-self (pathogen-associated molecular patterns, or PAMPs), damaged-self (damage-associated molecular pattern, or DAMPs) and altered-self molecules (e.g. tumour-associated molecular patterns, or TAMPs) (118). Sequence analysis of the BDCA-2 CRD showed the presence of the tripeptide motif EPN (Glu-Pro-Asn), predicting the selective binding to the equatorial configuration of the hydroxyl groups at C3 and C4 of mannose, glucose, N-acetylglucosamine and fucose (117). Crystallographic analysis of the core domain of BDCA-2 CRD showed that its basic architecture is coherent with a typical CLR constituted by two α-helices and five β-strands (119). Differently from the CRD of other CLRs, a long loop region connecting α2-helix to β3-strand suggests the formation of a domain-swapped dimer, devoid of carbohydrate-binding ability, which may represent a regulatory mechanism that preserves BDCA-2 binding to galactosylated proteins in the Golgi apparatus before membrane exposure (119).
The nature and identity of BDCA-2 ligands is eagerly being sought after. Curiously, in contrast with the predicted mannose, N-acetyl glucosamine and glucose residues (120), a glycan array identified asialo-oligosaccharides with terminal galactose as BDCA-2 ligands (121). The binding ability for galactose–terminated glycans was subsequently ascribed to the interaction with a secondary site rather with the primary calcium-dependent binding site (122). Because serum glycoproteins display asialo-galactose residues, these were hypothesized to represent BDCA-2 ligands. Indeed, IgG, IgA, IgM but also α2-macroglobulin were demonstrated to bind BDCA-2, even if with low affinity (123). In the lack of any evidence of pDC activation following this binding, it was speculated that serum glycoproteins could compete with other ligands to maintain circulating pDCs in a quiescent state. In line with this, altered IgG galactosylation was described in autoimmune diseases characterized by pDC activation such as rheumatoid arthritis, primary Sjogren’s syndrome, psoriatic arthritis, and systemic lupus erythematosus (SLE) (124). Among PAMPs, different molecules from both DNA and RNA virus were shown to bind BDCA-2, possibly contributing to pDC exhaustion observed in chronic infections and, albeit for short term, during the acute phase of LCMV, HSV-1, VSV and MCMV infections (106). Recently, HBsAg, HIVgp120 and non-structural-1 (NS1) glycosylated protein from Zyka virus were shown to bind BDCA-2 and activate its downstream signaling pathways, leading to impaired type I IFN production upon TLR7 or TLR9 triggering (125–127). Tumors exploit modifications of cell surface carbohydrates to increase cell adhesion and migration, thus promoting invasiveness and metastasizing, but also to elude effective immune responses (128–130). For instance, carbohydrate changes of the carcino-embryonic antigen expressed by human colorectal cancer cells trigger the CLR DC-SIGN, which inhibits DC maturation and antitumor T cell activation by (131). Similarly, experiments exploiting a fluorescent tetramer encoding the BDCA-2 CRD, showed that BDCA-2 binds to tumor cells (including ovarian, colon, pancreatic carcinoma and breast adenocarcinoma) but not to non-tumor cells such as primary B and T cells (121). Cells expressing BDCA-2 ligands impaired the production of IFN-α following TLR9 stimulation, while ligand-negative cells did not, unless pre-treated with neuraminidase, which unmasks BDCA-2 binding sites (121). These findings suggest that tumor cells may modulate the expression of glycoproteins as a mechanism to inhibit human pDCs via BDCA-2 triggering.
To date, the only known function of BDCA-2 is the inhibition of TLR-dependent pDC activation. However, given the lack of well-defined biological BDCA-2 ligands, most studies investigating the mechanisms of BDCA-2 activation in pDCs were performed with crosslinking antibodies. As a result, the nature, the affinity and the kinetics of BDCA-2 triggering by natural ligands remains largely unknown and need further elucidation. Available results indicate that BDCA-2 signal transduction relies on the association with the common gamma chain of the Fcϵ receptor (FcϵRIγ), driving the assembly of a B cell receptor-like signalosome (132, 133) (Figure 4, left panel). Indeed, BDCA-2 triggering promotes the activation of the tyrosine kinase Syk that recruits the adaptor protein SLP65, leading, in turn, to phospholipase Cγ2 (PLCγ2) activation with the release of inositol 1,4,5-triphosphate and diacyl-glycerol. These second messengers are required for diverse membrane functionality including calcium flux. BDCA2 engagement has also been associated to AKT and MEK1/2-ERK activation (134, 135). To date, the pathway leading to BDCA-2 inhibition of TLR-dependent NF-kB activation remains partially elucidated (133), but PLCγ2 activation and calcium mobilization were suggested to impair the recruitment of MyD88 to TLRs through the activation of the serine phosphatase calcineurin (128, 136) (Figure 4, left panel).
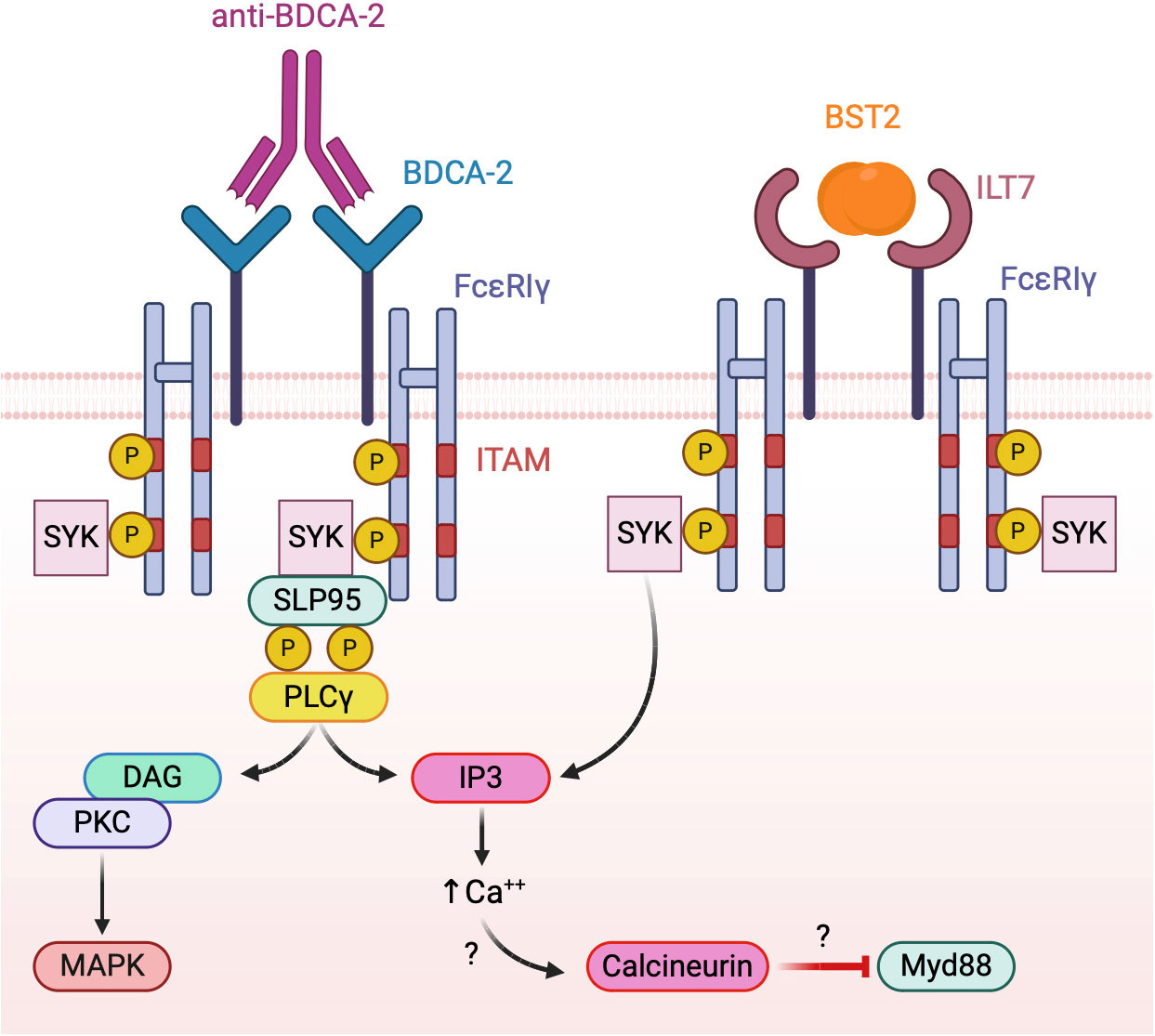
Figure 4 Model of BDCA-2 and ILT7 signaling. Both receptors associate with the ITAM-bearing adapter FcεRIγ chain. Receptor triggering activates a BCR-like signalosome leading to the inhibition of TLR signaling, possibly interfering with the TLR adapter MyD88.
Because of its specificity and inhibitory function, BDCA-2 is an attractive candidate for therapeutic strategies aimed at targeting pDCs or at modulating their activity. Bivalent binding by the F(ab)2 domain of anti-BDCA-2 antibodies is essential for BDCA-2 activation (137), while Fc region involvement seems to be dispensable. In fact, anti-BDCA-2 antibodies devoid of effector functions block the production of type I IFNs by TLR7/9-activated pDCs. However, the Fc region of a humanized monoclonal antibody against BDCA-2 appeared critical for inhibiting the production of type I IFN stimulated by immune complexes through internalization of CD32a (138). Regardless this difference, anti-BDCA-2 antibodies appeared as appealing tools for the treatment of SLE, where immune-complexes and type I IFNs play a pathogenetic role. A recent Phase II clinical trial involving patients with SLE demonstrated that Litifilimab (a humanized antibody against BDCA-2) could reduce cutaneous and joint involvement (LILAC ClinicalTrials.gov number NCT02847598) (139, 140). Another strategy to target BDCA-2 was the generation of a chimeric anti-BDCA-2 antibody (ch122A2), characterized by low fucose contents in order to increase its affinity for CD16/FcγRIIIa to activate the antibody-dependent cell-mediated cytotoxicity. In preclinical settings, ch122A2 induced an efficient and fast depletion of blood pDC in humanized mice (141). A proposed clinical application of this antibody is primarily the treatment of patients with pDC malignancy like BPDCN or pDC-AML; however, other hematological and solid cancers where pDC infiltration associates with a poor prognosis may be potential therapeutic targets.
ILT7/LILRA4/CD85g is the only other pDC-specific IR in humans (142–144) (Figure 3). Similarly to BDCA-2, its expression is upregulated by IFN-α (37) and downregulated when pDCs are activated by TLR agonists or treated with the survival cytokine IL-3 (142), possibly as a result of reduced transcriptional expression by activated pDCs (145).
The ILT (also known leukocyte Ig-like receptor-LILR- or monocyte Ig-like receptor-MIR-) gene family is composed of 11 transmembrane proteins characterized by two or four extracellular C2-type Immunoglobulin-like domains. ILTs are expressed by various population of antigen presenting cells in humans and primates but not in rodents (144, 146). Sequence analysis revealed the existence of separate subgroups of ILTs: one characterised by a long intracellular tail bearing immunoreceptor tyrosine-based inhibitory motifs (ITIMs), one devoid of any transmembrane domain, and one with short cytoplasmic tails without ITIMs but characterized by the presence of a charged residue in the transmembrane domain, which allows the association with signalling adapter molecules that possess ITAMs.
ILT7, characterized by four extracellular immunoglobulin domains, belongs to the latter subgroup, displaying a positively charged arginine residue at position 449 within the predicted transmembrane segment, through which it associates with FcϵRIγ, the same ITAM-bearing adapter used by BDCA-2 (142). Similar to BDCA-2, both Src family kinases and Syk are rapidly phosphorylated after ILT7 crosslinking in human primary pDCs indicating the onset of ITAM signaling together with prominent intracellular calcium mobilization (142) (Figure 4, right panel).
In the search for ligands, ILT7 reporter cells were found to be activated in the presence of human breast carcinoma cells and melanoma cell lines but not by common laboratory mammalian cell lines (143). Bone marrow stromal cell antigen 2 (BST2; CD317) was identified as the ILT7 ligand capable of inducing changes similar to those observed upon cross-linking by anti-ILT7 antibodies (143). When ILT7 was cross-linked by either anti–ILT7 antibodies or recombinant BST2 protein, pDCs stimulated by the TLR9 ligand CpG oligonucleotide and TLR7 ligand influenza virus produced less IFN-α and TNF-α. By contrast, the expression of the costimulatory molecules CD80 and CD86 was not affected (142, 143). Given that BST2 is robustly induced by IFNs and inflammatory cytokines, its interaction with ILT7 was identified as a negative feedback mechanism to prevent prolonged IFN production after viral infection (147). In accordance with original experiments showing constitutive expression of BST2 by cancer cells (143), more recent observations confirmed BST2 overexpression in myelomas, lung cancer, breast cancer, colorectal cancer, and pancreatic cancer (148), which could represent a strong predictor of tumor size, aggressiveness, and poor patient survival (149, 150). In vitro, BST2 expression by human breast cancer and melanoma cell lines could suppress the production of type I IFNs via ILT7 (143). Of interest, ILT7 was recently found to be modulated in tumor-infiltrating pDC of melanoma patients (151). These pieces of evidence strongly suggest that the interaction of BST2 with ILT7 may contribute to tumor immune suppression and pDC–tumor crosstalk (144).
Human pDCs express several other membrane receptors conveying inhibitory signals that, unlike BDCA-2 and ILT7, are also expressed by other immune or non-immune cells.
Two of these receptors, NKp44 and DCIR, are CLRs (group V and II, respectively) like BCDA-2, but both express intracellular ITIMs (Figure 3), which are normally involved in the inhibition of kinase-mediated signals by recruiting tyrosine phosphatases like Src homology region 2 domain-containing phosphatase (SHP)-1 or -2. The ITIM sequence of NKp44 was originally shown to be non-functional in the attenuation of NK-like cells activation (152), thus classifying it as a NK cell-triggering receptor. However, its ligation by the ligand proliferating cell nuclear antigen (PCNA) was later found to deliver ITIM-dependent inhibitory signals into NK cells (153). Thus, in NK cells NKp44 works as a dual function receptor, possibly depending on the streghth of its engagement as described for many CLRs (154). PCNA overexpression is a hallmark of cancer virulence and promotes cancer survival via several mechanisms, including immune evasion through inhibition of NKp44-mediated NK cell attack. Consistent with this view, downregulation of endogenous PCNA in pancreas, prostate, breast and brain tumor cell lines by a siRNA approach and the blockade of NKp44-PCNA interaction in triple negative breast cancer cells by a monoclonal antibody increased NK cytotoxicity and tumor killing (155). NKp44 is also constitutively expressed by a small subset of tonsil pDCs and can be induced in blood pDCs by IL-3 stimulation. In pDCs, NKp44 crosslinking by a specific antibody inhibited the production of IFN-α in response to TLR9 agonists via the association with the ITAM-bearing adaptor protein DAP12 (156). In PCNA+ human melanoma, infiltrating pDCs showed increased NKp44 levels, which correlated with a low activation level, suggesting that the interaction of NKp44 with PCNA expressed by melanoma cells could contribute to pDC dysfunctions typically observed in melanoma patients. In addition, melanoma patients displaying higher frequencies of NKp44+ pDCs in their blood were more likely to have worse clinical outcome (151). However, it was recently demonstrated that NKp44 engagement by dimers of platelet derived growth factor (PDGF-DD), another physiologic ligand, enhanced the secretion of IFN-α induced by a TLR9 ligand (157) suggesting that NKp44 possibly works as a dual function receptor also in pDCs and that its inhibitory role needs to be re-assessed in each specific tumor context.
DCIR (also known as CLEC4A) is expressed on a variety of immune cells such as cDCs, B cells and monocytes/macrophages in addition to pDCs. Due to the presence of an intracellular ITIM domain (Figure 3), it is generally regarded as an IR (158), although, like other CLRs (including the above mentioned NKp44) it can deliver activatory signals in certain cell types and conditions (154). In human pDCs, DCIR crosslinking inhibited TLR9-induced IFN production (53, 159). In respect with BDCA-2, pDC inhibition by DCIR was less effective and TLR9-specific since it could not be observed when pDCs were stimulated with TLR7 ligands (53). Very recently, asialo-biantennary N-glycans were shown to represent a DCIR functional ligand, capable to regulate DC functions in both humans and mice (160). However, DCIR was previously shown to interact with several ligands of both pathogenic and endogenous origin (129). In pDCs, DCIR binding by HCV glycoprotein E2 inhibited the production of type I IFNs by HCV particles through a rapid AKT and ERK1/2 phosphorylation (134). The recognition of self-glycans by DCIR prevented autoimmunity in murine models of rheumatoid arthritis (159). In cancers, DCIR could recognize aberrant glycosylation in prostatic, gastric and colon cancer human cell lines (129). In a mouse model of inflammation-induced colorectal cancer, the administration of antibodies blocking the interaction of DCIR with asialo-biantennary N-glycans reduced tumor incidence by reverting the DCIR-dependent blockade of alarmin recognition by TLRs, suggesting a crucial role for DCIR in the maintenance of the intestinal immune system functionality and that DCIR may represent a promising target for the treatment of colitis and colon cancers (161). Additionally, skin delivery of DCIR small hairpin RNA delayed tumor growth in mouse models of bladder and lung tumor by enhancing T cell mediated immunity and also potentiated the anti-tumor effects of a DNA vaccine (162).
ILT2, unlike ILT7, bears four intracellular ITIM motifs (Figure 3) and is broadly expressed on blood pDCs, monocytes, B cells, cDCs, NK cell subsets and T cells (163). ILT2 engagement significantly suppresses the ability of DC subsets, including pDCs, to produce cytokines, upregulate costimulatory molecules, and stimulate T-cell proliferation (164–166). In humans, whole blood stimulation with TLR4 and TLR7 agonists increased membrane expression of ILT2 in pDCs and, consistent with its immunosuppressive role, IL-10 treatment during TLR stimulation further increased ILT2 expression (167). ILT2 recognized pathogens as well as endogeous ligands (165, 166), particularly non-classical MHC class I molecules (163). Among them, HLA-G expression has been described in several tumor types, where it contributed to malignant progression by contrasting immune surveillance via the interaction with ILT2 and ILT4 (168). In accordance, anti-HLA strategies were recently proposed as novel immune checkpoint inhibition approaches in solid cancers (169). In chronic lymphocytic leukemia, ILT2 expression was significantly decreased on leukemic cells and increased on NK cells, particularly in patients with advanced disease and with poor prognostic features. ILT2 suppressed NK cell activity, which could be restored by ILT2 blockade: in combination with the immunomodulatory drug lenalidomide, ILT2 blockade potentiated the elimination of human leukemic cells (170). Disruption of ILT2 activation with blocking monoclonal antibodies increased NK cell-mediated IFN-γ production and cytotoxicity against human glioblastoma cell lines, partially reverting the immunosuppression linked to this malignancy. In addition, co-treatment with temozolomide strengthened the antitumor capacity of immune cells treated with anti-ILT2 (171). Also, Fc-silent antibodies against ILT2 significantly enhanced antibody-dependent phagocytosis of lymphoma cell lines when combined with both rituximab and blockade of CD47 (172). These findings suggest that the blocking of ILT2 may be an interesting strategy to improve tumor immunotherapy.
Leukocyte-Associated Ig-like Receptor-1 (LAIR1) is an ITIM-bearing immune-IR expressed by the majority of immune cells, including T cells, B cells, NK cells, monocyte/macrophages, neutrophils, pDCs, as well as by tumor cells (173). Crosslinking of LAIR1 in human pDCs inhibited TLR-dependent type I IFN production, displaying a coordinated regulatory function with NKp44 (156, 174). Four different types of ligands are described, including components of the complement system and collagens, suggesting a potential immune-regulatory function of the extracellular matrix (175, 176). In a retrospective study, LAIR1 expression was found to associate to poor prognosis in invasive breast carcinoma (177), but also to resistance to PD1/PD-L1 inhibition in patients (178). Of interest, LAIR1 blockade by antagonist antibodies inhibited tumor development in a humanized mouse model by affecting, among others, the recruitment of pro-tumorigenic pDCs (179). In addition to blocking antibody, LAIR1-inhibitory signaling can be blocked also by taking advantage of LAIR2, a natural agonist (180) as proposed by a work using a dimeric LAIR2 Fc fusion protein to target collagens in tumors and reverse immune suppression (181). The potential of LAIR1 blockade in cancer immunotherapy is currently emerging (179, 182). However, since LAIR1 is widely expressed also by tumor cells, where it may induce either proliferation or inhibition depending on the tumor type (173), its therapeutic exploitation needs to be carefully tailored to each specific cancer microenvironment.
T cell immunoglobulin and mucin domain-containing protein 3 (TIM3) is a member of the TIM family of immunoregulatory proteins expressed by pDCs, T cells, regulatory T cells, NK cells, and myeloid cells. TIM3 lacks intracellular inhibitory signaling motifs and the precise intracellular signalling mechanism remains poorly elucidated (183). Different mechanisms were proposed for TIM-3-induced suppression of IFN production in pDCs activated by nucleic acids. Chiba and colleagues highlighted the ability of TIM3 to bind and sequester HMGB1 away from TLR, thus avoiding the sensing of tumour-derived nucleic acids bound to HMGB1 itself. In contrast, Schwartz and colleagues suggested that TIM3 could act by recruiting IRF7 into acidic lysosomes, thus promoting the degradation of proteins important for IFN-α production (184, 185). Tumour cells can exert immunosuppression by expressing TIM3 ligands such as galectin-9 and CEACAM-1 (186, 187). Increased serum levels of galectin-9 was found in cancer patients and predicted poor response to treatment in high grade serous ovarian carcinoma and in adult leukemia patients (188). Consistent with a suppressive function in the tumour microenvironment, TIM3 was found upregulated in lung tumor-infiltrating pDCs (167, 184). One group showed that galectin-9 could block TLR-induced pDC activation in vitro and in a murine model also via the engagement of CD44 (189), a widely-expressed adhesion receptor involved in cancer metastasizing and regulation of T cell responses (190). In human pDCs, CD44 engagement by galectin-9 impaired mTOR-dependent TLR activation (189).
Finally, also the triggering of the CD300a/c glycoproteins by crosslinking antibodies was shown to decrease type I IFN and TNF-α secretion by human pDCs stimulated with TLR7 and TLR9 ligands (191). CD300 are a group of type I transmembrane receptors belonging to the B7 family with a single IgV-like extracellular domain containing 2 disulfide bonds (192). While CD300a contains several ITIMS in its long intracytoplasmic domain, CD300c associates with the ITAM-bearing adapters DAP12 and/or FcϵRIγ via a transmembrane glutamic acid residue (192) (Figure 3). Both receptors are expressed by virtually all leukocytes and possibly recognize lipids that are exposed on the outer leaflet of the plasma membrane of dead and activated cell. CD300 receptors are also highly expressed by human cancer cells, especially in acute myeloid leukemia (193). To date, their therapeutic potential in cancer immunotherapy remains to be elucidated.
Although being a minor population both in the circulation and in the tumor microenvironment, evidence described in Section 3 indicates pDCs as interesting targets for anticancer immunotherapy. Several therapeutic protocols have been developed to this end, mostly aiming at reverting the distinctive feature of immunosuppressive TA-pDCs, i.e. the impaired secretion of type I IFNs.
The most used approach to stimulate pDC production of type I IFNs is TLR7 and TLR9 stimulation, either individually or in combination (194). Indeed, TLR9 engagement by intralesional administration of CpG ODN nanorings gave promising results in a thymoma mouse model, where the increased production of IFN-α by pDCs associated to reduced tumor size and volume (195, 196). In a melanoma mouse model, CpG-activated pDCs were indispensable to induce CD8+ T cell antitumor response through cDC activation (197, 198). Single-stranded RNAs delivered by the positively charged protein protamine promoted T cell proliferation, demonstrating that protamine–RNA complexes can be used to stimulate human DC subsets ex vivo for future immunotherapeutic settings (199). The potent synthetic TLR7 agonist imiquimod, approved for the treatment of basal cell carcinoma (200), was shown to increase the infiltration of activated pDCs into melanoma lesions and its combination with monobenzone led to metastases regression in phase II clinical trial in late-stage melanoma patients (201). Vidutolimod (202), a virus like particle containing a TLR9 agonist known as G10, enhanced IFN-α production by pDCs showing high therapeutic efficacy when administered alone or in combination with an anti-PD-1 therapy in patients with melanoma (203, 204). The combined stimulation with TLR agonists and FLT3L, a growth factor of both cDCs and pDCs, enhanced cDC antigen presentation and T cell immunity in mouse models of melanoma (205) and glioma (206). In line with these observations, the combined administration of imiquimod, FLT3L and a peptide-based vaccine not only increased the number of peptide-specific CD8+ T cells but also prompted the mobilization of cDCs and pDCs in melanoma patients (207). However, selective delivery of TLR7/9 agonists to pDCs in vivo still needs improving.
IR blockade may represent an alternative pDC-boosting strategy, especially in tumors characterized by high levels of inhibitory ligands or when TLRs are desensitized by continuous stimulation with exogenous or endogenous ligands.
Many cancer types express de novo, or overexpress, IR ligands which often represent a strong predictor of poor outcome and metastasis, as observed for BST2 (148), PCNA (208), HLA-G (168), galectin-9 (186), CEACAM-1 (186, 187) and modifications of cell surface carbohydrates capable to trigger CLRs (128–130). It is conceivable that these ligands abundantly expressed in cancers contribute to pDC exhaustion by engaging IRs. Indeed, ligand-expressing tumor cells were found to block pDC activation by engaging BDCA-2 (121), ILT7 (143), TIM3 (167, 184) or CD44 (189). In this setting, therapeutic strategies preventing receptor engagement or signaling would revert TA-pDC blockade (Figure 1). Among such strategies, blocking antibodies were developed against LAIR1 (179), DCIR (161), ILT2 (170, 171), NKp44 (155) and TIM3 (2) (Figure 5A). As alternative strategies, LAIR1 inhibition was also achieved via an Fc fusion protein of LAIR2, a natural agonist capable of sequestering the ligands (181) (Figure 5A), while DCIR expression was decreased by skin delivery of specific small hairpin RNA (162) (Figure 5A). As a matter of facts, TIM3 and, to some extent, ILT2 are already promising emerging targets for checkpoint blockade (2). Also, DCIR blockade was recently shown to reduce the incidence of experimental inflammation-induced colon carcinoma (161) and the potential of LAIR1 blockade in cancer immunotherapy is rapidly emerging (179, 182). However, these IRs are expressed by different immune and tumor cells and in most studies the neat contribution of pDC rescue in the elicited antitumor response is difficult to deduce or not addressed at all. Unfortunately, blocking antibodies for pDC-specific IRs, namely BDCA-2 and ILT7, are currently unavailable. However, BDCA-2 signaling could be blocked in vitro by saturating ligand-expressing cells with a tetramer encoding the BDCA-2 CRD or by treating them with β-(1–4)-galactosidase which removes terminal galactose that are crucial for BDCA-2 triggering (121, 128–130). In addition, an anti-BDCA-2 monovalent Fab was unable to activate BDCA-2 and to inhibit type I IFN production (137) suggesting that low avidity, monovalent antibodies could be exploited as therapeutic strategy to block BDCA-2 activation in tumors. Finally, a BDCA-2-binding antibody engineered to favor the activation of cytotoxicity efficiently depleted blood pDCs in humanized mice (141), possibly representing a primary tool for the treatment of pDC malignancies but also of other cancers where the presence of pDCs associates with a poor prognosis (Figure 5B).
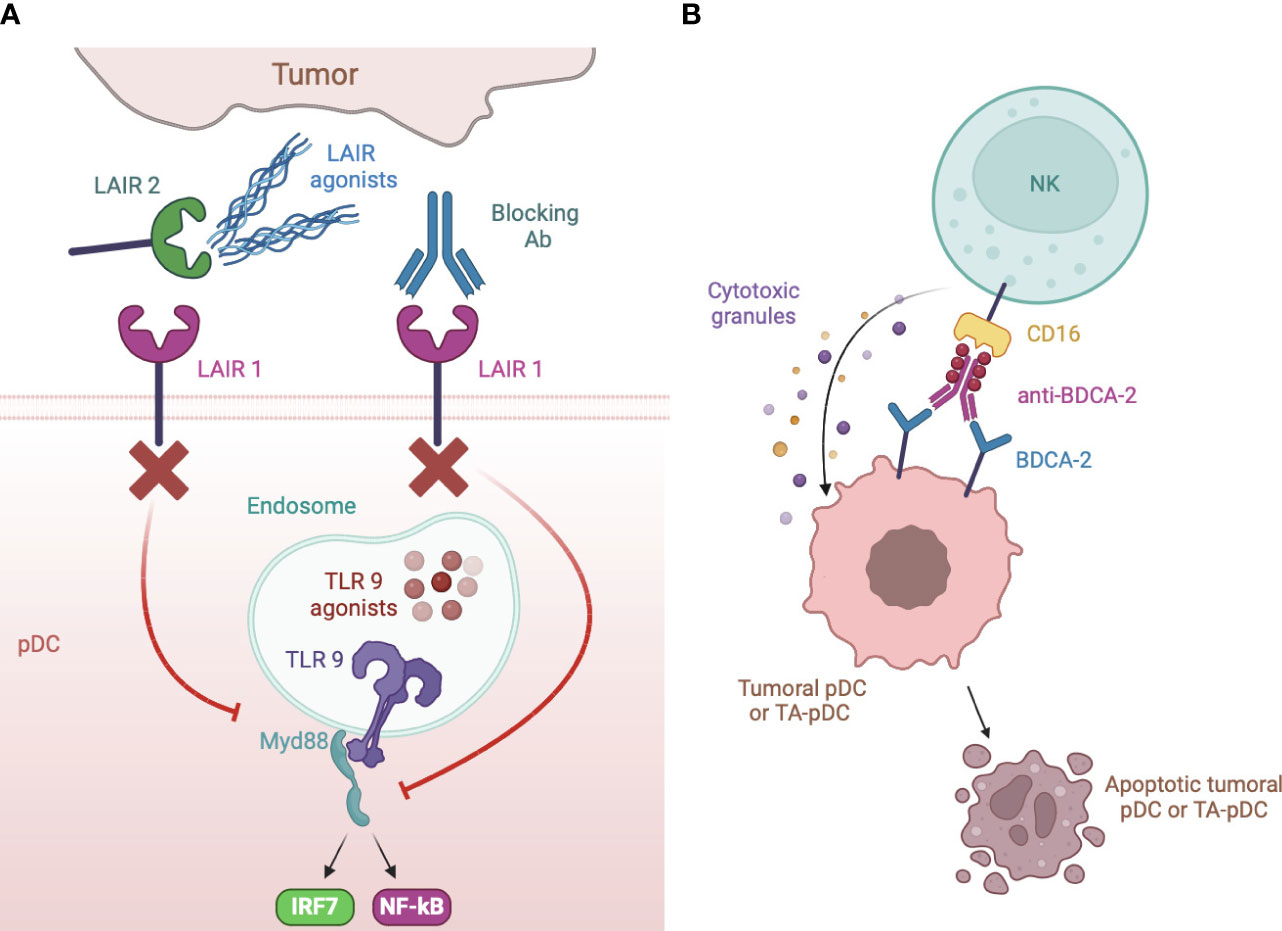
Figure 5 Strategies of IR targeting. (A) LAIR1 can be blocked to rescue type I IFN production by specific blocking antibodies or by a dimeric LAIR2 Fc fusion protein that sequesters ligands to membrane LAIR1. Blocking antibody were also developed against DCIR, ILT2, NKp44 and TIM3. (B) anti-BDCA-2 antibodies manipulated to increase their affinity for CD16 induce apoptotic cell death of neoplastic but also, possibly, TA- pDCs by NK cell activation.
This review summarizes the current knowledge on the pathophysiology of IRs expressed by pDCs, with a particular emphasis on their hijacking in cancer contexts, where pDC functions are generally reduced or abrogated. This evidence provides a proof of concept that the regulation of IFN secretion by pDCs may be regarded as an “innate checkpoint” which, similar to the “classical” adaptive immune checkpoint PD1 expressed in CD8+ T cells, restrains autoimmunity and immunopathology but may favor chronic infections and tumors. Accordingly, IRs may represent potential targets for innate and adaptive combined cancer immunotherapy to unleash T-cell–mediated tumor killing.
To date, however, the therapeutical exploitation of IR blockade to revert pDC exhaustion is hampered by several unanswered questions. First, the biology of pDCs in tumors is incompletely understood. For example, in the context of hepatic ischemia-reperfusion injury following surgical removal of hepatocellular carcinoma, tolerogenic pDCs associated to a better prognosis since type I IFNs crucially contributed to early tumor recurrence (209). Furthermore, OX40+ pDCs were found indispensable to activate cDCs and stimulate an efficient antitumor CD8+ T cells response in a mouse model of squamous carcinoma, which growth accelerated upon pDC depletion (210). pDC exhaustion may also be tumor- and even stage-specific, as recently shown in colon cancer where the presence of activated pDCs, as assessed by nuclear localization of IRF7, associated with increased patient survival (211). Type I IFNs themselves do play a dual role in cancer immunity, being protective in the early phases, while increasing the expression of PD1 and PD-L1 upon prolonged exposures (212). Thus, timing and duration of pDC activation may represent critical parameters in antitumor immune responses, requiring to be thoroughly understood and tightly regulated depending on the specific tumor context (69, 70). Also tumor-specific mechanisms of pDC suppression are incompletely known, not only in terms of IR ligand expression, but also concerning the distribution of IRs on pDC subsets and the possibility of their simultaneous engagement: in vitro, the engagement of one single IR is sufficient to block pDC activation, but no studies so far addressed the result of multiple engagement nor any possible hierarchical relationship among IRs. Finally, the expression patterns of “classical” immune checkpoint receptors (and ligands) on pDC subsets, poorly known to date, could affect the results of combined checkpoint inhibitor therapies.
We also mentioned that the avidity of IR engagement may influence to the final response of pDC (213). This is particularly relevant when IR inhibitory role is assessed by using crosslinking antibodies, that generally bind with high avidity, for example in the lack of specific ligands, but may hold true also for natural ligand endowed with different affinity. In the case of NKp44, PCNA overexpression by tumors sustained immune evasion through NKp44-mediated inhibition of both NK cells and pDCs (151), in accordance with the inhibitory role described by using crosslinking antibodies (155). In striking contrast, NKp44 engagement by PDGF-DD increased the production of type I IFNs by human pDCs activated with a TLR9 agonist (but, notably, not with a TLR7 agonist) (157). Thus, the role of IRs may differ in specific cancer context as well as their potential as therapeutic targets. However, such IR feature may also be exploited for the design of therapeutic tools, as demonstrated by the different activity of monovalent Fab fragment or cross-linking bivalent anti-BDCA-2 antibodies (137)
Last but not least, despite some IRs such as TIM3 and ILT2 are recognized targets for checkpoint blockade (2), they are widely expressed on immune and even tumor cells and the neat contribution of pDC exhaustion in tumor growth and the actual therapeutic significance of pDC rescue via IR blockade remains difficult to assess. By contrast, the anticancer potential of BDCA-2 and ILT7 blockade received little attention so far, partly depending on the lack of specific reagents. These IRs definitely deserve attention as targets in pathological conditions where pDC-specific modulation is required or pDC depletion can be advantageous.
In conclusion, despite encouraging evidence, more work is required to fully unravel the effects of IR engagement on pDC functions in specific tumor microenvironments and to uncover the beneficial role of therapeutic blockade of pDC-specific IRs in future immunotherapeutic strategies.
LT: Conceptualization, Writing – original draft. ML: Writing – original draft. GZ: Visualization, Writing – original draft. VS: Writing – review & editing. TS: Writing – review & editing. SS: Conceptualization, Funding acquisition, Supervision, Writing – review & editing. ADP: Conceptualization, Supervision, Writing – review & editing. DB: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Italian Association for Cancer Research (AIRC IG-2017/20776 to SS) and Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR, PRIN Prot. 2017/7J4E75 to SS, 2017/8ALPCM_005 to DB). ML was the recipient of a fellowship from AIRC (code 25307).
Figures in the article were drawn using BioRender.com (accessed on 14 February 2024).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The reviewer VL declared a shared affiliation with the authors ML, GZ and SS to the handling editor at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, et al. CD47 blockade by Hu5F9-G4 and rituximab in non-hodgkin's lymphoma. N Engl J Med. (2018) 379:1711–21. doi: 10.1056/NEJMoa1807315
2. Dutta S, Ganguly A, Chatterjee K, Spada S, Mukherjee S. Targets of immune escape mechanisms in cancer: basis for development and evolution of cancer immune checkpoint inhibitors. Biol (Basel). (2023) 12:218. doi: 10.3390/biology12020218
3. Stanczak MA, Läubli H. Siglec receptors as new immune checkpoints in cancer. Mol Aspects Med. (2023) 90:101112. doi: 10.1016/j.mam.2022.101112
4. Lentz RW, Colton MD, Mitra SS, Messersmith WA. Innate immune checkpoint inhibitors: the next breakthrough in medical oncology? Mol Cancer Ther. (2021) 20:961–74. doi: 10.1158/1535-7163.MCT-21-0041
5. Naimi A, Mohammed RN, Raji A, Chupradit S, Yumashev AV, Suksatan W, et al. Tumor immunotherapies by immune checkpoint inhibitors (ICIs); the pros and cons. Cell Commun Signal. (2022) 20:44. doi: 10.1186/s12964-022-00854-y
6. Mantovani A, Longo DL. Macrophage checkpoint blockade in cancer - back to the future. N Engl J Med. (2018) 379:1777–9. doi: 10.1056/NEJMe1811699
7. Musella M, Manic G, De Maria R, Vitale I, Sistigu A. Type-I-interferons in infection and cancer: Unanticipated dynamics with therapeutic implications. Oncoimmunology. (2017) 6:e1314424. doi: 10.1080/2162402X.2017.1314424
8. Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. (1999) 5:919–23. doi: 10.1038/11360
9. Ishikawa F, Niiro H, Iino T, Yoshida S, Saito N, Onohara S, et al. The developmental program of human dendritic cells is operated independently of conventional myeloid and lymphoid pathways. Blood. (2007) 110:3591–660. doi: 10.1182/blood-2007-02-071613
10. Reizis B, Idoyaga J, Dalod M, Barrat F, Naik S, Trinchieri G, et al. Reclassification of plasmacytoid dendritic cells as innate lymphocytes is premature. Nat Rev Immunol. (2023) 23:336–7. doi: 10.1038/s41577-023-00864-y
11. Ziegler-Heitbrock L, Ohteki T, Ginhoux F, Shortman K, Spits H. Reclassifying plasmacytoid dendritic cells as innate lymphocytes. Nat Rev Immunol. (2023) 23:1–2. doi: 10.1038/s41577-022-00806-0
12. Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. (2008) 135:37–48. doi: 10.1016/j.cell.2008.09.016
13. Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med. (2011) 365:127–38. doi: 10.1056/NEJMoa1100066
14. Cytlak U, Resteu A, Bogaert D, Kuehn HS, Altmann T, Gennery A, et al. Ikaros family zinc finger 1 regulates dendritic cell development and function in humans. Nat Commun. (2018) 9:1239. doi: 10.1038/s41467-018-02977-8
15. Musumeci A, Lutz K, Winheim E, Krug AB. What makes a pDC: recent advances in understanding plasmacytoid DC development and heterogeneity. Front Immunol. (2019) 10:1222. doi: 10.3389/fimmu.2019.01222
16. Grouard G, Rissoan MC, Filgueira L, Durand I, Banchereau J, Liu YJ. The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. J Exp Med. (1997) 185:1101–11. doi: 10.1084/jem.185.6.1101
17. Kennedy AJ, Davenport AP. International union of basic and clinical pharmacology CIII: chemerin receptors CMKLR1 (Chemerin. Pharmacol Rev. (2018) 70:174–96. doi: 10.1124/pr.116.013177
18. Sozzani S, Vermi W, Del Prete A, Facchetti F. Trafficking properties of plasmacytoid dendritic cells in health and disease. Trends Immunol. (2010) 31:270–7. doi: 10.1016/j.it.2010.05.004
19. Cella M, Facchetti F, Lanzavecchia A, Colonna M. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat Immunol. (2000) 1:305–10. doi: 10.1038/79747
20. Penna G, Sozzani S, Adorini L. Cutting edge: selective usage of chemokine receptors by plasmacytoid dendritic cells. J Immunol. (2001) 167:1862–6. doi: 10.4049/jimmunol.167.4.1862
21. Koda Y, Nakamoto N, Chu PS, Teratani T, Ueno A, Amiya T, et al. CCR9 axis inhibition enhances hepatic migration of plasmacytoid DCs and protects against liver injury. JCI Insight. (2022) 7:e159910. doi: 10.1172/jci.insight.159910
22. Albanesi C, Scarponi C, Bosisio D, Sozzani S, Girolomoni G. Immune functions and recruitment of plasmacytoid dendritic cells in psoriasis. Autoimmunity. (2010) 43:215–9. doi: 10.3109/08916930903510906
23. Tiberio L, Del Prete A, Schioppa T, Sozio F, Bosisio D, Sozzani S. Chemokine and chemotactic signals in dendritic cell migration. Cell Mol Immunol. (2018) 15:346–52. doi: 10.1038/s41423-018-0005-3
24. Schnurr M, Toy T, Shin A, Hartmann G, Rothenfusser S, Soellner J, et al. Role of adenosine receptors in regulating chemotaxis and cytokine production of plasmacytoid dendritic cells. Blood. (2004) 103:1391–7. doi: 10.1182/blood-2003-06-1959
25. Gutzmer R, Köther B, Zwirner J, Dijkstra D, Purwar R, Wittmann M, et al. Human plasmacytoid dendritic cells express receptors for anaphylatoxins C3a and C5a and are chemoattracted to C3a and C5a. J Invest Dermatol. (2006) 126:2422–9. doi: 10.1038/sj.jid.5700416
26. Devosse T, Guillabert A, D'Haene N, Berton A, De Nadai P, Noel S, et al. Formyl peptide receptor-like 2 is expressed and functional in plasmacytoid dendritic cells, tissue-specific macrophage subpopulations, and eosinophils. J Immunol. (2009) 182:4974–84. doi: 10.4049/jimmunol.0803128
27. Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. (2004) 5:1219–26. doi: 10.1038/ni1141
28. Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. (2005) 23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633
29. Ito T, Kanzler H, Duramad O, Cao W, Liu YJ. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood. (2006) 107:2423–31. doi: 10.1182/blood-2005-07-2709
30. Hoeffel G, Ripoche AC, Matheoud D, Nascimbeni M, Escriou N, Lebon P, et al. Antigen crosspresentation by human plasmacytoid dendritic cells. Immunity. (2007) 27:481–92. doi: 10.1016/j.immuni.2007.07.021
31. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. (2015) 15:471–85. doi: 10.1038/nri3865
32. Matsui T, Connolly JE, Michnevitz M, Chaussabel D, Yu CI, Glaser C, et al. CD2 distinguishes two subsets of human plasmacytoid dendritic cells with distinct phenotype and functions. J Immunol. (2009) 182:6815–23. doi: 10.4049/jimmunol.0802008
33. Bryant C, Fromm PD, Kupresanin F, Clark G, Lee K, Clarke C, et al. A CD2 high-expressing stress-resistant human plasmacytoid dendritic-cell subset. Immunol Cell Biol. (2016) 94:447–57. doi: 10.1038/icb.2015.116
34. Zhang H, Gregorio JD, Iwahori T, Zhang X, Choi O, Tolentino LL, et al. A distinct subset of plasmacytoid dendritic cells induces activation and differentiation of B and T lymphocytes. Proc Natl Acad Sci USA. (2017) 114:1988–93. doi: 10.1073/pnas.1610630114
35. Alculumbre SG, Saint-André V, Di Domizio J, Vargas P, Sirven P, Bost P, et al. Diversification of human plasmacytoid predendritic cells in response to a single stimulus. Nat Immunol. (2018) 19:63–75. doi: 10.1038/s41590-017-0012-z
36. Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity. (2010) 33:955–66. doi: 10.1016/j.immuni.2010.11.020
37. Chen Y, Yang JE, Tang JM, Mao QG, Zheng QZ, Zheng Y. Predictive value of plasmacytoid dendritic cells and Toll-like receptor-9 regarding the treatment efficacy of interferon-α in HBeAg-positive chronic hepatitis B patients. Exp Ther Med. (2019) 18:4541–6. doi: 10.3892/etm
38. Reizis B. Plasmacytoid dendritic cells: development, regulation, and function. Immunity. (2019) 50:37–50. doi: 10.1016/j.immuni.2018.12.027
39. Ciancanelli MJ, Huang SX, Luthra P, Garner H, Itan Y, Volpi S, et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science. (2015) 348:448–53. doi: 10.1126/science.aaa1578
40. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. (2020) 370:eabd4570. doi: 10.15252/embj.201798836
41. Tomasello E, Naciri K, Chelbi R, Bessou G, Fries A, Gressier E, et al. Molecular dissection of plasmacytoid dendritic cell activation. EMBO J. (2018) 37:e98836. doi: 10.15252/embj.201798836
42. Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, et al. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. (2005) 434:1035–40. doi: 10.1038/nature03547
43. Guiducci C, Ott G, Chan JH, Damon E, Calacsan C, Matray T, et al. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J Exp Med. (2006) 203:1999–2008. doi: 10.1084/jem.20060401
44. Szabo A, Magyarics Z, Pazmandi K, Gopcsa L, Rajnavolgyi E, Bacsi A. TLR ligands upregulate RIG-I expression in human plasmacytoid dendritic cells in a type I IFN-independent manner. Immunol Cell Biol. (2014) 92:671–8. doi: 10.1038/icb.2014.38
45. Deb P, Dai J, Singh S, Kalyoussef E, Fitzgerald-Bocarsly P. Triggering of the cGAS-STING pathway in human plasmacytoid dendritic cells inhibits TLR9-mediated IFN production. J Immunol. (2020) 205:223–36. doi: 10.4049/jimmunol.1800933
46. Hanabuchi S, Watanabe N, Wang YH, Ito T, Shaw J, Cao W, et al. Human plasmacytoid predendritic cells activate NK cells through glucocorticoid-induced tumor necrosis factor receptor-ligand (GITRL). Blood. (2006) 107:3617–23. doi: 10.1182/blood-2005-08-3419
47. Cederarv M, Söderberg-Nauclér C, Odeberg J. HCMV infection of PDCs deviates the NK cell response into cytokine-producing cells unable to perform cytotoxicity. Immunobiology. (2009) 214:331–41. doi: 10.1016/j.imbio.2008.10.009
48. Salvi V, Vermi W, Cavani A, Lonardi S, Carbone T, Facchetti F, et al. IL-21 may promote granzyme B-dependent NK/plasmacytoid dendritic cell functional interaction in cutaneous lupus erythematosus. J Invest Dermatol. (2017) 137:1493–500. doi: 10.1016/j.jid.2017.03.016
49. Villadangos JA, Young L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity. (2008) 29:352–61. doi: 10.1016/j.immuni.2008.09.002
50. Tel J, Schreibelt G, Sittig SP, Mathan TS, Buschow SI, Cruz LJ, et al. Human plasmacytoid dendritic cells efficiently cross-present exogenous Ags to CD8+ T cells despite lower Ag uptake than myeloid dendritic cell subsets. Blood. (2013) 121:459–67. doi: 10.1182/blood-2012-06-435644
51. Schnurr M, Chen Q, Shin A, Chen W, Toy T, Jenderek C, et al. Tumor antigen processing and presentation depend critically on dendritic cell type and the mode of antigen delivery. Blood. (2005) 105:2465–72. doi: 10.1182/blood-2004-08-3105
52. Benitez-Ribas D, Adema GJ, Winkels G, Klasen IS, Punt CJ, Figdor CG, et al. Plasmacytoid dendritic cells of melanoma patients present exogenous proteins to CD4+ T cells after Fc gamma RII-mediated uptake. J Exp Med. (2006) 203:1629–35. doi: 10.1084/jem.20052364
53. Meyer-Wentrup F, Benitez-Ribas D, Tacken PJ, Punt CJ, Figdor CG, de Vries IJ, et al. Targeting DCIR on human plasmacytoid dendritic cells results in antigen presentation and inhibits IFN-alpha production. Blood. (2008) 111:4245–53. doi: 10.1182/blood-2007-03-081398
54. Tel J, Sittig SP, Blom RA, Cruz LJ, Schreibelt G, Figdor CG, et al. Targeting uptake receptors on human plasmacytoid dendritic cells triggers antigen cross-presentation and robust type I IFN secretion. J Immunol. (2013) 191:5005–12. doi: 10.4049/jimmunol.1300787
55. Sepulveda-Toepfer JA, Pichler J, Fink K, Sevo M, Wildburger S, Mudde-Boer LC, et al. TLR9-mediated activation of dendritic cells by CD32 targeting for the generation of highly immunostimulatory vaccines. Hum Vaccin Immunother. (2019) 15:179–88. doi: 10.1080/21645515.2018.1514223
56. Yu CF, Peng WM, Oldenburg J, Hoch J, Bieber T, Limmer A, et al. Human plasmacytoid dendritic cells support Th17 cell effector function in response to TLR7 ligation. J Immunol. (2010) 184:1159–67. doi: 10.4049/jimmunol.0901706
57. Ruocco G, Rossi S, Motta C, Macchiarulo G, Barbieri F, De Bardi M, et al. T helper 9 cells induced by plasmacytoid dendritic cells regulate interleukin-17 in multiple sclerosis. Clin Sci (Lond). (2015) 129:291–303. doi: 10.1042/CS20140608
58. Miyahara Y, Chen H, Moriyama M, Mochizuki K, Kaneko N, Haque ASMR, et al. Toll-like receptor 9-positive plasmacytoid dendritic cells promote Th17 immune responses in oral lichen planus stimulated by epithelium-derived cathepsin K. Sci Rep. (2023) 13:19320. doi: 10.1038/s41598-023-46090-3
59. Jego G, Pascual V, Palucka AK, Banchereau J. Dendritic cells control B cell growth and differentiation. Curr Dir Autoimmun. (2005) 8:124–39. doi: 10.1159/000082101
60. Liang S, Wei H, Sun R, Tian Z. IFNalpha regulates NK cell cytotoxicity through STAT1 pathway. Cytokine. (2003) 23:190–9. doi: 10.1016/S1043-4666(03)00226-6
61. Pujantell M, Skenteris NT, Claussen JM, Grünhagel B, Thiele RJ, Altfeld M. Sex-dependent differences in type I IFN-induced natural killer cell activation. Front Immunol. (2023) 14:1277967. doi: 10.3389/fimmu.2023.1277967
62. Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med. (1999) 189:521–30. doi: 10.1084/jem.189.3.521
63. Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. (2005) 174:4465–9. doi: 10.4049/jimmunol.174.8.4465
64. U'Ren L, Guth A, Kamstock D, Dow S. Type I interferons inhibit the generation of tumor-associated macrophages. Cancer Immunol Immunother. (2010) 59:587–98. doi: 10.1007/s00262-009-0776-6
65. Oliveira IC, Sciavolino PJ, Lee TH, Vilcek J. Downregulation of interleukin 8 gene expression in human fibroblasts: unique mechanism of transcriptional inhibition by interferon. Proc Natl Acad Sci USA. (1992) 89:9049–53. doi: 10.1073/pnas.89.19.9049
66. Singh RK, Gutman M, Bucana CD, Sanchez R, Llansa N, Fidler IJ. Interferons alpha and beta down-regulate the expression of basic fibroblast growth factor in human carcinomas. Proc Natl Acad Sci USA. (1995) 92:4562–6. doi: 10.1073/pnas.92.10.4562
67. McCarty MF, Bielenberg D, Donawho C, Bucana CD, Fidler IJ. Evidence for the causal role of endogenous interferon-alpha/beta in the regulation of angiogenesis, tumorigenicity, and metastasis of cutaneous neoplasms. Clin Exp Metastasis. (2002) 19:609–15. doi: 10.1023/A:1020923326441
68. von Marschall Z, Scholz A, Cramer T, Schäfer G, Schirner M, Oberg K, et al. Effects of interferon alpha on vascular endothelial growth factor gene transcription and tumor angiogenesis. J Natl Cancer Inst. (2003) 95:437–48. doi: 10.1093/jnci/95.6.437
69. Aricò E, Castiello L, Capone I, Gabriele L, Belardelli F. Type I interferons and cancer: an evolving story demanding novel clinical applications. Cancers (Basel). (2019) 11:1943. doi: 10.3390/cancers11121943
70. Zhou L, Zhang Y, Wang Y, Zhang M, Sun W, Dai T, et al. A dual role of type I interferons in antitumor immunity. Adv Biosyst. (2020) 4:e1900237. doi: 10.1002/adbi.201900237
71. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. (2022) 36:1703–19. doi: 10.1038/s41375-022-01613-1
72. Vermi W, Soncini M, Melocchi L, Sozzani S, Facchetti F. Plasmacytoid dendritic cells and cancer. J Leukoc Biol. (2011) 90:681–90. doi: 10.1189/jlb.0411190
73. Zhou B, Lawrence T, Liang Y. The role of plasmacytoid dendritic cells in cancers. Front Immunol. (2021) 12:749190. doi: 10.3389/fimmu.2021.749190
74. Treilleux I, Blay JY, Bendriss-Vermare N, Ray-Coquard I, Bachelot T, Guastalla JP, et al. Dendritic cell infiltration and prognosis of early stage breast cancer. Clin Cancer Res. (2004) 10:7466–74. doi: 10.1158/1078-0432.CCR-04-0684
75. Labidi-Galy SI, Treilleux I, Goddard-Leon S, Combes JD, Blay JY, Ray-Coquard I, et al. Plasmacytoid dendritic cells infiltrating ovarian cancer are associated with poor prognosis. Oncoimmunology. (2012) 1:380–2. doi: 10.4161/onci.18801
76. Han N, Zhang Z, Liu S, Ow A, Ruan M, Yang W, et al. Increased tumor-infiltrating plasmacytoid dendritic cells predicts poor prognosis in oral squamous cell carcinoma. Arch Oral Biol. (2017) 78:129–34. doi: 10.1016/j.archoralbio.2017.02.012
77. Pang L, Ng KT, Liu J, Yeung WO, Zhu J, Chiu TS, et al. Plasmacytoid dendritic cells recruited by HIF-1α/eADO/ADORA1 signaling induce immunosuppression in hepatocellular carcinoma. Cancer Lett. (2021) 522:80–92. doi: 10.1016/j.canlet.2021.09.022
78. Sosa Cuevas E, Bendriss-Vermare N, Mouret S, De Fraipont F, Charles J, Valladeau-Guilemond J, et al. Diversification of circulating and tumor-infiltrating plasmacytoid DCs towards the P3 (CD80. Clin Transl Immunol. (2022) 11:e1382. doi: 10.1002/cti2.1382
79. Labidi-Galy SI, Sisirak V, Meeus P, Gobert M, Treilleux I, Bajard A, et al. Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer Res. (2011) 71:5423–34. doi: 10.1158/0008-5472.CAN-11-0367
80. Conrad C, Gregorio J, Wang YH, Ito T, Meller S, Hanabuchi S, et al. Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3(+) T-regulatory cells. Cancer Res. (2012) 72:5240–9. doi: 10.1158/0008-5472.CAN-12-2271
81. Sisirak V, Faget J, Gobert M, Goutagny N, Vey N, Treilleux I, et al. Impaired IFN-α production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res. (2012) 72:5188–97. doi: 10.1158/0008-5472.CAN-11-3468
82. Le Mercier I, Poujol D, Sanlaville A, Sisirak V, Gobert M, Durand I, et al. Tumor promotion by intratumoral plasmacytoid dendritic cells is reversed by TLR7 ligand treatment. Cancer Res. (2013) 73:4629–40. doi: 10.1158/0008-5472.CAN-12-3058
83. Sisirak V, Vey N, Goutagny N, Renaudineau S, Malfroy M, Thys S, et al. Breast cancer-derived transforming growth factor-β and tumor necrosis factor-α compromise interferon-α production by tumor-associated plasmacytoid dendritic cells. Int J Cancer. (2013) 133:771–8. doi: 10.1002/ijc.28072
84. Terra M, Oberkampf M, Fayolle C, Rosenbaum P, Guillerey C, Dadaglio G, et al. Tumor-derived TGFβ Alters the ability of plasmacytoid dendritic cells to respond to innate immune signaling. Cancer Res. (2018) 78:3014–26. doi: 10.1158/0008-5472.CAN-17-2719
85. Aspord C, Leccia MT, Charles J, Plumas J. Plasmacytoid dendritic cells support melanoma progression by promoting Th2 and regulatory immunity through OX40L and ICOSL. Cancer Immunol Res. (2013) 1:402–15. doi: 10.1158/2326-6066.CIR-13-0114-T
86. Huang XM, Liu XS, Lin XK, Yu H, Sun JY, Liu XK, et al. Role of plasmacytoid dendritic cells and inducible costimulator-positive regulatory T cells in the immunosuppression microenvironment of gastric cancer. Cancer Sci. (2014) 105:150–8. doi: 10.1111/cas.12327
87. Dey M, Chang AL, Miska J, Wainwright DA, Ahmed AU, Balyasnikova IV, et al. Dendritic cell-based vaccines that utilize myeloid rather than plasmacytoid cells offer a superior survival advantage in Malignant glioma. J Immunol. (2015) 195:367–76. doi: 10.4049/jimmunol.1401607
88. Pedroza-Gonzalez A, Zhou G, Vargas-Mendez E, Boor PP, Mancham S, Verhoef C, et al. Tumor-infiltrating plasmacytoid dendritic cells promote immunosuppression by Tr1 cells in human liver tumors. Oncoimmunology. (2015) 4:e1008355. doi: 10.1080/2162402X.2015.1008355
89. Zhou ZJ, Xin HY, Li J, Hu ZQ, Luo CB, Zhou SL. Intratumoral plasmacytoid dendritic cells as a poor prognostic factor for hepatocellular carcinoma following curative resection. Cancer Immunol Immunother. (2019) 68:1223–33. doi: 10.1007/s00262-019-02355-3
90. Yu H, Huang X, Liu X, Jin H, Zhang G, Zhang Q, et al. Regulatory T cells and plasmacytoid dendritic cells contribute to the immune escape of papillary thyroid cancer coexisting with multinodular non-toxic goiter. Endocrine. (2013) 44:172–81. doi: 10.1007/s12020-012-9853-2
91. Ray A, Das DS, Song Y, Richardson P, Munshi NC, Chauhan D, et al. Targeting PD1-PDL1 immune checkpoint in plasmacytoid dendritic cell interactions with T cells, natural killer cells and multiple myeloma cells. Leukemia. (2015) 29:1441–4. doi: 10.1038/leu.2015.11
92. Laheurte C, Seffar E, Gravelin E, Lecuelle J, Renaudin A, Boullerot L, et al. Interplay between plasmacytoid dendritic cells and tumor-specific T cells in peripheral blood influences long-term survival in non-small cell lung carcinoma. Cancer Immunol Immunother. (2023) 72:579–89. doi: 10.1007/s00262-022-03271-9
93. Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. (2007) 117:2570–82. doi: 10.1172/JCI31911
94. Curiel TJ, Cheng P, Mottram P, Alvarez X, Moons L, Evdemon-Hogan M, et al. Dendritic cell subsets differentially regulate angiogenesis in human ovarian cancer. Cancer Res. (2004) 64:5535–8. doi: 10.1158/0008-5472.CAN-04-1272
95. Sorrentino R, Terlizzi M, Di Crescenzo VG, Popolo A, Pecoraro M, Perillo G, et al. Human lung cancer-derived immunosuppressive plasmacytoid dendritic cells release IL-1α in an AIM2 inflammasome-dependent manner. Am J Pathol. (2015) 185:3115–24. doi: 10.1016/j.ajpath.2015.07.009
96. Sawant A, Hensel JA, Chanda D, Harris BA, Siegal GP, Maheshwari A, et al. Depletion of plasmacytoid dendritic cells inhibits tumor growth and prevents bone metastasis of breast cancer cells. J Immunol. (2012) 189:4258–65. doi: 10.4049/jimmunol.1101855
97. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. (2015) 36:265–76. doi: 10.1016/j.it.2015.02.008
98. van Vlerken-Ysla L, Tyurina YY, Kagan VE, Gabrilovich DI. Functional states of myeloid cells in cancer. Cancer Cell. (2023) 41:490–504. doi: 10.1016/j.ccell.2023.02.009
99. Bekeredjian-Ding I, Schäfer M, Hartmann E, Pries R, Parcina M, Schneider P, et al. Tumour-derived prostaglandin E and transforming growth factor-beta synergize to inhibit plasmacytoid dendritic cell-derived interferon-alpha. Immunology. (2009) 128:439–50. doi: 10.1111/j.1365-2567.2009.03134.x
100. Hartmann E, Wollenberg B, Rothenfusser S, Wagner M, Wellisch D, Mack B, et al. Identification and functional analysis of tumor-infiltrating plasmacytoid dendritic cells in head and neck cancer. Cancer Res. (2003) 63:6478–87.
101. Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med. (2012) 18:1224–31. doi: 10.1038/nm.2830
102. Bosisio D, Salvi V, Gagliostro V, Sozzani S. Angiogenic and antiangiogenic chemokines. Chem Immunol Allergy. (2014) 99:89–104. doi: 10.1159/000353317
103. Beckebaum S, Zhang X, Chen X, Yu Z, Frilling A, Dworacki G, et al. Increased levels of interleukin-10 in serum from patients with hepatocellular carcinoma correlate with profound numerical deficiencies and immature phenotype of circulating dendritic cell subsets. Clin Cancer Res. (2004) 10:7260–9. doi: 10.1158/1078-0432.CCR-04-0872
104. Demoulin S, Herfs M, Somja J, Roncarati P, Delvenne P, Hubert P. HMGB1 secretion during cervical carcinogenesis promotes the acquisition of a tolerogenic functionality by plasmacytoid dendritic cells. Int J Cancer. (2015) 137:345–58. doi: 10.1002/ijc.29389
105. Xu Z, Li P, Fan L, Wu M. The potential role of circRNA in tumor immunity regulation and immunotherapy. Front Immunol. (2018) 9:9. doi: 10.3389/fimmu.2018.00009
106. Greene TT, Jo YR, Zuniga EI. Infection and cancer suppress pDC derived IFN-I. Curr Opin Immunol. (2020) 66:114–22. doi: 10.1016/j.coi.2020.08.001
107. de la Cruz-López KG, Castro-Muñoz LJ, Reyes-Hernández DO, García-Carrancá A, Manzo-Merino J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front Oncol. (2019) 9:1143. doi: 10.3389/fonc.2019.01143
108. Raychaudhuri D, Bhattacharya R, Sinha BP, Liu CSC, Ghosh AR, Rahaman O, et al. Lactate induces pro-tumor reprogramming in intratumoral plasmacytoid dendritic cells. Front Immunol. (2019) 10:1878. doi: 10.3389/fimmu.2019.01878
109. Wu D, Sanin DE, Everts B, Chen Q, Qiu J, Buck MD, et al. Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity. (2016) 44:1325–36. doi: 10.1016/j.immuni.2016.06.006
110. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell. (2017) 169:570–86. doi: 10.1016/j.cell.2017.04.004
111. Sozzani S, Bosisio D, Scarsi M, Tincani A. Type I interferons in systemic autoimmunity. Autoimmunity. (2010) 43:196–203. doi: 10.3109/08916930903510872
112. Sozzani S, Del Prete A, Bosisio D. Dendritic cell recruitment and activation in autoimmunity. J Autoimmun. (2017) 85:126–40. doi: 10.1016/j.jaut.2017.07.012
113. Wu P, Wu J, Liu S, Han X, Lu J, Shi Y, et al. TLR9/TLR7-triggered downregulation of BDCA2 expression on human plasmacytoid dendritic cells from healthy individuals and lupus patients. Clin Immunol. (2008) 129:40–8. doi: 10.1016/j.clim.2008.06.004
114. Dzionek A, Sohma Y, Nagafune J, Cella M, Colonna M, Facchetti F, et al. BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J Exp Med. (2001) 194:1823–34. doi: 10.1084/jem.194.12.1823
115. Riboldi E, Daniele R, Cassatella MA, Sozzani S, Bosisio D. Engagement of BDCA-2 blocks TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. Immunobiology. (2009) 214:868–76. doi: 10.1016/j.imbio.2009.06.016
116. Taylor ME, Drickamer K. Mammalian sugar-binding receptors: known functions and unexplored roles. FEBS J. (2019) 286:1800–14. doi: 10.1111/febs.14759
117. Willment JA. Fc-conjugated C-type lectin receptors: Tools for understanding host-pathogen interactions. Mol Microbiol. (2022) 117:632–60. doi: 10.1111/mmi.14837
118. Fischer S, Stegmann F, Gnanapragassam VS, Lepenies B. From structure to function - Ligand recognition by myeloid C-type lectin receptors. Comput Struct Biotechnol J. (2022) 20:5790–812. doi: 10.1016/j.csbj.2022.10.019
119. Nagae M, Ikeda A, Kitago Y, Matsumoto N, Yamamoto K, Yamaguchi Y. Crystal structures of carbohydrate recognition domain of blood dendritic cell antigen-2 (BDCA2) reveal a common domain-swapped dimer. Proteins. (2014) 82:1512–8. doi: 10.1002/prot.24504
120. Lee RT, Hsu TL, Huang SK, Hsieh SL, Wong CH, Lee YC. Survey of immune-related, mannose/fucose-binding C-type lectin receptors reveals widely divergent sugar-binding specificities. Glycobiology. (2011) 21:512–20. doi: 10.1093/glycob/cwq193
121. Riboldi E, Daniele R, Parola C, Inforzato A, Arnold PL, Bosisio D, et al. Human C-type lectin domain family 4, member C (CLEC4C/BDCA-2/CD303) is a receptor for asialo-galactosyl-oligosaccharides. J Biol Chem. (2011) 286:35329–33. doi: 10.1074/jbc.C111.290494
122. Jégouzo SA, Feinberg H, Dungarwalla T, Drickamer K, Weis WI, Taylor ME. A novel mechanism for binding of galactose-terminated glycans by the C-type carbohydrate recognition domain in blood dendritic cell antigen 2. J Biol Chem. (2015) 290:16759–71. doi: 10.1074/jbc.M115.660613
123. Kim JW, Budzak J, Liu Y, Jégouzo SAF, Drickamer K, Taylor ME. Identification of serum glycoprotein ligands for the immunomodulatory receptor blood dendritic cell antigen 2. Glycobiology. (2018) 28:592–600. doi: 10.1093/glycob/cwy050
124. Dekkers G, Rispens T, Vidarsson G. Novel concepts of altered immunoglobulin G galactosylation in autoimmune diseases. Front Immunol. (2018) 9:553. doi: 10.3389/fimmu.2018.00553
125. Xu Y, Hu Y, Shi B, Zhang X, Wang J, Zhang Z, et al. HBsAg inhibits TLR9-mediated activation and IFN-alpha production in plasmacytoid dendritic cells. Mol Immunol. (2009) 46:2640–6. doi: 10.1016/j.molimm.2009.04.031
126. Dhamanage A, Thakar M, Paranjape R. Human immunodeficiency virus-1 impairs IFN-alpha production induced by TLR-7 agonist in plasmacytoid dendritic cells. Viral Immunol. (2017) 30:28–34. doi: 10.1089/vim.2016.0084
127. Bos S, Poirier-Beaudouin B, Seffer V, Manich M, Mardi C, Desprès P, et al. Zika virus inhibits IFN-α Response by human plasmacytoid dendritic cells and induces NS1-dependent triggering of CD303 (BDCA-2) signaling. Front Immunol. (2020) 11:582061. doi: 10.3389/fimmu.2020.582061
128. Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. (2009) 9:465–79. doi: 10.1038/nri2569
129. Bloem K, Vuist IM, van den Berk M, Klaver EJ, van Die I, Knippels LM, et al. DCIR interacts with ligands from both endogenous and pathogenic origin. Immunol Lett. (2014) 158:33–41. doi: 10.1016/j.imlet.2013.11.007
130. da Costa V, Freire T. Advances in the immunomodulatory properties of glycoantigens in cancer. Cancers (Basel). (2022) 14:1854. doi: 10.3390/cancers14081854
131. van Gisbergen KP, Aarnoudse CA, Meijer GA, Geijtenbeek TB, van Kooyk Y. Dendritic cells recognize tumor-specific glycosylation of carcinoembryonic antigen on colorectal cancer cells through dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin. Cancer Res. (2005) 65:5935–44. doi: 10.1158/0008-5472.CAN-04-4140
132. Cao W, Zhang L, Rosen DB, Bover L, Watanabe G, Bao M, et al. BDCA2/Fc epsilon RI gamma complex signals through a novel BCR-like pathway in human plasmacytoid dendritic cells. PloS Biol. (2007) 5:e248. doi: 10.1371/journal.pbio.0050248
133. Röck J, Schneider E, Grün JR, Grützkau A, Küppers R, Schmitz J, et al. CD303 (BDCA-2) signals in plasmacytoid dendritic cells via a BCR-like signalosome involving Syk, Slp65 and PLCgamma2. Eur J Immunol. (2007) 37:3564–75. doi: 10.1002/eji.200737711
134. Florentin J, Aouar B, Dental C, Thumann C, Firaguay G, Gondois-Rey F, et al. HCV glycoprotein E2 is a novel BDCA-2 ligand and acts as an inhibitor of IFN production by plasmacytoid dendritic cells. Blood. (2012) 120:4544–51. doi: 10.1182/blood-2012-02-413286
135. Janovec V, Aouar B, Font-Haro A, Hofman T, Trejbalova K, Weber J, et al. The MEK1/2-ERK pathway inhibits type I IFN production in plasmacytoid dendritic cells. Front Immunol. (2018) 9:364. doi: 10.3389/fimmu.2018.00364
136. Kang YJ, Kusler B, Otsuka M, Hughes M, Suzuki N, Suzuki S, et al. Calcineurin negatively regulates TLR-mediated activation pathways. J Immunol. (2007) 179:4598–607. doi: 10.4049/jimmunol.179.7.4598
137. Jähn PS, Zänker KS, Schmitz J, Dzionek A. BDCA-2 signaling inhibits TLR-9-agonist-induced plasmacytoid dendritic cell activation and antigen presentation. Cell Immunol. (2010) 265:15–22. doi: 10.1016/j.cellimm.2010.06.005
138. Pellerin A, Otero K, Czerkowicz JM, Kerns HM, Shapiro RI, Ranger AM, et al. Anti-BDCA2 monoclonal antibody inhibits plasmacytoid dendritic cell activation through Fc-dependent and Fc-independent mechanisms. EMBO Mol Med. (2015) 7:464–76. doi: 10.15252/emmm.201404719
139. Furie RA, van Vollenhoven RF, Kalunian K, Navarra S, Romero-Diaz J, Werth VP, et al. Trial of anti-BDCA2 antibody litifilimab for systemic lupus erythematosus. N Engl J Med. (2022) 387:894–904. doi: 10.1056/NEJMoa2118025
140. Werth VP, Furie RA, Romero-Diaz J, Navarra S, Kalunian K, van Vollenhoven RF, et al. Trial of anti-BDCA2 antibody litifilimab for cutaneous lupus erythematosus. N Engl J Med. (2022) 387:321–31. doi: 10.1056/NEJMoa2118024
141. Fournier N, Jacque E, Fontayne A, Derache D, Dupont G, Verhaeghe L, et al. Improved in vitro and in vivo activity against CD303-expressing targets of the chimeric 122A2 antibody selected for specific glycosylation pattern. MAbs. (2018) 10:651–63. doi: 10.1080/19420862.2018.1451283
142. Cao W, Rosen DB, Ito T, Bover L, Bao M, Watanabe G, et al. Plasmacytoid dendritic cell-specific receptor ILT7-Fc epsilonRI gamma inhibits Toll-like receptor-induced interferon production. J Exp Med. (2006) 203:1399–405. doi: 10.1084/jem.20052454
143. Cao W, Bover L, Cho M, Wen X, Hanabuchi S, Bao M, et al. Regulation of TLR7/9 responses in plasmacytoid dendritic cells by BST2 and ILT7 receptor interaction. J Exp Med. (2009) 206:1603–14. doi: 10.1084/jem.20090547
144. Cao W, Bover L. Signaling and ligand interaction of ILT7: receptor-mediated regulatory mechanisms for plasmacytoid dendritic cells. Immunol Rev. (2010) 234:163–76. doi: 10.1111/j.0105-2896.2009.00867.x
145. Nakajima H, Asai A, Okada A, Ping L, Hamajima F, Sata T, et al. Transcriptional regulation of ILT family receptors. J Immunol. (2003) 171:6611–20. doi: 10.4049/jimmunol.171.12.6611
146. Canavez F, Young NT, Guethlein LA, Rajalingam R, Khakoo SI, Shum BP, et al. Comparison of chimpanzee and human leukocyte Ig-like receptor genes reveals framework and rapidly evolving genes. J Immunol. (2001) 167:5786–94. doi: 10.4049/jimmunol.167.10.5786
147. Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, et al. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. (2008) 3:245–52. doi: 10.1016/j.chom.2008.03.001
148. Mahauad-Fernandez WD, Okeoma CM. The role of BST-2/Tetherin in host protection and disease manifestation. Immun Inflamm Dis. (2016) 4:4–23. doi: 10.1002/iid3.92
149. Mahauad-Fernandez WD, DeMali KA, Olivier AK, Okeoma CM. Bone marrow stromal antigen 2 expressed in cancer cells promotes mammary tumor growth and metastasis. Breast Cancer Res. (2014) 16:493. doi: 10.1186/s13058-014-0493-8
150. Mukai S, Oue N, Oshima T, Mukai R, Tatsumoto Y, Sakamoto N, et al. Overexpression of transmembrane protein BST2 is associated with poor survival of patients with esophageal, gastric, or colorectal cancer. Ann Surg Oncol. (2017) 24:594–602. doi: 10.1245/s10434-016-5100-z
151. Sosa Cuevas E, Valladeau-Guilemond J, Mouret S, Roubinet B, de Fraipont F, Landemarre L, et al. Unique CLR expression patterns on circulating and tumor-infiltrating DC subsets correlated with clinical outcome in melanoma patients. Front Immunol. (2022) 13:1040600. doi: 10.3389/fimmu.2022.1040600
152. Campbell KS, Yusa S, Kikuchi-Maki A, Catina TL. NKp44 triggers NK cell activation through DAP12 association that is not influenced by a putative cytoplasmic inhibitory sequence. J Immunol. (2004) 172:899–906. doi: 10.4049/jimmunol.172.2.899
153. Rosental B, Brusilovsky M, Hadad U, Oz D, Appel MY, Afergan F, et al. Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44. J Immunol. (2011) 187:5693–702. doi: 10.4049/jimmunol.1102267
154. Del Fresno C, Iborra S, Saz-Leal P, Martínez-López M, Sancho D. Flexible signaling of myeloid C-type lectin receptors in immunity and inflammation. Front Immunol. (2018) 9:804. doi: 10.3389/fimmu.2018.00804
155. Marrufo AM, Mathew SO, Chaudhary P, Malaer JD, Ahmed N, Vishwanatha JK, et al. Blocking PCNA interaction with NKp44 enhances primary natural killer cell-mediated lysis of triple-negative breast cancer cells. Am J Cancer Res. (2023) 13:1082–90.
156. Fuchs A, Cella M, Kondo T, Colonna M. Paradoxic inhibition of human natural interferon-producing cells by the activating receptor NKp44. Blood. (2005) 106:2076–82. doi: 10.1182/blood-2004-12-4802
157. Barrow AD, Cella M, Edeling MA, Khan MA, Cervantes-Barragan L, Bugatti M, et al. Cutting edge: PDGF-DD binding to NKp44 costimulates TLR9 signaling and proinflammatory cytokine secretion in human plasmacytoid dendritic cells. J Immunol. (2024) 212:369–74. doi: 10.4049/jimmunol.2200496
158. Bates EE, Fournier N, Garcia E, Valladeau J, Durand I, Pin JJ, et al. APCs express DCIR, a novel C-type lectin surface receptor containing an immunoreceptor tyrosine-based inhibitory motif. J Immunol. (1999) 163:1973–83. doi: 10.4049/jimmunol.163.4.1973
159. Fujikado N, Saijo S, Yonezawa T, Shimamori K, Ishii A, Sugai S, et al. Dcir deficiency causes development of autoimmune diseases in mice due to excess expansion of dendritic cells. Nat Med. (2008) 14:176–80. doi: 10.1038/nm1697
160. Kaifu T, Yabe R, Maruhashi T, Chung SH, Tateno H, Fujikado N, et al. DCIR and its ligand asialo-biantennary N-glycan regulate DC function and osteoclastogenesis. J Exp Med. (2021) 218:e20210435. doi: 10.1084/jem.20210435
161. Sun H, Tang C, Chung SH, Ye XQ, Makusheva Y, Han W, et al. Blocking DCIR mitigates colitis and prevents colorectal tumors by enhancing the GM-CSF-STAT5 pathway. Cell Rep. (2022) 40:111158. doi: 10.1016/j.celrep.2022.111158
162. Weng TY, Li CJ, Li CY, Hung YH, Yen MC, Chang YW, et al. Skin delivery of Clec4a small hairpin RNA elicited an effective antitumor response by enhancing CD8. Mol Ther Nucleic Acids. (2017) 9:419–27. doi: 10.1016/j.omtn.2017.10.015
163. Shiroishi M, Tsumoto K, Amano K, Shirakihara Y, Colonna M, Braud VM, et al. Human inhibitory receptors Ig-like transcript 2 (ILT2) and ILT4 compete with CD8 for MHC class I binding and bind preferentially to HLA-G. Proc Natl Acad Sci USA. (2003) 100:8856–61. doi: 10.1073/pnas.1431057100
164. Ju XS, Hacker C, Scherer B, Redecke V, Berger T, Schuler G, et al. Immunoglobulin-like transcripts ILT2, ILT3 and ILT7 are expressed by human dendritic cells and down-regulated following activation. Gene. (2004) 331:159–64. doi: 10.1016/j.gene.2004.02.018
165. Wagner CS, Walther-Jallow L, Buentke E, Ljunggren HG, Achour A, Chambers BJ. Human cytomegalovirus-derived protein UL18 alters the phenotype and function of monocyte-derived dendritic cells. J Leukoc Biol. (2008) 83:56–63. doi: 10.1189/jlb.0307181
166. Young NT, Waller EC, Patel R, Roghanian A, Austyn JM, Trowsdale J. The inhibitory receptor LILRB1 modulates the differentiation and regulatory potential of human dendritic cells. Blood. (2008) 111:3090–6. doi: 10.1182/blood-2007-05-089771
167. Carenza C, Calcaterra F, Oriolo F, Di Vito C, Ubezio M, Della Porta MG, et al. Costimulatory molecules and immune checkpoints are differentially expressed on different subsets of dendritic cells. Front Immunol. (2019) 10:1325. doi: 10.3389/fimmu.2019.01325
168. Martín-Villa JM, Vaquero-Yuste C, Molina-Alejandre M, Juarez I, Suárez-Trujillo F, López-Nares A, et al. HLA-G: too much or too little? Role in cancer and autoimmune disease. Front Immunol. (2022) 13:796054. doi: 10.3389/fimmu.2022.796054
169. Lin A, Yan WH. HLA-G/ILTs targeted solid cancer immunotherapy: opportunities and challenges. Front Immunol. (2021) 12:698677. doi: 10.3389/fimmu.2021.698677
170. Villa-Álvarez M, Sordo-Bahamonde C, Lorenzo-Herrero S, Gonzalez-Rodriguez AP, Payer AR, Gonzalez-Garcia E, et al. Ig-like transcript 2 (ILT2) blockade and lenalidomide restore NK cell function in chronic lymphocytic leukemia. Front Immunol. (2018) 9:2917. doi: 10.3389/fimmu.2018.02917
171. Lorenzo-Herrero S, Sordo-Bahamonde C, Martínez-Pérez A, Corte-Torres MD, Fernández-Vega I, Solís-Hernández MP, et al. Immunoglobulin-like transcript 2 blockade restores antitumor immune responses in glioblastoma. Cancer Sci. (2023) 114:48–62. doi: 10.1111/cas.15575
172. Zeller T, Lutz S, Münnich IA, Windisch R, Hilger P, Herold T, et al. Dual checkpoint blockade of CD47 and LILRB1 enhances CD20 antibody-dependent phagocytosis of lymphoma cells by macrophages. Front Immunol. (2022) 13:929339. doi: 10.3389/fimmu.2022.929339
173. Van Laethem F, Donaty L, Tchernonog E, Lacheretz-Szablewski V, Russello J, Buthiau D, et al. LAIR1, an ITIM-containing receptor involved in immune disorders and in hematological neoplasms. Int J Mol Sci. (2022) 23:16136. doi: 10.3390/ijms232416136
174. Bonaccorsi I, Cantoni C, Carrega P, Oliveri D, Lui G, Conte R, et al. The immune inhibitory receptor LAIR-1 is highly expressed by plasmacytoid dendritic cells and acts complementary with NKp44 to control IFNα production. PloS One. (2010) 5:e15080. doi: 10.1371/journal.pone.0015080
175. Lebbink RJ, de Ruiter T, Adelmeijer J, Brenkman AB, van Helvoort JM, Koch M, et al. Collagens are functional, high affinity ligands for the inhibitory immune receptor LAIR-1. J Exp Med. (2006) 203:1419–25. doi: 10.1084/jem.20052554
176. Olde Nordkamp MJ, Boross P, Yildiz C, Jansen JH, Leusen JH, Wouters D, et al. Inhibition of the classical and lectin pathway of the complement system by recombinant LAIR-2. J Innate Immun. (2014) 6:284–92. doi: 10.1159/000354976
177. Joseph C, Alsaleem MA, Toss MS, Kariri YA, Althobiti M, Alsaeed S, et al. The ITIM-containing receptor: leukocyte-associated immunoglobulin-like receptor-1 (LAIR-1) modulates immune response and confers poor prognosis in invasive breast carcinoma. Cancers (Basel). (2020) 13:80. doi: 10.3390/cancers13010080
178. Peng DH, Rodriguez BL, Diao L, Chen L, Wang J, Byers LA, et al. Collagen promotes anti-PD-1/PD-L1 resistance in cancer through LAIR1-dependent CD8. Nat Commun. (2020) 11:4520. doi: 10.1038/s41467-020-18298-8
179. Xie J, Gui X, Deng M, Chen H, Chen Y, Liu X, et al. Blocking LAIR1 signaling in immune cells inhibits tumor development. Front Immunol. (2022) 13:996026. doi: 10.3389/fimmu.2022.996026
180. Lebbink RJ, van den Berg MC, de Ruiter T, Raynal N, van Roon JA, Lenting PJ, et al. The soluble leukocyte-associated Ig-like receptor (LAIR)-2 antagonizes the collagen/LAIR-1 inhibitory immune interaction. J Immunol. (2008) 180:1662–9. doi: 10.4049/jimmunol.180.3.1662
181. Ramos MIP, Tian L, de Ruiter EJ, Song C, Paucarmayta A, Singh A, et al. Cancer immunotherapy by NC410, a LAIR-2 Fc protein blocking human LAIR-collagen interaction. Elife. (2021) 10:e62927. doi: 10.7554/eLife.62927
182. Horn LA, Chariou PL, Gameiro SR, Qin H, Iida M, Fousek K, et al. Remodeling the tumor microenvironment via blockade of LAIR-1 and TGF-β signaling enables PD-L1-mediated tumor eradication. J Clin Invest. (2022) 132:e155148. doi: 10.1172/JCI155148
183. Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol. (2020) 20:173–85. doi: 10.1038/s41577-019-0224-6
184. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. (2012) 13:832–42. doi: 10.1038/ni.2376
185. Schwartz JA, Clayton KL, Mujib S, Zhang H, Rahman AK, Liu J, et al. Tim-3 is a Marker of Plasmacytoid Dendritic Cell Dysfunction during HIV Infection and Is Associated with the Recruitment of IRF7 and p85 into Lysosomes and with the Submembrane Displacement of TLR9. J Immunol. (2017) 198:3181–94. doi: 10.4049/jimmunol.1601298
186. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. (2005) 6:1245–52. doi: 10.1038/ni1271
187. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. Corrigendum: CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. (2016) 536:359. doi: 10.1038/nature17421
188. Moar P, Tandon R. Galectin-9 as a biomarker of disease severity. Cell Immunol. (2021) 361:104287. doi: 10.1016/j.cellimm.2021.104287
189. Panda SK, Facchinetti V, Voynova E, Hanabuchi S, Karnell JL, Hanna RN, et al. Galectin-9 inhibits TLR7-mediated autoimmunity in murine lupus models. J Clin Invest. (2018) 128:1873–87. doi: 10.1172/JCI97333
190. Senbanjo LT, Chellaiah MA. CD44: A multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front Cell Dev Biol. (2017) 5:18. doi: 10.3389/fcell.2017.00018
191. Ju X, Zenke M, Hart DN, Clark GJ. CD300a/c regulate type I interferon and TNF-alpha secretion by human plasmacytoid dendritic cells stimulated with TLR7 and TLR9 ligands. Blood. (2008) 112:1184–94. doi: 10.1182/blood-2007-12-127951
192. Borrego F. The CD300 molecules: an emerging family of regulators of the immune system. Blood. (2013) 121:1951–60. doi: 10.1182/blood-2012-09-435057
193. Xu ZJ, Jin Y, Zhang XL, Xia PH, Wen XM, Ma JC, et al. Pan-cancer analysis identifies CD300 molecules as potential immune regulators and promising therapeutic targets in acute myeloid leukemia. Cancer Med. (2023) 12:789–807. doi: 10.1002/cam4.4905
194. Lim SN, Kuhn S, Hyde E, Ronchese F. Combined TLR stimulation with Pam3Cys and Poly I: C enhances Flt3-ligand dendritic cell activation for tumor immunotherapy. J Immunother. (2012) 35:670–9. doi: 10.1097/CJI.0b013e318270e135
195. Kim YH, Gratzinger D, Harrison C, Brody JD, Czerwinski DK, Ai WZ, et al. In situ vaccination against mycosis fungoides by intratumoral injection of a TLR9 agonist combined with radiation: a phase 1/2 study. Blood. (2012) 119:355–63. doi: 10.1182/blood-2011-05-355222
196. Gungor B, Yagci FC, Tincer G, Bayyurt B, Alpdundar E, Yildiz S, et al. CpG ODN nanorings induce IFNα from plasmacytoid dendritic cells and demonstrate potent vaccine adjuvant activity. Sci Transl Med. (2014) 6:235ra61. doi: 10.1126/scitranslmed.3007909
197. Lou Y, Liu C, Kim GJ, Liu YJ, Hwu P, Wang G. Plasmacytoid dendritic cells synergize with myeloid dendritic cells in the induction of antigen-specific antitumor immune responses. J Immunol. (2007) 178:1534–41. doi: 10.4049/jimmunol.178.3.1534
198. Nierkens S, den Brok MH, Garcia Z, Togher S, Wagenaars J, Wassink M, et al. Immune adjuvant efficacy of CpG oligonucleotide in cancer treatment is founded specifically upon TLR9 function in plasmacytoid dendritic cells. Cancer Res. (2011) 71:6428–37. doi: 10.1158/0008-5472.CAN-11-2154
199. Sköld AE, van Beek JJ, Sittig SP, Bakdash G, Tel J, Schreibelt G, et al. Protamine-stabilized RNA as an ex vivo stimulant of primary human dendritic cell subsets. Cancer Immunol Immunother. (2015) 64:1461–73. doi: 10.1007/s00262-015-1746-9
200. Lacarrubba F, Potenza MC, Gurgone S, Micali G. Successful treatment and management of large superficial basal cell carcinomas with topical imiquimod 5% cream: a case series and review. J Dermatolog Treat. (2011) 22:353–8. doi: 10.3109/09546634.2010.548503
201. Teulings HE, Tjin EPM, Willemsen KJ, van der Kleij S, Ter Meulen S, Kemp EH, et al. Anti-Melanoma immunity and local regression of cutaneous metastases in melanoma patients treated with monobenzone and imiquimod; a phase 2 a trial. Oncoimmunology. (2018) 7:e1419113. doi: 10.1080/2162402X.2017.1419113
202. Lemke-Miltner CD, Blackwell SE, Yin C, Krug AE, Morris AJ, Krieg AM, et al. Antibody opsonization of a TLR9 agonist-containing virus-like particle enhances in situ immunization. J Immunol. (2020) 204:1386–94. doi: 10.4049/jimmunol.1900742
203. Ribas A, Medina T, Kirkwood JM, Zakharia Y, Gonzalez R, Davar D, et al. Overcoming PD-1 blockade resistance with CpG-A toll-like receptor 9 agonist vidutolimod in patients with metastatic melanoma. Cancer Discov. (2021) 11:2998–3007. doi: 10.1158/2159-8290.CD-21-0425
204. Sabree SA, Voigt AP, Blackwell SE, Vishwakarma A, Chimenti MS, Salem AK, et al. Direct and indirect immune effects of CMP-001, a virus-like particle containing a TLR9 agonist. J Immunother Cancer. (2021) 9:e002484. doi: 10.1136/jitc-2021-002484
205. Kreiter S, Diken M, Selmi A, Diekmann J, Attig S, Hüsemann Y, et al. FLT3 ligand enhances the cancer therapeutic potency of naked RNA vaccines. Cancer Res. (2011) 71:6132–42. doi: 10.1158/0008-5472.CAN-11-0291
206. Candolfi M, King GD, Yagiz K, Curtin JF, Mineharu Y, Muhammad AK, et al. Plasmacytoid dendritic cells in the tumor microenvironment: immune targets for glioma therapeutics. Neoplasia. (2012) 14:757–70. doi: 10.1593/neo.12794
207. Shackleton M, Davis ID, Hopkins W, Jackson H, Dimopoulos N, Tai T, et al. The impact of imiquimod, a Toll-like receptor-7 ligand (TLR7L), on the immunogenicity of melanoma peptide vaccination with adjuvant Flt3 ligand. Cancer Immun. (2004) 4:9.
208. Lv Q, Zhang J, Yi Y, Huang Y, Wang Y, Zhang W. Proliferating cell nuclear antigen has an association with prognosis and risks factors of cancer patients: a systematic review. Mol Neurobiol. (2016) 53:6209–17. doi: 10.1007/s12035-015-9525-3
209. Pang L, Yeung OWH, Ng KTP, Liu H, Zhu J, Liu J, et al. Postoperative plasmacytoid dendritic cells secrete IFNα to promote recruitment of myeloid-derived suppressor cells and drive hepatocellular carcinoma recurrence. Cancer Res. (2022) 82:4206–18. doi: 10.1158/0008-5472.CAN-22-1199
210. Poropatich K, Dominguez D, Chan WC, Andrade J, Zha Y, Wray B, et al. OX40+ plasmacytoid dendritic cells in the tumor microenvironment promote antitumor immunity. J Clin Invest. (2020) 130:3528–42. doi: 10.1172/JCI131992
211. Kießler M, Plesca I, Sommer U, Wehner R, Wilczkowski F, Müller L, et al. Tumor-infiltrating plasmacytoid dendritic cells are associated with survival in human colon cancer. J Immunother Cancer. (2021) 9:e001813. doi: 10.1136/jitc-2020-001813
212. Constantinidou A, Alifieris C, Trafalis DT. Targeting Programmed Cell Death -1 (PD-1) and Ligand (PD-L1): A new era in cancer active immunotherapy. Pharmacol Ther. (2019) 194:84–106. doi: 10.1016/j.pharmthera.2018.09.008
Keywords: BDCA-2, ILT7, pDC exhaustion, C-type lectin receptor, TLR7, checkpoint inhibitors, interferon alpha
Citation: Tiberio L, Laffranchi M, Zucchi G, Salvi V, Schioppa T, Sozzani S, Del Prete A and Bosisio D (2024) Inhibitory receptors of plasmacytoid dendritic cells as possible targets for checkpoint blockade in cancer. Front. Immunol. 15:1360291. doi: 10.3389/fimmu.2024.1360291
Received: 22 December 2023; Accepted: 22 February 2024;
Published: 05 March 2024.
Edited by:
Jesse Haramati, University of Guadalajara, MexicoReviewed by:
Valeria Lucarini, Sapienza University of Rome, ItalyCopyright © 2024 Tiberio, Laffranchi, Zucchi, Salvi, Schioppa, Sozzani, Del Prete and Bosisio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Bosisio, ZGFuaWVsYS5ib3Npc2lvQHVuaWJzLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.