 Rodney Ogwang1,2
Rodney Ogwang1,2 Lewis Murugu1,3
Lewis Murugu1,3 Irene N. Nkumama4Lydia Nyamako1
Irene N. Nkumama4Lydia Nyamako1 Oscar Kai1Kennedy Mwai1Linda Murungi1
Oscar Kai1Kennedy Mwai1Linda Murungi1 Richard Idro2,5
Richard Idro2,5 Philip Bejon1
Philip Bejon1 James Tuju1,3
James Tuju1,3 Sam Muchina Kinyanjui1†Faith H. A. Osier6*†
Sam Muchina Kinyanjui1†Faith H. A. Osier6*†- 1Centre for Geographic Medicine Research (Coast), Kenya Medical Research Institute-Wellcome Trust Research Programme, Kilifi, Kenya
- 2College of Health Sciences, Makerere University, Kampala, Uganda
- 3Department of Biological Sciences, Pwani University, Kilifi, Kenya
- 4Centre of Infectious Diseases, Heidelberg University Hospital, Heidelberg, Germany
- 5Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom
- 6Department of Life Sciences, Imperial College London, London, United Kingdom
Background: Malaria remains a major global health priority, and monoclonal antibodies (mAbs) are emerging as potential new tools to support efforts to control the disease. Recent data suggest that Fc-dependent mechanisms of immunity are important mediators of protection against the blood stages of the infection, but few studies have investigated this in the context of mAbs. We aimed to isolate mAbs agnostic to cognate antigens that target whole merozoites and simultaneously induce potent neutrophil activity measured by the level of reactive oxygen species (ROS) production using an antibody-dependent respiratory burst (ADRB) assay.
Methods: We used samples from semi-immune adults living in coastal Kenya to isolate mAbs that induce merozoite-specific ADRB activity. We then tested whether modifying the expressed IgG1 isotype to an IgG–IgA Fc region chimera would enhance the level of ADRB activity.
Results: We isolated a panel of nine mAbs with specificity to whole merozoites. mAb J31 induced ADRB activity in a dose-dependent fashion. Compared to IgG1, our modified antibody IgG–IgA bi-isotype induced higher ADRB activity across all concentrations tested. Further, we observed a negative hook effect at high IgG1 mAb concentrations (i.e., >200 µg/mL), but this was reversed by Fc modification. We identified MSP3.5 as the potential cognate target of mAb J31.
Conclusions: We demonstrate an approach to engineer mAbs with enhanced ADRB potency against blood-stage parasites.
Introduction
Malaria remains a major public health priority. This is particularly true in Sub-Saharan Africa (SSA) where the burden of morbidity and mortality is the highest (1). Naturally acquired immunity (NAI) to Plasmodium falciparum is known to develop gradually after multiple malaria episodes (2). In areas with a high intensity of malaria transmission, epidemiological data show that the burden of severe and life-threatening malaria is the highest in early childhood, while uncomplicated malaria dominates in older children and young adults. These individuals remain susceptible to infection and are frequently found to have asymptomatic parasitemia. The acquisition of partial control of parasite growth following multiple episodes of malaria is considered semi-immunity (3, 4). Classical passive transfer studies demonstrated that purified immunoglobulins from such adults living in malaria-endemic areas can result in the reduction of parasitemia and resolution of clinical symptoms in non-immune patients (5). Although this evidences the existence of NAI, neither the specificity of the antibodies nor their mechanisms of action have been fully characterized or understood.
Monoclonal antibodies (mAbs) are emerging as potential and exciting new tools to support efforts to control malaria (6–9). Initial concerns about their widespread use in resource-constrained environments in SSA are increasingly being countered by emerging advances in technology that are leading to reduced costs (6), longer serum half-lives, and increased potency (6). Indeed, recent studies have shown that mAbs such as CIS43LS (8, 10) and L9LS (11) that target different epitopes of the P. falciparum circumsporozoite protein (PfCSP) show up to 80% protection in controlled human infection studies in naïve adults (8, 10, 11). Moreover, CIS43LS protected adults over a 6-month period spanning an entire malaria transmission season in Mali (8). However, all mAbs currently undergoing field evaluation target sporozoites, while those against merozoites in the blood stage of the parasite life cycle have not progressed to clinical development.
Blood-stage parasites are responsible for the clinical manifestations of malaria. Thus, the development of mAbs against merozoites may be useful for the treatment and prevention of malaria in infants, children, pregnant women, or naïve travelers visiting areas with ongoing malaria transmission. To date, mAbs targeting blood-stage parasites have shown low potency in vitro and thus require high concentrations of antibodies (12–14). For example, an approximate serum concentration of 100 mg/kg of reticulocyte-binding protein homolog 5 (PfRH-5)-specific antibody was thought to be necessary for protection from clinical episodes of malaria (15, 16). We hypothesized that this may be because mAbs targeting this life cycle stage were designed with a focus on antibody Fab-mediated neutralization as captured in the growth inhibition assay (GIA) and not on Fc-mediated effector functions that are increasingly understood to be important for immunity (17–22). The GIA measures the capability of antibodies in the absence of immune cells or complement to inhibit parasite replication in vitro (23–26). It is widely used as the reference assay for measuring the inhibitory activity of antibodies against blood-stage parasites. However, the correlation between GIA and vaccine efficacy has been relatively inconsistent (27–29). In addition, in studies investigating naturally acquired immunity using the human challenge model, GIA was not correlated with outcome (20). Therefore, a focus on mAbs that induce Fc-mediated function may be a powerful approach to improve their potency.
Neutrophils are the most abundant immune cell type in blood and are effective antimicrobial cells that play a vital role within the innate arm of the immune system (30). Indeed, it has been shown that individuals with lower neutrophil blood counts are predisposed to overwhelming infections (31, 32). In malaria, neutrophils may mediate protection via parasite clearance through phagocytosis (33, 34), the production of reactive oxygen species (ROS) (22, 35, 36), or the release of neutrophil extracellular traps (NETs) (34). High levels of neutrophil activity measured using an antibody-dependent respiratory burst (ADRB) assay have been associated with protection against malaria in multiple studies in different settings (33, 34). Here, we aimed to isolate mAbs that targeted merozoites and simultaneously induced antibody-dependent neutrophil function. We then tested whether this ADRB activity would be enhanced through an IgG–IgA bi-isotype approach that in theory would result in the engagement of a broader panel of Fc receptors (37–39). We first screened adults from malaria-endemic areas in coastal Kenya to identify individuals with total IgG antibodies that induced high ADRB levels against P. falciparum merozoites in vitro. We then used B cells from these individuals to isolate merozoite-specific mAbs with demonstrable ADRB activity. Thereafter, we applied a genetic engineering strategy to develop an IgG–IgA bi-isotype antibody and tested whether it enhanced the original ADRB activity.
Results
Prioritization of samples for the isolation of functional monoclonal antibodies
We began by testing all available serum samples (n = 217) for antibody binding to whole merozoites and the level of ADRB as previously described (19, 21). We then selected samples that had high levels of merozoite-specific antibodies and that induced high levels of ADRB (33, 34). Adults from Kilifi South (an area of moderate-to-high malaria transmission (40)) had significantly higher total merozoite antibodies than those from Kilifi North (an area of low transmission intensity (41)) (p < 0.0001) (Figure 1A). This was equally true for antibodies that induced ADRB (Figure 1B). There was a strong positive correlation between the level of total merozoite IgG and the level of ADRB targeting merozoites (r = 0.65, p < 0.0001) (Supplementary Figure 1). To down-select individuals for mAb isolation, we defined a threshold for high levels of ADRB using our positive control of pooled hyperimmune serum (PHIS). We classified samples with ADRB levels above the 25th percentile of that induced by PHIS as having high ADRB. Ten of 217 (4.6%) samples met these criteria, and we prioritized them for mAb isolation. All 10 samples were from Kilifi South.
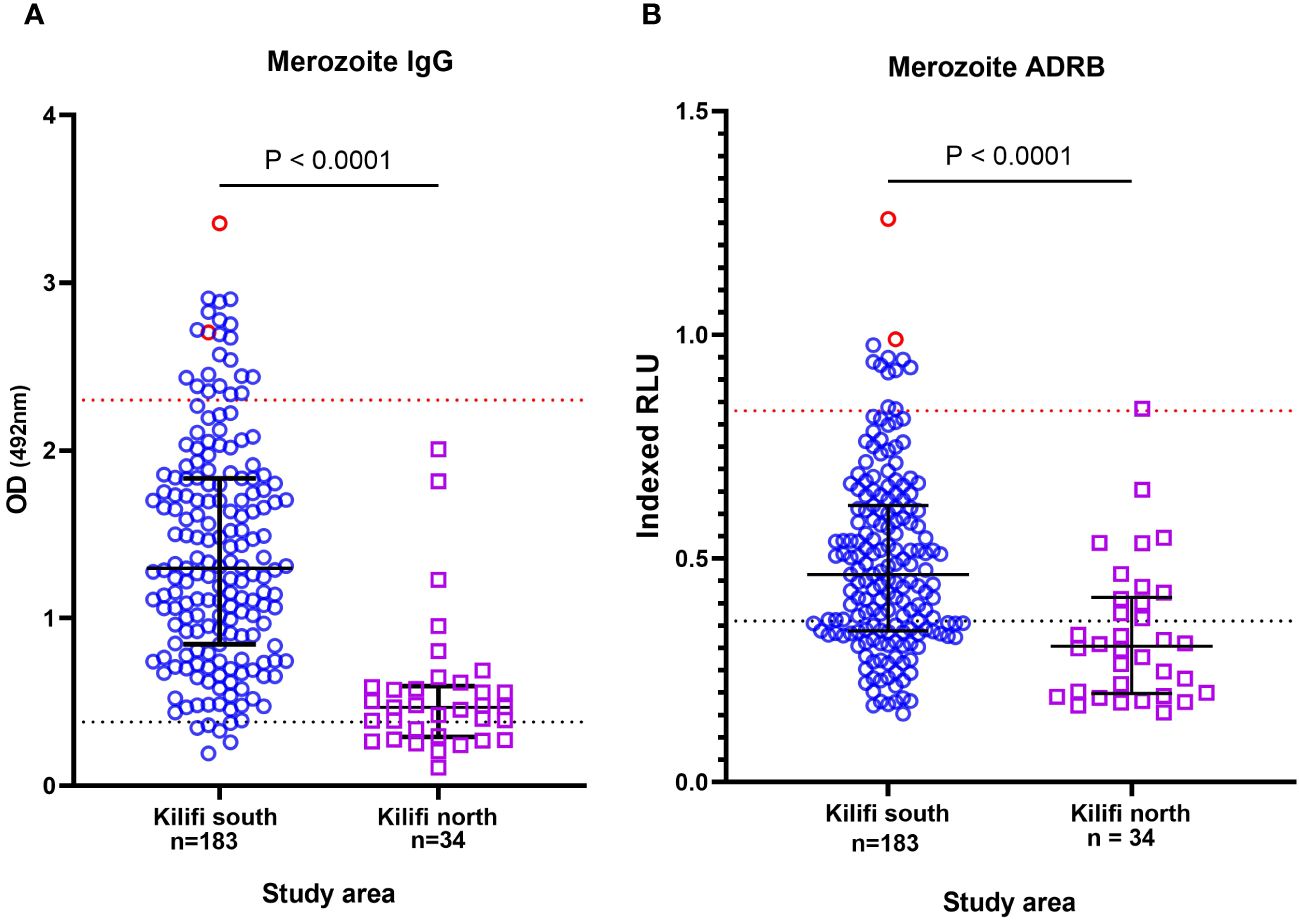
Figure 1 Prioritization of individuals with high levels of functional antibodies. Plot showing the level of (A) total anti-merozoite IgG and (B) antibody-dependent respiratory burst (ADRB) function. Each point represents an individual from Kilifi South in blue and Kilifi North in purple. The points in red show the individuals selected for monoclonal antibody (mAb) isolation. The blue dotted line represents 3 SD above the mean of negative control (serum from malaria-naïve individual, n = 10). The red line shows the 25th percentile of positive controls (pooled hyperimmune serum).
Antigen-agnostic isolation of merozoite-specific mAbs
Next, to generate a panel of merozoite-specific mAbs, an antigen-agnostic, negative selection strategy was employed to isolate memory B cells (CD19+/IgG+) from cryopreserved peripheral blood mononuclear cells (PBMCs) from two individuals with the highest ADRB activity (Figure 1B; Supplementary Figure 2). Memory B cells were sorted (4 cells/well) into 384-well plates that contained culture media supplemented with CD40L, IL-2, and IL-21 to enable proliferation and differentiation into antibody-producing cells (42). B-cell differentiation and proliferation were determined by total IgG ELISA of the culture supernatants after 12–13 days of culture. To select for merozoite-specific mAbs, supernatants from individual wells were screened using ELISA to whole merozoites of the laboratory-adapted P. falciparum 3D7 strain. From one individual, 54 wells of 760 had detectable levels of merozoite-specific IgG within culture supernatant, and of these, 17 wells that had the highest levels of anti-merozoite IgG were prioritized (Supplementary Figure 3). From the second individual, of the 464 wells with sorted and cultured cells, 14 wells showed low-level merozoite-specific IgG. Next, mRNA was extracted from B cells in the 31 selected wells expressing merozoite-specific IgG and cDNA synthesized. Thereafter, a published nested PCR protocol (43) was used to amplify the IgG variable region corresponding to heavy and light chain genes from the cDNA of the selected wells (Supplementary Figure 4A) (43). These were subsequently cloned upstream of the human IgG1 expression vectors and co-transfected into Expi293F cells. A total of 35 human mAbs were successfully expressed as shown by dot blots detecting human IgG following expression (Supplementary Figure 4B).
We then tested mAb binding to whole merozoites and observed that 9/35 (25.7%) bound to merozoites, which are shown in Supplementary Figure 4C. These nine mAbs showed variable levels of expression (Figure 2A), reactivity to whole merozoites (Figure 2B), and ADRB activity (Figure 2C). We selected mAb J31 for further characterization because it induced the highest relative ADRB activity with expression yields at approximately 193 µg/mL of culture. For mAb J31, ADRB activity increased in a dose-dependent pattern with an increase in concentration, although we observed a hook effect (a phenomenon where the effectiveness of antibody is decreased when antibody or antigen is in excess) at antibody concentrations above ~200 µg/mL (Figure 2D).
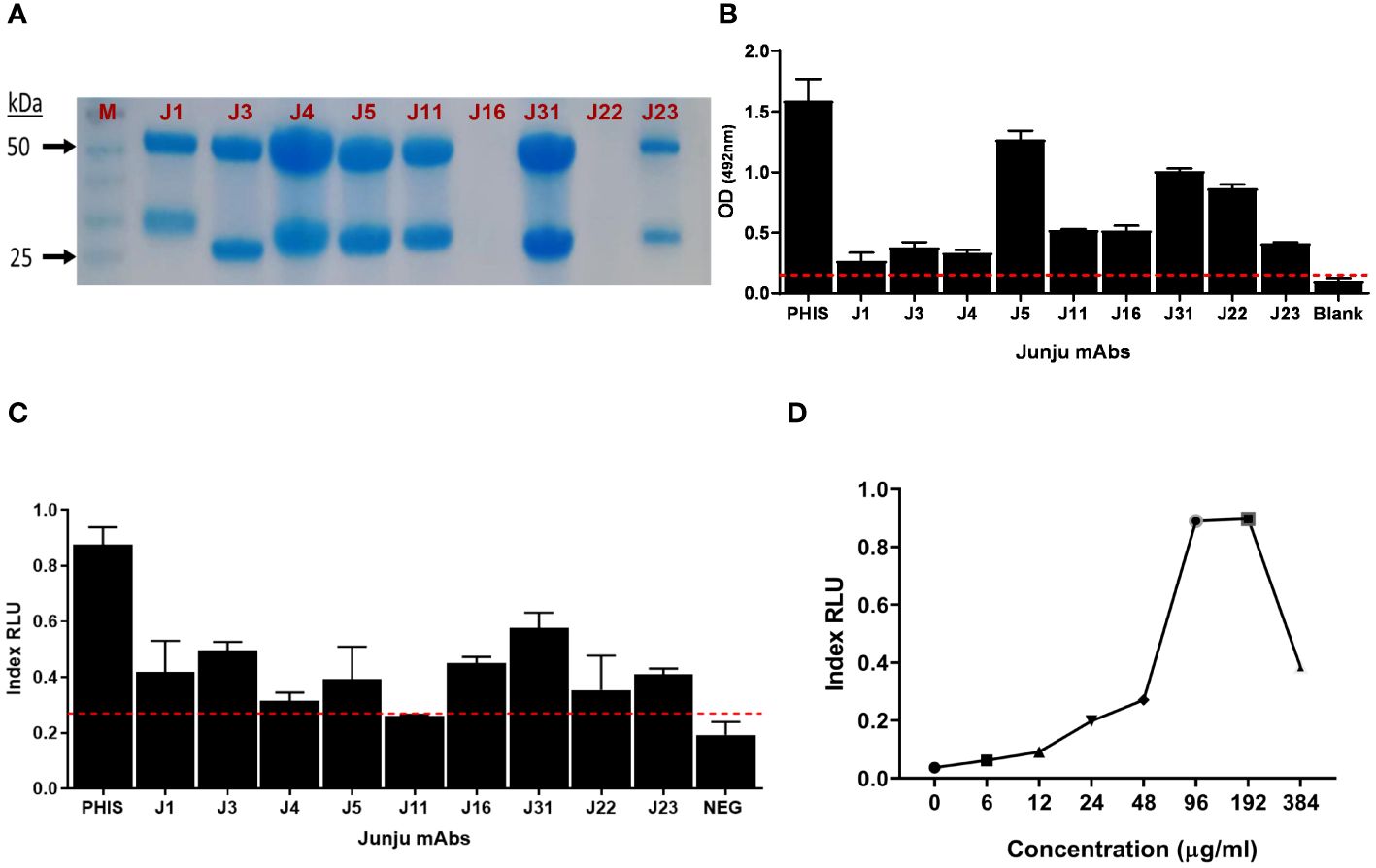
Figure 2 Isolation of merozoite-specific monoclonal antibodies (mAbs) with antibody-dependent respiratory burst (ADRB) activity. (A) Sodium dodecyl sulfate (SDS) gel showing nine selected mAbs. (B) The level of mAb reactivity to whole merozoites by ELISA. (C) The level of ADRB activity for the different mAbs. Error bars represent the interquartile range of two replicates tested at 100 µg/mL. The red line shows seropositivity cut-off of 3 SD above the mean of three negative control replicates. (D) Dose–response curve of the level of ADRB activity for mAb Junju 31. Each point represents a single replicate for antibody at each concentration.
The cognate target of mAb J31 is MSP3.5
Next, to identify the cognate target of mAb J31, we took advantage of our in-house protein microarray (KILchip 1.0), which consists of 111 correctly folded merozoite stage antigens prioritized as potential targets of naturally acquired immunity (44). We optimized the chip assay for mAb binding and determined that using a concentration < 3 µg/mL of mAb was the most appropriate. We applied a published analytical approach to investigate the specificity of J31. This involved creating an A score that represented the number of standard deviations above the background mean fluorescence intensity (MFI) of all antigens on the chip. An A score above 2.8 was considered a significant antibody–antigen interaction (45). We ranked all the A scores from the highest to lowest and found that J31 recognized MSP3.5 with the highest A score (Figure 3A). To confirm this, we then evaluated binding to the six antigens with the highest A scores from the microarray analysis by a more stringent ELISA. These were merozoite thrombospondin-related anonymous protein (MTRAP), merozoite surface protein-6 (MSP-6), and erythrocyte binding antigen-175 (EBA-175), erythrocyte binding antigen-140 (EBA-140), and secreted protein with altered thrombospondin repeat (SPATR). MSP3.5 remained the target antigen with the highest OD value (Figures 3B, C). Additional analysis using more specific assays, such as surface plasmon resonance (SPR), and with lower concentrations of the mAb J31 and antigen is needed to confirm the specificity of the interaction between mAb J31 and MSP3.5 and not other antigens.
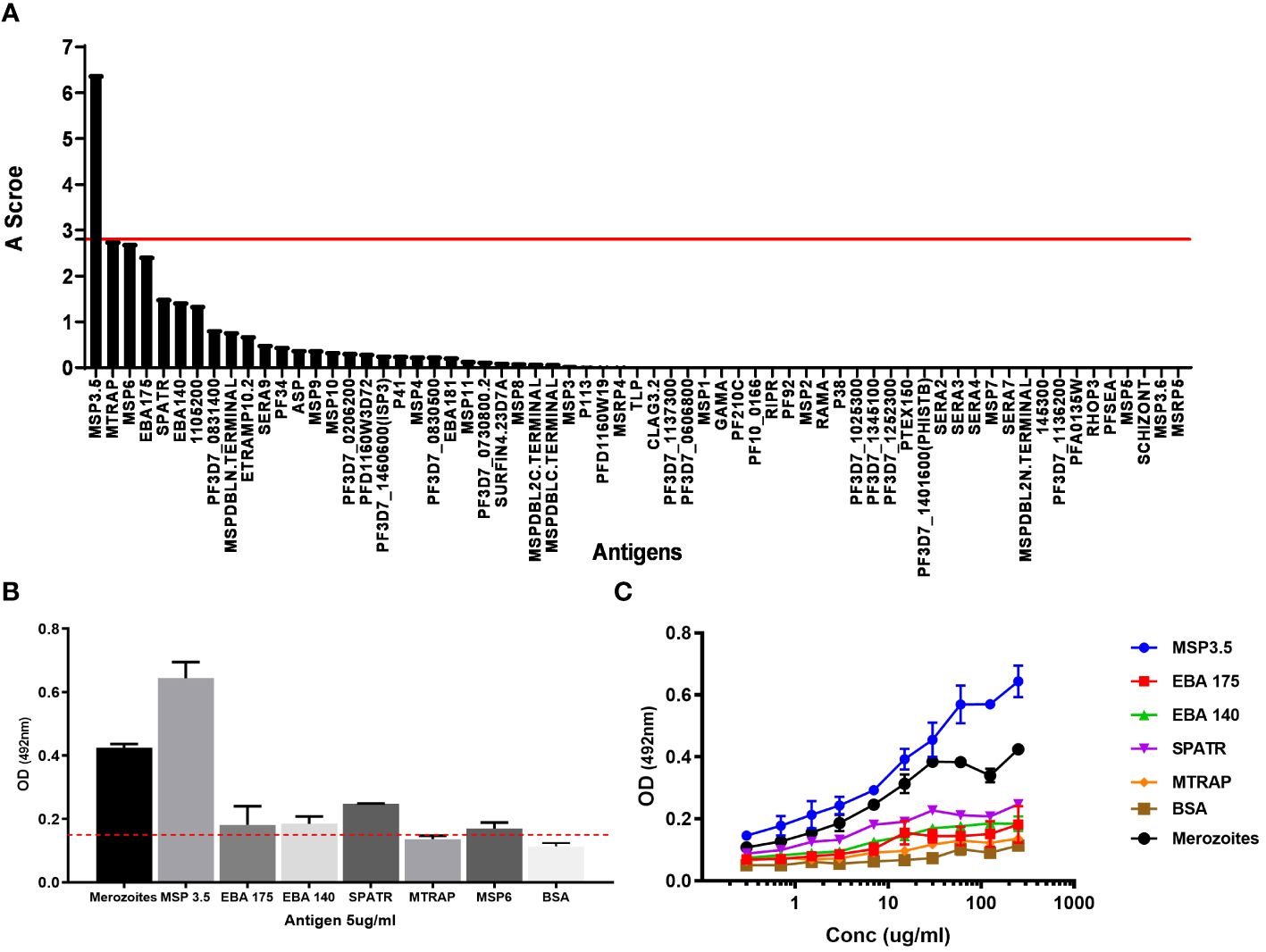
Figure 3 Cognate antigen identification for J31. (A) The affinity score of antigens recognized by the monoclonal antibody (mAb) J31 using KILchip microarray. The red line shows the positivity cut-off score (A = 2.8) above which significant binding is considered. (B) ELISA confirmation of cognate antigen. For ELISA, antigens were coated at 5 μg/mL, and mAb J31 was tested at 100 μg/mL. The red line shows reactivity cut-off of 3 SD above the mean of bovine serum albumin (BSA). (C) Serial dilution of mAb J31 to different antigens. MSP6 not included due to antigen availability. In both (B, C), whole merozoite extract was prepared using laboratory-adapted Plasmodium falciparum 3D7 stain included as positive control.
IgG–IgA “bi-isotype” enhanced mAb ADRB activity
Next, whether an Fc region cross isotype (IgG–IgA) consisting of IgG and IgA sequences would increase the ADRB activity of J31 was tested (Figure 4A). Codon-optimized sequences published by Kelton and colleagues (46) were adopted, our bi-isotype Fc portion was designed, and the construct was ordered from GeneArt. The IgG-IgA Fc region construct was cloned into the heavy chain plasmid to replace the entire IgG1 Fc portion of J31 using restriction sites NheI and DraIII. To confirm successful cloning, a restriction digest with enzyme PstI was conducted and showed three bands for the IgG–IgA heavy chain compared to two bands for the standard IgG1 (Supplementary Figure 5). The two mAb variants were then expressed using our standard antibody co-transfection protocol in expi293F cells.
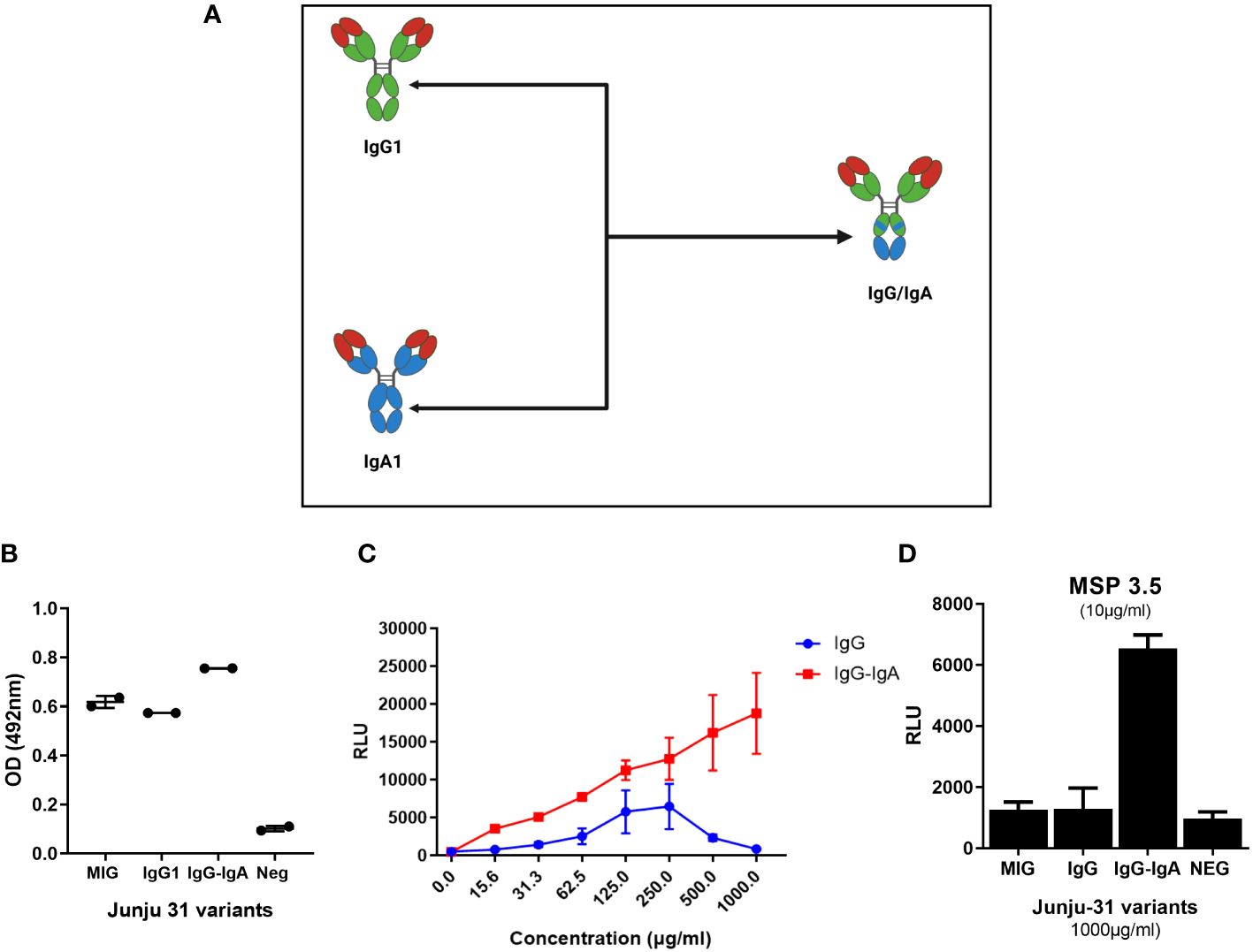
Figure 4 The IgG–IgA “bi-isotype” monoclonal antibody (mAb) enhanced antibody-dependent respiratory burst (ADRB) activity. (A) A schematic representation of IgG–IgA showing regions of the Fc carrying IgG1(green) and IgA (blue). (B) ELISA showing binding of antibody variants to MSP3.5 coated at 5 μg/mL. (C) The level of ADRB activity against whole merozoites of the different variants (red, J31-IgG–IgA; blue, J31-IgG1). (D) The level of antigen-specific ADRB activity of the different mAb variants. MSP3.5 was coated at 10 µg/mL, while antibodies were tested at 1 mg/μL.
To evaluate binding properties, we showed by ELISA that the engineered IgG–IgA cross isotype recognized MSP3.5 at a comparable level to native IgG1 J31 (Figure 4B). Thereafter, we compared the level of ADRB activity between the original and cross-isotype mAb against whole merozoites. The J31 IgG–IgA variant showed a twofold enhanced ADRB activity compared to IgG1. This was consistent across different antibody concentrations, and the level of activity increased in a dose-dependent fashion. Of interest, the hook effect observed with the IgG1 variant at higher antibody doses (Figures 2D, 4C, blue line) was not seen with the IgG–IgA variant (Figure 4C, red line). Next, we developed an antigen-specific ADRB assay using MSP3.5. In a similar fashion, IgG–IgA had higher levels of ADRB activity (Figure 4D).
Discussion
Passive transfer of mAbs is emerging as a potential tool that may support the malaria control effort (6, 7). Two mAbs, CIS43LS and L9LS, that target the circumsporozoite protein have shown greater than 80% protection in phase 1 controlled human infection studies (10, 11). Notably, among Malian adults living in an area with intense but highly seasonal malaria transmission, CIS43LS showed a ≤75% reduction in clinical episodes of malaria over 6 months at an intravenous (IV) dose of 10 mg/kg (8). All mAbs that have progressed to clinical evaluation target the sporozoite stages of the parasite life cycle, while mAbs targeting asexual stage merozoites that are responsible for the clinical manifestations of malaria have lagged. Here, we aimed to generate merozoite-specific mAbs with potent Fc-mediated functional activity, as this has been shown to be strongly correlated with protection in recent studies. Using an antigen-agnostic approach, we isolated a panel of functional mAbs targeting whole merozoites. Thereafter, as a proof of principle for Plasmodium-specific mAbs, we showed that a bioengineered IgG–IgA cross-isotype mAb can enhance the ADRB activity against merozoites twofold. Similar approaches may now be generalizable to improve the functional potency of future blood-stage mAbs.
Using a similar cross-isotype approach, antibody trastuzumab that targets the Her2 antigen expressed by Her2+ cancer cells was engineered and showed higher Fc-mediated function (46). The mechanism by which the IgG–IgA cross isotype may induce higher levels of ADRB is not completely understood. Our favored hypothesis is that the cross IgG–IgA isotype enhances activation of the immunoreceptor tyrosine-based activation motif (ITAM) in a similar fashion to IgA (39, 47–49). It is thought that since the stoichiometry of the IgA : FcαRI interaction is in a 1:2 ratio, a single cross-isotype mAb would result in the activation of four ITAM molecules, while an IgG : FcγRIIa interaction only engages a single ITAM (47–49). Another possibility is that the IgA : FcαRI interaction is qualitatively superior to IgG : FcγRIIa, resulting in higher activation (48). Important to note is the growing body of evidence that suggests a role for functional IgA antibodies in protection against malaria (50, 51). A post-hoc systems serology analysis within a controlled human infection trial evaluating reticulocyte-binding protein homolog 5 (RH5) showed that the magnitude of anti-RH5 IgA in plasma is associated with a delayed time to malaria diagnosis (50). Importantly, the authors propose that the IgA antibodies may be acting through a neutrophil activation pathway (50). Another independent study demonstrated a protective role for circulating IgA antibodies against pre-erythrocytic parasites (51). These strengthen the possible use case for a potent bi-isotype antibody. The clinical utility of the IgG–IgA cross isotype may be hindered due to the poor serum half-life suggested by low FcγRIIIa/CD16a and FcRn interaction observed with the IgG–IgA–trastuzumab cross isotype (46). However, other chimeric antibody strategies have been developed that may overcome this and potentially allow FcRn interaction. Examples include an anti-CD20 and FcαRI/CD89 IgG bi-specific antibody (37) and chimeras that express either Fcα-Fcγ or Fcγ-Fcα in tandem (38). In addition, despite the fact that mAbs are typically well tolerated, it is important to mention that bi-isotypes may be immunogenic in vivo leading to anti-drug antibody (ADA) responses that may lower their efficacy (52). Additional complications include low stability and the potential for adverse reactions such as acute anaphylaxis and serum sickness (53).
Our analysis suggests that MSP3.5 is the cognate target of mAb J31. Additional confirmatory experiments using SPR, immuno-precipitations (54), mass spectrometry (44), and structural studies were considered but not feasible in the prevailing circumstances. This is a major limitation to our antigen identification, which depends on antibody/antigen binding as determined using ELISA. In partial mitigation, we screened our antibody against >100 immunogenic merozoite surface antigens using an established protein microarray platform (44). Importantly, the conclusions made regarding engineering a more functionally active mAb would be supportable irrespective of antigen. MSP3.5 is part of a protein family consisting of eight proteins of which MSP3 (55–59), MSP6 (60, 61), H101, and H103 (62) have been investigated in sero-epidemiological studies and linked with protection against clinical episodes of malaria (63). Notably, two vaccine formulations based on MSP3 have been tested: a long synthetic peptide derived from a conserved region of MSP3 (MSP3-LSP) (64, 65) and a protein fusion consisting of two antigens, GLURP and MSP3 (GMZ2) (66, 67). Although both reported good safety data, they showed poor protective efficacy in phase 1 clinical trials. Members of the family retain a unique motif (a small stretch of amino acids NLRNA/NLRNG) at the N-terminus. However, unlike other members, MSP3.5 does not show high levels of sequence relatedness (≈32% identity and ≈54% similarity of amino acid residues) at the C-terminus (63). MSP3.5 has not yet been studied in the context of naturally acquired immunity or vaccine studies.
Only a limited number of mAbs targeting merozoite antigens have been isolated; however, none have progressed to clinical testing. The level of in vitro functions has been investigated primarily using the growth inhibition assay. For example, isolated mAbs targeting the highly conserved RH5 (68) and those targeting the merozoite surface protein 1 complex (MSP1) (69, 70) show >90% growth inhibition but at high concentrations of approximately 1 mg/mL. In vivo studies suggest that a dose of 100 mg/kg or higher may be required for clinical protection (15). However, these were modeled using the growth inhibition assay, which may not be the best correlate for protection (16). The development of mAbs to blood-stage antigens faces some challenges like antigenic variation (71, 72) and parasite load, in addition to a very short merozoite extracellular phase (12, 73). These challenges will require careful mAb design and optimization to achieve potent mAbs with clinical relevance. Importantly, our antigen-agnostic approach may increase the likelihood of identifying new antibodies.
Passive transfer studies clearly demonstrated the feasibility of using pooled polyclonal antibodies as a therapy (5, 74). This, therefore, allows optimism that with continued research, the development of potent antibody biologics for malaria treatment is achievable. Importantly, the potential clinical use of mAbs within Africa is becoming plausible with declining costs, improving technology, and growing infrastructure (75, 76). Potential use cases for blood stage-specific mAbs include chemoprophylaxis in regions with highly seasonal malaria transmission (8) and for naïve travelers visiting malaria-endemic regions (77). Additionally, mAbs may be useful for chemoprevention in special populations such as children with sickle cell anemia (78), children recovering from severe malaria who are at increased risk of repeat infection and poor outcomes (79), and pregnant women (80). We did not test the in vivo function of our antibodies in murine or non-human primates; therefore, the clinical utility of this mAb requires further investigation. In addition, to strengthen the use case of the cross IgG–IgA bi-isotype, a comparison with IgA variants would be relevant.
Conclusion
We show that an IgG–IgA cross-type mAb enhances ADRB activity against whole Plasmodium merozoites. Similar cross-isotype approaches may be used to enhance the in vitro activity of other anti-Plasmodium antibodies. Future studies should investigate the in vivo utility of this approach describing their efficacy and pharmacokinetic properties. The role of MSP3.5 in naturally acquired immunity should be explored in more detail. Antigen-agnostic approaches may enable the identification of novel vaccine targets.
Methods
Blood samples were collected from adults participating in studies of naturally acquired immunity to malaria conducted by the KEMRI-Wellcome Trust Research Programme. Samples from which both plasma and PBMCs (n = 217) were available were randomly selected from cross-sectional surveys conducted in 2015, 2017, and 2018. Study participants lived in either Kilifi North or South. These are sublocations within Kilifi County with differing levels of malaria transmission intensity. Kilifi North reports low-level transmission (41), while Kilifi South records moderate-to-high malaria transmission (40).
Ethical considerations
The samples from coastal Kenya were obtained under ethical approvals granted by the KEMRI Scientific and Ethics Review Unit, approval reference number No. 3149. All participants provided written informed consent. All data were anonymized and stored in secure KEMRI servers in Kilifi, Kenya, and managed by the Kilifi Data Governance Committee.
Merozoite ELISA
P. falciparum merozoites of the 3D7 strains were isolated as described previously (19). Parasites were thawed and cultured to obtain trophozoites at high parasitemia (8%–12%). The trophozoites were isolated by magnetic purification and cultured in a fresh complete medium to allow development to the early schizont stage. A protease inhibitor, trans-epoxysuccinyl-l-leucylamido(4-guanidino)butane (E64; Sigma-Aldrich, St. Louis, MO, USA), was added to allow maturation of the schizonts without rupture (81). Merozoites were harvested by filtration of the mature schizonts through 1.2-µm filters (Pall, Port Washington, NY, USA). The merozoites were stained using either 1 µg/mL ethidium bromide (EtBr, Thermo Fisher Scientific, Waltham, MA, USA) or 1× SYBR green dye (Thermo Fisher) for 30 min and counted against CountBright™ Absolute Counting Beads (Thermo Fisher) using BD FACS Canto II flow cytometer.
Next, 96-well plates (Thermo Fisher) were coated with 100 µL/well of 3D7 merozoites at 5 × 106 merozoites/mL at 4°C overnight. The plates were washed and blocked with 200 µL/well of blocking buffer, 1% casein (Thermo Fisher), for 2 hours at 37°C. Samples were diluted at 1:500 with blocking buffer, added at 200 µL/well, and incubated for 1 hour at 37°C. Plates were washed before 100 µL/well of respective secondary antibodies [horseradish peroxidase (HRP)-conjugated: rabbit anti-human IgG (Dako, Glostrup, Denmark)] was added and incubated for 1 hour at 37°C. The plates were then washed four times, and 100 µL/well of the substrate [o-phenylenediamine dihydrochloride (OPD); Sigma-Aldrich] was added and incubated for 20 min in the dark at room temperature (RT). The reaction was stopped with 30 µL of 1 M hydrochloric acid (HCl; Sigma-Aldrich), and the absorbance was measured at 492 nM using a BioTek Cytation 3 cell imaging multi-mode reader.
Antibody-dependent respiratory burst assay
We conducted the ADRB assay as previously described (33, 34) with minor modifications. The assay involves three major steps: i) parasite preparation, ii) neutrophil isolation, and iii) a chemiluminescence detection method.
Parasite preparation
P. falciparum trophozoites of the 3D7 strain were isolated by magnetic purification and cultured in a fresh complete medium to allow development to the early schizont stage. A protease inhibitor E64 (Sigma-Aldrich) was added to allow the maturation of the trophozoites for 8–12 hours while preventing schizont rupture (81). The schizonts were then quantified and stored at −80°C until use.
At the time of the assay, schizonts (10 × 105 schizonts/mL) or purified antigen (10 µg/mL) in phosphate-buffered saline (PBS) were coated into white opaque 96-well plates (Greiner, Frickenhausen, Germany) at 100 µL/well and incubated overnight at room temperature. The plates were then washed and blocked with 200 μL/well casein in PBS for 1 hour at room temperature. The plates were washed before the addition of 50 μL of diluted plasma (1:50 in 1× PBS) to each well, followed by a 1-hour incubation at 37°C.
Neutrophil isolation
Polymorphonuclear leucocytes (PMNs) were prepared from freshly collected whole blood for each assay using a standard procedure. Whole blood at a volume of 40 mL from healthy donors was mixed at a ratio of 1:1 with Hank’s Balanced Salt Solution (HBSS; Thermo Fisher). The diluted blood was carefully layered on 7.5 mL aliquots of Ficoll (GE Healthcare, Chicago, IL, USA). This was then centrifuged at 600 × g for 15 min. The supernatant was carefully removed without disturbing the red cell pellet. The pellet was resuspended in 5 mL of HBSS, then mixed with 3% dextran at a ratio of 1:2, and incubated at room temperature in the dark for 30 min. The supernatant was then carefully collected and centrifuged at 500 × g for 7 min at 4°C. The supernatant was discarded, and red cell contaminants were lysed. The cells were then centrifuged at 500 × g for 7 min at 4°C, and the pellet was resuspended in 1 mL of ice-cold PMN buffer [HBSS with 0.1% bovine serum albumin (BSA; Sigma-Aldrich), 1% d-glucose]. The PMN count was determined using a hemocytometer, and the concentration was adjusted using the PMN buffer to 1.0 × 107/mL. The cells were kept on ice.
Chemiluminescence detection
The schizont-coated plates were washed before the addition of 50 μL of PMNs (1 × 107 PMNs/mL) and 50 μL of 0.04 mg/mL isoluminol (Santa Cruz Biotechnology, Dallas, TX, USA) per well. The plates were immediately loaded onto a plate reader, and ADRB activity was quantified via chemiluminescence at 450 nM every 2 min for 1.5 hours, captured as the maximal relative light units (RLUs). The positive controls were PHIS and MIG (Purified human IgG from Malawian adults), while the negative controls were malaria-naïve sera from German donors as described under merozoite ELISAs above. To account for the variability of ADRB activity from PMNs from different individuals, each sample was tested in duplicate using PMNs from two independent donors (22, 36). To further minimize variability between donors, the RLU values were indexed based on the PHIS-positive controls in each plate (22, 36). Thus, the mean RLU for the PHIS control was used as the reference sample with an indexed RLU of 1.0. The indexed RLU value for each test sample was calculated as RLU of sample/RLU of PHIS. The mean indexed RLU values using PMNs from two donors were then calculated.
Memory B-cell isolation
PBMC samples were thawed and transferred into pre-warmed (37°C) media [Roswell Park Memorial Institute (RPMI) + 50% fetal bovine serum (FBS)]. The tube was then centrifuged at 400 × g for 5 min, the supernatant was discarded, and cells were resuspended in cold sterile FACS buffer [3 mL PBS, 2% fetal calf serum (FCS), 2 mM EDTA, and 25 mM HEPES]. Thawed cells were stained for memory B cells with primary fluorophore-conjugated antibodies to human CD3, CD8, CD14, CD19, CD20, CD27, IgG, and IgM (BD Pharmingen, San Diego, CA, USA) in equimolar ratios. Staining was performed for 30 min at 4°C in PBS with 1 mM EDTA and 1% FBS. Cells were selected for the phenotype (CD3−/CD8−/CD14−) while (CD19+/CD20+). Memory B cells were sorted four cells per well into 384-well plates containing feeder medium [complete Iscove’s Modified Dulbecco’s medium (IMDM), IL-2 (10,000 U/mL), IL-21 (100 μg/mL], and mitomycin C-treated feeder cell (3T3-msCD40L)] and cultured for 13–14 days (42).
Immunoglobulin gene cloning and generation of recombinant antibodies
From wells with Plasmodium-specific B cells of interest, RNA was extracted using TRIzol™ reagent (Thermo Fisher Scientific) following the manufacturer’s instructions with slight modifications. B cells from the select wells were transferred to a separate tube homogenized with TRIzol reagent (1,100 µL), followed by the addition of chloroform (200 µL, Sigma-Aldrich). The homogenate was then centrifuged at 1,400 × g for 35 min at 4°C. GlycoBlue 2 µL (Ambion, Austin, TX, USA) was added to a clean RNase-free tube followed by the homogenate aqueous phase and isopropanol (500 µL, Sigma-Aldrich), which were mixed and incubated overnight at 4°C. The next day, samples were centrifuged at 13,000 rpm for 30 min at room temperature, supernatants were discarded, and the pellet was washed gently with 500 µL of 75% ethanol. Thereafter, all ethanol was removed, and the pellet was air-dried and resuspended in RNAsecure (20 µL, Ambion) by heating for 10 min at 60°C. The RNA sample was mixed gently and stored at −80°C.
Gene cloning and recombinant antibody production were performed as previously described (43). Briefly, cDNA was generated from RNA samples using random hexamer primers (SuperScript III First-Strand Synthesis, Invitrogen, Carlsbad, CA, USA). Immunoglobulin (Ig) heavy and corresponding kappa or lambda L-chain gene transcripts were then amplified via semi-nested PCR using primers previously published (43). Amplified IgG variable regions were cloned into human Igh1 [AbVec-hIgKappa (FJ475056)] and Igk [AbVec-hIgG1 (FJ475055)] or Igl [PBR322 based IG-lambda expression vector (FJ517647)] expression vectors. This was conducted using high-fidelity restriction enzymes (New England Biolabs, Ipswich, MA, USA) in CutSmart buffer (AgeI HF, SalI HF for the heavy chain, AgeI-HF, BsiWI-HF for the kappa chain, and AgeI HF, XhoI-HF for the lambda chain). Expression plasmids were transformed into competent cells (One Shot™ Top10 Escherichia coli) by heat shock method (82) and plated on Luria–Bertani agar plates containing ampicillin (100 µg/mL). Single colonies were selected and expanded in Luria–Bertani broth containing ampicillin (100 µg/mL) overnight in a shaking incubator (270 rpm, 37°C). In instances where both kappa and lambda light chains were amplified from a single culture well, both variants were cloned and expressed as independent mAbs.
Antibody expression and purification
The overnight bacterial culture was harvested by centrifugation (6,000 × g, 4°C) for 15 min. Expression plasmid DNA was isolated by maxiprep purification following the manufacturer’s instructions (QIAGEN, Valencia, CA, USA) and thereafter used for antibody expression in HEK293F cells using the Expi293 Expression System (Invitrogen) according to the manufacturer’s instructions. Briefly, cells were cultured to a concentration of 3.0 × 106 cells/mL and co-transfected with the appropriate pair of antibody expression vectors using the ExpiFectamine 293 transfection reagent (Invitrogen). Cells were then incubated at 37°C with 8% CO2 in an orbital shaker at 125 rpm, and culture supernatants were harvested 6 days post-transfection (44). Antibodies were then purified using protein G resin (Amintra Protein G Resin, Expedeon, Cambridge, UK) following the manufacturer’s instructions. Briefly, clarified expression media were incubated together with G protein resin overnight at 4°C under slow rotation. The resin was then passed through spin columns (Bio-Rad, Hercules, CA, USA) pre-equilibrated with 1× PBS. The resin was washed twice, and the bound antibody was eluted with 0.1 M glycine, pH 3, and neutralized immediately with 1 M Tris, pH 8.
Maintaining and treatment of feeder cells 3T3-msCD40L
Frozen feeder cells (3T3-msCD40L) were thawed rapidly in a water bath at 37°C. The 3T3-msCD40L cells were added into the culture medium [IMDM with GlutaMAX (Gibco, Grand Island, NY, USA) made to 5% FBS and 1% penicillin-streptomycin (Gibco)]. Cells were seeded in a T-75 flask at 1.0 × 10.05 cells/mL. Cultures were maintained in an incubator at 37°C, with 5% CO2 for 3–4 days until confluency. At confluency, the culture supernatant was removed by settling. The attached cells were washed two times with 10 mL PBS-GLUCOSE (Sigma-Aldrich) and warmed to 37°C. Trypsin-EDTA (0.05% Trypsin and 0.53 mM EDTA-4Na (Gibco)] was added and incubated for 1 min at 37°C 5% CO2. To stop the reaction, 10 mL 5% FBS IMDM was added, and by gently tapping, attached cells were loosened, washed, reseeded, and cultured in fresh culture media at 37°C with 5% CO2. Cells were then treated with 50 mg/mL mitomycin C (Sigma-Aldrich) at a suspension of approximately 1 × 106 cells/mL in 5% FBS IMDM for 4 hours at 37°C. This incubation was performed in closed tubes to allow agitation within a rotation system. The cells were washed four times to remove all the mitomycin C and resuspended to an appropriate cell count for plating into 384-well plates (42).
Dot blots
Antibody expression was successfully confirmed using standard blots on a 0.2-µm nitrocellulose membrane. Here, 2 μL of culture supernatants was spotted onto the membrane and air-dried for 10 min. A positive control (pooled malaria hyperimmune sera) and negative control [mock transfection with polyethylenimine (PEI) alone] were also included. Subsequently, the membrane was blocked with 1% (milk powder/PBS) for 1 hour. Following blocking, the membrane was washed 3× with PBST (0.1% Tween-20 in 1× PBS) for 5 min. The membrane was then incubated with anti-human IgG-HRP diluted at 1:5,000 in blocking buffer for 1 hour at room temperature. The membrane was washed 3× using PBST. Meanwhile, the substrate (Novex® ECL Chemiluminescent substrate) was prepared by mixing 2 mL luminol (reagent A) with 2 mL enhancer (reagent B) in the dark. The membrane was then incubated with the substrate for 60 sec, and imaging was conducted using the ChemiDoc XRS+ system (Bio-Rad).
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel analysis
This was carried out as previously described. Briefly, the agarose gel was cast between two plates measuring (16 × 16 × 12 cm) with spacers measuring 1.0 mm. A 4-cm-high stacking gel (acrylamide 6% w/v, pH 6.8) was applied on top of the separating gel (acrylamide 10% w/v, pH 8.8). Subsequently, 10 µL of purified monoclonal antibody samples was mixed with the loading buffer [50 mM Tris-HC1, pH 6.8, containing sodium dodecyl sulfate (SDS) 2% w/v, glycerol 10% v/v, bromophenol blue 0.1% w/v] and heated for 10 min at 100°C. The denatured samples were then pipetted on separate wells on the gel. The rainbow protein ladder was also included in a separate well. Electrophoresis was performed at 100 V for 120 min in a chamber containing the running buffer (Tris-glycine chloride, pH 8.3). The gel was developed using InstantBlue® Coomassie Protein Stain.
Sanger sequencing and sequence analysis
Variable genes cloned into appropriate vectors were sequenced via Big Dye Terminator Cycle sequencing (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s recommendation. Briefly, a 10-µL reaction mix was prepared consisting of 0.5 µL BigDye terminator 3.1 ready mix, 1.75 µL 5× BigDye sequencing buffer, 4.75 µL of deionized water, 1 µL of 5 µM concentration sequencing primer, and 2 µL of plasmid diluted to 10 ng/mL. Cycle sequencing consists of the following: initial denaturation at 95°C for 10 sec, amplification of 25 cycles (96°C for 10 sec, annealing at 60°C for 5 sec, elongation at 60°C for 4 min), and final extension of 15°C for 10 min. The sequencing reaction was then purified using ethanol/EDTA precipitation, and capillary sequencing was performed using the 3730 DNA analyzer (Thermo Fisher, USA). Sequence analysis was then conducted with Geneious Prime® 2020.0.3, and IgG gene analyses used IGBLAST [a tool used for Ig and T-cell receptor (TR) V domain sequences].
Statistical analysis
Data were compiled using Microsoft Excel 2016 before being exported into GraphPad Prism version 6.05 for Windows and R studio (R version 4.1.1). The means of normally distributed data were compared using Student’s t-test as appropriate and presented as mean (SD) with a two-tailed p-value <0.05 considered significant. If data were not normally distributed, non-parametric methods like the Mann–Whitney U test were used and presented as median [interquartile range (IQR)]. All correlations were determined using Spearman’s rank correlation test with significance set at p-value <0.05. Antibody seropositivity was defined as a cut-off above the mean optical density (OD) plus 3 standard deviations of malaria-naïve European sera. Seropositivity for ADRB and opsonic phagocytosis assay (OPA) was defined as a cut-off above the mean RLU plus 3 standard deviations above RLU, respectively, of malaria-naïve European sera. The custom protein microarray KILchip was used to screen potential antigen targets of the mAbs. The microarray quantification software captures all antibody intensity parameters in a pre-prepared template referred to as a “.gal” file and stores the data in a “.gpr/.txt” file (44, 83). The analysis method was adopted from Jeong et al. (45). Briefly, an affinity score (A) indicates the number of standard deviations above background-corrected MFIs, and the specificity score (S) represents the difference between the A score of the two antigens with consecutive “A” scores when scores are ranked from the highest to lowest (Sn = An − An+1). “A” scores were determined as A = (I − M)/σ, where “I” is the mean tag subtracted MFI value for any given spot pair, “M” is the median value for “I” for all spots on the array, and “σ” is the standard deviation for “I”. All antigens with an “A” score above 2.8 were considered significant and selected as potential antigen candidates for ELISA analysis. S-scores greater than 3 over the next listed target were considered statistically significant and indicated antibody specificity (45, 83).
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by KEMRI Scientific and Ethics Review Unit. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
RO: Writing – Original draft, Conceptualization, Investigation, Visualization, Methodology, Writing – review & editing. LeM: Writing – review & editing, Methodology, Visualization, Investigation. INN: Writing – review & editing, Investigation, Methodology. LN: Writing – review & editing, Methodology, Investigation. OK: Writing – review & editing, Methodology. JT: Conceptualization, Investigation, Methodology, Writing – review & editing. PB: Writing – review & editing. KM: Writing – review & editing, Methodology, Formal analysis. LiM: Writing – review & editing, Investigation, Methodology. RI: Writing – review & editing. SK: Funding acquisition, Supervision, Writing – review & editing. FHAO: Writing – original draft, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. FHAO was supported by a Wellcome Discovery Award 226669/Z/22/Z. RO was funded through the DELTAS Africa Initiative Grant No. 107754/Z/15/Z-DELTAS Africa. The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)’s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust [107769/Z/10/Z] and the UK government. This work was supported in part by an NIHR Global Health Research Unit grant number 16/136/33; Tackling Infections to Benefit Africa (TIBA) and by a Sofja Kovalevskaja Award from the Alexander von Humboldt Foundation (3.2 - 1184811 - KEN - SKP) and an EDCTP Senior Fellowship (TMA 2015 SF1001) which is part of the EDCTP2 programme supported by the European Union awarded to FHAO.
Acknowledgments
We are very grateful to the study participants at the different study sites in Kilifi. In addition, we thank all the clinical, laboratory, and administrative teams that contributed to participant recruitment, sample collection, and processing. Thanks to James Nyagwange for his helpful comments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1360220/full#supplementary-material
References
2. Langhorne J, Ndungu FM, Sponaas AM, Marsh K. Immunity to malaria: more questions than answers. Nat Immunol. (2008) 9:725–32. doi: 10.1038/ni.f.205
3. Doolan DL, Dobaño C, Baird JK. Acquired immunity to malaria. Clin Microbiol Rev. (2009) 22:13–36. doi: 10.1128/CMR.00025-08
4. Rodriguez-Barraquer I, Arinaitwe E, Jagannathan P, Kamya MR, Rosenthal PJ, Rek JV, et al. Quantification of anti-parasite and anti-disease immunity to malaria as a function of age and exposure. Elife. (2018) 7:e35832. doi: 10.7554/eLife.35832
5. Cohen S, McGregor I, Carrington S. Gamma-globulin and acquired immunity to human malaria. Nature. (1961) 192:733–7. doi: 10.1038/192733a0
6. Macintyre F, Ramachandruni H, Burrows JN, Holm R, Thomas A, Mohrle JJ, et al. Injectable anti-malarials revisited: discovery and development of new agents to protect against malaria. Malaria J. (2018) 17:402–2. doi: 10.1186/s12936-018-2549-1
7. Wells T, Donini C. Monoclonal antibodies for malaria. New Engl J Med. (2022) 387:462–5. doi: 10.1056/NEJMe2208131
8. Kayentao K, Ongoiba A, Preston AC, Healy SA, Doumbo S, Doumtabe D, et al. Safety and efficacy of a monoclonal antibody against malaria in Mali. New Engl J Med. (2022) 387:1833–42. doi: 10.1056/NEJMoa2206966
9. Daily JP. Monoclonal antibodies — A different approach to combat malaria. New Engl J Med. (2022) 387:460–1. doi: 10.1056/NEJMe2207865
10. Gaudinski MR, Berkowitz NM, Idris AH, Coates EE, Holman LA, Mendoza F, et al. A monoclonal antibody for malaria prevention. New Engl J Med. (2021) 385:803–14. doi: 10.1056/NEJMoa2034031
11. Wu RL, Idris AH, Berkowitz NM, Happe M, Gaudinski MR, Buettner CV, et al. Low-dose subcutaneous or intravenous monoclonal antibody to prevent malaria. N Engl J Med. (2022) 387:397–407. doi: 10.1056/NEJMoa2203067
12. Alanine DGW, Quinkert D, Kumarasingha R, Mehmood S, Donnellan FR, Minkah NK, et al. Human antibodies that slow erythrocyte invasion potentiate malaria-neutralizing antibodies. Cell. (2019) 178:216–228.e21. doi: 10.1016/j.cell.2019.05.025
13. Julien J-P, Wardemann H. Antibodies against Plasmodium falciparum malaria at the molecular level. Nat Rev Immunol. (2019) 19:761–75. doi: 10.1038/s41577-019-0209-5
14. Knudsen AS, Bjornsson KH, Bassi MR, Walker MR, Kok A, Cristinoi BV, et al. Strain-dependent inhibition of erythrocyte invasion by monoclonal antibodies against plasmodium falciparum cyRPA. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.716305
15. Douglas AD, Baldeviano GC, Jin J, Miura K, Diouf A, Zenonos ZA, et al. A defined mechanistic correlate of protection against Plasmodium falciparum malaria in non-human primates. Nat Commun. (2019) 10:1953. doi: 10.1038/s41467-019-09894-4
16. Douglas AD, Baldeviano GC, Lucas CM, Lugo-Roman LA, Crosnier C, Bartholdson SJ, et al. A PfRH5-Based Vaccine Is Efficacious against Heterologous Strain Blood-Stage Plasmodium falciparum Infection in Aotus Monkeys. Cell Host Microbe. (2015) 17:130–9. doi: 10.1016/j.chom.2014.11.017
17. Musasia FK, Nkumama IN, Frank R, Kipkemboi V, Schneider M, Mwai K, et al. Phagocytosis of Plasmodium falciparum ring-stage parasites predicts protection against malaria. Nat Commun. (2022) 13:4098. doi: 10.1038/s41467-022-31640-6
18. Boyle MJ, Reiling L, Feng G, Langer C, Osier FH, Aspeling-Jones H, et al. Human Antibodies Fix Complement to Inhibit Plasmodium falciparum Invasion of Erythrocytes and Are Associated with Protection against Malaria. Immunity. (2015) 42:580–90. doi: 10.1016/j.immuni.2015.02.012
19. Osier FHA, Feng G, Boyle MJ, Langer C, Zhou J, Richards JS, et al. Opsonic phagocytosis of Plasmodium falciparummerozoites: mechanism in human immunity and a correlate of protection against malaria. BMC Med. (2014) 12:108. doi: 10.1186/1741-7015-12-108
20. Nkumama IN, Odera D, Musasia F, Mwai K, Nyamako L, Murungi L, et al. Breadth of Fc-mediated effector function delineates grades of clinical immunity following human malaria challenge. bioRxiv. (2022). 2022.10.11.511755. doi: 10.1101/2022.10.11.511755
21. Odera DO, Tuju J, Mwai K, Nkumama IN, Furle K, Chege T, et al. Anti-merozoite antibodies induce natural killer cell effector function and are associated with immunity against malaria. Sci Trans Med. (2023) 15:eabn5993. doi: 10.1126/scitranslmed.abn5993
22. Murungi LM, Sonden K, Llewellyn D, Rono J, Guleid F, Williams AR, et al. Targets and mechanisms associated with protection from severe plasmodium falciparum malaria in Kenyan children. Infect Immun. (2016) 84:950–63. doi: 10.1128/IAI.01120-15
23. Teo A, Feng G, Brown GV, Beeson JG, Rogerson SJ. Functional antibodies and protection against blood-stage malaria. Trends Parasitol. (2016) 32:887–98. doi: 10.1016/j.pt.2016.07.003
24. Persson KE, Lee CT, Marsh K, Beeson JG. Development and optimization of high-throughput methods to measure Plasmodium falciparum-specific growth inhibitory antibodies. J Clin Microbiol. (2006) 44:1665–73. doi: 10.1128/JCM.44.5.1665-1673.2006
25. Wilson DW, Crabb BS, Beeson JG. Development of fluorescent Plasmodium falciparum for in vitro growth inhibition assays. Malar J. (2010) 9:152. doi: 10.1186/1475-2875-9-152
26. Bergmann-Leitner ES, Duncan EH, Burge JR, Spring M, Angov E. Miniaturization of a high-throughput pLDH-based Plasmodium falciparum growth inhibition assay for small volume samples from preclinical and clinical vaccine trials. Am J Trop Med Hyg. (2008) 78:468–71. doi: 10.4269/ajtmh.2008.78.468
27. McCallum FJ, Persson KE, Mugyenyi CK, Fowkes FJ, Simpson JA, Richards JS, et al. Acquisition of Growth-Inhibitory Antibodies against Blood-Stage Plasmodium falciparum. PloS One. (2008) 3:e3571. doi: 10.1371/journal.pone.0003571
28. Dent AE, Bergmann-Leitner ES, Wilson DW, Tisch DJ, Kimmel R, Vulule J, et al. Antibody-mediated growth inhibition of plasmodium falciparum: relationship to age and protection from parasitemia in Kenyan children and adults. PloS One. (2008) 3:e3557. doi: 10.1371/journal.pone.0003557
29. Duncan CJ, Hill AV, Ellis RD. Can growth inhibition assays (GIA) predict blood-stage malaria vaccine efficacy? Hum Vaccin Immunother. (2012) 8:706–14. doi: 10.4161/hv.19712
30. Burn GL, Foti A, Marsman G, Patel DF, Zychlinsky A. The neutrophil. Immunity. (2021) 54:1377–91. doi: 10.1016/j.immuni.2021.06.006
31. Newburger PE. Autoimmune and other acquired neutropenias. Hematol Am Soc Hematol Educ Program. (2016) 2016:38–42. doi: 10.1182/asheducation-2016.1.38
32. Skokowa J, Dale DC, Touw IP, Zeidler C, Welte K. Severe congenital neutropenias. Nat Rev Dis Primers. (2017) 3:17032. doi: 10.1038/nrdp.2017.32
33. Garcia-Senosiain A, Kana IH, Singh S, Das MK, Dziegiel MH, Hertegonne S, et al. Neutrophils dominate in opsonic phagocytosis of P. falciparum blood-stage merozoites and protect against febrile malaria. Commun Biol. (2021) 4:984. doi: 10.1038/s42003-021-02511-5
34. Knackstedt SL, Georgiadou A, Apel F, Abu-Abed U, Moxon CA, Cunnington AJ, et al. Neutrophil extracellular traps drive inflammatory pathogenesis in malaria. Sci Immunol. (2019) 4(40). doi: 10.1126/sciimmunol.aaw0336
35. Babatunde KA, Adenuga OF. Neutrophils in malaria: A double-edged sword role. Front Immunol. (2022) 13:922377. doi: 10.3389/fimmu.2022.922377
36. Llewellyn D, Miura K, Fay MP, Williams AR, Murungi LM, Shi J, et al. Standardization of the antibody-dependent respiratory burst assay with human neutrophils and Plasmodium falciparum malaria. Sci Rep. (2015) 5:14081. doi: 10.1038/srep14081
37. Heemskerk N, Gruijs M, Temming AR, Heineke MH, Gout DY, Hellingman T, et al. Augmented antibody-based anticancer therapeutics boost neutrophil cytotoxicity. J Clin Invest. (2021) 131(6). doi: 10.1172/JCI134680
38. Borrok MJ, Luheshi NM, Beyaz N, Davies GC, Legg JW, Wu H, et al. Enhancement of antibody-dependent cell-mediated cytotoxicity by endowing IgG with FcαRI (CD89) binding. MAbs. (2015) 7:743–51. doi: 10.1080/19420862.2015.1047570
39. Cottignies-Calamarte A, Tudor D, Bomsel M. Antibody Fc-chimerism and effector functions: When IgG takes advantage of IgA. Front Immunol. (2023) 14:1037033. doi: 10.3389/fimmu.2023.1037033
40. Mwangi TW, Ross A, Snow RW, Marsh K. Case definitions of clinical malaria under different transmission conditions in Kilifi District, Kenya. J Infect Dis. (2005) 191:1932–9. doi: 10.1086/430006
41. O'Meara WP, Mwangi TW, Williams TN, McKenzie FE, Snow RW, Marsh K. Relationship between exposure, clinical malaria, and age in an area of changing transmission intensity. Am J Trop Med Hyg. (2008) 79:185–91. doi: 10.4269/ajtmh.2008.79.185
42. Huang J, Doria-Rose NA, Longo NS, Laub L, Lin CL, Turk E, et al. Isolation of human monoclonal antibodies from peripheral blood B cells. Nat Protoc. (2013) 8:1907–15. doi: 10.1038/nprot.2013.117
43. Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J Immunol Methods. (2008) 329:112–24. doi: 10.1016/j.jim.2007.09.017
44. Kamuyu G, Tuju J, Kimathi R, Mwai K, Mburu J, Kibinge N, et al. KILchip v1.0: A Novel Plasmodium falciparum Merozoite Protein Microarray to Facilitate Malaria Vaccine Candidate Prioritization. Front Immunol. (2018) 9:2866. doi: 10.3389/fimmu.2018.02866
45. Jeong JS, Jiang L, Albino E, Marrero J, Rho HS, Hu J, et al. Rapid identification of monospecific monoclonal antibodies using a human proteome microarray*. Mol Cell Proteomics. (2012) 11:O111.016253. doi: 10.1074/mcp.O111.016253
46. Kelton W, Mehta N, Charab W, Lee J, Lee CH, Kojima T, et al. IgGA: a "cross-isotype" engineered human Fc antibody domain that displays both IgG-like and IgA-like effector functions. Chem Biol. (2014) 21:1603–9. doi: 10.1016/j.chembiol.2014.10.017
47. Saunders KO. Conceptual approaches to modulating antibody effector functions and circulation half-life. Front Immunol. (2019) 10. doi: 10.3389/fimmu.2019.01296
48. Brandsma AM, Bondza S, Evers M, Koutstaal R, Nederend M, Jansen JHM, et al. Potent fc receptor signaling by igA leads to superior killing of cancer cells by neutrophils compared to igG. Front Immunol. (2019) 10:704. doi: 10.3389/fimmu.2019.00704
49. Herr AB, White CL, Milburn C, Wu C, Bjorkman PJ. Bivalent binding of IgA1 to FcalphaRI suggests a mechanism for cytokine activation of IgA phagocytosis. J Mol Biol. (2003) 327:645–57. doi: 10.1016/S0022-2836(03)00149-9
50. Minassian AM, Silk SE, Barrett JR, Nielsen CM, Miura K, Diouf A, et al. Reduced blood-stage malaria growth and immune correlates in humans following RH5 vaccination. Med. (2021) 2:701–719.e19. doi: 10.1016/j.medj.2021.03.014
51. Tan J, Cho H, Pholcharee T, Pereira LS, Doumbo S, Doumtabe D, et al. Functional human IgA targets a conserved site on malaria sporozoites. Sci Transl Med. (2021) 13(599). doi: 10.1126/scitranslmed.abg2344
52. Vaisman-Mentesh A, Rosenstein S, Yavzori M, Dror Y, Fudim E, Ungar B, et al. Molecular landscape of anti-drug antibodies reveals the mechanism of the immune response following treatment with TNFα Antagonists. Front Immunol. (2019) 10:2921. doi: 10.3389/fimmu.2019.02921
53. Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discovery. (2010) 9:325–38. doi: 10.1038/nrd3003
54. Paul G, Deshmukh A, Kaur I, Rathore S, Dabral S, Panda A, et al. A novel Pfs38 protein complex on the surface of Plasmodium falciparum blood-stage merozoites. Malaria J. (2017) 16:79. doi: 10.1186/s12936-017-1716-0
55. Oeuvray C, Bouharoun-Tayoun H, Gras-Masse H, Bottius E, Kaidoh T, Aikawa M, et al. Merozoite surface protein-3: a malaria protein inducing antibodies that promote Plasmodium falciparum killing by cooperation with blood monocytes. Blood (1994) 84(5):1594–602. doi: 10.1182/blood.V84.5.1594.bloodjournal8451594
56. Roussilhon C, Oeuvray C, Müller-Graf C, Tall A, Rogier C, Trape J-F, et al. Long-term clinical protection from falciparum malaria is strongly associated with IgG3 antibodies to merozoite surface protein 3. PloS Med. (2007) 4:e320. doi: 10.1371/journal.pmed.0040320
57. Soe S, Theisen M, Roussilhon C, Aye K-S-, Druilhe P. Association between protection against clinical malaria and antibodies to merozoite surface antigens in an area of hyperendemicity in Myanmar: complementarity between responses to merozoite surface protein 3 and the 220-kilodalton glutamate-rich protein. Infect Immun. (2004) 72:247–52. doi: 10.1128/IAI.72.1.247-252.2004
58. Singh S, Soe S, Mejia JP, Roussilhon C, Theisen M, Corradin G, et al. Identification of a conserved region of Plasmodium falciparum MSP3 targeted by biologically active antibodies to improve vaccine design. J Infect Dis. (2004) 190:1010–8. doi: 10.1086/423208
59. Osier F, Polley SD, Mwangi T, Lowe B, Conway DJ, Marsh K. Naturally acquired antibodies to polymorphic and conserved epitopes of Plasmodium falciparum merozoite surface protein 3. Parasite Immunol. (2007) 29:387–94. doi: 10.1111/j.1365-3024.2007.00951.x
60. Trucco C, Fernandez-Reyes D, Howell S, Stafford WH, Scott-Finnigan TJ, Grainger M, et al. The merozoite surface protein 6 gene codes for a 36 kDa protein associated with the Plasmodium falciparum merozoite surface protein-1 complex. Mol Biochem Parasitol. (2001) 112:91–101. doi: 10.1016/S0166-6851(00)00350-9
61. Singh S, Soe S, Roussilhon C, Corradin G, Druilhe P. Plasmodium falciparum merozoite surface protein 6 displays multiple targets for naturally occurring antibodies that mediate monocyte-dependent parasite killing. Infect Immun. (2005) 73:1235–8. doi: 10.1128/IAI.73.2.1235-1238.2005
62. Pearce JA, Mills K, Triglia T, Cowman AF, Anders RF. Characterisation of two novel proteins from the asexual stage of Plasmodium falciparum, H101 and H103. Mol Biochem Parasitol. (2005) 139:141–51. doi: 10.1016/j.molbiopara.2004.09.012
63. Singh S, Soe S, Weisman S, Barnwell JW, Perignon JL, Druilhe P. A Conserved Multi-Gene Family Induces Cross-Reactive Antibodies Effective in Defense against Plasmodium falciparum. PloS One. (2009) 4:e5410. doi: 10.1371/journal.pone.0005410
64. Sirima SB, Nebie I, Ouedraogo A, Tiono AB, Konate AT, Gansane A, et al. Safety and immunogenicity of the Plasmodium falciparum merozoite surface protein-3 long synthetic peptide (MSP3-LSP) malaria vaccine in healthy, semi-immune adult males in Burkina Faso, West Africa. Vaccine. (2007) 25:2723–32. doi: 10.1016/j.vaccine.2006.05.090
65. Sirima SB, Tiono AB, Ouedraogo A, Diarra A, Ouedraogo AL, Yaro JB, et al. Safety and immunogenicity of the malaria vaccine candidate MSP3 long synthetic peptide in 12-24 months-old Burkinabe children. PloS One. (2009) 4:e7549. doi: 10.1371/journal.pone.0007549
66. Esen M, Kremsner PG, Schleucher R, Gassler M, Imoukhuede EB, Imbault N, et al. Safety and immunogenicity of GMZ2 - a MSP3-GLURP fusion protein malaria vaccine candidate. Vaccine. (2009) 27:6862–8. doi: 10.1016/j.vaccine.2009.09.011
67. Dejon-Agobe JC, Ateba-Ngoa U, Lalremruata A, Homoet A, Engelhorn J, Nouatin OP, et al. Controlled human malaria infection of healthy adults with lifelong malaria exposure to assess safety, immunogenicity, and efficacy of the asexual blood stage malaria vaccine candidate GMZ2. Clin Infect Dis. (2019) 69:1377–84. doi: 10.1093/cid/ciy1087
68. Wright KE, Hjerrild KA, Bartlett J, Douglas AD, Jin J, Brown RE, et al. Structure of malaria invasion protein RH5 with erythrocyte basigin and blocking antibodies. Nature. (2014) 515:427–30. doi: 10.1038/nature13715
69. Pleass RJ, Ogun SA, McGuinness DH, van de Winkel JG, Holder AA, Woof JM. Novel antimalarial antibodies highlight the importance of the antibody Fc region in mediating protection. Blood. (2003) 102:4424–30. doi: 10.1182/blood-2003-02-0583
70. Patel PN, Dickey TH, Hopp CS, Diouf A, Tang WK, Long CA, et al. Neutralizing and interfering human antibodies define the structural and mechanistic basis for antigenic diversion. Nat Commun. (2022) 13:5888. doi: 10.1038/s41467-022-33336-3
71. Biggs BA, Gooze L, Wycherley K, Wollish W, Southwell B, Leech JH, et al. Antigenic variation in Plasmodium falciparum. Proc Natl Acad Sci. (1991) 88:9171–4. doi: 10.1073/pnas.88.20.9171
72. Rénia L, Goh YS. Malaria parasites: the great escape. Front Immunol. (2016) 7. doi: 10.3389/fimmu.2016.00463
73. Gilson PR, Crabb BS. Morphology and kinetics of the three distinct phases of red blood cell invasion by Plasmodium falciparum merozoites. Int J Parasitol. (2009) 39:91–6. doi: 10.1016/j.ijpara.2008.09.007
74. McGregor I, Carrington S, Cohen S. Treatment of East African P. falciparum malaria with West African human γ-globulin. Trans R Soc Trop Med Hygiene. (1963) 57:170–5. doi: 10.1016/0035-9203(63)90058-0
75. Kelley B. Industrialization of mAb production technology: the bioprocessing industry at a crossroads. MAbs. (2009) 1(5):443–52. doi: 10.4161/mabs.1.5.9448
76. Klutz S, Holtmann L, Lobedann M, Schembecker G. Cost evaluation of antibody production processes in different operation modes. Chem Eng Sci. (2016) 141:63–74. doi: 10.1016/j.ces.2015.10.029
77. Kisalu NK, Idris AH, Weidle C, Flores-Garcia Y, Flynn BJ, Sack BK, et al. A human monoclonal antibody prevents malaria infection by targeting a new site of vulnerability on the parasite. Nat Med. (2018) 24:408–16. doi: 10.1038/nm.4512
78. Oniyangi O, Omari AAA. Malaria chemoprophylaxis in sickle cell disease. Cochrane Database systematic Rev. (2006) 2006:CD003489–CD003489. doi: 10.1002/14651858
79. Kwambai TK, Dhabangi A, Idro R, Opoka R, Watson V, Kariuki S, et al. Malaria chemoprevention in the postdischarge management of severe anemia. New Engl J Med. (2020) 383:2242–54. doi: 10.1056/NEJMoa2002820
80. Pham-Huy A, Top KA, Constantinescu C, Seow CH, El-Chaar D. The use and impact of monoclonal antibody biologics during pregnancy. Cmaj. (2021) 193:E1129–e1136. doi: 10.1503/cmaj.202391
81. Boyle MJ, Wilson DW, Richards JS, Riglar DT, Tetteh KK, Conway DJ, et al. Isolation of viable Plasmodium falciparum merozoites to define erythrocyte invasion events and advance vaccine and drug development. Proc Natl Acad Sci U.S.A. (2010) 107:14378–83. doi: 10.1073/pnas.1009198107
82. Froger A, Hall JE. Transformation of plasmid DNA into E. coli using the heat shock method. J Vis Exp. (2007) 2007(6):253. doi: 10.3791/253
Keywords: malaria, monoclonal antibodies, neutrophils, semi-immune, Kenya
Citation: Ogwang R, Murugu L, Nkumama IN, Nyamako L, Kai O, Mwai K, Murungi L, Idro R, Bejon P, Tuju J, Kinyanjui SM and Osier FHA (2024) Bi-isotype immunoglobulins enhance antibody-mediated neutrophil activity against Plasmodium falciparum parasites. Front. Immunol. 15:1360220. doi: 10.3389/fimmu.2024.1360220
Received: 22 December 2023; Accepted: 19 March 2024;
Published: 08 April 2024.
Edited by:
Takafumi Tsuboi, Ehime University, JapanReviewed by:
Julius Clemence Hafalla, University of London, United KingdomBernard Kanoi, Mount Kenya University, Kenya
Copyright © 2024 Ogwang, Murugu, Nkumama, Nyamako, Kai, Mwai, Murungi, Idro, Bejon, Tuju, Kinyanjui and Osier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Faith H. A. Osier, Zi5vc2llckBpbXBlcmlhbC5hYy51aw==
†These authors have contributed equally to this work and share last authorship