 Renuka Ramalingam Manoharan
Renuka Ramalingam Manoharan Ankush Prasad
Ankush Prasad Pavel Pospíšil
Pavel Pospíšil Julia Kzhyshkowska
Julia Kzhyshkowska
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 07 March 2024
Sec. Molecular Innate Immunity
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1359600
This article is part of the Research Topic Reactive Oxygen Species (ROS) Signaling and Immune Diseases. View all 9 articles
The innate immune response represents the first-line of defense against invading pathogens. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) have been implicated in various aspects of innate immune function, which involves respiratory bursts and inflammasome activation. These reactive species widely distributed within the cellular environment are short-lived intermediates that play a vital role in cellular signaling and proliferation and are likely to depend on their subcellular site of formation. NADPH oxidase complex of phagocytes is known to generate superoxide anion radical (O2•−) that functions as a precursor for antimicrobial hydrogen peroxide (H2O2) production, and H2O2 is utilized by myeloperoxidase (MPO) to generate hypochlorous acid (HOCl) that mediates pathogen killing. H2O2 modulates the expression of redox-responsive transcriptional factors, namely NF-kB, NRF2, and HIF-1, thereby mediating redox-based epigenetic modification. Survival and function of immune cells are under redox control and depend on intracellular and extracellular levels of ROS/RNS. The current review focuses on redox factors involved in the activation of immune response and the role of ROS in oxidative modification of proteins in macrophage polarization and neutrophil function.
In biology, reactive species formed by redox reaction or electronic excitation hold a growing interest due to their significant impact on the spectrum of pathological processes, including inflammation and aging (1). Based on the nature of reactive atoms, they are named reactive oxygen species (oxygen), reactive nitrogen species (nitrogen) and reactive sulfur species (sulfur), respectively. Produced by nearly all organisms and cells, ROS consists of two subclasses: highly reactive radical and non-radical species. Free oxygen radicals include superoxide anion (O2•-), hydroxyl (HO•), peroxyl (ROO•), and alkoxyl (RO•), while hydrogen peroxide (H2O2), singlet oxygen (1O2), hypochlorite anion and ozone represent the non-radical species (2) (Figure 1). ROS subcellular origin and its levels within the cellular environment defines its role. Its function as physiological secondary messengers in signal transduction becoming increasingly apparent; for example, ROS oxidize sulfhydryl (SH) groups of cysteine residues in protein kinases, including protein kinase A(PKA), protein kinase C (PKC), Calcium–calmodulin (CaM)-dependent protein kinase II (CaMKII) and receptor tyrosine kinase (RTK), which activate and phosphorylate their protein targets involved in signaling (3–5).
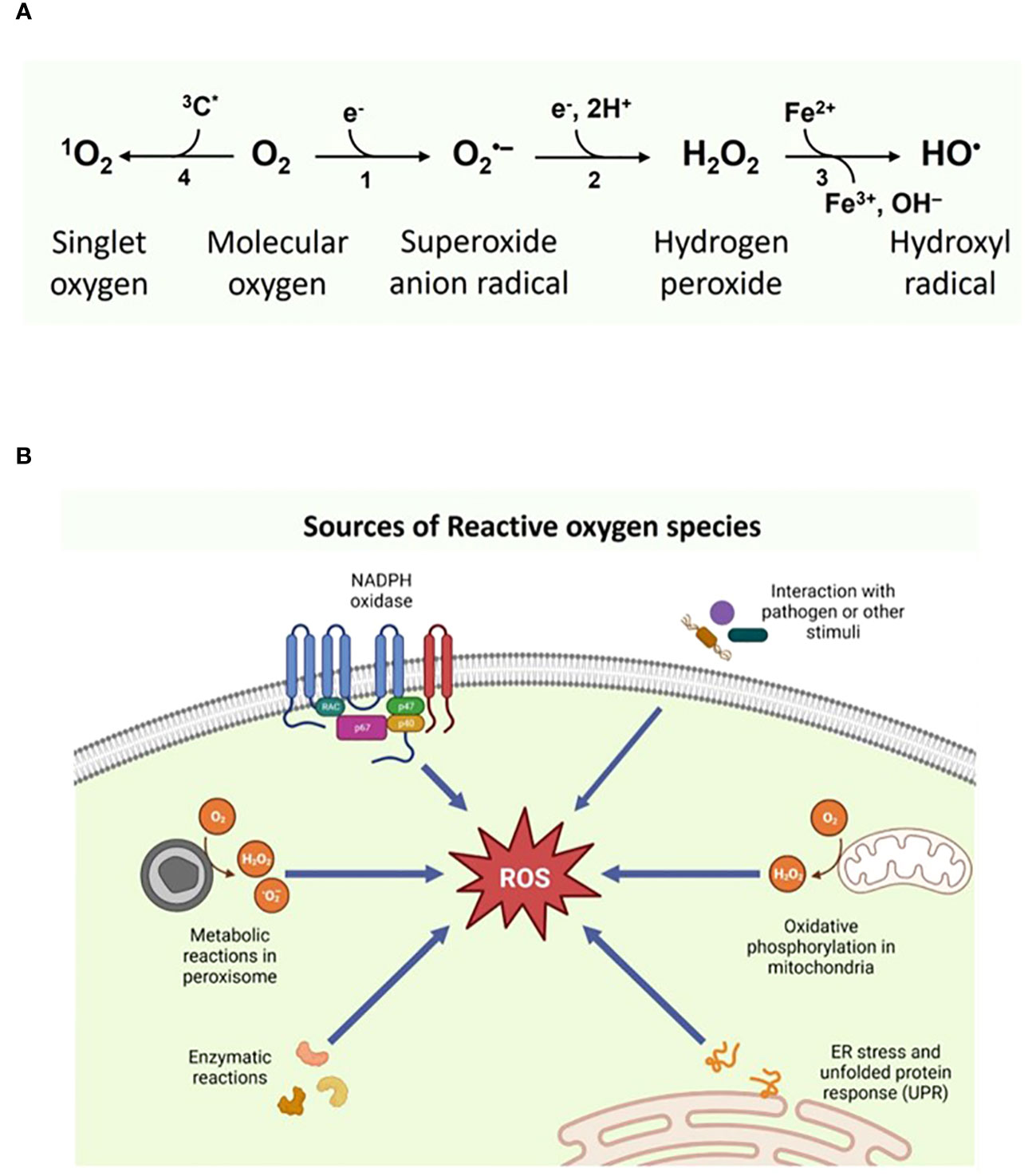
Figure 1 (A) Types of Reactive oxygen species and their (B) intracellular sources. Figure 1B was Created with BioRender.com.
Reported as the major subspecies within cells, O2•- and H2O2 highly differ in their chemical parameter and functions. An increase in cellular O2•- levels remains closely associated with oxidative stress and cellular damage, wherein the oxidation of biological macromolecules and irreversible protein inactivation disturb cellular signaling events (6). H2O2 is highly diffusible and relatively stable compared to O2•- and is considered a pleiotropic physiological signaling agent. In general, H2O2 at physiological pH mediates signaling via oxidation of cysteine residues, wherein the exposed thiol group (Cys-SH) are deprotonated to the thiolate group (Cys-S-) and susceptible to oxidation. H2O2-dependent signaling occurs with a lower intracellular concentration of approximately 1-100 nM, mediating reversible oxidation of the thiolate group to a sulfenic group (Cys-SOH) and covalent linkage of cysteine residues by disulfide bonds (Cys-S-S-Cys) altering its activity and localization (7). An antioxidant defense system can reverse such protein oxidation and thus serve as essential redox switches in various cellular processes. Nevertheless, excessive production of H2O2 mediates irreversible nonspecific oxidation of proteins, resulting in a state referred to as oxidative stress (8). This inherent duality of ROS serving as a beneficial secondary messenger in signaling and causing harmful effects through the accumulation of protein adducts signifies its antagonistic pleiotropy (6).
ROS regulate cellular homeostasis and signaling events within a cellular environment, thus serving as primary modulators of cellular dysfunction and contributing to disease pathology. In almost every subcellular organelle, including cytoplasm, endoplasmic reticulum (ER), mitochondria and peroxisomes, ROS are generated as byproducts as a part of its basal metabolic function (9, 10). An imbalance in ROS generation and scavenging by antioxidants results in pathological conditions, including cancer, neurodegenerative disorders and atherosclerosis. Studies carried out so far substantiate the role of ROS in various metabolic processes like mitochondrial ROS promoting monocyte migration, wherein NADPH oxidase (NOX)-mediated cytosolic ROS production aids in innate immunity (11). Depending on cell type, tissue environment and source, ROS participates in normal physiological processes or contribute to metabolic dysfunction and inflammatory signaling in sterile and infectious inflammation.
During the initial stage of an inflammatory process, immune cells activated in response to invading pathogens or agents release various inflammatory mediators, aiding in increased vascular permeability and leukocyte migration towards the tissue injury site (12). ROS regulates various intracellular adhesion molecules, including ICAM-1, VCAM-1 and selectin expression, ensuring their interaction with leukocytes and transendothelial migration. On the contrary, enhanced expression of superoxide dismutase reduces leukocyte binding to endothelium by decreasing the expression of adhesion molecules (13). Additionally, a gradient increase in H2O2 at the tissue injury site is an early factor in leukocyte recruitment (14). Cytokines, including tumor necrosis factor (TNF), vascular endothelial growth factor (VEGF), and NOX stimulate cell migration and adhesion (15).
Metabolic reprogramming, which includes changes in the activity of fatty acid oxidation (FAO), tricarboxylic acid (TCA) cycle, glycolysis levels, involvement of pentose phosphate pathway and mitochondrial respiration mediates the phenotypical and functional role of myeloid cells.
Over the past two decades, immune cell functions, namely proliferation and differentiation, have been linked to various metabolic pathways; thus, immunometabolism is inseparable from redox reactions (11). Thus, bioenergetic and biosynthetic demands for T and B cell response, macrophages and dendritic cells are regulated by metabolic pathway and their resulting breakdown products (16). In aerobic organisms, during oxidative phosphorylation in mitochondria, the electron transport chain shuttles electrons to molecular oxygen (O2), producing free oxygen radicals. ROS are well known for their role in inflammation through respiratory bursts, wherein macrophages and neutrophils phagocytize pathogens and cellular debris. The process activates the NOX assembly and activation, which triggers further ROS production (17). NOXs play a vital role in inflammatory response through respiratory burst and neutrophil extracellular trap formation. NOX-derived ROS are reportedly involved in angiogenesis, a process of the growth of new vessels and a key event in the proliferative phase of inflammation. It has been reported that NOX remains localized at the leading edge of migrating cells and is linked to actin and IQGAP1 protein (Ras GTPase-activating-like protein), and any disruption at this binding site results in impaired migration of endothelial cells (18).
Nevertheless, enhanced production of reactive species has to be regulated to avoid oxidation of biomolecules leading to cellular toxicity and cell death. Alterations in redox balance are often associated with chronic immune activation, such as autoimmune disorders and neurodegenerative diseases. Therefore, the redox state is an intrinsic cellular and systemic homeostasis indicator.
The release of local damage-associated molecular patterns (DAMPs) in response to tissue damage elicits enhanced cytokine production via sterile inflammation. Cytokines belonging to the interleukin-1(IL-1) family have been proposed to be essential drivers of sterile inflammation, which recruits neutrophils and macrophages to the damaged site. During sterile inflammation, host-derived DAMPs released by stressed cells contribute to macrophage polarization that aids in resolving sterile inflammation and restores homeostasis (19). In endothelial cells, under hyperglycemic conditions, an excessive glucose load triggers ROS formation in mitochondria, impairing its functions and causing cellular damage through interaction with various cellular constituents, including DNA, proteins and lipids (20). These ROS trigger the activation of pro-inflammatory transcription factors, namely NFκB and activating protein-1(AP-1), resulting in enhanced inflammatory cytokines/chemokines expression. In addition, activated endothelial cells attract monocytes that promote inflammation and macrovascular and microvascular injury (21).
Macrophages, especially adipose tissue macrophages (ATM), drive diabetic pathology. In healthy individuals, adipocytes secrete adiponectin, which induces M2-like polarization of ATM and suppresses ROS and its related pathway genes (22). On the contrary, in obesity conditions, a reduction in adiponectin levels induces M1-like polarized macrophages, thereby enhancing glucose consumption through GLUT1 (glucose transporter 1). Thus, an interplay between obesity and hyperglycemia promotes ROS formation, glycolytic metabolism, and the release of pro-inflammatory cytokine mediators by macrophages (23). Hence, focusing more on metabolic reprogramming during macrophage polarization events is essential to identify potential targets for treating inflammation and metabolic disorders. A mitochondrial reactive oxidative stress increase was reported in mice subjected to ventilator-induced lung injury, activating NLRP3 to produce IL-1β and lung inflammation in combination with TLR4 signaling (24).
Though the primary source of ROS in vivo is through aerobic respiration, cellular events, including peroxisomal β-oxidation of fatty acids, arginine metabolism, tissue-specific cellular enzymes and phagocytosis stimulation by pathogens also contributed to ROS production (3). Based on its source, cell type and environment, ROS signaling contributes to either normal physiological processes or metabolic dysfunction through inflammatory signaling. Diseased conditions, including diabetes mellitus, atherosclerosis and stroke, are known to be associated with redox balance (25).
The NADPH oxidase (NOX) family of proteins remains the primary cytosolic source of ROS, consist of seven different isoforms and comprises membrane and cytosolic components that are actively involved in the host response to various stimuli including bacterial and viral infections, cellular signaling and regulation of gene expression. Among the isoforms, NOX2 is well characterized for its role in phagocytic functions. Both NOX and inducible nitric oxide synthase (iNOS) are vital in generating enhanced ROS levels within phagocytes via oxidative burst to kill invading pathogens (26). In comparison to mitochondria, O2•- produced by NOX is dismutated into H2O2 by superoxide dismutase1 (SOD1), whereas nitric oxide produced by inducible nitric oxide synthase (iNOS) reacts with O2•- resulting in peroxynitrite production (19). For example, ROS drive hypoxia-inducible factor 1α (HIF1α) mediated GLUT1 expression, hexokinase activity, and resultant glycolysis in response to low oxygen tension as part of the angiogenic response (27) The co-localization of neutrophil phosphofructokinase 2 with NOX2 leads to its activation, resulting in NADP+ production as its byproduct, facilitating an enhanced glycolytic rate. The increase in glycolysis rate and enhancement of NOX2 activity and the relation between these processes are still under study (28).
Mitochondria are considered as the redox-active compartment within the cell, accounting for nearly 90% of oxygen utilization (1). They serve as a significant contributor to ROS in the form of O2•-. Mitochondrial SOD converts O2•- into H2O2, which in turn gets converted into HO• through the Fenton reaction, which in turn oxidizes biomolecules. SOD1 is constitutively expressed and regulates cytosolic O2•- levels (29). Factors like hyperoxia, oxidative stress, and inflammatory cytokines induce SOD2, whereas SOD3 are cell and tissue-specific and likely of significant importance in protection against stress factors from the extracellular environment. In addition, Grx (glutaredoxin), glutathione, and Trx (thioredoxin) systems play a predominant role in mitochondrial ROS buffering, like SOD (30).
Under stress conditions, the ER tubular network holds a unique oxidizing environment, wherein redox signaling mediators play a vital role in ROS generation and mediate protein folding. Protein folding is highly sensitive to ER redox status, and dysregulation of disulfide bond formation in response to ER stress increases luminal oxidative stress, leading to a decline in ER function. During protein folding, protein disulfide isomerase (PDI) and endoplasmic reticulum oxidoreductase 1 (ERO1) introduce disulphide bonds into folded proteins, resulting in H2O2 formation (31). Protein disulfide isomerase introduces disulfide bonds onto protein substrates through thiol oxidation, resulting in a reduced state. However, PDI is reoxidized through ERO1, which transfers electrons from O2 through the flavin adenine dinucleotide cofactor, forming H2O2 (32). In addition to the PD1/ERO1 pathway, ROS are produced through NOX4, NADPH-P450 reductase (NPR), and GSH (33). NOX4 is reported to be consistently associated with ER, and NOX4 associated with p22phox utilizes NADH or NADPH as an electron donor to produce O2•-. They are also reported to interact with PDI, whereas the absence of PDI results in cell death (34). In macrophages, the interaction between p22phox and PDI was observed (26).
Peroxisomes, like mitochondria, are vital organelles that regulate crucial processes such as α- and β-oxidation, amino acid catabolism, glyoxylate metabolism, ketogenesis, polyamine oxidation and isoprenoid and cholesterol metabolism (35). Peroxisomal electron transfer leads to free electrons rather than ATP, which are transferred to H2O to form H2O2. In peroxisomes, H2O2 is produced by various oxygen-consuming oxidases, including D-amino acid oxidase, xanthine oxidase, d-aspartate oxidase, polyamine oxidase and acyl-CoA oxidase. In addition, peroxisomal oxidases and xanthine oxidases generate ROS and nitric oxide (36). The lysosomal electron transport chain generates HO• via proton translocation to maintain an optimal pH for acidic hydrolases (9).
Thus, ROS are produced as a byproduct of cellular events wherein the NOX family of proteins mediate the reduction of O2 to O2•- and phagocytes NOX accounts for an increased amount of O2•- and H2O2 production by respiratory burst (3). Upon stimulation, the membrane-bound catalytic core assembles with proteins from the cytosol (p47 phox, p67 phox and small G protein Rac), activating O2•- production. •NO produced by Nitric oxide synthase (NOS) can migrate through the cell membrane via diffusion and mediate several signaling pathways in a dose-dependent manner (37). However, inflammatory activation of iNOS by cytokines or lipopolysaccharides enhances cellular levels of •NO and results in inflammatory diseases and septic shock (38). Both oxidative and nitrosative stress can hinder the functioning of intracellular redox buffer systems, resulting in decreased antioxidant capacity of affected cells (39). Thus, a proper balance between ROS-RNS is essential in regulating immunological response. A schematic representation of types and sources of ROS are presented in Figures 1A, B, respectively.
Sulfur-containing biomolecules are crucial in protein folding, deactivation of reactive species, enzymes, redox signaling and other biochemical functions. Remarkably, most of the functions are associated with proteins and protein adducts, whereas its functions can be traced back to two amino acids, cysteine and methionine and their respective thiol or thioether functionality (40, 41). Oxidative stress targets the sulfhydryl group of cysteine and the methionine thioether group, resulting in increased post-translational modification events. Being sensitive to redox transformations, thiol, the side chain of cysteine, acquires different oxidation states. While thiol and disulfide are commonly known, growing evidence of protein modification also reports other oxygen derivatives, including sulfenic, sulfinic and sulfonic derivatives. Disulfide formation remains the most common thiol oxidation wherein the disulfide bonds are reasonably stable and stabilize protein structures via intra- and intermolecular disulfide bridges. Cysteine thiyl radical and sulfenic acid formation is reversible, and both intermediates are highly unstable. Both serve as precursors for several oxidized cysteine modifications (42). Sulfenic acids are highly reactive and play a prominent role in enzyme catalysis and cell signaling. They remain as key intermediates to other oxidation states, namely sulfinic and sulfonic acids. During the inflammatory process, immune cells, thiol or thiolate anion reaction with hypochlorous acid (HOCl) result in the formation of sulfenic acids (43).
Approximately 5% of cellular proteins remain in either sulfinic or sulfonic acid forms. The functional role of sulfinic acid modification has been reported mainly with the peroxiredoxin (Prxs) family (44), which reduces H2O2 and alkyl peroxides to water and alcohol. Under physiological conditions, in contrast to sulfenic acids, sulfinic derivatives do not react with thiols or undergo self-condensation reactions. Sulfinic acids are stable intermediates but oxidize readily to sulfonic acid (RSO3H), the most highly oxidized species of thiols and disulfides. Potent oxidizing agents, halogens, H2O2, and nitric acid can generate sulfonic acids from thiols (20). Introducing highly oxidized sulfur species can result in protein structural changes or inhibit enzyme activity that requires thiolate for catalysis. Alternatively, these reported cysteine oxidation products also serve as a prerequisite for proper protein function. Thus, the irreversible oxidation of cysteine to sulfinic and sulfonic acid can influence cellular homeostasis and protein functions in multiple ways. Oxidation of cysteine to sulfenic and sulfinic acid modifications can be reversed by S-glutathionylation, wherein glutaredoxin mediates sulfinic acid reduction, conjugation of sulfenic acid via S-glutathionylation, and deglutathionylation by glutaredoxin or sulfiredoxin. As noted by the N-end rule pathway, irreversible cysteine oxidation can also target a protein for degradation; for example, oxidation of N-terminal cysteine residues to sulfinic and sulfonic acid in specific mammalian proteins, such as GTPase-activating proteins (RGS) is required for arginylation by ATE1 R-transferases and subsequent ubiquitin-dependent degradation. Thus, the overoxidation of cysteine to sulfonic acid cannot be reversed, and the damaged proteins have to be degraded by the proteasome (45). Apart from cysteine, the other sulfur-containing amino acids that undergo oxidative modification include methionine, which is reduced to methionine sulfoxide by methionine sulfoxide reductases. Methionine sulfone formation resulting from further oxidation events is considered a stable modification.
Concerning aromatic amino acids, tyrosine remains the primary target of protein oxidation events due to its redox-active structure. Its phenolic side chain gets oxidized easily, forming an intermediary tyrosyl radical. Upon reaction with HO•, these radicals form 3-hydroxylysine, a neurotransmitter analogue 3,4 dihydroxyphenylalanine (DOPA) and, in interaction with another tyrosyl radical, forms a fluorescent protein crosslink dityrosine. Hydroxyl radicals, upon interaction with tryptophan and histidine, form hydroxytryptophan and 2-oxohistidine, respectively (46).
Reported as a spontaneous non-enzymatic reaction, glycation involves the response of free-reducing sugars with lysine and arginine amino acid residues, DNA and lipids forming Amadori products. This product, in turn, undergoes irreversible rearrangement and dehydration reaction, leading to the formation of advanced glycation end products (AGEs) (47). Introduced by Louis-Camille Maillard in 1912, glycation results in loss of protein function and impaired tissue elasticity in the skin, blood vessels and tendons. Glycation reactions are reported to be enhanced during oxidative stress and hyperglycemia conditions, thus playing a pivotal role in the pathogenesis of diabetic complications and aging. The formation of AGEs does not entirely rely on oxidative conditions; more specifically, only selected AGEs are generated by oxidation. Formed by a combination of glycation and oxidation, a subset population of AGEs is termed glycoxidation products. Excessive generation of ROS from glucose autoxidation and covalent attachment of glucose molecules to circulating proteins results in the formation of AGEs (48). They serve as biomarkers for both oxidative and carbonyl stress. Carboxymethyl lysine (CML), reported as the most abundant AGEs in vivo, is formed by oxidative degradation of Amadori product fructoselysine. An alternative non-oxidative mechanism involves the reaction of α-dicarbonyl compound glyoxal and lysine, leading to CML formation via an isomerization mechanism. The oxidative degradation of carbohydrates, lipids, nucleotides, and serine mainly forms the precursor, glyoxal. Further oxidation reaction of this glyoxal results in the formation of α-oxoamide AGE glyoxylyl lysine. As it relies on oxidative processes, glyoxylyl lysine is considered an even more sensitive marker than CML. Apart from oxidatively formed glycoxidation products, some AGEs are formed by precursors generated by oxidation, for example, glucose oxidation or Amadori products to glucosone. Lysine-mediated cleavage of glucosone results in formyl lysine formation, and reports from recent studies confirm high levels of formaldehyde metabolism products (formyl lysine, formyl phosphate) in murine tissues (49).
An increase in levels of ROS can impose direct damage to lipids. The most prevalent ROS reported to affect lipids profoundly include HO• and hydroperoxyl (HO2•). In biological systems, the Fenton reaction forms HO• through redox cycling. Hydroperoxyl radicals play an essential role in lipid peroxidation, wherein this protonated form of O2•- produces H2O2, which can react with redox-active metals, yielding HO•. Hydroperoxyl radicals (HO2•) are reported to be much stronger than O2•- and could initiate the oxidation of polyunsaturated phospholipids, thereby impairing membrane function (28). The lipid peroxidation process involves hydrogen abstraction from carbon with oxygen insertion, resulting in lipid peroxyl radicals and hydroperoxides (LOOH). More specifically, free oxygen radicals target lipids containing carbon-carbon double bond(s), especially polyunsaturated fatty acids (PUFAs). Under physiological or subtoxic lipid peroxidation rates, cells survive by upregulation of antioxidant pathways and proteins, whereas, at toxic concentration levels, cells induce apoptosis, eventually leading to cellular damage (32). Thus, lipid peroxidation events might facilitate disease progression and aging. During the process of lipid oxidation, several reactive carbonyl species (RCS) are formed, which include LOOH and different aldehydes formed as secondary products, such as malondialdehyde (MDA), propanal, hexanal, and 4-hydroxynonenal (4-HNE) (4).
Malondialdehyde is reported to be the most abundant secondary aldehyde generated by the decomposition of arachidonic acid and larger PUFAs. The reactivity of MDA is pH-dependent; thus, at physiological pH, it exists as an enolate ion with low reactivity. Upon a pH decrease, MDA enolizes to β-hydroxy acrolein with increased reactivity. Malondialdehyde initial reaction with proteins generates Schiff-base adducts referred to as advanced lipid peroxidation end-products (ALEs). Under oxidative stress conditions, acetaldehyde in the presence of MDA generates highly immunogenic malondialdehyde acetaldehyde (MAA) adducts. These adducts are of biological importance as they can alter the functional properties of biomolecules, resulting in disease progression. Protein Kinase C (PKC) plays a vital role in the intracellular signal transduction process, which involves cell proliferation and differentiation, inflammation and cytoskeletal organization. The binding of MAA adducts induces activation of PKC-α, a specific isoform in hepatic cells, resulting in increased secretion of urokinase-type plasminogen activator, causative of hepatic fibrosis (44). Another important example of α/β unsaturated RCS is 4-hydroxy-2- nonenal (4-HNE). They are reported to be highly reactive, wherein nucleophilic attack of cysteine and histidine forms stable Michael adducts (45). 4-HNE protein adducts can contribute to protein crosslinking and induce carbonyl stress. For example, 4-HNE is reported to modify membrane-associated protein, G-protein signaling 4 (RGS4) at cysteine residue during oxidative stress, thereby altering signaling events in stressed cells (46). However, based on their cellular level and pathways involved in lipid peroxidation products, MDA and 4-HNE pose a dual behaviour of either enhancing cell survival or promoting cell death.
ROS-mediated protein carbonylation events are characterized as the most common type of non-enzymatic post-translational modification (PTM). This stable modification is achieved by either direct oxidation of protein-bound amino acids, oxidative cleavage of the protein backbone and incorporation of carbonyls from glycoxidation or lipoxidation. ROS/reactive intermediates such as H2O2 and lipid hydroperoxides interact with specific amino acids, arginine, lysine, proline or threonine, causing protein-protein cross-linkages, resulting in protein denaturation and loss of activity. Various oxidation products have been reported so far, which include tryptophan forms kynurenine, nitrotryptophan; Phenylalanine forms 2,3- 2-, 3-, and 4-hydroxyphenylalanine, Dihydroxyphenylalanine; Histidine forms 2-Oxohistidine; Arginine and proline forms glutamic semialdehyde (50). Superoxide anion radical formation from O2 or HO• by the interaction of H2O2 with free iron (Fe2+) through the Fenton reaction results in the interaction of ROS with the amino acids mentioned above. Superoxide anion radical generated from O2 is converted by SOD to H2O2 and later into H2O by catalase, glutathione peroxidase or peroxiredoxin (47). The direct oxidation of amino acids, aminoadipic and glutamic semialdehyde contribute to approximately 60% of total protein carbonylation in the liver. Hydroxyl radical-mediated abstraction of hydrogen located next to the N6-amino function of lysine, metal-catalyzed oxidation of the carbon-centered radical, and hydrolysis of the resulting imine mediates aminoadipic semialdehyde formation. Concerning oxidative cleavage of the protein backbone, O2•- facilitates RO• formation at α-carbon next to a peptide bond. The RO• fragments either through the diamide pathway (homolytic cleavage of carbon-carbon bond) or the α-amidation pathway (carbon-nitrogen bond) (50). Protein carbonylation remains a valuable biomarker in aging and diseases, wherein they are shown to impair protein structure and function. In a carbonylated protein profiling study from lean and obese individuals with or without type 2 diabetes (T2D), 36 out of 158 unique carbonylated proteins were reported to be present only in obese patients with T2D. These identified proteins were found to play a vital role in intracellular signaling and angiogenesis, cell adhesion and cytoskeletal remodeling (51).
Highly oxidized proteins appear to be relatively poor substrates for degradation by ubiquitination. Thus, dysfunctional carbonylated proteins accumulate as covalently crossed protein aggregates, making them highly resistant to proteolysis, thereby affecting the functional integrity of cells during the aging and disease processes. On the contrary, proteins that have undergone mild oxidation are highly susceptible to proteasomal degradation due to exposure to hydrophobic amino acids by unfolding targeted protein domains. Hydrophobic surface patches remain the central motif recognized by the proteasome. Remarkably, 19S and 20S proteasome subunits are highly susceptible to carbonylation and HNE modification, suppressing their proteolytic activities (52). Protein carbonylation may also be beneficial by regulating and activating signaling pathways involved in antioxidant defense and cellular homeostasis. Carbonylation depends on the cellular redox environment, ROS abundance and its proximity to the proteins.
Nitric oxide (NO), also referred to as Nitric oxide radical (•NO), remains an important signaling molecule that exhibits pleiotropic functions like vasodilation, neurotransmission and pro-inflammatory signaling. Nitric oxide synthase (NOS) utilizes L-arginine as a substrate and, along with oxygen, produces citrulline and NO. They are reported to mediate both anti- and pro-oxidant mechanisms. In immune cells, nitric oxide limits ROS production via NADPH oxidase and pretreatment of cells with NO protects them against oxidative stress. Recent clinical trials support the beneficial effects of NO pretreatment in ischemia–reperfusion-mediated tissue injury (53). On the contrary, NO readily reacts with O2•- forming RNS, peroxynitrite (ONOO−) and nitrogen dioxide (NO2). In vivo, these RNS are reported to be potent oxidizing agents that can directly or reversibly modify cysteine residues through S-nitrosylation. Nitrogen dioxide radicals interact at the ortho-position of the tyrosine aromatic ring, resulting in the formation of irreversible modification of 3-nitrotyrosine. Both peroxynitrite and protein tyrosine nitration is reportedly involved in aging. Protein tyrosine nitration serves as a potential biomarker of disease progression. While initial experiments demonstrated the involvement of endogenous peroxynitrite and protein tyrosine nitration in apoptosis of motoneurons in culture, further work in ALS animal models confirmed the formation of 3-nitrotyrosine and protein-derived radicals in spinal cord motor neurons during disease progression (54, 55). Various protein oxidation events used as biomarkers in clinical settings are presented in Table 1.
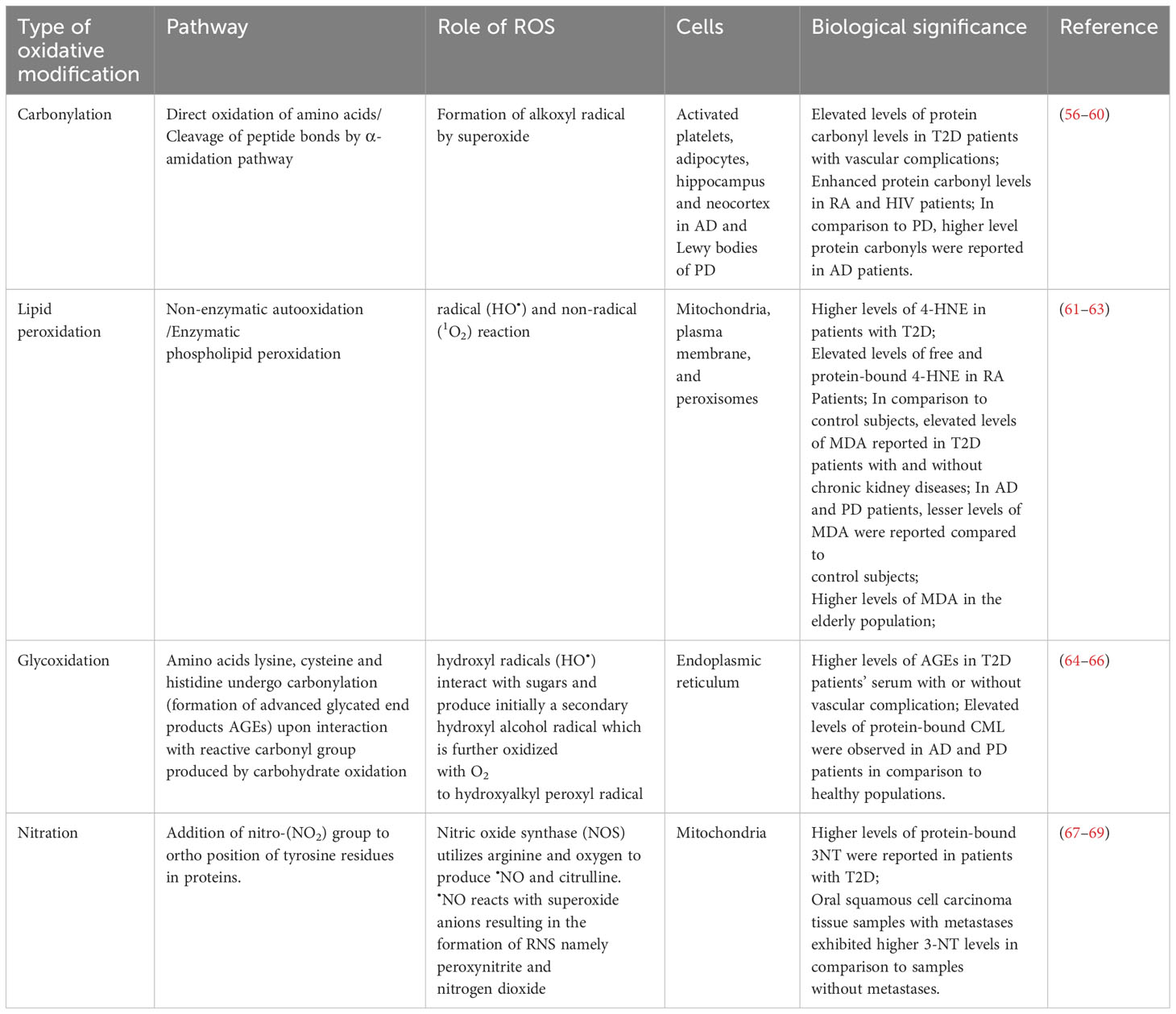
Table 1 Oxidative modification of proteins as biomarkers in clinical studies.
Macrophages are a heterogeneous population of immune cells that play a vital role in tissue homeostasis in response to pathogen infection by phagocytosis and mediate tissue repair during injury. They rapidly recognize, engulf and destroy pathogens or apoptotic cells, which can be attributed to their plasticity and heterogeneity (70). Through polarization events, macrophages adopt either a pro-inflammatory phenotype classified as M1 macrophage or an anti-inflammatory M2 phenotype that mediates wound healing and inflammation resolution (71). Recently, it has been observed that excess molecular stimuli induce diverse and partially overlapping macrophage phenotypes that are distinct from M1 and M2. Macrophage activation by ROS, cytokines and commensal lipopolysaccharide (LPS) results in the activation of NF-κB and PI3K/AKT signaling pathways (72, 73). Thus, upregulated NF-κB increases pro-inflammatory chemokines and cytokine transcription, inducible NO synthase (iNOS) and HIF1 α. These signaling pathways, enzymes, and transcription factors are essential in maintaining macrophage activation and M1 polarization by driving metabolic reprogramming (17).
ROS regulate the intracellular signalosome within a constantly evolving cellular microenvironment, thus underlying its role in polarization and the specialized function of immune cell populations (74). M1 and M2 macrophages differ from resting macrophages in their phenotype and exhibit distinct metabolic profiles (75). M1 macrophage metabolism is characterized by aerobic glycolysis, changes in pentose phosphate pathway (PPP), FAS, and truncated TCA cycle, while M2 mostly depends on oxidative metabolism, fatty acid oxidation (FAO) and decreased glycolysis (76) (Figure 2).
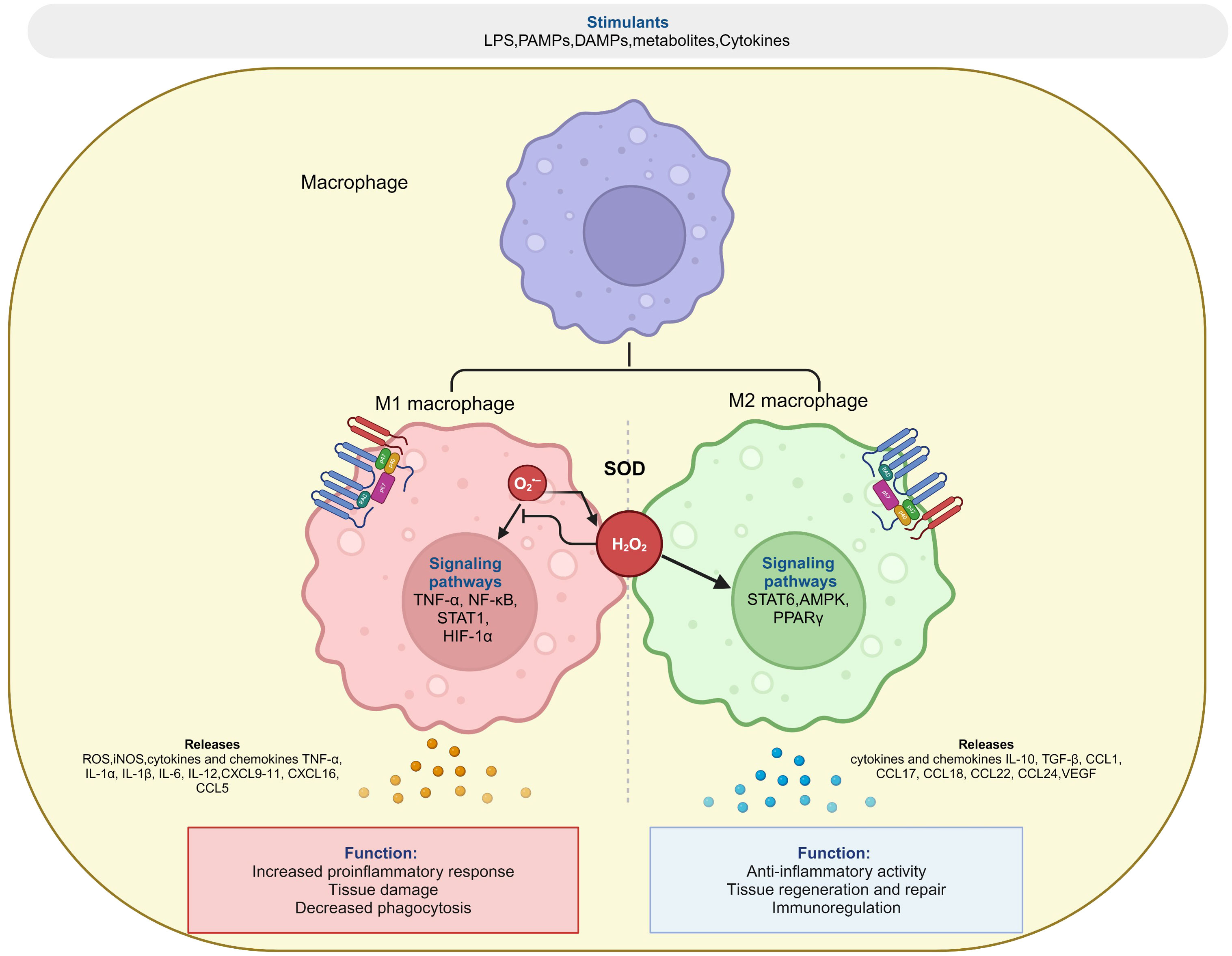
Figure 2 Redox regulation of macrophage polarization. Superoxide (O2•-) generated by NOX or mitochondrial electron transfer chain is converted into H2O2 by superoxide dismutase (SOD), which balance between both pro-inflammatory (M1) and anti-inflammatory (M2) response of macrophage during polarization events. The figure was Created with BioRender.com.
Glycolysis is a crucial metabolic event for M1 macrophages, and its inhibition affects typical functions of their inflammatory phenotype, including phagocytosis, ROS production, and pro-inflammatory cytokine secretion. Lipopolysaccharides and Toll-like receptor (TLRs) mediated differentiation of M1 macrophages are associated with a metabolic shift towards glycolysis, blocking it by glucose derivate like 2-deoxyglucose impairs both ROS and pro-inflammatory cytokine production. Numerous transcription factors are known to be involved in maintaining metabolic changes associated with M1 macrophages, and notable factors were discussed in detail. Reports demonstrated the involvement of HIF1α in activating inflammatory macrophage through the glycolysis mechanism (77). They act as modulators of the methylation status of hypoxia-responsive elements in the promoter regions. Enhanced expression of HIF1α reduces mitochondrial activity by suppressing electron transport chain enzymes, resulting in mitochondrial autophagy. HIF1α exaggerates glycolytic flux, thereby increasing the expression of glucose transporters (GLUT1 and GLUT3) and inflammatory mediators. More specifically, M1 macrophages rely on glycolysis and accumulation of succinate from the TCA cycle to stabilize HIF1α, which in turn activates the transcription of glycolytic genes sustaining glycolytic metabolism in M1 macrophages (36). Increased levels of HIF1α were evident along the differentiation of monocytes into tissue macrophages and play a prominent role in the uptake of bacteria by macrophages under hypoxic conditions and expression of tumour necrosis factor (TNFα) and nitric oxide (NO) through inducible NO synthetase (iNOS). In macrophages, overexpression of glucose receptors enhances glycolysis, which induces ROS production and pro-inflammatory mediators (78). In M1 macrophages, aerobic glycolysis induction depends entirely on redox-sensitive transcription factor HIF1α activated by the NF-κB pathway during inflammation. HIF1α interacts with pyruvate kinase isoenzyme M2 (PKM2), thereby mediating the transcription of glycolytic enzymes and inflammatory factors like IL-1β. ROS-like NO-mediated prolyl hydroxylases (PHDs) inhibition induces HIF1α (79). Further, NO reduces oxidative phosphorylation by nitrosylation, inhibiting proteins involved in the mitochondrial electron transport chain. Thus, ROS production positively regulates and maintains the shift towards aerobic glycolysis in M1 macrophages.
NADPH generated by PPP regulates the inflammatory response of M1 macrophages. They mediate ROS and NO production through NOX and iNOS, respectively, to kill invading pathogens and sustain the functionality of TRX and GSH antioxidant systems. Results presented by Nguyen and co-workers demonstrate that deletion of the TRX1 system impairs NLRP3 inflammasome formation and binding of NF- κB to target DNA in monocytes and macrophages (10). It leads to defective production of pro-inflammatory cytokines and ROS accumulation. Additionally, mitochondrial ROS-induced DNA damage aid in a significant drop in NAD+ levels in M1 macrophages. Suppression of PPP in macrophages attenuates LPS-induced inflammatory and oxidative stress response. Compared to glycolysis, intermediates of the TCA cycle, namely succinate and citrate, support biosynthesis in M1 macrophages (11). In LPS-stimulated macrophages, two breaks in the TCA cycle result in the accumulation of succinate and citrate, stabilizing HIF-1α and the subsequent increase in IL-1β transcription. Furthermore, succinate dehydrogenase-mediated oxidation of succinate and increased mitochondrial membrane potential drive ROS production (72). Another TCA cycle metabolite, citrate, gets transported into the cytosol and utilized for fatty acid synthesis to support membrane biogenesis and synthesis of pro-inflammatory lipid mediators, namely prostaglandins. In LPS-activated macrophages, ROS-dependent oxidation of unsaturated phospholipids results in glutamine utilization to feed the TCA cycle and lead to cytoplasmic accumulation of oxaloacetate. These metabolites stabilize HIF-1α, enhancing IL-1β secretion in atherosclerosis (13).
In M1 macrophages, ROS-mediated activation of NRF2 is essential for PPP maintenance and NADPH production, which is necessary for FAS, TRX, and GSH systems. IL-1β is produced as an inactive precursor in response to pathogens, and thus, further processing of it into a biologically active form requires the formation of multiprotein complexes termed inflammasomes. Mitochondrial ROS and its oxidation products are known to play a prominent role in inflammasome activation, whereas the role of NOX is highly dispensable (14).
Unlike M1 macrophages, M2 macrophages hold an intact TCA cycle and enhanced mitochondrial OXPHOS. CD36 internalizes circulating lipoproteins and fatty acids, mediating fatty acid uptake and fueling OXPHOS. The increased cellular concentration of IL-4 and IL-13 drives M1 macrophages towards anti-inflammatory and healing phenotypes described as M2 macrophages. Tyrosine phosphorylation and signal transducer and activator of transcription 6 (STAT6) activation mediate polarization of macrophages into the M2 phase. IL-4 and IL-13 suppress pro-inflammatory cytokine production by upregulating transforming growth factor beta (TGF-β) activity (70). Adenosine 5′-monophosphate-activated protein kinase (AMPK) and peroxisome proliferator-activated receptor (PPAR) are found to play a vital role in the transition of macrophage polarization states through IL-13 and IL-4. AMPK inhibits NF-κB and stimulates OXPHOS and FAO, reducing HIF1α levels and inflammation and terminating aerobic glycolysis (80). AMPK negatively regulates LPS-induced inflammatory response in macrophages by inhibiting NF-κB activity and activating the PI3K/Akt signaling pathway. Enhanced IL-10 expression promotes TAM activation by PPARα/β, resulting in a polarization of M2 macrophages (12). The importance of FAO in M2 polarization was highlighted in several studies wherein blocking FAO with inhibitors against mitochondrial carnitine palmitoyl-transferase 1 inhibited the activation of M2 macrophages. Results from various studies confirm the PPARγ-mediated activation of M2 signature genes either through oleic acid and IL-4 stimulation or by promoting glutamine oxidation fueling OXPHOS (81). Modification in arginine metabolism emphasizes the transition from M1 to M2 polarization. Increased activity of iNOS mediates arginine metabolism to produce NO, which maintains the switch towards aerobic glycolysis in M1 macrophages. In the case of M2 macrophages, arginine gets metabolized into ornithine and urea due to increased transcription of arginase-1. Both urea and ornithine are essential in M2 macrophage proliferation and survival. Additionally, glutamine metabolism is particularly interesting since glutamine oxidation depletes extracellular glucose levels in an inflammatory environment, maintaining TCA activity and activating the glutamine–UDP-N-acetylglucosamine (GlcNAc) pathway to reinforce M2 polarization (82).
Neutrophils are the first responders against invading pathogens through innate and humoral immunity. Activated neutrophils rely on glycolysis as their primary source of energy under both physiological and inflammatory environments; however, they can regulate their metabolism to carry out its effector functions, namely phagocytosis, oxidative burst, degranulation, extracellular trap formation and chemotaxis (83). Neutrophil extracellular trap (NETs) formation by neutrophils relies on glycolysis and PPP as a source of NADPH, resulting in free oxygen radical production. Superoxide anion radical, thus produced, further induces the formation of ROS and HOCl used by neutrophils in oxidative bursts following phagocytosis of invading pathogens. ROS promote several steps of NETosis, including releasing neutrophil elastase from granules by increasing membrane permeability and degradation of H1 linker and core histones resulting in chromatin decondensation (84). The morphological changes associated with NETosis were promoted by ROS, which in turn inactivated caspases to block apoptosis and trigger autophagy. Secondary oxidants, namely HOCl, mediate PMA-induced NETosis, and it entirely depends on NOX activity (Figure 3). The absence of extracellular Cl-, a substrate for Myeloperoxidase (MPO) in vitro results in decreased NET production (85). On the contrary, calcium ionophores induced NETosis are NOX independent and rely on mtROS. NOX-independent Netosis depends on calcium which in turn activates peptidyl arginase deiminases resulting in cellular hypercitrullination (86).
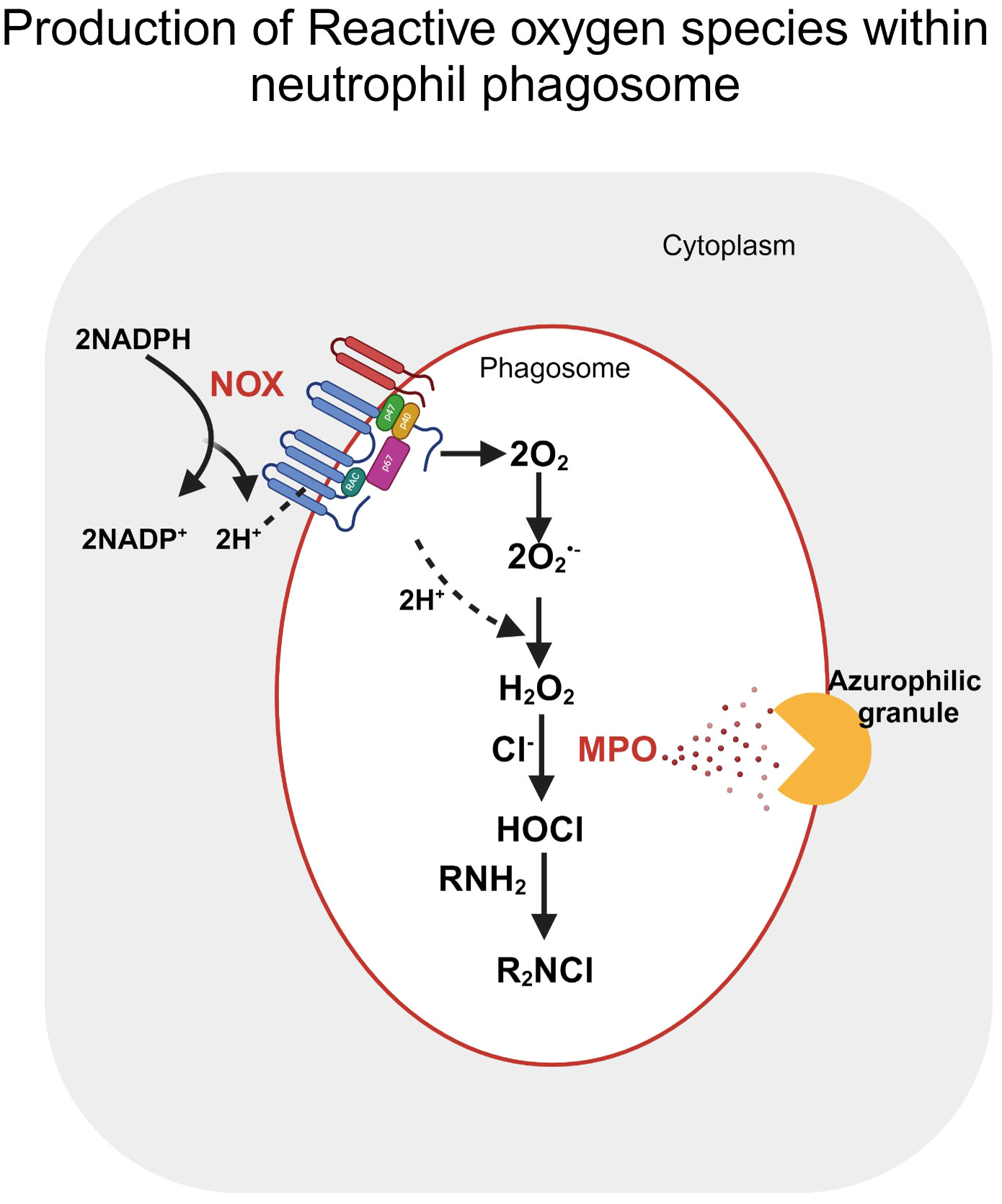
Figure 3 Reactive oxygen species production within the neutrophil phagosome. Post stimulation, oxygen reduction by NOX in the presence of NADPH produces superoxide (O2•-) within the phagosome. H2O2 produced by either the enzymatic or spontaneous dismutation of Superoxide further stimulates neutrophil granules, releasing myeloperoxidase (MPO) within the phagosome. MPO catalyses the oxidation of halides (Cl-) by interaction with H2O2, forming HOCl. The figure was Created with BioRender.com.
Neutrophil functions are highly influenced by cellular redox status, which includes both ROS/RNS production and cellular antioxidant systems (87). Enhanced ROS production is reported to comprise phagocytosis, resulting in dysregulated oxidative burst events and NET production. ROS levels determine the sensing of pathogens by neutrophils and their subsequent activation of NLRP3 inflammasome and cytokine synthesis (88). Additionally, chronically upregulated ROS and cytokine production lowers neutrophil migration by internalizing CXCR2, a membrane chemokine receptor. Neutrophil functions, namely oxidative burst and NET formation, were sustained by the glutathione system (GSH). Its basal activity was reported to be lower in neutrophils compared to other myeloid cells. Prolonged neutrophil activity and excessive production of MPO due to chronic nitrooxidative stress and inflammation lead to the depletion in GSH levels (89). Thus, depleted GSH levels in neutrophils affect their chemotaxis, transmigration and cytoskeletal reorganization, resulting in early apoptosis and impaired degranulation. Redox factors involved in macrophage and neutrophil function are presented in Table 2.
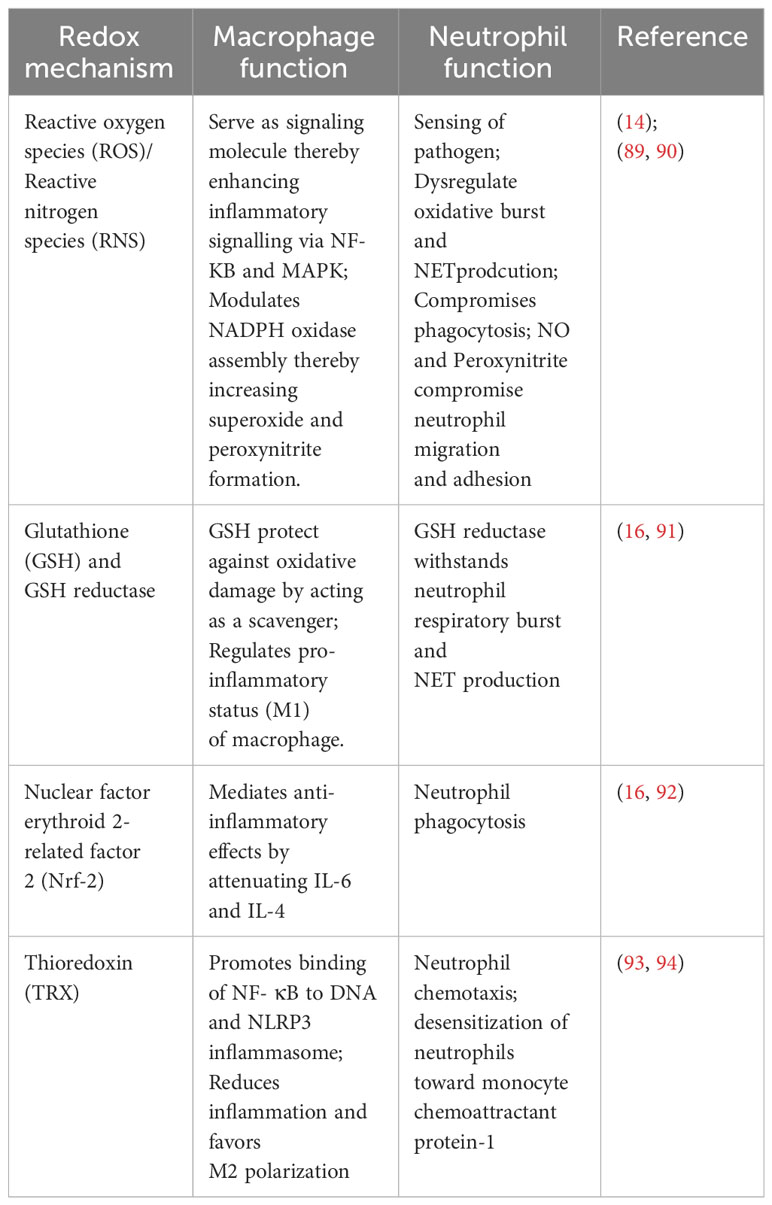
Table 2 Redox mechanism influencing macrophage and neutrophil functions.
Multiple transcriptional and epigenetic modification factors are known to be involved in macrophage differentiation and its activation. Epigenetic changes within macrophages allow them to switch between cellular programs affecting phenotype plasticity. DNA demethylation is known to be involved in the process of monocyte-to-macrophage differentiation. DNA demethylation affects specific genes that regulate actin cytoskeleton and phagocytosis (19). ROS-mediated oxidation of amino acid residues in histone H3 results in chromatin relaxation and accumulation of transcription factors. Cysteine residues at histone H3 sense redox changes and mediate further opening of chromatin structures (95). In LPS-stimulated macrophages, lipid peroxidation products are reported to form lysine adducts with H2, H3 and H4, including H3K23 and H3K27. These modifications at histone acetylation and methylation sites are known to be associated with the epigenetic patterning of cardiovascular diseases (96). LPS stimulation and TLR-4-dependent activation of inflammatory genes primarily depend on H3 and H4 acetylation. ROS-mediated post-translational modification of both class I and II histone deacetylases impair its enzymatic function, resulting in open chromatin structure.
Additionally, lipid peroxidation products mediate the carbonylation of HDACs, resulting in ubiquitination and proteasomal degradation of HDAC function, increased acetylation of histones in macrophages and release of pro-inflammatory cytokines. HDACs are predominant in regulating immunological pathways, more precisely in M1 activation. The difference in the expression pattern of almost all HDAC classes was observed in cells stimulated with LPS. Studies concerning macrophage stimulation with LPS result in an initial decrease in HDAC 4,5 and 7 expression, thereby leading to cyclooxygenase-2 gene activation (97). On the contrary, HDAC6 aids in the expression of pro-inflammatory genes in macrophages stimulated with LPS and thus, its inhibition limits macrophage activation. Concerning the link between DNA methylation and LPS stimulation, SOCS1, a negative regulator of cytokine signals, has been found. DNMT1-mediated hypermethylation of SOCS1 results in the loss of its activity, thereby enhancing the expression of LPS-induced Pro-inflammatory cytokines, namely TNF-α and IL-6. DNMT1 has also been reported to improve the demethylation and trimethylation events of H3K9 in regulator proteins like Notch1 and KIF4 and mediate their polarization towards M1 macrophages. Furthermore, DNA methyltransferase (DNMTs) is known to be involved in M2 differentiation and phenotypic regulation. Individuals with atherosclerosis and apolipoprotein E knockout mice fed an atherogenic diet displayed enhanced DNMT levels in macrophages (98). Since the activity of PPAR-γ is reduced, the macrophage transition from M1 to M2 is affected, and thus, the progression of atherosclerosis is marked by increased pro-inflammatory cytokine production. Oxidative stress is linked to increased histone acetyltransferase (HATs) activity of p300/CBP along with NFĸB DNA binding, promoting pro-inflammatory gene expression. In endothelial cells and hyperglycemic adipocytes, enhanced expression of HAT GCN5 and H3 acetylation is associated with increased ROS production, as confirmed in diabetic models. p300/CBP mediated acetylation of H3K9 at NOX2 promoter encourages ROS generation underlying complexity of epigenetic modifications in ROS balance and response (16).
Upon activation, neutrophils generate a range of ROS and O2•- generated were reported to damage proteins and are limited to the compartment site it has been generated as its rate of dismutation was enhanced by SOD to H2O2. Neutrophil heme protein myeloperoxidase utilizes H2O2 to oxidize the halides chloride (Cl-), iodide (I-), and bromide (Br-), or pseudohalide anion thiocyanate (SCN-), into hypochlorous acid (HOCl) (99). Being a potent oxidant, HOCl poses a high reactivity towards biological macromolecules targeting free and protein-associated cysteine, methionine residues and low molecular weight thiols, and its ability to diffuse from the site of generation is very short. Thus, HOCl reacts with amines and forms chloramines, which are less reactive than HOCl and diffuse further from the generation site. The reaction of HOCl and chloramines with cytosine produces 5-chlorocytosine (5-clC), directly incorporated into DNA as a chlorinated nucleotide (100). Studies carried out reported gene silencing, and no significant changes were observed in global methylation levels due to the incorporation of 5-clC. Chloramines are also reported to interact with histone amine groups, thereby preventing methylation or acetylation events (101). However, the lack of in vivo experimental evidence makes it unclear how 5-clC and chloramine levels are regulated under physiological conditions. Neutrophil oxidants can react with cellular targets, including small molecules and redox-sensitive components of epigenetic pathways. Intracellular availability of methionine and ascorbate was depleted by neutrophil oxidants and reported to impact methylation by disrupting S-adenosylmethionine (SAM) levels (102). Mass spectrometry analysis to investigate the oxidant effects of methylation reported impaired cytosine methylation on newly replicated DNA in the Jurkat T-lymphoma cell line upon sub-lethal level exposure to glycine chloramines (103). The study reported DNMT1 inhibition and depletion of SAM levels at doses, which had minimal effect on cell proliferation. Though H2O2 treatment inhibited DNMT1, it did not reduce SAM or global methylation levels (104). Further experimentation is required to determine whether the methylation and demethylation effects observed are heritable to subsequent generations.
A sustained pro-oxidant cellular environment mediates the development and progression of various pathological conditions due to redox imbalance. Dysregulation in these redox environments decreases the activity of mitochondria, TCA cycle and immune cell metabolism (30). ROS and RNS are ubiquitous byproducts of cellular metabolism, and any disparity between their generation and degradation in aging and diseases results in oxidative and nitrosative stress. Oxidative stress can irreversibly damage cellular structures, including membrane lipids or lipoproteins, forming oxidation-specific epitopes (OSE) on damaged cells (105). This damage-associated molecular pattern is recognized and removed by innate immune cells, including macrophages and neutrophils, enabling cellular homeostasis. Excessive accumulation of these oxidation products triggers chronic inflammation and metabolic disorders, including atherosclerosis, diabetes and age-related macular degeneration (106). Immune cells function and survival are regulated by various redox factors, including the intracellular and extracellular concentration of ROS/RNS and cellular antioxidants, namely glutathione, thioredoxin and Nrf-2 (107). Considering diabetes, metabolic imbalance in these conditions is characterized by increased glycolytic flux, and ROS act as a secondary messenger and mediates metabolic shift towards pro-inflammatory macrophage phenotype. ROS were also reported to activate multiple pro-inflammatory signaling pathways, including MAPK, NLRP3 and NFκB, resulting in an epigenetic modification in hyperglycemic conditions (108). A crosstalk between these immune cells and endothelial cells in diseased conditions is reported to stimulate increased ROS formation and inflammatory phenotypes further. Thus, consideration should be paved towards ROS generated by macrophages and neutrophils to suppress inflammation in metabolic disorders.
The functioning of individual immune cells is under redox control and reported to be sensitive to intracellular and extracellular concentrations of ROS and influenced by the activity of cellular antioxidants. Redox mechanisms regulate and modulate various immune functions, including metabolic reprogramming of dendritic cells (DCs), T cells, B cells, and natural killer cells (NK), aiding in its activation and regulation (109). ROS are reported to be involved in diverse biological events, including Epithelial–mesenchymal transition (EMT). This transdifferentiation process is vital in invasion and metastasis phenomena during neoplastic progression. ROS regulate the integrin arrangement and urokinase plasminogen activator (uPA) pathway in extracellular matrix remodelling (110). ROS can influence the function of various proteins involved in the EMT process through reversible or irreversible oxidative modification of protein on free cysteine residues (111). Thus, targeting redox regulation to prevent EMT and tumor metastasis is promising.
Mechanistic insight into the specific immune response generated for oxidation-specific epitopes at functional levels should be studied, which can aid in understanding oxidative stress and its associated chronic inflammations. Though M1 and M2 represent the two extreme phenotype characteristics of macrophage activation stages regulated by redox metabolism, tissue-resident macrophages comprise a distinct subset and hold tissue-specific functions dependent on oxygen and nutrient supply, which can certainly influence redox status and metabolism (112). Thus, studies focusing on the characterization of redox proteome during an immune response are essential. Methodology focusing on in vivo localization and visualization of ROS and their sources will aid in a better understanding of this complex redox metabolism in health and diseases.
RRM: Conceptualization, Writing – original draft, Writing – review & editing. AP: Conceptualization, Project administration, Supervision, Writing – original draft, Writing – review & editing. PP: Writing – review & editing. JK: Conceptualization, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by [1] the European Regional Development Fund project “Plants as a tool for sustainable global development” (CZ.02.1.01/0.0/0.0/16_019/0000827); [2] state contract of the Ministry of Science and Higher Education of the Russian Federation “Genetic and epigenetic editing of tumor cells and microenvironment in order to block metastasis” no. 075-15-2021-1073 (to JK); [3] Tomsk State University Development Programme (Priority-2030).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
3-NT, 3-Nitrotyrosine; 4-HNE, 4-Hydroxy-2-nonenal; AD, Alzheimer’s disease; PD, Parkinson disease; CML, Carboxymethyl lysine; AGEs, Advanced glycation endproduct; DNPH 2,4-Dinitrophenylhydrazine; RA, Rheumatoid arthritis; RCS, Reactive carbonyl species; RNS, Reactive nitrogen species; ROS, Reactive oxygen species; T2D, Type 2 diabetes; VCAM1 Vascular cell adhesion protein 1; ICAM1, Intercellular Adhesion Molecule 1.
1. Checa J, Aran JM. Reactive oxygen species: drivers of physiological and pathological processes. JIR. (2020) 13:1057–73. doi: 10.2147/JIR.S275595
2. Phaniendra A, Jestadi DB, Periyasamy L. Free radicals: properties, sources, targets, and their implication in various diseases. Ind J Clin Biochem. (2015) 30:11–26. doi: 10.1007/s12291-014-0446-0
3. Prasad A, Manoharan RR, Sedlářová M, Pospíšil P. Free radical-mediated protein radical formation in differentiating monocytes. IJMS. (2021) 22:9963. doi: 10.3390/ijms22189963
4. Manoharan RR, Sedlářová M, Pospíšil P, Prasad A. Detection and characterization of free oxygen radicals induced protein adduct formation in differentiating macrophages. Biochim Biophys Acta (BBA) Gen Subj. (2023) 1867:130324. doi: 10.1016/j.bbagen.2023.130324
5. Sharifi-Rad M, Anil Kumar NV, Zucca P, Varoni EM, Dini L, Panzarini E, et al. Lifestyle, oxidative stress, and antioxidants: back and forth in the pathophysiology of chronic diseases. Front Physiol. (2020) 11:694. doi: 10.3389/fphys.2020.00694
6. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signaling agents. Nat Rev Mol Cell Biol. (2020) 21:363–83. doi: 10.1038/s41580-020-0230-3
7. Lambeth JD. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Radical Biol Med. (2007) 43:332–47. doi: 10.1016/j.freeradbiomed.2007.03.027
8. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. (2014) 94:909–50. doi: 10.1152/physrev.00026.2013
9. Fransen M, Nordgren M, Wang B, Apanasets O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim Biophys Acta (BBA) Mol Basis Dis. (2012) 1822:1363–73. doi: 10.1016/j.bbadis.2011.12.001
10. Nguyen GT, Green ER, Mecsas J. Neutrophils to the ROScue: mechanisms of NADPH oxidase activation and bacterial resistance. Front Cell Infect Microbiol. (2017) 7:373. doi: 10.3389/fcimb.2017.00373
11. Morris G, Gevezova M, Sarafian V, Maes M. Redox regulation of the immune response. Cell Mol Immunol. (2022) 19:1079–101. doi: 10.1038/s41423-022-00902-0
12. Muri J, Thut H, Feng Q, Kopf M. Thioredoxin-1 distinctly promotes NF-κB target DNA binding and NLRP3 inflammasome activation independently of Txnip. eLife. (2020) 9:e53627. doi: 10.7554/eLife.53627
13. Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. (2014) 6:411–21. doi: 10.1038/nrm3801
14. Van der Vliet A, Janssen-Heininger YM. Hydrogen peroxide as a damage signal in tissue injury and inflammation: murderer, mediator, or messenger? J Cell Biochem. (2014) 3:427–35. doi: 10.1002/jcb.24683
15. Hattori H, Subramanian KK, Sakai J, Jia Y, Li Y, Porter TF, et al. Small-molecule screen identifies reactive oxygen species as key regulators of neutrophil chemotaxis. Proc Natl Acad Sci USA. (2010) 107:3546–51. doi: 10.1073/pnas.0914351107
16. Gostner JM, Becker K, Fuchs D, Sucher R. Redox regulation of the immune response. Redox Rep. (2013) 18:88–94. doi: 10.1179/1351000213Y.0000000044
17. Rendra E, Riabov V, Mossel DM, Sevastyanova T, Harmsen MC, Kzhyshkowska J. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology. (2019) 224:242–53. doi: 10.1016/j.imbio.2018.11.010
18. Ikeda S, Yamaoka-Tojo M, Hilenski L, Patrushev NA, Anwar GM, Quinn MT, et al. IQGAP1 regulates reactive oxygen species-dependent endothelial cell migration through interacting with Nox2. Arteriosc Thromb Vasc Biol. (2005) 11:2295–300. doi: 10.1161/01.ATV.0000187472.55437.af
19. Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. (2018) 18:e27. doi: 10.4110/in.2018.18.e27
20. Pitocco D, Tesauro M, Alessandro R, Ghirlanda G, Cardillo C. Oxidative stress in diabetes: implications for vascular and other complications. Int J Mol Sci. (2013) 14:21525–50. doi: 10.3390/ijms141121525
21. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a Pro-inflammatory phenotype. J Biol Chem. (2014) 289:7884–96. doi: 10.1074/jbc.M113.522037
22. Yao J, Wu D, Qiu Y. Adipose tissue macrophage in obesity-associated metabolic diseases. Front Immunol. (2022) 13:977485. doi: 10.3389/fimmu.2022.977485
23. Wu J, Yan Z, Schwartz DE, Yu J, Malik AB, Hu G. Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. J Immunol. (2013) 190:3590–9. doi: 10.4049/jimmunol.1200860
24. Reczek CR, Chandel NS. ROS-dependent signal transduction. Curr Opin Cell Biol. (2015) 33:8–13. doi: 10.1016/j.ceb.2014.09.010
25. Hasheminasabgorji E, Jha JC. Dyslipidemia, diabetes and atherosclerosis: role of inflammation and ROS-redox-sensitive factors. Biomedicines. (2021) 11:1602. doi: 10.3390/biomedicines9111602
26. Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. (2015) 1:5–23. doi: 10.1038/cmi.2014.89
27. Shen N, Wang Y, Sun X, Bai X, He J, Cui Q, et al. Expression of hypoxia-inducible factor 1α, glucose transporter 1, and hexokinase 2 in primary central nervous system lymphoma and the correlation with the biological behaviors. Brain Behav. (2020) 10:e01718. doi: 10.1002/brb3.1718
28. Baillet A, Hograindleur M, El Benna J, Grichine A, Berthier S, Morel F, et al. Unexpected function of the phagocyte NADPH oxidase in supporting hyperglycolysis in stimulated neutrophils: key role of 6-phosphofructo-2-kinase. FASEB J. (2017) 31:663–73. doi: 10.1096/fj.201600720R
29. Tirichen H, Yaigoub H, Xu W, Wu C, Li R, Li Y. Mitochondrial reactive oxygen species and their contribution in chronic kidney disease progression through oxidative stress. Front Physiol. (2021) 12:627837. doi: 10.3389/fphys.2021.627837
30. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. (2018) 6:877–902. doi: 10.1161/CIRCRESAHA.117.311401
31. Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal. (2014) 3:396–413. doi: 10.1089/ars.2014.5851
32. Espinosa-Diez C, Miguel V, Mennerich D, Kietzmann T, Sánchez-Pérez P, Cadenas S, et al. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. (2015) 6:183–97. doi: 10.1016/j.redox.2015.07.008
33. Zeeshan HM, Lee GH, Kim HR, Chae HJ. Endoplasmic reticulum stress and associated ROS. Int J Mol Sci. (2016) 3:327. doi: 10.3390/ijms17030327
34. Prior KK, Wittig I, Leisegang MS, Groenendyk J, Weissmann N, Michalak M, et al. The endoplasmic reticulum chaperone calnexin is a NADPH oxidase NOX4 interacting protein. J Biol Chem. (2016) 13:7045–59. doi: 10.1074/jbc.M115.710772
35. Fransen M, Lismont C, Walton P. The peroxisome-mitochondria connection: how and why? Int J Mol Sci. (2017) 6:1126. doi: 10.3390/ijms18061126
36. Wanders RJ, Waterham HR, Ferdinandusse S. Metabolic interplay between peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Front Cell Dev Biol. (2016) 3:83. doi: 10.3389/fcell.2015.00083
37. Vermot A, Petit-Härtlein I, Smith SME, Fieschi F. NADPH oxidases (NOX): an overview from discovery, molecular mechanisms to physiology and pathology. Antioxid (Basel). (2021) 6:890. doi: 10.3390/antiox10060890
38. Zamora R, Vodovotz Y, Billiar TR. Inducible nitric oxide synthase and inflammatory diseases. Mol Med. (2000) 6:347–73. doi: 10.1007/BF03401781
39. Santos CXC, Stolf BS, Takemoto PVA, Amanso AM, Lopes LR, Souza EB, et al. Protein disulfide isomerase (PDI) associates with NADPH oxidase and is required for phagocytosis of Leishmania chagasi promastigotes by macrophages. J Leuk Biol. (2009) 86:989–98. doi: 10.1189/jlb.0608354
40. Reddie KG, Carroll KS. Expanding the functional diversity of proteins through cysteine oxidation. Curr Opin Chem Biol. (2008) 12:746–54. doi: 10.1016/j.cbpa.2008.07.028
41. Trujillo M, Alvarez B, Radi R. One- and two-electron oxidation of thiols: mechanisms, kinetics and biological fates. Free Radical Res. (2016) 50:150–71. doi: 10.3109/10715762.2015.1089988
42. Nagy P, Ashby MT. Reactive sulfur species: kinetics and mechanisms of the oxidation of cysteine by hypohalous acid to give cysteine sulfenic acid. J Am Chem Soc. (2007) 129:14082–91. doi: 10.1021/ja0737218
43. Vivancos AP, Castillo EA, Biteau B, Nicot C, Ayté J, Toledano MB, et al. A cysteine-sulfinic acid in peroxiredoxin regulates H 2 O 2 -sensing by the antioxidant Pap1 pathway. Proc Natl Acad Sci USA. (2005) 102:8875–80. doi: 10.1073/pnas.0503251102
44. Zabel R, Weber G. Comparative study of the oxidation behavior of sulfur-containing amino acids and glutathione by electrochemistry-mass spectrometry in the presence and absence of cisplatin. Anal Bioanal Chem. (2016) 408:1237–47. doi: 10.1007/s00216-015-9233-x
45. Xiong Y, Uys JD, Tew KD, Townsend DM. S-glutathionylation: from molecular mechanisms to health outcomes. Antioxid Redox Signaling. (2011) 15:233–70. doi: 10.1089/ars.2010.3540
46. Rodríguez-García A, García-Vicente R, Morales ML, Ortiz-Ruiz A, Martínez-López J, Linares M. Protein carbonylation and lipid peroxidation in hematological Malignancies. Antioxidants. (2020) 9:1212. doi: 10.3390/antiox9121212
47. Kehm R, Baldensperger T, Raupbach J, Höhn A. Protein oxidation - Formation mechanisms, detection and relevance as biomarkers in human diseases. Redox Biol. (2021) 42:2213–317. doi: 10.1016/j.redox.2021.101901
48. Kim C-S, Park S, Kim J. The role of glycation in the pathogenesis of aging and its prevention through herbal products and physical exercise. JENB. (2017) 21:55–61. doi: 10.20463/jenb.2017.0027
49. Baldensperger T, Sanzo SD, Ori A, Glomb MA. Quantitation of reactive acyl-CoA species mediated protein acylation by HPLC–MS/MS. Anal Chem. (2019) 91:12336–43. doi: 10.1021/acs.analchem.9b02656
50. McCaskill ML, Kharbanda KK, Tuma DJ, Reynolds JD, DeVasure JM, Sisson JH, et al. Hybrid malondialdehyde and acetaldehyde protein adducts form in the lungs of mice exposed to alcohol and cigarette smoke: MAA ADDUCTS IN LUNG. Alcoholism: Clin Exp Res. (2011) 35:1106–13. doi: 10.1111/j.1530-0277.2011.01443.x
51. Gonos ES, Kapetanou M, Sereikaite J, Bartosz G, Naparło K, Grzesik M, et al. Origin and pathophysiology of protein carbonylation, nitration and chlorination in age-related brain diseases and aging. Aging. (2018) 10:868–901. doi: 10.18632/aging.101450
52. Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longevity. (2014) 2014:1–31. doi: 10.1155/2014/360438
53. Monroy CA, Doorn JA, Roman DL. Modification and functional inhibition of regulator of G-protein signaling 4 (RGS4) by 4-hydroxy-2-nonenal. Chem Res Toxicol. (2013) 26:1832–9. doi: 10.1021/tx400212q
54. Aiken CT, Kaake RM, Wang X, Huang L. Oxidative stress-mediated regulation of proteasome complexes. Mol Cell Proteomics. (2011) 10:5. doi: 10.1074/mcp.M110.006924
55. Curtis JM, Hahn WS, Long EK, Burrill JS, Arriaga EA, Bernlohr DA. Protein carbonylation and metabolic control systems. Trends Endocrinol Metab. (2012) 23:399–406. doi: 10.1016/j.tem.2012.05.008
56. Stringfellow HM, Jones MR, Green MC, Wilson AK, Francisco JS. Selectivity in ROS-induced peptide backbone bond cleavage. J Phys Chem A. (2014) 118:11399–404. doi: 10.1021/jp508877m
57. Bollineni RC, Fedorova M, Blüher M, Hoffmann R. Carbonylated plasma proteins as potential biomarkers of obesity induced type 2 diabetes mellitus. J Proteome Res. (2014) 13:5081–93. doi: 10.1021/pr500324y
58. Cassinadio JS, Jones BR, Chamberlain GR, Baxter GF. Nitric oxide treatments as adjuncts to reperfusion in acute myocardial infarction: a systematic review of experimental and clinical studies. Basic Res Cardiol. (2016) 111:23. doi: 10.1007/s00395-016-0540-y
59. Radi R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc Natl Acad Sci USA. (2018) 115:5839–48. doi: 10.1073/pnas.1804932115
60. Cassina P, Cassina A, Pehar M, Castellanos R, Gandelman M, de Leon A, et al. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci. (2008) 28:4115–22. doi: 10.1523/JNEUROSCI.5308-07.2008
61. Wang Z, Wang Y, Liu H, Che Y, Xu Y, E L. Age-related variations of protein carbonyls in human saliva and plasma: is saliva protein carbonyls an alternative biomarker of aging? AGE. (2015) 37:48. doi: 10.1007/s11357-015-9781-1
62. Sharma A, Weber D, Raupbach J, Dakal TC, Fließbach K, Ramirez A, et al. Advanced glycation end products and protein carbonyl levels in plasma reveal sex-specific differences in Parkinson’s and Alzheimer’s disease. Redox Biol. (2020) 34:101546. doi: 10.1016/j.redox.2020.101546
63. Almogbel E, Rasheed N. Elevated levels of protein carbonylation in patients with diabetic nephropathy: therapeutic and diagnostic prospects. Am J Med Sci. (2019) 358:26–32. doi: 10.1016/j.amjms.2019.03.01
64. Lou B, Boger M, Bennewitz K, Sticht C, Kopf S, Morgenstern J, et al. Elevated 4-hydroxynonenal induces hyperglycaemia via Aldh3a1 loss in zebrafish and associates with diabetes progression in humans. Redox Biol. (2020) 37:101723. doi: 10.1016/j.redox.2020.101723
65. Neelofar K, Arif Z, Arafat MY, Alam K, Ahmad J. A study on correlation between oxidative stress parameters and inflammatory markers in type 2 diabetic patients with kidney dysfunction in north Indian population. J Cell Biochem. (2019) 120:4892–902. doi: 10.1002/jcb.27763
66. Yavuzer H, Yavuzer S, Cengiz M, Erman H, Doventas A, Balci H, et al. Biomarkers of lipid peroxidation related to hypertension in aging. Hypertens Res. (2016) 39:342–8. doi: 10.1038/hr.2015.156
67. Stankova TR, Delcheva GT, Maneva AI, Vladeva SV. Serum levels of carbamylated LDL, nitrotyrosine and soluble lectin-like oxidized low-density lipoprotein receptor-1 in poorly controlled type 2 diabetes mellitus. Folia Med. (2019) 61:419–25. doi: 10.3897/folmed.61.e39343
68. Silva Servato JP, Ueira Vieira C, De Faria PR, Cardoso SV, Loyola AM. The importance of inducible nitric oxide synthase and nitrotyrosine as prognostic markers for oral squamous cell carcinoma. J Oral Pathol Med. (2019) 48:967–75. doi: 10.1111/jop.12942
69. Hawkins CL, Davies MJ. Detection, identification, and quantification of oxidative protein modifications. J Biol Chem. (2019) 294:19683–708. doi: 10.1074/jbc.REV119.006217
70. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
71. Canton M, Sánchez-Rodríguez R, Spera I, Venegas FC, Favia M, Viola A, et al. Reactive oxygen species in macrophages: sources and targets. Front Immunol. (2021) 12:734229. doi: 10.3389/fimmu.2021.734229
72. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. (2015) 21:65–80. doi: 10.1016/j.cmet.2014.12.005
73. Gordon S, Mantovani A. Diversity and plasticity of mononuclear phagocytes. Eur J Immunol. (2011) 41:2470–2. doi: 10.1002/eji.201141988
74. Strizova Z, Benesova I, Bartolini R, Novysedlak R, Cecrdlova E, Foley LK, et al. M1/M2 macrophages and their overlaps - myth or reality? Clin Sci (Lond). (2023) 15:1067–93. doi: 10.1042/CS20220531
75. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
76. Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front Immunol. (2017) 8:289. doi: 10.3389/fimmu.2017.00289
77. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. (2014) 40:274–88. doi: 10.1016/j.immuni.2014.01.006
78. Nelson VL, Nguyen HCB, Garcìa-Cañaveras JC, Briggs ER, Ho WY, DiSpirito JR, et al. PPARγ is a nexus controlling alternative activation of macrophages via glutamine metabolism. Genes Dev. (2018) 32:1035–44. doi: 10.1101/gad.312355.118
79. Wang T, Liu H, Lian G, Zhang S-Y, Wang X, Jiang C. HIF1 α -induced glycolysis metabolism is essential to the activation of inflammatory macrophages. Mediators Inflammation. (2017) 17:1–10. doi: 10.1155/2017/9029327
80. Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. (2009) 9:609–17. doi: 10.1038/nri2607
81. Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab. (2018) 28:463–475.e4. doi: 10.1016/j.cmet.2018.08.012
82. Wang S, Liu G, Li Y, Pan Y. Metabolic reprogramming induces macrophage polarization in the tumor microenvironment. Front Immunol. (2022) 13:840029. doi: 10.3389/fimmu.2022.840029
83. Jeon J-H, Hong C-W, Kim EY, Lee JM. Current understanding on the metabolism of neutrophils. Immune Netw. (2020) 20:e46. doi: 10.4110/in.2020.20.e46
84. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. (2010) 191:677–91. doi: 10.1083/jcb.201006052
85. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci USA. (2015) 112:2817–22. doi: 10.1073/pnas.1414055112
86. Mullen L, Mengozzi M, Hanschmann E-M, Alberts B, Ghezzi P. How the redox state regulates immunity. Free Radical Biol Med. (2020) 157:3–14. doi: 10.1016/j.freeradbiomed.2019.12.022
87. Sônego F, Castanheira FVES, Ferreira RG, Kanashiro A, Leite CAVG, Nascimento DC, et al. Paradoxical roles of the neutrophil in sepsis: protective and deleterious. Front Immunol. (2016) 7:155. doi: 10.3389/fimmu.2016.00155
88. Carrera-Quintanar L, Funes L, Herranz-López M, Martínez-Peinado P, Pascual-García S, Sempere JM, et al. Antioxidant supplementation modulates neutrophil inflammatory response to exercise-induced stress. Antioxidants. (2020) 9:1242. doi: 10.3390/antiox9121242
89. Akong-Moore K, Chow OA, Von Köckritz-Blickwede M, Nizet V. Influences of chloride and hypochlorite on neutrophil extracellular trap formation. PloS One. (2012) 7:e42984. doi: 10.1371/journal.pone.0042984
90. Clemen R, Arlt K, Miebach L, von Woedtke T, Bekeschus S. Oxidized proteins differentially affect maturation and activation of human monocyte-derived cells. Cells. (2022) 11:3659. doi: 10.3390/cells11223659
91. Diotallevi M, Checconi P, Palamara AT, Celestino I, Coppo L, Holmgren A, et al. Glutathione fine-tunes the innate immune response toward antiviral pathways in a macrophage cell line independently of its antioxidant properties. Front Immunol. (2017) 8:1239. doi: 10.3389/fimmu.2017.01239
92. Helou DG, Noël B, Gaudin F, Groux H, El Ali Z, Pallardy M, et al. Cutting edge: Nrf2 regulates neutrophil recruitment and accumulation in skin during contact hypersensitivity. J Immunol. (2019) 202:2189–94. doi: 10.4049/jimmunol.180106
93. Luo J-F, Shen X-Y, Lio CK, Dai Y, Cheng C-S, Liu J-X, et al. Activation of Nrf2/HO-1 pathway by nardochinoid C inhibits inflammation and oxidative stress in lipopolysaccharide-stimulated macrophages. Front Pharmacol. (2018) 9:911. doi: 10.3389/fphar.2018.00911
94. Rugemalira E, Roine I, Kuligowski J, Sánchez-Illana Á, Piñeiro-Ramos JD, Andersson S, et al. Protein oxidation biomarkers and myeloperoxidase activation in cerebrospinal fluid in childhood bacterial meningitis. Antioxidants. (2019) 8:441. doi: 10.3390/antiox8100441
95. García-Giménez JL, Òlaso G, Hake SB, Bönisch C, Wiedemann SM, Markovic J, et al. Histone h3 glutathionylation in proliferating mammalian cells destabilizes nucleosomal structure. Antioxid Redox Signaling. (2013) 19:1305–20. doi: 10.1089/ars.2012.5021
96. Tyurin VA, Balasubramanian K, Winnica D, Tyurina YY, Vikulina AS, He RR, et al. Oxidatively modified phosphatidylserines on the surface of apoptotic cells are essential phagocytic ‘eat-me’ signals: cleavage and inhibition of phagocytosis by Lp-PLA2. Cell Death Differ. (2014) 21:825–35. doi: 10.1038/cdd.2014.1
97. Wu C, Li A, Hu J, Kang J. Histone deacetylase 2 is essential for LPS-induced inflammatory responses in macrophages. Immunol Cell Biol. (2019) 97:72–84. doi: 10.1111/imcb.12203
98. Chen S, Yang J, Wei Y, Wei X. Epigenetic regulation of macrophages: from homeostasis maintenance to host defense. Cell Mol Immunol. (2020) 17:36–49. doi: 10.1038/s41423-019-0315-0
99. Clements MK, Siemsen DW, Swain SD, Hanson AJ, Nelson-Overton LK, Rohn TT, et al. Inhibition of actin polymerization by peroxynitrite modulates neutrophil functional responses. J Leuk Biol. (2003) 73:344–55. doi: 10.1189/jlb.080240
100. Łuczaj W, Gindzienska-Sieskiewicz E, Jarocka-Karpowicz I, Andrisic L, Sierakowski S, Zarkovic N, et al. The onset of lipid peroxidation in rheumatoid arthritis: consequences and monitoring. Free Radical Res. (2016) 50:304–13. doi: 10.3109/10715762.2015.1112901
101. Hecker M, Wagner AH. Role of protein carbonylation in diabetes. J Inherit Metab Dis. (2018) 41:29–38. doi: 10.1007/s10545-017-0104-9
102. Kondo A, Morita H, Nakamura H, Kotani K, Kobori K, Ito S, et al. Influence of fibrate treatment on malondialdehyde-modified LDL concentration. Clinica Chimica Acta. (2004) 339:97–103. doi: 10.1016/j.cccn.2003.09.005
103. Kietzmann T, Petry A, Shvetsova A, Gerhold JM, Görlach A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system: ROS and epigenetic modifications. Br J Pharmacol. (2017) 174:1533–54. doi: 10.1111/bph.13792
104. O’Connor KM, Das AB, Winterbourn CC, Hampton MB. Inhibition of DNA methylation in proliferating human lymphoma cells by immune cell oxidants. J Biol Chem. (2020) 295:7839–48. doi: 10.1074/jbc.RA120.013092
105. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. (2016) 8:485–97. doi: 10.1038/nri.2016.63
106. Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, et al. Oxidative stress, aging, and diseases. Clin Interventions Aging. (2018) 13:757–72. doi: 10.2147/CIA.S158513
107. Kurutas EB. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr J. (2016) 1:71. doi: 10.1186/s12937-016-0186-5
108. Yuan T, Yang T, Chen H, Fu D, Hu Y, Wang J, et al. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. (2019) 20:247–60. doi: 10.1016/j.redox.2018.09.025
109. Mulder WJM, Ochando J, Joosten LAB, Fayad ZA, Netea MG. Therapeutic targeting of trained immunity. Nat Rev Drug Discovery. (2019) 18:553–66. doi: 10.1038/s41423-022-00902-0
110. Jiang J, Wang K, Chen Y, Chen H, Nice EC, Huang C. Redox regulation in tumor cell epithelial–mesenchymal transition: Molecular basis and therapeutic strategy. Signal Transduct Target Ther. (2017) 1:1–12. doi: 10.1038/sigtrans.2017.36
111. Jiang J, Wang K, Nice EC, Zhang T, Huang C. High-throughput screening of cellular redox sensors using modern redox proteomics approaches. Expert Rev Proteomics. (2015) 12:543–55. doi: 10.1586/14789450.2015.1069189
Keywords: oxidative stress, reactive oxygen species, inflammation, innate immune response, macrophage, neutrophils, protein oxidation
Citation: Manoharan RR, Prasad A, Pospíšil P and Kzhyshkowska J (2024) ROS signaling in innate immunity via oxidative protein modifications. Front. Immunol. 15:1359600. doi: 10.3389/fimmu.2024.1359600
Received: 21 December 2023; Accepted: 22 February 2024;
Published: 07 March 2024.
Edited by:
Colin E. Murdoch, University of Dundee, United KingdomReviewed by:
Kavita Pal, Research and Education in Cancer (ACTREC), IndiaCopyright © 2024 Manoharan, Prasad, Pospíšil and Kzhyshkowska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ankush Prasad, YW5rdXNoLnByYXNhZEB1cG9sLmN6; Julia Kzhyshkowska, SnVsaWEua3poeXNoa293c2thQG1lZG1hLnVuaS1oZWlkZWxiZXJnLmRl
†ORCID: Renuka Ramalingam Manoharan, orcid.org/0000-0002-9198-496X
Ankush Prasad, ocid.org/0000-0002-2009-8987
Pavel Pospíšil, orcid.org/0000-0001-9126-2011
Julia Kzhyshkowska, orcid.org/0000-0003-0898-3075
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.