 Mikhail Y. Salnikov
Mikhail Y. Salnikov Katelyn M. MacNeil
Katelyn M. MacNeil Joe S. Mymryk
Joe S. Mymryk- 1Department of Microbiology and Immunology, Western University, London, ON, Canada
- 2Department of Oncology, Western University, London, ON, Canada
- 3Department of Otolaryngology, Western University, London, ON, Canada
- 4Lawson Health Research Institute, London, ON, Canada
Epstein-Barr virus (EBV) is a pathogen known to cause a number of malignancies, often taking years for them to develop after primary infection. EBV-associated gastric cancer (EBVaGC) is one such malignancy, and is an immunologically, molecularly and pathologically distinct entity from EBV-negative gastric cancer (EBVnGC). In comparison with EBVnGCs, EBVaGCs overexpress a number of immune regulatory genes to help form an immunosuppressive tumor microenvironment (TME), have improved prognosis, and overall have an “immune-hot” phenotype. This review provides an overview of the histopathology, clinical features and clinical outcomes of EBVaGCs. We also summarize the differences between the TMEs of EBVaGCs and EBVnGCs, which includes significant differences in cell composition and immune infiltration. A list of available EBVaGC and EBVnGC gene expression datasets and computational tools are also provided within this review. Finally, an overview is provided of the various chemo- and immuno-therapeutics available in treating gastric cancers (GCs), with a focus on EBVaGCs.
1 Introduction
Epstein-Barr virus (EBV) is a gamma-herpesvirus most known for influencing B lymphocyte proliferation and differentiation to a plasmablast/early plasma cell-like phenotype (1, 2), as well as causing cytopathic effects within mucosal epithelial cells (3). More rarely, EBV infects T and NK cells, causing lymphoproliferative disorders (4). By evading both the innate and adaptive immune systems, EBV can establish lifelong, latent infections within B lymphocytes, which can lead to reactivation and further infections (5). In fact, EBV is so successful that it is estimated that over 90% of the world population may be infected with it (6).
EBV is also associated with a number of different cancers, which include nasopharyngeal carcinomas (NPCs), EBV-associated gastric cancers (EBVaGCs), as well as Burkitt and other lymphomas (7, 8). Overall, EBV infections account for 1.5% of all cancers globally (9), with 84.6% (10), 8.8% (11), and 50% (10) of NPCs, gastric cancers (GCs), and Hodgkin’s lymphomas attributable to EBV infections, respectively. Currently, there are no approved vaccines against EBV, but there are a number of prophylactic and therapeutic vaccines currently undergoing trials (12–14). However, there are several approved therapies targeting EBV-associated cancers with various levels of effectiveness (9, 15).
Gastric cancer is the fifth most diagnosed cancer, with over a million new cases annually, and is the third most common cause of cancer-related death (16). GCs have a number of risk factors, which include Helicobacter pylori (H. pylori) and EBV infections, genetic and dietary factors, as well as smoking status and alcohol consumption (17–19). The etiology of EBV in GCs was first identified in 1990 by Burke et al. (20), with Shibata and Weiss demonstrating the presence of the EBV genome within cancerous and dysplastic cells (21). EBVaGCs have been established as molecularly and clinically distinct from EBV-negative GCs (EBVnGCs) by The Cancer Genome Atlas (TCGA), which defined 4 different subtypes of EBVnGCs: microsatellite-instable (MSI) tumors, genomically stable (GS) tumors, tumors with chromosomal instability (CIN), and tumors with DNA polymerase epsilon mutations (POLE) (22).
2 EBV infection of gastric epithelial cells
EBV transmission between individuals is typically mediated by saliva (23). Indeed, EBV causes infectious mononucleosis, which is sometimes referred to as the “kissing disease”. Primary infection of a naïve individual typically occurs when incoming EBV virions directly infect susceptible B cells in the tonsillar crypt (Figure 1). Alternatively, an incoming virus may directly infect tonsillar epithelial cells, which produce progeny viruses that later infect B cells present in the crypts. EBV establishes latency in the infected B cells, creating a reservoir of virus-infected B cells that persist throughout the life of the infected individual. Latently infected B cells occasionally undergo reactivation, producing viruses that reinfects oral epithelial cells. These cells then shed viruses into the saliva at high titre, allowing transmission to naïve individuals periodically throughout the lifetime of the infected individual (23). Interestingly, viruses released from infected B cells preferentially infect epithelial cells, while viruses released from infected epithelial cells preferentially infect B cells. The molecular basis for this switch in tropism is related to changes in the glycoprotein composition in the envelopes of B cell-derived and epithelial cell-derived viruses and helps to reinforce alternate replication between the two cell types (24).
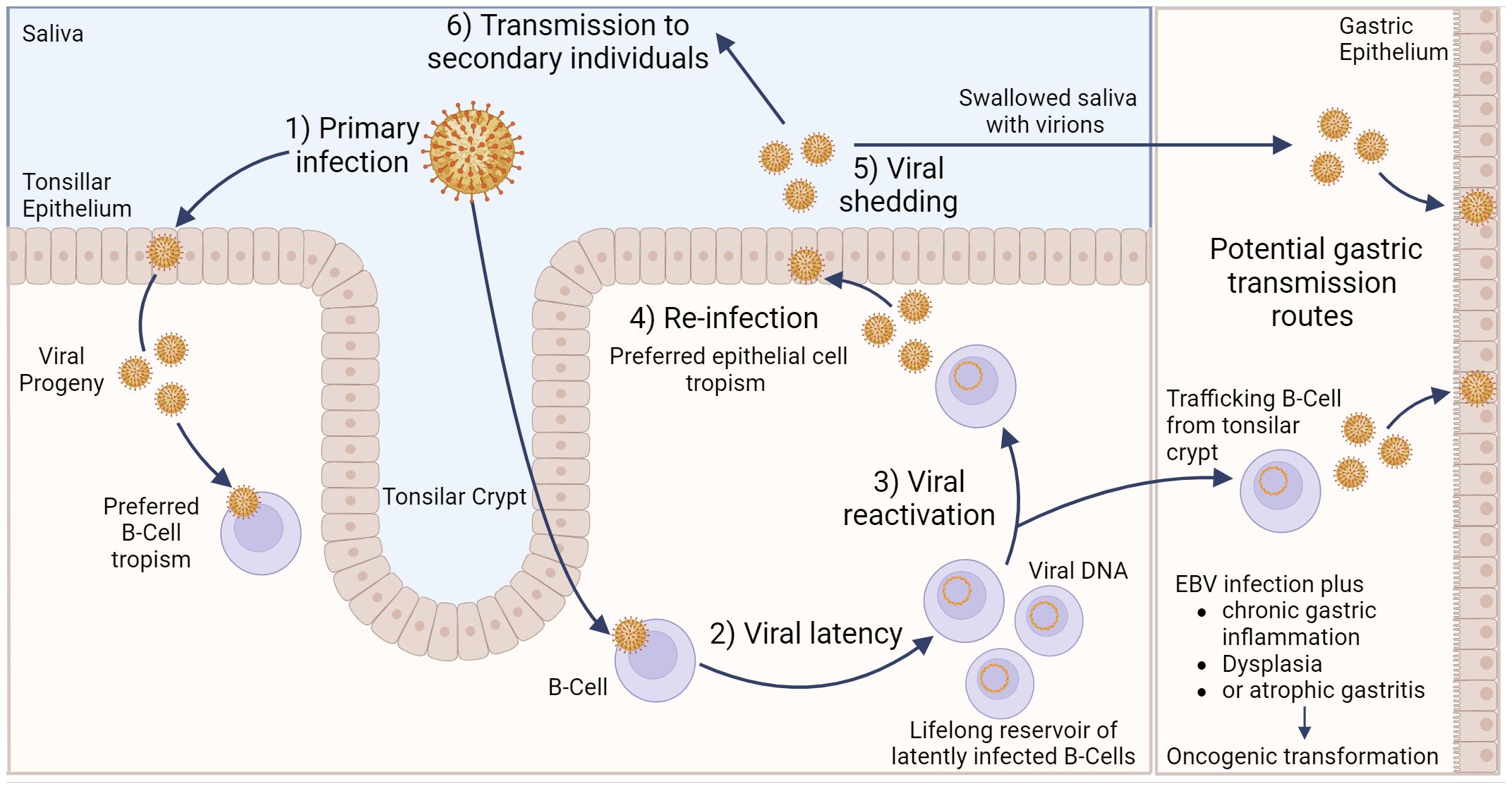
Figure 1 Model of Epstein–Barr virus infection in relation to gastric cancer. EBV can infect tonsillar epithelium or B cells located in the tonsillar crypt, with the resulting viral progeny having a preferred tropism for the opposite cell type. Following B cell infection, EBV can establish life-long latency in pools of B cells. Occasionally, these cells can become reactivated, resulting in subsequent release of viral progeny that exhibit a preferred epithelial cell tropism. Viral shedding from epithelial cells into salvia can result in EBV transmission to secondary individuals. Alternatively, salvia containing viral progeny can be swallowed, resulting in infection of gastric epithelial cells. Likewise, latently infected, trafficking B cells can undergo reactivation, and the resulting viral progeny can then infect gastric epithelial cells. Either of these potential routes could lead to gastric epithelial cell infection, which may subsequently lead to EBVaGC. Created with BioRender.com.
Direct infection of gastric epithelial cells could occur from swallowing saliva containing EBV virions shed from their own infected oral epithelium (Figure 1). This process would be akin to virus transmission from person to person, as described above, except that the infection is spreading from one anatomical location to another in the same individual. Alternatively, a productive EBV infection could be reactivated in latently infected B cells trafficking through the gastric mucosa and released to infect neighbouring epithelial cells (24). In support of this route of infection, coculture of epithelial cells with EBV-positive lymphocyte cells is approximately 800-fold more efficient than cell-free infection, suggesting the possibility of direct cell-to-cell mediated virus infection (25). A definitive answer to the exact mechanism by which gastric epithelial cells become infected by EBV remains to be determined.
3 EBVaGC histopathology and clinical features
EBVaGCs are considered to be molecularly and pathologically distinct entities from EBVnGCs (9, 26–28). This section will highlight a number of known differences, both macroscopic and microscopic, molecular and morphological, as well as differences related to clinical features and patient outcomes between EBVaGCs and EBVnGCs.
3.1 Histomorphology and histopathology
EBVaGCs preferentially develop in the proximal region of the stomach, which includes the cardia, fundus, and body (29), with lymphoepithelioma-like carcinoma and Crohn’s disease-like lymphocytic reaction being the dominant histological subtypes (30). In fact, over 90% of lymphoepithelioma-like carcinomas are EBV-positive (31), where tumor cells are outnumbered by tumor-infiltration lymphocytes (TILs) (29). In contrast, EBVnGCs predominate within the antrum region of the stomach (29) and exhibit a lower infiltration of TILs (32). Gastritis cystica profunda, a precancerous lesion associated with increased proliferation activity and cystic gastric glands within the submucosa, is also frequently associated with a positive EBV-status (33). Immunophenotyping of EBVaGCs indicates an even split between a gastric-like phenotype, expressing both the MUC5AC and MUC6 mucins, and a null phenotype, expressing neither gastric-like, nor intestinal-like phenotypes (34). A detailed description of the histology and immunophenotype of gastric differentiation markers in EBVaGC has been summarized previously (34).
Even though there is a disparity of TILs between EBVaGCs and EBVnGCs, increased infiltration by lymphocytes can lead to the formation of tertiary lymphoid structures in both types of GCs, which often develop their own germinal centers and correlates with CD4+ and CD8+ T cell infiltration (35, 36). Tertiary lymphoid structures are generally associated with better outcomes for most types of cancer (35), including GCs, where the presence of these structures is associated with higher levels of TILs and increased overall survival (32). Further exploration of the histomorphology and histopathology, in conjunction with the immune landscape, of EBVaGCs and EBVnGCs may yield deeper insights into the differences in tumor initiation and progression, as well as structural feature changes associated with these carcinomas.
3.2 Identification, clinical features and general outcomes
EBVaGC is commonly identified by in situ hybridization with a probe that specifically anneals to the small non-coding EBER1 viral RNA, which is abundantly expressed in EBV infected and transformed cells (37). These probes enable accurate detection of EBV-infected cells with high specificity in formaldehyde-fixed and paraffin-embedded GC samples (38). This is especially relevant, as EBV infections are associated with an 18-fold increased risk of developing GC (11).
Both EBVaGCs and EBVnGCs have a higher incidence in men as compared to women (39, 40). In contrast, EBVaGCs have a better prognosis and greater median survival time (30, 41, 42), an increased presence of TILs (43, 44), increased promotor hypermethylation (45, 46), and higher levels of MHC-I and MHC-II expression (47, 48) compared to EBVnGCs. EBVaGCs are also found more often among young individuals as compared to EBVnGCs (49, 50). A lower density of TILs is also associated with increased presence of lymph node metastasis, an important negative determinant of disease progression in GCs (51) that is also associated with poorer patient outcomes (28). Additionally, co-infections of H. pylori and EBV may result in earlier and more aggressive GC progression (52).
3.3 Cell markers associated with patient outcomes in EBVaGC
There are a number of differentially expressed cellular genes associated with differences in outcomes for patients with EBVaGC. Programmed death-ligand 1 (PD-L1), an immune checkpoint protein that binds to its receptor PD-1 on T cells and other immune cells, is overexpressed in EBVaGCs. Indeed, in a recent meta-analysis of 43 publications encompassing a total of 11,327 patients, there was a very clear increase in the association between PD-L1 expression and EBVaGC (OR = 6.36, 95% CI 3.91-10.3, p < 0.001) (53). This increase in PD-L1 likely occurs as a response to higher intratumoral levels of IFN-γ via activation of IRF3 and is associated with worse patient outcomes (54, 55). Poorer patient outcomes have also been associated with increased expression of indoleamine 2,3-dioxygenase 1 (IDO1), another potent immune suppressor (56, 57). Human epidermal growth factor receptor 2 (HER2) is a proto-oncogene that is downregulated by LMP2A, an EBV-encoded protein, in some cases of EBVaGC (49, 58). However, the increased expression of HER2 is associated with lower overall patient survival in EBVaGC (59). The identification of additional prognostic markers for EBVaGC will help develop a greater diversity of personalized and targeted therapies (60, 61).
3.4 Impact of H. pylori infection on EBVaGC
In addition to EBV, chronic inflammation and tissue damage induced by H. pylori infection increases the risk of developing GC (62). Multiple studies suggest that in the context of co-infections, these microorganisms may cooperate to promote infection, inflammation and possibly carcinogenesis (63–66). The mechanisms controlling the interactions between these two infectious agents are not entirely known and have been reviewed in detail by others (67). However, in a recent large cohort study, EBV and H. pylori co-infection was not identified as a clinically significant independent prognostic factor for the development of gastric cancer (68). Similarly, EBV and H. pylori co-infection did not significantly affect overall survival rate compared to those with EBV alone (68). It should also be noted that co-infection is clearly not essential for carcinogenesis, as EBVaGCs can still occur many years after successful eradication of H. pylori (69) and only a fraction of EBVaGCs are co-infected with H. pylori (22, 52).
4 Role of EBV-encoded proteins in EBVaGC tumor progression
EBV-encoded proteins play a major role in shaping the immune microenvironment of EBVaGCs and promoting tumor growth. EBV encoded latency protein Epstein–Barr nuclear antigen 1 (EBNA1) is uniformly expressed in EBVaGCs, with variable expression of latent membrane protein 1 (LMP1) and latent membrane protein 2A (LMP2A) (70, 71).
4.1 The functions of EBNA1 in EBVaGC
EBNA1 is expressed in all forms of EBV latency in proliferating cells and in all EBV-associated tumours (72). It performs critical roles in maintaining the persistence of latent EBV genomes in the nucleus as episomes and also plays a role in transcriptional activation of viral genes (73). Additionally, mounting evidence suggests that EBNA1 functions more directly to impact cell survival and oncogenesis (72). These include antagonism of the Tumor protein P53 (TP53) pathway (74), degradation of promyelocytic leukemia (PML) tumor suppressor protein (75), modulation of various signal transduction pathways (76), and increased oxidative stress (77). Most recently, a previously unknown link between EBV and genomic instability between EBNA1-induced breakage at 11q23 and the acquisition of chromosome 11 structural variations was identified, but this has yet to be specifically confirmed in EBVaGC (78).
Interestingly, specific amino acid changes in EBNA1 have been identified that were strongly associated with viruses isolated from EBVaGCs and NPCs, but not isolates from lymphoma and healthy individuals (71). One of these mutations (Thr85Ala) results in a gain of function interaction with the procollagen-lysine,2-oxoglutarate 5-dioxygenase (PLOD) 1 and PLOD3 lysyl hydroxylases (79). PLOD family proteins are strongly linked to multiple cancers, and several PLODs are recognized as a prognostic marker of gastric carcinoma (80). Future work will reveal whether these EBNA1-PLOD family interactions impact GC development and prognosis.
4.2 The functions of LMP1 and LMP2A in EBVaGC
Variable expression of both LMP1 and LMP2A can be detected in a subset of EBVaGC (70, 71). Both are transmembrane proteins located in the plasma membrane that function as constitutively active growth factor receptors. Together, in EBV infected B cells, they mimic normal antigen-derived signals that force differentiation into memory B cells, creating a long-lasting reservoir of EBV-infected cells and contributing to B cell oncogenesis (81, 82). The roles of LMP1 and LMP2A in EBVaGC are less clear, but are thought to similarly impact signal transduction pathways involved in epithelial cell growth and survival. Both have been recently reviewed in depth, although much remains to be discovered about their roles in EBVaGC (83, 84).
5 Role of EBV miRNAs in EBVaGC tumor progression
In addition to the two non-coding EBER viral RNAs expressed in all cells harbouring EBV, the EBV genome encodes up to 48 mature micro RNAs (miRNAs) (85). These viral miRNAs impact a variety of cellular functions, which include remodelling the cellular transcriptome, interference with immune signalling, and contribute to tumor progression and immune escape (86–88). Additionally, EBVs are known to produce long noncoding RNAs (lncRNAs), some of which play roles in hijacking human miRNAs and assisting viral replication, amongst other roles (89, 90). This section will give an overview of a number of miRNAs and lncRNAs produced by EBV and their association with EBVaGCs and other EBV-associated cancers.
5.1 The role of BART and BHRF1 miRNAs in EBVaGCs
The most highly transcribed viral RNAs in EBVaGCs map within the BamHI-A region of the genome (22), including the BamH1-A rightward transcripts (BARTs) (Figure 2). These encode 44 intronic viral miRNAs from 22 precursor hairpins (91). The EBV-encoded BART miRNAs are a group of small regulatory RNAs under 100 nucleotides long and are often highly expressed in EBV malignancies (92), suggesting that they may play a role in tumorigenesis (93). Unlike most cellular miRNAs, both the 5’ and 3’ sides of the miR-BART precursor hairpins are efficiently loaded into the RNA-induced silencing complex (94, 95). Roughly 99% of all virally derived polyadenylated transcripts in EBVaGCs are from the 44 miR-BARTs (92, 95). The viral miR-BARTs are also highly expressed, representing >10% of the total pool of miRNAs in EBVaGCs (96).
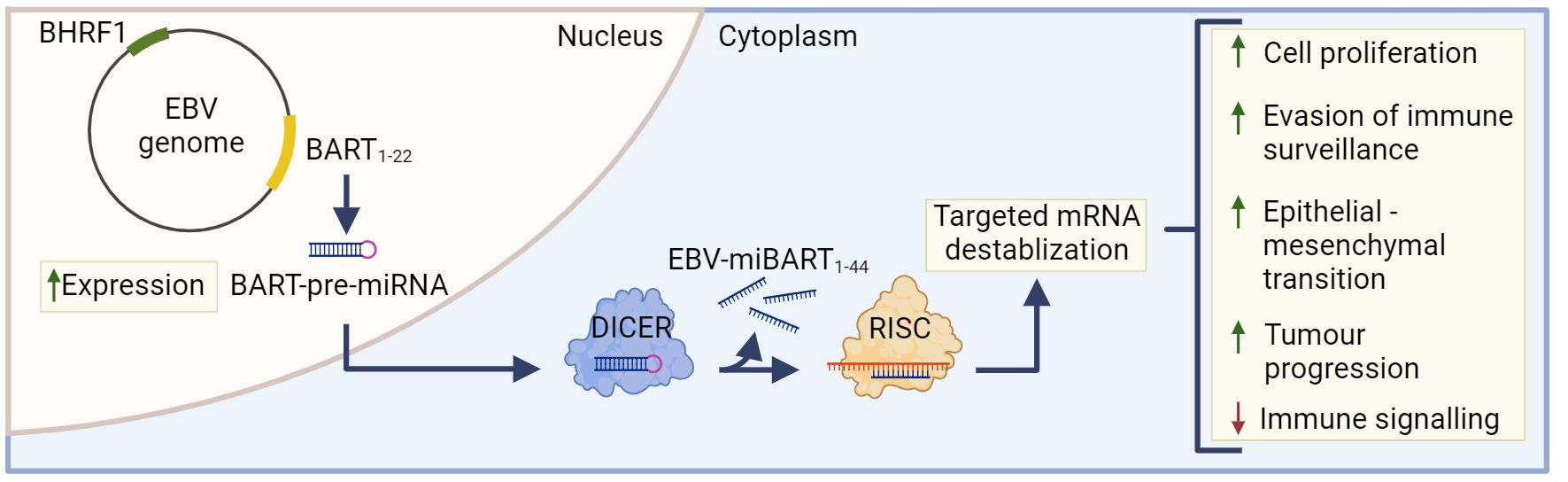
Figure 2 The role of EBV-miBARTS in EBV associated gastric cancer. The EBV genome encodes 22 BART-pre-miRNAs, which are highly expressed in EBVaGC. Following nuclear translocation, the BART pre-miRNAs are processed by Dicer, resulting in 44 EBV-miBARTs. EBV-miBARTs then guide the RISC complex to partially complementary sequences within target mRNAs, leading to targeted mRNA destabilization. This EBV-miBART-mediated regulation of gene expression promotes the expression of genes involved in tumor progression and survival, while decreases the expression of genes involved in immune signalling processes. Hypermethylation of the BHRF1 promotor leads to almost undetectable expression of BHRF1 miRNA in EBVaGC. Created with BioRender.com.
These viral miRNAs regulate gene expression post-transcriptionally by guiding the RISC complex to partially complementary sequences within target mRNAs, leading to mRNA destabilization (Figure 2) (97). Although the current understanding of EBV miRNA function is far from complete, many viral miRNAs target viral and cellular factors involved in host cell growth, survival, signalling pathways, metabolism and anti-viral immune responses (91, 98–102). Much of the existing work has been done in lymphoid cells and the exact roles that most EBV miR-BARTs play during the processes of gastric carcinogenesis are virtually unknown (85). The only comprehensive experimental study to identify cellular mRNAs targeted by miR-BARTs utilized photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) in latently infected lymphoblastoid cell lines. This identified 531 sites of interaction between seed sequences within EBV miRNAs and cellular 3’UTRs. This large number of interactions suggests that EBV miRNAs have profound and widespread effects on cellular gene expression (103).
Although in their infancy, existing studies of the roles of BART miRNAs in a variety of EBV malignancies have identified numerous functions, including the targeting of specific cellular transcripts (87), impairing NK cell-mediated recognition of infected cells (104, 105), decreased pathogen-recognition receptor (PRR)-mediated signalling and interferon induction (106), and impairment of antigen presentation pathways (101, 103).
Of note, EBV-miR-BART7-3p is one of the most highly expressed miRNAs in EBVaGCs (107) and plays a role in inducing cell proliferation and epithelial-to-mesenchymal transition (108). Additionally, EBV-miRNAs have been shown to modulate viral gene expression, often for the purposes of latent infections and evasion of immune surveillance (5, 109). Specifically, miR-BART5-5p and miR-BART19-5p are able to downregulate LMP1, an activator of many cellular immune signalling pathways (109, 110). Upregulated expression of EBV miRNA is associated with a 3-times higher mortality risk in NPCs and GCs (111), with miR-BART20-5p specifically associated with worse recurrence-free survival in EBVaGCs (112).
Although BamH1 fragment H rightward facing 1 (BHRF1) miRNAs are known to play a role in B cell transformation (113, 114), they are almost undetectable in GCs and NPCs (115). Indeed, the BHRF1 promoter is hypermethylated in almost all EBVaGC tissue, and this epigenetic modification likely contributes to its lack of expression (Figure 2) (116).
5.2 The role of lncRNAs in EBVaGCs
Long non-coding RNAs are defined as RNAs over 200 nucleotides long and lack protein-coding abilities (117). Both EBV and their host cells are known to produce a number of lncRNAs (89, 117, 118). In particular, several host cell lncRNAs are differentially expressed in EBVaGC, including small nucleolar RNA host gene 8 (SNHG8), which modulate the expression of a number of cellular and viral genes, as well induces cellular proliferation and invasion (119, 120). Though several EBV BART-encoded lncRNAs have shown to downregulate a number of genes in vitro (93), their in vivo functionality has not yet been confirmed.
6 Impact of genotype and polymorphisms on EBVaGC
Most EBV associated cancers, including NPC, Burkitt lymphoma and T/NK lymphomas, display distinct and non-overlapping geographic distribution patterns. In contrast, the frequency of EBVaGC shows no such regional variation (121). This suggests that regional environmental or host genetic differences may play a lesser role in EBVaGC compared to other EBV-associated cancers (121). It remains an open question whether genetic differences between EBV subtypes are associated with increased risk of developing EBVaGC post EBV infection.
There are two major genotypes of EBV, type 1 and 2, which differ in the sequence of a number of important viral genes (122). Type 1 EBV is the predominant strain in Western and Asian countries while type 2 EBV is frequently found in Africa. Significantly, EBV-1 and EBV-2 differ in their ability to transform B lymphocytes and epithelial cells into a state of continuous proliferation, as well as their association with both cancerous and non-cancerous disease (123, 124). Few studies have assessed if these genotypes, or the myriad of more subtle variants, contribute differently to the burden of EBVaGC (125). The possibility that functional differences between genetically distinct EBV variants contribute to CG risk clearly warrants further investigation on a large scale.
7 Hallmarks of cancer in EBVaGCs
All cancer features can be divided into “hallmark characteristics”, which include genomic instability, sustained proliferative signalling, replicative immortality, and angiogenesis (126, 127). Some of these hallmarks of cancer differ between EBVaGCs and EBVnGCs (34, 128–130). This section will explore the changes in the host genome, cell signalling pathways, and cell cycle regulation in EBVaGCs, and how such changes impact tumor progression and growth, as well as patient outcomes.
7.1 Genetic changes
EBV has been associated with a number of genetic changes within EBVaGCs (Figure 3). In particular, EBVaGCs are strongly associated with somatic mutations of PIK3CA and ARID1A, a feature it shares with the MSI GC subtype (131, 132). Mutations in PIK3CA are associated with increased activation of the Akt pathway, tumor growth, and invasiveness (133, 134), with the overexpression of PIK3CA being associated with poorer patient outcomes (Figure 3A) (135). Furthermore, PIK3CA can serve as a marker of tumor differentiation and morphology, with increased PIK3CA expression being associated with low grade tumor histology and intestinal-type GCs (135–137). Similarly, loss of ARID1A expression is associated with poorer outcomes in non-MSI EBVnGCs (138, 139), with decreased expression associated with TNM stage and depth of invasion (140) (Figure 3A). Furthermore, loss of ARID1A is also associated with the deficiency of the mismatch repair pathway and is correlated with the MSI subtype of GC (141, 142). Apart from PIK3CA and ARID1A, genes such as PTEN, SMAD4, CTNNB1, and NOTCH1 that are involved in Wnt and Notch signalling pathways, or cell cycle and chromatin regulation, are also frequently mutated in EBVaGCs (Figure 3A) (130). Interestingly, mutations of the tumor suppressor gene TP53 are rare in EBVaGC, albeit frequently mutated in other human cancers (143). Apart from mutations, EBVaGCs are also associated with amplification of 9p24.1, which contains the genes encoding JAK2, as well as PD-L1 and PD-L2, both of which are T cell exhaustion ligands (Figure 3A) (83, 130).
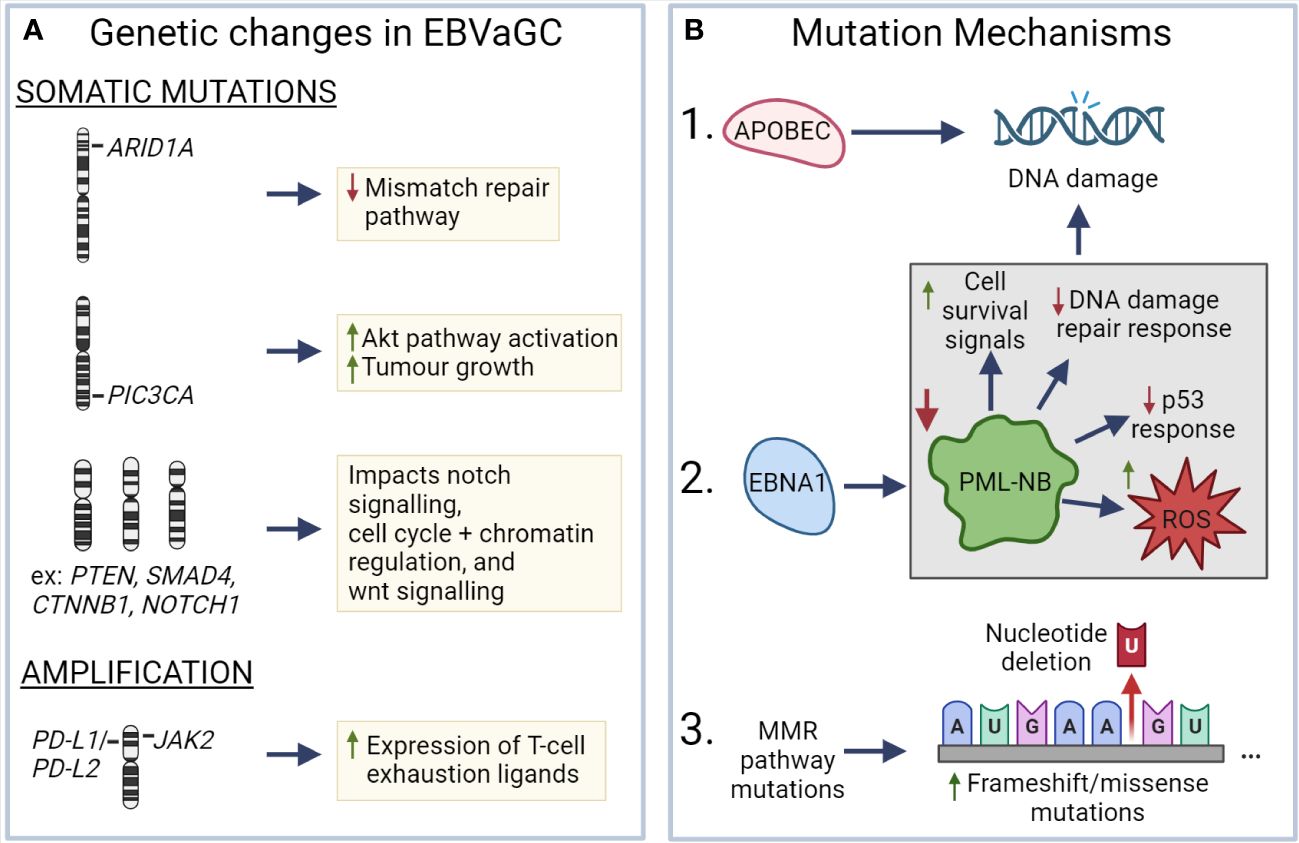
Figure 3 Known mechanisms that lead to genetic changes in EBV associated gastric cancer (A) ARID1A, PIC3CA, PTEN, SMAD4, CTNNB1, and NOTCH1 exhibit frequent somatic mutations in EBVaGC, resulting in deficiencies in the mismatch repair pathway, increased AKT pathway activation, impacts on wnt and notch signalling, as well as cell cycle and chromatin regulation. EBVaGCs are also associated with amplification of the chromosomal regions encoding JAK2, PD-L1 and PD-L2, which contributes to the increased expression of T cell exhaustion markers. (B) APOBEC and EBNA1, via downregulation of promyelocytic leukemia nuclear bodies (PML-NB), contributes to DNA damage. Frequent MMR pathway mutations result in an increase of frameshift and missense mutations. These different mechanisms can lead to genetic changes in EBVaGC. Created with BioRender.com.
There are a number of mechanisms which may be responsible for the aforementioned genetic changes (Figure 3B). One such mechanism is APOBEC-mediated DNA damage in EBVaGCs, where it is associated with activating mutations in the PIK3CA kinase (144). EBNA1, an EBV-associated protein, is also associated with DNA damage via the downregulation of promyelocytic leukemia nuclear bodies, resulting in impaired DNA repair responses, reduced p53 responses, promotion of cell survival signals, and increased reactive oxygen species (ROS) production and accumulation (75, 145). Additionally, many of the DNA mismatch repair pathway genes are mutated in EBVaGCs (146), resulting in an increased number of frameshift and missense mutations in both oncogenes and tumor suppressor genes (147), though such a highly mutated phenotype is often associated with positive responses to immune checkpoint inhibitor therapy, as they can lead to the generation of tumor neoantigens (148–150).
7.2 Epigenetic changes
Apart from genetic changes, EBVaGCs often feature epigenetic dysregulation, whether through altered DNA methylation patterns, the expression of EBV-associated miRNAs, or variable regulation of human miRNAs. Based on the CpG-island methylator phenotype (CIMP), EBVaGCs belong to the high (CIMP-H) category (129, 151). In fact, over 1000 genes have been found to be differentially methylated by EBV, with hypermethylated genes enriched in Wnt and protein kinase signalling pathways, among other cancer-associated pathways (152–154). EBV is also noted to express a number of miRNAs, with said miRNAs functioning to inhibit pro-apoptotic genes and promote cell survival, as well as regulate the expression of EBV-associated genes (115, 155). Additionally, EBV is known to downregulate a number of human miRNAs with tumor suppressor functions, particularly members of the let-7 and miR-200 families (156).
8 Tumor microenvironment
The TME is the ecosystem of the tumor and surrounding regions, allowing the tumor to persist, expand, and spread metastatically (157). Due to the molecular and pathology-associated differences between EBVaGCs and EBVnGCs, variations within the TME are also expected and are of potential interest. In addition to cellular neoantigens derived from tumor-specific DNA alterations that give rise to novel protein sequences, EBVaGCs express foreign viral antigens that represent excellent targets for T cell responses (158, 159). The presence of these non-self viral antigens in EBVaGCs is likely a key factor influencing the overall anti-tumor immune response and altered tumor immune microenvironment as compared to EBVnGCs (160). This section will give an overview of the known differences present between EBVaGCs and EBVnGCs, as well as various aspects of the TME in GCs.
8.1 Immune infiltration and immune cell composition
EBVnGCs are considered to be “immune-cold” tumors, with little to no T cell infiltration (161–163). EBVaGCs, on the other hand, are “immune-hot”, with high infiltration of immune cells, including CD8+, CD4+, and dendritic cells (DCs) (164, 165). The high lymphocytic infiltration of EBVaGCs is a common defining histological characteristic compared to EBVnGCs (20). Indeed, in a recent cohort study of 421 primary GCs, the density of CD3+ T cells in the tumor epithelium was 3.27 times higher (P<0.0001) than the next highest GC subtype (166). In EBVaGCs, the ratio of CD8+ to CD4+ T cells is 10:1, indicating increased cytotoxic antitumoral activity (167), with RNA-seq analysis of GCs showing that cytotoxic CD8+ T cell (CTL) signatures are strongly correlated with EBV viral load, which is expected given that viral antigens will be recognized as non-self by the host immune system (168). Though the level of infiltrating DCs is higher in EBVaGCs as compared to EBVnGCs (43, 165, 167), few are CD1a+, resulting in tolerogenic responses of T cells against tumor cells (164) (169, 170). EBV-infected epithelial cells help facilitate this process by secreting exosomes that inhibit DC maturation and contributes to tumor progression (165). Additionally, the TME of EBVaGC promotes CD4+ differentiation to Foxp3+ regulatory T cells via PD-1/PD-L1 signalling, contributing to the immunosuppressive environment of the TME (171, 172). Genes related to Th17 differentiation are also differentially expressed in EBVaGCs (173), with accumulation of Th17 cells associated with tumor progression and metastases via IL-17 secretion (174), as well as poorer patient outcomes for GCs (175, 176). Furthermore, a unique subpopulation of biphenotypic B cells, expressing both B and T cell markers, have been previously identified in EBVaGCs, though their role has not been deciphered (177).
In addition to the beneficial immune cells within the TME, there are also cells actively contributing to the immunosuppressive TME. One such group of immunosuppressive cells are the tumor-associated macrophages (TAMs), which are most often associated with the anti-inflammatory M2 phenotype and contribute to tumor cell proliferation and angiogenesis (178, 179). EBVaGCs, in particular, have a greater proportion of pro-inflammatory M1 macrophages compared to EBVnGCs (180). Even so, the role of macrophages within EBVaGCs may depend on the proportion of M1/M2 macrophages, with M1 TAMs dominating at early stages of the tumor and M2 TAMs during later stages (181–183). Another group of immunosuppressive cells are myeloid-derived suppressor cells (MDSCs), which are an adverse prognostic factor in GCs (184, 185) and are known to inhibit adaptive and innate immune antitumoral responses via a variety of mechanisms (186). MDSCs arise from myeloid progenitor cells that have not terminally differentiated into mature granulocytes or macrophages. MDSCs are also associated with increased regulatory T cell presence within the tumor and the Th2 cytokine, IL-13 (185). Interestingly, Th2 skewing of the CD4+ T cell helper response makes it the predominant phenotype in GCs (187). However, there is conflicting information, with Th2 shown to display both anti-tumor activity and benefitting tumor growth (188, 189). Even so, higher ratios of Th1/Th2 cells are associated with better outcomes (190).
8.2 Differences in antigen presentation
Effective T cell-specific anti-tumor responses require the presentation of a tumor-associated antigens in either the context of major histocompatibility complex class-I (MHC-I) or class-II (MHC-II) (191). Surveilling antigen-presenting cells (APCs) acquire exogenous peptides and present them in the context of MHC-II on the APC cell surface to activate antigen-specific CD4+ helper T cells (192). In conjunction with the ligation of co-stimulatory molecules between the APC and T cell, this two step process triggers proliferation and survival of antigen specific T cells (193). These activated CD4+ helper T cells subsequently stimulate CD8+ cytotoxic T cells specific for the same peptide antigen. This CTL response targets and lyses tumor cells displaying that specific, endogenously-derived antigenic peptide in the context of cell surface MHC-I (194, 195).
A prevalent mechanism of tumor cell evasion of the CTL response is the loss or down-regulation of the presentation of tumor antigens by the cancer cell in the context of MHC-I (196). Intriguingly, EBVaGCs express higher levels of MHC-I than other GC subtypes (47, 197). Indeed, EBVaGCs display high mRNA levels for all MHC-I components, including heavy and light chains, as well as factors required for loading, in comparison to both normal control tissues or EBVnGCs (47). This is likely a consequence of higher IFN-γ levels in EBVaGCs (168, 198). Intratumoral IFN-γ is produced by tumor-infiltrating lymphocytes and is a known inducer of transcription of the genes encoding MHC-I and components of the antigen loading complex (199). Interestingly, EBVaGCs were recently reported as exhibiting the highest IFN-γ gene response signature of all the GC subtypes (200, 201). Thus, EBVaGCs may more effectively display endogenously-derived antigenic peptides compared to EBVnGCs, enhancing their detection and lysis by CTLs.
As mentioned above, presentation of viral or tumor-derived neoantigens occurs in the context of MHC-II molecules, which are primarily expressed by professional APCs, such as DCs, macrophages and B cells (202). However, exposure of epithelial cells to IFN-γ also induces expression of MHC-II. These epithelial cells can subsequently function as accessory APCs to present antigens and stimulate an effective CTL response (203). Increased levels of MHC-II proteins on epithelial cells should enhance the presentation of exogenously-derived viral- and tumor-specific peptide antigens, enhancing CTL responses (204). Indeed, the underappreciated role for tumor cell derived MHC-II in anti-tumor immunity is becoming apparent, with numerous reports suggesting that tumor-specific MHC-II expression is correlated with favorable outcomes in many cancer types, including GCs (205, 206).
Interestingly, EBVaGCs display high mRNA levels for virtually all MHC-II genes, as well as the MHC-II-like α- and β-chains, and the invariant chain encoded by CD74 as compared to EBVnGCs (48, 198). The coordinated upregulation of MHC-II pathway genes by IFN-γ is clearly illustrated by strong global correlations in transcript abundance (48). These conclusions are supported by single cell RNA sequencing (scRNA-seq) analysis that conclusively shows much higher expression of MHC-II mRNAs in malignant epithelial cells isolated from an EBVaGC case compared to EBV-negative GCs (207). Immunohistochemical analysis also confirms higher protein expression of various MHC-II classes in EBVaGCs (167, 207, 208).
The coordinated upregulation of the components of both the MHC-I and MHC-II antigen presentation pathways by higher IFN-γ levels, combined with the expression of exogenous viral antigens, may help explain why the clinical outcomes for EBVaGCs are superior to EBVnGCs. An analogous situation is present in the TME of human papillomavirus-associated head and neck cancers, which similarly display upregulated MHC-I and II pathway components and better patient outcomes compared to those without a viral etiology (209, 210).
8.3 Molecular mechanisms of immunosuppression
As compared to EBVnGCs, a number of immunosuppressive genes are upregulated in the TME of EBVaGCs. One such gene is IDO1, a potent inhibitor of immune cells, allowing virus-associated tumors to persist in opposition to the increased local immune cell concentration (164, 168). IDO1 functions by exhausting tryptophan within the TME, producing kynurenine in the process. Reduced environmental tryptophan results in reduced proliferation, immune cell function, and contributes to tumor escape and metastasis (211). Programmed cell death protein 1 (PD-1), encoded by PDCD1, and its ligand PD-L1, encoded by cluster of differentiation 274 (CD274), are also upregulated in EBVaGCs (53, 212). Typically, PD-1 is expressed on T cells, and PD-L1 on TAMs and tumor cells within the TME (213). The interaction of PD-1 and PD-L1 inhibits T cell proliferation and a number of effector functions (214, 215). Additionally, PD-L1 has been shown to play roles in modulating epithelial-to-mesenchymal transition and chemoresistance in many types of cancers, including GCs (216). A number of other immune checkpoint molecules are also expressed by activated immune cells in EBVaGCs. These include hepatitis A virus cellular receptor 2/Tcell immunoglobulin and mucin domain 3 (TIM-3), encoded by HAVCR2/TIM-3 (217), lymphocyte activation gene 3 (LAG-3), encoded by LAG3 (218), and cytotoxic T lymphocyte associated protein 4 (CTLA-4), encoded by CTLA4 (219). These collectively modulate the magnitude and duration of immune response (220). The activation of these checkpoints has been noted in many other types of cancers and normally serves to protect the host from the negative consequences of sustained immune activation during infection or cancer (221). The overexpression of TIM-3, LAG-3, and CTLA-4 is also associated with non-response to immune checkpoint blockade monotherapy for PD-1/PD-L1 (222). Combined inhibition of immune checkpoints augments antitumor immunity and results in enhanced tumor clearance (223).
9 Immunotherapy and other treatments
Due to the differences in the TME and cellular targets present within EBVaGCs and EBVnGCs, there are also differences in overall treatment effectiveness and patient responses (40, 161, 164, 224). A number of therapies have been shown to be effective within EBVaGCs and related cancers, with this section highlighting those currently approved, those still undergoing testing, and potential immuno- and chemo-therapeutics (225–227).
9.1 Immunotherapeutics
EBVaGCs are known to exhibit differential gene expression compared to EBVnGCs, with some of these gene products serving as targets for immunotherapeutics. Both PD-1 and PD-L1 are overexpressed in EBVaGCs (53) and are associated with worse patient outcomes (228). Pembrolizumab, an anti-PD-1 antibody, is an immunotherapeutic approved for treating a wide range of cancers, with a favourable benefit-to-risk profile and impressive antitumoral activity (229, 230). In a ground-breaking study by Kim et al., an overall response rate of 100% was demonstrated in EBVaGCs, raising hopes that this therapeutic would revolutionize treatment for these cancers (231). It was also demonstrated that response-rate was related to PD-L1 expression, thus making EBVaGCs prime candidates for the testing of anti-PD-1 immunotherapeutic targets (232). Pembrolizumab is currently undergoing clinical trials for EBVaGCs (NCT04795661, NCT05166577). Camrelizumab, also an anti-PD-1 antibody, has shown inconclusive results, possibly due to low PD-L1 positivity and small sample size (233). Avelumab, another anti-PD-L1 antibody, has been considered for maintenance therapy for GCs as an alternative to first-line chemotherapeutic treatments, with it demonstrating increased safety, but no effect on overall survival (234, 235). Trastuzumab, an anti-HER2 antibody, has been tested in conjunction with chemotherapy, resulting in higher overall survival as compared to chemotherapy alone (236). The identification of a wider range of targets, as well as improved EBVaGC patient stratification likely to respond to these targeted agents, would increase effectiveness and diversity of options for monoclonal antibody therapy (237, 238).
Apart from antibodies, a number of other immunotherapeutic treatment options are being considered for treatment of EBVaGCs (239–241). One such option is chimeric antigen receptor (CAR) T cell therapy, with multiple clinical trials recruiting GC patients for these studies (NCT05583201, NCT05393986, NCT05396300, NCT04650451). Studies have shown that a number of antigens, which include HER2, CEA, EpCAM, Claudin 18.2, and NKG2D, could be potential targets for CAR T cell-based therapy (242, 243). CAR T cell therapy has been reported to provide 5 more incremental quality-adjusted life-years compared to 4.6 years for nonpharmaceutical approaches (surgery, radiation therapy, stem cell transplants), at a similar level of cost-effectiveness (244). However, CAR T cell therapy has a number of downsides, which include the side effects of targeting antigens present on tissues besides the tumor (245, 246), cytokine storm syndrome (247, 248) and decreased effectiveness due to the immunosuppressive environment often present within cancers (249). These issues can be partially or fully resolved with engineering CAR T cells to have inducible rather than constitutive cytokine production (250, 251) and employing CAR T cells in conjunction with immune checkpoint inhibitor therapy (252, 253). As an alternative to CAR T cell therapy, CAR natural killer cell (CAR NK-cell) therapy has shown promising results with human GC cell lines in mouse models (254). CAR NK-cell therapy appears to have several advantages, including a reduced risk of alloreactivity and graft-versus-host disease, and the ability to kill cancer cells through non-MHC-restrictive effects (255, 256).
Cancer vaccines represent another potential avenue for EBVaGC immunotherapy given the expression of “non-self”, virally encoded proteins, which should be highly antigenic. Such vaccines come in many flavors, including whole tumor cell vaccines, DNA vaccines, DC vaccines, peptide vaccines, cancer stem cell vaccines, and neoantigen-based vaccines (257). A number of these vaccines have been approved and been successfully employed in various cancers (258–261). In GCs and related cancers, a candidate vaccine against the gastrointestinal peptide gastrin inhibits tumor growth and metastases in addition to modifying the TME when given alone or alongside immune checkpoint inhibitors (262, 263). Currently, a DNA vaccine specific for EBVaGC and other EBV-associated cancers is currently undergoing preclinical trials (264). This vaccine targets the BARF1 antigen of EBV, which is highly expressed in EBVaGCs (265), and has been found to promote cell proliferation and block apoptosis (266, 267). BARF1 has previously been identified as a potential therapeutic target (268, 269). Results of the preclinical trial indicate that the vaccine elicits high titers of antibodies specific for BARF1 and supresses tumor growth via a CD8+ T cell mechanism (264).
The well-established ability of EBV to actively evade both intrinsic and adaptive immune responses via a plethora of different mechanism could compromise anti-tumor vaccines (5). Specifically, viral interference with the recognition of antigenic viral peptides via downregulation of MHC-I-based antigen loading and presentation during infection has been well described and could greatly blunt the effectiveness of MHC dependent T cell responses (5, 270). However, this may not be as significant of an impediment as anticipated, as recent studies have shown strong MHC-I and -II expression in EBVaGC, likely due to upregulation by the inflammatory cytokines in the TME (47, 48).
9.2 Chemotherapeutics
EBVaGCs are resistant to a number of chemotherapeutics, including 5-fluorouracil (271) and docetaxel (272), both of which are effective in EBVnGCs (273, 274). However, other chemotherapeutic options may effectively treat EBVaGCs. These include combination therapies, which exhibit improved effectiveness over monotherapies, with examples such as 5-fluorouracil with LY294002, a selective inhibitor of PI3K (271), the PI3K/mTOR dual inhibitor CMG002 with chloroquine (275), and ganciclovir with gemcitabine (276). Additionally, there is also the option of adjuvant therapy, in which surgical intervention is followed by a monotherapy or combination therapy. One such study employed gastrectomy followed by either capecitabine and oxaliplatin or S-1 monotherapy, although no difference in outcomes was observed between EBVaGCs and EBVnGCs (277). Additional studies were performed to identify better combinations of chemotherapeutics, although only limited results were achieved (278, 279).
Since EBVaGCs are often associated with hypermethylation of promoters within the genome, particularly those of tumor suppressors and negative regulators of the cell cycle (280, 281), drugs targeting DNA methylation using either DNA methyltransferase or histone deacetylase inhibitors have been suggested to have therapeutic potential. Currently, two DNA methyltransferase inhibitors, azacitidine and decitabine, have been approved for acute myeloid leukemia and myelodysplastic syndrome (282), but may also be employed in EBVaGCs. Another drug currently undergoing clinical trials, zebularine, was previously shown to demethylate the STING promoter, which regulates expression of the cGAS/STING innate immune pathway, resulting in increased CD8+ T and NK infiltration, as well as reduced tumor burden in GC cell line-based models (283). The benefits of these classes of drugs include the reversibility of effects, due to epigenetic changes, as well the possibility of reversing changes in the TME (154, 284). Downsides of such drugs include the potential of non-specific gene activation as a result of demethylation, which may include oncogenes, as well as the lack of studies exploring the long-term effects of epigenetic therapies (129). Further research in this class of therapeutics is necessary to determine their ultimate value as new treatment options for EBVaGC.
9.3 Drugs stimulating EBV reactivation from latency
EBV persistence in the infected individual is lifelong and largely mediated by the maintenance of the latent state of viral episomes in infected cells (285). Reactivation of latent virus could provide a means of killing EBVaGC cells, akin to the “shock and kill” strategies suggested for curing HIV (286, 287). In this way, reactivation of EBV infection from latency activates viral transcription, protein expression and virion production, potentially triggering cytolysis, immune-mediated clearance, and sensitivity to antiviral drugs such as ganciclovir and valganciclovir (288).
Reactivation of latent EBV infection is commonly referred to as lytic-induction therapy. This treatment approach typically involves a combination of a lytic inducer and nucleoside analogue antiviral pro-drugs (289). Reactivation of the EBV lytic cycle leads to the expression of numerous additional viral proteins not expressed during latency. These include the viral BGLF4 encoded protein kinase, which converts antiviral prodrugs like ganciclovir into their cytotoxic forms, consequently killing the infected cell (290) and adjacent cells via “bystander killing” (291). These additional viral products also potentially represent non-self antigens that may trigger improved anti-tumor responses or could be useful targets for CAR T cell-based therapy.
Multiple approaches for EBV lytic reactivation have been reported, primarily representing epigenetic-targeted therapies, such as histone deacetylase inhibitors (292, 293). Intriguingly, chemotherapy itself appears to trigger some level of EBV reactivation, which can be further enhanced by epigenetic modulators (294, 295). Although limited, existing studies assessing lytic induction therapy suggest potential utility in EBVaGC (276, 296), with ongoing screens searching for more effective small molecule inducers of EBV reactivation (297).
10 Datasets and tools in EBVaGC
This section serves to highlight public datasets and tools available for the exploration of EBVaGCs. The availability of said tools and datasets are most beneficial when performing in vitro or in vivo research, providing additional sources of validation and comparison. They can also assist in the process of hypothesis generation, by helping narrow down possible research targets that could be of interest when exploring oncogenesis, as well as potential biomarkers and therapeutic targets.
10.1 TCGA and TCGA-derived datasets
The Cancer Gene Atlas (TCGA) has a large collection of stomach adenocarcinoma (STAD) tissue samples, including bulk tumor gene expression data for up to 30 EBV-positive and 353 EBV-negative GC samples (22). The original dataset features patient clinical information, expression data for 20,531 cellular mRNAs and 1046 cellular miRNA genes, and methylation across 395,405 unique genomic probes. The aforementioned datasets can be accessed through the Broad Institute GDAC Firehose web portal (https://gdac.broadinstitute.org/) or other tools. Furthermore, additional sequencing of TCGA GC tissue samples have yielded datasets for expression levels of EBV-encoded mRNAs (201) and miRNAs (87), compilation of immune landscape features (298), and standardized patient outcome data (299). As one example of the utility of this data, bioinformatic comparisons confirmed that the EBVaGC TME exhibits many aspects of a T cell-inflamed phenotype, with greater T and NK cell infiltration, increased expression of many immune checkpoint markers (BTLA, CD96, CTLA4, LAG3, PD1, TIGIT and TIM3), and multiple T cell effector molecules in comparison with EBVnGCs (300).
10.2 EBVaGC-centric tools
The computational nature of working with molecular expression datasets may present a barrier to researchers not well-versed in computational techniques, which limits the accessibility of such datasets. While a number of viral-centric tools featuring molecular gene expression have been developed (301, 302), they are often few and far between. The same has been true for EBVaGCs up until recently; the development of the EBV-GCR web suite of tools (303) represents a useful addition. This tool set allows users to quickly query both viral and cellular gene expression data from single cell and bulk tumor GC mRNA sequencing data. Users can also easily obtain data regarding correlations between viral and cellular gene expression, genome-wide methylation, immune landscape features, and patient outcomes.
11 Perspectives and discussion
Even with the great wealth of existing knowledge related to EBVaGCs, there is still much to discover and understand to improve treatment options and patient outcomes. In particular, employing scRNA-seq for cancers can provide a deeper understanding of the TME composition, immune heterogeneity in tumors, evaluation of tumor escape mechanisms, as well as mechanisms of drug or therapy resistance (304). Currently, there are few studies leveraging scRNA-seq data for comparing EBVaGCs with EBVnGCs (207). Further, larger scale studies would be beneficial in understanding the various dimensions of EBVaGCs and their differences compared to EBVnGCs. Additionally, therapeutics targeting EBV-associated miRNAs are relatively unexplored (111) and given the roles for these virally encoded miRNAs in EBV-positive malignancies (100), could make for effective targets. An example of such a therapeutic was in a recent study in which researchers employed anti-miRNAs to silence miR-BART7-3p, reducing tumor growth in tested animal models (305). Another study found that circRNAome miRNA sponges of EBV can decrease the expression of multiple host miRNAs, thus helping drive carcinogenesis (306). In turn, microRNAs targeting EBV miRNAs could be developed in order to downregulate their expression. Advances in precision- and immuno-therapeutics should result in improved treatment options and patient outcomes.
Author contributions
MS: Conceptualization, Writing – original draft, Writing – review & editing. KM: Conceptualization, Visualization, Writing – review & editing. JM: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was financially supported by a grant from the Canadian Institutes of Health Research (PJT-173496).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mrozek-Gorska P, Buschle A, Pich D, Schwarzmayr T, Fechtner R, Scialdone A, et al. Epstein-Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc Natl Acad Sci U.S.A. (2019) 116:16046–55. doi: 10.1073/pnas.1901314116
2. Tsang CM, Deng W, Yip YL, Zeng MS, Lo KW, Tsao SW. Epstein-Barr virus infection and persistence in nasopharyngeal epithelial cells. Chin J Cancer. (2014) 33:549–55. doi: 10.5732/cjc.014.10169
3. Temple RM, Zhu J, Budgeon L, Christensen ND, Meyers C, Sample CE. Efficient replication of Epstein-Barr virus in stratified epithelium in vitro. Proc Natl Acad Sci U.S.A. (2014) 111:16544–9. doi: 10.1073/pnas.1400818111
4. Kim WY, Montes-Mojarro IA, Fend F, Quintanilla-Martinez L. Epstein-barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. (2019) 7:71. doi: 10.3389/fped.2019.00071
5. Ressing ME, van Gent M, Gram AM, Hooykaas MJ, Piersma SJ, Wiertz EJ. Immune evasion by epstein-barr virus. Curr Top Microbiol Immunol. (2015) 391:355–81. doi: 10.1007/978-3-319-22834-1_12
6. Tzellos S, Farrell PJ. Epstein-barr virus sequence variation-biology and disease. Pathogens. (2012) 1:156–74. doi: 10.3390/pathogens1020156
7. Farrell PJ. Epstein-barr virus and cancer. Annu Rev Pathol. (2019) 14:29–53. doi: 10.1146/annurev-pathmechdis-012418-013023
8. Umakanthan S, Bukelo MM. Molecular genetics in epstein-barr virus-associated Malignancies. Life (Basel). (2021) 11:593. doi: 10.3390/life11070593
9. Han S, Tay JK, Loh CJL, Chu AJM, Yeong JPS, Lim CM, et al. Epstein-barr virus epithelial cancers-A comprehensive understanding to drive novel therapies. Front Immunol. (2021) 12:734293. doi: 10.3389/fimmu.2021.734293
10. de Martel C, Georges D, Bray F, Ferlay J, Clifford GM. Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. Lancet Glob Health. (2020) 8:e180–90. doi: 10.1016/S2214-109X(19)30488-7
11. Tavakoli A, Monavari SH, Solaymani Mohammadi F, Kiani SJ, Armat S, Farahmand M. Association between Epstein-Barr virus infection and gastric cancer: a systematic review and meta-analysis. BMC Cancer. (2020) 20:493. doi: 10.1186/s12885-020-07013-x
12. Cui X, Snapper CM. Epstein barr virus: Development of vaccines and immune cell therapy for EBV-associated diseases. Front Immunol. (2021) 12:734471. doi: 10.3389/fimmu.2021.734471
13. Escalante GM, Mutsvunguma LZ, Muniraju M, Rodriguez E, Ogembo JG. Four decades of prophylactic EBV vaccine research: A systematic review and historical perspective. Front Immunol. (2022) 13:867918. doi: 10.3389/fimmu.2022.867918
14. Cohen JI. Vaccine development for epstein-barr virus. Adv Exp Med Biol. (2018) 1045:477–93. doi: 10.1007/978-981-10-7230-7_22
15. Pei Y, Wong JHY, Robertson ES. Targeted therapies for epstein-barr virus-associated lymphomas. Cancers (Basel). (2020) 12:2565. doi: 10.3390/cancers12092565
16. Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. (2020) 396:635–48. doi: 10.1016/S0140-6736(20)31288-5
17. Cheng XJ, Lin JC, Tu SP. Etiology and prevention of gastric cancer. Gastrointest Tumors. (2016) 3:25–36. doi: 10.1159/000443995
18. Yusefi AR, Bagheri Lankarani K, Bastani P, Radinmanesh M, Kavosi Z. Risk factors for gastric cancer: A systematic review. Asian Pac J Cancer Prev. (2018) 19:591–603. doi: 10.22034/APJCP.2018.19.3.591
19. Machlowska J, Baj J, Sitarz M, Maciejewski R, Sitarz R. Gastric cancer: Epidemiology, risk factors, classification, genomic characteristics and treatment strategies. Int J Mol Sci. (2020) 21:4012. doi: 10.3390/ijms21114012
20. Burke AP, Yen TS, Shekitka KM, Sobin LH. Lymphoepithelial carcinoma of the stomach with Epstein-Barr virus demonstrated by polymerase chain reaction. Mod Pathol. (1990) 3:377–80.
21. Shibata D, Weiss LM. Epstein-Barr virus-associated gastric adenocarcinoma. Am J Pathol. (1992) 140:769–74.
22. Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. (2014) 513:202–9. doi: 10.1038/nature13480
23. Dunmire SK, Verghese PS, Balfour HH Jr. Primary Epstein-Barr virus infection. J Clin Virol. (2018) 102:84–92. doi: 10.1016/j.jcv.2018.03.001
24. Hutt-Fletcher LM. The long and complicated relationship between epstein-barr virus and epithelial cells. J Virol. (2017) 91:e01677-16. doi: 10.1128/JVI.01677-16
25. Imai S, Nishikawa J, Takada K. Cell-to-cell contact as an efficient mode of Epstein-Barr virus infection of diverse human epithelial cells. J Virol. (1998) 72:4371–8. doi: 10.1128/JVI.72.5.4371-4378.1998
26. Ribeiro J, Oliveira A, Malta M, Oliveira C, Silva F, Galaghar A, et al. Clinical and pathological characterization of Epstein-Barr virus-associated gastric carcinomas in Portugal. World J Gastroenterol. (2017) 23:7292–302. doi: 10.3748/wjg.v23.i40.7292
27. Yang J, Liu Z, Zeng B, Hu G, Gan R. Epstein-Barr virus-associated gastric cancer: A distinct subtype. Cancer Lett. (2020) 495:191–9. doi: 10.1016/j.canlet.2020.09.019
28. Kang BW, Seo AN, Yoon S, Bae HI, Jeon SW, Kwon OK, et al. Prognostic value of tumor-infiltrating lymphocytes in Epstein-Barr virus-associated gastric cancer. Ann Oncol. (2016) 27:494–501. doi: 10.1093/annonc/mdv610
29. Song HJ, Kim KM. Pathology of epstein-barr virus-associated gastric carcinoma and its relationship to prognosis. Gut Liver. (2011) 5:143–8. doi: 10.5009/gnl.2011.5.2.143
30. Song HJ, Srivastava A, Lee J, Kim YS, Kim KM, Ki Kang W, et al. Host inflammatory response predicts survival of patients with Epstein-Barr virus-associated gastric carcinoma. Gastroenterology. (2010) 139:84–92.e2. doi: 10.1053/j.gastro.2010.04.002
31. Murphy G, Pfeiffer R, Camargo MC, Rabkin CS. Meta-analysis shows that prevalence of Epstein-Barr virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology. (2009) 137:824–33. doi: 10.1053/j.gastro.2009.05.001
32. Cheng N, Li P, Cheng H, Zhao X, Dong M, Zhang Y, et al. Prognostic value of tumor-infiltrating lymphocytes and tertiary lymphoid structures in epstein-barr virus-associated and -negative gastric carcinoma. Front Immunol. (2021) 12:692859. doi: 10.3389/fimmu.2021.692859
33. Itami H, Morita K, Nakai T, Uchiyama T, Sugimoto S, Sasaki S, et al. Gastritis cystica profunda is associated with aberrant p53 and Epstein-Barr virus in gastric cancer: A clinicopathological, immunohistochemical and in situ hybridization study. Pathol Int. (2021) 71:42–50. doi: 10.1111/pin.13039
34. Shinozaki-Ushiku A, Kunita A, Fukayama M. Update on Epstein-Barr virus and gastric cancer (review). Int J Oncol. (2015) 46:1421–34. doi: 10.3892/ijo.2015.2856
35. Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. (2019) 19:307–25. doi: 10.1038/s41568-019-0144-6
36. Goc J, Germain C, Vo-Bourgais TK, Lupo A, Klein C, Knockaert S, et al. Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res. (2014) 74:705–15. doi: 10.1158/0008-5472.CAN-13-1342
37. Lerner MR, Andrews NC, Miller G, Steitz JA. Two small RNAs encoded by Epstein-Barr virus and complexed with protein are precipitated by antibodies from patients with systemic lupus erythematosus. Proc Natl Acad Sci U.S.A. (1981) 78:805–9. doi: 10.1073/pnas.78.2.805
38. Rowlands DC, Ito M, Mangham DC, Reynolds G, Herbst H, Hallissey MT, et al. Epstein-Barr virus and carcinomas: rare association of the virus with gastric adenocarcinomas. Br J Cancer. (1993) 68:1014–9. doi: 10.1038/bjc.1993.472
39. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
40. Chen JN, He D, Tang F, Shao CK. Epstein-Barr virus-associated gastric carcinoma: a newly defined entity. J Clin Gastroenterol. (2012) 46:262–71. doi: 10.1097/MCG.0b013e318249c4b8
41. Camargo MC, Kim WH, Chiaravalli AM, Kim KM, Corvalan AH, Matsuo K, et al. Improved survival of gastric cancer with tumour Epstein-Barr virus positivity: an international pooled analysis. Gut. (2014) 63:236–43. doi: 10.1136/gutjnl-2013-304531
42. Park JH, Kim EK, Kim YH, Kim JH, Bae YS, Lee YC, et al. Epstein-Barr virus positivity, not mismatch repair-deficiency, is a favorable risk factor for lymph node metastasis in submucosa-invasive early gastric cancer. Gastric Cancer. (2016) 19:1041–51. doi: 10.1007/s10120-015-0565-1
43. Gong LP, Chen JN, Xiao L, He Q, Feng ZY, Zhang ZG, et al. The implication of tumor-infiltrating lymphocytes in Epstein-Barr virus-associated gastric carcinoma. Hum Pathol. (2019) 85:82–91. doi: 10.1016/j.humpath.2018.11.002
44. He HC, Han R, Xu BH, Huang C, Li MM, He CY, et al. Circulating Epstein-Barr virus DNA associated with hepatic impairment and its diagnostic and prognostic role in patients with gastric cancer. Front Med (Lausanne). (2022) 9:1028033. doi: 10.3389/fmed.2022.1028033
45. Stanland LJ, Luftig MA. The role of EBV-induced hypermethylation in gastric cancer tumorigenesis. Viruses. (2020) 12:1222. doi: 10.3390/v12111222
46. Leong MML, Lung ML. The impact of epstein-barr virus infection on epigenetic regulation of host cell gene expression in epithelial and lymphocytic Malignancies. Front Oncol. (2021) 11:629780. doi: 10.3389/fonc.2021.629780
47. Ghasemi F, Gameiro SF, Tessier TM, Maciver AH, Mymryk JS. High levels of class I major histocompatibility complex mRNA are present in epstein-barr virus-associated gastric adenocarcinomas. Cells. (2020) 9:499. doi: 10.3390/cells9020499
48. Ghasemi F, Tessier TM, Gameiro SF, Maciver AH, Cecchini MJ, Mymryk JS. High MHC-II expression in Epstein-Barr virus-associated gastric cancers suggests that tumor cells serve an important role in antigen presentation. Sci Rep. (2020) 10:14786. doi: 10.1038/s41598-020-71775-4
49. Moore A, Hikri E, Goshen-Lago T, Barkan T, Morgenstern S, Brook E, et al. Young-onset gastric cancer and Epstein-Barr Virus (EBV) - a major player in the pathogenesis? BMC Cancer. (2020) 20:34. doi: 10.1186/s12885-020-6517-0
50. Jia X, Guo T, Li Z, Zhang M, Feng Y, Dong B, et al. Clinicopathological and immunomicroenvironment characteristics of epstein-barr virus-associated gastric cancer in a chinese population. Front Oncol. (2021) 10:586752. doi: 10.3389/fonc.2020.586752
51. Kinami S, Saito H, Takamura H. Significance of lymph node metastasis in the treatment of gastric cancer and current challenges in determining the extent of metastasis. Front Oncol. (2021) 11:806162. doi: 10.3389/fonc.2021.806162
52. Rihane FE, Erguibi D, Elyamine O, Abumsimir B, Ennaji MM, Chehab F. Helicobacter pylori co-infection with Epstein-Barr virus and the risk of developing gastric adenocarcinoma at an early age: Observational study infectious agents and cancer. Ann Med Surg (Lond). (2021) 68:102651. doi: 10.1016/j.amsu.2021.102651
53. Lima A, Sousa H, Medeiros R, Nobre A, MaChado M. PD-L1 expression in EBV associated gastric cancer: a systematic review and meta-analysis. Discovery Oncol. (2022) 13:19. doi: 10.1007/s12672-022-00479-0
54. Nakano H, Saito M, Nakajima S, Saito K, Nakayama Y, Kase K, et al. PD-L1 overexpression in EBV-positive gastric cancer is caused by unique genomic or epigenomic mechanisms. Sci Rep. (2021) 11:1982. doi: 10.1038/s41598-021-81667-w
55. Gu L, Chen M, Guo D, Zhu H, Zhang W, Pan J, et al. PD-L1 and gastric cancer prognosis: A systematic review and meta-analysis. PLoS One. (2017) 12:e0182692. doi: 10.1371/journal.pone.0182692
56. Sawada L, Vallinoto ACR, Brasil-Costa I. Regulation of the immune checkpoint indoleamine 2,3-dioxygenase expression by epstein-barr virus. Biomolecules. (2021) 11:1792. doi: 10.3390/biom11121792
57. Liu WL, Lin YH, Xiao H, Xing S, Chen H, Chi PD, et al. Epstein-Barr virus infection induces indoleamine 2,3-dioxygenase expression in human monocyte-derived macrophages through p38/mitogen-activated protein kinase and NF-kappaB pathways: impairment in T cell functions. J Virol. (2014) 88:6660–71. doi: 10.1128/JVI.03678-13
58. Zhang YW, Zhao XX, Tan C, Zhang ZG, Jiang Y, Chen JN, et al. Epstein-Barr virus latent membrane protein 2A suppresses the expression of HER2 via a pathway involving TWIST and YB-1 in Epstein-Barr virus-associated gastric carcinomas. Oncotarget. (2015) 6:207–20. doi: 10.18632/oncotarget.v6i1
59. Kurokawa Y, Matsuura N, Kimura Y, Adachi S, Fujita J, Imamura H, et al. Multicenter large-scale study of prognostic impact of HER2 expression in patients with resectable gastric cancer. Gastric Cancer. (2015) 18:691–7. doi: 10.1007/s10120-014-0430-7
60. Qian Y, Daza J, Itzel T, Betge J, Zhan T, Marme F, et al. Prognostic cancer gene expression signatures: current status and challenges. Cells. (2021) 10:648. doi: 10.3390/cells10030648
61. He Y, Song H, Jiang Y, Ren W. Identification of immune-related prognostic markers in gastric cancer. J Healthc Eng. (2022) 2022:7897274. doi: 10.1155/2022/7897274
62. Wroblewski LE, Peek RM Jr., Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. (2010) 23:713–39. doi: 10.1128/CMR.00011-10
63. Pandey S, Jha HC, Shukla SK, Shirley MK, Robertson ES. Epigenetic regulation of tumor suppressors by helicobacter pylori enhances EBV-induced proliferation of gastric epithelial cells. mBio. (2018) 9:e00649-18. doi: 10.1128/mBio.00649-18
64. Kashyap D, Baral B, Jakhmola S, Singh AK, Jha HC. Helicobacter pylori and Epstein-Barr Virus Coinfection Stimulates Aggressiveness in Gastric Cancer through the Regulation of Gankyrin. mSphere. (2021) 6:e0075121. doi: 10.1128/mSphere.00751-21
65. Saju P, Murata-Kamiya N, Hayashi T, Senda Y, Nagase L, Noda S, et al. Host SHP1 phosphatase antagonizes Helicobacter pylori CagA and can be downregulated by Epstein-Barr virus. Nat Microbiol. (2016) 1:16026. doi: 10.1038/nmicrobiol.2016.26
66. Cardenas-Mondragon MG, Carreon-Talavera R, Camorlinga-Ponce M, Gomez-Delgado A, Torres J, Fuentes-Panana EM. Correction: epstein barr virus and helicobacter pylori co-infection are positively associated with severe gastritis in pediatric patients. PLoS One. (2013) 8:e62850. doi: 10.1371/annotation/865eaad7-8547-49ac-a42d-47e9d0755bb3
67. Singh S, Jha HC. Status of epstein-barr virus coinfection with helicobacter pylori in gastric cancer. J Oncol. (2017) 2017:3456264. doi: 10.1155/2017/3456264
68. Noh JH, Shin JY, Lee JH, Park YS, Lee IS, Kim GH, et al. Clinical significance of epstein-barr virus and helicobacter pylori infection in gastric carcinoma. Gut Liver. (2023) 17:69–77. doi: 10.5009/gnl210593
69. Nishikawa J, Shuto T, Yanagi A, Takagi T, Ogawa R, Sasaki S, et al. Epstein-Barr virus-associated gastric carcinomas developed after successful eradication of Helicobacter pylori. Clin J Gastroenterol. (2020) 13:506–11. doi: 10.1007/s12328-020-01094-8
70. Luo B, Wang Y, Wang XF, Liang H, Yan LP, Huang BH, et al. Expression of Epstein-Barr virus genes in EBV-associated gastric carcinomas. World J Gastroenterol. (2005) 11:629–33. doi: 10.3748/wjg.v11.i5.629
71. Borozan I, Zapatka M, Frappier L, Ferretti V. Analysis of epstein-barr virus genomes and expression profiles in gastric adenocarcinoma. J Virol. (2018) 92:e01239-17. doi: 10.1128/JVI.00330-18
72. Frappier L. Contributions of Epstein-Barr nuclear antigen 1 (EBNA1) to cell immortalization and survival. Viruses. (2012) 4:1537–47. doi: 10.3390/v4091537
73. Leight ER, Sugden B. EBNA-1: a protein pivotal to latent infection by Epstein-Barr virus. Rev Med Virol. (2000) 10:83–100. doi: 10.1002/(ISSN)1099-1654
74. Chatterjee K, Das P, Chattopadhyay NR, Mal S, Choudhuri T. The interplay between Epstein-Bar virus (EBV) with the p53 and its homologs during EBV associated Malignancies. Heliyon. (2019) 5:e02624. doi: 10.1016/j.heliyon.2019.e02624
75. Sivachandran N, Dawson CW, Young LS, Liu FF, Middeldorp J, Frappier L. Contributions of the Epstein-Barr virus EBNA1 protein to gastric carcinoma. J Virol. (2012) 86:60–8. doi: 10.1128/JVI.05623-11
76. Luo Y, Liu Y, Wang C, Gan R. Signaling pathways of EBV-induced oncogenesis. Cancer Cell Int. (2021) 21:93. doi: 10.1186/s12935-021-01793-3
77. Gruhne B, Sompallae R, Marescotti D, Kamranvar SA, Gastaldello S, Masucci MG. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc Natl Acad Sci U.S.A. (2009) 106:2313–8. doi: 10.1073/pnas.0810619106
78. Li JSZ, Abbasi A, Kim DH, Lippman SM, Alexandrov LB, Cleveland DW. Chromosomal fragile site breakage by EBV-encoded EBNA1 at clustered repeats. Nature. (2023) 616:504–9. doi: 10.1038/s41586-023-05923-x
79. Shire K, Marcon E, Greenblatt J, Frappier L. Characterization of a cancer-associated Epstein-Barr virus EBNA1 variant reveals a novel interaction with PLOD1 and PLOD3. Virology. (2021) 562:103–9. doi: 10.1016/j.virol.2021.07.009
80. Li SS, Lian YF, Huang YL, Huang YH, Xiao J. Overexpressing PLOD family genes predict poor prognosis in gastric cancer. J Cancer. (2020) 11:121–31. doi: 10.7150/jca.35763
81. Caldwell RG, Wilson JB, Anderson SJ, Longnecker R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity. (1998) 9:405–11. doi: 10.1016/S1074-7613(00)80623-8
82. Gires O, Zimber-Strobl U, Gonnella R, Ueffing M, Marschall G, Zeidler R, et al. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J. (1997) 16:6131–40. doi: 10.1093/emboj/16.20.6131
83. Morales-Sanchez A, Fuentes-Panana EM. Epstein-barr virus-associated gastric cancer and potential mechanisms of oncogenesis. Curr Cancer Drug Targets. (2017) 17:534–54. doi: 10.2174/1568009616666160926124923
84. Huang X, Zhang M, Zhang Z. The role of LMP1 in epstein-barr virus-associated gastric cancer. Curr Cancer Drug Targets. (2024) 24:127–41. doi: 10.2174/1568009623666230512153741
85. De Re V, Caggiari L, De Zorzi M, Fanotto V, Miolo G, Puglisi F, et al. Epstein-Barr virus BART microRNAs in EBV- associated Hodgkin lymphoma and gastric cancer. Infect Agent Cancer. (2020) 15:42. doi: 10.1186/s13027-020-00307-6
86. Zidovec Lepej S, Matulic M, Grskovic P, Pavlica M, Radmanic L, Korac P. miRNAs: EBV mechanism for escaping host’s immune response and supporting tumorigenesis. Pathogens. (2020) 9:353. doi: 10.3390/pathogens9050353
87. Ungerleider N, Bullard W, Kara M, Wang X, Roberts C, Renne R, et al. EBV miRNAs are potent effectors of tumor cell transcriptome remodeling in promoting immune escape. PLoS Pathog. (2021) 17:e1009217. doi: 10.1371/journal.ppat.1009217
88. Zebardast A, Tehrani SS, Latifi T, Sadeghi F. Critical review of Epstein-Barr virus microRNAs relation with EBV-associated gastric cancer. J Cell Physiol. (2021) 236:6136–53. doi: 10.1002/jcp.30297
89. Zhang J, Li X, Hu J, Cao P, Yan Q, Zhang S, et al. Long noncoding RNAs involvement in Epstein-Barr virus infection and tumorigenesis. Virol J. (2020) 17:51. doi: 10.1186/s12985-020-01308-y
90. Torres K, Landeros N, Wichmann IA, Polakovicova I, Aguayo F, Corvalan AH. EBV miR-BARTs and human lncRNAs: Shifting the balance in competing endogenous RNA networks in EBV-associated gastric cancer. Biochim Biophys Acta Mol Basis Dis. (2021) 1867:166049. doi: 10.1016/j.bbadis.2020.166049
91. Skalsky RL, Cullen BR. EBV noncoding RNAs. Curr Top Microbiol Immunol. (2015) 391:181–217. doi: 10.1007/978-3-319-22834-1_6
92. Edwards RH, Marquitz AR, Raab-Traub N. Epstein-Barr virus BART microRNAs are produced from a large intron prior to splicing. J Virol. (2008) 82:9094–106. doi: 10.1128/JVI.00785-08
93. Marquitz AR, Mathur A, Edwards RH, Raab-Traub N. Host gene expression is regulated by two types of noncoding RNAs transcribed from the epstein-barr virus bamHI A rightward transcript region. J Virol. (2015) 89:11256–68. doi: 10.1128/JVI.01492-15
94. Medley JC, Panzade G, Zinovyeva AY. microRNA strand selection: Unwinding the rules. Wiley Interdiscip Rev RNA. (2021) 12:e1627. doi: 10.1002/wrna.1627
95. Chen SJ, Chen GH, Chen YH, Liu CY, Chang KP, Chang YS, et al. Characterization of Epstein-Barr virus miRNAome in nasopharyngeal carcinoma by deep sequencing. PLoS One. (2010) 5:e12745. doi: 10.1371/journal.pone.0012745
96. Hooykaas MJ, Kruse E, Wiertz EJ, Lebbink RJ. Comprehensive profiling of functional Epstein-Barr virus miRNA expression in human cell lines. BMC Genomics. (2016) 17:644. doi: 10.1186/s12864-016-2978-6
98. Zuo L, Yue W, Du S, Xin S, Zhang J, Liu L, et al. An update: Epstein-Barr virus and immune evasion via microRNA regulation. Virol Sin. (2017) 32:175–87. doi: 10.1007/s12250-017-3996-5
99. Kincaid RP, Sullivan CS. Virus-encoded microRNAs: an overview and a look to the future. PLoS Pathog. (2012) 8:e1003018. doi: 10.1371/journal.ppat.1003018
100. Wang M, Gu B, Chen X, Wang Y, Li P, Wang K. The function and therapeutic potential of epstein-barr virus-encoded microRNAs in cancer. Mol Ther Nucleic Acids. (2019) 17:657–68. doi: 10.1016/j.omtn.2019.07.002
101. Albanese M, Tagawa T, Bouvet M, Maliqi L, Lutter D, Hoser J, et al. Epstein-Barr virus microRNAs reduce immune surveillance by virus-specific CD8+ T cells. Proc Natl Acad Sci U.S.A. (2016) 113:E6467–75. doi: 10.1073/pnas.1605884113
102. Tagawa T, Albanese M, Bouvet M, Moosmann A, Mautner J, Heissmeyer V, et al. Epstein-Barr viral miRNAs inhibit antiviral CD4+ T cell responses targeting IL-12 and peptide processing. J Exp Med. (2016) 213:2065–80. doi: 10.1084/jem.20160248
103. Skalsky RL, Corcoran DL, Gottwein E, Frank CL, Kang D, Hafner M, et al. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. (2012) 8:e1002484. doi: 10.1371/journal.ppat.1002484
104. Wong TS, Chen S, Zhang MJ, Chan JY, Gao W. Epstein-Barr virus-encoded microRNA BART7 downregulates major histocompatibility complex class I chain-related peptide A and reduces the cytotoxicity of natural killer cells to nasopharyngeal carcinoma. Oncol Lett. (2018) 16:2887–92. doi: 10.3892/ol
105. Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe. (2009) 5:376–85. doi: 10.1016/j.chom.2009.03.003
106. Lu Y, Qin Z, Wang J, Zheng X, Lu J, Zhang X, et al. Epstein-barr virus miR-BART6-3p inhibits the RIG-I pathway. J Innate Immun. (2017) 9:574–86. doi: 10.1159/000479749
107. Shinozaki-Ushiku A, Kunita A, Isogai M, Hibiya T, Ushiku T, Takada K, et al. Profiling of virus-encoded microRNAs in epstein-barr virus-associated gastric carcinoma and their roles in gastric carcinogenesis. J Virol. (2015) 89:5581–91. doi: 10.1128/JVI.03639-14
108. Cai LM, Lyu XM, Luo WR, Cui XF, Ye YF, Yuan CC, et al. EBV-miR-BART7-3p promotes the EMT and metastasis of nasopharyngeal carcinoma cells by suppressing the tumor suppressor PTEN. Oncogene. (2015) 34:2156–66. doi: 10.1038/onc.2014.341
109. Wang M, Yu F, Wu W, Wang Y, Ding H, Qian L. Epstein-Barr virus-encoded microRNAs as regulators in host immune responses. Int J Biol Sci. (2018) 14:565–76. doi: 10.7150/ijbs.24562
110. Riley KJ, Rabinowitz GS, Yario TA, Luna JM, Darnell RB, Steitz JA. EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J. (2012) 31:2207–21. doi: 10.1038/emboj.2012.63
111. Abusalah MAH, Irekeola AA, Hanim Shueb R, Jarrar M, Yean Yean C. Prognostic Epstein-Barr Virus (EBV) miRNA biomarkers for survival outcome in EBV-associated epithelial Malignancies: Systematic review and meta-analysis. PLoS One. (2022) 17:e0266893. doi: 10.1371/journal.pone.0266893
112. Kang BW, Choi Y, Kwon OK, Lee SS, Chung HY, Yu W, et al. High level of viral microRNA-BART20-5p expression is associated with worse survival of patients with Epstein-Barr virus-associated gastric cancer. Oncotarget. (2017) 8:14988–94. doi: 10.18632/oncotarget.v8i9
113. Haar J, Contrant M, Bernhardt K, Feederle R, Diederichs S, Pfeffer S, et al. The expression of a viral microRNA is regulated by clustering to allow optimal B cell transformation. Nucleic Acids Res. (2016) 44:1326–41. doi: 10.1093/nar/gkv1330
114. Feederle R, Haar J, Bernhardt K, Linnstaedt SD, Bannert H, Lips H, et al. The members of an Epstein-Barr virus microRNA cluster cooperate to transform B lymphocytes. J Virol. (2011) 85:9801–10. doi: 10.1128/JVI.05100-11
115. Zhang J, Huang T, Zhou Y, Cheng ASL, Yu J, To KF, et al. The oncogenic role of Epstein-Barr virus-encoded microRNAs in Epstein-Barr virus-associated gastric carcinoma. J Cell Mol Med. (2018) 22:38–45. doi: 10.1111/jcmm.13354
116. Li J, Liu W, Che K, Zhang Y, Zhao Z, Luo B. The methylation status and expression of epstein-barr virus early genes BARF1 and BHRF1 in epstein-barr virus-associated gastric carcinomas. Gastroenterol Res Pract. (2017) 2017:3804146. doi: 10.1155/2017/3804146
117. Silva JMC, Teixeira EB, Mourao R, Ferraz RS, Moreira FC, de Assumpcao PP, et al. The landscape of lncRNAs in gastric cancer: from molecular mechanisms to potential clinical applications. Front Pharmacol. (2023) 14:1237723. doi: 10.3389/fphar.2023.1237723
118. Liu Y, Hu Z, Zhang Y, Wang C. Long non-coding RNAs in Epstein-Barr virus-related cancer. Cancer Cell Int. (2021) 21:278. doi: 10.1186/s12935-021-01986-w
119. Huang T, Ji Y, Hu D, Chen B, Zhang H, Li C, et al. SNHG8 is identified as a key regulator of epstein-barr virus(EBV)-associated gastric cancer by an integrative analysis of lncRNA and mRNA expression. Oncotarget. (2016) 7:80990–1002. doi: 10.18632/oncotarget.v7i49
120. Zou C, Liao J, Hu D, Su Y, Lin H, Lin K, et al. SNHG8 Promotes the Progression of Epstein-Barr Virus-Associated Gastric Cancer via Sponging miR-512-5p and Targeting TRIM28. Front Oncol. (2021) 11:734694. doi: 10.3389/fonc.2021.734694
121. Kanda T, Yajima M, Ikuta K. Epstein-Barr virus strain variation and cancer. Cancer Sci. (2019) 110:1132–9. doi: 10.1111/cas.13954
122. Sample J, Young L, Martin B, Chatman T, Kieff E, Rickinson A, et al. Epstein-Barr virus types 1 and 2 differ in their EBNA-3A, EBNA-3B, and EBNA-3C genes. J Virol. (1990) 64:4084–92. doi: 10.1128/jvi.64.9.4084-4092.1990
123. Romero-Masters JC, Huebner SM, Ohashi M, Bristol JA, Benner BE, Barlow EA, et al. B cells infected with Type 2 Epstein-Barr virus (EBV) have increased NFATc1/NFATc2 activity and enhanced lytic gene expression in comparison to Type 1 EBV infection. PLoS Pathog. (2020) 16:e1008365. doi: 10.1371/journal.ppat.1008365
124. Singh DR, Nelson SE, Pawelski AS, Cantres-Velez JA, Kansra AS, Pauly NP, et al. Type 1 and Type 2 Epstein-Barr viruses induce proliferation, and inhibit differentiation, in infected telomerase-immortalized normal oral keratinocytes. PLoS Pathog. (2022) 18:e1010868. doi: 10.1371/journal.ppat.1010868
125. Re V, Brisotto G, Repetto O, De Zorzi M, Caggiari L, Zanussi S, et al. Overview of epstein-barr-virus-associated gastric cancer correlated with prognostic classification and development of therapeutic options. Int J Mol Sci. (2020) 21:9400. doi: 10.3390/ijms21249400
126. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discovery. (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
127. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
128. Ignatova E, Seriak D, Fedyanin M, Tryakin A, Pokataev I, Menshikova S, et al. Epstein-Barr virus-associated gastric cancer: disease that requires special approach. Gastric Cancer. (2020) 23:951–60. doi: 10.1007/s10120-020-01095-z
129. Yau TO, Tang CM, Yu J. Epigenetic dysregulation in Epstein-Barr virus-associated gastric carcinoma: disease and treatments. World J Gastroenterol. (2014) 20:6448–56. doi: 10.3748/wjg.v20.i21.6448
130. Chen ZH, Yan SM, Chen XX, Zhang Q, Liu SX, Liu Y, et al. The genomic architecture of EBV and infected gastric tissue from precursor lesions to carcinoma. Genome Med. (2021) 13:146. doi: 10.1186/s13073-021-00963-2
131. Cristescu R, Lee J, Nebozhyn M, Kim KM, Ting JC, Wong SS, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. (2015) 21:449–56. doi: 10.1038/nm.3850
132. Rodriquenz MG, Roviello G, D’Angelo A, Lavacchi D, Roviello F, Polom K. MSI and EBV positive gastric cancer’s subgroups and their link with novel immunotherapy. J Clin Med. (2020) 9:1427. doi: 10.3390/jcm9051427
133. Kim JW, Lee HS, Nam KH, Ahn S, Kim JW, Ahn SH, et al. PIK3CA mutations are associated with increased tumor aggressiveness and Akt activation in gastric cancer. Oncotarget. (2017) 8:90948–58. doi: 10.18632/oncotarget.v8i53
134. Boger C, Kruger S, Behrens HM, Bock S, Haag J, Kalthoff H, et al. Epstein-Barr virus-associated gastric cancer reveals intratumoral heterogeneity of PIK3CA mutations. Ann Oncol. (2017) 28:1005–14. doi: 10.1093/annonc/mdx047
135. Jang SH, Kim KJ, Oh MH, Lee JH, Lee HJ, Cho HD, et al. Clinicopathological significance of elevated PIK3CA expression in gastric cancer. J Gastric Cancer. (2016) 16:85–92. doi: 10.5230/jgc.2016.16.2.85
136. Wen F, He S, Sun C, Li T, Wu S. PIK3CA and PIK3CB expression and relationship with multidrug resistance in colorectal carcinoma. Int J Clin Exp Pathol. (2014) 7:8295–303.
137. Ishibashi T, Nakayama K, Razia S, Ishikawa M, Nakamura K, Yamashita H, et al. High frequency of PIK3CA mutations in low-grade serous ovarian carcinomas of Japanese patients. Diagnostics (Basel). (2019) 10:13. doi: 10.3390/diagnostics10010013
138. Luchini C, Veronese N, Solmi M, Cho H, Kim JH, Chou A, et al. Prognostic role and implications of mutation status of tumor suppressor gene ARID1A in cancer: a systematic review and meta-analysis. Oncotarget. (2015) 6:39088–97. doi: 10.18632/oncotarget.v6i36
139. Wang DD, Chen YB, Pan K, Wang W, Chen SP, Chen JG, et al. Decreased expression of the ARID1A gene is associated with poor prognosis in primary gastric cancer. PLoS One. (2012) 7:e40364. doi: 10.1371/journal.pone.0040364
140. Ashizawa M, Saito M, Min AKT, Ujiie D, Saito K, Sato T, et al. Prognostic role of ARID1A negative expression in gastric cancer. Sci Rep. (2019) 9:6769. doi: 10.1038/s41598-019-43293-5
141. Wang R, Chen M, Ye X, Poon K. Role and potential clinical utility of ARID1A in gastrointestinal Malignancy. Mutat Res Rev Mutat Res. (2021) 787:108360. doi: 10.1016/j.mrrev.2020.108360
142. Inada R, Sekine S, Taniguchi H, Tsuda H, Katai H, Fujiwara T, et al. ARID1A expression in gastric adenocarcinoma: clinicopathological significance and correlation with DNA mismatch repair status. World J Gastroenterol. (2015) 21:2159–68. doi: 10.3748/wjg.v21.i7.2159
143. Ribeiro J, Malta M, Galaghar A, Silva F, Afonso LP, Medeiros R, et al. P53 deregulation in Epstein-Barr virus-associated gastric cancer. Cancer Lett. (2017) 404:37–43. doi: 10.1016/j.canlet.2017.07.010
144. Bobrovnitchaia I, Valieris R, Drummond RD, Lima JP, Freitas HC, Bartelli TF, et al. APOBEC-mediated DNA alterations: A possible new mechanism of carcinogenesis in EBV-positive gastric cancer. Int J Cancer. (2020) 146:181–91. doi: 10.1002/ijc.32411
145. Kim SM, Hur DY, Hong SW, Kim JH. EBV-encoded EBNA1 regulates cell viability by modulating miR34a-NOX2-ROS signaling in gastric cancer cells. Biochem Biophys Res Commun. (2017) 494:550–5. doi: 10.1016/j.bbrc.2017.10.095
146. He CY, Qiu MZ, Yang XH, Zhou DL, Ma JJ, Long YK, et al. Classification of gastric cancer by EBV status combined with molecular profiling predicts patient prognosis. Clin Transl Med. (2020) 10:353–62. doi: 10.1002/ctm2.32
147. Ratti M, Lampis A, Hahne JC, Passalacqua R, Valeri N. Microsatellite instability in gastric cancer: molecular bases, clinical perspectives, and new treatment approaches. Cell Mol Life Sci. (2018) 75:4151–62. doi: 10.1007/s00018-018-2906-9
148. Muro K, Chung HC, Shankaran V, Geva R, Catenacci D, Gupta S, et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol. (2016) 17:717–26. doi: 10.1016/S1470-2045(16)00175-3
149. Pietrantonio F, Randon G, Di Bartolomeo M, Luciani A, Chao J, Smyth EC, et al. Predictive role of microsatellite instability for PD-1 blockade in patients with advanced gastric cancer: a meta-analysis of randomized clinical trials. ESMO Open. (2021) 6:100036. doi: 10.1016/j.esmoop.2020.100036
150. Kang YK, Boku N, Satoh T, Ryu MH, Chao Y, Kato K, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2017) 390:2461–71. doi: 10.1016/S0140-6736(17)31827-5
151. Kusano M, Toyota M, Suzuki H, Akino K, Aoki F, Fujita M, et al. Genetic, epigenetic, and clinicopathologic features of gastric carcinomas with the CpG island methylator phenotype and an association with Epstein-Barr virus. Cancer. (2006) 106:1467–79. doi: 10.1002/cncr.21789
152. Zhao J, Liang Q, Cheung KF, Kang W, Lung RW, Tong JH, et al. Genome-wide identification of Epstein-Barr virus-driven promoter methylation profiles of human genes in gastric cancer cells. Cancer. (2013) 119:304–12. doi: 10.1002/cncr.27724
153. Zhao Z, Liu W, Liu J, Wang J, Luo B. The effect of EBV on WIF1, NLK, and APC gene methylation and expression in gastric carcinoma and nasopharyngeal cancer. J Med Virol. (2017) 89:1844–51. doi: 10.1002/jmv.24863
154. Zhang L, Wang R, Xie Z. The roles of DNA methylation on the promotor of the Epstein-Barr virus (EBV) gene and the genome in patients with EBV-associated diseases. Appl Microbiol Biotechnol. (2022) 106:4413–26. doi: 10.1007/s00253-022-12029-3
155. Kang D, Skalsky RL, Cullen BR. EBV BART microRNAs target multiple pro-apoptotic cellular genes to promote epithelial cell survival. PLoS Pathog. (2015) 11:e1004979. doi: 10.1371/journal.ppat.1004979
156. Marquitz AR, Mathur A, Chugh PE, Dittmer DP, Raab-Traub N. Expression profile of microRNAs in Epstein-Barr virus-infected AGS gastric carcinoma cells. J Virol. (2014) 88:1389–93. doi: 10.1128/JVI.02662-13
157. de Visser KE, Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. (2023) 41:374–403. doi: 10.1016/j.ccell.2023.02.016
158. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. (2015) 348:69–74. doi: 10.1126/science.aaa4971
159. Aleksic M, Liddy N, Molloy PE, Pumphrey N, Vuidepot A, Chang KM, et al. Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur J Immunol. (2012) 42:3174–9. doi: 10.1002/eji.201242606
160. Zeng D, Li M, Zhou R, Zhang J, Sun H, Shi M, et al. Tumor microenvironment characterization in gastric cancer identifies prognostic and immunotherapeutically relevant gene signatures. Cancer Immunol Res. (2019) 7:737–50. doi: 10.1158/2326-6066.CIR-18-0436
161. Ma J, Li J, Hao Y, Nie Y, Li Z, Qian M, et al. Differentiated tumor immune microenvironment of Epstein-Barr virus-associated and negative gastric cancer: implication in prognosis and immunotherapy. Oncotarget. (2017) 8:67094–103. doi: 10.18632/oncotarget.v8i40
162. Ren F, Zhao Q, Zhao M, Zhu S, Liu B, Bukhari I, et al. Immune infiltration profiling in gastric cancer and their clinical implications. Cancer Sci. (2021) 112:3569–84. doi: 10.1111/cas.15057
163. Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold tumors: A therapeutic challenge for immunotherapy. Front Immunol. (2019) 10:168. doi: 10.3389/fimmu.2019.00168
164. Cho J, Kang MS, Kim KM. Epstein-barr virus-associated gastric carcinoma and specific features of the accompanying immune response. J Gastric Cancer. (2016) 16:1–7. doi: 10.5230/jgc.2016.16.1.1
165. Hinata M, Kunita A, Abe H, Morishita Y, Sakuma K, Yamashita H, et al. Exosomes of epstein-barr virus-associated gastric carcinoma suppress dendritic cell maturation. Microorganisms. (2020) 8:1776. doi: 10.3390/microorganisms8111776
166. Lu S, Wang LJ, Lombardo K, Kwak Y, Kim WH, Resnick MB. Expression of indoleamine 2, 3-dioxygenase 1 (IDO1) and tryptophanyl-tRNA synthetase (WARS) in gastric cancer molecular subtypes. Appl Immunohistochem Mol Morphol. (2020) 28:360–8. doi: 10.1097/PAI.0000000000000761
167. van Beek J, zur Hausen A, Snel SN, Berkhof J, Kranenbarg EK, van de Velde CJ, et al. Morphological evidence of an activated cytotoxic T-cell infiltrate in EBV-positive gastric carcinoma preventing lymph node metastases. Am J Surg Pathol. (2006) 30:59–65. doi: 10.1097/01.pas.0000176428.06629.1e
168. Strong MJ, Xu G, Coco J, Baribault C, Vinay DS, Lacey MR, et al. Differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: implications for possible immune adjuvant therapy. PLoS Pathog. (2013) 9:e1003341. doi: 10.1371/journal.ppat.1003341
169. Dudek AM, Martin S, Garg AD, Agostinis P. Immature, semi-mature, and fully mature dendritic cells: toward a DC-cancer cells interface that augments anticancer immunity. Front Immunol. (2013) 4:438. doi: 10.3389/fimmu.2013.00438
170. Mellman I. Dendritic cells: master regulators of the immune response. Cancer Immunol Res. (2013) 1:145–9. doi: 10.1158/2326-6066.CIR-13-0102
171. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. (2009) 206:3015–29. doi: 10.1084/jem.20090847
172. Zitvogel L, Kroemer G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology. (2012) 1:1223–5. doi: 10.4161/onci.21335
173. Deng SZ, Wang XX, Zhao XY, Bai YM, Zhang HM. Exploration of the Tumor Immune Landscape and Identification of Two Novel Immunotherapy-Related Genes for Epstein-Barr virus-associated Gastric Carcinoma via Integrated Bioinformatics Analysis. Front Surg. (2022) 9:898733. doi: 10.3389/fsurg.2022.898733
174. Iida T, Iwahashi M, Katsuda M, Ishida K, Nakamori M, Nakamura M, et al. Tumor-infiltrating CD4+ Th17 cells produce IL-17 in tumor microenvironment and promote tumor progression in human gastric cancer. Oncol Rep. (2011) 25:1271–7. doi: 10.3892/or
175. Li Q, Li Q, Chen J, Liu Y, Zhao X, Tan B, et al. Prevalence of Th17 and Treg cells in gastric cancer patients and its correlation with clinical parameters. Oncol Rep. (2013) 30:1215–22. doi: 10.3892/or.2013.2570
176. Yamada Y, Saito H, Ikeguchi M. Prevalence and clinical relevance of Th17 cells in patients with gastric cancer. J Surg Res. (2012) 178:685–91. doi: 10.1016/j.jss.2012.07.055
177. Salnikov MY, Fonseca GJ, Mymryk JS. Differences in the tumor microenvironment of EBV-associated gastric cancers revealed using single-cell transcriptome analysis. Cancers (Basel). (2023) 15:3178. doi: 10.3390/cancers15123178
178. Yamaguchi T, Fushida S, Yamamoto Y, Tsukada T, Kinoshita J, Oyama K, et al. Tumor-associated macrophages of the M2 phenotype contribute to progression in gastric cancer with peritoneal dissemination. Gastric Cancer. (2016) 19:1052–65. doi: 10.1007/s10120-015-0579-8
179. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol. (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
180. Panda A, Mehnert JM, Hirshfield KM, Riedlinger G, Damare S, Saunders T, et al. Immune activation and benefit from avelumab in EBV-positive gastric cancer. J Natl Cancer Inst. (2018) 110:316–20. doi: 10.1093/jnci/djx213
181. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. (2010) 141:39–51. doi: 10.1016/j.cell.2010.03.014
182. Jaynes JM, Sable R, Ronzetti M, Bautista W, Knotts Z, Abisoye-Ogunniyan A, et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci Transl Med. (2020) 12:eaax6337. doi: 10.1126/scitranslmed.aax6337
183. Yang M, McKay D, Pollard JW, Lewis CE. Diverse functions of macrophages in different tumor microenvironments. Cancer Res. (2018) 78:5492–503. doi: 10.1158/0008-5472.CAN-18-1367
184. Choi HS, Ha SY, Kim HM, Ahn SM, Kang MS, Kim KM, et al. The prognostic effects of tumor infiltrating regulatory T cells and myeloid derived suppressor cells assessed by multicolor flow cytometry in gastric cancer patients. Oncotarget. (2016) 7:7940–51. doi: 10.18632/oncotarget.v7i7
185. Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. (2011) 60:1419–30. doi: 10.1007/s00262-011-1028-0
186. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
187. Yang P, Qiu G, Wang S, Su Z, Chen J, Wang S, et al. The mutations of Th1 cell-specific T-box transcription factor may be associated with a predominant Th2 phenotype in gastric cancers. Int J Immunogenet. (2010) 37:111–5. doi: 10.1111/j.1744-313X.2010.00899.x
188. Ellyard JI, Simson L, Parish CR. Th2-mediated anti-tumour immunity: friend or foe? Tissue Antigens. (2007) 70:1–11. doi: 10.1111/j.1399-0039.2007.00869.x
189. Schreiber S, Hammers CM, Kaasch AJ, Schraven B, Dudeck A, Kahlfuss S. Metabolic interdependency of th2 cell-mediated type 2 immunity and the tumor microenvironment. Front Immunol. (2021) 12:632581. doi: 10.3389/fimmu.2021.632581
190. Ubukata H, Motohashi G, Tabuchi T, Nagata H, Konishi S, Tabuchi T. Evaluations of interferon-gamma/interleukin-4 ratio and neutrophil/lymphocyte ratio as prognostic indicators in gastric cancer patients. J Surg Oncol. (2010) 102:742–7. doi: 10.1002/jso.21725
191. Rock KL, Reits E, Neefjes J. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol. (2016) 37:724–37. doi: 10.1016/j.it.2016.08.010
192. Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol. (2015) 15:203–16. doi: 10.1038/nri3818
193. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
194. Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. (2011) 35:161–8. doi: 10.1016/j.immuni.2011.07.010
195. Luckheeram RV, Zhou R, Verma AD, Xia B. CD4(+)T cells: differentiation and functions. Clin Dev Immunol. (2012) 2012:925135. doi: 10.1155/2012/925135
196. DhatChinamoorthy K, Colbert JD, Rock KL. Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol. (2021) 12:636568. doi: 10.3389/fimmu.2021.636568
197. Park Y, Koh J, Kwak Y, Ahn SH, Park DJ, Kim HH, et al. Clinicopathologic significance of human leukocyte antigen class I expression in patients with stage II and III gastric cancer. Cancer Immunol Immunother. (2019) 68:1779–90. doi: 10.1007/s00262-019-02410-z
198. Kim SY, Park C, Kim HJ, Park J, Hwang J, Kim JI, et al. Deregulation of immune response genes in patients with Epstein-Barr virus-associated gastric cancer and outcomes. Gastroenterology. (2015) 148:137–147.e9. doi: 10.1053/j.gastro.2014.09.020
199. Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. (1997) 15:749–95. doi: 10.1146/annurev.immunol.15.1.749
200. Liu Y, Sethi NS, Hinoue T, Schneider BG, Cherniack AD, Sanchez-Vega F, et al. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell. (2018) 33:721–735.e8. doi: 10.1016/j.ccell.2018.03.010
201. Chakravorty S, Yan B, Wang C, Wang L, Quaid JT, Lin CF, et al. Integrated pan-cancer map of EBV-associated neoplasms reveals functional host-virus interactions. Cancer Res. (2019) 79:6010–23. doi: 10.1158/0008-5472.CAN-19-0615
202. van den Elsen PJ, Holling TM, Kuipers HF, van der Stoep N. Transcriptional regulation of antigen presentation. Curr Opin Immunol. (2004) 16:67–75. doi: 10.1016/j.coi.2003.11.015
203. Reith W, LeibundGut-Landmann S, Waldburger JM. Regulation of MHC class II gene expression by the class II transactivator. Nat Rev Immunol. (2005) 5:793–806. doi: 10.1038/nri1708
204. Wosen JE, Mukhopadhyay D, Macaubas C, Mellins ED. Epithelial MHC class II expression and its role in antigen presentation in the gastrointestinal and respiratory tracts. Front Immunol. (2018) 9:2144. doi: 10.3389/fimmu.2018.02144
205. Axelrod ML, Cook RS, Johnson DB, Balko JM. Biological consequences of MHC-II expression by tumor cells in cancer. Clin Cancer Res. (2019) 25:2392–402. doi: 10.1158/1078-0432.CCR-18-3200
206. Ma XC, Hattori T, Kushima R, Terata N, Kodama M. Expression of HLA-class II antigen in gastric carcinomas. Its relationship to histopathological grade, lymphocyte infiltration and five-year survival rate. Acta Oncol. (1994) 33:187–90. doi: 10.3109/02841869409098403
207. Zhang M, Hu S, Min M, Ni Y, Lu Z, Sun X, et al. Dissecting transcriptional heterogeneity in primary gastric adenocarcinoma by single cell RNA sequencing. Gut. (2020) 70:464–75. doi: 10.1136/gutjnl-2019-320368
208. Chapel F, Fabiani B, Davi F, Raphael M, Tepper M, Champault G, et al. Epstein-Barr virus and gastric carcinoma in Western patients: comparison of pathological parameters and p53 expression in EBV-positive and negative tumours. Histopathology. (2000) 36:252–61. doi: 10.1046/j.1365-2559.2000.00843.x
209. Gameiro SF, Zhang A, Ghasemi F, Barrett JW, Nichols AC, Mymryk JS. Analysis of class I major histocompatibility complex gene transcription in human tumors caused by human papillomavirus infection. Viruses. (2017) 9:252. doi: 10.3390/v9090252
210. Gameiro SF, Ghasemi F, Barrett JW, Nichols AC, Mymryk JS. High level expression of MHC-II in HPV+ Head and neck cancers suggests that tumor epithelial cells serve an important role as accessory antigen presenting cells. Cancers (Basel). (2019) 111129. doi: 10.3390/cancers11081129
211. Xiang Z, Li J, Song S, Wang J, Cai W, Hu W, et al. A positive feedback between IDO1 metabolite and COL12A1 via MAPK pathway to promote gastric cancer metastasis. J Exp Clin Cancer Res. (2019) 38:314. doi: 10.1186/s13046-019-1318-5
212. Sasaki S, Nishikawa J, Sakai K, Iizasa H, Yoshiyama H, Yanagihara M, et al. EBV-associated gastric cancer evades T-cell immunity by PD-1/PD-L1 interactions. Gastric Cancer. (2019) 22:486–96. doi: 10.1007/s10120-018-0880-4
213. Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. (2020) 10:727–42.
214. Ji M, Liu Y, Li Q, Li XD, Zhao WQ, Zhang H, et al. PD-1/PD-L1 pathway in non-small-cell lung cancer and its relation with EGFR mutation. J Transl Med. (2015) 13:5. doi: 10.1186/s12967-014-0373-0
215. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. (2006) 439:682–7. doi: 10.1038/nature04444
216. Dong P, Xiong Y, Yue J, Hanley SJB, Watari H. Tumor-intrinsic PD-L1 signaling in cancer initiation, development and treatment: Beyond immune evasion. Front Oncol. (2018) 8:386. doi: 10.3389/fonc.2018.00386
217. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. (2017) 276:97–111. doi: 10.1111/imr.12520
218. He Y, Rivard CJ, Rozeboom L, Yu H, Ellison K, Kowalewski A, et al. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci. (2016) 107:1193–7. doi: 10.1111/cas.12986
219. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
220. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
221. Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. (2018) 11:39. doi: 10.1186/s13045-018-0582-8
222. Bai Y, Xie T, Wang Z, Tong S, Zhao X, Zhao F, et al. Efficacy and predictive biomarkers of immunotherapy in Epstein-Barr virus-associated gastric cancer. J Immunother Cancer. (2022) 10:e004080. doi: 10.1136/jitc-2021-004080
223. Mimura K, Kua LF, Xiao JF, Asuncion BR, Nakayama Y, Syn N, et al. Combined inhibition of PD-1/PD-L1, Lag-3, and Tim-3 axes augments antitumor immunity in gastric cancer-T cell coculture models. Gastric Cancer. (2021) 24:611–23. doi: 10.1007/s10120-020-01151-8
224. Pikula A, Kwietniewska M, Rawicz-Pruszynski K, Cisel B, Skorzewska M, Geca K, et al. The importance of Epstein-Barr virus infection in the systemic treatment of patients with gastric cancer. Semin Oncol. (2020) 47:127–37. doi: 10.1053/j.seminoncol.2020.04.001
225. Naseem M, Barzi A, Brezden-Masley C, Puccini A, Berger MD, Tokunaga R, et al. Outlooks on Epstein-Barr virus associated gastric cancer. Cancer Treat Rev. (2018) 66:15–22. doi: 10.1016/j.ctrv.2018.03.006
226. Kang BW, Baek DW, Kang H, Baek JH, Kim JG. Novel therapeutic approaches for epstein-barr virus associated gastric cancer. Anticancer Res. (2019) 39:4003–10. doi: 10.21873/anticanres.13555
227. Zheng X, Huang Y, Li K, Luo R, Cai M, Yun J. Immunosuppressive tumor microenvironment and immunotherapy of epstein-barr virus-associated Malignancies. Viruses. (2022) 14:1017. doi: 10.3390/v14051017
228. Seo AN, Kang BW, Kwon OK, Park KB, Lee SS, Chung HY, et al. Intratumoural PD-L1 expression is associated with worse survival of patients with Epstein-Barr virus-associated gastric cancer. Br J Cancer. (2017) 117:1753–60. doi: 10.1038/bjc.2017.369
229. Shitara K, Van Cutsem E, Bang YJ, Fuchs C, Wyrwicz L, Lee KW, et al. Efficacy and safety of pembrolizumab or pembrolizumab plus chemotherapy vs chemotherapy alone for patients with first-line, advanced gastric cancer: The KEYNOTE-062 phase 3 randomized clinical trial. JAMA Oncol. (2020) 6:1571–80. doi: 10.1001/jamaoncol.2020.3370
230. Kamath SD, Kalyan A, Benson AB. 3rd, Pembrolizumab for the treatment of gastric cancer. Expert Rev Anticancer Ther. (2018) 18:1177–87. doi: 10.1080/14737140.2018.1526084
231. Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med. (2018) 24:1449–58. doi: 10.1038/s41591-018-0101-z
232. Derks S, Liao X, Chiaravalli AM, Xu X, Camargo MC, Solcia E, et al. Abundant PD-L1 expression in Epstein-Barr Virus-infected gastric cancers. Oncotarget. (2016) 7:32925–32. doi: 10.18632/oncotarget.v7i22
233. Sun YT, Guan WL, Zhao Q, Wang DS, Lu SX, He CY, et al. PD-1 antibody camrelizumab for Epstein-Barr virus-positive metastatic gastric cancer: a single-arm, open-label, phase 2 trial. Am J Cancer Res. (2021) 11:5006–15.
234. Moehler M, Ryu MH, Dvorkin M, Lee KW, Coskun HS, Wong R, et al. Maintenance avelumab versus continuation of first-line chemotherapy in gastric cancer: JAVELIN Gastric 100 study design. Future Oncol. (2019) 15:567–77. doi: 10.2217/fon-2018-0668
235. Moehler M, Dvorkin M, Boku N, Ozguroglu M, Ryu MH, Muntean AS, et al. Phase III trial of avelumab maintenance after first-line induction chemotherapy versus continuation of chemotherapy in patients with gastric cancers: Results from JAVELIN gastric 100. J Clin Oncol. (2021) 39:966–77. doi: 10.1200/JCO.20.00892
236. Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. (2010) 376:687–97. doi: 10.1016/S0140-6736(10)61121-X
237. Korfer J, Lordick F, Hacker UT. Molecular targets for gastric cancer treatment and future perspectives from a clinical and translational point of view. Cancers (Basel). (2021) 13:5216. doi: 10.3390/cancers13205216
238. Reddavid R, Dagatti S, Franco C, Puca L, Tomatis M, Corso S, et al. Molecularly targeted therapies for gastric cancer. State Art. Cancers (Basel). (2021) 13:4094. doi: 10.3390/cancers13164094
239. Agnarelli A, Vella V, Samuels M, Papanastasopoulos P, Giamas G. Incorporating immunotherapy in the management of gastric cancer: Molecular and clinical implications. Cancers (Basel). (2022) 14:4378. doi: 10.3390/cancers14184378
240. Olnes MJ, Martinson HA. Recent advances in immune therapies for gastric cancer. Cancer Gene Ther. (2021) 28:924–34. doi: 10.1038/s41417-021-00310-y
241. Jin X, Liu Z, Yang D, Yin K, Chang X. Recent progress and future perspectives of immunotherapy in advanced gastric cancer. Front Immunol. (2022) 13:948647. doi: 10.3389/fimmu.2022.948647
242. Yang L, Wang Y, Wang H. Use of immunotherapy in the treatment of gastric cancer. Oncol Lett. (2019) 18:5681–90. doi: 10.3892/ol
243. Caruso HG, Heimberger AB, Cooper LJN. Steering CAR T cells to distinguish friend from foe. Oncoimmunology. (2019) 8:e1271857. doi: 10.1080/2162402X.2016.1271857
244. Baumgardner JR, Brauer MS, Zhang J, Hao Y, Liu Z, Lakdawalla DN. CAR-T therapy and historical trends in effectiveness and cost-effectiveness of oncology treatments. J Comp Eff Res. (2020) 9:327–40. doi: 10.2217/cer-2019-0065
245. Zhang H, Ye ZL, Yuan ZG, Luo ZQ, Jin HJ, Qian QJ. New strategies for the treatment of solid tumors with CAR-T cells. Int J Biol Sci. (2016) 12:718–29. doi: 10.7150/ijbs.14405
246. Xia AL, Wang XC, Lu YJ, Lu XJ, Sun B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: challenges and opportunities. Oncotarget. (2017) 8:90521–31. doi: 10.18632/oncotarget.v8i52
247. Santomasso B, Bachier C, Westin J, Rezvani K, Shpall EJ. The other side of CAR T-cell therapy: Cytokine release syndrome, neurologic toxicity, and financial burden. Am Soc Clin Oncol Educ Book. (2019) 39:433–44. doi: 10.1200/EDBK_238691
248. Frey N, Porter D. Cytokine release syndrome with chimeric antigen receptor T cell therapy. Biol Blood Marrow Transplant. (2019) 25:e123–7. doi: 10.1016/j.bbmt.2018.12.756
249. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) 11:69. doi: 10.1038/s41408-021-00459-7
250. Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev. (2014) 257:83–90. doi: 10.1111/imr.12125
251. He H, Liao Q, Zhao C, Zhu C, Feng M, Liu Z, et al. Conditioned CAR-T cells by hypoxia-inducible transcription amplification (HiTA) system significantly enhances systemic safety and retains antitumor efficacy. J Immunother Cancer. (2021) 9:e002755. doi: 10.1136/jitc-2021-002755
252. Hosseinkhani N, Derakhshani A, Kooshkaki O, Abdoli Shadbad M, Hajiasgharzadeh K, Baghbanzadeh A, et al. Immune checkpoints and CAR-T cells: The pioneers in future cancer therapies? Int J Mol Sci. (2020) 21:8305. doi: 10.3390/ijms21218305
253. Andrea AE, Chiron A, Mallah S, Bessoles S, Sarrabayrouse G, Hacein-Bey-Abina S. Advances in CAR-T cell genetic engineering strategies to overcome hurdles in solid tumors treatment. Front Immunol. (2022) 13:830292. doi: 10.3389/fimmu.2022.830292
254. Cao B, Liu M, Huang J, Zhou J, Li J, Lian H, et al. Development of mesothelin-specific CAR NK-92 cells for the treatment of gastric cancer. Int J Biol Sci. (2021) 17:3850–61. doi: 10.7150/ijbs.64630
255. Pan K, Farrukh H, Chittepu V, Xu H, Pan CX, Zhu Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res. (2022) 41:119. doi: 10.1186/s13046-022-02327-z
256. Daher M, Rezvani K. Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr Opin Immunol. (2018) 51:146–53. doi: 10.1016/j.coi.2018.03.013
257. Qin H, Sheng J, Zhang D, Zhang X, Liu L, Li B, et al. New strategies for therapeutic cancer vaccines. Anticancer Agents Med Chem. (2019) 19:213–21. doi: 10.2174/1871520618666181109151835
258. Sondak VK, Sosman JA. Results of clinical trials with an allogenic melanoma tumor cell lysate vaccine: Melacine. Semin Cancer Biol. (2003) 13:409–15. doi: 10.1016/j.semcancer.2003.09.004
259. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. (2010) 363:411–22. doi: 10.1056/NEJMoa1001294
260. Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. (2011) 364:2119–27. doi: 10.1056/NEJMoa1012863
261. Trimble CL, Morrow MP, Kraynyak KA, Shen X, Dallas M, Yan J, et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: a randomised, double-blind, placebo-controlled phase 2b trial. Lancet. (2015) 386:2078–88. doi: 10.1016/S0140-6736(15)00239-1
262. Smith JP, Cao H, Chen W, Mahmood K, Phillips T, Sutton L, et al. Gastrin vaccine alone and in combination with an immune checkpoint antibody inhibits growth and metastases of gastric cancer. Front Oncol. (2021) 11:788875. doi: 10.3389/fonc.2021.788875
263. Osborne N, Sundseth R, Gay MD, Cao H, Tucker RD, Nadella S, et al. Vaccine against gastrin, a polyclonal antibody stimulator, decreases pancreatic cancer metastases. Am J Physiol Gastrointest Liver Physiol. (2019) 317:G682–93. doi: 10.1152/ajpgi.00145.2019
264. Zhu X, Perales-Puchalt A, Wojtak K, Xu Z, Yun K, Bhojnagarwala PS, et al. DNA immunotherapy targeting BARF1 induces potent anti-tumor responses against Epstein-Barr-virus-associated carcinomas. Mol Ther Oncolytics. (2022) 24:218–29. doi: 10.1016/j.omto.2021.12.017
265. A. zur Hausen AA, Craanen ME, Middeldorp JM, Meijer CJ, van den Brule AJ. Unique transcription pattern of Epstein-Barr virus (EBV) in EBV-carrying gastric adenocarcinomas: expression of the transforming BARF1 gene. Cancer Res. (2000) 60:2745–8.
266. Sall A, Caserta S, Jolicoeur P, Franqueville L, de Turenne-Tessier M, Ooka T. Mitogenic activity of Epstein-Barr virus-encoded BARF1 protein. Oncogene. (2004) 23:4938–44. doi: 10.1038/sj.onc.1207607
267. Wei MX, de Turenne-Tessier M, Decaussin G, Benet G, Ooka T. Establishment of a monkey kidney epithelial cell line with the BARF1 open reading frame from Epstein-Barr virus. Oncogene. (1997) 14:3073–81. doi: 10.1038/sj.onc.1201128
268. Lo AK, Dawson CW, Lung HL, Wong KL, Young LS. The therapeutic potential of targeting BARF1 in EBV-associated Malignancies. Cancers (Basel). (2020) 12:1940. doi: 10.3390/cancers12071940
269. Turrini R, Merlo A, Martorelli D, Fae DA, Sommaggio R, Montagner IM, et al. A BARF1-specific mAb as a new immunotherapeutic tool for the management of EBV-related tumors. Oncoimmunology. (2017) 6:e1304338. doi: 10.1080/2162402X.2017.1304338
270. Ressing ME, Horst D, Griffin BD, Tellam J, Zuo J, Khanna R, et al. Epstein-Barr virus evasion of CD8(+) and CD4(+) T cell immunity via concerted actions of multiple gene products. Semin Cancer Biol. (2008) 18:397–408. doi: 10.1016/j.semcancer.2008.10.008
271. Shin JY, Kim JO, Lee SK, Chae HS, Kang JH. LY294002 may overcome 5-FU resistance via down-regulation of activated p-AKT in Epstein-Barr virus-positive gastric cancer cells. BMC Cancer. (2010) 10:425. doi: 10.1186/1471-2407-10-425
272. Shin HJ, Kim DN, Lee SK. Association between Epstein-Barr virus infection and chemoresistance to docetaxel in gastric carcinoma. Mol Cells. (2011) 32:173–9. doi: 10.1007/s10059-011-0066-y
273. Xu ZY, Tang JN, Xie HX, Du YA, Huang L, Yu PF, et al. 5-Fluorouracil chemotherapy of gastric cancer generates residual cells with properties of cancer stem cells. Int J Biol Sci. (2015) 11:284–94. doi: 10.7150/ijbs.10248
274. Tetzlaff ED, Cheng JD, Ajani JA. Review of docetaxel in the treatment of gastric cancer. Ther Clin Risk Manag. (2008) 4:999–1007. doi: 10.2147/TCRM
275. Kim MY, Kruger AJ, Jeong JY, Kim J, Shin PK, Kim SY, et al. Combination therapy with a PI3K/mTOR dual inhibitor and chloroquine enhances synergistic apoptotic cell death in epstein-barr virus-infected gastric cancer cells. Mol Cells. (2019) 42:448–59. doi: 10.14348/molcells.2019.2395
276. Lee HG, Kim H, Kim EJ, Park PG, Dong SM, Choi TH, et al. Targeted therapy for Epstein-Barr virus-associated gastric carcinoma using low-dose gemcitabine-induced lytic activation. Oncotarget. (2015) 6:31018–29. doi: 10.18632/oncotarget.v6i31
277. Baek DW, Kang BW, Kim JG. The predictive value of epstein-barr virus-positivity in patients undergoing gastrectomy followed by adjuvant chemotherapy. Chonnam Med J. (2018) 54:173–7. doi: 10.4068/cmj.2018.54.3.173
278. Dank M, Zaluski J, Barone C, Valvere V, Yalcin S, Peschel C, et al. Randomized phase III study comparing irinotecan combined with 5-fluorouracil and folinic acid to cisplatin combined with 5-fluorouracil in chemotherapy naive patients with advanced adenocarcinoma of the stomach or esophagogastric junction. Ann Oncol. (2008) 19:1450–7. doi: 10.1093/annonc/mdn166
279. Luo D, Wang L, Chen X, Xiong Y, Yi F, Ding J, et al. Comparison of platinum/S-1 and platinum/5-fluorouracil as first-line chemotherapy for advanced gastric or gastroesophageal junction cancer: A meta-analysis based on randomized controlled trials. Chemotherapy. (2020) 65:11–20. doi: 10.1159/000506671
280. Kang GH, Lee S, Kim WH, Lee HW, Kim JC, Rhyu MG, et al. Epstein-barr virus-positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype-positive gastric carcinoma. Am J Pathol. (2002) 160:787–94. doi: 10.1016/S0002-9440(10)64901-2
281. Chong JM, Sakuma K, Sudo M, Ushiku T, Uozaki H, Shibahara J, et al. Global and non-random CpG-island methylation in gastric carcinoma associated with Epstein-Barr virus. Cancer Sci. (2003) 94:76–80. doi: 10.1111/j.1349-7006.2003.tb01355.x
282. Kuo HK, Griffith JD, Kreuzer KN. 5-Azacytidine induced methyltransferase-DNA adducts block DNA replication in vivo. Cancer Res. (2007) 67:8248–54. doi: 10.1158/0008-5472.CAN-07-1038
283. Lai J, Fu Y, Tian S, Huang S, Luo X, Lin L, et al. Zebularine elevates STING expression and enhances cGAMP cancer immunotherapy in mice. Mol Ther. (2021) 29:1758–71. doi: 10.1016/j.ymthe.2021.02.005
284. Zhu M, Liang Q, Chen T, Kong Q, Ye G, Yu S, et al. Identification and validation of methylated differentially expressed miRNAs and immune infiltrate profile in EBV-associated gastric cancer. Clin Epigenet. (2021) 13:22. doi: 10.1186/s13148-020-00989-0
285. Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer. (2004) 4:757–68. doi: 10.1038/nrc1452
287. Kim Y, Anderson JL, Lewin SR. Getting the “Kill” into “Shock and kill”: strategies to eliminate latent HIV. Cell Host Microbe. (2018) 23:14–26. doi: 10.1016/j.chom.2017.12.004
288. Wildeman MA, Novalic Z, Verkuijlen SA, Juwana H, Huitema AD, Tan IB, et al. Cytolytic virus activation therapy for Epstein-Barr virus-driven tumors. Clin Cancer Res. (2012) 18:5061–70. doi: 10.1158/1078-0432.CCR-12-0574
289. Yiu SPT, Dorothea M, Hui KF, Chiang AKS. Lytic induction therapy against epstein-barr virus-associated Malignancies: Past, present, and future. Cancers (Basel). (2020) 12:2142. doi: 10.3390/cancers12082142
290. Meng Q, Hagemeier SR, Fingeroth JD, Gershburg E, Pagano JS, Kenney SC. The Epstein-Barr virus (EBV)-encoded protein kinase, EBV-PK, but not the thymidine kinase (EBV-TK), is required for ganciclovir and acyclovir inhibition of lytic viral production. J Virol. (2010) 84:4534–42. doi: 10.1128/JVI.02487-09
291. Freeman SM, Abboud CN, Whartenby KA, Packman CH, Koeplin DS, Moolten FL, et al. The “bystander effect”: tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. (1993) 53:5274–83.
292. Nehme Z, Pasquereau S, Herbein G. Control of viral infections by epigenetic-targeted therapy. Clin Epigenet. (2019) 11:55. doi: 10.1186/s13148-019-0654-9
293. Kerr JR. Epstein-Barr virus (EBV) reactivation and therapeutic inhibitors. J Clin Pathol. (2019) 72:651–8. doi: 10.1136/jclinpath-2019-205822
294. Feng WH, Israel B, Raab-Traub N, Busson P, Kenney SC. Chemotherapy induces lytic EBV replication and confers ganciclovir susceptibility to EBV-positive epithelial cell tumors. Cancer Res. (2002) 62:1920–6.
295. Feng WH, Kenney SC. Valproic acid enhances the efficacy of chemotherapy in EBV-positive tumors by increasing lytic viral gene expression. Cancer Res. (2006) 66:8762–9. doi: 10.1158/0008-5472.CAN-06-1006
296. Hui KF, Cheung AK, Choi CK, Yeung PL, Middeldorp JM, Lung ML, et al. Inhibition of class I histone deacetylases by romidepsin potently induces Epstein-Barr virus lytic cycle and mediates enhanced cell death with ganciclovir. Int J Cancer. (2016) 138:125–36. doi: 10.1002/ijc.29698
297. Tikhmyanova N, Paparoidamis N, Romero-Masters J, Feng X, Mohammed FS, Reddy PAN, et al. Development of a novel inducer for EBV lytic therapy. Bioorg Med Chem Lett. (2019) 29:2259–64. doi: 10.1016/j.bmcl.2019.06.034
298. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The immune landscape of cancer. Immunity. (2018) 48:812–830.e14. doi: 10.1016/j.immuni.2018.03.023
299. Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. (2018) 173:400–416.e11. doi: 10.1016/j.cell.2018.02.052
300. Salnikov M, Prusinkiewicz MA, Lin S, Ghasemi F, Cecchini MJ, Mymryk JS. Tumor-infiltrating T cells in EBV-associated gastric carcinomas exhibit high levels of multiple markers of activation, effector gene expression, and exhaustion. Viruses. (2023) 15:176. doi: 10.3390/v15010176
301. Tang Z, Fan W, Li Q, Wang D, Wen M, Wang J, et al. MVIP: multi-omics portal of viral infection. Nucleic Acids Res. (2022) 50:D817–27. doi: 10.1093/nar/gkab958
302. Salnikov M, Gameiro SF, Zeng PYF, Barrett JW, Nichols AC, Mymryk JS. The HPV induced cancer resource (THInCR): a suite of tools for investigating HPV-dependent human carcinogenesis. mSphere. (2022) 7:e0031722. doi: 10.1128/msphere.00317-22
303. Salnikov MY, Wang E, Christensen E, Prusinkiewicz MA, Shooshtari P, Mymryk JS. The EBV gastric cancer resource (EBV-GCR): A suite of tools for investigating EBV-associated human gastric carcinogenesis. Viruses. (2023) 15:853. doi: 10.3390/v15040853
304. Zhang Y, Wang D, Peng M, Tang L, Ouyang J, Xiong F, et al. Single-cell RNA sequencing in cancer research. J Exp Clin Cancer Res. (2021) 40:81. doi: 10.1186/s13046-021-01874-1
305. Cai L, Li J, Zhang X, Lu Y, Wang J, Lyu X, et al. Gold nano-particles (AuNPs) carrying anti-EBV-miR-BART7-3p inhibit growth of EBV-positive nasopharyngeal carcinoma. Oncotarget. (2015) 6:7838–50. doi: 10.18632/oncotarget.v6i10
Keywords: Epstein-Barr virus, gastric cancer, EBVaGC, tumor microenvironment, immune landscape, therapeutics, tumor virus, immunotherapy
Citation: Salnikov MY, MacNeil KM and Mymryk JS (2024) The viral etiology of EBV-associated gastric cancers contributes to their unique pathology, clinical outcomes, treatment responses and immune landscape. Front. Immunol. 15:1358511. doi: 10.3389/fimmu.2024.1358511
Received: 19 December 2023; Accepted: 14 March 2024;
Published: 26 March 2024.
Edited by:
Hem Chandra Jha, Indian Institute of Technology Indore, IndiaReviewed by:
Bing Luo, Qingdao University, ChinaDharmendra Kashyap Kashyap, Fox Chase Cancer Center, United States
Copyright © 2024 Salnikov, MacNeil and Mymryk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joe S. Mymryk, am15bXJ5a0B1d28uY2E=