 Ruiyu Tao1†
Ruiyu Tao1† Zhengkai Li
Zhengkai Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 21 February 2024
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1354825
This article is part of the Research Topic Emerging Strategies to Overcome Current Roadblocks in CAR T Cell Immunotherapy View all 7 articles
CAR-T cell therapy, a novel immunotherapy, has made significant breakthroughs in clinical practice, particularly in treating B-cell-associated leukemia and lymphoma. However, it still faces challenges such as poor persistence, limited proliferation capacity, high manufacturing costs, and suboptimal efficacy. CRISPR/Cas system, an efficient and simple method for precise gene editing, offers new possibilities for optimizing CAR-T cells. It can increase the function of CAR-T cells and reduce manufacturing costs. The combination of CRISPR/Cas9 technology and CAR-T cell therapy may promote the development of this therapy and provide more effective and personalized treatment for cancer patients. Meanwhile, the safety issues surrounding the application of this technology in CAR-T cells require further research and evaluation. Future research should focus on improving the accuracy and safety of CRISPR/Cas9 technology to facilitate the better development and application of CAR-T cell therapy. This review focuses on the application of CRISPR/Cas9 technology in CAR-T cell therapy, including eliminating the inhibitory effect of immune checkpoints, enhancing the ability of CAR-T cells to resist exhaustion, assisting in the construction of universal CAR-T cells, reducing the manufacturing costs of CAR-T cells, and the security problems faced. The objective is to show the revolutionary role of CRISPR/Cas9 technology in CAR-T cell therapy for researchers.
The increasing incidence and mortality of cancer have become a major global problem, necessitating the expeditious development of efficacious therapeutic drugs or methodologies. Chimeric antigen receptor (CAR)-T cell therapy opens up a new road for cancer treatment. The principle is to engineer patients’ T cells in vitro to express specific CAR proteins on the T cell surface. These T cells expressing CAR proteins can recognize and target the corresponding tumor antigens, and ultimately kill tumor cells. The concept of “CAR” was first proposed in 1989 (1, 2), and it has developed into the fifth generation CAR after more than two decades of development (Figure 1). There has been a rapid advancement in CAR-T cell therapy since 2012 (3). Up to now, several CAR-T cell therapies for hematological malignancies have been marketed, and the related targets include CD19 and B cell mature antigen (BCMA). Furthermore, a substantial number of clinical trials pertaining to this therapy continue to be conducted and registered annually. Over 200 studies related to CAR-T cell therapy were registered in 2023 (ClinicalTrials.gov, last accessed on December 3, 2023).
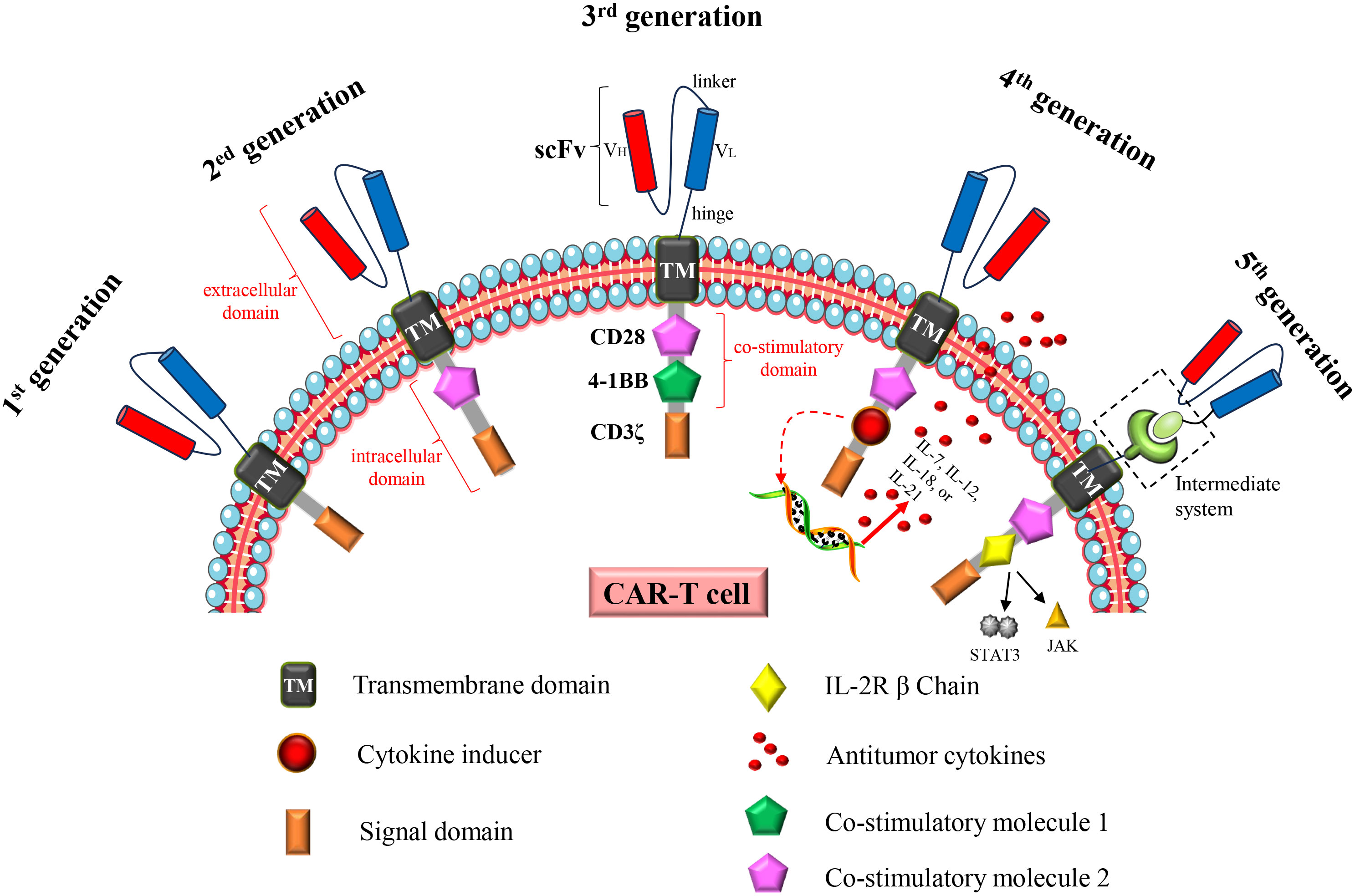
Figure 1 Structural changes to CAR constructs from first to fifth generation. The first generation CAR consists of a single-chain variable fragment (scFv), a transmembrane domain (TM) and an intracellular signaling domain (CD3ζ). The second generation CAR adds a costimulatory molecule (CD28 or 4-1BB) to the intracellular domain. The third generation CAR adds two costimulatory molecules (CD28 and 4-1BB) to the intracellular domain; The fourth generation CAR is based on the second generation CAR or the third generation CAR, and the cytokine receptor genes are added to the intracellular domain. The fifth generation CAR is to add an intermediate system in the extracellular domain, separating the scFv from the transmembrane domain in the extracellular domain; or insert an IL2 RB chain fragment between the costimulatory domain (CD28) and CD3ζ, inducing activation of the JAK-STAT pathway, thereby promoting cell proliferation. In addition, universal CAR-T cells generated using universal T cells (TCR gene and/or HLA class I locus of allogeneic T cells have been destroyed) also belong to the fifth generation CAR-T cells.
Despite the efficacy of the therapy in certain hematological malignancies, several challenges persist, including issues related to persistence, drug resistance, immune evasion, and toxic reactions (4–7). Additionally, CAR-T cells have limited effectiveness in treating solid tumors. Consequently, researchers have endeavored to enhance both the efficacy and safety of the therapy by optimizing CAR structure or employing combination therapies (8, 9), such as combined radiotherapy (10), chemotherapy (11), immune checkpoint inhibitor (12), and oncolytic virus (13). These strategies have demonstrated some improvement in the effectiveness and safety of CAR-T cells; however, they have not fully resolved the aforementioned issues. Moreover, the considerable expense associated with this therapy has somewhat impeded its widespread adoption in clinical settings (14). Hence, there remains a need to further refine CAR-T cells to improve their effectiveness and safety, while also reducing the costs associated with their manufacturing.
The clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 technology, a groundbreaking gene editing tool, has gained significant traction in the field of life science research. In 2012, Jennifer Doudna and Emmanuelle Charpentier first proved the potential of CRISPR/Cas9 in gene editing in prokaryotes (15). Subsequently, Zhang Feng et al. (16) demonstrated for the first time that CRISPR/Cas9 technology can also perform gene editing in human cells or mammalian cells. This technology utilizes the CRISPR/Cas system found in the natural immune system of bacteria and achieves precise gene cutting and editing through the Cas9 protein and guide RNA. It offers a simple, efficient, and cost-effective approach to editing almost any gene. Recently, researchers have started to optimize and modify CAR-T cells using CRISPR/Cas9 technology to enhance their targeting, persistence, antitumor activity, and safety. Exploring the application of this technology in CAR-T cells has become a prominent research focus in the field (17).
Taking advantage of CAR-T cells for cancer treatment is a promising strategy, particularly for patients who have not responded to traditional therapies. However, this therapy also faces some urgent problems. Addressing these challenges and optimizing CAR-T cells to enhance their therapeutic effectiveness and safety is a crucial area of research in this field. CRISPR/Cas9 technology has made designing and optimizing CAR-T cells easier and more convenient. In recent years, the application of CRISPR/Cas9 technology in the research of CAR-T cells has been steadily increasing. At present, several clinical trials of engineered CAR-T cells based on CRISPR/Cas9 technology are underway or have been completed, see Table 1. These trials highlight the immense potential of utilizing this technology to optimize and improve CAR-T cells.
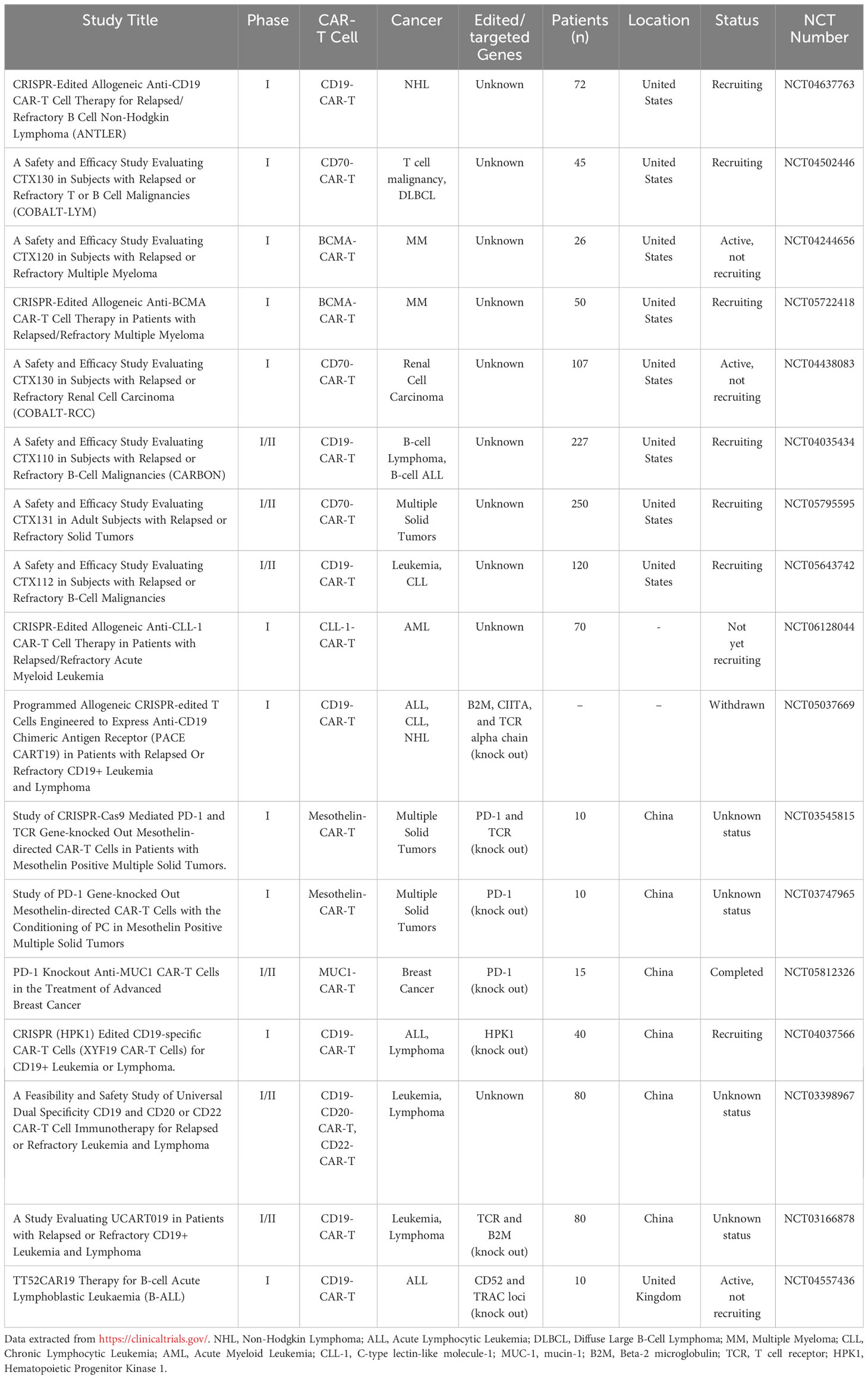
Table 1 Clinical trials of engineering CAR-T cells based on CRISPR/Cas9 technology.
Immune checkpoints play a crucial role as regulatory molecules within the immune system, exerting a negative influence on immune cell activation and impeding immune cell activity to prevent autoimmune overreaction. On the other hand, this mechanism also enables cancer cells to evade immune attacks. Among them, PD-1 and CTLA-4 are the most deeply studied immune checkpoint molecules. By obstructing the binding between immune checkpoints and their respective ligands, immune checkpoint inhibitors can effectively restore T cell immune function. To mitigate the immune checkpoint inhibition of CAR-T cells, a research team sought to enhance the efficacy of cancer treatment by combining PD-1 inhibitors with CAR-T cells. Specifically, the team engineered Mesothelin-CAR-T cells for the purpose of treating pleural mesothelioma in mice, and administered them in conjunction with a PD-1 inhibitor. The findings demonstrated that this combined therapeutic approach effectively extended the duration of CAR-T cell activity, significantly impeded tumor progression, and significantly prolonged the median survival time (18). Subsequently, the research team successfully validated the feasibility of employing CAR-T cells in combination with PD-1 inhibitors for treating malignant pleural mesothelioma in one clinical trial (19). Given that blocking immune checkpoints has the potential to reactivate T cells, a key question of interest would be whether selective reduction or elimination of immune checkpoint genes on T cells effectively maintain or enhance CAR-T cell function? Research has proved that employing CRISPR/Cas9 to delete PD-1 resulted in enhanced long-term persistence and activity of CAR-T cells (preclinical study) (20), and similarly, the deletion of CTLA-4 using CRISPR/Cas9 improved the proliferation and activity of CAR-T cells (preclinical study) (21). Numerous investigations conducted thus far have demonstrated that the abrogation of immune checkpoint inhibition can effectively enhance and optimize the performance of CAR-T cells across various dimensions (Table 2). Furthermore, this table presents the associated research pertaining to the manipulation of specific genes through the utilization of CRISPR/Cas9 technology, encompassing both gene knockout and gene knock-in approaches.
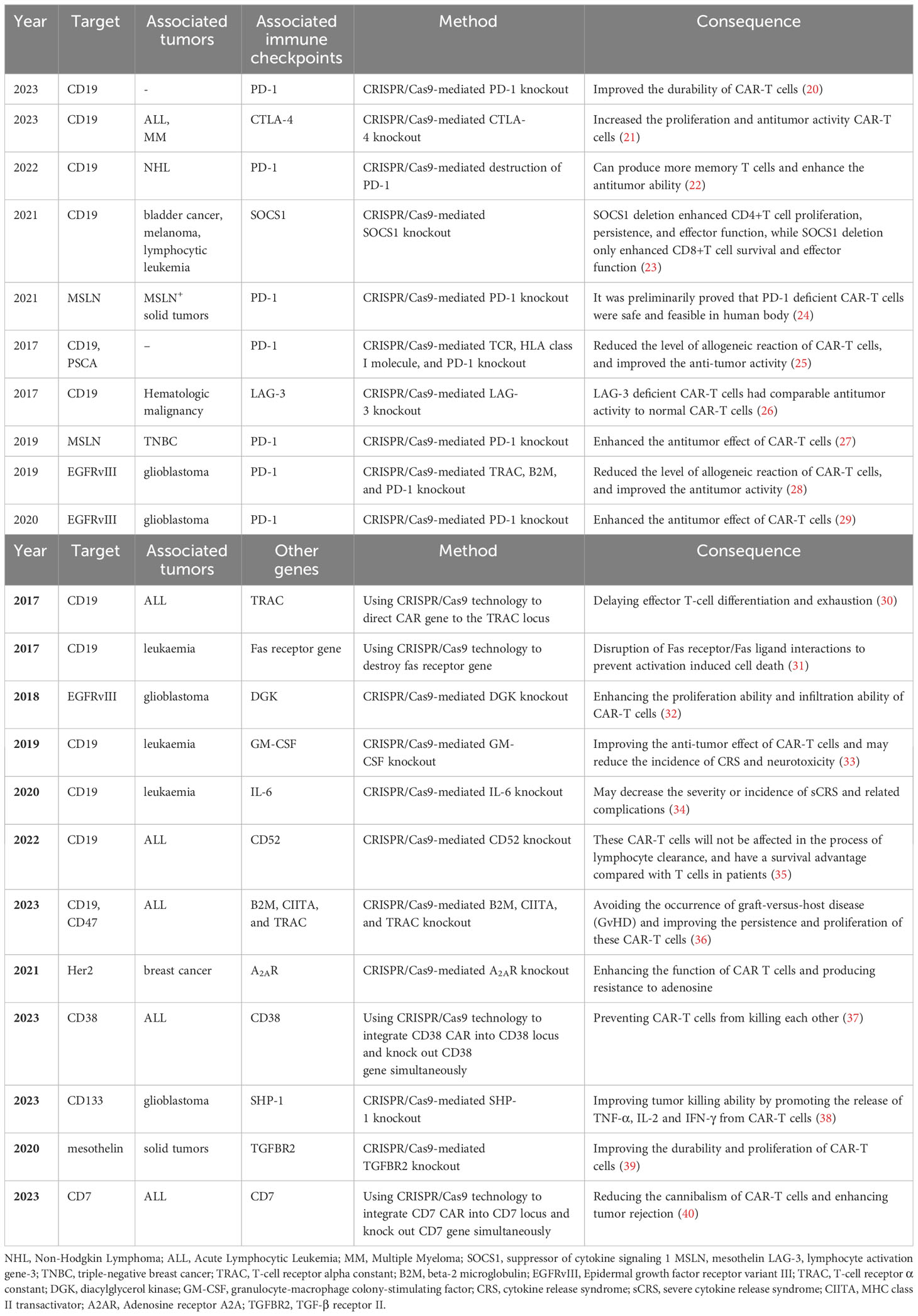
Table 2 CRISPR/CAS9-based knockout of immune checkpoint and other genes to improve CAR-T cell function.
Currently, the majority of extensively researched immune checkpoints are situated on the surface of cells. However, scientists have also identified certain checkpoints within cells, including phosphatase 1B (PTP1B). The utilization of CRISPR/Cas9 technology to eliminate PTPN1 has the potential to induce STAT5 signal activation, thereby facilitating the proliferation and activation of T cells (preclinical study) (41). Hence, the utilization of CRISPR/Cas9 technology to eliminate extracellular or intracellular immune checkpoints has the potential to augment the activation of CAR-T cells and bolster their antitumor efficacy. Banta et al. (42) discovered that the simultaneous inhibition of PD-1 and TIGIT leads to a more pronounced restoration of CD226 co-activation signaling compared to single inhibition, resulting in enhanced immune activity of CD8+ T cells (preclinical study). The finding establishes a theoretical foundation for the utilization of dual blockade of PD-1 and TIGIT in cancer immunotherapy. Moreover, some clinical trials have demonstrated the feasibility of utilizing multi-site CRISPR-Cas9 edited engineered T cells in humans (24, 43). This finding provides a theoretical basis for the application of CRISPR/Cas9 technology in knocking out multiple immune checkpoints and achieving efficient editing efficiency.
A significant challenge encountered in cancer treatment with CAR-T cell therapy pertains to the persistence of CAR-T cells. The persistence exhibits a positive correlation with the anti-tumor effect (44, 45), and plays a crucial role in preventing tumor recurrence. However, the duration of CAR-T cell activity and functionality within the body is constrained. Enhancing the survival duration of CAR-T cells and augmenting the survival rate can effectively enhance their long-term effectiveness. Several factors affect the persistence of CAR-T cells in vivo, including the structure of CARs, the quality and differentiation status of T cells, the exhaustion of CAR-T cells, and the differentiation phenotype of CAR-T cells (46–49).
T cell exhaustion refers to the progressive decline in the effector function of T cells as a result of long-term exposure to persistent antigens or chronic inflammation. Persistent antigen exposure will lead to the depletion of CAR-T cells and promote the transformation of CD8+ T cells into NK-like T cells (50). The exhaustion of CAR-T cells is a significant hindrance to their effectiveness in the treatment of solid tumors. CAR-T cells in an exhausted state are typically characterized by high expression of their immune checkpoints, including PD-1, TIM-3, and CTLA-4 (46, 51). By inhibiting these immune checkpoints, the exhaustion of CAR-T cells can be mitigated (18). Agarwal et al. (21) employed CRISPR/Cas9 technology to delete CTLA-4 in CAR-T cells, resulting in enhanced proliferation, persistence, and cytolytic activity of CAR-T cells (preclinical study). Moreover, the elimination of specific genes in T cells through CRISPR/Cas9 technology can also enhance the persistence of CAR-T cells. For instance, disrupting PR domain zinc finger protein 1 (PRDM1) through CRISPR/Cas9 technology ultimately enhances the persistence by promoting the expansion of memory CAR-T cells (preclinical study) (52). Furthermore, in order to prevent exhaustion, it is possible to enhance the resistance of CAR-T cells to inhibitory cytokines, in addition to targeting immune checkpoint genes and specific genes. Within the tumor microenvironment (TME), TGF-β serves as a crucial inhibitory cytokine. When TGF-β binds to TGF-β receptor II (TGβRII), it hinders the functionality of T cells (53). Alishah et al. (54) utilized CRISPR/Cas9 technology to knock out the TGFβRII gene in CAR-T cells, resulting in improved resistance to exhaustion and enhanced durability of these cells (preclinical study).
To mitigate the exhaustion of CAR-T cells, it is proposed to disrupt specific regulatory factors associated with T cell exhaustion. A study showed that the transcription factors ID3 and Sox4 were elevated in CAR-T cell dysfunction and exerted control over exhaustion-related genes (50). Employing CRISPR/Cas9 technology, researchers successfully knocked out ID3 and Sox4 in T cells, subsequently constructing CAR-T cells. The outcomes demonstrated that the downregulation of ID3 and SOX4 expression considerably postponed CAR-T cell dysfunction and enhanced their tumor-killing ability (preclinical study) (50). Furthermore, the incorporation of cytokine genes into CAR-T cells has been demonstrated to enhance the durability of CAR-T cells. The development of fourth-generation CAR-T cells builds upon the foundation of second or third-generation CAR-T cells by introducing additional anti-tumor cytokine receptor genes within the intracellular domain. Notably, research has indicated that various anti-tumor cytokines, including IL-7 (55), IL-12 (56), IL-15 (57), IL-18 (58), and IL-21 (59), can augment the functionality of CAR-T cells. Consequently, the utilization of fourth-generation CAR-T cells engineered with these cytokines can further enhance the persistence, proliferation, and cytotoxicity of CAR-T cells. CRISPR/Cas9 technology can more easily and accurately realize the construction of the fourth generation CAR sequence.
Enhancing the resistance of CAR-T cells to exhaustion holds significant importance in augmenting treatment durability, ameliorating treatment response rates, mitigating the risk of tumor recurrence, and facilitating the implementation of individualized treatment strategies. Presently, extensive research is being conducted on the persistence of CAR-T cells. The advent of CRISPR/Cas9 technology has furnished a precise and efficient instrument for optimizing and enhancing CAR-T cells. Nevertheless, additional research and experimentation are necessary to assess the effect of CRISPR/Cas9 technology on enhancing the anti-exhaustion ability of CAR-T cells, thereby promoting their clinical transformation.
Currently, the predominant form of CAR-T cells employed in clinical therapy is autologous CAR-T cells, wherein the primary source of T cells is derived from the patients themselves. This approach offers the advantage of circumventing the risk of immune rejection. However, autologous CAR-T cells are not without their drawbacks, such as the protracted duration required for their manufacturing to achieve a ready-to-use state, the limited quantity and quality of T cells contingent upon the patients’ health status, and the substantial financial burden incurred. Of utmost significance is the necessity to repeat the long and costly process of CAR-T cell manufacturing should the target tumor antigens undergo mutation or loss. To address the scarcity of autologous CAR-T cells, scholars conducted research and clinical experiments on allogeneic CAR-T cells utilizing CRISPR/Cas9 technology (60). Allogeneic T cell-derived CAR-T cells, also known as off-the-shelf or universal CAR-T cells, were developed as a solution to the shortage of autologous CAR-T cells. Nonetheless, the primary drawback of universal CAR-T cells lies in their potential to induce graft-versus-host disease (GvHD) and host-versus-graft response (HvGR). A comparison between autologous CAR-T cells and universal CAR-T cells is presented in Table 3.
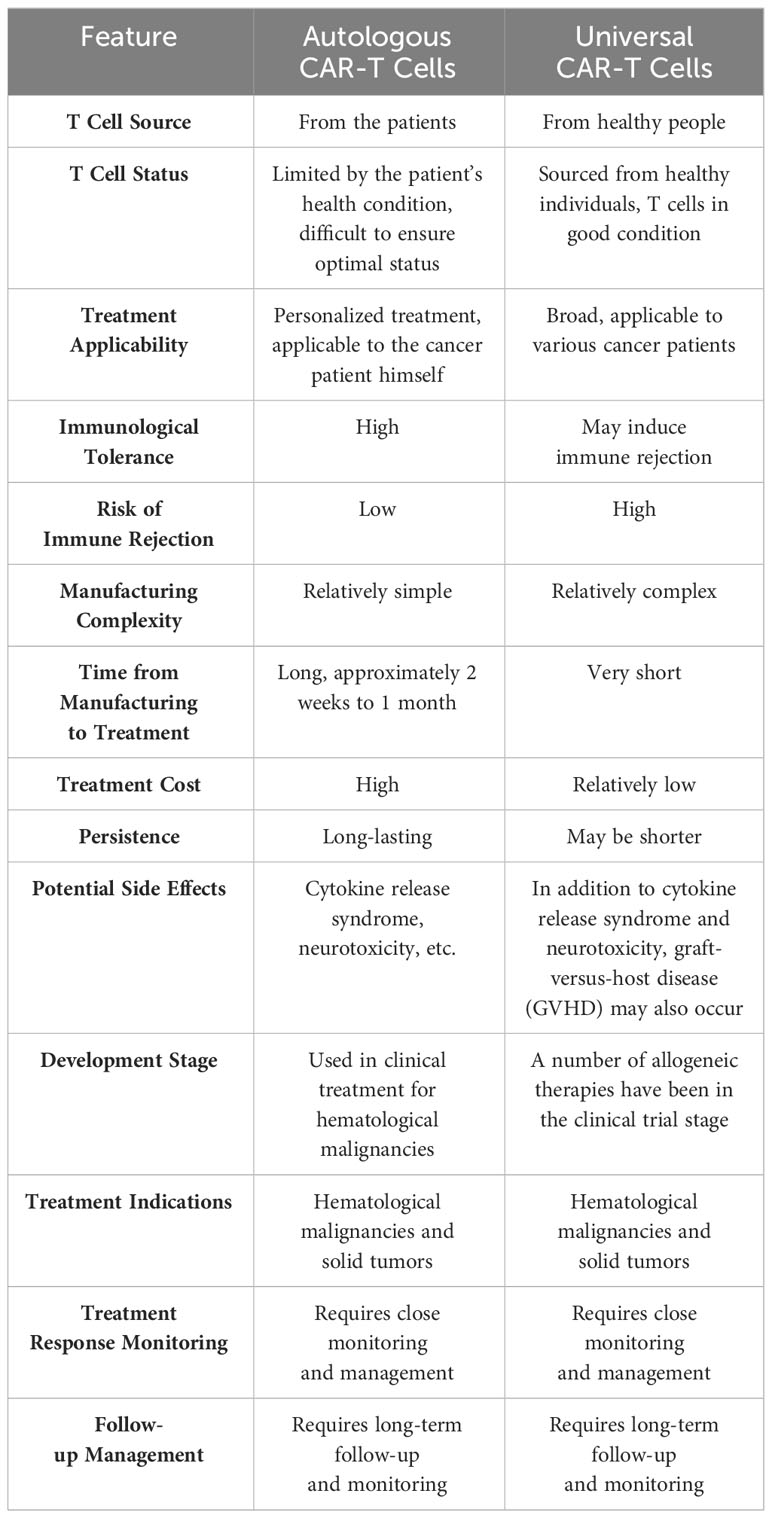
Table 3 Comparison between autologous CAR-T cells and universal CAR-T cells.
The researchers found that autologous CAR-T cells demonstrated superior efficacy, persistence, and reduced side effects compared to allogeneic CAR-T cells (61). Therefore, in recent times, researchers have continuously optimized the allogeneic CAR-T cells. For instance, Li et al. (62) employed CRISPR/Cas9 technology to eliminate T-cell receptor (TCR) and human leucocyte antigen (HLA)-I/II genes from CAR-T cells, while incorporating exogenous expression of HLA-E (preclinical study). The findings demonstrated that this approach effectively prevented rejection and enhanced the durability of CAR-T cells. Furthermore, an alternative approach for the production of an inexhaustible reservoir of universal allogeneic CAR-T cells for cancer immunotherapy involves the utilization of induced pluripotent stem cells (iPSCs) as raw materials. This method holds considerable appeal due to the inherent characteristics of iPSCs, which possess limitless proliferative capacity and the capability to differentiate into various cell lineages in the body. Iriguchi et al. (63) demonstrated that iPSCs can serve as a source for generating mature T cells, which can be utilized for off-the-shelf T cell immunotherapy (preclinical study). Ensuring the robust proliferative capacity of these iPSCs-derived T cells is crucial, and the researchers observed an approximately 200-fold expansion of these T cells during subsequent culture. Wang et al. (64) employed CRISPR/Cas9 technology to integrate the CAR gene into the endogenous TCRα constant (TRAC) locus chain, resulting in the successful development of CAR-T cells with diminished immunogenicity derived from iPSCs (preclinical study). The findings demonstrate that these iPSCs-derived CAR-T cells exhibit enhanced tumor cytotoxicity, prolonged survival duration, and decreased likelihood of eliciting an allogeneic response in the host. In addition, it is also feasible to modify some genes with CRISPR/Cas9 technology to enhance the therapeutic effect of iPSCs-derived CAR-T cells. Ueda et al. (65) successfully optimized iPSC-derived CAR-T cells through gene modification using CRISPR/Cas9 technology (preclinical study). Specifically, they achieved enhanced proliferation capacity and longevity of these CAR-T cells by disrupting the diacylglycerol kinase gene and introducing transduced interleukin-15 (IL-15) and its receptor subunit (IL-15Rα) gene expression.
Presently, the investigation into universal CAR-T cells is in its nascent phase, necessitating additional research and clinical trials to validate their viability and efficacy. Notably, 35 allogeneic CAR-T cell-related clinical trials (Phase I- Phase II) are ongoing (66). Undeniably, the utilization of CRISPR/Cas9 technology offers a crucial means and approach for the development and further optimization of universal CAR-T cells.
CAR-T cell therapy is limited in its clinical application to some extent by its high cost. This is mainly due to the complicated manufacturing process of CAR-T cells and the high cost of raw materials. Although there are options for automated manufacturing of CAR-T cells such as the CliniMacs Prodigy and Lonza Cocoon system, manual and modular methods are still largely used by CAR-T developers which inevitably contributes to increased production cost (14). CAR-T cell manufacturing costs can be reduced by 1) further optimizing CAR-T cell manufacturing and production processes to improve production efficiency and yield; and 2) establishing strict quality control systems to ensure CAR-T cell quality and consistency, reducing unnecessary waste and duplication.
The manufacturing cycle of CAR-T cells is usually about two weeks. To expedite this process, researchers have employed the transduction of non-activated resting T cells with lentiviral vectors. This enables the direct conversion of extracted T cells from patients into CAR-T cells, resulting in a more rapid manufacturing time. Under the condition that the function was not affected, the manufacturing time could be shortened to 24 hours (67). Furthermore, novel CAR-T cell manufacturing platforms can also shorten CAR-T cell manufacturing time. The Novartis T-charge platform represents an advanced platform of CAR-T cell therapy manufacturing, with the primary objective of reducing in vitro culture time and enhancing T cell proliferation potential. Dickinson et al. (68) utilized the T-charge platform to generate a novel autologous CD19-CAR-T cell therapy (YTB323), which shared the same CAR construct as tisagenleucel (clinical phase I/II). Notably, the findings demonstrated that YTB323 could be manufactured within 2 days, while preserving T cell stemness, and exhibited enhanced in vivo expansion and anti-tumor efficacy at a lower dose. The FasTCAR platform of Gracell Biotechnologies can shorten the CAR-T manufacturing time from two weeks to one day. Currently, there are ongoing clinical trials (NCT04638270, NCT05840107, and NCT04935580) evaluating the efficacy and safety of CAR-T cells produced using the FasTCAR platform. Moreover, The CRISPR/Cas9 technology exhibits high efficacy in the editing of genes and is capable of achieving precise modifications within a comparatively brief timeframe. Zhang et al. (22) used CRISPR/Cas9 technology to target the target sequence (clinical phase I). They introduced the CAR sequence template by electroporation, and inserted the CAR sequence into the target gene by homologous recombination repair mechanism, thus producing CAR-T cells independent of the traditional viral based transduction approach. This method simplifies the manufacturing process and shortens the manufacturing time. These methods reduce manufacturing costs indirectly by shortening the manufacturing time.
It should be pointed out that CRISPR/Cas9 editing technology itself cannot directly reduce the manufacturing cost of CAR-T cells. Because the technology is mainly used for gene editing, it does not directly affect other cost factors in the CAR-T cell manufacturing process. The cost of CAR-T cell p manufacturing involves many aspects, including cell culture, quality control, equipment, and manpower. CRISPR/Cas9 editing technology can indirectly reduce the cost of CAR-T cell manufacturing. Firstly, the utilization of CRISPR/Cas9 technology has the potential to enhance the functionality and activity of CAR-T cells, thereby augmenting their anti-tumor effectiveness. This, in turn, can lead to a reduction in the dose and duration of subsequent treatments, consequently mitigating the financial burden associated with therapy. Secondly, the application of CRISPR/Cas9 technology can enhance the proliferative and survival capabilities of CAR-T cells by modifying genes associated with cellular expansion and persistence. This can shorten the expansion time of CAR-T cells and reduce the culture costs. To summarize, the utilization of CRISPR/Cas9 technology has the potential to indirectly mitigate the expenses associated with the production of CAR-T cells by optimizing the function of CAR-T cells, enhancing their proliferation and persistence.
Despite the fact that CRISPR/Cas9 technology is an effective gene editing tool, there are still some safety problems to be solved in its application. These concerns encompass off-target effects, DNA damage, and immunogenicity (69–71). Some studies have demonstrated that the utilization of CRISPR technology for gene editing in human embryos or cells frequently results in the elimination of substantial segments of chromosomes (72–74). The employment of this technology also has the potential to activate p53, leading to DNA damage, while the activated p53 subsequently hampers the efficiency of the CRISPR-Cas9 system for gene editing (70, 75). Investigations involving the editing of human embryonic stem cells and other cell types have revealed that the Cas9 protein can directly interact with the DNA-PK complex, impeding the DNA repair pathway and potentially heightening the risk of chromosome fragmentation (76). Moreover, the utilization of the CRISPR-Cas system has the potential to induce unforeseen structural variations (SVs), including deletions, duplications, and insertions (77). Consequently, CRISPR/Cas9 technology may raise some potential safety issues for CAR-T cell therapy that need to be carefully considered and addressed.
June et al. (43) proved for the first time in a phase I clinical trial that it was feasible to modify T cells with CRISPR/Cas9 technology. The researchers used CRISPR-Cas9 to knock out the PD-1 gene and two genes encoding the T cell receptor (TCR) (TRAC and TRBC) of T cells. The outcomes revealed that the three patients who received T cells engineered with CRISPR-Cas9 did not exhibit cytokine release syndrome (CRS), while the modified T cells exhibited enduring survival and expansion. Hamilton et al. (78) highly praised this research and put forward some thoughts in this field, including 1) it is not clear whether the cells edited by Cas9 are immunogenic; 2) Whether the residual Cas9 will trigger the immune response; 3) Whether the engineered T cells edited by CRISPR are effective for advanced cancer. Furthermore, genome editing by CRISPR/Cas9 has the potential to induce chromosomal deletions and cause chromosomal instability in the preclinical production of CAR-T cells (79, 80). Tsuchida et al. (80) discovered that the utilization of CRISPR technology to disrupt the TRAC gene results in a range of 4%-22% of T cells exhibiting significant chromosome deletion (clinical phase I). To ascertain the generalizability of this occurrence, the investigators conducted CRISPR screening using a gRNA library encompassing all chromosomes and subsequently analyzed the chromosome deletions through experimental means. The findings indicate that 55% of gRNA sequences induce chromosome deletion, and any chromosome is susceptible to substantial fragment or complete chromosome deletion as a consequence of gene editing. Moreover, gRNA sequences located in proximity to the centromere exhibit a higher propensity for chromosome deletion. Finally, the researchers revealed that DNA double strand breaks (DSB) caused by CRISPR gene editing and the expression level of p53 in cells were the key factors leading to chromosome deletion. They tweaked the CAR-T cell manufacturing process by first performing Cas9-mediated gene editing on T cells and then activating those T cells. This method effectively reduces the frequency of chromosome deletions. Furthermore, the utilization of the CRISPR system may result in the enrichment of p53 mutant cells, thereby potentially instigating the progression of cancer (75). Jiang et al. found that transient inhibition of p53 may be a new strategy to reduce the enrichment of mutant cells (81).
Despite the safety concerns associated with its application, CRISPR/Cas9 technology undeniably holds significant potential for optimizing and enhancing CAR-T cell therapy. To address these safety issues, scientists and researchers are actively refining CRISPR-Cas9 technology and conducting meticulous preclinical and clinical trials to assess its safety and efficacy. In 2023, the Food and Drug Administration (FDA) approved the inaugural gene editing therapy, known as Casgevy, utilizing the CRISPR technology. Casgevy represents an in vitro gene editing therapy that employs CRISPR/Cas9 genome editing to alter the hematopoietic stem cells of patients, thereby offering a potential treatment for sickle cell disease. This significant achievement marks a noteworthy advancement in the field of biotechnology. When the BCL11A gene undergoes mutation, certain adults have the ability to generate fetal hemoglobin. Casgevy conducted a simulation of this mutation by employing gene editing technology to precisely cleave the BCL11A gene in hematopoietic stem cells of patients, thereby inducing the release of fetal hemoglobin (82). Thus far, certain phase I clinical trials have substantiated the safety and feasibility of utilizing CRISPR/Cas9-based CAR-T cells in human trials (22, 24, 35, 83).
The CRISPR/Cas9 technology, possessing immense potential, has proven to be highly beneficial in the realm of life sciences. It provides an important tool for researchers and clinicians in this field to carry out innovative research and develop treatment methods. Moreover, the CRISPR/Cas9 technology holds significant importance in the advancement of CAR-T cell optimization. The initial demonstration of CRISPR/Cas9 technology in 2012 showcased its potential for gene editing in prokaryotes, and subsequent studies confirmed its efficacy in gene editing in human and mammalian cells (15, 16). After 2012, CAR-T cell technology began to develop rapidly. Eventually, these two major technologies formed a robust alliance. In recent years, this strategy has been favored by experts in the CAR-T cell field and achieved rapid development. The application of CRISPR/Cas9 technology in CAR-T cell therapy holds significant potential. Additionally, more advanced CRISPR/Cas systems, such as Cas13, Cas14, Cas12a (Cpf1), and nickase Cas9 (nCas9), may be more efficient, more accurate, or safer than Cas9 system in gene editing (84, 85). For instance, Cas13system can directly edit single-stranded RNA without transcribing RNA into DNA, which simplifies the editing process and has high editing efficiency and specificity (86). In comparison to Cas9 and Cas13system, Cas14 system possesses a smaller size, facilitates easier transfer and expression within cells, and demonstrates enhanced specificity in gene editing (87, 88). These CRISPR/Cas systems may help to overcome the current obstacles and barriers faced by CAR-T cells, and may pave the way for a substantial breakthrough in treating solid tumors with CAR-T cells in the future.
RT: Conceptualization, Data curation, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing. XB: Investigation, Software, Visualization, Writing – original draft. XH: Data curation, Investigation, Visualization, Writing – original draft. JY: Investigation, Methodology, Writing – original draft. YM: Investigation, Validation, Writing – original draft. WC: Investigation, Writing – original draft. DZ: Investigation, Writing – original draft. ZL: Conceptualization, Resources, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by Clinical Study on Sleeve Gastrectomy Combined with Catgut Embedding at Acupoints for Obesity Complicated with Metabolic Diseases (Gansu Province Health Industry Research Plan, GSWSKY2022-88).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Gross G, Gorochov G, Waks T, Eshhar Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant Proc. (1989) 21:127–30.
2. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA. (1989) 86:10024–8. doi: 10.1073/pnas.86.24.10024
3. Miao L, Zhang J, Zhang Z, Wang S, Tang F, Teng M, et al. A bibliometric and knowledge-map analysis of CAR-T cells from 2009 to 2021. Front Immunol. (2022) 13:840956. doi: 10.3389/fimmu.2022.840956
4. Ruella M, Korell F, Porazzi P, Maus MV. Mechanisms of resistance to chimeric antigen receptor-T cells in haematological Malignancies. Nat Rev Drug Discovery. (2023) 22:976–95. doi: 10.1038/s41573-023-00807-1
5. Maalej KM, Merhi M, Inchakalody VP, Mestiri S, Alam M, Maccalli C, et al. CAR-cell therapy in the era of solid tumor treatment: current challenges and emerging therapeutic advances. Mol Cancer. (2023) 22:20. doi: 10.1186/s12943-023-01723-z
6. Liu Q, Hu T, Li H, Shen Y, Wu D, Ye B. Prolonged haematologic toxicity in CAR-T-cell therapy: A review. J Cell Mol Med. (2023) 27:3662–71. doi: 10.1111/jcmm.17930
7. Peters DT, Savoldo B, Grover NS. Building safety into CAR-T therapy. Hum Vaccines Immunotherapeutics. (2023) 19:2275457. doi: 10.1080/21645515.2023.2275457
8. Yin L, Wan Z, Sun P, Shuai P, Liu Y. Time to abandon CAR-T monotherapy for solid tumors. Biochim Biophys Acta Rev Cancer. (2023) 1878:188930. doi: 10.1016/j.bbcan.2023.188930
9. Miao L, Zhang J, Huang B, Zhang Z, Wang S, Tang F, et al. Special chimeric antigen receptor (CAR) modifications of T cells: A review. Front Oncol. (2022) 12:832765. doi: 10.3389/fonc.2022.832765
10. Fan J, Adams A, Sieg N, Heger JM, Gödel P, Kutsch N, et al. Potential synergy between radiotherapy and CAR T-cells - a multicentric analysis of the role of radiotherapy in the combination of CAR T cell therapy. Radiotherapy Oncol J Eur Soc Ther Radiol Oncol. (2023) 183:109580. doi: 10.1016/j.radonc.2023.109580
11. Wang AX, Ong XJ, D'Souza C, Neeson PJ, Zhu JJ. Combining chemotherapy with CAR-T cell therapy in treating solid tumors. Front Immunol. (2023) 14:1140541. doi: 10.3389/fimmu.2023.1140541
12. Wang A, Lv T, Song Y. Tandem CAR-T cells targeting MUC1 and PSCA combined with anti-PD-1 antibody exhibit potent preclinical activity against non-small cell lung cancer. Cell Immunol. (2023) 391-392:104760. doi: 10.1016/j.cellimm.2023.104760
13. Evgin L, Kottke T, Tonne J, Thompson J, Huff AL, van Vloten J, et al. Oncolytic virus-mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice. Sci Trans Med. (2022) 14:eabn2231. doi: 10.1126/scitranslmed.abn2231
14. Miao L, Zhang Z, Ren Z, Tang F, Li Y. Obstacles and coping strategies of CAR-T cell immunotherapy in solid tumors. Front Immunol. (2021) 12:687822. doi: 10.3389/fimmu.2021.687822
15. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Sci (New York NY). (2012) 337:816–21. doi: 10.1126/science.1225829
16. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Sci (New York NY). (2013) 339:819–23. doi: 10.1126/science.1231143
17. Ghaffari S, Khalili N, Rezaei N. CRISPR/Cas9 revitalizes adoptive T-cell therapy for cancer immunotherapy. J Exp Clin Cancer Res CR. (2021) 40:269. doi: 10.1186/s13046-021-02076-5
18. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. (2016) 126:3130–44. doi: 10.1172/jci83092
19. Adusumilli PS, Zauderer MG, Rivière I, Solomon SB, Rusch VW, O'Cearbhaill RE, et al. A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with Malignant pleural disease, in combination with the anti-PD-1 agent pembrolizumab. Cancer Discovery. (2021) 11:2748–63. doi: 10.1158/2159-8290.Cd-21-0407
20. Dötsch S, Svec M, Schober K, Hammel M, Wanisch A, Gökmen F, et al. Long-term persistence and functionality of adoptively transferred antigen-specific T cells with genetically ablated PD-1 expression. Proc Natl Acad Sci United States America. (2023) 120:e2200626120. doi: 10.1073/pnas.2200626120
21. Agarwal S, Aznar MA, Rech AJ, Good CR, Kuramitsu S, Da T, et al. Deletion of the inhibitory co-receptor CTLA-4 enhances and invigorates chimeric antigen receptor T cells. Immunity. (2023) 56:2388–2407.e9. doi: 10.1016/j.immuni.2023.09.001
22. Zhang J, Hu Y, Yang J, Li W, Zhang M, Wang Q, et al. Non-viral, specifically targeted CAR-T cells achieve high safety and efficacy in B-NHL. Nature. (2022) 609:369–74. doi: 10.1038/s41586-022-05140-y
23. Sutra Del Galy A, Menegatti S, Fuentealba J, Lucibello F, Perrin L, Helft J, et al. In vivo genome-wide CRISPR screens identify SOCS1 as intrinsic checkpoint of CD4(+) T(H)1 cell response. Sci Immunol. (2021) 6:eabe8219. doi: 10.1126/sciimmunol.abe8219
24. Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu Y, et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol Immunol. (2021) 18:2188–98. doi: 10.1038/s41423-021-00749-x
25. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res an Off J Am Assoc Cancer Res. (2017) 23:2255–66. doi: 10.1158/1078-0432.Ccr-16-1300
26. Zhang Y, Zhang X, Cheng C, Mu W, Liu X, Li N, et al. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front Med. (2017) 11:554–62. doi: 10.1007/s11684-017-0543-6
27. Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer immunology immunotherapy CII. (2019) 68:365–77. doi: 10.1007/s00262-018-2281-2
28. Choi BD, Yu X, Castano AP, Darr H, Henderson DB, Bouffard AA, et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J immunotherapy Cancer. (2019) 7:304. doi: 10.1186/s40425-019-0806-7
29. Nakazawa T, Natsume A, Nishimura F, Morimoto T, Matsuda R, Nakamura M, et al. Effect of CRISPR/cas9-mediated PD-1-disrupted primary human third-generation CAR-T cells targeting EGFRvIII on in vitro human glioblastoma cell growth. Cells. (2020) 9:998. doi: 10.3390/cells9040998
30. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. (2017) 543:113–27. doi: 10.1038/nature21405
31. Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget. (2017) 8:17002–11. doi: 10.18632/oncotarget.15218
32. Jung IY, Kim YY, Yu HS, Lee M, Kim S, Lee J. CRISPR/cas9-mediated knockout of DGK improves antitumor activities of human T cells. Cancer Res. (2018) 78:4692–703. doi: 10.1158/0008-5472.Can-18-0030
33. Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. (2019) 133:697–709. doi: 10.1182/blood-2018-10-881722
34. Kang L, Tang X, Zhang J, Li M, Xu N, Qi W, et al. Interleukin-6-knockdown of chimeric antigen receptor-modified T cells significantly reduces IL-6 release from monocytes. Exp Hematol Oncol. (2020) 9:11. doi: 10.1186/s40164-020-00166-2
35. Ottaviano G, Georgiadis C, Gkazi SA, Syed F, Zhan H, Etuk A, et al. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci Trans Med. (2022) 14:eabq3010. doi: 10.1126/scitranslmed.abq3010
36. Hu X, Manner K, DeJesus R, White K, Gattis C, Ngo P, et al. Hypoimmune anti-CD19 chimeric antigen receptor T cells provide lasting tumor control in fully immunocompetent allogeneic humanized mice. Nat Commun. (2023) 14:2020. doi: 10.1038/s41467-023-37785-2
37. Liao C, Wang Y, Huang Y, Duan Y, Liang Y, Chen J, et al. CD38-specific CAR integrated into CD38 locus driven by different promoters causes distinct antitumor activities of T and NK cells. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2023) 10:e2207394. doi: 10.1002/advs.202207394
38. Liu M, Zhang L, Zhong M, Long Y, Yang W, Liu T, et al. CRISPR/Cas9-mediated knockout of intracellular molecule SHP-1 enhances tumor-killing ability of CD133-targeted CAR T cells in vitro. Exp Hematol Oncol. (2023) 12:88. doi: 10.1186/s40164-023-00450-x
39. Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. (2020) 5:e133977. doi: 10.1172/jci.insight.133977
40. Jiang J, Chen J, Liao C, Duan Y, Wang Y, Shang K, et al. Inserting EF1α-driven CD7-specific CAR at CD7 locus reduces fratricide and enhances tumor rejection. Leukemia. (2023) 37:1660–70. doi: 10.1038/s41375-023-01948-3
41. Wiede F, Lu KH, Du X, Zeissig MN, Xu R, Goh PK, et al. PTP1B is an intracellular checkpoint that limits T-cell and CAR T-cell antitumor immunity. Cancer Discovery. (2022) 12:752–73. doi: 10.1158/2159-8290.Cd-21-0694
42. Banta KL, Xu X, Chitre AS, Au-Yeung A, Takahashi C, O'Gorman WE, et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8(+) T cell responses. Immunity. (2022) 55:512–526.e9. doi: 10.1016/j.immuni.2022.02.005
43. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Sci (New York NY). (2020) 367:eaba7365. doi: 10.1126/science.aba7365
44. Jafarzadeh L, Masoumi E, Fallah-Mehrjardi K, Mirzaei HR, Hadjati J. Prolonged persistence of chimeric antigen receptor (CAR) T cell in adoptive cancer immunotherapy: challenges and ways forward. Front Immunol. (2020) 11:702. doi: 10.3389/fimmu.2020.00702
45. Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature. (2022) 602:503–9. doi: 10.1038/s41586-021-04390-6
46. Pietrobon V, Todd LA, Goswami A, Stefanson O, Yang Z, Marincola F. Improving CAR T-cell persistence. Int J Mol Sci. (2021) 22:10828. doi: 10.3390/ijms221910828
47. López-Cantillo G, Urueña C, Camacho BA, Ramírez-Segura C. CAR-T cell performance: how to improve their persistence? Front Immunol. (2022) 13:878209. doi: 10.3389/fimmu.2022.878209
48. Liu Y, An L, Huang R, Xiong J, Yang H, Wang X, et al. Strategies to enhance CAR-T persistence. biomark Res. (2022) 10:86. doi: 10.1186/s40364-022-00434-9
49. Wei W, Chen ZN, Wang K. CRISPR/cas9: A powerful strategy to improve CAR-T cell persistence. Int J Mol Sci. (2023) 24:12317. doi: 10.3390/ijms241512317
50. Good CR, Aznar MA, Kuramitsu S, Samareh P, Agarwal S, Donahue G, et al. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell. (2021) 184:6081–6100.e26. doi: 10.1016/j.cell.2021.11.016
51. Hsu CL, Ou DL, Bai LY, Chen CW, Lin L, Huang SF, et al. Exploring markers of exhausted CD8 T cells to predict response to immune checkpoint inhibitor therapy for hepatocellular carcinoma. Liver Cancer. (2021) 10:346–59. doi: 10.1159/000515305
52. Yoshikawa T, Wu Z, Inoue S, Kasuya H, Matsushita H, Takahashi Y, et al. Genetic ablation of PRDM1 in antitumor T cells enhances therapeutic efficacy of adoptive immunotherapy. Blood. (2022) 139:2156–72. doi: 10.1182/blood.2021012714
53. Derynck R, Budi EH. Specificity, versatility, and control of TGF-β family signaling. Sci Signaling. (2019) 12:eaav5183. doi: 10.1126/scisignal.aav5183
54. Alishah K, Birtel M, Masoumi E, Jafarzadeh L, Mirzaee HR, Hadjati J, et al. CRISPR/Cas9-mediated TGFβRII disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells in vitro. J Trans Med. (2021) 19:482. doi: 10.1186/s12967-021-03146-0
55. Li F, Zhao S, Wei C, Hu Y, Xu T, Xin X, et al. Development of Nectin4/FAP-targeted CAR-T cells secreting IL-7, CCL19, and IL-12 for Malignant solid tumors. Front Immunol. (2022) 13:958082. doi: 10.3389/fimmu.2022.958082
56. Yang Y, Yang H, Alcaina Y, Puc J, Birt A, Vedvyas Y, et al. Inducible expression of interleukin-12 augments the efficacy of affinity-tuned chimeric antigen receptors in murine solid tumor models. Nat Commun. (2023) 14:2068. doi: 10.1038/s41467-023-37646-y
57. Bell M, Gottschalk S. Engineered cytokine signaling to improve CAR T cell effector function. Front Immunol. (2021) 12:684642. doi: 10.3389/fimmu.2021.684642
58. Jaspers JE, Khan JF, Godfrey WD, Lopez AV, Ciampricotti M, Rudin CM, et al. IL-18-secreting CAR T cells targeting DLL3 are highly effective in small cell lung cancer models. J Clin Invest. (2023) 133:e166028. doi: 10.1172/jci166028
59. Štach M, Ptáčková P, Mucha M, Musil J, Klener P, Otáhal P. Inducible secretion of IL-21 augments anti-tumor activity of piggyBac-manufactured chimeric antigen receptor T cells. Cytotherapy. (2020) 22:744–54. doi: 10.1016/j.jcyt.2020.08.005
60. Hu Y, Zhou Y, Zhang M, Ge W, Li Y, Yang L, et al. CRISPR/cas9-engineered universal CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia. Clin Cancer Res an Off J Am Assoc Cancer Res. (2021) 27:2764–72. doi: 10.1158/1078-0432.Ccr-20-3863
61. DiNofia AM, Grupp SA. Will allogeneic CAR T cells for CD19(+) Malignancies take autologous CAR T cells 'off the shelf'? Nat Rev Clin Oncol. (2021) 18:195–6. doi: 10.1038/s41571-021-00485-1
62. Li W, Zhu X, Xu Y, Chen J, Zhang H, Yang Z, et al. Simultaneous editing of TCR, HLA-I/II and HLA-E resulted in enhanced universal CAR-T resistance to allo-rejection. Front Immunol. (2022) 13:1052717. doi: 10.3389/fimmu.2022.1052717
63. Iriguchi S, Yasui Y, Kawai Y, Arima S, Kunitomo M, Sato T, et al. A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy. Nat Commun. (2021) 12:430. doi: 10.1038/s41467-020-20658-3
64. Wang B, Iriguchi S, Waseda M, Ueda N, Ueda T, Xu H, et al. Generation of hypoimmunogenic T cells from genetically engineered allogeneic human induced pluripotent stem cells. Nat Biomed Eng. (2021) 5:429–40. doi: 10.1038/s41551-021-00730-z
65. Ueda T, Shiina S, Iriguchi S, Terakura S, Kawai Y, Kabai R, et al. Optimization of the proliferation and persistency of CAR T cells derived from human induced pluripotent stem cells. Nat Biomed Eng. (2023) 7:24–37. doi: 10.1038/s41551-022-00969-0
66. Verma M, Obergfell K, Topp S, Panier V, Wu J. The next-generation CAR-T therapy landscape. Nat Rev Drug Discovery. (2023) 22:776–7. doi: 10.1038/d41573-023-00140-7
67. Ghassemi S, Durgin JS, Nunez-Cruz S, Patel J, Leferovich J, Pinzone M, et al. Rapid manufacturing of non-activated potent CAR T cells. Nat Biomed Eng. (2022) 6:118–28. doi: 10.1038/s41551-021-00842-6
68. Dickinson MJ, Barba P, Jäger U, Shah NN, Blaise D, Briones J, et al. A novel autologous CAR-T therapy, YTB323, with preserved T-cell stemness shows enhanced CAR T-cell efficacy in preclinical and early clinical development. Cancer Discovery. (2023) 13:1982–97. doi: 10.1158/2159-8290.Cd-22-1276
69. Zhang XH, Tee LY, Wang XG, Huang QS, Yang SH. Off-target effects in CRISPR/cas9-mediated genome engineering. Mol Ther Nucleic Acids. (2015) 4:e264. doi: 10.1038/mtna.2015.37
70. Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med. (2018) 24:939–46. doi: 10.1038/s41591-018-0050-6
71. Charlesworth CT, Deshpande PS, Dever DP, Camarena J, Lemgart VT, Cromer MK, et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med. (2019) 25:249–54. doi: 10.1038/s41591-018-0326-x
72. Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. (2018) 36:765–71. doi: 10.1038/nbt.4192
73. Zuccaro MV, Xu J, Mitchell C, Marin D, Zimmerman R, Rana B, et al. Allele-specific chromosome removal after cas9 cleavage in human embryos. Cell. (2020) 183:1650–1664.e15. doi: 10.1016/j.cell.2020.10.025
74. Alanis-Lobato G, Zohren J, McCarthy A, Fogarty NME, Kubikova N, Hardman E, et al. Frequent loss of heterozygosity in CRISPR-Cas9-edited early human embryos. Proc Natl Acad Sci USA. (2021) 118:e2004832117. doi: 10.1073/pnas.2004832117
75. Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. (2018) 24:927–30. doi: 10.1038/s41591-018-0049-z
76. Leibowitz ML, Papathanasiou S, Doerfler PA, Blaine LJ, Sun L, Yao Y, et al. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat Genet. (2021) 53:895–905. doi: 10.1038/s41588-021-00838-7
77. Hunt JMT, Samson CA, Rand AD, Sheppard HM. Unintended CRISPR-Cas9 editing outcomes: a review of the detection and prevalence of structural variants generated by gene-editing in human cells. Hum Genet. (2023) 142:705–20. doi: 10.1007/s00439-023-02561-1
78. Hamilton JR, Doudna JA. Knocking out barriers to engineered cell activity. Sci (New York NY). (2020) 367:976–7. doi: 10.1126/science.aba9844
79. Cullot G, Boutin J, Toutain J, Prat F, Pennamen P, Rooryck C, et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat Commun. (2019) 10:1136. doi: 10.1038/s41467-019-09006-2
80. Tsuchida CA, Brandes N, Bueno R, Trinidad M, Mazumder T, Yu B, et al. Mitigation of chromosome loss in clinical CRISPR-Cas9-engineered T cells. Cell. (2023) 186:4567–4582.e20. doi: 10.1016/j.cell.2023.08.041
81. Jiang L, Ingelshed K, Shen Y, Boddul SV, Iyer VS, Kasza Z, et al. CRISPR/cas9-induced DNA damage enriches for mutations in a p53-linked interactome: implications for CRISPR-based therapies. Cancer Res. (2022) 82:36–45. doi: 10.1158/0008-5472.Can-21-1692
82. Frangoul H, Altshuler D, Cappellini MD, Chen YS, Domm J, Eustace BK, et al. CRISPR-cas9 gene editing for sickle cell disease and β-thalassemia. New Engl J Med. (2021) 384:252–360. doi: 10.1056/NEJMoa2031054
83. Hu Y, Zu C, Zhang M, Wei G, Li W, Fu S, et al. Safety and efficacy of CRISPR-based non-viral PD1 locus specifically integrated anti-CD19 CAR-T cells in patients with relapsed or refractory Non-Hodgkin's lymphoma: a first-in-human phase I study. EClinicalMedicine. (2023) 60:102010. doi: 10.1016/j.eclinm.2023.102010
84. Hillary VE, Ceasar SA. A review on the mechanism and applications of CRISPR/cas9/cas12/cas13/cas14 proteins utilized for genome engineering. Mol Biotechnol. (2023) 65:311–25. doi: 10.1007/s12033-022-00567-0
85. Qin L, Li J, Wang Q, Xu Z, Sun L, Alariqi M, et al. High-efficient and precise base editing of C•G to T•A in the allotetraploid cotton (Gossypium hirsutum) genome using a modified CRISPR/Cas9 system. Plant Biotechnol J. (2020) 18:45–56. doi: 10.1111/pbi.13168
86. Liu L, Li X, Wang J, Wang M, Chen P, Yin M, et al. Two distant catalytic sites are responsible for C2c2 RNase activities. Cell. (2017) 168:121–34. doi: 10.1016/j.cell.2016.12.031
87. Harrington LB, Burstein D, Chen JS, Paez-Espino D, Ma E, Witte IP, et al. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Sci (New York NY). (2018) 362:839–42. doi: 10.1126/science.aav4294
Keywords: CAR-T cell, CRISPR/Cas9, immune checkpoints, durability, cost, security
Citation: Tao R, Han X, Bai X, Yu J, Ma Y, Chen W, Zhang D and Li Z (2024) Revolutionizing cancer treatment: enhancing CAR-T cell therapy with CRISPR/Cas9 gene editing technology. Front. Immunol. 15:1354825. doi: 10.3389/fimmu.2024.1354825
Received: 13 December 2023; Accepted: 01 February 2024;
Published: 21 February 2024.
Edited by:
Claudia Arndt, Helmholtz Center Dresden-Rossendorf, GermanyReviewed by:
Jaichandran Sivalingam, Bioprocessing Technology Institute (A*STAR), SingaporeCopyright © 2024 Tao, Han, Bai, Yu, Ma, Chen, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhengkai Li, bGl6a2x6dTEyM0AxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.