 Qingyu Li
Qingyu Li Jingya Li
Jingya Li Menglan Zhou
Menglan Zhou Ying Ge
Ying Ge Zhengyin Liu
Zhengyin Liu Taisheng Li
Taisheng Li Li Zhang4*
Li Zhang4*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 19 April 2024
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1354349
This article is part of the Research Topic Case Reports in Autoimmune and Autoinflammatory Disorders View all 42 articles
Antiphospholipid antibodies (aPL) are both laboratory evidence and causative factors for a broad spectrum of clinical manifestations of antiphospholipid syndrome (APS), with thrombotic and obstetric events being the most prevalent. Despite the aPL-triggered vasculopathy nature of APS, vasculitic-like manifestations rarely exist in APS and mainly appear associated with other concurrent connective tissue diseases like systemic lupus erythematous. Several studies have characterized pulmonary capillaritis related to pathogenic aPL, suggesting vasculitis as a potential associated non-thrombotic manifestation. Here, we describe a 15-year-old girl who develops hepatic infarction in the presence of highly positive aPL, temporally related to prior non-severe COVID-19 infection. aPL-related hepatic vasculitis, which has not been reported before, contributes to liver ischemic necrosis. Immunosuppression therapy brings about favorable outcomes. Our case together with retrieved literature provides supportive evidence for aPL-related vasculitis, extending the spectrum of vascular changes raised by pathogenic aPL. Differentiation between thrombotic and vasculitic forms of vascular lesions is essential for appropriate therapeutic decision to include additional immunosuppression therapy. We also perform a systematic review to characterize the prevalence and clinical features of new-onset APS and APS relapses after COVID-19 for the first time, indicating the pathogenicity of aPL in a subset of COVID-19 patients.
Antiphospholipid syndrome (APS) is a systemic autoimmune disorder characteristic of arterial, venous, or microvascular thrombosis, obstetric morbidity, and well-defined non-thrombotic manifestations in the setting of persistent antiphospholipid antibodies (aPL) (1, 2). aPL, composed of a diverse family of acquired autoantibodies, are recognized as causative factors for clinical manifestations of APS (3). Both genetic and environmental elements could exert as precipitating factors for aPL production, with infection being the most prevalent trigger (4–6). In the recent COVID-19 pandemic, the observed high prevalence of aPL has been reported, yet the potential pathogenicity of these antibodies remains uncertain and controversial (7). The molecular mimicry between severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral proteins and native tissues and the neoepitope caused by SARS-CoV-2-induced oxidative stress probably contribute to aPL generation (8). In addition to widely reported aPL-related thrombosis, associated non-thrombotic manifestations are emerging with considerable evidence (9). Compared with adult patients, aPL-related non-thrombotic complications, both criteria and non-criteria, are more frequently presented in pediatric patients (10). aPL-related vasculitis is characterized as the inflammation of vessel walls and is only well-confirmed in pulmonary capillaries as diffuse alveolar hemorrhage (DAH) (11). This rare manifestation can result in occlusion of vessel lumen in the absence of thrombus, which might make it clinically indistinguishable to thrombotic events. Differentiation between thrombotic and vasculitic causes of aPL-related vascular damage is essential for proper therapeutic decision to adequately include immunosuppression therapy (12). Here, we describe an uncommon case of a young girl with aPL-related vasculitis-induced liver infarction after non-severe COVID-19 infection, providing valuable information for development of pathogenic aPL in infectious diseases and aPL-related vasculitic manifestations.
A 15-year-old girl presented to the emergency department with hyperpyrexia and abdominal pain persisting for over a month. Initial laboratory results revealed elevated inflammation markers, liver dysfunction, and prolonged activated partial thromboplastin time (aPTT) (62.2 s; normal: 25–37 s) and prothrombin time (PT) (17.8 s; normal: 11–14 s). There were no bleeding signs clinically. Abdominal CT demonstrated liver “abscess-like” lesions (Figures 1A, B) as well as possible cholecystitis (Figures 1C, D), and the histopathological examination of liver biopsy specimens confirmed the acute hepatic necrosis. An empiric anti-infective therapy was initiated with intravenous ertapenem and changed to meropenem and metronidazole later. Vitamin K and plasma transfusion were applied for correction of coagulation disorders but turned out to be ineffective. Possible pathogens were under intense exploration, but all proved negative after following tests: traditional microbiologic culture and metagenomic next-generation sequencing of peripheral blood samples and liver biopsy specimens; serology screenings for fungi, SASR-CoV-2, hepatitis viruses, TORCH pathogens, Leishmania, and mycobacteria tuberculosis; Epstein–Barr virus DNA analysis; and microscopic examination of parasites in stool samples. In addition, tumor marker analysis, bone marrow examination, specific staining of liver biopsy specimens, and ceruloplasmin test showed no abnormalities. The efficacy of anti-infective therapy was undetermined with fluctuating inflammation markers and unrelieved abdominal pain. Elevated D-dimer (14.87 mg/L; normal: 0–0.55 mg/L), fibrin and fibrinogen degradation products (FDP) (27.6 μg/mL; normal: 0–5 μg/mL), and fibrinogen (7.52 g/mL; normal: 1.8–3.5 g/mL) were also indicated. Repeated CT demonstrated enlargement or reduction of some liver lesions, as well as the emergence of new lesions. The antibiotics were improved to intravenous ertapenem and vancomycin after the re-elevation of C-reactive protein. Unexpectedly, her symptoms worsened with a re-elevated fever peak and persistent coagulation disorders (Figure 2).
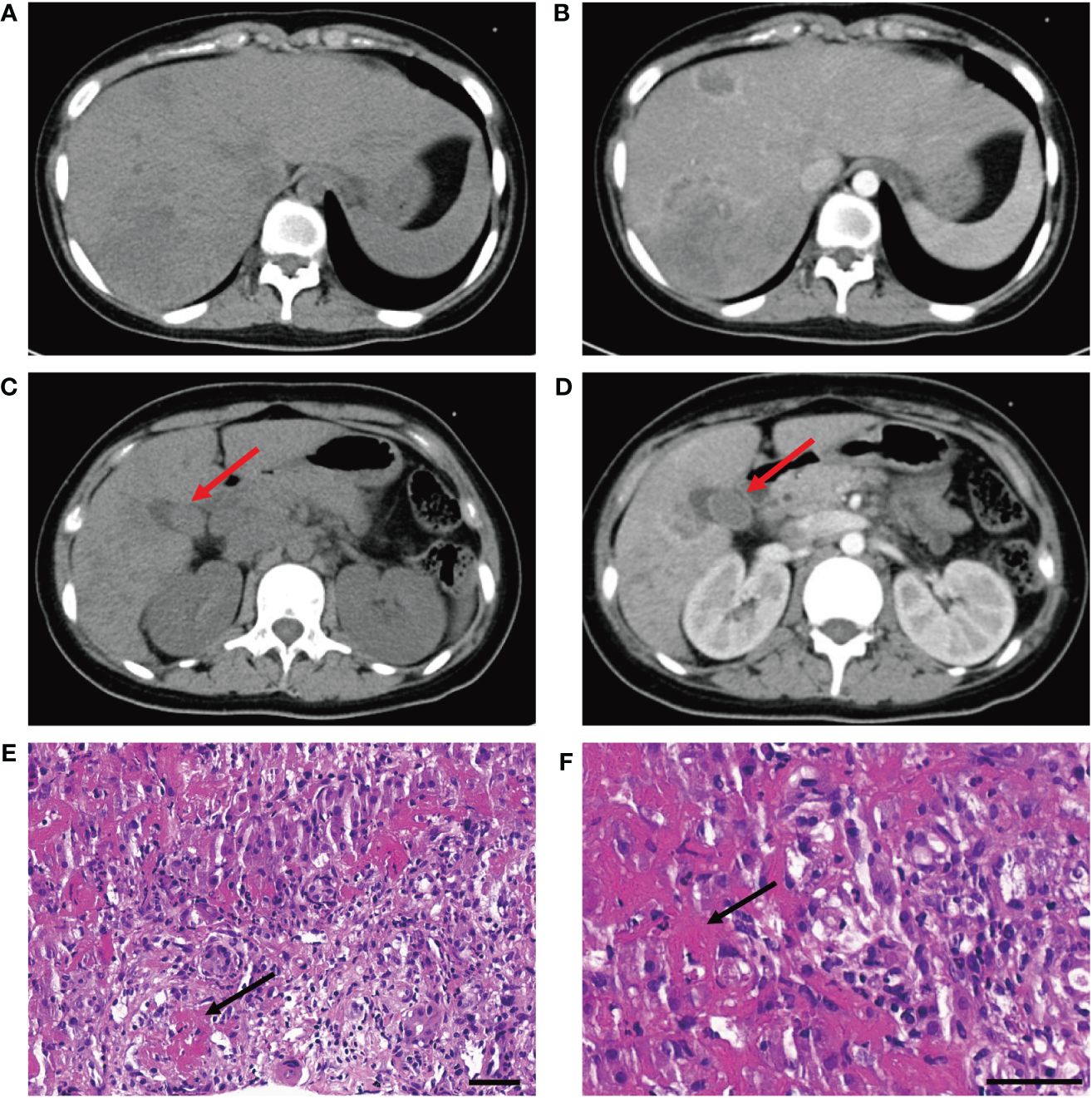
Figure 1 (A, B) CT scan without and with contrast demonstrates liver "abscess-like" lesions. (C, D) CT scan without and with contrast demonstrates possible cholecystitis (red arrowhead). (E, F) Histopathological examination reveals vasculitis of hepatic arteries and resultant liver infarction (black arrowhead) (bar is 50 μm).
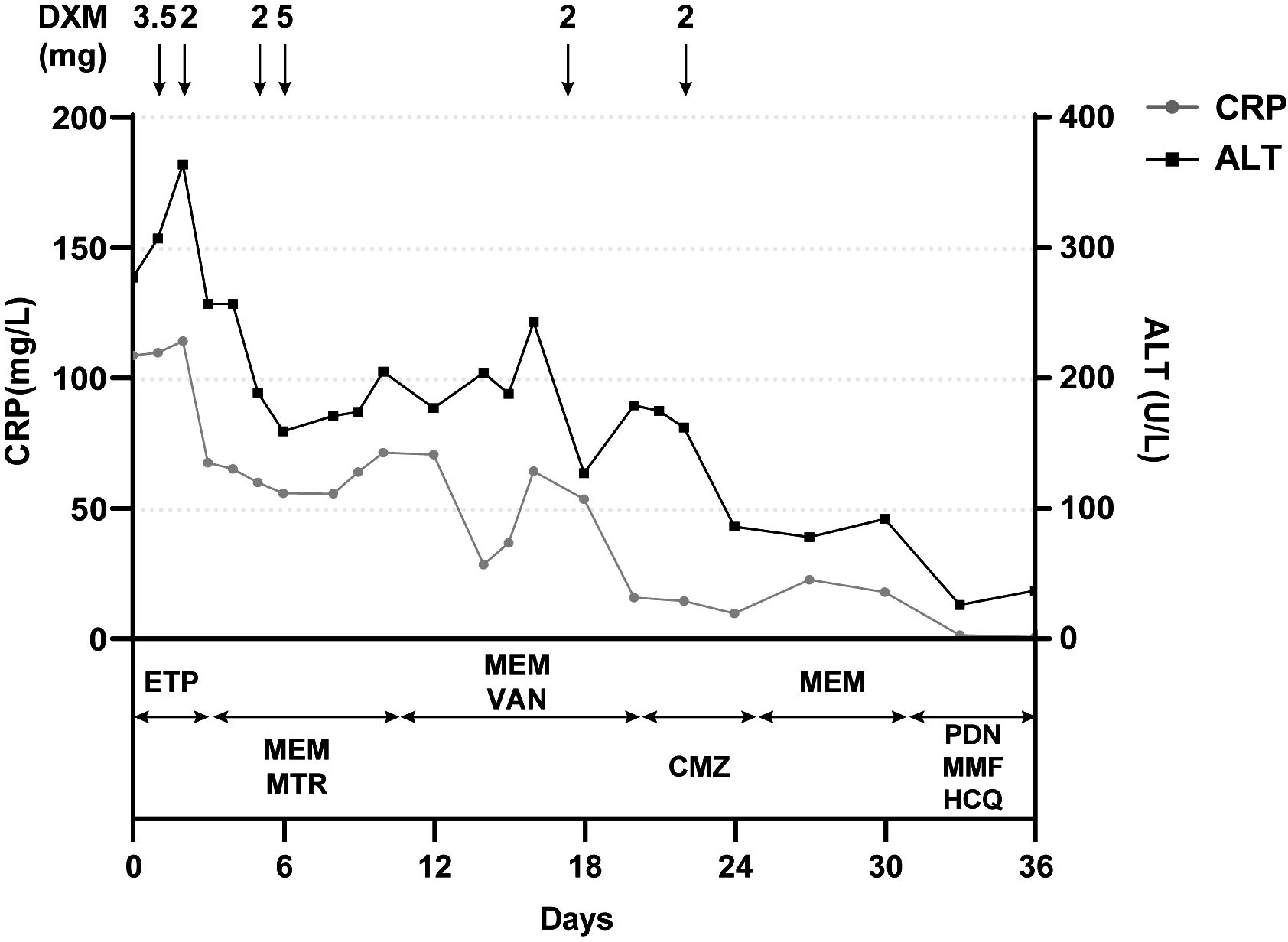
Figure 2 Positive correlation between clinical course and the administration of dexamethasone but not antibiotics. Laboratory reference range for indicators: CRP<3.0 mg/L; ALT: 9–50 U/L. CRP, C-reactive protein; ALT, alanine aminotransferase; DXM, dextromethorphan; ETP, ertapenem; MEM, meropenem; MTR, metronidazole; VAN, vancomycin; CMZ, cefmetazole; PDN, prednisone; MMF, mycophenolate mofetil; HCQ, hydroxychloroquine.
The young girl had a medical history of mild SARS-CoV-2 Omicron variant infection, presented with only nasal congestion and fatigue. The onset of abdominal pain occurred 7 days after testing negative for SARS-CoV-2 antigen and complete remission of COVID-19 symptoms. As the efficacy of low-dose dexamethasone given during plasma transfusion could not be excluded for transient improvement of the patient (Figure 2), an immune dysregulation secondary to infection was considered. Serologic testing of antinuclear antibodies and autoimmune hepatitis antibodies only revealed the presence of low titers of antinuclear antibody (1:80) and smooth muscle antibody (1:80). Autoimmune liver diseases were ruled out. Antibodies associated with other systemic autoimmune diseases were comprehensively evaluated, and the presence of IgG anticardiolipin (aCL) and IgG anti-β2-glycoprotein I (anti-β2GPI) antibodies, as well as lupus anticoagulant (LA), was demonstrated (Table 1). Other autoantibodies were undetected. The aPTT and PT correction tests showed negative results. Consecutive examinations demonstrated disease progression following changes of aPL titers, and hepatic lesions were considered to be related to aPL. Considering the hypercoagulable state of pathogenic aPL, the patient underwent CT scan, MRI, MRA, and vascular ultrasound and multiple small acute to subacute cerebral infarcts were indicated. No thrombus and other abnormalities were detected. The liver biopsy specimen was histologically re-evaluated. Immune infiltration with a large number of neutrophils and fibrinoid necrosis were evident in some small- and medium-sized arteries, and adjacent hepatic tissues underwent ischemic infarction consequently (Figures 1D–F). The patient traversed the most severe phase because of administered low-dose dexamethasone. Further therapy included 40 mg prednisone once a day, 500 mg mycophenolate mofetil (MMF) twice a day, and 200 mg hydroxychloroquine twice a day for immunosuppression, along with 100 mg aspirin once a day to prevent future thrombosis. The patient were discharged from the hospital with reduced inflammation markers, diminished abdominal pain, and healed liver lesions as shown in CT examination. The administration of MMF, hydroxychloroquine and aspirin remained unchanged after discharge, whereas prednisone was gradually tapered. The dose reduction proceeded at 5 mg per week until reaching a daily dose of 20 mg, followed by a weekly reduction of 2.5 mg until reaching a daily dose of 15 mg. Scheduled follow-up appointments were conducted. Six months after discharge, the patient discontinued medication autonomously and subsequently experienced a relapsed right-upper quadrant pain with re-elevated aPL titers and significantly prolonged aPTT (Table 1). D-dimer, FDP, and fibrinogen were within normal ranges. Resumption of treatment yielded amelioration. Considering the persistence of medium-to-highly positive aCL and LA for over 12 weeks, as well as aPL-related hepatic vasculitis and cerebral infarction, the diagnosis was made as highly probable APS with vasculitis as a non-criteria manifestation.
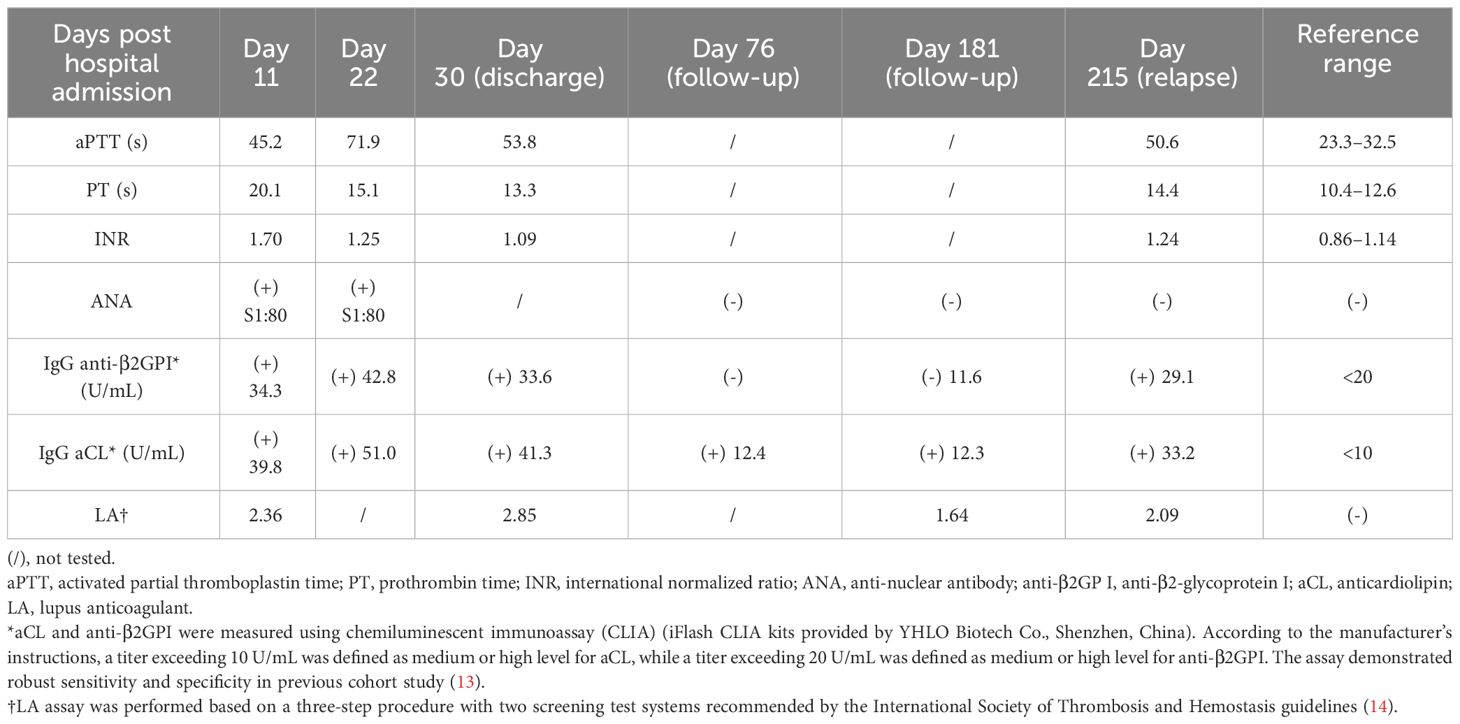
Table 1 Disease parameters and titers of autoantibodies.
Compared with the updated Sapporo criteria with only vascular thrombosis and pregnancy morbidity as diagnostic manifestations of APS (1), the new 2023 ACR/EULAR criteria has introduced several well-defined non-thrombotic manifestations into the clinical criteria for APS classification, including microvascular diseases, cardiac valve diseases, and thrombocytopenia (2). A progression and advancement of the comprehension of aPL-related clinical manifestations is indicated. However, limitations still exist as patients with criteria aPL and comparatively uncommon non-thrombotic manifestations and patients with fulfillment of clinical criteria but seronegative conventional aPL might be inadequately excluded. These conditions are therefore suggested to be referred as “probable APS” or “non-criteria APS” (15). Our case met the laboratory criteria based on persistence of medium-to-highly positive aCL and LA. The complete and sustained remission of hepatic vasculitis was achieved only when aPL were managed at lower titers with pathogenic effects effectively suppressed. The development of cerebral infarction happened in the setting of highly positive aPL and in the absence of other vascular risk factors. Hepatic vasculitis and cerebral infarction were therefore considered to be associated manifestations. The pathophysiology of cerebral infarcts was undetermined, yet the remarkably elevated D-dimer and FDP suggested a possibility of thrombotic events. Accordingly, our case was assessed as highly probable APS with aPL-related hepatic vasculitis as a non-criteria manifestation, and the development of pathogenic aPL was associated with prior COVID-19 infection.
Infections have been implicated in induction of autoimmunity including aPL production (16), with the recent COVID-19 pandemic being no exception (7). A large number of studies have reported high prevalence of aPL (5%–71%), both criteria and non-criteria types, in COVID-19 patients (7, 17). Several potential mechanisms have been proposed but require further validation (8). Molecular mimicry supposes that the S1 and S2 subunits of the SARS-CoV-2 viral S protein might form a phospholipid-like epitope shared with native tissues, triggering aPL production and provoking an immunogenic response (18–20). The neoepitope model posits that oxidative stress induced by SARS-CoV-2 can alter the conformation of β2GPI (21, 22) and create a neoepitope for antibody generation (23).
Despite the observed high prevalence, the pathogenicity of COVID-19-associated aPL remains uncertain and controversial. To explore the potential roles of aPL, numerous studies have analyzed the correlations of aPL and clinical manifestations in COVID-19 patients, yet a consensus could not be reached. COVID-19-associated aPL were demonstrated to be natural or nonpathogenic in most studies (24–50), which was also shown in the largest meta-analysis published in 2021 (51). Additionally, anti-β2GPI in COVID-19 was reported to rarely (5%) recognize domain I of β2GPI, the molecular region most commonly associated with pathogenicity (24). On the contrary, associations of aPL with disease severity and thrombosis in COVID-19 patients were also reported (52–67). Notably, the largest cohort study demonstrated a correlation between the presence of aCL or IgA anti-β2GPI and thrombotic events (65). IgG antibodies purified from COVID-19 patients with high aPL titers were found to trigger neutrophil extracellular trap release and potentiate thrombosis in mice, similarly to IgG isolated from individuals with definite APS (59). Additionally, infections have been reported as the most common causative factor of catastrophic APS (CAPS), suggesting that infection-induced aPL could exhibit biological activity in a subset of patients (6). Several theories have been proposed to explain the heterogeneity. The “two hits” theory holds that aPL (first hit) induce a thrombophilic state, but clotting requires additional thrombophilic condition (second hit), often involving an innate immunity activator like inflammation, infection, or surgery (3). Furthermore, infections are proposed to more likely trigger APS in individuals with genetic propensity, immune defects, or hormonal abnormalities (16). Therefore, the pathogenicity of aPL exhibits heterogeneity across COVID-19 patients and susceptible individuals with predisposing factors might present aPL-related manifestations in the presence of COVID-19-associated aPL.
Albeit the intense exploration of aPL in COVID-19 patients by multiple studies, most of them neither specify the duration of aPL positivity nor subgroup patients according to antibody levels. All COVID-19 patients with positive aPL were incorporated, and individuals with or without pathogenic aPL were merged together for characterization and analysis, contributing to the debatable pathogenicity of aPL. Systematic analyses based on these studies could not reveal the prevalence and features of patients developing pathogenic aPL after COVID-19. Conversely, new-onset APS in COVID-19 patients have also been reported, wherein persistently high-titer aPL, associated thrombotic and non-thrombotic manifestations, and recovery following treatments based on APS management guidelines substantially indicate the pathogenicity of aPL. Therefore, new-onset APS cases could be a narrow representative of COVID-19 patients with pathogenic aPL. We systematically reviewed the literature of relevant cases up to February 2024 using PubMed and EMBASE to analyze the APS onset after COVID-19 for the first time. The cases reported as APS after COVID-19 infection with sufficient information to meet the updated Sapporo criteria or the new 2023 ACR/EULAR criteria (1, 2) or with limited information but compelling evidence to support the diagnosis of APS were included. The former and the latter were annotated as definite APS and highly probable APS respectively. Nine cases (68–76) were identified and evaluated together with our case (Table 2). The patients ranged from 15 to 89 (mean = 41.10) years of age and most of the patients were female, consistent with the epidemiology of APS that is more common in middle-aged women (82). According to WHO-issued guidelines (83), COVID-19 severity of these cases encompassed a spectrum from non-severe to critical, indicating that aPL-related manifestations did not merely develop on the basis of cytokine storms in critical patients. The time interval from COVID-19 to the onset of APS varied from 7 to 41 days (mean = 18 days) and a probably more frequent occurrence during the convalescent period was suggested. Definite or probable CAPS was reported in three cases (30%), significantly higher than the approximate 1% incidence of CAPS in all APS patients (84). Thrombosis and corresponding organ infarctions (80%) were the most common manifestation, followed by thrombocytopenia (30%). There were 12 patients mentioned in four cohort studies who also fulfilled the inclusion criteria but were not included due to the lack of individualized information (50, 64, 67, 77–81, 85). In addition to newly diagnosed APS, five cases have reported relapses of completely remitted APS following COVID-19 infections (Table 2), reiterating SARS-CoV-2 as a potential trigger for pathogenic effects of aPL and exacerbation of APS in some patients. Here, we report the first case of developing pathogenic aPL in a juvenile after non-severe COVID-19, diagnosed as highly probable APS. Notably, the absence of any medical history in the patient alerts the possibility of developing severe aPL-related symptoms following non-severe COVID-19 infection in previously healthy individuals, which was also indicated in a healthy woman developing obstetric APS (OAPS) after non-severe COVID-19 infection (73).
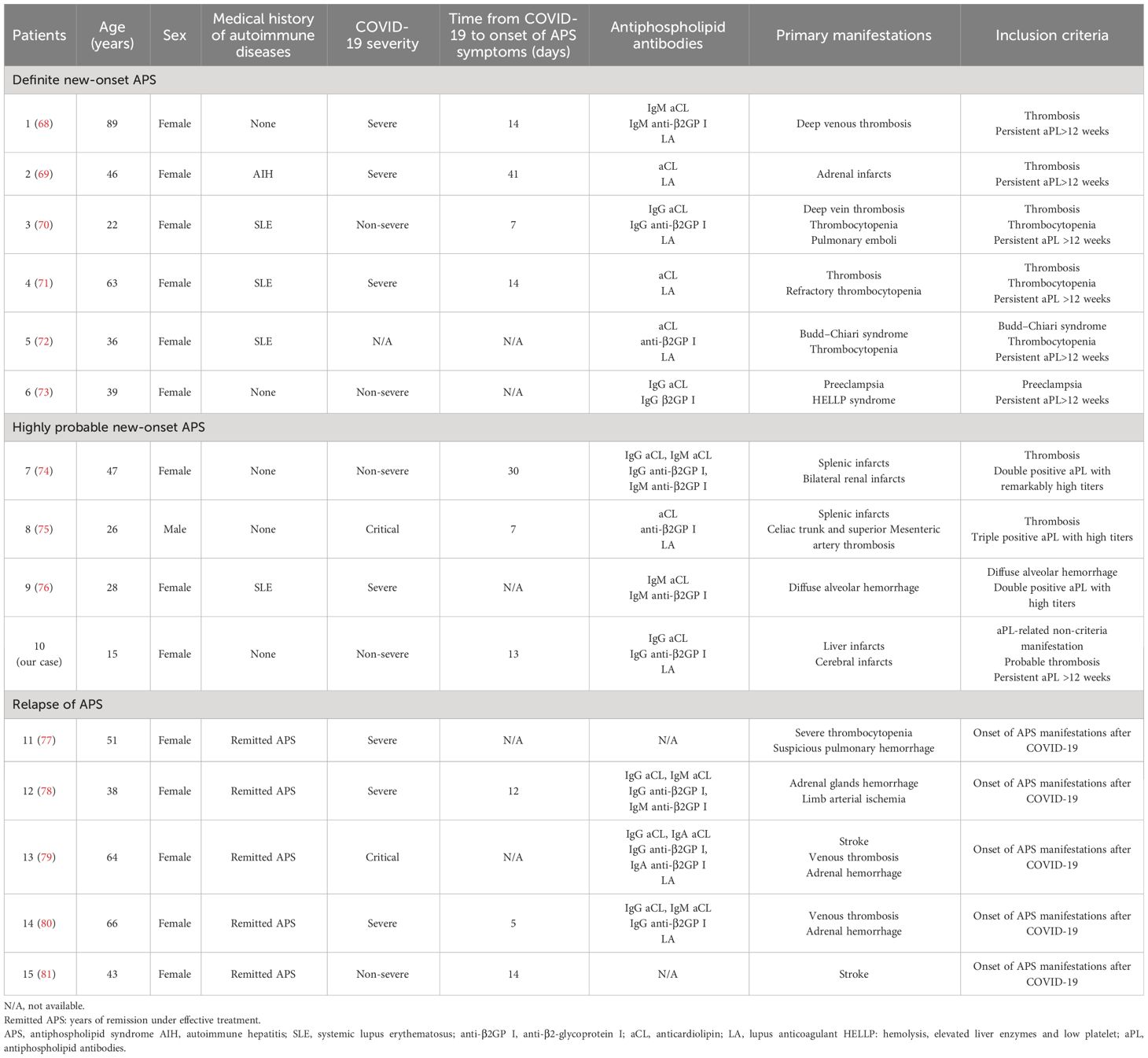
Table 2 Demographic and clinical characteristics of APS after COVID-19.
Differences in distribution, clinical presentations, and outcomes exist between pediatric and adult APS (86). Compared with adult patients, juvenile patients more frequently exhibit non-thrombotic aPL-related manifestations (10). A study including 121 juveniles fulfilling the updated Sapporo criteria demonstrated a high prevalence of associated non-thrombotic manifestations with neurologic, hematologic, and skin disorders being the most common (87). Non-thrombotic manifestations sometimes precede later thrombotic events (88), leaving pediatric patients with isolated non-thrombotic manifestations being inadequately excluded from APS patient population. Accordingly, diagnostic criteria for definite APS are inapplicable in juveniles. Recommendations for management of pediatric APS published by SHARE initiative advocated for the incorporation of non-criteria manifestations into classification criteria for pediatric APS (86). Therefore, recent studies in pediatric APS have concentrated mainly on pathogenic aPL and associated manifestations rather than definite APS. A study of pediatric APS including definite and probable cases revealed high percentage of hematologic and skin disorders (89). Moreover, another analysis of children with medium or highly positive aPL suggested that more than half exhibited non-thrombotic aPL-related manifestations alone (90).
In our case, the histopathology of liver biopsy specimens revealed immune infiltration and fibrinoid necrosis of arteries without granulomatosis, indicating the existence of hepatic vasculitis that has not been reported in association with pathogenic aPL before. The resultant occlusion of arteries gave rise to liver ischemic necrosis in the absence of any notable thrombus or microthrombus. The patient was successfully treated with immunosuppression, further supporting a vasculitic other than thrombotic etiology.
Although debatable, vascular lesions raised by aPL could be inflammatory. DAH, characterized by bleeding into the alveolar space resulting from disruption and injury of pulmonary microcirculation, represents a genuine inflammatory complication of APS and has been included into clinical criteria for APS in the 2023 ACR/EULAR criteria (2, 91). Several studies have investigated the primary APS-associated DAH in recent years (11, 92–94). Surgical or transbronchial biopsies were performed in 20 cases and capillaritis without thrombus or microthrombus was histologically documented in 11 of them (55%), indicating an isolated inflammatory vasculopathy in DAH development. The recommended and efficient treatment of DAH in APS with glucocorticoids and immunomodulatory agents re-emphasizes an inflammatory instead of thrombotic etiopathology of DAH (91). Additionally, mesenteric vasculitis is considered to be one of aPL-related microvascular manifestations as well (95). Sporadic cases with authentic associated vasculitic manifestations have also been reported in cerebral (96), renal (97), aortic (98), and cutaneous (99) vasculature, and no local thrombus or microthrombus was noted in these inflammatory lesions.
Therapy for APS is diverse and individualized based on a broad spectrum of manifestations. Long-term oral anticoagulants like warfarin are recommended for thrombotic APS (100), and alternative therapies such as extended therapeutic dose of low-molecular-weight heparin can be utilized for patients with recurrent thrombotic events despite warfarin (101). For aPL carriers with high-risk profiles or OAPS patients, low-dose aspirin is proposed for primary thrombosis prevention, particularly in individuals with additional vascular risk factors (100, 102). Glucocorticoids; immunomodulatory agents including MMF, cyclophosphamide, and azathioprine; and B-cell-modulating agents like rituximab and belimumab, are recommended in cases with non-thrombotic manifestations (103, 104). Notably, these recommendations, based on adult-derived studies, might be improper for pediatric populations due to differences in physiological conditions, metabolic capacities and duration of medication. Additionally, the low prevalence and heterogeneity of APS in juveniles impede the formation and limit the strength of evidence-based guidelines (86, 105), contributing to substantial variations in treatment regimens that are mostly based on physicians’ experience or observational studies.
In our case, aspirin was administered without anticoagulants. The decision was made based on vasculitis-induced hepatic infarction as the major clinical presentation, repair of cerebral lesions with indefinite pathology before systemic treatment, impaired liver synthetic function for coagulation factors, and the absence of other thrombosis risk factors. As concurrent thrombosis risk factors like arterial hypertension, hyperlipidemia, atherosclerosis and smoking are rarely observed in younger subjects, long-term anticoagulation therapy is not indicated in pediatric thrombotic APS patients harboring discontinuous aPL (106–108). Likewise, immunosuppressive therapy in our case reduced aPL titers close to baseline levels and suppressed their pathogenicity, reminiscent of patients with discontinuous aPL. Combined together, anticoagulants were not administered temporarily. However, the patient underwent intensive and regular follow-up to monitor for emergence of any additional thrombosis risk factors, in which scenario, anticoagulants would be introduced as a replacement of aspirin.
aPL-related thrombosis and vasculitis can cause similar clinical presentations including organ infarctions, whereas the treatment decision is different due to the underlying pathologies. The histopathologic results helped us to confirm the inflammatory vasculopathy and guided the treatment to adequately include immunosuppression comprising glucocorticoids and immunomodulatory agents. Therefore, when no thrombus is detected by non-invasive examinations, biopsy for confirmation of the underlying vasculopathy is suggested in APS, if possible and especially when liver is involved.
Our treatments were individualized based on an atypical case. Although the outcome was favorable, the efficacy and safety of aspirin without anticoagulants require further validation during extended follow-up. We merely recommend the addition of immunosuppressants to conventional therapy for managing aPL-related vasculitis.
Given the perplexing and contentious nature of aPL produced during infections, the COVID-19 pandemic provides a distinctive opportunity to comprehensively assess this issue. The literature review and analysis evaluate the onset and relapse of APS after COVID-19 infection, suggesting that SARS-CoV-2-triggered aPL may exert pathogenic effects in a subset of COVID-19 patients.
Altogether, we endorse the hypothesis that pathogenic aPL can raise vascular damage manifested as vasculitis other than thrombosis, conveying distinct therapeutic considerations to include immunosuppression therapy. In addition to vasculitis, other forms of vascular lesions including proliferative vascular diseases have also been described in APS (109), extending the spectrum of vascular changes associated with pathogenic aPL. Such cumulative evidence supports the statement that the nature of APS should be extended to both thrombophilia and vasculopathy.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
QL: Conceptualization, Formal analysis, Investigation, Writing – original draft. JL: Conceptualization, Formal analysis, Investigation, Writing – original draft. MZ: Writing – review & editing. YG: Writing – review & editing. ZL: Funding acquisition, Writing – review & editing. TL: Funding acquisition, Writing – review & editing. LZ: Resources, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National High Level Hospital Clinical Research Funding (2022-PUMCH-B-043) and the National Natural Science Foundation of China (82202541).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. (2006) 4:295–306. doi: 10.1111/j.1538-7836.2006.01753.x
2. Barbhaiya M, Zuily S, Naden R, Hendry A, Manneville F, Amigo M-C, et al. 2023 ACR/EULAR antiphospholipid syndrome classification criteria. Ann Rheumatic Dis. (2023) 82:1258–70. doi: 10.1002/art.42624
3. Meroni PL, Borghi MO, Raschi E, Tedesco F. Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat Rev Rheumatol. (2011) 7:330–9. doi: 10.1038/nrrheum.2011.52
4. Martirosyan A, Aminov R, Manukyan G. Environmental triggers of autoreactive responses: induction of antiphospholipid antibody formation. Front Immunol. (2019) 10:1609. doi: 10.3389/fimmu.2019.01609
5. Iuliano A, Galeazzi M, Sebastiani GD. Antiphospholipid syndrome's genetic and epigenetic aspects. Autoimmun Rev. (2019) 18:102352. doi: 10.1016/j.autrev.2019.102352
6. Rodríguez-Pintó I, Moitinho M, Santacreu I, Shoenfeld Y, Erkan D, Espinosa G, et al. Catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis of 500 patients from the International CAPS Registry. Autoimmun Rev. (2016) 15:1120–4. doi: 10.1016/j.autrev.2016.09.010
7. Serrano M, Espinosa G, Serrano A, Cervera R. COVID-19 and the antiphospholipid syndrome. Autoimmun Rev. (2022) 21:103206. doi: 10.1016/j.autrev.2022.103206
8. Tung ML, Tan B, Cherian R, Chandra B. Anti-phospholipid syndrome and COVID-19 thrombosis: connecting the dots. Rheumatol Adv Pract. (2021) 5:rkaa081. doi: 10.1093/rap/rkaa081
9. Koike T, Bohgaki M, Amengual O, Atsumi T. Antiphospholipid antibodies: lessons from the bench. J Autoimmun. (2007) 28:129–33. doi: 10.1016/j.jaut.2007.02.009
10. Avcin T, Cimaz R, Rozman B. The Ped-APS Registry: the antiphospholipid syndrome in childhood. Lupus. (2009) 18:894–9. doi: 10.1177/0961203309106917
11. Deane KD, West SG. Antiphospholipid antibodies as a cause of pulmonary capillaritis and diffuse alveolar hemorrhage: a case series and literature review. Semin Arthritis Rheumatol. (2005) 35:154–65. doi: 10.1016/j.semarthrit.2005.05.006
12. Lally L, Sammaritano LR. Vasculitis in antiphospholipid syndrome. Rheum Dis Clin North Am. (2015) 41:109–23, ix. doi: 10.1016/j.rdc.2014.09.009
13. Hu C, Li S, Xie Z, You H, Jiang H, Shi Y, et al. Comparison of different test systems for the detection of antiphospholipid antibodies in a Chinese cohort. Front In Immunol. (2021) 12:648881. doi: 10.3389/fimmu.2021.648881
14. Devreese KMJ, de Groot PG, de Laat B, Erkan D, Favaloro EJ, Mackie I, et al. Guidance from the Scientific and Standardization Committee for lupus anticoagulant/antiphospholipid antibodies of the International Society on Thrombosis and Haemostasis: Update of the guidelines for lupus anticoagulant detection and interpretation. J Thromb Haemostasis. (2020) 18:2828–39. doi: 10.1111/jth.15047
15. Pires da Rosa G, Bettencourt P, Rodríguez-Pintó I, Cervera R, Espinosa G. "Non-criteria" antiphospholipid syndrome: A nomenclature proposal. Autoimmun Rev. (2020) 19:102689. doi: 10.1016/j.autrev.2020.102689
16. Dalekos GN, Zachou K, Liaskos C. The antiphospholipid syndrome and infection. Curr Rheumatol Rep. (2001) 3:277–85. doi: 10.1007/s11926-001-0031-4
17. Hasan Ali O, Bomze D, Risch L, Brugger SD, Paprotny M, Weber M, et al. Severe Coronavirus disease 2019 (COVID-19) is associated with elevated serum immunoglobulin (Ig) A and antiphospholipid IgA antibodies. Clin Infect Dis. (2021) 73:e2869–e74. doi: 10.1093/cid/ciaa1496
18. Gharavi AE, Pierangeli SS. Origin of antiphospholipid antibodies: induction of aPL by viral peptides. Lupus. (1998) 7 Suppl 2:S52–S4. doi: 10.1177/096120339800700213
19. Al-Beltagi M, Saeed NK, Bediwy AS. COVID-19 disease and autoimmune disorders: A mutual pathway. World J Methodol. (2022) 12:200–23. doi: 10.5662/wjm.v12.i4.200
20. Amin NM. Antiphospholipid syndromes in infectious diseases. Hematol Oncol Clin North Am. (2008) 22:131–43. doi: 10.1016/j.hoc.2007.10.001
21. Buinitskaya Y, Gurinovich R, Wlodaver CG, Kastsiuchenka S. Highlights of COVID-19 pathogenesis. Insights into Oxidative Damage. (2020). doi: 10.6084/m9.figshare.12121575.v10
22. Delgado-Roche L, Mesta F. Oxidative stress as key player in severe acute respiratory syndrome Coronavirus (SARS-CoV) infection. Arch Med Res. (2020) 51:384–7. doi: 10.1016/j.arcmed.2020.04.019
23. López-Pedrera C, Barbarroja N, Jimenez-Gomez Y, Collantes-Estevez E, Aguirre MA, Cuadrado MJ. Oxidative stress in the pathogenesis of atherothrombosis associated with anti-phospholipid syndrome and systemic lupus erythematosus: new therapeutic approaches. Rheumatol (Oxford England). (2016) 55:2096–108. doi: 10.1093/rheumatology/kew054
24. Borghi MO, Beltagy A, Garrafa E, Curreli D, Cecchini G, Bodio C, et al. Anti-phospholipid antibodies in COVID-19 are different from those detectable in the anti-phospholipid syndrome. Front In Immunol. (2020) 11:584241. doi: 10.3389/fimmu.2020.584241
25. Gazzaruso C, Mariani G, Ravetto C, Malinverni L, Tondelli E, Cerrone M, et al. Lupus anticoagulant and mortality in patients hospitalized for COVID-19. J Thromb Thrombolysis. (2021) 52:85–91. doi: 10.1007/s11239-020-02335-w
26. Siguret V, Voicu S, Neuwirth M, Delrue M, Gayat E, Stépanian A, et al. Are antiphospholipid antibodies associated with thrombotic complications in critically ill COVID-19 patients? Thromb Res. (2020) 195:74–6. doi: 10.1016/j.thromres.2020.07.016
27. Bowles L, Platton S, Yartey N, Dave M, Lee K, Hart DP, et al. Lupus anticoagulant and abnormal coagulation tests in patients with Covid-19. New Engl J Med. (2020) 383:288–90. doi: 10.1056/NEJMc2013656
28. Trahtemberg U, Rottapel R, Dos Santos CC, Slutsky AS, Baker A, Fritzler MJ. Anticardiolipin and other antiphospholipid antibodies in critically ill COVID-19 positive and negative patients. Ann Rheumatic Diseases. (2021) 80:1236–40. doi: 10.1136/annrheumdis-2021-220206
29. Galeano-Valle F, Oblitas CM, Ferreiro-Mazón MM, Alonso-Muñoz J, Del Toro-Cervera J, di Natale M, et al. Antiphospholipid antibodies are not elevated in patients with severe COVID-19 pneumonia and venous thromboembolism. Thromb Res. (2020) 192:113–5. doi: 10.1016/j.thromres.2020.05.017
30. Cristiano A, Fortunati V, Cherubini F, Bernardini S, Nuccetelli M. Anti-phospholipids antibodies and immune complexes in COVID-19 patients: a putative role in disease course for anti-annexin-V antibodies. Clin Rheumatol. (2021) 40:2939–45. doi: 10.1007/s10067-021-05580-3
31. Lerma LA, Chaudhary A, Bryan A, Morishima C, Wener MH, Fink SL. Prevalence of autoantibody responses in acute coronavirus disease 2019 (COVID-19). J Transl Autoimmun. (2020) 3:100073. doi: 10.1016/j.jtauto.2020.100073
32. Gatto M, Perricone C, Tonello M, Bistoni O, Cattelan AM, Bursi R, et al. Frequency and clinical correlates of antiphospholipid antibodies arising in patients with SARS-CoV-2 infection: findings from a multicentre study on 122 cases. Clin Exp Rheumatol. (2020) 38:754–9.
33. Gendron N, Dragon-Durey M-A, Chocron R, Darnige L, Jourdi G, Philippe A, et al. Lupus anticoagulant single positivity during the acute phase of COVID-19 is not associated with venous thromboembolism or in-hospital mortality. Arthritis Rheumatol (Hoboken NJ). (2021) 73:1976–85. doi: 10.1002/art.41777
34. Vlachoyiannopoulos PG, Magira E, Alexopoulos H, Jahaj E, Theophilopoulou K, Kotanidou A, et al. Autoantibodies related to systemic autoimmune rheumatic diseases in severely ill patients with COVID-19. Ann Rheumatic Diseases. (2020) 79:1661–3. doi: 10.1136/annrheumdis-2020-218009
35. Rosales-Castillo A, Sabio JM. Assessment of antiphospholipid antibodies during the follow-up of patients after SARS-CoV-2 infection. Med Clin (Engl Ed). (2022) 158:437–8. doi: 10.1016/j.medcle.2021.09.015
36. Atalar E, Erden A, Guven SC, Armagan B, Ates İ, Küçüksahin O, et al. The clinical significance of antiphospholipid antibodies in COVID-19 infection. J Infect Dev Ctries. (2022) 16:276–82. doi: 10.3855/jidc.15423
37. Gutiérrez López de Ocáriz X, Castro Quismondo N, Vera Guerrero E, Rodríguez Rodríguez M, Ayala Díaz R, Martínez López J. Thrombosis and antiphospholipid antibodies in patients with SARS-COV-2 infection (COVID-19). Int J Lab Hematol. (2020) 42:e280–e2. doi: 10.1111/ijlh
38. Previtali G, Seghezzi M, Moioli V, Sonzogni A, Cerutti L, Marozzi R, et al. The pathogenesis of thromboembolic disease in covid-19 patients: Could be a catastrophic antiphospholipid syndrome? Thromb Res. (2020) 194:192–4. doi: 10.1016/j.thromres.2020.06.042
39. Ferrari E, Sartre B, Squara F, Contenti J, Occelli C, Lemoel F, et al. High prevalence of acquired thrombophilia without prognosis value in patients with Coronavirus disease 2019. J Am Heart Assoc. (2020) 9:e017773. doi: 10.1161/JAHA.120.017773
40. Sciascia S, Radin M, Bazzan M, Montaruli B, Cosseddu D, Norbiato C, et al. Antiphospholipid antibodies and infection: non nova Sed Nove. Front In Immunol. (2021) 12:687534. doi: 10.3389/fimmu.2021.687534
41. Tvito A, Ben-Chetrit E, Zimmerman FS, Asher E, Helviz Y. Lupus anticoagulant in patients with COVID-19. Int J Lab Hematol. (2021) 43:e17–e8. doi: 10.1111/ijlh.13334
42. Oba S, Hosoya T, Kaneshige R, Kawata D, Yamaguchi T, Mitsumura T, et al. Thrombosis and antiphospholipid antibodies in Japanese COVID-19: based on propensity score matching. Front In Immunol. (2023) 14:1227547. doi: 10.3389/fimmu.2023.1227547
43. Nosrati A, Torabizadeh Z, Kheirkhah D, Vahedi Larijani L, Alizade-Navaei R, Mobini M, et al. Evaluation of antiphospholipid antibodies in COVID-19 patients with coagulopathy. Tanaffos. (2022) 21:45–53.
44. Bnina AB, Dhia RB, Gnaba S, Annabi A, Chouchane S, Naija W, et al. Assessment of antiphospholipid antibodies profiles based on severity of COVID-19 pneumonia. Pan Afr Med J. (2022) 42:110. doi: 10.11604/pamj.2022.42.110.33020
45. Kahlon N, Shazadeh Safavi P, Abuhelwa Z, Sheikh T, Burmeister C, Doddi S, et al. Prevalence and clinical significance of antiphospholipid antibodies in hospitalized patients with COVID-19 infection. Cureus. (2022) 14:e27862. doi: 10.7759/cureus.27862
46. Amezcua-Guerra LM, Rojas-Velasco G, Brianza-Padilla M, Vázquez-Rangel A, Márquez-Velasco R, Baranda-Tovar F, et al. Presence of antiphospholipid antibodies in COVID-19: a case series study. Ann Rheumatic Diseases. (2021) 80:e73. doi: 10.1136/annrheumdis-2020-218100
47. Gasparini G, Canepa P, Verdiani S, Carmisciano L, Cozzani E, De Grazia D, et al. A retrospective study on the prevalence of anti-phospholipid antibodies, thrombotic events and cutaneous signs of vasculopathy in 173 hospitalized COVID-19 patients. Int J Immunopathol Pharmacol. (2021) 35:20587384211042115. doi: 10.1177/20587384211042115
48. Devreese KMJ, Linskens EA, Benoit D, Peperstraete H. Antiphospholipid antibodies in patients with COVID-19: A relevant observation? J Thromb Haemostasis JTH. (2020) 18:2191–201. doi: 10.1111/jth.14994
49. Espinosa G, Zamora-Martínez C, Pérez-Isidro A, Neto D, Bravo-Gallego LY, Prieto-González S, et al. Persistent antiphospholipid antibodies are not associated with worse clinical outcomes in a prospective cohort of hospitalised patients with SARS-CoV-2 infection. Front In Immunol. (2022) 13:911979. doi: 10.3389/fimmu.2022.911979
50. Zlatković-Švenda M, Ovuka M, Ogrič M, Čučnik S, Žigon P, Radivčev A, et al. Antiphospholipid antibodies and vascular thrombosis in patients with severe forms of COVID-19. Biomedicines. (2023) 11:3117. doi: 10.20944/preprints202308.0991.v1
51. Taha M, Samavati L. Antiphospholipid antibodies in COVID-19: a meta-analysis and systematic review. RMD Open. (2021) 7:e001580. doi: 10.1136/rmdopen-2021-001580
52. Pascolini S, Vannini A, Deleonardi G, Ciordinik M, Sensoli A, Carletti I, et al. COVID-19 and immunological dysregulation: can autoantibodies be useful? Clin Transl Sci. (2021) 14:502–8. doi: 10.1111/cts.12908
53. Serrano M, Espinosa G, Lalueza A, Bravo-Gallego LY, Diaz-Simón R, Garcinuño S, et al. Beta-2-glycoprotein-I deficiency could precipitate an antiphospholipid syndrome-like prothrombotic situation in patients with Coronavirus disease 2019. ACR Open Rheumatol. (2021) 3:267–76. doi: 10.1002/acr2.11245
54. Bertin D, Brodovitch A, Beziane A, Hug S, Bouamri A, Mege JL, et al. Anticardiolipin IgG autoantibody level is an independent risk factor for COVID-19 severity. Arthritis Rheumatol (Hoboken NJ). (2020) 72:1953–5. doi: 10.1002/art.41409
55. Gazzaruso C, Carlo Stella N, Mariani G, Nai C, Coppola A, Naldani D, et al. High prevalence of antinuclear antibodies and lupus anticoagulant in patients hospitalized for SARS-CoV2 pneumonia. Clin Rheumatol. (2020) 39:2095–7. doi: 10.1007/s10067-020-05180-7
56. Anaya J-M, Monsalve DM, Rojas M, Rodríguez Y, Montoya-García N, Mancera-Navarro LM, et al. Latent rheumatic, thyroid and phospholipid autoimmunity in hospitalized patients with COVID-19. J Transl Autoimmun. (2021) 4:100091. doi: 10.1016/j.jtauto.2021.100091
57. Sadeghi A, Hasanlu M, Feyzi A, Mansori K, Ghodrati S, Parsamanesh N. Evaluating the relationship between antiphospholipid antibodies and COVID-19 severity. DNA Cell Biol. (2023) 42:65–71. doi: 10.1089/dna.2022.0293
58. Frapard T, Hue S, Rial C, de Prost N, Mekontso Dessap A. Antiphospholipid Autoantibodies and Thrombosis in Patients With COVID-19: Comment on the Article by Bertin et al. Arthritis Rheumatol (Hoboken NJ). (2021) 73:897–9. doi: 10.1002/art.41634
59. Zuo Y, Estes SK, Ali RA, Gandhi AA, Yalavarthi S, Shi H, et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci Transl Med. (2020) 12:eabd3876. doi: 10.1126/scitranslmed.abd3876
60. Le Joncour A, Frere C, Martin-Toutain I, Gougis P, Ghillani-Dalbin P, Maalouf G, et al. Antiphospholipid antibodies and thrombotic events in COVID-19 patients hospitalized in medicine ward. Autoimmun Rev. (2021) 20:102729. doi: 10.1016/j.autrev.2020.102729
61. Fan S, Xiao M, Han F, Xia P, Bai X, Chen H, et al. Neurological manifestations in critically ill patients with COVID-19: A retrospective study. Front Neurol. (2020) 11:806. doi: 10.3389/fneur.2020.00806
62. Reyes Gil M, Barouqa M, Szymanski J, Gonzalez-Lugo JD, Rahman S, Billett HH. Assessment of lupus anticoagulant positivity in patients with Coronavirus disease 2019 (COVID-19). JAMA Netw Open. (2020) 3:e2017539. doi: 10.1001/jamanetworkopen.2020.17539
63. Lee SJ, Yoon T, Ha JW, Kim J, Lee KH, Lee JA, et al. Prevalence, clinical significance, and persistence of autoantibodies in COVID-19. Virol J. (2023) 20:236. doi: 10.1186/s12985-023-02191-z
64. Vollmer O, Tacquard C, Dieudonné Y, Nespola B, Sattler L, Grunebaum L, et al. Follow-up of COVID-19 patients: LA is transient but other aPLs are persistent. Autoimmun Rev. (2021) 20:102822. doi: 10.1016/j.autrev.2021.102822
65. Gil-Etayo FJ, Garcinuño S, Lalueza A, Díaz-Simón R, García-Reyne A, Pleguezuelo DE, et al. Anti-phospholipid antibodies and COVID-19 thrombosis: A co-star, not a supporting actor. Biomedicines. (2021) 9:899. doi: 10.3390/biomedicines9080899
66. Xiao M, Zhang Y, Zhang S, Qin X, Xia P, Cao W, et al. Antiphospholipid antibodies in critically ill patients with COVID-19. Arthritis Rheumatol (Hoboken NJ). (2020) 72:1998–2004. doi: 10.1002/art.41425
67. Arcani R, Cauchois R, Suchon P, Weber S, Jean R, Jarrot P-A, et al. "True" Antiphospholipid syndrome in COVID-19: contribution of the follow-up of antiphospholipid autoantibodies. Semin Thromb Hemost. (2023) 49:97–102. doi: 10.1055/s-0042-1758118
68. Khan FA. UNMASKING TRIPLE POSITIVE ANTIPHOSPHOLIPID SYNDROME IN COVID-19 INFECTION: A CASE REPORT. J Gen Internal Med. (2023) 38:S561. doi: 10.1007/s11606-023-08226-z
69. MaChado IFR, Menezes IQ, Figueiredo SR, Coelho FMA, Terrabuio DRB, Ramos DV, et al. Primary adrenal insufficiency due to bilateral adrenal infarction in COVID-19: a case report. J Clin Endocrinol Metab. (2022) 107:e394–400. doi: 10.1210/clinem/dgab557
70. Aguirre-Alastuey ME, Suárez-Díaz S, Rodríguez-Jerez F, Coto-Hernández R, Caminal-Montero L. Venous thrombosis in a systemic lupus erythematosus patient with antiphospholipid antibodies coinciding with mild Covid-19. Lupus. (2021) 30:172–4. doi: 10.1177/0961203320967407
71. Freeman-Beman L, Ratner S, Kabani N, Neculiseanu E, Ginzler E. COVID-19 coagulopathy in a patient with systemic lupus erythematosus and antiphospholipid antibodies. J Clin Rheumatol. (2021) 27:e60–e1. doi: 10.1097/RHU.0000000000001599
72. La Mura V, Gualtierotti R, Martinelli I, Ferrari B, Ierardi AM, Bitto N, et al. Acute liver necrosis in a SARS-CoV-2 positive patient with triple positive antiphospholipid syndrome. Res Pract Thromb Haemostasis. (2021) 5:231. doi: 10.1002/rth2.12589
73. Gozzoli GI, Piovani E, Negri B, Mascherpa M, Orabona R, Zanardini C, et al. Frequency of positive antiphospholipid antibodies in pregnant women with SARS-CoV-2 infection and impact on pregnancy outcome: A single-center prospective study on 151 pregnancies. Front In Immunol. (2022) 13:953043. doi: 10.3389/fimmu.2022.953043
74. Bitterman L, Solhjoo M, Shah V, Kwon SM, Torralba K, Kazbour H. Catastrophic antiphospholipid syndrome as a complication of COVID-19 infection. J Investig Med High Impact Case Rep. (2023) 11:23247096231165736. doi: 10.1177/23247096231165736
75. Tay V, Habibah M, Mollyza M, Shereen SC, Azmillah R, Lau I. Case report: Covid-19 and disseminated tuberculosis coinfection unveiling catastrophic antiphospholipid antibody syndrome: An association or de novo phenomenon. Int J Rheumatic Diseases. (2023) 26:337.
76. Nahidi SM, Garg Y, Mahadeo DS, Sharma M, Acosta C, Seetharam K, et al. Cardiac arrest in the setting of probable catastrophic antiphospholipid syndrome in young patient with a history of COVID infection and polyglandular disorder-Case report. SAGE Open Med Case Rep. (2023) 11:2050313X231220803. doi: 10.1177/2050313X231220803
77. Hayden A, Vyas-Lahar A, Rella V, Rudinskaya A. Severe refractory thrombocytopenia in a woman positive for coronavirus disease 2019 with lupus and antiphospholipid syndrome. Lupus. (2020) 29:1472–4. doi: 10.1177/0961203320940389
78. Maria ATJ, Diaz-Cau I, Benejean J-M, Nutz A, Schiffmann A, Biron-Andreani C, et al. Flare of antiphospholipid syndrome in the course of COVID-19. TH Open. (2020) 4:e207–e10. doi: 10.1055/s-0040-1716735
79. Chidharla A, Syed SB, Chatterjee T, Tarantino MD. A case report of COVID-associated catastrophic antiphospholipid syndrome successfully treated with eculizumab. J Blood Med. (2021) 12:929–33. doi: 10.2147/JBM.S324873
80. Frankel M, Feldman I, Levine M, Frank Y, Bogot NR, Benjaminov O, et al. Bilateral adrenal hemorrhage in Coronavirus disease 2019 patient: A case report. J Clin Endocrinol Metab. (2020) 105:dgaa487. doi: 10.1210/clinem/dgaa487
81. Kincaid KJ, Simpkins AN. Failure of anticoagulation to prevent stroke in context of lupus-associated anti-phospholipid syndrome and mild COVID-19. J Stroke Cerebrovascular Dis. (2021) 30:105817. doi: 10.1016/j.jstrokecerebrovasdis.2021.105817
82. Dabit JY, Valenzuela-Almada MO, Vallejo-Ramos S, Duarte-García A. Epidemiology of antiphospholipid syndrome in the general population. Curr Rheumatol Rep. (2022) 23:85. doi: 10.1007/s11926-021-01038-2
83. World Health Organization. Clinical management of COVID-19: interim guidance 2020 (2023). Available at: https://www.who.int/publications/i/item/WHO-2019-nCoV-clinical-2021-1.
84. Cervera R, Piette J-C, Font J, Khamashta MA, Shoenfeld Y, Camps MT, et al. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheumatol. (2002) 46:1019–27. doi: 10.1002/art.10187
85. Ogrič M, Žigon P, Sodin-Semrl S, Zlatković-Švenda M, Zdravković M, Ovuka M, et al. Longitudinal analysis of antiphospholipid antibody dynamics after infection with SARS-CoV-2 or vaccination with BNT162b2. Int J Mol Sci. (2022) 24:211. doi: 10.3390/ijms24010211
86. Groot N, de Graeff N, Avcin T, Bader-Meunier B, Dolezalova P, Feldman B, et al. European evidence-based recommendations for diagnosis and treatment of paediatric antiphospholipid syndrome: the SHARE initiative. Ann Rheum Dis. (2017) 76:1637–41. doi: 10.1136/annrheumdis-2016-211001
87. Avcin T, Cimaz R, Silverman ED, Cervera R, Gattorno M, Garay S, et al. Pediatric antiphospholipid syndrome: clinical and immunologic features of 121 patients in an international registry. Pediatrics. (2008) 122:e1100–7. doi: 10.1542/peds.2008-1209
88. Islabão AG, Trindade VC, da Mota LMH, Andrade DCO, Silva CA. Managing antiphospholipid syndrome in children and adolescents: current and future prospects. Paediatr Drugs. (2022) 24:13–27. doi: 10.1007/s40272-021-00484-w
89. Ma J, Song H, Wei M, He Y. Clinical characteristics and thrombosis outcomes of paediatric antiphospholipid syndrome: analysis of 58 patients. Clin Rheumatol. (2018) 37:1295–303. doi: 10.1007/s10067-017-3776-5
90. Rozic M, Trampus-Bakija A, Rener-Primec Z, Kitanovski L, Kveder T, Avcin T. PReS-FINAL-2349: Spectrum of thrombotic and non-thrombotic manifestations in 159 children with positive antiphospholipid antibodies. Pediatr Rheumatol. (2013) 11:P339. doi: 10.1186/1546-0096-11-S2-P339
91. Stoots SA, Lief L, Erkan D. Clinical insights into diffuse alveolar hemorrhage in antiphospholipid syndrome. Curr Rheumatol Rep. (2019) 21:56. doi: 10.1007/s11926-019-0852-7
92. Cartin-Ceba R, Peikert T, Ashrani A, Keogh K, Wylam ME, Ytterberg S, et al. Primary antiphospholipid syndrome-associated diffuse alveolar hemorrhage. Arthritis Care Res (Hoboken). (2014) 66:301–10. doi: 10.1002/acr.22109
93. Sangli SS, Ryu JH, Baqir M. Diffuse alveolar hemorrhage in primary versus secondary antiphospholipid syndrome. J Clin Rheumatol. (2021) 27:e297–301. doi: 10.1097/RHU.0000000000001358
94. Yachoui R, Sehgal R, Amlani B, Goldberg JW. Antiphospholipid antibodies-associated diffuse alveolar hemorrhage. Semin Arthritis Rheumatol. (2015) 44:652–7. doi: 10.1016/j.semarthrit.2014.10.013
95. Zhao Y, Qi W, Huang C, Zhou Y, Wang Q, Tian X, et al. Serum calprotectin as a potential predictor of microvascular manifestations in patients with antiphospholipid syndrome. Rheumatol Ther. (2023) 10:1769–83. doi: 10.1007/s40744-023-00610-9
96. Quintero M, Mirza N, Chang H, Perl A. Antiphospholipid antibody syndrome associated with primary angiitis of the central nervous system: report of two biopsy proven cases. Ann Rheum Dis. (2006) 65:408–9. doi: 10.1136/ard.2005.040444
97. Almeshari K, Alfurayh O, Akhtar M. Primary antiphospholipid syndrome and self-limited renal vasculitis during pregnancy: case report and review of the literature. Am J Kidney Dis. (1994) 24:505–8. doi: 10.1016/S0272-6386(12)80909-7
98. Escoda T, George J, Jarrot PA, Jean R, Mazodier K, Sanderson F, et al. Aortitis is an under-recognized manifestation of antiphospholipid syndrome: A case report and literature review. Lupus. (2022) 31:744–53. doi: 10.1177/09612033221091142
99. Sheth K, Parke A. Cutaneous vasculitis in a patient with antiphospholipid antibody syndrome. Conn Med. (2016) 80:75–9.
100. Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet. (2010) 376:1498–509. doi: 10.1016/S0140-6736(10)60709-X
101. Garcia D, Erkan D. Diagnosis and management of the antiphospholipid syndrome. N Engl J Med. (2018) 378:2010–21. doi: 10.1056/NEJMra1705454
102. Tektonidou MG, Andreoli L, Limper M, Amoura Z, Cervera R, Costedoat-Chalumeau N, et al. EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann Rheumatic Diseases. (2019) 78:1296–304. doi: 10.1136/annrheumdis-2019-215213
103. Cohen H, Cuadrado MJ, Erkan D, Duarte-Garcia A, Isenberg DA, Knight JS, et al. 16th international congress on antiphospholipid antibodies task force report on antiphospholipid syndrome treatment trends. Lupus. (2020) 29:1571–93. doi: 10.1177/0961203320950461
104. Xourgia E, Tektonidou MG. Management of non-criteria manifestations in antiphospholipid syndrome. Curr Rheumatol Rep. (2020) 22:51. doi: 10.1007/s11926-020-00935-2
105. Tarango C, Palumbo JS. Antiphospholipid syndrome in pediatric patients. Curr Opin Hematol. (2019) 26:366–71. doi: 10.1097/MOH.0000000000000523
106. Coloma Bazán E, Donate López C, Moreno Lozano P, Cervera R, Espinosa G. Discontinuation of anticoagulation or antiaggregation treatment may be safe in patients with primary antiphospholipid syndrome when antiphospholipid antibodies became persistently negative. Immunol Res. (2013) 56:358–61. doi: 10.1007/s12026-013-8407-x
107. Criado-García J, Fernández-Puebla RA, Jiménez LL, Velasco F, Santamaría M, Blanco-Molina A. [Anticoagulation treatment withdrawal in primary antiphospholipid syndrome when anticardiolipin antibodies become negative]. Rev Clin Esp. (2008) 208:135–7. doi: 10.1157/13115821
108. Ravelli A, Martini A. Antiphospholipid antibody syndrome in pediatric patients. Rheum Dis Clin North Am. (1997) 23:657–76. doi: 10.1016/S0889-857X(05)70351-3
Keywords: antiphospholipid antibodies, COVID-19, pediatrics, vasculitis, non-thrombotic manifestation, vasculopathy
Citation: Li Q, Li J, Zhou M, Ge Y, Liu Z, Li T and Zhang L (2024) Antiphospholipid antibody-related hepatic vasculitis in a juvenile after non-severe COVID-19: a case report and literature review. Front. Immunol. 15:1354349. doi: 10.3389/fimmu.2024.1354349
Received: 12 December 2023; Accepted: 29 March 2024;
Published: 19 April 2024.
Edited by:
Mattia Bellan, University of Eastern Piedmont, ItalyReviewed by:
Tommaso Bucci, University of Liverpool, United KingdomCopyright © 2024 Li, Li, Zhou, Ge, Liu, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Zhang, c2VhMTk4MjUxNUAxNjMuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.