
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 23 February 2024
Sec. Immunological Tolerance and Regulation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1351898
This article is part of the Research Topic Immunology at the feto-maternal interface View all 19 articles
 Hadida Yasmin1†
Hadida Yasmin1† Chiara Agostinis2†
Chiara Agostinis2† Miriam Toffoli3
Miriam Toffoli3 Tamali Roy1
Tamali Roy1 Silvia Pegoraro2
Silvia Pegoraro2 Andrea Balduit2
Andrea Balduit2 Gabriella Zito2
Gabriella Zito2 Nicoletta Di Simone4,5
Nicoletta Di Simone4,5 Giuseppe Ricci2,3
Giuseppe Ricci2,3 Taruna Madan6
Taruna Madan6 Uday Kishore7,8*
Uday Kishore7,8* Roberta Bulla9*
Roberta Bulla9*Pregnancy is an immunologically regulated, complex process. A tightly controlled complement system plays a crucial role in the successful establishment of pregnancy and parturition. Complement inhibitors at the feto-maternal interface are likely to prevent inappropriate complement activation to protect the fetus. In the present study, we aimed to understand the role of Factor H (FH), a negative regulator of complement activation, in normal pregnancy and in a model of pathological pregnancy, i.e. preeclampsia (PE). The distribution and expression of FH was investigated in placental tissues, various placental cells, and in the sera of healthy (CTRL) or PE pregnant women via immunohistochemistry, RT-qPCR, ELISA, and Western blot. Our results showed a differential expression of FH among the placental cell types, decidual stromal cells (DSCs), decidual endothelial cells (DECs), and extravillous trophoblasts (EVTs). Interestingly, FH was found to be considerably less expressed in the placental tissues of PE patients compared to normal placental tissue both at mRNA and protein levels. Similar results were obtained by measuring circulating FH levels in the sera of third trimester CTRL and PE mothers. Syncytiotrophoblast microvesicles, isolated from the placental tissues of PE and CTRL women, downregulated FH expression by DECs. The present study appears to suggest that FH is ubiquitously present in the normal placenta and plays a homeostatic role during pregnancy.
Pregnancy is a delicate biological process where the maternal immune system undergoes several immunomodulatory changes involving both innate and adaptive immunity to ensure tolerance at the feto-maternal interface. Complement is a crucial player in the tightly regulated immunological mechanisms for successful implantation, placental development, and parturition (1–3), displaying also non-canonical functions. Several years after its discovery, the contribution of the complement system to a successful pregnancy has increasingly been recognised (4).
Complement is a pivotal component of humoral innate immunity, which is made up of over 50 fluid-phase, membrane-bound and intracellular proteins that participate in three independent but collaborative activation pathways: classical, lectin and alternative. These three pathways converge on the activation of common component C3, and on the generation of anaphylatoxins such as C3a, C4a, C5a, and C5a-desArg, opsonins such as, C3b, iC3b, C3d, C4b, and cytolytic membrane attack complex (MAC) (5). Complement activation must be tightly regulated since complement dysregulation or over-activation can turn it from a homeostatic to a pathological effector driving several inflammatory conditions.
Several studies have demonstrated that dysregulation of complement activation predisposes to the development of pregnancy-related complications, such as pre-term birth, spontaneous abortion, recurrent miscarriage, and preeclampsia (PE) (6–9). PE is a common pregnancy disorder characterized by high blood pressure with proteinuria or end-organ injury. It is characterized by abnormal placentation, which may be related to defective decidualization, implantation, and angiogenesis (10–12). Besides shallow trophoblast invasion and inadequate remodeling of uterine arteries, dysregulation of immune and non-immune system is also a focal point in the development of PE. Excessive increase in the apoptosis of villous trophoblast can also cause over-activation of complement, enhancing pro-inflammatory response at the feto-maternal interface as observed in the case of PE (13). Moreover, increased levels of complement components and their activation-dependent cleaved products, including C3a, C5a, and C5b-9 complex, have been reported in the circulation of PE women (14), even though their exact contribution to PE pathophysiology is not fully understood.
Complement activation is tightly controlled through several complement regulatory proteins, Factor H (FH) being one of them in pregnancy. FH is 155 kDa soluble protein. Its gene is located on human chromosome 1 at the RAC (regulators of complement activation) gene cluster (15–17). Under physiological conditions, FH levels in the plasma range widely from 116 to 562 μg/ml, depending on genetic and environmental factors (18). FH is the main negative regulator of the complement alternative pathway in solution phase, but it is also involved in the inhibition of complement amplification on target cells and in the extracellular matrix of host tissues. It is a versatile innate immune molecule that protects host tissues by attenuating C3b attached to self surfaces and by restricting the formation of MAC. FH regulates enzymatic cleavage of C3 via its decay accelerating activity as well as cofactor activity for factor I–mediated C3b cleavage (19–22). Thus, by acting as a central negative regulator of the alternative pathway, FH limits the complement-mediated tissue damage leading to several diseases (23–25). FH can bind to molecules/ligands exposed on apoptotic and necrotic cells, including DNA, histones, and annexin II (and possibly the altered glycocalyx), in order to minimize the downstream pro-inflammatory effects of complement activation and the release of autoantigens that would potentially trigger autoimmunity (26–28).
Recent reports suggest that the FH level rises in maternal circulation during the first trimester of normal pregnancy (7), whereas it appears to decrease in severe PE (29). The presence of FH in placental tissue is largely attributed to the circulating blood in the placental vessels; whether it is synthesized locally by the major cell types constituting the placenta has not been investigated. The current study investigates the expression of FH in serum, placental tissues as well as various placental cells in normal pregnancy. FH levels were also measured in PE women to establish its contribution to the mechanisms underlying complement-dependent PE pathophysiology.
For this study, multiple cohorts of pregnant women at different stages of pregnancy were included (Supplementary Figure 1). For each cohort, PE was defined as the new onset of hypertension (systolic blood pressure ≥ 140 mmHg or diastolic ≥ 90 mmHg) on at least two occasions within 24 hours, along with the new onset of proteinuria (≥ 300 mg in a 24-hour urine collection, 50 mg/mmol protein/creatinine ratio, or at least 2+ on dipstick testing following two consecutive measurements).
A first cohort of patients was enrolled at the Institute for Maternal and Child Health, IRCCS “Burlo Garofolo” (Trieste, Italy) between October 2007 and April 2009, as described by Di Lorenzo and colleagues (30). From this serum bank, we selected first trimester sera of women who subsequently developed PE (n = 20) and sera of healthy pregnant women (n = 20) matched for maternal age (32 ± 4).
The third-trimester sera cohort was collected at the IRCCS “Policlinico Agostino Gemelli” (Rome, Italy). In particular, PE patients (n = 31) and CTRL (n = 26) had singleton pregnancies with undetected fetal abnormality and were matched for gestation week (32.7 ± 3.7 vs. 32.9 ± 4.5) (Table 1). A random selection from this cohort of samples was made to obtain pooled sera for cell stimulation. Blood samples were centrifuged at 500 xg for 7 min and the serum was immediately stored at -80°C.
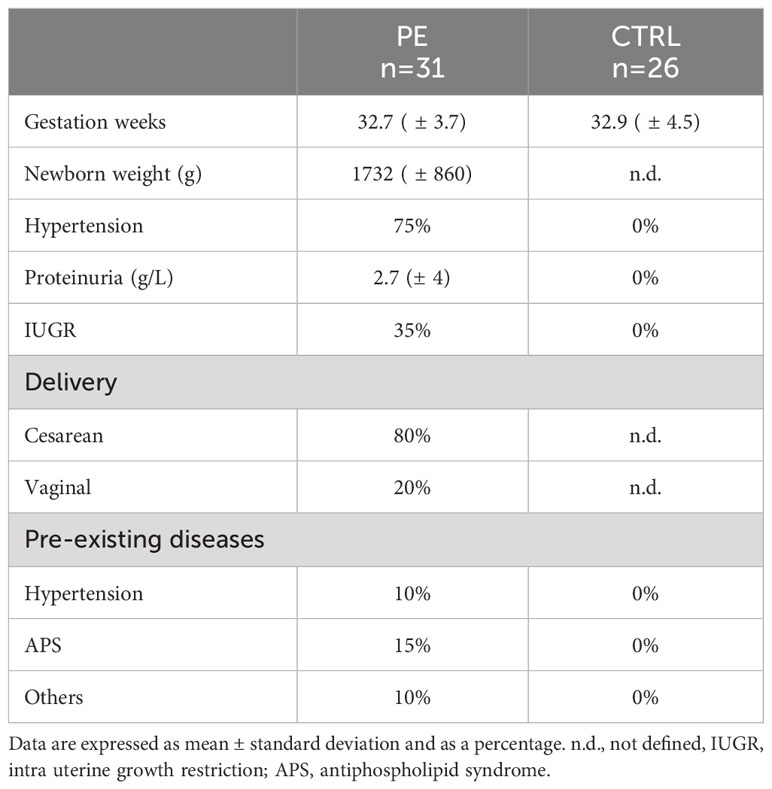
Table 1 Characteristics of the PE patients’ cohort enrolled at Institute “Policlinico Agostino Gemelli” (Rome, Italy).
Tissue samples of first-trimester placenta and decidua were obtained from healthy women undergoing elective termination of pregnancy at 8-12 weeks of gestation at the Institute for Maternal and Child Health, IRCCS “Burlo Garofolo” (Trieste, Italy). Within the same institute, term placenta samples were collected from both PE patients (n = 7) and healthy women (n = 8). A portion of these samples was fixed in 10%v/v buffered formalin for histological investigations, while another portion was shredded, added to 5 mL TRIzol (ThermoFisher), and stored at -80°C for total mRNA isolation.
The study was reviewed and approved by the Regional Ethical Committee of FVG (CEUR-2020-Os-156; Prot. 0022668/P/GEN/ARCS and CEUR-2019-Sper-17) and adhered to the principles of the Declaration of Helsinki. The written consent was obtained from all the participants.
Syncytiotrophoblast derived microvesicles (STBMs), prepared via dual-placental perfusion of PE and healthy placentae, were obtained from the Nuffield Department of Obstetrics and Gynaecology, John Radcliffe Hospital University of Oxford, United Kingdom, as previously described (31).
Endothelial and stromal cells were isolated from first trimester decidual biopsy specimens, as previously described, with minor modifications (32, 33). Briefly, decidual tissues were digested with 0.25% trypsin (Sigma-Aldrich) and 50 μg/mL DNase I (Roche, Milan, Italy) in PBS, overnight at 4°C, and then treated with 3 mg/mL collagenase type I (Worthington Biochemical, DBA, Milano, Italy), for 30 min at 37°C. Following Ficoll-Paque Plus density gradient (GE Healthcare, Euroclone, Milan, Italy) centrifugation, decidual human endothelial cells (DECs) were isolated by positive selection using Dynabeads M-450 (Dynal, Oslo, Norway) coated with lectin Ulex europaeus 1 (Sigma-Aldrich). DECs were cultured in a flask pre-coated with 5 μg/cm2 fibronectin (Roche) using Human Endothelial Serum Free Medium (HESFM; Gibco, Life Technologies, Milan, Italy), supplemented with 20 ng/mL basic Fibroblast Growth Factor (bFGF; Immunological Sciences), 10 ng/mL Epidermal Growth Factor (EGF; Immunological Sciences), and penicillin (50 U/mL)/streptomycin (50 μg/mL) (PS; Sigma-Aldrich).
Decidual stromal cells (DSCs) were obtained by culturing the endothelial-negative cell fraction in RPMI plus 10% fetal bovine serum (FBS), without growth factors. Non-adherent cells were removed via extensive washing; adherent cells were used only when the resulting cell population was negative for CD14, CD45, Cytokeratin 8/18, von Willebrand Factor (vWF), or CD31. The purity of DECs and DSCs isolated from first trimester decidua was routinely assessed by cytofluorimetric and qPCR analysis, as previously reported (34).
Extravillous trophoblasts (EVTs) were purified from placental tissues incubated with Hanks’ Balanced Salt Solution (HBSS) containing 0.25% trypsin and 0.2 mg/mL DNase I (Roche) for 20 min at 37°C, following the published procedure (35). Cells were seeded in 25 cm2-flask pre-coated with 5 μg/cm2 fibronectin (Roche) in RPMI (Gibco, Life Technologies) supplemented with 10% FBS to remove non-adherent leucocytes and syncytiotrophoblasts, and used within 12 h. The purity of EVTs isolated from first trimester placenta was routinely assessed by immunofluorescence and qPCR analysis, as previously reported (36).
Human umbilical vein endothelial cells (HUVECs) were isolated from umbilical cords of healthy placentae, following a published protocol (37), and cultured in HESFM supplemented with 20 ng/mL EGF, 10 ng/mL bFGF, 1% PS, and 10% FBS. Immortalized human hepatocytes (IHH, kindly provided by Dr. Cristina Bellarosa, Italian Liver Foundation, Trieste, Italy) were cultured in Dulbecco’s modified Eagle medium (Gibco), supplemented with non-essential amino acids, 2mM L-glutamine (Gibco). 100 μg/mL of penicillin/streptomycin (Gibco), and 10% FBS at 37°C and 5% CO2.
Term placental tissues from PE and healthy women were fixed in 10% v/v buffered formalin, paraffin-embedded and stored at 4°C. Tissue sections of 3-4 µm were deparaffinized with xylene and rehydrated with decreasing concentrations of ethanol (100%, 95%, 70%) and dH2O. The antigen unmasking technique was performed using citrate buffer (pH 6), for 20 min at 98°C in thermostatic bath. After neutralization of the endogenous peroxidase activity with 3% v/v H2O2 for 5 min, sections were incubated with PBS + 2% bovine serum albumin (BSA) for 30 min to block non-specific binding. Next, rabbit anti-human FH polyclonal antibody (1:100, #PA5-83957, Invitrogen) was incubated at 4°C. Staining was revealed via anti-rabbit horseradish peroxidase (HRP)-conjugate (1:500, Sigma), incubated for 30 min at room temperature, and 3-amino-9-ethylcarbazole (AEC, Vector Laboratories). Sections were counterstained with Mayer Haematoxylin (DiaPath, Italy) and examined under a Leica DM 2000 optical microscope. Kidney was stained as positive control tissue. Images were collected using a Leica DFC 7000 T digital camera (Leica Microsystems, Wetzlar, Germany). To quantify the staining, we utilized an immunoreactive score (38), which is commonly used for immunohistochemical evaluation. For each slide, we blindly analyzed three different visual fields of the microscope, attributing the following scores: 0 = no color reaction, 1 = mild reaction, 2 = moderate reaction, 3 = intense reaction.
Total RNA was extracted from placental tissues of PE and healthy (CTRL) women as well as from isolated placental cells. The isolation of RNA from tissue samples was carried out using Phenol-Chloroform extraction method. Placental cells (EVTs, DECs, and DSCs) and IHH were stimulated for 24 h with 10% sera of PE or CTRL women (5 pooled sera), 100 ng/mL TNF-α, 10 ng/mL IL-1β or 50 μg/mL STBMs derived from PE or CTRL women. After incubation, cells were lysed and the total RNA was isolated. For each placental cell type (EVTs, DECs or DSCs), the experiments were repeated using at least four different cell populations. Total RNA was isolated using an RNA purification kit (Norgen Biotek Company, Thorold, Canada). RNA quantification was performed via NanoDrop® spectrometer (NanoDrop). RNA was converted to cDNA using SensiFAST™ cDNA Synthesis Kit (Meridian Bioscience, Memphis, TN, USA).
For Real-Time quantitative PCR (RT-qPCR), SYBR Select Master Mix (Applied Biosystems, ThermoFisher) was used. The reaction was performed by the Rotor-Gene 6000 (Corbett, Explera), via 45 cycles of denaturation (10 sec at 95°C), and annealing (45 sec at 60°C, melting temperature of the primers). Following primers were used: CFH (forward 5’- ACC GTG AAT GTG ACA CAG ATG G-3’; reverse 5’- ATT CCC GAT CTG GTT CCA TTG C-3’), GAPDH (forward 5’-GAT CAT CAG CAA TGC CTC CT-3’; reverse 5’- GT GGT CAT GAG TCC TTC CA-3’). The mRNA expression level of FH was evaluated with a comparative quantification provided with Rotor Gene 1.7 Software (Corbett Research) and normalized with the expression of a housekeeping gene (GAPDH).
Placental STBMs, isolated from CTRL (n = 4) or PE patients (n = 4), were lysed. After boiling, 20 µg of STBMs, diluted in Laemmli buffer, were subjected to a 10% v/v SDS-PAGE under reducing conditions. Proteins were transferred to a nitrocellulose membrane using the wet system of Mini Blot Module (Thermo Fisher Scientific), at a constant voltage for 60 min. An hour of blocking step with 5% skimmed milk in Tris-Buffered Saline + Tween 20 (TBST; 10 mM Tris, pH 8.0, 150 mM NaCl, 0.5% Tween 20) was followed by an overnight incubation at 4°C with primary antibodies: anti- FH (1:1000; #PA5-83957, Invitrogen) and anti-actin (1:1000; sc-8432, Santa Cruz). The following day, the membrane was incubated with anti-rabbit LI-COR IRDye 800CW and anti-mouse LI-COR IRDye 680CW (1:10,000; LI-COR Biosciences, Lincoln, NE, USA), for 1 h at room temperature. The fluorescence intensity was acquired in the Odyssey® CLx near-infrared scanner (LI‐COR Biosciences, Lincoln, NE, USA) and normalized for anti-actin. Image acquisition, processing and data analysis were performed via Image Studio Ver 5.2 (LI-COR Biosciences).
Human Complement FH DuoSet ELISA kit (#DY4779; R&D Systems, Inc., Minneapolis, Canada) was used to quantify FH in serum samples, following the manufacturer's instructions. The plate was read by the PowerWave X Microplate Reader (Bio-Tek Instruments) spectrophotometer at 450 nm.
Data obtained from in vitro cell stimulation assays were analyzed by unpaired non-parametric test (Wilcoxon) comparing resting and treated conditions, while data obtained from patient samples were analyzed by unpaired t-test comparing PE vs controls healthy. STBM analysis was statistically evaluated using Mann-Whitney test. Results were expressed as mean ± standard deviations. p-values (p) < 0.05 were considered statistically significant. All statistical analyses were performed using GraphPad Prism software 9.0 (GraphPad Software Inc., La Jolla, CA, USA).
To investigate the physiological role of FH in placenta, we first assessed the basal expression of FH in placental tissues and cells. We analyzed FH tissue distribution by immunohistochemistry (IHC) using first trimester and term placental sections with rabbit anti-human FH antibodies. As shown in Figures 1A, B, the positivity for FH staining in first trimester placentae was particularly intense in decidual spiral arteries and in the syncytiotrophoblast monolayer, whereas in third trimester placenta, FH was present in the inner of villi vessels and at the level of intervillous spaces (Figures 1C, D). In order to confirm the local synthesis of FH, we performed RT-qPCR using placental tissue mRNA isolated from first and third trimester normal placenta. In placental tissues, FH expression was observed both in the first (n = 4) as well as third (n = 4) trimester samples derived from healthy pregnancies, where both the decidua and the fetal villi expressed FH (Figure 1E). The expression levels were widely variable among samples. No significant difference could be observed between first and third trimester.
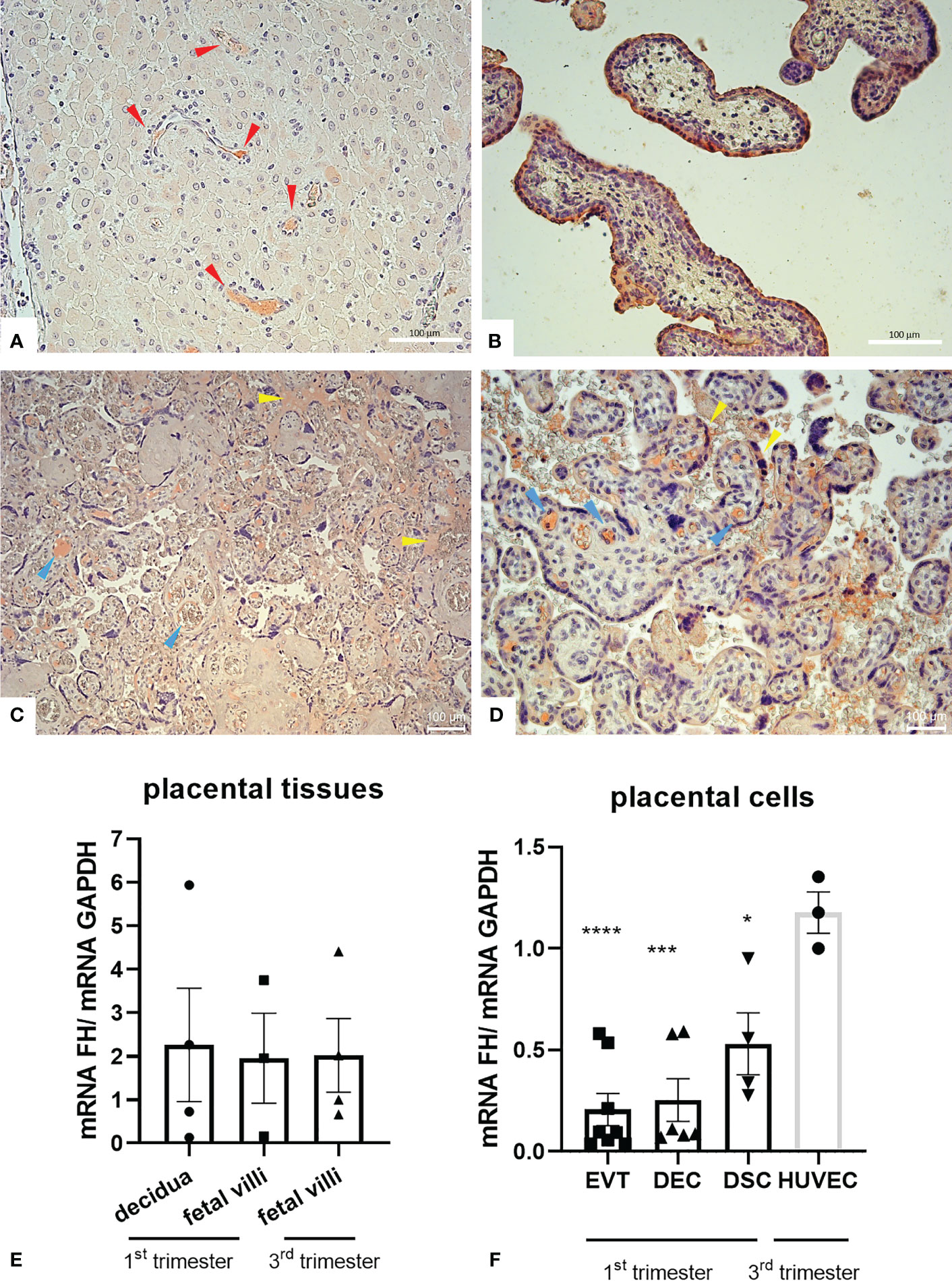
Figure 1 (A–D) Immunohistochemical analysis for the distribution of Factor H (FH) in placental tissues. First trimester [(A, B) n = 3] and term human placental [(C, D) n = 8] sections were stained with anti-FH polyclonal antibody. Staining was detected via 3-amino-9-ethylcarbazole (AEC) substrate chromogen. Nuclei were stained with Mayer’s Hematoxylin. The positivity for FH staining in first trimester placentae was particularly intense in decidual vessels (red arrows) and in the syncytiotrophoblast monolayer, whereas in villi vessels (yellow arrows) and in intervillous spaces and syncythium surface (blue arrows) for term placentae. Magnification, (A, B, D) 200x, (C) 200x. Scale bar, 100 µm. (E, F) mRNA expression levels of FH were measured by RT-qPCR in placental tissues (E) and placental cells (F) isolated from normal pregnancies. FH was expressed in all placental tissues (n = 4 first trimester decidua samples, n = 3 first trimester villous samples and n = 4 term villous samples) and placental cells examined, i.e., extravillous trophoblast (EVT, n = 8), decidual endothelial cells (DECs, n = 6), and decidual stromal cells (DSCs, n = 4). FH gene expression was analyzed using HUVECs as a calibrator, since they showed the highest FH expression. GAPDH was used as a housekeeping gene to normalize gene expression results. Data are expressed as mean ± SD of two independent experiments performed in triplicate. *p < 0.05, ***p < 0.001, ****p < 0.0001, as compared to HUVECs (Mann-Whitney test).
To identify cells that were responsible for the local production of FH, we performed RT-qPCR on isolated first trimester placental cells (EVTs, DECs, and DSCs). FH gene expression was analyzed using HUVECs as a calibrator, since they showed the highest FH mRNA expression. FH was expressed at similar levels by all the three placental cell investigated (DSCs, DECs, and EVTs) (Figure 1F).
To better elucidate the role of FH in pregnancy, we first investigated its expression levels in a pathological condition that affects placental development, such as PE. Thus, we compared the local distribution of FH in CTRL and PE placental tissues by IHC. Placentae from CTRL women (n = 8) had higher FH expression than PE pregnancies (n = 7) (Figure 2, Supplementary Figure 2). FH was almost undetectable in PE placentae (Figures 2C, D), making it difficult to define its possible localization, even though villi vessels were slightly positive; FH in CTRL placentae was mainly localized in the fetal villous endothelium (blue arrows) and intervillous spaces (red arrows) (Figures 2A, B). The IHC quantitation demonstrated a significantly lower level of FH in PE placentae (Figure 2E). Our results were partially confirmed by RT-qPCR analysis of placental tissue. Although no statistical significance was reached, the results showed lower FH gene expression in PE placental tissue compared to CTRL placenta (Figure 2F). When dividing patients into late-onset (LO) and early-onset EOPE, we found that EOPE patients had the lowest FH levels (Figure 2F), suggesting the involvement of FH in PE-associated pathologic mechanisms.
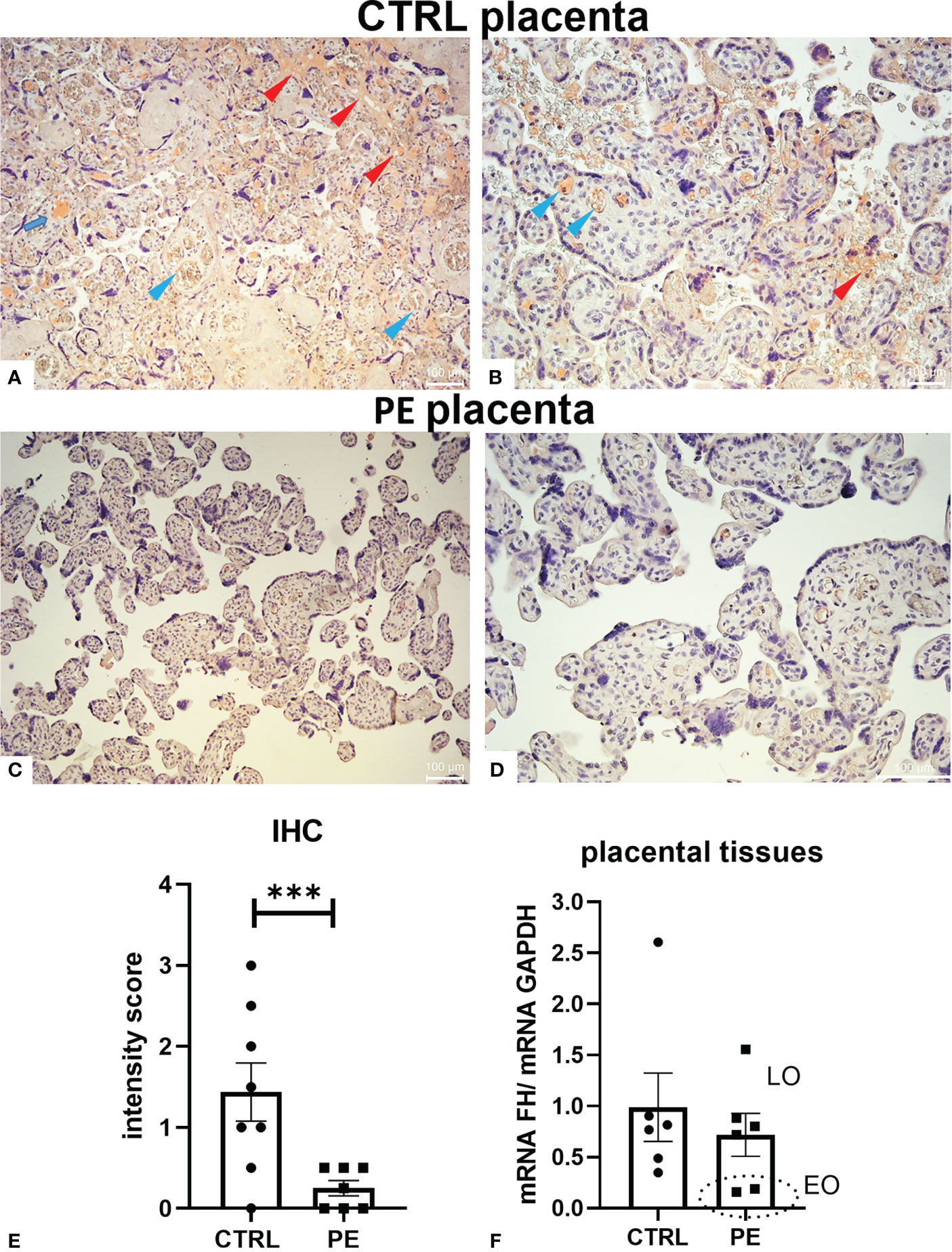
Figure 2 Localization of Factor H (FH) in placental tissues from normal (CTRL) and preeclamptic (PE) tissue. (A–D) The samples representative of CTRL [(A) original magnification 10 X; (B) original magnification 20X] or PE [(C) original magnification 10X; (D) original magnification 20 X] pregnancies were stained with anti-FH antibody. FH staining was detected in all the normal placental tissues, being mainly localized in fetal villi endothelium (blue arrows) and intervillous spaces (red arrows), whereas it was almost undetectable in PE placentae (C, D). Staining was detected via 3-amino-9-ethylcarbazole (AEC) substrate chromogen. Nuclei were stained with Mayer’s Hematoxylin. Scale bars, 100 µm. (E) Quantitation of IHC staining using an immunoreactive score (PE n = 7, CTRL n = 8). For each slide, three different visual fields of the microscope, were analyzed giving a score as described in Materials and Methods section. ***p < 0.001 (t-test). (F) FH mRNA expression levels in term placental tissues of CTRL (n = 6) and PE [further divided into early onset, (EO, n = 2), and late onset, (LO, n = 4)] pregnancies were assessed by RT-qPCR. FH expression was lower in PE placentae compared to CTRL placentae; interestingly, EOPE samples exhibited minimal expression (marked with a circle). GAPDH was used as a housekeeping gene to normalize gene expression levels.
Based on previous observations, we hypothesized that microenvironmental factors, including pro-inflammatory cytokines present in the PE placentae, could be responsible for the decreased FH expression by placental cells. Thus, DECs and DSCs isolated from first trimester placentae were stimulated with a pool of 5 different CTRL or PE sera, and analyzed for FH mRNA expression levels. As shown in Figures 3A, B, FH expression was significantly reduced in DECs and DSCs following incubation with PE sera compared to cells incubated with matched CTRL sera. We also investigated the ability of PE sera to modulate FH expression using an immortalized hepatocyte cell line (IHH), but we did not observe any significant modulation of FH transcript levels (Figure 3C).
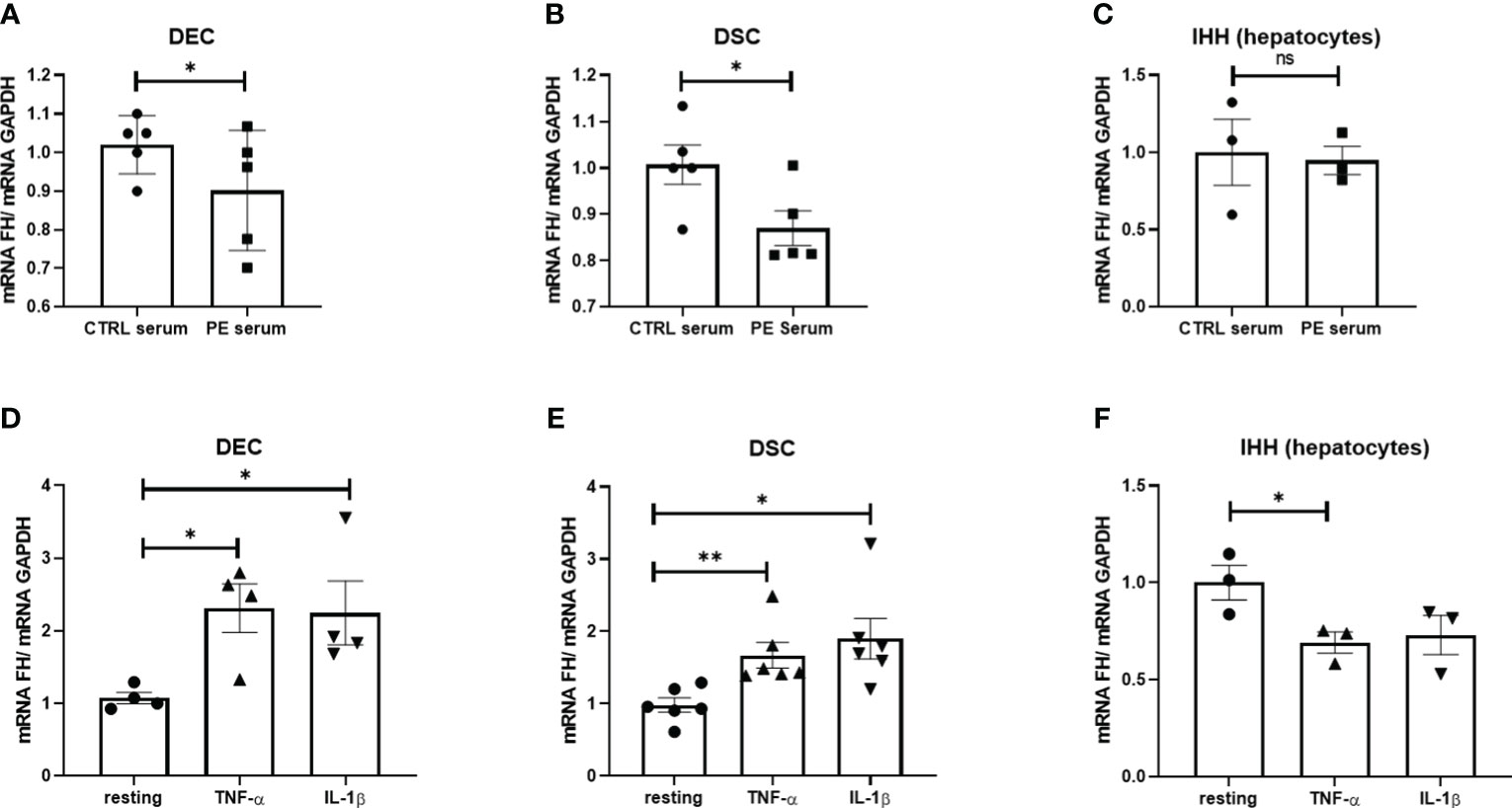
Figure 3 (A–C) Levels of Factor H (FH) mRNA expression in DECs (A), DSCs (B) and IHH (D) following in vitro stimulation with serum from normal (CTRL) and PE women for 24 h. Pooled PE (n = 5) or CTRL (n = 5) serum samples were used. (D–F) Levels of FH mRNA expression in DECs (D), DSCs (E) or IHH (F) stimulated with TNF-α or IL-1β for 24h. FH mRNA levels were significantly increased in DECs and DSCs after stimulation with TNF-α and IL-1β, whereas in IHH, FH transcripts were down-regulated with only TNF-α. Data are expressed as mean ± SD of two independent experiments performed in triplicate. *p < 0.05, **p < 0.01, as compared to resting (Mann-Whitney test). ns, non significant.
To determine which humoral factors in PE sera might be responsible for modulating FH expression, placental cells and IHH were stimulated with key pro-inflammatory cytokines known to be elevated in PE sera, such as TNF-α (39) and IL-1β (40). Surprisingly, none of these cytokines were able to induce a decrease in FH expression by the placental cells, but rather we observed their upregulation (Figures 3D, E). In contrast, IHH showed a significant downregulation of FH expression, especially after stimulation with TNF-α (Figure 3F).
Out of a range of immunomodulatory properties exerted by the placenta, the shedding of STBMs may contribute significantly to pregnancy success (41, 42). Interestingly, STBM levels are elevated in women with PE, possibly contributing to vascular endothelial dysfunction (43). Therefore, DECs were incubated for 24 h with STBMs isolated from the placenta of PE and CTRL women. Consistent with the earlier study, STBMs isolated from PE placentae were able to downregulate FH expression (Supplementary Figure 3); however, we were unable to obtain a statistically significant result.
To examine whether the observations gathered at the placental level might also reflect at the systemic level, circulating FH levels were measured by ELISA in the sera of PE women collected at the time of diagnosis and in the sera of matched CTRL women.
In agreement with the results we obtained using placenta, we found lower circulating FH levels in the sera of PE women compared to CTRL (healthy) women (Figure 4). Therefore, we investigated whether the differences in FH levels between PE patients and CTRL women could be used to define FH as an early marker for PE. Thus, we quantified FH in serum samples collected between 11 and 13 weeks of gestation from pregnant women who later developed PE and in sera of matched CTRL women. Compared to PE, FH concentrations were significantly higher in the sera of CTRL women during the third trimester of pregnancy (Figure 4A, CTRL: 556.95 µg/mL ± SD 191.52 vs PE: 324.21 µg/mL ± SD 131.04), and, to a lesser extent, in the first trimester (Figure 4B CTRL: 624.06 µg/mL ± SD 273.05 vs PE: 450.97 µg/mL ± SD 226.16). Since a significant difference in FH levels was observed between PE and CTRL sera, we hypothesized that FH may bind to apoptotic bodies of the placenta. Therefore, we investigated the presence of FH deposition on STBMs, a method previously described by Tannetta et al. (44). We performed Western blot analysis using solubilized STBMs from PE (n = 4) and CTRL placentas (n = 4), and measured FH protein expression levels. As shown in Figures 4C, D, FH was present in STBMs from both PE placenta and CTRL, with significantly higher levels in PE.
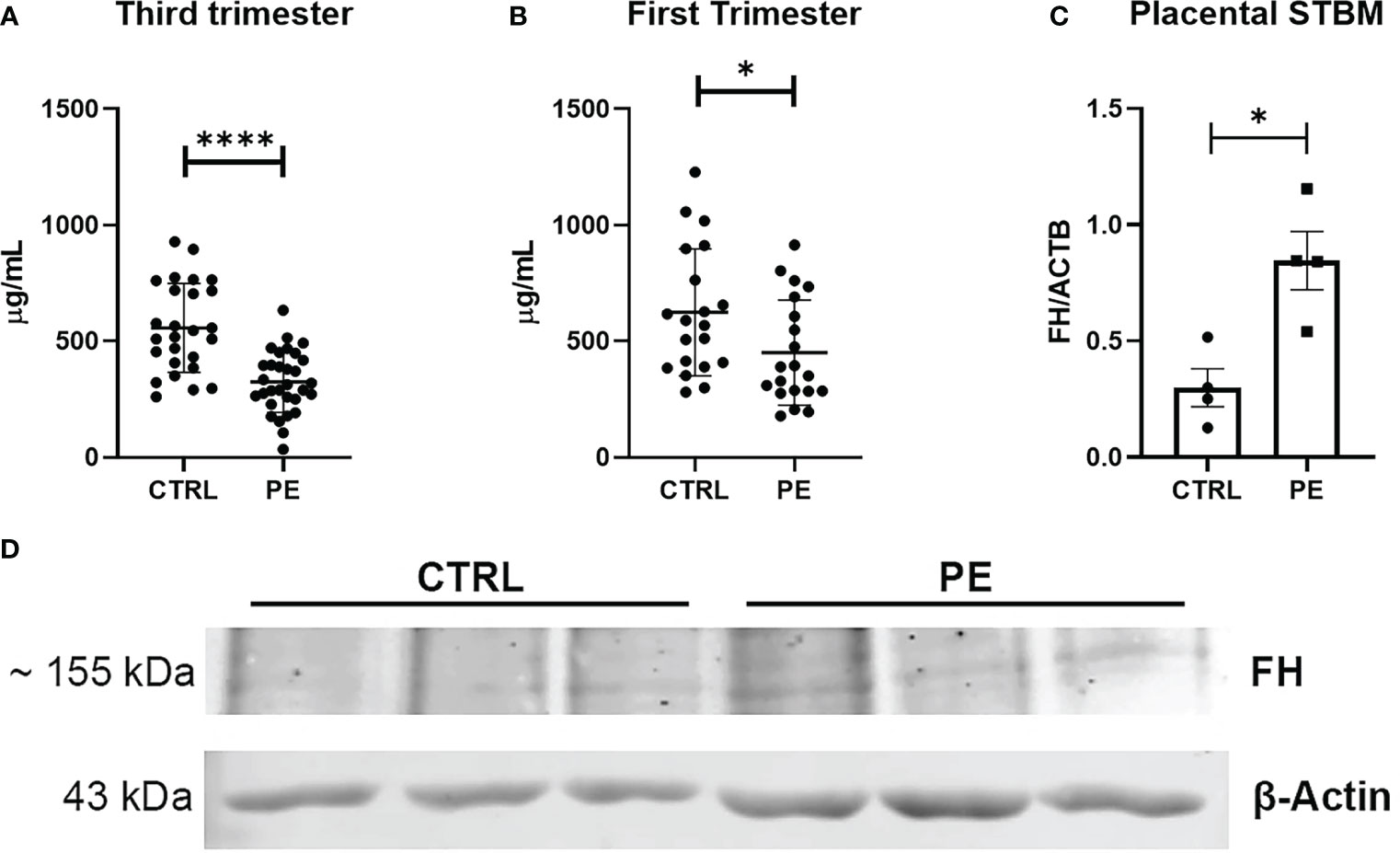
Figure 4 (A, B) Measurement of circulating Factor H (FH) levels in sera of normal (CTRL) and preeclamptic (PE) women, collected during third (A) or first (B) trimester, by ELISA. During both the first trimester and the third trimester lower levels of FH were observed in PE pregnancies compared to CTRL, showing a highly significant difference (n = 26 PE vs n =31 CTRL sera were compared for third trimester of pregnancy; n = 20 PE vs n = 20 CTRL for first trimester of pregnancy). *p < 0.05, ****p < 0.0001 (t-test). (C, D) Evaluation of FH deposition on placental STBMs via Western blot. STBMs derived from PE (n = 4) and CTRL (n = 4) were solubilized and separated by SDS-PAGE under reducing conditions. After transfer, the membrane was probed with anti-FH antibody and IRDye 800CW secondary antibody. Signal intensity was detected using an Odyssey CLx near-infrared scanner (LI-COR Biosciences, Lincoln, NE, USA). Image acquisition, processing and data analysis were performed with Image Studio 5.2 (LI-COR Biosciences). Histograms represent the mean ± SD of two independent experiments performed in duplicate. Beta-actin (ACTB) was used to normalize the results. *p < 0.05 (Mann-Whitney test).
Pregnancy is a complex biological process in which the maternal immune system undergoes several changes that are necessary for the protection of the mother and the fetus from pathogenic challenges and for a successful delivery of the fetus (4). The complement system is critical for a successful pregnancy (1, 2), thus dysregulation along the complement cascade may play a major role in pregnancy-associated complications, including PE. In particular, complement regulators impose strict control on complement activation. FH, a negative regulator of complement alternative pathway, is a very multifaceted molecule which is able to bind a wide range of target ligands and fulfil different functions.
Aiming to investigate the role of FH in pregnancy, we first explored its expression pattern during different trimesters of the pregnancy. Our results confirmed a local production of FH at the feto-maternal interface, both in the decidua and in the villi, but showed no differences in FH transcript levels between the first and third trimester of pregnancy. IHC staining pattern was very different between the first trimester and term placenta: in first-trimester double-layered syncytium, FH staining was widespread; conversely, in term placenta monolayered villi, FH was present in maternal intervillous space and into the fetal villous vessels. Placental tissue may present intravillous fibrin deposits, which are expected to stain for FH (45). This is likely to represent the maternal FH, while the expected endothelial staining of villous vessels is the fetal FH; thus, the two display the expression of FH molecules of different origins.
To determine which placental cells were responsible for the local production of FH, we analyzed the cells isolated from first trimester placental tissue by RT-qPCR. All the placental cell types (DSCs, DECs, and EVTs) expressed FH to a similar degree, although the highest FH expression was observed in HUVECs, which we used as a positive control based on an earlier study by Rayes et al. (46).
Compared to PE, placentae from CTRL women had higher FH staining, which allowed us to conclude that FH was mainly localized in the fetal villous endothelium and intervillous spaces. Conversely, FH in PE placenta was almost undetectable. The presence of FH in healthy placenta has been previously reported with a significantly variable staining pattern, being uniformly localized in the syncytiotrophoblast layer in 70% of the samples, but not in the villous vessels (45). However, in agreement with our results, most of PE placentae remained negative for FH staining (45). Lokki and Heikkinen-Eloranta demonstrated intracellular localization of FH in the placental syncytiotrophoblast of PE placenta, and suggested that FH may exacerbate thrombotic microangiopathy in PE independent of complement activation (47). However, the existence of an association between FH and PE should be further confirmed by analyzing FH localization and synthesis via IHC and RT-qPCR using a greater number of cases, via an accurate clustering of the patients based on severity and pathogenesis of the disease. The results of Belmonte et al. demonstrated the involvement of the lectin pathway in complement deposition in PE placenta (48). This evidence is in accordance with our results that failed to detect FH deposition in PE placentae. However, we cannot exclude that FH staining was undetectable due to the damage and the dysmorphia typical of the PE villi.
Under physiological conditions, plasma FH levels can vary between 116 and 562 μg/mL depending on genetic and environmental factors, but they may increase in pregnant women (18). There are several reports about an increase in the circulating FH levels during pregnancy (7, 14, 49). Our study also suggests that soluble FH levels remain high during pregnancy both in the first and the third trimester. Interestingly, we observed a decrease in circulating FH levels when comparing PE with healthy pregnancy, particularly in the third trimester. Our results also highlighted a statistically significant difference between PE and healthy controls from the prospective study cohort. In the first trimester of pregnancy, patients who subsequently (after the 20th week of gestation) developed PE had lower FH levels, indicating FH as a potential predictive marker of PE, although further studies are needed. Previous studies have already reported lower circulating levels of FH in PE patients (29, 50), in manner independent of the levels of anti-FH autoantibodies (50). In a prospective study, serum levels of FH were shown to increase in the middle and late stages of pregnancy, whereas FH was reduced in both EOPE and LOPE compared to control groups (29). However, these differences were not observed in the second and third trimesters of pregnancy. Another study pointed out that FH concentrations in the serum of PE patients were higher during the first trimester than in a normal pregnancy; subsequently, these values were similar to those in a normal pregnancy (7). Ari et al. found that serum C3 and FH levels in patients with HELLP (Hemolysis, Elevated Liver enzymes and Low Platelets) syndrome were not significantly different from those of PE patients and healthy pregnant women (51).
Insufficient levels of FH can be attributed to decreased FH expression due to mutations. Five rare variants (L3V, R127H, R166Q, C1077S, and N1176K) in the FH gene have been found to predispose to the occurrence of severe PE (52). Moreover, FH reduction can also be due to the presence of anti-FH autoantibodies, consumption of FH, or by the fact that FH is overwhelmed by a massive disease burden (53).
The observation of both local and systemic lower FH levels in PE led us to consider that microenvironmental factors may be responsible for reduced FH production by placental cells. After stimulation with pooled PE or control sera, we thus analyzed FH mRNA expression in DECs and DSCs. Our results showed a significantly reduced FH expression in cells stimulated with PE sera compared to control sera. The downregulation of FH gene may be explained by the particular conditions of PE, where the placenta is characterized by increased oxidative stress and complement activation. Thus, upregulation of the FH gene probably protects the cells by inhibiting complement activation under physiological conditions, while its downregulation can potentially lead to adverse pregnancy outcomes.
To identify the factors present in PE sera that might modulate FH expression, cells were incubated with pro-inflammatory cytokines known to be increased in PE women’s blood, such as TNF-α and IL-1β (39, 54–59). Previously, Daha’s group demonstrated that stimulation of HUVECs with IL-1 led to a decrease in FH expression (60). Unexpectedly, FH mRNA expression was increased in DECs and DSCs after stimulation with pro-inflammatory cytokines. Due to the multifactorial nature of PE (61), the in vitro stimulation with these two cytokines may likely be insufficient to mimic a PE-like scenario in placental cells. Moreover, the results obtained with IHH suggested that the selected amount of TNF-α (100 ng/mL) and IL-1β (10 ng/mL) was higher compared to their levels in PE sera. We can hypothesize differential levels of cytokine sensitivity between placental cells and hepatocytes. Since the potent pro-inflammatory properties of TNF-α and IL-1β can trigger excessive inflammation, and directly or indirectly, activate the complement system, placental cells upregulate FH expression possibly to control complement activation.
STBMs, released into the maternal circulation to ensure feto-maternal communication, are significantly increased in PE and can contribute to the local pro-inflammatory properties by carrying a variety of placental proteins into the maternal circulation (62). Tanetta et al. found 538 unique proteins in STBMs of PE, including several complement proteins and regulators (e.g., C1q, C3, CD55, CD59, and vitronectin) (63). In another study, Baig et al. identified 25 differentially expressed proteins in PE, with CD59 being downregulated (64). Our results showed that STBMs, isolated from PE placentae, downregulated FH gene expression in DECs, suggesting that binding of STBMs to DECs may have an inhibitory effect on FH production. Surprisingly, we also found higher levels of FH in STBMs isolated from PE samples than in control samples. Despite this high proportion of FH in STBMs, complement activation is dysregulated/poorly controlled in PE. One possibility is that FH expressed by/on the placental cells is sequestered into STBMs, which may act as decoy substrates. A previous study in tumour models demonstrated that FH can successfully be carried by extracellular vesicles (65). Thus, FH may not be available to the cells for complement regulation, or alternatively, its ability to inhibit complement activation may be masked. In fact, it is widely recognized that FH must be in soluble form to be biologically effective, which is not the case in PE, consistent with the lower circulating FH levels observed in PE mothers when analyzing serum, tissues, and isolated cells. Therefore, it is important to investigate the immunomodulatory properties of FH-loaded STBMs in PE immunopathology.
Under physiological conditions, FH is locally expressed at the feto-maternal interface in all trimesters of pregnancy. However, it can be localized in syncytiotrophoblasts in the first trimester and in villous endothelium in the third trimester placenta. Decidual cells (DECs, EVTs, and DSCs) appear to synthesize FH. PE placentae are almost negative for FH staining. Circulating levels of FH are significantly lower in the PE sera compared to controls. The causes for this reduction could be multiple (e.g., mutations, consumption, less synthesis). PE sera contain humoral factors that can cause downregulation of FH expression by placental cells, possibly due to the presence of STBMs highly loaded with FH in PE. Further studies are required to understand the mechanisms linking FH and PE onset.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The study was reviewed and approved by the Regional Ethical Committee of FVG (CEUR-2020-Os-156; Prot. 0022668/P/GEN/ARCS and CEUR-2019-Sper-17) and adhered to the principles of the Declaration of Helsinki. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
HY: Data curation, Formal analysis, Investigation, Methodology, Project administration, Writing – original draft. CA: Formal analysis, Investigation, Methodology, Validation, Writing – original draft. MT: Investigation, Methodology, Software, Writing – review & editing. TR: Investigation, Methodology, Validation, Writing – review & editing. SP: Data curation, Investigation, Software, Writing – review & editing. AB: Data curation, Methodology, Software, Writing – review & editing. GZ: Formal analysis, Methodology, Software, Writing – review & editing. ND: Data curation, Investigation, Methodology, Writing – review & editing. GR: Formal analysis, Investigation, Methodology, Writing – review & editing. TM: Conceptualization, Investigation, Writing – review & editing. UK: Conceptualization, Formal analysis, Resources, Supervision, Writing – review & editing. RB: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Ministry of Health, Rome -Italy, in collaboration with the Institute for Maternal and Child Health IRCCS Burlo Garofolo, Trieste -Italy (RC24/19 to GR, RC09/21, RC01/23 to CA, and RC03/23 to AB), DST-SERB-TARE project, Government of India (Sanction No. TAR/2022/000626), to HY and UAEU Start Up grant #12F043 and UPAR grant #12F061 to U.K.
We thank Prof. Ian Sargent and his group of the Nuffield Department of Obstetrics and Gynaecology, University of Oxford (John Radcliffe Hospital, Oxford, UK) for providing STBM samples, and Dr. Cristina Bellarosa (Liver Foundation, Trieste, Italy) for providing IHH cell line.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1351898/full#supplementary-material
Supplementary Figure 1 | Graphical description of the multiple cohorts of pregnant women at different stages of pregnancy.
Supplementary Figure 2 | IHC analysis of FH in placental tissues from three normal (CTRL) and three preeclamptic (PE) tissues. Original magnification 10X. FH staining was detected in all the normal placental tissues (left panels), whereas it was almost undetectable in PE placentae (right panels). Staining was detected via 3-amino-9-ethylcarbazole (AEC) substrate chromogen. Nuclei were stained with Mayer’s Hematoxylin. Scale bars, 100 µm.
Supplementary Figure 3 | Levels of FH mRNA expression in DECs stimulated for 24h with 50 μg/mL STBMs of PE or CTRL placentae. Data are expressed as mean ± SD of two independent experiments performed in triplicate.
1. Bulla R, Bossi F, Tedesco F. The complement system at the embryo implantation site: friend or foe? Front Immunol (2012) 3:55. doi: 10.3389/fimmu.2012.00055
2. Madhukaran SP, Alhamlan FS, Kale K, Vatish M, Madan T, Kishore U. Role of collectins and complement protein C1q in pregnancy and parturition. Immunobiology (2016) 221(11):1273–88. doi: 10.1016/j.imbio.2016.06.002
3. Girardi G, Lingo JJ, Fleming SD, Regal JF. Essential role of complement in pregnancy: from implantation to parturition and beyond. Front Immunol (2020) 11:1681. doi: 10.3389/fimmu.2020.01681
4. Mor G, Cardenas I. The immune system in pregnancy: a unique complexity. Am J Reprod Immunol (2010) 63(6):425–33. doi: 10.1111/j.1600-0897.2010.00836.x
5. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol (2015) 6:262. doi: 10.3389/fimmu.2015.00262
6. Buurma A, Cohen D, Veraar K, Schonkeren D, Claas FH, Bruijn JA, et al. Preeclampsia is characterized by placental complement dysregulation. Hypertension (2012) 60(5):1332–7. doi: 10.1161/HYPERTENSIONAHA.112.194324
7. He YD, Xu BN, Wang ML, Wang YQ, Yu F, Chen Q, et al. Dysregulation of complement system during pregnancy in patients with preeclampsia: A prospective study. Mol Immunol (2020) 122:69–79. doi: 10.1016/j.molimm.2020.03.021
8. Regal JF, Gilbert JS, Burwick RM. The complement system and adverse pregnancy outcomes. Mol Immunol (2015) 67(1):56–70. doi: 10.1016/j.molimm.2015.02.030
9. Teirilä L, Heikkinen-Eloranta J, Kotimaa J, Meri S, Lokki AI. Regulation of the complement system and immunological tolerance in pregnancy. Semin Immunol (2019) 45:101337. doi: 10.1016/j.smim.2019.101337
10. Laresgoiti-Servitje E. A leading role for the immune system in the pathophysiology of preeclampsia. J Leukoc Biol (2013) 94(2):247–57. doi: 10.1189/jlb.1112603
11. Jung E, Romero R, Yeo L, Gomez-Lopez N, Chaemsaithong P, Jaovisidha A, et al. The etiology of preeclampsia. Am J Obstet Gynecol (2022) 226(2S):S844–66. doi: 10.1016/j.ajog.2021.11.1356
12. Magee LA, Nicolaides KH, von Dadelszen P. Preeclampsia. N Engl J Med (2022) 386(19):1817–32. doi: 10.1056/NEJMra2109523
13. Raguema N, Moustadraf S, Bertagnolli M. Immune and apoptosis mechanisms regulating placental development and vascularization in preeclampsia. Front Physiol (2020) 11:98. doi: 10.3389/fphys.2020.00098
14. Derzsy Z, Prohászka Z, Rigó J, Füst G, Molvarec A. Activation of the complement system in normal pregnancy and preeclampsia. Mol Immunol (2010) 47(7-8):1500–6. doi: 10.1016/j.molimm.2010.01.021
15. Sim RB, DiScipio RG. Purification and structural studies on the complement-system control protein beta 1H (Factor H). Biochem J (1982) 205(2):285–93. doi: 10.1042/bj2050285
16. Rodriguez de Cordoba S, Lublin DM, Rubinstein P, Atkinson JP. Human genes for three complement components that regulate the activation of C3 are tightly linked. J Exp Med (1985) 161(5):1189–95. doi: 10.1084/jem.161.5.1189
17. Rodriguez de Cordoba S, Rubinstein P. Quantitative variations of the C3b/C4b receptor (CR1) in human erythrocytes are controlled by genes within the regulator of complement activation (RCA) gene cluster. J Exp Med (1986) 164(4):1274–83. doi: 10.1084/jem.164.4.1274
18. Esparza-Gordillo J, Soria JM, Buil A, Almasy L, Blangero J, Fontcuberta J, et al. Genetic and environmental factors influencing the human factor H plasma levels. Immunogenetics (2004) 56(2):77–82. doi: 10.1007/s00251-004-0660-7
19. Whaley K, Ruddy S. Modulation of the alternative complement pathways by beta 1 H globulin. J Exp Med (1976) 144(5):1147–63. doi: 10.1084/jem.144.5.1147
20. Weiler JM, Daha MR, Austen KF, Fearon DT. Control of the amplification convertase of complement by the plasma protein beta1H. Proc Natl Acad Sci U.S.A. (1976) 73(9):3268–72. doi: 10.1073/pnas.73.9.3268
21. Morgan HP, Schmidt CQ, Guariento M, Blaum BS, Gillespie D, Herbert AP, et al. Structural basis for engagement by complement factor H of C3b on a self surface. Nat Struct Mol Biol (2011) 18(4):463–70. doi: 10.1038/nsmb.2018
22. Kouser L, Abdul-Aziz M, Nayak A, Stover CM, Sim RB, Kishore U. Properdin and factor h: opposing players on the alternative complement pathway "see-saw". Front Immunol (2013) 4:93. doi: 10.3389/fimmu.2013.00093
23. Ferreira VP, Pangburn MK, Cortés C. Complement control protein factor H: the good, the bad, and the inadequate. Mol Immunol (2010) 47(13):2187–97. doi: 10.1016/j.molimm.2010.05.007
24. Józsi M. Factor H family proteins in complement evasion of microorganisms. Front Immunol (2017) 8:571. doi: 10.3389/fimmu.2017.00571
25. Meri S, Haapasalo K. Function and dysfunction of complement factor H during formation of lipid-rich deposits. Front Immunol (2020) 11:611830. doi: 10.3389/fimmu.2020.611830
26. Parente R, Clark SJ, Inforzato A, Day AJ. Complement factor H in host defense and immune evasion. Cell Mol Life Sci (2017) 74(9):1605–24. doi: 10.1007/s00018-016-2418-4
27. Leffler J, Herbert AP, Norström E, Schmidt CQ, Barlow PN, Blom AM, et al. Annexin-II, DNA, and histones serve as factor H ligands on the surface of apoptotic cells. J Biol Chem (2010) 285(6):3766–76. doi: 10.1074/jbc.M109.045427
28. Ferluga J, Kouser L, Murugaiah V, Sim RB, Kishore U. Potential influences of complement factor H in autoimmune inflammatory and thrombotic disorders. Mol Immunol (2017) 84:84–106. doi: 10.1016/j.molimm.2017.01.015
29. Jia K, Ma L, Wu S, Yang W. Serum levels of complement factors C1q, Bb, and H in normal pregnancy and severe pre-eclampsia. Med Sci Monit (2019) 25:7087–93. doi: 10.12659/MSM.915777
30. Di Lorenzo G, Ceccarello M, Cecotti V, Ronfani L, Monasta L, Vecchi Brumatti L, et al. First trimester maternal serum PIGF, free β-hCG, PAPP-A, PP-13, uterine artery Doppler and maternal history for the prediction of preeclampsia. Placenta (2012) 33(6):495–501. doi: 10.1016/j.placenta.2012.03.003
31. Agostinis C, Stampalija T, Tannetta D, Loganes C, Vecchi Brumatti L, De Seta F, et al. Complement component C1q as potential diagnostic but not predictive marker of preeclampsia. Am J Reprod Immunol (2016) 76(6):475–81. doi: 10.1111/aji.12586
32. Agostinis C, Rami D, Zacchi P, Bossi F, Stampalija T, Mangogna A, et al. Pre-eclampsia affects procalcitonin production in placental tissue. Am J Reprod Immunol (2018) 79(4):e12823. doi: 10.1111/aji.12823
33. Agostinis C, Masat E, Bossi F, Ricci G, Menegazzi R, Lombardelli L, et al. Transcriptomics and immunological analyses reveal a pro-angiogenic and anti-inflammatory phenotype for decidual endothelial cells. Int J Mol Sci (2019) 20(2):1604. doi: 10.3390/ijms20071604
34. Carlino C, Stabile H, Morrone S, Bulla R, Soriani A, Agostinis C, et al. Recruitment of circulating NK cells through decidual tissues: a possible mechanism controlling NK cell accumulation in the uterus during early pregnancy. Blood (2008) 111(6):3108–15. doi: 10.1182/blood-2007-08-105965
35. Bulla R, Agostinis C, Bossi F, Rizzi L, Debeus A, Tripodo C, et al. Decidual endothelial cells express surface-bound C1q as a molecular bridge between endovascular trophoblast and decidual endothelium. Mol Immunol (2008) 45(9):2629–40. doi: 10.1016/j.molimm.2007.12.025
36. Bulla R, Bossi F, Agostinis C, Radillo O, Colombo F, De Seta F, et al. Complement production by trophoblast cells at the feto-maternal interface. J Reprod Immunol (2009) 82(2):119–25. doi: 10.1016/j.jri.2009.06.124
37. Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest (1973) 52(11):2745–56. doi: 10.1172/JCI107470
38. Fedchenko N, Reifenrath J. Different approaches for interpretation and reporting of immunohistochemistry analysis results in the bone tissue - a review. Diagn Pathol (2014) 9:221. doi: 10.1186/s13000-014-0221-9
39. Lau SY, Guild SJ, Barrett CJ, Chen Q, McCowan L, Jordan V, et al. Tumor necrosis factor-alpha, interleukin-6, and interleukin-10 levels are altered in preeclampsia: a systematic review and meta-analysis. Am J Reprod Immunol (2013) 70(5):412–27. doi: 10.1111/aji.12138
40. Kalinderis M, Papanikolaou A, Kalinderi K, Ioannidou E, Giannoulis C, Karagiannis V, et al. Elevated serum levels of interleukin-6, interleukin-1β and human chorionic gonadotropin in pre-eclampsia. Am J Reprod Immunol (2011) 66(6):468–75. doi: 10.1111/j.1600-0897.2011.01019.x
41. Chaouat G. Placental immunoregulatory factors. J Reprod Immunol (1987) 10(3):179–88. doi: 10.1016/0165-0378(87)90085-4
42. Mincheva-Nilsson L, Baranov V. Placenta-derived exosomes and syncytiotrophoblast microparticles and their role in human reproduction: immune modulation for pregnancy success. Am J Reprod Immunol (2014) 72(5):440–57. doi: 10.1111/aji.12311
43. Redman CW, Sargent IL. Immunology of pre-eclampsia. Am J Reprod Immunol (2010) 63(6):534–43. doi: 10.1111/j.1600-0897.2010.00831.x
44. Tannetta DS, Dragovic RA, Gardiner C, Redman CW, Sargent IL. Characterisation of syncytiotrophoblast vesicles in normal pregnancy and pre-eclampsia: expression of Flt-1 and endoglin. PloS One (2013) 8(2):e56754. doi: 10.1371/journal.pone.0056754
45. Lokki AI, Heikkinen-Eloranta J, Jarva H, Saisto T, Lokki ML, Laivuori H, et al. Complement activation and regulation in preeclamptic placenta. Front Immunol (2014) 5:312. doi: 10.3389/fimmu.2014.00312
46. Rayes J, Roumenina LT, Dimitrov JD, Repessé Y, Ing M, Christophe O, et al. The interaction between factor H and VWF increases factor H cofactor activity and regulates VWF prothrombotic status. Blood (2014) 123(1):121–5. doi: 10.1182/blood-2013-04-495853
47. Lokki AI, Heikkinen-Eloranta J. Pregnancy induced TMA in severe preeclampsia results from complement-mediated thromboinflammation. Hum Immunol (2021) 82(5):371–8. doi: 10.1016/j.humimm.2021.03.006
48. Belmonte B, Mangogna A, Gulino A, Cancila V, Morello G, Agostinis C, et al. Distinct roles of classical and lectin pathways of complement in preeclamptic placentae. Front Immunol (2022) 13:882298. doi: 10.3389/fimmu.2022.882298
49. Savaris RF, Becker C, E. e. P. Guedes Neto: Maternal plasma levels of complement Factor H in miscarriage and in normal pregnancy: A cohort study. J Reprod Immunol (2016) 114:1–5. doi: 10.1016/j.jri.2016.01.002
50. Dijkstra DJ, Lokki AI, Gierman LM, Borggreven NV, van der Keur C, Eikmans M, et al. Circulating levels of anti-C1q and anti-factor H autoantibodies and their targets in normal pregnancy and preeclampsia. Front Immunol (2022) 13:842451. doi: 10.3389/fimmu.2022.842451
51. Ari E, Yilmaz Y, Gul A, Alahdab YO, Kedrah AE, Macunluoglu B, et al. Human serum complement C3 and factor H in the syndrome of hemolysis, elevated liver enzymes, and low platelet count. Am J Reprod Immunol (2009) 62(4):238–42. doi: 10.1111/j.1600-0897.2009.00731.x
52. Lokki AI, Ren Z, Triebwasser M, Daly E, Perola M, Auro K, et al. and FINNPEC: Identification of complement factor H variants that predispose to pre-eclampsia: A genetic and functional study. BJOG (2023) 130(12):1473–1482. doi: 10.1111/1471-0528.17529
53. Cortes C, Desler C, Mazzoli A, Chen JY, Ferreira VP. The role of properdin and Factor H in disease. Adv Immunol (2022) 153:1–90. doi: 10.1016/bs.ai.2021.12.001
54. Cemgil Arikan D, Aral M, Coskun A, Ozer A. Plasma IL-4, IL-8, IL-12, interferon-γ and CRP levels in pregnant women with preeclampsia, and their relation with severity of disease and fetal birth weight. J Matern Fetal Neonatal Med (2012) 25(9):1569–73. doi: 10.3109/14767058.2011.648233
55. Spence T, Allsopp PJ, Yeates AJ, Mulhern MS, Strain JJ, McSorley EM. Maternal serum cytokine concentrations in healthy pregnancy and preeclampsia. J Pregnancy (2021) 2021:6649608. doi: 10.1155/2021/6649608
56. Trisnawati E, Nontji W, Nurasni S. Tumour necrosis factor-α (TNF-α) serum levels in preeclampsia pregnant women and pregnant women at risk with preeclampsia. Enfermería Clínica (2020) 30:27–30. doi: 10.1016/j.enfcli.2019.07.021
57. Mosimann B, Wagner M, Poon LC, Bansal AS, Nicolaides KH. Maternal serum cytokines at 30-33 weeks in the prediction of preeclampsia. Prenat Diagn (2013) 33(9):823–30. doi: 10.1002/pd.4129
58. Szarka A, Rigó J, Lázár L, Beko G, Molvarec A. Circulating cytokines, chemokines and adhesion molecules in normal pregnancy and preeclampsia determined by multiplex suspension array. BMC Immunol (2010) 11:59. doi: 10.1186/1471-2172-11-59
59. Sharma D, Singh A, Trivedi SS, Bhattacharjee J. Role of endothelin and inflammatory cytokines in pre-eclampsia - A pilot North Indian study. Am J Reprod Immunol (2011) 65(4):428–32. doi: 10.1111/j.1600-0897.2010.00903.x
60. Brooimans RA, van der Ark AA, Buurman WA, van Es LA, Daha MR. Differential regulation of complement factor H and C3 production in human umbilical vein endothelial cells by IFN-gamma and IL-1. J Immunol (1990) 144(10):3835–40.
61. Valenzuela FJ, Pérez-Sepúlveda A, Torres MJ, Correa P, Repetto GM, Illanes SE. Pathogenesis of preeclampsia: the genetic component. J Pregnancy (2012) 2012:632732. doi: 10.1155/2012/632732
62. Tannetta D, Masliukaite I, Vatish M, Redman C, Sargent I. Update of syncytiotrophoblast derived extracellular vesicles in normal pregnancy and preeclampsia. J Reprod Immunol (2017) 119:98–106. doi: 10.1016/j.jri.2016.08.008
63. Tannetta D, Mackeen M, Kessler B, Sargent I, Redman C. Multi-dimensional protein identification technology analysis of syncytiotrophoblast vesicles released from perfused preeclampsia placentas. Pregnancy Hypertens (2012) 2(3):201–2. doi: 10.1016/j.preghy.2012.04.046
64. Baig S, Kothandaraman N, Manikandan J, Rong L, Ee KH, Hill J, et al. Proteomic analysis of human placental syncytiotrophoblast microvesicles in preeclampsia. Clin Proteomics (2014) 11(1):40. doi: 10.1186/1559-0275-11-40
Keywords: factor H, complement system, pregnancy, placenta, preeclampsia, microvesicles
Citation: Yasmin H, Agostinis C, Toffoli M, Roy T, Pegoraro S, Balduit A, Zito G, Di Simone N, Ricci G, Madan T, Kishore U and Bulla R (2024) Protective role of complement factor H against the development of preeclampsia. Front. Immunol. 15:1351898. doi: 10.3389/fimmu.2024.1351898
Received: 07 December 2023; Accepted: 30 January 2024;
Published: 23 February 2024.
Edited by:
Mahmood Jeddi-Tehrani, Culture and Research, IranCopyright © 2024 Yasmin, Agostinis, Toffoli, Roy, Pegoraro, Balduit, Zito, Di Simone, Ricci, Madan, Kishore and Bulla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Uday Kishore, dWtpc2hvcmVAaG90bWFpbC5jb20=; dWRheS5raXNob3JlQHVhZXUuYWMuYWU=; Roberta Bulla, cmJ1bGxhQHVuaXRzLml0
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.