
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 20 March 2024
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1351405
 Selina Wachtendorf1,2‡
Selina Wachtendorf1,2‡ Fitriasari Jonin1,2‡
Fitriasari Jonin1,2‡ Aaron Ochel1
Aaron Ochel1 Fabian Heinrich1†
Fabian Heinrich1† Astrid M. Westendorf3
Astrid M. Westendorf3 Gisa Tiegs1,2
Gisa Tiegs1,2 Katrin Neumann1,2*
Katrin Neumann1,2*Introduction: The alarmin IL-33 has been implicated in the pathology of immune-mediated liver diseases. IL-33 activates regulatory T cells (Tregs) and type 2 innate lymphoid cells (ILC2s) expressing the IL-33 receptor ST2. We have previously shown that endogenous IL-33/ST2 signaling activates ILC2s that aggravate liver injury in murine immune-mediated hepatitis. However, treatment of mice with exogenous IL-33 before induction of hepatitis ameliorated disease severity. Since IL-33 induces expression of amphiregulin (AREG) crucial for Treg function, we investigated the immunoregulatory role of the ST2+ Treg/AREG axis in immune-mediated hepatitis.
Methods: C57BL/6, ST2-deficient (Il1rl1-/-) and Areg-/- mice received concanavalin A to induce immune-mediated hepatitis. Foxp3Cre+ x ST2fl/fl mice were pre-treated with IL-33 before induction of immune-mediated hepatitis. Treg function was assessed by adoptive transfer experiments and suppression assays. The effects of AREG and IL-33 on ST2+ Tregs and ILC2s were investigated in vitro. Immune cell phenotype was analyzed by flow cytometry.
Results and discussion: We identified IL-33-responsive ST2+ Tregs as an effector Treg subset in the murine liver, which was highly activated in immune-mediated hepatitis. Lack of endogenous IL-33 signaling in Il1rl1-/- mice aggravated disease pathology. This was associated with reduced Treg activation. Adoptive transfer of exogenous IL-33-activated ST2+ Tregs before induction of hepatitis suppressed inflammatory T-cell responses and ameliorated disease pathology. We further showed increased expression of AREG by hepatic ST2+ Tregs and ILC2s in immune-mediated hepatitis. Areg-/- mice developed more severe liver injury, which was associated with enhanced ILC2 activation and less ST2+ Tregs in the inflamed liver. Exogenous AREG suppressed ILC2 cytokine expression and enhanced ST2+ Treg activation in vitro. In addition, Tregs from Areg-/- mice were impaired in their capacity to suppress CD4+ T-cell activation in vitro. Moreover, application of exogenous IL-33 before disease induction did not protect Foxp3Cre+ x ST2fl/fl mice lacking ST2+ Tregs from immune-mediated hepatitis. In summary, we describe an immunoregulatory role of the ST2+ Treg/AREG axis in immune-mediated hepatitis, in which AREG suppresses the activation of hepatic ILC2s while maintaining ST2+ Tregs and reinforcing their immunosuppressive capacity in liver inflammation.
Immune-mediated liver diseases such as autoimmune hepatitis (AIH) are characterized by hepatic inflammation, tissue destruction and development of fibrosis. The relevance of immunological pathways in disease pathology has been highlighted by genome-wide association studies, identifying risk loci that host genes involved in innate and adaptive immunity (1). However, immune-mediated processes leading to liver inflammation and immunoregulation are still poorly defined.
IL-33 functions as an alarmin, which is released as a local danger signal to alert the immune system (2). IL-33 is the only ligand of the suppression of tumorigenicity (ST)2 receptor (3), and activates ST2-expressing immune cells such as type 2 innate lymphoid cells (ILC2s) and regulatory T cells (Tregs) (4). IL-33-activated ILC2s express type 2 cytokines (5) and the epidermal growth factor amphiregulin (AREG), that contributes to tissue regeneration (6–8). IL-33 further induces activation of ST2+ Tregs, an effector Treg subset (9) that predominantly localizes in non-lymphoid tissue (10). IL-33 stimulates expression of AREG in Tregs and both, IL-33 and AREG ensure maintenance of Treg function during inflammation (11, 12). Whether ST2+ Tregs and their effector molecule AREG are important for immunoregulation in immune-mediated hepatitis is not known so far.
Elevated IL-33 levels have been described in patients with acute and chronic liver disease (13), including AIH (14), suggesting that IL-33 contributes to liver disease pathology. In the murine model of concanavalin (Con)A-induced immune-mediated hepatitis, which shares some similarities with acute AIH (15), hepatocytes have been shown to increase expression of IL-33 (16). We have recently demonstrated that formation of necrotic lesions in immune-mediated hepatitis was associated with the release of IL-33 in liver tissue and an aggravation of the disease through activation of hepatic ILC2s (17). Moreover, blockage of endogenous IL-33 was found to ameliorate liver injury (18), further supporting the pro-inflammatory role of IL-33 in immune-mediated hepatitis. In contrast, IL-33- and ST2-deficient mice developed more severe liver injury (19, 20), pointing to a protective effect of endogenous IL-33/ST2 in immune-mediated hepatitis. We have further shown that targeting the IL-33/ST2 axis by application of exogenous IL-33 suppressed immune-mediated hepatitis, which correlated with an expansion of ST2+ Tregs in the liver (17).
In this study, we investigated the role of the ST2+ Treg/AREG axis in the pathogenicity of immune-mediated hepatitis. IL-33-responsive ST2+ Tregs constituted a hepatic effector Treg subset that suppressed liver inflammation upon activation with exogenous IL-33. Hepatic ILC2s and ST2+ Tregs expressed AREG in immune-mediated hepatitis. Areg-/- mice developed more severe liver injury, that was associated with increased ILC2 activation and less ST2+ Tregs in the inflamed liver. Treatment of mice with exogenous IL-33 elevated AREG expression in ILC2s and ST2+ Tregs and led to reduced effector T-cell activation in immune-mediated hepatitis. The immunosuppressive effect of exogenous IL-33 was abrogated in mice lacking ST2+ Tregs. Thus, the ST2+ Treg/AREG axis regulates inflammatory immune responses in immune-mediated hepatitis.
Areg-/- mice (Aregtm1Dle/J) were provided by Matias A. Avila (Hepatology Program, CIMA, Universidad de Navarra, Pamplona, Spain). Il1rl1-/- mice (Il1rl1tm1Anjm) were provided by Max Löhning (Experimental Immunology and Osteoarthritis Research, Department of Rheumatology and Clinical Immunology, Charité-Universitätsmedizin Berlin, Berlin, Germany). Foxp3Cre x ST2fl/fl mice (B6.Il1rl1tm1c(KOMP)WtsixFoxp3tm1(cre)Saka) (21) were provided by Astrid M. Westendorf (Infection Immunology, Institute of Medical Microbiology, University Hospital Essen, University of Duisburg-Essen, Essen, Germany). C57BL/6 mice, FIR-tiger (Foxp3-IRES-mRFP [FIR]xIL-10-IRES-GFP [tiger]) (22) and all other transgenic mice were bred in the animal facility of the University Medical Center Hamburg-Eppendorf (Hamburg, Germany) according to the Federation of European Laboratory Animal Science Association guidelines. All mice received human care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals”. The experiments were approved by the institutional review board (Behörde für Justiz und Verbraucherschutz, Hamburg, Germany) and carried out according to the German animal protection law. Mice were housed in IVC cages under controlled conditions (22°C, 55% humidity, and 12-hour day-night rhythm) and fed a standard laboratory chow (LASvendi, Altromin, Germany). We have used male mice for the in vivo and a mixture of male and female mice for the in vitro studies.
Mice intraperitoneally received recombinant murine (rm)IL-33 (0.3 µg/mouse; BioLegend, San Diego, CA) on three or four consecutive days. To induce immune-mediated hepatitis, ConA (7 mg/kg; Sigma-Aldrich, München, Germany) was intravenously injected and mice were analyzed 24 hours later. For adoptive transfer experiments, FIR-tiger mice were treated with rmIL-33 (0.3 µg/mouse) or PBS on three consecutive days. Thereafter, Foxp3+ (mRFP+) CD4+ Tregs were isolated by FACS. 3x105 Tregs were transferred into C57BL/6 recipient mice, which received ConA (7 mg/kg) 24 hours later. Mice were analyzed one day after induction of hepatitis. Heart blood was drawn from individual mice and liver injury was quantified by automated measurement of plasma activities of alanine transaminase (ALT) using a COBAS Mira System (Roche Diagnostic, Mannheim, Germany).
Liver samples were fixed with 4% formalin and embedded in paraffin. 3 μm liver sections were cut, stained with hematoxylin and eosin (H&E) following standard protocols, and analyzed by light microscopy.
To expand hepatic ILC2s for isolation, C57BL/6 mice were treated with rmIL-33 (0.3 µg/mouse) on four consecutive days. Hepatic non-parenchymal cells were isolated by Percoll density gradient centrifugation. Single non-parenchymal cells were stained with lineage antibodies (anti-CD3, 145-2C11; anti-CD11b, M1/70; anti-CD45R/B220, RA3-6B2; anti-TER-119, TER-119; anti-GR1, RB6-8C5; all APC; all BioLegend), anti-Sca-1 (D7, Pacific Blue) and anti-ST2 (RMST2-2, PE-Cy7; both BioLegend) antibodies. Lineage-negative (lin-) cells were enriched by MACS and ST2+ Sca-1+ lin- ILC2s were isolated by FACS (BD FACSAriaTM III sorter; BD Biosciences, Heidelberg, Germany) (23).
CD4+ T cells were isolated from spleen and lymph nodes using the CD4+ T Cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. For Treg isolation, CD4+ T cells were stained with an anti-CD25-PE antibody (PC61, BioLegend) and anti-PE MicroBeads (Miltenyi Biotec), and CD25+ CD4+ Tregs were isolated by MACS.
2x104 CD25+ CD4+ Tregs were cultured with rmIL-2 (100 U/ml; R&D Systems, Minneapolis, MN) in presence of rmIL-33 (10 ng/ml; BioLegend), rmAREG (10 ng/ml; R&D Systems) or both (10 ng/ml each) for 1.5 days. 1.5x104 hepatic ILC2s were cultured with rmIL-2 (1 U/ml) in presence of rmIL-33 (10 ng/ml), rmAREG (10 ng/ml) or both (10 ng/ml each) for 3.5 days.
CD4+ T cells were isolated from spleen and lymph nodes of C57BL/6 mice and stained with an anti-CD25-PE antibody and anti-PE MicroBeads. CD25- CD4+ responder T cells were isolated by MACS and labeled with carboxyfluorescein succinimidyl ester (CFSE; CFSE Cell Division Tracker Kit, Biolegend) to assess T-cell proliferation. CD25+ CD4+ Tregs were isolated from spleen and lymph nodes of C57BL/6 and Areg-/- mice by MACS. 1x105 responder CD4+ T cells were cultured with 1x105 Tregs for 2.5 days. For responder CD4+ T-cell activation, co-cultures were done with Dynabeads (Dynabeads Mouse T-Activator CD3/CD28; ThermoFisher Scientific).
The percentage of inhibition was calculated as .
Cells were incubated with anti-CD16/32 antibody (93; BioLegend) prior to antibody staining in order to prevent unspecific binding. LIVE/DEAD Fixable Staining Kits (Thermo Fisher Scientific) were used to exclude dead cells. For cell surface analysis, cells were stained with fluorochrome-labelled antibodies listed in Supplementary Table S1. For intracellular and intranuclear staining, cells were re-stimulated with phorbol myristate acetate (20 ng/ml) and ionomycin (1 µg/ml) for 4 hours with the addition of brefeldin A (1 µg/ml; all Sigma Aldrich) and monensin (2 µM; BioLegend) after 30 min. Thereafter, cells were fixed using the Transcription Factor Staining Buffer Set (ThermoFisher Scientific, Waltham, MA) and incubated in Permeabilization buffer with antibodies listed in Supplementary Table S2. Data were acquired using a BD LSRFortessa II (BD Biosciences) and analyzed by FlowJo software (Tree Star, Ashland, OR, USA).
Plasma AREG levels were analyzed using the Mouse Amphiregulin ELISA DuoSets (R&D Systems) and the Infinite M200 plate reader (Tecan, Männedorf, Switzerland) according to the manufacturer’s instructions.
Total RNA was isolated from shock-frozen liver tissue using the NucleoSpin RNA Midi Kit (Macherey-Nagel, Düren, Germany). Genomic DNA was digested using the DNA-free Kit (ThermoFisher Scientific). 1 µg RNA was transcribed into cDNA using the Verso cDNA Synthesis Kit (ThermoFisher Scientific) and the Biometra TAdvanced thermal cycler (Analytik Jena, Jena, Germany). Quantitative RT-PCR was performed using the PowerUp SYBR Green Master Mix (ThermoFisher Scientific) and the QuantStudio 7 Flex (Applied Biosystems, Waltham, USA). Relative mRNA levels were calculated using the ΔΔCT method after normalization to the reference gene β-actin. Quantification was shown in x-fold changes to the corresponding control cDNA. Primers were designed for detection of exon overlapping amplicons and were obtained from Metabion (Martinsried, Germany). Sequences of the primers are listed in Supplementary Table S3.
Data were analyzed using the GraphPad Prism software (GraphPad software, San Diego, CA). For comparisons between two groups, a non-parametric Mann-Whitney U test and for more than two groups, a one-way ANOVA with Tukey’s post-hoc test were used. Data were shown as medians. A p value of less than 0.05 was considered statistically significant with the following ranges *p< 0.05, **p< 0.01, ***p< 0.001, and ****p< 0.0001.
We analyzed the phenotype of hepatic ST2+ Foxp3+ Tregs in immune-mediated hepatitis to assess their impact on liver disease pathology. ST2+ Tregs have been described as a tissue-associated Treg subset crucial for immunosuppression and organ homeostasis (10, 24). In line with this, we determined an elevated frequency of ST2+ Foxp3+ Tregs in naïve liver compared to spleen (Figure 1A, Supplementary Figure 1A). Hepatic ST2+ Foxp3+ Tregs showed enhanced expression of the tissue residency markers CD103 and CD69 compared to ST2- Foxp3+ Tregs, while they expressed less CCR7, a chemokine receptor required for lymph node migration (Figure 1B, Supplementary Figure 1B). Phenotypically, hepatic ST2+ Foxp3+ Tregs showed stronger expression of their key transcription factor Foxp3, the activation marker killer cell lectin like receptor G1 (KLRG1) and inducible T cell co-stimulator (ICOS), of the proliferation marker Ki-67, and of the inhibitory molecules cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), IL-10, programmed death ligand 1 (PD-L1) and T cell immunoreceptor with Ig and ITIM domains (TIGIT) than ST2- Foxp3+ Tregs in liver homeostasis (Figures 1C, Supplementary Figure 1B). In immune-mediated hepatitis, liver injury and formation of necrotic lesions (Figure 1D) were associated with an elevated frequency of hepatic ST2+ Foxp3+ Tregs (Figure 1E). Expression of Foxp3 was not further increased in ST2+ and ST2- Foxp3+ Tregs in liver inflammation (Supplementary Figure 1C). Both, hepatic ST2+ and ST2- Tregs were activated in immune-mediated hepatitis and enhanced expression of inhibitory molecules. In fact, by calculating the fold change in frequencies, we found stronger activation of ST2- Tregs compared to ST2+ Tregs in immune-mediated hepatitis (KLRG1: ST2- 3.4 ± 1.0, ST2+ 1.2 ± 0.1; ICOS: ST2- 4.9 ± 1.2, ST2+ 2.3 ± 0.2). Expression of inhibitory molecules was also stronger induced in ST2- Tregs (CTLA-4: ST2- 1.7 ± 0.2, ST2+ 1.3 ± 0.1; IL-10: ST2- 2.5 ± 0.9, ST2+ 1.1 ± 0.5; PD-L1: ST2- 9.7 ± 8.8, ST2+ 3.6 ± 0.7; TIGIT: ST2- 2.3 ± 0.8, ST2+ 1.5 ± 0.1). We believe that due to the elevated basal activation and inhibitory molecule expression of ST2+ Tregs, they do not become activated as strong as ST2- Tregs in immune-mediated hepatitis. However, activation and inhibitory molecule expression were still elevated in ST2+ compared to ST2- Tregs (Figure 1F). Thus, ST2+ Foxp3+ Tregs constituted an activated effector Treg subset in the murine liver.
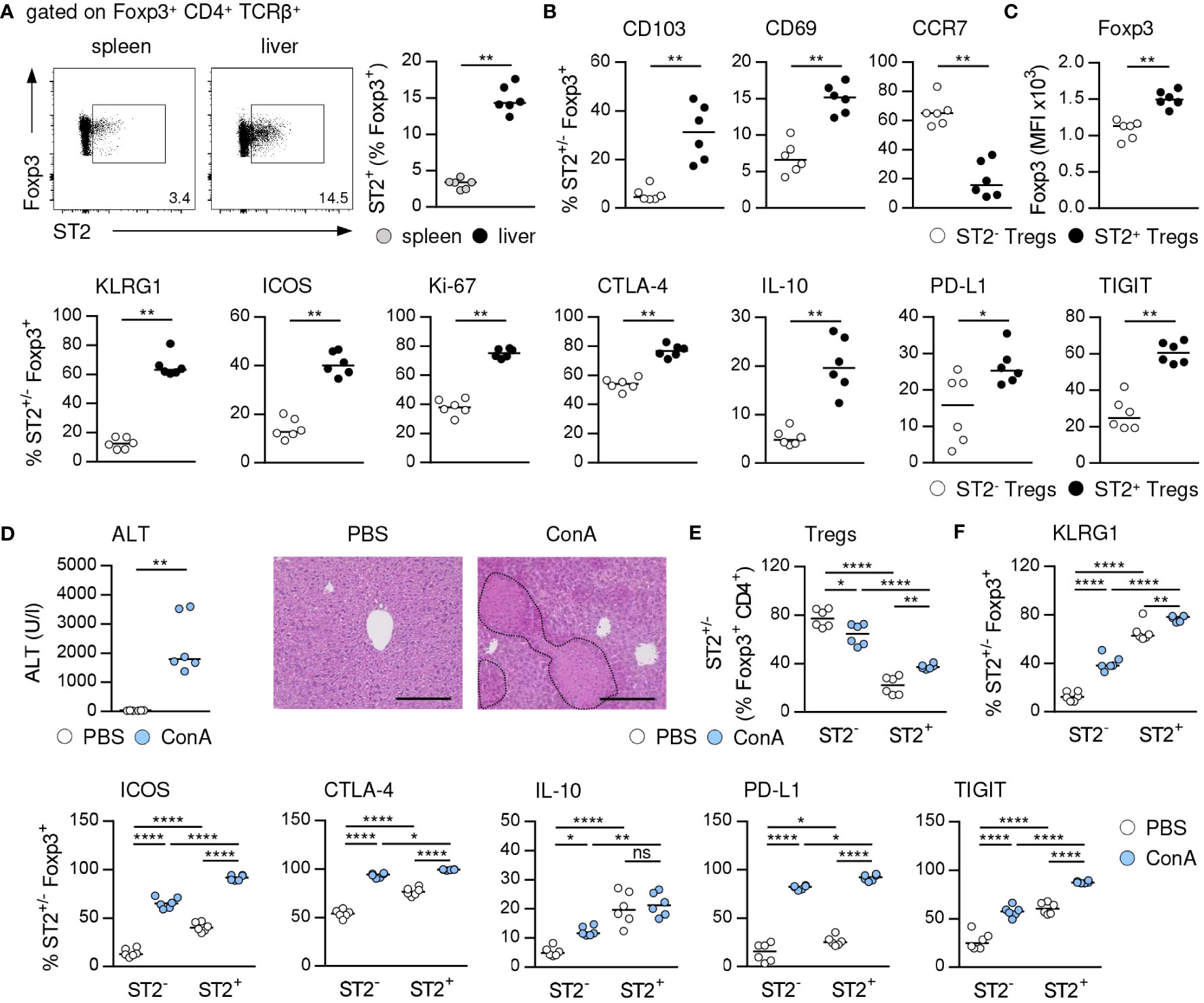
Figure 1 Phenotype of ST2+ Foxp3+ Tregs in liver homeostasis and immune-mediated hepatitis. (A) Frequency of ST2+ Tregs was determined in liver and spleen of naïve WT mice. Representative dot plots are shown. (B, C) Phenotype of hepatic ST2+ and ST2- Tregs was analyzed in naïve WT mice. (D) WT mice received ConA and were analyzed 24 hours later. Plasma ALT levels were determined. Liver sections were stained with H&E to visualize necrotic areas (dotted line). (E) Frequency and (F) phenotype of hepatic ST2+ and ST2- Tregs were analyzed. Bars represent 200 µm. Medians of two independent experiments with 3 mice per group and experiment are shown. *p< 0.05; **p< 0.01; ****p< 0.0001; ns, not significant; MFI, mean fluorescent intensity.
IL-33 is the only known ligand of ST2 and has been described as a potent activator of Tregs (4). We demonstrated that treatment with exogenous IL-33, which did not induce liver injury and tissue damage (Figure 2A), selectively increased the frequency of hepatic ST2+ Foxp3+ Tregs in C57BL/6 wild-type (WT) mice and their activation and expression of inhibitory molecules. ST2+ Foxp3+ Tregs did not enhance expression of Foxp3 in response to exogenous IL-33. We determined elevated Foxp3 expression by Foxp3+ Tregs from IL-33-treated WT compared to Il1rl1-/- mice, probably due to lack of ST2+ Tregs in Il1rl1-/- mice (Figures 2B, C, Supplementary Figure 2A). Moreover, Foxp3+ Tregs of IL-33-treated WT mice were stronger activated and showed enhanced expression of inhibitory molecules than Foxp3+ Tregs from IL-33-treated Il1rl1-/- mice (Figure 2D). Thus, targeting the IL-33/ST2 axis by application of exogenous IL-33 selectively induced ST2+ Treg activation and expansion in the liver. In addition, culture of WT Tregs in presence of IL-33 enhanced their expansion, activation and expression of inhibitory molecules, whereas Tregs from Il1rl1-/- mice were not stimulated by IL-33 in vitro. Similar to the in vivo data, IL-33-stimulated Il1rl1-/- Foxp3+ Tregs showed decreased Foxp3 expression compared to WT Foxp3+ Tregs in vitro (Supplementary Figures 2B, C). We further showed that ST2+ but not ST2- Foxp3+ Tregs from WT mice were activated by IL-33 in vitro (Supplementary Figure 2D).
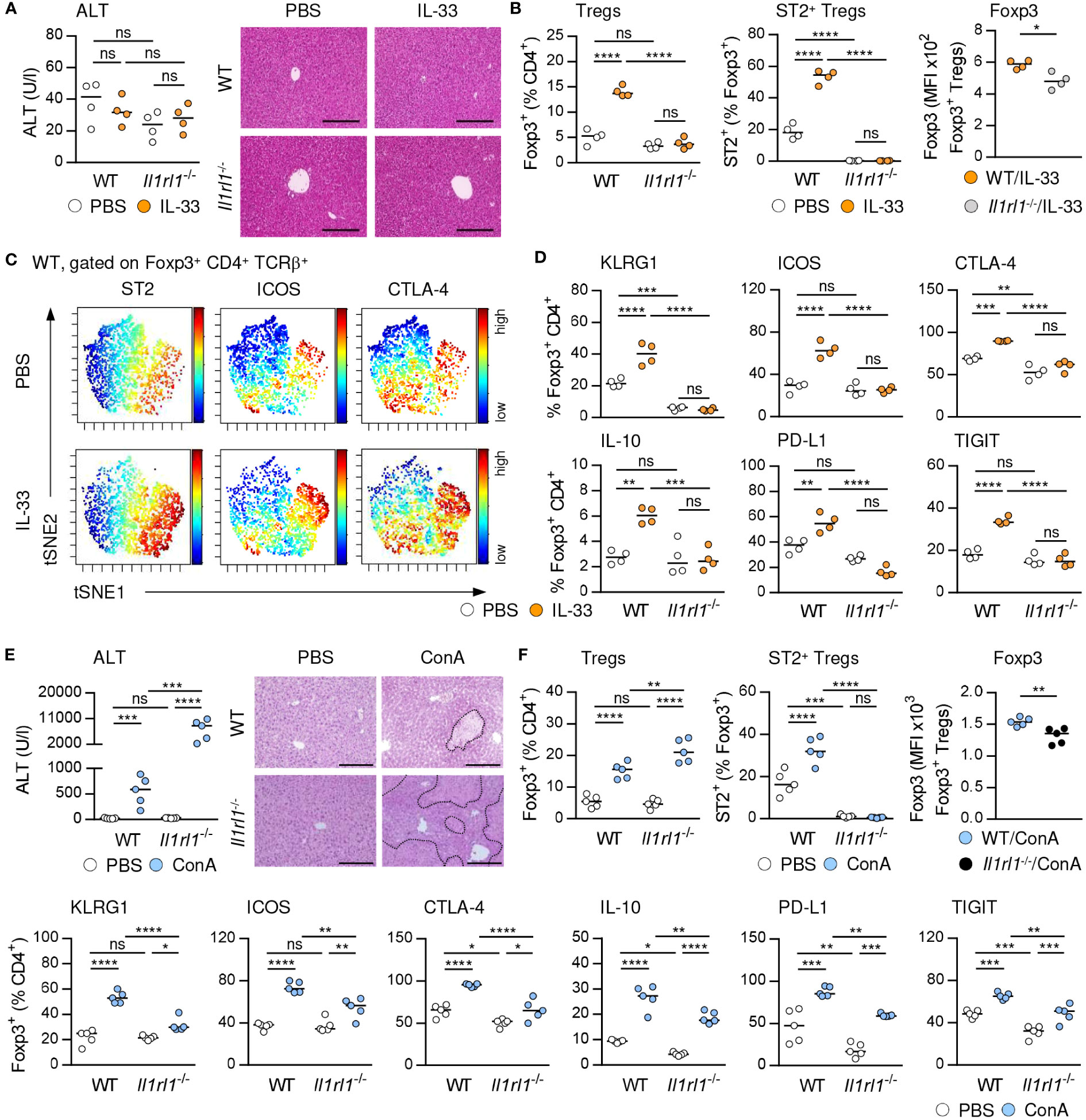
Figure 2 IL-33-induced activation of hepatic ST2+ Foxp3+ Tregs. (A) WT and Il1rl1-/- mice were treated with IL-33 on four consecutive days. Plasma ALT levels were determined. Liver sections were stained with H&E. (B) Frequencies and MFI of Foxp3 in hepatic Tregs were analyzed. (C) ViSNE analysis of Tregs from WT mice is shown. (D) Phenotype of Tregs from WT and Il1rl1-/- mice was analyzed. (E) WT and Il1rl1-/- mice received ConA and were analyzed 24 hours later. Plasma ALT levels were determined. Liver sections were stained with H&E to visualize necrotic areas (dotted line). (F) Frequency and phenotype of hepatic Tregs were analyzed. Bars represent 200 µm. Medians of one out of two independent experiments with 4-5 mice per group and experiment are shown. *p< 0.05; **p< 0.01; ***p< 0.001; ****p< 0.01; ns, not significant; MFI, mean fluorescent intensity.
We and others have shown that ST2-deficient (Il1rl1-/-) mice developed more severe liver injury and tissue damage than WT mice (Figure 2E) (20), suggesting an immunoregulatory role of endogenous IL-33 in immune-mediated hepatitis. Liver pathology was aggravated in Il1rl1-/- mice despite the absence of hepatic ILC2s (Supplementary Figure 3) and an elevated frequency of Foxp3+ Tregs in the inflamed liver (Figure 2F). However, Il1rl1-/- mice lacked ST2+ Foxp3+ Tregs and the remaining Foxp3+ Treg population showed reduced Foxp3 expression, activation and expression of inhibitory molecules in immune-mediated hepatitis compared to Foxp3+ Tregs in WT mice (Figure 2F). Thus, endogenous IL-33 selectively activated hepatic ST2+ Foxp3+ Tregs and lack of this Treg subset in Il1rl1-/- mice resulted in an overall reduced Treg activation in immune-mediated hepatitis.
To assess the immunosuppressive capacity of Tregs activated by exogenous IL-33, we isolated Foxp3+ Tregs from IL-33-treated FIR-tiger mice and adoptively transferred them into WT mice one day before induction of immune-mediated hepatitis. As control, naïve Foxp3+ Tregs from PBS-treated FIR-tiger mice were transferred. We showed reduced liver injury and formation of necrotic lesions after transfer of IL-33-activated Foxp3+ Tregs (Figures 3A, B). This was associated with decreased expression of the pro-inflammatory cytokines interferon (IFN)γ and tumor necrosis factor (TNF)α by hepatic CD8+ and CD4+ T cells (Figure 3C). In contrast, transfer of naïve Foxp3+ Tregs did not alter disease pathology and immune cell phenotype (Figures 3A-C), demonstrating the high immunosuppressive potential of Tregs that were pre-activated through exogenous IL-33 in immune-mediated hepatitis.
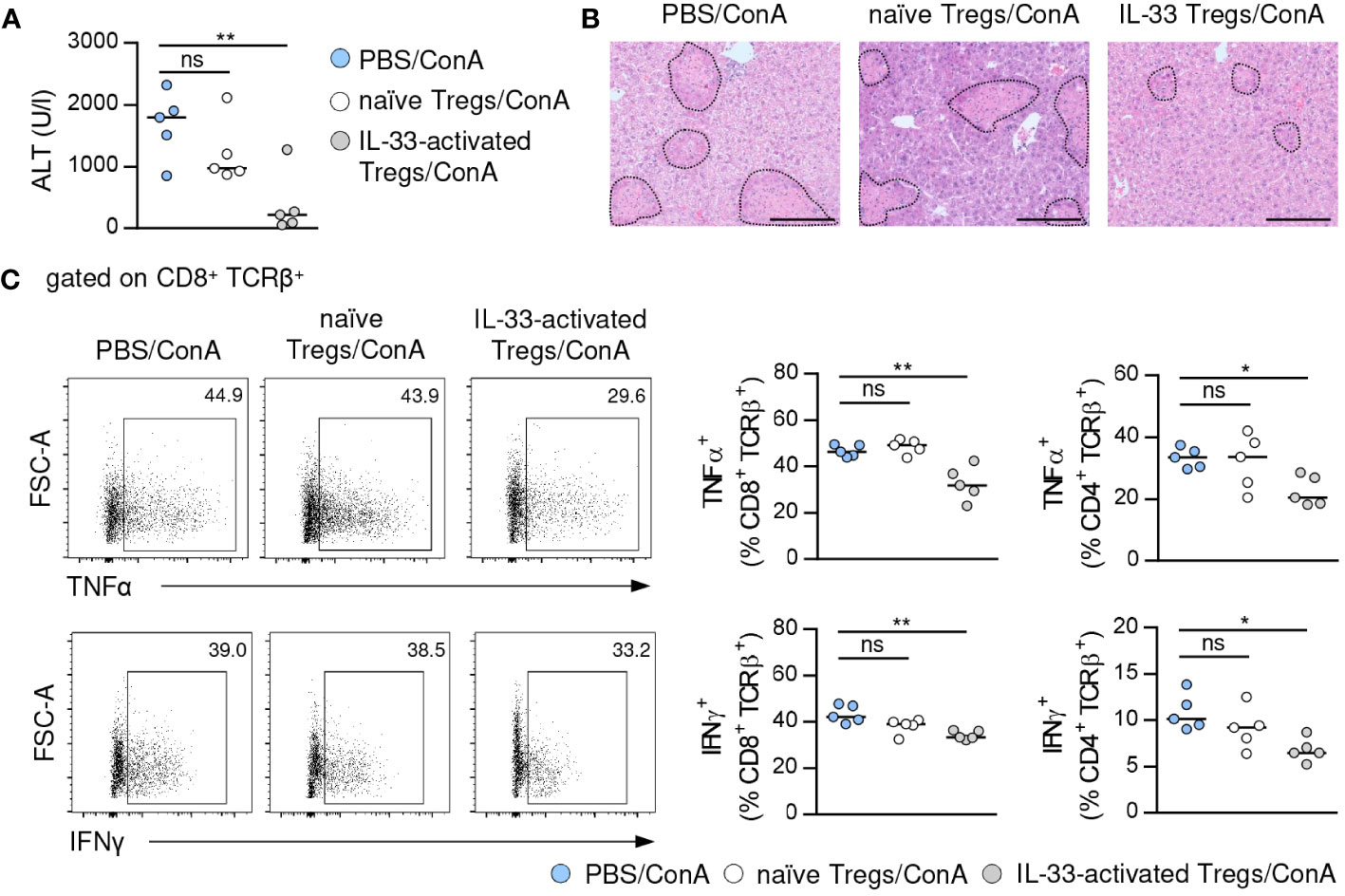
Figure 3 Immunosuppressive potential of IL-33-activated Tregs. (A) FIR-tiger mice were treated with IL-33 or PBS on three consecutive days. Foxp3+ Tregs were isolated by FACS and transferred into WT mice one day before ConA challenge. WT mice were analyzed 24 hours after induction of immune-mediated hepatitis. Plasma ALT activity was determined. (B) Liver samples were stained with H&E to visualize necrotic areas (dotted line). (C) Phenotype of hepatic CD4+ and CD8+ T cells was analyzed. Representative dot plots are shown. Bars represent 200 µm. Medians of one out of two independent experiments with 5 mice per group and experiment are shown. *p< 0.05; **p< 0.01; ns, not significant.
IL-33 has been described to induce AREG expression in Tregs and ILC2s thereby supporting Treg function and tissue repair (6, 12). Indeed, we showed increased AREG expression in hepatic ST2+ Foxp3+ Tregs and ILC2s upon treatment with exogenous IL-33 (Supplementary Figure 4A). IL-33 also enhanced expression of AREG in ST2+ Foxp3+ Tregs (Supplementary Figure 4B) and hepatic ILC2s (Supplementary Figures 5A, B) in vitro.
In immune-mediated hepatitis, liver Areg mRNA and serum levels of AREG were elevated (Figure 4A), and we determined enhanced expression of AREG in hepatic ST2+ Foxp3+ Tregs and ILC2s (Figure 4B). In Il1rl1-/- mice, Foxp3+ Tregs showed reduced expression of AREG compared to Foxp3+ Tregs from WT mice, suggesting that endogenous IL-33 induced AREG expression in ST2+ Tregs in liver inflammation (Supplementary Figure 4C). In Areg-/- mice, ST2+ Foxp3+ Tregs and ILC2s did not express AREG (Supplementary Figure 4C). Interestingly, Areg-/- mice developed more severe liver injury and tissue damage than WT mice (Figure 4C). Hepatic Il33 mRNA expression was not altered in Areg-/- compared to WT mice in immune-mediated hepatitis (Supplementary Figure 4C), indicating that the elevated liver injury shown in Areg-/- mice did not correlate with increased Il33 mRNA induction. Lack of AREG resulted in a reduced frequency of hepatic ST2+ Foxp3+ Tregs in immune-mediated hepatitis (Figure 4D), whereas their phenotype was not altered compared to ST2+ Foxp3+ Tregs from WT mice. Notably, Areg-/- ST2+ Foxp3+ Tregs up-regulated expression of Foxp3 and the anti-apoptotic protein B cell lymphoma 2 (Bcl-2) during liver inflammation (Supplementary Figure 4D), probably to increase their stability in absence of AREG. Functionally, we showed an impaired capacity of Areg-/- CD25+ Tregs to inhibit activation and subsequent proliferation of CD25- CD4+ T cells in vitro (Figure 4E).
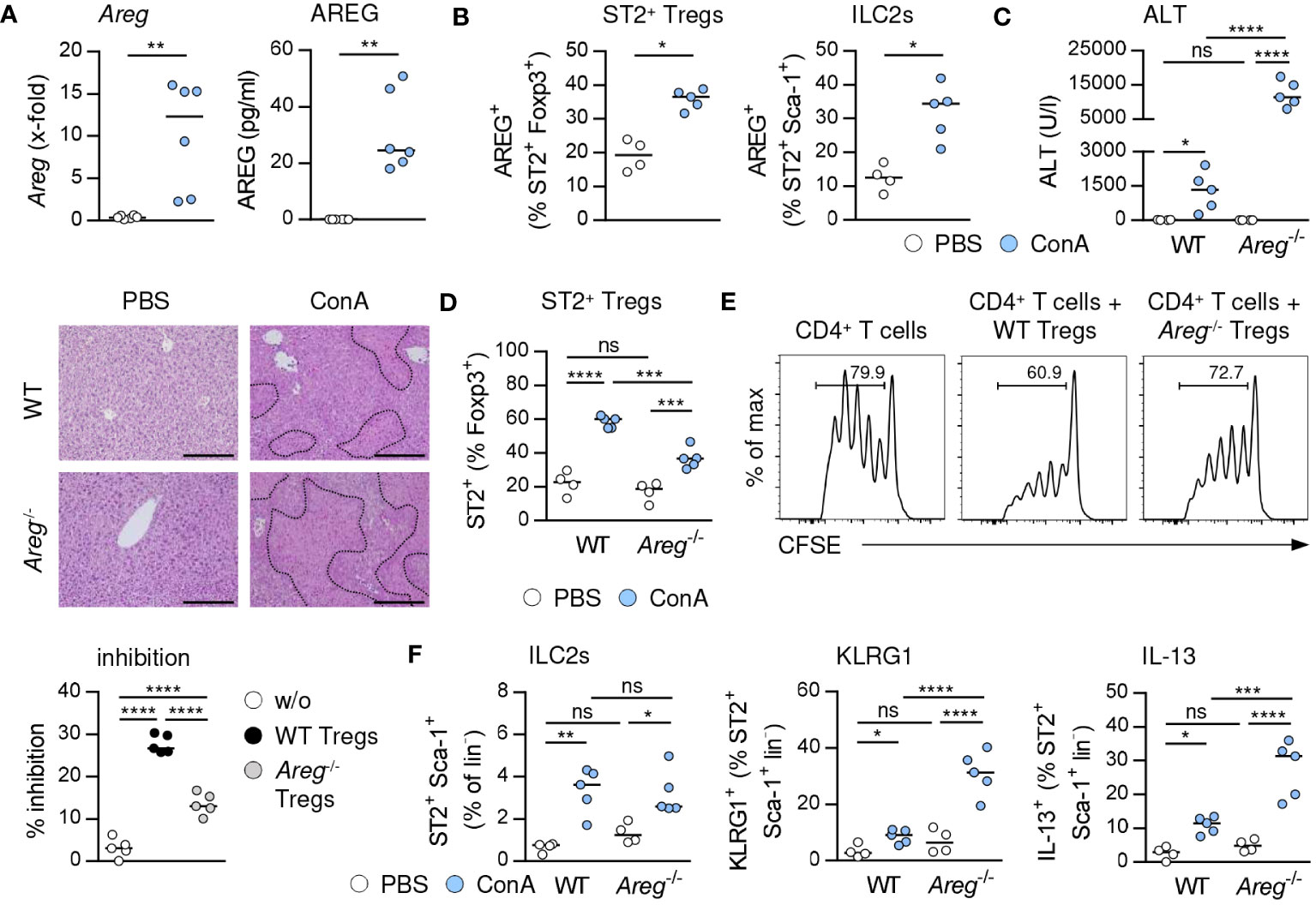
Figure 4 Immunoregulatory role of AREG in immune-mediated hepatitis. (A) WT and Areg-/- mice received ConA and were analyzed 24 hours later. Areg mRNA expression and plasma AREG levels were analyzed in WT mice. (B) AREG expression was determined in hepatic ST2+ Tregs and ILC2s. (C) Plasma ALT levels were determined. Liver samples were stained with H&E to visualize necrotic areas (dotted line). (D) Frequency of hepatic ST2+ Tregs was analyzed. (E) CFSE-labeled CD25- CD4+ T cells were activated in presence or absence of CD25+ CD4+ Tregs from WT and Areg-/- mice. Histograms show frequencies of proliferating CD4+ T cells. Percent inhibition of CD4+ T cell proliferation is depicted. (F) Frequency and phenotype of hepatic ILC2s were analyzed in WT and Areg-/- mice. Bars represent 200 µm. Medians of one out of two independent experiments with 4-5 mice per group and experiment are shown. *p< 0.05; **p< 0.01; ***p< 0.001; ****p< 0.01; ns, not significant; CSFE, carboxyfluorescein succinimidyl ester; MFI, mean fluorescent intensity.
The frequency of hepatic ST2+ Foxp3+ Tregs was also reduced in Areg-/- mice treated with exogenous IL-33. Again, their phenotype was comparable to ST2+ Foxp3+ Tregs from IL-33-treated WT mice, but they showed elevated expression of Foxp3 as well as Ki-67 (Supplementary Figure 4E), indicating stronger proliferation. In vitro, IL-33 induced a lower expansion of ST2+ Foxp3+ Tregs from Areg-/- mice, and the IL-33-induced expression of inhibitory molecules was reduced in Areg-/- compared to WT ST2+ Foxp3+ Tregs (Supplementary Figure 4F).
We further demonstrated similar frequencies of hepatic ILC2s in Areg-/- and WT mice in immune-mediated hepatitis. However, lack of AREG resulted in an enhanced ILC2 activation and IL-13 expression (Figure 4F). In contrast, activation and type 2 cytokine expression of ILC2s were not altered in Areg-/- compared to WT mice upon treatment with exogenous IL-33 (Supplementary Figure 5C). Thus, AREG exerted an immunoregulatory function in immune-mediated hepatitis.
To further assess the effect of AREG on immune cell phenotype, we cultured Tregs and hepatic ILC2s in presence of AREG. Exogenous AREG induced an activation and expansion of ST2+ Foxp3+ Tregs. They increased expression of inhibitory molecules, whereas Bcl-2 expression was not altered. Moreover, AREG stimulated its own expression in ST2+ Foxp3+ Tregs. In contrast, ST2- Foxp3+ Tregs were not activated by AREG (Figure 5A). We did also not observe an effect of AREG on activation of Foxp3+ Tregs from Il1rl1-/- mice (Supplementary Figure 6A), demonstrating that exogenous AREG selectively activated ST2+ Foxp3+ Tregs.
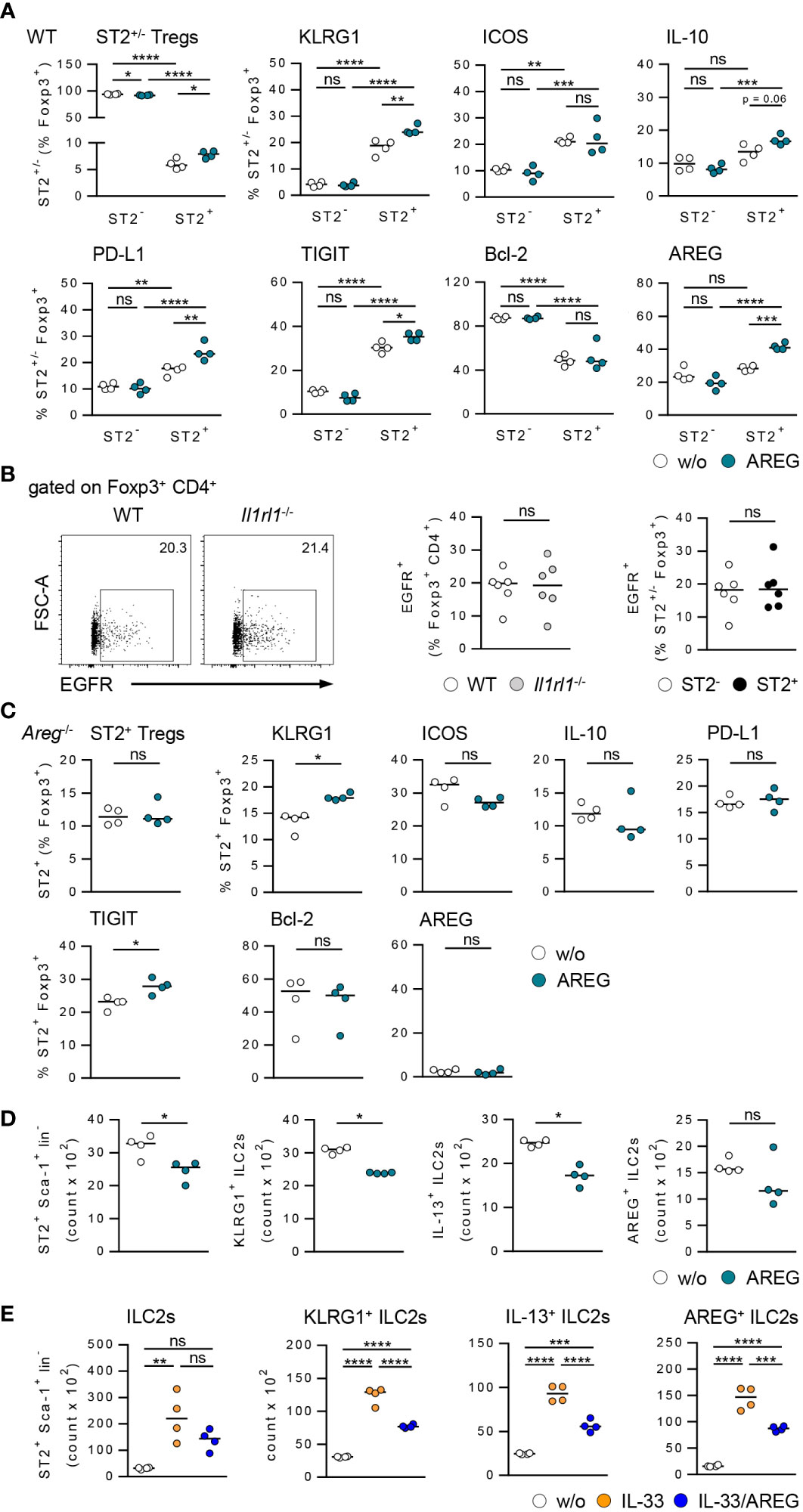
Figure 5 Effects of AREG on activation of ST2+ Foxp3+ Tregs and ILC2s in vitro. (A) CD25+ CD4+ Tregs were isolated from WT mice and cultured in presence of AREG for 1.5 days. Phenotype of ST2+ and ST2- Tregs was analyzed. (B) EGFR expression was determined in Foxp3+ Tregs from WT and Il1rl1-/- mice as well as ST2+ and ST2- Tregs from WT mice. 6 mice per group from two individual experiments are shown. (C) CD25+ CD4+ Tregs were isolated from Areg-/- mice and cultured in presence of AREG for 1.5 days. Phenotype of ST2+ Tregs was analyzed. (D, E) Hepatic ILC2s were isolated from IL-33-treated WT mice and cultured in presence of (D) AREG and (E) IL-33 or IL-33/AREG for 3.5 days. ILC2 number and phenotype were determined. Medians of one out of two independent experiments are shown. 4 wells per sample and experiment were seeded into culture plates. *p< 0.05; **p< 0.01; ***p< 0.001; ****p< 0.01; ns, not significant; w/o, without.
Since AREG is a ligand of the epidermal growth factor receptor (EGFR), we analyzed EGFR expression by Tregs from WT and Il1rl1-/- mice. The frequency of EGFR+ Foxp3+ Tregs was not altered in Il1rl1-/- compared to WT mice. ST2+ and ST2- Foxp3+ Tregs from WT mice did also not differ in their EGFR expression (Figure 5B), indicating an EGFR-independent effect of AREG on ST2+ Foxp3+ Tregs in vitro. We also showed that exogenous AREG did not induce an expansion of ST2+ Foxp3+ Tregs from Areg-/- mice. With the exception of TIGIT, AREG-deficient ST2+ Foxp3+ Tregs did not up-regulate expression of inhibitory molecules or Bcl-2 in response to AREG (Figure 5C).
Culture of hepatic ILC2s in presence of AREG resulted in a reduced ILC2 number and decreased their activation and IL-13 expression (Figure 5D). Moreover, exogenous AREG reduced the IL-33-induced activation of ILC2s and their expression of IL-13 and AREG (Figure 5E). In contrast, AREG did not affect the activation of WT or Areg-/- ST2+ Foxp3+ Tregs by IL-33 in vitro (Supplementary Figures 6B, C). Thus, exogenous AREG differentially regulated the activation of ST2+ Foxp3+ Tregs and hepatic ILC2s in vitro.
Previously, we have shown that treatment of WT mice with exogenous IL-33 before ConA challenge potently suppressed immune-mediated hepatitis (17). In contrast, liver injury and tissue damage were not improved in Il1rl1-/- mice (Figure 6A), demonstrating that targeting the IL-33/ST2 axis by exogenous IL-33 ameliorated disease pathology. In WT mice, we observed reduced frequencies of hepatic CD4+ and CD8+ T cells expressing IFNγ and TNFα in immune-mediated hepatitis after pre-treatment with exogenous IL-33 (Figure 6B). The frequencies of hepatic ST2+ Foxp3+ Tregs and ILC2s were increased and both up-regulated expression of AREG (Figure 6C). Moreover, Foxp3 expression and activation of ST2+ Foxp3+ Tregs but not their expression of inhibitory molecules was further enhanced by treatment with exogenous IL-33 (Supplementary Figure 7).
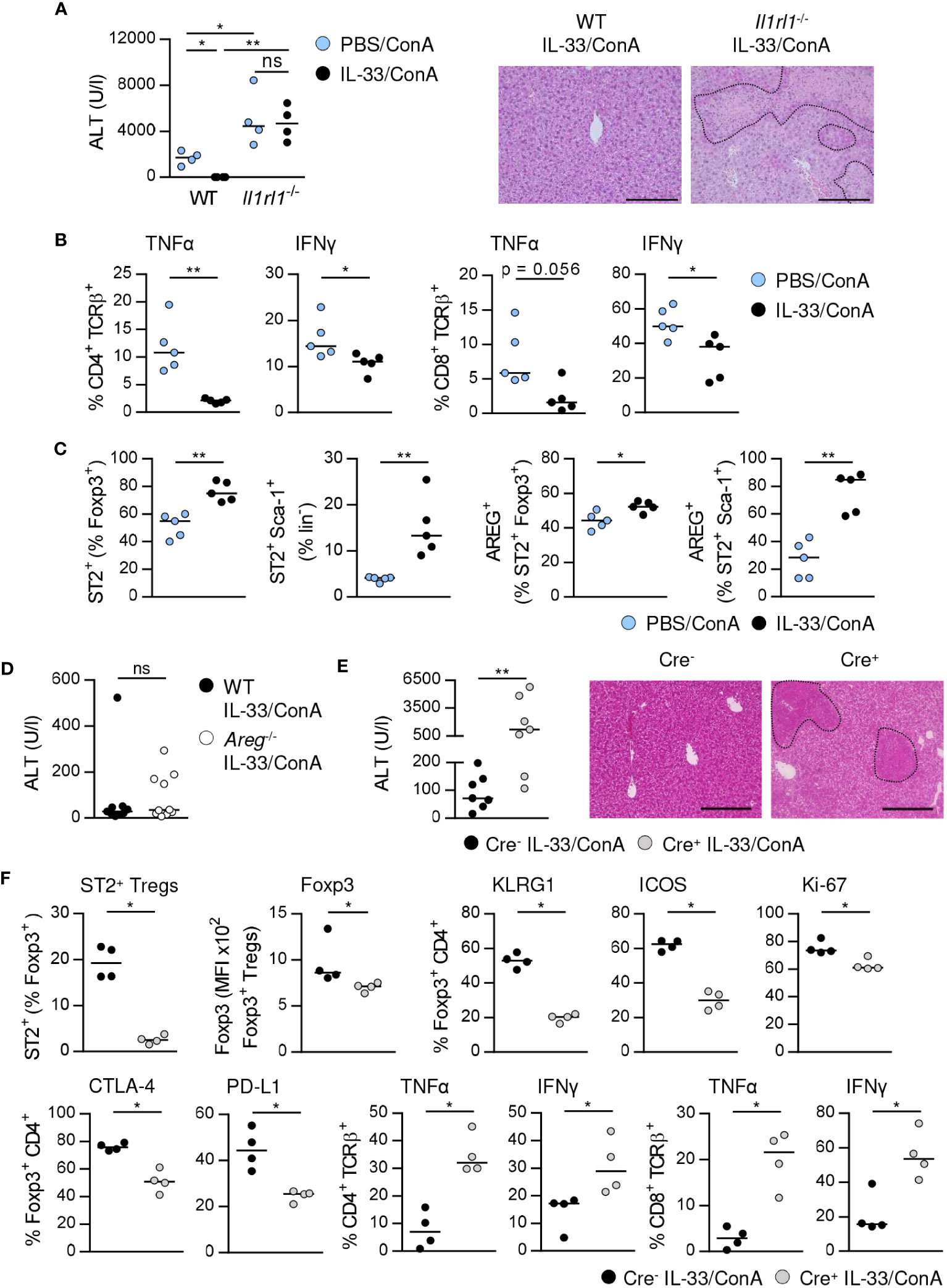
Figure 6 ST2+ Treg-dependent immunosuppressive effect of IL-33 on liver disease pathology. (A) WT and Il1rl1-/- mice were treated with IL-33 on three consecutive days. On the fourth day, mice received ConA and were analyzed 24 hours later. Plasma ALT levels were determined. Liver sections were stained with H&E to visualize necrotic areas (dotted line). (B) Expression of TNFα and IFNγ by hepatic CD4+ and CD8+ T cells was determined. (C) Frequencies of hepatic ST2+ Foxp3+ Tregs and ILC2s and their expression of AREG were analyzed. (D) WT and Areg-/- mice were treated with IL-33 for three days. At day four, mice received ConA and were analyzed one day later. Plasma ALT levels were determined. 9-10 mice per group from two individual experiments are shown. (E) Foxp3Cre+ x ST2fl/fl and Foxp3Cre- x ST2fl/fl mice were treated with IL-33 for three days. At day four, mice received ConA and were analyzed 24 hours later. ALT levels were determined. Liver sections were stained with H&E to visualize necrotic areas (dotted line). (F) Frequency of hepatic ST2+ Foxp3+ Tregs was determined. Phenotype of hepatic Foxp3+ Tregs and CD4+ and CD8+ T cells was analyzed. Bars represent 200 µm. Medians of one out of two independent experiments with 4-5 mice per group and experiment are shown. *p< 0.05; **p< 0.01; ns, not significant; MFI, mean fluorescent intensity.
We analyzed whether exogenous IL-33 mediates its immunosuppressive effect via induction of AREG. Therefore, we treated Areg-/- mice with IL-33 before ConA challenge. However, ALT levels were not significantly altered in Areg-/- compared to WT mice (Figure 6D). To assess whether activation of ST2+ Tregs by exogenous IL-33 is critical for the immunosuppressive effect of this cytokine on liver disease pathology, we treated Foxp3Cre x ST2fl/fl mice (21) with IL-33 before induction of hepatitis. Foxp3Cre+ x ST2fl/fl mice developed more severe liver injury and tissue damage than Foxp3Cre- x ST2fl/fl mice after treatment with exogenous IL-33 (Figure 6E). This correlated with a strongly reduced frequency of hepatic ST2+ Tregs in Foxp3Cre+ x ST2fl/fl mice compared to Foxp3Cre- x ST2fl/fl mice (Figure 6F). Moreover, we showed decreased Foxp3 expression, activation, proliferation and inhibitory molecule expression by hepatic Foxp3+ Tregs as well as elevated expression of TNFα and IFNγ by hepatic CD4+ and CD8+ T cells in Foxp3Cre+ x ST2fl/fl mice in immune-mediated hepatitis after pre-treatment with IL-33 (Figure 6F). Thus, the immunosuppressive effect of exogenous IL-33 depends on ST2+ Treg activation.
Immune-mediated liver diseases such as AIH are live-threatening diseases with limited therapeutic options so far. Thus, understanding cellular and molecular mechanisms in hepatic inflammation will provide the opportunity to selectively target pathways involved in disease pathology. In this study, we described the immunoregulatory role of AREG in immune-mediated hepatitis by differentially affecting two liver-residing immune cell populations, ST2+ Tregs and ILC2s. The main findings are summarized in Figure 7.
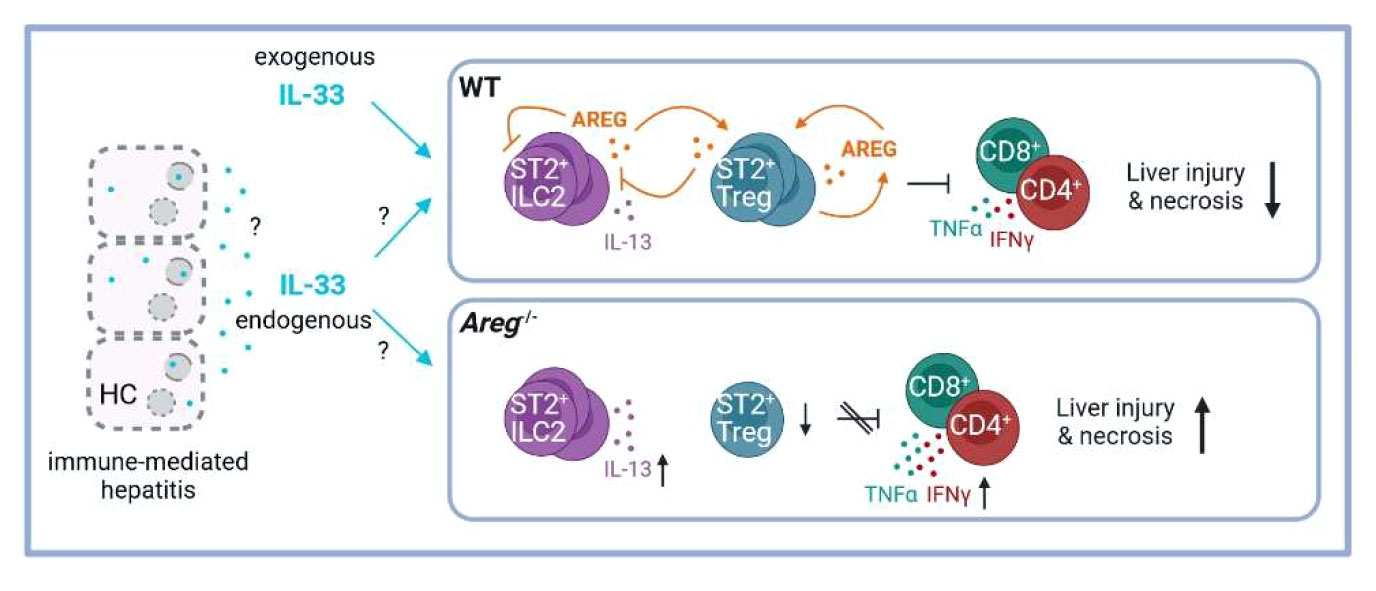
Figure 7 Scheme of the proposed ST2+ Treg/AREG axis in immune-mediated hepatitis. IL-33 induces activation of hepatic ST2+ Tregs and ILC2s, as well as their expression of AREG in immune-mediated hepatitis. Paracrine AREG further increases activation and AREG production of ST2+ Tregs, while suppressing activation and effector cytokine expression by hepatic ILC2s. Disease pathology of immune-mediated hepatitis was aggravated in absence of AREG, which correlated with enhanced ILC2 activation and effector cytokine expression and reduced ST2+ Tregs in the inflamed liver. Created with BioRender.com. HC, hepatocytes.
Our Treg phenotype analysis revealed ST2+ Tregs as an activated Treg subset residing in the murine liver, which was characterized by higher expression of the Treg-defining transcription factor Foxp3 and inhibitory molecules crucial for immunosuppressive function than ST2- Tregs. This is in line with other studies describing ST2+ Tregs as a tissue-associated effector Treg subset (9, 10). Moreover, ST2+ Tregs were shown to be superior to ST2- Tregs in suppressing CD4+ T-cell activation in vitro through enhanced expression of inhibitory molecules (9). In immune-mediated hepatitis, ST2+ and ST2- Tregs were activated, however, activation and inhibitory molecule expression was further elevated in ST2+ Tregs in liver inflammation. Evidence for an immunosuppressive function of ST2+ Tregs and their activation through endogenous IL-33 in hepatic inflammation derived from Il1rl1-/- mice, which developed more severe immune-mediated hepatitis despite an elevated frequency of Tregs in the liver. However, due to lack of ST2+ Tregs in Il1rl1-/- mice, the remaining Tregs were less activated and showed decreased expression of inhibitory molecules in immune-mediated hepatitis. Another study also describing increased liver injury in Il1rl1-/- mice showed enhanced T-cell numbers and IFNγ expression (20), which might be a consequence of impaired Treg-mediated suppression of inflammatory T-cell responses in mice lacking ST2+ Tregs. In addition, one study using Il33-/- mice correlated impaired activation of ST2+ Tregs in absence of endogenous IL-33 with enhanced hepatic infiltration of NK cells and an aggravated liver injury (19). Taken together, these studies highlight the immunoregulatory role of endogenous IL-33 in immune-mediated hepatitis through inhibiting inflammatory immune cell activation and recruitment. Nevertheless, divergent effects of the IL-33/ST2 axis have been described in immune-mediated hepatitis. We have previously demonstrated that hepatic release of IL-33 induced activation of ILC2s that aggravated liver injury by recruitment of inflammatory eosinophils (17). Invariant natural killer T (iNKT) cells also became activated in immune-mediated hepatitis and induced strong nuclear IL-33 expression in hepatocytes (16, 25). Blockage of endogenous IL-33 signaling through an anti-IL-33 antibody reduced iNKT-cell activation and ameliorated disease pathology further supporting a pro-inflammatory role of IL-33 in immune-mediated hepatitis (18). Endogenous IL-33 has also been identified as a disease-driving factor in liver ischemia/reperfusion (I/R) injury, another disease with hepatic lesions. Here, Il33-/- (26, 27) and Il1rl1-/- mice (26) were shown to develop less severe liver I/R injury, which correlated with reduced infiltration of neutrophils (27) and formation of neutrophil extracellular traps in liver sinusoids (26). Moreover, iNKT cells have been implicated in the initiation of I/R injury and by using Il33-/- mice, it has been shown that their hepatic recruitment depended on release of IL-33 (28). Moreover, blockage of endogenous IL-33/ST2 signaling in Il1rl1-/- mice reduced acetaminophen-induced liver injury, in which massive necrosis and release of IL-33 correlated with hepatic recruitment of neutrophils (29). Thus, endogenous IL-33 aggravated hepatic I/R and drug-induced injury by recruitment of inflammatory immune cells to the liver. In contrast to the findings in Il1rl1-/- mice, one study demonstrated that IL-33 deficiency in Il33-/- mice exacerbated acetaminophen-induced liver injury by disrupting M2 macrophage polarization and activating hepatocyte autophagy. This demonstrates a protective effect of endogenous IL-33 in drug-induced liver injury through modulating macrophage polarization and hepatocyte autophagy (30). In conclusion, endogenous IL-33 can exert opposite effects on liver disease pathology depending on the targeted immune cell populations.
Our adoptive transfer experiments provided evidence for a superior immunosuppressive function of ST2+ Tregs activated by exogenous IL-33 in immune-mediated hepatitis and demonstrated an inhibitory effect on effector T-cell activation resulting in reduced expression of TNFα and IFNγ, two mediators of liver damage in immune-mediated hepatitis (31, 32). Since transfer of naïve Tregs did not ameliorate disease severity, this reveals the potential of exogenous IL-33 to selectively promote the anti-inflammatory effect of IL-33-responsive ST2+ Tregs in immune-mediated hepatitis. This is further supported by the finding that treatment with exogenous IL-33 protected Foxp3Cre –x ST2fl/fl but not Foxp3Cre+ x ST2fl/fl mice lacking ST2+ Tregs from immune-mediated hepatitis. Importantly, expression of TNFα and IFNγ by hepatic CD4+ and CD8+ T cells was enhanced in absence of ST2+ Tregs, further demonstrating that ST2+ Tregs pre-activated with exogenous IL-33 suppress inflammatory T-cell responses in immune-mediated hepatitis. Since one recent study demonstrated that Treg depletion resulted in enhanced iNKT-cell activation and liver injury in immune-mediated hepatitis, which was not the case in iNKT cell-deficient mice (33), it might conceivable that activated ST2+ Tregs also suppress inflammatory iNKT-cell activation.
The role of AREG in liver disease is not well understood. Elevated hepatic AREG expression have been described in patients with liver cirrhosis (34) and cholestatic liver disease (35). In mice, AREG were found to protect from cholestatic injury (35), and to promote liver regeneration after partial hepatectomy (34). In contrast, AREG aggravated murine liver fibrosis (36) and impaired anti-viral immune responses in murine hepatitis B virus infection (37). We here demonstrated a beneficial role of AREG in immune-mediated hepatitis, a well-established preclinical model that shares some similarities with acute AIH (15).
We focused our analyses to the acute inflammatory phase of the disease, where processes of liver regeneration have not started yet (38). In this disease phase, ST2+ Tregs and ILC2s, two liver-residing, IL-33-responsive immune cell subsets increased expression of AREG. That hepatic ILC2s can express AREG has also been shown in patients with various liver diseases (32). However, ST2+ Tregs and ILC2s were differentially influenced by AREG. ST2+ Tregs depended on intrinsic expression of AREG for their survival and function as shown by reduced ST2+ Treg numbers in Areg-/- mice despite enhanced expression of Foxp3 and anti-apoptotic Bcl-2 or stronger proliferation, and an impaired capacity of Areg-/- Tregs to suppress CD4+ T-cell activation. Interestingly, we found that exogenous AREG selectively activated ST2+ Tregs although ST2- Tregs also expressed the AREG receptor EGFR. AREG has been described to promote the immunosuppressive activity of EGFR-expressing Tregs in vitro (12, 37). However, EGFR-independent mechanisms of AREG function need to be elucidated. Importantly, the activating effect of AREG was lost in Areg-/- ST2+ Tregs, indicating that exogenous AREG cannot compensate for the loss of intrinsic AREG expression in ST2+ Tregs. The finding of an impaired activation of Areg-/- ST2+ Tregs by exogenous IL-33 further highlights that AREG expression by ST2+ Tregs acts in an autocrine loop to promote their activation and function. Thus, exogenous IL-33 and AREG stimulate AREG expression in ST2+ Tregs thereby supporting their maintenance and function in immune-mediated hepatitis.
In contrast to the stimulating effect on ST2+ Tregs, exogenous AREG inhibited hepatic ILC2 activation and cytokine expression and also reduced IL-33-mediated activation of ILC2s in vitro. In line with this, we observed increased ILC2 activation and cytokine expression in immune-mediated hepatitis in absence of AREG. Thus, AREG-expressing ILC2s might support ST2+ Treg function and inhibit their own effector function in immune-mediated hepatitis. Since ST2+ Tregs also express AREG, they might contribute to the regulation of ILC2 activation in liver injury. Consistently, a stimulating effect of ILC2s on the suppressive capacity of Tregs has been shown in vitro (39). Moreover, Tregs were found to inhibit ILC2 cytokine expression in vitro and in vivo (40). Our data further showed that in contrast to ST2+ Tregs, ILC2s did not depend on intrinsic AREG expression for function since activation of ILC2s by exogenous IL-33 was not altered in Areg-/- mice.
AREG-expressing ST2+ Tregs and ILC2s may also exert a tissue protective effect in immune-mediated hepatitis as it was shown for Tregs in muscle repair (41) and viral infection (42) as well as for ILC2s in biliary atresia (43), lung infection (6) and intestinal inflammation (8). In the liver, AREG has been identified as mitogen for hepatocytes thereby supporting hepatic regeneration (34). A recent studies further showed that AREG promoted epithelial homeostasis and repair in experimental biliary atresia (43).
There are some limitations of the study. The functional relevance of AREG specifically expressed by ST2+ Tregs or ILC2s in immune-mediated hepatitis is still not known. Adoptive transfer experiments with Tregs or hepatic ILC2s isolated from Areg-/- mice would be possible to assess disease pathology of immune-mediated hepatitis if Tregs or hepatic ILC2s do not express AREG. Moreover, the effect of exogenous AREG on ST2+ Tregs and hepatic ILC2s has been investigated in vitro. To strengthen these data, treatment of mice with exogenous AREG and subsequent ST2+ Treg and ILC2 phenotype analysis could be done. Additionally, we compared frequencies of the different immune cells in our analyses. Cell numbers would support these data and therefore, counting beads should be included in the analyses to calculate cell numbers in future studies. In the end, this is a murine study, which allows functional analyses to identify novel immunological pathways involved in the pathogenesis of immune-mediated hepatitis. However, the findings presented here need to be further addressed in patients with immune-mediated liver disease.
In summary, this study reveals the high immunosuppressive capacity of ST2+ Tregs in immune-mediated hepatitis, which depend on intrinsic AREG as well as exogenous IL-33 for their maintenance, activation and function in liver inflammation. It further identifies AREG as inhibitor of hepatic ILC2 effector function. The fact that the ST2+ Treg/AREG axis promotes immunoregulation and inhibits immunity in immune-mediated hepatitis provides important insights into the cellular and molecular mechanisms through which hepatic immune responses are regulated in immune-mediated hepatitis.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The animal study was approved by Behörde für Justiz und Verbraucherschutz, Hamburg, Germany. The study was conducted in accordance with the local legislation and institutional requirements.
SW: Formal analysis, Investigation, Methodology, Writing – review & editing. FJ: Formal analysis, Investigation, Methodology, Writing – review & editing. AO: Investigation, Writing – review & editing. FH: Investigation, Writing – review & editing. AW: Methodology, Writing – review & editing. GT: Conceptualization, Writing – review & editing. KN: Conceptualization, Funding acquisition, Supervision, Visualization, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work received financial support from the Deutsche Forschungsgemeinschaft (DFG) priority program SPP 1937 “Innate Lymphoid Cells” granted to KN and Landesforschungsförderung (LFF) Hamburg granted to KN.
The authors thank the excellent technical assistance of Elena Tasika and Carsten Rothkegel and the members of the Cytometry und Cell Sorting Core Unit for cell sorting.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1351405/full#supplementary-material
Tregs, regulatory T cells; ILC2s, type 2 innate lymphoid cells; AREG, amphiregulin; concanavalin A, ConA; AIH, autoimmune hepatitis; ST2, suppression of tumorigenicity 2; rm, recombinant murine; ALT, alanine transaminase; H&E, hematoxylin and eosin; lin, lineage; CFSE, carboxyfluorescein succinimidyl ester; KLRG1, killer cell lectin like receptor G1; ICOS, inducible T cell co-stimulator; CTLA-4, cytotoxic T lymphocyte-associated protein 4; PD-L1, programmed death ligand 1; TIGIT, T cell immunoreceptor with Ig and ITIM domains; WT, wild-type; IFN, interferon; TNF, tumor necrosis factor; Bcl-2, B cell lymphoma 2; iNKT, invariant natural killer T; I/R, ischemia reperfusion.
1. Carbone M, Neuberger JM. Autoimmune liver disease, autoimmunity and liver transplantation. J Hepatol. (2014) 60:210–23. doi: 10.1016/j.jhep.2013.09.020
2. Cayrol C, Girard JP. IL-33: An alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. (2014) 31:31–7. doi: 10.1016/j.coi.2014.09.004
3. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
4. Griesenauer B PS. The ST2/IL-33 axis in immune cells during inflammatory diseases. Front Immunol. (2017) 8:475. doi: 10.3389/fimmu.2017.00475
5. Kabata H, Moro K, Koyasu S. The group 2 innate lymphoid cell (ILC2) regulatory network and its underlying mechanisms. Immunol Rev. (2018) 286:37–52. doi: 10.1111/imr.12706
6. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. (2011) 12:1045–54. doi: 10.1038/ni.2131
7. Zaiss DMW, Gause WC, Osborne LC Artis D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity. (2015) 42:216–26. doi: 10.1016/j.immuni.2015.01.020
8. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A. (2015) 112:10762–7. doi: 10.1073/pnas.1509070112
9. Siede J, Fröhlich A, Datsi A, Hegazy AN, Varga DV, Holecska V, et al. IL-33 receptor-expressing regulatory t cells are highly activated, Th2 biased and suppress CD4 T Cell proliferation through IL-10 and TGFβ Release. PloS One. (2016) 11:e0161507. doi: 10.1371/journal.pone.0161507
10. Delacher M, Imbusch CD, Weichenhan D, Breiling A, Hotz-Wagenblatt A, Träger U, et al. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat Immunol. (2017) 18:1160–72. doi: 10.1038/ni.3799
11. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. (2014) 513:564–8. doi: 10.1038/nature13577
12. Zaiss DM, van Loosdregt J, Gorlani A, Bekker CP, Gröne A, Sibilia M, et al. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity. (2013) 38:275–84. doi: 10.1016/j.immuni.2012.09.023
13. Neumann K, Schiller B, Tiegs G. NLRP3 inflammasome and IL-33: novel players in sterile liver inflammation. Int J Mol Sci. (2018) 19:2732. doi: 10.3390/ijms19092732
14. Abe K, Takahashi A, Fujita M, Hayashi M, Okai K, Nozawa Y, et al. Interleukin-33/ST2-mediated inflammation plays a critical role in the pathogenesis and severity of type I autoimmune hepatitis. Hepatol Commun. (2019) 3:670–84. doi: 10.1002/hep4.1326
15. Erhardt A, Tiegs G. Tolerance induction in response to liver inflammation. Dig Dis. (2010) 28:86–92. doi: 10.1159/000282069
16. Arshad MI, Rauch M, L’helgoualc’h A, Julia V, Leite-de-Moraes MC, Lucas-Clerc C, et al. NKT cells are required to induce high IL-33 expression in hepatocytes during ConA-induced acute hepatitis. Eur J Immunol. (2011) 41:2341–8. doi: 10.1002/eji.201041332
17. Neumann K, Karimi K, Meiners J, Voetlause R, Steinmann S, Dammermann W, et al. A proinflammatory role of type 2 innate lymphoid cells in murine immune-mediated hepatitis. J Immunol. (2017) 198:128–37. doi: 10.4049/jimmunol.1600418
18. Chen J, Duan L, Xiong A, Zhang H, Zheng F, Tan Z, et al. Blockade of IL-33 ameliorates Con A-induced hepatic injury by reducing NKT cell activation and IFN-γ production in mice. J Mol Med (Berl). (2012) 90:1505–15. doi: 10.1007/s00109-012-0938-4
19. Noel G, Arshad MI, Filliol A, Genet V, Rauch M, Lucas-Clerc C, et al. Ablation of interaction between IL-33 and ST2 + regulatory T cells increases immune cell-mediated hepatitis and activated NK cell liver infiltration. Am J Physiol Gastrointest Liver Physiol. (2016) 311:G313–23. doi: 10.1152/ajpgi.00097.2016
20. Volarevic V, Mitrovic M, Milovanovic M, Zelen I, Nikolic I, Mitrovic S, et al. Protective role of IL-33/ST2 axis in Con A-induced hepatitis. J Hepatol. (2012) 56:26–33. doi: 10.1016/j.jhep.2011.03.022
21. Pastille E, Wasmer MH, Adamczyk A, Vu VP, Mager LF, Phuong NNT, et al. The IL-33/ST2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer. Mucosal Immunol. (2019) 12:990–1003. doi: 10.1038/s41385-019-0176-y
22. Kamanaka M, Kim ST, Wan YY, Sutterwala FS, Lara-Tejero M, Galán JE, et al. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity. (2006) 25:941–52. doi: 10.1016/j.immuni.2006.09.013
23. Steinmann S, Schoedsack M, Heinrich F, Breda PC, Ochel A, Tiegs G, et al. Hepatic ILC2 activity is regulated by liver inflammation-induced cytokines and effector CD4+ T cells. Sci Rep. (2020) 10:1071. doi: 10.1038/s41598-020-57985-w
24. Zhang C, Li L, Feng K, Fan D, Xue W, Lu J. “Repair” Treg cells in tissue injury. Cell Physiol Biochem. (2017) 43:2155–69. doi: 10.1159/000484295
25. Arshad MI, Piquet-Pellorce C, L’Helgoualc’h A, Rauch M, Patrat-Delon S, Ezan F, et al. TRAIL but not FasL and TNFα, regulates IL-33 expression in murine hepatocytes during acute hepatitis. Hepatology. (2012) 56:2353–62. doi: 10.1002/hep.25893
26. Yazdani HO, Chen HW, Tohme S, Tai S, van der Windt DJ, Loughran P, et al. IL-33 exacerbates liver sterile inflammation by amplifying neutrophil extracellular trap formation. J Hepatol. (2017) 68:130–139. doi: 10.1016/j.jhep.2017.09.010
27. Barbier L, Robin A, Sindayigaya R, Ducousso H, Dujardin F, Thierry A, et al. Endogenous interleukin-33 acts as an alarmin in liver ischemia-reperfusion and is associated with injury after human liver transplantation. Front Immunol. (2021) 12:744927. doi: 10.3389/fimmu.2021.744927
28. Robin A, Mackowiak C, Bost R, Dujardin F, Barbarin A, Thierry A, et al. Early activation and recruitment of invariant natural killer T cells during liver ischemia-reperfusion: the major role of the alarmin interleukin-33. Front Immunol. (2023) 14:1099529. doi: 10.3389/fimmu.2023.1099529
29. Antunes MM, Araújo AM, Diniz AB, Pereira RVS, Alvarenga DM, David BA, et al. IL-33 signalling in liver immune cells enhances drug-induced liver injury and inflammation. Inflammation Res. (2018) 67:77–88. doi: 10.1007/s00011-017-1098-3
30. Wang Z, Wu L, Pan B, Chen Y, Zhang T, Tang N. Interleukin 33 mediates hepatocyte autophagy and innate immune response in the early phase of acetaminophen-induced acute liver injury. Toxicology. (2021) 456:152788. doi: 10.1016/j.tox.2021.152788
31. Gantner F, Leist M, Lohse AW, Germann PG, Tiegs G. Concanavalin A-induced T-cell-mediated hepatic injury in mice: the role of tumor necrosis factor. Hepatology. (1995) 21:190–8. doi: 10.1002/(ISSN)1527-3350
32. Küsters S, Gantner F, Künstle G, Tiegs G. Interferon gamma plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gatroenterology. (1996) 111:462–71. doi: 10.1053/gast.1996.v111.pm8690213
33. Venken K, Decruy T, Sparwasser T ED. Tregs protect against invariant NKT cell-mediated autoimmune colitis and hepatitis. Immunology. (2024) 171:277–85. doi: 10.1111/imm.13718
34. Berasain C, García-Trevijano ER, Castillo J, Erroba E, Lee DC, Prieto J, et al. Amphiregulin: an early trigger of liver regeneration in mice. Gastroenterology. (2005) 128:424–32. doi: 10.1053/j.gastro.2004.11.006
35. Santamaría E, Rodríguez-Ortigosa CM, Uriarte I, Latasa MU, Urtasun R, Alvarez-Sola G, et al. The epidermal growth factor receptor ligand amphiregulin protects from cholestatic liver injury and regulates bile acids synthesis. Hepatology. (2019) 69:1632–47. doi: 10.1002/hep.30348
36. Perugorria MJ, Latasa MU, Nicou A, Cartagena-Lirola H, Castillo J, Goñi S, et al. The epidermal growth factor receptor ligand amphiregulin participates in the development of mouse liver fibrosis. Hepatology. (2008) 48:1251–61. doi: 10.1002/hep.22437
37. Dai K, Huang L, Chen J, Yang L, Gong Z. Amphiregulin promotes the immunosuppressive activity of intrahepatic CD4+ regulatory T cells to impair CD8+ T-cell immunity against hepatitis B virus infection. Immunology. (2015) 144:506–17. doi: 10.1111/imm.12400
38. Trautwein C, Rakemann T, Malek NP, Plümpe J, Tiegs G, Manns MP. Concanavalin A-induced liver injury triggers hepatocyte proliferation. J Clin Invest. (1998) 101:1960–9. doi: 10.1172/JCI504
39. Rauber S, Luber M, Weber S, Maul L, Soare A, Wohlfahrt T, et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat Med. (2017) 23:938–44. doi: 10.1038/nm.4373
40. Rigas D, Lewis G, Aron JL, Wang B, Banie H, Sankaranarayanan I, et al. Type 2 innate lymphoid cell suppression by regulatory T cells attenuates airway hyperreactivity and requires inducible T-cell costimulator-inducible T-cell costimulator ligand interaction. J Allergy Clin Immunol. (2017) 139:1468–1477.e2. doi: 10.1016/j.jaci.2016.08.034
41. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell. (2013) 155:1282–95. doi: 10.1016/j.cell.2013.10.054
42. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell. (2015) 27:1078–89. doi: 10.1016/j.cell.2015.08.021
Keywords: Treg maintenance and function, ST2+ Tregs, ILC2s, amphiregulin, hepatic immunoregulation, liver inflammation
Citation: Wachtendorf S, Jonin F, Ochel A, Heinrich F, Westendorf AM, Tiegs G and Neumann K (2024) The ST2+ Treg/amphiregulin axis protects from immune-mediated hepatitis. Front. Immunol. 15:1351405. doi: 10.3389/fimmu.2024.1351405
Received: 06 December 2023; Accepted: 29 February 2024;
Published: 20 March 2024.
Edited by:
Pushpa Pandiyan, Case Western Reserve University, United StatesReviewed by:
Alice Barbarin, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2024 Wachtendorf, Jonin, Ochel, Heinrich, Westendorf, Tiegs and Neumann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katrin Neumann, a2F0Lm5ldW1hbm5AdWtlLmRl
†Present address: Fabian Heinrich, Institute of Legal Medicine, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
Center for Data and Statistical Science for Health, London School of Hygiene and Tropical Medicine, London, United Kingdom
‡These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.