 Roy P. M. Snijckers
Roy P. M. Snijckers Amanda C. Foks
Amanda C. Foks- Division of BioTherapeutics, Leiden Academic Centre for Drug Research, Leiden University, Leiden, Netherlands
Adaptive immunity plays a profound role in atherosclerosis pathogenesis by regulating antigen-specific responses, inflammatory signaling and antibody production. However, as we age, our immune system undergoes a gradual functional decline, a phenomenon termed “immunosenescence”. This decline is characterized by a reduction in proliferative naïve B- and T cells, decreased B- and T cell receptor repertoire and a pro-inflammatory senescence associated secretory profile. Furthermore, aging affects germinal center responses and deteriorates secondary lymphoid organ function and structure, leading to impaired T-B cell dynamics and increased autoantibody production. In this review, we will dissect the impact of aging on adaptive immunity and the role played by age-associated B- and T cells in atherosclerosis pathogenesis, emphasizing the need for interventions that target age-related immune dysfunction to reduce cardiovascular disease risk.
1 Introduction
Aging leads to an overall impairment of physiological systems due to a gradual decline of cells in terms of function and viability (1). Aging of the immune system, termed “immunosenescence”, induces a skewing of hematopoietic stem cells (HSCs) towards myelopoiesis, diminishing the lymphoid cell pool (2). Alongside a reduction in proliferative naïve T- and B cells and relative increase in central memory cells, the B- and T cell receptor repertoire is reduced upon aging, resulting in decreased antigen responses (3, 4). Furthermore, the adaptation of a senescence associated secretory profile (SASP) by immune cells further contributes to a chronic low-level inflammation termed “inflammaging” (5). Collectively, immunosenescence impairs the response to physiological stress in the aging population while increasing susceptibility to infections and inflammatory disease (6, 7). Aging has additionally emerged as one of the most prominent risk factors for cardiovascular disease (CVD). Specifically, in 2019, the annual rate of cardiovascular-related death was increased by more than 80-fold when comparing age groups of 15-49 years to 70 years and older, whilst being responsible for more than half of all deaths in the latter (8). Atherosclerosis, a lipid-driven chronic inflammatory disease, lies at the heart of CVD pathogenesis (9). Whereas initiation of atherosclerotic plaques often occurs upon damage to the endothelium and subsequent infiltration of lipids into the vessel wall, its progression is marked by the infiltration of immune components leading to chronic inflammation of the plaque (10). Over time, the formation of necrotic debris, plaque destabilization and eventual rupture drive potentially fatal acute cardiovascular events such as a myocardial infarction or stroke (11). In light of the gradual functional decline of the aging immune system, it comes as no surprise that the incidence of acute cardiovascular events also greatly increases with age, even though atherosclerotic vascular changes already start occurring during early adolescence (12, 13). Understanding the interplay between aging, adaptive immunity, and atherosclerosis has significant implications for understanding and addressing age-related cardiovascular complications. Preclinical research has paved the way for this understanding, providing mechanistic insights into the role of adaptive immunity in atherosclerosis pathogenesis. Nevertheless, the majority of preclinical research into atherosclerosis is performed using young adult mice models of 3-6 months of age, equivalent to 20-30 years in humans (14). This subsequently poses translational problems to the actual atherosclerotic cardiovascular disease patient population seeking treatment at advanced age. The characterization of atherosclerotic development in available aging mouse models has shown promising potential in bridging this gap of knowledge. It has been shown previously that aging promotes atherosclerosis development in low density lipoprotein receptor (Ldlr) deficient mice as well as in Apolipoprotein E (ApoE) deficient mice (15–17). Similarly, C57BL/6 mice treated with PCSK9-AAV to induce hypercholesterolemia at advanced age, show increased atherosclerosis development compared to their young counterparts (18). Particularly in chow-diet fed aged Ldlr-/- mice, plaques gradually developed in the context of mild hypercholesterolemia, and show human-like plaque features such as fibrosis and calcification (15). In addition, the immune system ages in these mice, displaying both immunosenescence and inflammaging, providing a clinically relevant setting for elucidating the impact of the aging immune system on atherosclerosis progression (19). With a rising life expectancy and a rapidly increasing aging population, enhancing our understanding of age-associated immunity has developed into a major public health priority.
In this review, we provide a comprehensive overview on the current state of knowledge regarding the changing role of adaptive immunity in atherosclerotic plaque development upon aging. First, the scene will be set through a discussion on the importance of adaptive immunity in atherosclerosis pathogenesis and the hallmarks of immunosenescence. Subsequently, atherosclerosis specific T- and B cell dynamics will be dissected in the context of the aging cell and deteriorating lymphoid tissues. Finally, the potential use of immunoaging as a biomarker or therapeutic target to combat atherosclerotic CVD will be discussed.
2 Adaptive immunity in atherosclerosis
The hallmark feature of atherosclerotic plaque initiation is considered to be the accumulation of low density lipoproteins (LDL) in the tunica intima. This can occur due to a "leaky" endothelial cell layer of the vessel wall in response to damage, for example at sites of shear stress (20). Modification of LDL, primarily oxidation (oxLDL), promotes the recruitment and infiltration of monocytes into the vessel wall, and the subsequent accumulation of cholesterol-enriched foam cells that contribute to plaque growth and necrotic core formation (21, 22). Adaptive immune responses, carried out by T- and B cells, play a crucial role in atherosclerosis progression (23, 24).
2.1 T cell immunity in atherosclerosis
Distinct subsets of T cells, both effector memory and regulatory T cells (Tregs), influence plaque development and stability. Notably, interferon gamma (IFNγ) secreting T helper (Th) 1 cells are the most common T cells found in atherosclerotic plaques. Th1 cells are considered pro-atherogenic, partially due to their role in stimulating macrophage polarization towards pro-inflammatory M1 effector cells (25). Conversely, interleukin (IL) 10 and transforming growth factor-β secreting Tregs exhibit clear anti-atherogenic functions, such as the inhibition of pro-atherogenic effector T cells as well as the stimulation of efferocytosis by macrophages (26, 27). This atheroprotective role of Tregs may however evolve into maladaptive immunity as Tregs autoreactive to Apolipoprotein B 100 (ApoB100), the main protein in LDL particles, can lose the expression of CD25 and forkhead box P3 (FoxP3) in hyperlipidemic ApoE-/- mice. In atherosclerosis, these so-called exTregs have been shown to exhibit a pro-inflammatory Th1/Th17 phenotype with cytotoxic properties (28, 29). A fraction of exTregs can additionally convert to T follicular helper (Tfh) cells during atherogenesis dependent on the accumulation of intracellular cholesterol (30). The role of Th2 and Th17 cells can be either pro- or anti-atherogenic, dependent on the cytokines they secrete (31–33). Similarly, cytotoxic CD8+ T cells display pro-atherogenic effects through IFNγ secretion and stimulating monopoiesis but can additionally exert an atheroprotective function by regulating Tfh cell mediated germinal center (GC) B cell responses as well as contributing to plaque stability (34–36). Advances in single cell technology further support the importance of adaptive immunity in atherosclerosis and revealed T-cells to be the most abundant leukocyte present in human carotid atherosclerotic plaques, outnumbering myeloid populations (37). Additionally, T cell receptor (TCR) sequencing has exposed plaque specific clonal expansion of CD4+ effector T cells with transcriptome profiles indicative of recent antigen-mediated T cell activation, thus suggesting an autoimmune component in atherosclerosis pathology (38).
The contribution of T cells to pathogenic immunity is not only restricted to the atherosclerotic plaque. Rather, their involvement in the activation of B cells at secondary lymphoid organs (SLOs), such as the lymph nodes or the spleen, plays a significant role (22, 39). Here, naïve T cells are primed by follicular dendritic cells (FDCs) in T cell zones, leading to their differentiation towards pro-atherogenic Tfh cells (40–42). C-X-C Motif Chemokine Ligand (CXCL) 13 dependent localization of Tfh cells towards the T-B cell border allows for their direct interaction with B cells (43–45). Symbiotic IL-21/IL21R and CD40/CD40L signaling further solidifies commitment to the Tfh lineage and drives FO B cell differentiation towards GC B cells (42, 46). Through this interaction, Tfh cells drive the humoral response by aiding in antibody affinity maturation through stimulation of somatic hypermutation and class switch recombination (42). Upon differentiation into plasmablasts and plasma cells, B cells influence atherosclerosis progression via the production of auto-antibodies targeting oxLDL and necrotic debris (47). Tfh cells are counteracted by follicular regulatory T helper cells (Tfr) which express both Tfh markers such as Bcl-6 and CXCR5 for migration on the CXCL13 gradient as well as the Treg marker FoxP3 (48). Initially, Tfr also express the Treg marker CD25, but down regulate its expression upon maturation and migration towards the germinal center, favoring the expression of Tfh related genes (49). The adoptive transfer of Tfr cells in ApoE-/- mice has resulted in a decrease of atherosclerotic plaques alongside a decrease in infiltrating macrophages as well as a decrease in the amount of splenic Tfh cells whilst stimulating the expansion of atheroprotective regulatory B cells (50).
2.2 B cell immunity in atherosclerosis
Research concerning the atherogenicity of B cells has highlighted the existence of both pro-atherogenic and atheroprotective B cell subsets. B2 cells are the most prominent subset present within the B cell population of the circulation and SLOs. B2 cells are subdivided into follicular B cells (FO B cells) and marginal zone B cells (MZ B cells) which either stimulate or inhibit plaque progression, depending on the cell type. FO B cells mainly contribute to the germinal center response through their interaction with Tfh cells, differentiating into GC B cells to produce high affinity immunoglobulin (Ig) G autoantibodies against (neo) self-antigens (51). Previously, a 50% decrease in FO B cells was achieved through stimulation of the B- and T-lymphocyte attenuator (BTLA) pathway, which significantly diminished atherosclerotic plaque size in Ldlr-/- mice (52). Additionally, a lack of FO B cells induced by CD23-specific Blimp-1 or Pax5 deletion leads to a reduction in IgG and atherosclerotic lesion size, while IgG from atherosclerotic mice, compared to IgG derived from non-atherosclerotic mice, increased plaque size when injected into Ldlr-/- mice lacking endogenous IgG (53, 54). IgG autoantibodies targeting oxidation specific epitopes (OSEs) such as heat shock protein (HSP) 60/65 overexpressed by stressed endothelial cells as well as products of oxLDL breakdown such as malondialdehyde, 4-hydroxynenal and phosphocholine-containing oxidized phospholipids have furthermore been shown to increase due to hyperlipidemia (54, 55). As the polar opposite to FO B cells, MZ B cells inhibit Tfh cells, suppressing their atherogenic functionality while producing atheroprotective IgM in a T-cell dependent manner (56, 57). B1 cells, composed of B1a and B1b cells, similarly stand among the anti-atherogenic B cell subsets by producing substantial amounts of germline encoded IgM natural antibodies targeting OSEs. Through OSE binding, these natural antibodies facilitate efficient phagocytosis of oxLDL and apoptotic cells in non-immunogenic ways (58). Lastly, IL-10+ regulatory B cells have been shown to inversely correlate with plaque severity (59). Nevertheless, whether the atheroprotective role of regulatory B cells is IL-10 dependent is unclear as B cell–specific deficiency in IL-10 has not been shown to affect lesion size or composition in Ldlr-/- mice (60).
Summarizing, adaptive immunity significantly impacts atherosclerosis pathogenesis, with T- and B cells playing pivotal roles. T cell subsets such as Th1 cells are pro-atherogenic, while Tregs exhibit anti-atherogenic functions but can become maladaptive in hyperlipidemic conditions. Additionally, T cells facilitate humoral responses through interacting with B cells in SLOs, aiding in the production and affinity maturation of IgG auto-antibodies against (neo) self-antigens. B cell subsets have dichotomous effects on plaque progression, with FO B cells promoting it and Bregs as well as OSE-specific IgM producing MZ B cells and B1 cells offering atheroprotection. Understanding the nuances of adaptive immunity in atherosclerosis is crucial for developing targeted therapeutic interventions.
3 Immunosenescence
As we age, the components of our immune system undergo significant changes in terms of viability and function, potentially impacting the role they play in CVD. Several immunological changes can be included among the cardinal features of immunosenescence such as an increase in central memory cells alongside a decrease in peripheral naïve T- and B cells, increased autoimmune responses by cells of the adaptive immune system and a reduction in the capacity for antigen stimulated immune responses, severely impacting the susceptibility for infectious disease and age-associated inflammatory diseases in the elderly population (3–7). Additionally, immunosenescence induces an increase in chronic systemic inflammation caused by changes in the secretory profile of senescent immune cells, i.e. the SASP (5). To understand these aging-associated changes, it is however of vital importance to first discuss the most commonly established mechanisms underlying immunosenescence, before diving deeper into the effects of aging on T- and B cells in atherosclerosis.
3.1 The “first hit” paradigm
The “first hit” paradigm, as postulated by Franceschi et al. in 1997, encapsulates the concept of chronic exposure to stressors leading to a gradual increase in pro-inflammatory status over time, thereby augmenting the risk of age-related diseases (61). These stressors are represented by physical, chemical and biological agents which constantly assault organisms. Among these stressors, exogenous factors, such as x-rays, ultraviolet light, and a variety of chemicals; as well as endogenous reactive metabolites consistently impede on cellular viability (62, 63). Protection is granted through the DNA damage response system, antioxidants and the immune system while the individual variety in genetic disposition conveys variable susceptibility to specific stressors, aging and disease. This chronic exposure to stressors can be partially attributed to natural chronological aging, representing the passage of time. As such, some features of immunosenescence are inherent to chronological aging, including both thymic involution and replicative immunosenescence which will be discussed in detail in the next sections (64, 65). Nevertheless, biological aging, encompassing the multifaceted interplay of various agents influencing the aging process, is profoundly affected by lifestyle factors such as diet, exercise, and smoking, as well as various comorbidities which can induce premature biological aging in terms of stress-induced senescence, cholesterol-induced changes in hematopoiesis and excessive DNA damage (66, 67). Similarly, chronic infections, for example with cytomegalovirus (CMV), can lead to immunosenescence, increasing senescent CD4+ CD28- T cells, as well as increasing CVD risk (68–70). An increased risk for complications related to ischemic CVD has similarly been observed to present itself in a cohort of older individuals following infections with SARS-CoV-2 (71). Chronic infections with HIV or human gamma herpes viruses, such as Epstein-Barr virus, furthermore lead to an increase in age-associated immune subsets, such as age-associated B cells (ABC), which could contribute to inflammatory diseases (72).
3.2 Aging-induced changes in hematopoiesis
Immunosenescence is a main contributor to the “first hit” by driving inflammation. One of the most prominent features of immunosenescence becomes apparent through aging-induced changes in the hematopoietic output of primary lymphoid tissues, namely the bone marrow and thymus. The bone marrow has a critical function in producing common lymphoid progenitor cells (CLPs) during hematopoiesis and serves as the primary site for B cell development (73). However, with age, the quality of HSCs drops in terms of regenerative capacity, impacting self-renewal. This in turn leads to a reduction in the output of naïve T- and B cells from the bone marrow (74).
Additionally, HSCs from aged (22-24 months) mice have been shown to down-regulate genes involved in lymphopoiesis while upregulating myeloid genes when compared to young mice (2-3 months), resulting in a skewing of hematopoiesis from lymphoid cells towards a myeloid output (75) (Figure 1). Hematopoietic myeloid skewing upon aging has furthermore been validated in the human hematopoietic system on a cellular level as well as through gene expression profiling (76). Notably, myelopoiesis can also be regulated through cholesterol efflux pathways and is promoted through hypercholesteremia (66, 67). Various studies have concluded both hypercholesteremia and hyperglycemia induced myelopoiesis to accelerate atherosclerosis and impair its resolution (67, 77, 78).
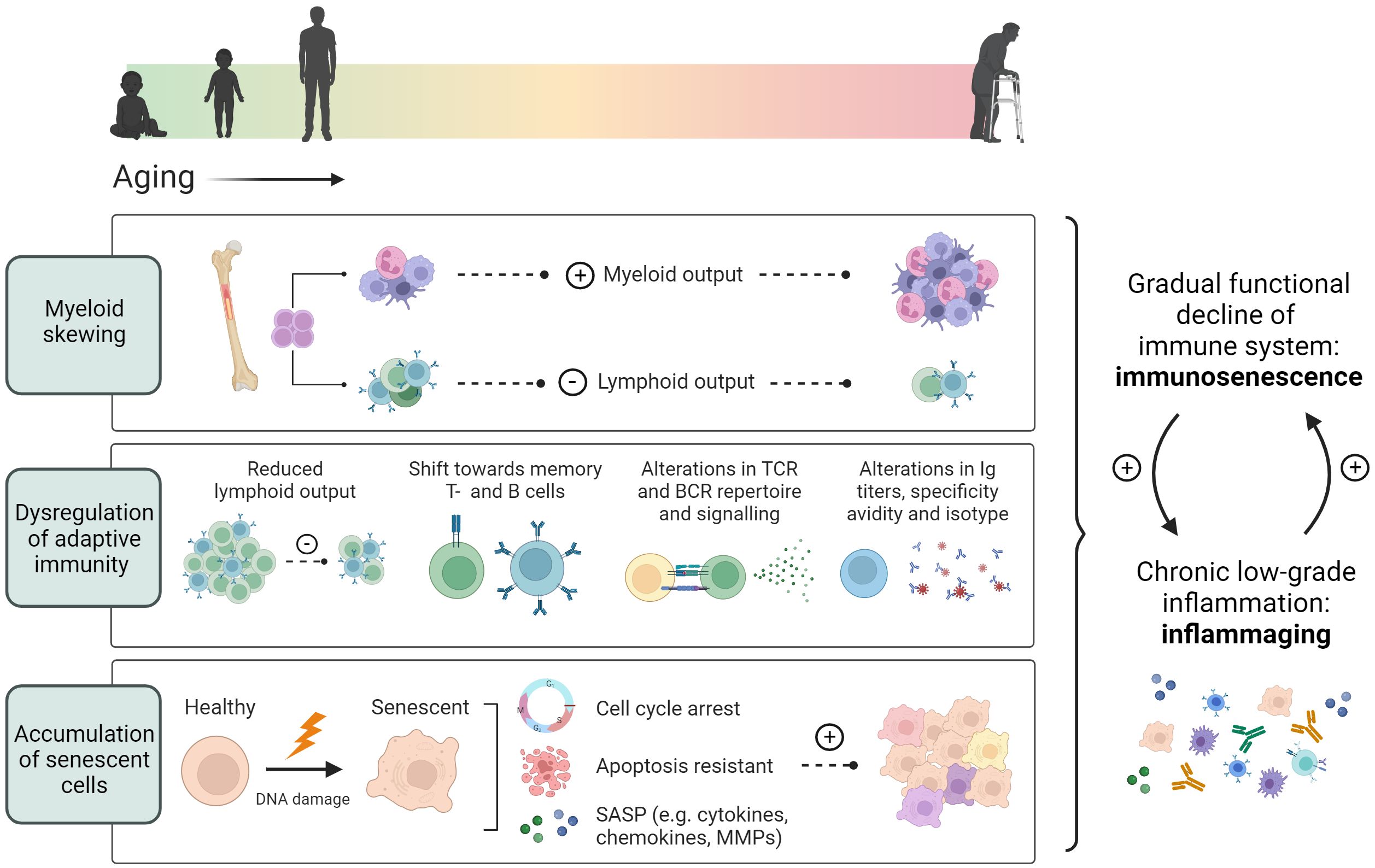
Figure 1 Age-associated immune alterations. Aging promotes skewing of hematopoietic stem cells towards myelopoiesis, resulting in a reduced lymphoid output from the bone marrow. In addition, thymic involution causes a decrease in the peripheral naïve T cell pool, while memory T- and B cells increase during aging. An age-associated reduction in TCR and BCR repertoire, and alterations in antibody secretion, specificity and avidity, further contribute to dysregulated adaptive immunity. Additionally, aging induces cellular senescence characterized by an induction of cell cycle arrest, apoptotic resistance and the production of a senescence associated secretory profile. These age-associated changes lead to an overall functional decline of the immune system termed immunosenescence and a chronic low-grade inflammation termed inflammaging.
Aging additionally stimulates the clonal hematopoiesis of indeterminate potential (CHIP) which refers to the expansion of HSCs containing mutations unaffiliated to the development of hematopoietic malignancies (79). Notably, Jaiswal et al. have confirmed a 1.9-fold elevated risk of coronary artery disease in carriers of CHIP compared to non-carriers alongside an increase in coronary artery calcification and a 4-fold greater risk for early onset myocardial infarction (80). The risk of coronary artery disease positively associated with CHIP mutations of DNMT3A, ASXL1, JAK2 and TET2 as determined by whole genome sequencing.
Following the hematopoietic production of CLPs in the bone marrow, CLPs relocate to the thymus where they develop into competent and immunotolerant naïve T cells. However, the thymus already becomes fully developed in the early postnatal phase and shrinks with age, a process termed thymic involution. This deterioration of the thymus is associated with a gain of adipose tissue and severely affects the output of naïve T cells into the periphery, a prominent hallmark of immunosenescence (65, 81).
3.3 Accumulation of senescent cells
Immune cells can acquire a senescent phenotype upon aging as characterized by an irreversible loss of proliferative capacity, resistance to apoptotic cell death, and the acquisition of a senescence-associated secretory phenotype (82, 83) (Figure 1). This is partially due to the fact that most cells cannot divide indefinitely. With each replication cycle that occurs, telomeres shorten. When they become of insufficient length to support cell division, cells go into replicative senescence (64). In addition to classical replicative senescence induced through telomere shortening, driving factors working towards replicative senescence have been described as the accumulation of DNA damage and an overactive DNA damage response (82). The buildup of DNA damage with age is stimulated through the prolonged assault on the genetic code by stressors, resulting in genomic instability (63). The accumulation of DNA damage confers cytotoxicity, activating cell cycle arrest checkpoints, which serve as a signal to send cells down apoptotic pathways in order to protect them from damaged or misincorporated nucleotides becoming mutagenic (82). However, with prolonged cell cycle arrest, cells can become senescent, acquiring a resistance against apoptosis in the process (5, 84). The link between DNA damage and atherosclerosis has long been known, with coronary artery disease patients exhibiting significantly increased accumulation of DNA damage in peripheral blood lymphocytes, correlating with atherosclerosis severity and risk factors such as diabetes and hypercholesterolemia (85–87).
It must however be noted that the identification of senescent cells has proven a significant hurdle due to the lack of a single specific marker. The expression of cyclin-dependent kinase inhibitor p16Ink4a and senescence-associated β-galactosidase (SA-β-gal) have for instance been extensively used in previous research as markers of cellular senescence (88–91). However, atherosclerotic plaques from human endarterectomy samples display an increase in p16Ink4a concomitant to macrophage content (92). Additionally, a subclass of p16Ink4a and SA-β-gal expressing macrophages has been observed as distinct from bona fide senescent cells induced through genotoxic stress in vitro (93). Expression of p16Ink4a becomes upregulated upon monocyte differentiation while both SA-β-gal and p16Ink4a exert control over macrophage polarization in response to immune stimuli (94, 95). p16Ink4a deficiency in bone marrow derived macrophages additionally leads to a reduction in pro-inflammatory signaling (96). Therefore, it would not be advised to rely solely on SA-β-gal and p16Ink4a as markers of senescence, especially in light of the prevalent role played by pro-inflammatory macrophages in plaque development. Similarly, expression of p53 and p21cip1 are often used as markers of senescence to address cell cycle arrest, proteins such as ataxia-telangiectasia mutated protein, ataxia telangiectasia and Rad3-related protein and phosphorylated histone H2AX are used as indicators of excessive DNA damage and an overactive DNA damage response, while the loss of the cell’s capacity to proliferate in its senescent state is attributed to reduced Ki-67 levels (82). However, on their own, none of these markers are specific for senescence. Because of this, it becomes paramount to apply them collectively to comprehensively investigate alterations in overarching systems typically dysregulated upon aging.
3.4 Pro-inflammatory senescence associated secretory profile
The SASP comprises of several pro-inflammatory factors, such as interleukins, chemokines, secreted proteases, and secreted extracellular matrix components (97). SASP acquisition by senescent cells is stimulated through nuclear factor kappa B (NFκB) signaling and the transcription factor CCAAT/enhancer-binding protein beta which is in turn epigenetically regulated by arginine methyltransferase 4 (98, 99). Vascular smooth muscle cells, endothelial cells and macrophages from human atherosclerotic plaques have been found to contain increased levels of activated NFκB (100, 101). The activation of NFκB can lead to the production of SASP factors such as Tumor necrosis factor α (TNFα), IL-1 β, IL-6 and IL-8, as well as various matrix metalloproteinases (MMP) (102). Eventually, the induction of both local and systemic chronic inflammation by the SASP can lead to tissue damage and fibrosis (97). An increased secretion of MMP-9 as well as a decrease in collagen production by senescent smooth muscle cells can furthermore lead to plaque instability through remodeling of the vessel wall (103, 104). Additionally, NFκB-mediated production of the chemokine monocyte chemoattractant protein-1 and the leukocyte receptors intercellular adhesion molecule 1 and vascular cell adhesion molecule 1 can subsequently drive the recruitment of monocytes (97). The SASP is also able to induce senescence in neighboring cells, exacerbating the accumulation of senescent cells with age (105). It has been speculated that the accumulation of senescent cells in the subendothelial space of atherosclerotic plaques contributes to the initial stages of atherosclerosis through the production of SASP markers such as IL-1α, TNFα and MMPs (91). However, in addition to the use of SA-β-gal and p16Ink4a as the main markers of senescence in this study, it must be noted that non-senescent macrophages can similarly express IL-1α, TNFα and MMPs in a high capacity.
Thus, as we age, our immune system undergoes significant changes, including aging-induced alterations in hematopoiesis, accumulation of senescent cells, and the development of a pro-inflammatory senescence-associated secretory profile (SASP). These changes contribute to immunosenescence and can potentially increase CVD risk. Hematopoietic myeloid skewing and thymic involution impact immune cell production and functionality, while senescent cells exhibit an irreversible loss of proliferative capacity and contribute to chronic inflammation through adapting a SASP.
4 Aged adaptive immunity in atherosclerosis
4.1 T cell aging
The T cell compartment is strongly affected by aging. T cell numbers decrease as a consequence of reduced lymphoid output from the bone marrow and reduced generation of naïve T cells by the thymus (81). T cells additionally exhibit canonical features of cellular aging such as telomeric shortening, with CD4+ T cell telomere length decreasing by ~3000 bp between ages 20-60 years (7, 106). Accelerated telomere shortening has furthermore been observed in autoimmune disease, as well as in circulating lymphocytes of CVD patients (107–109). Simultaneously, as T cells age, their differentiation and secretory profile becomes skewed towards more extreme phenotypes evident from highly clonal regulatory and effector memory T cell compartments (15, 38). These factors can further contribute to alterations in the TCR repertoire observed in the T cell memory pool, impacting their capacity to elicit an immune response to a broad range of antigens upon aging (110). Age-associated TCR diversity is additionally affected by the persistent selection of T cells with a specificity against either antigens of viruses with lifelong retention (e.g. CMV and Epstein–Barr virus) or auto-antigens (81). Furthermore, this phenomenon can potentially serve as a driver of senescence as CMV seropositive octogenarians display a loss of CD27 and CD28 by CD4+ T cells, and a loss of CD45RO and CD27 in CD8+ T cells, as is characteristic for senescent T cells (69, 111, 112). Atherosclerotic plaques from patients who suffered a myocardial infarction as well as those with unstable angina similarly contain autoreactive CD4+ CD28- T cells, while the expansion of CD4+ CD28- T cells has been attributed to an increased cytotoxic affinity towards endothelial cells in acute coronary syndromes (113–116). Moreover, CD4+ CD28- T cells in coronary artery disease express increased levels of granzymes (Gzm) A and B, perforin, granulysin and IFNγ upon ex vivo stimulation, indicative of a highly cytotoxic and inflammatory phenotype (117). In addition to expansion of CD4+ CD28- T cells, CD8+ T cells with high expression of CD57 have been observed by Grievink et al. in both elderly healthy populations and coronary artery disease patients as compared to the young healthy population (118). Increased expression of senescent markers CD57 and killer cell lectin-like receptor subfamily G member 1 (KLRG1) drive terminal differentiation and have also been found to occur in terminally differentiated effector T memory cells re-expressing CD45RA (TEMRA) in the peripheral blood of CVD patients (38). TEMRA cells express cytotoxic and inflammatory genes driven by p38 MAPK signaling as well as a senescent phenotype characterized by impaired proliferation and mitochondrial dysfunction (119, 120). Furthermore, the association of cell membrane bound CD153 (CD30L) with the TCR/CD3 on senescent T cells has been found to stimulate T cell signaling and activation, while CD153-/- mice exhibit a reduction in aging-associated T- and B cells (121). The blocking of CD153 in atherosclerotic Ldlr-/- mice using monoclonal antibodies has additionally been shown to lead to a reduction in lesion size and adventitial T cells (122). Interestingly, aging also results in accumulation of cholesterol in the T cell membrane (123). This may promote cellular senescence, as T cells from middle-aged Ldlr-/- mice with a specific deficiency in cholesterol efflux transporters ABCA1 and ABCG1, showed upregulated Cyclin dependent kinase inhibitor 1a (Cdkn1a) and impaired proliferative capacity (124). At the same time, accumulation of cholesterol in T cells from T cell-specific Abca1/Abcg1 deficient middle-aged mice increased apoptosis, contributing to a reduction in the peripheral T cell pool and atherosclerotic plaque development.
The shift towards more extreme T cell phenotypes upon aging becomes evident in the immune compartments of aged atherosclerotic Ldlr-/- mice which contain increased pro-atherogenic T cells producing IFNγ as well as expanded regulatory and effector T cell phenotypes (15). Additionally, an age-associated increase in pro-inflammatory CD8+ GzmK+ T cells was observed within the atherosclerotic aorta of Ldlr-/- mice, as well as in atherosclerotic plaques from CVD patients (38, 125). Atherosclerotic murine CD8+ GzmK+ T cells primarily displayed an effector and memory phenotype by increased expression of Ccl5, Nkg7, Cd52, and Id2 whilst the expression of exhaustion markers Tox-1, Eomes and Lag3 was more prevalent in the CD8+ GzmK+ T cell population from aged Ldlr-/- mice compared to young atherosclerotic mice (15). Extracellular GzmK has furthermore been shown to exacerbate secretion of aging related inflammatory factors such as IL-6, chemokine C-C motif ligand (CCL) 2, and chemokine CXCL1 in fibroblasts both in the presence of IFNγ and without (126). Moreover, in human carotid artery plaques, GzmK expressing CD8+ T cells were shown to exhibit plaque specific clonal expansion and displayed high levels of genes involved in T cell activation such as CD69, FOS and FOSB (38). Nevertheless, whether activation and clonal expansion of CD8+ GzmK+ T cells in human atherosclerotic plaques is achieved through stimulation of TCRs specific against known atherosclerosis associated auto-antigens such as oxLDL, ApoB100 or HSP60/65, and whether CD8+ GzmK+ are causally associated with atherosclerotic CVD remains to be investigated.
Additionally, deterioration of CD8+ T cell tolerance has furthermore been suggested in aged ApoE-/- mice (78–85 weeks) on a standard chow diet due to decreased expression of Ccr7, S1pr1 and Sell, involved in regulating lymphocyte egress from SLOs, as well as an upregulation of genes involved in cytotoxic activity and cytokine production such as Slamf7, S100a6, Gzmk, Nkg7, Ccl5, Ifng and Prf1, and a dysregulation of exhaustion associated genes such as Pdcd1, Tigit, Lag3 and Havcr2 (127). Integration of the mouse data with single-cell RNA sequencing (scRNAseq) data from human coronary and carotid plaques showed similar transcription profiles between species concerning pathways involved in the egress from lymphoid organs and activation of CD8+ T cells.
Aging not only induces the expansion of pro-inflammatory and cytotoxic T cell subsets, but also stimulates an increase in T cells with regulatory phenotypes. An overall increase of Tregs was observed in the atherosclerotic aorta of aged Ldlr-/- mice alongside a heightened expression of functional Treg markers such as Ctla4, Lag3, and Tnfrsf4 (OX40) and genes encoding for the IL-35 cytokine as compared to young Ldlr-/- mice (15). Similar upregulation of genes indicative of Treg activity was demonstrated in plaques from human endarterectomy specimens as compared to patient PBMCs (38). Moreover, Tregs show clonal expansion in the human carotid plaque. Previously, it has been reported that Treg functionality can decrease upon aging (128, 129). Whether aging also impacts the immunosuppressive capacity of Tregs in the atherosclerotic environment, remains to be elucidated. It may be that conversion from Tregs towards exTregs as described before, can significantly alter their function, for instance causing the acquisition of a cytotoxic phenotype. But while some studies describe such a maladaptive Treg conversion (28, 29), others showed that Tregs in human plaques did not show any co-expression of FOXP3 and RORC nor showed any evidence of Treg-Th17 conversion by complementary RNA velocity analysis (38), warranting further research into the function of Tregs in the atherosclerotic microenvironment.
4.2 B cell aging
Aging is associated with significant changes in the B cell compartment including changes in B cell subsets and the emergence of age-associated B cells (ABCs), as well as alterations in the BCR repertoire and antibody responses. In addition to an aging-associated decrease in self renewal of HSCs in the bone marrow, a reduction in the differentiation of progenitor B cells, precursor B cells and immature B cells has for instance been correlated to the presence of aged bone marrow stroma in aged mouse models (73, 130). This impaired differentiation can be partially attributed to a reduced capacity of the murine progenitor B cells to respond to stromal derived IL-7 on which they depend for their survival (131, 132). Similar to T cells, a reduction in B cell precursors in lymphoid organs could conceivably impact the diversity of the BCR repertoire. However, research concerning a potential age-related deterioration of BCR repertoire diversity in humans has given rise to conflicting results. Although a reduced BCR repertoire has been demonstrated in the peripheral blood and lymph nodes of the elderly population, an increase in BCR diversity has for example been observed in aged spleens obtained from archived human biopsy samples (133–135). In mice, it was recently shown that the BCR repertoire diversity decreases upon aging. Interestingly, dietary restriction has been shown to attenuate the observed aging-associated decrease in BCR diversity in C3B6F1 mice, achieving a similar retention of BCR repertoire in mice started on a restrictive diet during mid- life as compared to mice on a chronic diet (136, 137).
The effect of aging on B cell subsets has been shown to extend to B2 cells by decreased levels of MZ B cells in aged BALB/c mice (138). MZ B cells originating from aged C57BL/6J mice additionally display an impaired ability to generate antibodies in response to T cell independent activation by trinitrophenyl-lipopolysaccharide (139). Nevertheless, aging induced changes in MZ B cell frequency and function are poorly understood in the context of atherosclerosis. However, an age-associated decrease in the plasma levels of B cell activating factor (BAFF), which is essential for the survival of both FO- and MZ B2 cells, but not peritoneal B1 cells, has been observed in healthy individuals (140–142). In models of experimental atherosclerosis, B2 cell depletion achieved through the antibody-mediated blocking of the BAFF receptor has resulted in decreased B cell zones in the spleen of hyperlipidemic ApoE-/- mice while limiting the progression of atherosclerotic plaques (143). However, neutralization of BAFF itself has unexpectedly led to the promotion of atherosclerotic plaque progression even though both FO- and MZ B2 cells were similarly depleted (144). This study suggests BAFF to convey atheroprotection through the binding of transmembrane activator and CAML interactor (TACI), inhibiting the production of proatherogenic CXCL10 by IRF7-dependent toll-like receptor (TLR) 9 signaling. Aging furthermore impedes on class switch recombination and somatic mutation in B2 cells through a reduction in the expression of E47 transcription factor and activation-induced cytidine deaminase as well as through a decrease in their DNA binding capacity (145). Nevertheless, whether these aging associated reductions are specific to a particular subset of B2 cells is unclear as this study magnetically sorted murine splenic B cells using anti-B220 and did not differentiate between FO- and MZ B2 cells.
Multiple studies have demonstrated the atheroprotective effects of B1 cells through the secretion of IgM targeting OSEs (143, 146). Additionally, the levels of B1 cells and IgM specific for either ApoB100 or various OSEs, have been shown to negatively correlate with CVD risk (147–151). However, in the peripheral blood of the elderly population an overall decrease in the frequency of B1 cells can be observed as well as decreased levels of circulating atheroprotective oxLDL-specific IgM compared to young individuals, which shows an additional reduction in age-matched coronary artery disease patients (118, 152). Whether aging also impacts the efficacy of oxLDL-specific IgM antibodies is not yet known. The aging-associated reduction of B1 cell frequency in CVD patients has been correlated to the inhibition of XBP-1 and Blimp-1 transcription factors and an increased presence of Pax5, a marker expressed by most B cells but which is lost upon plasma cell differentiation (152). Furthermore, the expression of C-X-C chemokine receptor type (CXCR) 4 by B1 cells has been demonstrated to correlate to levels of OSE specific IgM antibodies in a cohort of CVD patients (151). This correlation has mechanistically been demonstrated in aged ApoE−/− mice by CXCR4 dependent production of IgM by B1a cells (151). However, a comparison of B1a and B1b cells through adoptive transfer experiments in aged ApoE-/- Rag1-/- mice has shown B1b cells to be the most efficient producers of OSE specific IgM (153). B1b cells additionally migrate to the spleen more efficiently than B1a cells through increased expression of C-C chemokine receptor type (CCR) 6.
Expansion of a late memory IgD- CD27- double negative (DN) B cells has furthermore been described in the peripheral blood of the elderly population as well as in autoimmune diseases (154–156) but their role in atherosclerosis is poorly understood. DN B cells display senescent features indicated by telomeric shortening, a reduced proliferative capacity and the expression of SASP markers such as IL-6, TNFα, p16INK4 and pro-inflammatory micro-RNAs (154, 157, 158). Late memory DN B cells are a heterogenous population which contain CD21low CD11c expressing age-associated B cells (ABCs) (159, 160). ABCs can produce high levels of autoantibodies and pro-inflammatory cytokines, such as IFNγ, TNFα, IL-6 and IL-1β, following activation via TLR7 and TLR9 (161). Similar to DN B cells, ABCs accumulate during aging and in autoimmune diseases, such as rheumatoid arthritis and multiple sclerosis (159, 162–164). Aging-induced formation of fat-associated lymphoid clusters likewise display an accumulation of ABCs in a manner dependent on the NOD-, LRR- and pyrin domain-containing protein 3 inflammasome (165). This finding could prove relevant for atherosclerotic pathogenesis due to the presence of perivascular adipose tissue involved in the local stimulation of plaque development (166). In light of this, the presence of ABCs has recently been uncovered in atherosclerotic aortas of aged Ldlr-/- mice using scRNAseq and flow cytometry and in carotid plaques obtained from CVD patients (15). In comparison to other B cell clusters observed in the aortic arches of atherosclerotic mice, ABCs displayed a distinctive expression profile characterized by upregulated Tbx21, Itgam, Itgax (encoding CD11c), Fas, Zbtb20, and Ighg3. The expansion of ABCs was additionally shown to be more prominent in aortic arches of Ldlr-/- and ApoE-/- mice as compared to healthy age-matched C57BL/6 mice. In line with these research findings, Pattarabanjird et al. describe an age-associated increase of CD11c expressing B cells, containing ABCs, in the spleen and bone marrow of ApoE-/- mice concomitant with an increase in aortic plaque size (167). Furthermore, ABCs have been demonstrated as highly potent antigen presenting cells in vivo through transcriptome analysis of aorta-enriched ABCs from Ldlr-/- mice (15), as well as in vitro by antigen-specific T cell proliferation assays and through the use of multiphoton microscopy in which ABCs display an increased stability of antigen specific B-T cell interactions (15, 168). There have been indications of ABCs being able to skew T cell differentiation towards a Th17 phenotype through their antigen presenting capability, but whether this also occurs in atherosclerosis context remains to be investigated (15, 169). Interestingly, the frequency of CD11c expressing B cells positively correlated to plaque severity in cardiac catheterization patients assessed through coronary angiography (167).
Overall, aging severely affects the role played by adaptive immunity in atherosclerosis development. In addition to an increase in circulatory pro-inflammatory cytokines and reduced OSE specific IgM, this has become particularly evident from the emergence of maladaptive subsets in both the T- and B cell compartments (i.e. CD4+ CD28- T cells, CD8+ GzmK+ T cells and ABCs) as observed upon aging as well as in CVD (Figure 2).
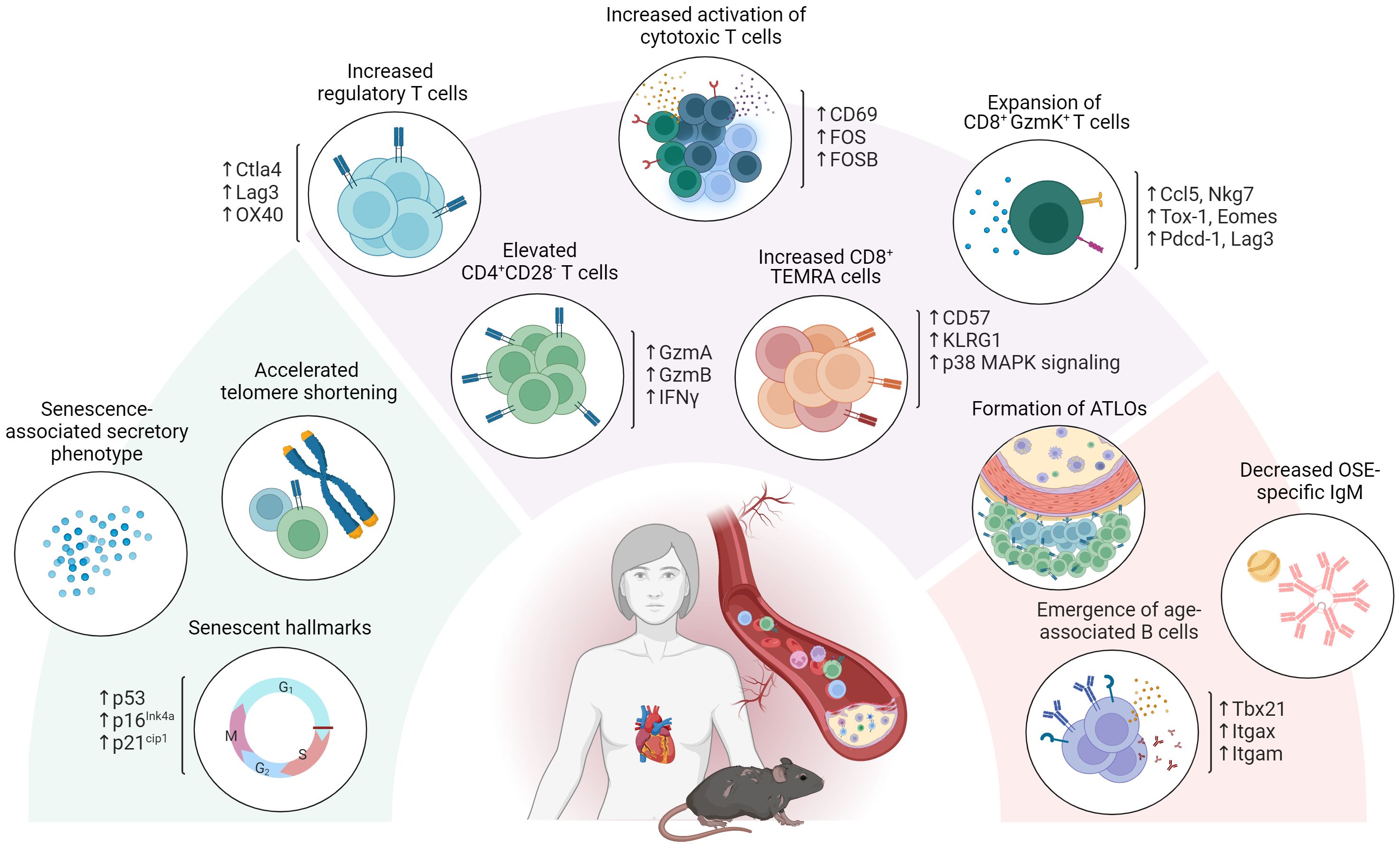
Figure 2 Age-associated adaptive immunity in atherosclerosis. Aging severely affects the role of adaptive immunity in atherosclerosis development. In addition to exhibiting senescent hallmarks such as telomeric shortening, cell cycle arrest and adopting a senescence associated secretory profile, atherosclerosis specific T- and B cell dynamics are affected through the emergence of maladaptive age-associated subsets including CD4+ CD28- T cells, CD8+ GzmK+ T cells and age-associated B cells (ABCs). Atherosclerosis specific humoral immunity additionally shows a decrease in OSE specific IgM and an increase in BAFF, essential for B2 cell survival upon aging.
4.3 T-B cell interactions and deteriorating secondary lymphoid tissues with age
Aging stimulates early Tfh differentiation in mice as evident from increased numbers of pre-cursor Tfh cells in draining lymph nodes through modulating expression of RBPJ, the Notch-associated transcription factor (170). Additionally, an increase in activated memory Tfh cells has been observed in aged atherosclerotic mice compared to their younger counterparts (34). The formation of spontaneous germinal centers and autoantibody production has furthermore been reported in relation to CD30L expressing Tfh cells with high expression of the SASP factor osteopontin and senescence-associated heterochromatic foci in the nuclei, contributing to autoimmunity in lupus mouse models (171–173). Similar overactivation of the Tfh–GC B cell axis has been previously shown to exacerbate atherogenesis in ApoE-/- mice deficient for Qa-1–restricted CD8+ regulatory T cells, a phenotype subsequently rescued by antibody treatment specific against inducible costimulator-ligand (ICOSL) to inhibit Tfh activity (34). In contrast, a reduction in Tfh expansion following influenza vaccination has been observed in the elderly population as compared to individuals aged 18-36 years (174). In line with this finding, Silva-Cayetano et al. have shown that following immunization with NP-KLH, a model antigen to assess vaccine responses, an aging-associated decrease in germinal center formation and quality of antibody responses was observed through spatial dysregulation of Tfh cells in BALB/c mice (175). Aged Tfh cells overexpress CXCR4, impeding on their colocalization with FDCs necessary for their priming. This impairs effective Tfh-B cell interactions and Tfh support of B cell survival, proliferation and differentiation towards GC B cells. Notably, disruptions in T-B cell interactions impact Tfh cell proliferation as well, as evident by the accumulation of Tfh cells in the spleens of irradiated Ldlr-/- mice reconstituted with MZ B cell deficient bone marrow (57). This study showed that MZ B cells exert control over the Tfh response to a high cholesterol diet in a PD-L1 and ATF3-dependent manner. In absence of MZB cells, atherosclerosis was aggravated. In line with this, it has been shown that inhibition of the Tfh-GC B cell axis through PD-L1 expressing B cells protects against atherosclerosis (52).
In addition to an altered function of aged Tfh cells, SLOs display age-related deterioration, decreasing in both size and complexity of their architecture (176). Firstly, the trafficking of T cells through the SLO scaffolding becomes impeded by an aging induced reduction in fibroblastic reticular cells which lay down the extracellular matrix (177, 178). Additionally, this reduction affects the deposition of IL-7 on the extracellular matrix which is essential for the preservation of naïve T cells (179). Similarly, SLOs experience a decrease in the area of the light zone alongside the therein contained FDCs necessary for priming Tfh cells (175, 180). Overall, these age-related changes impede the correct function and localization of cells from the adaptive immune system and can lead to aberrant germinal center responses. Nevertheless, how the aging induced deterioration of SLO architecture and function might impact either pathogenic or protective humoral responses relating to atherosclerotic CVD risk is still understudied. This is especially true as the development of artery tertiary lymphoid organs (ATLOs) have been reported in aged atherosclerotic ApoE-/- mice (181, 182). Though ATLOs occur at sites of chronic inflammation, they include dedicated T cell zones, B cell follicles and germinal centers and are capable of regulating B cell responses in a manner similar to that of SLOs (183). However, ATLOs exhibit significant angiogenesis and high endothelial venules which facilitate the recruitment of naïve T- and B cells to the lymphoid tissue (182, 184). As such, ATLOs have been suggested to be involved the pathogenesis of advanced atherosclerosis by stimulation of primary immune responses (183).
5 Immunoaging as biomarker or therapeutic target to combat atherosclerotic CVD
Currently few biomarkers exist that can indicate risk of major adverse cardiovascular events (MACE). Increased plasma levels of low-density lipoprotein cholesterol (LDL-C), non–high-density lipoprotein cholesterol (non-HDL-C), IL-18, and TNFα, IL-6 and high-sensitivity C-reactive protein (hsCRP) have previously been associated with poor cardiovascular outcomes (185–187). However, inflammatory risk assessed by circulating hsCRP is for example unlikely to represent the full extent of atherosclerosis associated inflammation, as hsCRP is solely driven by circulating IL-6 levels (24). With an aging population at increased risk for CVD and cardiovascular related death, it is of the utmost importance to discover new biomarkers for assessing atherosclerotic disease state to inform timely and appropriate clinical intervention. As increased incidence of atherosclerotic MACE occurs in parallel to aging, the breakdown of immunotolerance and increased susceptibility for the development of autoimmune responses in the elderly population could provide valuable biomarkers for this exact purpose (127, 188–190). As such, the systemic expansion of atherogenic age-associated immune cells and their secreted products (e.g. autoantibodies), could be explored to stratify individuals at risk for atherosclerosis driven MACE (118, 154, 191). Illustratively, the trajectory of immune aging by the algorithmic summary of elements of immunosenescence, termed the IMM-AGE score, has been found to predict all-cause mortality more accurately than an individual’s chronological age and established risk factors (192). Additionally, an increased IMM-AGE score was observed for patients diagnosed with cardiovascular disease in the Framingham Heart Study. Nevertheless, the predictive clinical value of the IMM-AGE score for risk of MACE has not yet been determined in a prospective cohort.
The current treatment of CVD relies on risk factor management, whilst no treatments directly attacking inflammatory processes in the vessel wall are currently available. The necessity for such treatments is illustrated by the persistence of cardiovascular events as well as residual inflammatory risk in patients recruited in contemporary lipid lowering trials (193–195). Current strategies therefore additionally focus on targeting the underlying immunity involved in CVD pathogenesis. The blocking of IL-1β with Canakinumab (CANTOS trial) as well as the Low-Dose Colchicine-2 (LoDoCo2) trial have illustrated the effectiveness of modulating inflammation for the prevention of atherosclerosis induced CVD events, independent of lipid levels (196, 197). However, the increased rate of fatal sepsis in these trials highlights the need for atherosclerosis specific immune modulation in contrast to a general systemic reduction in inflammation. As such, the safety and biological efficacy of Treg expansion by low dose IL-2 in patients with ischemic heart disease was confirmed in the LILACS study, while its application for the reduction of vascular inflammation is currently under investigation in the IVORY trial (198, 199). Similarly, reduction of mature B cells by Rituximab, a monoclonal antibody against CD20, in the RITA-MI study did not report any drug related safety issues (200). Administration of anti-CD20 in mice has been shown to deplete pro-atherogenic B2 cells as opposed to B1 cells, attenuating atherosclerosis (201, 202).
With an increased life expectancy, the aging population similarly increases, which additionally emphasizes the need for strategies to delay or reverse age-associated immunosenescence. Significant attention goes towards clearance of senescent cells by a class of drugs called senolytics. In atherosclerotic mice, treatment with either Quercetin or Navitoclax which target anti-apoptotic Bcl-2 family proteins, resulted in decreased atherosclerotic lesion size and circulating pro-inflammatory factors (91, 203–205). However, through the targeting of apoptotic pathways, Quercetin and Navitoclax can non-specifically affect healthy cells as well, resulting in severe thrombopenia and neutropenia (206–208). Though the use of proteolysis-targeting chimera technology has been shown to ameliorate the platelet toxicity of Bcl-2 inhibitors, and next generation senolytics with higher selectivity are emerging, the use of senolytics for the clinical treatment of CVD still seems a ways off (209). As such, specific depletion of maladaptive age-associated immune cell subsets warrants further investigation. However, a first step in guiding this process would be high dimensional characterization of aged immunity in atherosclerosis in order to uncover more specific markers for targeting through therapeutic intervention, an effort currently led by single cell technologies.
6 The external validity of immunological findings from mice studies
Age matching laboratory mice to the actual atherosclerotic CVD patient population is invaluable for improving the external validity of research concerning the immune components at play in atherosclerotic plaque progression. However, laboratory mice are not without their limitations. Laboratory mice are kept within highly standardized environments in which the temperature, humidity, light cycles and diets (among others) are regulated. This is especially true in immunological research in which specific pathogen-free (SPF) mice have become the standard. SPF husbandry impacts the gut microbiome heterogeneity and maturation of the murine immune system, limiting its translatability to human immunity matured through exposure to both acute and chronic pathogens (210, 211). A noteworthy development to circumvent an immature immune system caused by limited exposure to pathogens can be found in “wildling” mice born through the transfer of lab-strain embryos into wild mice (212). The immune landscape of wildling mice closely resembles that of wild mice. Additionally, for both human clinical trials targeting the adaptive immune system through administration of a CD28-superagonist (CD28SA) or the innate immune system by anti-TNF-α treatment in sepsis, Rosshart et al. have found C57BL/6 wildling mice to be better predictors of clinical outcome as compared to conventional laboratory mice (212). However, whether research into the inflammatory aspects of atherosclerotic pathogenesis could benefit from using wildling mice on an atherosclerosis prone background to increase their external validity is currently unclear. Moreover, intensifying research efforts into immunosenescence in CVD patients may contribute to the identification of biomarkers for the stratification of patients as well as the identification of therapeutic targets specific to the aged immune landscape at play in CVD in order to mitigate disease burden.
7 Conclusion and future perspectives
Given the demographic shift towards an elderly population, it has become paramount to elucidate the role of age-associated maladaptive immunity in the progression of atherosclerotic CVD occurring in parallel to aging. Current advances in single cell technology have shed light on the age-associated dysregulation of atherosclerosis specific T-B cell dynamics leading to the expansion of atherogenic, pro-inflammatory and cytotoxic cellular subsets. In the future, this information could contribute to the identification and therapeutic depletion of maladaptive immunity to reduce atherosclerotic CVD risk in the elderly population. However, the in-depth characterization of age-associated maladaptive immunity involved in atherosclerotic CVD pathogenesis, as well as the age-specific immune cell organization across the plaque environment and lymphoid tissues are an ongoing process. Both these factors will need to be explored further to improve upon the classical stratification of the atherosclerotic CVD patient population seeking treatment at advanced age.
Author contributions
RPMS: Conceptualization, Visualization, Writing – original draft. ACF: Conceptualization, Supervision, Visualization, Writing – review & editing, Funding acquisition.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Dutch Heart Foundation grant number 2018T051 to ACF, and the ERA-CVD B-eatATHERO consortium; Dutch Heart Foundation grant number 2019T107 to ACF. This work was supported by the project ‘B-specific’ funded by the European Union under Grant Agreement No. 101115159.
Acknowledgments
Figures in this review were created using BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part III: cellular and molecular clues to heart and arterial aging. Circulation. (2003) 107:490–7. doi: 10.1161/01.CIR.0000048894.99865.02
2. Ho YH, Del Toro R, Rivera-Torres J, Rak J, Korn C, Garcia-Garcia A, et al. Remodeling of bone marrow hematopoietic stem cell niches promotes myeloid cell expansion during premature or physiological aging. Cell Stem Cell. (2019) 25:407–18.e6. doi: 10.1016/j.stem.2019.06.007
3. Salam N, Rane S, Das R, Faulkner M, Gund R, Kandpal U, et al. T cell ageing: effects of age on development, survival & function. Indian J Med Res. (2013) 138:595–608.
4. Nikolich-Žugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol. (2018) 19:10–9. doi: 10.1038/s41590-017-0006-x
5. Kumari R, Jat P. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol. (2021) 9:645593. doi: 10.3389/fcell.2021.645593
6. Nikolich-Žugich J. Aging of the T cell compartment in mice and humans: from no naive expectations to foggy memories. J Immunol. (2014) 193:2622–9. doi: 10.4049/jimmunol.1401174
7. Weyand CM, Goronzy JJ. Aging of the immune system. Mechanisms and therapeutic targets. Ann Am Thorac Soc. (2016) 13 Suppl 5:S422–S8. doi: 10.1513/AnnalsATS.201602-095AW
8. Ritchie H, Spooner F, Roser M. Causes of death. Our world in data (2018). Availabloe at: https://ourworldindata.org/causes-of-death.
9. Frostegard J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. (2013) 11:117. doi: 10.1186/1741-7015-11-117
10. Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. (2006) 6:508–19. doi: 10.1038/nri1882
11. Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. (2013) 38:1092–104. doi: 10.1016/j.immuni.2013.06.009
12. Raitakari O, Pahkala K, Magnussen CG. Prevention of atherosclerosis from childhood. Nat Rev Cardiol. (2022) 19:543–54. doi: 10.1038/s41569-021-00647-9
13. Mortality GBD, Causes of Death C. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. (2016) 388:1459–544. doi: 10.1016/S0140-6736(16)31012-1
14. Flurkey K, Currer JM, Harrison D. Mouse models in aging research. Mouse Biomed research: Elsevier;. (2007) p:637–72. doi: 10.1016/B978-012369454-6/50074-1
15. Smit V, de Mol J, Schaftenaar FH, Depuydt MAC, Postel RJ, Smeets D, et al. Single-cell profiling reveals age-associated immunity in atherosclerosis. Cardiovasc Res. (2023) 119:2508–21. doi: 10.1093/cvr/cvad099
16. Liu M, Zhang W, Li X, Han J, Chen Y, Duan Y. Impact of age and sex on the development of atherosclerosis and expression of the related genes in apoE deficient mice. Biochem Biophys Res Commun. (2016) 469:456–62. doi: 10.1016/j.bbrc.2015.11.064
17. Caligiuri G, Nicoletti A, Zhou X, Tornberg I, Hansson GK. Effects of sex and age on atherosclerosis and autoimmunity in apoE-deficient mice. Atherosclerosis. (1999) 145:301–8. doi: 10.1016/S0021-9150(99)00081-7
18. Gogulamudi VR, Durrant JR, Adeyemo AO, Ho HM, Walker AE, Lesniewski LA. Advancing age increases the size and severity of spontaneous atheromas in mouse models of atherosclerosis. Geroscience. (2023) 45:1913–31. doi: 10.1007/s11357-023-00776-8
19. Maue AC, Yager EJ, Swain SL, Woodland DL, Blackman MA, Haynes L. T-cell immunosenescence: lessons learned from mouse models of aging. Trends Immunol. (2009) 30:301–5. doi: 10.1016/j.it.2009.04.007
20. Zhang X, Sessa WC, Fernandez-Hernando C. Endothelial transcytosis of lipoproteins in atherosclerosis. Front Cardiovasc Med. (2018) 5:130. doi: 10.3389/fcvm.2018.00130
21. Gimbrone MA Jr., Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
22. Bjorkegren JLM, Lusis AJ. Atherosclerosis: recent developments. Cell. (2022) 185:1630–45. doi: 10.1016/j.cell.2022.04.004
23. Engelen SE, Robinson AJB, Zurke YX, Monaco C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: how to proceed? Nat Rev Cardiol. (2022) 19:522–42. doi: 10.1038/s41569-021-00668-4
24. Mallat Z, Binder CJ. The why and how of adaptive immune responses in ischemic cardiovascular disease. Nat Cardiovasc Res. (2022) 1:431–44. doi: 10.1038/s44161-022-00049-1
25. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
26. Roy P, Orecchioni M, Ley K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat Rev Immunol. (2022) 22:251–65. doi: 10.1038/s41577-021-00584-1
27. Foks AC, Lichtman AH, Kuiper J. Treating atherosclerosis with regulatory T cells. Arterioscler Thromb Vasc Biol. (2015) 35:280–7. doi: 10.1161/ATVBAHA.114.303568
28. Freuchet A, Roy P, Armstrong SS, Oliaeimotlagh M, Kumar S, Orecchioni M, et al. Identification of human exT(reg) cells as CD16(+)CD56(+) cytotoxic CD4(+) T cells. Nat Immunol. (2023) 24:1748–61. doi: 10.1038/s41590-023-01589-9
29. Wolf D, Gerhardt T, Winkels H, Michel NA, Pramod AB, Ghosheh Y, et al. Pathogenic autoimmunity in atherosclerosis evolves from initially protective apolipoprotein B(100)-reactive CD4(+) T-regulatory cells. Circulation. (2020) 142:1279–93. doi: 10.1161/CIRCULATIONAHA.119.042863
30. Gaddis DE, Padgett LE, Wu R, McSkimming C, Romines V, Taylor AM, et al. Apolipoprotein AI prevents regulatory to follicular helper T cell switching during atherosclerosis. Nat Commun. (2018) 9:1095. doi: 10.1038/s41467-018-03493-5
31. Silveira A, McLeod O, Strawbridge RJ, Gertow K, Sennblad B, Baldassarre D, et al. Plasma IL-5 concentration and subclinical carotid atherosclerosis. Atherosclerosis. (2015) 239:125–30. doi: 10.1016/j.atherosclerosis.2014.12.046
32. Binder CJ, Hartvigsen K, Chang MK, Miller M, Broide D, Palinski W, et al. IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J Clin Invest. (2004) 114:427–37. doi: 10.1172/JCI200420479
33. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. (2019) 50:892–906. doi: 10.1016/j.immuni.2019.03.021
34. Clement M, Guedj K, Andreata F, Morvan M, Bey L, Khallou-Laschet J, et al. Control of the T follicular helper-germinal center B-cell axis by CD8(+) regulatory T cells limits atherosclerosis and tertiary lymphoid organ development. Circulation. (2015) 131:560–70. doi: 10.1161/CIRCULATIONAHA.114.010988
35. Schafer S, Zernecke A. CD8(+) T cells in atherosclerosis. Cells. (2020) 10:37. doi: 10.3390/cells10010037
36. van Duijn J, Kritikou E, Benne N, van der Heijden T, van Puijvelde GH, Kroner MJ, et al. CD8+ T-cells contribute to lesion stabilization in advanced atherosclerosis by limiting macrophage content and CD4+ T-cell responses. Cardiovasc Res. (2019) 115:729–38. doi: 10.1093/cvr/cvy261
37. Depuydt MAC, Prange KHM, Slenders L, Ord T, Elbersen D, Boltjes A, et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ Res. (2020) 127:1437–55. doi: 10.1161/CIRCRESAHA.120.316770
38. Depuydt MAC, Schaftenaar F, Prange KHM, Boltjes A, Hemme E, Delfos L, et al. Single-cell T-cell receptor sequencing of paired human atherosclerotic plaques and blood reveals autoimmune-like features of expanded effector T-cells. Nat Cardiovasc Res. (2023) 379:112–25. doi: 10.1038/s44161-022-00208-4
39. Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. (2020) 17:387–401. doi: 10.1038/s41569-020-0352-5
40. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science. (2009) 325:1001–5. doi: 10.1126/science.1176676
41. Baptista D, Mach F, Brandt KJ. Follicular regulatory T cell in atherosclerosis. J Leukoc Biol. (2018) 104:925–30. doi: 10.1002/JLB.MR1117-469R
42. Zhu Y, Zou L, Liu YC. T follicular helper cells, T follicular regulatory cells and autoimmunity. Int Immunol. (2016) 28:173–9. doi: 10.1093/intimm/dxv079
43. Hardtke S, Ohl L, Förster R. Balanced expression of CXCR5 and CCR7 on follicular T helper cells determines their transient positioning to lymph node follicles and is essential for efficient B-cell help. Blood. (2005) 106:1924–31. doi: 10.1182/blood-2004-11-4494
44. Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. (2000) 192:1553–62. doi: 10.1084/jem.192.11.1553
45. Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, et al. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J Exp Med. (2011) 208:1377–88. doi: 10.1084/jem.20102065
46. Crotty S. T follicular helper cell biology: A decade of discovery and diseases. Immunity. (2019) 50:1132–48. doi: 10.1016/j.immuni.2019.04.011
47. Wu JT, Wu LL. Auto antibodies against oxidized LDL: A potential marker for atherosclerosis. Clinics Lab Med. (1997) 17:595–604. doi: 10.1016/S0272-2712(18)30213-0
48. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. (2011) 17:975–82. doi: 10.1038/nm.2425
49. Wing JB, Kitagawa Y, Locci M, Hume H, Tay C, Morita T, et al. A distinct subpopulation of CD25(-) T-follicular regulatory cells localizes in the germinal centers. Proc Natl Acad Sci U S A. (2017) 114:E6400–E9. doi: 10.1073/pnas.1705551114
50. Burger F, Miteva K, Baptista D, Roth A, Fraga-Silva RA, Martel C, et al. Follicular regulatory helper T cells control the response of regulatory B cells to a high-cholesterol diet. Cardiovasc Res. (2021) 117:743–55. doi: 10.1093/cvr/cvaa069
51. Sage AP, Tsiantoulas D, Binder CJ, Mallat Z. The role of B cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:180–96. doi: 10.1038/s41569-018-0106-9
52. Douna H, Amersfoort J, Schaftenaar FH, Kroner MJ, Kiss MG, Slutter B, et al. B- and T-lymphocyte attenuator stimulation protects against atherosclerosis by regulating follicular B cells. Cardiovasc Res. (2020) 116:295–305. doi: 10.1093/cvr/cvz129
53. Tay C, Liu YH, Kanellakis P, Kallies A, Li Y, Cao A, et al. Follicular B cells promote atherosclerosis via T cell-mediated differentiation into plasma cells and secreting pathogenic immunoglobulin G. Arterioscler Thromb Vasc Biol. (2018) 38:e71–84. doi: 10.1161/ATVBAHA.117.310678
54. Centa M, Prokopec KE, Garimella MG, Habir K, Hofste L, Stark JM, et al. Acute loss of apolipoprotein E triggers an autoimmune response that accelerates atherosclerosis. Arterioscler Thromb Vasc Biol. (2018) 38:e145–e58. doi: 10.1161/ATVBAHA.118.310802
55. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. (2016) 16:485–97. doi: 10.1038/nri.2016.63
56. Harrison J, Newland SA, Jiang W, Giakomidi D, Zhao X, Clement M, et al. Marginal Zone B cells produce ‘natural’atheroprotective IgM antibodies in a T-cell-dependent manner. Cardiovasc Res. (2024) 120(3):318–28. doi: 10.21203/rs.3.rs-1753743/v1
57. Nus M, Sage AP, Lu Y, Masters L, Lam BYH, Newland S, et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat Med. (2017) 23:601–10. doi: 10.1038/nm.4315
58. Tsiantoulas D, Gruber S, Binder CJ. B-1 cell immunoglobulin directed against oxidation-specific epitopes. Front Immunol. (2012) 3:415. doi: 10.3389/fimmu.2012.00415
59. Douna H, Amersfoort J, Schaftenaar FH, Kroon S, van Puijvelde GHM, Kuiper J, et al. Bidirectional effects of IL-10(+) regulatory B cells in Ldlr(-/-) mice. Atherosclerosis. (2019) 280:118–25. doi: 10.1016/j.atherosclerosis.2018.11.019
60. Sage AP, Nus M, Baker LL, Finigan AJ, Masters LM, Mallat Z. Regulatory B cell-specific interleukin-10 is dispensable for atherosclerosis development in mice. Arterioscler Thromb Vasc Biol. (2015) 35:1770–3. doi: 10.1161/ATVBAHA.115.305568
61. Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
62. Tubbs A, Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell. (2017) 168:644–56. doi: 10.1016/j.cell.2017.01.002
63. Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. (2014) 16:2–9. doi: 10.1038/ncb2897
64. Chang E, Harley CB. Telomere length and replicative aging in human vascular tissues. Proc Natl Acad Sci U S A. (1995) 92:11190–4. doi: 10.1073/pnas.92.24.11190
65. Liang Z, Dong X, Zhang Z, Zhang Q, Zhao Y. Age-related thymic involution: Mechanisms and functional impact. Aging Cell. (2022) 21:e13671. doi: 10.1111/acel.13671
66. Murphy AJ, Dragoljevic D, Tall AR. Cholesterol efflux pathways regulate myelopoiesis: a potential link to altered macrophage function in atherosclerosis. Front Immunol. (2014) 5:490. doi: 10.3389/fimmu.2014.00490
67. Seijkens T, Hoeksema MA, Beckers L, Smeets E, Meiler S, Levels J, et al. Hypercholesterolemia-induced priming of hematopoietic stem and progenitor cells aggravates atherosclerosis. FASEB J. (2014) 28:2202–13. doi: 10.1096/fj.13-243105
68. Roberts ET, Haan MN, Dowd JB, Aiello AE. Cytomegalovirus antibody levels, inflammation, and mortality among elderly Latinos over 9 years of follow-up. Am J Epidemiol. (2010) 172:363–71. doi: 10.1093/aje/kwq177
69. Spyridopoulos I, Martin-Ruiz C, Hilkens C, Yadegarfar ME, Isaacs J, Jagger C, et al. CMV seropositivity and T-cell senescence predict increased cardiovascular mortality in octogenarians: results from the Newcastle 85+ study. Aging Cell. (2016) 15:389–92. doi: 10.1111/acel.12430
70. Simanek AM, Dowd JB, Pawelec G, Melzer D, Dutta A, Aiello AE. Seropositivity to cytomegalovirus, inflammation, all-cause and cardiovascular disease-related mortality in the United States. PloS One. (2011) 6:e16103. doi: 10.1371/journal.pone.0016103
71. Eberhardt N, Noval MG, Kaur R, Amadori L, Gildea M, Sajja S, et al. SARS-CoV-2 infection triggers pro-atherogenic inflammatory responses in human coronary vessels. Nat Cardiovasc Res. (2023) 2:899–916. doi: 10.1038/s44161-023-00336-5
72. Mouat IC, Allanach JR, Fettig NM, Fan V, Girard AM, Shanina I, et al. Gammaherpesvirus infection drives age-associated B cells toward pathogenicity in EAE and MS. Sci Adv. (2022) 8:eade6844. doi: 10.1126/sciadv.ade6844
73. Chinn IK, Blackburn CC, Manley NR, Sempowski GD. Changes in primary lymphoid organs with aging. Semin Immunol. (2012) 24:309–20. doi: 10.1016/j.smim.2012.04.005
74. Linton PJ, Dorshkind K. Age-related changes in lymphocyte development and function. Nat Immunol. (2004) 5:133–9. doi: 10.1038/ni1033
75. Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. (2005) 102:9194–9. doi: 10.1073/pnas.0503280102
76. Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci U S A. (2011) 108:20012–7. doi: 10.1073/pnas.1116110108
77. Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. (2013) 17:695–708. doi: 10.1016/j.cmet.2013.04.001
78. Flynn MC, Kraakman MJ, Tikellis C, Lee MKS, Hanssen NMJ, Kammoun HL, et al. Transient intermittent hyperglycemia accelerates atherosclerosis by promoting myelopoiesis. Circ Res. (2020) 127:877–92. doi: 10.1161/CIRCRESAHA.120.316653
79. Tyrrell DJ, Goldstein DR. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat Rev Cardiol. (2021) 18:58–68. doi: 10.1038/s41569-020-0431-7
80. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. (2017) 377:111–21. doi: 10.1056/NEJMoa1701719
81. Mittelbrunn M, Kroemer G. Hallmarks of T cell aging. Nat Immunol. (2021) 22:687–98. doi: 10.1038/s41590-021-00927-z
82. Yousefzadeh M, Henpita C, Vyas R, Soto-Palma C, Robbins P, Niedernhofer L. DNA damage-how and why we age? Elife. (2021) 10:e62852. doi: 10.7554/eLife.62852
83. Ghamar Talepoor A, Doroudchi M. Immunosenescence in atherosclerosis: A role for chronic viral infections. Front Immunol. (2022) 13:945016. doi: 10.3389/fimmu.2022.945016
84. Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. (2014) 15:1139–53. doi: 10.15252/embr.201439245
85. Manfredi S, Masetti S, Botto N, Colombo MG, Terrazzi M, Vassalle C, et al. P53 codon 72 polymorphism in coronary artery disease: no evidence for association with increased risk or micronucleus frequency. Environ Mol Mutagen. (2002) 40:110–5. doi: 10.1002/em.10098
86. Andreassi MG, Botto N, Rizza A, Colombo MG, Palmieri C, Berti S, et al. Deoxyribonucleic acid damage in human lymphocytes after percutaneous transluminal coronary angioplasty. J Am Coll Cardiol. (2002) 40:862–8. doi: 10.1016/S0735-1097(02)02042-9
87. Botto N, Rizza A, Colombo MG, Mazzone AM, Manfredi S, Masetti S, et al. Evidence for DNA damage in patients with coronary artery disease. Mutat Res. (2001) 493:23–30. doi: 10.1016/S1383-5718(01)00162-0
88. Itahana K, Campisi J, Dimri GP. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol. (2007) 371:21–31. doi: 10.1007/978-1-59745-361-5_3
89. Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. (2011) 286:36396–403. doi: 10.1074/jbc.M111.257071
90. Valieva Y, Ivanova E, Fayzullin A, Kurkov A, Igrunkova A. Senescence-associated beta-galactosidase detection in pathology. Diagnostics (Basel). (2022) 12:2309. doi: 10.3390/diagnostics12102309
91. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. (2016) 354:472–7. doi: 10.1126/science.aaf6659
92. Holdt LM, Sass K, Gabel G, Bergert H, Thiery J, Teupser D. Expression of Chr9p21 genes CDKN2B (p15(INK4b)), CDKN2A (p16(INK4a), p14(ARF)) and MTAP in human atherosclerotic plaque. Atherosclerosis. (2011) 214:264–70. doi: 10.1016/j.atherosclerosis.2010.06.029
93. Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, et al. Aging of mice is associated with p16(Ink4a)- and beta-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY). (2016) 8:1294–315. doi: 10.18632/aging.v8i7
94. Fuentes L, Wouters K, Hannou SA, Cudejko C, Rigamonti E, Mayi TH, et al. Downregulation of the tumour suppressor p16 INK4A contributes to the polarisation of human macrophages toward an adipose tissue macrophage (ATM)-like phenotype. Diabetologia. (2011) 54:3150–6. doi: 10.1007/s00125-011-2324-0
95. Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, et al. p16(Ink4a) and senescence-associated beta-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging (Albany NY). (2017) 9:1867–84. doi: 10.18632/aging.v9i8
96. Cudejko C, Wouters K, Fuentes L, Hannou SA, Paquet C, Bantubungi K, et al. p16INK4a deficiency promotes IL-4-induced polarization and inhibits proinflammatory signaling in macrophages. Blood. (2011) 118:2556–66. doi: 10.1182/blood-2010-10-313106
97. Sun Y, Wang X, Liu T, Zhu X, Pan X. The multifaceted role of the SASP in atherosclerosis: from mechanisms to therapeutic opportunities. Cell Biosci. (2022) 12:74. doi: 10.1186/s13578-022-00815-5
98. Salminen A, Kauppinen A, Kaarniranta K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal. (2012) 24:835–45. doi: 10.1016/j.cellsig.2011.12.006
99. Kowenz-Leutz E, Pless O, Dittmar G, Knoblich M, Leutz A. Crosstalk between C/EBPβ phosphorylation, arginine methylation, and SWI/SNF/Mediator implies an indexing transcription factor code. EMBO J. (2010) 29:1105–15. doi: 10.1038/emboj.2010.3
100. Bourcier T, Sukhova G, Libby P. The nuclear factor κ-B signaling pathway participates in dysregulation of vascular smooth muscle cells in vitroand in human atherosclerosis. J Biol Chem. (1997) 272:15817–24. doi: 10.1074/jbc.272.25.15817
101. Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, et al. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J Clin Invest. (1996) 97:1715–22. doi: 10.1172/JCI118598
102. Monaco C, Andreakos E, Kiriakidis S, Mauri C, Bicknell C, Foxwell B, et al. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc Natl Acad Sci U S A. (2004) 101:5634–9. doi: 10.1073/pnas.0401060101
103. Mistriotis P, Andreadis ST. Vascular aging: Molecular mechanisms and potential treatments for vascular rejuvenation. Ageing Res Rev. (2017) 37:94–116. doi: 10.1016/j.arr.2017.05.006
104. Gardner SE, Humphry M, Bennett MR, Clarke MC. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1α–dependent senescence-associated secretory phenotype. Arteriosclerosis thrombosis Vasc Biol. (2015) 35:1963–74. doi: 10.1161/ATVBAHA.115.305896
105. Khosla S, Farr JN, Tchkonia T, Kirkland JL. The role of cellular senescence in ageing and endocrine disease. Nat Rev Endocrinol. (2020) 16:263–75. doi: 10.1038/s41574-020-0335-y
106. Hodes RJ, Hathcock KS, Weng NP. Telomeres in T and B cells. Nat Rev Immunol. (2002) 2:699–706. doi: 10.1038/nri890
107. Koetz K, Bryl E, Spickschen K, O’Fallon WM, Goronzy JJ, Weyand CM. T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci U S A. (2000) 97:9203–8. doi: 10.1073/pnas.97.16.9203
108. Fujii H, Shao L, Colmegna I, Goronzy JJ, Weyand CM. Telomerase insufficiency in rheumatoid arthritis. Proc Natl Acad Sci U S A. (2009) 106:4360–5. doi: 10.1073/pnas.0811332106
109. Samani NJ, Boultby R, Butler R, Thompson JR, Goodall AH. Telomere shortening in atherosclerosis. Lancet. (2001) 358:472–3. doi: 10.1016/S0140-6736(01)05633-1
110. Lanfermeijer J, Borghans JAM, van Baarle D. How age and infection history shape the antigen-specific CD8(+) T-cell repertoire: Implications for vaccination strategies in older adults. Aging Cell. (2020) 19:e13262. doi: 10.1111/acel.13262
111. Aguilera MO, Delgui LR, Romano PS, Colombo MI. Chronic infections: A possible scenario for autophagy and senescence cross-talk. Cells. (2018) 7:162. doi: 10.3390/cells7100162
112. Akbar AN, Henson SM, Lanna A. Senescence of T lymphocytes: implications for enhancing human immunity. Trends Immunol. (2016) 37:866–76. doi: 10.1016/j.it.2016.09.002
113. Schmidt D, Goronzy JJ, Weyand CM. CD4+ CD7- CD28- T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J Clin Invest. (1996) 97:2027–37. doi: 10.1172/JCI118638
114. Liuzzo G, Goronzy JJ, Yang H, Kopecky SL, Holmes DR, Frye RL, et al. Monoclonal T-cell proliferation and plaque instability in acute coronary syndromes. Circulation. (2000) 101:2883–8. doi: 10.1161/01.CIR.101.25.2883
115. Liuzzo G, Kopecky SL, Frye RL, O’Fallon WM, Maseri A, Goronzy JJ, et al. Perturbation of the T-cell repertoire in patients with unstable angina. Circulation. (1999) 100:2135–9. doi: 10.1161/01.CIR.100.21.2135
116. Nakajima T, Schulte S, Warrington KJ, Kopecky SL, Frye RL, Goronzy JJ, et al. T-cell-mediated lysis of endothelial cells in acute coronary syndromes. Circulation. (2002) 105:570–5. doi: 10.1161/hc0502.103348
117. Teo FH, de Oliveira RT, Mamoni RL, Ferreira MC, Nadruz W Jr., Coelho OR, et al. Characterization of CD4+CD28null T cells in patients with coronary artery disease and individuals with risk factors for atherosclerosis. Cell Immunol. (2013) 281:11–9. doi: 10.1016/j.cellimm.2013.01.007
118. Grievink HW, Smit V, Huisman BW, Gal P, Yavuz Y, Klerks C, et al. Cardiovascular risk factors: The effects of ageing and smoking on the immune system, an observational clinical study. Front Immunol. (2022) 13:968815. doi: 10.3389/fimmu.2022.968815
119. Perl A, Morel L. Expanding scope of TEMRA in autoimmunity. EBioMedicine. (2023) 90:104520. doi: 10.1016/j.ebiom.2023.104520
120. Callender LA, Carroll EC, Beal RWJ, Chambers ES, Nourshargh S, Akbar AN, et al. Human CD8(+) EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell. (2018) 17:e12675. doi: 10.1111/acel.12675
121. Fukushima Y, Sakamoto K, Matsuda M, Yoshikai Y, Yagita H, Kitamura D, et al. cis interaction of CD153 with TCR/CD3 is crucial for the pathogenic activation of senescence-associated T cells. Cell Rep. (2022) 40:111373. doi: 10.1016/j.celrep.2022.111373
122. Foks AC, Bot I, Frodermann V, de Jager SC, Ter Borg M, van Santbrink PJ, et al. Interference of the CD30-CD30L pathway reduces atherosclerosis development. Arterioscler Thromb Vasc Biol. (2012) 32:2862–8. doi: 10.1161/ATVBAHA.112.300509
123. Larbi A, Dupuis G, Khalil A, Douziech N, Fortin C, Fulop T Jr. Differential role of lipid rafts in the functions of CD4+ and CD8+ human T lymphocytes with aging. Cell Signal. (2006) 18:1017–30. doi: 10.1016/j.cellsig.2005.08.016
124. Bazioti V, La Rose AM, Maassen S, Bianchi F, de Boer R, Halmos B, et al. T cell cholesterol efflux suppresses apoptosis and senescence and increases atherosclerosis in middle aged mice. Nat Commun. (2022) 13:3799. doi: 10.1038/s41467-022-31135-4
125. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. (2019) 25:1576–88. doi: 10.1038/s41591-019-0590-4
126. Mogilenko DA, Shpynov O, Andhey PS, Arthur L, Swain A, Esaulova E, et al. Comprehensive profiling of an aging immune system reveals clonal GZMK(+) CD8(+) T cells as conserved hallmark of inflammaging. Immunity. (2021) 54:99–115.e12. doi: 10.1016/j.immuni.2020.11.005
127. Wang Z, Zhang X, Lu S, Zhang C, Ma Z, Su R, et al. Pairing of single-cell RNA analysis and T cell antigen receptor profiling indicates breakdown of T cell tolerance checkpoints in atherosclerosis. Nat Cardiovasc Res. (2023) 2:290–306. doi: 10.1038/s44161-023-00218-w
128. Zhao L, Sun L, Wang H, Ma H, Liu G, Zhao Y. Changes of CD4+CD25+Foxp3+ regulatory T cells in aged Balb/c mice. J Leukoc Biol. (2007) 81:1386–94. doi: 10.1189/jlb.0506364
129. Guo Z, Wang G, Wu B, Chou WC, Cheng L, Zhou C, et al. DCAF1 regulates Treg senescence via the ROS axis during immunological aging. J Clin Invest. (2020) 130:5893–908. doi: 10.1172/JCI136466
130. Labrie JE 3rd, Sah AP, Allman DM, Cancro MP, Gerstein RM. Bone marrow microenvironmental changes underlie reduced RAG-mediated recombination and B cell generation in aged mice. J Exp Med. (2004) 200:411–23. doi: 10.1084/jem.20040845
131. Stephan RP, Lill-Elghanian DA, Witte PL. Development of B cells in aged mice: decline in the ability of pro-B cells to respond to IL-7 but not to other growth factors. J Immunol. (1997) 158:1598–609. doi: 10.4049/jimmunol.158.4.1598
132. Stephan RP, Reilly CR, Witte PL. Impaired ability of bone marrow stromal cells to support B-lymphopoiesis with age. Blood. (1998) 91:75–88. doi: 10.1182/blood.V91.1.75.75_75_88
133. Muggen AF, de Jong M, Wolvers-Tettero ILM, Kallemeijn MJ, Teodosio C, Darzentas N, et al. The presence of CLL-associated stereotypic B cell receptors in the normal BCR repertoire from healthy individuals increases with age. Immun Ageing. (2019) 16:22. doi: 10.1186/s12979-019-0163-x
134. Tabibian-Keissar H, Hazanov L, Schiby G, Rosenthal N, Rakovsky A, Michaeli M, et al. Aging affects B-cell antigen receptor repertoire diversity in primary and secondary lymphoid tissues. Eur J Immunol. (2016) 46:480–92. doi: 10.1002/eji.201545586
135. Gibson KL, Wu YC, Barnett Y, Duggan O, Vaughan R, Kondeatis E, et al. B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell. (2009) 8:18–25. doi: 10.1111/j.1474-9726.2008.00443.x
136. Riley RL, Khomtchouk K, Blomberg BB. Inflammatory immune cells may impair the preBCR checkpoint, reduce new B cell production, and alter the antibody repertoire in old age. Exp Gerontol. (2018) 105:87–93. doi: 10.1016/j.exger.2018.01.023
137. Monzo C, Gkioni L, Beyer A, Valenzano DR, Gronke S, Partridge L. Dietary restriction mitigates the age-associated decline in mouse B cell receptor repertoire diversity. Cell Rep. (2023) 42:112722. doi: 10.1016/j.celrep.2023.112722
138. Birjandi SZ, Ippolito JA, Ramadorai AK, Witte PL. Alterations in marginal zone macrophages and marginal zone B cells in old mice. J Immunol. (2011) 186:3441–51. doi: 10.4049/jimmunol.1001271
139. Turner VM, Mabbott NA. Ageing adversely affects the migration and function of marginal zone B cells. Immunology. (2017) 151:349–62. doi: 10.1111/imm.12737
140. Mongini PK, Inman JK, Han H, Fattah RJ, Abramson SB, Attur M. APRIL and BAFF promote increased viability of replicating human B2 cells via mechanism involving cyclooxygenase 2. J Immunol. (2006) 176:6736–51. doi: 10.4049/jimmunol.176.11.6736
141. Jin R, Kaneko H, Suzuki H, Arai T, Teramoto T, Fukao T, et al. Age-related changes in BAFF and APRIL profiles and upregulation of BAFF and APRIL expression in patients with primary antibody deficiency. Int J Mol Med. (2008) 21:233–8. doi: 10.3892/ijmm
142. Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, et al. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science. (2001) 293:2111–4. doi: 10.1126/science.1061964
143. Kyaw T, Cui P, Tay C, Kanellakis P, Hosseini H, Liu E, et al. BAFF receptor mAb treatment ameliorates development and progression of atherosclerosis in hyperlipidemic ApoE(-/-) mice. PloS One. (2013) 8:e60430. doi: 10.1371/journal.pone.0060430
144. Tsiantoulas D, Sage AP, Goderle L, Ozsvar-Kozma M, Murphy D, Porsch F, et al. B cell-activating factor neutralization aggravates atherosclerosis. Circulation. (2018) 138:2263–73. doi: 10.1161/CIRCULATIONAHA.117.032790
145. Frasca D, Nguyen D, Riley RL, Blomberg BB. Decreased E12 and/or E47 transcription factor activity in the bone marrow as well as in the spleen of aged mice. J Immunol. (2003) 170:719–26. doi: 10.4049/jimmunol.170.2.719
146. Rosenfeld SM, Perry HM, Gonen A, Prohaska TA, Srikakulapu P, Grewal S, et al. B-1b cells secrete atheroprotective igM and attenuate atherosclerosis. Circ Res. (2015) 117:e28–39. doi: 10.1161/CIRCRESAHA.117.306044
147. de Faire U, Su J, Hua X, Frostegård A, Halldin M, Hellenius M-L, et al. Low levels of IgM antibodies to phosphorylcholine predict cardiovascular disease in 60-year old men: effects on uptake of oxidized LDL in macrophages as a potential mechanism. J autoimmunity. (2010) 34:73–9. doi: 10.1016/j.jaut.2009.05.003
148. Su J, Hua X, Vikstrom M, Leander K, Gigante B, Hellenius ML, et al. Low levels of IgM antibodies to oxidized cardiolipin increase and high levels decrease risk of cardiovascular disease among 60-year olds: a prospective study. BMC Cardiovasc Disord. (2013) 13:1. doi: 10.1186/1471-2261-13-1
149. Thiagarajan D, Frostegard AG, Singh S, Rahman M, Liu A, Vikstrom M, et al. Human igM antibodies to malondialdehyde conjugated with albumin are negatively associated with cardiovascular disease among 60-year-olds. J Am Heart Assoc. (2016) 5:e004415. doi: 10.1161/JAHA.116.004415
150. Engelbertsen D, Vallejo J, Quach TD, Fredrikson GN, Alm R, Hedblad B, et al. Low levels of igM antibodies against an advanced glycation endproduct-modified apolipoprotein B100 peptide predict cardiovascular events in nondiabetic subjects. J Immunol. (2015) 195:3020–5. doi: 10.4049/jimmunol.1402869
151. Upadhye A, Srikakulapu P, Gonen A, Hendrikx S, Perry HM, Nguyen A, et al. Diversification and CXCR4-dependent establishment of the bone marrow B-1a cell pool governs atheroprotective igM production linked to human coronary atherosclerosis. Circ Res. (2019) 125:e55–70. doi: 10.1161/CIRCRESAHA.119.315786
152. Rodriguez-Zhurbenko N, Quach TD, Hopkins TJ, Rothstein TL, Hernandez AM. Human B-1 cells and B-1 cell antibodies change with advancing age. Front Immunol. (2019) 10:483. doi: 10.3389/fimmu.2019.00483
153. Srikakulapu P, Pattarabanjird T, Upadhye A, Bontha SV, Osinski V, Marshall MA, et al. B-1b cells have unique functional traits compared to B-1a cells at homeostasis and in aged hyperlipidemic mice with atherosclerosis. Front Immunol. (2022) 13:909475. doi: 10.3389/fimmu.2022.909475
154. Colonna-Romano G, Bulati M, Aquino A, Pellicano M, Vitello S, Lio D, et al. A double-negative (IgD-CD27-) B cell population is increased in the peripheral blood of elderly people. Mech Ageing Dev. (2009) 130:681–90. doi: 10.1016/j.mad.2009.08.003
155. Huang W, Sinha J, Newman J, Reddy B, Budhai L, Furie R, et al. The effect of anti-CD40 ligand antibody on B cells in human systemic lupus erythematosus. Arthritis Rheumatol. (2002) 46:1554–62. doi: 10.1002/art.10273
156. Fraussen J, Marquez S, Takata K, Beckers L, Montes Diaz G, Zografou C, et al. Phenotypic and ig repertoire analyses indicate a common origin of igD(-)CD27(-) double negative B cells in healthy individuals and multiple sclerosis patients. J Immunol. (2019) 203:1650–64. doi: 10.4049/jimmunol.1801236
157. Beckers L, Somers V, Fraussen J. IgD(-)CD27(-) double negative (DN) B cells: Origins and functions in health and disease. Immunol Lett. (2023) 255:67–76. doi: 10.1016/j.imlet.2023.03.003
158. Frasca D, Diaz A, Romero M, Blomberg BB. Human peripheral late/exhausted memory B cells express a senescent-associated secretory phenotype and preferentially utilize metabolic signaling pathways. Exp Gerontol. (2017) 87:113–20. doi: 10.1016/j.exger.2016.12.001
159. Cancro MP. Age-associated B cells. Annu Rev Immunol. (2020) 38:315–40. doi: 10.1146/annurev-immunol-092419-031130
160. Bulati M, Caruso C, Colonna-Romano G. From lymphopoiesis to plasma cells differentiation, the age-related modifications of B cell compartment are influenced by “inflamm-ageing”. Ageing Res Rev. (2017) 36:125–36. doi: 10.1016/j.arr.2017.04.001
161. Phalke S, Rivera-Correa J, Jenkins D, Flores Castro D, Giannopoulou E, Pernis AB. Molecular mechanisms controlling age-associated B cells in autoimmunity. Immunol Rev. (2022) 307:79–100. doi: 10.1111/imr.13068
162. Russell Knode LM, Naradikian MS, Myles A, Scholz JL, Hao Y, Liu D, et al. Age-associated B cells express a diverse repertoire of V(H) and vkappa genes with somatic hypermutation. J Immunol. (2017) 198:1921–7. doi: 10.4049/jimmunol.1601106
163. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood. (2011) 118:1305–15. doi: 10.1182/blood-2011-01-331462
164. Manni M, Gupta S, Ricker E, Chinenov Y, Park SH, Shi M, et al. Regulation of age-associated B cells by IRF5 in systemic autoimmunity. Nat Immunol. (2018) 19:407–19. doi: 10.1038/s41590-018-0056-8
165. Camell CD, Lee A, Günther P, Goldberg EL, Spadaro O, Youm Y-H, et al. Aging induces Nlrp3 inflammasome dependent adipose B cell expansion to impair metabolic homeostasis. bioRxiv. (2019) 607192. doi: 10.1101/607192
166. Verhagen SN, Visseren FL. Perivascular adipose tissue as a cause of atherosclerosis. Atherosclerosis. (2011) 214:3–10. doi: 10.1016/j.atherosclerosis.2010.05.034
167. Pattarabanjird T, Srikakulapu P, Ransegnola B, Marshall MA, Ley K. Single-cell profiling of CD11c+ B cells in atherosclerosis. Front Immunol. (2024) 14:1296668. doi: 10.3389/fimmu.2023.1296668
168. Rubtsov AV, Rubtsova K, Kappler JW, Jacobelli J, Friedman RS, Marrack P. CD11c-expressing B cells are located at the T cell/B cell border in spleen and are potent APCs. J Immunol. (2015) 195:71–9. doi: 10.4049/jimmunol.1500055
169. Hao Y, O’Neill P, Naradikian MS, Scholz JL, Cancro MP. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood. (2011) 118:1294–304. doi: 10.1182/blood-2011-01-330530
170. Webb LMC, Fra-Bido S, Innocentin S, Matheson LS, Attaf N, Bignon A, et al. Ageing promotes early T follicular helper cell differentiation by modulating expression of RBPJ. Aging Cell. (2021) 20:e13295. doi: 10.1111/acel.13295
171. Lefebvre JS, Masters AR, Hopkins JW, Haynes L. Age-related impairment of humoral response to influenza is associated with changes in antigen specific T follicular helper cell responses. Sci Rep. (2016) 6:25051. doi: 10.1038/srep25051
172. Keszei M, Detre C, Castro W, Magelky E, O’Keeffe M, Kis-Toth K, et al. Expansion of an osteopontin-expressing T follicular helper cell subset correlates with autoimmunity in B6.Sle1b mice and is suppressed by the H1-isoform of the Slamf6 receptor. FASEB J. (2013) 27:3123–31. doi: 10.1096/fj.12-226951
173. Tahir S, Fukushima Y, Sakamoto K, Sato K, Fujita H, Inoue J, et al. A CD153+CD4+ T follicular cell population with cell-senescence features plays a crucial role in lupus pathogenesis via osteopontin production. J Immunol. (2015) 194:5725–35. doi: 10.4049/jimmunol.1500319
174. Stebegg M, Bignon A, Hill DL, Silva-Cayetano A, Krueger C, Vanderleyden I, et al. Rejuvenating conventional dendritic cells and T follicular helper cell formation after vaccination. Elife. (2020) 9:e52473. doi: 10.7554/eLife.52473
175. Silva-Cayetano A, Fra-Bido S, Robert PA, Innocentin S, Burton AR, Watson EM, et al. Spatial dysregulation of T follicular helper cells impairs vaccine responses in aging. Nat Immunol. (2023) 24:1124–37. doi: 10.1038/s41590-023-01519-9
176. Thompson HL, Smithey MJ, Surh CD, Nikolich-Zugich J. Functional and homeostatic impact of age-related changes in lymph node stroma. Front Immunol. (2017) 8:706. doi: 10.3389/fimmu.2017.00706
177. Becklund BR, Purton JF, Ramsey C, Favre S, Vogt TK, Martin CE, et al. The aged lymphoid tissue environment fails to support naive T cell homeostasis. Sci Rep. (2016) 6:30842. doi: 10.1038/srep30842
178. Brown FD, Turley SJ. Fibroblastic reticular cells: organization and regulation of the T lymphocyte life cycle. J Immunol. (2015) 194:1389–94. doi: 10.4049/jimmunol.1402520
179. Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, et al. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol. (2007) 8:1255–65. doi: 10.1038/ni1513
180. Turner VM, Mabbott NA. Structural and functional changes to lymph nodes in ageing mice. Immunology. (2017) 151:239–47. doi: 10.1111/imm.12727
181. Hu D, Mohanta SK, Yin C, Peng L, Ma Z, Srikakulapu P, et al. Artery tertiary lymphoid organs control aorta immunity and protect against atherosclerosis via vascular smooth muscle cell lymphotoxin β receptors. Immunity. (2015) 42:1100–15. doi: 10.1016/j.immuni.2015.05.015
182. Grabner R, Lotzer K, Dopping S, Hildner M, Radke D, Beer M, et al. Lymphotoxin beta receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE-/- mice. J Exp Med. (2009) 206:233–48. doi: 10.1084/jem.20080752
183. Mohanta SK, Yin C, Peng L, Srikakulapu P, Bontha V, Hu D, et al. Artery tertiary lymphoid organs contribute to innate and adaptive immune responses in advanced mouse atherosclerosis. Circ Res. (2014) 114:1772–87. doi: 10.1161/CIRCRESAHA.114.301137
184. Lee Y, Chin RK, Christiansen P, Sun Y, Tumanov AV, Wang J, et al. Recruitment and activation of naive T cells in the islets by lymphotoxin beta receptor-dependent tertiary lymphoid structure. Immunity. (2006) 25:499–509. doi: 10.1016/j.immuni.2006.06.016
185. Kaptoge S, Seshasai SR, Gao P, Freitag DF, Butterworth AS, Borglykke A, et al. Inflammatory cytokines and risk of coronary heart disease: new prospective study and updated meta-analysis. Eur Heart J. (2014) 35:578–89. doi: 10.1093/eurheartj/eht367
186. Li L, Liu S, Zhang Z, Zhou L, Zhang Z, Xiong Y, et al. Prognostic value of high-sensitivity C-reactive protein in patients undergoing percutaneous coronary intervention with different glycemic metabolism status. Cardiovasc Diabetol. (2023) 22:223. doi: 10.1186/s12933-023-01932-2
187. Zhang Y, Wu NQ, Li S, Zhu CG, Guo YL, Qing P, et al. Non-HDL-C is a better predictor for the severity of coronary atherosclerosis compared with LDL-C. Heart Lung Circ. (2016) 25:975–81. doi: 10.1016/j.hlc.2016.04.025
188. Goronzy JJ, Li G, Yang Z, Weyand CM. The janus head of T cell aging - autoimmunity and immunodeficiency. Front Immunol. (2013) 4:131. doi: 10.3389/fimmu.2013.00131
189. Weksler ME, Szabo P. The effect of age on the B-cell repertoire. J Clin Immunol. (2000) 20:240–9. doi: 10.1023/A:1006659401385
190. Kogut I, Scholz JL, Cancro MP, Cambier JC. B cell maintenance and function in aging. Semin Immunol. (2012) 24(5):342–9. doi: 10.1016/j.smim.2012.04.004
191. Nakazato R, Arsanjani R, Achenbach S, Gransar H, Cheng VY, Dunning A, et al. Age-related risk of major adverse cardiac event risk and coronary artery disease extent and severity by coronary CT angiography: results from 15 187 patients from the International Multisite CONFIRM Study. Eur Heart J Cardiovasc Imaging. (2014) 15:586–94. doi: 10.1093/ehjci/jet132
192. Alpert A, Pickman Y, Leipold M, Rosenberg-Hasson Y, Ji X, Gaujoux R, et al. A clinically meaningful metric of immune age derived from high-dimensional longitudinal monitoring. Nat Med. (2019) 25:487–95. doi: 10.1038/s41591-019-0381-y
193. Ridker PM. How common is residual inflammatory risk? Circ Res. (2017) 120:617–9. doi: 10.1161/CIRCRESAHA.116.310527
194. Lim S, Park YM, Sakuma I, Koh KK. How to control residual cardiovascular risk despite statin treatment: focusing on HDL-cholesterol. Int J Cardiol. (2013) 166:8–14. doi: 10.1016/j.ijcard.2012.03.127
195. Han SH, Nicholls SJ, Sakuma I, Zhao D, Koh KK. Hypertriglyceridemia and cardiovascular diseases: revisited. Korean Circ J. (2016) 46:135–44. doi: 10.4070/kcj.2016.46.2.135
196. Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, et al. Colchicine in patients with chronic coronary disease. N Engl J Med. (2020) 383:1838–47. doi: 10.1056/NEJMoa2021372
197. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
198. Sriranjan R, Zhao TX, Tarkin J, Hubsch A, Helmy J, Vamvaka E, et al. Low-dose interleukin 2 for the reduction of vascular inflammation in acute coronary syndromes (IVORY): protocol and study rationale for a randomised, double-blind, placebo-controlled, phase II clinical trial. BMJ Open. (2022) 12:e062602. doi: 10.1136/bmjopen-2022-062602
199. Zhao TX, Sriranjan RS, Tuong ZK, Lu Y, Sage AP, Nus M, et al. Regulatory T-cell response to low-dose interleukin-2 in ischemic heart disease. NEJM Evid. (2022) 1:EVIDoa2100009. doi: 10.1056/EVIDoa2100009
200. Zhao TX, Aetesam-Ur-Rahman M, Sage AP, Victor S, Kurian R, Fielding S, et al. Rituximab in patients with acute ST-elevation myocardial infarction: an experimental medicine safety study. Cardiovasc Res. (2022) 118:872–82. doi: 10.1093/cvr/cvab113
201. Ait-Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L, et al. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. (2010) 207:1579–87. doi: 10.1084/jem.20100155
202. Kyaw T, Tay C, Khan A, Dumouchel V, Cao A, To K, et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol. (2010) 185:4410–9. doi: 10.4049/jimmunol.1000033
203. Jiang Y-H, Jiang L-Y, Wang Y-C, Ma D-F, Li X. Quercetin attenuates atherosclerosis via modulating oxidized LDL-induced endothelial cellular senescence. Front Pharmacol. (2020) 11:512. doi: 10.3389/fphar.2020.00512
204. Primikyri A, Chatziathanasiadou MV, Karali E, Kostaras E, Mantzaris MD, Hatzimichael E, et al. Direct binding of Bcl-2 family proteins by quercetin triggers its pro-apoptotic activity. ACS Chem Biol. (2014) 9:2737–41. doi: 10.1021/cb500259e
205. Kleemann R, Verschuren L, Morrison M, Zadelaar S, van Erk MJ, Wielinga PY, et al. Anti-inflammatory, anti-proliferative and anti-atherosclerotic effects of quercetin in human in vitro and in vivo models. Atherosclerosis. (2011) 218:44–52. doi: 10.1016/j.atherosclerosis.2011.04.023
206. Hwang HV, Tran DT, Rebuffatti MN, Li CS, Knowlton AA. Investigation of quercetin and hyperoside as senolytics in adult human endothelial cells. PloS One. (2018) 13:e0190374. doi: 10.1371/journal.pone.0190374
207. Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. (2015) 7:279ra40. doi: 10.1126/scitranslmed.aaa4642
208. Hubbard GP, Wolffram S, Lovegrove JA, Gibbins JM. Ingestion of quercetin inhibits platelet aggregation and essential components of the collagen-stimulated platelet activation pathway in humans. J Thromb Haemost. (2004) 2:2138–45. doi: 10.1111/j.1538-7836.2004.01067.x
209. He Y, Zhang X, Chang J, Kim HN, Zhang P, Wang Y, et al. Using proteolysis-targeting chimera technology to reduce navitoclax platelet toxicity and improve its senolytic activity. Nat Commun. (2020) 11:1996. doi: 10.1038/s41467-020-15838-0
210. Tao L, Reese TA. Making mouse models that reflect human immune responses. Trends Immunol. (2017) 38:181–93. doi: 10.1016/j.it.2016.12.007
211. Dobson GP, Letson HL, Biros E, Morris J. Specific pathogen-free (SPF) animal status as a variable in biomedical research: have we come full circle? EBioMedicine. (2019) 41:42–3. doi: 10.1016/j.ebiom.2019.02.038
Keywords: adaptive immunity, atherosclerosis, aging, immunosenescence, inflammaging
Citation: Snijckers RPM and Foks AC (2024) Adaptive immunity and atherosclerosis: aging at its crossroads. Front. Immunol. 15:1350471. doi: 10.3389/fimmu.2024.1350471
Received: 05 December 2023; Accepted: 28 March 2024;
Published: 15 April 2024.
Edited by:
Klaus Ley, Augusta University, United StatesReviewed by:
Jan Nilsson, Lund University, SwedenPayel Roy, La Jolla Institute for Immunology (LJI), United States
Copyright © 2024 Snijckers and Foks. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amanda C. Foks, YS5jLmZva3NAbGFjZHIubGVpZGVudW5pdi5ubA==