 Yuxin Niu
Yuxin Niu Qin Ning
Qin Ning Tao Chen
Tao Chen- Department of Infectious Diseases, Tongji Hospital, Tongji Medical College and State Key Laboratory for Diagnosis and Treatment of Severe Zoonostic Infectious Disease, Huazhong University of Science and Technology, Wuhan, Hubei, China
Dabie Banda virus (DBV), a tick-borne pathogen, was first identified in China in 2009 and causes profound symptoms including fever, leukopenia, thrombocytopenia and multi-organ dysfunction, which is known as severe fever with thrombocytopenia syndrome (SFTS). In the last decade, global incidence and mortality of SFTS increased significantly, especially in East Asia. Though previous studies provide understandings of clinical and immunological characteristics of SFTS development, comprehensive insight of antiviral immunity response is still lacking. Here, we intensively discuss the antiviral immune response after DBV infection by integrating previous ex- and in-vivo studies, including innate and adaptive immune responses, anti-viral immune responses and long-term immune characters. A comprehensive overview of potential immune targets for clinical trials is provided as well. However, development of novel strategies for improving the prognosis of the disease remains on challenge. The current review may shed light on the establishment of immunological interventions for the critical disease SFTS.
1 Introduction
Severe fever with thrombocytopenia syndrome (SFTS) was discovered in China in 2009 (1) as a burgeoning severe hemorrhagic fever disease caused by severe fever with thrombocytopenia syndrome virus (SFTSV) infection, which was formally renamed family Phenuiviridae, genus Bandavirus, species Dabie bandavirus (DBV) in 2019 (2). SFTS has subsequently emerged in Japan, South Korea, and Vietnam. SFTS generally occurs between March and November, with the highest incidence from May through July (3, 4). The tick species Haemaphysalis longicornis is a major vector of SFTS and is responsible for exposing over 40% of China’s population in 1140 counties (5, 6). The detection of DBV-specific antibodies and RNA in some animals besides ticks suggests that they may also participate in disease transmission (7–10).
The existing body of research on the clinical manifestations of the disease reveals that patients with SFTS develop a fever with temperature higher than 38°C, thrombocytopenia, lymphadenectasis, gastrointestinal discomfort, and lymphadenopathy. In severe cases, acute multiple organ failure leads to mortality. The disease typically follows a course consisting of several stages, including the incubation period, febrile stage, multiple organ failure, remission, and convalescence (3, 4, 11–14). Respiratory distress, signs of bleeding, neurological symptoms, increased levels of lactate dehydrogenase (LDH), aspartate transaminase/alanine transaminase (AST/ALT) ratio, interleukin 6 (IL-6), neutrophil percentage, C-reactive protein/lymphocyte ratio, activated partial thromboplastin time (APTT), and thrombin time (TT) are correlated with poor prognosis and high mortality in individuals diagnosed with SFTS (13, 15–18). Thus, it can be inferred that SFTS is a multifaceted systemic ailment characterized by hepatic impairment, inflammatory response, and anomalous coagulation (3).
2 Trigger of antiviral immunity: DBV invasion of the body
Ticks carrying DBV transmit the virus to humans through infestation. The virus attaches to the surface of host cells through interactions between glycoproteins and membrane factors, such as Dendritic cell-specific ICAM-3 grabbing non-integrin (DC-SIGN), and subsequently enters cells through the clathrin-dependent pathway (4, 19–21). (Figure 1A). Recently, CCR2 has been discovered as DBV entry binding receptor to virus glycoprotein N (22). Skin-resident cells surrounded the bite location, including keratinocytes, mast cells, immature Langerhans cells and epidermal dendritic cells, are hypothesized to be the first target cell for DBV infection and transmission. Mast cells in skin mucous membranes are found as sentinels for tissue injury, DBV invasion (23) and local responses by degranulating and releasing pre-stored histamine and other bioactive substances within minutes or de novo synthesized mediators and lipid mediators within hours (24, 25).
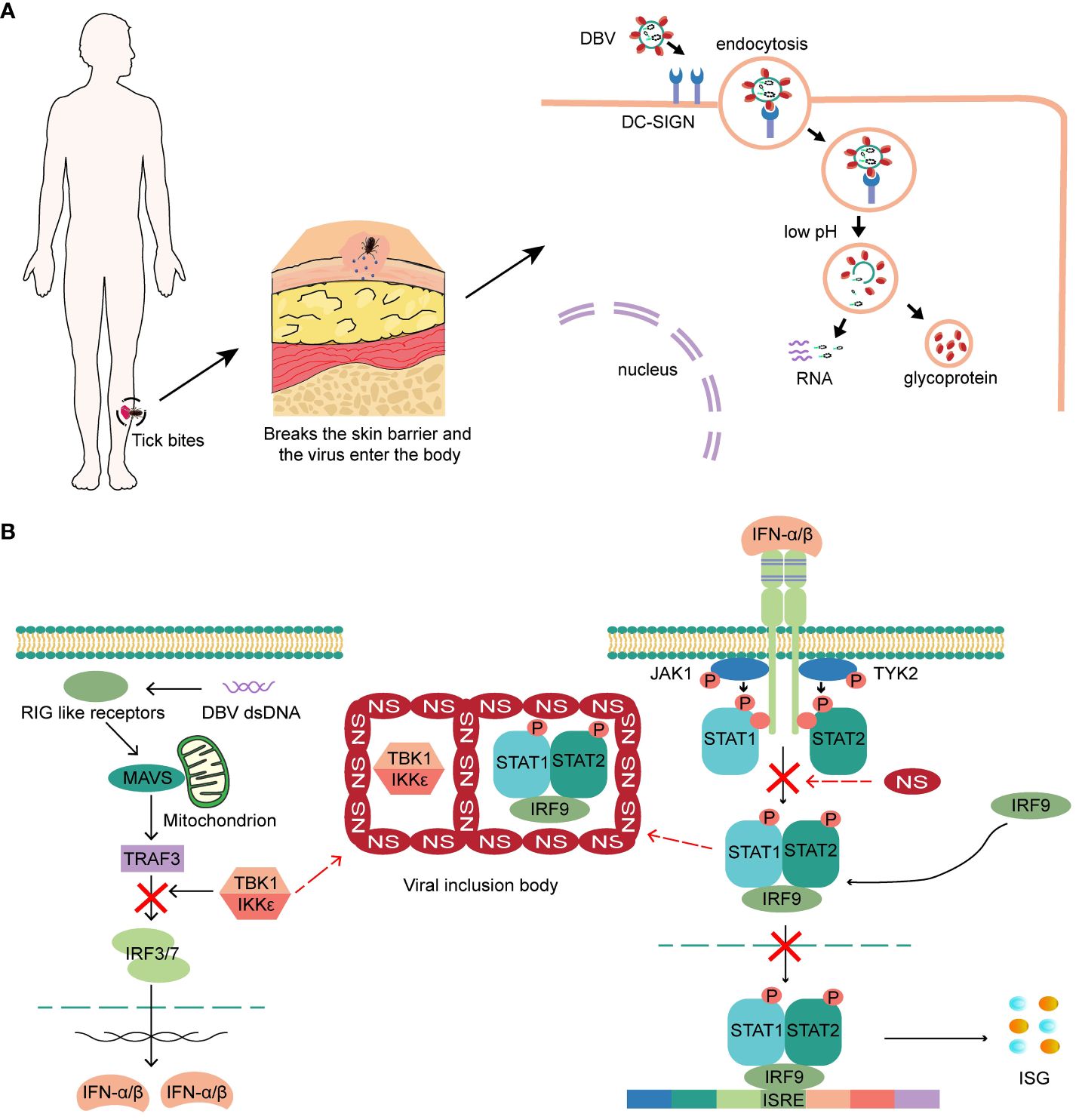
Figure 1 (A) The virus attaches to the surface of host cells, and subsequently enters cells. Low pH triggers the fusion of the virus with the cell membrane and releases the viral ribonucleic protein complex into the cytoplasm. (B) NSs of DBV can hinder the production and response of intracellular IFN by encapsulating the STAT heterodimer and TBK1/IKKϵ, and block the phosphorylation of STAT2 and nuclear translocation of STAT, ultimately leading to a complete blockade of the IFN response.
Both live or UV-inactivated DBV virions can stimulate mast cells for releasing bioactive mediators to enhance endothelial and vascular permeability (24). Loss of tight junction between endothelial cells allows a huge amount of white blood cells to exude the site of viral invasion with enhanced immune response. In the early stages of SFTS resulting from tick bites, mast cells play a crucial role in boosting the body’s defense response by producing various bioactive chemicals.
3 Innate immunity in DBV infection
The host innate immune response exhibits an initial response to pathogenic microorganisms and acts as a crucial component of the adaptive immune response. Interferon, monocytes, macrophages, and NK cells are identified as critical responders of antiviral innate immunity.
3.1 DBV NSs block the interferon response
Interferon performs various roles in innate and acquired immunity, exhibiting potent and universal antiviral activity (26). After infecting epithelial cells, fibroblasts, and pDCs surround the bite location, the immune system responds by secreting interferon-α (IFN-α) and IFN-β. IFN-γ is produced by activated macrophages and NK cells in the early stages or by activated Th1 cells in the later stages, thus inducing an antiviral state. DBV binds to pattern recognition receptors on the cell surface and activates critical kinases that initiate type I interferon (IFN-I) production (27). Secreted IFN then binds to IFNARs on the cell surface, activating JAK-STAT signaling and increasing the expression of antiviral interferon-stimulated genes (ISGs) (28). However, the nonstructural proteins (NSs) of DBV can hinder the production and response of intracellular IFN by encapsulating the STAT heterodimer and TBK1/IKKϵ (29, 30) that are integral molecules of the IFN-I production pathway to form viral inclusion bodies. NSs can also block the phosphorylation of STAT2 and nuclear translocation of STAT, ultimately leading to a complete blockade of the IFN response (4, 31, 32) (Figure 1B). Recently discovered a higher prevalence of exacerbated IFN-I signaling pathways and increased ISGs expression in patients with fatal SFTS, and a direct correlation between enhanced ISGs expression and disease severity. Therefore, an increased IFN-I response may be detrimental rather than beneficial (33, 34). The blockade of the interferon response seems to occur only in DBV-infected cells, whereas the majority of cells in SFTS patients still show a high enrichment of the IFN-I signaling pathway, which may contribute to the exacerbation of the disease.
3.2 Intermediate phenotype transition of monocytes
Peripheral monocytes are derived from bone marrow and express a variety of receptors that exert strong phagocytosis and pathogen-clearing abilities. They are believed playing pivotal role during expansion of DBV replication via being directly infected by the virus (22, 35, 36). In the early stages of infection, monocytes suffer extensive apoptosis and result in viral particle release (37), which might be contributed by immune surveillance to clear virus-infected target cells. Previous analysis has revealed that intermediate monocytes are more prone to DBV infection than classical monocytes. In vitro, transition of monocytes from the classical to the intermediate phenotype was observed by DBV stimulated peripheral blood mononuclear cells of patients with SFTS, especially in deceased patients (33). The expression of RNA virus infection acquired gene CTSL and CTSB was found significantly increase in intermediate monocytes (33, 38, 39), which may explain why intermediate monocytes are more susceptible to DBV infection. Meanwhile, intermediate monocytes exhibit enhanced expression of ISGs and IFN-dependent chemokines, as well as complement activation. Overactivation of type I IFN response and complement cascade reaction by intermediate monocytes is the hallmark of DBV induced organism injury and indicator of poor prognosis (33). Furthermore, monocytes isolated from individuals with acute SFTS produce significantly lower TNF-α levels by LPS stimulation. This might indicate that monocytes are not major source of TNF-α in SFTS patients (35).
3.3 Macrophages M2 phenotype differentiation
Circulating monocytes can migrate to inflammatory tissues for differentiating to macrophages during viral infections, which further activate and release proinflammatory mediators. The function of macrophages could be significantly promoted by IFN-γ with expressing iNOS enzyme and NO to kill virus-infected cells. Based on the specific staining of DBV-positive macrophages of spleen, macrophages are hypothesized to be one of important target cells for DBV infection. Consistently, replication of DBV is observed in primary mouse macrophages in vitro infection model, without affecting phagocytic activity to platelets (40). Another study reported that DBV infection significantly increased the expression of miR-146a and miR-146b in macrophages and facilitated their differentiation into M2 phenotypes (41). M2 macrophages can enhance phagocytic activity but suppress the production of proinflammatory cytokines and reduce pathogen-killing capability (42), which may promote the expansion of DBV (4, 43). In the early stage of DBV infection, elevated expression of IFN-γ encourages macrophages M1 phenotype transition with proinflammatory role via STAT1 pathway, while the elevated DBV replication and IL10 levels promote macrophage M2 differentiation in the late stage of infection (4, 41, 44).
3.4 Activation and exhaustion of NK cells
NK cells are one of primary responders of innate immunity to viral infections (4). The surface expression of major histocompatibility complex class I molecules on infected host cells is generally downregulated during viral infection, which can be recognized by NK cells, leading to the direct elimination of infected cells. In addition, NK cells secrete proinflammatory cytokines and aid in the early elimination of the virus. Recent studies have highlighted a negative association between NK cell depletion and severe SFTS, particularly during the early stages of infection (45, 46). CD56dimCD16+ NK cells are the primary subset of cytotoxic NK cells and their decrease in patients with SFTS may impair the clearance of virus-infected cells and immunomodulation. Furthermore, the activation and functional enhancement of CD56dimCD16+ NK cells have also been observed in the acute phase of SFTS, with high expression of Ki-67 and GZMB and relatively low expression of NKG2A (47, 48).
3.5 Upregulated complement-related gene expression but downregulated protein levels
Activation of the complement system and coagulation disorders can increase the risk of infection caused by various pathogens (49). Complement activation is well known in patients with disseminated intravascular coagulation, and there is a connection between the complement and coagulation systems (50). A recent study examining serum proteins in SFTS patients found a decrease in complement system proteins in those who passed away, except for MASP2. Conversely, complement proteins C4a, C4b, C1s, and C1R were elevated, while C6 and C7 proteins were reduced in recovering patients (51, 52). Therefore, the fatal patients are thought to have deficiencies in their innate immune responses, including the down-regulation of the complement system that leads to the progression of DIC characterized by coagulation dysfunction. Interestingly, another study reported upregulated expression of complement-related genes in patients who died of SFTS (33). This phenomenon may be caused by excessive activation of the complement system and subsequent depletion of complement components. Complement-dependent cytotoxicity undoubtedly assists the body in clearing viruses.
4 Adaptive immunity in DBV infection
Patients with SFTS experience decreased lymphocyte blood counts with a significant portion of their lymphocytes are activated and exhibit enhanced functionality, suggesting that these patients mount a robust adaptive immune response against DBV (53). The destruction of antiviral immunity in these patients occurs due to the combined damage inflicted on B and T cells.
4.1 B cells activated by DBV act as target cells for viral replication, leading to systemic dissemination
Pathological examination of fatal SFTS cases revealed that large hematopoietic cells in lymphoid organs (lymph nodes, spleen, and bone marrow) comprise the majority of the cells infected by the virus. Additionally, mature lymphocytes are particularly sensitive to DBV infection in fatal cases, with a notable presence of DBV+ mononuclear cells found in the capillaries of non-lymphoid organs in deceased patients. The infected cells can be recognized through the detection of plasmablast markers (MUM1 and CD38). However, unlike B cells infected with DBV, DBV+ cells in these capillaries do not express CD20 (54). CD20 is a familiar marker for B cells that starts to express in late pre-B lymphocytes (but not in pre-B lymphocytes) and is absent from terminally differentiated plasmablasts and plasma cells (55). Another study showed that most mature B cells infected with DBV in the lymph nodes were activated and had immunophenotypes similar to those of plasmablasts (54, 56). Based on these findings, it can be inferred that B cells at different stages of plasma cell differentiation serve as sites for DBV replication and spread the virus to non-lymphoid organs (54, 57). Elevated proportions of plasmablasts with functional impairments have also been observed (33), resulting in the absence of DBV-specific IgG antibodies against glycoprotein and nucleocapsid protein. Various factors, including impaired DC differentiation and antigen presentation function, failed follicular helper T cell differentiation and B cell antibody class switching, lead to comprehensive impairment of humoral immunity and affect virus clearance (37, 58).
Although DBV infection can activate B cells and cause them to replicate at a low level, most activated B cells are not infected with DBV. Despite limited viral infection and replication efficiency in peripheral blood B cells, the B cell lineage contributes to B cells activation (56). The sirtuin and IFN signaling pathways exhibit broad induction in the whole B cell population of patients who die of the disease, whereas the infected B cell population shows a specific reduction in these pathways compared to the uninfected population (57). This may be due to uninfected immune cells attempting to activate the IFN and sirtuin pathways to against virus. DBV infection of B cells induces uninfected B cells to produce cytokines and chemokines, such as IL-6 in vitro (56). However, DBV-infected B cells can block their signaling pathways and hamper paracrine effects on neighboring uninfected cells in vivo ultimately allowing high viral replication (33, 57). This may help DBV evade the host immune system (59).
4.2 Decreased T cell counts accompanied by highly active and exhausted phenotypes
DBV infection impairs DCs differentiation and function (58) hindering the development of T follicular helper cells and compromising the effectiveness of virus-specific humoral response (37). T-cell depletion is prevalent during the initial stages of DBV infection, particularly in patients with severe underlying conditions (46). Moreover, changes in T lymphocytes are similar to the overall dynamics of lymphocytes. CD4+ T cell deficiency and Th2 and Th17 bias are strongly correlated with SFTS severity (60). The significant decrease of peripheral T cells is revealed as its apoptosis via the Fas/FasL pathway in patients with SFTS (53). High levels of PD-1 expression are detected in exhausted T cells (61–63) in some chronic viral infections. Approximately 20% of CD4+ and CD8+ T cells display increased PD-1 expression, enhanced cytotoxicity, and effector functions in the early stages of DBV infection (53, 64). Additionally, CD8+ T cells that are similar to the response of antigen-specific T cells during acute viral infections exhibit a stronger response than CD4+ T cells (65). They can be activated rapidly and are recruited mainly by intermediate monocytes via CXCL10 (66). The differentiation from classical to intermediate monocytes in SFTS patients may lead to enhanced CD8+ T cell recruitment and an extensive immunological response. Cellular immunity is critical for virus clearance on antiviral immunity. However, T cell apoptosis, exhaustion, and decreased numbers occur with disease progression, indicating potential impairment of cellular immunity (64).
5 DBV replication and clearance and patient prognosis of SFTS
After the virus enters the human body, monocytes, macrophages and mature B cells are promptly infected which lead to extensive viral replication and expansion (22, 54). Subsequently, classical monocytes differentiate into intermediate monocytes with a decrease in CCR2 expression (33, 67). Severe apoptosis of monocytes in the early stages of the infection may result in the release of large amounts of viral particles, leading to the persistent elevation of the viral load in the peripheral blood. At the same time, B cells may experience differentiation blockage under the infection of DBV, and significantly elevated number of plasmablasts with defective antibody secretion function developed which consistently result in humoral immunity insufficient and low level of viral clearance (37). The DBV is eventually cleared from the body through the combined activation of the complement system, NK cells and T cells within 30 days of onset in recovered patients (68). However, patients with advanced age, high viral loads, and concomitant severe underlying diseases and comorbidities are more likely to succumb. Patients recovering from SFTS rarely experience long-term complications, and the levels of DBV-specific IgM antibodies in their bodies decrease over time, becoming undetectable after six months. In contrast, DBV-specific IgG antibodies peaked six months after recovery, then steadily declined but remained detectable for more than eight years. IgG antibodies are found maintaining high levels for a long period and play protective roles against DBV reinfection (68). These findings align with the only one reported case of DBV reinfection. The patient’s second attack was less severe than the first, possibly because the patient had low levels of DBV IgG antibodies compared to other patients treated at the same hospital (69). Individuals who recover from DBV infection typically generate protective IgG antibodies that are unique to the virus. However, low levels of these antibodies diminish their protective effects and lead to reinfection in rare cases.
6 Discussion
A complete description of the immune response in DBV-infected people is required to identify possible targets for therapeutic intervention. In conclusion, cells at the site of infection begin antiviral immunity. DBV can influence macrophage activation and differentiation (41), activate NK cells but reduce their numbers (47), and alter the monocyte phenotype into intermediate monocytes. B cells might be prevented from differentiating into plasma cells, increasing the frequency of plasmablasts with IFN-I response-related pathways. The function of CD4+ and CD8+ T cells is enhanced in SFTS patients accompanied with apoptosis via the Fas/FasL pathway (53) (Figure 2). Based on this, we proposed some potential intervention strategies, including lowering the susceptibility of monocytes, macrophages, and mature B cells to DBV, inhibiting viral replication within these cells, and improving T cell apoptosis and depletion to facilitate the body’s virus clearance, which requires a clear understanding of the specific mechanisms by which DBV enters different cells. Currently, it is accepted that DBV can interact with CCR2 to enter monocytes, and in vitro experiments have shown that CCR2 inhibitors can inhibit viral replication (22). The mechanisms by which the virus infects macrophages and B cells and T cell exhaustion remain unknown, making treatment intervention for SFTS a significant challenge.
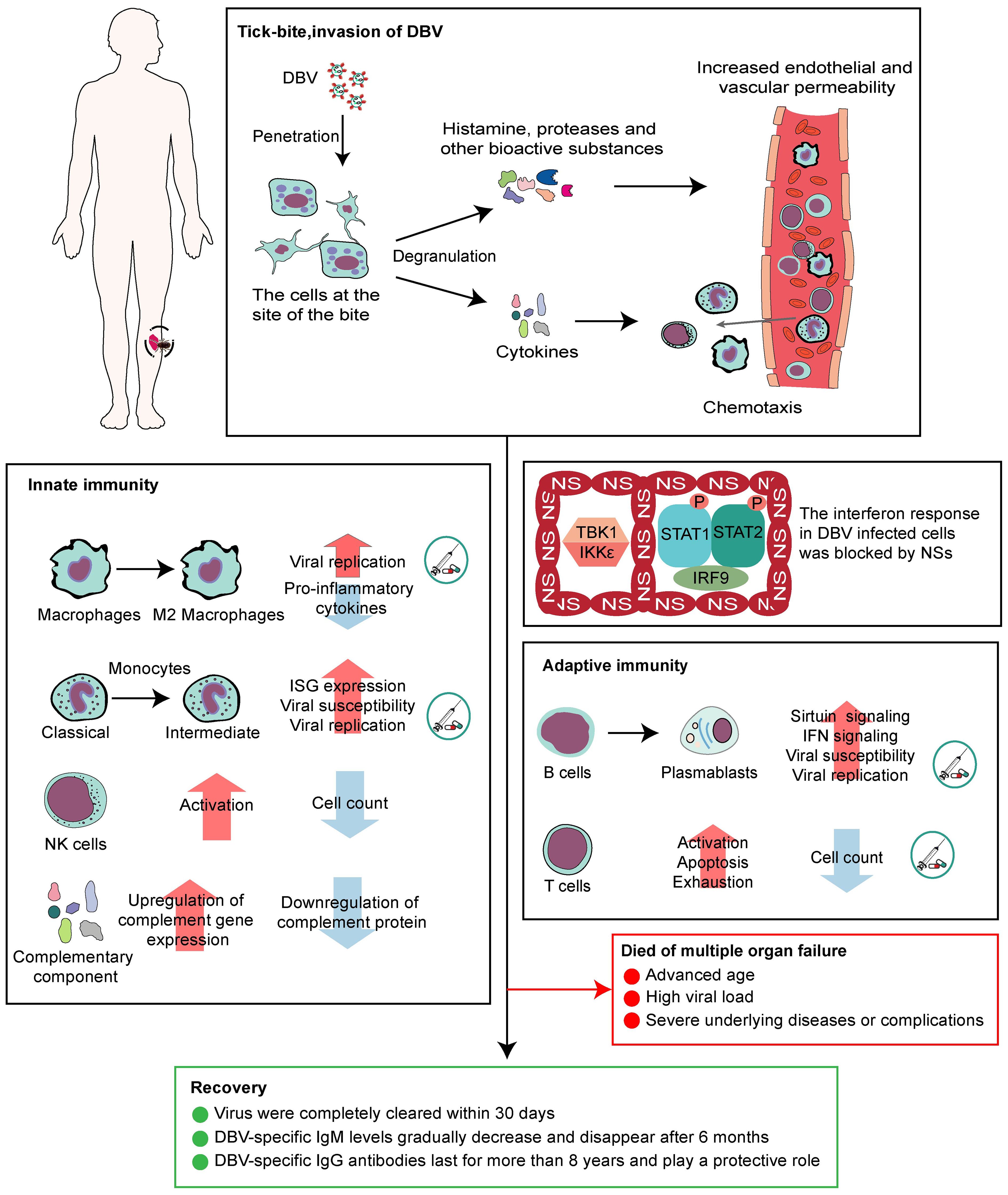
Figure 2 Antiviral immunity and patient prognosis of SFTS.
Available clinical trials of SFTS have mainly focused on broad-spectrum antivirals (Supplementary Material). Favipiravir and ribavirin may be more effective in patients with low viral loads (70, 71), suggesting the need for the early administration of antiviral agents. In addition, favipiravir has not shown any benefit in patients older than 70 years (70). Retrospective studies have demonstrated that IFN-α therapy has no significant therapeutic effect on SFTS (33). Although arginine therapy may boost T cell activity by stimulating the restoration of CD3-ζ chain expression, its clinical application merely accelerates platelet and AST/ALT normalization without enhancing patient survival rates (72). Most clinical trials have shown limited efficacy in treating SFTS patients, underscoring the importance of developing targeted therapies for SFTS patients and the need for further investigation into potential treatment options. It is crucial to continue advancing our understanding of SFTS and to develop effective clinical interventions to improve patient outcomes.
Author contributions
YN: Writing – original draft. YL: Investigation, Writing – review & editing. LH: Investigation, Writing – review & editing. WL: Investigation, Writing – review & editing. QC: Investigation, Writing – review & editing. TL: Investigation, Writing – review & editing. QN: Writing – review & editing. TC: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Key Research and Development Program of China (grant number 2021YFC2600200).
Acknowledgments
The Figures were partly generated from Servier Medical Art, licensed under a Creative Common Attribution 4.0 Generic. License.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1348836/full#supplementary-material
References
1. Yu X-J, Liang M-F, Zhang S-Y, Liu Y, Li J-D, Sun Y-L, et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. N Engl J Med. (2011) 364:1523–32. doi: 10.1056/NEJMoa1010095
2. Kuhn JH, Adkins S, Alioto D, Alkhovsky SV, Amarasinghe GK, Anthony SJ, et al. 2020 Taxonomic update for phylum negarnaviricota (Riboviria: Orthornavirae), including the large orders bunyavirales and mononegavirales. Arch Virol. (2020) 165:3023–72. doi: 10.1007/s00705-020-04731-2
3. Liu Q, He B, Huang S-Y, Wei F, Zhu X-Q. Severe fever with thrombocytopenia syndrome, an emerging tick-borne zoonosis. Lancet Infect Dis. (2014) 14:763–72. doi: 10.1016/S1473-3099(14)70718-2
4. Li J, Li S, Yang L, Cao P, Lu J. Severe fever with thrombocytopenia syndrome virus: A highly lethal bunyavirus. Crit Rev Microbiol. (2021) 47:112–25. doi: 10.1080/1040841X.2020.1847037
5. Liu S, Chai C, Wang C, Amer S, Lv H, He H, et al. Systematic review of severe fever with thrombocytopenia syndrome: Virology, epidemiology, and clinical characteristics. Rev Med Virol. (2014) 24(2):90–102. doi: 10.1002/rmv.1776
6. Zhao G-P, Wang Y-X, Fan Z-W, Ji Y, Liu M-J, Zhang W-H, et al. Mapping ticks and tick-borne pathogens in China. Nat Commun. (2021) 12:1075. doi: 10.1038/s41467-021-21375-1
7. Niu G, Li J, Liang M, Jiang X, Jiang M, Yin H, et al. Severe fever with thrombocytopenia syndrome virus among domesticated animals, China. Emerg Infect Dis. (2013) 19:756–63. doi: 10.3201/eid1905.120245
8. Tian H, Yu P, Chowell G, Li S, Wei J, Tian H, et al. Severe fever with thrombocytopenia syndrome virus in humans, domesticated animals, ticks, and mosquitoes, Shaanxi Province, China. Am J Trop Med Hyg. (2017) 96:1346–9. doi: 10.4269/ajtmh.16-0333
9. Wang G-S, Wang J-B, Tian F-L, Zhang H-J, Yin F-F, Xu C, et al. Severe fever with thrombocytopenia syndrome virus infection in minks in China. Vector Borne Zoonotic Dis. (2017) 17:596–8. doi: 10.1089/vbz.2017.2115
10. Huang X-Y, Du Y-H, Wang H-F, You A-G, Li Y, Su J, et al. Prevalence of severe fever with thrombocytopenia syndrome virus in animals in Henan Province, China. Infect Dis Poverty. (2019) 8:56. doi: 10.1186/s40249-019-0569-x
11. Li D. A highly pathogenic new bunyavirus emerged in China. Emerg Microbes Infect. (2013) 2:e1. doi: 10.1038/emi.2013.1
12. Zhan J, Wang Q, Cheng J, Hu B, Li J, Zhan F, et al. Current status of severe fever with thrombocytopenia syndrome in China. Virol Sin. (2017) 32:51–62. doi: 10.1007/s12250-016-3931-1
13. Ding YP, Liang MF, Jb Ye, Qh L, Ch X, Long B, et al. Prognostic value of clinical and immunological markers in acute phase of sfts virus infection. Clin Microbiol Infect. (2014) 20:O870–O8. doi: 10.1111/1469-0691.12636
14. Guang C, Tao C, Sainan S, Ke M, Xiaojing W, Di W, et al. Expert consensus on diagnosis and treatment of severe fever with thrombocytopenia syndrome. Chin J Clin Infect Dis. (2022) 15:253–63. doi: 10.3760/cma.j.issn.1674-2397.2022.04.003
15. Zhang Y-Z, He Y-W, Dai Y-A, Xiong Y, Zheng H, Zhou D-J, et al. Hemorrhagic fever caused by a novel bunyavirus in China: Pathogenesis and correlates of fatal outcome. Clin Infect Dis. (2012) 54:527–33. doi: 10.1093/cid/cir804
16. Wang L, Xu Y, Zhang S, Bibi A, Xu Y, Li T. The ast/alt ratio (De ritis ratio) represents an unfavorable prognosis in patients in early-stage sfts: An observational cohort study. Front Cell Infect Microbiol. (2022) 12:725642. doi: 10.3389/fcimb.2022.725642
17. Qian F, Zhou W, Liu Y, Ge Z, Lai J, Zhao Z, et al. High C-reactive protein to lymphocyte ratio predicts mortality outcomes of patients with severe fever with thrombocytopenia syndrome: A multicenter study in China. J Med Virol. (2023) 95:e28546. doi: 10.1002/jmv.28546
18. Sun Y, Jin C, Zhan F, Wang X, Liang M, Zhang Q, et al. Host cytokine storm is associated with disease severity of severe fever with thrombocytopenia syndrome. J Infect Dis. (2012) 206:1085–94. doi: 10.1093/infdis/jis452
19. Hofmann H, Li X, Zhang X, Liu W, Kühl A, Kaup F, et al. Severe fever with thrombocytopenia virus glycoproteins are targeted by neutralizing antibodies and can use dc-sign as a receptor for ph-dependent entry into human and animal cell lines. J Virol. (2013) 87:4384–94. doi: 10.1128/JVI.02628-12
20. Yuan F, Zheng A. Entry of severe fever with thrombocytopenia syndrome virus. Virol Sin. (2017) 32:44–50. doi: 10.1007/s12250-016-3858-6
21. Tani H, Shimojima M, Fukushi S, Yoshikawa T, Fukuma A, Taniguchi S, et al. Characterization of glycoprotein-mediated entry of severe fever with thrombocytopenia syndrome virus. J Virol. (2016) 90:5292–301. doi: 10.1128/JVI.00110-16
22. Zhang L, Peng X, Wang Q, Li J, Lv S, Han S, et al. Ccr2 is a host entry receptor for severe fever with thrombocytopenia syndrome virus. Sci Adv. (2023) 9:eadg6856. doi: 10.1126/sciadv.adg6856
23. Marshall JS, Portales-Cervantes L, Leong E. Mast cell responses to viruses and pathogen products. Int J Mol Sci. (2019) 20(17):4241. doi: 10.3390/ijms20174241
24. Wang Y-N, Zhang Y-F, Peng X-F, Ge H-H, Wang G, Ding H, et al. Mast cell-derived proteases induce endothelial permeability and vascular damage in severe fever with thrombocytopenia syndrome. Microbiol Spectr. (2022) 10:e0129422. doi: 10.1128/spectrum.01294-22
25. Elieh Ali Komi D, Wöhrl S, Bielory L. Mast cell biology at molecular level: A comprehensive review. Clin Rev Allergy Immunol. (2020) 58:342–65. doi: 10.1007/s12016-019-08769-2
26. Chyuan IT, Tzeng H-T, Chen J-Y. Signaling pathways of type I and type iii interferons and targeted therapies in systemic lupus erythematosus. Cells. (2019) 8(9):963–. doi: 10.3390/cells8090963
27. Min Y-Q, Ning Y-J, Wang H, Deng F. A rig-I-like receptor directs antiviral responses to a bunyavirus and is antagonized by virus-induced blockade of trim25-mediated ubiquitination. J Biol Chem. (2020) 295:9691–711. doi: 10.1074/jbc.RA120.013973
28. Darnell JE, Kerr IM, Stark GR. Jak-stat pathways and transcriptional activation in response to ifns and other extracellular signaling proteins. Science. (1994) 264:1415–21. doi: 10.1126/science.8197455
29. Ning Y-J, Wang M, Deng M, Shen S, Liu W, Cao W-C, et al. Viral suppression of innate immunity via spatial isolation of tbk1/ikkϵ from mitochondrial antiviral platform. J Mol Cell Biol. (2014) 6:324–37. doi: 10.1093/jmcb/mju015
30. Wu X, Qi X, Qu B, Zhang Z, Liang M, Li C, et al. Evasion of Antiviral Immunity through Sequestering of Tbk1/Ikkϵ/Irf3 into Viral Inclusion Bodies. J Virol. (2014) 88:3067–76. doi: 10.1128/JVI.03510-13
31. Ning Y-J, Feng K, Min Y-Q, Cao W-C, Wang M, Deng F, et al. Disruption of type I interferon signaling by the nonstructural protein of severe fever with thrombocytopenia syndrome virus via the hijacking of stat2 and stat1 into inclusion bodies. J Virol. (2015) 89:4227–36. doi: 10.1128/JVI.00154-15
32. Yoshikawa R, Sakabe S, Urata S, Yasuda J. Species-specific pathogenicity of severe fever with thrombocytopenia syndrome virus is determined by anti-stat2 activity of nss. J Virol. (2019) 93(10):e02226-18. doi: 10.1128/JVI.02226-18
33. Li H, Li X, Lv S, Peng X, Cui N, Yang T, et al. Single-cell landscape of peripheral immune responses to fatal sfts. Cell Rep. (2021) 37:110039. doi: 10.1016/j.celrep.2021.110039
34. Trinchieri G. Type I interferon: Friend or foe? J Exp Med. (2010) 207:2053–63. doi: 10.1084/jem.20101664
35. Peng C, Wang H, Zhang W, Zheng X, Tong Q, Jie S, et al. Decreased monocyte subsets and tlr4-mediated functions in patients with acute severe fever with thrombocytopenia syndrome (Sfts). Int J Infect Dis. (2016) 43:37–42. doi: 10.1016/j.ijid.2015.12.009
36. Qu B, Qi X, Wu X, Liang M, Li C, Cardona CJ, et al. Suppression of the interferon and nf-Κb responses by severe fever with thrombocytopenia syndrome virus. J Virol. (2012) 86:8388–401. doi: 10.1128/JVI.00612-12
37. Song P, Zheng N, Liu Y, Tian C, Wu X, Ma X, et al. Deficient humoral responses and disrupted B-cell immunity are associated with fatal sftsv infection. Nat Commun. (2018) 9:3328. doi: 10.1038/s41467-018-05746-9
38. Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the ebola virus glycoprotein is necessary for infection. Science. (2005) 308:1643–5. doi: 10.1126/science.1110656
39. Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U.S.A. (2005) 102:11876–81. doi: 10.1073/pnas.0505577102
40. Jin C, Liang M, Ning J, Gu W, Jiang H, Wu W, et al. Pathogenesis of emerging severe fever with thrombocytopenia syndrome virus in C57/bl6 mouse model. Proc Natl Acad Sci U.S.A. (2012) 109:10053–8. doi: 10.1073/pnas.1120246109
41. Zhang L, Fu Y, Wang H, Guan Y, Zhu W, Guo M, et al. Severe fever with thrombocytopenia syndrome virus-induced macrophage differentiation is regulated by mir-146. Front Immunol. (2019) 10:1095. doi: 10.3389/fimmu.2019.01095
42. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: An immunologic functional perspective. Annu Rev Immunol. (2009) 27:451–83. doi: 10.1146/annurev.immunol.021908.132532
43. Gordon S. Alternative activation of macrophages. Nat Rev Immunol. (2003) 3:23–35. doi: 10.1038/nri978
44. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. Functional differentiation. Front Immunol. (2014) 5:514. doi: 10.3389/fimmu.2014.00514
45. Sun L, Hu Y, Niyonsaba A, Tong Q, Lu L, Li H, et al. Detection and evaluation of immunofunction of patients with severe fever with thrombocytopenia syndrome. Clin Exp Med. (2014) 14:389–95. doi: 10.1007/s10238-013-0259-0
46. Lu Q-B, Cui N, Hu J-G, Chen W-W, Xu W, Li H, et al. Characterization of immunological responses in patients with severe fever with thrombocytopenia syndrome: A cohort study in China. Vaccine. (2015) 33:1250–5. doi: 10.1016/j.vaccine.2015.01.051
47. Li M, Xiong Y, Li M, Zhang W, Liu J, Zhang Y, et al. Depletion but activation of cd56cd16 Nk cells in acute infection with severe fever with thrombocytopenia syndrome virus. Virol Sin. (2020) 35:588–98. doi: 10.1007/s12250-020-00224-3
48. Fu B, Tian Z, Wei H. Subsets of human natural killer cells and their regulatory effects. Immunology. (2014) 141:483–9. doi: 10.1111/imm.12224
49. Ramlall V, Thangaraj PM, Meydan C, Foox J, Butler D, Kim J, et al. Immune complement and coagulation dysfunction in adverse outcomes of Sars-Cov-2 infection. Nat Med. (2020) 26:1609–15. doi: 10.1038/s41591-020-1021-2
50. Kurosawa S, Stearns-Kurosawa DJ. Complement, thrombotic microangiopathy and disseminated intravascular coagulation. J Intensive Care. (2014) 2:65. doi: 10.1186/s40560-014-0061-4
51. Lee S-Y, Yun SH, Lee H, Lee YG, Seo G, Kim NH, et al. Serum proteomics of severe fever with thrombocytopenia syndrome patients. Clin Proteomics. (2022) 19:32. doi: 10.1186/s12014-022-09368-8
52. Gai Z-T, Zhang Y, Liang M-F, Jin C, Zhang S, Zhu C-B, et al. Clinical progress and risk factors for death in severe fever with thrombocytopenia syndrome patients. J Infect Dis. (2012) 206:1095–102. doi: 10.1093/infdis/jis472
53. Li M-M, Zhang W-J, Liu J, Li M-Y, Zhang Y-F, Xiong Y, et al. Dynamic changes in the immunological characteristics of T lymphocytes in surviving patients with severe fever with thrombocytopenia syndrome (Sfts). Int J Infect Dis. (2018) 70:72–80. doi: 10.1016/j.ijid.2018.03.010
54. Suzuki T, Sato Y, Sano K, Arashiro T, Katano H, Nakajima N, et al. Severe fever with thrombocytopenia syndrome virus targets B cells in lethal human infections. J Clin Invest. (2020) 130:799–812. doi: 10.1172/JCI129171
55. Pavlasova G, Mraz M. The regulation and function of cd20: An "Enigma" of B-cell biology and targeted therapy. Haematologica. (2020) 105:1494–506. doi: 10.3324/haematol.2019.243543
56. Wada Y, Miyamoto S, Iida S, Sano K, Sato Y, Ainai A, et al. Propagation of activated B cells by in vitro severe fever with thrombocytopenia syndrome virus infection of human peripheral blood mononuclear cells. J Infect Dis. (2022) 225:269–81. doi: 10.1093/infdis/jiab343
57. Park A, Park S-J, Jung KL, Kim SM, Kim E-H, Kim Y-I, et al. Molecular signatures of inflammatory profile and B-cell function in patients with severe fever with thrombocytopenia syndrome. mBio. (2021) 12(1):e02583-20. doi: 10.1128/mBio.02583-20
58. Song P, Zheng N, Zhang L, Liu Y, Chen T, Bao C, et al. Downregulation of interferon-Β and inhibition of tlr3 expression are associated with fatal outcome of severe fever with thrombocytopenia syndrome. Sci Rep. (2017) 7:6532. doi: 10.1038/s41598-017-06921-6
59. Wang T, Xu L, Zhu B, Wang J, Zheng X. Immune escape mechanisms of severe fever with thrombocytopenia syndrome virus. Front Immunol. (2022) 13:937684. doi: 10.3389/fimmu.2022.937684
60. Li M-M, Zhang W-J, Weng X-F, Li M-Y, Liu J, Xiong Y, et al. Cd4 T cell loss and th2 and th17 bias are associated with the severity of severe fever with thrombocytopenia syndrome (Sfts). Clin Immunol. (2018) 195:8–17. doi: 10.1016/j.clim.2018.07.009
61. Keir ME, Francisco LM, Sharpe AH. Pd-1 and its ligands in T-cell immunity. Curr Opin Immunol. (2007) 19:309–14. doi: 10.1016/j.coi.2007.04.012
62. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted cd8 T cells during chronic viral infection. Nature. (2006) 439:682–7. doi: 10.1038/nature04444
63. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. Pd-1 expression on hiv-specific T cells is associated with T-cell exhaustion and disease progression. Nature. (2006) 443:350–4. doi: 10.1038/nature05115
64. Huang M, Wang T, Huang Y, Wang Y, Wu S, Wang F, et al. The clinical and immunological characteristics in fatal severe fever with thrombocytopenia syndrome virus (Sftsv) infection. Clin Immunol. (2023) 248:109262. doi: 10.1016/j.clim.2023.109262
65. McElroy AK, Akondy RS, Davis CW, Ellebedy AH, Mehta AK, Kraft CS, et al. Human ebola virus infection results in substantial immune activation. Proc Natl Acad Sci U.S.A. (2015) 112:4719–24. doi: 10.1073/pnas.1502619112
66. Zong L, Yang F, Liu S, Gao Y, Xia F, Zheng M, et al. Cd8+ T cells mediate antiviral response in severe fever with thrombocytopenia syndrome. FASEB J. (2023) 37:e22722. doi: 10.1096/fj.202201343RR
67. Thomas GD, Hamers AAJ, Nakao C, Marcovecchio P, Taylor AM, McSkimming C, et al. Human blood monocyte subsets: A new gating strategy defined using cell surface markers identified by mass cytometry. Arterioscler Thromb Vasc Biol. (2017) 37:1548–58. doi: 10.1161/ATVBAHA.117.309145
68. Hu L, Kong Q, Liu Y, Li J, Bian T, Ma X, et al. Time course of severe fever with thrombocytopenia syndrome virus and antibodies in patients by long-term follow-up study, China. Front Microbiol. (2021) 12:744037. doi: 10.3389/fmicb.2021.744037
69. Lv S-M, Yuan C, Zhang L, Wang Y-N, Dai Z-N, Yang T, et al. Severe fever with thrombocytopenia syndrome with re-infection in China: A case report. Infect Dis Poverty. (2021) 10:90. doi: 10.1186/s40249-021-00877-6
70. Yuan Y, Lu Q-B, Yao W-S, Zhao J, Zhang X-A, Cui N, et al. Clinical efficacy and safety evaluation of favipiravir in treating patients with severe fever with thrombocytopenia syndrome. EBioMedicine. (2021) 72:103591. doi: 10.1016/j.ebiom.2021.103591
71. Li H, Lu Q-B, Xing B, Zhang S-F, Liu K, Du J, et al. Epidemiological and clinical features of laboratory-diagnosed severe fever with thrombocytopenia syndrome in China, 2011-17: A prospective observational study. Lancet Infect Dis. (2018) 18:1127–37. doi: 10.1016/S1473-3099(18)30293-7
Keywords: SFTS, antiviral immunity, immune cells, immune response, clinical treatment
Citation: Niu Y, Liu Y, Huang L, Liu W, Cheng Q, Liu T, Ning Q and Chen T (2024) Antiviral immunity of severe fever with thrombocytopenia syndrome: current understanding and implications for clinical treatment. Front. Immunol. 15:1348836. doi: 10.3389/fimmu.2024.1348836
Received: 06 December 2023; Accepted: 21 March 2024;
Published: 05 April 2024.
Edited by:
Pei-Hui Wang, Shandong University, ChinaReviewed by:
Rui Song, Capital Medical University, ChinaHong Du, Air Force Medical University, China
Cheng Zhang, Hebei Agricultural University, China
Copyright © 2024 Niu, Liu, Huang, Liu, Cheng, Liu, Ning and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qin Ning, cW5pbmdAdmlwLnNpbmEuY29t; Tao Chen, Y2hlbnRhb190amhAdmlwLnNpbmEuY29t