 Abdullah Al-Danakh1†
Abdullah Al-Danakh1† Mohammed Safi2†
Mohammed Safi2† Yuli Jian3†Linlin Yang1†Xinqing Zhu1
Yuli Jian3†Linlin Yang1†Xinqing Zhu1 Qiwei Chen1Kangkang Yang4
Qiwei Chen1Kangkang Yang4 Shujing Wang3*Jianjun Zhang2*
Shujing Wang3*Jianjun Zhang2* Deyong Yang1,5*
Deyong Yang1,5*- 1Department of Urology, First Affiliated Hospital of Dalian Medical University, Dalian, Liaoning, China
- 2Department of Thoracic/Head and Neck Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 3Department of Biochemistry and Molecular Biology, Institute of Glycobiology, Dalian Medical University, Dalian, China
- 4Institute for Genome Engineered Animal Models of Human Diseases, National Center of Genetically Engineered Animal Models for International Research, Dalian Medical University, Dalian, Liaoning, China
- 5Department of Surgery, Healinghands Clinic, Dalian, Liaoning, China
Older patients with cancer, particularly those over 75 years of age, often experience poorer clinical outcomes compared to younger patients. This can be attributed to age-related comorbidities, weakened immune function, and reduced tolerance to treatment-related adverse effects. In the immune checkpoint inhibitors (ICI) era, age has emerged as an influential factor impacting the discovery of predictive biomarkers for ICI treatment. These age-linked changes in the immune system can influence the composition and functionality of tumor-infiltrating immune cells (TIICs) that play a crucial role in the cancer response. Older patients may have lower levels of TIICs infiltration due to age-related immune senescence particularly T cell function, which can limit the effectivity of cancer immunotherapies. Furthermore, age-related immune dysregulation increases the exhaustion of immune cells, characterized by the dysregulation of ICI-related biomarkers and a dampened response to ICI. Our review aims to provide a comprehensive understanding of the mechanisms that contribute to the impact of age on ICI-related biomarkers and ICI response. Understanding these mechanisms will facilitate the development of treatment approaches tailored to elderly individuals with cancer.
1 Introduction
As life expectancy has increased, geriatric oncology is becoming more critical due to the ever-increasing number of older cancer patients, which has so far concentrated mainly on traditional therapy tolerance (1–3). The correlation between incidence of malignancy and aging has been well established, and age-related immunological deterioration has been acknowledged for even longer (4, 5). It has been demonstrated that age-related accumulation of mutations and DNA methylation contribute to the development of cancer (6–8). Further, recent report have emphasized particular alterations that are contributing to the aging-associated decline of the individual’s immune system (9, 10). Immunosenescence describe the process through which the immune system is assumed to become less capable of performing its functions properly (11, 12). There is growing evidence that the immune response of the elderly to cancer may be diminished or compromised for the following reasons: (1) they have fewer naïve B and T cells, which could leave gaps in their repertoire for neoantigens; (2) their memory T cells, which are capable of recognizing tumor cells, have become exhausted; (3) their immune systems have more immune-suppressive cells (13, 14), and (4) alteration of response of macrophages, and neutrophils with age that are necessary for T cell activation in order to eliminate cancer (15) (Figure 1).
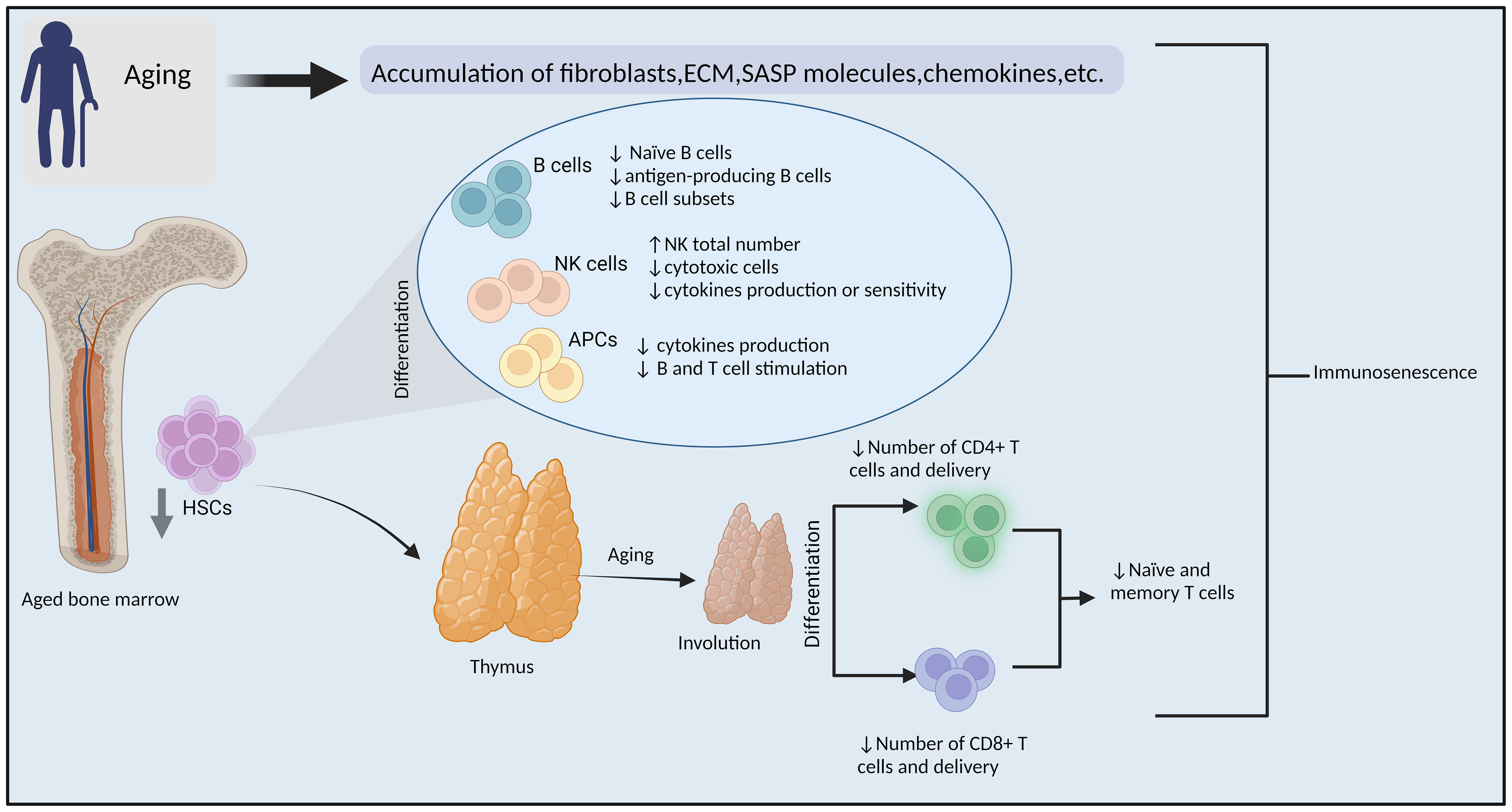
Figure 1 Immunosenescence. Mechanisms of aging-related impairment of the immune system and tumorigenesis. (ECM: extracellular matrix, SASP: senescence-associated secretory phenotype, HSCs: hematopoietic stem cells, NK cells, natural killer cells, APCs: antigen-presenting cells, ↓= Decreased ↑=increased). By BioRender (https://app.biorender.com/).
The past 2 decades have critically advanced our understanding of immune checkpoints. The interaction between T-cell immune checkpoint proteins and their partners on cancer cells (and other cells) transmits "off" signals to T cells upon binding, suppressing the immune response and aiding cancer cell escape. Novel immune-oncology medications known as immune checkpoint inhibitors (ICI) are designed to obstruct this interaction between cancer cells and immune cells, essentially establishing a barrier that thwarts the inhibitory effects. These medications do not stimulate the immune system; rather, they counteract the suppression cancer cells exert on immune cells (Figure 2).
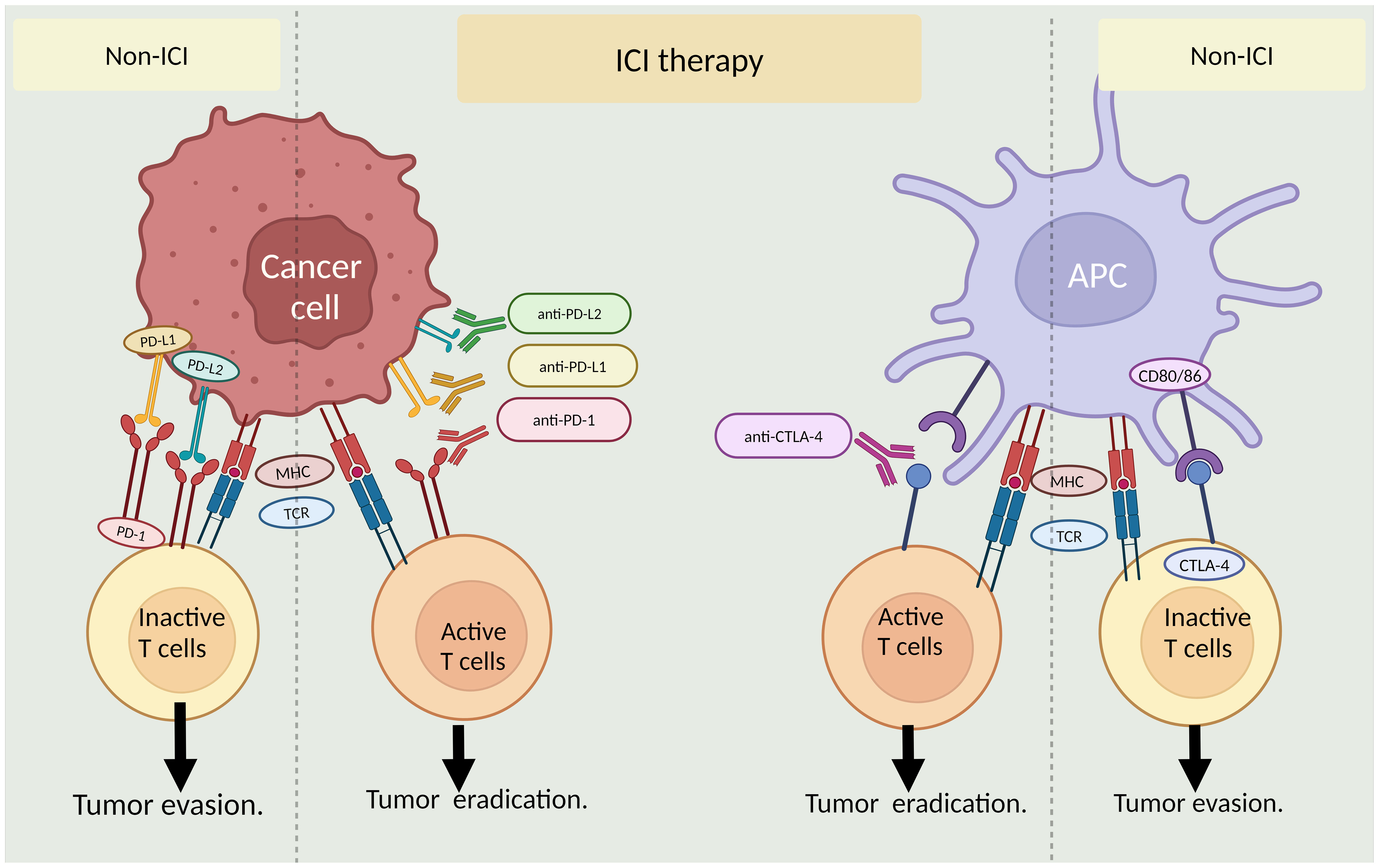
Figure 2 Role of immune checkpoint inhibitors (ICI) in cancer treatment. The left panel shows ICI interaction between cancer cells and T-cells with or without ICI therapy. The right panel illustrates ICI interaction between antigen-presenting cells (APCs)and T-cells in the presence and absence of ICI therapy. MHC: major histocompatibility complex, TCR: T-cell receptor. By BioRender (https://app.biorender.com/).
Recently, ICI has been applied in many cancer types as a promising treatment option. However, it is essential to comprehend the influence of aging on how the immune system responds to tumors in order to make informed decisions on the creation and implementation of ICI in elderly individuals. With the introduction of groundbreaking antibodies that modulate the immune system to eliminate tumors, immunosenescence has gained attention in oncology (16–19).
Also critical to the implementation of ICI therapy in an elderly population is the role of immune checkpoint gene expression. The expression levels of some immune checkpoint genes and their corresponding ligands serve as biological markers for different ICI responses. The expression of a widely recognized biomarker, programmed death-ligand 1(PD-L1), shows demonstrated credibility in predicting the patient’s response to anti-programmed death 1 (PD-1)/PD-L1 treatments (20–24). Other biomarkers associated with ICI response are cytotoxic T lymphocyte antigen 4 (CTLA-4), programmed death ligand 2 (PD-L2), cluster of differentiation 80 (CD80), Janus kinase 2 (JAK2), lymphocyte activation gene 3 (LAG-3), hepatitis A virus cellular receptor 2 (HAVCR2), transforming growth factor-B1 (TGF-B1), C-X-C motif chemokine ligand 9 (CXCL9), and cluster of differentiation 86 (CD86) (25–27).
The impact of aging on immune checkpoint gene expression holds considerable significance for the use of ICI. However, the investigation of the correlation between immune regulatory gene expression and aging has not been systematically explored. Age-related alterations in the immune system impact the expression and regulation of significant immune genes, such as PD-L1, as well as the composition and functionality of the tumor-infiltrating lymphocytes (TILs). Elderly individuals may demonstrate distinct PD-L1 expression, diverse degrees of TIL infiltration, and heightened exhaustion of T cells as distinguished by the up-regulation of inhibitory receptors such as PD-1, T-cell immunoglobulin and mucin-domain containing-3 (Tim-3), and LAG-3 (28). These modifications in immune checkpoint genes and TILs that occur with age may have an impact on the effectiveness of ICI and the general response to immunotherapy in elderly individuals (29). Alterations in immune checkpoint receptors, such as CTLA-4 and indoleamine 2,3-dioxygenase (IDO), may also influence the immune response and the efficacy of immunotherapeutic approaches. Although the precise effect of aging on CTLA-4 expression and function remains incompletely understood, research has indicated a rise in CTLA-4 expression in elderly individuals, which may play a role in immune dysregulation (28).
Comprehending the intricate relationship between immune checkpoint genes and age-related immune alterations is imperative in developing tailored and efficacious immunotherapeutic approaches for elderly individuals with cancer. Customizing treatment strategies to consider the distinct immune profiles and features of elderly individuals may augment treatment efficacy within this demographic. Additional investigation is necessary to elucidate the fundamental mechanisms responsible for age-related alterations in immune checkpoint genes. Furthermore, it is imperative to discover new targets that can be utilized to achieve effective immunotherapy in elderly individuals. These pursuits will ultimately enhance the effectiveness and practicality of immunotherapeutic interventions in the context of aging and cancer (Figure 2).
2 Immune-related genes and aging
2.1 Identification of immune biomarkers related to aging
The aging process plays a crucial role in contributing to and predicting cancer development (30). Aging biomarkers are a combination of biological parameters that hold diagnostic as well as therapeutic values in age-related diseases, including cancer (31). In most cancer tissues, age is linked with changes in somatic mutations, DNA damage repair signatures, and somatic copy-number alterations in most cancer tissues (32–38). Different cancer forms have age-related mutation patterns in well-recognized cancer driver genes. These include mutational signatures linked to APOBEC cytidine deaminase activity, isocitrate dehydrogenase 1 (IDH1), ATRX, POLE/POLD1, PIK3CA, TP53, GATA3, CDH1, ARID1A, KRAS, BRAF V600, CTNNB1, and growth factor signaling pathways (32–35, 39–46). The estrogen receptor-positive (ER+) subtype, associated with a favorable prognosis, is more frequently diagnosed in older individuals. In contrast, the aggressive human epidermal growth factor receptor 2-positive (HER2+) subtype is more prevalent in younger patients (47, 48). Additional genes linked with aging in general include the telomerase reverse transcriptase (TERT), APOE, KL, the FOXO family, SIRT6, the VEGF family, and components of the mitochondrial signaling pathway, chronic inflammation, metabolism, and genes interacting with numerous other pathways such as NF-κB, AMPK, mTOR, P53, and PGC1α (49–57). Finally, among the most altered genes in cancer with age are those of the immune microenvironment, which plays an important role in cancer prognosis and therapy. Hence, identifying and validating immune-related biomarkers associated with aging in cancer holds immense potential for clinical applications (Figure 3) (19, 28, 58, 59).
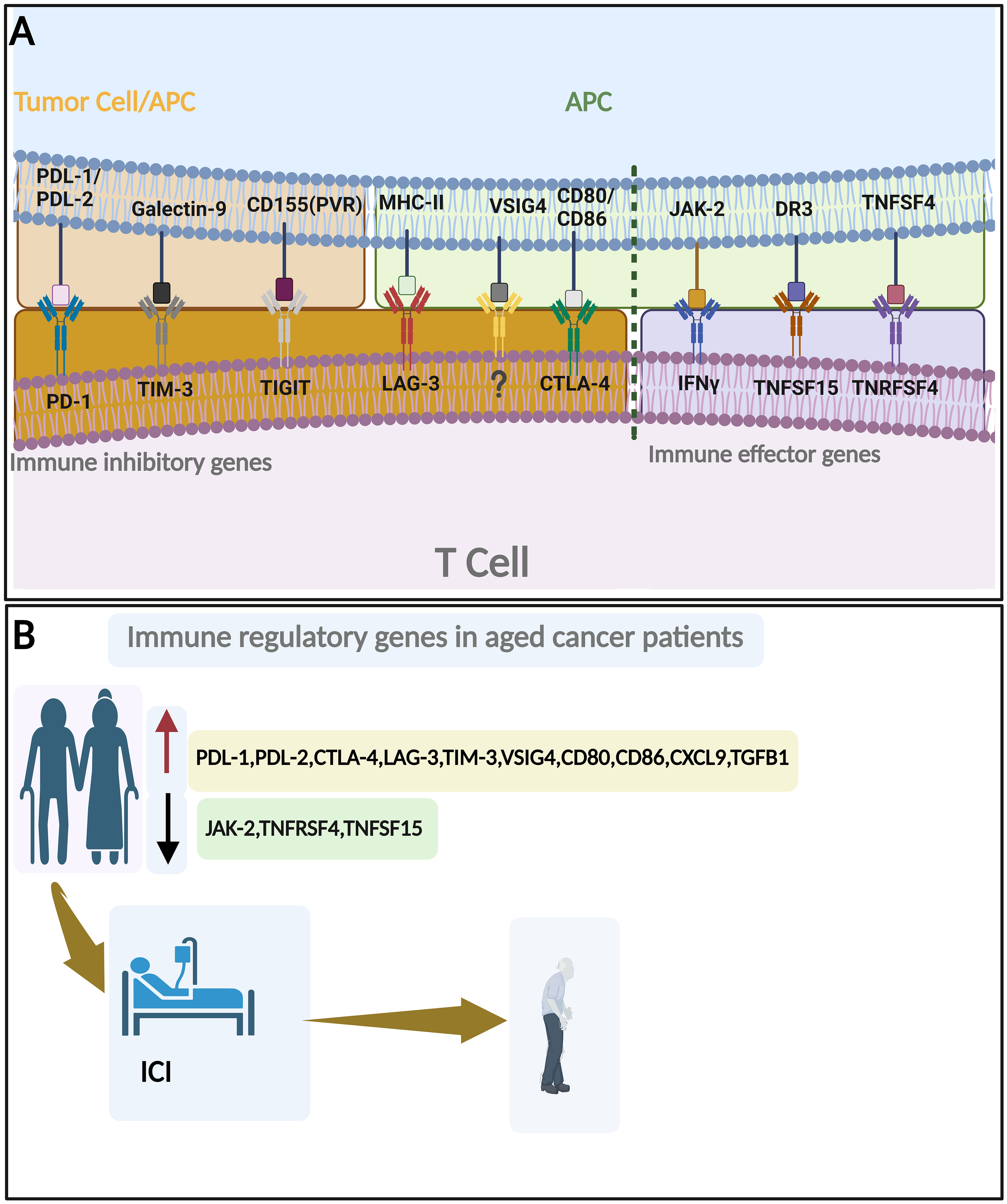
Figure 3 Immune regulatory genes and aging. (A) Important immune inhibitory genes (left to the broken line) and immune effector genes (Right to the broken line). (B) Change of immune regulatory genes with aging in cancer patients and their roles in ICI response. By BioRender (https://app.biorender.com/).
We constructed a protein-protein interaction network using a set of immunological genes associated with aging. Cytoscape software (60) confirmed that all of these genes had robust interactions with one another (Figure 4). Then, we identified the biological pathways and processes underlying this group of genes using the ShinyGO 0.80 web-based tool (61). The associations between these genes and the enrichment of pathways associated with aging and immunology, as documented in the Kyoto Encyclopedia of Genes and Genomes and Gene Ontology, are illustrated in Supplementary Figure 1 and Supplementary Table 1.
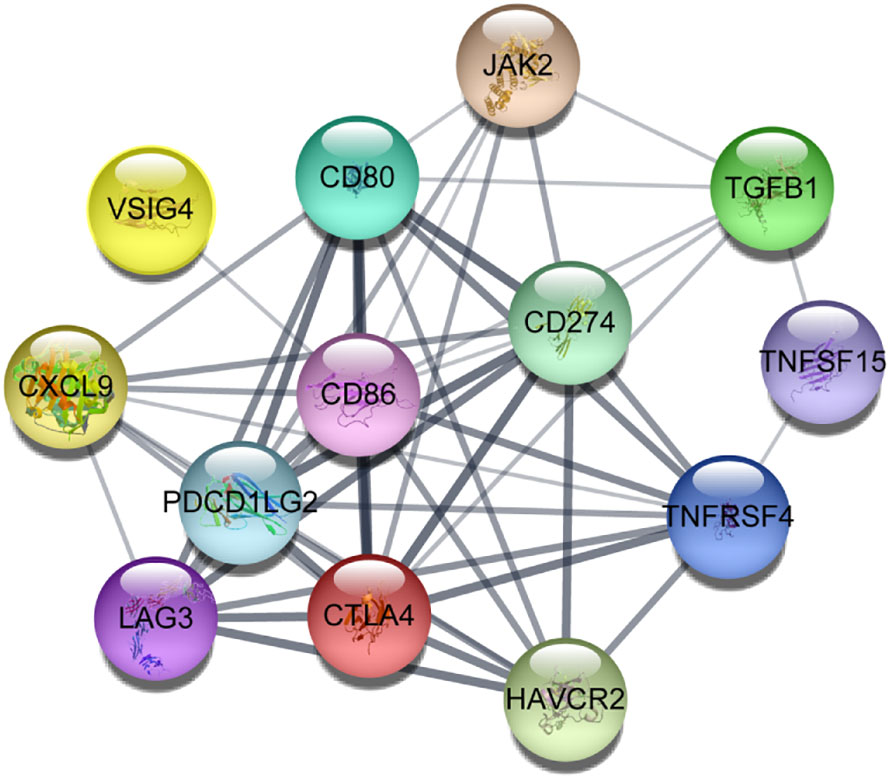
Figure 4 Protein-protein interactions between aging-related critical immune genes in which darker lines represent more robust connections visualized using Cytoscape software.
These biomarkers may aid in cancer risk stratification, treatment selection, and the development of targeted immunotherapeutic strategies tailored to the unique immune profiles of older individuals. Furthermore, immune-related biomarkers could also serve as endpoints in clinical trials, allowing the assessment of treatment response and the evaluation of interventions aimed at modulating age-related immune dysfunction (62). By unraveling the molecular underpinnings of age-related immune dysfunction and its implications for cancer, researchers strive to improve cancer prevention, diagnosis, and treatment approaches for the growing population of older individuals (Figure 3; Table 1). In the rest of this section, we will discuss each aging-related immune biomarker in detail.
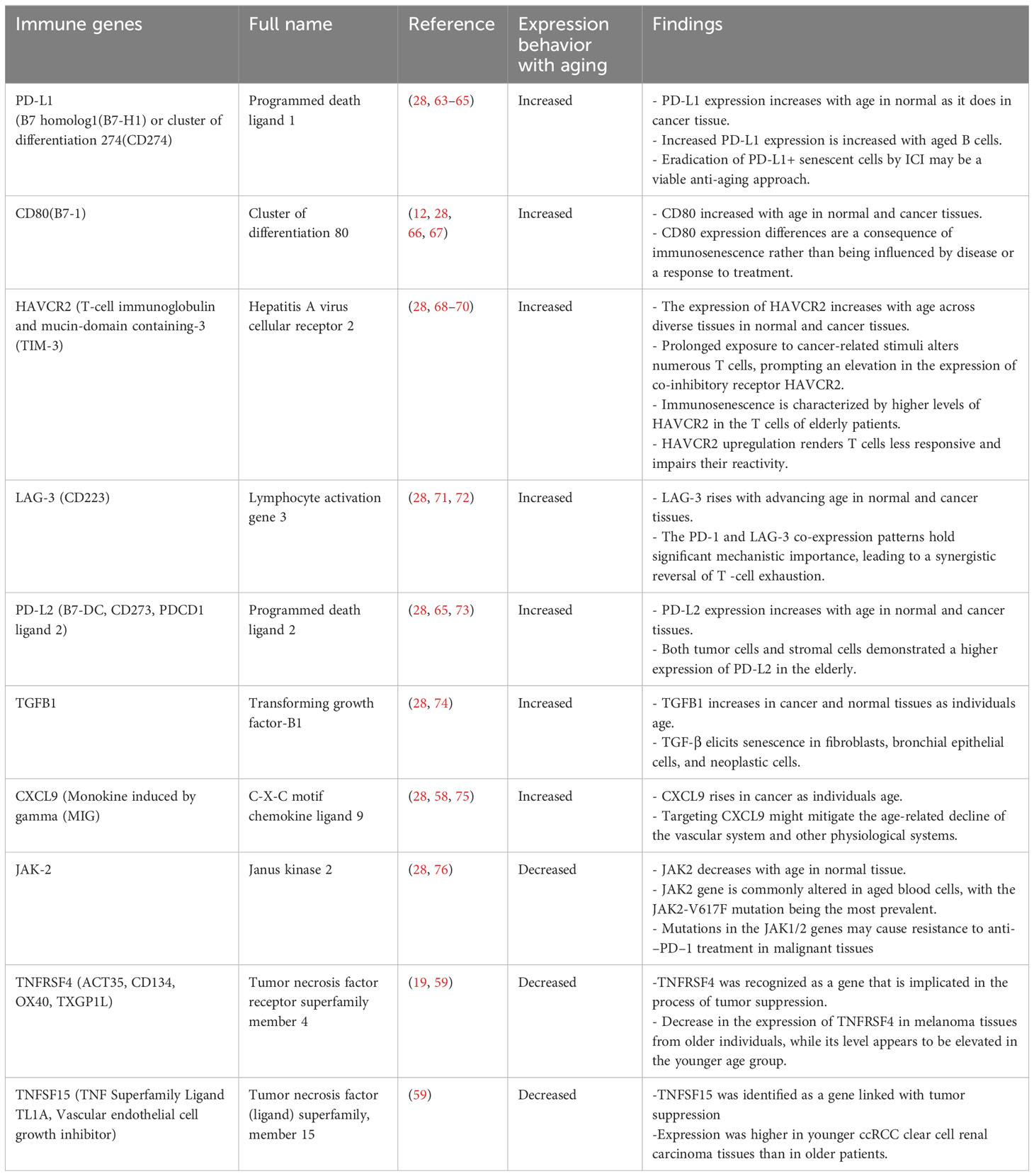
Table 1 Overview of the fluctuations in immune gene expression as a consequence of the aging process.
2.2 PD-L1
PD-L1, also known as CD274, is a cell surface protein that plays a significant role in immune response regulation by binding to T cells through PD-1, thereby suppressing cancer immunity, and serving as a signal to evade detection (77, 78). The PD-1 interacted with PD-L1 on the surface of T-cells dephosphorylates T-cell receptors, specifically involving SHP-1/2. The observed effect of this phenomenon is the inhibition of T-cell mediated cytotoxicity toward malignant cells, which is attributed to the reduction of T-cell proliferation and activity (77, 79). ICI, specifically monoclonal antibodies targeting PD-1 and/or PD-L1, have been employed in various cancer therapies (80). Despite the considerable clinical advantages of PD-1/PD-L1 blockade therapy for various cancer types, the response rates of patients remain below 40%, and the underlying mechanism is not entirely understood (77). Still, distinct types of cancers exhibit high PD-L1 levels that facilitate immune evasion by cancer cells (81).
The intricate modulation of PD-L1 expression in malignant cells encompasses a multifaceted interplay of intricate signaling cascades, including but not limited to nuclear factor-κB (NF-κB), mitogen-activated protein kinase (MAPK), molecular target of rapamycin (mTOR) signal transducer and activator of transcription (STAT), and cellular myelocytomatosis (c-myc). Additionally, the degradation of PD-L1 protein occurs by many pathways, including proteasomes and lysosomes. These mechanisms contribute to the enhanced efficacy of cancer immunotherapy but may be limited by changes related to aging (77, 82). Multiple studies have reported a higher level of PD-L1 expression within the geriatric population compared to the younger cohort (28, 63, 64). It has been observed that senescent cells increase the expression of PD-L1, thereby leading to the deactivation of immune cells. The upregulation of PD-L1 during senescence is contingent upon the activation of the proinflammatory pathway (63). In addition, the factors secreted by cells undergoing senescence can induce an increase in PD-L1 expression in non-senescent cells, which occurs through the JAK-STAT signaling pathway. Furthermore, the research (63) indicates that the intervention of prolongevity through rapamycin reduces PD-L1 expression in aging cells. Finally, it was discovered that PD-L1 expression is increased in multiple tissues in mice that have undergone natural aging, as well as in the lungs of patients with idiopathic pulmonary fibrosis (63). Collectively, the findings indicate that the process of senescence and aging is linked to an increase in the expression of PD-L1. Hence, the targeting of PD-L1 presents a promising avenue for developing new therapeutic interventions aimed at addressing the pathophysiological processes linked to senescence and age-related disorders, including malignancies (63).
The process of surveillance by the immune system, which involves the recognition and elimination of aberrant cells, for instance, cancer cells, is subject to negative regulation by immunological checkpoints, including those associated with senescent cells. In order to investigate this inquiry, scholars conducted an analysis of the manifestation of numerous immune-regulated checkpoint molecules in human fibroblast cell line HCA2, which is recognized as an in vitro representation of senescent fibroblasts. The overall concentration of PD-L1 was shown to be dramatically and particularly elevated in nutlin3a-induced senescent cells(n-Sen) and DNA damage-induced senescent cells(d-Sen) when compared to starvation-induced quiescent cells (64, 83, 84). The findings additionally demonstrated that senescent cells exhibit heterogeneous in the expression of the immunological checkpoint protein PD-L1. Furthermore, it is seen that there was an age-related accumulation of PD-L1+ senescent cells in vivo. Cells expressing PD-L1 are resistant to T cell surveillance, even when a senescence-associated secretory phenotypes (SASP) is present, whereas cells not expressing PD-L1 are vulnerable to T cell surveillance. “P16 positive cells” are those containing observable amounts of the p16 protein, a key player in regulating the cell cycle and frequently associated with cancer. Moreover, p16 is among the regulatory factors engaged in initiating and sustaining cellular senescence (85).
The in vivo examination of senescent cells (namely, p16+ cells) using single-cell analysis demonstrated a positive association between PD-L1 and elevated levels of the SASP. In accordance, the administration of PD-1 antibody to mice that are naturally aging, as well as to a mouse model with normal livers or induced nonalcoholic steatohepatitis, results in a decrease in the overall quantity of p16+ cells in vivo, as well as a reduction in the PD-L1+ population in an activated CD8+ T cell–dependent manner (85). Thus, this intervention effectively improved several aging-related characteristics The findings of this study indicate that the varied expression of PD-L1 plays a crucial role in the buildup of senescent cells and inflammation linked to the aging process. Furthermore, the removal of PD-L1+ senescent cells by immune checkpoint inhibition shows promise as a potential approach for anti-aging treatment (64, 83). Recently, researchers found that PD-L1 expression of CD8+ effector T cells was significantly higher in aged mice than younger mice and anti-PD-L1 immunotherapy led to a decrease in cell proliferation in vitro and a reduction in anti-tumor immune response in older hosts, as compared to their effects in young mouse lymphoma models (86, 87). A separate investigation observed that the levels of PD-L1 and IDO1 were elevated in the brains of healthy adult humans as they aged. Additionally, the aging process was associated with increased circulating regulatory T cell numbers and decreased CD8+ T cells (88). While all of the evidence suggests that cancer patients with high PD-L1 expression benefit more from ICI, whether older patients with cancer who express PD-L1 will respond better to ICI is not completely known. Patients over 65 or even 70 are sometimes ineligible to participate in various ICI investigations; this group is generally too small to evaluate. However, because people are frequently diagnosed with cancer around the age of 65, studies on the efficacy and safety of ICI in older patients are required (28, 58, 89).
2.3 CD80
The cluster of differentiation 80 (CD80), also known as B7-1, is a cell surface protein expressed on tumor cells or APCs (90). It interacts with both con-inhibitory CTLA-4 and co-stimulatory CD28 receptors, thereby playing a vital function in immune response regulation (91–93). The competitive binding of CD28 and CTLA-4 to CD80 is subject to regulation by various factors, including affinity to CD80 and the expression kinetics of CD28 and CTLA-4 in T cells (93). The affinity of CTLA-4 to CD80 is approximately ten times greater than its affinity to CD28. Also, CTLA-4 is predominantly expressed on T cells that have been activated, whereas CD28 is expressed on T cells in a constitutive manner (94). Prior studies have indicated that the expression level of CD80 might influence the pro-/anti-oncogenic function of CD80 on neoplastic cells (93–97). The downregulation of CD80 expression is a mechanism tumors employ to evade immune surveillance because CTLA-4 exhibits a higher affinity for CD80 than CD28, does resulting in preferential binding to the CTLA-4. Conversely, the upregulation of CD80 induces T-cell activation and enhances tumor rejection. Moreover, research has demonstrated that a soluble variant of CD80 has the capability to attach to PD-L1, thereby impeding the PD-1/PD-L1 axis (98). Furthermore, the interaction between PD-L1 and CD80 in a cis configuration, rather than a trans configuration, hinders the immunosuppressive axis of PD-1/PD-L1 and CTLA-4/CD80 (99, 100). Antibodies that block PD-L1 prevent the interaction between CD80 and PD-L1 on dendritic cells that are associated with tumors, thereby promoting the anti-tumor immune response mediated by CD80 (93, 101). The expression behavior of CD80 appears altered with aging. Furthermore, it has been observed that the expression levels of immune checkpoint molecules, including Tim-3, T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT), and CTLA-4, exhibit a rising pattern with advancing age (12). Studies investigating the relationship between aging and CD80 expression in the era of ICI have demonstrated a positive correlation between advanced age and elevated CD80 levels (28, 58). A group of researchers found that the population of CD14+ monocytes remained stable; however, there was an age-related increase in the expression of major histocompatibility complex (MHC) II cell surface receptor (HLA-DR), CD80, and CD86 by monocytes. Nevertheless, no discernible distinctions were observed upon juxtaposing Alzheimer disease (AD) patients with age-matched healthy controls or subsequent to administering rivastigmine treatment to AD patients (67). The findings suggest that alterations in the manifestation of HLA-DR, CD80, and CD86 are attributable to immunosenescence rather than AD pathology or the administration of rivastigmine to AD patients. A recent investigation demonstrated that in the context of aging, neurodegenerative disease, central nervous system inflammation, and injury, antigen-presenting cells, such as microglia or infiltrating macrophages, increase the expression of immunomodulatory proteins, such as CD80 (67, 102). A universal deficiency in the enhancement of the co-stimulatory molecule CD80 on monocytes in relation to age was noted across all TLR ligands that were examined, including TLR1/2, TLR2/6, TLR4, TLR5, and TLR8 (66, 103). The profound connection between the upregulation of CD80 expression upon TLR engagement and the development of a protective antibody response to influenza vaccination has significant implications for adaptive immunity. This observation diverges from the trends observed in other findings, highlighting the nuanced role of CD80 and the complexity of its regulation in the aging process and cancer. Although the CD80-encoded protein plays a role in tumorigenesis and progression, especially when interacting with CTLA-4, there is evidence to suggest that patients with elevated CD80 expression may experience advantages when treated with ICI-like antibodies targeting CTLA4 or PD-1/PD-L1 (104). However, the response to ICI treatment in older cancer patients expressing CD80 remains dichotomous and requires additional research to elucidate.
2.4 HAVCR2
The T-cell immunoglobulin and mucin domain 3(TIM-3) protein, which is encoded by the HAVCR2 gene, is a cell surface molecule containing both immunoglobulin and mucin domains. Its initial identification was as a cell surface marker that exhibits specificity towards interferon (IFN-γ)-producing CD4+ T helper 1(Th1) and CD8+ cytotoxic 1(Tc1) cells (105). Tim-3 is a member of the Tim gene family, which is located in syntenic chromosomal regions in humans (5q33.2) and mice (11B1.1) that have been linked to allergies and autoimmune diseases (106, 107). The research conducted by Monney et al. provided initial evidence of Tim-3's inhibitory role as a checkpoint receptor on T cells by introducing Tim-3 monoclonal antibodies in vivo, which worsened symptoms in an experimental autoimmune encephalomyelitis model (108). Subsequent to the aforementioned events, two separate investigations concluded that interfering with the interactions between Tim-3 and its ligand, using Tim-3-Ig or Tim-3 mAb, led to increased Th1 responses and promoted the development of autoimmune diabetes in nonobese diabetic mice (109, 110). Despite these findings, the lack of a classical inhibitory signaling motif in the cytoplasmic tail of Tim-3 cast doubt on the protein’s inhibitory function. The association between germline loss of function mutations in HAVCR2 and two diseases resulting from hyperactivated T and myeloid cells, hemophagocytic lymphohistocytosis (HLH) and subcutaneous panniculitis-like-T-cell(SPTCL), establish Tim-3 as a negative regulator or “immune checkpoint” (111, 112). Tim-3 expression is simultaneously controlled and expressed on CD4+ and CD8+ T cells in conjunction with other immune checkpoint receptors, including PD-1, LAG-3, and TIGIT (113, 114). In general, with aging, Tim-3 demonstrates increased expression. Senescent T cells have been found to possess atypical characteristics, including reduced expression of CD27 and CD28 and increased expression of CD57, killer cell lectin-like receptor subfamily G, Tim-3, TIGIT, and cytotoxic T-lymphocyte-associated protein 4. These molecular alterations have been linked to the formation of malignant tumors (12, 115, 116). Certain researchers contend that the dysfunction of T-cells with aging is distinct from the exhaustion of T-cells, which is a condition of reduced cell response that occurs as a result of prolonged exposure to situations such as viral infection and cancers (69). Continuous antigen activation leads to T cell depletion and a steady increase in the levels of inhibitory checkpoint receptors [such as PD-1, CTLA-4, LAG-3, inducible co-stimulator (ICOS), Tim-3, and killer cell lectin-like receptor G1(KLRG-1)] on CD4+ T cell that consequently downregulate T cell receptor (TCR)-induced intracellular signaling (117, 118). Tim-3 expression uniquely identifies the most dysfunctional or terminally exhausted subset of CD8+ T cells in malignancies (119, 120). Targeting of Tim-3 is a potential therapeutic opportunity. Co-blockade of the Tim-3 and PD-1 pathways has demonstrated exceptional efficacy in preclinical cancer models for both solid and hematologic tumors (121, 122). Current clinical trials investigate anti-Tim-3 in conjunction with anti-PD-1 for treating solid malignancies due to their persistent antigen T-cell stimulation (120, 123). Considered a detrimental regulatory factor of T-cell death, TIM-3 is expressed on the Th1 cells' surface and achieves its function by binding to its ligand, galectin-9 (124). The inhibitory effects of TIM-3 extend to innate and adaptive immunological reactions, particularly those involving CD8+ and Th17 cells, as well as tolerant acquisition mediated by regulatory T cells (Tregs) (105, 125). HAVCR2 expression was higher in most tumors, including glioblastoma and low-grade glioma (GBMLGG), lung adenocarcinoma (LUAD), prostatic adenocarcinoma (PRAD), and sarcoma (SARC) (70). It has also been shown that age leads to an increase in Tim-3+ murine T cells, which in turn decreases T cell’s proliferative capacity (126, 127). A prior study has indicated that overexpression of immune exhaustion markers like TIM-3 can result in resistance to ICI therapy. Consequently, targeting this exhaustion marker on its own or in conjunction with PD-1 may lead to more favorable clinical responses (128). Despite TIM-3 heightened expression in the elderly population, the impact of this expression on ICI response in older cancer patients warrants further investigation (28).
2.5 LAG-3
The LAG-3 protein is classified as a type I transmembrane cell-surface protein that operates in the immune system as an inhibitory co-receptor, analogous to the PD-1 and CTLA-4 proteins (129). The identification of the LAG-3 encoding gene dates back to 1990 when complementary DNA clones expressed in an IL-2-dependent CD3-negative natural killer-cell line were analyzed (130). Researchers observed that the LAG3 gene encodes a membrane protein of four extracellular domains belonging to the immunoglobulin superfamily (131). It has been shown that exhausted CD4+ T lymphocytes have an increased copy number of the LAG3 gene, which subsequently downregulates the TCR-induced intracellular signaling (69, 118). Comparably, LAG-3 upregulation is also observed in dysfunctional immunosenescent cells (69, 118, 127, 132). Recent research has shown that as individuals age, there is a drop in the production of the proapoptotic protein Bim, which leads to an increase in the lifetime of naïve CD4+ T cells. This finding has led to the notion that the extended lifespan of these cells makes them more susceptible to accumulating defects (72). Additionally, naïve T cells have the potential to develop a’semi-memory’ phenotype as they mature, signifying their entry into differentiation pathways. In turn, aged naïve T cells have shown expression of molecules typically associated with prolonged stimulation and exhaustion, including PD-1 and LAG-3 (71). T-cell exhaustion can result in a reduction of cytokine production that is responsible for tumor-killing (132). Hence, elevated levels of LAG-3 restrict the activation of T-cells, so diminishing their capacity to target and eliminate cancerous cells. Conversely, suppression of LAG-3 reestablishes the normal functioning of T-cells (133).
LAG-3 shows potential as a therapeutic target in cancer. Recent work has demonstrated a significant association between the expression of LAG-3 and the presence of LAG-3-positive cells within tumors, and adverse clinical outcomes, tumor advancement, and unfavorable prognosis across various cancer types such as lymphomas, renal cell carcinoma, colorectal cancer, breast cancer, and head and neck squamous cell carcinoma (HNSCC) (134). The investigators’ initial focus was to examine the effects of LAG-3 inhibition as a standalone treatment; however, it was shown that employing a single-agent approach yielded minimal effectiveness. In contrast, the simultaneous inhibition of LAG-3 and PD-1 receptors had a synergistic effect, leading to a reduction in tumor growth and the enhancement of anti-tumor immune responses, which may be attributable to differences in the inhibitory mechanisms and or expression patterns of PD-1 and LAG-3 (129). Moreover, the protein expression levels of LAG-3, PD-1, and TIM-3 in NSCLC tissue were evaluated in a previous study that concluded a negative association between LAG-3 and anti PD-1 therapy response (135). In March 2022, the FDA approved the combined use of anti–PD-1 antibody nivolumab and the LAG-3 blocker relatlimab-rmbw (Opdualag) for the therapy of individuals aged 12 and above who have unresectable or advanced melanoma (136, 137). Importantly, a potential factor in resistance to anti–PD-1/anti–PD-L1 immunotherapies in cancer is LAG-3 and PD-1 co-expression, which may lead to significant T-cell dysfunctionality (138). In accordance with this, PD-1 and LAG-3 co-blockades increase many T-cell anti-tumor activities (138). In a nutshell, LAG-3 has emerged as a viable option for cancer immunotherapy, but more study of these strategies is critical, particularly in the older population, where LAG-3 expression is significantly higher than in younger people (28, 58, 139).
2.6 PD-L2
PD-L2, which is alternatively referred to as B7-DC or CD273, is categorized within the B7 family members. This protein’s extracellular region comprises an immunoglobulin (Ig)-like V-type domain and an Ig-like C2-type domain; it is classified as a type I transmembrane protein. The expression of PD-L2 has been observed in both neoplastic and immunological cells, and its elevated expression has been established to be a significant factor in both tumor formation and evasion of immune surveillance (140). PD-L2 overexpression was seen to correlate with a negative prognosis in individuals diagnosed with HNSCC, adenoid cystic carcinoma (ACC), esophageal cancer, and other related conditions (141). Moreover, a study conducted on colon cancer revealed that the expression of PD-L2 exhibited a positive correlation with neuroinvasion and a negative correlation with CD8+ tumor-infiltrating lymphocyte density (142). PD-L2 has been identified as a significant independent prognostic factor in the context of anti-PD-1 immunotherapy. The proposition put forth by researchers is that PD-L2 has a significant function in circumventing anti-tumor immunity, similar to PD-L1. This implies that for effective immunotherapy in cancers that express PD-L2, such as HNSCC, RCC, and LUSC, it is imperative to take into account the blockade of PD-1/PD-L2 (141).
In one investigation, the tumors of aged mice exhibited a greater abundance of PD-L2+ B16 tumor and stromal cells, as well as a higher PD-L2 mean fluorescence intensity, compared to tumors of young mice (65). Variations in the expression of PD-L2 and PD-1 may shed light on the more effective response of aged mice to PD-1 inhibitors compared to PD-L1 inhibitors. This enhanced efficacy can be attributed to the ability of anti-PD-1 therapies to block the inhibitory signals originating from both PD-L2 and PD-L1 simultaneously. This dual blockade potentially offers a more comprehensive therapeutic approach, especially in contexts where both these ligands are expressed. Prior research has indicated that PD-L2 levels in B cells rise with the aging of individuals in many organs, including blood, bone marrow, and spleen. Furthermore, aged PD-L2+ B cells modulate CD4+ T-cell activity differently from their younger counterparts, and this modulation depends on PD-L2. Additionally, PD-L2+ B cells impede the growth of subcutaneous MC38 colon cancer. Similarly, age was significantly associated with PD-L2 level (73).
It is worth noting that both the overproduction and inadequate removal of senescent cells could potentially play a role in the development of pathological aging. Efforts are underway to ascertain the mechanisms by which senescent cells evade immune effector-mediated destruction. These mechanisms entail the expression of immunosuppressive molecules, such as PD-L1 and PD-L2, on the surface of cells to bind to PD-1 receptors in T cells, and tolerogenic variants of MHC class-I are also involved. Furthermore, senescent cells can release certain substances that can potentially attract immunosuppressive and pro-inflammatory cells toward the surrounding microenvironment (143). Therapeutic intervention could be targeted towards each immune evasion mechanism, such as obstructing the interaction between PD-1 and PD-L1 or PD-L2, enhancing the expression of immunogenic MHC class-I molecules, and eradicating immunosuppressive cell populations. The study conducted by Chaib et al. revealed a noteworthy finding wherein the expression of PD-L2 was observed to be heightened in various cellular models of senescence, while PD-L1 expression remained unaffected. Notably, eradicating tumors induced by doxorubicin treatment can be achieved through anti PD-L2 treatment by decreasing the presence of senescent cells within the tumor and limiting the recruitment of immunosuppressive cells (144). The suggestion is that PD-L2, much like PD-L1, plays a vital function in evading the body’s anti-tumor immune mechanisms. This implies that to achieve the most effective immunotherapy in PD-L2 expressing cancers like HNSCC, RCC, and LUSC, it’s essential to consider blocking the PD-1/PD-L2 interaction (141). However, our understanding of the regulatory mechanisms governing PD-L2 remains somewhat unclear, and as of now, there are no established clinical practices or trials for immunotherapy strategies targeting PD-L2. This knowledge gap is especially pronounced when it comes to elderly patients who have high PD-L2 expression, making it imperative for further in-depth research in both areas (58, 68).
2.7 TGFB1
Transforming growth factor-β (TGF-β) is a group of related proteins produced by all cells and functions through a complex cell surface receptor system. Since its identification as a secreted factor that triggers reversible phenotypic changes in specific fibroblast cell lines, TGF-β has been the subject of extensive research regarding its involvement in malignancy development and progression (145). Three groups of findings (145, 146) propelled the development of these studies: (1) Tumor cell lines and tissues frequently exhibit elevated levels of TGF-β expression in comparison to healthy cells or tissue; (2) The growth inhibitory effect of TGF-β observed in normal epithelial cells is frequently compromised in carcinomas; and (3) The autocrine and paracrine TGF-β signaling pathways in tumor cells regulate significant alterations in gene expression, which suppress the epithelial phenotype, facilitate invasion and dissemination, foster stem cell characteristics, release immunosuppressive cytokines, and contribute to cancer drug resistance (145, 146). In order to achieve sustained remission, therapeutic strategies that target TGF-β signaling ought to concentrate on cancer cells as well as immunological and stromal components of the tumor microenvironment (TME) (146). Increased TGF-β signaling has been correlated with resistance to many anti-cancer treatments, including molecularly targeted medicines and chemotherapy as well as immunotherapies (147). Consequently, only a subset of individuals among those with diminished TGF-β signaling patterns exhibit a positive response to anti-PD-1 or -PD-L1, anti-CTLA-4, and CART cell therapies. The potential benefits of therapeutic repression of TGF-β signaling to improve response to anti-PD-1/-PD-L1 are expected to be substantial. Unlike chemotherapy, immunotherapy, including TGF-β blockade, can yield delayed or unpredictable responses partly due to the cascade of immune events required for a productive anti-tumor cytotoxic response (148–152).
Although the effect of TGF-β signaling varies significantly depending on the specific cell kind and cellular environment, several growth-inhibiting processes are thought to play a role in the emergence of senescent characteristics. According to a multitude of findings (74) pertaining to pathology associated with aging, the influence of TGF-β signaling can be broadly categorized into two dimensions: (1) the compromise of TGF-β signaling in specific cellular populations, as evidenced by the decline in neuroprotective functions in AD and the attenuation of TGF-β-mediated suppression of growth in tumor, and (2) the persistent rise of TGF-β signaling that is associated with tissue fibrosis, continual inflammation, diminished regenerative capability, and metabolic dysfunction reported in AD, wasting of muscles, weight gain, and various other pathological conditions. Alterations in TGFB1 influenced by the aging process affect Smad2/3 interactions, notably with transcriptional intermediary factor 1 (Tif1γ), and shift the activin receptor-like kinase (ALK)1/ALK5 ratio, favoring Smad 1/5/8 pathways (74, 153–155). This change underlies aging-associated dysregulation in processes like metabolism and angiogenesis, contributing to cancer progression where the TGF-β plays a pivotal role. TGF-β's role in inducing senescence, affecting the tumor microenvironment (TME), and modulating PD-L1 expression highlights its impact on the efficacy of ICI therapy (156, 157). A plethora of studies have highlighted the numerous regulatory mechanisms behind the context-dependent influence of TGF-β signaling in the past few years (158). Nonetheless, the intricate relationship between TGFB1 signaling and aging remains enigmatic within various pathological contexts. Further investigations are necessary to assess the response to ICI therapy in elderly cancer patients characterized by elevated levels of TGFB1 (28).
2.8 CXCL9
CXCL9 is a member of the ELR-negative CXC chemokine subfamily that is activated by interferon-γ (IFN-γ) and released by tumor-associated dendritic cells (159). The chemokine CXCL9 facilitates the infiltration of lymphocytes with tumor-suppressive properties into solid tumors by activating its receptor CXCR3 (160). In preclinical cancer models, CXCR3 chemokine activity was shown to be crucial for effective immune checkpoint inhibition, owing to CXCR3's role in both T-cell recruitment and T-cell activation (161–163). A recent report on over 1000 patients with various tumor types indicated that CXCL9 gene expression and tumor mutational burden are highly effective markers for predicting the response to ICI (164). CXCR3 serves as the co-receptor for CXCL9, CXCL10, and CXCL11. Within the TME, the CXCL9/10/11-CXCR3 signaling pathway has the potential to elicit anti-tumor immunity through various mechanisms. These mechanisms include facilitating the chemotactic migration of CXCR3-activated immune cells to tumor sites and activating the STAT and phosphoinositide 3-kinase (PI3K)-Akt signaling pathways. This activation results in the upregulation of PD-L1 expression, which is typically indicative of a favorable response to ICI (165, 166). Elevated levels of CXCL9 and CXCL10 have been observed to be associated with heightened tumor CD8+ T cell density and enhanced patient survival across various cancer types (58, 167, 168).
In the context of aging other than cancer cells, research has identified that the most significant factor influencing the inflammatory aging clock (iAge)was the chemokine CXCL9, which has been implicated in the process of cardiac aging, unfavorable cardiac remodeling, and impaired vascular function (75). In that study, the elderly group exhibited elevated expression levels of immune genes linked to chemokines, specifically CCL5, CXCL9, and CXCL10. In addition, it has been observed that aging endothelial cells in both humans and mice exhibited a decline in functionality, cellular senescence, and characteristic phenotypes of arterial stiffness. Notably, these effects could be mitigated through the inhibition of CXCL9. Ultimately, the authors ascertain a significant function of CXCL9 in chronic inflammation associated with aging and established a multimorbidity measure that can promptly detect age-related clinical phenotypes (75). Indeed, Furman and his colleagues have recently published a study demonstrating that iAge exhibits a correlation with various diseases and remarkable longevity. By employing deep learning techniques in examining the blood immunome of a cohort comprising 1,001 individuals, a modifiable chemokine CXCL9 was identified as being linked to cardiac aging. This discovery holds promise for the early detection of age-related pathology and presents a potential target for therapeutic interventions. In light of research has predominantly indicated that CXCL9 is linked to the response to ICI in advanced cancer patients, the observed increase in CXCL9 expression in older individuals, as a response marker for emerging immune-oncology treatments in this population (28, 58, 169).
2.9 JAK2
Janus kinase 2 (JAK2) is a cytoplasmic non-receptor tyrosine kinase that plays aa vital role in the JAK/STAT signaling pathway, a crucial regulator of, differentiation, survival, and immune response. Indeed, previous studies unveiled that the classical inflammatory factor JAK2 was associated with PD-L1 (170, 171). Somatic mutations that occur with advancing age in hematopoietic stem cells result in clonal sub-populations forming in the bloodstream of elderly individuals. This phenomenon is commonly linked to chronic inflammatory alterations, cardiovascular disease, and malignant progression. JAK2 is one of the genes that frequently undergo mutations during the aging process. In line with the conditions associated with these clonal modifications, the aberrant activation of JAK2 in myeloproliferative neoplasms is associated with the excessive proliferation of myeloid precursor cells, the aberrant release of inflammatory cytokines, and an increase in agglutination and thrombosis (76). The JAK2 p.V617F mutation is frequently detected in myeloproliferative neoplasms and has been associated with various complications (172).
Researchers have revealed that resistance to T-cell based therapies across both in vitro and in vivo models of breast cancer is predominantly due to the disruption of JAK2, a kinase integral to the interferon-gamma signaling pathway. This discovery highlights a new resistance mechanism to such therapies and suggests that monitoring JAK2 and interferon-gamma responses could effectively indicate patient response to immunotherapy treatments (173). Loss-of-function mutations in the JAK1/2 genes may cause resistance to anti-PD-1 treatment in certain malignant tissues (174). This instance emphasizes the potential importance of amplifying the PD-L1, PD-L2, and JAK2 gene clusters as a biomarker for sensitivity to chemotherapy (175). Significantly, the enhancement of this group of genes seems to make individuals vulnerable to nivolumab, irrespective of the existence of rearranged during transfection (RET) rearrangement, a well-known mutation that stimulates non-small cell lung cancer (NSCLC). This prompts the inquiry of whether including regular screening for rare but powerful markers of ICI sensitivity might aid in choosing the first systemic treatment for patients with NSCLC (175). The tumor cell membrane has a transmembrane-attached peptide-MHC class I receptor maintained by beta-2-microglobulin(B2M). Additionally, it contains the PD-L1 ligand and IFNGR1/IFNGR2 heterodimer, which are bound by JAK1 and JAK2 (176). The activation of phospho-STAT downstream of JAK1/JAK2 is known to facilitates the regulation of various targets, including MHC class I and PD-L1 expression, through its interaction with tumor cell DNA (176). According to a previous study, the activation of the oncogenic JAK2 gene increases the expression of PD-L1 on numerous cell types, including monocytes, megakaryocytes, MDSCs, and platelets. This upregulation is mediated by the JAK2-STAT3 and JAK2-STAT5 signaling pathways, which elicited a substantial response to ICI therapy (177). Overall, the role of the JAK2 gene in cancer treatment highlights its potential as a target, while its relevance to older cancer patients requires further investigation (28).
2.10 TNFRSF4
Tumor necrosis factor receptor superfamily member 4 (TNFRSF4), also known as CD134 or OX40, is a cell surface receptor protein that belongs to the tumor necrosis factor receptor superfamily and is predominantly expressed on activated T cells (178). Previous studies showed that TNFRSF4 facilitates the activation of the NF-kappa-B pathway by mediating tumor necrosis factor receptor-associated factor 2 (TRAF2) and TRAF5 (179, 180). The downstream signaling pathways of TNFRSF4 have also been identified to include the NFAT and P13K/PKB pathways (181, 182). The principal role of TNFRSF4 is to enhance the replication, growth, and survival of T cells as well as the production of cytokines through promoting the activity of the abovementioned signaling processes (179). The TNFRSF4 molecule has been identified as a co-stimulatory agent that may significantly promote effective antitumoral responses in sarcoma, melanoma, and breast cancer, as suggested by preclinical research (183, 184). The available data indicate that the amplification of anti-PD-1 therapy can be enhanced by targeting TNFRSF4, owing to the ability of TNFRSF4 agonism to amplify the expression of PD-L1 (185). Furthermore, tumor cytotoxicity can be augmented by upregulating TNFRSF4 within chimeric antigen receptor T cells via transfection and synergism with ICI (186). The existing literature presents a comprehensive analysis of the recently discovered gene TNFRSF4, encompassing its clinical effectiveness and potential correlation with immune cells (187). Although the activation of TNFRSF4-signaling in leukemic stem cells did not lead to the reduction of regulatory T cells, it impeded the ability of regulatory T cells to protect leukemic stem cells against CD8+ cytotoxic T lymphocyte-induced death. The levels of TNFRSF4 mRNA were notably elevated in the bone marrow of individuals recently diagnosed with chronic myeloid leukemia. These levels were found to be linked to the expression of the transcription factor FOXP3, which is specifically restricted to regulatory T cells (188). A series of research studies have explored the role of TNFRSF4 as a therapeutic agent in preclinical tumor models, highlighting its significant contribution to immunotherapy (189, 190). Recent findings indicate that TP53 mutations enhance the immunogenicity of breast cancer, and there is a correlation between elevated TNFRSF4 expression and TP53 mutations (191). Conversely, in patients with relapsed acute myeloid leukemia, TNFRSF4 expression is significantly increased in CD8+ T cells and regulatory T cells (Tregs) when compared to healthy donors (192).TNFRSF4 has been identified as a novel molecule in endometrial cancer (EC), and its increased expression is associated with favorable clinical outcomes. This elevated expression is closely linked to the abundance of CD4+ and CD8+ T cells. As a secondary immune checkpoint, TNFRSF4 has potential implications for endometrial cancer patients’ prognosis and immunomodulation (193). We previously introduced a novel prognostic model that significantly predicted survival outcomes based on TNFRSF4 and its association with age. TNFRSF4 exhibited a noteworthy survival association and negative correlation with age. Senescence and immunological-related pathways were highly correlated with TNFRSF4 expression (19). Given its significance in prediction and therapy, clinical agents aimed at TNFRSF4 present a promising avenue for managing tumor progression. This approach becomes increasingly attainable with the advent of newly developed direct and/or broad-spectrum small molecule inhibitors targeting checkpoint proteins. However, further research is needed, especially in the context of elderly patients.
2.11 TNFSF15
Tumor necrosis factor ligand superfamily member 15(TNFSF15), also known as TNF-like ligand 1A (TL1A) or vascular endothelial growth inhibitor (VEGI) which belongs to the TNF ligand family and is stimulated by TNF and IL-1α. It serves as an immune checkpoint gene, regulates the immune system, maintains cellular viability, and modulates inflammatory responses (194). The expression of the TNFSF15 protein is predominantly observed in immune cells, including dendritic cells, monocytes, and T cells, and has been associated with diverse immune-mediated mechanisms (195). The TNFSF15 gene interacts with its corresponding receptor, death receptor 3 (DR3) or TNFRSF25, which exhibits a preponderant expression on T cells (194). This interaction facilitates the transmission of signals that exert an impact on T-cell proliferation, cytokine production, and differentiation.
Preclinical models of autoimmune diseases and cancer have demonstrated promising results in blocking the TNFSF15-DR3 pathway, indicating that targeting TNFSF15 could potentially mitigate abnormal immune responses and alleviate inflammation. Maintaining vascular and lymphatic vessel homeostasis, especially in the presence of diseases such as cancer and stroke, relies heavily on its pivotal function, making it a plausible therapeutic target (196–199).
TNFSF15 is a cytokine that can inhibit the formation of new blood vessels and may also stimulate the development and specialization of macrophages into the M1 phenotype, which is known for its ability to destroy tumors. Scientists discovered (200) that the development of Lewis lung carcinoma cells in mice was significantly slowed when those cells had higher levels of TNFSF15. Further, TNFSF15 may simultaneously decrease the expression of VEGFR1 on the cell membrane and increase the synthesis of soluble VEGFR1, shifting the VEGF/VEGFR1 signaling pathway from promoting angiogenesis to inhibiting angiogenesis (200). Our team previously revealed that TNFSF15 gene expression was downregulated in clear cell renal cell carcinoma of geriatric with negative association between the TNFSF15 gene expression and age (59). Moreover, the protein-protein interaction between TNFSF15, other immune checkpoints, and proteins associated with aging demonstrates TNFSF15’s involvement in the aging mechanism. Furthermore, it has been established that the TNFSF15 gene is involved in several biological processes, such as senescence, cellular senescence, immune system, immune cell infiltration, and immunological function. An additional investigation demonstrated an age-dependent correlation of TNFSF15, which was observable not solely in individuals with cancer but also in those with other ailments related to the immune system (201, 202). It’s important to note that the role of TNFSF15 in immunotherapy is still an active area of investigation, and its significance may vary depending on the cancer type and individual patient characteristics (203–206). As our understanding of TNFSF15’s role in the immune response and its interactions with immunotherapies continues to evolve, it may become a more prominent factor in tailoring personalized treatment strategies, particularly in the elderly group.
2.12 VSIG4
V-set and Ig domain-containing 4(VSIG4), also known as CRIg or Z39Ig is a newly discovered B-7 related protein (207). The VSIG4 molecule serves as a receptor for complement C3, facilitating the elimination of pathogens. Additionally, it possesses the ability to impede the proliferation of CD4+ and CD8+ T-cells, as well as the production of IL-2, through the binding of an unknown receptor on T-cells (208, 209). The mRNA of VSIG4 exhibits significant expression levels across multiple tissues, with notable prevalence in the liver, lung, placenta, and potentially the central nervous system. However, the protein manifestation of VSIG4 is limited to the macrophage surface (209). According to recent research, VSIG4+ cells have been detected in the tissues of individuals with rheumatoid arthritis, atherosclerosis, and chronic hepatitis B virus infected livers, implying that VSIG4 may serve as a contributing factor to the development of such inflammatory conditions (210). The protein VSIG4 has garnered attention in inflammatory disease and cancer due to its multifaceted checkpoint properties. Additional research conducted in vitro and in vivo has shown that anti-VSIG4 antibodies can repolarize M2 macrophages and trigger an immunological response that ultimately activates T cells (211). Upregulating VSIG4 enhanced the production of pro-inflammatory cytokines in M2 macrophages as well as pro-inflammatory cytokines originating from myeloid cells and T cells (211). VSIG4 has been identified as a novel biomarker of aging macrophages in tissues that are linked to age-related systemic inflammation and immunosenescence (212). The accumulation of VSIG4 in male mice exhibited a significant correlation with heightened physiological frailty, suggesting its connection to a fundamental mechanism of the natural aging process of organisms (212). Upregulating VSIG4 enhances the production of pro-inflammatory cytokines in M2 macrophages as well as pro-inflammatory cytokines originating from myeloid cells and T cells (212). Researchers have discovered that gut microbial DNA and the immunological checkpoint gene VSIG4 play crucial opposing roles in both healthy aging and the development of hypertension and diabetes associated with aging (213). VSIG4 also has a vital function in promoting “healthy aging” by inhibiting insulin resistance and hypertension, which are often linked to the aging process. Although the exact role of VSIG4 in advancing cancerous growth is not yet fully understood, recent research has shown that VSIG4 promotes cell proliferation, migration, and resistance to immune attacks. This identifies VSIG4 as a potential target for cancer treatment, particularly for patients with a positive prognosis (214, 215). In summary, VSIG4 shows promise as a potential therapeutic target for ICI in cancer therapy (216). However, it’s crucial to emphasize that ongoing research is investigating the role of VSIG4 in the response to ICI, and its significance may differ depending on the specific cancer type and patient groups. Further studies are necessary to gain a deeper understanding of how VSIG4 expression impacts ICI responses and to explore the potential of combining VSIG4-targeted therapies with ICI. Consequently, the clinical utility of VSIG4 as either a predictive biomarker or a therapeutic target in immunotherapy is a field in continual development including older patients.
3 Conclusion and perspectives
Tumors derived from individuals belonging to distinct age groups exhibit varying responses to ICI, which may be attributed to the expression profile of age-related immune genes. This observation accompanies the documented rise in cancer occurrence and mortality with advancing age (32). However, only anecdotal evidence suggests that elderly individuals treated with PD-1, PD-L1, and CTLA-4 antibodies do not have inferior responses or have increased toxicity as a consequence of their advanced age, which is encouraging (217, 218). Still, the age issue will become critical as new medicines are approved and novel combinations are explored; broader and more extensive studies concentrating on the age question will become increasingly important, and they should be considered when conducting clinical trials. It’s crucial to note that not all genes associated with aging hold the potential to serve as viable targets. The attainment of satisfactory efficacy relies on combining these genes with an adaptive operational strategy and efficient carriers. Moreover, certain age-related genes may function solely as prognostic indicators or have the capability to identify effective medications for specific diseases (219–222). It is also vital to have a strong synergy with other genes and signaling pathways to prevent excessive signaling cascades (223, 224) that will lead to uncontrolled, unfavorable effects.
The secretion of SASP by cancer cells in a state of senescence attracts immune cells and inhibits the proliferation of neighboring tumor cells. Nevertheless, the persistent and prolonged buildup of SASP would facilitate the proliferation of tumors and their spread to other parts of the body (225). Hence, an optimal approach for eradicating malignant cells can include a two-step process: senescence-inducing treatment and senolytic therapy (226–228). Senescence of malignant cells can be induced by a variety of mechanisms, including chemotherapeutic agents (e.g., inhibitors of topoisomerase I and II, platinum-based therapy, alkylating agents, and microtubule inhibitors), upregulation of cyclin-dependent kinases (CDK), telomere attrition, and epigenetic region modulation (227, 229–239). Moreover, CDK inhibitors, including those for CDK4/6, CDK2/4/6, CDK7, and CDK12, are capable of halting the progression of malignant cell cycles in various types of cancer (240–243). Recent research has also demonstrated that cancer cell senescence can be increased by inhibiting telomerase (e.g., with the inhibitors GRN163L and BIBR15), DNA methyltransferase (decitabine), and histone deacetylase (GCN5, PCAF, p300/CBP, and Tip60) (232, 244–247). We expect further trials and products to emerge in the aging process, contributing to a comprehensive understanding of targetability.
Immunosenescence, a process characterized by the immune system’s adaptive responses to previous challenges encountered over an individual’s lifetime, has been associated with unfavorable clinical outcomes (18, 248, 249). In circumstances unrelated to tumors, it is commonly observed that older people tend to exhibit less efficient immune responses when facing diseases like the new pandemic due to coronavirus disease 2019 (COVID-19) (4, 250). This phenomenon is typically linked to the systemic immune senescence. Specifically, age-related immune alterations, such as a reduction in TCR diversity, a decline in the cytotoxic cell’s capability, and an increase in inflammatory signaling, and others (Figure 5) (251–254).
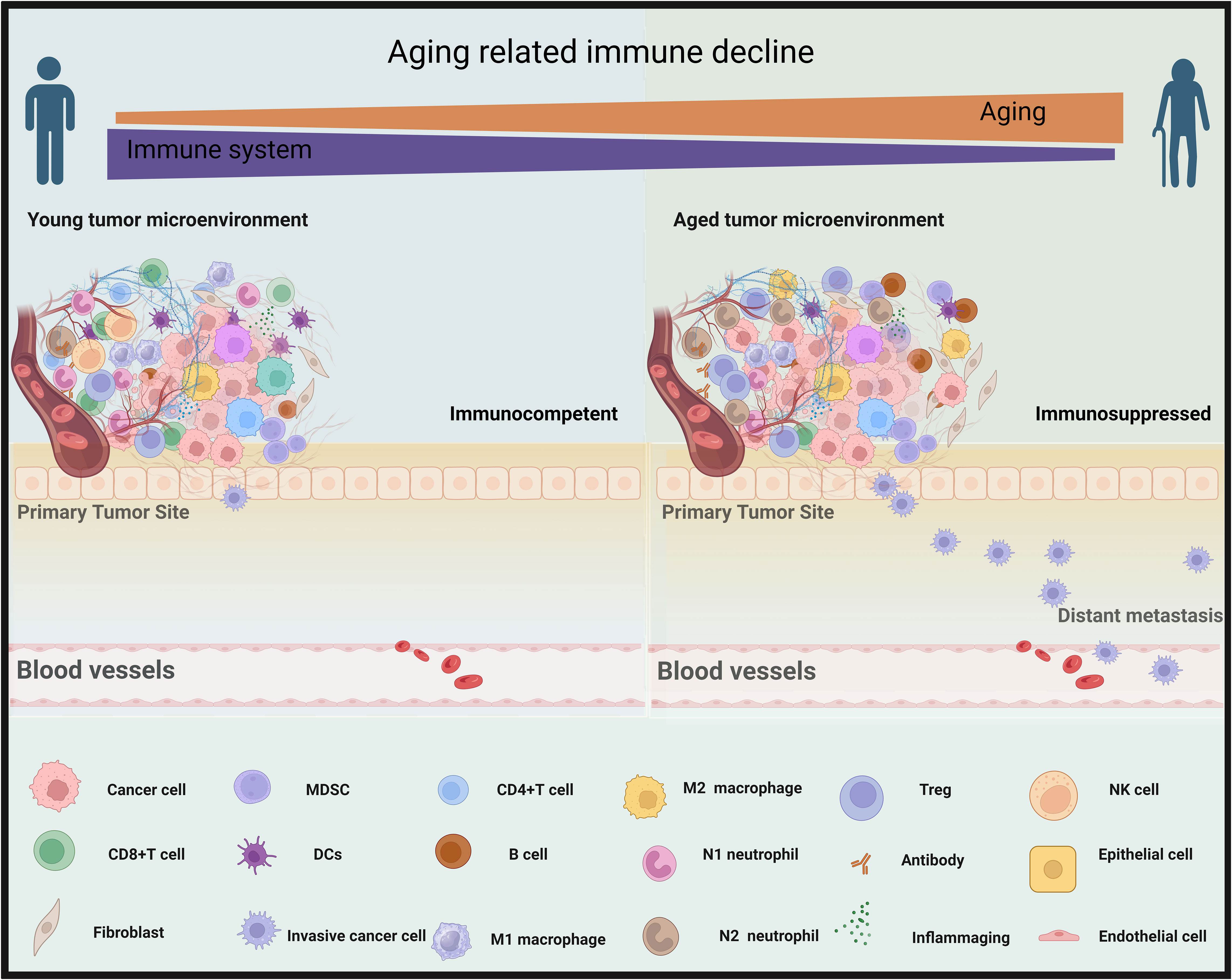
Figure 5 Aging contributes to a decline in immune function within tumor microenvironments. In the left panel, the tumor microenvironment of a young person is immunocompetent, with an abundance of cancer-fighting immune responses (CD8+T cells, M1 macrophages, natural killer (NK) cells, CD4+ T cells, N1 neutrophil, and dendritic cells [DCs]) that counteract the cancer growth and metastasis. Right, elderly cancer patients, as depicted in the right panel, exhibit an immunosuppressed microenvironment with a prevalence of less effective immune components (M2 macrophages, myeloid-derived suppressor cells (MDSC), B cells, T regulatory T cells [Treg], N2 neutrophils, and (inflammaging which is an age-related increase in the levels of pro-inflammatory markers in blood and tissues), that are less equipped to combat metastasizing cancer cells effectively. By BioRender (https://app.biorender.com/).
The aforementioned findings highlighted the potential importance of immunological aging and its effect on malignancy genesis and progression (255). However, in order to apply this knowledge to cancer treatment and patient management, it is necessary to conduct a more thorough analysis of the relationship between the overall immune system and the specific immunological milieu within tumors, which is influenced by the aging process, especially in the ICI era. Despite the often observed reduction in immune function that accompanies the aging process, a significant body of evidence from clinical trial analysis indicates that older individuals derive comparable or even enhanced benefits from ICI therapy when compared to their younger counterparts (62, 68, 256, 257). In reconciling inconsistencies and establishing a consensus regarding the impact of age on immunotherapy response, recent studies indicated that age-related impairments in the immune system did not influence the efficacy and toxicity of ICI in geriatric patients compared to their younger counterparts (217, 258–263). Therefore, regardless of chronological age, ICI can be considered for elderly patients. Even so, there remains ongoing debate regarding this matter, and it is worth noting that older individuals with cancer may receive less frequent utilization of ICI compared to young cancer patients (62, 264, 265).
In summary, aging and cancer are closely intertwined, as aging is a risk factor for the development of cancer. Nevertheless, advancements in cancer treatment, particularly ICI therapy, offer new hope for patients by leveraging the body’s immune system’s ability to overcome cancer. Continued research and innovation in this field are crucial to improving patient outcomes, expanding the application of ICI, and ultimately finding more effective and personalized approaches to combating cancer in the context of aging.
Author contributions
AA: Validation, Visualization, Writing – original draft, Writing – review & editing. MS: Conceptualization, Visualization, Writing – original draft. YJ: Validation, Visualization, Writing – review & editing. LY: Investigation, Writing – original draft, Writing – review & editing. SW: Project administration, Supervision, Writing – review & editing. XZ: Data curation, Investigation, Writing – original draft. QC: Funding acquisition, Validation, Writing – review & editing. KY: Visualization, Writing – review & editing. JZ: Conceptualization, Resources, Writing – review & editing. DY: Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82203730) and Dalian Medical University, Interdisciplinary Research Cooperation Project Team Funding (JCHZ2023021).
Acknowledgments
The utilization of BioRender was acknowledged for creating the figures of this work. The authors also acknowledge Dr Wardah Khaled for visualization and designing article figures. The manuscript was edited by Sarah Bronson of the Research Medical Library at The University of Texas MD Anderson Cancer Center.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1348189/full#supplementary-material
Glossary

References
1. Braithwaite D, Anton S, Mohile S, DeGregori J, Gillis N, Zhou D, et al. Cancer and aging: A call to action. Aging Cancer (2022) 3(2):87–94. doi: 10.1002/aac2.12055
2. Hsu T, Elias R, Swartz K, Chapman A. Developing sustainable cancer and aging programs. Am Soc Clin Oncol Educ Book (2023) 43):e390980. doi: 10.1200/EDBK_390980
3. Goede V. Frailty and cancer: current perspectives on assessment and monitoring. Clin Interv Aging (2023) 18:505–21. doi: 10.2147/CIA.S365494
4. Gardner ID. The effect of aging on susceptibility to infection. Rev Infect Dis (1980) 2(5):801–10. doi: 10.1093/clinids/2.5.801
5. Trofatter KF Jr. Immune responses and aging. Clin Obstet Gynecol (1986) 29(2):384–96. doi: 10.1097/00003081-198606000-00020
6. Klutstein M, Moss J, Kaplan T, Cedar H. Contribution of epigenetic mechanisms to variation in cancer risk among tissues. Proc Natl Acad Sci USA. (2017) 114(9):2230–4. doi: 10.1073/pnas.1616556114
7. Xie W, Kagiampakis I, Pan L, Zhang YW, Murphy L, Tao Y, et al. DNA methylation patterns separate senescence from transformation potential and indicate cancer risk. Cancer Cell (2018) 33(2):309–321.e5. doi: 10.1016/j.ccell.2018.01.008
8. Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science (2017) 355(6331):1330–4. doi: 10.1126/science.aaf9011
9. Dewan SK, Zheng SB, Xia SJ, Bill K. Senescent remodeling of the immune system and its contribution to the predisposition of the elderly to infections. Chin Med J (Engl) (2012) 125(18):3325–31.
10. Weiskopf D, Weinberger B, Grubeck-Loebenstein B. The aging of the immune system. Transpl Int (2009) 22(11):1041–50. doi: 10.1111/j.1432-2277.2009.00927.x
11. Liu Z, Liang Q, Ren Y, Guo C, Ge X, Wang L, et al. Immunosenescence: molecular mechanisms and diseases. Signal Transduct Target Ther (2023) 8(1):200. doi: 10.1038/s41392-023-01451-2
12. Lian J, Yue Y, Yu W, Zhang Y. Immunosenescence: a key player in cancer development. J Hematol Oncol (2020) 13(1):151. doi: 10.1186/s13045-020-00986-z
13. Pawelec G. Does patient age influence anti-cancer immunity? Semin Immunopathol (2019) 41(1):125–31. doi: 10.1007/s00281-018-0697-6
14. Remon J, Levy A, Singh P, Hendriks LEL, Aldea M, Arrieta O. Current challenges of unresectable stage III NSCLC: are we ready to break the glass ceiling of the PACIFIC trial? Ther Adv Med Oncol (2022) 14:17588359221113268. doi: 10.1177/17588359221113268
15. Jackaman C, Tomay F, Duong L, Abdol Razak NB, Pixley FJ, Metharom P, et al. Aging and cancer: The role of macrophages and neutrophils. Ageing Res Rev (2017) 36:105–16. doi: 10.1016/j.arr.2017.03.008
16. Liu S, Sun Q, Ren X. Novel strategies for cancer immunotherapy: counter-immunoediting therapy. J Hematol Oncol (2023) 16(1):38. doi: 10.1186/s13045-023-01430-8
17. Crooke SN, Ovsyannikova IG, Poland GA, Kennedy RB. Immunosenescence: A systems-level overview of immune cell biology and strategies for improving vaccine responses. Exp Gerontol (2019) 124:110632. doi: 10.1016/j.exger.2019.110632
18. Aiello A, Farzaneh F, Candore G, Caruso C, Davinelli S, Gambino CM, et al. Immunosenescence and its hallmarks: how to oppose aging strategically? A review of potential options for therapeutic intervention. Front Immunol (2019) 10:2247. doi: 10.3389/fimmu.2019.02247
19. Safi M, Jin C, Aldanakh A, Feng P, Qin H, Alradhi M, et al. Immune checkpoint inhibitor (ICI) genes and aging in Malignant melanoma patients: a clinicogenomic TCGA study. BMC Cancer (2022) 22(1):978. doi: 10.1186/s12885-022-09860-2
20. Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther (2015) 14(4):847–56. doi: 10.1158/1535-7163.MCT-14-0983
21. Davis AA, Patel VG. The role of PD-L1 expression as a predictive biomarker: an analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J Immunother Cancer (2019) 7(1):278. doi: 10.1186/s40425-019-0768-9
22. Alradhi M, Zhang Z, Safi M, Al-danakh A, Aldhbi M, Baldi S, et al. A novel nomogram and prognostic factor for metastatic renal cell carci noma survival in the era of immune checkpoint inhibitors (ICIs). Front Pharmacol 13. doi: 10.3389/fphar.2022.996404
23. Safi M, Al-Azab M, Jin C, Trapani D, Baldi S, Adlat S, et al. Age-Based Disparities in Metastatic Melanoma Patients Treated in the I mmune Checkpoint Inhibitors (ICI) Versus Non-ICI Era: A Populat ion-Based Study. Front Immunol 12:609728. doi: 10.3389/fimmu.2021.609728
24. Safi M, Ahmed H, Al-Azab M, Xia Y-L, Shan X, Al-Radhi M, et al. PD-1/PDL-1 inhibitors and cardiotoxicity; molecular, etiological and M anagement outlines. J Adv Res 29:45–54. doi: 10.1016/j.jare.2020.09.006
25. Taube JM. Unleashing the immune system: PD-1 and PD-Ls in the pre-treatment tumor microenvironment and correlation with response to PD-1/PD-L1 blockade. Oncoimmunology (2014) 3(11):e963413. doi: 10.4161/21624011.2014.963413
26. Wu SP, Liao RQ, Tu HY, Wang WJ, Dong ZY, Huang SM, et al. Stromal PD-L1-Positive Regulatory T cells and PD-1-Positive CD8-Positive T cells Define the Response of Different Subsets of Non-Small Cell Lung Cancer to PD-1/PD-L1 Blockade Immunotherapy. J Thorac Oncol (2018) 13(4):521–32. doi: 10.1016/j.jtho.2017.11.132
27. Conway JR, Kofman E, Mo SS, Elmarakeby H, Van Allen E. Genomics of response to immune checkpoint therapies for cancer: implications for precision medicine. Genome Med (2018) 10(1):93. doi: 10.1186/s13073-018-0605-7
28. Erbe R, Wang Z, Wu S, Xiu J, Zaidi N, La J, et al. Evaluating the impact of age on immune checkpoint therapy biomarkers. Cell Rep (2021) 36(8):109599. doi: 10.1016/j.celrep.2021.109599
29. Bai R, Lv Z, Xu D, Cui J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. biomark Res (2020) 8(1):34. doi: 10.1186/s40364-020-00209-0
30. Cui Y, Wang K, Jiang D, Jiang Y, Shi D, DeGregori J, et al. Promoting longevity with less cancer: The 2022 International Conferenc e on Aging and Cancer. Aging Cancer 4(3-4):111–20. doi: 10.1002/aac2.12068
31. Aging Biomarker C, Bao H, Cao J, Chen M, Chen M, Chen W, et al. Biomarkers of aging. Sci China Life Sci 66(5):893–1066. doi: 10.1007/s11427-023-2305-0
32. Chatsirisupachai K, Lagger C, de Magalhães JP. Age-associated differences in the cancer molecular landscape. Trends Cancer (2022) 8(11):962–71. doi: 10.1016/j.trecan.2022.06.007
33. Chatsirisupachai K, Lesluyes T, Paraoan L, Van Loo P, de Magalhães JP. An integrative analysis of the age-associated multi-omic landscape acr oss cancers. Nat Commun 12(1):2345. doi: 10.1038/s41467-021-22560-y
34. Lee W, Wang Z, Saffern M, Jun T, Huang K-L. Genomic and molecular features distinguish young adult cancer from lat er-onset cancer. Cell Rep 37(7):110005. doi: 10.1016/j.celrep.2021.110005
35. Li CH, Haider S, Boutros PC. Age influences on the molecular presentation of tumours. Nat Commun (2022) 13(1):208. doi: 10.1038/s41467-021-27889-y
36. Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, et al. Clock-like mutational processes in human somatic cells. Nat Genet (2015) 47(12):1402–7. doi: 10.1038/ng.3441
37. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med (2017) 9:1–14. doi: 10.1186/s13073-017-0424-2
38. Milholland B, Auton A, Suh Y, Vijg J. Age-related somatic mutations in the cancer genome. Oncotarget (2015) 6(28):24627. doi: 10.18632/oncotarget.5685
39. Gerhauser C, Favero F, Risch T, Simon R, Feuerbach L, Assenov Y, et al. Molecular evolution of early-onset prostate cancer identifies molecular risk markers and clinical trajectories. Cancer Cell (2018) 34(6):996–1011.e8.
40. Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell (2016) 164(3):550–63. doi: 10.1016/j.cell.2015.12.028
41. Mealey NE, O’Sullivan DE, Pader J, Ruan Y, Wang E, Quan ML, et al. Mutational landscape differences between young-onset and older-onset breast cancer patients. BMC Cancer (2020) 20(1):1–18. doi: 10.1186/s12885-020-6684-z
42. Waks AG, Kim D, Jain E, Snow C, Kirkner GJ, Rosenberg SM, et al. Somatic and germline genomic alterations in very young women with breast cancer. Clin Cancer Res (2022) 28(11):2339–48. doi: 10.1158/1078-0432.CCR-21-2572
43. Shah Y, Verma A, Marderstein AR, White J, Bhinder B, Medina JSG, et al. Pan-cancer analysis reveals molecular patterns associated with age. Cell Rep (2021) 37(10). doi: 10.1016/j.celrep.2021.110100
44. Liao S, Hartmaier RJ, McGuire KP, Puhalla SL, Luthra S, Chandran UR, et al. The molecular landscape of premenopausal breast cancer. Breast Cancer Res (2015) 17(1):1–13. doi: 10.1186/s13058-015-0618-8
45. Kan Z, Ding Y, Kim J, Jung HH, Chung W, Lal S, et al. Multi-omics profiling of younger Asian breast cancers reveals distinct ive molecular signatures. Nat Commun 9(1):1725. doi: 10.1038/s41467-018-04129-4
46. Berger AC, Korkut A, Kanchi RS, Hegde AM, Lenoir W, Liu W, et al. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell (2018) 33(4):690-705.e9.
47. Lieu CH, et al. Comprehensive genomic landscapes in early and later onset colorectal cancer. Clin Cancer Res (2019) 25(19):5852–8. doi: 10.1158/1078-0432.CCR-19-0899
48. Berg M, et al. DNA sequence profiles of the colorectal cancer critical gene set KRAS-BRAF-PIK3CA-PTEN-TP53 related to age at disease onset. PloS One (2010) 5(11):e13978. doi: 10.1371/journal.pone.0013978
49. Nilsson PM. Early vascular aging (EVA): consequences and prevention. Vasc Health Risk Manage (2008) 4(3):547–52. doi: 10.2147/VHRM.S1094
50. Yu J, Li T, Zhu J. Gene therapy strategies targeting aging-related diseases. Aging Dis 14(2):398–417. doi: 10.14336/AD.2022.00725
52. Sebastiani P, Gurinovich A, Nygaard M, Sasaki T, Sweigart B, Bae H, et al. APOE alleles and extreme human longevity. J Gerontol: Ser A (2019) 74(1):44–51. doi: 10.1093/gerona/gly174
53. Willcox BJ, Donlon TA, He Q, Chen R, Grove JS, Yano K, et al. FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci (2008) 105(37):13987–92. doi: 10.1073/pnas.0801030105
54. Chen C, Zhou M, Ge Y, Wang X. SIRT1 and aging related signaling pathways. Mech Ageing Dev (2020) 187:111215. doi: 10.1016/j.mad.2020.111215
55. Korotkov A, Seluanov A, Gorbunova V. Sirtuin 6: linking longevity with genome and epigenome stability. Trends Cell Biol (2021) 31(12):994–1006. doi: 10.1016/j.tcb.2021.06.009
56. Ylä-Herttuala S, Baker AH. Cardiovascular gene therapy: past, present, and future. Mol Ther (2017) 25(5):1095–106. doi: 10.1016/j.ymthe.2017.03.027
57. Hegde PS, Wallin JJ, Mancao C. Predictive markers of anti-VEGF and emerging role of angiogenesis inhibitors as immunotherapeutics. Semin Cancer Biol (2018) 52(Pt 2):117–24. doi: 10.1016/j.semcancer.2017.12.002
58. Guan R, Lyu Q, Lin A, Liang J, Ding W, Cao M, et al. Influence of different age cutoff points on the prediction of prognosis of cancer patients receiving ICIs and potential mechanistic exploration. Front Oncol (2021) 11:670927. doi: 10.3389/fonc.2021.670927
59. Al-Danakh A, Safi M, Alradhi M, Chen Q, Baldi S, Zhu X, et al. Immune checkpoint inhibitor (ICI) genes and aging in clear cell renal cell carcinoma (ccRCC): clinical and genomic study. Cells (2022) 11(22):3641. doi: 10.3390/cells11223641
60. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res (2003) 13(11):2498–504. doi: 10.1101/gr.1239303
61. Ge SX, Jung D, Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36(8):2628–9. doi: 10.1093/bioinformatics/btz931
62. Jain V, Hwang WT, Venigalla S, Nead KT, Lukens JN, Mitchell TC, et al. Association of age with efficacy of immunotherapy in metastatic melanoma. Oncologist (2020) 25(2):e381–5. doi: 10.1634/theoncologist.2019-0377
63. Onorati A, Havas AP, Lin B, Rajagopal J, Sen P, Adams PD, et al. Upregulation of PD-L1 in senescence and aging. Mol Cell Biol (2022) 42(10):e0017122. doi: 10.1128/mcb.00171-22
64. Wang TW, Johmura Y, Suzuki N, Omori S, Migita T, Yamaguchi K, et al. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature (2022) 611(7935):358–64. doi: 10.1038/s41586-022-05388-4
65. Garcia MG, Deng Y, Murray C, Reyes RM, Padron A, Bai H, et al. Immune checkpoint expression and relationships to anti-PD-L1 immune checkpoint blockade cancer immunotherapy efficacy in aged versus young mice. Aging Cancer (2022) 3(1):68–83. doi: 10.1002/aac2.12045
66. Shaw AC, Joshi S, Greenwood H, Panda A, Lord JM. Aging of the innate immune system. Curr Opin Immunol (2010) 22(4):507–13. doi: 10.1016/j.coi.2010.05.003
67. Busse S, Steiner J, Alter J, Dobrowolny H, Mawrin C, Bogerts B, et al. Expression of HLA-DR, CD80, and CD86 in healthy aging and Alzheimer's disease. J Alzheimers Dis (2015) 47(1):177–84. doi: 10.3233/JAD-150217
68. Erbe R, Wang Z, Wu S, Xiu J, Zaidi N, Lippman ME, et al. Analysis of immune checkpoint blockade biomarkers in elderly patients using large-scale cancer genomics data. J Clin Oncol (2021) 39(15_suppl):2543–3. doi: 10.1200/JCO.2021.39.15_suppl.2543
69. Cunha LL, Perazzio SF, Azzi J, Cravedi P, Riella LV. Remodeling of the immune response with aging: immunosenescence and its potential impact on COVID-19 immune response. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.01748
70. Li H, Yang D, Hao M, Liu H. Differential expression of HAVCR2 gene in pan-cancer: A potential biomarker for survival and immunotherapy. Front Genet (2022) 13:972664. doi: 10.3389/fgene.2022.972664
71. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
72. Tsukamoto H, Clise-Dwyer K, Huston GE, Duso DK, Buck AL, Johnson LL, et al. Age-associated increase in lifespan of naive CD4 T cells contributes to T-cell homeostasis but facilitates development of functional defects. Proc Natl Acad Sci USA. (2009) 106(43):18333–8. doi: 10.1073/pnas.0910139106
73. Okadome K, Baba Y, Nomoto D, Yagi T, Kalikawe R, Harada K, et al. Prognostic and clinical impact of PD-L2 and PD-L1 expression in a cohort of 437 oesophageal cancers. Br J Cancer (2020) 122(10):1535–43. doi: 10.1038/s41416-020-0811-0
74. Tominaga K, Suzuki HI. TGF-β Signaling in cellular senescence and aging-related pathology. Int J Mol Sci (2019) 20(20). doi: 10.3390/ijms20205002
75. Sayed N, Huang Y, Nguyen K, Krejciova-Rajaniemi Z, Grawe AP, Gao T, et al. An inflammatory aging clock (iAge) based on deep learning tracks multimorbidity, immunosenescence, frailty and cardiovascular aging. Nat Aging (2021) 1:598–615. doi: 10.1038/s43587-021-00082-y
76. Perner F, Perner C, Ernst T, Heidel FH. Roles of JAK2 in aging, inflammation, hematopoiesis and Malignant transformation. Cells (2019) 8(8). doi: 10.3390/cells8080854
77. Gou Q, Dong C, Xu H, Khan B, Jin J, Liu Q, et al. PD-L1 degradation pathway and immunotherapy for cancer. Cell Death Dis (2020) 11(11):955. doi: 10.1038/s41419-020-03140-2
78. Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol (2012) 24(2):207–12. doi: 10.1016/j.coi.2011.12.009
79. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med (2012) 209(6):1201–17. doi: 10.1084/jem.20112741
80. Iwai Y, Hamanishi J, Chamoto K, Honjo T. Cancer immunotherapies targeting the PD-1 signaling pathway. J BioMed Sci (2017) 24(1):26. doi: 10.1186/s12929-017-0329-9
81. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27(4):450–61. doi: 10.1016/j.ccell.2015.03.001
82. Ritprajak P, Azuma M. Intrinsic and extrinsic control of expression of the immunoregulatory molecule PD-L1 in epithelial cells and squamous cell carcinoma. Oral Oncol (2015) 51(3):221–8. doi: 10.1016/j.oraloncology.2014.11.014
83. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
84. Johmura Y, Shimada M, Misaki T, Naiki-Ito A, Miyoshi H, Motoyama N, et al. Necessary and sufficient role for a mitosis skip in senescence induction. Mol Cell (2014) 55(1):73–84. doi: 10.1016/j.molcel.2014.05.003
85. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature (2016) 530(7589):184–9. doi: 10.1038/nature16932
86. Mirza N, Duque MA, Dominguez AL, Schrum AG, Dong H, Lustgarten J. B7-H1 expression on old CD8+ T cells negatively regulates the activation of immune responses in aged animals. J Immunol (2010) 184(10):5466–74. doi: 10.4049/jimmunol.0903561
87. Zhao B, Wu B, Feng N, Zhang X, Zhang X, Wei Y, et al. Aging microenvironment and antitumor immunity for geriatric oncology: the landscape and future implications. J Hematol Oncol (2023) 16(1):28. doi: 10.1186/s13045-023-01426-4
88. Ladomersky E, Scholtens DM, Kocherginsky M, Hibler EA, Bartom ET, Otto-Meyer S, et al. The coincidence between increasing age, immunosuppression, and the incidence of patients with glioblastoma. Front Pharmacol (2019) 10:200. doi: 10.3389/fphar.2019.00200
89. Zhang S, Bai X, Shan F. The progress and confusion of anti-PD1/PD-L1 immunotherapy for patients with advanced non-small cell lung cancer. Int Immunopharmacol (2020) 80:106247. doi: 10.1016/j.intimp.2020.106247
90. Hajihassan Z, Afsharian NP, Ansari-Pour N. In silico engineering a CD80 variant with increased affinity to CTLA-4 and decreased affinity to CD28 for optimized cancer immunotherapy. J Immunol Methods (2023) 513:113425. doi: 10.1016/j.jim.2023.113425
91. Lenschow DJ, Su GH, Zuckerman LA, Nabavi N, Jellis CL, Gray GS, et al. Expression and functional significance of an additional ligand for CTLA-4. Proc Natl Acad Sci USA. (1993) 90(23):11054–8. doi: 10.1073/pnas.90.23.11054
92. Sansom DM. CD28, CTLA-4 and their ligands: who does what and to whom? Immunology (2000) 101(2):169–77. doi: 10.1046/j.1365-2567.2000.00121.x
93. Vackova J, Polakova I, Johari SD, Smahel M. CD80 expression on tumor cells alters tumor microenvironment and efficacy of cancer immunotherapy by CTLA-4 blockade. Cancers (Basel) (2021) 13(8). doi: 10.3390/cancers13081935
94. Lindsten T, Lee KP, Harris ES, Petryniak B, Craighead N, Reynolds PJ, et al. Characterization of CTLA-4 structure and expression on human T cells. J Immunol (1993) 151(7):3489–99. doi: 10.4049/jimmunol.151.7.3489
95. Ganesan PL, Alexander SI, Watson D, Logan GJ, Zhang GY, Alexander IE. Robust anti-tumor immunity and memory in Rag-1-deficient mice following adoptive transfer of cytokine-primed splenocytes and tumor CD80 expression. Cancer Immunol Immunother (2007) 56(12):1955–65. doi: 10.1007/s00262-007-0339-7
96. Feng XY, Lu L, Wang KF, Zhu BY, Wen XZ, Peng RQ, et al. Low expression of CD80 predicts for poor prognosis in patients with gastric adenocarcinoma. Future Oncol (2019) 15(5):473–83. doi: 10.2217/fon-2018-0420
97. Tirapu I, Huarte E, Guiducci C, Arina A, Zaratiegui M, Murillo O, et al. Low surface expression of B7-1 (CD80) is an immunoescape mechanism of colon carcinoma. Cancer Res (2006) 66(4):2442–50. doi: 10.1158/0008-5472.CAN-05-1681
98. Ostrand-Rosenberg S, Horn LA, Alvarez JA. Novel strategies for inhibiting PD-1 pathway-mediated immune suppression while simultaneously delivering activating signals to tumor-reactive T cells. Cancer Immunol Immunother (2015) 64(10):1287–93. doi: 10.1007/s00262-015-1677-5
99. Chaudhri A, Xiao Y, Klee AN, Wang X, Zhu B, Freeman GJ. PD-L1 binds to B7-1 only in Cis on the same cell surface. Cancer Immunol Res (2018) 6(8):921–9. doi: 10.1158/2326-6066.CIR-17-0316
100. Zhao Y, Lee CK, Lin CH, Gassen RB, Xu X, Huang Z, et al. PD-L1:CD80 cis-heterodimer triggers the co-stimulatory receptor CD28 while repressing the inhibitory PD-1 and CTLA-4 pathways. Immunity (2019) 51(6):1059–1073.e9. doi: 10.1016/j.immuni.2019.11.003
101. Mayoux M, Roller A, Pulko V, Sammicheli S, Chen S, Sum E, et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci Transl Med (2020) 12(534). doi: 10.1126/scitranslmed.aav7431
102. Morano NC, Smith RS, Danelon V, Schreiner R, Patel U, Herrera NG, et al. Human immunomodulatory ligand B7-1 mediates synaptic remodeling via the p75 neurotrophin receptor. J Clin Invest (2022) 132(22). doi: 10.1172/JCI157002
103. van Duin D, Allore HG, Mohanty S, Ginter S, Newman FK, Belshe RB, et al. Prevaccine determination of the expression of costimulatory B7 molecules in activated monocytes predicts influenza vaccine responses in young and older adults. J Infect Dis (2007) 195(11):1590–7. doi: 10.1086/516788
104. Feng W, He Z, Shi L, Zhu Z, Ma H. Significance of CD80 as a prognostic and immunotherapeutic biomarker in lung adenocarcinoma. Biochem Genet (2023) 61(5):1937–66. doi: 10.1007/s10528-023-10343-7
105. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature (2002) 415(6871):536–41. doi: 10.1038/415536a
106. McIntire JJ, Umetsu SE, Akbari O, Potter M, Kuchroo VK, Barsh GS, et al. Identification of Tapr (an airway hyperreactivity regulatory locus) and the linked Tim gene family. Nat Immunol (2001) 2(12):1109–16. doi: 10.1038/ni739
107. Lee J, Phong B, Egloff AM, Kane LP. TIM polymorphisms–genetics and function. Genes Immun (2011) 12(8):595–604. doi: 10.1038/gene.2011.75
108. Nandini A, Catherine S-P, Ana Carrizosa A. Tim-3 finds its place in the cancer immunotherapy landscape. J ImmunoTher Cancer (2020) 8(1):e000911.
109. Sabatos CA, Chakravarti S, Cha E, Schubart A, Sánchez-Fueyo A, Zheng XX, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol (2003) 4(11):1102–10. doi: 10.1038/ni988
110. Sánchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol (2003) 4(11):1093–101. doi: 10.1038/ni987
111. Gayden T, Sepulveda FE, Khuong-Quang D-A, Pratt J, Valera ET, Garrigue A, et al. Germline HAVCR2 mutations altering TIM-3 characterize subcutaneous panniculitis-like T cell lymphomas with hemophagocytic lymphohistiocytic syndrome. Nat Genet (2018) 50(12):1650–7. doi: 10.1038/s41588-018-0251-4
112. Polprasert C, Takeuchi Y, Kakiuchi N, Yoshida K, Assanasen T, Sitthi W, et al. Frequent germline mutations of HAVCR2 in sporadic subcutaneous panniculitis-like T-cell lymphoma. Blood Adv (2019) 3(4):588–95. doi: 10.1182/bloodadvances.2018028340
113. DeLong JH, O’Hara Hall A, Rausch M, Moodley D, Perry J, Park J, et al. IL-27 and TCR stimulation promote T cell expression of multiple inhibitory receptors. Immunohorizons (2019) 3(1):13–25. doi: 10.4049/immunohorizons.1800083
114. Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature (2018) 558(7710):454–9. doi: 10.1038/s41586-018-0206-z
115. Huff WX, Kwon JH, Henriquez M, Fetcko K, Dey M. The evolving role of CD8(+)CD28(-) immunosenescent T cells in cancer immunology. Int J Mol Sci (2019) 20(11). doi: 10.3390/ijms20112810
116. Mondal AM, Horikawa I, Pine SR, Fujita K, Morgan KM, Vera E, et al. p53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J Clin Invest (2013) 123(12):5247–57. doi: 10.1172/JCI70355
117. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25(21):9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
118. Anderson AC, Joller N, Kuchroo VK. Lag-3, tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44(5):989–1004. doi: 10.1016/j.immuni.2016.05.001
119. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J Exp Med (2010) 207(10):2175–86. doi: 10.1084/jem.20100637
120. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207(10):2187–94. doi: 10.1084/jem.20100643
121. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood J Am Soc Hematol (2011) 117(17):4501–10. doi: 10.1182/blood-2010-10-310425
122. Ngiow SF, Von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-γ–mediated antitumor immunity and suppresses established tumorsMechanism of anti-TIM3. Cancer Res (2011) 71(10):3540–51. doi: 10.1158/0008-5472.CAN-11-0096
123. Ngiow SF, Von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-γ–mediated antitumor immunity and suppresses established tumors. Cancer Res (2011) 71(10):3540–51. doi: 10.1158/0008-5472.CAN-11-0096
124. Li G, Huang R, Fan W, Wang D, Wu F, Zeng F, et al. Galectin-9/TIM-3 as a key regulator of immune response in gliomas with chromosome 1p/19q codeletion. Front Immunol (2021) 12:800928. doi: 10.3389/fimmu.2021.800928
125. Tang R, Rangachari M, Kuchroo VK. Tim-3: A co-receptor with diverse roles in T cell exhaustion and tolerance. Semin Immunol (2019) 42:101302. doi: 10.1016/j.smim.2019.101302
126. Lee KA, Shin KS, Kim GY, Song YC, Bae EA, Kim IK, et al. Characterization of age-associated exhausted CD8+ T cells defined by increased expression of Tim-3 and PD-1. Aging Cell (2016) 15(2):291–300. doi: 10.1111/acel.12435
127. Goronzy JJ, Fang F, Cavanagh MM, Qi Q, Weyand CM. Naive T cell maintenance and function in human aging. J Immunol (2015) 194(9):4073–80. doi: 10.4049/jimmunol.1500046
128. Roy AM, George S. Emerging resistance vs. losing response to immune check point i nhibitors in renal cell carcinoma: two differing phenomena. Cancer Drug Resist 6(3):642–55. doi: 10.20517/cdr.2023.47
129. Maruhashi T, Sugiura D, Okazaki I-M, Okazaki T. LAG-3: from molecular functions to clinical applications. J Immunother Cancer 8(2):e001014. doi: 10.1136/jitc-2020-001014
130. Ruffo E, Wu RC, Bruno TC, Workman CJ, Vignali DAA. Lymphocyte-activation gene 3 (LAG3): The next immune checkpoint receptor. Semin Immunol (2019) 42:101305. doi: 10.1016/j.smim.2019.101305
131. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med 171(5):1393–405. doi: 10.1084/jem.171.5.1393
133. Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in muri ne self- and tumor-tolerance systems. J Clin Invest 117(11):3383–92. doi: 10.1172/JCI31184
134. Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P, Zeng Y, et al. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer (2018) 9(5-6):176–89. doi: 10.18632/genesandcancer.180
135. He Y, Yu H, Rozeboom L, Rivard CJ, Ellison K, Dziadziuszko R, et al. LAG-3 protein expression in non-small cell lung cancer and its relationship with PD-1/PD-L1 and tumor-infiltrating lymphocytes. J Thorac Oncol (2017) 12(5):814–23. doi: 10.1016/j.jtho.2017.01.019
136. Paik J. Nivolumab plus relatlimab: first approval. Drugs (2022) 82(8):925–31. doi: 10.1007/s40265-022-01723-1
137. Sordo-Bahamonde C, Lorenzo-Herrero S, González-Rodríguez AP, Payer Á R, González-García E, López-Soto A, et al. LAG-3 blockade with relatlimab (BMS-986016) restores anti-leukemic responses in chronic lymphocytic leukemia. Cancers (Basel) (2021) 13(9). doi: 10.3390/cancers13092112
138. Chocarro L, Blanco E, Arasanz H, Fernández-Rubio L, Bocanegra A, Echaide M, et al. Clinical landscape of LAG-3-targeted therapy. Immuno Oncol Technol (2022) 14:100079. doi: 10.1016/j.iotech.2022.100079
139. Cai L, Li Y, Tan J, Xu L, Li Y. Targeting LAG-3, TIM-3, and TIGIT for cancer immunotherapy. J Hematol Oncol (2023) 16(1):101. doi: 10.1186/s13045-023-01499-1
140. Jian Y, Yang K, Sun X, Zhao J, Huang K, Aldanakh A, et al. Current advance of immune evasion mechanisms and emerging immunotherap ies in renal cell carcinoma. Front Immunol 12:639636. doi: 10.3389/fimmu.2021.639636
141. Wang Y, Du J, Gao Z, Sun H, Mei M, Wang Y, et al. Evolving landscape of PD-L2: bring new light to checkpoint immunotherapy. Br J Cancer (2023) 128(7):1196–207. doi: 10.1038/s41416-022-02084-y
142. Huang KC, Chiang SF, Chen TW, Chen WT, Yang PC, Ke TW, et al. Prognostic relevance of programmed cell death 1 ligand 2 (PDCD1LG2/PD-L2) in patients with advanced stage colon carcinoma treated with chemotherapy. Sci Rep (2020) 10(1):22330. doi: 10.1038/s41598-020-79419-3
143. Giannoula Y, Kroemer G, Pietrocola F. Cellular senescence and the host immune system in aging and age-related disorders. BioMed J (2023) 46(3):100581. doi: 10.1016/j.bj.2023.02.001
144. Chaib S, López-Domínguez JA, Lalinde M, Prats N, Marín I, Meyer K, et al. The efficacy of chemotherapy is limited by intratumoural senescent cells that persist through the upregulation of PD-L2. bioRxiv (2022), 2022.11. 04.501681.
145. Nixon BG, Gao S, Wang X, Li MO. TGFβ control of immune responses in cancer: a holistic immuno-oncology perspective. Nat Rev Immunol (2023) 23(6):346–62. doi: 10.1038/s41577-022-00796-z
146. Derynck R, Turley SJ, Akhurst RJ. TGFβ biology in cancer progression and immunotherapy. Nat Rev Clin Oncol (2021) 18(1):9–34. doi: 10.1038/s41571-020-0403-1
147. Huang S, Hölzel M, Knijnenburg T, Schlicker A, Roepman P, McDermott U, et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-β receptor signaling. Cell (2012) 151(5):937–50. doi: 10.1016/j.cell.2012.10.035
148. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer (2009) 45(2):228–47. doi: 10.1016/j.ejca.2008.10.026
149. Gulley JL, Madan RA, Pachynski R, Mulders P, Sheikh NA, Trager J, et al. Role of antigen spread and distinctive characteristics of immunotherapy in cancer treatment. J Natl Cancer Inst (2017) 109(4). doi: 10.1093/jnci/djw261
150. Dodagatta-Marri E, Ma H-Y, Liang B, Li J, Meyer DS, Sun K-H, et al. Integrin αvβ8 on T cells is responsible for suppression of anti-tumor immunity in syngeneic models and is a promising target for tumor immunotherapy. (2020).
151. Seymour L, Bogaerts J, Perrone A, Ford R, Schwartz LH, Mandrekar S, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol (2017) 18(3):e143–52. doi: 10.1016/S1470-2045(17)30074-8
152. Holmgaard RB, Schaer DA, Li Y, Castaneda SP, Murphy MY, Xu X, et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J Immunother Cancer (2018) 6:1–15. doi: 10.1186/s40425-018-0356-4
153. Quéré R, Saint-Paul L, Carmignac V, Martin RZ, Chrétien ML, Largeot A, et al. Tif1γ regulates the TGF-β1 receptor and promotes physiological aging of hematopoietic stem cells. Proc Natl Acad Sci USA. (2014) 111(29):10592–7. doi: 10.1073/pnas.1405546111
154. van der Kraan PM. The changing role of TGFβ in healthy, ageing and osteoarthritic joints. Nat Rev Rheumatol (2017) 13(3):155–63. doi: 10.1038/nrrheum.2016.219
155. Stanilova S, Stanilov N, Julianov A, Manolova I, Miteva L. Transforming growth factor-β1 gene promoter -509C/T polymorphism in association with expression affects colorectal cancer development and depends on gender. PloS One (2018) 13(8):e0201775. doi: 10.1371/journal.pone.0201775
156. Matsuda S, Revandkar A, Dubash TD, Ravi A, Wittner BS, Lin M, et al. TGF-β in the microenvironment induces a physiologically occurring immune-suppressive senescent state. Cell Rep (2023) 42(3):112129. doi: 10.1016/j.celrep.2023.112129
157. Mortezaee K, Majidpoor J. Transforming growth factor-β signalling in tumour resistance to the anti-PD-(L)1 therapy: Updated. J Cell Mol Med (2023) 27(3):311–21. doi: 10.1111/jcmm.17666
158. David CJ, Massagué J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat Rev Mol Cell Biol (2018) 19(7):419–35. doi: 10.1038/s41580-018-0007-0
159. Xiu W, Luo J. CXCL9 secreted by tumor-associated dendritic cells up-regulates PD-L1 expression in bladder cancer cells by activating the CXCR3 signaling. BMC Immunol (2021) 22(1):3. doi: 10.1186/s12865-020-00396-3
160. Ozga AJ, Chow MT, Luster AD. Chemokines and the immune response to cancer. Immunity (2021) 54(5):859–74. doi: 10.1016/j.immuni.2021.01.012
161. Chen IX, Newcomer K, Pauken KE, Juneja VR, Naxerova K, Wu MW, et al. A bilateral tumor model identifies transcriptional programs associated with patient response to immune checkpoint blockade. Proc Natl Acad Sci USA. (2020) 117(38):23684–94. doi: 10.1073/pnas.2002806117
162. House IG, Savas P, Lai J, Chen AXY, Oliver AJ, Teo ZL, et al. Macrophage-derived CXCL9 and CXCL10 are required for antitumor immune responses following immune checkpoint blockade. Clin Cancer Res (2020) 26(2):487–504. doi: 10.1158/1078-0432.CCR-19-1868
163. Seitz S, Dreyer TF, Stange C, Steiger K, Bräuer R, Scheutz L, et al. CXCL9 inhibits tumour growth and drives anti-PD-L1 therapy in ovarian cancer. Br J Cancer (2022) 126(10):1470–80. doi: 10.1038/s41416-022-01763-0
164. Litchfield K, Reading JL, Puttick C, Thakkar K, Abbosh C, Bentham R, et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell (2021) 184(3):596–614.e14. doi: 10.1016/j.cell.2021.01.002
165. Zhang C, Li Z, Xu L, Che X, Wen T, Fan Y, et al. CXCL9/10/11, a regulator of PD-L1 expression in gastric cancer. BMC Cancer (2018) 18(1):462. doi: 10.1186/s12885-018-4384-8
166. Shahabuddin S, Ji R, Wang P, Brailoiu E, Dun N, Yang Y, et al. CXCR3 chemokine receptor-induced chemotaxis in human airway epithelial cells: role of p38 MAPK and PI3K signaling pathways. Am J Physiol Cell Physiol (2006) 291(1):C34–9. doi: 10.1152/ajpcell.00441.2005
167. Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, et al. 12-Chemokine gene signature identifies lymph node-like structures in melanoma: potential for patient selection for immunotherapy? Sci Rep (2012) 2:765. doi: 10.1038/srep00765
168. Stoll G, Pol J, Soumelis V, Zitvogel L, Kroemer G. Impact of chemotactic factors and receptors on the cancer immune infiltrate: a bioinformatics study revealing homogeneity and heterogeneity among patient cohorts. Oncoimmunology (2018) 7(10):e1484980. doi: 10.1080/2162402X.2018.1484980
169. Rossi N, Lee KA, Bermudez MV, Visconti A, Thomas AM, Bolte LA, et al. Circulating inflammatory proteins associate with response to immune checkpoint inhibition therapy in patients with advanced melanoma. eBioMedicine (2022) 83. doi: 10.1016/j.ebiom.2022.104235
170. Peng J, Xiao L-S, Dong Z-Y, Li W-W, Wang K-Y, Wu D-H, et al. Potential predictive value of JAK2 expression for Pan-cancer response to PD-1 blockade immunotherapy. Trans Cancer Res (2018) 7(3):462–71. doi: 10.21037/tcr.2018.04.09
171. Van Roosbroeck K, Ferreiro JF, Tousseyn T, van der Krogt J-A, Michaux L, Pienkowska-Grela B, et al. Genomic alterations of the JAK2 and PDL loci occur in a broad spectrum of lymphoid Malignancies. Genes Chromosomes Cancer (2016) 55(5):428–41. doi: 10.1002/gcc.22345
172. Hundal J, Lopetegui-Lia N, Vredenburgh J. Discovery, significance, and utility of JAK2 mutation in squamous cell carcinoma of the lung. Cureus 14(6):e25913.
173. Lahuerta EJA, Martínez-Sabadell A, Ruiz IR, Alonso MR, Escorihuela M, Luque A, et al. Abstract 1690: JAK2 downmodulation leads to interferon gamma deficient response and resistance to immunotherapy in breast cancer. Cancer Res (2021) 81(13_Supplement):1690–0. doi: 10.1158/1538-7445.AM2021-1690
174. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov (2017) 7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223
175. Phillips WJ, Awan A, Lo B, Redway A, Nicholas G. The role of concurrent amplification of PD-L1, PD-L2 and JAK2 in metastatic lung adenocarcinoma as a biomarker of immune checkpoint inhibitor response: a case report. Precis Cancer Med (2023) 6. doi: 10.21037/pcm-22-39
176. Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell (2020) 37(4):443–55. doi: 10.1016/j.ccell.2020.03.017
177. Yadav R, Hakobyan N, Wang J-C. Role of next generation immune checkpoint inhibitor (ICI) therapy in Philadelphia negative classic myeloproliferative neoplasm (MPN): review of the literature. Int J Mol Sci (2023) 24(15):12502. doi: 10.3390/ijms241512502
178. Gamaleldin MA, Imbaby SAE. The role of tumor necrosis factor receptor superfamily member 4 (TNFRSF4) gene expression in diagnosis and prognosis of acute myeloid leukemia. Mol Biol Rep (2021) 48(10):6831–43. doi: 10.1007/s11033-021-06682-6
179. Gu S, Zi J, Han Q, Song C, Ge Z. Elevated TNFRSF4 gene expression is a predictor of poor prognosis in non-M3 acute myeloid leukemia. Cancer Cell Int (2020) 20(1):146. doi: 10.1186/s12935-020-01213-y
180. Kawamata S, Hori T, Imura A, Takaori-Kondo A, Uchiyama T. Activation of OX40 signal transduction pathways leads to tumor necrosis factor receptor-associated factor (TRAF) 2- and TRAF5-mediated NF-κB activation *. J Biol Chem (1998) 273(10):5808–14. doi: 10.1074/jbc.273.10.5808
181. Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol Rev (2009) 229(1):173–91. doi: 10.1111/j.1600-065X.2009.00766.x
182. Watts TH. TNF/TNFR FAMILY MEMBERS IN COSTIMULATION OF T CELL RESPONSES. Annu Rev Immunol (2005) 23(1):23–68. doi: 10.1146/annurev.immunol.23.021704.115839
183. Kjaergaard J, Tanaka J, Kim JA, Rothchild K, Weinberg A, Shu S. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res (2000) 60(19):5514–21.
184. Weinberg AD, Rivera MM, Prell R, Morris A, Ramstad T, Vetto JT, et al. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J Immunol (2000) 164(4):2160–9. doi: 10.4049/jimmunol.164.4.2160
185. Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood (2005) 105(7):2845–51. doi: 10.1182/blood-2004-07-2959
186. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol (2020) 20(11):651–68. doi: 10.1038/s41577-020-0306-5
187. Li K, Ma L, Sun Y, Li X, Ren H, Tang S-C, et al. The immunotherapy candidate TNFSF4 may help the induction of a promising immunological response in breast carcinomas. Sci Rep (2021) 11(1):18587. doi: 10.1038/s41598-021-98131-4
188. Hinterbrandner M, Rubino V, Stoll C, Forster S, Schnüriger N, Radpour R, et al. Tnfrsf4-expressing regulatory T cells promote immune escape of chronic myeloid leukemia stem cells. JCI Insight (2021) 6(23). doi: 10.1172/jci.insight.151797
189. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol (2015) 5:34. doi: 10.3389/fonc.2015.00034
190. Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria JC, Zitvogel L, Marabelle A. Rationale for anti-OX40 cancer immunotherapy. Eur J Cancer (2016) 52:50–66. doi: 10.1016/j.ejca.2015.08.021
191. Liu Z, Jiang Z, Gao Y, Wang L, Chen C, Wang X. TP53 mutations promote immunogenic activity in breast cancer. J Oncol (2019) 2019:5952836. doi: 10.1155/2019/5952836
192. Williams P, Basu S, Garcia-Manero G, Hourigan CS, Oetjen KA, Cortes JE, et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer (2019) 125(9):1470–81. doi: 10.1002/cncr.31896
193. Ma H, Feng PH, Yu SN, Lu ZH, Yu Q, Chen J. Identification and validation of TNFRSF4 as a high-profile biomarker for prognosis and immunomodulation in endometrial carcinoma. BMC Cancer (2022) 22(1):543. doi: 10.1186/s12885-022-09654-6
194. Zwolak A, Chan SR, Harvilla P, Mahady S, Armstrong AA, Luistro L, et al. A stable, engineered TL1A ligand co-stimulates T cells via specific binding to DR3. Sci Rep (2022) 12(1):20538. doi: 10.1038/s41598-022-24984-y
195. Liu W, Zhan C, Cheng H, Kumar PR, Bonanno JB, Nathenson SG, et al. Mechanistic basis for functional promiscuity in the TNF and TNF receptor superfamilies: structure of the LIGHT:DcR3 assembly. Structure (2014) 22(9):1252–62. doi: 10.1016/j.str.2014.06.013
196. Zhang Z, Li LY. TNFSF15 modulates neovascularization and inflammation. Cancer Microenviron (2012) 5(3):237–47. doi: 10.1007/s12307-012-0117-8
197. Deng W, Gu X, Lu Y, Gu C, Zheng Y, Zhang Z, et al. Down-modulation of TNFSF15 in ovarian cancer by VEGF and MCP-1 is a pre-requisite for tumor neovascularization. Angiogenesis (2012) 15(1):71–85. doi: 10.1007/s10456-011-9244-y
198. Yang G-L, Zhao Z, Qin T-T, Wang D, Chen L, Xiang R, et al. TNFSF15 inhibits VEGF-stimulated vascular hyperpermeability by inducin g VEGFR2 dephosphorylation. FASEB J 31(5):2001–12. doi: 10.1096/fj.201600800R
199. Qin T-T, Xu G-C, Qi J-W, Yang G-L, Zhang K, Liu H-L, et al. Tumour necrosis factor superfamily member 15 (Tnfsf15) facilitates lym phangiogenesis via up-regulation of Vegfr3 gene expression in lymphati c endothelial cells. J Pathol 237(3):307–18. doi: 10.1002/path.4577
200. Yang G-L, Li L-Y. Counterbalance: modulation of VEGF/VEGFR activities by TNFSF15. Signal Transduct Target Ther (2018) 3(1):21. doi: 10.1038/s41392-018-0023-8
201. Fava VM, Sales-Marques C, Alcaïs A, Moraes MO, Schurr E. Age-dependent association of TNFSF15/TNFSF8 variants and leprosy type 1 reaction. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.00155
202. Zhu L, Wang F, Huang J, Wang H, Wang G, Jiang J, et al. Inflammatory aging clock: A cancer clock to characterize the patients’ subtypes and predict the overall survival in glioblastoma. Front Genet (2022) 13. doi: 10.3389/fgene.2022.925469
203. Zhao C-C, Han Q-J, Ying H-Y, Gu X-X, Yang N, Li L-Y, et al. TNFSF15 facilitates differentiation and polarization of macrophages toward M1 phenotype to inhibit tumor growth. OncoImmunology (2022) 11(1):2032918. doi: 10.1080/2162402X.2022.2032918
204. You J, Bian J, Chen J, Xia T, Deng A, Zhang M, et al. TNFSF15 and MIA variant associated with immunotherapy and prognostic evaluation in esophageal cancer. J Oncol (2023) 2023:1248024. doi: 10.1155/2023/1248024
205. Ma B, Cao L, Li Y. A novel 10-gene immune-related lncRNA signature model for the prognosis of colorectal cancer. Math Biosci Eng (2021) 18(6):9743–60. doi: 10.3934/mbe.2021477
206. Huang Y, Zhang W, Li Q, Wang Z, Yang X. Identification of m6A/m5C/m1A-associated LncRNAs for prognostic assessment and immunotherapy in pancreatic cancer. Sci Rep (2023) 13(1):3661. doi: 10.1038/s41598-023-30865-9
207. Liao Y, Guo S, Chen Y, Cao D, Xu H, Yang C, et al. VSIG4 expression on macrophages facilitates lung cancer development. Lab Invest (2014) 94(7):706–15. doi: 10.1038/labinvest.2014.73
208. Vogt L, Schmitz N, Kurrer MO, Bauer M, Hinton HI, Behnke S, et al. VSIG4, a B7 family-related protein, is a negative regulator of T cell activation. J Clin Invest (2006) 116(10):2817–26. doi: 10.1172/JCI25673
209. Helmy KY, Katschke KJ Jr, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell (2006) 124(5):915–27. doi: 10.1016/j.cell.2005.12.039
210. Guo S, Yang C, Mei F, Wu S, Luo N, Fei L, et al. Down-regulation of Z39Ig on macrophages by IFN-gamma in patients with chronic HBV infection. Clin Immunol (2010) 136(2):282–91. doi: 10.1016/j.clim.2010.03.007
211. Hall BM, Gleiberman AS, Strom E, Krasnov PA, Frescas D, Vujcic S, et al. Immune checkpoint protein VSIG4 as a biomarker of aging in murine adipose tissue. Aging Cell (2020) 19(10):e13219. doi: 10.1111/acel.13219
212. Sazinsky S, Nguyen P, Zafari M, Phennicie R, Wahle J, Komoroski V, et al. 886 Targeting VSIG4, a novel macrophage checkpoint, repolarizes suppressive macrophages which induces an inflammatory response in primary cell in vitro assays and fresh human tumor cultures. J ImmunoTher Cancer (2021) 9(Suppl 2):A928–8. doi: 10.1136/jitc-2021-SITC2021.886
213. Liu MA, Shahabi S, Jati S, Tang K, Gao H, Jin Z, et al. Gut microbial DNA and immune checkpoint gene Vsig4/CRIg are key antagonistic players in healthy aging and age-associated development of hypertension and diabetes. Front Endocrinol (Lausanne) (2022) 13:1037465. doi: 10.3389/fendo.2022.1037465
214. Xu T, Jiang Y, Yan Y, Wang H, Lu C, Xu H, et al. VSIG4 is highly expressed and correlated with poor prognosis of high-grade glioma patients. Am J Transl Res (2015) 7(6):1172–80.
215. Jiang Y, Han L, Yang J, Yang M, Zhang J, Xue M, et al. Identification of a novel immune checkpoint molecule V-set immunoglobulin domain-containing 4 that leads to impaired immunity infiltration in pancreatic ductal adenocarcinoma. Cancer Immunol Immunother (2023) 72(8):2701–16. doi: 10.1007/s00262-023-03438-y
216. Liu B, Cheng L, Gao H, Zhang J, Dong Y, Gao W, et al. The biology of VSIG4: Implications for the treatment of immune-mediated inflammatory diseases and cancer. Cancer Lett (2023) 553:215996. doi: 10.1016/j.canlet.2022.215996
217. Kim CM, Lee JB, Shin SJ, Ahn JB, Lee M, Kim HS. The efficacy of immune checkpoint inhibitors in elderly patients: a meta-analysis and meta-regression. ESMO Open (2022) 7(5):100577. doi: 10.1016/j.esmoop.2022.100577
218. Stoff R, Grynberg S, Asher N, Laks S, Steinberg Y, Schachter J, et al. Efficacy and toxicity of Ipilimumab-Nivolumab combination therapy in elderly metastatic melanoma patients. Front Oncol (2022) 12:1020058. doi: 10.3389/fonc.2022.1020058
219. Oh E, Kim JH, Um J, Jung DW, Williams DR, Lee H. Genome-wide transcriptomic analysis of non-tumorigenic tissues reveals aging-related prognostic markers and drug targets in renal cell carcinoma. Cancers (Basel) (2021) 13(12). doi: 10.3390/cancers13123045
220. Xiao G, Zhang X, Zhang X, Chen Y, Xia Z, Cao H, et al. Aging-related genes are potential prognostic biomarkers for patients with gliomas. Aging (Albany NY) (2021) 13(9):13239–63. doi: 10.18632/aging.203008
221. Chen X, Wang L, Hong L, Su Z, Zhong X, Zhou H, et al. Identification of aging-related genes associated with clinical and prognostic features of hepatocellular carcinoma. Front Genet (2021) 12:661988. doi: 10.3389/fgene.2021.661988
222. Yang J, Jiang Q, Liu L, Peng H, Wang Y, Li S, et al. Identification of prognostic aging-related genes associated with immunosuppression and inflammation in head and neck squamous cell carcinoma. Aging (Albany NY) (2020) 12(24):25778–804. doi: 10.18632/aging.104199
223. Dai W, Xu X, Wang D, Wu J, Wang J. Cancer therapy with a CRISPR-assisted telomerase-activating gene expression system. Oncogene (2019) 38(21):4110–24. doi: 10.1038/s41388-019-0707-8
224. Vaiserman A, De Falco E, Koliada A, Maslova O, Balistreri CR. Anti-ageing gene therapy: Not so far away? Ageing Res Rev (2019) 56:100977. doi: 10.1016/j.arr.2019.100977
225. Wang B, Kohli J, Demaria M. Senescent cells in cancer therapy: friends or foes? Trends Cancer 6(10):838–57. doi: 10.1016/j.trecan.2020.05.004
226. Wyld L, Bellantuono I, Tchkonia T, Morgan J, Turner O, Foss F, et al. Senescence and cancer: A review of clinical implications of senescence and senotherapies. Cancers (Basel) (2020) 12(8). doi: 10.3390/cancers12082134
227. Guo J, Huang X, Dou L, Yan M, Shen T, Tang W, et al. Aging and aging-related diseases: from molecular mechanisms to interventions and treatments. Signal Transduct Target Ther (2022) 7(1):391. doi: 10.1038/s41392-022-01251-0
228. Wang L, Lankhorst L, Bernards R. Exploiting senescence for the treatment of cancer. Nat Rev Cancer (2022) 22(6):340–55. doi: 10.1038/s41568-022-00450-9
229. Prasanna PG, Citrin DE, Hildesheim J, Ahmed MM, Venkatachalam S, Riscuta G, et al. Therapy-induced senescence: opportunities to improve anticancer therap y. J Natl Cancer Institute 113(10):1285–98. doi: 10.1093/jnci/djab064
230. Bury M, Le Calvé B, Ferbeyre G, Blank V, Lessard F. New Insights into CDK Regulators: Novel Opportunities for Cancer Therapy. Trends Cell Biol 31(5):331–44. doi: 10.1016/j.tcb.2021.01.010
231. Mengual Gomez DL, Armando RG, Cerrudo CS, Ghiringhelli PD, Gomez DE. Telomerase as a cancer target. Development of new molecules. Curr Topics Med Chem 16(22):2432–40. doi: 10.2174/1568026616666160212122425
232. Ramaiah MJ, Tangutur AD, Manyam RR. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci (2021) 277:119504. doi: 10.1016/j.lfs.2021.119504
233. Zhu H, Blake S, Kusuma FK, Pearson RB, Kang J, Chan KT. Oncogene-induced senescence: From biology to therapy. Mech Ageing Dev (2020) 187:111229. doi: 10.1016/j.mad.2020.111229
234. Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer (2009) 9(5):338–50. doi: 10.1038/nrc2607
235. Cattrini C, Capaia M, Boccardo F, Barboro P. Etoposide and topoisomerase II inhibition for aggressive prostate cancer: Data from a translational study. Cancer Treat Res Commun (2020) 25:100221. doi: 10.1016/j.ctarc.2020.100221
236. Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer (2006) 6(10):789–802. doi: 10.1038/nrc1977
237. Skok Ž, Zidar N, Kikelj D, Ilaš J. Dual inhibitors of human DNA topoisomerase II and other cancer-related targets. J Med Chem 63(3):884–904. doi: 10.1021/acs.jmedchem.9b00726
238. Xiang M, Colevas AD, Holsinger FC, Le Q-TX, Beadle BM. Survival after definitive chemoradiotherapy with concurrent cisplatin or carboplatin for head and neck cancer. J Natl Compr Canc Netw (2019) 17(9):1065–73. doi: 10.6004/jnccn.2019.7297
239. Klein LE, Freeze BS, Smith Iii AB, Horwitz SB. The Microtubule Stabilizing Agent Discodermolide is a Potent Inducer o f Accelerated Cell Senescence. Cell Cycle 4(3):501–7. doi: 10.4161/cc.4.3.1550
240. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol (2016) 13(7):417–30. doi: 10.1038/nrclinonc.2016.26
241. Kumarasamy V, Vail P, Nambiar R, Witkiewicz AK, Knudsen ES. Functional determinants of cell cycle plasticity and sensitivity to CDK4/6 inhibition. Cancer Res (2021) 81(5):1347–60. doi: 10.1158/0008-5472.CAN-20-2275
242. Sava GP, Fan H, Coombes RC, Buluwela L, Ali S. CDK7 inhibitors as anticancer drugs. Cancer Metastasis Rev (2020) 39(3):805–23. doi: 10.1007/s10555-020-09885-8
243. Liu H, Liu K, Dong Z. Targeting CDK12 for cancer therapy: function, mechanism, and drug discovery. Cancer Res (2021) 81(1):18–26. doi: 10.1158/0008-5472.CAN-20-2245
244. Pascolo E, Wenz C, Lingner J, Hauel N, Priepke H, Kauffmann I, et al. Mechanism of human telomerase inhibition by BIBR1532, a synthetic, non -nucleosidic drug candidate. J Biol Chem 277(18):15566–72. doi: 10.1074/jbc.M201266200
245. Phatak P, Burger AM. Telomerase and its potential for therapeutic intervention. Br J Pharmacol (2007) 152(7):1003–11. doi: 10.1038/sj.bjp.0707374
246. Burchett KM, Yan Y, Ouellette MM. Telomerase inhibitor Imetelstat (GRN163L) limits the lifespan of human pancreatic cancer cells. PloS One 9(1):e85155. doi: 10.1371/journal.pone.0085155
247. Dhillon S. Decitabine/cedazuridine: first approval. Drugs (2020) 80(13):1373–8. doi: 10.1007/s40265-020-01389-7
248. Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence is both functional/adaptive and dysfunctional/maladaptive. Semin Immunopathol (2020) 42(5):521–36. doi: 10.1007/s00281-020-00818-9
249. Palacios-Pedrero MÁ, Jansen JM, Blume C, Stanislawski N, Jonczyk R, Molle A, et al. Signs of immunosenescence correlate with poor outcome of mRNA COVID-19 vaccination in older adults. Nat Aging (2022) 2(10):896–905. doi: 10.1038/s43587-022-00292-y
250. Bartleson JM, Radenkovic D, Covarrubias AJ, Furman D, Winer DA, Verdin E, et al. SARS-CoV-2, COVID-19 and the aging immune system. Nat Aging (2021) 1(9):769–82. doi: 10.1038/s43587-021-00114-7
251. Britanova OV, Putintseva EV, Shugay M, Merzlyak EM, Turchaninova MA, Staroverov DB, et al. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J Immunol (2014) 192(6):2689–98. doi: 10.4049/jimmunol.1302064
252. Solana R, Mariani E. NK and NK/T cells in human senescence. Vaccine (2000) 18(16):1613–20. doi: 10.1016/S0264-410X(99)00495-8
253. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
254. Rodriguez IJ, Lalinde Ruiz N, Llano León M, Martínez Enríquez L, Montilla Velásquez MDP, Ortiz Aguirre JP, et al. Immunosenescence study of T cells: A systematic review. Front Immunol (2020) 11:604591. doi: 10.3389/fimmu.2020.604591
255. Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol (2017) 8:1960. doi: 10.3389/fimmu.2017.01960
256. Kugel CH 3rd, Douglass SM, Webster MR, Kaur A, Liu Q, Yin X, et al. Age correlates with response to anti-PD1, reflecting age-related differences in intratumoral effector and regulatory T-cell populations. Clin Cancer Res (2018) 24(21):5347–56. doi: 10.1158/1078-0432.CCR-18-1116
257. Elias R, Giobbie-Hurder A, McCleary NJ, Ott P, Hodi FS, Rahma O. Efficacy of PD-1 & PD-L1 inhibitors in older adults: a meta-analysis. J Immunother Cancer (2018) 6(1):26. doi: 10.1186/s40425-018-0336-8
258. Wong SK, Nebhan CA, Johnson DB. Impact of patient age on clinical efficacy and toxicity of checkpoint inhibitor therapy. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.786046
259. Yan X, Tian X, Wu Z, Han W. Impact of age on the efficacy of immune checkpoint inhibitor-based combination therapy for non-small-cell lung cancer: A systematic review and meta-analysis. Front Oncol (2020) 10. doi: 10.3389/fonc.2020.01671
260. O’Rourke K. Study shows that patients older than 80 years benefit from immunotherapy. Cancer (2022) 128(6):1155–6. doi: 10.1002/cncr.34138
261. Samani A, Zhang S, Spiers L, Suwaidan AA, Merrick S, Tippu Z, et al. Impact of age on the toxicity of immune checkpoint inhibition. J ImmunoTher Cancer (2020) 8(2):e000871. doi: 10.1136/jitc-2020-000871
262. Kanesvaran R, Cordoba R, Maggiore R. Immunotherapy in older adults with advanced cancers: implications for clinical decision-making and future research. Am Soc Clin Oncol Educ Book (2018) 38):400–14. doi: 10.1200/EDBK_201435
263. Wu Q, Wang Q, Tang X, Xu R, Zhang L, Chen X, et al. Correlation between patients' age and cancer immunotherapy efficacy. Oncoimmunology (2019) 8(4):e1568810. doi: 10.1080/2162402x.2019.1568810
264. Daste A, Domblides C, Gross-Goupil M, Chakiba C, Quivy A, Cochin V, et al. Immune checkpoint inhibitors and elderly people: A review. Eur J Cancer (2017) 82:155–66. doi: 10.1016/j.ejca.2017.05.044
Keywords: aging, immunosenescence, neoplasm, immune biomarkers, immune checkpoint inhibitors
Citation: Al-Danakh A, Safi M, Jian Y, Yang L, Zhu X, Chen Q, Yang K, Wang S, Zhang J and Yang D (2024) Aging-related biomarker discovery in the era of immune checkpoint inhibitors for cancer patients. Front. Immunol. 15:1348189. doi: 10.3389/fimmu.2024.1348189
Received: 02 December 2023; Accepted: 29 January 2024;
Published: 15 March 2024.
Edited by:
Mohd Wajid Ali Khan, University of Hail, Saudi ArabiaReviewed by:
Subhash Kumar Tripathi, Seattle Children’s Research Institute, United StatesSaravanan Rajendrasozhan, University of Hail, Saudi Arabia
Copyright © 2024 Al-Danakh, Safi, Jian, Yang, Zhu, Chen, Yang, Wang, Zhang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deyong Yang, eWFuZ2RleW9uZ0BkbXUuZWR1LmNu; Jianjun Zhang, anpoYW5nMjBAbWRhbmRlcnNvbi5vcmc=; Shujing Wang, d2FuZ3NodWppbmdAZG11LmVkdS5jbg==
†These authors have contributed equally to this work