 Mohammad Haque1†Lauren Kaminsky1Raz Abdulqadir1Jessica Engers1Evgeny Kovtunov1Manmeet Rawat1Rana Al-Sadi2*†‡Thomas Y. Ma1‡
Mohammad Haque1†Lauren Kaminsky1Raz Abdulqadir1Jessica Engers1Evgeny Kovtunov1Manmeet Rawat1Rana Al-Sadi2*†‡Thomas Y. Ma1‡- 1Department of Medicine, Penn State Milton S. Hershey Medical Center, Hershey, PA, United States
- 2Department of Internal Medicine, University of Nebraska Medical Center, Omaha, NE, United States
Background: Defective intestinal epithelial tight junction (TJ), characterized by an increase in intestinal TJ permeability, has been shown to play a critical role in the pathogenesis of inflammatory bowel disease (IBD). Tumor necrosis factor-α (TNF-α) is a key pro-inflammatory cytokine involved in the immunopathology of IBD and has been shown to cause an increase in intestinal epithelial TJ permeability. Although TNF-α antibodies and other biologics have been advanced for use in IBD treatment, these therapies are associated with severe side effects and have limited efficacy, and there is an urgent need for therapies with benign profiles and high therapeutic efficacy. Probiotic bacteria have beneficial effects and are generally safe and represent an important class of potential therapeutic agents in IBD. Lactobacillus acidophilus (LA) is one of the most used probiotics for wide-ranging health benefits, including in gastrointestinal, metabolic, and inflammatory disorders. A specific strain of LA, LA1, was recently demonstrated to have protective and therapeutic effects on the intestinal epithelial TJ barrier. However, the mechanisms of actions of LA1 remain largely unknown.
Methods: The primary aim of this study was to investigate microbial-epithelial interactions and novel signaling pathways that regulate the effect of LA1 on TNF-α-induced increase in intestinal epithelial TJ permeability, using cell culture and animal model systems.
Results and Conclusion: Pre-treatment of filter-grown Caco-2 monolayers with LA1 prevented the TNF-α-induced increase in intestinal epithelial TJ permeability by inhibiting TNF-α-induced activation of NF-κB p50/p65 and myosin light chain kinase (MLCK) gene and kinase activity in a TLR-2-dependent manner. LA1 produced a TLR-2- and MyD88-dependent activation of NF-κB p50/p65 in immune cells; however, LA1, in intestinal cells, inhibited the NF-κB p50/p65 activation in a TLR-2-dependent but MyD88-independent manner. In addition, LA1 inhibition of NF-κB p50/p65 and MLCK gene was mediated by TLR-2 pathway activation of phosphatidylinositol 3-kinase (PI3K) and IKK-α phosphorylation. Our results demonstrated novel intracellular signaling pathways by which LA1/TLR-2 suppresses the TNF-α pathway activation of NF-κB p50/p65 in intestinal epithelial cells and protects against the TNF-α-induced increase in intestinal epithelial TJ permeability.
1 Introduction
Inflammatory bowel disease (IBD) includes Crohn’s disease (CD) and ulcerative colitis (UC) and is characterized by recurrent intestinal inflammation primarily affecting the gastrointestinal tract (1–4). IBD is triggered in part by an inappropriate immune response to gut microbial antigens (5–7). A single cell layer of columnar intestinal epithelial cells connected by tight junctions (TJs) coats the entire intestinal mucosal surface and serves as a protective barrier against the mucosal penetration of luminal contents (2, 8, 9). TJs are located at the apical-most points on the basolateral membrane and form a physical and functional barrier against the paracellular permeation of noxious substances present in the intestinal lumen (including bacteria, bacterial toxins, bacterial by-products, digestive enzymes, and degraded food products) (10–12). The disruption of the intestinal TJ barrier allows paracellular permeation of bacterial antigens and bacterial by-products into the intestinal tissue, leading to the immune activation (1, 13). Patients with IBD have a defective intestinal TJ barrier manifested by an increase in intestinal permeability (14–16). Clinical studies have shown that the phenomenon of “leaky gut” correlates with poor clinical outcomes, whereas therapeutic re-tightening of the intestinal TJ barrier is associated with more rapid clinical improvement and prolonged clinical remission (17–19).
During the active phase of IBD, pro-inflammatory cytokines are released into the gut mucosal compartment, including tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, and interferon (IFN)-γ (20–26). TNF-α plays a central role in the pathogenesis of intestinal inflammation in patients with IBD and, at clinically relevant concentrations, TNF-α causes a marked increase in intestinal permeability (27–34). The inhibition of the TNF-α-induced increase in intestinal permeability has been shown to prevent intestinal inflammation and promote healing in animal models of IBD (23, 35–39). Anti-TNF-α antibody therapy is widely used to treat intestinal inflammation associated with IBD and to maintain clinical remission (40–43). However, these therapies are associated with severe side effects and frequent clinical relapses, and thus, there remains an urgent need for alternative treatment modalities with minimum side effects and high efficacy (44–47). Probiotic bacteria are health-promoting commensal bacteria naturally occurring in the gut with excellent safety profiles and represent an important potential therapeutic option (48–52). The beneficial actions of probiotics may be exerted by up-regulation or maintenance of intestinal epithelial TJ barrier, mucin secretion, modification of gut microbiota composition and activity, protection against oxidative stress and the capability to decrease the risk of accumulation of reaction oxygen metabolites, and reduction of immune response and subsequently reduction of inflammation (53–58). Despite the potential promise, essential limitations related to the use of probiotics exist due to the lack of detailed preclinical and mechanistic studies and precise identification of the specific probiotic strains that have anti-inflammatory biological actions (52). The beneficial effects of gut microbiota are bacterial strain and host-specific (59). Although there is an important recognition that some commensal bacteria have a beneficial role in gut homeostasis by preserving or promoting the intestinal barrier function, it remains unclear as to which probiotic species and strains produce a persistent, predictable enhancement in TJ barrier and which could be advanced to treat intestinal inflammation by targeting the TJ barrier. For example, published studies have indicated that Lactobacillus acidophilus, L. casei, L. plantarum, or L. rhamnosus cause a modest enhancement in intestinal epithelial TJ barrier, while others have been shown to have minimal, disruptive, or no effect (60–70). A recent study from our laboratory reported that a particular strain of probiotic bacteria Lactobacillus acidophilus (LA), referred to as LA1, caused a unique, marked enhancement of the intestinal epithelial TJ barrier and prevented the development of dextran sulfate sodium (DSS)-induced colitis by preserving the intestinal epithelial TJ barrier (71). In contrast, LA strain LA3 did not affect the intestinal TJ barrier function or protect against the DSS-induced colitis (71). Previous studies have shown that the TNF-α-induced increase in intestinal epithelial TJ permeability is mediated by NF-κB p50/p65 activation of myosin light chain kinase (MLCK) gene and kinase activity in both cell culture and animal model systems (27–30, 32, 33, 72). The major aim of this study was to investigate the strain-specific TJ barrier protective effects of LA1 against the TNF-α-induced increase in intestinal epithelial TJ permeability and to delineate the intracellular mechanisms involved using in vitro intestinal epithelial cell and live animal model systems.
Previous studies have shown that the stimulation of the Toll-like receptor-2 (TLR-2) signaling pathway leads to the MyD88-dependent NF-κB p50/p65 activation in immune cells and other cell types in response to bacterial infections (73–76). In the studies described herein, we provide potentially paradigm-shifting finding that in intestinal epithelial cells, and unlike in immune cells, the probiotic bacterial strain LA1 protects the intestinal epithelial TJ barrier by inhibiting NF-κB p50/p65 activation in a TLR-2-dependent and MyD88-independent intracellular signaling process. Our results suggest that the probiotic bacteria in the intestinal mucosal surface may suppress enterocyte NF-κB activation and preserve the intestinal epithelial TJ barrier function by accessing the TLR-2 complex on the enterocyte apical membrane surface.
2 Materials and methods
2.1 Reagents
Cell culture media [Dulbecco’s Modified Eagle Medium (DMEM)], trypsin, fetal bovine serum (FBS), glutamine, penicillin, streptomycin, and phosphate-buffered saline (PBS) were purchased from GIBCO-BRL (Grand Island, NY). Polyinosinic-polycytidylic acid [poly(I:C)] was purchased from Invivogen (Sand Diego, CA). Peptidoglycan from Bacillus subtilis, Anti-NIK, p-IKK-α, p-IKK-β, p-IκB-α, pMLC, p-NF-κBp65, MLCK, pPI3K were obtained from Sigma-Aldrich (St. Louis, MO). Anti-β-actin-HRP tagged antibody was obtained from Santa Cruz Biotechnology (Dallas, TX). Anti-TLR-2 antibodies were purchased from Abcam (Cambridge, MA). Horseradish peroxidase-conjugated secondary antibodies for Western blot analysis were purchased from Invitrogen (San Francisco, CA). Human and mouse recombinant TNF-α were purchased from R&D systems (Minneapolis, MN).
2.2 Determination of Caco-2 epithelial monolayer resistance and paracellular permeability
Caco-2 cells (passage 18) were purchased from the American Type Culture Collection (ATCC) and maintained at 37°C in a culture medium as previously described (27, 71). For filter growth, Caco-2 cells were plated on Transwell plates for 3-4 weeks until the transepithelial resistance (TER) reached 400-500 Ω*cm2. Caco-2 paracellular permeability was assessed by measuring the luminal-to-serosal flux rate of a paracellular probe, fluorescein isothiocyanate-labeled FITC dextran 10 kDa (mol wt.: 10,000 g/mol). For determination of mucosal-to-serosal flux rates, a known concentration (25 μg/ml) of FITC dextran 10 kDa was added to the apical solution at the beginning of each experiment. After each experimental period, the solution from the basolateral chamber was collected and measured in a fluorescence microplate reader SpectraMax iD3 (Molecular Devices, San Jose, CA).
2.3 Preparation of bacterial culture and cell-free culture supernatant
The Lactobacillus strains were purchased from ATCC. With shaking, these bacteria were grown overnight in MRS broth (Difco, Detroit, MI) at 37°C. Live bacteria were spun down by centrifuging at 12,000 × g for 10 min, and pellets were stored at −80°C for further experiments. For treating Caco-2 monolayers, the bacterial pellet was suspended in DMEM and diluted to OD600nm = 0.135 (1x108 colony-forming units (CFU)) in the same media and applied to the apical surface of cell monolayers (71).
2.4 Determination of mouse small intestinal permeability in vivo
The Penn State University Animal Care and Use Committee approved the animal studies. Wild-type (WT) mice (C57BL/6 background), TLR-2 deficient mice (TLR-2-/-), and MyD88 deficient mice (MyD88-/-) were obtained from The Jackson Laboratory (Bar Harbor, Maine). Mice were treated with 5 μg of TNF-α by intraperitoneal injection (i.p.) (27, 28) and the LA effect on intestinal permeability was determined in vivo using a re-cycling intestinal perfusion method (77, 78). For in vivo studies, 1 × 109 CFU of LA in 200 μl PBS was administered 24 hr before TNF-α treatment by oral-gastric gavage, and mouse intestinal permeability was measured after 24 hr of TNF-α treatment. Mice were anesthetized, and a 6-10 cm segment of mouse small intestine was isolated and cannulated with a small diameter plastic tube and continuously perfused with 5 ml Krebs-phosphate saline buffer for a 2 hr perfusion period. An external recirculating pump was used to recirculate the perfusate at a constant flow rate (0.75 ml/min). The intestinal permeability was assessed by measuring the luminal-to-serosal flux rate of the paracellular probe Texas Red-labeled dextran 10kDa (MW = 10,000 g/mol).
2.5 Western blot analysis
Caco-2 monolayers were treated with 10 ng/ml TNF-α for various time periods. At the end of the experimental period, Caco-2 monolayers were immediately rinsed with ice-cold PBS, and cells were lysed with RIPA lysis buffer (Sigma-Aldrich, St. Louis, MO). Laemmli gel loading buffer was added to the lysate that contained 20 to 50 µg of protein and boiled for 7 minutes, after which time proteins were separated on SDS-PAGE gel. Proteins from the gel were transferred to a membrane (Trans-Blot Transfer Medium, Nitrocellulose Membrane; Bio-Rad Laboratories) overnight. The membrane was incubated for 2 hours in a blocking solution (5% dry milk in tris-buffered saline/Tween 20 buffer). The membrane was incubated with proper primary antibodies in a blocking solution. After being washed in tris-buffered saline/1% Tween buffer, the membrane was incubated in appropriate secondary antibodies and developed with the Biorad ChemiDoc Imaging System (BioRad, Hercules, California).
2.6 Immunofluorescence study
Intestinal tissues were collected from the mice at the indicated time points. Samples were collected in embedding cassettes and blocked with 10% neutral buffered formalin. Samples were infiltrated with wax, and infiltrated tissues were embedded into wax blocks. For immunostaining, sections were de-parafinized with 3 washes of xylene. Caco-2 monolayers were fixed on filters with ice-cold methanol. Antigen unmasking was done according to manufacturer protocol, and samples were incubated for 2 hr with anti-NF-κB p65 antibody (Santa Cruz) or anti-pMLC antibody (Cell Signaling Technology, Danvers, MA), Alexa conjugated, or Cy3 secondary antibody (Cell Signaling Technology; Danvers, MA) was used to detect the primary antibodies. All the primary and secondary antibodies were used at the concentrations suggested by the manufacturers. ProLong Gold antifade reagent (Invitrogen), containing DAPI as a nuclear stain, was used to mount the samples on glass slides. Samples were imaged using the C2+ confocal microscope system (Nikon) and ZEISS Axio Imager.M2 Microscope with images processed with ZEN 3.2 (Blue edition) software.
2.7 Immunostaining of splenocytes
Spleens were harvested from wild-type (WT), TLR-2-/-, and MyD88-/- mice. Splenocytes were processed into single-cell suspensions and plated on coverslips in 6-well plates. Following adherence to coverslips, splenocytes were treated with LA or TNF-α for 30 minutes, then fixed in methanol for 20 minutes at -20°C. Cells were permeabilized with 0.1% Triton X-100 in PBS for 10 minutes and blocked in 10% NDS for 1 hr. Staining with anti-NF-κB p65 antibody (1:300; Santa Cruz Biotechnology; Dallas, TX) in 1% NDS for 1 hour was followed by incubation with secondary antibody, goat anti-mouse IgG-FITC (1:1000; Invitrogen; Waltham, MA) in 1% NDS for 1 hr. ProLong Gold Antifade reagent was used to mount coverslips on slides. Sections were imaged using the C2+ confocal microscope system (Nikon) or with Nikon Fluorescent microscope with images minimally processed using NIS Elements software and were processed with ZEN 3.2 (Blue edition) software.
2.8 RNA isolation and reverse transcription
Caco-2 cells (5 × 105/filter) were seeded into 6-well transwell permeable inserts and grown to confluence. Filter-grown Caco-2 cells were treated with proper experimental reagents for desired periods. At the end of the experimental period, cells were washed twice with ice-cold PBS. Total RNA was isolated using Qiagen RNeasy Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. Total RNA concentration was calculated at 260 nm by NanoDrop One Spectrophotometer (Thermofisher Scientific, Waltham, MA). The reverse transcription (RT) was carried out using the GeneAmp Gold RNA PCR core kit (Applied Biosystems, Foster City, CA). Two micrograms of total RNA from each sample were reverse transcribed into cDNA in a 40-μl reaction containing 1× RT-PCR buffer, 2.5 mM MgCl2, 250 μM of each dNTP, 20 U RNase inhibitor, 10 mM DL-Dithiothreitol (DTT), 1.25 μM random hexamer and 30 U multiscribe RT. The RT reactions were performed in a thermocycler (PTC-100, MJ Research, Waltham, MA, USA) at 25°C for 10 min., 42°C for 30 min., and 95°C for 5 min.
2.9 Quantification of gene expression using real-time PCR
The real-time PCRs were carried out using Taqman universal PCR master mix kit (Applied Biosystems, Branchburg, NJ, USA) as previously described (9). Each real-time PCR reaction contained 10 μl RT reaction mix, 25 μl 2× Taqman universal PCR master mix, 0.2 μM probe and 0.6 μM primers. Primer and probe design for the real-time PCR was made with Primer Express version 2 from Applied Biosystems. [The MLCK specific primer pairs consisted of 5′-AGGAAGGCAGCATTGAGGTTT-3′[forward], 5′-GCTTTCAGCAGGCAGAGGTAA-3′[reverse]; probe specific for MLCK consisted of FAM 5′-TGAAGATGCTGGCTCC-3′ TAMRA; the internal control glyceraldehyde 3-phosphate dehydrogenase (GAPDH)-specific primer pairs consisted of 5′-CCACCCATGGCAAATTCC-3′[forward], 5′-TGGGATTTCCATTGATGACCAG-3′[reverse]; probe specific for GAPDH consisted of JOE 5′-TGGCACCGTCAAGGCTGAGAACG-3′ TAMRA].
2.10 Cloning of the full-length MLCK promoter region and luciferase assay
The full-length (FL) MLCK promoter region (2091-bp) (accession# DQ090939) was cloned using Genome Walker system (Clontech, Mountain View, CA, USA) (9). FL MLCK promoter reporter plasmids were transfected using the pGL-3 basic luciferase reporter vector. The primers used for cloning the FL MLCK promoter construct are 5’GCCGGTACCGAGAAGCAGGAGAGTATTAAATG3’. DNA construct of MLCK promoters was transiently transfected into Caco-2 cells using transfection reagent lipofectamine 2000 (Life Technologies). Renilla luciferase vector (pRL-TK, Promega) was cotransfected with the plasmid construct as an internal control, as previously described (9, 33). After specific treatment, Caco-2 cells were washed twice with 1 ml ice-cold PBS, followed by the addition of 400 μl 1× passive lysis buffer, incubated at room temperature for 15 min, scraped and transferred into an Eppendorf tube, and centrifuged for 15 s at 13,000 rpm in a microcentrifuge. Luciferase activity was determined using the dual luciferase assay kit (Promega). Twenty microliters of the supernatant were used for each assay. Luciferase values were determined, and the value of reporter luciferase activities was then divided by that of Renilla luciferase activities to normalize for differences in transfection efficiencies. The average activity value of the control samples was set to 1.0. The luciferase activity of the MLCK promoter in treated samples was determined relative to the control samples.
2.11 Statistical analysis
The statistical significance of differences between mean values was assessed using students’ t-tests for unpaired data and analysis of variance analysis whenever needed. All reported significance levels represent two-tailed P values. P < 0.05 was used to show statistical significance. All in vitro experiments that used Caco-2 monolayers, including assessment of the TJ barrier function and biochemical and molecular studies, were performed in triplicates or quadruplicates and were repeated at least three times for reproducibility. The immunoblot analysis and cell imaging studies were repeated three to four times. The densitometry analysis was performed using ImageJ 1.54g. The animal studies were performed individually, and each experimental group consisted of three to six animals.
3 Results
3.1 Lactobacillus acidophilus strain LA1 inhibits the TNF-α-induced increase in Caco-2 TJ permeability
The effect of Lactobacillus acidophilus (LA1 or LA3) on TNF-α-induced increase in intestinal epithelial TJ permeability was examined in filter-grown Caco-2 monolayers. TNF-α (10 ng/ml) addition to the basolateral compartment produced a drop in Caco-2 TER (~25% drop) and an increase (~5-fold increase) in trans-epithelial flux of paracellular marker, FITC-labeled dextran 10 kDa, in filter-grown Caco-2 monolayers over the 24-hr experimental period (Figures 1A, B). As previously reported (71), LA1 (1x108 CFU/ml) addition to the apical membrane resulted in an enhancement in Caco-2 TJ barrier function with an 80% increase in Caco-2 TER and a corresponding decrease in luminal-to-serosal dextran flux (45% decrease) while LA3 had no effect (data not shown). Based on previous studies that found the beneficial concentrations of probiotic bacterial species to range from 1 × 107 to 1 × 109 CFU/ml, bacterial concentration of 1 × 108 CFU/ml were used in Caco-2 cells in the current studies (71, 79, 80). We also performed a dose-dependent effect of LA in Caco-2 monolayers to verify the most beneficial dose of LA on the intestinal TJ barrier. We found that a 1x108 CFU/ml bacterial concentration exerted the maximal effect on Caco-2 TJ barrier (data not shown). The LA1 addition inhibited the TNF-α-induced decrease in Caco-2 TER and increase in dextran flux (Figures 1A, B). In contrast, LA3 did not inhibit the TNF-α effect on Caco-2 TER or dextran flux (Figures 1C, D), indicating that LA1 causes a strain-specific inhibition of TNF-α-induced increase in Caco-2 TJ permeability.
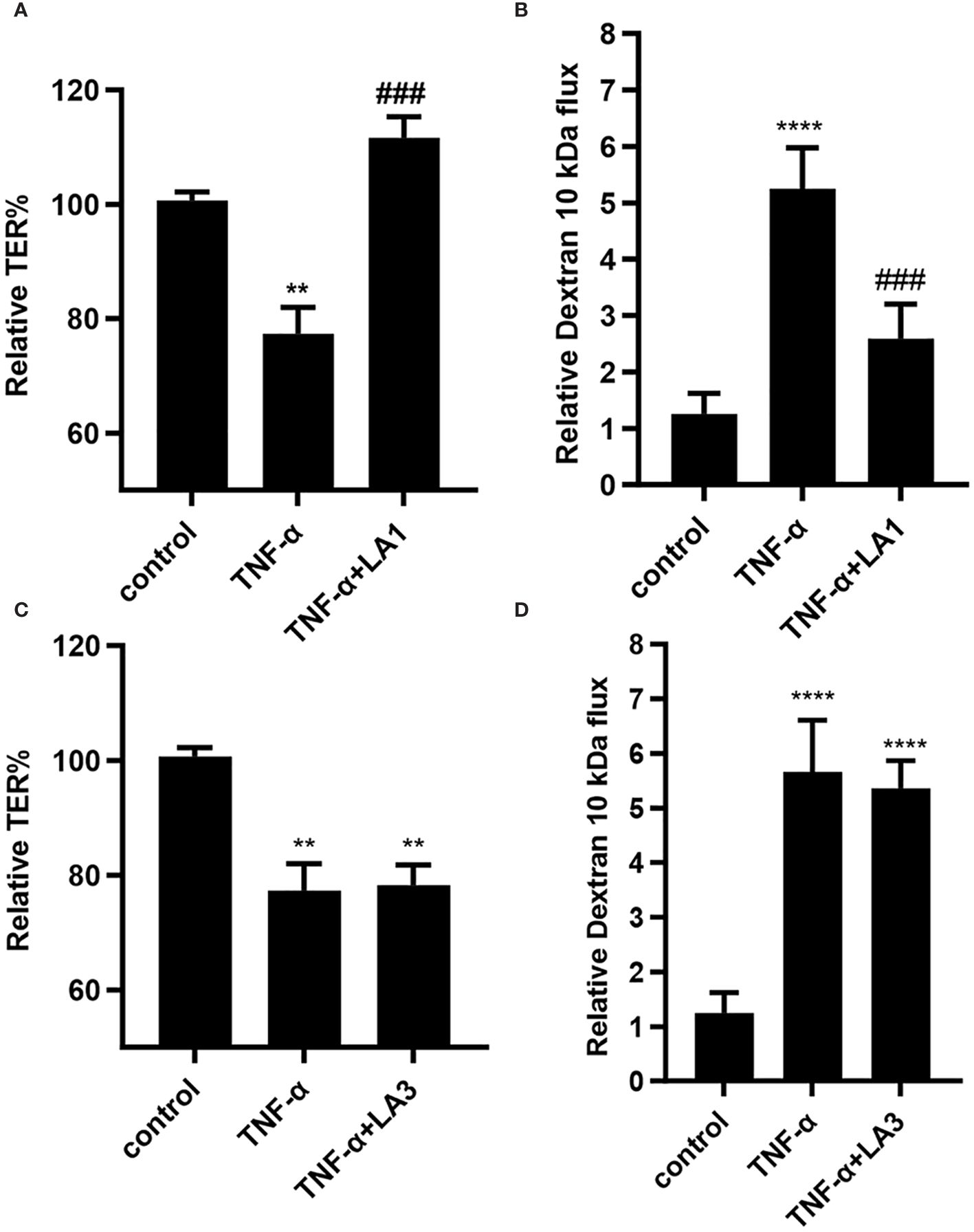
Figure 1 Effect of Lactobacillus acidophilus strains, LA1 and LA3, on TNF-α-induced increase in filter-grown Caco-2 TJ permeability. (A) Strain LA1 prevented the TNF-α-induced drop in Caco-2 TER. LA1 (1x108 CFU/ml for 24 hr); TNF-α (10 ng/ml for 24 hr); LA1 pre-treated 2 hr prior to TNF-α treatment. **, P<0.01 vs. control; ###, P<0.001 vs. TNF-α treatment. (B) LA1 prevented the TNF-α-induced increase in mucosal-to-serosal flux of paracellular marker dextran 10 kDa; ****, P<0.0001 vs. control; ###, P<0.001 vs. TNF-α treatment. (C) Strain LA3 did not affect the TNF-α-induced drop in Caco-2 TER or (D) the TNF-α-induced increase in Caco-2 TJ permeability to dextran 10 kDa. **, P<0.01 vs. control ****, P<0.0001 vs. control.
3.2 LA1 prevents the TNF-α-induced increase in Caco-2 TJ permeability by suppressing NF-κB p50/p65 activation
The TNF-α-induced increase in intestinal epithelial TJ permeability is mediated by a canonical pathway activation of nuclear transcription factor NF-κB p50/p65 (27, 28, 33, 81, 82). The activation of NF-κB p50/p65 requires inhibitory-κB kinase (IKK) induced phosphorylation and proteasomal degradation of IκB-α, leading to NF-κB p50/p65 activation and nuclear translocation (83–86). In the following studies, the effect of LA1 on TNF-α-induced activation of NF-κB p50/p65 was examined. The TNF-α treatment caused degradation of IκB-α, phosphorylation of NF-κB p65 subunit (Ser536) (Figure 2A), and subsequent nuclear translocation of NF-κB p65 in Caco-2 monolayers (Figure 2B). As shown in the immunostaining studies, TNF-α treatment caused NF-κB p65 translocation to the nucleus (Figure 2B). LA1 treatment by itself did not activate the basal level of IκB-α or NF-κB activation (Figure 2B). However, pre-treatment with LA1 almost completely inhibited the TNF-α-induced degradation of IκB-α and NF-κB activation (Figure 2). LA3 did not prevent the TNF-α-induced activation of NF-κB p65 as measured by the phosphorylation of NF-κB p65 and degradation of IκB-α, (Figure 2A).
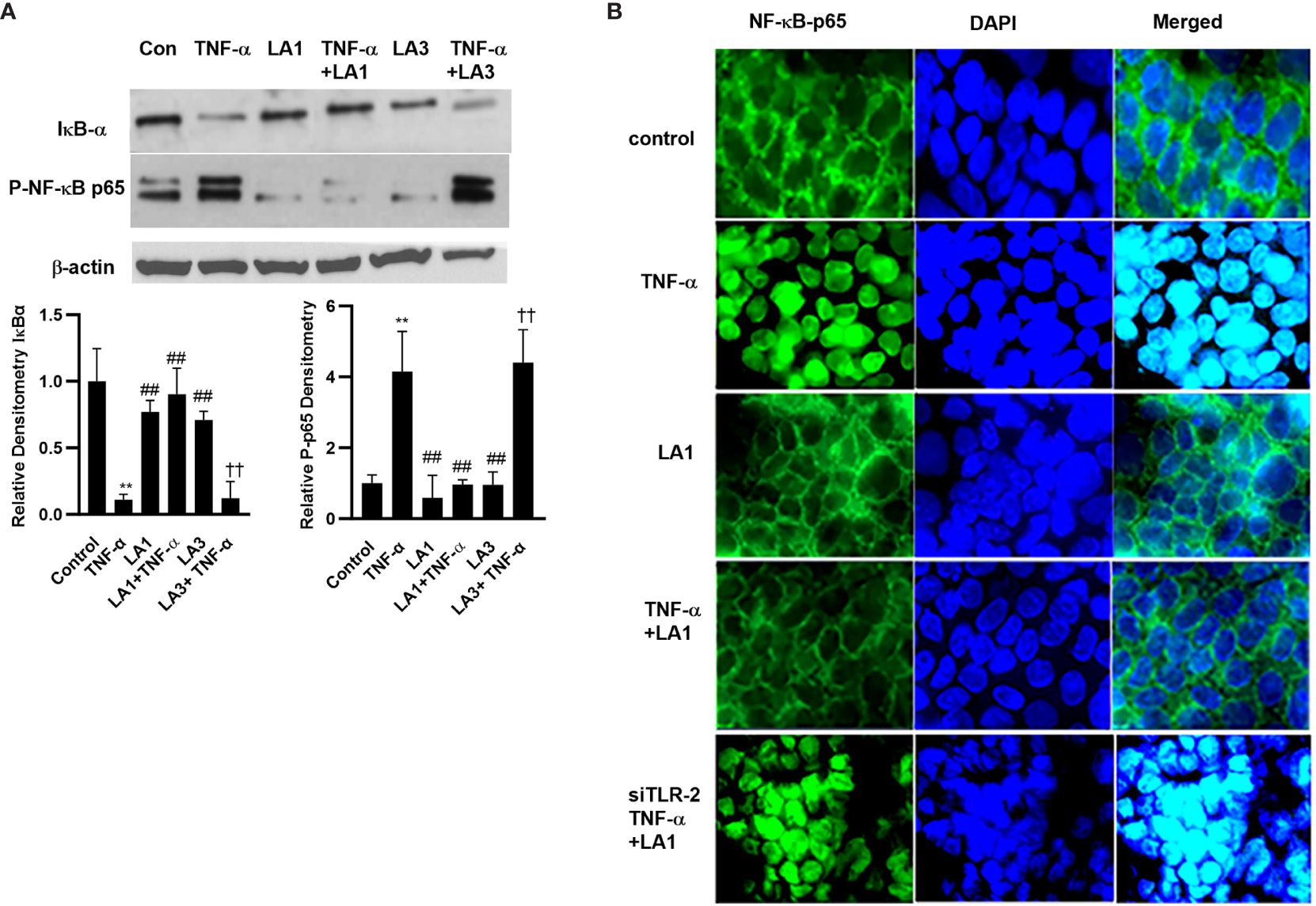
Figure 2 Effect of LA1 on TNF-α activation of NF-κB p65 in Caco-2 monolayers. (A) LA1 treatment (1x108 CFU/ml) did not affect the activation of NF-κB p65 as assessed by degradation of IκB-α and phospho-p65 expression in Caco-2 monolayers (30-min experimental period). TNF-α (10 ng/ml) caused a rapid increase in phospho-p65 expression and degradation of IκB-α in Caco-2 monolayers (30-min experimental period). LA1, but not LA3, prevented the TNF-α-induced activation of NF-κB p65. Relative densitometry of IκB-α and p-p65 indicated below; **, P<0.01 vs. control; ##, P<0.01 vs. TNF-α treatment; ††, P<0.01 vs. LA3 treatment. (B) LA1 did not affect the cytoplasmic-to-nuclear translocation of NF-κB p65 in Caco-2 monolayers but inhibited the TNF-α-induced nuclear translocation of NF-κB p65 (30-min experimental period). TLR-2 siRNA transfection (48 hr) prevented the LA1 inhibition of TNF-α-induced nuclear translocation of NF-κB p65 as determined by immunofluorescence imaging. NF-κBp65: green; Nuclei: blue; 60x (Nikon fluorescent microscopy).
3.3 LA1 stimulates NF-κB p50/p65 activation in mouse immune cells in a TLR-2-dependent manner
Expanding on the above studies showing LA1 inhibition of TNF-α-induced activation of NF-κB p50/p65 in intestinal epithelial cells, in the next studies, we examined the effect of LA1 on NF-κB activation in immune cells. Toll-like receptors (TLRs) have a crucial role in immune response against bacterial invasion (87, 88). In response to pathogen invasion, plasma membrane-associated TLRs, including TLR-2, TLR-4, and TLR-5, recognize pathogen-associated molecular patterns (PAMPs) and cause NF-κB p50/p65 activation via a MyD88-dependent process (74–76). In the following studies, mouse immune cells were obtained from freshly resected mouse spleen. The splenocytes (consisting of lymphocytes, monocytes, and macrophages) were isolated from the mouse spleen as described in the methods section. The LA1 effect on NF-κB p50/p65 activation in mouse splenocytes was assessed by NF-κB p65 nuclear translocation. The CD3+ T-cells were identified by antibody labeling and immunostaining. In the untreated control immune cells, NF-κB p65 was localized in the cytoplasm (Figure 3A). The LA1 addition caused a rapid (within 30 minutes) cytoplasmic-to-nuclear translocation of NF-κB p65 in both CD3− immune cells and CD3+ T-cells (Figure 3A), indicating NF-κB activation. The TNF-α treatment of splenocytes also caused nuclear translocation of NF-κB p65 in both CD3− immune cells and CD3+ T cells (Figure 3A). Next, the regulatory requirement of TLR-2 or MyD88 in LA1 activation of NF-κB was determined in TLR-2 or MyD88 deficient immune cells (obtained from global TLR-2 or MyD-88 knock-out mice, respectively). LA1 did not induce NF-κB activation in immune cells obtained from TLR-2 -/- or MyD88 -/- mice (Figure 3B), confirming that the LA1 induced NF-κB p50/p65 nuclear translocation in immune cells was dependent on TLR-2 and MyD88. Together, these data indicated that LA1 activates NF-κB p50/p65 in immune cells in a TLR-2- and MyD88-dependent manner. On the other hand, TNF-α is known to activate NF-κB by binding to its receptors TNFRI and II, but not to TLRs (89, 90). The signaling pathway triggered by TNF-α is known to activate NF-κB in a TRAF/TRADD-dependent pathway and MyD88-independent pathway. TNF-α treatment caused NF-κB activation in immune cells obtained from TLR-2 -/- and MyD88 -/- mice (data not shown), indicating that TNF-α activation of NF-κB in immune cells is independent on TLR-2 and/or MyD88. We also showed that peptidoglycan (PGN), an activator of NF-κB in a TLR-2 and MyD88-dependent manner (91, 92), caused nuclear translocation of NF-κB p65 in immune cells obtained from WT mice, but not in in immune cells obtained from TLR-2 -/- and MyD88 -/- mice (Figure 3C). In contrast, polyinosinic-polycytidylic acid [poly(I:C)], a PAMP known to activate NF-κB in a TLR-2 and MyD88-independent manner (93, 94), caused NF-κB p65 nuclear translocation in immune cells obtained from WT, TLR-2-/- and MyD88-/- mice (Figure 3D).
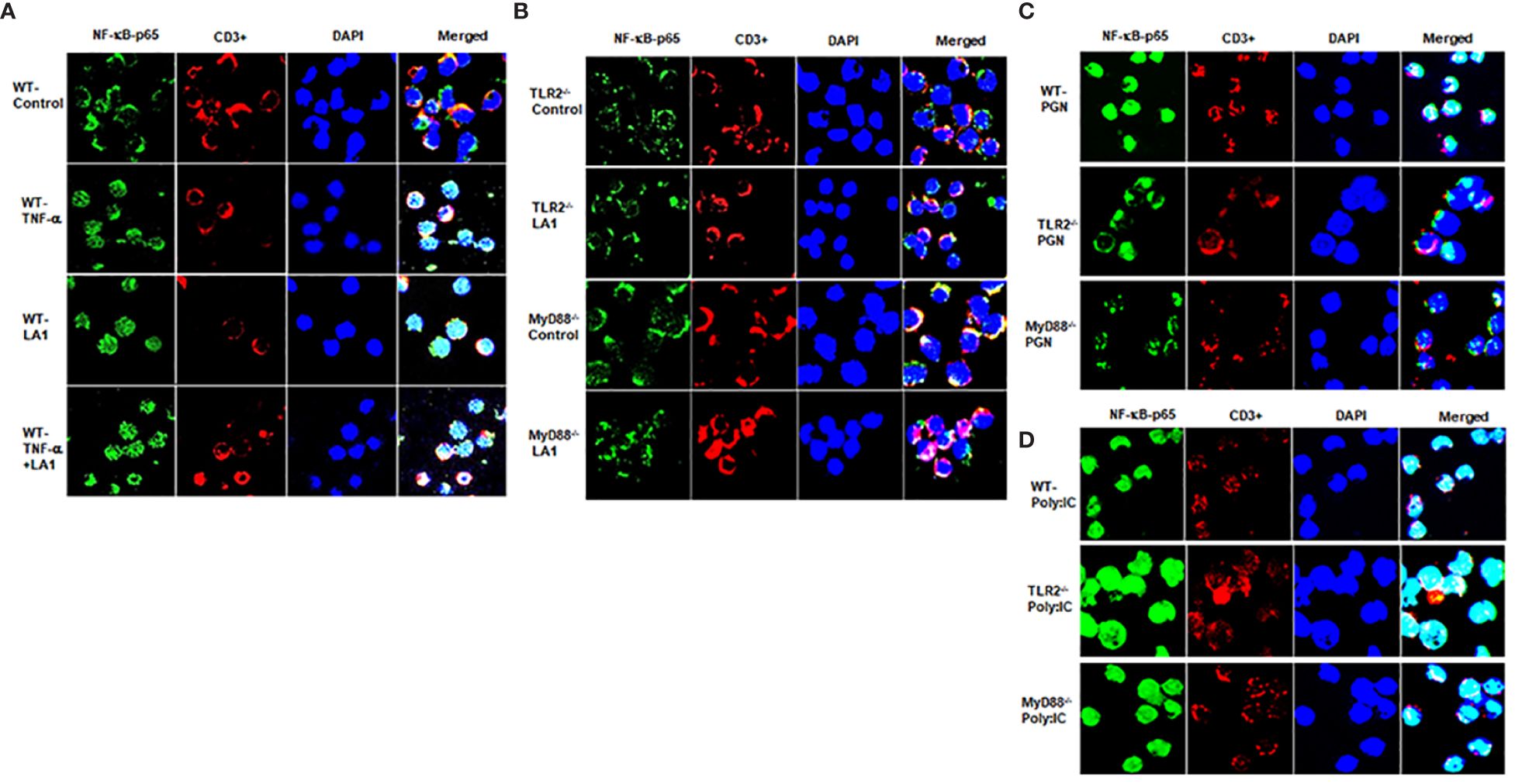
Figure 3 Effect of LA1 on NF-κB activation in mouse splenocytes isolated from wild-type (WT), TLR-2-/- and MyD88-/- mice. Immunofluorescence images showing: (A) TNF-α (10 ng/ml) and LA1 (1x108 CFU/ml) treatments caused cytoplasmic-to-nuclear translocation of NF-κB p65 (30-min experimental period) in splenocytes isolated from WT mice (WT, wild-type); (B) LA1 did not cause nuclear translocation of NF-κB p65 in immune cells from TLR-2-/- or MyD88-/- mice (30-min experimental period).; (C) Peptidoglycan (PGN) (30 µg/ml) caused nuclear translocation of NF-kB p65 in immune cells from WT, but not in immune cells from TLR-2-/- or MyD88-/- mice (30-min experimental period); (D) Poly: IC (100 μg/ml) caused nuclear translocation of NF-kB p65 in immune cells from WT, TLR-2-/- and MyD88-/- mice (30-min experimental period).NF-κBp65: green; T-cell marker CD3: red; Nuclei: blue; 60x (Nikon fluorescent microscopy).
3.4 LA1 inhibits the NF-κB p50/p65 activation in intestinal epithelial cells in a TLR-2-dependent manner
In the following studies, the knock-down of TLR-2 by siRNA on LA1 inhibition of NF-κB p50/p65 activation in Caco-2 monolayers was determined. As shown in Figure 2, LA1 inhibited the TNF-α-activation of NF-κB in Caco-2 intestinal epithelial cells. The silencing of TLR-2 by siRNA (Figure 4A) prevented the LA1 inhibition of TNF-α-induced IκB-α degradation, NF-κB p65 phosphorylation (Figure 4B) and NF-κB p65 nuclear translocation (Figure 2B) as well as the increase in Caco-2 TJ permeability (Figures 4C, D). Interestingly, knocking-down MyD88 (Figure 4E) did not alter the LA1 inhibition of NF-κB p50/p65 activation (Figure 4E) or the increase in Caco-2 intestinal epithelial TJ permeability (Figures 4F, G), suggesting that the TLR-2 pathway inhibition of NF-κB activation in Caco-2 cells was MyD88-independent. In combination, these findings suggested that LA1 activates NF-κB p50/p65 in immune cells in a TLR-2 and MyD88-dependent process, whereas LA1 inhibits the TNF-α induced activation of NF-κB p50/p65 in intestinal epithelial cells in a TLR-2-dependent-but MyD88-independent process.
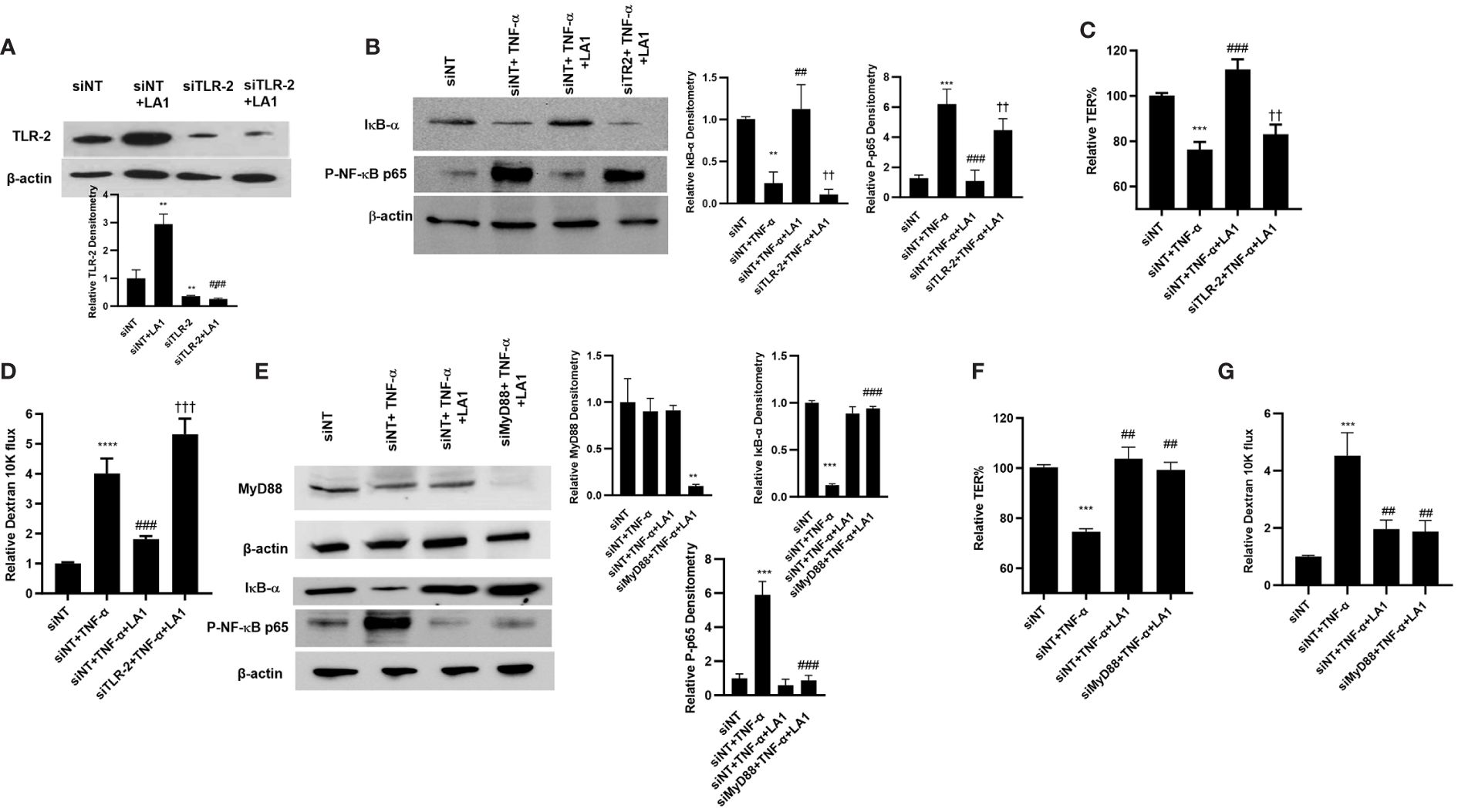
Figure 4 Intracellular mechanisms involved in LA1 protection against TNF-α-induced increase in Caco-2 intestinal epithelial TJ permeability. (A) TLR-2 siRNA transfection (48 hr) prevented the LA1-induced increase in TLR-2 protein expression (24 hr treatment). (siNT: siRNA non-target transfection). SiRNA transfection was performed for 48 hr prior to LA1 or TNF-α treatments. Relative densitometry of TLR-2 indicated below; **, P<0.01 vs. siNT; ###, P<0.001 vs. LA1 treatment. (B) TLR-2 siRNA transfection prevented the LA1 inhibition of TNF-α-induced degradation of IκB-α and phosphorylation of NF-κB p65 (30-min experimental period). Relative densitometry of IκB-α and p-p65 indicated right; **, P<0.01 vs. siNT; ##, P<0.01 vs. TNF-α treatment; ††, P<0.01 vs. TNF-α+LA1 treatment. (C) TLR-2 siRNA transfection prevented the LA1 inhibition of TNF-α-induced decrease in Caco-2 TER (24hr experimental period), ***, P<0.001 vs. control; ###, P<0.001 vs. TNF-α treatment; ††, P<0.01 vs TNF-α with LA1 pre-treatment) and (D) increase to dextran 10 kDa flux (24hr experimental period); **, P<0.01 vs. control; ###, P<0.001 vs. LA1 treatment; **** P<0.0001 vs control; †††, P<0.001 vs. TNF-α with LA1 pre-treatment. (E) Knocking down MyD88 by siRNA transfection (48 hr) caused a significant depletion of MyD88 expression, but did not prevent the LA1 inhibition of TNF-α-induced degradation of IκB-α and phosphorylation of NF-κB p65 (30-min experimental period). Relative densitometry of MyD88, IκB-α and p-p65 indicated right; **, P<0.01 vs. siNT, TNF-α, and TNF-α+LA1 treatments; ***, P<0.001 vs. siNT; ###, P<0.001 vs. TNF-α treatment. (F) SiRNA MyD88 transfection did not prevent the LA1 inhibition of TNF-α-induced decrease in Caco-2 TER or (G) increase to dextran 10 kDa flux (24 hr experimental period); ***, P<0.001 vs. control; ##, P<0.01 vs. TNF-α treatment.
3.5 PI3 kinase mediates the TLR-2 pathway inhibition of NF-κB activation
Next, we investigated the crosstalk between TLR-2 and TNF-α signal transduction pathways and the protein kinase that mediates the LA1 inhibition of TNF-α effect on NF-κB activation and on intestinal TJ permeability. The TNF-α activation of NF-κB p50/p65 is regulated up-stream by mitogen-activated protein-3 kinase (MAP3K) NF-κB-inducing kinase (NIK) phosphorylation and activation of IκB kinases, IKK-α and IKK-β (28). TNF-α caused activation and phosphorylation of NIK (Thr-559) in Caco-2 cells as assessed by phospho-NIK immunoblotting (Figure 5A); LA1 did not prevent the TNF-α activation of NIK, suggesting that LA1 does not inhibit NF-κB p50/p65 activation at the level of NIK. TNF-α also caused activation of both IKK-α and IKK-β catalytic subunits (Figure 5B); LA1 prevented the TNF-α-induced activation and phosphorylation of IKK-α (Ser-176), but not IKK-β phosphorylation (Ser-180) (Figure 5B), demonstrating that LA1 inhibits the IKK-α phosphorylation without affecting IKK-β phosphorylation. Phosphatidylinositol 3-kinase (PI3K) is known to be activated by the TLR-2 signal transduction pathway (95–101). Next, the possibility that PI3K acts as an upstream protein kinase that mediates the LA1/TLR-2 inhibition of TNF-α-induced IKK-α and NF-κB activation was examined. LA1caused PI3K (Ser-361) phosphorylation in Caco-2 monolayers, while LA3 did not (Figure 5C). Silencing PI3K by siRNA (Figure 5D) prevented the LA1 inhibition of TNF-α-induced IKK-α phosphorylation and IκB-α degradation (Figure 5E) and decrease in Caco-2 TER (Figure 5F).
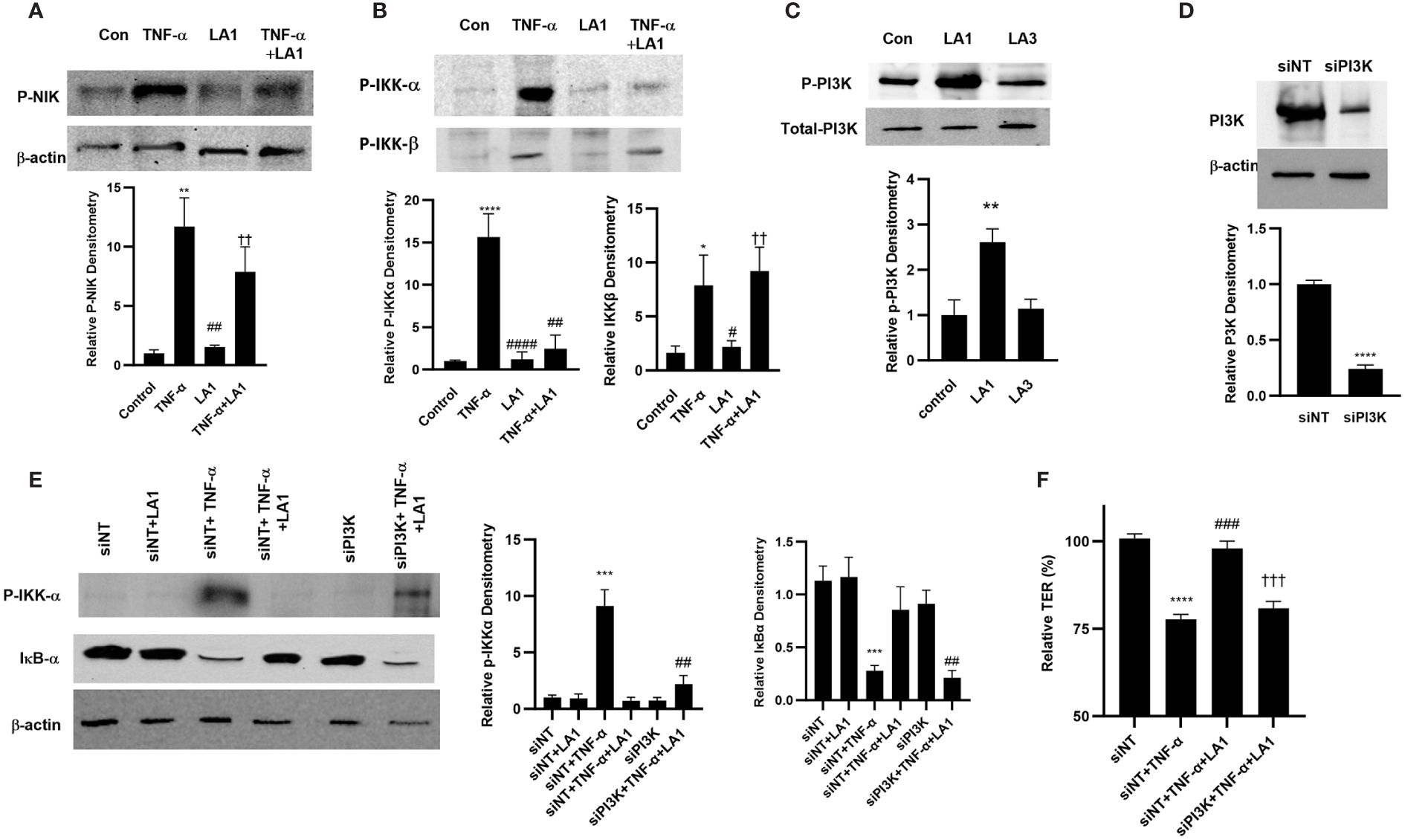
Figure 5 Effect of LA1 inhibition on TNF-α activation of upstream signaling kinases in Caco-2 monolayers. (A) LA1 did not prevent the TNF-α-induced activation of NIK as assessed by phospho-NIK (30-min experimental period). Relative densitometry of P-NIK indicated below; **, P<0.01 vs. control; ##, P<0.01 vs. TNF-α treatment; ††, P<0.01 vs. LA1 treatment. (B) LA1 prevented the TNF-α-induced activation of IKK-α, but not IKK-β, as assessed by phospho-IKK-α/IKK-β (30-min experimental period). Relative densitometry of p-IKK-α and p-IKK-β indicated below; *, P<0.05 vs. control ****, P<0.0001 vs. control; #, P<0.05; ##, P<0.01; ####, P<0.0001 TNF-α treatment;††, P<0.01 vs. LA1 treatment. (C) LA1, but not LA3, caused activation of PI3K as assessed by phospho-PI3K (30-min experimental period). Relative densitometry of p-PI3K indicated below; **, P<0.01 vs. control. (D) SiRNA-induced knock-down of PI3K resulted in a significant depletion of PI3K expression (48 hr). Relative densitometry of PI3K indicated below; ****, P<0.0001 vs. siNT. (E) Knocking down PI3K by siRNA transfection prevented the LA1 inhibition of TNF-α-induced phosphorylation of IKK-α or degradation of IκB-α. SiRNA transfection was performed for 48 hr prior to LA1 treatment for 30 min. Relative densitometry of p-IKK-α and IκB-α indicated right; ***, P<0.001 vs. siNT; ##, P<0.01 vs.TNF-α +LA1 treatment. (F) Knocking PI3K by siRNA prevented the LA1 inhibition of TNF-α induced drop in Caco-2 TER. SiRNA transfection was performed for 48 hr prior to LA1 treatment for 24 hr; ****, P<0.0001 vs. control; ###, P<0.001 vs. LA1 or TNF-α treatment; †††, P<0.001 vs TNF-α with LA1 pre-treatment.
3.6 LA1 inhibits the MLCK gene activity and kinase activity induced by TNF-α
MLCK is a TJ effector protein that plays a central role in the regulation of intestinal TJ barrier (102, 103). The increase in intestinal TJ permeability caused by TNF-α has been shown to be regulated in part by NF-κB p50/p65 activation of MLCK gene and increase in MLCK activity in the intestinal epithelial cells, leading to the opening and disruption of the intestinal TJ barrier (28, 30, 32, 33, 72, 102–109). The effect of LA1 on increase in MLCK gene activity, protein expression, and kinase activity in filter-grown Caco-2 monolayers caused by TNF-α was investigated. TNF-α caused an upregulation of MLCK gene activity as evidenced by the increase in the full-length MLCK promoter activity (2,091 bp promoter region, accession # DQ090939) as assessed by the luciferase reporter gene, and an upregulation in mRNA levels (Figures 6A, B). TNF-α also caused an upregulation in MLCK expression and kinase activity, as assessed by Caco-2 MLC phosphorylation (Figures 6C, D). As seen in Figure 6D, TNF-α produced a marked increase in phosphorylated-MLC near the per-junctional regions of filter-grown Caco-2 monolayers. The LA1 pre-treatment inhibited the upregulation of MLCK promoter activity, mRNA and protein expression, and MLC phosphorylation (Figures 6A–D). The inhibition of PI3K by siRNA abolished the LA1 inhibition of TNF-α increase in MLCK expression (Figure 6E).
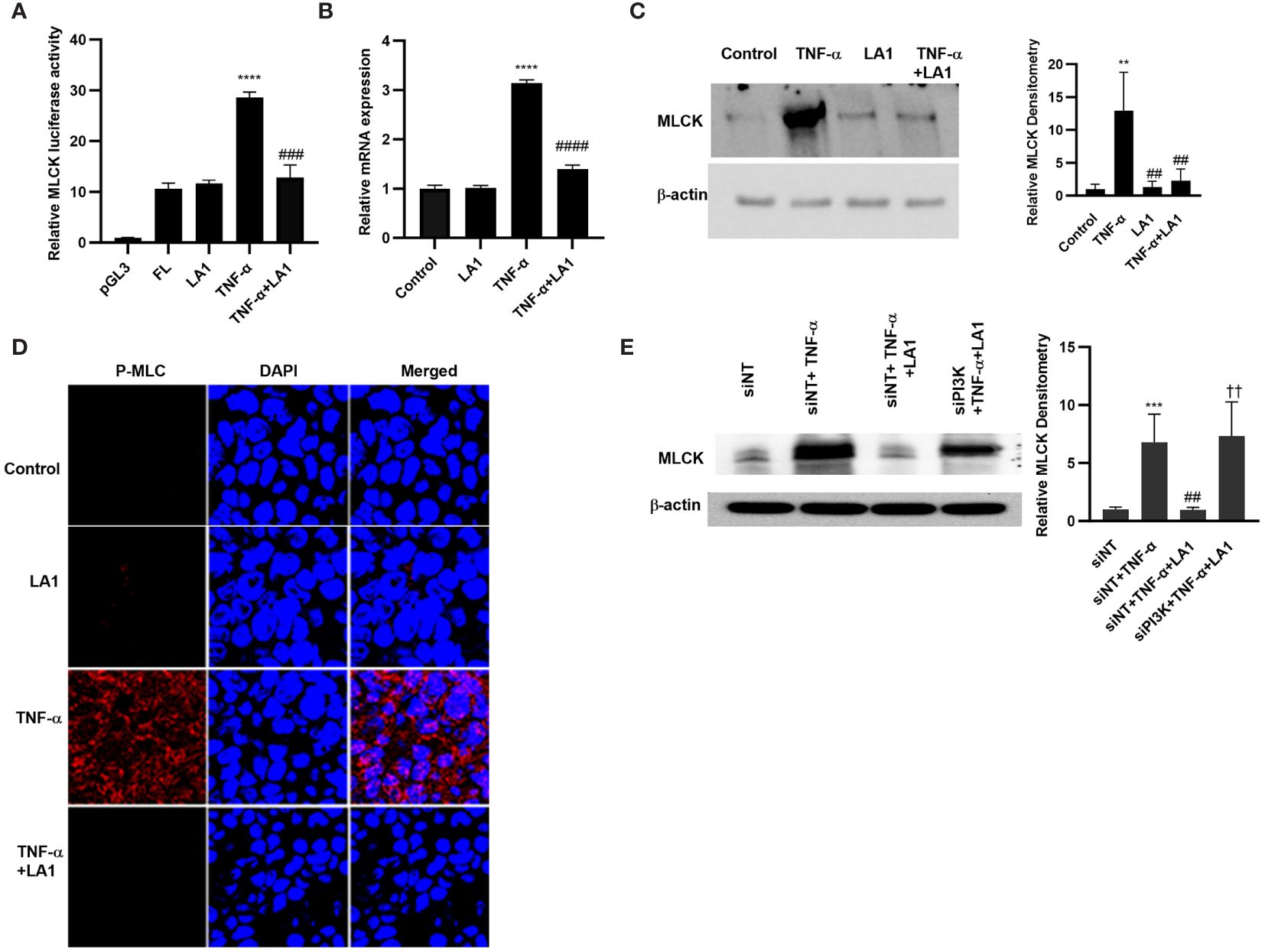
Figure 6 Effect of LA1 on TNF-α activation of MLCK gene, protein expression and activity in Caco-2 monolayers. (A) LA1 (1x108 CFU/ml) prevented the TNF-α-(10 ng/ml) induced increase in MLCK gene. pGL-3 basic vector containing the MLCK promoter region was transfected into the filter-grown Caco-2 cells. The MLCK promoter activity was determined by the luciferase assay and expressed as relative luciferase activity; ****P < 0.0001 vs. Full-Length (FL); ###, P<0.001 vs. TNF-α treatment. (B) LA1 inhibited the MLCK TNF-α induced increase in MLCK mRNA levels; ****, P < 0.0001 vs. control; ####, P<0.0001 vs. TNF-α treatment, (C) protein expression (Relative densitometry of MLCK indicated right; **, P<0.01 vs. control; ##, P<0.01 vs.TNF-α) and (D) MLCK kinase activity as assessed by phospho-MLC (P-MLC) immunofluorescence imaging (24 hr experimental period). P-MLC: red; Nuclei: blue; 63x (Confocal microscopy). (E) Knocking down PI3K by siRNA transfection (48 hr) prevented the LA1 inhibition of TNF-α-induced increase in MLCK protein expression. SiRNA transfection was performed for 48 hr prior to LA1 or TNF-α treatments. Relative densitometry of MLCK indicated right; ***, P<0.001 vs. siNT; ##, P<0.01 vs.TNF-α; ††, P<0.01 vs. TNF-α+LA1 treatment.
3.7 LA1 inhibits the TNF-α–induced increase in mouse intestinal permeability in vivo
In these animal studies, we assessed the LA1 effect on TNF-α-induced increase in intestinal epithelial TJ permeability in mice. The effects of LA1 and TNF-α administration on mouse small intestinal permeability were determined in vivo by “recycling intestinal perfusion of an isolated small intestinal segment (6-8 cm in linear length) in live mice using Texas Red- dextran 10 kDa as the paracellular marker”, as described previously by us (71). In these studies, LA1 was administered by oral-gastric gavage 24 hr prior to the intraperitoneal (i.p.) treatment of TNF-α (5 μg for 24 hr). The TNF-α treatment caused an increase in mouse small intestinal permeability to dextran 10 kDa by the end of the 24hr treatment period (27) (Figure 7A). A dose-dependent effect of LA1 on mouse intestinal permeability was examined to verify the most beneficial dose of LA1 on the intestinal TJ barrier. The bacterial concentration of 1x109 CFU/ml exerted the maximal effect on mouse intestinal TJ barrier (data not shown). The pre-treatment with LA1, but not LA3, inhibited the TNF-α increase in mouse intestinal permeability (Figure 7A). The TNF-α caused downregulation of IκB-α and an increase in NF-κB p65 phosphorylation in mouse intestinal tissue (Figure 7B), and LA1 pre-treatment inhibited the IκB-α degradation and NF-κB p65 phosphorylation (Figure 7B). The images of the mouse intestinal tissue showed that NF-κB p65 is present in the cytoplasm of the intestinal epithelial cells in the control mice (Figure 7C). Following TNF-α administration, NF-κB p65 translocated to the nuclei in mouse enterocytes (Figure 7C); the LA1 pre-treatment prevented the nuclear translocation of NF-κB p65 (Figure 7C). Additionally, TNF-α caused an upregulation in MLCK expression in intestinal tissues (Figure 8A) and an increase in phosphorylated MLC in the mouse enterocytes as shown by the immunostaining of the intestinal mucosal surface (Figure 8B). The requirement of TLR-2 in LA1 protective effect against the TNF-α-induced increase in mouse intestinal permeability was also determined in TLR-2−/− mice. The TNF-α-induced increase in intestinal permeability was inhibited in LA1 pre-treated wild-type mice (Figure 7A); the LA1 inhibition of TNF-α-induced increase in mouse intestinal permeability was prevented in the TLR-2−/− mice (Figure 8C), suggesting that TLR-2 was required for the LA1 intestinal barrier protective effect. On the other hand, LA1-induced decrease in mouse intestinal permeability was not prevented in MyD88-/- mice (Figure 8D). And, LA1 pre-treatment prevented the TNF-α induced increase in mouse intestinal permeability in MyD88-/- mice, suggesting that the LA1 effect on mouse intestinal permeability was MyD88-independent.
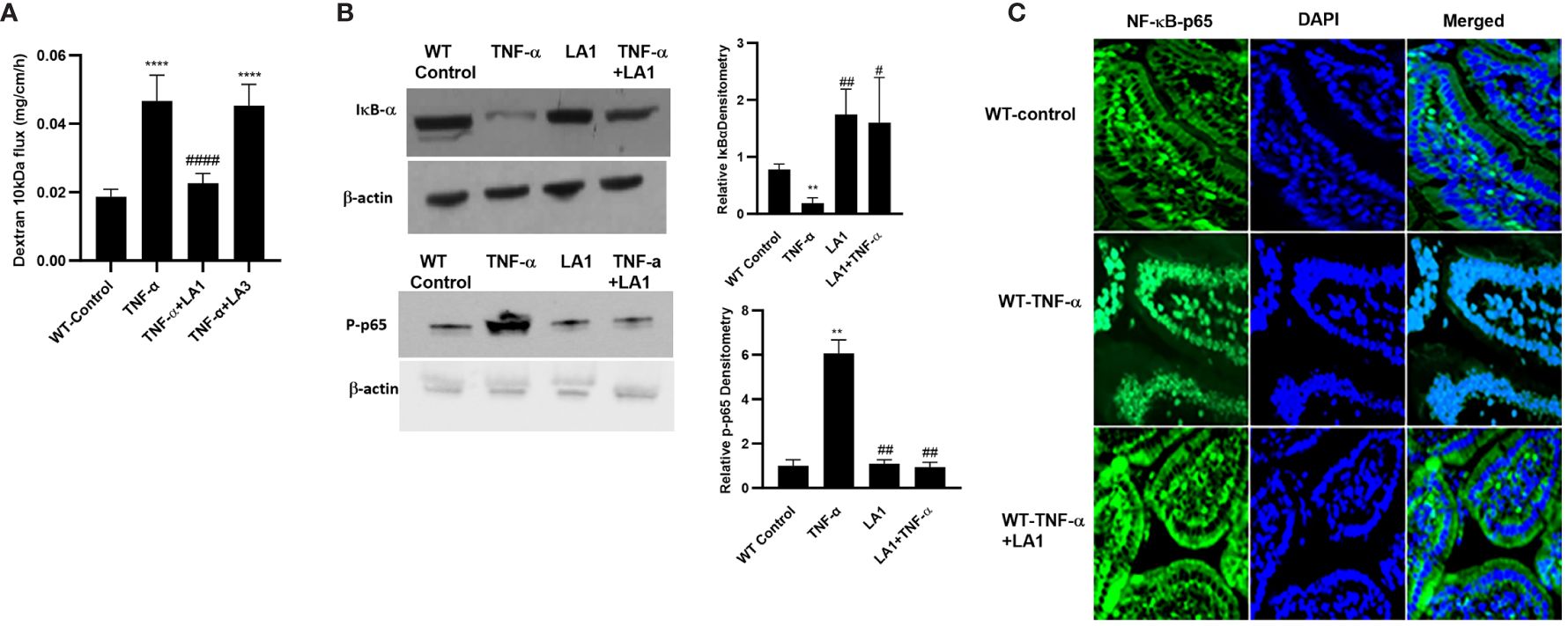
Figure 7 Effect of LA1 on TNF-α induced increase in mouse intestinal permeability in-vivo. (A) Oral gavage of LA1, but not LA3, (1 x 109 CFU/ml) prevented the TNF-α -induced increase in dextran 10 kDa flux in mouse intestinal tissues; ****, P<0.0001 vs. control; ####, P<0.00001 vs. TNF-α treatment. (TNF-α (5 μg) was administered intraperitoneally). Relative densitometry of IκB-α and p-p65 indicated right; **, P<0.01 vs. WT control; #, P<0.05; ##, P<0.01 vs.TNF-α; (B) Intraperitoneal injection of TNF-α (5 μg) caused degradation of IκB-α and increase in phospho-p65, and LA1 prevented the TNF-α-induced activation of NF-κB (2-hr experimental period). (C) LA1 inhibited the TNF-α-induced cytoplasmic-to-nuclear translocation of NF-κB p65 (2-hr experimental period) in small intestinal tissues as determined by immunofluorescence imaging, NF-κBp65: green; Nuclei: blue; 60x (Nikon fluorescent microscopy).
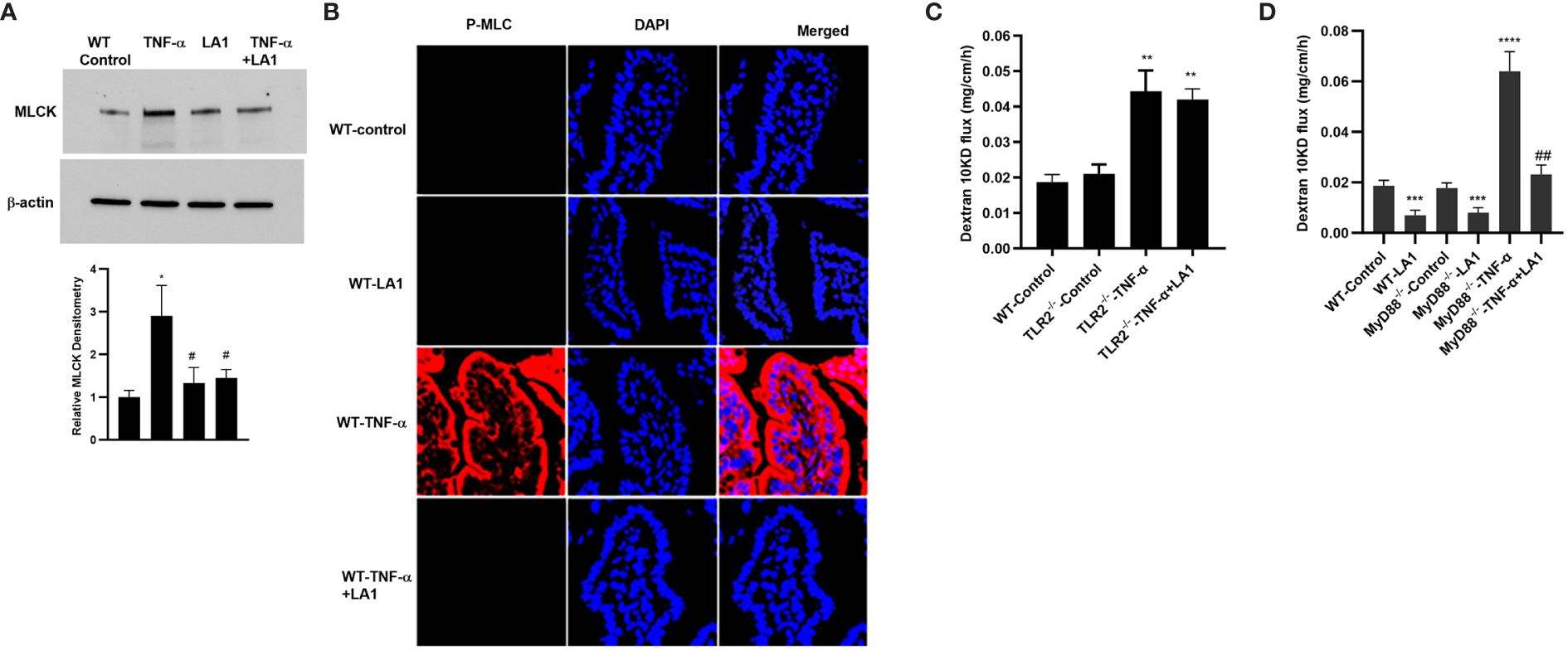
Figure 8 Effect of oral gavage of LA1 (1x109 CFU/ml) on TNF-α-induced increase in MLCK expression and activity in-vivo. (A) LA1 prevented the TNF-α-induced increase in MLCK protein expression in mouse small intestinal tissue (24-hr experimental period). Relative densitometry of MLCK indicated below; *, P<0.05 vs. WT control; #, P<0.05 vs.TNF-α treatment. (B) LA1 inhibited the TNF-α-induced increase in MLCK activity as assessed by phospho-MLC immunostaining (24-hr experimental period); P-MLC: red; Nuclei: blue; 63x (Confocal microscopy). (C) LA1 did not prevent the TNF-α-induced increase in mouse intestinal permeability to dextran10 kDa in TLR-2-/- mice; **, P<0.001 vs. wild-type (WT) control and TLR-2-/- control (24-hr experimental period). (D) LA1 caused a decrease in mouse intestinal permeability to dextran10 kDa in wild-type (WT) and in MyD88-/- mice (24-hr experimental period), and prevented the TNF-α induced increase in mouse intestinal permeability in WT and MyD88-/- mice. ***, P<0.001 vs. WT and vs. MyD88-/- control; ****, P<0.0001 vs. control; ##, P<0.01 vs. TNF-α treatment.
4 Discussion
Defective intestinal epithelial TJ barrier has been shown to play a role in the pathogenesis of IBD, and, in patients with IBD, therapeutic tightening of the intestinal TJ barrier correlates with rapid resolution of intestinal inflammation and prolonged clinical remission (110). Pro-inflammatory cytokine TNF-α is highly expressed in patients with IBD and plays a crucial role in the initiation, prolongation, and persistence of intestinal inflammation, and anti-TNF-α antibody therapy has been shown to be highly effective in the treatment of CD and UC (40, 42, 43). TNF-α produces a rapid and prolonged increase in intestinal TJ permeability by activating MLCK gene in intestinal epithelial cells (27, 31–33–72, 104). Intestinal microflora plays an important role in maintaining a healthy gut microenvironment and also in initiating or promoting exaggerated inflammatory responses in various disease states (87, 111, 112). Probiotic bacteria are gut bacteria that provide health benefits, including the restoration of intestinal barrier function (48, 50, 59). Most probiotic bacteria studied to date have no or modest effect on intestinal epithelial TJ barrier (113, 114). Although Lactobacillus species are the most widely studied probiotic bacterial species in relation to health benefits, most of the Lactobacillus species studied have “had a modest effect on intestinal TJ barrier” (60–63, 66–68, 70). Hsieh et al. tested over 30 probiotic bacterial species and strains, including Lactobacillus and Enterococcus, and found that the majority had no effect or caused a decrease in intestinal TJ barrier function and that Bifidobacterium strains produced a modest increase (115). Lepine et al. showed that L. acidophilus strain (W37) caused a modest enhancement in Caco-2 TJ barrier (64), and Guo et al. found that another strain of Lactobacillus, L. acidophilus strain (ATCC No. 53103) produced a 25% increase in Caco-2 TER (62). Madsen et al. (116) and Mennigen et al. (113) showed that probiotic mixture VSL#3 (containing nine probiotic bacterial species, including L. acidophilus) also caused a modest enhancement in mice and human intestinal epithelial barrier function. Similarly, in our previous studies, we also found that many of the over 20 probiotic species tested, including Lactobacillus plantarum, L. brevis, L. helveticus, L. casei, and Bifidobacterium longum had no effect on Caco-2 TJ barrier and several probiotic species, including L. johnsonii, L. rhamnosus and B. breve had a modest effect (10-25% increase) on Caco-2 TER. However, there was one particular strain of LA that caused an optimal enhancement in intestinal epithelial TJ barrier function, while other LA strains tested “had a modest or no effect on the intestinal TJ barrier” (71). We found two strains of LA, referred to as LA1 and LA3, that had distinct effects on intestinal epithelial TJ barrier in Caco-2 monolayers and in mice small intestine (71). LA1 caused a marked enhancement in intestinal epithelial TJ barrier function, while LA3 had no effect on the intestinal TJ barrier (71). LA1 concentration of 1x108 CFU/ml and 1x109 CFU/ml exerted the maximal enhancement of intestinal TJ barrier in Caco-2 cells and in mouse intestinal tissues, respectively. Additionally, LA1, but not LA3, inhibited the development of DSS-induced colitis and prevented intestinal inflammation by preserving the intestinal barrier (71). The anti-inflammatory and antioxidant mechanisms involved in different strains of LA related to the enhancement of the intestinal barrier have been examined (57, 117–120). For example, a study by Chorawala et al. showed that LA preserve the intestinal TJ barrier by exerting anti-inflammatory response against LPS-induced colitis (117). The study also showed the antioxidant effect of LA and the enhancement of the barrier on colonic cells in mouse model of colitis. The colonic C-reactive protein, nitric oxide and pro-inflammatory cytokines were all reduced after exposure to LA and other probiotic strains (117). De Matuoaka et al. also showed the potential role of LA strains as protective agents against arsenic toxicity and arsenic-induced intestinal barrier disruption in Caco-2 cells (118). The main aim of the present study was to investigate a novel intracellular mechanism that mediate the LA1 inhibition of TNF-α-induced increase in intestinal epithelial TJ permeability. The findings in the present study expand on our previous studies showing that “LA1 causes a unique, strain-specific enhancement of intestinal barrier function” (71) and provide new insight into the signaling pathways and the mechanisms by which LA1 inhibits the TNF-α-induced increase in intestinal TJ permeability. Our results suggest for the first time that LA1 inhibits the TNF-α increase in intestinal TJ permeability via a novel intracellular process in which LA1 suppresses the NF-κB p50/p65 activation in a TLR-2-dependent and MyD88-independent mechanisms in the intestinal epithelial cells.
NF-κB p50/p65 is a ubiquitous pro-inflammatory nuclear transcription factor that mediates the activation of immune response and plays a central role in promoting intestinal inflammation in IBD and other inflammatory conditions of the gut (7, 83, 84). The NF-κB activation is separated into canonical and non-canonical pathways: the canonical pathway leads to the activation of NF-κB p50/p65 (RelA) heterodimer, and the non-canonical pathway leads to the activation of NF-κB p52/RelB heterodimer (28, 85, 121). The canonical pathway activation of NF-kB p50/p65 is known to be regulated by the IKK complex consisting of the catalytic subunits IKK-α and IKKβ and the regulatory subunit IKK-γ (122–124). The IKK catalytic subunits enzymatically catalyze the phosphorylation, ubiquitination, and degradation of IκB-α protein and the concomitant NF-κB p50/p65 activation (124, 125). The non-canonical pathway is classically regulated by the up-stream NIK activation of IKK-α homodimer and the subsequent phosphorylation and proteolytic processing of p100 and production of p52 and RelB (122, 124, 126). The TNF-α induced increase in intestinal TJ permeability has been shown to be mediated by the activation of the canonical pathway NF-κB p50/p65 in intestinal epithelial cells via a newly described intracellular mechanism, in which the NIK (typically associated with the non-canonical pathway p52/RelB activation) sequentially activates IKK-α and NF-κB p50/p65 (28). In the studies herein, we show that LA1 by itself does not affect the basal levels of NF-kB p50/65 activation in intestinal epithelial cells but inhibits the TNF-α activation of NF-κB p50/p65 and the subsequent increase in intestinal TJ permeability in a TLR-2 dependent mechanism. Toll-like receptors (TLRs) are pattern recognition receptors (PRRs) that play a crucial role in innate immunity by recognizing various PAMPs in response to microbe invasion (127). The current scientific paradigm is that, in response to a microbe invasion, TLRs, including TLR-2, TLR-4, and TLR-5, recruit and employ adaptor protein MyD88 to activate NF-κB p50/p65 and stimulate immune response (76, 128–130). In our studies herein, LA1 also produced a rapid activation of NF-κB p50/p65 in immune cells in a TLR-2 and MyD88-dependent manner (Figure 3). Although the prior studies have uniformly found TLR signal transduction pathways, including TLR-2, TLR-4, and TLR-5, to activate NF-κB p50/p65 in immune cells and other cell types, our results in intestinal epithelial cells demonstrated that LA1 inhibits the NF-κB p50/p65 activation in a TLR-2 dependent process and that TLR-2 knock-down abolished the LA1 suppression of NF-kB activity (Figure 4). This shows a unique mechanism of LA effect on intestinal epithelial cells distinct from the immune response. It is also interesting to note that the LA1/TLR-2 pathway suppression of intestinal epithelial NF-κB p50/p65 was independent of MyD88 (Figure 4E), a known master regulator of NF-κB activation (128–132). In combination, our results showed a novel finding that TLR-2 complex has a differential regulatory role in NF-κB activation depending on the cell-type, and that LA1/TLR-2 signal transduction pathway activates NF-κB p50/p65 in immune cells, whereas it inhibits the NF-κB activation in intestinal epithelial cells. Since intestinal epithelial cells are at the interface of the outside and the internal milieu of the body and serve as a physical and functional barrier that selectively regulates the transport of needed nutrients yet simultaneously restricts the trans-epithelial uptake of noxious substances and gut bacteria in the intestinal lumen, it would be phylogenetically disadvantageous and detrimental in the evolution of organisms to have luminally accessible apical membrane receptors that trigger inflammatory response or lead to the disruption of the intestinal barrier function in the presence of gut bacteria. On the other hand, having apical membrane receptors such as TLR-2 accessible to commensal or probiotic bacteria and which can be regulated to enhance the intestinal TJ barrier or suppress inflammatory response (up-regulate immune tolerance) would be evolutionally advantageous (133, 134).
As for the crosstalk between TLR-2 and TNF-α signal transduction pathways and the intermediatory protein kinase that modulates the NF-κB activation process, our data demonstrated that LA1 caused PI3K activation in intestinal epithelial cells in a TLR-2-dependent process, and that PI3K plays an important regulatory role in NF-κB p50/p65 inhibition by interfering with IKK-α phosphorylation (Figure 5E). The increase in intestinal TJ permeability caused by TNF-α has been found to be mediated by NF-κB p50/p65 binding and activation of enterocyte MLCK promoter and MLCK-induced opening of the TJ barrier (28–31, 34, 104). Our results herein also show that LA1 inhibits the TNF-α induced increase in intestinal permeability by PI3K-dependent inhibition of MLCK gene activation and MLCK activity in Caco-2 cells and mouse enterocytes (Figures 6, 8). These results suggest that TLR-2 complex and PI3K are potential therapeutic targets to inhibit enterocyte NF-κB p50/p65 activation and MLCK activity and to preserve the intestinal TJ barrier in various intestinal inflammatory disorders.
In conclusion, our results show that LA1 inhibits the TNF-α-induced increase in intestinal epithelial TJ permeability via a TLR-2-dependent inhibition of NF-κB p50/p65 activation. LA1 produces a TLR-2- and MyD88-dependent activation of NF-κB p50/p65 in immune cells and TLR-2-dependent, but MyD88-independent, suppression of NF-κB activation in intestinal epithelial cells, demonstrating that LA1 activation of TLR-2 signal-transduction pathway has a cell-type specific and differential regulatory effect on NF-κB p50/p65 activation. The LA1/TLR-2 and TNF-α/TNF-α-receptor signaling pathway crosstalk was mediated in part by PI3K inhibition at the level of IKK-α and the subsequent down-stream inhibition of enterocyte NF-κB p50/p65 and MLCK gene activity. In composite, these data provide important and novel insight into the role TLR-2 and PI3K play in cell-type specific inhibition of TNF-α-induced activation of NF-κB p50/p65 and increase in intestinal TJ permeability.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used. The animal study was approved by Penn State University Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
MH: Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – review & editing. LK: Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – review & editing. RA: Formal analysis, Methodology, Writing – review & editing. JE: Methodology, Writing – review & editing, Validation. EK: Methodology, Formal analysis, Validation, Writing – review & editing. MR: Methodology, Writing – review & editing. RA-S: Conceptualization, Project administration, Supervision, Validation, Writing – review & editing, Methodology, Formal analysis, Investigation, Visualization, Writing – original draft. TM: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research project was supported by the National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-121073.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ahmad R, Sorrell MF, Batra SK, Dhawan P, Singh AB. Gut permeability and mucosal inflammation: bad, good or context dependent. Mucosal Immunol. (2017) 10(2):307–17. doi: 10.1038/mi.2016.128
2. Ma TYN, Al-Sadi R. Tight junctions and the intestinal barrier. In: Physiology of the gastrointestinal tract, 6th ed. Burlington, MA: Elsevier Academic Press (2018).
3. Martini E, Krug SM, Siegmund B, Neurath MF, Becker C. Mend your fences: The epithelial barrier and its relationship with mucosal immunity in inflammatory bowel disease. Cell Mol Gastroenterol Hepatol. (2017) 4(1):33–46. doi: 10.1016/j.jcmgh.2017.03.007 508
4. Oligschlaeger Y, Yadati T, Houben T, Condello Olivan CM, Shiri-Sverdlov R. Inflammatory bowel disease: A stressed "Gut/Feeling". Cells. (2019) 8(7):659. doi: 10.3390/cells8070659
5. Alexander M, Ang QY, Nayak RR, Bustion AE, Sandy M, Zhang B, et al. Human gut bacterial metabolism drives Th17 activation and colitis. Cell Host Microbe. (2022) 30(1):17–30.e9. doi: 10.1016/j.chom.2021.11.001 514
6. Morgan NN, Duck LW, Wu J, Rujani M, Thomes PG, Elson CO, et al. Crohn's disease patients uniquely contain inflammatory responses to flagellin in a CD4 effector memory subset. Inflammation Bowel Dis. (2022) 28(12):1893–903. doi: 10.1093/ibd/izac146
7. Nissim-Eliraz E, Nir E, Marsiano N, Yagel S, Shpigel NY. NF-kappa-B activation unveils the presence of inflammatory hotspots in human gut xenografts. PloS One. (2021) 16(5):e0243010. doi: 10.1371/journal.pone.0243010
8. Al-Sadi R, Khatib K, Guo S, Ye D, Youssef M, Ma T. Occludin regulates macromolecule flux across the intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. (2011) 300(6):G1054–64. doi: 10.1152/ajpgi.00055.2011
9. Al-Sadi R, Ye D, Said HM, Ma TY. Cellular and molecular mechanism of interleukin-1beta modulation of caco-2 intestinal epithelial tight junction barrier. J Cell Mol Med. (2011) 15(4):970–82. doi: 10.1111/j.1582-4934.2010.01065.x
10. Fasano A. Regulation of intercellular tight junctions by zonula occludens toxin and its eukaryotic analogue zonulin. Ann N Y Acad Sci. (2000) 915:214–22. doi: 10.1111/j.1749-6632.2000.tb05244.x
11. Turner JR. 'Putting the squeeze' on the tight junction: understanding cytoskeletal regulation. Semin Cell Dev Biol. (2000) 11(4):301–8. doi: 10.1006/scdb.2000.0180
12. Watts TL, Fasano A. Modulation of intestinal permeability: a novel and innovative approach for the oral delivery of drugs, macromolecules and antigens. Biotechnol Genet Eng Rev. (2000) 17:433–53. doi: 10.1080/02648725.2000.10648001
13. Fukui H. Increased intestinal permeability and decreased barrier function: Does it really influence the risk of inflammation? Inflammation Intest Dis. (2016) 1(3):135–45. doi: 10.1159/000447252
14. Barbara G, Barbaro MR, Fuschi D, Palombo M, Falangone F, Cremon C, et al. Inflammatory and microbiota-related regulation of the intestinal epithelial barrier. Front Nutr. (2021) 8:718356. doi: 10.3389/fnut.2021.718356
15. Miner-Williams WM, Moughan PJ. Intestinal barrier dysfunction: implications for chronic inflammatory conditions of the bowel. Nutr Res Rev. (2016) 29(1):40–59. doi: 10.1017/S0954422416000019
16. Miyakawa M, Konno T, Kohno T, Kikuchi S, Tanaka H, Kojima T. Increase in epithelial permeability and cell metabolism by high mobility group box 1, inflammatory cytokines and TPEN in caco-2 cells as a novel model of inflammatory bowel disease. Int J Mol Sci. (2020) 21(22):8434. doi: 10.3390/ijms21228434
17. Kiesslich R, Duckworth CA, Moussata D, Gloeckner A, Lim LG, Goetz M, et al. Local barrier dysfunction identified by confocal laser endomicroscopy predicts relapse in inflammatory bowel disease. Gut. (2012) 61(8):1146–53. doi: 10.1136/gutjnl-2011-300695
18. Neurath MF, Travis SP. Mucosal healing in inflammatory bowel diseases: a systematic review. Gut. (2012) 61(11):1619–35. doi: 10.1136/gutjnl-2012-302830
19. Osterman MT, Gordon IO, Davis EM, Ciorba M, Glover SC, Abraham B, et al. Mucosal biomarker of innate immune activation predicts response to vedolizumab in crohn's disease. Inflammation Bowel Dis. (2020) 26(10):1554–61. doi: 10.1093/ibd/izz222
20. Brandtzaeg P. Inflammatory bowel disease: clinics and pathology. do inflammatory bowel disease and periodontal disease have similar immunopathogeneses? Acta Odontol Scand. (2001) 59(4):235–43. doi: 10.1080/00016350152509265
21. Noguchi M, Hiwatashi N, Liu Z, Toyota T. Secretion imbalance between tumour necrosis factor and its inhibitor in inflammatory bowel disease. Gut. (1998) 43(2):203–9. doi: 10.1136/gut.43.2.203
22. Actis GC, Rosina F, Mackay IR. Inflammatory bowel disease: beyond the boundaries of the bowel. Expert Rev Gastroenterol Hepatol. (2011) 5(3):401–10. doi: 10.1586/egh.11.23
23. Katsanos KH, Papadakis KA. Inflammatory bowel disease: Updates on molecular targets for biologics. Gut Liver. (2017) 11(4):455–63. doi: 10.5009/gnl16308
24. Nikolaus S, Bauditz J, Gionchetti P, Witt C, Lochs H, Schreiber S. Increased secretion of pro-inflammatory cytokines by circulating polymorphonuclear neutrophils and regulation by interleukin 10 during intestinal inflammation. Gut. (1998) 42(4):470–6. doi: 10.1136/gut.42.4.470
25. Seegert D, Rosenstiel P, Pfahler H, Pfefferkorn P, Nikolaus S, Schreiber S. Increased expression of IL-16 in inflammatory bowel disease. Gut. (2001) 48(3):326–32. doi: 10.1136/gut.48.3.326
26. Tsianos EV, Katsanos K. Do we really understand what the immunological disturbances in inflammatory bowel disease mean? World J Gastroenterol. (2009) 15(5):521–5. doi: 10.3748/wjg.15.521
27. Al-Sadi R, Guo S, Ye D, Ma TY. TNF-alpha modulation of intestinal epithelial tight junction barrier is regulated by ERK1/2 activation of elk-1. Am J Pathol. (2013) 183(6):1871–576 84. doi: 10.1016/j.ajpath.2013.09.001
28. Al-Sadi R, Guo S, Ye D, Rawat M, Ma TY. TNF-alpha modulation of intestinal tight junction permeability is mediated by NIK/IKK-alpha axis activation of the canonical NF-kappaB pathway. Am J Pathol. (2016) 186(5):1151–65. doi: 10.1016/j.ajpath.2015.12.016
29. Clayburgh DR, Musch MW, Leitges M, Fu YX, Turner JR. Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J Clin Invest. (2006) 116(10):2682–94. doi: 10.1172/JCI29218
30. Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-alpha modulation of caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol. (2005) 288(3):G422–30. doi: 10.1152/ajpgi.00412.2004
31. Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, et al. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa b activation. Am J Physiol Gastrointest Liver Physiol. (2004) 286(3):G367–76. doi: 10.1152/ajpgi.00173.2003
32. Weber CR, Raleigh DR, Su L, Shen L, Sullivan EA, Wang Y, et al. Epithelial myosin light chain kinase activation induces mucosal interleukin-13 expression to alter tight junction ion selectivity. J Biol Chem. (2010) 285(16):12037–46. doi: 10.1074/jbc.M109.064808
33. Ye D, Ma TY. Cellular and molecular mechanisms that mediate basal and tumour necrosis factor-alpha-induced regulation of myosin light chain kinase gene activity. J Cell Mol Med. (2008) 12(4):1331–46. doi: 10.1111/j.1582-4934.2008.00302.x
34. Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, et al. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology. (2002) 123(1):163–72. doi: 10.1053/gast.2002.34235
35. Bruewer M, Samarin S, Nusrat A. Inflammatory bowel disease and the apical junctional complex. Ann N Y Acad Sci. (2006) 1072:242–52. doi: 10.1196/annals.1326.017
36. Theiss AL, Jenkins AK, Okoro NI, Klapproth JM, Merlin D, Sitaraman SV. Prohibitin inhibits tumor necrosis factor alpha-induced nuclear factor-kappa b nuclear translocation via the novel mechanism of decreasing importin alpha3 expression. Mol Biol Cell. (2009) 20(20):4412–23. doi: 10.1091/mbc.e09-05-0361
37. Khare V, Krnjic A, Frick A, Gmainer C, Asboth M, Jimenez K, et al. Mesalamine and azathioprine modulate junctional complexes and restore epithelial barrier function in intestinal inflammation. Sci Rep. (2019) 9(1):2842. doi: 10.1038/s41598-019-39401-0
38. Yuan SN, Wang MX, Han JL, Feng CY, Wang M, Wang M, et al. Improved colonic inflammation by nervonic acid via inhibition of NF-kappaB signaling pathway of DSS-induced colitis mice. Phytomedicine. (2023) 112:154702. doi: 10.1016/j.phymed.2023.154702
39. Zhou Z, Wang L, Feng P, Yin L, Wang C, Zhi S, et al. Inhibition of epithelial TNF-alpha receptors by purified fruit bromelain ameliorates intestinal inflammation and barrier dysfunction in colitis. Front Immunol. (2017) 8:1468. doi: 10.3389/fimmu.2017.01468
40. Ardizzone S, Bianchi Porro G. Biologic therapy for inflammatory bowel disease. Drugs. (2005) 65(16):2253–86. doi: 10.2165/00003495-200565160-00002
41. Bachmann C, Klibanov AL, Olson TS, Sonnenschein JR, Rivera-Nieves J, Cominelli F, et al. Targeting mucosal addressin cellular adhesion molecule (MAdCAM)-1 to noninvasively image experimental crohn's disease. Gastroenterology. (2006) 130(1):8–16. doi: 10.1053/j.gastro.2005.11.009
42. Shetty A, Forbes A. Pharmacogenomics of response to anti-tumor necrosis factor therapy in patients with crohn's disease. Am J Pharmacogenomics. (2002) 2(4):215–21. doi: 10.2165/00129785-200202040-00001
43. Souza RF, Caetano MAF, Magalhaes HIR, Castelucci P. Study of tumor necrosis factor receptor in the inflammatory bowel disease. World J Gastroenterol. (2023) 29(18):2733–46. doi: 10.3748/wjg.v29.i18.2733
44. Langhorst J, Wulfert H, Lauche R, Klose P, Cramer H, Dobos GJ, et al. Systematic review of complementary and alternative medicine treatments in inflammatory bowel diseases. J Crohns Colitis. (2015) 9(1):86–106. doi: 10.1093/ecco-jcc/jju007
45. Schwermer M, Fetz K, Langler A, Ostermann T, Zuzak TJ. Complementary, alternative, integrative and dietary therapies for children with crohn's disease - a systematic review. Complement Ther Med. (2020) 52:102493. doi: 10.1016/j.ctim.2020.102493
46. Bashir NS, Hughes A, Ungar WJ. Infliximab pricing in international economic evaluations in inflammatory bowel disease to inform biologic and biosimilar access policies: A systematic review. MDM Policy Pract. (2023) 8(1):23814683231156433. doi: 10.1177/23814683231156433
47. Ng JY, Liu H, Wang MC. Complementary and alternative medicine mention and recommendations in inflammatory bowel disease guidelines: systematic review and assessment using AGREE II. BMC Complement Med Ther. (2023) 23(1):230. doi: 10.1186/s12906-023-04062-0
48. Chandler M, Wollins E, Toles A, Borum M, Doman DB. The emerging therapeutic role of probiotics in inflammatory bowel disease. Gastroenterol Hepatol (N Y). (2008) 4(9):634–40.
49. Lin SC, Cheifetz AS. The use of complementary and alternative medicine in patients with inflammatory bowel disease. Gastroenterol Hepatol (N Y). (2018) 14(7):415–25.
50. Darb Emamie A, Rajabpour M, Ghanavati R, Asadolahi P, Farzi S, Sobouti B, et al. The effects of probiotics, prebiotics and synbiotics on the reduction of IBD complications, a periodic review during 2009-2020. J Appl Microbiol. (2021) 130(6):1823–38. doi: 10.1111/jam.14907
51. Kim DY, Lee TS, Jung DH, Song EJ, Jang AR, Park JY, et al. Oral administration of lactobacillus sakei CVL-001 improves recovery from dextran sulfate sodium-induced colitis in mice by microbiota modulation. Microorganisms. (2023) 11(5):1359. doi: 10.3390/microorganisms11051359
52. Le A, Mantel M, Marchix J, Bodinier M, Jan G, Rolli-Derkinderen M. Inflammatory bowel disease therapeutic strategies by modulation of the microbiota: how and when to introduce pre-, pro-, syn-, or postbiotics? Am J Physiol Gastrointest Liver Physiol. (2022) 323(6):G523–G53. doi: 10.1152/ajpgi.00002.2022
53. Bibiloni R, Fedorak RN, Tannock GW, Madsen KL, Gionchetti P, Campieri M, et al. VSL3 probiotic-mixture induces remission in patients with active ulcerative colitis. Am J Gastroenterol. (2005) 100(7):1539–46. doi: 10.1111/j.1572-0241.2005.41794.x
54. Huynh HQ, deBruyn J, Guan L, Diaz H, Li M, Girgis S, et al. Probiotic preparation VSL3 induces remission in children with mild to moderate acute ulcerative colitis: a pilot study. Inflammation Bowel Dis. (2009) 15(5):760–8. doi: 10.1002/ibd.20816
55. Guandalini S. Are probiotics or prebiotics useful in pediatric irritable bowel syndrome or inflammatory bowel disease? Front Med (Lausanne). (2014) 1:23. doi: 10.3389/fmed.2014.00023
56. Guo S, Chen S, Ma J, Ma Y, Zhu J, Ma Y, et al. Escherichia coli nissle 1917 protects intestinal barrier function by inhibiting NF-kappaB-Mediated activation of the MLCK-P-MLC signaling pathway. Mediators Inflamm. (2019) 2019:5796491. doi: 10.1155/2019/5796491
57. Amaretti A, di Nunzio M, Pompei A, Raimondi S, Rossi M, Bordoni A. Antioxidant properties of potentially probiotic bacteria: in vitro and in vivo activities. Appl Microbiol Biotechnol. (2013) 97(2):809–17. doi: 10.1007/s00253-012-4241-7
58. Averina OV, Poluektova EU, Marsova MV, Danilenko VN. Biomarkers and utility of the antioxidant potential of probiotic lactobacilli and bifidobacteria as representatives of the human gut microbiota. Biomedicines. (2021) 9(10):1340. doi: 10.3390/biomedicines9101340
59. Grover S, Rashmi HM, Srivastava AK, Batish VK. Probiotics for human health –new innovations and emerging trends. Gut Pathog. (2012) 4(1):15. doi: 10.1186/1757-4749-4-15
60. Blackwood BP, Yuan CY, Wood DR, Nicolas JD, Grothaus JS, Hunter CJ. Probiotic lactobacillus species strengthen intestinal barrier function and tight junction integrity in experimental necrotizing enterocolitis. J Probiotics Health. (2017) 5(1):159. doi: 10.4172/2329-8901.1000159
61. Eun CS, Kim YS, Han DS, Choi JH, Lee AR, Park YK. Lactobacillus casei prevents impaired barrier function in intestinal epithelial cells. APMIS. (2011) 119(1):49–56. doi: 10.1111/j.1600-0463.2010.02691.x
62. Guo S, Gillingham T, Guo Y, Meng D, Zhu W, Walker WA, et al. Secretions of bifidobacterium infantis and lactobacillus acidophilus protect intestinal epithelial barrier function. J Pediatr Gastroenterol Nutr. (2017) 64(3):404–12. doi: 10.1097/MPG.0000000000001310
63. Hummel S, Veltman K, Cichon C, Sonnenborn U, Schmidt MA. Differential targeting of the e-Cadherin/beta-Catenin complex by gram-positive probiotic lactobacilli improves epithelial barrier function. Appl Environ Microbiol. (2012) 78(4):1140–7. doi: 10.1128/AEM.06983-11
64. Lepine AFP, de Wit N, Oosterink E, Wichers H, Mes J, de Vos P. Lactobacillus acidophilus attenuates salmonella-induced stress of epithelial cells by modulating tight-junction genes and cytokine responses. Front Microbiol. (2018) 9:1439. doi: 10.3389/fmicb.2018.01439
65. Montalto M, Maggiano N, Ricci R, Curigliano V, Santoro L, Di Nicuolo F, et al. Lactobacillus acidophilus protects tight junctions from aspirin damage in HT-29 cells. Digestion. (2004) 69(4):225–8. doi: 10.1159/000079152
66. Mujagic Z, de Vos P, Boekschoten MV, Govers C, Pieters HH, de Wit NJ, et al. The effects of lactobacillus plantarum on small intestinal barrier function and mucosal gene transcription; a randomized double-blind placebo controlled trial. Sci Rep. (2017) 7:40128. doi: 10.1038/srep40128
67. Parassol N, Freitas M, Thoreux K, Dalmasso G, Bourdet-Sicard R, Rampal P. Lactobacillus casei DN-114 001 inhibits the increase in paracellular permeability of enteropathogenic escherichia coli-infected T84 cells. Res Microbiol. (2005) 156(2):256–62. doi: 10.1016/j.resmic.2004.09.013
68. Resta-Lenert S, Barrett KE. Live probiotics protect intestinal epithelial cells from the effects of infection with enteroinvasive escherichia coli (EIEC). Gut. (2003) 52(7):988–97. doi: 10.1136/gut.52.7.988
69. Wang H, Zhang Q, Niu Y, Zhang X, Lu R. Surface-layer protein from lactobacillus acidophilus NCFM attenuates tumor necrosis factor-alpha-induced intestinal barrier dysfunction and inflammation. Int J Biol Macromol. (2019) 136:27–34. doi: 10.1016/j.ijbiomac.2019.06.04
70. Zakostelska Z, Kverka M, Klimesova K, Rossmann P, Mrazek J, Kopecny J, et al. Lysate of probiotic lactobacillus casei DN-114 001 ameliorates colitis by strengthening the gut barrier function and changing the gut microenvironment. PloS One. (2011) 6(11):e27961. doi: 10.1371/journal.pone.0027961
71. Al-Sadi R, Nighot P, Nighot M, Haque M, Rawat M, Ma TY. Lactobacillus acidophilus induces a strain-specific and toll-like receptor 2-dependent enhancement of intestinal epithelial tight junction barrier and protection against intestinal inflammation. Am J Pathol. (2021) 191(5):872–84. doi: 10.1016/j.ajpath.2021.02.003
72. Marchiando AM, Shen L, Graham WV, Edelblum KL, Duckworth CA, Guan Y, et al. The epithelial barrier is maintained by in vivo tight junction expansion during pathologic intestinal epithelial shedding. Gastroenterology. (2011) 140(4):1208–18 e1-2. doi: 10.1053/j.gastro.2011.01.004
73. Bagheri V, Askari A, Arababadi MK, Kennedy D. Can toll-like receptor (TLR) 2 be considered as a new target for immunotherapy against hepatitis b infection? Hum Immunol. (2014) 75(6):549–54. doi: 10.1016/j.humimm.2014.02.018
74. Nemati M, Larussa T, Khorramdelazad H, Mahmoodi M, Jafarzadeh A. Toll-like receptor 2: An important immunomodulatory molecule during helicobacter pylori infection. Life Sci. (2017) 178:17–29. doi: 10.1016/j.lfs.2017.04.006
75. Sabroe I, Dower SK, Whyte MK. The role of toll-like receptors in the regulation of neutrophil migration, activation, and apoptosis. Clin Infect Dis. (2005) 41 Suppl 7:S421–6. doi: 10.1086/431992
76. Takeuchi O, Akira S. Toll-like receptors; their physiological role and signal transduction system. Int Immunopharmacol. (2001) 1(4):625–35. doi: 10.1016/s1567-5769(01)00010-8
77. Al-Sadi R, Youssef M, Rawat M, Guo S, Dokladny K, Haque M, et al. MMP-9-induced increase in intestinal epithelial tight permeability is mediated by p38 kinase signaling pathway activation of MLCK gene. Am J Physiol Gastrointest Liver Physiol. (2019) 316(2):G278–G90. doi: 10.1152/ajpgi.00126.2018
78. Nighot P, Al-Sadi R, Rawat M, Guo S, Watterson DM, Ma T. Matrix metalloproteinase 9-induced increase in intestinal epithelial tight junction permeability contributes to the severity of experimental DSS colitis. Am J Physiol Gastrointest Liver Physiol. (2015) 309(12):G988–97. doi: 10.1152/ajpgi.00256.2015
79. Laparra JM, Glahn RP, Miller DD. Assessing potential effects of inulin and probiotic bacteria on fe availability from common beans (Phaseolus vulgaris l.) to caco-2 cells. J Food Sci. (2009) 74(2):H40–6. doi: 10.1111/j.1750-3841.2008.01027.x
80. Makras L, Triantafyllou V, Fayol-Messaoudi D, Adriany T, Zoumpopoulou G, Tsakalidou E, et al. Kinetic analysis of the antibacterial activity of probiotic lactobacilli towards salmonella enterica serovar typhimurium reveals a role for lactic acid and other inhibitory compounds. Res Microbiol. (2006) 157(3):241–7. doi: 10.1016/j.resmic.2005.09.002
81. Boivin MA, Ye D, Kennedy JC, Al-Sadi R, Shepela C, Ma TY. Mechanism of glucocorticoid regulation of the intestinal tight junction barrier. Am J Physiol Gastrointest Liver Physiol. (2007) 292(2):G590–8. doi: 10.1152/ajpgi.00252.2006
82. Al-Sadi R, Ye D, Dokladny K, Ma TY. Mechanism of IL-1beta-induced increase in intestinal epithelial tight junction permeability. J Immunol. (2008) 180(8):5653–61.
83. Jobin C, Sartor RB. The i kappa B/NF-kappa b system: a key determinant of mucosalinflammation and protection. Am J Physiol Cell Physiol. (2000) 278(3):C451–62. doi: 10.1152/ajpcell.2000.278.3.C451
84. Kondylis V, Kumari S, Vlantis K, Pasparakis M. The interplay of IKK, NF-kappaB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol Rev. (2017) 277(1):113–27. doi: 10.1111/imr.12550
85. Neurath MF, Becker C, Barbulescu K. Role of NF-kappaB in immune and inflammatory responses in the gut. Gut. (1998) 43(6):856–60. doi: 10.1136/gut.43.6.856
86. Wang B, Shen J. NF-kappaB inducing kinase regulates intestinal immunity and homeostasis. Front Immunol. (2022) 13:895636. doi: 10.3389/fimmu.2022.895636
87. Larabi A, Barnich N, Nguyen HTT. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy. (2020) 16(1):38–51. doi: 10.1080/15548627.2019.1635384
88. Seya T, Funami K, Taniguchi M, Matsumoto M. Antibodies against human toll-like receptors (TLRs): TLR distribution and localization in human dendritic cells. J Endotoxin Res. (2005) 11(6):369–74. doi: 10.1179/096805105X67292
89. Cabal-Hierro L, Rodriguez M, Artime N, Iglesias J, Ugarte L, Prado MA, et al. TRAF-mediated modulation of NF-kB AND JNK activation by TNFR2. Cell Signal. (2014) 26(12):2658–66. doi: 10.1016/j.cellsig.2014.08.011
90. Bardag-Gorce F, Oliva J, Lin A, Li J, French BA, French SW. SAMe prevents the up regulation of toll-like receptor signaling in mallory-denk body forming hepatocytes. Exp Mol Pathol. (2010) 88(3):376–9. doi: 10.1016/j.yexmp.2010.02.004
91. Im J, Baik JE, Lee D, Park OJ, Park DH, Yun CH, et al. Bacterial lipoproteins induce BAFF production via TLR2/MyD88/JNK signaling pathways in dendritic cells. Front Immunol. (2020) 11:564699. doi: 10.3389/fimmu.2020.564699
92. Yang HL, Sun YZ, Hu X, Ye JD, Lu KL, Hu LH, et al. Bacillus pumilus SE5 originated PG and LTA tuned the intestinal TLRs/MyD88 signaling and microbiota in grouper (Epinephelus coioides). Fish Shellfish Immunol. (2019) 88:266–71. doi: 10.1016/j.fsi.2019.03.005
93. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. (2003) 301(5633):640–3. doi: 10.1126/science.1087262
94. Zheng H, Heiderscheidt CA, Joo M, Gao X, Knezevic N, Mehta D, et al. MYD88-dependent and -independent activation of TREM-1 via specific TLR ligands. Eur J Immunol. (2010) 40(1):162–71. doi: 10.1002/eji.200839156
95. Bono C, Guerrero P, Erades A, Jordan-Pla A, Yanez A, Gil ML. Direct TLR2 signaling through mTOR and TBK1 induces C/EBPbeta and IRF7-dependent macrophage differentiation in hematopoietic stem and progenitor cells. Stem Cells. (2022) 40(10):949–62. doi: 10.1093/stmcls/sxac053
96. Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology. (2007) 132(4):1359–74. doi: 10.1053/j.gastro.2007.02.056
97. Huang Y, Zhang X, Wang P, Li Y, Yao J. Identification of hub genes and pathways in colitis-associated colon cancer by integrated bioinformatic analysis. BMC Genom Data. (2022) 23(1):48. doi: 10.1186/s12863-022-01065-7
98. Jiang X, Fang L, Wu H, Mei X, He F, Ding P, et al. TLR2 regulates allergic airway inflammation and autophagy through PI3K/Akt signaling pathway. Inflammation. (2017) 40(4):1382–92. doi: 10.1007/s10753-017-0581-x
99. Li B, Xi P, Wang Z, Han X, Xu Y, Zhang Y, et al. PI3K/Akt/mTOR signaling pathway participates in streptococcus uberis-induced inflammation in mammary epithelial cells in concert with the classical TLRs/NF-kB pathway. Vet Microbiol. (2018) 227:103–11. doi: 10.1016/j.vetmic.2018.10.031
100. Liu YD, Ji CB, Li SB, Yan F, Gu QS, Balic JJ, et al. Toll-like receptor 2 stimulation promotes colorectal cancer cell growth via PI3K/Akt and NF-kappaB signaling pathways. Int Immunopharmacol. (2018) 59:375–83. doi: 10.1016/j.intimp.2018.04.033
101. Sun PP, Li D, Su M, Ren Q, Guo WP, Wang JL, et al. Cell membrane-bound toll-like receptor-1/2/4/6 monomers and -2 heterodimer inhibit enterovirus 71 replication by activating the antiviral innate response. Front Immunol. (2023) 14:1187035. doi: 10.3389/fimmu.2023.1187035
102. Shen L. Tight junctions on the move: molecular mechanisms for epithelial barrier regulation. Ann N Y Acad Sci. (2012) 1258:9–18. doi: 10.1111/j.1749-6632.2012.06613.x
103. Yao Y, Feng Q, Shen J. Myosin light chain kinase regulates intestinal permeability of mucosal homeostasis in crohn's disease. Expert Rev Clin Immunol. (2020) 16(12):1127–41. doi: 10.1080/1744666X.2021.1850269
104. Graham WV, He W, Marchiando AM, Zha J, Singh G, Li HS, et al. Intracellular MLCK1 diversion reverses barrier loss to restore mucosal homeostasis. Nat Med. (2019) 25(4):690–700. doi: 10.1038/s41591-019-0393-7
105. He F, Peng J, Deng XL, Yang LF, Camara AD, Omran A, et al. Mechanisms of tumor necrosis factor-alpha-induced leaks in intestine epithelial barrier. Cytokine. (2012) 59(2):264–72. doi: 10.1016/j.cyto.2012.04.008
106. Odenwald MA, Turner JR. Intestinal permeability defects: is it time to treat? Clin Gastroenterol Hepatol. (2013) 11(9):1075–83. doi: 10.1016/j.cgh.2013.07.001
107. Savkovic SD, Ramaswamy A, Koutsouris A, Hecht G. EPEC-activated ERK1/2 participate in inflammatory response but not tight junction barrier disruption. Am J Physiol Gastrointest Liver Physiol. (2001) 281(4):G890–8. doi: 10.1152/ajpgi.2001.281.4.G890
108. Shen Y, Zhou M, Yan J, Gong Z, Xiao Y, Zhang C, et al. miR-200b inhibits TNF-alpha-induced IL-8 secretion and tight junction disruption of intestinal epithelial cells in vitro. Am J Physiol Gastrointest Liver Physiol. (2017) 312(2):G123–G32. doi: 10.1152/ajpgi.00316.2016
109. Suzuki M, Nagaishi T, Yamazaki M, Onizawa M, Watabe T, Sakamaki Y, et al. Myosin light chain kinase expression induced via tumor necrosis factor receptor 2 signaling in the epithelial cells regulates the development of colitis-associated carcinogenesis. PloS One. (2014) 9(2):e88369. doi: 10.1371/journal.pone.0088369
110. Fries W, Belvedere A, Vetrano S. Sealing the broken barrier in IBD: intestinal permeability, epithelial cells and junctions. Curr Drug Targets. (2013) 14(12):1460–70. doi: 10.2174/1389450111314120011
111. DuPont AW, DuPont HL. The intestinal microbiota and chronic disorders of the gut. Nat Rev Gastroenterol Hepatol. (2011) 8(9):523–31. doi: 10.1038/nrgastro.2011.133
112. Shadnoush M, Hosseini RS, Khalilnezhad A, Navai L, Goudarzi H, Vaezjalali M. Effects of probiotics on gut microbiota in patients with inflammatory bowel disease: A double-blind, placebo-controlled clinical trial. Korean J Gastroenterol. (2015) 65(4):215–21. doi: 10.4166/kjg.2015.65.4.215
113. Mennigen R, Nolte K, Rijcken E, Utech M, Loeffler B, Senninger N, et al. Probiotic mixture VSL3 protects the epithelial barrier by maintaining tight junction protein expression and preventing apoptosis in a murine model of colitis. Am J Physiol Gastrointest Liver Physiol. (2009) 296(5):G1140–9. doi: 10.1152/ajpgi.90534.2008
114. di Vito R, Conte C, Traina G. A multi-strain probiotic formulation improves intestinal barrier function by the modulation of tight and adherent junction proteins. Cells. (2022) 11(16):2617. doi: 10.3390/cells11162617
115. Hsieh CY, Osaka T, Moriyama E, Date Y, Kikuchi J, Tsuneda S. Strengthening of the intestinal epithelial tight junction by bifidobacterium bifidum. Physiol Rep. (2015) 3(3):e12327. doi: 10.14814/phy2.12327
116. Madsen K, Cornish A, Soper P, McKaigney C, Jijon H, Yachimec C, et al. Probiotic bacteria enhance murine and human intestinal epithelial barrier function. Gastroenterology. (2001) 121(3):580–91. doi: 10.1053/gast.2001.27224
117. Chorawala MR, Chauhan S, Patel R, Shah G. Cell wall contents of probiotics (Lactobacillus species) protect against lipopolysaccharide (LPS)-induced murine colitis by limiting immuno-inflammation and oxidative stress. Probiotics Antimicrob Proteins. (2021) 13(4):1005–17. doi: 10.1007/s12602-020-09738-4
118. de Matuoka ECG, Monedero V, Zuniga M, Velez D, Devesa V. In vitro evaluation of the protective role of lactobacillus StrainsAgainst inorganic arsenic toxicity. Probiotics Antimicrob Proteins. (2020) 12(4):1484–91. doi: 10.1007/s12602-020-09639-6
119. Toumi R, Abdelouhab K, Rafa H, Soufli I, Raissi-Kerboua D, Djeraba Z, et al. Beneficial role of the probiotic mixture ultrabiotique on maintaining the integrity of intestinal mucosal barrier in DSS-induced experimental colitis. Immunopharmacol Immunotoxicol. (2013) 35(3):403–9. doi: 10.3109/08923973.2013.790413
120. Zhang P, Han X, Zhang X, Zhu X. Lactobacillus acidophilus ATCC 4356 alleviates renal ischemia-reperfusion injury through antioxidant stress and anti-inflammatory responses and improves intestinal microbial distribution. Front Nutr. (2021) 8:667695. doi: 10.3389/fnut.2021.667695
121. Al-Sadi R, Ye D, Said HM, Ma TY. IL-1beta-induced increase in intestinal epithelial tight junction permeability is mediated by MEKK-1 activation of canonical NF-kappaB pathway. Am J Pathol. (2010) 177(5):2310–22. doi: 10.2353/ajpath.2010.100371
122. Thu YM, Richmond A. NF-kappaB inducing kinase: a key regulator in the immune system and in cancer. Cytokine Growth Factor Rev. (2010) 21(4):213–26. doi: 10.1016/j.cytogfr.2010.06.002
123. Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. (2002) 296(5573):1634–5. doi: 10.1126/science.1071924
124. Brasier AR. The NF-kappaB regulatory network. Cardiovasc Toxicol. (2006) 6(2):111–30. doi: 10.1385/ct:6:2:111
125. Funder JW. Glucocorticoid and mineralocorticoid receptors: biology and clinical relevance. Annu Rev Med. (1997) 48:231–40. doi: 10.1146/annurev.med.48.1.231
126. Razani B, Reichardt AD, Cheng G. Non-canonical NF-kappaB signaling activation and regulation: principles and perspectives. Immunol Rev. (2011) 244(1):44–54. doi: 10.1111/j.1600-065X.2011.01059.x
127. Swerdlow MP, Kennedy DR, Kennedy JS, Washabau RJ, Henthorn PS, Moore PF, et al. Expression and function of TLR2, TLR4, and Nod2 in primary canine colonic epithelial cells. Vet Immunol Immunopathol. (2006) 114(3-4):313–9. doi: 10.1016/j.vetimm.2006.09.003
128. Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, et al. Toll-like receptor 2-8 mediated NF-kappa b activation requires a Rac1-dependent pathway. Nat Immunol. (2000) 1(6):533–40. doi: 10.1038/82797
129. Price AE, Shamardani K, Lugo KA, Deguine J, Roberts AW, Lee BL, et al. A map of toll-like receptor expression in the intestinal epithelium reveals distinct spatial, cell type-specific, and temporal patterns. Immunity. (2018) 49(3):560–75.e6. doi: 10.1016/j.immuni.2018.07.016
130. Wells JM, Rossi O, Meijerink M, van Baarlen P. Epithelial crosstalk at the microbiota-mucosal interface. Proc Natl Acad Sci USA. (2011) 108 Suppl 1(Suppl 1):4607–14. doi: 10.1073/pnas.1000092107
131. Cronin JG, Turner ML, Goetze L, Bryant CE, Sheldon IM. Toll-like receptor 4 and MYD88-dependent signaling mechanisms of the innate immune system are essential for the response to lipopolysaccharide by epithelial and stromal cells of the bovine endometrium. Biol Reprod. (2012) 86(2):51. doi: 10.1095/biolreprod.111.092718
132. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. (2010) 11(5):373–84. doi: 10.1038/ni.1863
133. Cristofori F, Dargenio VN, Dargenio C, Miniello VL, Barone M, Francavilla R. Anti-inflammatory and immunomodulatory effects of probiotics in gut inflammation: A door to the body. Front Immunol. (2021) 12:578386. doi: 10.3389/fimmu.2021.578386
Keywords: Lactobacillus acidophilus, TNF-α, NF-κB, TLR-2, MLCK, PI3K, intestinal tight junction barrier
Citation: Haque M, Kaminsky L, Abdulqadir R, Engers J, Kovtunov E, Rawat M, Al-Sadi R and Ma TY (2024) Lactobacillus acidophilus inhibits the TNF-α-induced increase in intestinal epithelial tight junction permeability via a TLR-2 and PI3K-dependent inhibition of NF-κB activation. Front. Immunol. 15:1348010. doi: 10.3389/fimmu.2024.1348010
Received: 01 December 2023; Accepted: 25 June 2024;
Published: 16 July 2024.
Edited by:
Juandy Jo, University of Pelita Harapan, IndonesiaReviewed by:
Hoda Ibrahim Bahr, Suez Canal University, EgyptGustavo Pedraza-Alva, Universidad Nacional Autónoma de México, Mexico
Copyright © 2024 Haque, Kaminsky, Abdulqadir, Engers, Kovtunov, Rawat, Al-Sadi and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rana Al-Sadi, ralsadi@unmc.edu
†These authors have contributed equally to this work
‡These authors have contributed equally to this work and share senior authorship