 Hendrik Gassmann1,2*
Hendrik Gassmann1,2* Melanie Thiede1Jennifer Weiß1Emilie Biele1Luisa Flohé1Helena Lachermaier1Carolin Prexler1Valentina Evdokimova3
Melanie Thiede1Jennifer Weiß1Emilie Biele1Luisa Flohé1Helena Lachermaier1Carolin Prexler1Valentina Evdokimova3 Laszlo Radvanyi3
Laszlo Radvanyi3 Irfan Akhtar1
Irfan Akhtar1 Mina N. F. Morcos1
Mina N. F. Morcos1 Franziska Auer1
Franziska Auer1 Sebastian J. Schober1Julia Hauer1,2
Sebastian J. Schober1Julia Hauer1,2 Uwe Thiel1
Uwe Thiel1 Kristina von Heyking1*
Kristina von Heyking1*- 1Department of Pediatrics, Children’s Cancer Research Center, Kinderklinik München Schwabing, TUM School of Medicine, Technical University of Munich, Munich, Germany
- 2Cancer Consortium (DKTK), partner site Munich, a partnership between DKFZ and University TUM School of Medicine, Technical University of Munich, Munich, Germany
- 3Ontario Institute for Cancer Research, Toronto, ON, Canada
Introduction: Pediatric sarcomas, including osteosarcoma (OS), Ewing sarcoma (EwS) and rhabdomyosarcoma (RMS) carry low somatic mutational burden and low MHC-I expression, posing a challenge for T cell therapies. Our previous study showed that mediators of monocyte maturation sensitized the EwS cell line A673 to lysis by HLA-A*02:01/CHM1319-specific allorestricted T cell receptor (TCR) transgenic CD8+ T cells (CHM1319 CD8+ T cells).
Methods: In this study, we tested a panel of monocyte maturation cytokines for their ability to upregulate immunogenic cell surface markers on OS, EwS and RMS cell lines, using flow cytometry. xCELLigence, SRB and ELISpot assays were used to assess whether TNF pretreatment increases CD8+ T cell cytotoxicity.
Results: We observed that TNF and IL-1β upregulated MHC class I, ICAM-1 as well as CD83 and PD-L1 on the surface of pediatric sarcoma cell lines, while IL-4, GM-CSF, IL-6 and PGE2 failed to induce respective effects. Although pretreatment of pediatric sarcoma cell lines with TNF did not improve unspecific peripheral blood mononuclear cells (PBMCs) cytotoxicity, TNF enhanced specific lysis of 1/3 HLA-A2+ EwS cell lines by CHM1319 CD8+ T cells depending on MHC-I expression and ICAM-1 upregulation.
Discussion: Our study supports utilization of TNF or TNF-inducing regimens for upregulation of MHC-I and costimulatory surface molecules on pediatric sarcoma cells and for enhancing recognition of responsive HLA-A2+ EwS tumor cells by antigen-specific CD8+ T cells.
1 Introduction
Pediatric sarcomas account for 10% of all childhood cancers, of which osteosarcoma (OS) and Ewing sarcoma (EwS) are the two most frequent bone cancers, while rhabdomyosarcoma (RMS) is the most common soft tissue tumor (1). Pediatric sarcomas are considered non-immunogenic tumors with low mutational burden (2), low MHC-I expression (3–6) and scarce T cell infiltration (7–9). Thus far immune checkpoint blockade failed to show clinical benefit in pediatric sarcomas (9–12). Therefore, immunogenic conditioning of pediatric sarcomas prior to T cell mediated immunotherapy is of particular importance.
We previously generated HLA-A*02:01/CHM1319-specific allorestricted CD8+ T cells (CHM1319 CD8+ T cells) targeting EwS, which exerted anti-tumoral effects in vitro and in vivo. Furthermore, CHM1319 CD8+ T cells were associated with a partial response in at least one out of three treatment-refractory EwS patients (13–15). We also observed that the pretreatment of the EwS cell line A673 with mediators of monocyte cell maturation, including GM-CSF and IL-4 for 5 days followed by, IL-1β, IL-6, TNF and PGE2 for 2 days, enhanced the recognition and cytotoxicity of CHM1319 CD8+ T cells against A673 cells in vitro (16). Yet, for the clinical setting, the administration or induction of six different cytokines is impracticable.
In this study, we hypothesized that individual monocyte maturation mediators induce immunogenicity of pediatric sarcoma cells, thereby enhancing their recognition and killing by healthy donor peripheral blood mononuclear cells (PBMCs) or CHM1319 CD8+ T cells. Among six cytokines tested, we identified TNF as the most prominent MHC-I and ICAM-1 inducer and CHM1319 CD8+ T cell sensitizer. As such, our study provides the basis for further evaluating TNF or TNF-inducing conditioning regimes prior to adoptive T cell therapies in pediatric sarcomas.
2 Materials and methods
2.1 Cell lines and cell culture conditions
OS (SJSA-1, U2OS), EwS cell lines (A673, CHLA10, EW7, TC32, TC71, TTC466, TTC633, SB-KMS-KS1/SBSR-AKS and SKNMC) and RMS cell lines (RD, RH30) were used in this study. Cells were purchased from American Type Culture Collection (ATCC): A673, MRC5 fibroblasts, RD, SJSA-1 and U2OS, or the German Collection of Microorganisms and Cell Cultures (DSMZ): RH30, SKNMC and TC71. SB-KMS-KS1 was established at the Children’s Cancer Research Center, Kinderklinik Schwabing (Technical University Munich, Germany). CHLA10, TC32, TTC466 and TTC633 were kindly provided by Prof. Poul Sorensen (University of British Columbia, Canada), which were originally obtained from the Childhood Cancer Repository (CCR, Alex’s Lemonade Stand Foundation, Children’s Oncology Group, COG). EW7 was kindly provided by Prof. Olivier Delattre (Institut Curie, Paris, France). OS cell lines, EwS cell lines and RH30 were cultured in RPMI 1640 medium (Life Technologies Limited, Paisley, UK). RD was cultured in DMEM (Life Technologies Limited, Paisley, UK), and CHLA10 was cultured in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 4 mM L-Glutamine and 1X ITS (5 μg/ml insulin, 5 μg/ml transferrin, 5 ng/ml selenous acid). MRC5 and TTC633 were cultured in DMEM/F12 (Life Technologies Limited Paisley, UK). Growth medium of all cell lines was supplemented with 10% fetal bovine serum (FBS) (Life Technologies Limited, Paisley, UK), 100 U/ml penicillin and 100 µg/ml streptomycin (Life Technologies Corporation, Grand Island, NY, USA). All cell lines were cultured at 37°C with 5% CO2 and regularly tested for mycoplasma contamination (Lonza).
For cytokine treatment, 0.005 –2 x 105 pediatric sarcoma cells were seeded in flat bottom 6-well plates (Falcon) or 96-well plates (Sartorius, Germany) in 3 ml or 200 µl of growth medium, respectively. Next day, the medium was replaced and supplemented with either 800 U/ml GM-CSF (Sanofi, Paris, France), 1000 U/ml IL-4, 10 ng/ml IL-1β, 1000 U/ml IL-6, 1-100 ng/ml TNF or 1 µg/ml PGE2 (all from R&D Systems, Minneapolis, MN, USA). As a positive control, cells were treated with 100 U/ml IFNγ (R&D Systems, Minneapolis, MN, USA). Cells were treated for 72 h or 96 h and analyzed by flow cytometry. For western blotting, cells were treated with 10 ng/ml IL-1β (#IL038, Millipore), 10 ng/ml IL-6 (#GF338, Millipore) or 10 ng/ml TNF (#PHC3011, Thermo Fisher), or 10 ng/ml IFNα (#8927, Cell Signaling) as positive control. Where indicated, EwS cells were preincubated with monocyte maturation mediators (DC cytokines), as described previously for the generation of monocytic-derived dendritic cells (16, 17). Briefly, EwS cells were incubated with 800 U/ml GM-CSF and 1000 U/ml IL-4 for 5 days, with medium renewal at day 3. At day 6, fresh medium supplemented with IL-1β, IL-6, TNF (10 ng/ml) and PGE2 (concentrations as above) was added, and cells were harvested for analysis at day 7.
2.2 PBMC isolation
The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Commission of the Medical Faculty of the Technical University of Munich (2562/09). Healthy donor buffy coats were acquired from the DRK-Blutspendedienst (Ulm, Germany) or, after informed consent, venous EDTA-blood was obtained from healthy donors. Density-gradient centrifugation with Ficoll-Paque (GE Healthcare, Uppsala, Sweden) was used to isolate PBMCs as reported previously (18). The ACK Lysis Buffer (Thermo Fisher Scientific) was used to remove contaminating erythrocytes.
2.3 Generation of HLA-A*02:01/CHM1319-specific allorestricted T-cell receptor transgenic CD8+ T cells
HLA-A*02:01/CHM1319-specific allorestricted T-cell receptor (TCR) transgenic CD8+ T cells (CHM1319 CD8+ T cells) were generated as described previously (14, 18, 19). Briefly, CD8+ T cells were negatively isolated (untouched, Miltenyi Biotech, Germany) from healthy donor PBMCs and activated with anti-CD3/CD28 Dynabeads™ (Thermo Fisher Scientific BalticsUAB, Vilnius, Lithuania) according to the supplier’s instructions and 100 U/ml recombinant human (rh) IL-2 (Novartis) for 2 days. At day 3 and 4, T cells were retrovirally transduced with the CHM1319 TCR transgene using the retroviral vector pMP-71 (14). Transduction efficacy was evaluated by flow cytometry and T cells were selected using anti-PE microbeads (Miltenyi Biotech, Germany) bound to PE anti-mouse TCRβ chain antibody (H57-587, BioLegend) recognizing the murinized constant region of the CHM1319 CD8+ T cells. T cells were cultured in AIM-V medium (Life Technologies Limited, Paisley, UK) supplemented with 5% human AB serum (Sigma-Aldrich, Germany), 100 U/ml IL-2, 2 ng/ml IL15 (R&D Systems) and antibiotics, as described above.
2.4 Flow cytometry
To assess the surface expression of immunogenic markers, 0.5-2 x 105 cells were stained in 96-well plates in 50 µl PBS with 1:100 diluted fluorochrome-conjugated antibodies against CD86 (REA968, VioBlue), CD83 (REA714, APC-Vio770), PD-L1 (REA1197, VioBright FITC), MHC-I (HLA-A,B,C; REA230, APC), MHC-II (HLA-DR, DP, DQ; REA332, PE-Vio770), PD-1 (REA1165, APC), CD152/CTLA-4 (BNI3, APC), CD223/LAG-3 (REA351, APC), CD366/TIM-3 (REA635, APC) or respective isotype controls (mouse IgG2a-FITC, mouse IgG1-APC/IS5-21F5 or REA293), at 4°C protected from light. All antibodies were obtained from Miltenyi Biotech, Germany, except for HLA-A2 (BB7.2) and CD152/CTLA-4 (BNI3), both BD Biosciences. Transduction of CHM1319 CD8+ T cells was confirmed by staining the murinized part of the TCR with an anti-mouse β TCR chain antibody (H57-597, PE), obtained from BioLegend. After 15 min-incubation, cells were washed with 200 µl PBS at 300 g for 5 min, resuspended in 100 µl PBS and acquired on the MACSQuant Analyzer 10 (Miltenyi Biotech, Germany). Single stainings of respective cells were used for compensation of multicolor staining panels. Data were analyzed using FlowJo™ 10.1 (BD Bioscience, Heidelberg, Germany) and applying the following gating strategy (1): doublet discrimination (FSC-H vs. FSC-A) (2), gating on cells (SSC-A vs. FSC-A) (3), dead cell exclusion using PI (B3 vs. B2; Supplementary Figure 1).
2.5 Western blotting
To quantify the total protein expression of immunogenic markers, cells were lysed on the plates with RIPA buffer (#20-188, Millipore) supplemented with protease inhibitor cocktail (Roche Diagnostics). Western blotting was performed as described previously (20). In brief, cell extracts were normalized for protein concentration using the Pierce BCA Protein Assay kit (#23225; Thermo Fisher), diluted 5 times in Laemmli buffer, boiled and resolved by sodium dodecyl sulfate (SDS) gel electrophoresis in 10% polyacrylamide gels. After transferring to 0.45 µm nitrocellulose membranes (#1620115; Bio-Rad), the membranes were blocked for 1 h with 5% milk-TBST (20 mM Tris-HCl, pH 7.8, 150 mM NaCl, 0.1% Tween-20) and incubated overnight with primary antibodies diluted at 1:1,000 in 0.5% milk-TBST. The membranes were then washed three times with TBST, followed by the 1 h incubation with the secondary peroxidase-linked anti-mouse or anti-rabbit IgGs (Cell Signaling) diluted at 1:5,000 in 0.5% milk-TBST. After washing the membrane with TBST, the chemiluminescent signals were detected with the ECL solution (RPN2232; Amersham) using the ChemiDoc XRS+ imaging instrument. The primary antibody against MHC-I (W6/32, sc-32235), was obtained from Santa Cruz Biotechnology. GAPDH (ab9485) was purchased from Abcam.
2.6 xCELLigence real-time cell growth assay
For the xCELLigence growth assay, 1 x 104 TNF-pretreated cells (A673, TC32, SJSA-1, U2OS, RD and RH30) were seeded in 150 µl medium/well in 96-well E-plates (Roche Diagnostics, Penzberg, Germany). To measure cytotoxic activity of PBMCs, freshly isolated healthy donor-derived PBMCs were added 24 h or 48 h later, at effector to target ratios (E:T) of 0.5:1, 1:1, 2:1, 5:1 or 10:1, to a final volume of 250 µl/well. Real-time cell growth was monitored in 15 min intervals for a maximum of 120 h using the impedance-based xCELLigence RTCA SP system (Roche Diagnostics, Penzberg, Germany).
2.7 Sulforhodamine B assay
SRB assay was performed as described previously (21). Briefly, 1 x 104 TNF-pretreated EwS cells were seeded in 96-well plates, in a total volume of 150 µl/well. Next day, CHM1319 CD8+ T cells were added at an E:T ratio of 1:1 or as indicated in the figure legends. After 24 h incubation, the supernatant was collected, centrifuged at 300 g for 5 min to remove cells and stored at -80°C. Cells were fixated with ice-cold 10% trichloroacetic acid overnight at 4°C and stained with 0.5% SRB (Sigma-Aldrich) in 1% acetic acid for 30 min. The cytotoxic effect was quantified by solubilizing the cell-bound SRB in 10 mM Tris buffer (pH 10) and measuring the optical density at 510 nm using the absorbance plate reader (Infinite M Nano, Tecan).
2.8 ELISpot assay
96-well mixed cellulose ester plates (MultiScreen-HA Filter Plate, 0,45 μm, Millipore, Germany) were coated with 10 μg/ml IFNγ capture antibody (mAB 1-D1K) or 10 µg/ml Granzyme B capture antibody (mAB GB10) in 50 µl PBS/well at 4°C over night. Next day, plates were washed 4x with 200 μl PBS for 10 min each at 4°C, and then blocked with 150 μl of T cell medium/well at 37°C for 1 h. Unspecific or CHM1319 CD8+ T cells were seeded onto the coated 96-well mixed cellulose ester plates (5,000-20,000 cells/well) in 50 µl T cell medium/well. After 30 min, 20,000 EwS cells in 50 µl T cell medium/well were carefully added. For MHC-I and ICAM-1 blockade experiments, EwS cells were preincubated either with 1-100 µg/ml W6/32 antibody (Helmholtz Zentrum München, Germany), or an anti-ICAM-1 antibody (0.1-20 µg/ml; clone P2A4, MAB2146Z, Millipore) or IgG1 control (20 µg/ml; CBL610; Millipore) at 37°C for 30 min. For positive and negative controls, 1 x 107 T2 cells/ml were pulsed with 35 µg/ml of a CHM1319 or Influenza peptide (GILGFVFTL; both Thermo Fisher) at 37°C for 2 h, washed and added at a concentration of 20,000 cells/50 µl/well (22). Cells were co-cultured for 20 h at 37°C. Next day, plates were washed 6x with 0.05% Tween (Sigma) in PBS and dried briefly. The wells were incubated with 2 μg/ml IFNγ detection antibody (7B61-Biotin) or 2µg/ml Granzyme B detection antibody (GB11-Biotin) in 0.5% BSA-PBS/well at 37°C for 2 h. After washing 6x with 0.05% Tween in PBS, plates were dried and incubated with 100 μl of Streptavidin-HRP at a 1:100 dilution in 0.5% BSA-PBS at room temperature and protected from light for 1 h. Plates were washed 3x with 0.05% Tween in PBS, 3x with PBS, dried briefly and developed using developing solution freshly prepared by adding 25 μl of 30% (w/w) H2O2 solution (Sigma) per 10 ml of 3-Amino-9-ethylcarbazole solution (Sigma); 100 μl/well. Plates were incubated for up to 8 min, washed with running tap water and air dried thoroughly. Spots were counted using the ELISpot reader (AID iSpot, light source AID Xenon Light Source ELR0024, AID Autoimmun Diagnostika GmbH).
2.9 Quantitative RT-PCR
Total RNA of A673 and TC32 EwS cell lines was isolated using the RNeasy Plus Mini Kit (Quiagen) after three days of incubation with 0, 1, 10 or 100 ng/ml TNF and reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Differential gene expression of CHM1 (HS00170877_m1) was then analyzed by qRT-PCR using TaqMan Universal PCR Master Mix and fluorescence detection with a QuantStudio6Pro (Applied Biosystems). Gene expression was normalized to glyceraldehyde-3- phosphate dehydrogenase (GAPDH; Hs99999905_m1) using the ΔΔCT method (all Applied Biosystems).
2.10 Statistical analysis
Statistical analyses were performed and graphs plotted using GrapPadPrism 9 (GraphPad Software, San Diego, CA, USA). One-way or two-way ANOVA with multiple comparison Tukey test, Dunnett´s test or Šidák test were performed to calculate p values. ns not significant, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001. The illustrations were created with Biorender.com.
3 Results
3.1 TNF and IL-1β upregulate immunogenic surface markers on pediatric sarcoma cell lines
We assessed a panel of pediatric sarcoma cell lines, including OS (SJSA-1, U2OS), RMS (RD, RH30) and EwS (A673, TC32, TC71 and SKNMC) for their basal expression of the immunogenic surface markers CD86, CD83, PD-L1, ICAM-1, MHC-I and MHC-II in comparison to healthy donor-derived PBMCs. Flow cytometry showed a variable expression of MHC-I on all tested cell lines, with the highest levels detected in the OS cell lines SJSA-1 and U2OS as well as the EwS cell line A673, and the lowest in TC71 and SKNMC (Figure 1, Supplementary Figure 2A). Compared to healthy donor PBMCs, MHC-I was significantly lower on all pediatric sarcoma cell lines, except for the OS cell line SJSA (Figure 1, Supplementary Figure 2B). Notably, SJSA-1 and U2OS cells showed PD-L1 expression (Figure 1, Supplementary Figure 2A), demonstrating a mixed expression of immunogenic and immunosuppressive markers among pediatric sarcomas; which is in accordance with partial PD-L1 expression in osteosarcomas (5, 23, 24). SJSA-1 was the only pediatric sarcoma cell line with basal ICAM-1 surface expression. None of the tested cell lines was positive for CD83, CD86 or MHC-II, which is in line with previous observations in EwS (3).
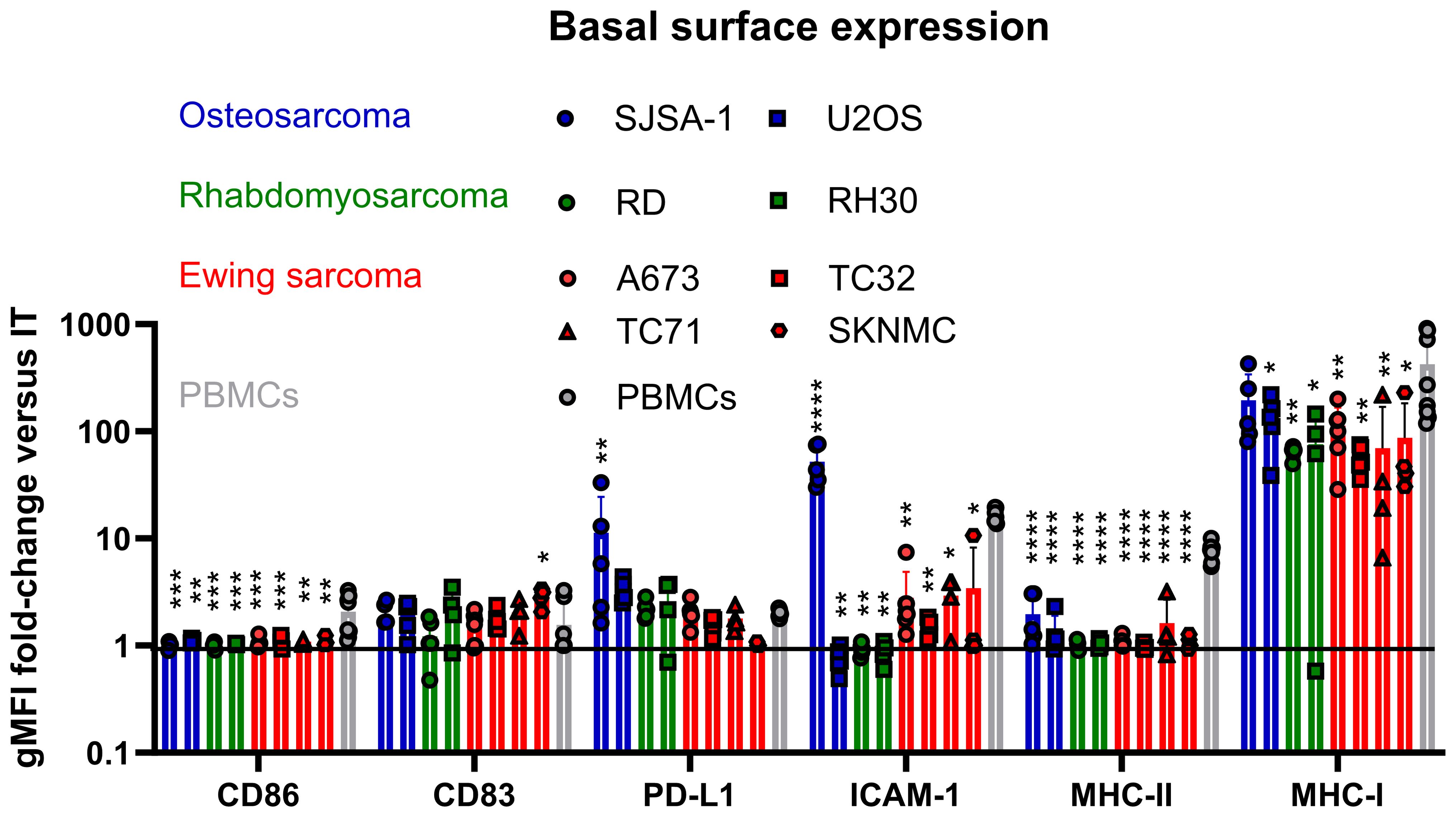
Figure 1. Pediatric sarcomas exhibit lower MHC-I expression and lack co-stimulatory surface molecules compared to healthy donor PBMCs. Flow cytometry of osteosarcoma (SJSA-1, U2OS), rhabdomyosarcoma (RD, RH30) and Ewing sarcoma (A673, TC32, TC71 and SKNMC) cell lines compared to healthy donor PBMCs. Surface expression is displayed as ratio of the geometric mean fluorescence intensities (gFMI) of the specific staining and isotype control. Ratio of 1 indicates lack of surface expression. Each symbol represents an independent experiment or donor with at least two replicates. Data are presented as mean ± SD. One-way ANOVA with multiple comparison Dunnett`s test was used to calculate p values. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
To test if the surface expression of immunogenic markers can be enhanced, pediatric sarcoma cell lines were incubated for 96 h with individual cytokines commonly used together for the differentiation and maturation of monocytic-derived dendritic cells (DC), including IL-4, GM-CSF, IL-1β, IL-6, TNF and PGE2 (13, 16). As these cytokines exert pleiotropic and context-dependent functions, we tested their individual effect on pediatric sarcoma cells in an unbiased manner. Flow cytometry analyses showed that IL-1β and TNF were the most efficient inducers of ICAM-1 and MHC-I in the majority of cell lines, while IL-4, GM-CSF, IL-6 and PGE2 had weak to no effects (Figure 2, Supplementary Figures 3 and 4A). The effects of IL-1β and TNF were partly comparable to the positive control IFNγ, which strongly induced MHC-I on TC71 and SKNMC, and slightly upregulated ICAM-1 on A673, TC32, TC71 and SKNMC, which is in line with previous reports (25). Western blots confirmed that IL-1β, TNF and IFNα (used as positive control) were inducers of MHC-I in A673 and TC32 (Supplementary Figure 4B), with TNF showing the strongest MHC-I stimulation in A673. In some cell lines (most notably, A673), IL-1β and TNF also induced CD83 (but not CD86 or MHC-II) and PD-L1. In RMS RD cells, IL-1β induced higher ICAM-1 expression than TNF (Supplementary Figures 5, 6). Overall, A673 cells were the strongest responders and TNF was the most potent inducer of immunogenic surface markers. These markers, including ICAM-1, MHC-I and CD83 were variably induced by TNF in the pediatric sarcoma cell lines tested here.
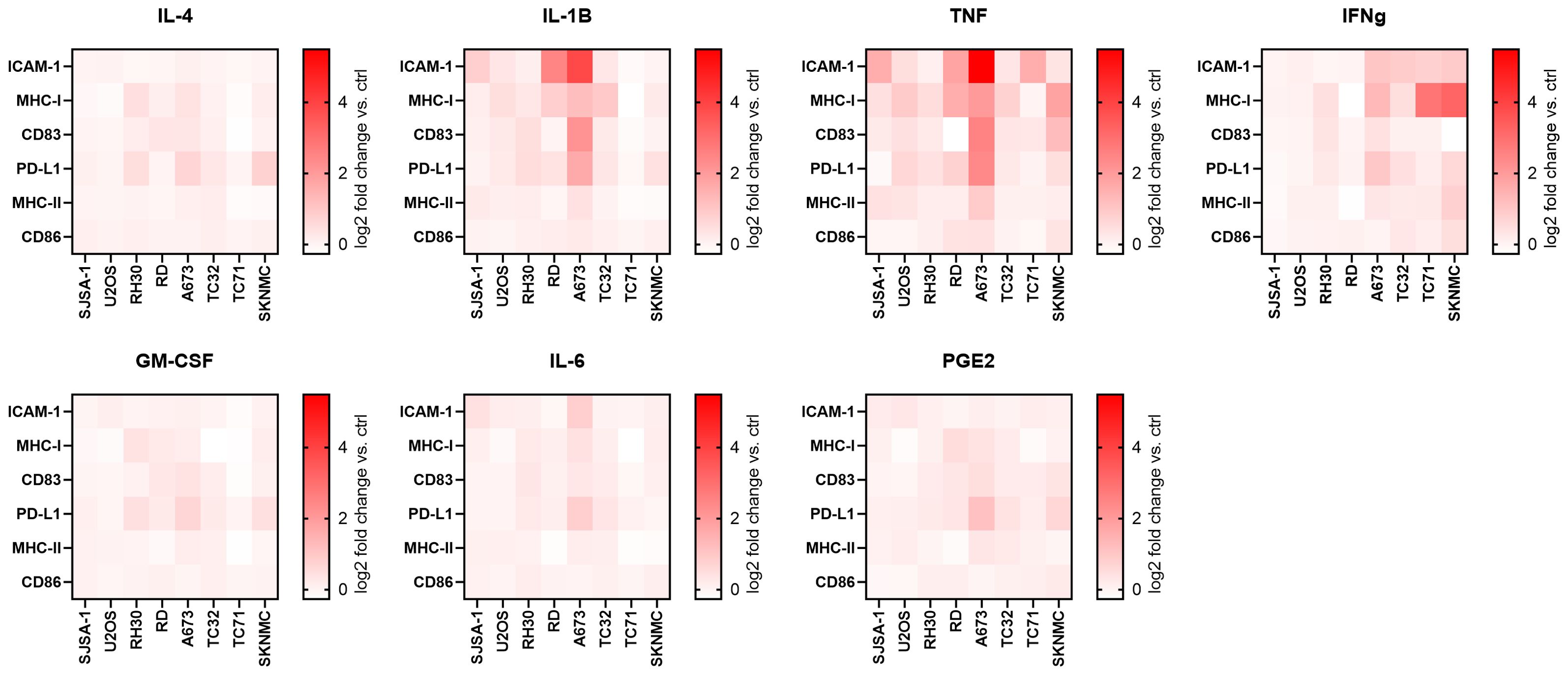
Figure 2. TNF and IL-1β upregulate immunogenic surface markers on pediatric sarcoma cell lines. Flow cytometry of eight pediatric sarcoma cell lines after 96 h treatment with indicated cytokines or left untreated (ctrl). IFNγ-treatment was used as positive ctrl. Shown are heatmaps for the log2 fold change of the geometric mean fluorescence intensities (gMFI) of the specific staining minus gMFI of the respective isotype ctrl of the cytokine-treated versus ctrl cells. Results from two (SJSA-1, U2OS, RH30, RD, A673 and TC32) or three (TC71, SKNMC) independent experiments with three replicates each are shown.
Importantly, TNF-mediated upregulation of MHC-I and ICAM-1 on RD, SJSA-1 and A673 was dose-dependent (Figure 3), suggesting physiological relevance. However, their upregulation was less prominent on RH30 and TC32 (≤ 2-fold change vs control), and U2OS showed no change for ICAM-1 (Figure 3). In some cell lines, including U2OS, RH30 and A673, TNF also slightly upregulated CD83 as well as PD-L1 (Supplementary Figures 7-9). Using the additional EwS cell lines CHLA10 and EW7, we confirmed the dose-dependent upregulation of both MHC-I and ICAM-1 in response to TNF, although ICAM-1 upregulation was weaker compared to A673 cells (Supplementary Figure 9).
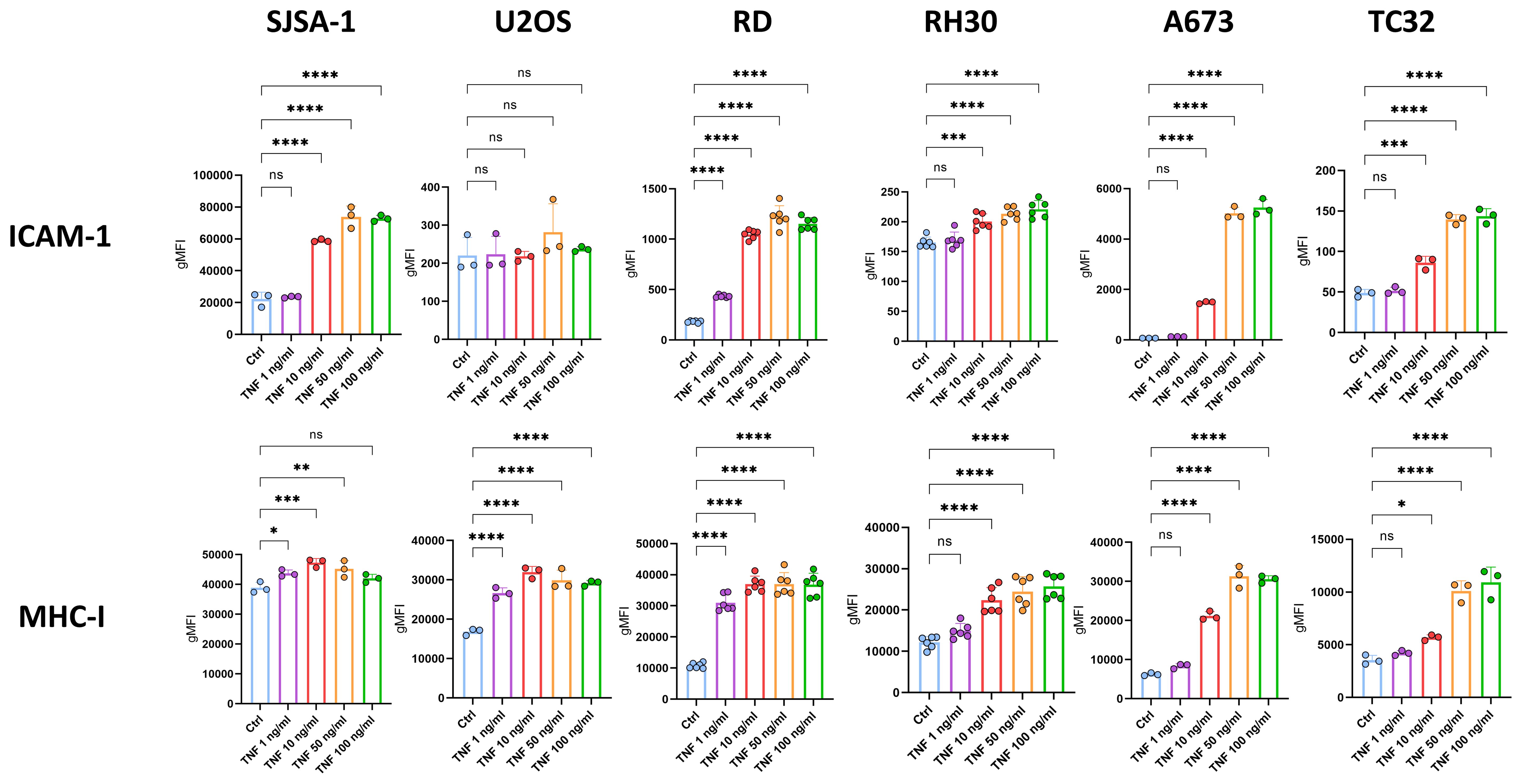
Figure 3. TNF dose-dependently upregulates ICAM-1 and MHC-I on pediatric sarcoma cell lines. Flow cytometry of pediatric sarcoma cell lines after 96 h treatment with indicated doses of TNF or left untreated as control (ctrl). Expression of ICAM-1 and MHC-I is shown as geometric mean fluorescence intensities (gMFI) minus gMFI of the respective isotype ctrl. Representative results from three independent experiments with three (SJSA-1, U2OS, A673, TC32) or six (RD, RH30) replicates are shown. Data are presented as mean ± SD. One-way ANOVA with multiple comparison Dunnett´s test was used to calculate p values. ns = not significant, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.001.
Due to the response to TNF, surface expression of its two receptors TNFR1 and TNFR2 was evaluated on pediatric sarcoma cell lines using flow cytometry. All pediatric sarcoma cell lines tested showed modest to strong TNFR1 expression, while TNFR2 was weakly expressed. RD was the only cell line with strong TNFR2 expression, at similar levels compared to healthy donor PBMCs (Supplementary Figure 10).
Associated with the strong upregulation of immunogenic surface markers on RD and A673 cells, TNF inhibited their growth, while RH30, TC32 and the OS cell lines SJSA-1 and U2OS were not affected (Figure 4). Therefore, TNF is capable of upregulating ICAM-1, MHC-I and, to a lesser extent, CD83 and PD-L1 on multiple pediatric sarcoma cell lines without promoting tumor growth in vitro, and thus was further assessed for its immunostimulatory activity.
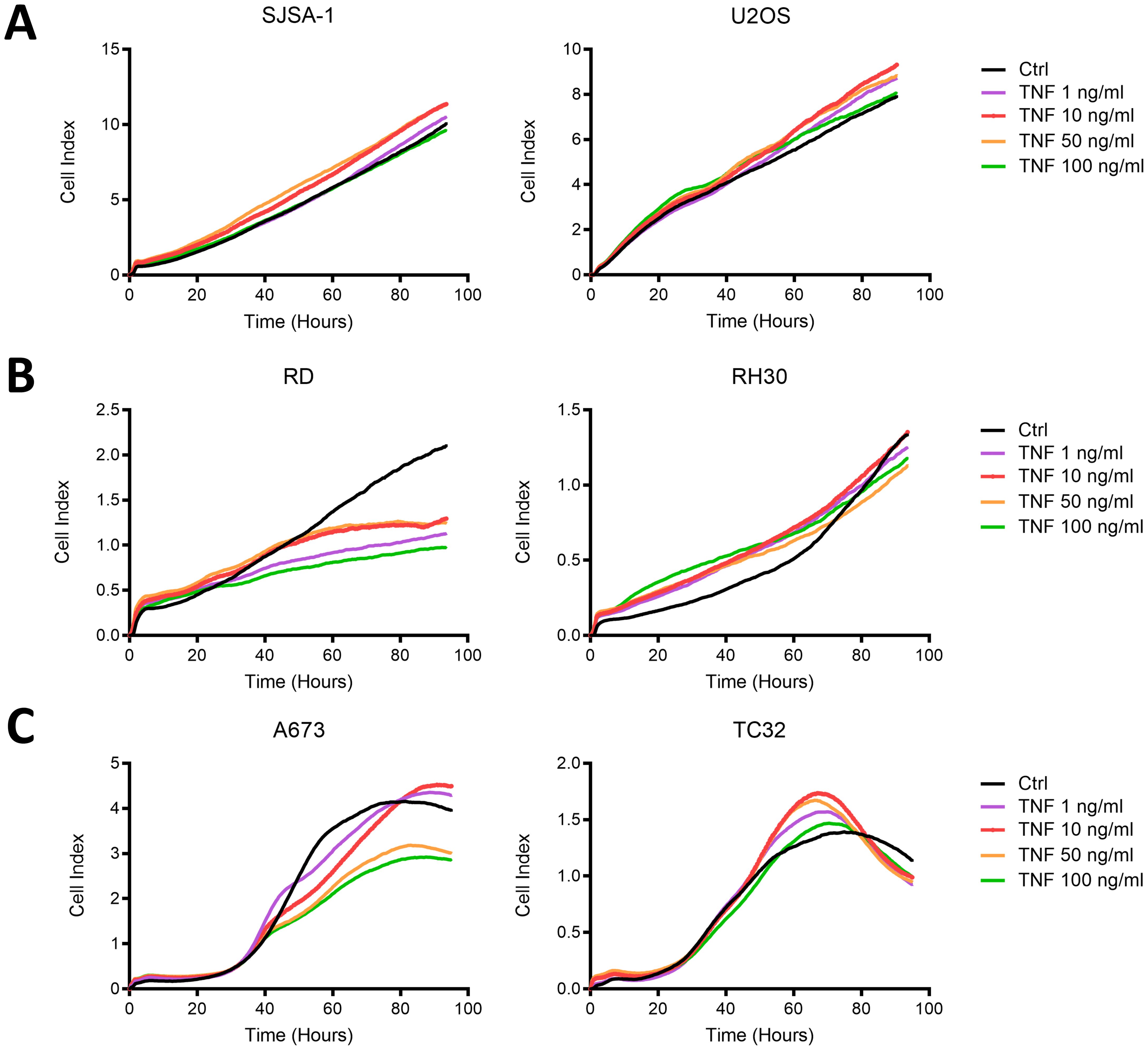
Figure 4. TNF inhibits the growth of RMS cell line RD and EwS cell line A673, but not OS cell lines or EwS TC32 in vitro. Live cell growth assay xCELLigence of (A) osteosarcoma (OS) cell lines, (B) rhabdomyosarcoma (RMS) cell lines or (C) Ewing sarcoma (EwS) cell lines in the presence of indicated doses of TNF. Representative results from two (U2OS, SJSA-1, A673, TC32) or three (RD, RH30) independent experiments with three replicates are shown.
3.2 TNF sensitizes A673 cells to EwS antigen-specific CD8+ T cells depending on MHC-I and ICAM-1
As TNF induced expression of co-stimulatory molecules on OS, RMS and EwS cell lines (Figures 2, 3), we tested whether pretreatment with TNF sensitizes pediatric sarcoma cell lines towards killing by HLA-mismatched healthy donor-derived PBMCs. Using the xCELLigence assay, we observed that HLA-mismatched PBMCs exerted partial tumor control against SJSA-1 at effector to target ratios (E:T) of 1:1 and 5:1 and against TC32 at E:T 5:1 and 10:1 (Figures 5A, C), but failed to control growth of U2OS, RD, RH30 and A673 (Figures 5A–C). However, pretreatment of these cell lines with a range of TNF concentrations did not improve the anti-tumor activity of HLA-mismatched PBMCs against these pediatric sarcoma cell lines (Figure 5), indicating that the upregulated co-stimulatory molecules by TNF do not promote the anti-tumor activity of unspecific PBMCs. This could be due to the resting state or the low antigen specificity of HLA-mismatched PBMCs.
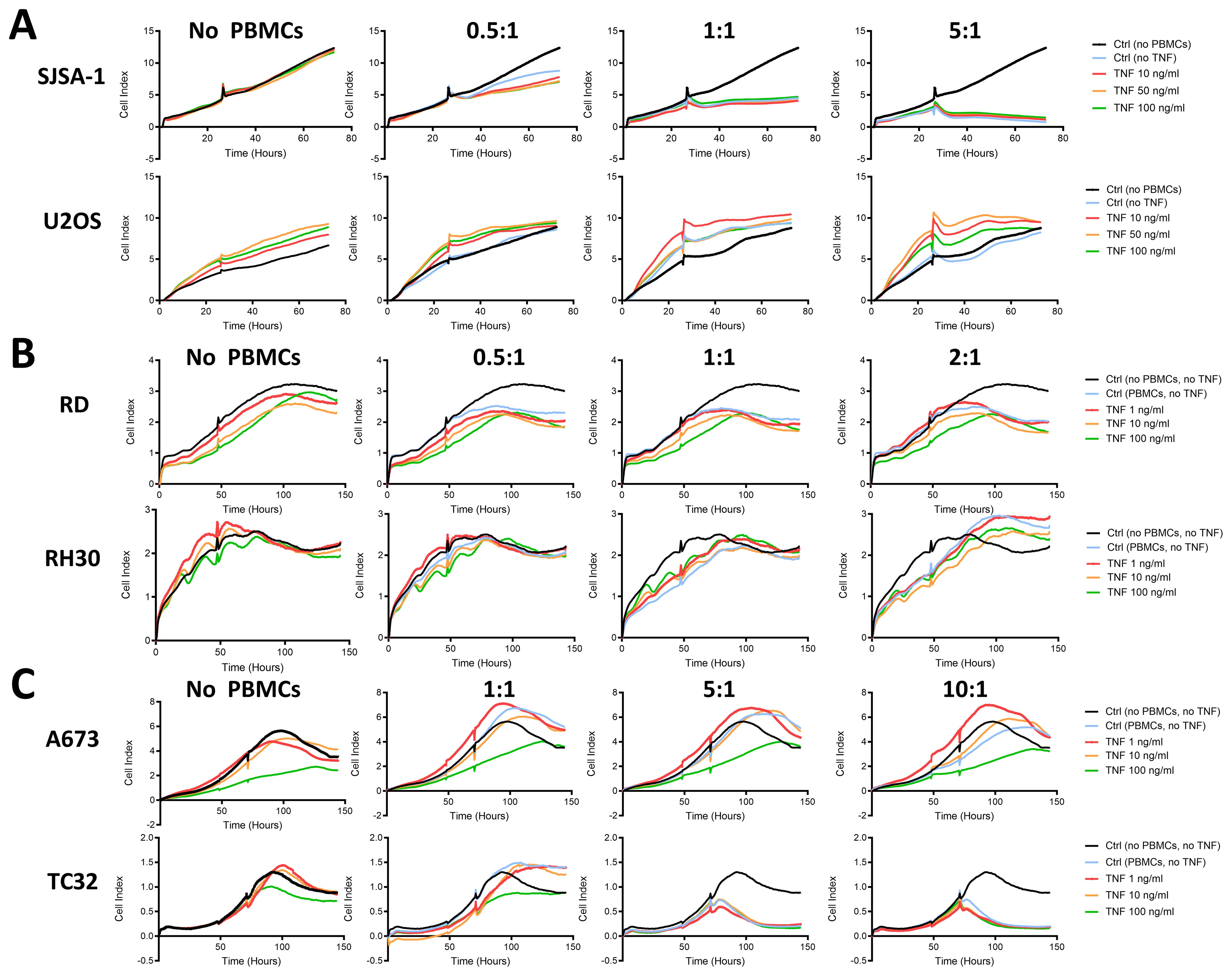
Figure 5. TNF-pretreatment of pediatric sarcoma cell lines fails to enhance cytotoxicity of HLA-mismatched PBMCs. Live cell growth assay xCELLigence of (A) osteosarcoma (SJSA-1, U2OS), (B) rhabdomyosarcoma (RD, RH30) and (C) Ewing sarcoma (A673, TC32) cell lines pretreated with indicated amount of TNF for 96 h prior to seeding. Healthy donor-derived, HLA-mismatched PBMCs were added at the indicated effector to target ratios to tumor cells after 48 h or when the cell index reached 1-2 (small notch represents addition of PBMCs). Representative results from three independent experiments with three independent donors in triplicates are shown.
Because TNF-pretreatment of pediatric sarcoma cell lines did not promote anti-tumor activity of unspecific PBMCs, we tested whether pretreatment with TNF sensitizes EwS cell lines to killing by antigen-specific CHM1319 CD8+ T cells. As shown by SRB assay, CHM1319 CD8+ T cells lysed HLA-A2+ (A673, TC32, EW7), but not HLA-A2- (CHLA10) EwS cell lines (Figure 6A, Supplementary Figure 11), in line with our previous findings (26). The pretreatment of EwS cell lines with increasing doses of TNF did not further improve tumor cell lysis by CHM1319 CD8+ T cells at the E:T of 1:1 (Figure 6A), apart from A673, which showed a tendency to be sensitized by the increasing doses of TNF. However, at increased E:T of 1.25:1, 2.5:1 and 5:1, the pretreatment with 10 ng/ml TNF significantly improved killing of A673 by CHM1319 CD8+ T cells, while TC32 and EW7 cells remained unaffected (Figure 6B). The lysis of HLA-A2- EwS cell line CHLA10 was not altered by pretreatment with TNF (Figure 6B), confirming specificity of CHM1319 CD8+ T cells towards HLA-A2-positive cells. Granzyme B ELISpot showed that TNF-pretreatment of A673, but not TC32, increased Granzyme B release from CHM1319 CD8+ T cells at E:T of 0.625:1 and 2.5:1 (Figure 6C), confirming increased lysis of A673 after TNF-pretreatment.
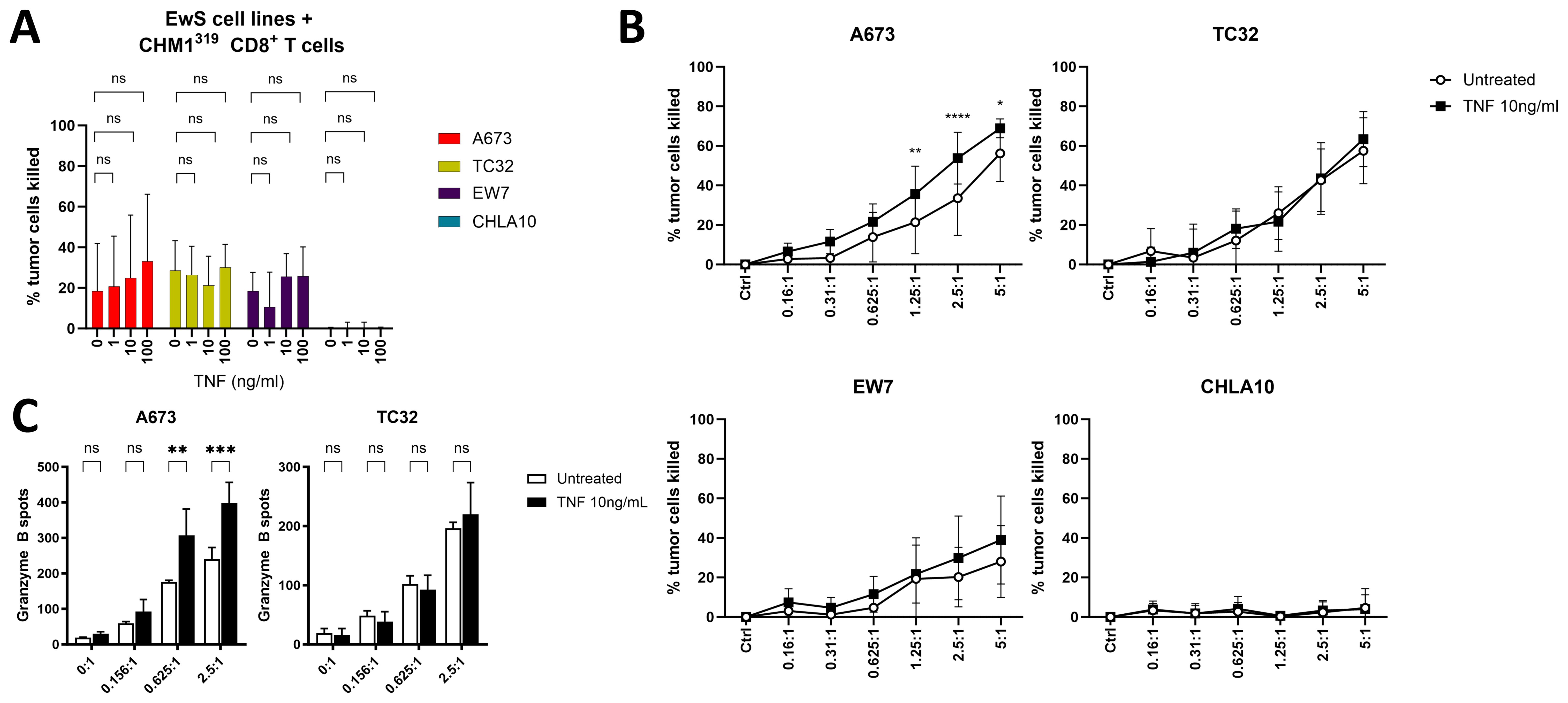
Figure 6. TNF-pretreatment partly sensitizes EwS cells against antigen-specific CD8+ T cells. (A, B) SRB assay showing cell growth of four EwS cell lines pretreated with different amounts of TNF or left untreated for 4 days. HLA-A*02:01/CHM1319-specific allorestricted TCR transgenic CD8+ T cells (CHM1319 CD8+ T cells) were added at an effector to target ratio of 1:1 (A) or as indicated (B) and analyzed after 24 h. (C) Granzyme B ELISpot of CHM1319 CD8+ T cells co-cultured at indicated effector to target rations with untreated or TNF-pretreated (10ng/ml, 72 h) A673 and TC32 for 24 h. (A) Combined results from three (EW7, CHLA10) or four (A673, TC32) independent experiments with three or four independent donors in quadruplicates are shown. Combined results from four (B) or representative results from three (C) independent experiments with four (B) or three (C) independent donors in triplicates are shown. Data are presented as mean ± SD. Two-way ANOVA with Dunnett´s (A), Šídák´s (B) or Tukey´s (C) multiple comparisons test was used to calculate p values. ns = not significant, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
To investigate the potential long-term effect of TNF pretreatment on CHM1319 CD8+ T cells, it was evaluated, if TNF-pretreatment of EwS cell lines promotes exhaustion of CHM1319 CD8+ T cells using flow cytometry. The exhaustion markers TIM-3, PD-1, CTLA-4 and LAG-3 were not significantly increased on CHM1319 CD8+ T cells after 96 h-coculture with TNF-pretreated compared to untreated A673 and TC32 cells (Supplementary Figure 12). PD-1 and LAG-3 were significantly upregulated on CHM1319 CD8+ T cells after co-culture with EwS cell lines in comparison to CHM1319 CD8+ T cells only, indicating the induction of exhaustion in CHM1319 CD8+ T cells after antigen stimulation by EwS cell lines independent of TNF-pretreatment. Overall, PD-1 and LAG-3 expression levels showed inter-donor heterogeneity (data not shown). Therefore, TNF-pretreatment of EwS cell lines A673 and TC32 does not enhance an exhaustion phenotype in antigen-specific CD8+ T cells in vitro.
To mechanistically examine if TNF-induced upregulation of MHC-I on EwS cells is important for the enhanced T cell recognition and killing, the recognition of TNF-pretreated EwS cells was assessed in the presence of MHC-I blocking antibody (W6/32). The IFNγ release of CHM1319 CD8+ T cells was dose-dependently abrogated after blockade of MHC-I on A673 and TC32 using the anti-MHC-I antibody W6/32 (Figures 7A, B, top panels), confirming MHC-I specificity of CHM1319 CD8+ T cells. The pretreatment of A673 and TC32 with TNF significantly increased the IFNγ release from CHM1319 CD8+ T cells. This increased IFNγ release remained under partial MHC-I blockade on A673, but not TC32, thus indicating that TNF enhances T cell recognition of EwS cells by upregulating MHC-I. As recognition of TNF-pretreated TC32 did not remain increased under partial MHC-I blockade, although TNF dose-dependently upregulated MHC-I on TC32 (Figure 3), additional co-stimulatory surface molecules may contribute to the enhanced recognition of TNF-pretreated A673 by CHM1319 CD8+ T cells. In line with TNF-pretreatment of pediatric sarcoma cell lines not promoting anti-tumor activity of unspecific PBMCs (Figure 5), TNF-pretreatment of A673 and TC32 did not enhance IFNγ release of unspecific CD8+ T cells (Figures 7A, B, bottom panels), further indicating that TNF does not promote MHC-I independent cytotoxicity.
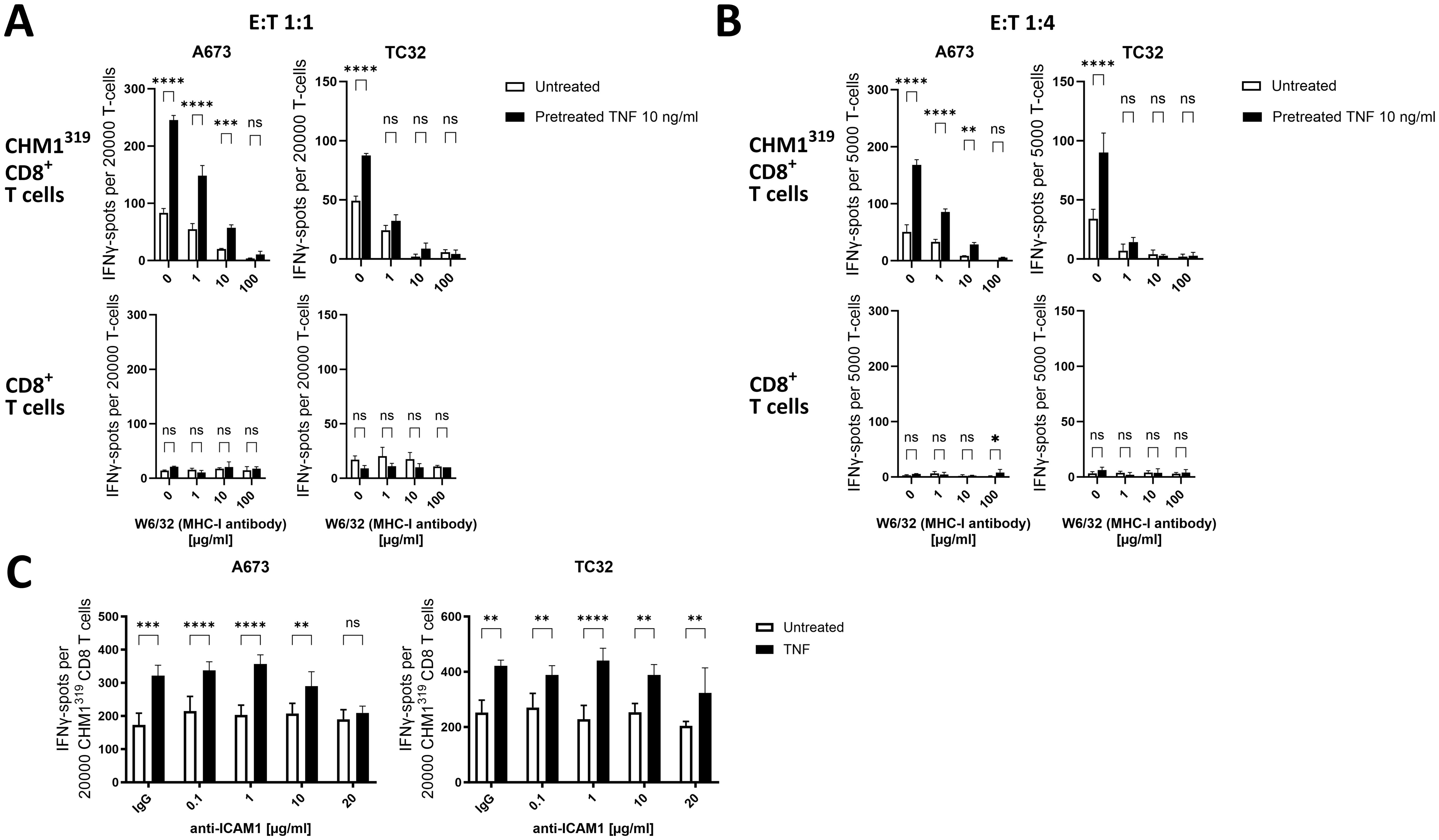
Figure 7. TNF-pretreatment sensitizes EwS cell lines to antigen-specific CD8+ T cells depending on MHC-I and ICAM-1 upregulation. (A–C) ELISpot assay showing count of IFNγ spots. Ewing sarcoma (EwS) cells A673 and TC32 were pretreated with 10ng/ml TNF for 72 h and afterwards incubated for 30 min with (A, B) MHC-I blocking antibody W6/32 at indicated doses or (C) with an IgG control antibody (20 µg/ml) or ICAM-1 blocking antibody at indicated doses. Then EwS cells were co-cultured with HLA-A*02:01/CHM1319-specific allorestricted TCR transgenic CD8+ T cells (CHM1319 CD8+ T cells) or unspecific CD8+ T cells at effector to target ratios of (A, C) 1:1 and (B) 1:4 for 24 h. Representative results from three independent experiments with three independent donors in (A, B) triplicates or (C) quadruplicates are displayed. Data are presented as mean ± SD. Two-way ANOVA with multiple comparison Šidák test was used to calculate p values. ns = not significant, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
Since recognition of TNF-pretreated A673 remained enhanced under partial MHC-I blockade and TNF also strongly upregulated ICAM-1 on A673, we tested if membranous ICAM-1 contributes to the improved recognition by EwS antigen-specific transgenic T cells. Indeed, ICAM-1 blockade with 20µg/ml of the anti ICAM-1 antibody (clone P2A4) reduced the recognition of A673 pretreated with TNF by CHM1319 CD8+ T cells (Figure 7C), suggesting that ICAM-1 is involved in the enhanced recognition by antigen-specific CD8+ T cells. In line with the minor upregulation of ICAM-1 on TC32 by TNF pretreatment, ICAM-1 blockade did not affect recognition of TC32 regardless of whether the cells were pre-treated with TNF or not (Figure 7C). In addition to upregulation of immunogenic surface markers, altered antigen expression can also affect the recognition of EwS cell lines by CHM1319 CD8+ T cells. Therefore, we measured the mRNA expression of CHM1 in A673 and TC32 EwS cell lines after TNF pretreatment. As TNF significantly reduced mRNA expression of CHM1 in A673, but not in TC32 (Supplementary Figure 13), antigen expression and peptide loading on MHC-I may not be involved in the enhanced recognition and lysis of A673 cells after pretreatment with TNF.
Together, these results indicate that TNF sensitizes the EwS cell line A673 depending on the presence of MHC-I and upregulation of ICAM-1 for the recognition and lysis by CHM1319 CD8+ T cells without enhancing exhaustion.
3.3 A673 is the most responsive EwS cell line to pro-inflammatory cytokines
Since A673 cells showed superior sensitivity to lysis by EwS antigen-specific CD8+ T cells after TNF-pretreatment, we analyzed additional EwS cell lines, to identify those which can be sensitized using five different cytokine treatment combinations, including TNF. However, among a panel of six EwS cell lines tested by flow cytometry, A673 cells stood out as the strongest responders to treatment with TNF, TNF+IFNγ, DC and DC+IFNγ, especially in upregulating CD83, ICAM-1 and PD-L1 (Figure 8A). In contrast, MHC-I was upregulated at rather equal levels on the six EwS cell lines by the five treatment conditions. Prominent responses were observed in combinational treatments of IFNγ with TNF or the DC cocktail, upregulating ICAM-1, MHC-I and CD83 on three (A673, TC32 and CHLA10) and MHC-I on all six EwS cell lines (Figure 8A). However, these treatments also resulted in a strong upregulation of PD-L1.
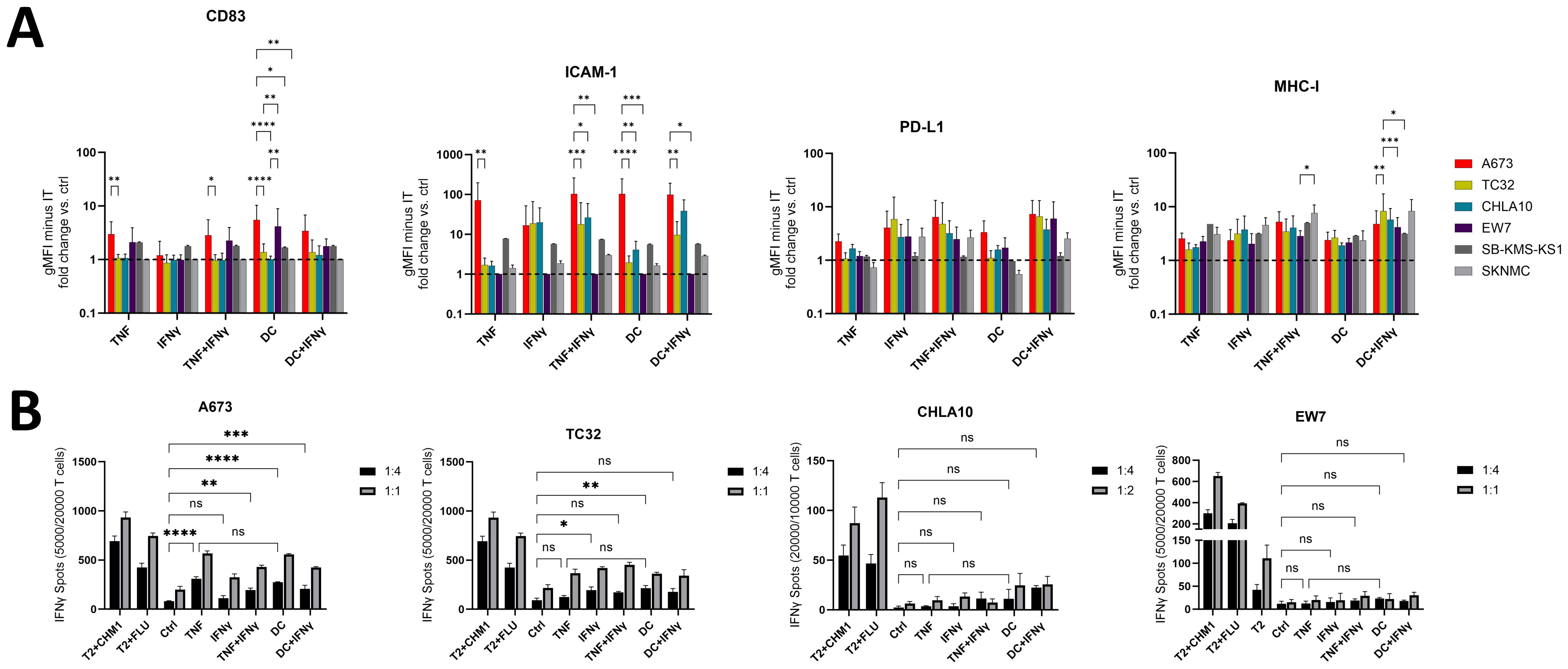
Figure 8. Pretreatment with TNF or monocyte maturation mediators sensitizes A673 and partly TC32 for the recognition by CHM1319 CD8+ T cells. (A) Flow cytometry of Ewing sarcoma (EwS) cell lines A673, TC32 and EW7 (all HLA-A2+) as well as CHLA10, SKNMC and SB-KMS-KS1 (all HLA-A2-) after pretreatment with TNF (10 ng/ml, 3 days) +/- IFNγ (100 U/ml, 1 day) or monocyte maturation mediators (DC cocktail: IL-4, GM-CSF for 5 days, followed by IL-1β, IL-6, TNF and PGE2 for 2 days) +/- IFNγ (100 U/ml, 1 day) or IFNγ (100 U/ml, 1 day) alone. Control (ctrl) cells were left untreated. Fold change over ctrl cells of the geometric mean fluorescence intensities (gMFI) minus the gMFI of the respective isotype ctrl is shown. Combined results from one (SB-KMS-KS1 and SKNMC), four (CHLA10), five (EW7), eight (A673) or nine (TC32) independent experiments with triplicates are shown. (B) ELISpot assay showing IFNγ spot count. EwS cells from (A) were co-cultured at indicated effector to target ratios with CHM1319-specific TCR-transgenic CD8+ T cells (CHM1319 CD8+ T cells) for 24 h. Representative results from two (CHLA10), four (A673, EW7) or five (TC32) independent experiments with triplicates are shown. Two-way ANOVA with multiple comparison Tukey´s test was used to calculate p values. ns = not significant, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.001.
Further analysis of HLA-A2+ (A673, TC32 and EW7) and HLA-A2- (CHLA10, SB-KMS-KS1 and SKNMC; Supplementary Figure 11) EwS cell lines by IFNγ ELISpots showed that pretreatment with TNF reliably enhanced the recognition of A673 by CHM1319 CD8+ T cells, while recognition of TC32 was improved at E:T ratios of 1:1. In contrast, EW7 and the HLA-A2- cell lines CHLA10, SB-KMS-KS1 and SKNMC failed to respond (Figure 8B, Supplementary Figure 14). TNF efficiency was comparable to that of the DC cytokine cocktail, which enhanced sensitivity of A673 and partly TC32 (3/5 independent experiments; Figure 8B).
In summary, pretreatment of EwS cell lines with TNF, IFNγ, DC maturation mediators or their combinations with IFNγ induce distinct patterns of immunogenic and immunosuppressive cell surface markers. TNF appears to be the strongest and most advantageous as monotreatment, based upon its ability to firmly induce the immunostimulatory surface markers MHC-I, ICAM-1 and CD83. Among three HLA-positive EwS cell lines tested, A673 cells exhibited a defined response, including upregulation of immunostimulatory markers and enhanced sensitivity to antigen-specific T cells, while TC32 cells showed partial response and EW7 were non-responders. The responsiveness of A673 depends on MHC-I and partly ICAM-1, providing a rational to induce MHC-I and ICAM-1 expression to improve the efficacy of adoptive cellular therapies in pediatric sarcomas (Figure 9).
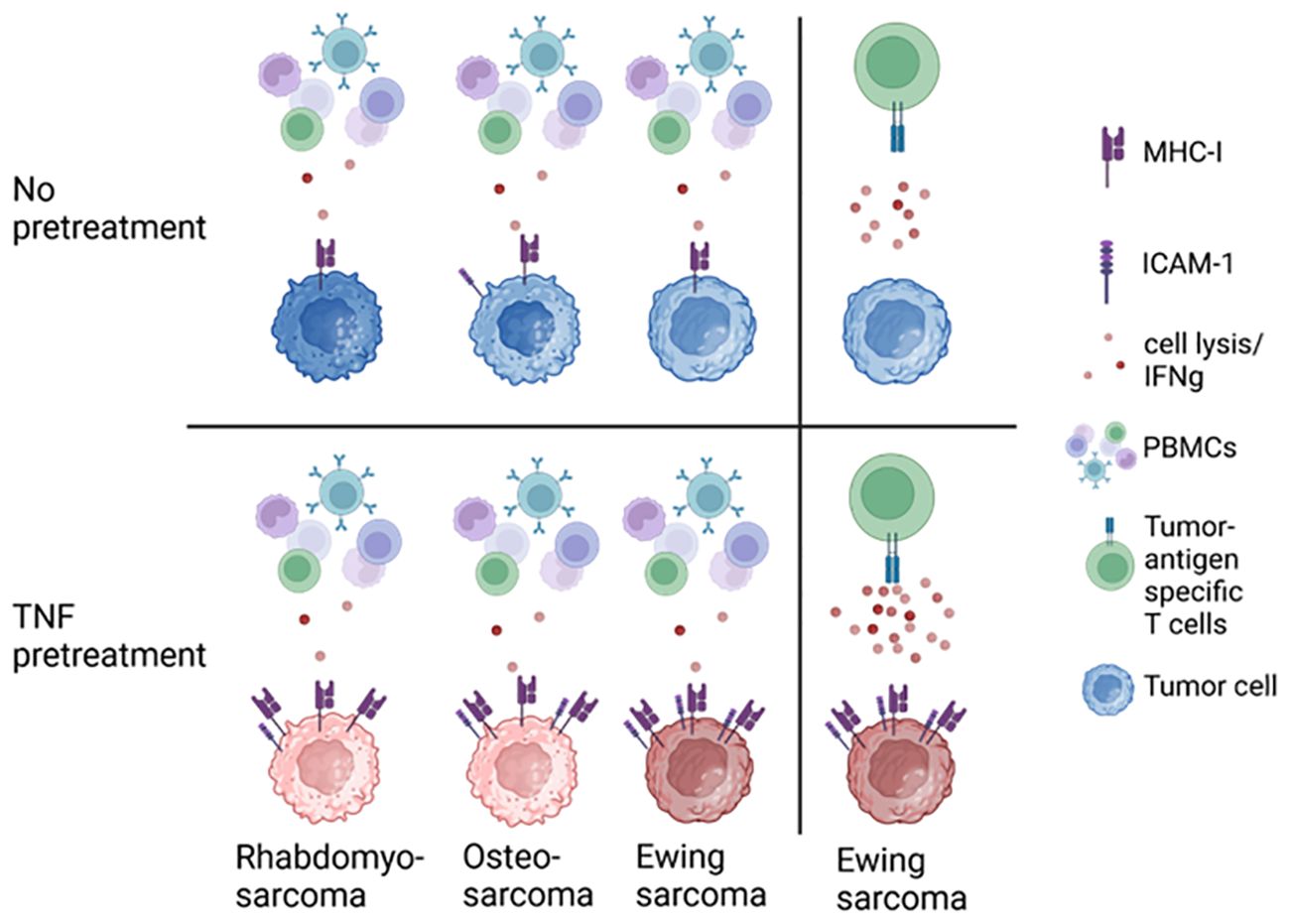
Figure 9. TNF pretreatment upregulates MHC-I and ICAM-1 expression on pediatric sarcoma cell lines, sensitizing Ewing sarcoma to tumor antigen-specific CD8+ T cells.
4 Discussion
Most pediatric sarcomas are non-T cell-inflamed tumors, with 5-year survival rates less than 20-30% in patients with metastatic disease. So far, immunotherapies have not yielded strong clinical responses in pediatric sarcomas, despite their success in T cell-infiltrated immunogenic tumors (9). In this work, we sought to test strategies to enhance immunogenicity in general and specifically the sensitivity of pediatric sarcomas for adoptive T cell transfer by inducing immunogenic cell surface markers. We used a panel of EwS, RMS and OS pediatric sarcoma cell lines to test six cytokines, used for monocyte differentiation and DC maturation (IL-4, GM-CSF, IL-1β, IL-6, TNF and PGE2), for their ability to induce cancer cell immunogenicity and sensitivity to tumor antigen-specific T cells.
Applying flow cytometry, immunoblotting, tumor killing and ELISpot assays, we identified TNF and IL-1β to be the strongest inducers of MHC-I, and partly ICAM-1 and CD83 on the majority of pediatric sarcoma cell lines tested. Especially TNF was superior for upregulating immunostimulatory cell surface molecules and sensitizing EwS cells to tumor antigen-specific T cells. In contrast to TNF and IL-1β, the other tested monocyte maturation mediators, including IL-4, GM-CSF, IL-6 and PGE2, weakly affected expression of immunogenic surface markers on pediatric sarcoma cells. This is in accordance with clinical trials, which failed to show clinical benefit in response to administration of GM-CSF in EwS and OS (27–29). Despite expression of IL-6R and IL-6ST on EwS cell lines (30), IL-6 did not modulate expression of immunogenic markers on pediatric sarcoma cell lines. This suggests that IL-6R-pSTAT3 signaling is not involved in the upregulation of immunogenic markers in pediatric sarcomas but prevents apoptosis and promotes migration in pediatric sarcomas (30–32).
TNF enhanced the recognition of EwS cell line A673 and to a lesser extent TC32 by CHM1319 CD8+ T cells, but not to HLA-mismatched healthy donor-derived PBMCs, indicating that MHC-I peptide specific TCR repertoires are required for the effect of TNF. Indeed, MHC-I blockade by the MHC-I blocking antibody W6/32 demonstrated that the enhanced recognition of antigen-specific CD8+ T cells against A673 and TC32 depends on TNF-induced MHC-I upregulation. However, even in the specific TCR context, TNF only improved the lysis of one out of three HLA-A2-positive EwS cells against CHM1319 CD8+ T cells despite MHC-I upregulation. The blockade of ICAM-1 showed that TNF-induced membranous ICAM-1 contributes to the enhanced recognition of A673 by CHM1319 CD8+ T cells. This is in line with the membranous ICAM-1 promoting T cell binding and immune synapse formation (33, 34), as well as enhanced cytotoxicity of antigen-specific T cells (35). Yet, further experiments, including ICAM-1 knockout experiments, are required for a final conclusion regarding the role of ICAM-1 in the recognition of TNF-pretreated EwS cells by CHM1319 CD8+ T cells. As TNF downregulated the expression of the target antigen CHM1, altered antigen expression, processing and loading on MHC-I may have not contributed to the TNF-induced enhanced lysis of A673 by CHM1319 CD8+ T cells. Other potential mechanisms may include differential regulation of TNF signaling, receptors of the TNF superfamily (36), or other regulatory genes or pathways, variations in EWS-FLI1 protein levels (25) or metabolic states, each of which requires further investigation. The most interesting possibility would be if A673 represents a subset of EwS tumors, which are more susceptible for adoptive T cell transfer and other types of immunotherapy.
Although the efficacy of TNF and IFNγ in upregulating MHC-I on OS and EwS was previously reported (37, 38), our study demonstrates that TNF may be advantageous for T cell-based immunotherapies. As a potential mechanism, TNF signals through TNFR1 to activate the NFκB pathway, which can induce MHC-I and ICAM-1 expression (39, 40). In turn, upregulation of MHC-I improves recognition of tumor cells by cytotoxic T cells (40, 41). In the tumor microenvironment, the interplay of TNF and IFNγ can be a prerequisite for controlling tumor development (42). Indeed, in OS and EwS patients tumor infiltrating CD14+CD16+ myeloid cells show a TNF signaling signature (43). However, despite their presence in the TME, the T cell tumor infiltration is low (7, 8) and response to immune checkpoint blockade is poor, indicating that TNF and IFNγ present in the TME are insufficient to stimulate an effective anti-tumor immunity.
The systemic administration of TNF in humans showed systemic toxicity and low anti-tumor response (44–47), while localized administration in the form of isolated limb perfusion in combination with chemotherapeutics exerts clinical responses and improves limb salvage (48). Here, TNF sensitized the EwS cell line A673 to antigen-specific CD8+ T cells. Hence, a translational approach could be to stimulate the local release of TNF from tumor cells or the tumor microenvironment (49, 50) shortly before adoptive T cell transfer, enhancing tumor cell immunogenicity and T cell therapy efficacy. TNF exerts pleiotropic effects on T cells depending on their cell state, including TCR-dependent activation, cytokine production, proliferation, apoptosis or promoting immunosuppression (51). Here, we did not assess the direct effect of TNF on antigen-specific CD8+ T cells. As the next step, the effects of TNF-inducing regimen on the T cell infiltration, phenotype, activation, anti-tumor cytotoxicity and exhaustion, as well as the tumor microenvironment should be evaluated in suitable in vivo models.
The findings of this study highlight that established pediatric sarcomas differ in their immunological properties even within a tumor entity, emphasizing the importance of selecting a panel of cell lines in the functional testing of immunotherapies, including T cell therapies. This is in line with EwS cell lines showing a stable but variable degree of genome, transcriptome and phenotype (52). In summary, this study identifies TNF as a potential treatment opportunity for enhancing immunogenicity of pediatric sarcomas without promoting tumor growth. Among all pediatric sarcoma cell lines tested, A673 EwS cells were the most responsive and the TNF-induced upregulation of MHC-I and ICAM-1 sensitized A673 for the lysis by CHM1319 CD8+ T cells. Further studies should evaluate the role of TNF and TNF-inducing regimens in vivo as a neoadjuvant sensitizing upfront adoptive T cell transfer in pediatric sarcomas.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Ethics Commission of the Medical Faculty of the Technical University of Munich. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
HG: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. MT: Formal analysis, Investigation, Writing – review & editing. JE: Investigation, Writing – review & editing, Validation. EB: Methodology, Writing – review & editing. HL: Investigation, Writing – review & editing. VE: Formal analysis, Investigation, Writing – original draft, Writing – review & editing. LR: Methodology, Writing – review & editing. IA: Methodology, Visualization, Writing – review & editing. MM: Methodology, Visualization, Writing – review & editing. FA: Methodology, Visualization, Writing – review & editing. SS: Writing – review & editing, Conceptualization, Methodology. JH: Methodology, Writing – review & editing. UT: Conceptualization, Funding acquisition, Methodology, Supervision, Writing – original draft, Writing – review & editing. KH: Conceptualization, Formal analysis, Investigation, Methodology, Supervision, Visualization, Writing – original draft, Writing – review & editing. LF: Investigation, Writing – review & editing. CP: Formal analysis, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. HG received funding from the KKF clinician-scientist program of the School of Medicine, Technical University of Munich and the German Cancer Consortium (DKTK), partner site Munich. UT is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project number 501830041, the “Zukunft Gesundheit e.V.” and the Dr. Sepp and Hanne Sturm Memorial Foundation. UT and HG are funded by the Dr. Robert Pfleger-Foundation, and UT and SS by the Wilhelm Sander-Foundation (2018.072.1. and 2021.007.1.). We also thank the Dr. Robert Pfleger-Stiftung for funding this study.
Acknowledgments
We thank Dr. Nadine Kliese for the excellent administration of this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1347404/full#supplementary-material
References
1. Parkin DM, Stiller CA, Draper GJ, Bieber CA. The international incidence of childhood cancer. Int J Cancer. (1988) 42:511–20. doi: 10.1002/ijc.2910420408
2. Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature. (2018) 555:321–7. doi: 10.1038/nature25480
3. Berghuis D, de Hooge AS, Santos SJ, Horst D, Wiertz EJ, van Eggermond MC, et al. Reduced human leukocyte antigen expression in advanced-stage Ewing sarcoma: implications for immune recognition. J pathology. (2009) 218:222–31. doi: 10.1002/path.v218:2
4. Yabe H, Tsukahara T, Kawaguchi S, Wada T, Torigoe T, Sato N, et al. Prognostic significance of HLA class I expression in Ewing’s sarcoma family of tumors. J Surg Oncol. (2011) 103:380–5. doi: 10.1002/jso.v103.5
5. Sundara YT, Kostine M, Cleven AH, Bovée JV, Schilham MW, Cleton-Jansen AM. Increased PD-L1 and T-cell infiltration in the presence of HLA class I expression in metastatic high-grade osteosarcoma: a rationale for T-cell-based immunotherapy. Cancer immunology immunotherapy: CII. (2017) 66:119–28. doi: 10.1007/s00262-016-1925-3
6. Tsukahara T, Kawaguchi S, Torigoe T, Asanuma H, Nakazawa E, Shimozawa K, et al. Prognostic significance of HLA class I expression in osteosarcoma defined by anti-pan HLA class I monoclonal antibody, EMR8-5. Cancer science. (2006) 97:1374–80. doi: 10.1111/j.1349-7006.2006.00317.x
7. MaChado I, López-Guerrero JA, Scotlandi K, Picci P, Llombart-Bosch A. Immunohistochemical analysis and prognostic significance of PD-L1, PD-1, and CD8+ tumor-infiltrating lymphocytes in Ewing’s sarcoma family of tumors (ESFT). Virchows Archiv: an Int J pathology. (2018) 472:815–24. doi: 10.1007/s00428-018-2316-2
8. van Erp AEM, Versleijen-Jonkers YMH, Hillebrandt-Roeffen MHS, van Houdt L, Gorris MAJ, van Dam LS, et al. Expression and clinical association of programmed cell death-1, programmed death-ligand-1 and CD8(+) lymphocytes in primary sarcomas is subtype dependent. Oncotarget. (2017) 8:71371–84. doi: 10.18632/oncotarget.19071
9. Evdokimova V, Gassmann H, Radvanyi L, Burdach SEG. Current state of immunotherapy and mechanisms of immune evasion in ewing sarcoma and osteosarcoma. Cancers. (2022) 15. doi: 10.3390/cancers15010272
10. Tawbi HA, Burgess M, Bolejack V, Van Tine BA, Schuetze SM, Hu J, et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. (2017) 18:1493–501. doi: 10.1016/S1470-2045(17)30624-1
11. D’Angelo SP, Mahoney MR, Van Tine BA, Atkins J, Milhem MM, Jahagirdar BN, et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. (2018) 19:416–26. doi: 10.1016/S1470-2045(18)30006-8
12. Davis KL, Fox E, Merchant MS, Reid JM, Kudgus RA, Liu X, et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): a multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol. (2020) 21:541–50. doi: 10.1016/S1470-2045(20)30023-1
13. Thiel U, Pirson S, Müller-Spahn C, Conrad H, Busch DH, Bernhard H, et al. Specific recognition and inhibition of Ewing tumour growth by antigen-specific allo-restricted cytotoxic T cells. Br J Cancer. (2011) 104:948–56. doi: 10.1038/bjc.2011.54
14. Blaeschke F, Thiel U, Kirschner A, Thiede M, Rubio RA, Schirmer D, et al. Human HLA-A*02:01/CHM1+ allo-restricted T cell receptor transgenic CD8+ T cells specifically inhibit Ewing sarcoma growth in vitro and in vivo. Oncotarget. (2016) 7:43267–80. doi: 10.18632/oncotarget.v7i28
15. Thiel U, Schober SJ, Einspieler I, Kirschner A, Thiede M, Schirmer D, et al. Ewing sarcoma partial regression without GvHD by chondromodulin-I/HLA-A*02:01-specific allorestricted T cell receptor transgenic T cells. Oncoimmunology. (2017) 6:e1312239–e. doi: 10.1080/2162402X.2017.1312239
16. Biele E, Schober SJ, Prexler C, Thiede M, Heyking KV, Gassmann H, et al. Monocyte maturation mediators upregulate CD83, ICAM-1 and MHC class 1 expression on ewing’s sarcoma, enhancing T cell cytotoxicity. Cells. (2021) 10. doi: 10.3390/cells10113070
17. Gassmann H, Schneider K, Evdokimova V, Ruzanov P, Schober SJ, Xue B, et al. Ewing sarcoma-derived extracellular vesicles impair dendritic cell maturation and function. Cells. (2021) 10:2081. doi: 10.3390/cells10082081
18. Schober SJ, Thiede M, Gassmann H, Prexler C, Xue B, Schirmer D, et al. MHC class I-restricted TCR-transgenic CD4(+) T cells against STEAP1 mediate local tumor control of ewing sarcoma in vivo. Cells. (2020) 9. doi: 10.3390/cells9071581
19. Schirmer D, Grünewald TG, Klar R, Schmidt O, Wohlleber D, Rubío RA, et al. Transgenic antigen-specific, HLA-A*02:01-allo-restricted cytotoxic T cells recognize tumor-associated target antigen STEAP1 with high specificity. Oncoimmunology. (2016) 5:e1175795. doi: 10.1080/2162402X.2016.1175795
20. Evdokimova V, Tognon C, Ng T, Ruzanov P, Melnyk N, Fink D, et al. Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell. (2009) 15:402–15. doi: 10.1016/j.ccr.2009.03.017
21. Schober SJ, Schoening C, Eck J, Middendorf C, Lutsch J, Knoch P, et al. The oncolytic adenovirus XVir-N-31 joins forces with CDK4/6 inhibition augmenting innate and adaptive antitumor immunity in ewing sarcoma. Clin Cancer Res. (2023) 29:1996–2011. doi: 10.1158/1078-0432.CCR-22-1961
22. Thiel U, Schober SJ, Ranft A, Gassmann H, Jabar S, Gall K, et al. No difference in survival after HLA mismatched versus HLA matched allogeneic stem cell transplantation in Ewing sarcoma patients with advanced disease. Bone Marrow Transplant. (2021) 56:1550–1557. doi: 10.1038/s41409-020-01200-x
23. Koirala P, Roth ME, Gill J, Piperdi S, Chinai JM, Geller DS, et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci Rep. (2016) 6:30093. doi: 10.1038/srep30093
24. Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun. (2017) 8:1751. doi: 10.1038/s41467-017-01883-9
25. Bailey KM, Julian CM, Klinghoffer AN, Bernard H, Lucas PC, McAllister-Lucas LM. EWS-FLI1 low Ewing sarcoma cells demonstrate decreased susceptibility to T-cell-mediated tumor cell apoptosis. Oncotarget. (2019) 10:3385–99. doi: 10.18632/oncotarget.v10i36
26. Kirschner A, Thiede M, Grünewald TG, Alba Rubio R, Richter GH, Kirchner T, et al. Pappalysin-1 T cell receptor transgenic allo-restricted T cells kill Ewing sarcoma in vitro and in vivo. Oncoimmunology. (2017) 6:e1273301. doi: 10.1080/2162402X.2016.1273301
27. Luksch R, Massimino M, Cefalo G, Lombardi F, Ferrari A, Casanova M, et al. Effects of recombinant human granulocyte-macrophage colony-stimulating factor in an intensive treatment program for children with Ewing’s sarcoma. Haematologica. (2001) 86:753–60.
28. Anderson PM, Markovic SN, Sloan JA, Clawson ML, Wylam M, Arndt CA, et al. Aerosol granulocyte macrophage-colony stimulating factor: a low toxicity, lung-specific biological therapy in patients with lung metastases. Clin Cancer research: an Off J Am Assoc Cancer Res. (1999) 5:2316–23.
29. Arndt CA, Koshkina NV, Inwards CY, Hawkins DS, Krailo MD, Villaluna D, et al. Inhaled granulocyte-macrophage colony stimulating factor for first pulmonary recurrence of osteosarcoma: effects on disease-free survival and immunomodulation. a report from the Children’s Oncology Group. Clin Cancer research: an Off J Am Assoc Cancer Res. (2010) 16:4024–30. doi: 10.1158/1078-0432.CCR-10-0662
30. Lissat A, Joerschke M, Shinde DA, Braunschweig T, Meier A, Makowska A, et al. IL6 secreted by Ewing sarcoma tumor microenvironment confers anti-apoptotic and cell-disseminating paracrine responses in Ewing sarcoma cells. BMC Cancer. (2015) 15:552. doi: 10.1186/s12885-015-1564-7
31. Gross AC, Cam H, Phelps DA, Saraf AJ, Bid HK, Cam M, et al. IL-6 and CXCL8 mediate osteosarcoma-lung interactions critical to metastasis. JCI Insight. (2018) 3. doi: 10.1172/jci.insight.99791
32. Xiao H, Bid HK, Chen X, Wu X, Wei J, Bian Y, et al. Repositioning Bazedoxifene as a novel IL-6/GP130 signaling antagonist for human rhabdomyosarcoma therapy. PloS One. (2017) 12:e0180297. doi: 10.1371/journal.pone.0180297
33. Franciszkiewicz K, Le Floc’h A, Boutet M, Vergnon I, Schmitt A, Mami-Chouaib F. CD103 or LFA-1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T-cell effector functions. Cancer Res. (2013) 73:617–28. doi: 10.1158/0008-5472.CAN-12-2569
34. Petit A-E, Demotte N, Scheid B, Wildmann C, Bigirimana R, Gordon-Alonso M, et al. A major secretory defect of tumour-infiltrating T lymphocytes due to galectin impairing LFA-1-mediated synapse completion. Nat Commun. (2016) 7:12242. doi: 10.1038/ncomms12242
35. Herzfeldt A-K, Gamez MP, Martin E, Boryn LM, Baskaran P, Huber HJ, et al. Complementary CRISPR screen highlights the contrasting role of membrane-bound and soluble ICAM-1 in regulating antigen-specific tumor cell killing by cytotoxic T cells. eLife. (2023) 12:e84314. doi: 10.7554/eLife.84314.sa2
36. Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. (2009) 9:271–85. doi: 10.1038/nri2526
37. Borowski A, van Valen F, Ulbrecht M, Weiss EH, Blasczyk R, Jorgens H, et al. Monomorphic HLA class I-(non-A, non-B) expression on Ewing’s tumor cell lines, modulation by TNF-α and IFN-γ. Immunobiology. (1999) 200:1–20. doi: 10.1016/S0171-2985(99)80029-1
38. Scotlandi K, Baldini N, Campanacci M, Lollini PL, Picci P, Serra M. Induction of HLA class II antigens in osteosarcoma cells by interferons and tumor necrosis factor alpha. Anticancer Res. (1992) 12:767–72.
39. Roebuck KA, Finnegan A. Regulation of intercellular adhesion molecule-1 (CD54) gene expression. J leukocyte Biol. (1999) 66:876–88. doi: 10.1002/jlb.66.6.876
40. Cornel AM, Mimpen IL, Nierkens S. MHC class I downregulation in cancer: underlying mechanisms and potential targets for cancer immunotherapy. Cancers. (2020) 12. doi: 10.3390/cancers12071760
41. Hazini A, Fisher K, Seymour L. Deregulation of HLA-I in cancer and its central importance for immunotherapy. J immunotherapy Cancer. (2021) 9:e002899. doi: 10.1136/jitc-2021-002899
42. Müller-Hermelink N, Braumüller H, Pichler B, Wieder T, Mailhammer R, Schaak K, et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell. (2008) 13:507–18. doi: 10.1016/j.ccr.2008.04.001
43. Cillo AR, Mukherjee E, Bailey NG, Onkar S, Daley J, Salgado C, et al. Ewing sarcoma and osteosarcoma have distinct immune signatures and intercellular communication networks. Clin Cancer Res. (2022) 28(22):4968–82. doi: 10.1158/1078-0432.CCR-22-1471
44. Kimura K, Taguchi T, Urushizaki I, Ohno R, Abe O, Furue H, et al. Phase I study of recombinant human tumor necrosis factor. Cancer chemotherapy Pharmacol. (1987) 20:223–9. doi: 10.1007/BF00570490
45. Whitehead RP, Fleming T, Macdonald JS, Goodman PJ, Neefe J, Braun TJ, et al. A phase II trial of recombinant tumor necrosis factor in patients with metastatic colorectal adenocarcinoma: a Southwest Oncology Group study. J Biol response modifiers. (1990) 9:588–91.
46. Brown TD, Goodman P, Fleming T, Macdonald JS, Hersh EM, Braun TJ. A phase II trial of recombinant tumor necrosis factor in patients with adenocarcinoma of the pancreas: a Southwest Oncology Group study. J immunotherapy: Off J Soc Biol Ther. (1991) 10:376–8. doi: 10.1097/00002371-199110000-00010
47. Muggia FM, Brown TD, Goodman PJ, Macdonald JS, Hersh EM, Fleming TR, et al. High incidence of coagualopathy in phase II studies of recombinant tumor necrosis factor in advanced pancreatic and gastric cancers. Anti-cancer Drugs. (1992) 3:211–7. doi: 10.1097/00001813-199206000-00002
48. Jakob J, Hohenberger P. Role of isolated limb perfusion with recombinant human tumor necrosis factor α and melphalan in locally advanced extremity soft tissue sarcoma. Cancer. (2016) 122:2624–32. doi: 10.1002/cncr.v122.17
49. Rübe CE, van Valen F, Wilfert F, Palm J, Schuck A, Willich N, et al. Ewing’s sarcoma and peripheral primitive neuroectodermal tumor cells produce large quantities of bioactive tumor necrosis factor-alpha (TNF-alpha) after radiation exposure. Int J Radiat oncology biology physics. (2003) 56:1414–25. doi: 10.1016/s0360-3016(03)00418-8
50. van Valen F, Kentrup-Lardong V, Truckenbrod B, Rübe C, Winkelmann W, Jürgens H. Regulation of the release of tumour necrosis factor (TNF)α and soluble TNF Receptor by γ irradiation and interferon γ in Ewing’s sarcoma/peripheral primitive neuroectodermal tumour cells. J Cancer Res Clin Oncol. (1997) 123:245–52. doi: 10.1007/BF01208634
51. Mehta AK, Gracias DT, Croft M. TNF activity and T cells. Cytokine. (2018) 101:14–8. doi: 10.1016/j.cyto.2016.08.003
Keywords: pediatric sarcomas, osteosarcoma, Ewing sarcoma, rhabdomyosarcoma, immunotherapy, adoptive T cell transfer, MHC-I, TNF
Citation: Gassmann H, Thiede M, Weiß J, Biele E, Flohé L, Lachermaier H, Prexler C, Evdokimova V, Radvanyi L, Akhtar I, Morcos MNF, Auer F, Schober SJ, Hauer J, Thiel U and von Heyking K (2024) Cytokine screening identifies TNF to potentially enhance immunogenicity of pediatric sarcomas. Front. Immunol. 15:1347404. doi: 10.3389/fimmu.2024.1347404
Received: 30 November 2023; Accepted: 04 October 2024;
Published: 11 December 2024.
Edited by:
Xiaowen Liu, Fudan University, ChinaCopyright © 2024 Gassmann, Thiede, Weiß, Biele, Flohé, Lachermaier, Prexler, Evdokimova, Radvanyi, Akhtar, Morcos, Auer, Schober, Hauer, Thiel and von Heyking. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hendrik Gassmann, aGVuZHJpay5nYXNzbWFubkB0dW0uZGU=; Kristina von Heyking, a3Jpc3RpbmEuaGV5a2luZ0B0dW0uZGU=