 Selen Ünlü1,2
Selen Ünlü1,2 Blanca G. Sánchez Navarro3†
Blanca G. Sánchez Navarro3† Elif Cakan4Daniel Berchtold3†Rafael Meleka Hanna3
Elif Cakan4Daniel Berchtold3†Rafael Meleka Hanna3 Secil Vural1,5Atay Vural1,6
Secil Vural1,5Atay Vural1,6 Andreas Meisel3
Andreas Meisel3 Miriam L. Fichtner1,3*†
Miriam L. Fichtner1,3*†- 1Koç University Research Center for Translational Medicine (KUTTAM), İstanbul, Türkiye
- 2Koç University School of Medicine, Istanbul, Türkiye
- 3Department of Neurology with Experimental Neurology, Integrated Myasthenia Gravis Center, Neuroscience Clinical Research Center, Charité Universitätsmedizin Berlin, Berlin, Germany
- 4Ragon Institute of Massachusetts General Hospital, Massachusetts Institute of Technology and Harvard University, Cambridge, MA, United States
- 5Department of Dermatology and Venereology, Koç University School of Medicine, İstanbul, Türkiye
- 6Department of Neurology, Koç University School of Medicine, İstanbul, Türkiye
IgG4 subclass antibodies represent the rarest subclass of IgG antibodies, comprising only 3-5% of antibodies circulating in the bloodstream. These antibodies possess unique structural features, notably their ability to undergo a process known as fragment-antigen binding (Fab)-arm exchange, wherein they exchange half-molecules with other IgG4 antibodies. Functionally, IgG4 antibodies primarily block and exert immunomodulatory effects, particularly in the context of IgE isotype-mediated hypersensitivity reactions. In the context of disease, IgG4 antibodies are prominently observed in various autoimmune diseases combined under the term IgG4 autoimmune diseases (IgG4-AID). These diseases include myasthenia gravis (MG) with autoantibodies against muscle-specific tyrosine kinase (MuSK), nodo-paranodopathies with autoantibodies against paranodal and nodal proteins, pemphigus vulgaris and foliaceus with antibodies against desmoglein and encephalitis with antibodies against LGI1/CASPR2. Additionally, IgG4 antibodies are a prominent feature in the rare entity of IgG4 related disease (IgG4-RD). Intriguingly, both IgG4-AID and IgG4-RD demonstrate a remarkable responsiveness to anti-CD20-mediated B cell depletion therapy (BCDT), suggesting shared underlying immunopathologies. This review aims to provide a comprehensive exploration of B cells, antibody subclasses, and their general properties before examining the distinctive characteristics of IgG4 subclass antibodies in the context of health, IgG4-AID and IgG4-RD. Furthermore, we will examine potential therapeutic strategies for these conditions, with a special focus on leveraging insights gained from anti-CD20-mediated BCDT. Through this analysis, we aim to enhance our understanding of the pathogenesis of IgG4-mediated diseases and identify promising possibilities for targeted therapeutic intervention.
1 Introduction
A prerequisite for selecting effective therapies is a profound understanding of the underlying (immuno-) pathology. Many existing therapies, while exhibiting efficacy, are often expensive and may induce side effects that significantly compromise the overall quality of life for patients (1). Therefore, a deeper understanding of the immunopathology is necessary to make informed treatment decisions and to identify therapies that are both effective and efficient in a personal-tailored manner. To gain deeper insights into immunopathology, a valuable approach is to employ reverse translational medicine. In reverse translational medicine, scientific discoveries are informed by clinical observations (2). The clinical observation this review is based on is the remarkable effect of anti-CD20-mediated B cell depletion therapy (BCDT) in disorders with a prevalence of IgG4 subclass antibodies (3–12). This effect is not exclusive for IgG4-mediated diseases where antibodies are the major effectors of pathology like in IgG4 autoimmune diseases (IgG4-AID); IgG4-related disease (IgG4-RD) also responds well to anti-CD20-mediated BCDT (13). In this review will first explore B cells, antibody subclasses, and their properties in general, before we specifically highlight the unique features of IgG4 subclass antibodies in the context of IgG4-AID and IgG4-RD. In the concluding segment of this review, we will examine potential therapies for these diseases, with a particular focus on exploring insights derived from anti-CD20-mediated BCDT.
2 Insights into B cell functions and antibody diversity
In autoimmune diseases, the immune system malfunctions, targeting the body’s own structures. While the immune response involves a variety of cells, some of these autoimmune diseases are characterized by a prominent role of B cells and their effector molecules - the autoantibodies. B cells originate in the bone marrow and undergo several stages of development before maturing into antibody-secreting cells (ASCs), namely plasmablasts and plasma cells (14, 15). The distinct developmental stages of B cell subsets can be identified by surface markers that are expressed at varying levels throughout the maturation process (16). These surface markers are the basis for several B cell targeting therapies, which we will further explore in the section on therapeutic interventions.
In addition to their function as ASCs, B cells play diverse roles in the immune system. They contribute to antigen presentation, cytokine secretion, and the regulation of immune responses (17–19). B cells play a crucial role in modulating T cell responses in both health and disease. Both B and T cells originate from common precursors in the bone marrow, but T cells undergo their final maturation in the thymus (20). B and T cells constitute the adaptive immune system. While B cells contribute to the humoral immune response, T cells serve as the effectors of the cellular response (15). Through antigen presentation, B cells contribute to the negative selection of autoreactive T cells in the thymus, regulate the extent of primary CD4+ T cell responses, and contribute to T cell memory generation (21–23). Antigen presentation by B cells contributes to the immunopathology in several T cell-mediated diseases, including autoimmune hepatitis, rheumatoid arthritis and multiple sclerosis (24–28). Furthermore, B cells are essential for the formation and maintenance of humoral immunological memory, which is crucial for a rapid and effective response upon re-exposure to pathogens (29–31). These memory B cells constitute a crucial reservoir for the generation of ASCs and the corresponding antibody repertoire (14, 31). Throughout the progression of an immune response to an external antigen, B cells undergo affinity maturation, enhancing the affinity of antibodies they produce (32–34). This heightened affinity arises from the interplay of clonal selection and the somatic hypermutation (SHM) process, leading to the gradual accumulation of antibodies with successively greater affinities.
Within the human serum, there is a constant presence of approximately ≈ 9-15 g/L of various IgG subclass antibodies circulating throughout the body (35). These antibodies are polyclonal which means they display diverse specificities, recognizing individual distinct antigens. The interaction between an antibody and its corresponding antigen is highly specific. The segment of the antibody directly involved in this antibody-antigen interaction is termed the variable region (Figure 1A), whereas the constant region - the basis for the categorization of antibodies into isotypes and subclasses - is linked to the antibody’s effector and pathogenic function as well as maturation state (Figures 1B, C) (36). Antibodies are categorized into IgD, IgM, IgE, IgA, and IgG isotypes (37). IgD and IgM are primarily linked to B cells during the naive stage, while antigen-experienced B cells predominantly employ the other isotypes (38).
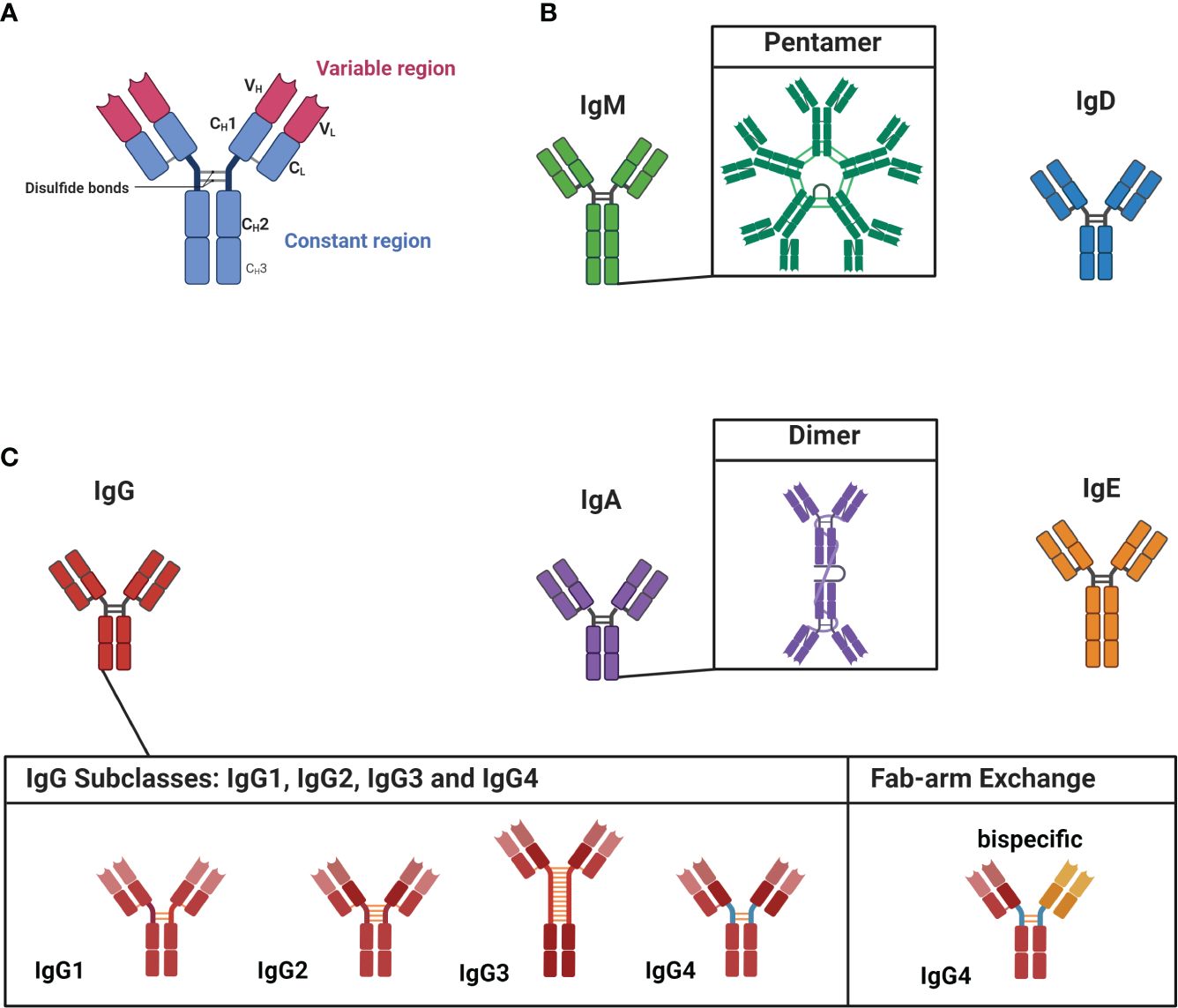
Figure 1 Structure of antibodies, isotypes and IgG subclasses. (A) Antibodies are dimeric structures that consist of two identical antigen-binding sites, known as the variable region (VH and VL). This region, called the fragment-antigen binding (Fab) region, is formed by the combination of the light chain and the heavy chain and is the antigen binding site of the antibody. The hinge region connects the Fab region to the constant region (Fc region, CH1-CH3), which determines the antibody’s isotype and function. (B) The isotypes of IgM and IgD are associated with less experienced, or “naïve,” cells. IgM is found as a pentamer. (C) IgG, IgA, and IgE are linked to more matured antibodies. IgA has the ability to form dimeric structures, while IgG has four subclasses: IgG1, IgG2, IgG3, and IgG4. IgG4 subclass antibodies can undergo Fab-arm exchange, allowing them to become bispecific. Figure created with Biorender.com.
3 The divergent properties of IgG1-4 subclass autoantibodies and unique features of IgG4
The diseases highlighted in this review predominantly involve autoantibodies belonging to the IgG subclass, which can be further divided into IgG1, IgG2, IgG3 and IgG4 (37). Each subclass has distinct structural and functional properties, leading to varied effector functions including complement activation, opsonization (= presenting antigens to phagocytes), antibody-dependent cell-mediated cytotoxicity (ADCC) and neutralization of toxins (37). The most abundant subclass is IgG1 (≈ 65-70%) (39); opsonization, ADCC and complement activation are among the predominant effector functions of IgG1 antibodies (37, 40). IgG2 (≈ 20-25%) antibodies, in contrast, are less effective in complement activation, but potent in opsonization of encapsulated bacteria (39–41). The third most common subclass is IgG3 (≈ 5-8%); IgG3 antibodies are very potent effectors of the immune system and activate the complement cascade, induce ADCC and neutralize toxins (39, 41, 42).
Notably, the least common subclass IgG4 (≈ 3-5%) stands out among these subclasses due to its unique ability to undergo the process of fragment antigen binding (Fab)-arm exchange (39, 43). IgG subclass antibodies are normally dimers with two identical binding sites (Figure 1A). Fab-arm exchange enables IgG4 subclass antibodies to exchange (half)-molecules with other IgG4 antibodies, resulting in bispecific antibodies with two distinct variable regions (Figure 1C; on the left). Fab-arm exchange requires the CH3-domain of the IgG4 antibody (see Figure 1A) and a reducing environment. As this reaction does not require any additional proteins or co-factors, it results in a highly dynamic process during which IgG4 antibodies constantly exchange arms in vivo which generates an ever-changing repertoire of hetero-bivalent IgG4 antibodies (43). Fab-arm exchanged-bispecific IgG4 antibodies cannot cross-link antigens and do not form immune complexes (37). In addition, structural differences in the CH2 domain of IgG4 antibodies are responsible for its low affinity toward C1q from the complement cascade and activating Fcγ receptors (44, 45). However, a recent study revealed that elevated levels of IgG4 antibodies can activate the complement cascade (46). Both variable and constant region glycosylation were found to influence this process. Nonetheless, the biological relevance of this finding remains uncertain. Thus, IgG4 subclass antibodies are generally considered immunomodulatory and referred to as ‘blocking antibodies’ and might function as ‘antigen sink’ (45, 47–49). The blocking function is evident in the context of insect venom or house dust mite hypersensitivity. Antigen-specific IgG4 block the binding of sensitizing IgE to the allergens and consequently prevent allergic symptoms (50). In a similar manner, IgG4 can also prevent hypersensitivity reactions during chronic helminth infections (51, 52). The role of IgG4 subclass antibodies in IgG4-AID and IgG4-RD is further delineated for each specific condition in the subsequent chapter.
The generation of IgG4 most frequently occurs in situations of chronic antigen exposure and might be associated with a continuous germinal center reaction and consecutive rounds of class switch recombination that eventually terminates with a class switch toward IgG4 (53–55). This distinctive property renders IgG4 antibodies particularly prevalent in conditions involving recurrent antigen exposure, as evident in beekeepers and antibodies recognizing bee-related toxins (47, 56–59). The process of class switch to IgG4, similar to the generation of IgE, is regulated by the cytokines IL-4 and IL-13 and necessitates co-stimulation by CD40 (60). Importantly, IL-10 has been shown to reduce IgE secretion and increase IgG4 production and might be directly involved in regulating class switch recombination toward IgG4 (60). More recently, a specialized subset of follicular T helper (Tfh) cells was discovered in tertiary lymphoid tissues of patients suffering from IgG4-related disease. These Tfh cells express the markers BCL6, CXCR5, and ICOS as well as the cytokines IL-4, IL-21, and IL-10 and therefore might crucially regulate the switch to IgG4 (61).
4 Features of autoantibodies in IgG4-mediated diseases
IgG4 subclass antibodies are notably predominant in IgG4-AID and IgG4-RD (3–10). A summary of the clinical and epidemiological characteristics of the diseases can be found in Table 1. This review focuses on the immunopathology of these diseases and the properties of the corresponding antibodies in the context of disease.

Table 1 Comparison of characteristics of IgG4 subtypes of MG, CIDP, Pemphigus and IgG4-RD.
4.1 MuSK myasthenia gravis
The autoantibodies in myasthenia gravis (MG) target structures at the muscle endplate of the neuromuscular junction (NMJ) hindering neuromuscular transmission (100, 101). These autoantibodies disrupt the interaction between the ligand acetylcholine and the acetylcholine receptor (AChR) at the NMJ. They achieve this by directly targeting the AChR or structures within the muscle-specific tyrosine kinase (MuSK)/Low Density Lipoprotein Receptor-Related Protein 4 (LRP4) pathway, critical for the proper clustering and functionality of AChRs (102). The consequence of this interference is the disruption of signaling from nerves to muscles and patients present clinically with increased fatigability and muscle weakness (100, 101). MG is a very heterogeneous disease consisting of several different subtypes. These subtypes are partly categorized by the target antigen that the antibodies detect. So far, three main target antigens have been identified and validated: AChR (103, 104), MuSK (105), and LRP4 (106, 107). Although all of these subtypes are unified under the term MG, each disease has distinct clinical and immunological features (59). The immunological differences are further apparent in the subclass usage of each subtype. While AChR, and LRP4 autoantibodies are mostly of the IgG1, IgG2 and IgG3 subclass (59, 106, 108), MuSK is predominantly IgG4 (4–6). Interestingly, antibodies of the IgG1-3 subclasses in MuSK MG impact the clustering of AChRs (109, 110), resulting in a reduction of clustered AChRs. The precise mechanism underlying this effect remains to be fully elucidated and seems to be divergent from IgG4 MuSK autoantibodies (110).
The isolation and characterization of monoclonal autoantibodies (mAbs) against MuSK has significantly advanced our understanding of MuSK-MG (12, 111–114). The knowledge that plasmablasts are a source for MuSK autoantibodies and the development of mechanisms for the enrichment of this specific B cell population (111–114), were instrumental for the generation of MuSK mAbs (111, 113, 114). The ectodomain of MuSK consists of three Ig-like (Ig1-3) domains and a frizzled domain (115, 116). Previous findings revealed that the majority of MuSK autoantibodies recognize the Ig-like domain 1 on the MuSK receptor, as observed with human polyclonal sera (117). These polyclonal autoantibodies were proven to be pathogenic in vitro and in vivo via passive transfer (118–120), directly inhibiting the interaction between MuSK and LRP4 (109, 117). More recently, mAbs detecting the Ig-like domain 2 were also shown to have pathogenic capacities (113). The impact of valency on the pathogenicity after Fab-arm exchange of MuSK autoantibodies was initially demonstrated in sera-based experiments (121) and subsequently confirmed with human MuSK mAbs (112, 122, 123). It has further been shown that affinity maturation plays a crucial role in the pathogenic development of MuSK mAbs and that a high affinity coupled with monovalency is essential to reach a pathogenic threshold necessary for the potent disruption of AChR clusters at the NMJ (122). MuSK autoantibody titer correlates well with disease severity in MuSK MG (5, 6, 124–126), might be a potential biomarker to detect relapse in MuSK MG (114, 127) and treatment success with BCDT (114, 124).
4.2 Nodo-paranodopathies with autoantibodies targeting NF155, CNTN1, and CASPR1
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a progressive autoimmune peripheral neuropathy, where the main target is the myelin sheath of peripheral nerves (128, 129). Over the last decade, antibodies targeting the proteins located in the nodes of Ranvier and the paranodal region have been identified and reported to be present in 2-15% of patients clinically diagnosed with CIDP (10, 130). Although these patients are generally diagnosed clinically as CIDP, there is a growing consensus on classifying seropositive CIDP patients under nodo-paranodopathies instead of CIDP, as there are distinct immunopathological and clinical characteristics between IgG4-mediated nodo-paranodopathies and CIDP (131–133).
These antibodies target mainly neurofascin 155 (NF155), neurofascin 186 (NF186), neurofascin 140 (NF140), contactin-1 (CNTN1), and contactin-associated protein 1 (CASPR1) (8, 10, 134–138). In the majority of patients with nodo-paranodopathy, the dominant immunoglobulin subclass is IgG4, particularly for anti-NF155, anti-CNTN1, and anti-CASPR1 (8, 10). These proteins function as cell adhesion molecules. NF155 proteins are located mainly on the myelin loops of Schwann cells and bind to the CNTN1-CASPR1 complexes that are found on the axolemma. The resulting tripartite protein complexes attach the myelin loops strongly to the axon in the paranodes, resulting in formation of the largest junctions known in the body. Antibodies against CNTN1 and CASPR1 bind to the epitopes found in domains that interact with the partner proteins, therefore abolishing the formation of tripartite complexes and leading to disruption of paranodal junctions, resulting in conduction deficits (10, 139). A distinguished feature of anti-NF155 IgG4 antibodies is that these antibodies do not prevent the interaction of its target protein with its partners. Instead, binding of these antibodies leads to the formation of NF155 clusters on Schwann cell surface, resulting in depletion of proteins necessary for the formation of the paranodal complex (140). In summary, unlike classical CIDP, IgG4 antibodies against paranodal antigens do not cause inflammation or demyelination, but rather cause paranodal detachment and disturbance of nodal electrophysiology leading to conduction blocks and potentially axonal degeneration.
Further examination of patient sera with anti-NF155 antibodies revealed that pathogenic monospecific bivalent IgG4 antibodies are present in the sera. The effect of monovalency on pathogenicity is variable in nodo-paranodopathies. In contrast to MuSK MG, the decreased valency of anti–Neurofascin-155 IgG4 subclass autoantibodies resulting from Fab-arm exchange strongly diminished the effect of pathogenic antibodies (141). In CNTN1 autoantibodies, however, monovalency showed similar pathogenic capacities in comparison to their divalent counterparts (142). Similar to MuSK MG, the autoantibodies titers in nodo-paranodopathies correlate well with clinical diseases severity (137, 143, 144).
4.3 Pemphigus with autoantibodies targeting Dsg1 and Dsg2
Pemphigus is a group of autoimmune bullous diseases characterized by a pathogenic autoimmune response, primarily driven by autoantibodies targeting two key desmosomal adhesion proteins (145). Desmogleins are Ca2+-dependent transmembrane proteins situated in keratinocyte desmosomes and play a crucial role in maintaining the integrity and cohesion of keratinocytes within the epidermis (146). Dsg1 is primarily located in superficial layers, while Dsg3 is found in basal and parabasal layers of the skin (147). The two main subgroups of pemphigus are pemphigus vulgaris (PV) and pemphigus foliaceus (PF), the latter being less common (3, 7, 9).
Pathogenic autoantibodies in pemphigus are of the IgG1 and IgG4 sublcass. These antibodies specifically identify epitopes situated within the EC1 and EC2 domains of Dsg1 and Dsg3 (148). The interaction of these autoantibodies results in the obstruction of cell-cell adhesion, leading to the development of skin blisters (3, 149). This pathological mechanism has been substantiated in vitro, wherein the antibodies prompt the separation of cell sheets in cultured human keratinocytes and human skin explants (150–152). Clinical evidence underscores the pathogenicity of IgG4 in comparison to other serum IgG fractions, as eliminating IgG4 from PV sera has been observed to lead to a 81% reduction in dissociation in keratinocyte assays (7). In cases of active disease, both PV and PF patients typically manifest enriched desmoglein reactive IgG4 and IgG1, while individuals in remission and certain healthy relatives of pemphigus patients may solely exhibit IgG1 (149, 153–155). Additionally, the passive transfer of maternal antibodies to the fetus induces a transient neonatal form of pemphigus [201]. Polyclonality and epitope specificity affect the pathogenic effect of autoantibodies in PV (156). Beyond impeding cell-cell adhesion, the autoantibodies can also modulate signal transduction pathways that influence cytoskeleton rearrangement and cell adhesion in keratinocytes (152, 157, 158) For instance, the activation of p38 mitogen-activated protein kinase (p38MAPK) plays a vital role in causing the loss of cell cohesion. Blocking p38MAPK in the human epidermis has been shown to prevent blistering. Therefore, the specific morphological alterations induced by pathogenic IgG in mucocutaneous PV such as widening between desmosomes and the decrease in desmosome size are at least partially associated with p38MAPK signaling (152, 157). Monovalency resulting from Fab-arm exchange was shown to increase the pathogenic effect of patient-derived autoantibodies in PF (159). The effect of valency in PV seems less pronounced (160, 161). Monovalent single-chain variable-region fragments of autoantibodies derived from PV patients’ demonstrated pathogenic capacity in vivo (151) as well as Fabs of patient-derived mAbs (161). In pemphigus, there is no direct correlation of autoantibody titer to disease severity and as such, the titer cannot be used to monitor disease activity directly (162, 163).
4.4 LGI1/CASPR2-antibody Encephalitis
Antibody-mediated encephalitis is a heterogeneous group of disorders caused by more than 21 different antibodies (164). Among these, anti-LGI1 and anti-CASPR2 encephalitis are related to IgG4 as the dominant immunoglobulin subtype (165). LGI1 is a synaptic protein that binds to presynaptic metalloproteinase domain-containing protein 23 (ADAM23) and postsynaptic ADAM22. ADAM23 positions voltage-gated potassium channels in the presynaptic terminal and AMPA receptor in the postsynaptic membrane. Antibody binding disrupts LGI1-ADAM22/ADAM23 complexes on the cell surface leading to diminished AMPAR and voltage-gated potassium channel (VGKC) clusters. Loss of inhibitory VGKC complexes heightens neuron excitability, while AMPAR loss impairs long-term potentiation. The AMPAR loss is believed to directly contribute to the memory deficits observed in individuals with anti-LGI1 encephalitis (166, 167). CASPR2 serves as a transmembrane cell adhesion protein that interacts with Kv1.1 and Kv1.2 VGKCs in the juxtaparanodal region of myelinated peripheral nerves. In addition to its distribution in the nodes of Ranvier across the central and peripheral nervous systems, CASPR2 is also found in the synapses of the limbic system and basal ganglia (164). In mice, intrathecal infusion of anti-CNTN2 IgG, comprising a mixture of IgG1 and IgG4, purified from individuals with anti-CASPR2 encephalitis, were observed to induce memory deficits. This effect was attributed to hindering CASPR2/TAG1 interaction and reducing the surface levels of CASPR2, Kv1.1, and AMPAR, similar to LGI1 antibodies (168). CASPR2 antibodies exert their pathogenicity mainly through blocking (169), while studies showing the effect of monovalency on pathogenicity are currently missing. In LGI1 encephalitis the effect of valency on the pathogenic capacity has also not been investigated in detail yet. Similarly, the correlation of the autoantibody titer in the bloodstream with clinical disease severity is unknown. Levels of autoantibodies found in the cerebrospinal fluid might serve as a more accurate indicator in these pathologies of the central nervous system.
4.5 IgG4-RD
IgG4-RD is an immune-mediated systemic condition characterized by fibroinflammatory lesions in various organs; these lesions can mimic malignancies, infections, and inflammatory disorders, often accompanied by elevated IgG4 levels, though not always (170, 171). Recognized as a distinct disease only since 2003, early diagnoses of IgG4-related disease were often incidental findings during surgical resections of lesions initially suspected to be malignant (172, 173). IgG4-RD is characterized by three major histopathological findings: a dense lymphoplasmacytic infiltrate, fibrosis, at least focally in a storiform pattern, and obliterative phlebitis. Additionally, a diagnosis requires an increased number of IgG4 plasma cells in the tissue (174). The 2019 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) established comprehensive criteria for diagnosing and investigating IgG4-RD. These criteria include: (1) involvement of at least one of 11 possible organs, (2) a total of 32 exclusion criteria, and (3) eight weighted inclusion criteria (175).
Regarding the underlying immunopathology, CD4+ cytotoxic T lymphocytes play a pivotal role, constituting a major subset in both tissue and circulation. These cells secrete pro-fibrotic cytokines such as IL-1β, TGF-β1, and IFN-γ, along with cytolytic molecules like granzymes (176, 177). Among this population, the dominant effector subset is characterized by CD27lo CD28lo CD57hi cells with clonal expansion, and activated CD8+ T cells expressing granzyme-A are also observed (178). A recent study on tertiary lymphoid organs in IgG4-RD revealed a significant infiltration of a Tfh subset that is LAG3 and IL-10 positive (61). Activated B cells and plasmablasts may interact with these CD4+ T cell subsets, contributing to fibrosis and inflammation in IgG4-RD (176). Notably, a study highlighting dominant plasmablast clones identified galectin-3 autoantibodies, present in a subset of patients and correlated with galectin-3 plasma levels (179). Unlike autoantibody-mediated autoimmune disorders, the direct involvement of B cells in IgG4-RD pathology remains unclear and requires further investigation. The circulating IgG4 antibodies in IgG4-RD most likely are predominantly monovalent due to Fab-arm exchange. Interestingly, the levels of IgG4 subclass antibodies is not always elevated in these patients (170, 171).
5 Therapeutic Interventions
The standard treatment for autoimmune diseases previously involved broad immunosuppressive drugs (180). While these therapies improve symptoms, their effectiveness is often limited by adverse side effects. Moreover, not all patients respond to these conventional treatments. Interestingly, patients who do not respond to standard therapies have shown positive responses to treatments originally used in B cell malignancies, such as anti-CD20 mediated BCDT (3–12). Studying the efficacy of anti-CD20 mediated BCDT has provided valuable insights into potential therapies for B cell pathologies. Additionally, distinct therapies influence the B cell repertoire differently as observed in patients that received mycophenolate mofetil and anti-CD20-mediated BCDT (181), highlighting the potential benefits of combination therapy. In this chapter, we will first examine anti-CD20 mediated BCDT and then explore new therapeutic approaches.
5.1 Anti-CD20-mediated BCDT
Originally developed for treating B cell malignancies, anti-CD20 mediated BCDT has proven effective in managing diverse autoimmune conditions, including multiple sclerosis, PV, rheumatoid arthritis, CIDP, and MuSK MG (124, 135, 182–184). Anti-CD20 mediated BCDT exhibits remarkable efficiency in IgG4-AID and IgG4-RD (3–12).
Many MuSK MG patients enter stable remission for several years following anti-CD20 mediated BCDT, with significantly reduced or non-detectable MuSK autoantibody titers (124, 125, 185). However, relapses can occur in some patients over time (68, 185–187). In these cases, the MuSK autoantibody titer may increase months before clinical-detectable relapse (114, 125, 127). During relapse, frequencies of plasmablasts and memory B cells are elevated, with disease-related autoantibody-expressing B cells identified within these populations (111, 113, 114, 188–190). Consequently, MuSK MG patients treated with anti-CD20 mediated BCDT undergo cycles of remission followed by phases of clinical relapse. Most patients with IgG4-mediated nodo-paranodopathies are unresponsive to IVIg and steroids, unlike classical CIDP. As such, IVIg and steroid unresponsiveness should lead to antibody testing in CIDP patients. Anti-CD20 mediated BCDT shows beneficial effects in the majority of patients with IgG4-mediated nodo-paranodopathies, particularly those refractory to other therapies (191–195). In a large cohort of anti-NF155 antibody positive patients, 77% of patients responded to anti-CD20 mediated BCDT (194). In addition, serum neurofilament light chain (NfL) levels which are an indicator for axonal damage and anti-NF155 antibody titers were also decreased after therapy. Similar positive outcomes have been observed in pemphigus (196–198). High-dose anti-CD20 mediated BCDT therapy extended remission duration, although it did not impact the relapse rate (40%) compared to a low-dose regimen (197). Furthermore, treatment response to anti-CD20-mediated BCDT is good in LGI1/CASPR2-antibody encephalitis (11, 199). Interestingly, anti-CD20-mediated BCDT has emerged as a highly effective therapy option in IgG4-RD, underscoring the potential involvement of B cells in the pathophysiology (13).
However, it is crucial to emphasize that the response to anti-CD20 mediated BCDT varies and not all patients achieve remission (125, 197, 200, 201). In MuSK MG, non-response in patients might be associated with a prevalence of pathogenic IgG1-3 subclass antibodies (110). Additionally, in AChR MG, which was previously considered a less favorable target for anti-CD20 mediated BCDT, a small subset of patients’ benefits from anti-CD20 mediated BCDT. These positive responses have been observed in cases of severe refractory AChR MG (200, 202). Anti-CD20 mediated BCDT appears to yield better results when initiated during the early stages of AChR MG (203, 204). This variability in response is likely due to the heterogeneity of the underlying immunopathologies (59) and an increasing significance of long-lived plasma cells in the immunopathogenesis as the disease progresses over time. In some instances, disease progression occurred post-anti-CD20 mediated BCDT administration (205), potentially linked to the presence of anti-RTX antibodies, known to impede responsiveness of anti-CD20 mediated BCDT in IgG4-mediated nodo-paranodopathies (206). Ocrelizumab, another BCDT targeting CD19 with lower immunogenic potential, has proven to be a successful alternative in patients developing anti-RTX mAbs (207). The variation in response may, to some extent, be associated with IgG subclasses other than IgG4 playing significant roles in the immunopathology of these patients (110).
However, the complexity of CD20 expression should be acknowledged, extending beyond B cells to include T cells (Figure 2). Thus, anti-CD20 mediated BCDT does not exclusively affect B cells; subsets of T cells expressing CD20 on their surface are also impacted by this therapy (208–210). This depletion of CD20-expressing T cells has been observed in multiple sclerosis (208, 211). For B cells, CD20 is expressed at nearly all stages of B cell differentiation, excluding plasma cells, Pro-B-cells, and Pre-B-I (Figure 2) (16). The initial report of T cells with CD20dim expression (208, 212), was first widely considered a flow cytometry artifact (213). Subsequently, this population was identified as a distinct T cell subset with both immune-regulatory and proinflammatory activities (208, 214). The frequency of CD20+ T cells in the peripheral blood of healthy individuals is relatively low (1-4%). However, these cells are increased under inflammatory conditions and enriched in tissues including tonsils, thymus, bone marrow, and cerebrospinal fluid compared to the peripheral blood (208, 210, 215, 216). The origin of CD20-expressing T cells remains unclear (Figure 2). One proposed explanation is that T cells acquire CD20 through the process of trogocytosis, involving the simultaneous transfer of HLA-DR and CD20 from B cells (210, 216–218). However, the low expression of HLA-DR on T cells suggests that trogocytosis may not fully elucidate the origin of this subset (208, 217). Additionally, CD8+ T cells are known to interact mainly with cross-presenting dendritic cells during conditions such as viral infection, rather than B cells. As dendritic cells are not known to express CD20 on cell surface, the acquisition of CD20 by CD8+ T cells through trogocytosis remains questionable. Moreover, MS4A1, the gene that encodes CD20, was shown to be transcribed in T cells (208). Therefore, CD20 acquisition may occur during T cell activation (217, 219) or result from clonal expansion of CD20-expressing T cells (217, 220). Myelin-specific CD8+ CD20+ T cells were shown to be depleted together with B cells after CD20 monoclonal therapies (221). The major T cell subtypes that were lost included memory CD8+CD20+ and central memory CD8+ T cells but not CD4+CD20+ T cells and this may contribute to increased infection rate seen in these patients (222). Another study found that pretreatment levels of CD8+CD20+ T cells that have a proinflammatory phenotype have a significant inverse relationship with disease burden before treatment. Additionally, these cells were predictive of early disease activity following the initiation of anti-CD20 therapy (223). Therefore, RTX’s effectiveness in these autoimmune diseases may not only be the consequence of RTX’s effect on B cells.
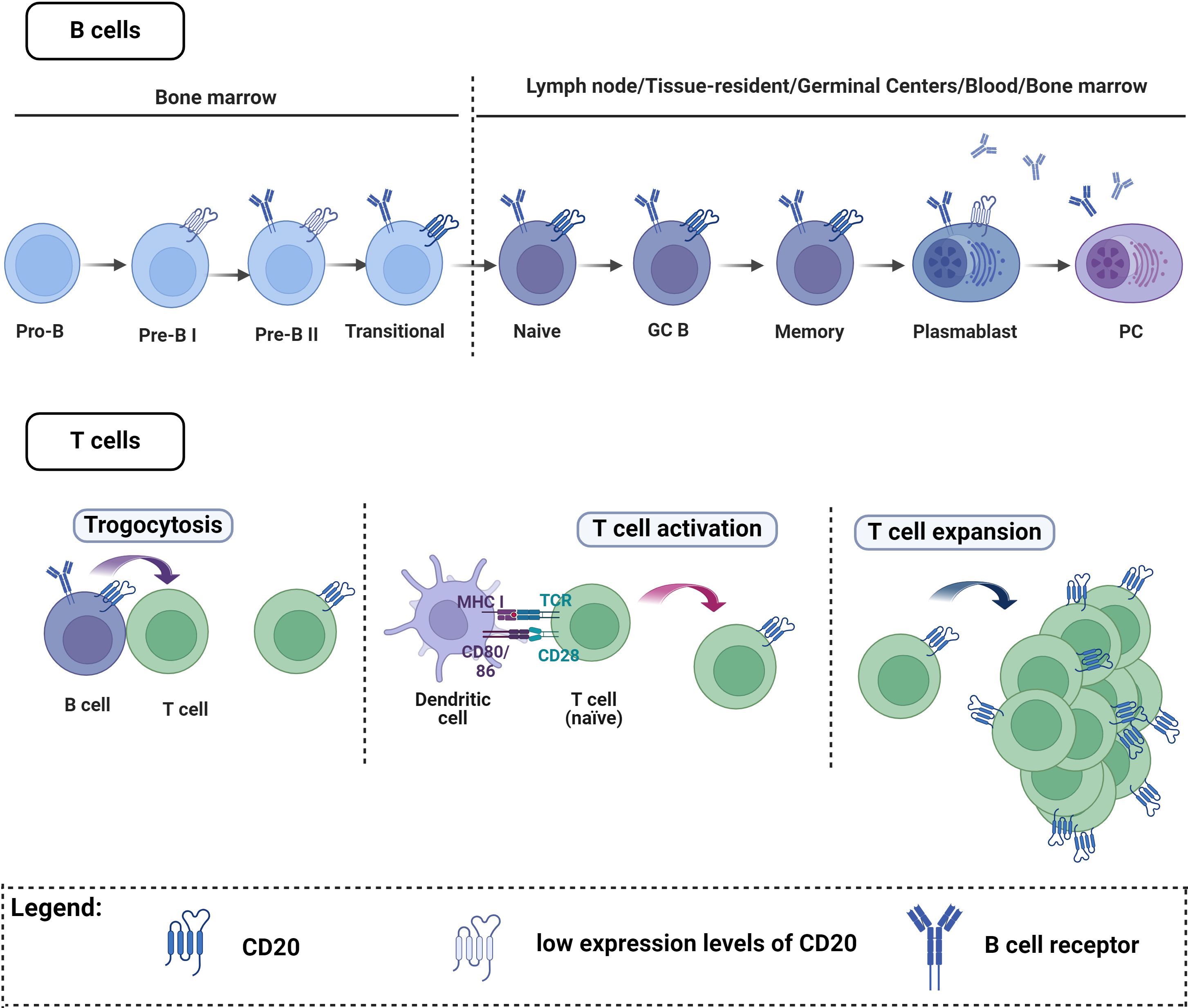
Figure 2 CD20 expression on B cells and T cells. B cells. CD20 is expressed at almost all stages of B cell maturation, with the exception of plasma cells, Pro-B cells, and Pre-B-I cells. T cells. A small number of T cells express CD20, and there are three main theories explaining how these cells acquire CD20: One explanation proposes that T cells may acquire CD20 through trogocytosis, involving simultaneous transfer from B cells. Alternatively, CD20 acquisition might occur during T cell activation or result from the clonal expansion of CD20-expressing T cells. Figure created with Biorender.com.
B cell depletion by anti-CD20 mediated BCDT is further limited, as disease-relevant B cells persist even after treatment (114, 190). The persistence of these B cell clones is not unique to MuSK MG; this phenomenon extends to various autoimmune disorders, including Sjögren’s syndrome, SLE, systemic sclerosis, and ANCA-associated vasculitis (181, 224, 225). These persistent B cells were found to reemerge in MuSK MG months before clinical detectable relapse simultaneously with an increase of autoantibody titer (114). Several characteristics distinguish persistent B cell clones. CD20 expression is crucial for the efficacy of anti-CD20-mediated B cell depletion therapy. Both persistent memory B cells and antibody-secreting cells (ASCs) express low CD20 levels (114, 190). Furthermore, persistent memory B cell subsets express genes associated with previous tissue homing (114, 188, 190, 226–228). Additionally, B cell memory clones that are highly expanded show a higher rates of persistence (190, 229–231). Alterations in the BAFF/APRIL system, including reduced BAFF-R expression and elevated TACI and BCMA levels, are observed in persistent clones and plasmablasts at the time of relapse (114, 190, 232). Nevertheless, it is evident that the remarkable effect of anti-CD20 mediated BCDT in IgG4-associated disorders suggests a direct impact on the reservoir of pathogenic cells in these diseases.
5.2 New therapeutic approaches
5.2.1 Anti-CD19 mediated BCDT
CD19 is a transmembrane glycoprotein from the immunoglobulin superfamily (233) and it is functionally associated with modulation of antigen-independent B cell differentiation and immunoglobulin-induced B cell activation (234). The expression profile of CD19 is broader than that of CD20, starting earlier at the stage of pro-B cells and extending into plasmablasts and plasma cells beyond the expression of CD20 (234, 235). Two anti-CD19 mAbs have reached clinical trials: XmAb5871 and Inebilizumab (236, 237). XmAb5871 is currently investigated in a phase II clinical trial (ClinicalTrials.gov Identifier: NCT02725476) and Inebilizumab (ClinicalTrials.gov Identifier: NCT04540497) in ongoing phase III clinical trials for patients with IgG4-RD. Inebilizumab is further tested in MG (MINT; ClinicalTrials.gov Identifier: NCT04524273). CD19 has gained attention in recent years as an alternative therapy to anti-CD20 mediated BCDT. Given its broader expression profile, especially in ASCs. Besides, it could be an interesting alternative for patients developing resistance to CD20 therapies, as it has already been reported in some non-responders (207).
5.2.2 Anti-CD38 mediated BCDT
The main expression of CD38 is observed in hematopoietic cells. It was first identified as a lymphocyte specific antigen, but current studies revealed that it is ubiquitously expressed (238, 239). Plasma cells and memory B cells express high levels of CD38, while it is expressed at low levels in normal lymph and bone marrow cells (240). CD38 is also expressed in B cell precursors, germinal center B cells, and plasma cells and in other immune cells like NK cells, neutrophils and myeloid cells (239). CD38 is a multifunctional, membrane-bound protein that serves as an antigen and as an enzyme. It catalyzes the metabolism of cyclic ADP-ribose (cADPR) and nicotinic acid adenine dinucleotide phosphate (NAADP). These are two different calcium second messengers, involved in several cell functions (238, 239). Besides this enzymatic function, CD38 can also act as a receptor for CD31, acting as an adhesion molecule for mediating selectin-like binding of hematopoietic cells to endothelial cells and facilitating their transmigration to tissue (239). The role of CD38 in immune cells ranges from immunomodulation to effector functions during inflammation, where it could regulate cell recruitment, cytokine release and NAD availability. This expression profile of CD38, together with its role in inflammatory processes, make CD38 an interesting target in the context of autoimmune diseases (239, 240). Antibodies targeting CD38 have been proposed as a potential therapeutic approach to eliminate plasma cells that produce autoantibodies. Daratumab and Isatuximab have already been established as an important therapeutic target for Multiple Myeloma (MM) (241). Isatuximab binds to an epitope that partially covers the catalytic site of CD38, without changing its configuration, while Daratumumab is binding outside the catalytic site (242). In SLE patients, Daratumumab was found to restore NK cytotoxic function which promoted the elimination of circulating plasma cells (240). Thus, anti-CD38 might be a potent drug to reduce the levels of ASCs.
5.2.3 Anti-CD40 mediated BCDT
CD40 is constitutively expressed on B lymphocytes, and CD40L is mainly expressed in the surface of activated CD4+ T cells, inducing activation, proliferation and cytokine production. Besides, they are found in other hematopoietic cells such as monocytes and dendritic cells as well as non-hematopoietic cells like mast cells, basophils, NK cells, macrophages, megakaryocytes and platelets (243). CD40 is a receptor of the TNF superfamily (243) and together with its ligand, CD40L (CD154), they form an important stimulatory immune checkpoint (244). CD40/CD40L interaction is essential for the formation of germinal centers and the production of class-switched antibodies. It is also important in humoral and cellular immunity, being involved in the activation of innate and adaptative immune cells, regulation of B cell, T cell and APC activation and immunological memory (243, 244).
This role in activation of immune responses makes it an attractive target for potential therapies, and recently, it has been the subject of multiple studies with this aim (244). Iscalimab is a human anti-CD40 monoclonal antibody (244) and it has been recently evaluated for safety and efficacy in moderate to severe symptomatic MG in a phase II clinical trial (245). Previously, a therapeutic benefit had been reported in patients with other autoimmune diseases such as Sjörgen Syndrome (246) and Graves´disease (247). In the case of Pemphigus, the interaction between CD40 and its ligand is important for the induction of pathogenic anti-Dsg3 IgG antibodies (248). Therefore, anti CD40L could be a potential therapeutic approach for Pemphigus (249). In summary, antibodies directed against the CD40/CD40L axis constitute a novel target with potential benefits for patients with autoimmune diseases, by targeting plasma cells involved in the production of pathogenic autoantibodies.
5.2.4 Targeting the BAFF/APRIL-system
The BAFF/APRIL-system plays a crucial role in regulating the survival and maintenance of B cells. This system consists of two key ligands, B-cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL), along with three receptors: the transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI), B cell maturation antigen (BCMA), and B-cell activating factor receptor (BAFF-R) (250). BAFF and APRIL ligands are primarily produced by myeloid and stromal cells, while their corresponding receptors are found on circulating B cells. These receptors are expressed at various stages of B cell development, suggesting a dynamic regulation of B cell survival and maintenance throughout maturation (16). Dysregulation within the BAFF/APRIL-system is associated with autoimmune diseases as well as cancer, allergies, transplants, infections, and immunodeficiencies (251). Elevated levels of BAFF/APRIL soluble receptors and ligands are linked to B cell pathologies, observed in conditions like MG, SLE, Sjögren´s syndrome, and RA, among others (251–255).
This pivotal role in autoimmune diseases suggests that targeting components of this system may offer a potential therapeutic avenue (251). Belimumab (GSK1550188), a recombinant monoclonal antibody targeting soluble BAFF, was the first FDA-approved biologic for SLE treatment, following positive safety and efficacy outcomes in phase III clinical trials (ClinicalTrials.gov Identifier: NCT00424476; ClinicalTrials.gov Identifier: NCT00410384) (251). Unexpectedly, Belimumab did not demonstrate significant efficacy in a phase II clinical trial for Myasthenia Gravis (AChR and MuSK) (ClinicalTrials.gov Identifier: NCT01480596) (256). Other BAFF-targeting agents such as Blisibimod and Tabalumab have undergone phase II/III clinical trials for diseases like SLE and MM (257–259). In recent years, research has focused on dual inhibitor agents, like Atacicept, which inhibit both BAFF and APRIL simultaneously. Atacicept is designed as the extracellular domain of the receptor TACI fused to the human Fc domain (260, 261). Atacicept has shown beneficial effects in SLE and RA patients by significantly depleting plasma cells (262–266) and Telitacicept, another dual inhibitor, has also demonstrated positive effects in autoimmune diseases (259). In a phase II clinical trial for SLE, Telitacicept exhibited beneficial effects (267). Additionally, there is an ongoing phase III clinical trial in China to evaluate the efficacy and safety of Telitacicept in the treatment of MG, currently recruiting (ClinicalTrials.gov Identifier: NCT04660565). Another ongoing phase IV clinical trial in China is assessing the use of Belimumab for the treatment of IgG4-RD, with results pending (ClinicalTrials.gov Identifier: NCT04660565). Although dual inhibtion of BAFF and APRIL seems to have beneficial effects in some autoimmune disorder, in multiple sclerosis a trial for atacicept had to be halted due to increased disease activity in patients compared to placebo (268–270). Thus, the complexity of the BAFF/APRIL-system has to be further investigated to better understand how therapy influences the underlying immunopathology of individual diseases.
5.2.5 Proteasome inhibitors
The ubiquitin/proteasome system constitutes one of the main pathways for intracellular protein degradation (271, 272). Therefore, it is a key component for maintaining a dynamic control over key signaling components of the immune response, as well as overall cell homeostasis. Malfunctioning of the proteasome system is associated with pathological conditions, like cancer or autoimmune disorders (272). Proteasome inhibition results in the accumulation of defective immunoglobulin chains, leading to stress in the endoplasmic reticulum, misfolding of proteins and ultimately cell apoptosis (271, 273). It has been proven to be critical for plasma cell function due to their high rate of antibody synthesis. Inhibiting the proteasome results in the apoptosis of plasma cells and a consequent decrease in antibody production. Besides, proteasome inhibition also hinders the production of pro-inflammatory cytokines (273). This makes the proteasome a promising novel target for antibody mediated autoimmune diseases, involving long-lived plasma cells, like SLE (273) or AChR-MG (59).
Bortezomib, initially approved for MM, is a dipeptide boronic acid derivative, that binds to the catalytic site of the proteasome with high affinity on plasma cells (272, 273). There have been several clinical trials for Bortezomib in SLE patients, that probe a remarkable efficacy of Bortezomib, especially in SLE patients that had been refractory to prior immunosuppression therapies (271, 274). Bortezomib and other proteasome inhibitors have shown beneficial effect in EAMG, a mouse model of MG (275, 276). In-vitro studies of thymic cell cultures derived from AChR-MG patients with Bortezomib could eliminate thymus-derived plasma cells and reduce IgG levels (277). A phase 2 clinical trial (ClinicalTrials.gov Identifier: NCT02102594) to investigate the use of Bortezomib on patients with therapy-refractory Myasthenia Gravis (generalized) or SLE or RA is currently still ongoing (278). Thus, Bortezomib is another good potential candidate for a more targeted immunotherapy in B cell pathologies.
5.2.6 FcRn inhibitors
Human IgG is one of the most abundant proteins in serum, probably because of the uniquely long half-life, for which neonatal fragment crystallizable (Fc) receptor (FcRn) plays an essential role (279, 280). The FcRn is a multifunctional atypical form of Fc-gamma receptor. It was first identified as the responsible of transporting IgG from the maternal to the fetal circulation. Later it was found that, among other functions, it plays an essential role in IgG recycling, by protecting IgG from intracellular degradation, therefore, expanding the half-life (280, 281). FcRn inhibitors enhance the catabolism of IgG by blocking the FcRn-mediated IgG recycling pathway leading to reduced IgG levels in serum and a decrease in pathogenic autoantibodies (282).
In recent years, FcRn inhibitors have emerged as promising targets for autoantibody-mediated autoimmune diseases. Efgartigimod (ARGX-113) binds to FcRn, preventing IgG recycling (282). A phase III trial in MG showed efficacy and tolerability (283) and an ongoing trial for pediatric MG patients is currently recruiting (ClinicalTrials.gov Identifier: NCT05374590), alongside a phase III trial assessing different dosing regimens (ClinicalTrials.gov Identifier: NCT04980495). SYNT001, another FcRn inhibitor, demonstrated efficacy in a phase I/II trial for Pemphigus (284). Rozanolixizumab, also an anti-FcRn monoclonal antibody, showed positive results in MG clinical trials (285). Several trials are ongoing for Rozanolixizumab in MG treatment, including a phase III trial (ClinicalTrials.gov Identifier: NCT04650854) and a self-administration study (ClinicalTrials.gov Identifier: NCT05681715). Additionally, a phase II/III trial for pediatric MG patients is ongoing (ClinicalTrials.gov Identifier: NCT06149559). Other FcRn inhibitors like Batoclimab are also in clinical trials, including a phase III trial for generalized MG (ClinicalTrials.gov Identifier: NCT05403541) and testing in patients with active CIDP (ClinicalTrials.gov Identifier: NCT05581199). Overall, FcRn inhibitors hold promise as therapy for IgG-driven autoimmune diseases like MG, offering targeted treatment with potentially fewer side effects than nonspecific therapies.
5.2.7 Complement inhibitors
The complement system comprises a network of over 30 proteins, which interact in a sequential and regulated manner, culminating in the formation of the membrane attack complex (MAC). The MAC inserts into cell membranes, leading to pore formation and cell damage (286). The classical complement pathway is activated by antibody-antigen complexes (see chapter 3). Not all antibody isotypes are capable of activating the complement cascade. Targeting the complement system in diseases with autoantibodies that are mainly of the IgG1 or IgG3 subclass has shown remarkable efficacy, especially in the context of AChR MG. AChR autoantibodies are mainly of the subclass IgG1 (287). Eculizumab, inhibiting complement activation by targeting C5, demonstrated beneficial outcomes in phase III trials (ClinicalTrials.gov Identifier: NCT02301624; ClinicalTrials.gov Identifier: NCT01997229) and is approved for treating AChR+ MG patients (288). Ravulizumab, another C5 inhibitor, is currently in phase III trials (ClinicalTrials.gov Identifier: NCT03920293) (289). However, complement inhibition may not provide clinical benefits for patients suffering from IgG4 mediated diseases.
5.2.8 BTK inhibitors
Bruton´s tyrosine kinase (BTK) is a multifunctional cytoplasmic protein member of the TEC kinase family (290), crucial in B-cell biology, involved in maintaining B-cell survival, proliferation, differentiation and activation (291). Apart from B cells, it is also expressed in mast cells, NK cells, T cells, macrophages, neutrophils, monocytes and basophils (291), highlighting its multifaceted role in the immune system. Among the functions of BTK, it integrates signaling to regulate B cell development through BCR (290). Besides it is also involved in TLR-mediated signaling, chemokine mediated homing of pre-B cells into lymphoid organs and mediates in inflammatory processes driven by IgG complexes (291). Due to its central role in B cell immunity, BTK inhibition therapies have been gaining attention as first-line therapy for B cell malignancies (292). Its involvement in B cell survival and differentiation has also linked BTK with autoimmune diseases (291). Indeed, studies have shown increased levels of BTK expression in B cells from patients suffering from autoimmune diseases, which appears to be correlated with autoantibody production (293). Therefore, BTK inhibitors represent a promising therapeutic strategy for autoimmune diseases. BTK inhibitors are small molecules that are able to downregulate various B cell functions like cell proliferation, differentiation, maturation and survival overall. Besides, they can inhibit the activity of macrophages, mast cells and eosinophils (249). Currently, inhibition of BTK is being investigated for SLE (271). Fenebrutinib, a BTK inhibitor, was investigated in a phase II clinical trial in SLE (ClinicalTrials.gov Identifier: NCT02908100). Despite effectively targeting the BTK pathway, it did not meet the trial’s primary endpoint for efficacy (294). In pemphigus, the BTK inhibitor PRN1008 has been studied in a phase III clinical trial (ClinicalTrials.gov Identifier: NCT03762265), but it was terminated based on lack of efficacy. Although, the efficacy of BTK inhibitors as standalone treatments is limited, it might still hold potential in combination therapies.
5.2.9 CAR and CAAR T cells
Chimeric antigen (CAR)-T cell therapies, initially designed for targeting tumor cells, involve priming T cells with anti-tumor activity and then reintroducing them into the patient (295–297). These modified T cells express chimeric antigen receptors (CARs), genetically engineered to have tumor antigen-specific binding sites and a T cell activating domain (298). This allows them to recognize and kill cells expressing the target antigen.
Anti-CD19 CAR T cell therapy gained FDA approval in 2017 for treating B cell malignancies (299), sparking interest in expanding CAR T cell applications beyond cancer, particularly in autoimmune therapies (300). CAR-T therapies aim to redirect T cells against autoantibody-secreting B cells. In addition to CAR T cells, chimeric autoantibody (CAAR) receptor T cell therapies were developed (301–305). CAAR T-cells, distinct from CAR T-cells, express chimeric autoantibody receptors targeting pathogenic antibodies from autoreactive B cells (306). These include anti-BCMA (301–304), investigated in the context of MG (ClinicalTrials.gov identifier: NCT04146051), anti-CD19 (303) (ClinicalTrials.gov identifier: NCT03030976), and most recently, anti-MuSK and anti-NMDA, which have shown initial beneficial effects in a mouse models (305, 307). Another phase I trial (ClinicalTrials.gov Identifier: NCT04422912) is assessing Dsg3-CAAR T cells in Pemphigus vulgaris (308). CAR and CAAR T-cell therapies constitute a promising approach for a more selective and personalized treatment of autoimmune diseases.
6 Conclusion and outlook
The reason why some autoimmune diseases are predominantly IgG4 and not another subclass is still not completely understood. Despite the shared characteristic of having autoantibodies belonging to the IgG4 subclass, these diseases exhibit numerous differences. No discernible pattern emerges concerning the site of pathology, age of onset, or specific genetic predispositions (Table 1). Generally, the pathogenesis of autoimmune disorders is the result of a combination of defects in the immune system together with environmental, and genetic factors similar to the observed pathogenesis of multiple sclerosis, SLE, and type 1 diabetes (309–313). Given the rarity of some of these diseases, our current understanding may be limited. Thus, it is highly probable that clear patterns are not yet discernible. Future investigations are likely to provide more clarity.
IgG4 antibodies play dual roles in health and disease (49). In health, they serve protective and regulatory functions by predominantly inhibiting the actions of IgE antibodies (45, 47–49). However, in autoimmune conditions, where IgE antibodies are not the primary effectors, IgG4 antibodies contribute directly to immunopathology. Similarly, Fab-arm exchange seems to be a coincidental aspect of IgG4-mediated diseases. Fab-arm exchange enhances the diversity of IgG4 antibodies and increases their potential to engage with antigens (45, 47–49). This intrinsic property of IgG4 antibodies can either enhance the pathogenicity of autoantibodies, as observed with MuSK autoantibodies (112, 121–123), without significantly affecting their effects as seen in CNTN1 autoantibodies and pemphigus (142, 151, 161) or diminish their pathogenic potential as seen with anti-Neurofascin-155 IgG4 (141). Therefore, there is a variability in the relevance of Fab-arm exchange for pathogenic capacity in these diseases. Nevertheless, the underlying immunopathology in these IgG4-mediated diseases appears to share similarities, as seen by the overall positive response to anti-CD20-mediated BCDT in these diseases (3–12). Hence, it is probable that comparable treatments will yield similar outcomes in these conditions.
Author contributions
SÜ: Writing – review & editing, Writing – original draft, Conceptualization. BS: Writing – review & editing. EÇ: Writing – review & editing, Writing – original draft, Conceptualization. DB: Writing – review & editing. RMH: Writing – review & editing. SV: Writing – review & editing, Writing – original draft, Conceptualization. AV: Writing – review & editing, Writing – original draft, Conceptualization. AM: Writing – review & editing, Writing – original draft, Conceptualization. MF: Writing – review & editing, Writing – original draft, Visualization, Supervision, Conceptualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. MF has received a pilot grant from the DGM (Deutsche Gesellschaft für Muskelkranke e.V. (DGM) and a DFG Research Fellowship (FI 2471/2-1).
Acknowledgments
The figures were created with biorender.com by MF.
Conflict of interest
MF has further received speaker honoraria from Alexion Pharmaceuticals and is a member of the Alexion-Akademie based in Germany. AM has received speaker or consultancy honoraria or financial research support (paid to his institution) from Alexion Pharmaceuticals, argenx, Axunio, Desitin, Grifols, Hormosan Pharma, Janssen, Merck, Octapharma, UCB, and Xcenda. He serves as medical advisory board chairman of the German Myasthenia Gravis Society.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Anand U, Dey A, Chandel AKS, Sanyal R, Mishra A, Pandey DK, et al. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. (2023) 10:1367–401. doi: 10.1016/j.gendis.2022.02.007
2. Ledford H. Translational Research: The full cycle. Nature. (2008) 453:843–5. doi: 10.1038/453843a
3. Futei Y, Amagai M, Ishii K, Kuroda-Kinoshita K, Ohya K, Nishikawa T. Predominant IgG4 subclass in autoantibodies of pemphigus vulgaris and foliaceus. J Dermatol Sci. (2001) 26:55–61. doi: 10.1016/S0923-1811(00)00158-4
4. Mcconville J, Farrugia ME, Beeson D, Kishore U, Metcalfe R, Newsom-Davis J, et al. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol. (2004) 55:580–4. doi: 10.1002/ana.20061
5. Ohta K, Shigemoto K, Fujinami A, Maruyama N, Konishi T, Ohta M. Clinical and experimental features of MuSK antibody positive MG in Japan. Eur J Neurol. (2007) 14:1029–34. doi: 10.1111/j.1468-1331.2007.01870.x
6. Niks EH, Van Leeuwen Y, Leite MI, Dekker FW, Wintzen AR, Wirtz PW, et al. Clinical fluctuations in MuSK myasthenia gravis are related to antigen-specific IgG4 instead of IgG1. J Neuroimmunol. (2008) 195:151–6. doi: 10.1016/j.jneuroim.2008.01.013
7. Funakoshi T, Lunardon L, Ellebrecht CT, Nagler AR, O’leary CE, Payne AS. Enrichment of total serum IgG4 in patients with pemphigus. Br J Dermatol. (2012) 167:1245–53. doi: 10.1111/j.1365-2133.2012.11144.x
8. Ng JK, Malotka J, Kawakami N, Derfuss T, Khademi M, Olsson T, et al. Neurofascin as a target for autoantibodies in peripheral neuropathies. Neurology. (2012) 79:2241–8. doi: 10.1212/WNL.0b013e31827689ad
9. Lo AS, Mao X, Mukherjee EM, Ellebrecht CT, Yu X, Posner MR, et al. Pathogenicity and epitope characteristics do not differ in igG subclass-switched anti-desmoglein 3 igG1 and igG4 autoantibodies in pemphigus vulgaris. PloS One. (2016) 11:e0156800. doi: 10.1371/journal.pone.0156800
10. Vural A, Doppler K, Meinl E. Autoantibodies against the node of ranvier in seropositive chronic inflammatory demyelinating polyneuropathy: diagnostic, pathogenic, and therapeutic relevance. Front Immunol. (2018) 9:1029. doi: 10.3389/fimmu.2018.01029
11. Dalakas MC. Autoimmune neurological disorders with igG4 antibodies: a distinct disease spectrum with unique igG4 functions responding to anti-B cell therapies. Neurotherapeutics. (2022) 19:741–52. doi: 10.1007/s13311-022-01210-1
12. Oconnor K, Fichtner M. The mechanisms of immunopathology underlying B cell depletion therapy-mediated remission and relapse in patients with MuSK MG: Immune mechanisms of MuSK MG. RRNMF Neuromuscular J. (2023) 4(12):e127167. doi: 10.17161/rrnmf.v4i3
13. Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PA, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. (2015) 74:1171–7. doi: 10.1136/annrheumdis-2014-206605
14. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. (2015) 15:160–71. doi: 10.1038/nri3795
15. Janeway C. Immunobiology : the immune system in health and disease; Chapter 8 The Development of B and T Lymphocytes. 9th ed. (2017) New York: Garland Science.
16. Krumbholz M, Derfuss T, Hohlfeld R, Meinl E. B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat Rev Neurol. (2012) 8:613–23. doi: 10.1038/nrneurol.2012.203
17. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. (2012) 30:221–41. doi: 10.1146/annurev-immunol-020711-074934
18. Shen P, Fillatreau S. Antibody-independent functions of B cells: a focus on cytokines. Nat Rev Immunol. (2015) 15:441–51. doi: 10.1038/nri3857
19. Ghosh D, Jiang W, Mukhopadhyay D, Mellins ED. New insights into B cells as antigen presenting cells. Curr Opin Immunol. (2021) 70:129–37. doi: 10.1016/j.coi.2021.06.003
20. Kumar BV, Connors TJ, Farber DL. Human T cell development, localization, and function throughout life. Immunity. (2018) 48:202–13. doi: 10.1016/j.immuni.2018.01.007
21. Bradley LM, Harbertson J, Biederman E, Zhang Y, Bradley SM, Linton PJ. Availability of antigen-presenting cells can determine the extent of CD4 effector expansion and priming for secretion of Th2 cytokines in vivo. Eur J Immunol. (2002) 32:2338–46. doi: 10.1002/1521-4141(200208)32:83.0.CO;2-R
22. Yamano T, Nedjic J, Hinterberger M, Steinert M, Koser S, Pinto S, et al. Thymic B cells are licensed to present self antigens for central T cell tolerance induction. Immunity. (2015) 42:1048–61. doi: 10.1016/j.immuni.2015.05.013
23. Adler LN, Jiang W, Bhamidipati K, Millican M, Macaubas C, Hung SC, et al. The other function: class II-restricted antigen presentation by B cells. Front Immunol. (2017) 8:319. doi: 10.3389/fimmu.2017.00319
24. Nakken B, Munthe LA, Konttinen YT, Sandberg AK, Szekanecz Z, Alex P, et al. B-cells and their targeting in rheumatoid arthritis–current concepts and future perspectives. Autoimmun Rev. (2011) 11:28–34. doi: 10.1016/j.autrev.2011.06.010
25. Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin-Doyer M, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med. (2013) 210:2921–37. doi: 10.1084/jem.20130699
26. Béland K, Marceau G, Labardy A, Bourbonnais S, Alvarez F. Depletion of B cells induces remission of autoimmune hepatitis in mice through reduced antigen presentation and help to T cells. Hepatology. (2015) 62:1511–23. doi: 10.1002/hep.27991
27. Jackson SW, Kolhatkar NS, Rawlings DJ. B cells take the front seat: dysregulated B cell signals orchestrate loss of tolerance and autoantibody production. Curr Opin Immunol. (2015) 33:70–7. doi: 10.1016/j.coi.2015.01.018
28. Fraussen J, Claes N, Van Wijmeersch B, Van Horssen J, Stinissen P, Hupperts R, et al. B cells of multiple sclerosis patients induce autoreactive proinflammatory T cell responses. Clin Immunol. (2016) 173:124–32. doi: 10.1016/j.clim.2016.10.001
29. Kurosaki T, Kometani K, Ise W. Memory B cells. Nat Rev Immunol. (2015) 15:149–59. doi: 10.1038/nri3802
30. Akkaya M, Kwak K, Pierce SK. B cell memory: building two walls of protection against pathogens. Nat Rev Immunol. (2020) 20:229–38. doi: 10.1038/s41577-019-0244-2
31. Inoue T, Kurosaki T. Memory B cells. Nat Rev Immunol. (2024) 24:5–17. doi: 10.1038/s41577-023-00897-3
32. Sarvas H, Makela O. Haptenated bacteriophage in the assay of antibody quantity and affinity: maturation of an immune response. Immunochemistry. (1970) 7:933–43. doi: 10.1016/0019-2791(70)90054-6
33. Rajewsky K. Clonal selection and learning in the antibody system. Nature. (1996) 381:751–8. doi: 10.1038/381751a0
34. Neuberger MS. Novartis Medal Lecture. Antibodies: a paradigm for the evolution of molecular recognition. Biochem Soc Trans. (2002) 30:341–50. doi: 10.1042/bst0300341
35. Oyeyinka GO, Salimonu LS, Williams AI, Johnson AO, Ladipo OA, Osunkoya BO. Range of normal serum immunoglobulin (IgG, IgA and IgM) values in Nigerians. Afr J Med Med Sci. (1984) 13:169–76.
36. Janeway C. Immunobiology : the immune system in health and disease; Chapter 4 Antigen Recognition by B-cell and T-cell Receptors. 9th ed. (2017) New York: Garland Science.
37. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. (2014) 5:520. doi: 10.3389/fimmu.2014.00520
38. Volkov M, Coppola M, Huizinga R, Eftimov F, Huizinga TWJ, van der Kooi AJ, et al. Comprehensive overview of autoantibody isotype and subclass distribution. J Allergy Clin Immunol. (2022) 150:999–1010. doi: 10.1016/j.jaci.2022.05.023
39. Hjelholt A, Christiansen G, Sørensen US, Birkelund S. IgG subclass profiles in normal human sera of antibodies specific to five kinds of microbial antigens. Pathog Dis. (2013) 67:206–13. doi: 10.1111/2049-632X.12034
40. De Taeye SW, Rispens T, Vidarsson G. The ligands for human igG and their effector functions. Antibodies (Basel). (2019) 8. doi: 10.3390/antib8020030
41. Thomson CA. IgG structure and function. In: Ratcliffe MJH, editor. Encyclopedia of immunobiology, vol. 15-22 . Academic Press, Oxford (2016).
42. Damelang T, Rogerson SJ, Kent SJ, Chung AW. Role of igG3 in infectious diseases. Trends Immunol. (2019) 40:197–211. doi: 10.1016/j.it.2019.01.005
43. Van Der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martinez-Martinez P, Vermeulen E, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. (2007) 317:1554–7. doi: 10.1126/science.1144603
44. Chappel MS, Isenman DE, Everett M, Xu YY, Dorrington KJ, Klein MH. Identification of the Fc gamma receptor class I binding site in human IgG through the use of recombinant IgG1/IgG2 hybrid and point-mutated antibodies. Proc Natl Acad Sci U.S.A. (1991) 88:9036–40. doi: 10.1073/pnas.88.20.9036
45. Pillai S. Is it bad, is it good, or is IgG4 just misunderstood? Sci Immunol. (2023) 8:eadg7327. doi: 10.1126/sciimmunol.adg7327
46. Oskam N, Damelang T, Streutker M, Ooijevaar-De Heer P, Nouta J, Koeleman C, et al. Factors affecting IgG4-mediated complement activation. Front Immunol. (2023) 14:1087532. doi: 10.3389/fimmu.2023.1087532
47. Nakagawa T, Miyamoto T. The role of IgG4 as blocking antibodies in asthmatics and in bee keepers. Int Arch Allergy Appl Immunol. (1985) 77:204–5. doi: 10.1159/000233787
48. James LK, Till SJ. Potential mechanisms for igG4 inhibition of immediate hypersensitivity reactions. Curr Allergy Asthma Rep. (2016) 16:23. doi: 10.1007/s11882-016-0600-2
49. Rispens T, Huijbers MG. The unique properties of IgG4 and its roles in health and disease. Nat Rev Immunol. (2023) 23:763–78. doi: 10.1038/s41577-023-00871-z
50. Vercelli D, De Monte L, Monticelli S, Di Bartolo C, Agresti A. To E or not to E? Can an IL-4-induced B cell choose between IgE and IgG4? Int Arch Allergy Immunol. (1998) 116:1–4. doi: 10.1159/000023918
51. Hussain R, Ottesen EA. IgE responses in human filariasis. IV. Parallel antigen recognition by IgE and IgG4 subclass antibodies. J Immunol. (1986) 136:1859–63. doi: 10.4049/jimmunol.136.5.1859
52. Rihet P, Demeure CE, Dessein AJ, Bourgois A. Strong serum inhibition of specific IgE correlated to competing IgG4, revealed by a new methodology in subjects from a S. mansoni endemic area. Eur J Immunol. (1992) 22:2063–70. doi: 10.1002/eji.1830220816
53. Aalberse RC, van der Gaag R, Van Leeuwen J. Serologic aspects of IgG4 antibodies. I. Prolonged immunization results in an IgG4-restricted response. J Immunol. (1983) 130:722–6. doi: 10.4049/jimmunol.130.2.722
54. Van Der Zee JS, Van Swieten P, Aalberse RC. Serologic aspects of IgG4 antibodies. II. IgG4 antibodies form small, nonprecipitating immune complexes due to functional monovalency. J Immunol. (1986) 137:3566–71. doi: 10.4049/jimmunol.137.11.3566
55. Irrgang P, Gerling J, Kocher K, Lapuente D, Steininger P, Habenicht K, et al. Class switch toward noninflammatory, spike-specific IgG4 antibodies after repeated SARS-CoV-2 mRNA vaccination. Sci Immunol. (2023) 8:eade2798. doi: 10.1126/sciimmunol.ade2798
56. Garcia-Robaina JC, de la Torre-Morin F, Vazquez-Moncholi C, Fierro J, Bonnet-Moreno C. The natural history of Apis-specific IgG and IgG4 in beekeepers. Clin Exp Allergy. (1997) 27:418–23. doi: 10.1046/j.1365-2222.1997.1350806.x
57. Varga EM, Kausar F, Aberer W, Zach M, Eber E, Durham SR, et al. Tolerant beekeepers display venom-specific functional IgG4 antibodies in the absence of specific IgE. J Allergy Clin Immunol. (2013) 131:1419–21. doi: 10.1016/j.jaci.2012.08.037
58. Trampert DC, Hubers LM, Van De Graaf SFJ, Beuers U. On the role of IgG4 in inflammatory conditions: lessons for IgG4-related disease. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1401–9. doi: 10.1016/j.bbadis.2017.07.038
59. Fichtner ML, Jiang R, Bourke A, Nowak RJ, O’connor KC. Autoimmune pathology in myasthenia gravis disease subtypes is governed by divergent mechanisms of immunopathology. Front Immunol. (2020) 11:776. doi: 10.3389/fimmu.2020.00776
60. Jeannin P, Lecoanet S, Delneste Y, Gauchat JF, Bonnefoy JY. IgE versus IgG4 production can be differentially regulated by IL-10. J Immunol. (1998) 160:3555–61. doi: 10.4049/jimmunol.160.7.3555
61. Munemura R, Maehara T, Murakami Y, Koga R, Aoyagi R, Kaneko N, et al. Distinct disease-specific Tfh cell populations in 2 different fibrotic diseases: IgG(4)-related disease and Kimura disease. J Allergy Clin Immunol. (2022) 150:440–455.e417. doi: 10.1016/j.jaci.2022.03.034
62. Phillips LH. The epidemiology of myasthenia gravis. Semin Neurol. (2004) 24:17–20. doi: 10.1055/s-2004-829593
63. Bubuioc AM, Kudebayeva A, Turuspekova S, Lisnic V, Leone MA. The epidemiology of myasthenia gravis. J Med Life. (2021) 14:7–16. doi: 10.25122/jml-2020-0145
64. Broers MC, Bunschoten C, Nieboer D, Lingsma HF, Jacobs BC. Incidence and prevalence of chronic inflammatory demyelinating polyradiculoneuropathy: A systematic review and meta-analysis. Neuroepidemiology. (2019) 52:161–72. doi: 10.1159/000494291
65. Rosi-Schumacher M, Baker J, Waris J, Seiffert-Sinha K, Sinha AA. Worldwide epidemiologic factors in pemphigus vulgaris and bullous pemphigoid. Front Immunol. (2023) 14:1159351. doi: 10.3389/fimmu.2023.1159351
66. Muñoz-Sánchez G, Planagumà J, Naranjo L, Couso R, Sabater L, Guasp M, et al. The diagnosis of anti-LGI1 encephalitis varies with the type of immunodetection assay and sample examined. Front Immunol. (2022) 13:1069368. doi: 10.3389/fimmu.2022.1069368
67. Wallace ZS, Miles G, Smolkina E, Petruski-Ivleva N, Madziva D, Cook C, et al. Incidence, prevalence and mortality of IgG4-related disease in the USA: a claims-based analysis of commercially insured adults. Ann Rheum Dis. (2023) 82:957–62. doi: 10.1136/ard-2023-223950
68. Guptill JT, Sanders DB, Evoli A. Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve. (2011) 44:36–40. doi: 10.1002/mus.22006
69. Rodolico C, Bonanno C, Toscano A, Vita G. MuSK-associated myasthenia gravis: clinical features and management. Front Neurol. (2020) 11:660. doi: 10.3389/fneur.2020.00660
70. Vallat JM, Sommer C, Magy L. Chronic inflammatory demyelinating polyradiculoneuropathy: diagnostic and therapeutic challenges for a treatable condition. Lancet Neurol. (2010) 9:402–12. doi: 10.1016/S1474-4422(10)70041-7
71. Kieseier BC, Mathey EK, Sommer C, Hartung HP. Immune-mediated neuropathies. Nat Rev Dis Primers. (2018) 4:31. doi: 10.1038/s41572-018-0027-2
72. Meyer N, Misery L. Geoepidemiologic considerations of auto-immune pemphigus. Autoimmun Rev. (2010) 9:A379–382. doi: 10.1016/j.autrev.2009.10.009
73. Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, Payne AS, et al. Pemphigus. Nat Rev Dis Primers. (2017) 3:17026. doi: 10.1038/nrdp.2017.26
74. Lepe K, Yarrarapu SNS, Zito PM. Pemphigus foliaceus. In: StatPearls. StatPearls Publishing Copyright © 2023, StatPearls Publishing LLC, Treasure Island (FL (2023).
75. Ghimire P, Khanal UP, Gajurel BP, Karn R, Rajbhandari R, Paudel S, et al. Anti-LGI1, anti-GABABR, and Anti-CASPR2 encephalitides in Asia: A systematic review. Brain Behav. (2020) 10:e01793. doi: 10.1002/brb3.1793
76. Guo K, Liu X, Lin J, Gong X, Li A, Liu Y, et al. Clinical characteristics, long-term functional outcomes and relapse of anti-LGI1/Caspr2 encephalitis: a prospective cohort study in Western China. Ther Adv Neurol Disord. (2022) 15:17562864211073203. doi: 10.1177/17562864211073203
77. Chiò A, Cocito D, Bottacchi E, Buffa C, Leone M, Plano F, et al. Idiopathic chronic inflammatory demyelinating polyneuropathy: an epidemiological study in Italy. J Neurol Neurosurg Psychiatry. (2007) 78:1349–53. doi: 10.1136/jnnp.2007.114868
78. Rajabally YA, Simpson BS, Beri S, Bankart J, Gosalakkal JA. Epidemiologic variability of chronic inflammatory demyelinating polyneuropathy with different diagnostic criteria: study of a UK population. Muscle Nerve. (2009) 39:432–8. doi: 10.1002/mus.21206
79. Hafsteinsdottir B, Olafsson E. Incidence and natural history of idiopathic chronic inflammatory demyelinating polyneuropathy: A population-based study in Iceland. Eur Neurol. (2016) 75:263–8. doi: 10.1159/000445884
80. Kridin K. Pemphigus group: overview, epidemiology, mortality, and comorbidities. Immunol Res. (2018) 66:255–70. doi: 10.1007/s12026-018-8986-7
81. Kridin K, Schmidt E. Epidemiology of pemphigus. JID Innov. (2021) 1:100004. doi: 10.1016/j.xjidi.2021.100004
82. Binks SNM, Klein CJ, Waters P, Pittock SJ, Irani SR. LGI1, CASPR2 and related antibodies: a molecular evolution of the phenotypes. J Neurol Neurosurg Psychiatry. (2018) 89:526–34. doi: 10.1136/jnnp-2017-315720
83. Niks EH, Kuks JB, Roep BO, Haasnoot GW, Verduijn W, Ballieux BE, et al. Strong association of MuSK antibody-positive myasthenia gravis and HLA-DR14-DQ5. Neurology. (2006) 66:1772–4. doi: 10.1212/01.wnl.0000218159.79769.5c
84. Bartoccioni E, Scuderi F, Augugliaro A, Chiatamone Ranieri S, Sauchelli D, Alboino P, et al. HLA class II allele analysis in MuSK-positive myasthenia gravis suggests a role for DQ5. Neurology. (2009) 72:195–7. doi: 10.1212/01.wnl.0000339103.08830.86
85. Alahgholi-Hajibehzad M, Yilmaz V, Gülsen-Parman Y, Aysal F, Oflazer P, Deymeer F, et al. Association of HLA-DRB1∗14, -DRB1∗16 and -DQB1∗05 with MuSK-myasthenia gravis in patients from Turkey. Hum Immunol. (2013) 74:1633–5. doi: 10.1016/j.humimm.2013.08.271
86. Ehsan S, Amirzargar A, Yekaninejad MS, Mahmoudi M, Mehravar S, Moradi B, et al. Association of HLA class II (DRB1, DQA1, DQB1) alleles and haplotypes with myasthenia gravis and its subgroups in the Iranian population. J Neurol Sci. (2015) 359:335–42. doi: 10.1016/j.jns.2015.11.021
87. Nikolic AV, Andric ZP, Simonovic RB, Rakocevic Stojanovic VM, Basta IZ, Bojic SD, et al. High frequency of DQB1*05 and absolute absence of DRB1*13 in muscle-specific tyrosine kinase positive myasthenia gravis. Eur J Neurol. (2015) 22:59–63. doi: 10.1111/ene.12525
88. Kanai T, Uzawa A, Kawaguchi N, Sakamaki T, Yoshiyama Y, Himuro K, et al. HLA-DRB1*14 and DQB1*05 are associated with Japanese anti-MuSK antibody-positive myasthenia gravis patients. J Neurol Sci. (2016) 363:116–8. doi: 10.1016/j.jns.2016.02.031
89. Hong Y, Li HF, Romi F, Skeie GO, Gilhus NE. HLA and MuSK-positive myasthenia gravis: A systemic review and meta-analysis. Acta Neurol Scand. (2018) 138:219–26. doi: 10.1111/ane.2018.138.issue-3
90. Blum S, Mccombe PA. Genetics of Guillain-Barré syndrome (GBS) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP): current knowledge and future directions. J Peripher Nerv Syst. (2014) 19:88–103. doi: 10.1111/jns5.12074
91. Moraes ME, Fernandez-Vina M, Lazaro A, Diaz LA, Filho GH, Friedman H, et al. An epitope in the third hypervariable region of the DRB1 gene is involved in the susceptibility to endemic pemphigus foliaceus (fogo selvagem) in three different Brazilian populations. Tissue Antigens. (1997) 49:35–40. doi: 10.1111/j.1399-0039.1997.tb02707.x
92. Loiseau P, Lecleach L, Prost C, Lepage V, Busson M, Bastuji-Garin S, et al. HLA class II polymorphism contributes to specify desmoglein derived peptides in pemphigus vulgaris and pemphigus foliaceus. J Autoimmun. (2000) 15:67–73. doi: 10.1006/jaut.2000.0388
93. Vodo D, Sarig O, Sprecher E. The genetics of pemphigus vulgaris. Front Med (Lausanne). (2018) 5:226. doi: 10.3389/fmed.2018.00226
94. Schmidt E, Kasperkiewicz M, Joly P. Pemphigus. Lancet. (2019) 394:882–94. doi: 10.1016/S0140-6736(19)31778-7
95. Petzl-Erler ML. Beyond the HLA polymorphism: A complex pattern of genetic susceptibility to pemphigus. Genet Mol Biol. (2020) 43:e20190369. doi: 10.1590/1678-4685-gmb-2019-0369
96. Kim TJ, Lee ST, Moon J, Sunwoo JS, Byun JI, Lim JA, et al. Anti-LGI1 encephalitis is associated with unique HLA subtypes. Ann Neurol. (2017) 81:183–92. doi: 10.1002/ana.24860
97. Binks S, Varley J, Lee W, Makuch M, Elliott K, Gelfand JM, et al. Distinct HLA associations of LGI1 and CASPR2-antibody diseases. Brain. (2018) 141:2263–71. doi: 10.1093/brain/awy109
98. Muñiz-Castrillo S, Joubert B, Elsensohn MH, Pinto AL, Saint-Martin M, Vogrig A, et al. Anti-CASPR2 clinical phenotypes correlate with HLA and immunological features. J Neurol Neurosurg Psychiatry. (2020) 91:1076–84. doi: 10.1136/jnnp-2020-323226
99. Terao C, Ota M, Iwasaki T, Shiokawa M, Kawaguchi S, Kuriyama K, et al. IgG4-related disease in the Japanese population: a genome-wide association study. Lancet Rheumatol. (2019) 1:e14–22. doi: 10.1016/S2665-9913(19)30006-2
100. Vincent A. Unravelling the pathogenesis of myasthenia gravis. Nat Rev Immunol. (2002) 2:797–804. doi: 10.1038/nri916
102. Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, et al. Agrin acts via a MuSK receptor complex. Cell. (1996) 85:513–23. doi: 10.1016/S0092-8674(00)81252-0
103. Lindstrom JM, Engel AG, Seybold ME, Lennon VA, Lambert EH. Pathological mechanisms in experimental autoimmune myasthenia gravis. II. Passive transfer of experimental autoimmune myasthenia gravis in rats with anti-acetylcholine recepotr antibodies. J Exp Med. (1976) 144:739–53. doi: 10.1084/jem.144.3.739
104. Lindstrom JM, Seybold ME, Lennon VA, Whittingham S, Duane DD. Antibody to acetylcholine receptor in myasthenia gravis. Prevalence, clinical correlates, and diagnostic value. Neurology. (1976) 26:1054–9. doi: 10.1212/WNL.26.11.1054
105. Hoch W, Mcconville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. (2001) 7:365–8. doi: 10.1038/85520
106. Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol. (2011) 69:418–22. doi: 10.1002/ana.22312
107. Pevzner A, Schoser B, Peters K, Cosma NC, Karakatsani A, Schalke B, et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol. (2012) 259:427–35. doi: 10.1007/s00415-011-6194-7
108. Lazaridis K, Tzartos SJ. Autoantibody specificities in myasthenia gravis; implications for improved diagnostics and therapeutics. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.00212
109. Koneczny I, Cossins J, Waters P, Beeson D, Vincent A. MuSK myasthenia gravis IgG4 disrupts the interaction of LRP4 with MuSK but both IgG4 and IgG1-3 can disperse preformed agrin-independent AChR clusters. PloS One. (2013) 8:e80695. doi: 10.1371/journal.pone.0080695
110. Cao M, Liu WW, Maxwell S, Huda S, Webster R, Evoli A, et al. IgG1-3 muSK antibodies inhibit AChR cluster formation, restored by SHP2 inhibitor, despite normal muSK, DOK7, or AChR subunit phosphorylation. Neurol Neuroimmunol Neuroinflamm. (2023) 10(6):e200147. doi: 10.1212/NXI.0000000000200147
111. Stathopoulos P, Kumar A, Nowak RJ, O’connor KC. Autoantibody-producing plasmablasts after B cell depletion identified in muscle-specific kinase myasthenia gravis. JCI Insight. (2017) 2:e94263–75. doi: 10.1172/jci.insight.94263
112. Huijbers MG, Vergoossen DL, Fillie-Grijpma YE, Van Es IE, Koning MT, Slot LM, et al. MuSK myasthenia gravis monoclonal antibodies: Valency dictates pathogenicity. Neurol Neuroimmunol Neuroinflamm. (2019) 6:e547. doi: 10.1212/NXI.0000000000000547
113. Takata K, Stathopoulos P, Cao M, Mane-Damas M, Fichtner ML, Benotti ES, et al. Characterization of pathogenic monoclonal autoantibodies derived from muscle-specific kinase myasthenia gravis patients. JCI Insight. (2019) 4. doi: 10.1172/jci.insight.127167
114. Fichtner ML, Hoehn KB, Ford EE, Mane-Damas M, Oh S, Waters P, et al. Reemergence of pathogenic, autoantibody-producing B cell clones in myasthenia gravis following B cell depletion therapy. Acta Neuropathol Commun. (2022) 10:154. doi: 10.1186/s40478-022-01454-0
115. Jennings CG, Dyer SM, Burden SJ. Muscle-specific trk-related receptor with a kringle domain defines a distinct class of receptor tyrosine kinases. Proc Natl Acad Sci United States America. (1993) 90:2895–9. doi: 10.1073/pnas.90.7.2895
116. Otsuka K, Ito M, Ohkawara B, Masuda A, Kawakami Y, Sahashi K, et al. Collagen Q and anti-MuSK autoantibody competitively suppress agrin/LRP4/MuSK signaling. Sci Rep. (2015) 5:13928. doi: 10.1038/srep13928
117. Huijbers MG, Zhang W, Klooster R, Niks EH, Friese MB, Straasheijm KR, et al. MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc Natl Acad Sci U.S.A. (2013) 110:20783–8. doi: 10.1073/pnas.1313944110
118. Cole RN, Reddel SW, Gervásio OL, Phillips WD. Anti-MuSK patient antibodies disrupt the mouse neuromuscular junction. Ann Neurol. (2008) 63:782–9. doi: 10.1002/ana.21371
119. Klooster R, Plomp JJ, Huijbers MG, Niks EH, Straasheijm KR, Detmers FJ, et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain. (2012) 135:1081–101. doi: 10.1093/brain/aws025
120. Plomp JJ, Huijbers MG, van der Maarel SM, Verschuuren JJ. Pathogenic IgG4 subclass autoantibodies in MuSK myasthenia gravis. Ann N Y Acad Sci. (2012) 1275:114–22. doi: 10.1111/j.1749-6632.2012.06808.x
121. Koneczny I, Stevens JA, De Rosa A, Huda S, Huijbers MG, Saxena A, et al. IgG4 autoantibodies against muscle-specific kinase undergo Fab-arm exchange in myasthenia gravis patients. J Autoimmun. (2017) 77:104–15. doi: 10.1016/j.jaut.2016.11.005
122. Fichtner ML, Vieni C, Redler RL, Kolich L, Jiang R, Takata K, et al. Affinity maturation is required for pathogenic monovalent IgG4 autoantibody development in myasthenia gravis. J Exp Med. (2020) 217(12):e20200513. doi: 10.1084/jem.20200513
123. Vergoossen DLE, Plomp JJ, Gstöttner C, Fillié-Grijpma YE, Augustinus R, Verpalen R, et al. Functional monovalency amplifies the pathogenicity of anti-MuSK IgG4 in myasthenia gravis. Proc Natl Acad Sci U.S.A. (2021) 118(13):e2020635118. doi: 10.1073/pnas.2020635118
124. Diaz-Manera J, Martinez-Hernandez E, Querol L, Klooster R, Rojas-Garcia R, Suarez-Calvet X, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology. (2012) 78:189–93. doi: 10.1212/WNL.0b013e3182407982
125. Marino M, Basile U, Spagni G, Napodano C, Iorio R, Gulli F, et al. Long lasting rituximab-induced reduction of specific - but not of total- IgG4 in MuSK-positive myasthenia gravis. Front Immunol. (2020) 11:2020. doi: 10.3389/fimmu.2020.00613
126. Berger B, Schröter N. Changes in antibody titers and clinical course in myasthenia gravis retrospective study. Prog Neurol Psychiatry. (2022) 26:28–33. doi: 10.1002/pnp.738
127. Triplett JD, Hardy TA, Riminton DS, Chu SYK, Reddel SW. Association between musk antibody concentrations and the myasthenia gravis composite score in 3 patients: A marker of relapse? Muscle Nerve. (2019) 60:307–11. doi: 10.1002/mus.26609
128. Dyck PJB, Tracy JA. History, diagnosis, and management of chronic inflammatory demyelinating polyradiculoneuropathy. Mayo Clin Proc. (2018) 93:777–93. doi: 10.1016/j.mayocp.2018.03.026
129. Stino AM, Naddaf E, Dyck PJ, Dyck PJB. Chronic inflammatory demyelinating polyradiculoneuropathy-Diagnostic pitfalls and treatment approach. Muscle Nerve. (2021) 63:157–69. doi: 10.1002/mus.27046
130. Stathopoulos P, Alexopoulos H, Dalakas MC. Autoimmune antigenic targets at the node of Ranvier in demyelinating disorders. Nat Rev Neurol. (2015) 11:143–56. doi: 10.1038/nrneurol.2014.260.
131. Uncini A, Vallat JM. Autoimmune nodo-paranodopathies of peripheral nerve: the concept is gaining ground. J Neurol Neurosurg Psychiatry. (2018) 89:627–35. doi: 10.1136/jnnp-2017-317192
132. Khadilkar SV, Kamat S, Patel R. Nodo-paranodopathies: concepts, clinical implications, and management. Ann Indian Acad Neurol. (2022) 25:1001–8. doi: 10.4103/aian.aian_382_22
133. Uncini A. Autoimmune nodo-paranodopathies 10 years later: Clinical features, pathophysiology and treatment. J Peripher Nerv Syst. (2023) 28 Suppl 3:S23–s35. doi: 10.1111/jns.12569
134. Mathey EK, Derfuss T, Storch MK, Williams KR, Hales K, Woolley DR, et al. Neurofascin as a novel target for autoantibody-mediated axonal injury. J Exp Med. (2007) 204:2363–72. doi: 10.1084/jem.20071053
135. Querol L, Nogales-Gadea G, Rojas-Garcia R, Martinez-Hernandez E, Diaz-Manera J, Suarez-Calvet X, et al. Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol. (2012) 73(3):370–80. doi: 10.1002/ana.23794
136. Doppler K, Appeltshauser L, Villmann C, Martin C, Peles E, Krämer HH, et al. Auto-antibodies to contactin-associated protein 1 (Caspr) in two patients with painful inflammatory neuropathy. Brain. (2016) 139:2617–30. doi: 10.1093/brain/aww189
137. Delmont E, Manso C, Querol L, Cortese A, Berardinelli A, Lozza A, et al. Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Brain. (2017) 140:1851–8. doi: 10.1093/brain/awx124
138. Querol L, Siles AM, Alba-Rovira R, Jáuregui A, Devaux J, Faivre-Sarrailh C, et al. Antibodies against peripheral nerve antigens in chronic inflammatory demyelinating polyradiculoneuropathy. Sci Rep. (2017) 7:14411. doi: 10.1038/s41598-017-14853-4
139. Manso C, Querol L, Mekaouche M, Illa I, Devaux JJ. Contactin-1 IgG4 antibodies cause paranode dismantling and conduction defects. Brain. (2016) 139:1700–12. doi: 10.1093/brain/aww062
140. Manso C, Querol L, Lleixà C, Poncelet M, Mekaouche M, Vallat JM, et al. Anti-Neurofascin-155 IgG4 antibodies prevent paranodal complex formation in vivo. J Clin Invest. (2019) 129:2222–36. doi: 10.1172/JCI124694
141. Jentzer A, Attal A, Roué C, Raymond J, Lleixà C, Illa I, et al. IgG4 valency modulates the pathogenicity of anti–neurofascin-155 igG4 in autoimmune nodopathy. Neurol - Neuroimmunology Neuroinflamm. (2022) 9:e200014. doi: 10.1212/NXI.0000000000200014
142. Taieb G, Jentzer A, Vegezzi E, Lleixà C, Illa I, Querol L, et al. Effect of monovalency on anti-contactin-1 IgG4. Front Immunol. (2023) 14:1021513. doi: 10.3389/fimmu.2023.1021513
143. Appeltshauser L, Junghof H, Messinger J, Linke J, Haarmann A, Ayzenberg I, et al. Anti-pan-neurofascin antibodies induce subclass-related complement activation and nodo-paranodal damage. Brain. (2023) 146:1932–49. doi: 10.1093/brain/awac418
144. Bresciani L, Salvalaggio A, Vegezzi E, Visentin A, Fortuna A, Anglani M, et al. Caspr1 antibodies autoimmune paranodopathy with severe tetraparesis: Potential relevance of antibody titers in monitoring treatment response. J Peripher Nerv Syst. (2023) 28:522–7. doi: 10.1111/jns.12565
145. Malik AM, Tupchong S, Huang S, Are A, Hsu S, Motaparthi K. An updated review of pemphigus diseases. Medicina (Kaunas). (2021) 57(10):1080. doi: 10.3390/medicina57101080
146. Fuchs M, Kugelmann D, Schlegel N, Vielmuth F, Waschke J. Desmoglein 2 can undergo Ca2+-dependent interactions with both desmosomal and classical cadherins including E-cadherin and N-cadherin. Biophys J. (2022) 121:1322–35. doi: 10.1016/j.bpj.2022.02.023
147. Amagai M. Autoimmune and infectious skin diseases that target desmogleins. Proc Jpn Acad Ser B Phys Biol Sci. (2010) 86:524–37. doi: 10.2183/pjab.86.524
148. Di Zenzo G, Di Lullo G, Corti D, Calabresi V, Sinistro A, Vanzetta F, et al. Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface. J Clin Invest. (2012) 122:3781–90. doi: 10.1172/JCI64413
149. Rock B, Martins CR, Theofilopoulos AN, Balderas RS, Anhalt GJ, Labib RS, et al. The pathogenic effect of IgG4 autoantibodies in endemic pemphigus foliaceus (fogo selvagem). N Engl J Med. (1989) 320:1463–9. doi: 10.1056/NEJM198906013202206
150. Ishii K, Harada R, Matsuo I, Shirakata Y, Hashimoto K, Amagai M. In vitro keratinocyte dissociation assay for evaluation of the pathogenicity of anti-desmoglein 3 IgG autoantibodies in pemphigus vulgaris. J Invest Dermatol. (2005) 124:939–46. doi: 10.1111/j.0022-202X.2005.23714.x
151. Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, et al. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. (2005) 115:888–99. doi: 10.1172/JCI24185
152. Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, Payne AS, et al. Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PloS One. (2012) 7:e50696. doi: 10.1371/journal.pone.0050696
153. Bhol K, Natarajan K, Nagarwalla N, Mohimen A, Aoki V, Ahmed AR. Correlation of peptide specificity and IgG subclass with pathogenic and nonpathogenic autoantibodies in pemphigus vulgaris: a model for autoimmunity. Proc Natl Acad Sci U.S.A. (1995) 92:5239–43. doi: 10.1073/pnas.92.11.5239
154. Kricheli D, David M, Frusic-Zlotkin M, Goldsmith D, Rabinov M, Sulkes J, et al. The distribution of pemphigus vulgaris-IgG subclasses and their reactivity with desmoglein 3 and 1 in pemphigus patients and their first-degree relatives. Br J Dermatol. (2000) 143:337–42. doi: 10.1046/j.1365-2133.2000.03659.x
155. Warren SJ, Arteaga LA, Rivitti EA, Aoki V, Hans-Filho G, Qaqish BF, et al. The role of subclass switching in the pathogenesis of endemic pemphigus foliaceus. J Invest Dermatol. (2003) 120:104–8. doi: 10.1046/j.1523-1747.2003.12017.x
156. Yamamoto Y, Aoyama Y, Shu E, Tsunoda K, Amagai M, Kitajima Y. Anti-desmoglein 3 (Dsg3) monoclonal antibodies deplete desmosomes of Dsg3 and differ in their Dsg3-depleting activities related to pathogenicity. J Biol Chem. (2007) 282:17866–76. doi: 10.1074/jbc.M607963200
157. Egu DT, Walter E, Spindler V, Waschke J. Inhibition of p38MAPK signalling prevents epidermal blistering and alterations of desmosome structure induced by pemphigus autoantibodies in human epidermis. Br J Dermatol. (2017) 177:1612–8. doi: 10.1111/bjd.15721
158. Yoshida K, Ishii K, Shimizu A, Yokouchi M, Amagai M, Shiraishi K, et al. Non-pathogenic pemphigus foliaceus (PF) IgG acts synergistically with a directly pathogenic PF IgG to increase blistering by p38MAPK-dependent desmoglein 1 clustering. J Dermatol Sci. (2017) 85:197–207. doi: 10.1016/j.jdermsci.2016.12.010
159. Rock B, Labib RS, Diaz LA. Monovalent Fab’ immunoglobulin fragments from endemic pemphigus foliaceus autoantibodies reproduce the human disease in neonatal Balb/c mice. J Clin Invest. (1990) 85:296–9. doi: 10.1172/JCI114426
160. Mascaró JM Jr., España A, Liu Z, Ding X, Swartz SJ, Fairley JA, et al. Mechanisms of acantholysis in pemphigus vulgaris: role of IgG valence. Clin Immunol Immunopathol. (1997) 85:90–6. doi: 10.1006/clin.1997.4408
161. Tsunoda K, Ota T, Saito M, Hata T, Shimizu A, Ishiko A, et al. Pathogenic relevance of IgG and IgM antibodies against desmoglein 3 in blister formation in pemphigus vulgaris. Am J Pathol. (2011) 179:795–806. doi: 10.1016/j.ajpath.2011.04.015
162. Bhol K, Tyagi S, Natarajan K, Nagarwalla N, Ahmed AR. Use of recombinant pemphigus vulgaris antigen in development of ELISA and IB assays to detect pemphigus vulgaris autoantibodies. J Eur Acad Dermatol Venereol. (1998) 10:28–35. doi: 10.1111/j.1468-3083.1998.tb00924.x
163. Bracke S, Speeckaert R, Van Geel N, De Bacquer D, Lambert J. Evaluation of commercially available ELISA assays as a tool for monitoring and managing pemphigus patients: a prospective study. Eur J Dermatol. (2013) 23:33–9. doi: 10.1684/ejd.2012.1901
164. Ryding M, Mikkelsen AW, Nissen MS, Nilsson AC, Blaabjerg M. Pathophysiological effects of autoantibodies in autoimmune encephalitides. Cells. (2024) 13(1):15. doi: 10.3390/cells13010015
165. Seery N, Butzkueven H, O’brien TJ, Monif M. Contemporary advances in antibody-mediated encephalitis: anti-LGI1 and anti-Caspr2 antibody (Ab)-mediated encephalitides. Autoimmun Rev. (2022) 21:103074. doi: 10.1016/j.autrev.2022.103074
166. Toshika O, Yuko F, Miwako Y, Taisuke M, Norihiko Y, Hiroshi T, et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci. (2013) 33:18161. doi: 10.1523/JNEUROSCI.3506-13.2013
167. Petit-Pedrol M, Sell J, Planagumà J, Mannara F, Radosevic M, Haselmann H, et al. LGI1 antibodies alter Kv1.1 and AMPA receptors changing synaptic excitability, plasticity and memory. Brain. (2018) 141:3144–59. doi: 10.1093/brain/awy253
168. Joubert B, Petit-Pedrol M, Planagumà J, Mannara F, Radosevic M, Marsal M, et al. Human CASPR2 antibodies reversibly alter memory and the CASPR2 protein complex. Ann Neurol. (2022) 91:801–13. doi: 10.1002/ana.26345
169. Patterson KR, Dalmau J, Lancaster E. Mechanisms of Caspr2 antibodies in autoimmune encephalitis and neuromyotonia. Ann Neurol. (2018) 83:40–51. doi: 10.1002/ana.25120
170. Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet. (2015) 385:1460–71. doi: 10.1016/S0140-6736(14)60720-0
171. Perugino CA, Stone JH. IgG4-related disease: an update on pathophysiology and implications for clinical care. Nat Rev Rheumatol. (2020) 16:702–14. doi: 10.1038/s41584-020-0500-7
172. Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. (2003) 38:982–4. doi: 10.1007/s00535-003-1175-y
173. Stone JH, Zen Y, Deshpande V. IgG4-related disease. New Engl J Med. (2012) 366:539–51. doi: 10.1056/NEJMra1104650
174. Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. (2012) 25:1181–92. doi: 10.1038/modpathol.2012.72
175. Wallace ZS, Naden RP, Chari S, Choi H, Della-Torre E, Dicaire JF, et al. The 2019 american college of rheumatology/european league against rheumatism classification criteria for igG4-related disease. Arthritis Rheumatol. (2020) 72:7–19. doi: 10.1002/art.41120
176. Mattoo H, Stone JH, Pillai S. Clonally expanded cytotoxic CD4+ T cells and the pathogenesis of IgG4-related disease. Autoimmunity. (2017) 50:19–24. doi: 10.1080/08916934.2017.1280029
177. Pillai S, Perugino C, Kaneko N. Immune mechanisms of fibrosis and inflammation in IgG4-related disease. Curr Opin Rheumatol. (2020) 32:146–51. doi: 10.1097/BOR.0000000000000686
178. Perugino CA, Kaneko N, Maehara T, Mattoo H, Kers J, Allard-Chamard H, et al. CD4(+) and CD8(+) cytotoxic T lymphocytes may induce mesenchymal cell apoptosis in IgG(4)-related disease. J Allergy Clin Immunol. (2021) 147:368–82. doi: 10.1016/j.jaci.2020.05.022
179. Perugino CA, Alsalem SB, Mattoo H, Della-Torre E, Mahajan V, Ganesh G, et al. Identification of galectin-3 as an autoantigen in patients with IgG(4)-related disease. J Allergy Clin Immunol. (2019) 143:736–745.e736. doi: 10.1016/j.jaci.2018.05.01
180. Fugger L, Jensen LT, Rossjohn J. Challenges, progress, and prospects of developing therapies to treat autoimmune diseases. Cell. (2020) 181:63–80. doi: 10.1016/j.cell.2020.03.007
181. Bashford-Rogers RJM, Bergamaschi L, Mckinney EF, Pombal DC, Mescia F, Lee JC, et al. Analysis of the B cell receptor repertoire in six immune-mediated diseases. Nature. (2019) 574:122–6. doi: 10.1038/s41586-019-1595-3
182. Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. (2004) 350:2572–81. doi: 10.1056/NEJMoa032534
183. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. (2008) 358:676–88. doi: 10.1056/NEJMoa0706383
184. Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. (2017) 389:2031–40. doi: 10.1016/S0140-6736(17)30070-3
185. Nowak RJ, Dicapua DB, Zebardast N, Goldstein JM. Response of patients with refractory myasthenia gravis to rituximab: a retrospective study. Ther Adv Neurol Disord. (2011) 4:259–66. doi: 10.1177/1756285611411503
186. Blum S, Gillis D, Brown H, Boyle R, Henderson R, Heyworth-Smith D, et al. Use and monitoring of low dose rituximab in myasthenia gravis. J Neurol Neurosurg Psychiatry. (2011) 82:659–63. doi: 10.1136/jnnp.2010.220475
187. Robeson KR, Kumar A, Keung B, Dicapua DB, Grodinsky E, Patwa HS, et al. Durability of the rituximab response in acetylcholine receptor autoantibody-positive myasthenia gravis. JAMA Neurol. (2016) 74(1):60–6. doi: 10.1001/jamaneurol.2016.4190
188. Anolik JH, Barnard J, Owen T, Zheng B, Kemshetti S, Looney RJ, et al. Delayed memory B cell recovery in peripheral blood and lymphoid tissue in systemic lupus erythematosus after B cell depletion therapy. Arthritis Rheum. (2007) 56:3044–56. doi: 10.1002/art.22810
189. Adlowitz DG, Barnard J, Biear JN, Cistrone C, Owen T, Wang W, et al. Expansion of activated peripheral blood memory B cells in rheumatoid arthritis, impact of B cell depletion therapy, and biomarkers of response. PloS One. (2015) 10:e0128269. doi: 10.1371/journal.pone.0128269
190. Jiang R, Fichtner ML, Hoehn KB, Pham MC, Stathopoulos P, Nowak RJ, et al. Single-cell repertoire tracing identifies rituximab-resistant B cells during myasthenia gravis relapses. JCI Insight. (2020) 5(14):e136471. doi: 10.1172/jci.insight.136471
191. Querol L, Rojas-García R, Diaz-Manera J, Barcena J, Pardo J, Ortega-Moreno A, et al. Rituximab in treatment-resistant CIDP with antibodies against paranodal proteins. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e149. doi: 10.1212/NXI.0000000000000149
192. Roux T, Debs R, Maisonobe T, Lenglet T, Delorme C, Louapre C, et al. Rituximab in chronic inflammatory demyelinating polyradiculoneuropathy with associated diseases. J Peripheral Nervous Syst. (2018) 23:235–40. doi: 10.1111/jns.12287
193. Jiao L, Xiang Y, Li S, Zhang F, Ruan X, Guo S. Efficacy of low dose rituximab in treatment-resistant CIDP with antibodies against NF-155. J Neuroimmunol. (2020) 345:577280. doi: 10.1016/j.jneuroim.2020.577280
194. Martín-Aguilar L, Lleixà C, Pascual-Goñi E, Caballero-Ávila M, Martínez-Martínez L, Díaz-Manera J, et al. Clinical and laboratory features in anti-NF155 autoimmune nodopathy. Neurol Neuroimmunol Neuroinflamm. (2022) 9(1):e1098. doi: 10.1212/NXI.0000000000001098
195. Wang W, Liu C, Li W, Zhang D, Shan Y, Zheng J, et al. Clinical and diagnostic features of anti-neurofascin-155 antibody-positive neuropathy in Han Chinese. Ann Clin Trans Neurol. (2022) 9:695–706. doi: 10.1002/acn3.51550
196. Leuci S, Levine D, Zhang J, Razzaque Ahmed A. Response in patients with pemphigus vulgaris to rituximab therapy. Basis of the biology of B cells. G Ital Dermatol Venereol. (2009) 144:379–409.
197. Wang HH, Liu CW, Li YC, Huang YC. Efficacy of rituximab for pemphigus: a systematic review and meta-analysis of different regimens. Acta Derm Venereol. (2015) 95:928–32. doi: 10.2340/00015555-2116
198. Mahmoudi H, Tavakolpour S, Balighi K, Farid AS, Nili A, Jan D, et al. Rituximab in practice: Clinical evaluation of patients with pemphigus after rituximab administration. Dermatologic Ther. (2021) 34:e14633. doi: 10.1111/dth.14633
199. Thaler FS, Zimmermann L, Kammermeier S, Strippel C, Ringelstein M, Kraft A, et al. Rituximab treatment and long-term outcome of patients with autoimmune encephalitis: real-world evidence from the GENERATE registry. Neurol Neuroimmunol Neuroinflamm. (2021) 8(6):e1088. doi: 10.1212/NXI.0000000000001088
200. Bastakoti S, Kunwar S, Poudel S, Quinonez J, Bista S, Singh N, et al. Rituximab in the management of refractory myasthenia gravis and variability of its efficacy in anti-muSK positive and anti-AChR positive myasthenia gravis. Cureus. (2021) 13:e19416. doi: 10.7759/cureus.19416
201. Chaganti S, Hannaford A, Vucic S. Rituximab in chronic immune mediated neuropathies: a systematic review. Neuromuscul Disord. (2022) 32:621–7. doi: 10.1016/j.nmd.2022.05.013
202. Landon-Cardinal O, Friedman D, Guiguet M, Laforêt P, Heming N, Salort-Campana E, et al. Efficacy of rituximab in refractory generalized anti-AChR myasthenia gravis. J Neuromuscul Dis. (2018) 5:241–9. doi: 10.3233/JND-180300
203. Brauner S, Eriksson-Dufva A, Hietala MA, Frisell T, Press R, Piehl F. Comparison between rituximab treatment for new-onset generalized myasthenia gravis and refractory generalized myasthenia gravis. JAMA Neurol. (2020) 77:974–81. doi: 10.1001/jamaneurol.2020.0851
204. Piehl F, Eriksson-Dufva A, Budzianowska A, Feresiadou A, Hansson W, Hietala MA, et al. Efficacy and safety of rituximab for new-onset generalized myasthenia gravis: the RINOMAX randomized clinical trial. JAMA Neurol. (2022) 79:1105–12. doi: 10.1001/jamaneurol.2022.2887
205. Mahmoudi H, Balighi K, Tavakolpour S, Daneshpazhooh M. Unexpected worsening of pemphigus vulgaris after rituximab: A report of three cases. Int Immunopharmacol. (2019) 71:40–2. doi: 10.1016/j.intimp.2019.02.037
206. Bai Y, Li W, Yan C, Hou Y, Wang Q. Anti-rituximab antibodies in patients with refractory autoimmune nodopathy with anti-neurofascin-155 antibody. Front Immunol. (2023) 14. doi: 10.3389/fimmu.2023.1121705
207. Casertano S, Signoriello E, Rossi F, Di Pietro A, Tuccillo F, Bonavita S, et al. Ocrelizumab in a case of refractory chronic inflammatory demyelinating polyneuropathy with anti-rituximab antibodies. Eur J Neurol. (2020) 27:2673–5. doi: 10.1111/ene.14498
208. Schuh E, Berer K, Mulazzani M, Feil K, Meinl I, Lahm H, et al. Features of human CD3+CD20+ T cells. J Immunol. (2016) 197:1111–7. doi: 10.4049/jimmunol.1600089
209. Vlaming M, Bilemjian V, Freile J.Á., Lourens HJ, Van Rooij N, Huls G, et al. CD20 positive CD8 T cells are a unique and transcriptionally-distinct subset of T cells with distinct transmigration properties. Sci Rep. (2021) 11:20499. doi: 10.1038/s41598-021-00007-0
210. Lee AYS. CD20(+) T cells: an emerging T cell subset in human pathology. Inflammation Res. (2022) 71:1181–9. doi: 10.1007/s00011-022-01622-x
211. Palanichamy A, Jahn S, Nickles D, Derstine M, Abounasr A, Hauser SL, et al. Rituximab efficiently depletes increased CD20-expressing T cells in multiple sclerosis patients. J Immunol. (2014) 193:580–6. doi: 10.4049/jimmunol.1400118
212. Hultin LE, Hausner MA, Hultin PM, Giorgi JV. CD20 (pan-B cell) antigen is expressed at a low level on a subpopulation of human T lymphocytes. Cytometry. (1993) 14:196–204. doi: 10.1002/cyto.990140212
213. Henry C, Ramadan A, Montcuquet N, Pallandre JR, Mercier-Letondal P, Deschamps M, et al. CD3+CD20+ cells may be an artifact of flow cytometry: comment on the article by Wilk et al. Arthritis Rheum. (2010) 62:2561–2563; author reply 2563-2565. doi: 10.1002/art.27527
214. Pavlasova G, Mraz M. The regulation and function of CD20: an “enigma” of B-cell biology and targeted therapy. Haematologica. (2020) 105:1494–506. doi: 10.3324/haematol.2019.243543
215. Wilk E, Witte T, Marquardt N, Horvath T, Kalippke K, Scholz K, et al. Depletion of functionally active CD20+ T cells by rituximab treatment. Arthritis Rheum. (2009) 60:3563–71. doi: 10.1002/art.24998
216. De Bruyn M, Wiersma VR, Wouters MC, Samplonius DF, Klip HG, Helfrich W, et al. CD20(+) T cells have a predominantly Tc1 effector memory phenotype and are expanded in the ascites of patients with ovarian cancer. Oncoimmunology. (2015) 4:e999536. doi: 10.1080/2162402X.2014.999536
217. Chen Q, Yuan S, Sun H, Peng L. CD3(+)CD20(+) T cells and their roles in human diseases. Hum Immunol. (2019) 80:191–4. doi: 10.1016/j.humimm.2019.01.001
218. Ochs J, Nissimov N, Torke S, Freier M, Grondey K, Koch J, et al. Proinflammatory CD20(+) T cells contribute to CNS-directed autoimmunity. Sci Transl Med. (2022) 14:eabi4632. doi: 10.1126/scitranslmed.abi4632
219. Murayama Y, Mukai R, Sata T, Matsunaga S, Noguchi A, Yoshikawa Y. Transient expression of CD20 antigen (pan B cell marker) in activated lymph node T cells. Microbiol Immunol. (1996) 40:467–71. doi: 10.1111/j.1348-0421.1996.tb01096.x
220. Takami A, Saito M, Nakao S, Asakura H, Nozue T, Onoe Y, et al. CD20-positive T-cell chronic lymphocytic leukaemia. Br J Haematol. (1998) 102:1327–9. doi: 10.1046/j.1365-2141.1998.00884.x
221. Sabatino JJ Jr., Wilson MR, Calabresi PA, Hauser SL, Schneck JP, Zamvil SS. Anti-CD20 therapy depletes activated myelin-specific CD8(+) T cells in multiple sclerosis. Proc Natl Acad Sci U.S.A. (2019) 116:25800–7. doi: 10.1073/pnas.1915309116
222. Mathias A, Pantazou V, Perriot S, Canales M, Jones S, Oberholster L, et al. Ocrelizumab impairs the phenotype and function of memory CD8(+) T cells: A 1-year longitudinal study in patients with multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2023) 10(2):e200084. doi: 10.1212/NXI.0000000000200084
223. Shinoda K, Li R, Rezk A, Mexhitaj I, Patterson KR, Kakara M, et al. Differential effects of anti-CD20 therapy on CD4 and CD8 T cells and implication of CD20-expressing CD8 T cells in MS disease activity. Proc Natl Acad Sci U.S.A. (2023) 120:e2207291120. doi: 10.1073/pnas.2207291120
224. Hershberg U, Meng W, Zhang B, Haff N, St Clair EW, Cohen PL, et al. Persistence and selection of an expanded B-cell clone in the setting of rituximab therapy for Sjögren’s syndrome. Arthritis Res Ther. (2014) 16:R51. doi: 10.1186/ar4481
225. De Bourcy CFA, Dekker CL, Davis MM, Nicolls MR, Quake SR. Dynamics of the human antibody repertoire after B cell depletion in systemic sclerosis. Sci Immunol. (2017) 2(15):eaan8289. doi: 10.1126/sciimmunol.aan8289
226. Leandro MJ, Cambridge G, Ehrenstein MR, Edwards JCW. Reconstitution of peripheral blood B cells after depletion with rituximab in patients with rheumatoid arthritis. Arthritis Rheumatism. (2006) 54:613–20. doi: 10.1002/art.21617
227. Mamani-Matsuda M, Cosma A, Weller S, Faili A, Staib C, Garçon L, et al. The human spleen is a major reservoir for long-lived vaccinia virus–specific memory B cells. Blood. (2008) 111:4653–9. doi: 10.1182/blood-2007-11-123844
228. Ramwadhdoebe TH, Van Baarsen LGM, Boumans MJH, Bruijnen STG, Safy M, Berger FH, et al. Effect of rituximab treatment on T and B cell subsets in lymph node biopsies of patients with rheumatoid arthritis. Rheumatology. (2019) 58:1075–85. doi: 10.1093/rheumatology/key428
229. Nakou M, Katsikas G, Sidiropoulos P, Bertsias G, Papadimitraki E, Raptopoulou A, et al. Rituximab therapy reduces activated B cells in both the peripheral blood and bone marrow of patients with rheumatoid arthritis: depletion of memory B cells correlates with clinical response. Arthritis Res Ther. (2009) 11:R131. doi: 10.1186/ar2798
230. Colliou N, Picard D, Caillot F, Calbo S, Le Corre S, Lim A, et al. Long-term remissions of severe pemphigus after rituximab therapy are associated with prolonged failure of desmoglein B cell response. Sci Transl Med. (2013) 5:175ra130. doi: 10.1126/scitranslmed.3005166
231. Maurer MA, Tuller F, Gredler V, Berger T, Lutterotti A, Lünemann JD, et al. Rituximab induces clonal expansion of IgG memory B-cells in patients with inflammatory central nervous system demyelination. J Neuroimmunol. (2016) 290:49–53. doi: 10.1016/j.jneuroim.2015.11.006
232. Becerra E, de la Torre I, Leandro MJ, Cambridge G. B cell phenotypes in patients with rheumatoid arthritis relapsing after rituximab: expression of B cell-activating factor-binding receptors on B cell subsets. Clin Exp Immunol. (2017) 190:372–83. doi: 10.1111/cei.13024
233. Wang K, Wei G, Liu D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol. (2012) 1:36. doi: 10.1186/2162-3619-1-36
234. Forsthuber TG, Cimbora DM, Ratchford JN, Katz E, Stüve O. B cell-based therapies in CNS autoimmunity: differentiating CD19 and CD20 as therapeutic targets. Ther Adv Neurol Disord. (2018) 11:1756286418761697. doi: 10.1177/1756286418761697
235. Hofmann K, Clauder AK, Manz RA. Targeting B cells and plasma cells in autoimmune diseases. Front Immunol. (2018) 9:835. doi: 10.3389/fimmu.2018.00835
236. Chu SY, Yeter K, Kotha R, Pong E, Miranda Y, Phung S, et al. Suppression of rheumatoid arthritis B cells by XmAb5871, an anti-CD19 antibody that coengages B cell antigen receptor complex and Fcγ receptor IIb inhibitory receptor. Arthritis Rheumatol. (2014) 66:1153–64. doi: 10.1002/art.38334
237. Cree BA, Bennett JL, Sheehan M, Cohen J, Hartung HP, Aktas O, et al. Placebo-controlled study in neuromyelitis optica-Ethical and design considerations. Mult Scler. (2016) 22:862–72. doi: 10.1177/1352458515620934
238. Lee HC. Structure and enzymatic functions of human CD38. Mol Med. (2006) 12:317–23. doi: 10.2119/2006-00086.Lee
239. Piedra-Quintero ZL, Wilson Z, Nava P, Guerau-De-Arellano M. CD38: an immunomodulatory molecule in inflammation and autoimmunity. Front Immunol. (2020) 11:597959. doi: 10.3389/fimmu.2020.597959
240. Ye X, Zhao Y, Ma W, Ares I, Martínez M, Lopez-Torres B, et al. The potential of CD38 protein as a target for autoimmune diseases. Autoimmun Rev. (2023) 22:103289. doi: 10.1016/j.autrev.2023.103289
241. Bisht K, Fukao T, Chiron M, Richardson P, Atanackovic D, Chini E, et al. Immunomodulatory properties of CD38 antibodies and their effect on anticancer efficacy in multiple myeloma. Cancer Med. (2023) 12:20332–52. doi: 10.1002/cam4.6619
242. Leleu X, Martin T, Weisel K, Schjesvold F, Iida S, Malavasi F, et al. Anti-CD38 antibody therapy for patients with relapsed/refractory multiple myeloma: differential mechanisms of action and recent clinical trial outcomes. Ann Hematol. (2022) 101:2123–37. doi: 10.1007/s00277-022-04917-5
243. Karnell JL, Rieder SA, Ettinger R, Kolbeck R. Targeting the CD40-CD40L pathway in autoimmune diseases: Humoral immunity and beyond. Adv Drug Delivery Rev. (2019) 141:92–103. doi: 10.1016/j.addr.2018.12.005
244. Tang T, Cheng X, Truong B, Sun L, Yang X, Wang H. Molecular basis and therapeutic implications of CD40/CD40L immune checkpoint. Pharmacol Ther. (2021) 219:107709. doi: 10.1016/j.pharmthera.2020.107709
245. Gomezmancilla B, Meriggioli MN, Genge A, Roubenoff R, Espié P, Dupuy C, et al. Efficacy and safety of iscalimab, a novel anti-CD40 monoclonal antibody, in moderate-to-severe myasthenia gravis: A phase 2 randomized study. J Clin Neurosci. (2024) 119:76–84. doi: 10.1016/j.jocn.2023.11.013
246. Fisher BA, Szanto A, Ng WF, Bombardieri M, Posch MG, Papas AS, et al. Assessment of the anti-CD40 antibody iscalimab in patients with primary Sjögren’s syndrome: a multicentre, randomised, double-blind, placebo-controlled, proof-of-concept study. Lancet Rheumatol. (2020) 2:e142–52. doi: 10.1016/S2665-9913(19)30135-3
247. Kahaly GJ, Stan MN, Frommer L, Gergely P, Colin L, Amer A, et al. A novel anti-CD40 monoclonal antibody, iscalimab, for control of graves hyperthyroidism-A proof-of-concept trial. J Clin Endocrinol Metab. (2020) 105(3):dgz013. doi: 10.1210/clinem/dgz013
248. Aoki-Ota M, Kinoshita M, Ota T, Tsunoda K, Iwasaki T, Tanaka S, et al. Tolerance induction by the blockade of CD40/CD154 interaction in pemphigus vulgaris mouse model. J Invest Dermatol. (2006) 126:105–13. doi: 10.1038/sj.jid.5700016
249. Bilgic Temel A, Murrell DF. Pharmacological advances in pemphigus. Curr Opin Pharmacol. (2019) 46:44–9. doi: 10.1016/j.coph.2019.01.001
250. Cancro MP. BLyS ligands and receptors. Totowa, NJ: Humana Press (2010). doi: 10.1007/978-1-60327-013-7
251. Vincent FB, Saulep-Easton D, Figgett WA, Fairfax KA, Mackay F. The BAFF/APRIL system: emerging functions beyond B cell biology and autoimmunity. Cytokine Growth Factor Rev. (2013) 24:203–15. doi: 10.1016/j.cytogfr.2013.04.003
252. Kim JY, Yang Y, Moon JS, Lee EY, So SH, Lee HS, et al. Serum BAFF expression in patients with myasthenia gravis. J Neuroimmunol. (2008) 199:151–4. doi: 10.1016/j.jneuroim.2008.05.010
253. Guptill JT, Yi JS, Sanders DB, Guidon AC, Juel VC, Massey JM, et al. Characterization of B cells in muscle-specific kinase antibody myasthenia gravis. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e77. doi: 10.1212/NXI.0000000000000077
254. Kang SY, Kang CH, Lee KH. B-cell-activating factor is elevated in serum of patients with myasthenia gravis. Muscle Nerve. (2016) 54:1030–3. doi: 10.1002/mus.25162
255. Samy E, Wax S, Huard B, Hess H, Schneider P. Targeting BAFF and APRIL in systemic lupus erythematosus and other antibody-associated diseases. Int Rev Immunol. (2017) 36:3–19. doi: 10.1080/08830185.2016.1276903
256. Hewett K, Sanders DB, Grove RA, Broderick CL, Rudo TJ, Bassiri A, et al. Randomized study of adjunctive belimumab in participants with generalized myasthenia gravis. Neurology. (2018) 90:e1425–34. doi: 10.1212/WNL.0000000000005323
257. Raje NS, Moreau P, Terpos E, Benboubker L, Grząśko N, Holstein SA, et al. Phase 2 study of tabalumab, a human anti-B-cell activating factor antibody, with bortezomib and dexamethasone in patients with previously treated multiple myeloma. Br J Haematol. (2017) 176:783–95. doi: 10.1111/bjh.14483
258. Merrill JT, Shanahan WR, Scheinberg M, Kalunian KC, Wofsy D, Martin RS. Phase III trial results with blisibimod, a selective inhibitor of B-cell activating factor, in subjects with systemic lupus erythematosus (SLE): results from a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. (2018) 77:883–9. doi: 10.1136/annrheumdis-2018-213032
259. Shi F, Xue R, Zhou X, Shen P, Wang S, Yang Y. Telitacicept as a BLyS/APRIL dual inhibitor for autoimmune disease. Immunopharmacol Immunotoxicol. (2021) 43:666–73. doi: 10.1080/08923973.2021.1973493
260. Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. (2000) 404:995–9. doi: 10.1038/35010115
261. Gross JA, Dillon SR, Mudri S, Johnston J, Littau A, Roque R, et al. TACI-Ig neutralizes molecules critical for B cell development and autoimmune disease. impaired B cell maturation in mice lacking BLyS. Immunity. (2001) 15:289–302. doi: 10.1016/S1074-7613(01)00183-2
262. Dall’era M, Chakravarty E, Wallace D, Genovese M, Weisman M, Kavanaugh A, et al. Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating trial. Arthritis Rheum. (2007) 56:4142–50. doi: 10.1002/art.23047
263. Tak PP, Thurlings RM, Rossier C, Nestorov I, Dimic A, Mircetic V, et al. Atacicept in patients with rheumatoid arthritis: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating, single- and repeated-dose study. Arthritis Rheum. (2008) 58:61–72. doi: 10.1002/art.23178
264. Cogollo E, Silva MA, Isenberg D. Profile of atacicept and its potential in the treatment of systemic lupus erythematosus. Drug Des Devel Ther. (2015) 9:1331–9. doi: 10.2147/DDDT
265. Isenberg D, Gordon C, Licu D, Copt S, Rossi CP, Wofsy D. Efficacy and safety of atacicept for prevention of flares in patients with moderate-to-severe systemic lupus erythematosus (SLE): 52-week data (APRIL-SLE randomised trial). Ann Rheum Dis. (2015) 74:2006–15. doi: 10.1136/annrheumdis-2013-205067
266. Stohl W. Inhibition of B cell activating factor (BAFF) in the management of systemic lupus erythematosus (SLE). Expert Rev Clin Immunol. (2017) 13(6):623–33. doi: 10.1080/1744666X.2017.1291343
267. Di W, Jing L, Dong X, Joan TM, Ronald FVV, Yi L, et al. Telitacicept in patients with active systemic lupus erythematosus: results of a phase 2b, randomised, double-blind, placebo-controlled trial. Ann Rheumatic Dis. (2023). doi: 10.1136/ard-2023-224854
268. Hartung HP. [Atacicept: a new B lymphocyte-targeted therapy for multiple sclerosis]. Nervenarzt. (2009) 80:1462–72. doi: 10.1007/s00115-009-2838-6
269. Hartung HP, Kieseier BC. Atacicept: targeting B cells in multiple sclerosis. Ther Adv Neurol Disord. (2010) 3:205–16. doi: 10.1177/1756285610371146
270. Kappos L, Hartung HP, Freedman MS, Boyko A, Radu EW, Mikol DD, et al. Atacicept in multiple sclerosis (ATAMS): a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol. (2014) 13:353–63. doi: 10.1016/S1474-4422(14)70028-6
271. Bag-Ozbek A, Hui-Yuen JS. Emerging B-cell therapies in systemic lupus erythematosus. Ther Clin Risk Manag. (2021) 17:39–54. doi: 10.2147/TCRM.S252592
272. Wang J, Fang Y, Fan RA, Kirk CJ. Proteasome inhibitors and their pharmacokinetics, pharmacodynamics, and metabolism. Int J Mol Sci. (2021) 22(21):11595. doi: 10.3390/ijms222111595
273. Parodis I, Gatto M, Sjöwall C. B cells in systemic lupus erythematosus: Targets of new therapies and surveillance tools. Front Med (Lausanne). (2022) 9:952304. doi: 10.3389/fmed.2022.952304
274. Alexander T, Cheng Q, Klotsche J, Khodadadi L, Waka A, Biesen R, et al. Proteasome inhibition with bortezomib induces a therapeutically relevant depletion of plasma cells in SLE but does not target their precursors. Eur J Immunol. (2018) 48:1573–9. doi: 10.1002/eji.201847492
275. Gomez AM, Vrolix K, Martinez-Martinez P, Molenaar PC, Phernambucq M, van der Esch E, et al. Proteasome inhibition with bortezomib depletes plasma cells and autoantibodies in experimental autoimmune myasthenia gravis. J Immunol. (2011) 186:2503–13. doi: 10.4049/jimmunol.1002539
276. Liu R-T, Zhang P, Yang C-L, Pang Y, Zhang M, Zhang N, et al. ONX-0914, a selective inhibitor of immunoproteasome, ameliorates experimental autoimmune myasthenia gravis by modulating humoral response. J Neuroimmunology. (2017) 311:71–8. doi: 10.1016/j.jneuroim.2017.08.005
277. Gomez AM, Willcox N, Vrolix K, Hummel J, Nogales-Gadea G, Saxena A, et al. Proteasome inhibition with bortezomib depletes plasma cells and specific autoantibody production in primary thymic cell cultures from early-onset myasthenia gravis patients. J Immunol. (2014) 193:1055–63. doi: 10.4049/jimmunol.1301555
278. Kohler S, Märschenz S, Grittner U, Alexander T, Hiepe F, Meisel A. Bortezomib in antibody-mediated autoimmune diseases (TAVAB): study protocol for a unicentric, non-randomised, non-placebo controlled trial. BMJ Open. (2019) 9:e024523. doi: 10.1136/bmjopen-2018-024523
279. Simister NE, Story CM, Chen HL, Hunt JS. An IgG-transporting Fc receptor expressed in the syncytiotrophoblast of human placenta. Eur J Immunol. (1996) 26:1527–31. doi: 10.1002/eji.1830260718
280. Patel DD, Bussel JB. Neonatal Fc receptor in human immunity: Function and role in therapeutic intervention. J Allergy Clin Immunol. (2020) 146:467–78. doi: 10.1016/j.jaci.2020.07.015
281. Burmeister WP, Gastinel LN, Simister NE, Blum ML, Bjorkman PJ. Crystal structure at 2.2 A resolution of the MHC-related neonatal Fc receptor. Nature. (1994) 372:336–43. doi: 10.1038/372336a0
282. Zuercher AW, Spirig R, Baz Morelli A, Rowe T, Kasermann F. Next-generation Fc receptor-targeting biologics for autoimmune diseases. Autoimmun Rev. (2019) 18:102366. doi: 10.1016/j.autrev.2019.102366
283. Howard JF Jr., Bril V, Vu T, Karam C, Peric S, Margania T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. (2021) 20:526–36. doi: 10.1212/WNL.96.15_supplement.4520
284. Werth VP, Culton DA, Concha JSS, Graydon JS, Blumberg LJ, Okawa J, et al. Safety, tolerability, and activity of ALXN1830 targeting the neonatal fc receptor in chronic pemphigus. J Invest Dermatol. (2021) 141:2858–2865.e2854. doi: 10.1016/j.jid.2021.04.031
285. Bril V, Drużdż A, Grosskreutz J, Habib AA, Mantegazza R, Sacconi S, et al. Safety and efficacy of rozanolixizumab in patients with generalised myasthenia gravis (MycarinG): a randomised, double-blind, placebo-controlled, adaptive phase 3 study. Lancet Neurol. (2023) 22:383–94. doi: 10.1016/S1474-4422(23)00077-7
286. Kareem S, Jacob A, Mathew J, Quigg RJ, Alexander JJ. Complement: Functions, location and implications. Immunology. (2023) 170:180–92. doi: 10.1111/imm.13663
287. Rodgaard A, Nielsen FC, Djurup R, Somnier F, Gammeltoft S. Acetylcholine receptor antibody in myasthenia gravis: predominance of IgG subclasses 1 and 3. Clin Exp Immunol. (1987) 67:82–8.
288. Howard JF, Utsugisawa K, Benatar M, Murai H, Barohn RJ, Illa I, et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. (2017) 16:976–86. doi: 10.1016/S1474-4422(17)30369-1
289. Vu T, Ortiz S, Katsuno M, Annane D, Mantegazza R, Beasley KN, et al. Ravulizumab pharmacokinetics and pharmacodynamics in patients with generalized myasthenia gravis. J Neurol. (2023) 270:3129–37. doi: 10.1007/s00415-023-11617-1
290. Zarrin AA, Bao K, Lupardus P, Vucic D. Kinase inhibition in autoimmunity and inflammation. Nat Rev Drug Discovery. (2021) 20:39–63. doi: 10.1038/s41573-020-0082-8
291. Mendes-Bastos P, Brasileiro A, Kolkhir P, Frischbutter S, Scheffel J, Moñino-Romero S, et al. Bruton’s tyrosine kinase inhibition-An emerging therapeutic strategy in immune-mediated dermatological conditions. Allergy. (2022) 77:2355–66. doi: 10.1111/all.15261
292. Mcdonald C, Xanthopoulos C, Kostareli E. The role of Bruton’s tyrosine kinase in the immune system and disease. Immunology. (2021) 164:722–36. doi: 10.1111/imm.13416
293. Rip J, van der Ploeg EK, Hendriks RW, Corneth OBJ. The role of bruton’s tyrosine kinase in immune cell signaling and systemic autoimmunity. Crit Rev Immunol. (2018) 38:17–62. doi: 10.1615/CritRevImmunol.v38.i1
294. Isenberg D, Furie R, Jones NS, Guibord P, Galanter J, Lee C, et al. Efficacy, safety, and pharmacodynamic effects of the bruton’s tyrosine kinase inhibitor fenebrutinib (GDC-0853) in systemic lupus erythematosus: results of a phase II, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. (2021) 73:1835–46. doi: 10.1002/art.41811
295. Irving BA, Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell. (1991) 64:891–901. doi: 10.1016/0092-8674(91)90314-O
296. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. (2011) 3:95ra73. doi: 10.1126/scitranslmed.3002842
297. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. (2016) 353:179–84. doi: 10.1126/science.aaf6756
298. Labanieh L, Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature. (2023) 614:635–48. doi: 10.1038/s41586-023-05707-3
299. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) 11:69. doi: 10.1038/s41408-021-00459-7
300. Blache U, Tretbar S, Koehl U, Mougiakakos D, Fricke S. CAR T cells for treating autoimmune diseases. RMD Open. (2023) 9(4):e002907. doi: 10.1136/rmdopen-2022-002907
301. Lee L, Draper B, Chaplin N, Philip B, Chin M, Galas-Filipowicz D, et al. An APRIL-based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood. (2018) 131:746–58. doi: 10.1182/blood-2017-05-781351
302. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. (2019) 380:1726–37. doi: 10.1056/NEJMoa1817226
303. Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. (2019) 25:947–53. doi: 10.1038/s41591-019-0421-7
304. Zhang W, Feng J, Cinquina A, Wang Q, Xu H, Zhang Q, et al. Treatment of systemic lupus erythematosus using BCMA-CD19 compound CAR. Stem Cell Rev Rep. (2021) 17:2120–3. doi: 10.1007/s12015-021-10251-6
305. Oh S, Mao X, Manfredo-Vieira S, Lee J, Patel D, Choi EJ, et al. Precision targeting of autoantigen-specific B cells in muscle-specific tyrosine kinase myasthenia gravis with chimeric autoantibody receptor T cells. Nat Biotechnol. (2023) 41:1229–38. doi: 10.1038/s41587-022-01637-z
306. Menon D, Bril V. Pharmacotherapy of generalized myasthenia gravis with special emphasis on newer biologicals. Drugs. (2022) 82:865–87. doi: 10.1007/s40265-022-01726-y
307. Reincke SM, Von Wardenburg N, Homeyer MA, Kornau HC, Spagni G, Li LY, et al. Chimeric autoantibody receptor T cells deplete NMDA receptor-specific B cells. Cell. (2023) 186:5084–5097.e5018. doi: 10.1016/j.cell.2023.10.001
308. Lee J, Lundgren DK, Mao X, Manfredo-Vieira S, Nunez-Cruz S, Williams EF, et al. Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris. J Clin Invest. (2020) 130:6317–24. doi: 10.1172/JCI138416
309. Mok CC, Lau CS. Pathogenesis of systemic lupus erythematosus. J Clin Pathol. (2003) 56:481–90. doi: 10.1136/jcp.56.7.481
310. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. (2015) 15:545–58. doi: 10.1038/nri3871
311. Phillips WD, Vincent A. Pathogenesis of myasthenia gravis: update on disease types, models, and mechanisms. F1000Res. (2016) 5:F1000 Faculty Rev-1513. doi: 10.12688/f1000research
312. Paschou SA, Papadopoulou-Marketou N, Chrousos GP, Kanaka-Gantenbein C. On type 1 diabetes mellitus pathogenesis. Endocr Connect. (2018) 7:R38–r46. doi: 10.1530/EC-17-0347
Keywords: IgG4, IgG4-AID, IgG4-RD, immunotherapies, antibodies
Citation: Ünlü S, Sánchez Navarro BG, Cakan E, Berchtold D, Meleka Hanna R, Vural S, Vural A, Meisel A and Fichtner ML (2024) Exploring the depths of IgG4: insights into autoimmunity and novel treatments. Front. Immunol. 15:1346671. doi: 10.3389/fimmu.2024.1346671
Received: 29 November 2023; Accepted: 29 February 2024;
Published: 18 April 2024.
Edited by:
Inga Koneczny, Medical University of Vienna, AustriaReviewed by:
Michelangelo Maestri, University of Pisa, ItalyAnna Punga, Uppsala University, Sweden
Vuslat Yilmaz, Istanbul University, Türkiye
Copyright © 2024 Ünlü, Sánchez Navarro, Cakan, Berchtold, Meleka Hanna, Vural, Vural, Meisel and Fichtner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miriam L. Fichtner, bWlyaWFtLmZpY2h0bmVyQGNoYXJpdGUuZGU=
†ORCID: Blanca G. Sánchez Navarro, orcid.org/0009-0001-3245-722X
Daniel Berchtold, orcid.org/0000-0002-7865-4157
Miriam L. Fichtner, orcid.org/0000-0002-0107-4103