 He Wang1
He Wang1 Hai Zhao
Hai Zhao- 1Department of Neurosurgery, The Affiliated Hospital of Qingdao University, Qingdao, Shandong, China
- 2Department of Neurosurgery, Tianshui First People's Hospital, Tianshui, China
- 3Department of Breast Surgery, School of Medicine, Qingdao University, Qingdao, Shandong, China
- 4Department of Operating Room, The Affiliated Hospital of Qingdao University, Qingdao, Shandong, China
Glioma, as the most frequently occurring primary malignancy in the central nervous system, significantly impacts patients’ quality of life and cognitive abilities. Ferroptosis, a newly discovered form of cell death, is characterized by significant iron accumulation and lipid peroxidation. This process is fundamentally dependent on iron. Various factors inducing ferroptosis can either directly or indirectly influence glutathione peroxidase, leading to reduced antioxidant capabilities and an increase in lipid reactive oxygen species (ROS) within cells, culminating in oxidative cell death. Recent research indicates a strong connection between ferroptosis and a range of pathophysiological conditions, including tumors, neurological disorders, ischemia-reperfusion injuries, kidney damage, and hematological diseases. The regulation of ferroptosis to intervene in the progression of these diseases has emerged as a major area of interest in etiological research and therapy. However, the exact functional alterations and molecular mechanisms underlying ferroptosis remain to be extensively studied. The review firstly explores the intricate relationship between ferroptosis and glioma, highlighting how ferroptosis contributes to glioma pathogenesis and how glioma cells may resist this form of cell death. Then, we discuss recent studies that have identified potential ferroptosis inducers and inhibitors, which could serve as novel therapeutic strategies for glioma. We also examine the current challenges in targeting ferroptosis in glioma treatment, including the complexity of its regulation and the need for precise delivery methods. This review aims to provide a comprehensive overview of the current state of research on ferroptosis in glioma, offering insights into future therapeutic strategies and the broader implications of this novel cell death pathway in cancer biology.
Introduction
In 2003, Dolma and team identified a novel compound, erastin, with a unique ability to selectively kill cancer cells expressing the RAS gene, marking a departure from known cell death mechanisms (1). This process lacked traditional signs like nuclear morphological changes, DNA fragmentation, or caspase activation and was unaffected by caspase inhibitors. Further studies by Yang and Yagoda revealed that this type of cell death could be thwarted by iron chelators, and they also discovered RSL3, another compound capable of inducing the same cell death pattern (2, 3). In 2012, Dixon et al. coined the term “ferroptosis” for this form of cell death while investigating how erastin killed RAS-mutant cancer cells (4) (see Figure 1). Ferroptosis is characterized morphologically by decreased mitochondrial volume, denser mitochondrial bilayer membranes, and often diminished or absent mitochondrial cristae yet with an intact cell membrane, normal-sized nucleus, and no chromatin condensation (2, 4–6). Biochemically, it involves the depletion of intracellular glutathione (GSH) and a decrease in glutathione peroxidase 4 (GPX4) activity, leading to unmetabolized lipid peroxides (7–10). The presence of Fe2+ causes a Fenton-like oxidation of lipids, resulting in a surge of ROS that promotes ferroptosis (2, 11, 12). Genetically, it’s regulated by multiple genes, primarily those associated with iron homeostasis and lipid peroxidation metabolism, though the specific mechanisms are still under investigation.
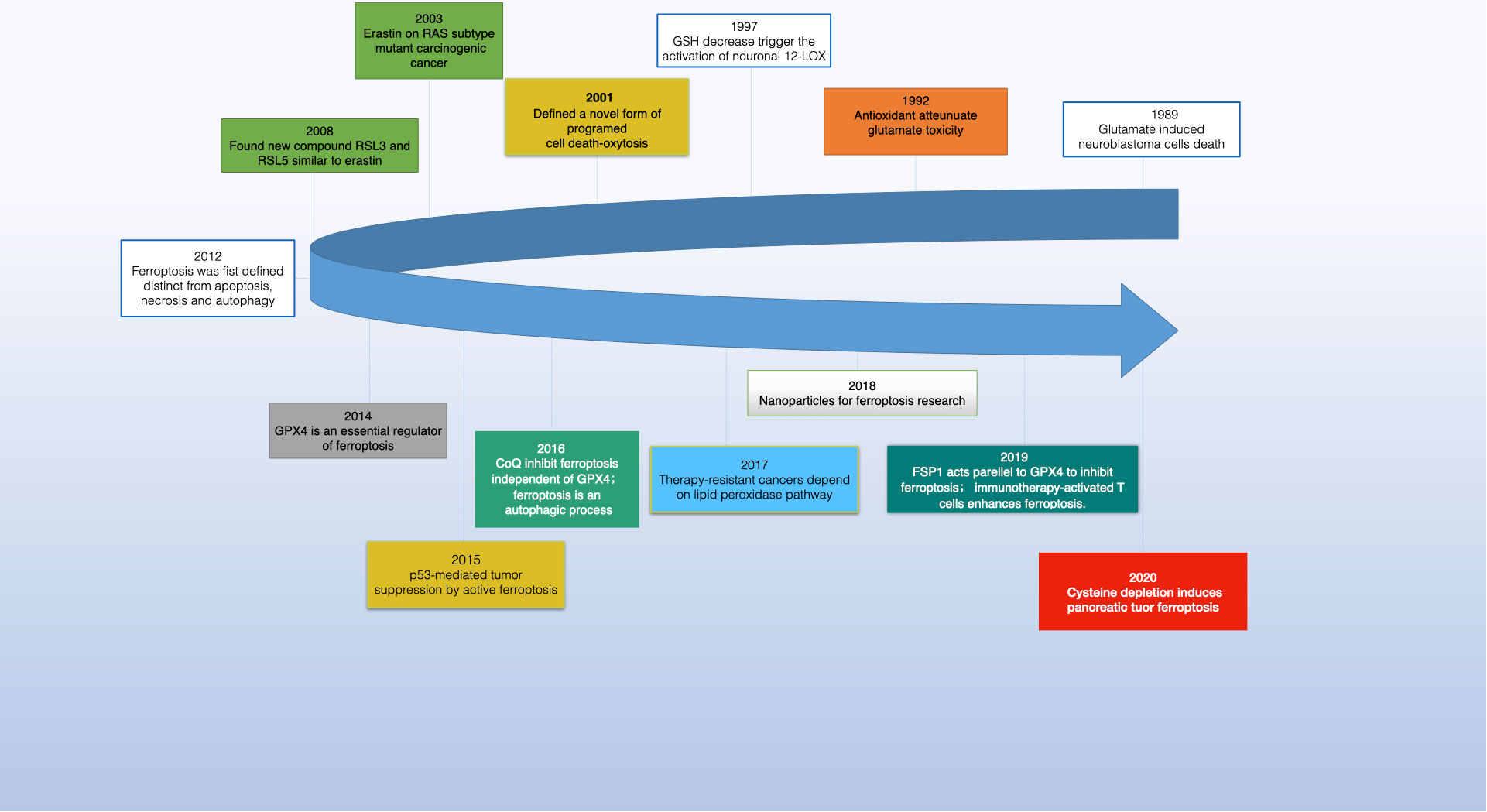
Figure 1 The development of ferroptosis in cancer research.
Ferroptosis inducers are categorized into four groups. The first includes erastin, which reduces GSH levels by inhibiting system Xc- and affects voltage-dependent anion channels (VDACs) for mitochondrial dysfunction. Erastin also enhances lysosomal-associated membrane protein 2a, promoting chaperone-mediated autophagy and GPX4 degradation (13–16). The second category, with members like RSL3 and DPI7, directly inhibits GPX4 (17, 18). The third category features FIN56, which induces ferroptosis by promoting GPX4 degradation and binding to squalene synthase, leading to coenzyme Q10 (COQ10) depletion and enhanced cell sensitivity to ferroptosis (19–21). Lastly, FINO2, resembling artemisinin, causes ferroptosis through labile iron oxidation and GPX4 inactivation (21–23). Research has also unearthed specific ferroptosis inhibitors like ferrostatin-1 (Fer-1), liproxstatin-1, and vitamin E, along with iron chelators, which prevent lipid peroxide formation. In 2014, Skouta et al. found that Fer-1 inhibited cell death in models of Huntington’s disease (HD), periventricular white matter (PVL), and renal insufficiency, highlighting the potential of ferrostatin in disease models and emphasizing the significance of ferroptosis beyond cell culture (24). The discovery of ferroptosis thus opens new avenues for understanding and treating various diseases (see Figure 2).
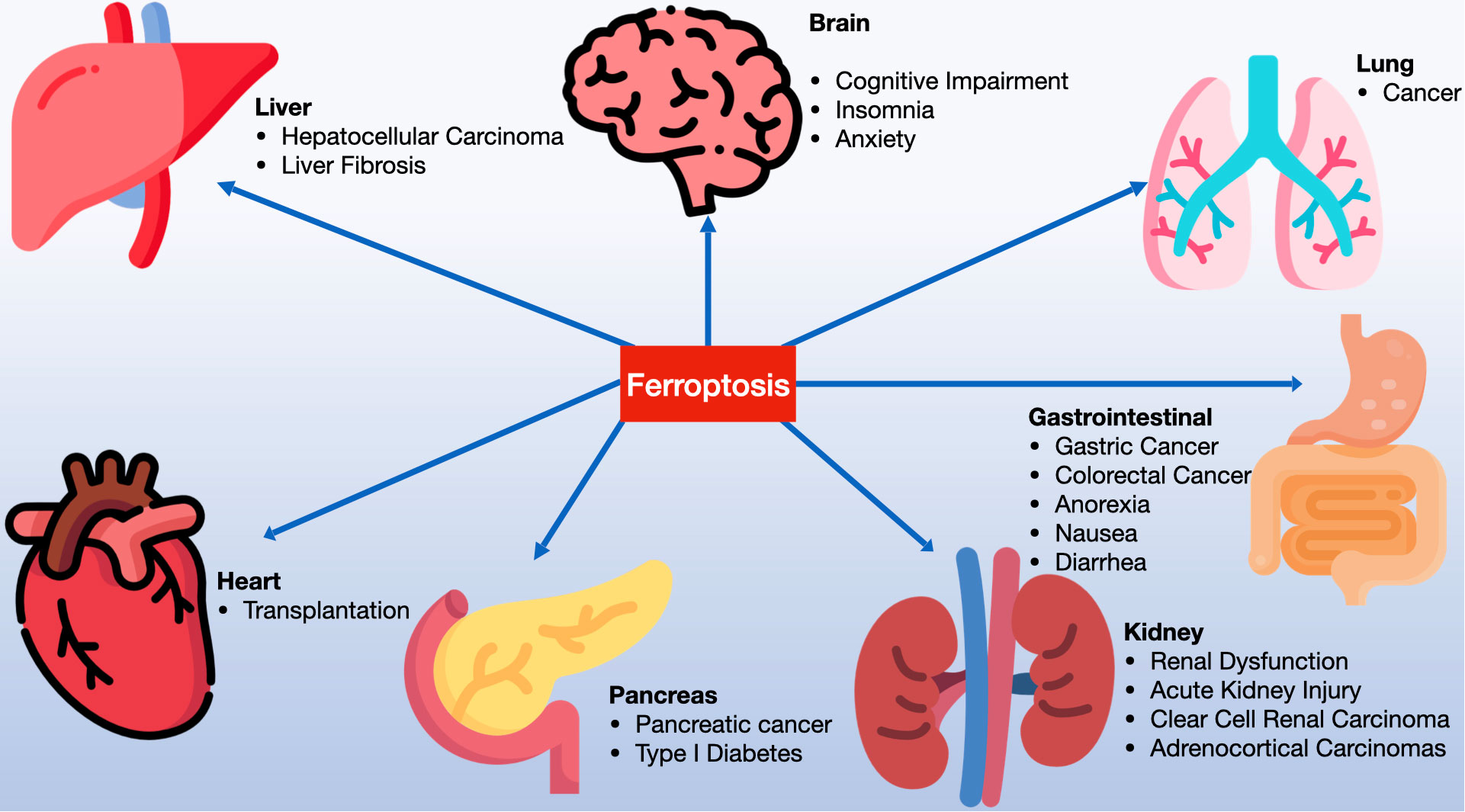
Figure 2 Ferroptosis is significantly involved in a variety of systemic diseases, including disorders of the nervous system, cardiac diseases, hepatic diseases, gastrointestinal conditions, pulmonary diseases, renal diseases, pancreatic disorders, among others.
Glioma, the most prevalent primary brain tumor, represents about 50-60% of central nervous system (CNS) tumors and roughly 81%’, of all intracranial malignancies (25, 26). Compared to other CNS tumors, gliomas exhibit notably higher recurrence rates. The World Health Organization (WHO) grades gliomas from 1 to 4, with grades 1 and 2 being low-grade and grades 3 and 4 as high-grade gliomas (27). The median overall survival (OS) for patients with low-grade gliomas is about 11.6 years, but this significantly drops to around three years for grade 3 gliomas and approximately 15 months for those with grade 4 gliomas (28, 29). Despite current treatments like surgical resection, radiotherapy, chemotherapy, novel molecular targeted therapies, and immunotherapy, patient outcomes remain largely unsatisfactory, with a dire prognosis. This underscores the urgent need for new therapeutic approaches and targets to enhance OS and life quality in glioma patients. Traditionally, cell death includes necrosis, apoptosis, autophagy, and pyroptosis (30). However, the recent focus has shifted to ferroptosis, a novel non-apoptotic form of cell death caused by iron-dependent lipid peroxidation, garnering increased research interest (4, 31, 32). In this review, we will primarily concentrate on the key molecular processes of ferroptosis and its possible effects on the development and treatment of gliomas, especially GBM. Additionally, we will offer a summary of the difficulties associated with using ferroptosis in glioma treatment and explore the therapeutic possibilities of leveraging ferroptosis to enhance treatment outcomes.
Discovery and characteristics of ferroptosis
The concept of “ferroptosis,” first introduced by Dixon and others in 2012, was preceded by a similar type of cell death termed “oxytosis” identified by Murphy and colleagues in 1989 (33). Oxytosis, caused by cysteine depletion in neurons, was linked to the inhibition of SLC7A11, a component crucial in ferroptosis. Both oxytosis and ferroptosis share similarities like gene expression patterns, lipoxygenase activity, and reactive oxygen species accumulation (34). By 2003, a unique form of cell death in RAS-expressing cancer cells, not prevented by caspase inhibitors but mitigated by iron-chelating agents, was observed (1, 3). This led to the discovery that RSL3, a RAS-selective lethal small molecule, could also induce this iron-dependent cell death, which Dixon et al. named ferroptosis (2).
Morphologically, ferroptosis is marked by mitochondrial changes such as shrinkage and decreased cristae. Biochemically, it involves oxidative stress and reduced antioxidative defense. The exact mechanisms, particularly how phospholipid peroxidation triggers ferroptosis and its specific epilipidomic patterns in different tissues and diseases, are still being researched. Emerging research has begun to reveal key regulators of ferroptosis. For instance, the glutathione/glutathione peroxidase 4 pathway, ACSL4, and selenium’s protective role against ferroptosis have been identified. The FSP1-CoQ10-NAD(P)H pathway and the GCH1-BH4 pathway were also recognized as GPX4-independent mechanisms influencing ferroptosis (35, 36). Recently, the DHODH-CoQ10 axis in the mitochondrial inner membrane and vitamin K’s role as an FSP1-dependent ferroptosis inhibitor were discovered, highlighting the expanding understanding of this cellular process (37).
Molecular mechanisms of ferroptosis
In 1984, J.M. Gutteridge discovered that iron salts can initiate lipid peroxidation by converting lipid peroxides into alkoxyl and peroxyl radicals (38). He also found that iron, when combined with ethylene diamine tetraacetic acid (EDTA), can start lipid peroxidation by reacting with hydrogen peroxide (H2O2) to create hydroxyl radicals (•OH) (38). This research laid the groundwork for understanding iron-dependent cell death. Additionally, it was observed that external glutamate can trigger cell death by inhibiting cystine uptake via xCT, leading to reduced glutathione production. This process, known as oxytosis, is reliant on oxidative stress and reactive oxygen species (ROS) production and set the stage for the concept of ferroptosis.
In 2012, Brent R. Stockwell defined ferroptosis as a new form of programmed cell death, specifically emphasizing its role in iron-dependent cell death in cancer cells. The molecular mechanisms of ferroptosis are distinct from other well-known forms of regulated cell death (RCD). Key biochemical events in ferroptosis include the accumulation of excess iron and ROS in cells, extensive lipid peroxidation, inactivation of xCT, and the depletion of glutathione and lipid repair enzymes. This understanding marks a significant advancement in the study of cell death and its implications in diseases like cancer (39–41).
Iron metabolism
Iron plays a crucial role in regulating ferroptosis, not only by triggering the non-enzymatic Fenton reaction that directly oxidizes polyunsaturated fatty acid-phospholipids (PUFA-PLs), but also by serving as a vital cofactor for enzymes involved in lipid peroxidation, such as ALOX and POR (42–45). Under normal circumstances, cells maintain a relatively stable level of labile iron through a finely tuned balance of iron absorption, use, storage, and export. However, disruptions in these iron metabolism processes can either accelerate or inhibit ferroptosis, depending on whether they increase or decrease the intracellular labile iron pool. For instance, most intracellular iron is stored as inert iron within ferritin. The autophagic breakdown of ferritin, known as ferritinophagy, releases iron from ferritin into the labile iron pool. Blocking NCOA4-mediated ferritinophagy lowers the labile iron pool and thus inhibits ferroptosis (46, 47). On the other hand, promoting ferritinophagy, such as by inhibiting cytosolic glutamate oxaloacetate transaminase 1 (GOT1), increases the labile iron pool and encourages ferroptosis. The exact mechanism by which GOT1 inhibition facilitates the release of labile iron through ferritinophagy is still under investigation (48–50). For a more comprehensive discussion on iron metabolism in the context of ferroptosis, interested readers are directed to recent detailed reviews on the subject (51).
Iron absorption and metabolism in the body can be summarized as following. Firstly, iron is a crucial trace element necessary for various biological functions in cells. Dietary iron is mainly categorized into non-heme and heme iron, with the latter being Fe2+ combined with protoporphyrin IX. Non-heme iron primarily exists as ferric salts in the diet, which are converted to Fe2+ by intestinal iron reductase. This Fe2+ is then absorbed by intestinal epithelial cells (IECs) through the DMT1 transporter and exits these cells via ferroportin 1 (FPN1) (52, 53). Once in the bloodstream, Fe2+ is oxidized to Fe3+ by ceruloplasmin (CP) and hephaestin (HP), and then binds to transferrin (Tf) for transport (54). Tf-Fe3+ attaches to cell membranes via the transferrin receptor (TfR), internalizes into cells as endosomes, and is reduced back to Fe2+ by STEAP3 in various cells before entering the cytoplasm through DMT1 on the endosomal membrane (55) (see Figure 3).
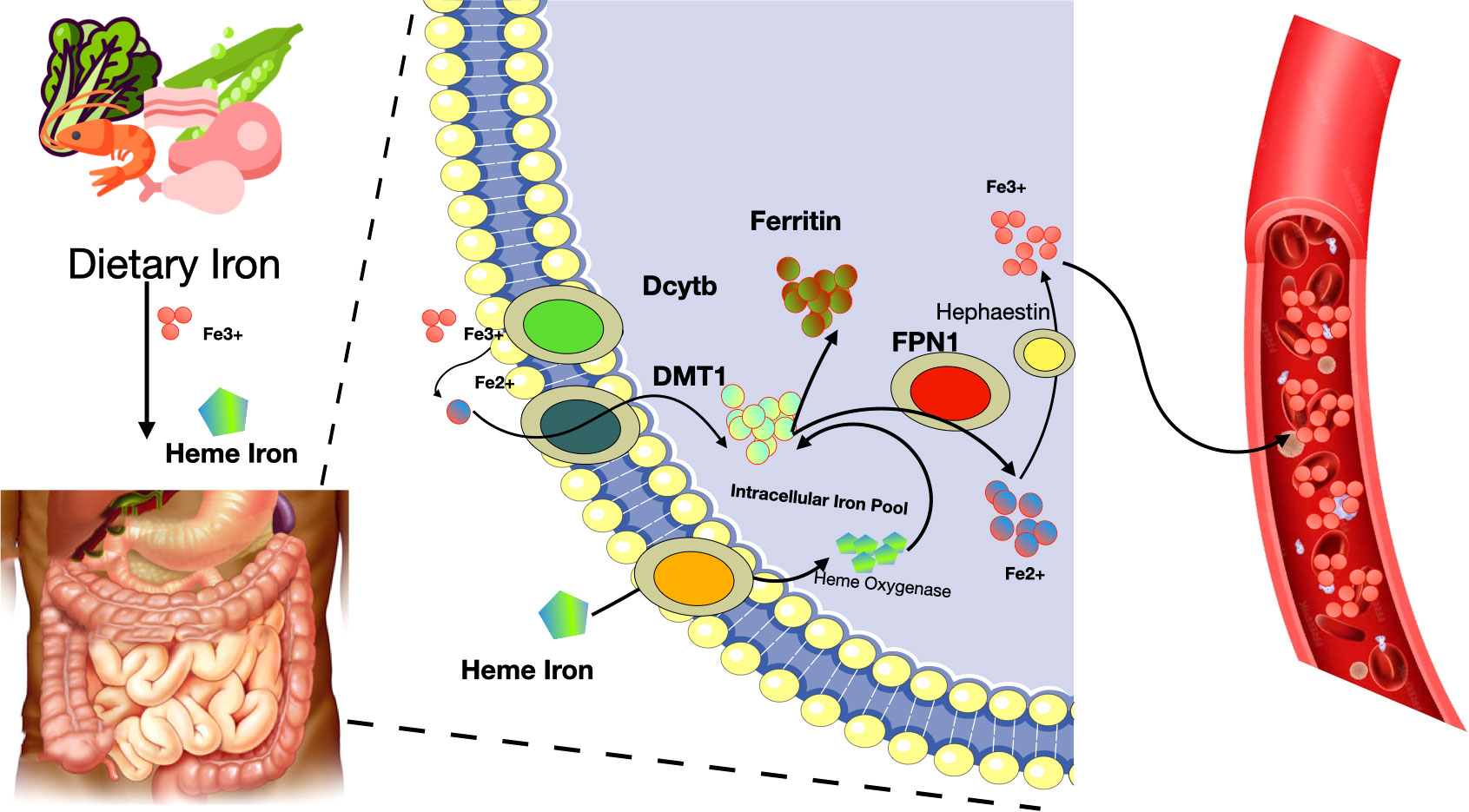
Figure 3 Iron absorption and metabolism in the body. Fe2+, ferrous cation; Fe3+, ferric cation; Dcytb, duodenal cytochrome b; DMT1, divalent metal transporter 1; FPN1, ferroportin 1.
Heme iron, a component of hemoglobin and myoglobin, has a less clear absorption mechanism. It’s believed that heme iron ingested is broken down by heme oxygenase in intestinal cells, releasing free ferric iron. Subsequently, a significant amount of Fe2+ accumulates in the cytoplasm, forming a labile iron pool crucial for processes like ferroptosis (56, 57). Excessive intracellular iron, in the presence of H2O2, triggers the Fenton reaction, leading to ROS formation such as •OH, which induces lipid peroxidation and hence ferroptosis (58–60). Iron responsive element binding protein 2 (IREB2) is an important regulator of iron metabolism and may influence sensitivity to ferroptosis (61–63). Autophagy also plays a role in iron regulation, affecting the recruitment of ferritin to autophagosomes for lysosomal degradation, thus releasing free iron (64–66). For example, ferritinophagy, facilitated by the cargo receptor NCOA4, specifically targets the ferritin heavy chain 1 (FTH1), releasing iron into autophagosomes for degradation (67). Conversely, a decrease in intracellular Fe2+ levels can hinder ferroptosis, as seen when erastin-induced ferroptosis is reduced following the knockout of autophagy-related genes ATG5 or ATG7 (68, 69). Therefore, iron metabolism is integral to the process of ferroptosis.
Lipid metabolism
Lipid peroxidation stands out as a key characteristic and the primary mechanism that drives ferroptosis. Reactive oxygen species (ROS) generated through the Fenton reaction interact with polyunsaturated fatty acids (PUFAs) in cell or organelle membranes, resulting in the formation of toxic phospholipid hydroperoxides (PLOOHs) and triggering ferroptosis (70–72). Various factors contribute to the process of lipid peroxidation, including enzymes such as acyl–coenzyme A synthetase long-chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3), and lipoxygenases (LOXs) (73–75). ACSL4, vital for lipid metabolism, and LPCAT3, key in catalyzing the reacylation of lysophospholipids to phospholipids, play crucial roles (76–78). They activate free long-chain PUFAs, facilitate the conversion of lysophosphatidylcholine (LPC) to lecithin, mediate the synthesis of oxidized phospholipids in cell membranes, and thus influence ferroptosis. ACSL4 specifically esterifies arachidonic acid (AA) into acyl-coenzyme A (acyl-CoA), essential for PUFA biosynthesis, which is pivotal in lipid peroxidation and ferroptosis. LOXs are critical regulators in this process. They significantly impact the onset of ferroptosis by promoting lipid autoxidation and are indicative of a cell’s sensitivity to ferroptosis (74, 79). Therefore, in the context of ferroptosis, lipid peroxidation leads to cell death by disrupting the lipid bilayer structure of cellular and organelle membranes (see Figure 4).
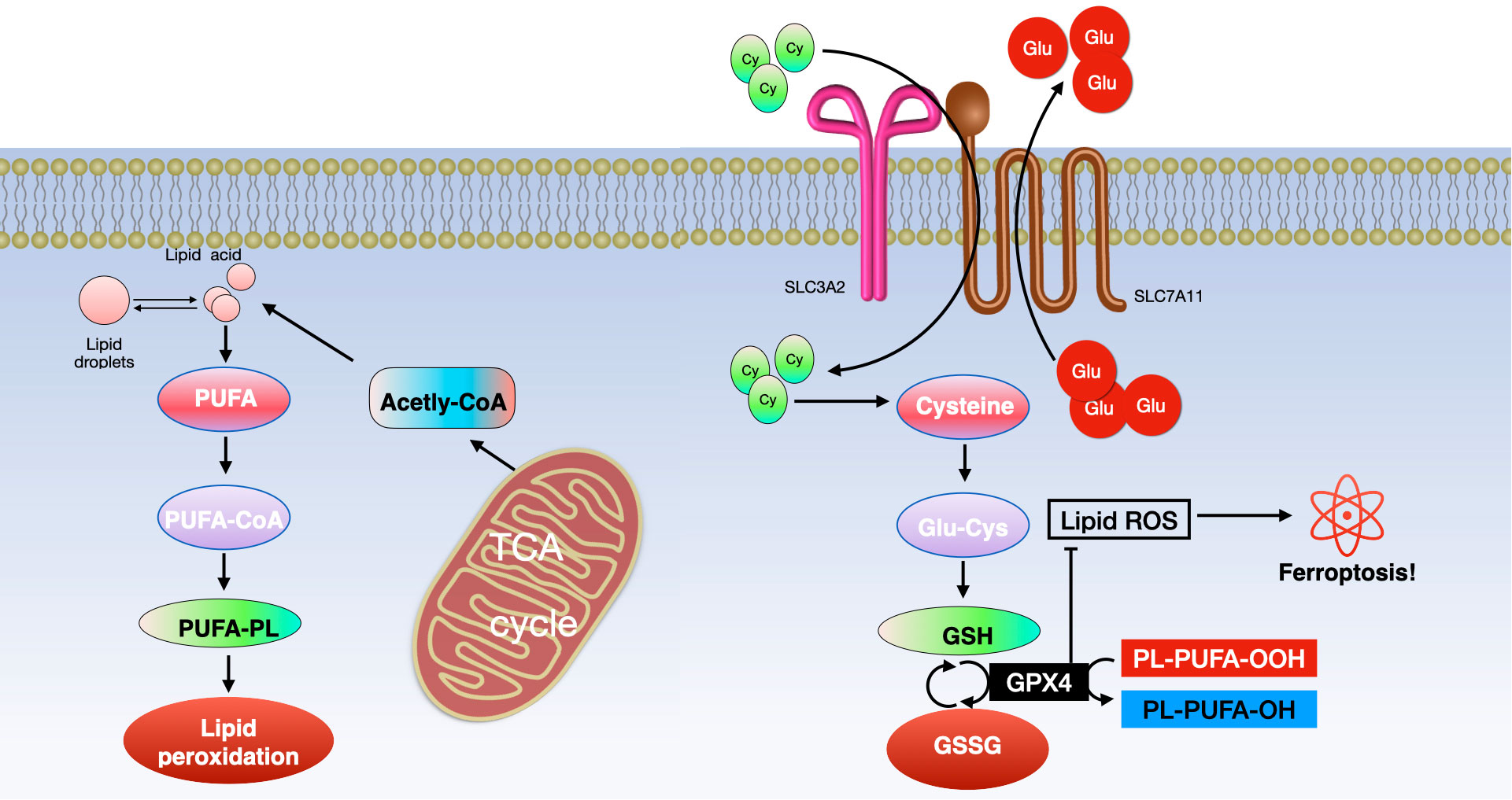
Figure 4 Ferroptosis involves two primary molecular pathways. (Left) The pathway of lipid metabolism and (Right) the xCT/GPX4 pathway. Key components include ACSL4 (long-chain fatty acid CoA ligase 4), PUFA (polyunsaturated fatty acid), LPCAT3 (lysophosphatidylcholine acyltransferase 3), PUFA-CoA (polyunsaturated fatty acid coenzyme A), ALOXs (lipoxygenases), PUFA-PL (phospholipid containing polyunsaturated fatty acid), TCA (tricarboxylic acid cycle), GSH (glutathione), GSSG (glutathione disulfide), and GPX4 (glutathione peroxidase 4).
Mitochondrial metabolism
Mitochondrial metabolic processes play a crucial role in initiating ferroptosis (80). One key aspect is the generation of mitochondrial reactive oxygen species (ROS), essential for lipid peroxidation and the onset of ferroptosis. Mitochondria are significant cellular sources of ROS, where electron leakage from complexes I and III of the electron transport chain leads to the formation of superoxides. These superoxides are then converted into hydrogen peroxide (H2O2) by superoxide dismutase (81). The H2O2 reacts with labile iron through the Fenton reaction to produce hydroxyl radicals, which in turn drive the peroxidation of polyunsaturated fatty acid-phospholipids (PUFA-PLs) (81, 82). Additionally, mitochondrial electron transport and proton pumping, crucial for ATP production, also play a role in promoting ferroptosis (83–86). Under conditions where ATP is depleted, AMP-activated protein kinase (AMPK) phosphorylates and inhibits acetyl-CoA carboxylase (ACC), leading to a suppression of PUFA-PL synthesis and thus blocking ferroptosis. Conversely, when ATP levels are sufficient, AMPK activation is inefficient, resulting in the activation of ACC and promotion of PUFA-PL synthesis and ferroptosis (85, 86). Furthermore, mitochondria’s role in biosynthetic pathways, such as the tricarboxylic acid (TCA) cycle and anaplerotic reactions (like glutaminolysis), contributes to ferroptosis (83). These processes likely drive ferroptosis through the promotion of ROS, ATP, and/or PUFA-PL generation (87–89). Hence, the multifaceted functions of mitochondria in bioenergetics, biosynthesis, and ROS generation are central to driving mitochondrial lipid peroxidation and ferroptosis (80).
The xCT and GPX4
Environmental stresses like high temperature and hypoxia can trigger iron reactions, necessitating cells to establish defense mechanisms against ferroptosis. The most classic defense against ferroptosis involves the antioxidant axis consisting of xCT, glutathione (GSH), and GPX4. xCT, a transmembrane protein, comprises the light-chain solute carrier family 7 member 11 (SLC7A11) and the heavy-chain solute carrier family 3 member 2 (SLC3A2, also known as CD98hc or 4F2hc) (90, 91). SLC7A11, the primary functional unit of xCT, regulates the intake of extracellular cysteine (Cys) into cells and the export of intracellular glutamic acid (Glu). SLC3A2 helps maintain xCT’s stability by anchoring and stabilizing SLC7A11 (92).
Cysteine is converted into reduced GSH alongside Glu and glycine (Gly) under the influence of glutamate cysteine ligase (GCL) and glutamylcysteine synthetase (GCS) (93, 94). Beclin 1 can suppress xCT activity, thereby promoting ferroptosis, by directly binding to SLC7A11 (95). GPX4, a crucial enzyme in preventing ferroptosis, reduces toxic phospholipid hydroperoxides (PLOOH) to non-toxic phospholipid alcohols (PLOH) in membranes, using GSH (96). The inhibitor 2-cyano-3,12-dioxooleana-1,9 (11)-dien-28-oic acid (CDDO) helps prevent the specific degradation of GPX4 through chaperone-mediated autophagy (CMA). It does so by affecting the interaction between heat shock protein 90 (HSP90) and lysosomes, thus inhibiting cell ferroptosis (13). However, this inhibition can be counteracted by suppressing the mammalian target of rapamycin (MTOR) pathway, which may lead to GPX4 degradation and promote ferroptosis (97). Consequently, xCT, GPX4, and GSH are key regulators and have a significant impact on the control of ferroptosis (see Figure 4).
FSP1-CoQH2 system
The FSP1-CoQH2 system has challenged the earlier belief that GPX4 was the sole defense mechanism against ferroptosis. Recent studies have shown that ferroptosis suppressor protein 1 (FSP1, also known as AIFM2) works independently of GPX4 to protect against ferroptosis (98, 99). FSP1, located on the plasma membrane and other subcellular compartments, is crucial in this role. Its presence on the plasma membrane is both necessary and sufficient for its function in ferroptosis suppression (98, 99). FSP1 acts as an NAD(P)H-dependent oxidoreductase, reducing ubiquinone (also known as coenzyme Q or CoQ) to ubiquinol (CoQH2). CoQH2, apart from its known role in mitochondrial electron transport, can intercept lipid peroxyl radicals, thus inhibiting lipid peroxidation and ferroptosis (98, 99). The generation of a non-mitochondrial CoQH2 pool by FSP1 as radical-trapping antioxidants plays a key role in its anti-ferroptosis activity (98, 99). Though CoQ is mainly produced in mitochondria, it is also found in non-mitochondrial membranes, including the plasma membrane (100–102). The exact sources of non-mitochondrial CoQ used by FSP1 in ferroptosis defense are yet to be determined.
DHODH-CoQH2 system
Additionally, the DHODH-CoQH2 system has been identified as another mitochondria-localized defense mechanism. This system, mediated by dihydroorotate dehydrogenase (DHODH), can compensate for the loss of GPX4 in detoxifying mitochondrial lipid peroxidation (36). DHODH, an enzyme in pyrimidine synthesis, reduces CoQ to CoQH2 in the inner mitochondrial membrane (36). When GPX4 is acutely inactivated, DHODH activity increases, leading to more CoQH2 production which neutralizes lipid peroxidation and protects against mitochondrial ferroptosis. Inactivating both mitochondrial GPX4 and DHODH leads to significant mitochondrial lipid peroxidation and ferroptosis (36). Notably, while mitochondrial GPX4 and DHODH can substitute for each other in reducing mitochondrial lipid peroxidation, cytosolic GPX4 and FSP1 cannot, likely due to their non-mitochondrial localization.
These findings suggest a model where ferroptosis defense systems are divided into two main categories: the GPX4 system and the CoQH2 system, each further divided into non-mitochondrial and mitochondrial components. For instance, cytosolic and mitochondrial GPX4 are part of the GPX4 system, while non-mitochondrial FSP1 and mitochondrial DHODH belong to the CoQH2 system. This division is presumably due to the need to mitigate lipid peroxides in mitochondria and the unique double-membrane structure of mitochondria, which limits the effectiveness of defense systems located in other compartments (103). However, further research is required to fully understand this compartmentalization model in ferroptosis regulation and reconcile it with some conflicting findings, such as the significant localization of cytosolic GPX4 in the intermembrane space of mitochondria (104). The role of cytosolic GPX4 in mitochondria and its potential impact on suppressing mitochondrial lipid peroxidation needs further investigation.
GCH1–BH4 system
Recent research has highlighted the role of GTP cyclohydrolase 1 (GCH1) as a key player in regulating ferroptosis (105, 106). GCH1 is involved in the rate-limiting step of the biosynthesis pathway for tetrahydrobiopterin (BH4), a cofactor for aromatic amino acid hydroxylases and other enzymes (107). BH4, aside from its role as a cofactor, is a radical-trapping antioxidant capable of capturing lipid peroxyl radicals, thus contributing to the inhibition of ferroptosis independently of its cofactor function (106). It is suggested that GCH1 helps prevent ferroptosis in two ways: firstly, through the generation of BH4 as a radical-trapping antioxidant, and secondly, via the GCH1-mediated production of coenzyme QH2 (CoQH2) and phospholipids (PLs) containing two polyunsaturated fatty acid (PUFA) tails. This is notable because most PUFA-PLs typically exhibit an asymmetric structure, with a PUFA tail at the sn-2 position and a saturated fatty acid tail at the sn-1 position (105, 106). The specific subcellular location where the GCH1-BH4 system functions, however, is still to be determined. This discovery adds another layer to the complex regulatory network of ferroptosis and underscores the multifaceted nature of cellular defense mechanisms against this form of cell death.
Ferroptosis in the tumor microenvironment
Recent studies have revealed the significant role of the tumor microenvironment (TME), particularly its immune cells, in determining the occurrence of tumor-cell ferroptosis (108). CD8+ cytotoxic T cells, key players in antitumor immunity within the TME, release interferon-γ (IFNγ), which inhibits cystine uptake in cancer cells by downregulating SLC7A11 expression (109). This action enhances lipid peroxidation and ferroptosis in tumors. Intriguingly, IFNγ also suppresses SLC7A11-mediated cystine transport in macrophages, indicating that IFNγ can regulate SLC7A11 in both cancer and non-cancer contexts. Furthermore, combining immune checkpoint inhibitors (ICIs) with cyst(e)inase intensifies T cell-mediated antitumor responses by synergistically promoting tumor ferroptosis. This suggests that ferroptosis plays a crucial role in T cell-mediated antitumor activity, and targeting SLC7A11 in conjunction with ICIs could be a promising cancer treatment strategy (108).
Ferroptotic cancer cells release various immunostimulatory signals like high mobility group box 1 (HMGB1), calreticulin, ATP, and phosphatidylethanolamine (110–112). These signals aid dendritic cell maturation, enhance macrophage phagocytosis of ferroptotic cancer cells, and boost the infiltration of CD8+ T cells into tumors. Early ferroptotic cells, after short-term treatment with a GPX4 inhibitor, can induce dendritic cell maturation and activate antitumor immunity, similar to a vaccination effect (111). This supports the notion of ferroptosis as a form of immunogenic cell death.
Ferroptosis induction in certain immunosuppressive cells also enhances antitumor immunity. Regulatory T (Treg) cells, which usually suppress tumor surveillance, are resistant to ferroptosis due to GPX4 induction (113, 114). However, inducing ferroptosis in Treg cells through Gpx4 deletion contributes to antitumor immunity. Similarly, targeting myeloid-derived suppressor cells (MDSCs) for ferroptosis, by inhibiting ASAH2-mediated suppression of the p53–haeme oxygenase 1 (HMOX1) axis, activates tumor-infiltrating cytotoxic CD8+ T cells and suppresses tumors (114, 115). Tumor-associated macrophages (TAMs) that exhibit an M2-like phenotype and suppress antitumor immunity are more vulnerable to ferroptosis induced by GPX4 inhibition than M1-like TAMs, which promote antitumor immunity (116). Inducing ferroptosis in M2-like TAMs without affecting M1-like TAMs could overcome the immunosuppressive TME and enhance cancer immunotherapy (117, 118) (see Figure 5).
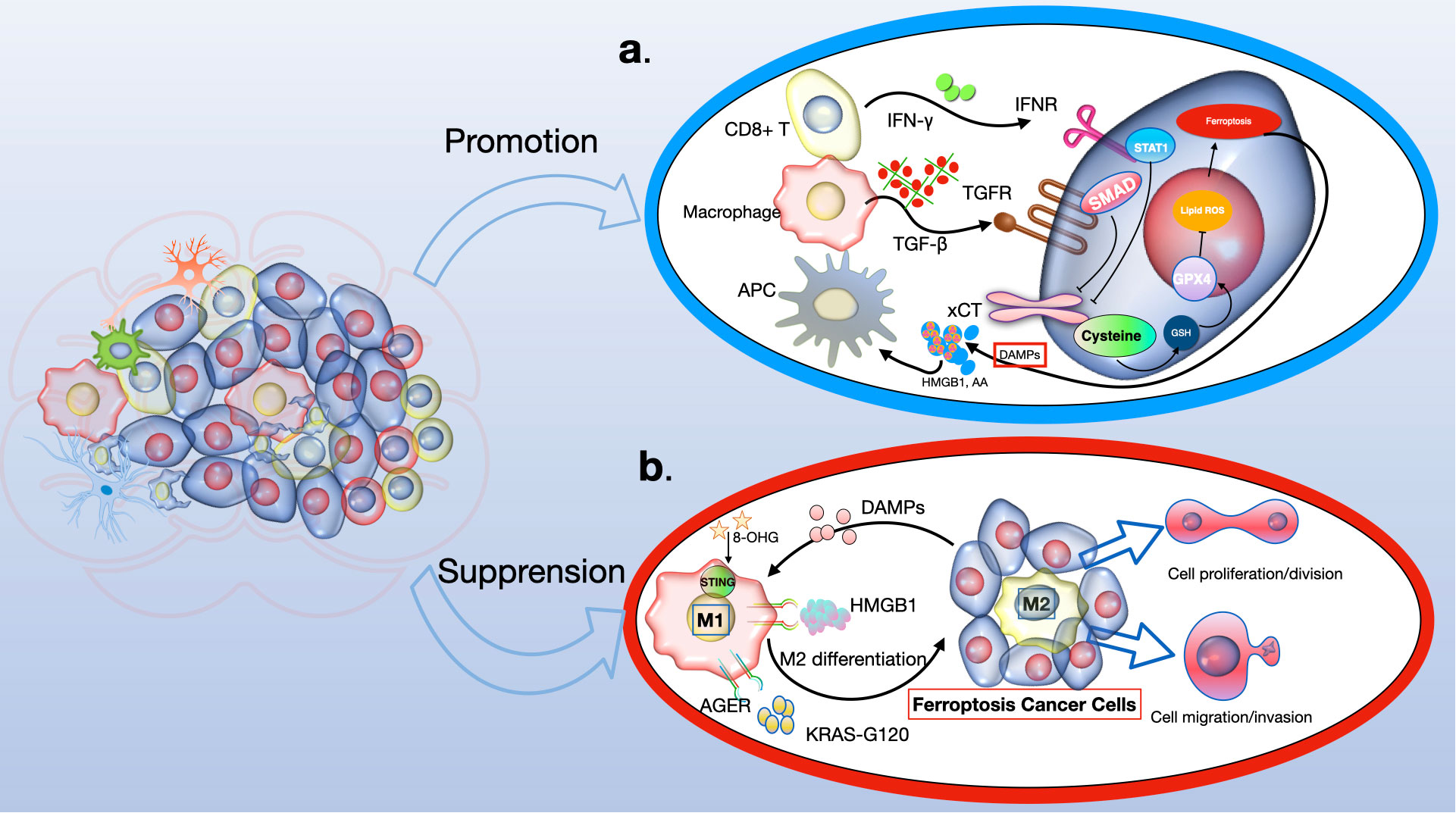
Figure 5 The role of ferroptosis in anti-tumour immunity. Ferroptosis has a dual role to play in antitumour immunity, dependent on the nature of the immune cell. (A) In the context of tumor immunity, CD8+ T cells secrete IFNγ, which activates the INFR pathway, leading to the suppression of SLC7A11 expression via STAT1. This process enhances ferroptosis in cancer cells. Additionally, macrophage-derived TGFβ1 suppresses the xCT system through SMAD proteins, further inducing lipid ROS-dependent ferroptosis through the GSH-GPX4 pathway. Consequently, ferroptotic glioma cells emit DAMPs (like HMGB1 and AA), which encourage the attraction and activation of immune cells. (B) Conversely, DAMPs such as HMGB1, KRAS-G12D, and 8-OHG influence the behavior of macrophages within the tumor milieu. Specifically, KRAS-G12D interacts with AGER on macrophage surfaces, leading to M2 macrophage polarization. This could potentially impede the effectiveness of the immune system's response against the tumor. IFN-γ, interferon-γ; INFR, interferon receptor; STAT1, signal transducer and activator of transcription 1; GSH, glutathione; GPX4, glutathione peroxidase 4; DAMPs, damage-associated molecular patterns; HMGB1, high mobility group protein B1; AA, arachidonic acid; 8-OHG, 8-hydroxyguanosine; AGER, advanced glycosylation end product-specific receptor.
However, evidence also suggests that ferroptosis can promote tumor growth in the context of tumor immunity. Gpx4-deficient T cells from specific knockout mice rapidly accumulate lipid peroxides upon activation, leading to ferroptosis (119). CD8+ T cells from tumors, but not from lymph nodes, show substantial lipid peroxide accumulation, indicating a vulnerability to ferroptosis. This could weaken antitumor immunity and aid tumor growth (120). CD36, which mediates fatty acid uptake in tumor-infiltrating CD8+ T cells, induces lipid peroxidation and ferroptosis, compromising antitumor immunity. Blocking CD36 or ferroptosis in CD8+ T cells restores their antitumor activity, while treating them with GPX4 inhibitors leads to impaired antitumor effects (121). Furthermore, T follicular helper (TFH) cells, a CD4+ T cell subset favoring antitumor immunity, are highly susceptible to ferroptosis, with GPX4 being crucial for their survival and function (122). The synergistic enhancement of antitumor immunity by targeting SLC7A11 along with ICIs may be due to the limited effect of Slc7a11 knockout or cystine deprivation on T cell viability or antitumor effects, possibly because of SLC7A11’s low expression and non-essential role in T cells (123). The contrasting effects of GPX4 deletion versus SLC7A11 deletion on T cell function are not fully understood but might be related to SLC7A11’s limited role in these cells (124, 125).
Therapeutic strategies targeting ferroptosis
Small molecules to induce ferroptosis in cancer
System Xc− plays a significant role in the survival and growth of many tumor cells, making it a potential target for cancer treatment. Erastin, known for inhibiting system Xc−, reduces glutathione (GSH) levels and induces ferroptosis. A notable discovery in Dixon’s 2012 study was that erastin led to ROS accumulation in NRAS-mutant HT-1080 fibrosarcoma cells, and this cell death was hindered by the iron chelator deferoxamine, indicating erastin’s role in inducing ferroptosis (4, 96, 126, 127). Further research highlighted the importance of the RAF/MEK/ERK signaling pathway in erastin-triggered ferroptosis in RAS-mutated cancers (128). Derivatives of erastin, like piperazine erastin and imidazole ketone erastin (IKE), have been developed to address erastin’s limitations, such as poor water solubility and unstable in-vivo metabolism. For instance, IKE has been effectively used in treating diffuse large B cell lymphoma (DLBCL) in the SUDHL6 xenograft animal model (129).
Sorafenib, a multi-kinase inhibitor used in treating advanced renal cell carcinoma, thyroid carcinoma, and hepatocellular carcinoma, is another inducer of ferroptosis. Its cytotoxicity to hepatocellular carcinoma was negated when treated with an iron chelator (130). However, some cancer cell lines have developed resistance to sorafenib, as seen in hepatocellular carcinoma cells with retinoblastoma (Rb) protein, where sorafenib-induced ferroptosis was inhibited (131). Additionally, the anti-inflammatory drug sulfasalazine (SAS) can induce ferroptosis in glioma cells by inhibiting system Xc− (132).
Some cancer cells induce ferroptosis through the transsulfuration pathway instead of system Xc−. GPX4 inactivation can eliminate these tumor cells, as seen with (1S, 3R)-RSL, which induces ferroptosis through direct GPX4 inhibition, and FIN56, which promotes ferroptosis by degrading GPX4 (133). Ferroptosis can also be induced by increasing the labile iron pool (LIP). Compounds like BAY 11-7085 can induce ferroptosis through the Nrf2-SLC7A11-HO-1 pathway, and overexpression of heme oxygenase-1 (HO-1), encoded by HMOX1, has been observed in MDA-MB-231 breast cancer and DBTRG-05MG glioblastoma cells (134). Increased TF expression and decreased FPN-1 expression, mediated by compounds like siramesine and lapatinib, can also induce ferroptosis. Autophagy contributes to this process by degrading ferritin in cancer cells. The cargo receptor nuclear receptor coactivator 4 (NCOA4) is significant in the autophagic turnover of ferritin in ferroptosis. In pancreatic cancer cells, NCOA4 overexpression inhibited FIH1 expression and promoted erastin-induced ferroptosis (47). Further research is necessary to discover novel molecules targeting NCOA4 for cancer treatment through ferroptosis. Besides these, more small molecules are being studied for their potential to induce ferroptosis in cancer therapy (see Figure 6).
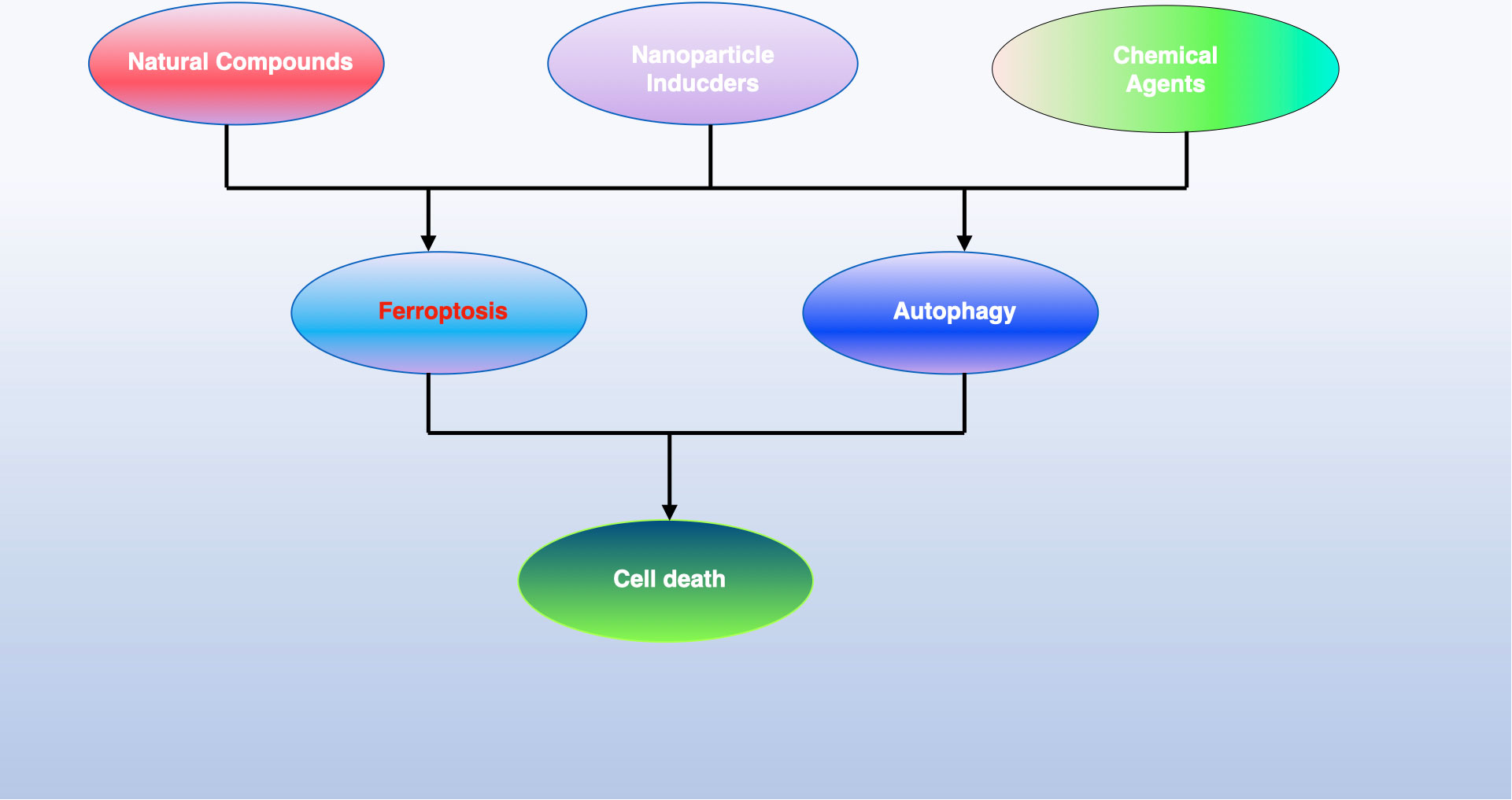
Figure 6 The iron maiden. Autophagy is an essential, ancient mechanism that preserves balance within cells by eliminating harmful components. It serves a dual function in determining cell fate, being implicated in both survival and death processes. Recent findings have shown that autophagy contributes to the initiation of ferroptosis, an iron-dependent form of cell death characterized by the build-up of toxic lipid peroxides. This review sheds light on the interaction between autophagy and ferroptosis and proposes that understanding this relationship could open new avenues for cancer treatment strategies.
Nanoparticle inducers of ferroptosis in cancer
Nanotechnology applications, particularly due to their unique physicochemical properties, have garnered significant interest in recent years. Many nanomaterials, including iron-containing nanoparticles, leverage the Fenton reaction for their functionality. For instance, Chen and colleagues developed a tumor-targeted nanoparticle called α‐enolase targeting peptide modified Pt-prodrug loaded Fe3O4 nanoparticles (ETP-PtFeNP). Treatment of tumor cells with ETP-PtFeNP resulted in increased ROS generation, enhanced immunogenicity, and a robust anti-tumor immune response (135). Additionally, a novel nanoparticle named SRF@FeIIITA (SFT) has been shown to be effective in inhibiting tumor progression. This nanoparticle combines photodynamic therapy (PDT) and ferroptosis by loading methylene blue (MB) into SFT. This is achieved by depositing tannic acid (TA) and Fe3+ onto SRF nanocrystals, enabling a dual-therapy approach (136).
Nanomaterials can also trigger ferroptosis through the manipulation of GSH metabolism. Arginine-capped manganese silicate nanobubbles (AMSNs), for example, have been developed to efficiently deplete GSH (137). This efficiency is attributed to their high surface area to volume ratio. In-vivo studies have demonstrated that AMSN treatment can suppress the growth of Huh7 xenograft tumors by downregulating GPX4. The effectiveness of AMSN-induced ferroptosis can be inhibited by the ferroptosis inhibitor liproxstatin-1, highlighting the potential of these nanomaterials in targeted cancer therapy through the induction of ferroptosis.
Ferroptosis modulation for tumor sensitization to anticancer therapies
Drug resistance presents a significant hurdle in chemotherapy treatment, but the use of ferroptosis inducers might offer a way to surmount this challenge. Incorporating ferroptosis agonists with chemotherapy drugs could emerge as an innovative approach to cancer therapy (137). Persister cells, which are cancer cells that survive after multiple rounds of chemotherapy, exhibit downregulated Nrf2-targeted genes (138). Accelerating ferroptosis in these cells can be achieved by inhibiting intracellular NF2 and the hippo signaling pathway (139). Moreover, persister cells typically show reduced levels of glutathione (GSH) and nicotinamide adenine dinucleotide phosphate (NADPH), making them more susceptible to lipid peroxidation. GPX4 inhibitors have been found to be particularly effective against these cells. Therefore, inducing ferroptosis in persister cells may be a promising strategy to overcome their drug resistance, offering a new avenue in the fight against cancer.
Ferroptosis-associated antitumor combination therapy
There have been limited studies on combining ferroptosis inducers with other anti-tumor therapies for clinical treatment, with many investigations still in the experimental phase. An important aspect in cancer cells, especially when the oxygen concentration is above 3–8% as found in most tissues, is the iron-sulfur cluster biosynthetic enzyme NFS1. Its high expression is often observed in well-differentiated adenocarcinomas. Research by Alvarez SW’s group suggested that inhibiting NFS1, along with suppressing cysteine transport, could induce ferroptosis in tumor cells (140). Interestingly, some tumors that are resistant to certain chemotherapy drugs show a heightened sensitivity to ferroptosis inducers. For instance, pancreatic cancer cells, known for their resistance to chemotherapy-induced apoptosis, demonstrate considerable sensitivity to artemisinin-induced ferroptosis. Thus, ferroptosis inducers emerge as a promising strategy for cancer therapies, particularly for types of cancer that are resistant to conventional treatments.
Targeting ferroptosis to prevent tumor metastasis
In terms of preventing tumor metastasis, targeting ferroptosis could be a potential approach. Clinical treatment of tumor metastasis is complex due to factors like tumor heterogeneity, oncogene activity, epithelial-mesenchymal transition (EMT), and the microenvironment of metastatic sites (141). Since cancer metastasis can be inhibited by high intracellular oxidative stress, ferroptosis, which involves the accumulation of such stress, may be an effective strategy. Nanoparticles present a substantial advantage in treating cancer metastasis due to their relatively low risk compared to locally injected agents (142). For example, a metal–organic network encapsulating the p53 plasmid (MON-p53) was developed using coordination between ferric iron (Fe3+) and tannic acid (TA). Treatment with MON-p53 showed the potential to suppress cancer cell migration in in-vitro wound healing assays, suggesting its possible role in inhibiting tumor metastasis (143, 144). Moreover, mesenchymal cancer cells, known for their metastatic potential and resistance to anti-cancer treatments, could be rendered more sensitive to chemotherapy and thus reduce metastasis. Antagonizing the NF2-YAP pathway, which promotes ferroptosis by up-regulating modulators like ACSL4 and transferrin receptor (TFRC), offers a new perspective on treating mesenchymal or metastatic cancer cells, which are highly sensitive to ferroptosis (139).
Challenges of ferroptosis in glioma
Ferroptosis plays a critical role in the growth and treatment of gliomas, but its mechanisms require further exploration (145). Unlike most regulated cell death (RCD) effector molecules, which are typically proteases or porins such as caspases in apoptosis and mixed lineage kinase domain-like protein (MLKL) in necroptosis, the effector molecules in ferroptosis are less clear (146–148). Phospholipid hydroperoxides (PLOOH) are currently considered the primary executors of ferroptosis, yet it’s uncertain if there are additional effector molecules downstream of PLOOH (103, 149). The interaction between ferroptosis and other RCD forms is also not fully understood. Some aspects of ferroptosis, like lipid peroxidation and regulators including GPX4 and SLC7A11, are also involved in other RCD types. Ferroptosis may influence the tumor immune microenvironment (TIME), affecting glioma development and treatment (see Figure 7).
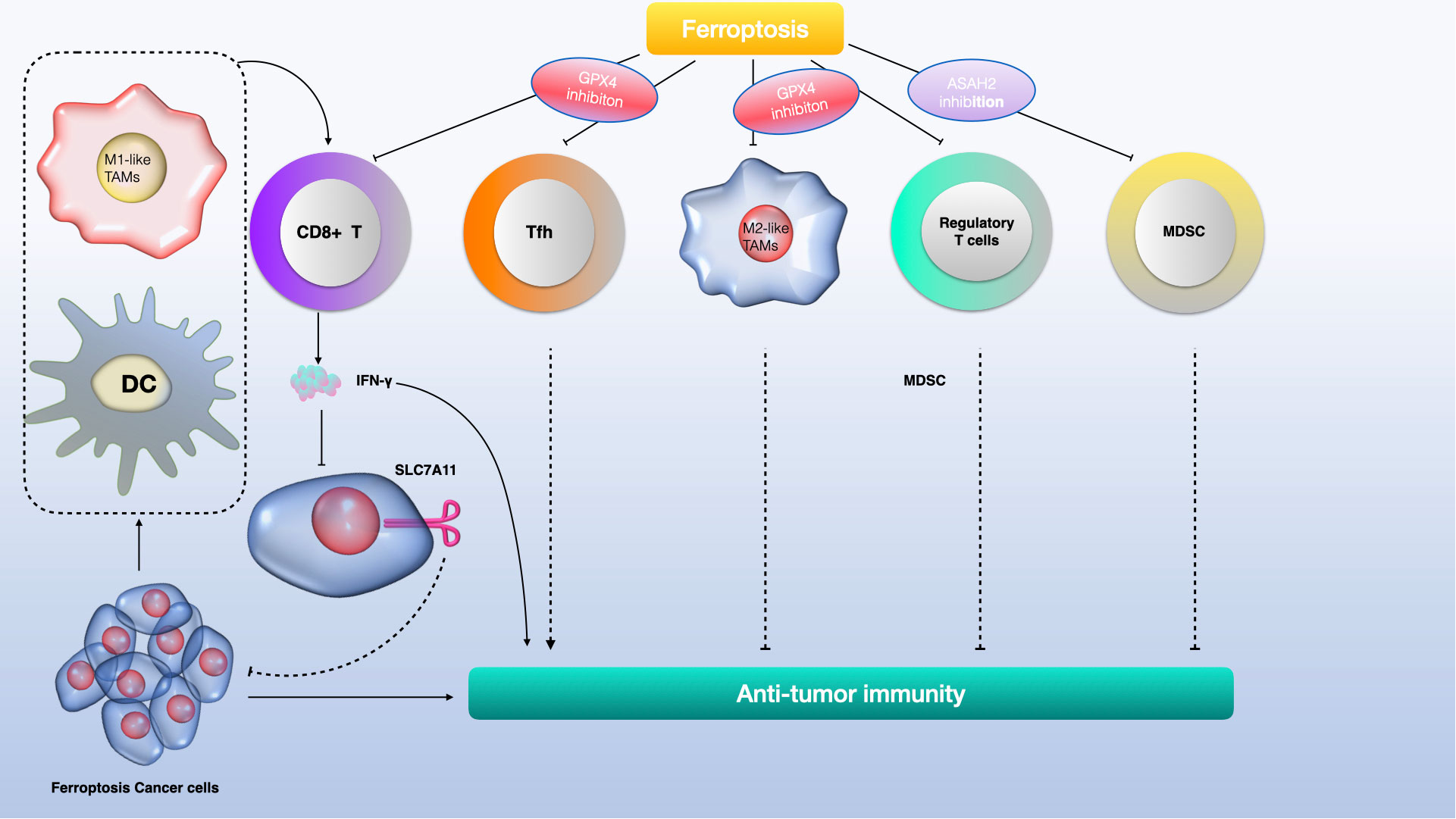
Figure 7 The role of ferroptosis in glioma immunity involves a complex interaction of immune cells and molecular pathways. CD8+ T cells produce interferon-gamma (IFNγ), activating the interferon receptor (INFR) which then suppresses SLC7A11 transcription through STAT1, promoting ferroptosis in tumor cells. Macrophages release TGFβ1, which via SMAD proteins, downregulates system xCT, leading to lipid ROS-induced ferroptosis through the glutathione (GSH) and glutathione peroxidase 4 (GPX4) axis. Ferroptotic glioma cells release damage-associated molecular patterns (DAMPs) like HMGB1 and arachidonic acid (AA), which attract and activate immune cells. Conversely, DAMPs like HMGB1, KRAS-G12D, and 8-hydroxyguanosine (8-OHG) can alter macrophage function in the tumor microenvironment. Notably, KRAS-G12D interaction with the AGER receptor on macrophages promotes M2 macrophage polarization, potentially hindering antitumor immunity.
Currently, biomarkers like GPX4, ACSL4, P53, and FTH1 are used in diagnosing and treating gliomas, but they are not definitive standards (35, 150). Reliable biomarkers that can accurately predict tumor response to ferroptosis induction, especially those detectable in-patient blood, urine, feces, and tumor tissue, are urgently needed. It’s also unclear which glioma patients are more susceptible to ferroptosis treatments. Assessments combining iron levels, gene expression, and mutations might help identify patients who would benefit most from ferroptosis, such as those with gliomas overexpressing SLC7A11 being potential targets for SLC7A11 inhibitors.
Ferroptosis has a dual role in tumor development and treatment. While it promotes glioma cell death, it can also diminish the efficacy of glioma treatments by increasing Treg cells, neutrophils, and M2-polarized macrophages in the TIME, thus suppressing antitumor immunity (151). Tumor cells undergoing ferroptosis might induce stress in surrounding tumor cells, enabling them to avoid ferroptosis by secreting cytokines (149). The nature of substances released by tumor cells post-ferroptosis and their effects on the surrounding cells and TIME require further investigation (128, 152). More evidence is needed to confirm whether cytokines released after ferroptosis enable surrounding glioma cells to evade immune surveillance and regulate TIME (152, 153).
Discussion
Targeting ferroptosis open up exciting avenues in cancer, but there are still unsolved questions in this field. Firstly, iron plays multiple roles in ferroptosis, and its function extends beyond redox reactions. A widely accepted model suggests iron is involved in generating lipid reactive oxygen species (ROS), either through Fenton chemistry or via iron-dependent oxidases (154). Iron’s necessity in ferroptosis might also reflect its role as a cofactor for various metabolic enzymes involved in ROS generation, such as the LOX family of enzymes or prolyl 4-hydroxylase isoform 1 (PHD1) (155, 156).
Secondly, the exact molecular executor of ferroptosis is not entirely clear. The oxidative fragmentation of polyunsaturated fatty acids (PUFAs) and resulting membrane lipid damage might be sufficient to induce cell death, possibly through plasma membrane permeabilization and damage to intracellular organelle membranes. Alternatively, the fragmented products of oxidized PUFAs, such as the toxic 4-hydroxynonenal (4-HNE), might promote death by reacting with and inactivating essential cellular proteins. The detoxification of 4-HNE by aldo-keto reductase family 1, member C (AKR1C), and its regulation by NRF2, indicates that 4-HNE accumulation may be a key ferroptotic driver (157, 158). However, the possibility of a specific death-inducing protein or protein complex activated downstream of lipid-ROS accumulation cannot be ruled out, warranting further research (159).
Thirdly, identifying molecular markers for cells undergoing ferroptosis remains challenging. Current methods mainly rely on observing increased cellular ROS and the effectiveness of ferroptosis inhibitors or iron chelators in preventing cell death. The mRNA expression of prostaglandin E synthase 2 (PTGS2) and ChaC glutathione-specific gamma-glutamylcyclotransferase 1 (CHAC1) has been found to elevate in cells undergoing ferroptosis (35, 159). However, these markers are not practical for use in live cells or intact tissues. An increase in heme oxygenase-1 (HO-1) expression upon erastin-mediated ferroptosis induction has been observed, but the universality of this marker across different cells and initiation pathways needs further validation (160). Thus, there is an ongoing search for more reliable ferroptosis markers for in vivo studies.
Fourthly, regarding the evolutionary aspect of ferroptosis, it poses an interesting question. Considering that iron-dependent oxidative metabolism has been a crucial part of life for billions of years, ferroptosis might indeed be one of the most ancient forms of programmed cell death. This suggests that evolutionary forces might have driven organisms to utilize this ROS/iron-driven cell death process for their own benefit, an intriguing area for further research and exploration.
In addition, recent studies have identified cuproptosis as a newly identified form of programmed cell death, which plays vital roles in tumorigenesis. Some researchers have shown that ferroptosis might interact with either cuproptosis or necroptosis. For example, it was demonstrated by Lifeng F. et al. that ferroptosis inducers enhance copper-induced cell death through depleting intracellular glutathione in liver cancer cells (161). While both ferroptosis and cuproptosis processes involve metal ions and contribute to cell death, their exact interrelationship is still under investigation. The distinct mechanisms suggest potential therapeutic targets in diseases where dysregulation of cell death is a factor. However, it’s important to note that research in these areas, especially cuproptosis, is still evolving, and the full extent of their relationship and implications in human health and disease are not yet fully understood.
Ferroptosis, as a novel mode of programmed cell death, is distinct from other types of regulated cell death due to its iron-dependent lipid peroxidation accumulation. This review highlights the regulatory mechanisms of ferroptosis and its multifaceted roles in the development and progression of glioma. Ferroptosis not only triggers the death of glioma cells but also influences their growth, invasion, migration, and resistance.
However, challenges such as poor blood-brain barrier (BBB) penetration and potential compensatory mechanisms limit the effectiveness of ferroptosis agents in glioma therapy. To address these challenges, it has been proposed that nanoengineered systems could improve the targeted delivery of drugs, thereby enhancing the effectiveness of glioma treatments. Despite the promising advantages of ferroptosis in treating glioma, there is a need for multidisciplinary collaboration to further investigate its benefits and drawbacks. Such research is crucial for evaluating the potential value of targeting ferroptosis in clinical applications.
Author contributions
HW: Software, Writing – original draft. YL: Software, Supervision, Investigation, Writing – original draft. SC: Software, Writing – original draft. XL: Methodology, Writing – original draft. DT: Validation, Writing – review & editing. SL: Supervision, Writing – review & editing. HZ: Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Shandong Province Natural Science Foundation grants ZR2022QH372.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell (2003) 3:285–96. doi: 10.1016/S1535-6108(03)00050-3
2. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol (2008) 15:234–45. doi: 10.1016/j.chembiol.2008.02.010
3. Yagoda N, Von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature (2007) 447:864–8. doi: 10.1038/nature05859
4. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
5. Ahola S, Langer T. Ferroptosis in mitochondrial cardiomyopathy. Trends Cell Biol (2023) 5:S0962-8924(23)00125-3. doi: 10.1016/j.tcb.2023.06.002
6. Liu Y, Lu S, Wu L, Yang L, Yang L, Wang J. The diversified role of mitochondria in ferroptosis in cancer. Cell Death Dis (2023) 14:519. doi: 10.1038/s41419-023-06045-y
7. Chen Q, Wang Y, Yue T, Wei H, Li S, Dong B. Fluorescence imaging of intracellular glutathione levels in the endoplasmic reticulum to reveal the inhibition effect of rutin on ferroptosis. Anal Chem (2023) 95:1949–57. doi: 10.1021/acs.analchem.2c04209
8. Ichihara G, Katsumata Y, Sugiura Y, Matsuoka Y, Maeda R, Endo J, et al. MRP1-dependent extracellular release of glutathione induces cardiomyocyte ferroptosis after ischemia-reperfusion. Circ Res (2023) 133:861–76. doi: 10.1161/CIRCRESAHA.123.323517
9. Li J, Liu J, Zhou Z, Wu R, Chen X, Yu C, et al. Tumor-specific GPX4 degradation enhances ferroptosis-initiated antitumor immune response in mouse models of pancreatic cancer. Sci Transl Med (2023) 15:eadg3049. doi: 10.1126/scitranslmed.adg3049
10. Xue Q, Yan D, Chen X, Li X, Kang R, Klionsky DJ, et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy (2023) 19:1982–96. doi: 10.1080/15548627.2023.2165323
11. Endale HT, Tesfaye W, Mengstie TA. ROS induced lipid peroxidation and their role in ferroptosis. Front Cell Dev Biol (2023) 11:1226044. doi: 10.3389/fcell.2023.1226044
12. Wang B, Wang Y, Zhang J, Hu C, Jiang J, Li Y, et al. ROS-induced lipid peroxidation modulates cell death outcome: mechanisms behind apoptosis, autophagy, and ferroptosis. Arch Toxicol (2023) 97:1439–51. doi: 10.1007/s00204-023-03476-6
13. Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci USA (2019) 116:2996–3005. doi: 10.1073/pnas.1819728116
14. Liu L, Luo C, Zheng D, Wang X, Wang R, Ding W, et al. TRPML1 contributes to antimony-induced nephrotoxicity by initiating ferroptosis via chaperone-mediated autophagy. Food Chem Toxicol (2023) 184:114378. doi: 10.1016/j.fct.2023.114378
15. Lee S, Hwang N, Seok BG, Lee S, Lee S-J, Chung SW. Autophagy mediates an amplification loop during ferroptosis. Cell Death Dis (2023) 14:464. doi: 10.1038/s41419-023-05978-8
16. Vélez EJ, Schnebert S, Goguet M, Balbuena-Pecino S, Dias K, Beauclair L, et al. Chaperone-mediated autophagy protects against hyperglycemic stress. Autophagy (2023) 5:1–17. doi: 10.1080/15548627.2023.2267415. Online ahead of print.
17. Xu L, Liu Y, Chen X, Zhong H, Wang Y. Ferroptosis in life: To be or not to be. BioMed Pharmacother (2023) 159:114241. doi: 10.1016/j.biopha.2023.114241
18. Cheff DM, Huang C, Scholzen KC, Gencheva R, Ronzetti MH, Cheng Q, et al. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol (2023) 62:102703. doi: 10.1016/j.redox.2023.102703
19. Golbashirzadeh M, Heidari HR, Talebi M, Khosroushahi AY. Ferroptosis as a potential cell death mechanism against cisplatin-resistant lung cancer cell line. Adv Pharm Bull (2023) 13:176. doi: 10.34172/apb.2023.019
20. Hu J, Cheng M, Jiang C, Liu L, He Z, Liu L, et al. Deferoxamine mitigates ferroptosis and inflammation in hippocampal neurons after subarachnoid hemorrhage by activating the nrf2/TXNRD1 axis. Mol Neurobiol (2023). doi: 10.1007/s12035-023-03525-2
21. Liang C, Zhang X, Yang M, Dong X. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater (2019) 31:e1904197. doi: 10.1002/adma.201904197
22. von Krusenstiern AN, Robson RN, Qian N, Qiu B, Hu F, Reznik E, et al. Identification of essential sites of lipid peroxidation in ferroptosis. Nat Chem Biol (2023) 19:719–30. doi: 10.1038/s41589-022-01249-3
23. Magtanong L, Mueller GD, Williams KJ, Billmann M, Chan K, Armenta DA, et al. Context-dependent regulation of ferroptosis sensitivity. Cell Chem Biol (2022) 29:1409–1418.e6. doi: 10.1016/j.chembiol.2022.06.004
24. Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc (2014) 136:4551–6. doi: 10.1021/ja411006a
25. Fang Y, Zhang Z. Arsenic trioxide as a novel anti-glioma drug: a review. Cell Mol Biol Lett (2020) 25:44. doi: 10.1186/s11658-020-00236-7
26. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2011-2015. Neuro Oncol (2018) 20:iv1–iv86. doi: 10.1093/neuonc/noy131
27. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
28. Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol (2005) 64(6):479–89. doi: 10.1093/jnen/64.6.479
29. Bleeker FE, Molenaar RJ, Leenstra S. Recent advances in the molecular understanding of glioblastoma. J Neurooncol (2012) 108:11–27. doi: 10.1007/s11060-011-0793-0
30. Aguirre JI, Castillo EJ, Kimmel DB. Biologic and pathologic aspects of osteocytes in the setting of medication-related osteonecrosis of the jaw (MRONJ). Bone (2021) 153:116168. doi: 10.1016/j.bone.2021.116168
31. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol (2021) 18:280–96. doi: 10.1038/s41571-020-00462-0
32. Kinowaki Y, Taguchi T, Onishi I, Kirimura S, Kitagawa M, Yamamoto K. Overview of ferroptosis and synthetic lethality strategies. Int J Mol Sci (2021) 22:9271. doi: 10.3390/ijms22179271
33. Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron (1989) 2:1547–58. doi: 10.1016/0896-6273(89)90043-3
34. Lewerenz J, Ates G, Methner A, Conrad M, Maher P. Oxytosis/ferroptosis—(Re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front Neurosci (2018) 12:214. doi: 10.3389/fnins.2018.00214
35. Yang WS, Sriramaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
36. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature (2021) 593:586–90. doi: 10.1038/s41586-021-03539-7
37. Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature (2022) 608:778–83. doi: 10.1038/s41586-022-05022-3
38. Gutteridge JMC. Lipid peroxidation initiated by superoxide-dependent hydroxyl radicals using complexed iron and hydrogen peroxide. FEBS Lett (1984) 172:245–9. doi: 10.1016/0014-5793(84)81134-5
39. Costa I, Barbosa DJ, Benfeito S, Silva V, Chavarria D, Borges F, et al. Molecular mechanisms of ferroptosis and their involvement in brain diseases. Pharmacol Ther (2023) 244:108373. doi: 10.1016/j.pharmthera.2023.108373
40. Latunde-Dada GO. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta - Gen Subj (2017) 1861:1893–900. doi: 10.1016/j.bbagen.2017.05.019
41. Kajarabille, Latunde-Dada. Programmed cell-death by ferroptosis: antioxidants as mitigators. Int J Mol Sci (2019) 20:4968. doi: 10.3390/ijms20194968
42. Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol (2019) 15:1137–47. doi: 10.1038/s41589-019-0408-1
43. Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun (2017) 482:419–25. doi: 10.1016/j.bbrc.2016.10.086
44. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA (2016) 113:E4966–75. doi: 10.1073/pnas.1603244113
45. Koppula P, Zhuang L, Gan B. Cytochrome P450 reductase (POR) as a ferroptosis fuel. Protein Cell (2021) 12:675–9. doi: 10.1007/s13238-021-00823-0
46. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res (2016) 26:1021–32. doi: 10.1038/cr.2016.95
47. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy (2016) 12:1425–8. doi: 10.1080/15548627.2016.1187366
48. Peng S, Sun C, Lai C, Zhang L. Exosomes derived from mesenchymal stem cells rescue cartilage injury in osteoarthritis through Ferroptosis by GOT1/CCR2 expression. Int Immunopharmacol (2023) 122:110566. doi: 10.1016/j.intimp.2023.110566
49. Yang C, Zhao Y, Wang L, Guo Z, Ma L, Yang R, et al. De novo pyrimidine biosynthetic complexes support cancer cell proliferation and ferroptosis defence. Nat Cell Biol (2023) 25:836–47. doi: 10.1038/s41556-023-01146-4
50. Kremer DM, Nelson BS, Lin L, Yarosz EL, Halbrook CJ, Kerk SA, et al. GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat Commun (2021) 12:4860. doi: 10.1038/s41467-021-24859-2
51. Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol (2020) 8:590226. doi: 10.3389/fcell.2020.590226
52. Anderson GJ, Frazer DM, McKie AT, Wilkins SJ, Vulpe CD. The expression and regulation of the iron transport molecules hephaestin and IREG1. Cell Biochem Biophys (2002) 36:137–46. doi: 10.1385/CBB:36:2-3:137
53. Hirayama T, Inden M, Tsuboi H, Niwa M, Uchida Y, Naka Y, et al. A Golgi-targeting fluorescent probe for labile Fe(ii) to reveal an abnormal cellular iron distribution induced by dysfunction of VPS35. Chem Sci (2019) 10:1514–21. doi: 10.1039/c8sc04386h
54. Yang Q, Liu W, Zhang S, Liu S. The cardinal roles of ferroportin and its partners in controlling cellular iron in and out. Life Sci (2020) 258:118135. doi: 10.1016/j.lfs.2020.118135
55. Guan W, Xia M, Ji M, Chen B, Li S, Zhang M, et al. Iron induces two distinct Ca2+ signalling cascades in astrocytes. Commun Biol (2021) 4:525. doi: 10.1038/s42003-021-02060-x
56. Yan H, Zou T, Tuo Q, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther (2021) 6:49. doi: 10.1038/s41392-020-00428-9
57. Khalil S, Holy M, Grado S, Fleming R, Kurita R, Nakamura Y, et al. A specialized pathway for erythroid iron delivery through lysosomal trafficking of transferrin receptor 2. Blood Adv (2017) 1:1181–94. doi: 10.1182/bloodadvances.2016003772
58. Volani C, Doerrier C, Demetz E, Haschka D, Paglia G, Lavdas AA, et al. Dietary iron loading negatively affects liver mitochondrial function. Metallomics (2017) 9:1634–44. doi: 10.1039/C7MT00177K
59. Liu J, Kang R, Tang D. Signaling pathways and defense mechanisms of ferroptosis. FEBS J (2022) 289:7038–50. doi: 10.1111/febs.16059
60. Paul BD, Snyder SH. The unusual amino acid L-ergothioneine is a physiologic cytoprotectant. Cell Death Differ (2010) 17:1134–40. doi: 10.1038/cdd.2009.163
61. Lee S, Kim WJ. Candidate genes for COPD: current evidence and research. Int J Chron Obstruct Pulmon Dis (2015) 10:2249. doi: 10.2147/COPD.S80227
62. Zumbrennen-Bullough KB, Becker L, Garrett L, Hölter SM, Calzada-Wack J, Mossbrugger I, et al. Abnormal brain iron metabolism in irp2 deficient mice is associated with mild neurological and behavioral impairments. PloS One (2014) 9:e98072. doi: 10.1371/journal.pone.0098072
63. Dobbyn A, Huckins LM, Boocock J, Sloofman LG, Glicksberg BS, Giambartolomei C, et al. Landscape of conditional eQTL in dorsolateral prefrontal cortex and co-localization with schizophrenia GWAS. Am J Hum Genet (2018) 102:1169–84. doi: 10.1016/j.ajhg.2018.04.011
64. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature (2014) 508:105–9. doi: 10.1038/nature13148
65. Ryu MS, Zhang D, Protchenko O, Shakoury-Elizeh M, Philpott CC. PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis. J Clin Invest (2017) 127:1786–97. doi: 10.1172/JCI90519
66. Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis (2019) 10:822. doi: 10.1038/s41419-019-2064-5
67. Fang Y, Chen X, Tan Q, Zhou H, Xu J, Gu Q. Inhibiting ferroptosis through disrupting the NCOA4–FTH1 interaction: A new mechanism of action. ACS Cent Sci (2021) 7:980–9. doi: 10.1021/acscentsci.0c01592
68. Zhang Z, Yao Z, Wang L, Ding H, Shao J, Chen A, et al. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy (2018) 14:2083–103. doi: 10.1080/15548627.2018.1503146
69. Chen F, Cai X, Kang R, Liu J, Tang D. Autophagy-dependent ferroptosis in cancer. Antioxid Redox Signal (2023) 39:79–101. doi: 10.1089/ars.2022.0202
70. Pope LE, Dixon SJ. Regulation of ferroptosis by lipid metabolism. Trends Cell Biol (2023) 33:1077–87. doi: 10.1016/j.tcb.2023.05.003
71. Xin S, Schick JA. PUFAs dictate the balance of power in ferroptosis. Cell Calcium (2023) 110:102703. doi: 10.1016/j.ceca.2023.102703
72. Do Q, Zhang R, Hooper G, Xu L. Differential contributions of distinct free radical peroxidation mechanisms to the induction of ferroptosis. JACS Au (2023) 3:1100–17. doi: 10.1021/jacsau.2c00681
73. Sha W, Hu F, Xi Y, Chu Y, Bu S. Mechanism of ferroptosis and its role in type 2 diabetes mellitus. J Diabetes Res (2021) 2021:1–10. doi: 10.1155/2021/9999612
74. Shah R, Shchepinov MS, Pratt DA. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent Sci (2018) 4:387–96. doi: 10.1021/acscentsci.7b00589
75. Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol (2019) 21:579–91. doi: 10.1038/s41556-019-0305-6
76. Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol (2017) 13:81–90. doi: 10.1038/nchembio.2238
77. Tang Y, Zhou J, Hooi SC, Jiang YM, Lu GD. Fatty acid activation in carcinogenesis and cancer development: Essential roles of long-chain acyl-coa synthetases (review). Oncol Lett (2018) 16:1390–6. doi: 10.3892/ol.2018.8843
78. Orlando UD, Garona J, Ripoll GV, Maloberti PM, Solano ÁR, Avagnina A, et al. The functional interaction between acyl-coA synthetase 4, 5-lipooxygenase and cyclooxygenase-2 controls tumor growth: A novel therapeutic target. PloS One (2012) 7:e40794. doi: 10.1371/journal.pone.0040794
79. Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian D, et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ (2019) 26:2284–99. doi: 10.1038/s41418-019-0299-4
80. Gan B. Mitochondrial regulation of ferroptosis. J Cell Biol (2021) 220:e202105043. doi: 10.1083/jcb.202105043
81. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J (2009) 417:1–13. doi: 10.1042/BJ20081386
82. Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab (2020) 32:920–37. doi: 10.1016/j.cmet.2020.10.011
83. Friedman JR, Nunnari J. Mitochondrial form and function. Nature (2014) 505:335–43. doi: 10.1038/nature12985
84. Vasan K, Werner M, Chandel NS. Mitochondrial metabolism as a target for cancer therapy. Cell Metab (2020) 32:341–52. doi: 10.1016/j.cmet.2020.06.019
85. Li C, Dong X, Du W, Shi X, Chen K, Zhang W, et al. LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduct Target Ther (2020) 5:187. doi: 10.1038/s41392-020-00297-2
86. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol (2020) 22:225–34. doi: 10.1038/s41556-020-0461-8
87. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell (2015) 59:298–308. doi: 10.1016/j.molcel.2015.06.011
88. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell (2019) 73:354–363.e3. doi: 10.1016/j.molcel.2018.10.042
89. Heldt H-W, Piechulla B. Lipids are membrane constituents and function as carbon stores. In: Plant biochemistry. Elsevier (2021). p. 335–71. doi: 10.1016/B978-0-12-818631-2.00015-5
90. Miladinovic T, Ungard RG, Linher-Melville K, Popovic S, Singh G. Functional effects of TrkA inhibition on system xC–mediated glutamate release and cancer-induced bone pain. Mol Pain (2018) 14:1744806918776467. doi: 10.1177/1744806918776467
91. Sakakura Y, Sato H, Shiiya A, Tamba M, Sagara J, Matsuda M, et al. Expression and function of cystine/glutamate transporter in neutrophils. J Leukoc Biol (2007) 81:974–82. doi: 10.1189/jlb.0606385
92. Yan Y, Teng H, Hang Q, Kondiparthi L, Lei G, Horbath A, et al. SLC7A11 expression level dictates differential responses to oxidative stress in cancer cells. Nat Commun (2023) 14:3673. doi: 10.1038/s41467-023-39401-9
93. Yang H, Ramani K, Xia M, Ko KS, Li TWH, Oh P, et al. Dysregulation of glutathione synthesis during cholestasis in mice: Molecular mechanisms and therapeutic implications. Hepatology (2009) 49:1982–91. doi: 10.1002/hep.22908
94. Desideri E, Filomeni G, Ciriolo MR. Glutathione participates in the modulation of starvation-induced autophagy in carcinoma cells. Autophagy (2012) 8:1769–81. doi: 10.4161/auto.22037
95. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system xc– activity. Curr Biol (2018) 28:2388–2399.e5. doi: 10.1016/j.cub.2018.05.094
96. Chen X, Kang R, Kroemer G, Tang D. Targeting ferroptosis in pancreatic cancer: a double-edged sword. Trends Cancer (2021) 7:891–901. doi: 10.1016/j.trecan.2021.04.005
97. Arias E, Koga H, Diaz A, Mocholi E, Patel B, Cuervo AM. Lysosomal mTORC2/PHLPP1/akt regulate chaperone-mediated autophagy. Mol Cell (2015) 59:270–84. doi: 10.1016/j.molcel.2015.05.030
98. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
99. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature (2019) 575:693–8. doi: 10.1038/s41586-019-1707-0
100. Kalén A, Norling B, Appelkvist EL, Dallner G. Ubiquinone biosynthesis by the microsomal fraction from rat liver. BBA - Gen Subj (1987) 926:70–8. doi: 10.1016/0304-4165(87)90183-8
101. Turunen M, Olsson J, Dallner G. Metabolism and function of coenzyme Q. Biochim Biophys Acta - Biomembr (2004) 1660:171–99. doi: 10.1016/j.bbamem.2003.11.012
102. Takahashi T, Okamoto T, Mori K, Sayo H, Kishi T. Distribution of ubiquinone and ubiquinol homologues in rat tissues and subcellular fractions. Lipids (1993) 28:803–9. doi: 10.1007/BF02536234
103. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer (2022) 22:381–96. doi: 10.1038/s41568-022-00459-0
104. Liang H, Yoo SE, Na R, Walter CA, Richardson A, Ran Q. Short form glutathione peroxidase 4 is the essential isoform required for survival and somatic mitochondrial functions. J Biol Chem (2009) 284:30836–44. doi: 10.1074/jbc.M109.032839
105. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci (2020) 6:41–53. doi: 10.1021/acscentsci.9b01063
106. Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol (2020) 16:1351–60. doi: 10.1038/s41589-020-0613-y
107. Thöny B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J (2000) 347:1. doi: 10.1042/0264-6021:3470001
108. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
109. Sato H, Fujiwara K, Sagara JI, Bannai S. Induction of cystine transport activity in mouse peritoneal macrophages by bacterial lipopolysaccharide. Biochem J (1995) 310:547–51. doi: 10.1042/bj3100547
110. Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun (2019) 510:278–83. doi: 10.1016/j.bbrc.2019.01.090
111. Efimova I, Catanzaro E, van der Meeren L, Turubanova VD, Hammad H, Mishchenko TA, et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J Immunother Cancer (2020) 8:e001369. doi: 10.1136/jitc-2020-001369
112. Yu B, Choi B, Li W, Kim D-H. Magnetic field boosted ferroptosis-like cell death and responsive MRI using hybrid vesicles for cancer immunotherapy. Nat Commun (2020) 11:3637. doi: 10.1038/s41467-020-17380-5
113. Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer (2020) 19:116. doi: 10.1186/s12943-020-01234-1
114. Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J, et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep (2021) 35:109235. doi: 10.1016/j.celrep.2021.109235
115. Zhu H, Klement JD, Lu C, Redd PS, Yang D, Smith AD, et al. Asah2 represses the p53–hmox1 axis to protect myeloid-derived suppressor cells from ferroptosis. J Immunol (2021) 206:1395–404. doi: 10.4049/jimmunol.2000500
116. Xia Y, Rao L, Yao H, Wang Z, Ning P, Chen X. Engineering macrophages for cancer immunotherapy and drug delivery. Adv Mater (2020) 32:e2002054. doi: 10.1002/adma.202002054
117. Luo X, Gong HB, Gao HY, Wu YP, Sun WY, Li ZQ, et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ (2021) 28:1971–89. doi: 10.1038/s41418-020-00719-2
118. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol (2020) 16:278–90. doi: 10.1038/s41589-019-0462-8
119. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med (2015) 212:555–68. doi: 10.1084/jem.20140857
120. Drijvers JM, Gillis JE, Muijlwijk T, Nguyen TH, Gaudiano EF, Harris IS, et al. Pharmacologic screening identifies metabolic vulnerabilities of CD8+ T cells. Cancer Immunol Res (2021) 9:184–99. doi: 10.1158/2326-6066.CIR-20-0384
121. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab (2021) 33:1001–1012.e5. doi: 10.1016/j.cmet.2021.02.015
122. Yao Y, Chen Z, Zhang H, Chen C, Zeng M, Yunis J, et al. Selenium–GPX4 axis protects follicular helper T cells from ferroptosis. Nat Immunol (2021) 22:1127–39. doi: 10.1038/s41590-021-00996-0
123. Arensman MD, Yang XS, Leahy DM, Toral-Barza L, Mileski M, Rosfjord EC, et al. Cystine–glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity. Proc Natl Acad Sci USA (2019) 116:9533–42. doi: 10.1073/pnas.1814932116
124. Gmünder H, Eck H-P, Dröge W. Low membrane transport activity for cystine in resting and mitogenically stimulated human lymphocyte preparations and human T cell clones. Eur J Biochem (1991) 201:113–7. doi: 10.1111/j.1432-1033.1991.tb16263.x
125. Pacheco R, Oliva H, Martinez-Navío JM, Climent N, Ciruela F, Gatell JM, et al. Glutamate released by dendritic cells as a novel modulator of T cell activation. J Immunol (2006) 177:6695–704. doi: 10.4049/jimmunol.177.10.6695
126. Mbah NE, Lyssiotis CA. Metabolic regulation of ferroptosis in the tumor microenvironment. J Biol Chem (2022) 298:101617. doi: 10.1016/j.jbc.2022.101617
127. Dixon SJ, Stockwell BR. The hallmarks of ferroptosis. Annu Rev Cancer Biol (2019) 3:35–54. doi: 10.1146/annurev-cancerbio-030518-055844
128. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ (2016) 23:369–79. doi: 10.1038/cdd.2015.158
129. Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol (2019) 26:623–633.e9. doi: 10.1016/j.chembiol.2019.01.008
130. Hou W, Xia H, Zhou S, Fan Z, Xu H, Gong Q, et al. The MEK inhibitors enhance the efficacy of sorafenib against hepatocellular carcinoma cells through reducing p-ERK rebound. Transl Cancer Res (2019) 8:1224–32. doi: 10.21037/tcr.2019.07.11
131. Louandre C, Marcq I, Bouhlal H, Lachaier E, Godin C, Saidak Z, et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett (2015) 356:971–7. doi: 10.1016/j.canlet.2014.11.014
132. Sehm T, Fan Z, Ghoochani A, Rauh M, Engelhorn T, Minakaki G, et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget (2016) 7:36021–33. doi: 10.18632/oncotarget.8651
133. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol (2016) 12:497–503. doi: 10.1038/nchembio.2079
134. Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC. Heme oxygenase-1 mediates BAY 11–7085 induced ferroptosis. Cancer Lett (2018) 416:124–37. doi: 10.1016/j.canlet.2017.12.025
135. Chen Q, Liu L, Lu Y, Chen X, Zhang Y, Zhou W, et al. Tumor microenvironment-triggered aggregated magnetic nanoparticles for reinforced image-guided immunogenic chemotherapy. Adv Sci (2019) 6:1802134. doi: 10.1002/advs.201802134
136. Liu T, Liu W, Zhang M, Yu W, Gao F, Li C, et al. Ferrous-supply-regeneration nanoengineering for cancer-cell-specific ferroptosis in combination with imaging-guided photodynamic therapy. ACS Nano (2018) 12:12181–92. doi: 10.1021/acsnano.8b05860
137. Wang S, Li F, Qiao R, Hu X, Liao H, Chen L, et al. Arginine-rich manganese silicate nanobubbles as a ferroptosis-inducing agent for tumor-targeted theranostics. ACS Nano (2018) 12:12380–92. doi: 10.1021/acsnano.8b06399
138. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell (2010) 141:69–80. doi: 10.1016/j.cell.2010.02.027
139. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2–YAP signalling. Nature (2019) 572:402–6. doi: 10.1038/s41586-019-1426-6
140. Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature (2017) 551:639–43. doi: 10.1038/nature24637
141. Efferth T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Semin Cancer Biol (2017) 46:65–83. doi: 10.1016/j.semcancer.2017.02.009
142. Cabral H, Makino J, Matsumoto Y, Mi P, Wu H, Nomoto T, et al. Systemic targeting of lymph node metastasis through the blood vascular system by using size-controlled nanocarriers. ACS Nano (2015) 9:4957–67. doi: 10.1021/nn5070259
143. Zheng DW, Lei Q, Zhu JY, Fan JX, Li CX, Li C, et al. Switching apoptosis to ferroptosis: Metal-organic network for high-efficiency anticancer therapy. Nano Lett (2017) 17:284–91. doi: 10.1021/acs.nanolett.6b04060
144. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature (2017) 547:453–7. doi: 10.1038/nature23007
145. Hadian K, Stockwell BR. SnapShot: ferroptosis. Cell (2020) 181:1188–1188.e1. doi: 10.1016/j.cell.2020.04.039
146. Fan Y, Bergmann A. Multiple mechanisms modulate distinct cellular susceptibilities toward apoptosis in the developing drosophila eye. Dev Cell (2014) 30:48–60. doi: 10.1016/j.devcel.2014.05.007
147. Bárány I, Berenguer E, Solís MT, Pérez-Pérez Y, Santamaría ME, Crespo JL, et al. Autophagy is activated and involved in cell death with participation of cathepsins during stress-induced microspore embryogenesis in barley. J Exp Bot (2018) 69:1387–402. doi: 10.1093/jxb/erx455
148. Banerjee SK, Chatterjee A, Gupta S, Nagar A. Activation and regulation of NLRP3 by sterile and infectious insults. Front Immunol (2022) 13:896353. doi: 10.3389/fimmu.2022.896353
149. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol (2021) 22:266–82. doi: 10.1038/s41580-020-00324-8
150. Li Y, Cao Y, Xiao J, Shang J, Tan Q, Ping F, et al. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ (2020) 27:2635–50. doi: 10.1038/s41418-020-0528-x
151. Liu T, Zhu C, Chen X, Guan G, Zou C, Shen S, et al. Ferroptosis, as the most enriched programmed cell death process in glioma, induces immunosuppression and immunotherapy resistance. Neuro Oncol (2022) 24:1113–25. doi: 10.1093/neuonc/noac033
152. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy (2020) 16:2069–83. doi: 10.1080/15548627.2020.1714209
153. Wang H, Xu T, Huang Q, Jin W, Chen J. Immunotherapy for Malignant glioma: current status and future directions. Trends Pharmacol Sci (2020) 41:123–38. doi: 10.1016/j.tips.2019.12.003
154. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol (2014) 10:9–17. doi: 10.1038/nchembio.1416
155. Kuhn H, Banthiya S, van Leyen K. Mammalian lipoxygenases and their biological relevance. Biochim Biophys Acta - Mol Cell Biol Lipids (2015) 1851:308–30. doi: 10.1016/j.bbalip.2014.10.002
156. Siddiq A, Aminova LR, Troy CM, Suh K, Messer Z, Semenza GL, et al. Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways. J Neurosci (2009) 29:8828–38. doi: 10.1523/JNEUROSCI.1779-09.2009
157. Burczynski ME, Sridhar GR, Palackal NT, Penning TM. The reactive oxygen species- and michael acceptor-inducible human aldo-keto reductase AKR1C1 reduces the α,β-unsaturated aldehyde 4-hydroxy-2-nonenal to 1,4-dihydroxy-2-nonene. J Biol Chem (2001) 276:2890–7. doi: 10.1074/jbc.M006655200
158. Lou H, Du S, Ji Q, Stolz A. Induction of AKR1C2 by phase II inducers: Identification of a distal consensus antioxidant response element regulated by NRF2. Mol Pharmacol (2006) 69:1662–72. doi: 10.1124/mol.105.019794
159. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife (2014) 3:e02523. doi: 10.7554/eLife.02523
160. Kwon MY, Park E, Lee SJ, Chung SW. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget (2015) 6:24393–403. doi: 10.18632/oncotarget.5162
Keywords: ferroptosis, glioma, GBM, reactive oxygen species, target therapy
Citation: Wang H, Liu Y, Che S, Li X, Tang D, Lv S and Zhao H (2024) Deciphering the link: ferroptosis and its role in glioma. Front. Immunol. 15:1346585. doi: 10.3389/fimmu.2024.1346585
Received: 29 November 2023; Accepted: 05 January 2024;
Published: 23 January 2024.
Edited by:
Greta Forlani, University of Insubria, ItalyReviewed by:
Liangxue Zhou, Sichuan University, ChinaDongliang Liu, Baylor College of Medicine, United States
Copyright © 2024 Wang, Liu, Che, Li, Tang, Lv and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hai Zhao, eWlkYW9AcWR1LmVkdS5jbg==